
Submitted:
19 October 2023
Posted:
20 October 2023
You are already at the latest version
Abstract
Clear cell renal cell carcinoma (ccRCC) is the most frequent form of kidney cancer. Metastatic stages of ccRCC reduce the five-year survival rate to 15%. In this report we analyze the ccRCC-induced remodeling of the five KEGG-constructed excretory functional pathways in a surgically removed right kidney and its metastasis in the chest wall from the perspective of the Genomic Fabric Paradigm (GFP). GFP characterizes every single gene in each region by the independent variables: average expression level (AVE), relative expression variability (REV) and expression correlation (COR) with each other gene. While traditional approach is limited to only AVE analysis, the novel REV analysis indicates genes whose correct expression level is critical for cell survival and proliferation. COR analysis determines the real gene networks responsible for functional pathways. The analyses covered the pathways: aldosterone-regulated sodium reabsorption, collecting duct acid secretion, endocrine and other factor-regulated sodium reabsorption, proximal tubule bicarbonate reclamation, and vasopressin-regulated water reabsorption. The present study confirms the conclusion of our previously published articles on prostate and kidney cancers that even equally graded cancer nodules from the same tumor have different transcriptomic topologies. Therefore, the personalization of the anti-cancer therapy should go beyond the individual to his/her major cancer nodules.
Keywords:
ADCY6
; aldosterone-regulated sodium reabsorption
; AP2A1
; AVP
; collecting duct acid secretion
; CREB3L4
; endocrine and other factor-regulated sodium reabsorption
; ESR1
; proximal tubule bicarbonate reclamation
; vasopressin-regulated water reabsorption.
1. Introduction: Limits of the Gene Biomarker Paradigm for Cancer Diagnostic and Therapy
Cancer is a major cause of death worldwide and likely the most funded researched group of lethal diseases. Depending on tumor localization, size and metastatic stage, treatment options in specialized clinics may include: surgery, chemotherapy, radiation therapy, hormone therapy, bone marrow transplantation, targeted therapy, and immunotherapy [1]. For smaller tumors, at early stages, the thermal ablation offers a low-risk and minimally invasive solution [2,3]. Nevertheless, in spite of all academia and industry efforts, we still do not have an efficient answer to cancer, suggesting the need for a novel approach.
According to the American Cancer Society, 52,360 men and 29,440 women are expected to be diagnosed with kidney and pelvis cancer in 2023, out of which 9,920 men and 4,970 women might die from this disease [4]. The prevalence of kidney cancer is strongly dependent on age (most diagnosed people are over 65 years old), sex (twice more frequent in men than in women) and race (African Americans, American Indians, and Alaska Natives are affected in higher percentages than other races). While as long the cancer is localized only in the kidney, the 5-year survival rate is good (93%), it declines rapidly (15%) when it spreads to lungs, brain or bones [4]. The vast majority of kidney cancers are clear cell subtypes of Renal Cell Carcinoma (ccRCC), characterized by high inter- and intra-tumor heterogeneity and strong crosstalk with the cellular microenvironment [5].
A very dynamic and promising avenue is provided by the gene therapy as an alternative to the cell transplant [6]. As of October 5th, 2023, PubMed lists 143,107 articles for “cancer gene therapy” published from 1966 on, out of which 12,818 were published in 2021 alone. The majority of these articles looked for gene biomarkers whose altered sequence and expression level were supposed responsible for triggering cancerization and whose restoration allegedly provides the cure. In most publications, the biomarkers were identified by comparing sequencing (e.g. [7,8,9] or/and transcription (e.g. [10,11,12]) data in tissues collected from cancer-stricken and healthy people. The potency of the gene therapy was also tested on standard human cancer cell cultures (e.g. [13,14,15,16]).
However, what are the real predictive values of gene biomarkers for cancer diagnosis and therapy? The 38.0 release (08/31/2023) of NIH-National Cancer Institute GDC Data Portal [17] containing genomic data collected from 88,991 cancer cases in 68 primary sites, reported a total of 2,903,037 mutations located in 22,588 genes. Importantly, the Portal reported mutations in almost all genes affecting each of the 68 primary sites. Table 1 summarizes GDC data for 14 primary sites by presenting the number of mutated genes found in the investigated cases, how many of the mutated genes are protein coding, and the total number of mutations detected so far for that site.
Moreover, almost every single gene was found as mutated in at least one case from any of the all 68 primary sites. For instance, with respect to the reported cancer cases from Table 1, titin (TTN) appeared mutated in the following percentages of reported cases: 12.41 bladder, 2.75 bonne marrow, 14.47 brain, 2.99 breast, 5.05 colorectal, 10.28 head and neck, 5.71 kidneys, 6.88 lungs, 2.17 pancreas, 2.60 prostate, 13.46 skin, 15.51 stomach, and 12.21 uterus. The percentages include all 13,073 distinct somatic mutations observed for this gene in the 4,512 out of the 88,991 cases included in the portal database. So, not only none of the distinct mutations, but even together all kinds of altered sequence of TTN as a whole do not exhibit statistically significant sensitivity and/or specificity for a particular form of cancer. The same lack of significance is carried by all other “regular suspects” like tumor protein p53 (TP53) with 1,341 mutations identified in 4,934 cases across 47 out of 82 projects, KRAS (1,500 cases, 125 distinct mutations identified in 43 projects), or PTEN (1,228 cases, 846 mutations across 38 projects). The most frequently mutated gene in kidney cancer is von Hippel-Lindau tumor suppressor (VHL) detected in 342 (9.77%) out of 3,501 cases. However, VHL was found mutated also in 50 cases of cancer of other organs.
