
Submitted:
19 October 2023
Posted:
20 October 2023
You are already at the latest version
Abstract
The effect of Localized Surface Plasmon (LSP) of 5 nm mean size Au particles deposited on TiO2 P25 was investigated during the photo-thermal water gas shift reaction (WGSR). The effects of CO concentration, excitation light flux and energy, and molecular oxygen addition during the reaction were investigated. The photocatalytic WGSR rate under light excitation with wavelengths extending from 320 to 1100 nm was found to be higher than the thermal reaction alone at the same temperature (85 oC). A ratio H2/CO2 of near unity was found at high concentrations of CO. Addition of molecular oxygen during the reaction resulted in a slight decrease of molecular hydrogen production while the rates of CO2 formation and that of CO consumption changed by one order of magnitude. More important, it was found that the WGSR rates were still high under only visible light excitation (600 - 700 nm). The results prove that Au LSP alone triggers this chemical reaction without the need to excite the semiconductor on which they are deposited.
Keywords:
Au/TiO2 P25
; Localized Surface Plasmon (LSP)
; photo-thermal water gas shift reaction (WGSR)
; hydrogen production
; preferential oxidation reaction (PROX)
1. Introduction
The water gas shift reaction (WGSR) is one of the important industrial processes to adjust the CO to H2 ratio for methanol synthesis and to produce high-purity H2 for ammonia synthesis. In the WGSR, CO(g) reacts with H2O(g) to form CO2(g) and H2(g).
CO(g) + H2O(g) → CO2(g) + H2(g) ∆H = - 41.1 kJ/mol
The WGSR is performed at low (190-250 °C) and high (400-500 °C) temperatures with Cu/ZnO and Fe2O3-based catalysts, respectively [1]. At high temperatures, the CO conversion is equilibrium-limited and at low temperatures, the reaction is kinetically limited.
In addition to iron and copper-based catalysts, precious metal (Au, Pt and, Pd) containing catalysts have been investigated for WGSR at low temperatures [1,2,3,4]. Previous studies found a much higher WGSR activity of gold nanoparticles on reducible supports such as TiO2 and CeO2 than that on Al2O3 and SiO2 [3,5]. Among these catalysts, Au/TiO2 showed a comparable WGSR activity to that of commercial Cu/ZnO/Al2O3 catalysts [2]. It has also been proposed that the WGSR on Au-based catalysts takes place at the interfacial sites [5,6]. The proposed active interfacial site on Au/TiO2 is Auδ−− OV −Ti3+ (OV: oxygen vacancy) where electron-enriched Auδ−species enhanced CO chemisorption, while OV−Ti3+ contributed to the dissociation of water [5].
Mainly, two reaction mechanisms, redox (also called regenerative) and associative, have been proposed for the WGSR [7]. In the redox mechanism, CO reacts with lattice O of the catalyst forming CO2 and creating vacant sites. Then H2O dissociates to fill the vacant sites whereas the two protons of water take the two electrons left upon the creation of the vacancy to make one hydrogen molecule. In the associative mechanism, intermediate species are formed from the reaction between CO and surface -OH species derived from the dissociation of water. The proposed intermediates in previous studies are formates species (HCOO(a)) [3,8], carbonate-like species and carboxyl (HOCO) [9], which decompose to form CO2 and H2. In some studies, formates and carbonate-like species have been reported as spectators [10,11]. To our knowledge, the carboxyl species has not been observed experimentally on Au/TiO2.
The redox mechanism usually takes place at high temperatures whereas the associative mechanism occurs at low temperatures. However, density functional theory (DFT) calculation results indicated that carboxyl species are difficult to form during WGSR on Au/TiO2 catalysts at low temperatures. It was also found that formates are too stable to release H2 and hence it was suggested that the redox mechanism is the primary reaction pathway at low temperatures [12]. Another DFT computation study on the WGSR mechanism over Au10, Au13, and Au20 clusters reported that the carboxyl mechanism occurred over Au10 and Au20 clusters, while the redox mechanism took place over the most active Au13 cluster [13]. Despite these theoretical studies, still, the associative mechanism is widely accepted as the pathway of WGSR at low temperatures.
On precious-metal/TiO2 catalysts such as Au/TiO2[14], Pt/TiO2[4,15] and Pd/TiO2 [16], not only thermal but also photo-assisted WGSR has been studied. Pphoto-catalytic WGSR at low or ambient temperatures offers economical and environmental advantages. In particular, H2/CO2 would be generated at a desired ratio via a cleaner process using sunlight as an energy source. It has been proposed that the photo-asssited WGSR on M(Au, Pd, Pt)/TiO2 occurs at the M/TiO2 interface [16], similar to the thermal WGSR [8]. However, the photocatalytic WGSR over Pt/TiO2 and Pd/TiO2 occurs efficiently only at very low concentrations of CO. At high concentrations of CO, the rates of H2 and CO2 production decrease. A net negative effect on the activity with increasing the concentration of CO due to the strong adsorption of CO on Pt or Pd[15,16]. Au/TiO2 may work without negative effect on the activity at higher CO concentrations due to the weak CO adsorption on Au particles.
The Au/TiO2 catalyst has also been reported to show catalytic activity for CO oxidation at low temperatures [17]. The thermal CO oxidation on Au/TiO2 has been proposed to occur predominantly through Au-assisted Mars−van Krevelen (MvK) mechanism for reaction temperatures of 80 °C and above [18]. In the MvK mechanism, first CO molecules adsorb on Au particles and then abstract TiO2 lattice oxygen at the Au/TiO2 interface and finally CO2 molecules desorb forming oxygen vacancies, a reduced Au/TiO2−x. The next step in the Au-assisted MvK mechanism is the re-oxidation of the previously reduced catalyst [18]. Green et al. also proposed that the CO oxidation on Au/TiO2 occurred on metal sites at the Au/TiO2 interface [19]. It has also been shown that O-O bond scission is activated by the formation of a CO-O2 complex at the Au/TiO2 interface [17,19]. Moreover, the performance of Au/TiO2 catalyst for CO oxidation improves in the presence of water [20]. A water-mediated reaction mechanism for room-temperature CO oxidation over Au/TiO2 catalysts has been proposed [21]. DFT calculations showed that proton transfer at the Au/TiO2 interface facilitates O2 activation and binding, which leads to form Au-OOH that readily reacts with adsorbed CO on Au to form Au-COOH. Au-COOH decomposes to form hydrogen and CO2.
Previous studies have also shown the preferential oxidation (PROX) of CO in the presence of H2 on Au/TiO2 [20,22,23,24,25]. The PROX reaction of CO on Au/TiO2 in the presence of light irradiation has also been reported previously [26,27,28]. The UV irradiation over Au/TiO2 promotes the preferential oxidation of CO in a H2-rich stream [27]. The chemisorption of CO on Au/TiO2 was enhanced by UV irradiation but the chemisorption of H2 was suppressed on both TiO2 and Au surfaces. It has been reported that PROX reaction rates were increased by up to a factor of 3, when Au/TiO2 was irradiated by visible or UV light [26]. Yoshida et al. reported that the PROX rates of CO on Au/TiO2 under dark conditions increased in the presence of UV–visible light due to the effect of charge separation, surface plasmon resonance and the promoted electron transfer to the adsorbed O2 [26]. It has also been proposed that the photo-generated electrons from TiO2 cause changes of the chemisorbed energy of CO, H2 and O2 on Au/TiO2, in a manner that promotes the preferential oxidation of CO in a H2-rich stream [27].
Overall there are many studies on thermal WGSR [5,10,29,30,31,32] and PROX of CO [20,22,24,33,34] on Au/TiO2 but only a few investigations reported on photo-assisted WGSR [14] and PROX of CO [26,27] and further studies are required for fundamental understanding of these reactions, in particular with a high loading of Au particles with uniform size to enhance their LSPR (and therefore shifts the light frequency requirement to the middle of the sunlight). In this study, we have investigated the photo-assisted WGSR and PROX of CO on 8 wt.% Au/TiO2 at 85 °C.
2. Experimental
2.1. Preparation and characterization of 8 wt.% Au/ P25 TiO2
The Au/P25 TiO2 photocatalyst was prepared as described previously [35]. Gold nanoparticles (8 wt.%) were deposited on Degussa P25 TiO2 by the deposition–precipitation with urea method described by Zanella et al. [36], with some modifications. Briefly, the HAuCl4-3H2O solution and urea were added to a glass Schott bottle and vigorously stirred. Degussa P25 TiO2 was then added, and the resulting suspension thermostated at 80 °C in a dark room for 8 h. After 8 h, the yellow Au(III) impregnated TiO2 powder was collected by vacuum filtration, washed repeatedly with milli-Q water, and then air-dried at 70 °C overnight. The sample was then calcined at 300 °C for 2 hours to thermally reduce surface Au(III) species on TiO2 to metallic gold.