Mimicking a human cancer phenotype on genetically engineered animals (e.g. [18,19,20,21]) provides disputable etiologies owing that together with the manipulated gene hundreds others are regulated as reported in many studies, ours included (e.g. [22]). The set of significantly regulated genes in tissues of genetically engineered animals with respect to their wild type counterparts depends on the profiled tissue (e.g. [23,24]), silencing method used (e.g. [25]) and the genetic background {e.g. [26]).
Owing to the unrepeatable combination of favoring factors (some of them changing in time): race, sex, age, medical history, diet, climate, exposure to toxins, stress and other external stimuli, each human is a DYNAMIC UNIQUE. The dynamic unicity requires time-sensitive personalized therapeutic approach. A very important, yet still neglected in many published papers and public repositories, is the tumor genomic, transcriptomic and proteomic heterogeneity [27,28,29,30]. Thus, histopathologically distinct cancer nodules from the same tumor most frequently have different characteristics. Therefore, the best REFERENCE for cancer related genomic alterations of an individual is not the tissue of the average healthy person of the same race, sex and age group but the quasi-normal tissue surrounding his/her cancer nodules [31]. With this reference in mind, the true goal of the anti-cancer therapy is to restore what is considered normal for that person, hence the need for a personalized approach.
While the diagnostic value of the gene biomarkers is disputable, let us see whether the restoration of the correct sequence and/or expression level of the biomarkers can provide the therapeutic answer for cancer. Since the biomarkers are selected from the most frequently altered genes in cancer patients, it means that their sequences and/or expression levels are poorly protected by the cellular homeostatic mechanisms like the minor players in the cell life. Therefore, their restoration might be of little consequence.
It is very surprising (and disappointing) that almost all gene expression studies use only 0.01% of the information provided by the high throughput transcriptomic platforms (RNA-sequencing, Agilent microarray, Affimetrix, Illumina BeadChip arrays etc.) as will be presented below. Traditional analysis considers ONLY the expression levels of the quantified genes whose comparison between conditions tells what gene was significantly up-/down-regulated (according to arbitrarily introduced cut-off for the absolute fold change) or turned on/off. The genes are eventually clustered according to their similar behavior across conditions (e.g. [32,33]), but similar regulation does not necessarily mean that the clustered genes are interacting to each other (they may have an up-stream common regulator or transcription factor).
Using publicly available software (based on text mining the peer-reviewed literature) such are: Ingenuity [34], DAVID [35], KEGG [36], the regulated genes might be organized into functional pathway. However, the topology of the pathways constructed using such software have three major flaws: universality, rigidity and unicity. They are universal for not discriminating with respect to strain/race, sex, age, hormonal activity etc. and even with respect to tissue (for Ca2+- and other signaling pathways common to several tissues). They are rigid for not changing in response to ageing, medical treatment, external stimuli, and progression of a disease or other dynamic influencing factors. Finally, each constructed pathway has a unique wiring of the genes and not a spectrum of several possible gene circuits. If two simple elements like hydrogen and carbon can combine in so many ways to form the infinite variety of hydrocarbons, how could we assume that tens of much more complicated units (the genes) network in a single way to accomplish a particular task? Therefore, we have used KEGG constructed pathways only for illustrative purposes and the coordination analysis to determine the real gene networking.
Because of the above discussed deficiencies of the biomarker approach, we switched our research from biomarker to the Genomic Fabric Paradigm (GFP, [37]). GFP offers the most theoretically possible comprehensive characterization of the transcriptome and personalized solutions for cancer gene therapy.
The present study complements a previously published article [37] with the GFP analyses of the remodeling of the five KEGG-constructed excretion system’s functional pathways affected by clear cell renal cell carcinoma (ccRCC).
2. Materials and methods
2.1. The best choice of tissue samples
Nevertheless, for statistical significance, a transcriptomic study should profile several biological replicas of the compared conditions. Most authors use three biological replicas but four is (in our view) the best compromise to get enough statistical relevance beyond the inherent technical noise of the profiling method. In the case of solid tumors, the best choice is to take a point biopsy from the center of a cancer nodule (or each cancer nodule if there are more) and another one from the surrounding (almost normal) tissue, split each biopsy into four parts and profile separately the resulted quarters. Thus, the reference for the patient cancer is not the abstract, racially blind, ageless and sexless model of the human body but his/her own normal tissue for his/her race, age and sex. This procedure is standard in our lab and was used in studies of surgically removed tumors from kidney [37], thyroid [38], and prostate [39] cancer patients.
In this study, we re-analyze transcriptomic data from our experiments on the surgically removed right kidney affected by ccRCC (two primary cancer nodules, PTA and PTB) and its metastasis in the chest wall (CWM) compared to the quasi-normal surrounding kidney tissue (NOR). Data were obtained using Agilent-026652 Whole Human Genome Microarrays 4x44K v2 and are publicly accessible [40].