The 8 wt.% Au/TiO2 photocatalyst was characterized with transmission electron microscopy (TEM), X-ray florescence (XRF) spectroscopy, energy dispersive X-ray (EDX) spectroscopy, X-ray diffraction (XRD), UV–visible spectroscopy, photoluminescence, and X-ray photoelectron spectroscopy (XPS) and results were reported in a previous publication [35]. The average Au particle size derived from TEM images is ~ 5.1 nm. The UV–visible absorbance spectrum for the 8 wt.% Au/TiO2 shows an intense absorption below 400 nm due to the P25 TiO2 support and a broad absorption feature ~560–570 nm for the localized surface plasmon resonance (LSPR) of Au nanoparticles supported on TiO2.
2.2. Photoreaction setup
Photoreactions were carried out in a 140 mL flat bottom glass reactor. A 250 mg of Au/TiO2 catalysts was dispersed on the bottom of the glass reactor. We have opted to use excess catalyst so the photocatalytic rate (that is also dependent on the number of photons hitting the catalyst) would have negligible rate variations from one run to the other. Water was added through a micro syringe over the catalyst powder. The reactor was then purged with N2 to remove O2 present in the system. After purging, there was always presence of a small amount of O2, ≈ 5-6 × 10-6 moles (wall outgazing), was present in the reactor. CO was then added to the reactor using a syringe to obtain the desired CO/H2O ratio. All the photo-assisted WGSR except one were performed at 85 oC in order to keep water in the vapor form. A Xenon lamp, Max 303 from Asahi spectra, was used as the light source for photoreactions. In some experiments, a CoolLED pE-4000 LED light source was used and delivered to the sample using fiber optics. The reactants and products were analyzed by Gas Chromatography (GC). Hydrogen was analyzed with a Haysep Q packed column at 45 oC and a Thermal Conductivity Detector (TCD) by using N2 as a carrier gas. Oxygen, CO and CO2 gases were analyzed with a molecular sieve 5A column at 80 oC and TCD detector by using He as a carrier gas. Fluxes for the different light used are given in Supporting Materials, Figures S1-S7. All work was conducted at the corporate research center of SABIC at KAUST.
3. Results and Discussion
We have previously studied a family of Au/TiO2 P25 catalysts in some details [37,38]. These studies included, core and valence level spectroscopy, XRD, photo-luminescence, UV-Vis absorbance, TEM and EXAFS. Here we give a brief description of the relevant information related to the present study. Figure 1 presents selected data replotted from ref. 38. Figure 1A presents the UV-Vis of TiO2 P25 without and with different gold loadings from 0.5 to 10 wt.%. The TiO2 P25 contains about 80-20 of anatase and rutile with their band gaps being 3.2 (385 nm) and 3.0 (410 nm) respectively; here only the edge is shown for simplicity. The absorbance due to gold plasmon (localized surface plasmon (LSP) of Au nanoparticles) starts about 100 nm after the band gap of TiO2. The LSP is centered at about 560 nm, its intensity increases non linearity with increasing Au coverage and becomes wider. The widening of the peak is seen at both energy sides (towards the IR and towards the UV regions). It is to be noted that particles even at 8-10 wt.% are not touching each other; they are still dispersed on the surface. The TEM images in the insets show largely round shaped particles from which an average size of 5.1 nm is extracted. Figure 1B shows XPS Au4f of the same series. The binding energies at about 83.6 and 87.3 eV are for Au4f7/2 and Au4f5/2 respectively. Their peak area increases linearly with increasing coverage and all have a small shift of about 0.2-0.4 eV to lower energy (when compared to bulk Au at 84.0 eV). The slight shift to lower energy might be due to interfacial charge transfer or band bending and has not been corrected. The 8 wt. % corresponded to about 3 at. %. Recall that XPS sees the surface and near surface atoms only and therefore the extracted number differs from that found for bulk. Figure 1C presents the valence band of the same series. It contains in addition to the O2p structure in the 3-9 eV range (for pure TiO2) the Au5d band when Au is present. The Fermi level is put at the Au6s band binding energy position. This is more pronounced for the high loaded catalysts. The catalyst used in this study is therefore composed of Au particles (5.1 nm ins average size) largely in a metallic state (XPS Au4f, Au6d binding energy position), with 3 at. % and with a pronounced plasmon centered at about 560 nm and extending from 400 to 900 nm.
In the following, we present the thermal and photo-thermal photocatalytic reactions of CO and H2O to CO2 and H2 over the 8 wt.% Au/TiO2 P25 catalyst. In doing so we have in particular, changed the excitation energy in order to probe into the effect of Au LSPR on the reaction rate. In supporting information Figures S1-S7 the fluxes and energy of the different light excitations used are given.
3.1. Photocatalytic water gas shift reaction at ≈ 25 °C (room temperature)
First, we have investigated the photocatalytic WGSR at ≈ 25 °C. At this temperature there is no contribution from thermal activity. The photons energy extended from 320 nm to 1100 nm and the light fluxes in the UV (320-400 nm), visible (400-800 nm) and IR (800-1100 nm) were 8, 67 and 62 mW/cm2. The rates of production of H2 and CO2 were found to be equal 3.6 × 10-8 and 4.6 × 10-8 moles/min, respectively. Similar results will be discussed in more details in Section 3.3.
3.2. Thermal water gas shift reaction
To identify the effect of temperature, the thermal WGSR at 85, 150, 200 and 250 °C was performed in the absence of light irradiation. In all experiments water was used in excess (ratio [H2O]/[CO] > 3) and [CO] was kept at 6.7 x 10-6 mol/mL. The H2 production rate was found to be about 4.9 × 10-7 mol/min at 85 °C whereas it is 40 times higher, ca. 2 × 10-5 mol/min, at 250 °C. In these experiments high concentrations of reactants were used so the production rate would be less affected by changes in their concentration. Figure 2A,B shows the production of H2, and CO2 and CO consumption as well as the Arrhenius plot for H2 production from which an activation energy of 32.5 kJ/mol was extracted. The activation energy for industrial low temperature shift and high temperature shift water gas shift reactions are reported to be 52 and 110kJ/mol over CuZnO and Fe3O4-Cr2O3 respectively [39].
In order to monitor more accurately the consumption of CO, the thermal WGSR was carried out with lower initial concentration of CO, 5 mL of CO (2.2 × 10-4 moles/reactor volume or about 2 × 10-6 mol/mL)), and 20 μL of water at 85 oC temperature. The initial ratio of H2O to CO in the gas phase was 3 and results are presented in Figure 2C. The data show that the amounts of H2 and CO2 formed increase whereas the amounts of CO decreases with time. The rate of consumption of CO was 4.8 × 10-7 mole/min and the initial rates of production of H2 and CO2 were 2.4 × 10-7 and 3.4 × 10-7 mole/min, respectively. The rate of CO consumption was found to be 1.4 times higher than the rate of production of CO2 indicating that a fraction of CO converts to form adsorbed species, which do not further react to form CO2. In addition, a small fraction of CO converts to CH4 as evidenced by the detection of a trace amount of CH4 by gas chromatography. However, the methanation of CO generally requires reaction temperatures greater than 573 K [26]. The initial ratio of H2 to CO2 production was about 0.85, which was calculated after subtracting the CO2 formed by CO oxidation with the residual O2 (~ 6 x10-6 moles) present after purging the reactor. The observed H2 to CO2 ratio is lower than the expected value of 1.0 showing that H2 is consumed by some side reactions such as methanation to form CH4 as discussed earlier and reaction with residual O2 to form H2O. A ratio of H2/CO2 of less than 1.0 was reported by others for the WGSR on Au/TiO2 [14] and Pt/TiO2 [4]. The previously reported ratio of H2 to CO2 for WGSR carried out on 1 wt. % Au/TiO2 was 0.78 whereas it was in the range of 0.7 - 1.0 on Pt/TiO2 with and without irradiation [4].
The thermal WGSR at 85 oC, most likely, takes place via associative mechanism described in the introduction section. In the associative mechanism, first intermediate species such as formates, are formed at the Au/TiO2 interface by the reaction between adsorbed CO and surface OH groups, derived from the dissociation of H2O, on TiO2 as reported previously [8]. Thus, formed intermediate species react to form H2 and CO2. Adsorbed formate species on catalyst surfaces has been detected in many studies by infrared spectroscopy and proposed as intermediate [3,8].
The formation and decomposition of formates on Au/TiO2 can be seen as follow.
CO (g) + H2O (g) + O(s) → CO(a) + OH(a) + OH(s)
Molecular and dissociative adsorption of CO and water, respectively.
CO(a) + OH(a) → HCOO(a)
Formate formation
HCOO(a) → CO2 + [H(a)]
Formate decomposition
The [ ] indicates that the species is a not stable intermediate
[H(a)] + OH(s) → H2(g) + O(s)
Hydrogen production
Where (g), (s), and (a) stand for gas, surface and adsorbed, respectively.