2.2. Data filtering and normalization
The hybridized microarray spots with foreground fluorescence less than twice the background in one biological replica profiled with microarrays are eliminated from the analysis of all to be compared samples owing to the non-negligible technical noise. With this filtering, every profiled sample from all to be compared conditions were reduced to the same number of distinct transcripts, here “N = 13,314”. The background subtracted forward fluorescence of the microarray hybridized spot(s) with transcript “i” from the biological replica “k” (k = 1, 2, 3, 4) of condition “c”, “ai(c;k)” were normalized to the expression of the median gene expression for that profiled sample.
For microarray experiments, ai(c;k) is the sum of the net fluorescence of all spots probing redundantly transcript “i” in the biological replica “k” of condition “c”.
2.3. Independent characteristics of gene expression
2.3.1. Normalized average expression level.
The filtered and normalized expression values of each transcript “i” are averaged over the biological replicas of each condition “c” as . is the genomic measure that everybody in the field uses to determine whether that transcript abundance was up-/down-regulated or turned on/off when comparing cancer with healthy samples.
2.3.2. Relative Expression Variability
When properly selected (as in splitting a point biopsy into four parts), the biological replicas may be considered as different instances of the same system subjected to distinct local (not significantly regulating) conditions. This apply to ccRCC samples owing to the strong crosstalk of cancer cells with the non-uniform microenvironment [5]. Thus, we can add as an independent feature of the transcript “i” in condition “c” the Relative Expression Variability, “REV i(c)”, computed as the mid chi-square (χ2) interval estimate of the coefficient of variation for “n = 4” biological replicas and “υi” spots probing redundantly transcript “i”.
REV provides an indirect estimate of the strength of the cellular homeostatic mechanisms to control the transcript abundances, with the smallest REV indicating the most stably expressed (i.e. the most controlled) gene and the largest REV pointing to the most variably expressed (i.e. the least controlled gene). Since more control means more energy spent by the cell, it is natural to assume that the right expressions of the most controlled genes were more important for the cell survival or/and proliferation in the multicellular tissue. As such, the REV analysis tells first-hand the investigator about the cell priorities.
2.3.3. Expression coordination
Genes are not single but team players in the cell life. Considering the high efficiency of the cellular phenomena, we have introduced the Postulate of the Transcriptomic Stoichiometry (PTS) [41,42] as an extension to gene networking in functional pathways of Proust’s Law of Definite Proportions from chemistry [43]. PTS states that: in any steady-state condition, expressions of genes whose encoded products are part of a functional pathway are coordinated to ensure the maximum efficiency of that functional pathway. This means that the involved genes are set to produce the transcripts at the right abundance proportions. PTS and the coordination analysis can be used to determine the real gene network responsible for a particular pathway in a given condition.
The most difficult question is how the genes are networked: in pairs (e.g. agonist-antagonist), in “ménage à trois” (e.g. agonist, antagonist and a modulator of both), or in more complex gene inter- coordination clusters? To answer this question, we adopted the formalism of correlation functions similar to that used to describe the structure of simple liquids (like liquid argon) [44]. Thus, the configuration function “F” of “N” distinct genes is considered as a superposition of virtual configurations in which the genes are: independently expressed “f1”, coordinately expressed in pairs, “f2”, coordinately expressed in triplets, “f3”, and so on until all “N” genes are coordinately expressed as a whole, “fN”. As shown in Appendix A, the distribution functions of higher than pair-correlated genes can be neglected, so that:
The problem is now to select the suitable algorithm for the gene pairing. There are several weighted and unweighted types of correlation algorithms (e.g., [45,46,47,48]) aiming to determine the networks of interconnected genes and molecular interactions based on meta-analysis of large sets of genomic data on healthy and cancer affected populations. By contrast, our aim is to determine the gene network in a particular condition (normal or cancerous) of only one individual, so none of these analyses is suitable for our endeavor.
The simplest gene pairing is by Pearson correlation coefficient (here after denoted by “COR” instead of the traditional “r”) between the log2 of the normalized expressions of two genes (“i” and “j”) in the four replicas of the same condition “c”:
Although Marbach et al. [49] has shown that Pearson’s correlation is not the strongest method to determine the gene networks, it is enough accurate when taking into account the technical noise of the gene expression profiling platform.
With four biological replicas, two genes are (p < 0.05) significantly synergistically expressed (i.e. positive correlation) if COR ≥ 0.951, antagonistically expressed (negative correlation) if COR ≤ - 0.95 and independently expressed (null correlation) if |COR| ≤ 0.05. For microarrays probing the same transcript with two spots (i.e. 8 paired values), the p < 0.05 significant cut-off for synergism/antagonism is |COR| ≥ 0.71, for three spots (12 paired values) |COR|≥ 0.58 and so on, Pearson cut-off decreasing when the number of probing spots increases [50].
In [51] we defined the coordination score “COORD” with p-value “p” of a pathway “Γ” in condition “c = NOR, PTA, PTB, CWM” as:
Where “SYN/ANT/IND” are the percentages of all gene pairs from the pathway “Γ” that are synergistically/antagonistically/independently expressed with statistical significance (p-value) “p” in condition “c”.
2.3.4. Topology of the transcriptome and the Gene Master Regulator
We use GFP to compare the transcriptome of a cancer nodule with that of the surrounding normal tissue in a tumor. Thus, in addition to determining what genes were significantly up-/down regulated in cancer, we learn also how much the homeostatic control of the transcript abundance was altered for each gene or groups of gene and how the gene networks were remodeled.
“REV” and “COR” can be used to determine the Gene Commanding Height (GCH) that establishes the importance hierarchy of the genes in each region. The top gene (highest GCH) is termed Gene Master Regulator (GMR) of that region. GMR is the highly protected gene (i.e. low REV, meaning critical for the cell survival) that has the strongest influence on the expression of other genes through expression coordination [52,53].
For the GFP users, the transcriptome is no longer a chaotic collection of transcripts but a multi-dimensional hierarchized mathematical entity subjected to dynamic sets of expression variability and expression correlations of its components.
2.4. Transcriptome alteration in cancer
2.4.1. Measures of expression regulation
Traditional analysis considers a gene as significantly regulated in cancer with respect to normal if the expression ratio “x” (negative for down-regulation) has an absolute fold-change |x| larger than an arbitrarily introduced cut-off (most frequently 1.5). In most cases, it adds also the condition that the p-value of the heteroscedastic t-test of mean expressions is less than 0.05. However, the cut-off fold-change could be too stringent for very stably expressed genes (leading to false negatives) or too lax for very unstably expressed ones (introducing false positives). Therefore, we use to determine for each gene the cut-off of the absolute fold-change, “CUT”, considering the combined contributions of its biological variability and the technical noises of the platform in the two profiled conditions. Thus, the gene “i” is considered as significantly regulated in “cancer” with respect to the normal tissue (NOR), if:
In many studies, the transcriptome alteration is presented as percentages of the significantly up- and down-regulated genes. Nonetheless, the percentage presentation implicitly assumes that all regulated genes are uniform (+1 or – 1) contributors to the overall transcriptome alteration, while neglecting the contributions of the not significantly regulated genes. A more accurate measure of the expression regulation is the Weighted Individual (gene) Regulation “WIR” whose absolute values can be averaged for the genes included in a functional pathway “Γ” as the Weighted Pathway Regulation “WPR” ([54]). In addition to being applied to all genes (including the not significantly regulated), WIR takes into account the absolute expression change and the statistical significance of the regulation.
2.4.2. Regulation of the control of transcript abundance
In a previous paper [55], we defined the Relative Expression Control, “REC,” of gene “i” in condition (here region) “c” as:
Positive RECs indicate more than the median controlled genes and negative values indicate less than the median controlled genes in that condition. As to be shown below (Figure 1), ccRCC altered the REVs of individual genes and by consequence, their hierarchy (illustrated in Figure 2 below). However, the interest is how much cancer altered the transcript control; therefore, we define the total ccRCC-induced regulation of the control of the gene “i” transcript abundance as:
In this report, we considered const = 1.
2.4.3. Regulation of expression coordination:
The regulation of expression coordination can be computed with regard to a single gene, a group of genes (like those involved in a particular pathway) or with regard to all quantified genes. In this report, the correlation analysis is limited to the five KEGG-constructed excretory system pathways.
In (12), positive values indicate overall increase of the expression coordination and negative value overall decrease of the expression coordination of gene “i” with expressions of the genes from the reference pathway “Γ”.
2.4.4. The transcriptomic distance
Nonetheless, the most comprehensive measure of the transcriptome alteration should include also the change in the expression control and coordination with all other genes. Therefore, we redefined the Transcriptomic Distance of Individual gene, “TDI”, separating the gene “i” expressed in a cancer nodule with respect to its expression in the normal tissue, as:
can be further averaged for all genes pertaining to a particular pathway “Γ”.
2.5. Functional pathways
In this report, we used our experimental data from the profiled samples of a man with metastatic ccRCC [40] to determine the topology and the ccRCC-induced remodeling of the five KEGG-constructed functional pathways for the excretory system. The samples (point biopsies split in four and profiled separately) were collected from a chest wall metastasis (CWM), and two primary cancer nodules (denoted as PTA and PTB) and the normal (NOR) surrounding tissue from the removed right kidney. The analyzed pathways were: hsa04960 Aldosterone-regulated sodium reabsorption [56], hsa04966 Collecting duct acid secretion [57], hsa04961 Endocrine and other factor-regulated calcium reabsorption [58], hsa04964 Proximal tubule bicarbonate reclamation [59], and hsa04962 Vasopressin-regulated water reabsorption [60].
3. Results
3.1. The global picture
Out of the 13,314 adequately quantified genes in all 16 samples profiled, 26 were included in the KEGG pathway “Aldosterone-regulated sodium reabsorption”, 16 in the “Collecting duct acid secretion” 37 in the “Endocrine and other factor-regulated calcium reabsorption”, 18 in the “Proximal tubule bicarbonate reclamation”, and 36 in the “Vasopressin-regulated water reabsorption”. Nonetheless, the pathways were not mutually exclusive, some genes being counted in two (e.g. ADCY9 in hsa04961 and hsa04964) or even three excretory pathways (e.g. ATP1A1 in hsa04960, hsa04961 and hsa04964) so that the total number of quantified distinct excretory genes was 108.