Deviation from stoichiometry between CO2 and H2 occurred at 85oC for both CO concentrations investigated. It was not found at higher temperatures (at 150, 200, and 250oC the ratio was ≈ 1). This might be linked to the hydrogen formation of the reaction (equation 4). The following sentences may explain the reason. Strictly, one hydrogen molecule originates from the reaction of a hydride (upon the dissociation of the C-H of a formate) and a proton (upon the dissociation of a molecule of water). In other words, this is a recombination reaction with two distinctly different species (one hydrogen with two electrons (a hydride) and a proton). It is to be noted that a hydride has not been seen on Ti cations of TiO2 (that is the reason the brackets on equation 3 are put, so it is considered a transition state intermediate). On the other hand, CO2 formation results from the direct decomposition of a formate species (equation 3). It is possible that during the recombination (of H-(a) and H+(a)) the hydride loses one or two electrons into the lattice. When the temperature is increased the species may have enough motion (thermal energy) to increase the reaction rate and prevent electron loss. These may explain the deviation of stoichiometry around the temperature of 85oC.
3.3. Photo-assisted water gas shift reaction at 85 oC; effect of CO concentration
Experiments were performed in the presence of light extending from the UV to the IR (320-400; 400-750; 750-1100 nm). First the effect of light intensity was studied while, the light energy was kept constant. This is shown in Figure 3A,B. The difference in the light flux between A and B is about two (for all light regions). WGSR shows higher activity compared to the thermal reaction, at the same temperature, in both cases (two to three times higher). However, doubling the light intensity increased the activity by about 30%. We make no attempt to study the light intensity effect because the set up may not be ideal for this. The practical point to extract from this result is that slight variations of light flux reaching the catalyst, from one experiment to the other (say by 10%) would not dramatically affect the comparative study presented next. Again, as in the thermal reaction the ratio H2 to CO2 was less than the stoichiometric one and saturates faster than that of CO2. Yet increasing light intensity improved the ratio. At higher light intensity the CO2/H2 ratio was closer to unity than at the low light intensity (about 0.9). Also, similar to the thermal WGSR, the rate of consumption of CO was 1.3 times higher than the rate of production of CO2 indicating that a fraction of CO converts to form adsorbed species, which do not all further dissociate to form CO2.
To identify the effect of higher initial CO concentrations, WGSR was carried out at higher CO concentration than that used in the experiments described above. In this experiment, the reactor was purged with CO instead of N2 to decrease any possible outgassing from the wall of the reactor. Figure 3C presents the results for the WGSR under the same light energy and flux to that of Figure 3B. In Figure 3C the initial concentration of CO was three times that of Figure 3B while that of water was the same. The initial rate of hydrogen doubled (2 × 10-6 mol/min) and became closer to that of CO2 (2.4 × 10-6 mol/min). Figure 3D presents similar results but with an initial CO concentration of 5.8 × 10-3 mol, about 26 times that of Figure 3B (CO to water ratio was near unity). Both CO2 and H2 production were virtually the same (ratio near unity) while the rate increased to 7 × 10-6 mol/min. To summarize the above, it appears that under high concertation of CO (with negligible O2 outgassing from the walls of the reactor) and when the ratio of CO/H2O is near unity or above, the ratio of CO2 to H2 is near unity too. It is most likely that experimental artefacts are behind the sub-stoichiometric ratios observed by others.
Based on others results [14] and ours one may describe the reaction as follows. 1. UV light excites the TiO2 catalyst and generates electron hole pairs, then photo-excited electrons transfer from the conduction band of TiO2 to Au particles, this is in line with some of our more recent work on TiO2 and ZnO [40,41,42,43,44] and reduction of H2O takes place on Au particles or at the Au/TiO2 interface to produce H2. 2. The photo-generated holes oxidize CO to form CO2 on TiO2 or at the Au/TiO2 interface; the oxygen atom, to make CO2, originates from dissociated water in the form of surface hydroxyls. Although both thermal and photocatalytic WGSR occurred since the experiment was performed at 85 oC under UV light irradiation; the former was much weaker.
3.4. Photo-assisted water gas shift reaction at 85 oC; effect of O2 concentration
The effect of injecting O2 during the reaction is presented in Figure 4. The objective here is to see the effect of any potential contamination of O2 on the reaction rates since both hydrogen and CO oxidation can take place under photon irradiation as well as at 85oC, in the presence the Au/TiO2 catalyst. The reaction is conducted with a ratio CO to water equal to 1 and with the same photon energy and flux as those used in Figure 3B-D. Initially only the thermal reaction is conducted at 85oC (the first 30 minutes or so in the figure) then light was turned on. The rates for H2 and CO2 production increase and are like those observed in Section 3.2 and 3.3, within experimental reproducibility. At about 200 minutes 2 mL of O2 was injected into the reactor. This resulted in a very fast increase of CO2 production and a very fast consumption of CO. The consumption of CO was however higher than the production of CO2. This indicates that only a fraction of surface species that consumed CO have reacted to CO2. This may point out to the buildup of formate species, and seems to indicate that their kinetics is too slow at the photon flux used. Also, the decay of H2 production rate is found to be mild, much less than the rise of CO2. This is unlike Pd and Pt as indicated in the introduction section where both are highly active for hydrogen oxidation. The results are in line with preferential oxidation activity of Au particles observed thermally, shown here under the effect of photons. After a few minutes of reaction in the presence of O2, the rates for both products start to rise again, they are however weaker. The rate of hydrogen production has decreased by half and that of CO consumption was about four times slower. Table 3 presents the rates of the three compounds before, during, and after O2 introduction. It is interesting to note that after O2 injection and possibly its total consumption, the three rates became identical indicating that the WGSR has become the sole reaction. This might be because the surface/gas has now reached equilibrium and the fraction of available sites for reactions has become constant. It is worth presenting in a few equations the possible reactions that have taken place.

Table 2.
Effect of Changing CO concentration on the H2/CO2 ratio during the photo-thermal WGSR over 8 wt. % Au/TiO2 P25.
Table 2.
Effect of Changing CO concentration on the H2/CO2 ratio during the photo-thermal WGSR over 8 wt. % Au/TiO2 P25.
| [CO] | [H2O] | Ratio [H2O]/[CO] | Initial r(H2) mol/min | Initial r(CO2) mol/min | Ratio [H2]/[CO2] |
|---|---|---|---|---|---|
| 0.00022 | 0.0011 | 5.0 | 1.2 × 10-6 | 1.5 ×10-6 | 0.8 |
| 0.00067 | 0.0011 | 1.6 | 2 × 10-6 | 2.4 × 10-6 | 0.9 |
| 0.0058 | 0.0055 | ≈ 1 | 7 × 10-6 | 6 × 10-6 | 1.1 |
Cumulative analytical and experimental errors in rates and concentrations are about 10%.
Table 3.
Effect of O2 addition (ca. 9 × 10-5 mol) on the reaction products and CO consumption during the photo-thermal WGSR over 8 wt. % Au/TiO2 P25.
Table 3.
Effect of O2 addition (ca. 9 × 10-5 mol) on the reaction products and CO consumption during the photo-thermal WGSR over 8 wt. % Au/TiO2 P25.
| Before r(H2) mol/min |
Before r(CO2) mol/min |
Before r(CO) mol/min |
During r(H2) mol/min | During r(CO2) mol/min |
During r(CO)mol/min |
After r(H2) mol/min |
After r(CO2) mol/min |
After r(CO)mol/min |
|---|---|---|---|---|---|---|---|---|
| 0.75 × 10-6 | 0.5 × 10-6 | -1.2 × 10-6 | -0.5 × 10-6 | 11 × 10-6 | -19 ×10-6 | 0.35 × 10-6 | 0.35 × 10-6 | -0.35 × 10-6 |
There are two centers for reactions on the catalyst in the presence of light, the semiconductor (TiO2) and the metal (Au particles), including their interface. We will first address both separately for simplicity.
Upon light excitation, electrons are transferred from the valence band (VB) to the conduction band (CB) of TiO2
hν → e(CB) + h(VB)
In the presence of gas phase O2, the latter reacts with e(CB) and become O2-. that may dissociate to O-. and an O atom.
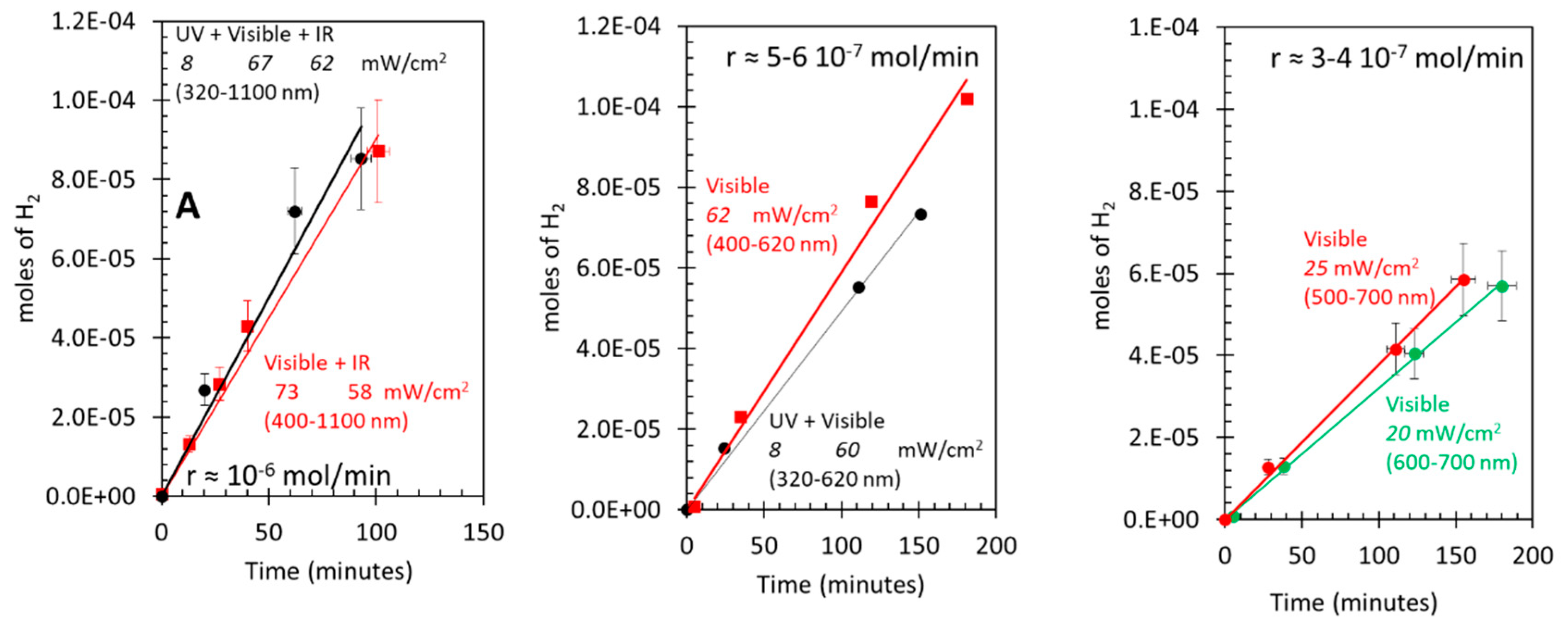
3.5. Photo-assisted water gas shift reaction at 85 oC; effect of light energy
Figure 5 presents the data for the WGSR under light excitation with different energies at 85 oC. These were (UV + visible + IR), (UV + visible), and visible. Figure 5A shows hydrogen production under (UV + visible + IR) and (visible + IR). The light flux was kept constant as well as all other parameters. The rates are virtually the same. Therefore, it is clear that light with wavelength below 400 nm (about 3 eV) is not needed. Since anatase TiO2 absorbs light only below 400 nm, it is not directly implicated in the reaction. However, since P25 contains about 20% rutile TiO2 it may still participate in the reaction. Figure 5B presents hydrogen production under similar light excitations but upon removing the late fraction of visible light and all IR light. The rates of hydrogen production under (UV + visible) (up to 620 nm) and visible excitation are very similar. Yet, in this case the rate has decreased by about 40%. It is thus clear that light with wavelength above 620 nm still affects the reaction rate. Figure 5C presents the same reaction only under visible light between 500 and 700 nm. This excites Au particles only. The rate of hydrogen production is about third of that of Figure 5A. However, when normalized to the number of photons used, the rate is higher than that obtained when using UV light to excite TiO2 in addition. Actually, using light above 600 nm the system still performs well. The straight forward conclusion is that a non-negligible fraction of the catalyst activity in the reaction is due to Au particles directly excited by visible light without the need to use UV light. Light with energy above 2 eV is not needed.
Based on the results found in this work, it appears that a Au/TiO2 P25 catalyst composed of Au particles with an average size of 5 nm, at high enough surface density (3 at. % based on XPS, and about 10% per particle size based on TEM), with a plasmon extending from 400 to 800 nm (centered at about 560 nm) is active for WGSR under visible light without the need of UV light to excite TiO2. The main barrier for the reaction is that of electron transfer (initially from CO) to make molecular hydrogen (from the protons of surface hydroxyls). Water dissociates readily on TiO2 and does not need the presence of Au particles. CO may adsorb on both TiO2 surface (adsorption energy is less than 0.5 eV (depending on the coverage – and on Au particles [48,49,50]. Upon visible light excitation LSPR of Au particles occurs. Energetically this is viewed as an electronic transition from 5d to 6s bands. Electrons at the 6s level have sufficient energy to reduce the protons of OH to atomic hydrogen while CO recombines with an adsorbed oxygen with a net result of injecting electrons into the 5d energy level. This is presented in Scheme 1 and by equations 11-15.
Au (LSPR) + visible light → 5d - 6s transfer
H2O(a) + 2 e- (6s) → 2H. + O2-(s)
O2- (a) + 2 h+ (5d) → O(a)
O(a) + CO(a) → CO2 (g) ΔHof = -283 kJ/mol
Total
H2O + CO → CO2 + H2 ΔHof = -41 kJ/mol
4. Conclusions
WGSR over 8 wt. % Au/TiO2 P25 (3 at. % Au/TiO2 P25) was investigated thermally and in photo-thermal conditions. The latter was studied under light with different energies and fluxes. This was to investigate the effect of Localized Surface Plasmon (LSP) of the 5 nm mean size Au particles deposited on TiO2 for the reaction, and de-couple it from that of the semiconductor support. In addition, the effects of CO concentration, and O2 addition during the reaction were investigated. The photocatalytic WGSR rate under light excitation with wavelengths extending from 320 to 1100 nm was found to be higher than the thermal reaction alone at the same temperature (85 oC). The expected ratio H2/CO2 of near unity was found only at high concentrations of CO. The rate of H2 production was much less affected by the addition of O2, than that of CO2 production indicating that Au/TiO2 performs well for both WGSR and PROX under photo-irradiation. Probably the most noticeable result observed is that Au particles of 5 nm in size and when present with a high density over the semiconductor oxide support (about 10 % particle density), show equal activity under visible light (600 – 700 nm) to when UV light was present in addition. Therefore, it is concluded that Au LSPR alone triggers this chemical reaction without the need to excite the semiconductor on which they are deposited.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
References
- Ratnasamy, C.; Wagner, J.P. Water Gas Shift Catalysis. Catal. Rev. 2009, 51, 325–440. [Google Scholar] [CrossRef]
- Sakurai H, Ueda A, Kobayashi T, Haruta M. Low-temperature water–gas shift reaction over gold depositedon TiO2. Chemical Communications 1997, 271–2.
- Sandoval, A.; Gómez-Cortés, A.; Zanella, R.; Díaz, G.; Saniger, J.M. Gold nanoparticles: Support effects for the WGS reaction. J. Mol. Catal. A: Chem. 2007, 278, 200–208. [Google Scholar] [CrossRef]
- Yixuan C, Zhaobin W, Yanxin C, Huaxin L, Zupei H, Huiqing L, et al. Metal-semiconductor catalyst: photocatalytic and electrochemical behavior of Pt-TiO2 for the water-gas shift reaction. Journal of Molecular Catalysis 1983, 21, 275–89.
- Tabakova, T. Recent Advances in Design of Gold-Based Catalysts for H2 Clean-Up Reactions. Front. Chem. 2019, 7, 517. [Google Scholar] [CrossRef]
- Fu, X.-P.; Guo, L.-W.; Wang, W.-W.; Ma, C.; Jia, C.-J.; Wu, K.; Si, R.; Sun, L.-D.; Yan, C.-H. Direct Identification of Active Surface Species for the Water–Gas Shift Reaction on a Gold–Ceria Catalyst. J. Am. Chem. Soc. 2019, 141, 4613–4623. [Google Scholar] [CrossRef]
- Shido, T.; Iwasawa, Y. Regulation of reaction intermediate by reactant in the water-gas shift reaction on CeO2, in relation to reactant-promoted mechanism. J. Catal. 1992, 136, 493–503. [Google Scholar] [CrossRef]
- Tabakova, T.; Boccuzzi, F.; Manzoli, M.; Sobczak, J.; Idakiev, V.; Andreeva, D. A comparative study of nanosized IB/ceria catalysts for low-temperature water-gas shift reaction. Appl. Catal. A: Gen. 2006, 298, 127–143. [Google Scholar] [CrossRef]
- Plata JJ, Romero-Sarria F, Suárez JA, Márquez AM, Laguna ÓH, Odriozola JA, et al. Improving the activity of gold nanoparticles for the water-gas shift reaction using TiO 2–Y 2 O 3: an example of catalyst design. Physical Chemistry Chemical Physics 2018, 20, 22076–83.
- Wang, J.; Kispersky, V.F.; Delgass, W.N.; Ribeiro, F.H. Determination of the Au active site and surface active species via operando transmission FTIR and isotopic transient experiments on 2.3wt.% Au/TiO2 for the WGS reaction. J. Catal. 2012, 289, 171–178. [Google Scholar] [CrossRef]
- Burch, R.; Goguet, A.; Meunier, F.C. A critical analysis of the experimental evidence for and against a formate mechanism for high activity water-gas shift catalysts. Appl. Catal. A: Gen. 2011, 409-410, 3–12. [Google Scholar] [CrossRef]
- Sun K, Kohyama M, Tanaka S, Takeda S. Reaction mechanism of the low-temperature water–gas shift reaction on Au/TiO2 catalysts. The Journal of Physical Chemistry C 2017, 121, 12178–87.