In PTA, we found the same percentage, 8.(3)%, of excretory genes being significantly up-regulated and down-regulated. In PTB the percentages were 3.70% down- and 15.74% up-regulated, while in CWM there were 7.41% down- and 6.48% up-regulated excretory genes. These results show than even closely located cancer nodules from the same tumor (PTA and PTB) may exhibit different gene alterations, questioning the validity of meta-analyses comparing ccRCC patients with healthy counterparts.
3.2. Independent characteristics of gene expression
Figure 1 illustrates the independence of the three types of expression characteristics for 37 genes involved in the KEGG-constructed pathway “Endocrine and other factor – regulated calcium reabsorption” [57] using our microarray data on Fuhrman grade 3 metastatic ccRCC samples [40]. For the COR analysis, we chose to represent the expression correlation with the estrogen receptor 1 (ESR1), owing to the kidney being one of “the most estrogen-responsive, not reproductive organs in the body” [61].
The independence of the three characteristics is visually evident. Therefore, each of the three types of analyses brings non-redundant information.
Of note are the differences between the normal tissue and the cancer nodules not only in the average expression levels of certain genes (AVE, as expected and reported by the traditional analysis) but also in the relative expression variability (REV) and correlation (COR) with other genes (here with ESR1). For instance, the significant expression antagonism (COR(NOR) = -0.96624, p = 0.0338) of AP2A1 (adaptor-related protein complex 2, alpha 1 subunit) with ESR1 in the normal tissue is switched into expression synergism in each of the three cancer nodules. We got the following correlation values (and statistical significances) between ESR1 and AP2A1 in the cancer samples: COR(PTA) = 0.9607 (p-val = 0.03093), COR(PTB) = 0.970449 (p-val = 0.02955), COR(CWM) = 0.958562 (p-val = 0.04144). This result means that in the normal tissue when expression of ESR1 goes up, that of AP2A1 goes down and when ESR1 goes down, AP2A1 goes up. By contrast, in cancer, expression of ESR1 oscillates in-phase with expression of AP2A1, so that expressions of both genes go up or down simultaneously.
3.3. ccRCC changed the gene hierarchy
Figure 2 presents the GCH scores for all quantified genes selected by KEGG as involved in the five excretory functional pathways. Of note are again the substantial inter-regional differences of the genes’ GCH scores, indicating distinct gene hierarchies within the corresponding pathways. For instance, the GCH of IGF1 (insulin-like growth factor 1 (somatomedin C)) from the “Aldosterone-regulated sodium reabsorption” pathway increased from 1.41 in NOR to 13.11 (9.28x) in PTA, although it remains practically unchanged in PTB (2.25) in CWM (2.38). CREB3L2 (cAMP responsive element binding protein 3-like 2) from “Vasopressin-regulated water reabsorption” exhibited a 9.57x increase of GCH in PTB with respect to PTA. The GCH of ATP1A2 (ATPase, Na+/K+ transporting, alpha 2 polypeptide) decreased by 16.85x in CWM with respect to PTB.
3.4. Measures of individual gene regulation
Figure 3 illustrates the six types of quantifying the ccRCC-induced alterations of the genes from the KEGG-constructed pathway hsa04961 “Endocrine and other factor-regulated calcium reabsorption”. The six types are: uniform +1/-1 for significant regulation, expression ratio (negative for the down-regulation), WIR (Weighted Individual (gene) Regulation, negative for down-regulation), regulation of the transcription control, regulation of the expression correlation with all other genes from the pathway, and TDI (Transcriptomic Distance of Individual gene).
There are notable differences among the three cancer nodules in all six measures. For instance, SLC8A1 (solute carrier family 8 (sodium/calcium exchanger), member 1), not regulated in PTA, was significantly up-regulated in PTB (x = 2.75) and significantly down-regulated in CWM (x = -1.84). Only one gene, ATP1B2 (ATPase, Na+/K+ transporting, beta 2 polypeptide), was significantly upregulated in both PTA (x = 1.91) and PTB (x = 2.67) but not in CWM. PLCB1 (phospholipase C, beta 1 (phosphoinositide-specific) was the single gene significantly down-regulated in PTB (x = - 2.60) and CWM (x = -2.01) but not in PTA.
As defined, WIR takes larger absolute values for highly expressed genes in the reference (here NOR) condition, sometimes even larger for not significantly regulated genes than for significantly regulated ones. For instance, DNM2 (dynamin2), a significantly down-regulated gene in PTB (x = - 2.47, CUT = 1.81, p = 0.0242) had WIR(PTB) = 4.73. DNM2 contribution to the transcriptomic alteration in PTB was substantially overpassed by that of the not statistically significantly regulated DNM1 (dynamin1: WIR = 95.74, x = -2.12, CUT = 1.66, p = 0.0969). The reason is that in NOR, AVEDNN1 = 94.30 >> 3.30 = AVEDNN2. Nonetheless, by considering the whole change in the expression level, WIR tells the investigator much more about the contribution of a gene to the transcriptome’s alteration that the uniform +1/-1 up-/down regulation.
Overall, the median expression control 100/<REV> increased from 2.58 in NOR to 4.27 in PTA, 2.92 in PTB and 3.96 in CWM. In the illustrated pathway, we found genes with substantial increase of the expression control and genes with substantial decrease of control in cancer. The most substantial increase of the expression control was for ADCY9 (adenylate cyclase 9) in PTB (ΔREC(PTB-NOR) = 12.38). It is remarkable that control of ADCY9 had modest change in PTA (ΔREC(PTA-NOR) = 2.08) but the largest decrease of all in CWM (ΔRECCWM-NOR) = -15.50). The largest decrease in PTA was exhibited by AP2A1 (adaptor-related protein complex 2, alpha 1 subunit): ΔREC(PTA-NOR) = -7.97, but with insignificant changes in the other nodules: ΔREC(PTB-NOR) = -0.17, ΔREC(CWM-NOR) = 0.26. Interestingly, there are genes (e.g. DNM2) whose control increased with respect to NOR in one cancer nodule (PTA, ΔREC(PTA-NOR) = 6.2) but decreased in the equally ranked other nodule from the same tumor (ΔREC(PTB-NOR) = -7.2), indicating shift in cell’s priorities. A substantial shift in cell priorities occurred also for VDR (vitamin D (1,25- dihydroxyvitamin D3) receptor) between nodules PTB (ΔREC(PTB-NOR) = 7.92) and CWM (ΔRECCWM-NOR) = -6.07).
We found genes such is ADCY6 with increased overall correlation with the other pathways genes with respect to NOR in one nodule (ΔCOR(PTA−NOR) = 7.39) but decreased in the other nodules (ΔCOR(PTB−NOR) = -6.04, ΔCOR(PTB−NOR) = -5.92), indicating profound remodeling of the gene networking. Interestingly, ADCY6 was significantly up-regulated in PTA (x = 1.54) and CWM (x = 1.96) but not in PTB (x = 1.29). A very similar behavior excepting that it was not significantly regulated in any of the three cancer nodules was exhibited by AP2A1 (ΔCOR(PTA−NOR) = 7.60, ΔCOR(PTB−NOR) = -7.02, ΔCOR(PTB−NOR) = -6.84).
Nevertheless, the most comprehensive measure that incorporates changes in all three types of characteristics is the transcriptomic distance of an individual gene (TDI) from its normal AVE, REV and COR (with all other genes within the pathway) in the normal tissue. From the TDI perspective, DNM1 (TDI = 95.95 in PTB) followed by FXYD2 (TDI = 80.21 in PTB) were the most altered genes within this set. Interestingly, with all differences at the individual gene level, the median TDI’s of the three nodules were close to each-other (5.72 for PTA, 5.73 for PTB and 5.37 for CWM).
3.5. Overall regulation of the excretory pathways
Table 2 presents the overall gene expression alterations of the five KEGG-constructed excretory pathways as percentages of the up- and down-regulated out of the quantified genes in the pathway and the weighted pathway regulation (WPR). With 23.08% in PTA and CWM and 30.77% in PTB total percentage of up- and down-regulated genes hsa04960 (Aldosterone-regulated sodium reabsorption) appears as the most altered pathway. However, from the more comprehensive WPR perspective, hsa04966 (Collecting duct acid secretion) is the most altered of the five pathways in all three cancer nodules.
Interestingly, all significantly regulated genes are up for hsa04964 (Proximal tubule bicarbonate reclamation) in all three cancer nodules, indicating a major activation of this
pathway in ccRCC. The results on the pathway hsa04960 (Aldosterone-regulated sodium reabsorption) are intriguing: while there are equal numbers of up- and down-regulated genes (3 = 11.54% of 26) in both PTA and CWM, in PTB all 8 (30.77% of 26) significantly regulated genes are over-expressed. This means that hsa04960 is balanced in PTA and CWM but strongly activated in PTB. The hsa04960 regulomes (sets of significantly regulated genes in this pathway) of the three cancer nodules are different, only one gene, PIK3R2 (phosphoinositide-3-kinase, regulatory subunit 2 (beta)), being significantly up-regulated in all three nodules.
3.6. Location of the regulated genes in the excretory system functional pathways
Figure 4 for hsa04962 “Vasopressin-regulated water reabsorption” and Figures S1–S4 from Supplementary Material for the other four KEGG-constructed excretory system pathways present for every profiled cancer nodule the localizations of the regulated genes. Of note are the inter-nodule differences in the subsets of the regulated genes.
We found that although the hsa0460 gene AVP (arginine vasopressin) was not significantly regulated in any of the three cancer nodules, expressions of several other genes were significantly altered (even not in the same way) in all profiled regions. For instance, AQP3 (aquaporin 3 (Gill blood group)) was found as down-regulated in PTA (x(PTA) = -2.808) and CWM (x(CWM) = -5.846) but up-regulated in PTB (x(PTB) = 2.034).
Unfortunately, the important AQP2 (aquaporin 2) and AVPR2 (arginine vasopressin receptor 2) were not quantified in this experiment.