- ZHANG X, XUE J, Yue M, QIAN M, XIA S, NI Z. Reaction mechanism of water gas shift over Aun clusters: a density functional theory study. Journal of Fuel Chemistry and Technology 2017, 45, 1473–80.
- Sastre, F.; Oteri, M.; Corma, A.; García, H. Photocatalytic water gas shift using visible or simulated solar light for the efficient, room-temperature hydrogen generation. Energy Environ. Sci. 2013, 6, 2211–2215. [Google Scholar] [CrossRef]
- Sato, S.; White, J.M. Photoassisted water-gas shift reaction over platinized titanium dioxide catalysts. J. Am. Chem. Soc. 1980, 102, 7206–7210. [Google Scholar] [CrossRef]
- Millard, L.; Bowker, M. Photocatalytic water-gas shift reaction at ambient temperature. J. Photochem. Photobiol. A: Chem. 2002, 148, 91–95. [Google Scholar] [CrossRef]
- Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M.J.; Delmon, B. Low-Temperature Oxidation of CO over Gold Supported on TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175–192. [Google Scholar] [CrossRef]
- Schlexer P, Widmann D, Behm RJ, Pacchioni G. CO oxidation on a Au/TiO2 nanoparticle catalyst via the Au-assisted Mars–van Krevelen mechanism. Acs Catalysis 2018, 8, 6513–25.
- Green IX, Tang W, Neurock M, Yates Jr JT. Spectroscopic observation of dual catalytic sites during oxidation of CO on a Au/TiO2 catalyst. Science 2011, 333, 736–9.
- Saavedra, J.; Whittaker, T.; Chen, Z.; Pursell, C.J.; Rioux, R.M.; Chandler, B.D. Controlling activity and selectivity using water in the Au-catalysed preferential oxidation of CO in H2. Nat. Chem. 2016, 8, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, J.; Doan, H.A.; Pursell, C.J.; Grabow, L.C.; Chandler, B.D. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science 2014, 345, 1599–1602. [Google Scholar] [CrossRef]
- Sangeetha P, Chang L-H, Chen Y-W. Preferential oxidation of CO in H2 stream on Au/TiO2 catalysts: effect of preparation method. Industrial & Engineering Chemistry Research 2009, 48, 5666–70.
- Leal, G.B.; Ciotti, L.; Watacabe, B.N.; da Silva, D.C.L.; Antoniassi, R.M.; Silva, J.C.M.; Linardi, M.; Giudici, R.; Vaz, J.M.; Spinacé, E.V. Preparation of Au/TiO2 by a facile method at room temperature for the CO preferential oxidation reaction. Catal. Commun. 2018, 116, 38–42. [Google Scholar] [CrossRef]
- Hartadi, Y.; Behm, R.J.; Widmann, D. Competition of CO and H2 for Active Oxygen Species during the Preferential CO Oxidation (PROX) on Au/TiO2 Catalysts. Catalysts 2016, 6, 21. [Google Scholar] [CrossRef]
- Bion, N.; Epron, F.; Moreno, M.; Mariño, F.; Duprez, D. Preferential Oxidation of Carbon Monoxide in the Presence of Hydrogen (PROX) over Noble Metals and Transition Metal Oxides: Advantages and Drawbacks. Top. Catal. 2008, 51, 76–88. [Google Scholar] [CrossRef]
- Yoshida Y, Izumi Y. Recent advances in the preferential thermal-/photo-oxidation of carbon monoxide: Noble versus inexpensive metals and their reaction mechanisms. Catalysis Surveys from Asia 2016, 20, 141–66.
- Dai, W.; Zheng, X.; Yang, H.; Chen, X.; Wang, X.; Liu, P.; Fu, X. The promoted effect of UV irradiation on preferential oxidation of CO in an H2-rich stream over Au/TiO2. J. Power Sources 2009, 188, 507–514. [Google Scholar] [CrossRef]
- Rodríguez-Aguado, E.; Infantes-Molina, A.; Talon, A.; Storaro, L.; León-Reina, L.; Rodríguez-Castellón, E.; Moretti, E. Au nanoparticles supported on nanorod-like TiO2 as catalysts in the CO-PROX reaction under dark and light irradiation: Effect of acidic and alkaline synthesis conditions. Int. J. Hydrogen Energy 2018, 44, 923–936. [Google Scholar] [CrossRef]
- Shekhar M, Wang J, Lee W-S, Williams WD, Kim SM, Stach EA, et al. Size and support effects for the water–gas shift catalysis over gold nanoparticles supported on model Al2O3 and TiO2. Journal of the American Chemical Society 2012, 134, 4700–8.
- Williams, W.D.; Shekhar, M.; Lee, W.-S.; Kispersky, V.; Delgass, W.N.; Ribeiro, F.H.; Kim, S.M.; Stach, E.A.; Miller, J.T.; Allard, L.F. Metallic Corner Atoms in Gold Clusters Supported on Rutile Are the Dominant Active Site during Water−Gas Shift Catalysis. J. Am. Chem. Soc. 2010, 132, 14018–14020. [Google Scholar] [CrossRef]
- Flytzani-Stephanopoulos, M. Gold Atoms Stabilized on Various Supports Catalyze the Water–Gas Shift Reaction. Accounts Chem. Res. 2013, 47, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Flytzani-Stephanopoulos, M. Design of single-atom metal catalysts on various supports for the low-temperature water-gas shift reaction. Catal. Today 2017, 298, 216–225. [Google Scholar] [CrossRef]
- Beck, A.; Horváth, A.; Stefler, G.; Scurrell, M.S.; Guczi, L. Role of Preparation Techniques in the Activity of Au/TiO2 Nanostructures Stabilised on SiO2: CO and Preferential CO Oxidation. Top. Catal. 2009, 52, 912–919. [Google Scholar] [CrossRef]
- Ma Z, Tao F, Gu X. Development of new gold catalysts for removing CO from H2. Heterogeneous Catalysis at Nanoscale for Energy Applications 2014, 217–38.
- Jovic, V.; Chen, W.-T.; Sun-Waterhouse, D.; Blackford, M.G.; Idriss, H.; Waterhouse, G.I. Effect of gold loading and TiO2 support composition on the activity of Au/TiO2 photocatalysts for H2 production from ethanol–water mixtures. J. Catal. 2013, 305, 307–317. [Google Scholar] [CrossRef]
- Zanella, R.; Giorgio, S.; Shin, C.-H.; Henry, C.R.; Louis, C. Characterization and reactivity in CO oxidation of gold nanoparticles supported on TiO2 prepared by deposition-precipitation with NaOH and urea. J. Catal. 2004, 222, 357–367. [Google Scholar] [CrossRef]
- Al-Azri ZH, Chen W-T, Chan A, Jovic V, Ina T, Idriss H, et al. The roles of metal co-catalysts and reaction media in photocatalytic hydrogen production: Performance evaluation of M/TiO2 photocatalysts (M= Pd, Pt, Au) in different alcohol–water mixtures. Journal of Catalysis 2015, 329, 355–67.
- Jovic, V.; Smith, K.E.; Idriss, H.; Waterhouse, G.I.N. Heterojunction Synergies in Titania-Supported Gold Photocatalysts: Implications for Solar Hydrogen Production. ChemSusChem 2015, 8, 2551–2559. [Google Scholar] [CrossRef] [PubMed]
- Keiski, R.; Desponds, O.; Chang, Y.-F.; Somorjai, G. Kinetics of the water-gas shift reaction over several alkane activation and water-gas shift catalysts. Appl. Catal. A: Gen. 1993, 101, 317–338. [Google Scholar] [CrossRef]
- Williams, O.B.J.; Katsiev, K.; Baek, B.; Harrison, G.; Thornton, G.; Idriss, H. Direct Visualization of a Gold Nanoparticle Electron Trapping Effect. J. Am. Chem. Soc. 2022, 144, 1034–1044. [Google Scholar] [CrossRef]
- Yim, C.-M.; Lamoureux, P.S.; Mellor, A.; Pang, C.L.; Idriss, H.; Pacchioni, G.; Thornton, G. Size and Shape Dependence of the Electronic Structure of Gold Nanoclusters on TiO2. J. Phys. Chem. Lett. 2021, 12, 8363–8369. [Google Scholar] [CrossRef]
- Katsiev K, Harrison G, Al-Salik Y, Thornton G, Idriss H. Gold cluster coverage effect on H2 production over rutile TiO2 (110). ACS Catalysis 2019, 9, 8294–305.