Only two excretory genes were similarly regulated in all three cancer nodules. The hsa04962 gene CREB3L4 (cAMP responsive element binding protein 3-like 4), was down-regulated: x(PTA) = -1.40 (CUT = 1.38, p = 0.040); x(PTB) = - 1.74 (CUT = 1.63, p = 0.031), x(CWM) = -1.95 (CUT = 1.48, p = 0.003). In contrast, the hsa04960 gene PIK3R2 (phosphoinositide-3-kinase, regulatory subunit 2 (beta)) was upregulated in all three cancer nodules: x(PTA) = 3.31 (CUT = 1.89, p = 0.013), x(PTB) = 1.85 (CUT = 1.82, p = 0.046), x(CWM) = 3.30 (CUT = 1.90, p = 0.032).
Two genes were oppositely regulated in the nodules PTB and CWM: the hsa04960 gene SFN (stratifin; x(PTB) = 2.03, x(CWM) = -1.95) and the hsa04961 gene SLC8A1 (solute carrier family 8 (sodium/calcium exchanger), member 1; x(PTB) = 2.75, x(CWM) = -1.84).
Three genes were similarly regulated in PTA and PTB: ATP1B2 (ATPase, Na+/K+ transporting, beta 2 polypeptide; x(PTA) = 1.91, x(PTB) = 2.67), DCTN2 (dynactin 2 (p50); x(PTA) = -1.51, x(PTB) = -2.06), SLC4A4 (solute carrier family 4 (sodium bicarbonate cotransporter), member 4; x(PTA) = 2.19, x(PTB) = 2.57).
Three regulated genes in PTA were similarly regulated in CWM: ADCY6 (adenylate cyclase 6; x(PTA) = 1.52, x(CWM) = 1.96), KRAS (Kirsten rat sarcoma viral oncogene homolog; x(PTA) = 2.21, x(CWM) = 2.77), PIK3CD (phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit delta; x(PTA) = -1.65, x(CWM) = -1.97).
Two genes were similarly regulated in PTB and CWM: CA2 (carbonic anhydrase II; x(PTB) = 9.94, x(CWM) = 3.44), PLCB1 (phospholipase C, beta 1 (phosphoinositide-specific), x(PTB) = - 2.60, x(CWM) = -2.01).
3.6. Tumor heterogeneity off the transcriptomic networks
Figure 5 presents the (p < 0.05) significant synergism, antagonism and independence among the genes from the hsa04961 pathway “Endocrine and other factor-regulated calcium reabsorption” [58]. It is interesting to observe that the percentage of the synergistic pairs increased from 12.28% in NOR to 26.90% in PTA, 20.76% in PTB and 16.96% in CWM. The percentage of the antagonistic pairs increased from 9.65% in NOR to 21.92% in PTA, 20.76% in PTB and 16.08% in CWM, while that of the independently expressed pairs decreased from 12.28% in NOR to 4.09% in PTA, 4.68% in PTB and 6.43% in CWM. Altogether, the coordination score COORD = % synergistic + % antagonistic - % independent increased from 9.65% in NOR to 44.74% in PTA, 36.84% in PTB and 26.61% in CWM. These results indicate a substantial ccRCC-triggered increase of inter-coordination of the genes involved in this pathway.
Of note are again the substantial differences between the PTA and PTB regions, indicating distinct wiring of the genes in the functional network. For instance, there are 7 genes (AP2S1, ATP1A2, ATP2B3, CLTCL1, DNM3, ESR1, PLCB2) whose significant correlations with the sodium/calcium exchanger SLC8A1 are opposite in the two kidney nodules, indicating major differences in the gene networking. Thus, ATP1A2, ATP2B3, CLTCL1, DNM3, and ESR1 are synergistically expressed with SLC8A1 in PTA but antagonistically expressed with SLC8A1 in PTB, while AP2S1 and PLCB2 are antagonistically expressed with SLC8A1 in PTA but synergistically expressed with SLC8A1 in PTB.
Figure 6 presents the (p < 0.05) statistically significant synergism, antagonism and independence of the quantified excretory genes with AVP in all four regions profiled. In the KEGG-constructed “Vasopressin-regulated water reabsorption” pathway, AVP is directly connected to AVPR2 (arginine vasopressin receptor 2; not quantified in the experiment) and indirectly connected to GNAS. Using the COR analysis, we found that in the normal kidney AVP is significantly connected to the CREB3L1, CREB3L4, DYNC1H1, and RAB5A.
The cancer reconfigures the hsa04962 networks, so that in PTA, AVP is significantly connected to CREB3L3, NSF, PRKACB, and RAB5B. In PTB, AVP is connected to: CREB3L2, RAB11A and RAB5B, while in CWM it is connected to no hsa04962 gene. Remarkably are also the hsa04962 genes with significant independence with respect to AVP: DYNC1H1 in NOR, CREB3 and RAB1A in PTA, ARHGDIB in PTB and ARHGDIA, DCTN1 and DYNC1H1 in CWM.