- Alsalik, Y.M.; Katsiev, K.; Idriss, H. Electron Transfer From a Semiconductor to a Metal and Its Implication on Photocatalysis for Hydrogen Production. J. Phys. Chem. C 2022, 126, 15184–15190. [Google Scholar] [CrossRef]
- Ziani, A.; Al-Taweel, S.; Nadeem, M.A.; Idriss, H. Effect of Gold Loading on Time-Resolved ps Photoluminescence of ZnO. J. Phys. Chem. C 2022, 126, 16148–16157. [Google Scholar] [CrossRef]
- Herring, C.J.; Montemore, M.M. Mechanistic Insights into Plasmonic Catalysis by Dynamic Calculations: O2 and N2 on Au and Ag Nanoparticles. Chem. Mater. 2023, 35, 1586–1593. [Google Scholar] [CrossRef]
- Roldán, A.; Ricart, J.M.; Illas, F. Origin of the size dependence of Au nanoparticles toward molecular oxygen dissociation. Theor. Chem. Accounts 2010, 128, 675–681. [Google Scholar] [CrossRef]
- Roldán, A.; González, S.; Ricart, J.M.; Illas, F. Critical Size for O2 Dissociation by Au Nanoparticles. Chemphyschem 2009, 10, 348–351. [Google Scholar] [CrossRef]
- Ramalho, J.P.P.; Illas, F.; Gomes, J.R.B. Adsorption of CO on the rutile TiO2(110) surface: a dispersion-corrected density functional theory study. Phys. Chem. Chem. Phys. 2016, 19, 2487–2494. [Google Scholar] [CrossRef]
- Pursell, C.J.; Chandler, B.D.; Manzoli, M.; Boccuzzi, F. CO Adsorption on Supported Gold Nanoparticle Catalysts: Application of the Temkin Model. J. Phys. Chem. C 2012, 116, 11117–11125. [Google Scholar] [CrossRef]
- Phala, N.S.; Klatt, G.; van Steen, E. A DFT study of hydrogen and carbon monoxide chemisorption onto small gold clusters. Chem. Phys. Lett. 2004, 395, 33–37. [Google Scholar] [CrossRef]
- Idriss, H. Oxygen vacancies role in thermally driven and photon driven catalytic reactions. Chem Catal. 2022, 2, 1549–1560. [Google Scholar] [CrossRef]
| 1 |
O2(g) + e(CB) → O2-. → O atom + O-.
(6)
The oxygen atom reacts with CO to give CO2, while the O-. may react with a proton of a surface OH group to give an OH radical. OH radicals are powerful oxidants. They then react with CO to give formates and inject an electron into the VB.
CO + O atom → CO2(g)
O-. + OH(s) → OH radical + O(s)
OH radical + CO(g) + h(VB) → HOCO(a)
The sum of the above equations is equation 10
2CO(g) + O2(g) + hν + OH(s) → CO2(g) + HOCO(a) + O(s)
Equations 7 and 9 may explain why the rate of disappearance of CO is twice that of the appearance of CO2, since largely only one oxygen atom of molecular oxygen has reacted to give the CO2 while the other gave a formate species (if the latter do not have a major role in the reaction at this temperature and light flux).
The other route is that related to Au particles and their plasmonic effect (LSP). O2 can dissociate on Au particles, yet this occurs on those with sizes below 2 nm or so [45,46,47].. Au particles of the catalyst used here are of mean particle size of 5 nm (about 3000 atoms), while defects on these and the possible presence of some much smaller particles (not identified by TEM) may still have activity for the dissociative adsorption of O2, their effect is neglected here. In particular, a recent time dependent DFT computational study of Au particles with different sizes has pointed out to two important observation that might be relevant to this work [45]. First, it seems that under light excitation particle size is not determinant and second, O2 dissociates largely because of the electric field effect and not by charge transfer. Based on these results, there are two distinct ways for O2 dissociation on Au/TiO2 either in the dark or under light excitation. 1. Dark dissociation seems to be on small Au particles (< 2nm or so). 2. Light induced dissociation can occur upon TiO2 excitation (UV) followed by charge transfer to Au particles and/or upon LSPR that directly excite Au particles (largely by electric field effect).
|
Figure 1.
A. UV-Vis-IR absorbance spectra of TiO2 P25 and Au/TiO2 P25 at the indicated atomic %. B. XPS Au4f binding energy region of the series of Au containing catalysts. All spectra show Au metallic state. C. Valence band region of the same series shown in A and B; in addition to the O2p region , signal related to 5d and 6s orbitals/bands appear at high at. % of Au. The 8 % Au/TiO2 catalyst used in this study is indicated. Figures are modified from ref. 38, with permission.
Figure 1.
A. UV-Vis-IR absorbance spectra of TiO2 P25 and Au/TiO2 P25 at the indicated atomic %. B. XPS Au4f binding energy region of the series of Au containing catalysts. All spectra show Au metallic state. C. Valence band region of the same series shown in A and B; in addition to the O2p region , signal related to 5d and 6s orbitals/bands appear at high at. % of Au. The 8 % Au/TiO2 catalyst used in this study is indicated. Figures are modified from ref. 38, with permission.
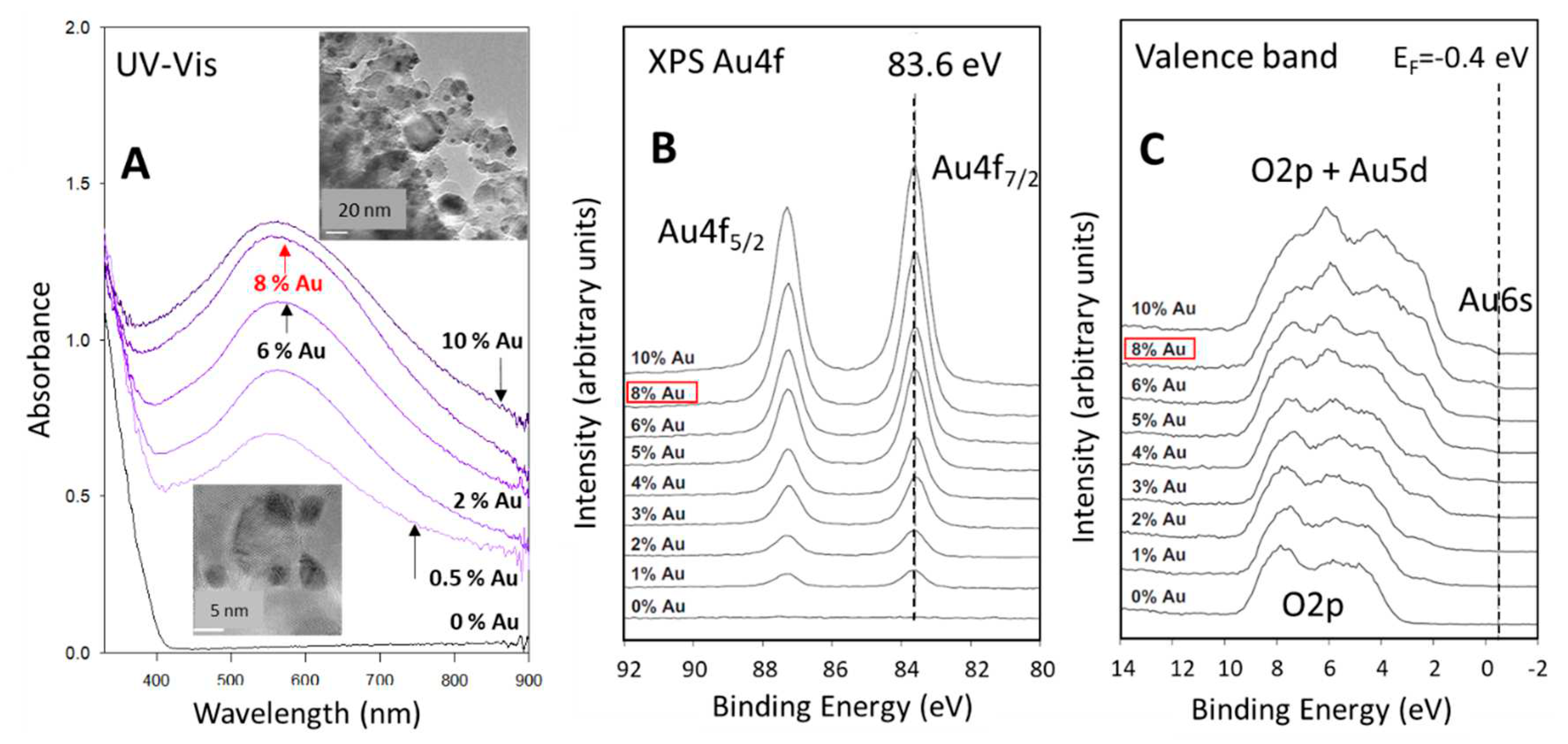
Figure 2.
A. CO consumption and H2 and CO2 production as a function of time at 150 oC during the WGSR reaction over 8 wt. % Au/TiO2 P25. B. Arrhenius plot for the hydrogen production rates of WGSR on the same catalyst. The rate constants are shown in Table 1. C. H2 and CO2 formation and CO consumption during thermal WGSR at 85 oC.
Figure 2.
A. CO consumption and H2 and CO2 production as a function of time at 150 oC during the WGSR reaction over 8 wt. % Au/TiO2 P25. B. Arrhenius plot for the hydrogen production rates of WGSR on the same catalyst. The rate constants are shown in Table 1. C. H2 and CO2 formation and CO consumption during thermal WGSR at 85 oC.
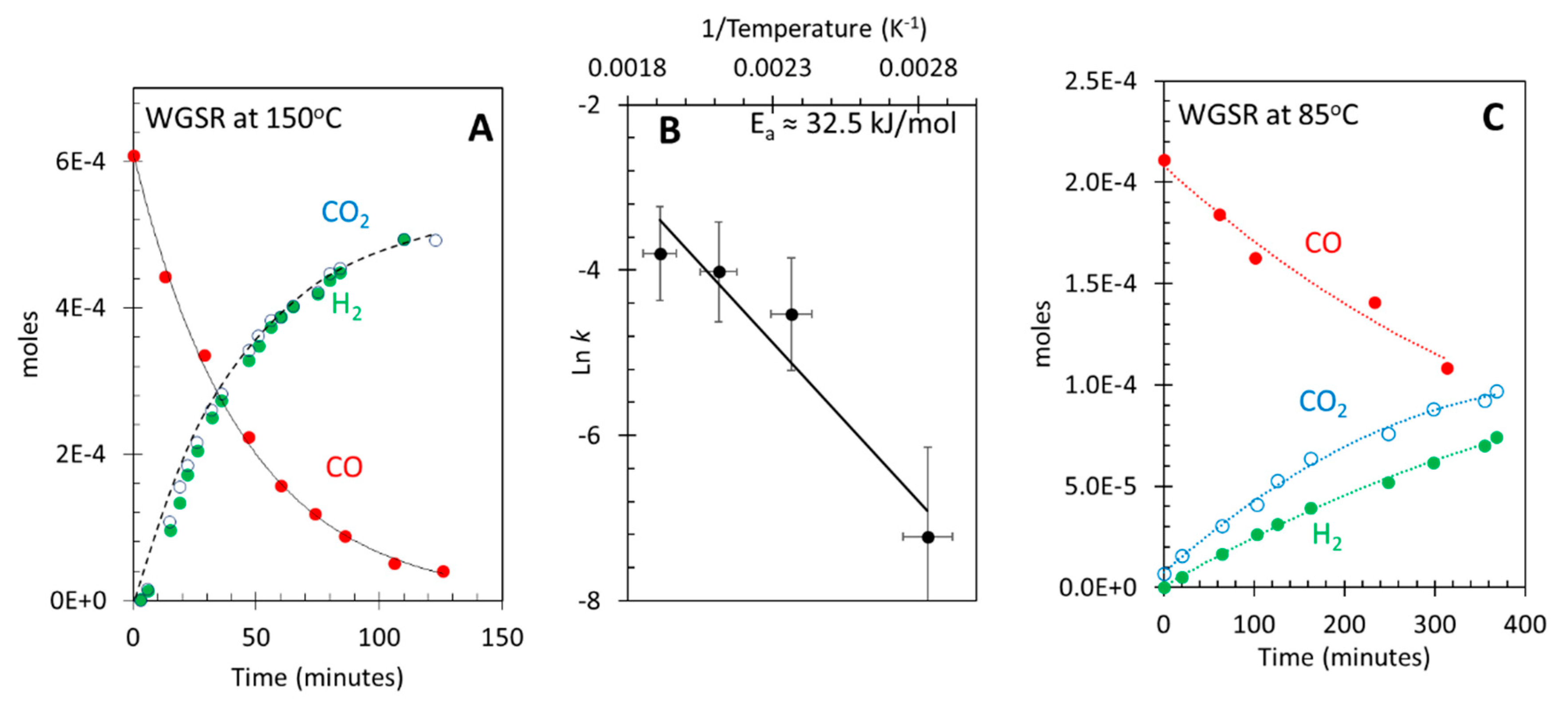
Figure 3.
Effect of changing CO concentration on the H2 to CO2 ratio during the Photo-thermal WGSR at 85oC over 8 wt. % Au/TiO2 P25. For all figures the same light energy distribution is used. A. CO2 and H2 production and CO consumption as a function of time with an initial CO concentration of ca. 2.5 × 10-4 mol. B. the same reaction except that the light flux was decreased. C. The same reaction as in B but with an increase of CO concentration to ca. 7.5 × 10-4 mol. D. The same reaction as in B but with a further increase of CO concentration to ca. 7.5 × 10-3 mol.
Figure 3.
Effect of changing CO concentration on the H2 to CO2 ratio during the Photo-thermal WGSR at 85oC over 8 wt. % Au/TiO2 P25. For all figures the same light energy distribution is used. A. CO2 and H2 production and CO consumption as a function of time with an initial CO concentration of ca. 2.5 × 10-4 mol. B. the same reaction except that the light flux was decreased. C. The same reaction as in B but with an increase of CO concentration to ca. 7.5 × 10-4 mol. D. The same reaction as in B but with a further increase of CO concentration to ca. 7.5 × 10-3 mol.
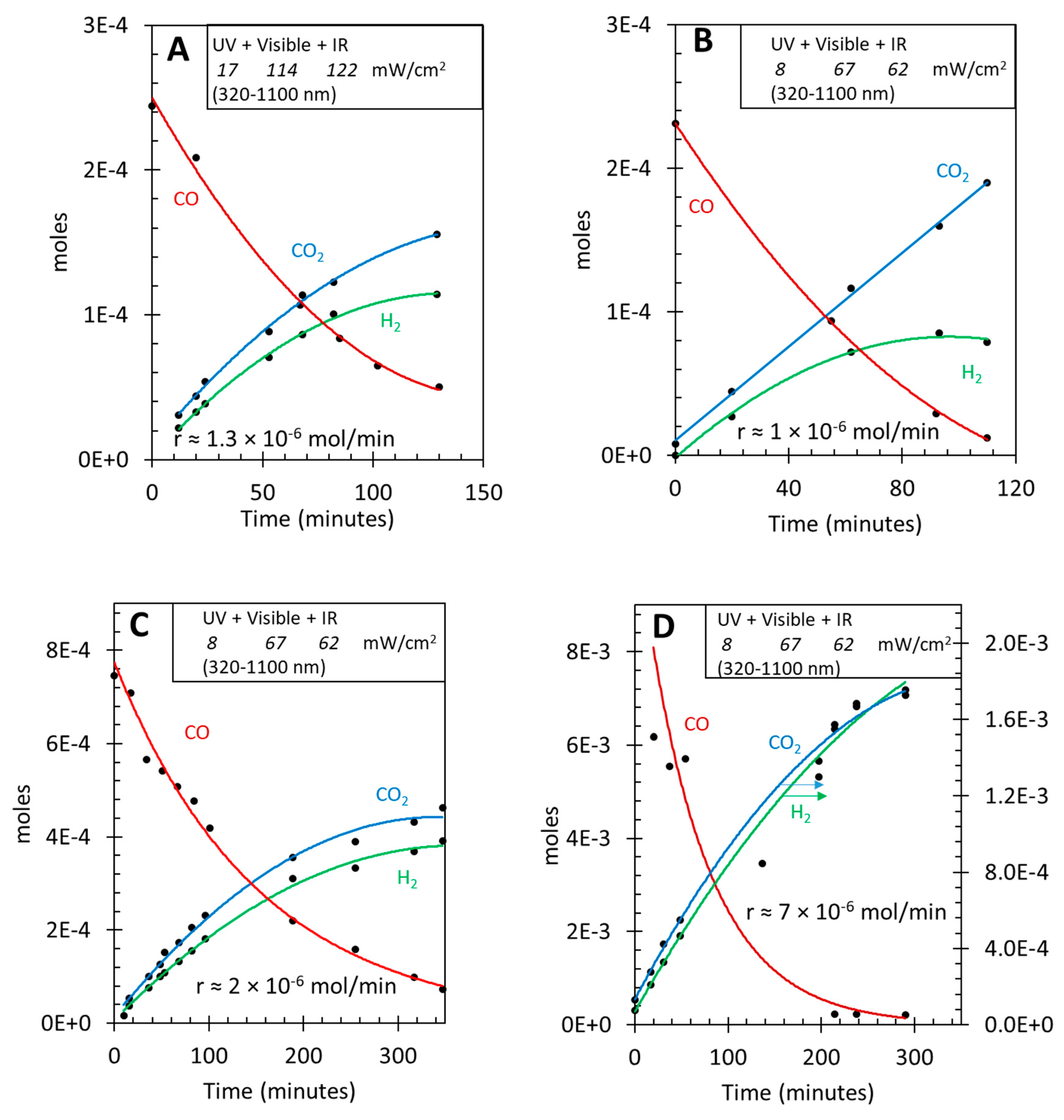
Figure 4.
Effect of oxygen addition on the Photo-thermal WGSR as a function of time over 8 wt.% Au/TiO2 P25. A. Hydrogen production. B. CO2 production. C. CO consumption.
Figure 4.
Effect of oxygen addition on the Photo-thermal WGSR as a function of time over 8 wt.% Au/TiO2 P25. A. Hydrogen production. B. CO2 production. C. CO consumption.
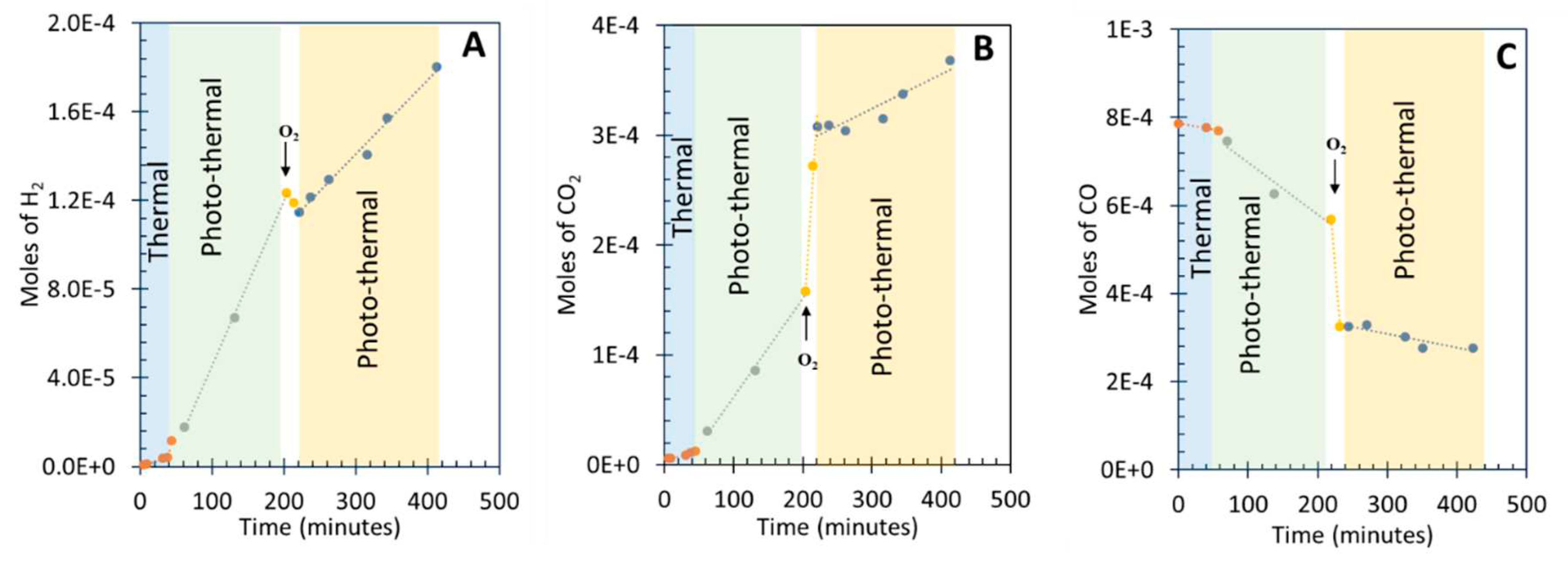
Figure 5.
(A) Hydrogen production over 8 wt. % Au/TiO2 P25, under “UV + visible + IR” and “visible + IR” with the indicated fluxes. (B) Hydrogen production over 8 wt. % Au/TiO2 P25, under “UV + visible” (400- 620 nm) and visible (400-620 nm) with the indicated fluxes. Hydrogen production over 8 wt. % Au/TiO2 P25, under visible light only in two regions (500-700 nm and 600-700 nm) with the indicated fluxes.
Figure 5.
(A) Hydrogen production over 8 wt. % Au/TiO2 P25, under “UV + visible + IR” and “visible + IR” with the indicated fluxes. (B) Hydrogen production over 8 wt. % Au/TiO2 P25, under “UV + visible” (400- 620 nm) and visible (400-620 nm) with the indicated fluxes. Hydrogen production over 8 wt. % Au/TiO2 P25, under visible light only in two regions (500-700 nm and 600-700 nm) with the indicated fluxes.
Scheme 1.
Schematic representation of Au LSPR effect on WGSR of a 8 wt.% Au/TiO2 P25 catalyst. Au mean particle size is 5 nm. Visible light excitation of about 500-700 nm may enter in resonance with the 5d-6s band excitation of Au atoms in the particles. These leave holes in the 5d and puts electrons in the 6s/Fermi level. At the interface Au/TiO2 electrons may be transferred to protons of adsorbed water while holes react with its oxygen anions. Not that CO oxidation is an exothermic reaction providing energy for the system. Therefore, the chemical input is provided from CO [51] (see equations 11-15).
Scheme 1.
Schematic representation of Au LSPR effect on WGSR of a 8 wt.% Au/TiO2 P25 catalyst. Au mean particle size is 5 nm. Visible light excitation of about 500-700 nm may enter in resonance with the 5d-6s band excitation of Au atoms in the particles. These leave holes in the 5d and puts electrons in the 6s/Fermi level. At the interface Au/TiO2 electrons may be transferred to protons of adsorbed water while holes react with its oxygen anions. Not that CO oxidation is an exothermic reaction providing energy for the system. Therefore, the chemical input is provided from CO [51] (see equations 11-15).
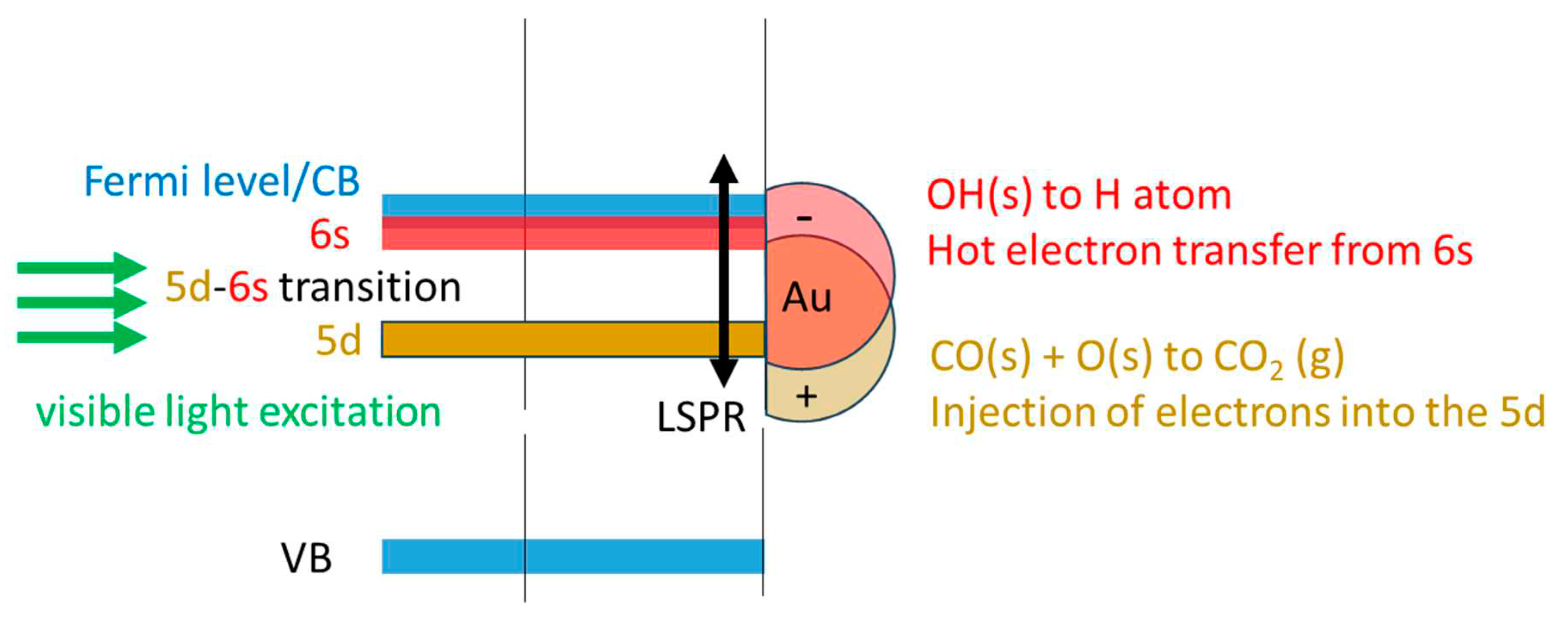
Table 1.
Hydrogen production rates at different temperatures during the WGSR over 8 wt. % Au/TiO2 P25.
Table 1.
Hydrogen production rates at different temperatures during the WGSR over 8 wt. % Au/TiO2 P25.
| T (oC) | T (K) | 1/T(K) | Rate (H2 moles/min) |
|---|---|---|---|
| 80 | 353 | 0.002832 | 4.87 × 10-7 |
| 150 | 423 | 0.002363 | 8.69 × 10-6 |
| 200 | 473 | 0.002113 | 9.00 × 10-6 |
| 250 | 523 | 0.001911 | 1.97 × 10-5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