4. Discussion
This study continues the analyses from a previous article [37] on four samples collected from the chest wall metastasis (CWM), two primary tumor regions (PTA and PTB) and the surrounding normal tissue (NOR) in the right kidney of a man with ccRCC. In [37] we analyzed the ccRCC impact on cyclins, cyclin kinases, and functional pathways: apoptosis, chemokine and VEGF signaling, oxidative phosphorylation, basal transcription factors, and RNA polymerase, while here we focused on the five KEGG-constructed functional pathways of the excretory system.
An important finding of the previous paper was that even equally Fuhrman-graded (3) closely located nodules from the same kidney exhibit substantial transcriptomic differences and are under the command of distinct gene master regulators. Substantial transcriptomic differences between equally graded cancer nodules were also reported by us in two prostate cancer studies [31,39]. Importantly, tumor heterogeneity is not limited to genes’ expression levels but encompasses also the control of transcripts’ abundances and the way the genes’ are interconnected networking in functional pathways. The tumor heterogeneity and the unrepeatable set of cancer favoring factors characterizing each human imposes the normal tissue surrounding the cancer nodule(s) in the tumor as the most trustable reference.
The novel findings on the alterations of the mechanisms controlling the transcript abundances and the remodeling of the functional pathways were possible by adopting the Genomic Fabric Paradigm (GFP) and use of its mathematically advanced algorithms and software (detailed in [52]). The independence of the three types of expression characteristics was illustrated here for genes involved in the “Endocrine and other factor-regulated calcium absorption” KEGG-constructed pathway. The independence of expression characteristics can be proven for any other gene subset (principle discussed in [51]). For instance, in previously published cancer genomics papers, we proved the independence for genes involved in: chemokine signaling [37], apoptosis [53], evading apoptosis [39], and mTOR signaling [31].
The gene networks constructed with COR analysis do not have the faults of universality, unicity and rigidity as those built with KEGG and other software. COR-determined networks are not universal but instead depend on the race, age and other personal characteristics of the patient that modulate his/her gene expression, as we proved in [31] for two prostate cancer patients and two standard human prostate cancer cell lines. They are not unique, cancer nodule(s) and normal tissue exhibiting different gene wiring and certainly not rigid but remodeling during ageing, progression of the disease and in response to a treatment and other external stimuli. Thus, the Postulate of the Transcriptomic Stoichiometry (PTS) also extends Dalton’s Law of Multiple Proportions from chemistry.
The analysis of the expression correlation among the genes of the “Endocrine and other factor-regulated calcium reabsorption” revealed interesting partnership with the estrogen receptor 1 (ESR1), a tumor driver and drug targeting factor in cancer [62] and regulator of age-related mitochondrial dysfunction and inflammation [63]. Thus, while its expression synergisms with ATP2B3 (ATPase, Ca++ transporting, plasma membrane 3) and KLK2 (kallikrein-related peptidase 2) in NOR are not modified by ccRCC, the antagonistic expression with AP2A1 is switched to synergistic one in all three cancer nodules. Because AP2A1 is important in intracellular membrane trafficking [64], our result indicates that cancer switched the type of the estrogen influence on the intracellular transport phenomena [67].
We found that the excretory genes rank much lower than the GMRs identified for each region in [37]. Thus, GNAQ (guanine nucleotide binding protein (G protein), q polypeptide), the top gene involved in the “Endocrine and other factor-regulated calcium reabsorption” in both NOR (GCH(NOR) = 9.46) and CWM (GCH(CWM) = 17.43) is far below the GMRs of these regions: DAPK3 (death-associated protein kinase 3, GCH(NOR) = 30.31) and ALG13 (UDP-N-acetylglucosaminyltransferase subunit, GCH(CWM) = 82.95). IGF1 (insulin-like growth factor 1 (somatomedin C), GCH(PTA) = 13.11) from the pathway “Aldosterone-regulated sodium reabsorption” is far below TASOR (transcription activation suppressor, GCH(PTA) = 63.97) in PTA. ATP1A2 (ATPase, Na+/K+ transporting, alpha 2 polypeptide, GCH(PTB) = 9.95) from the pathway “Proximal tubule bicarbonate reclamation” reclamation is below FAM27C (family with sequence similarity 27, member C, GCH(PTB) = 57.19) in PTB. The much lower GCH scores means that although excretory pathways were strongly regulated, no excretory gene is an efficient target for the gene therapy against any of the three cancer nodules.
The substantial decrease in homeostatic control of AP2A1 expression in PTA but not in PTB and CWM indicates that, although not significantly regulated (x(PTA-NOR) = 1.22, x(PTB-NOR) = 1.04, x(CWM-NOR) = 1.13), AP2A1 lost its importance for the cell homeostasis in PTA while keeping it at NOR level in the other nodules. The most interesting case for the expression control analysis was that of ADCY9, a biomarker for glioma [65], lung [66] and hepatocellular carcinoma [67], colorectal [68], bladder [69], and pancreatic cancers [70]. ADCY9 exhibited the largest increase of the homeostatic control in PTB and the largest decrease in CWM among all profiled excretory genes, while its control in PTA remain as in the normal tissue. These results indicate that while cancer had no effect on the homeostatic control of ADCY9 transcript abundance in one region (PTA) of the tumor, it made the right ADCY9 expression critical in another region (PTB) and totally irrelevant in the metastases (CWM). Since expression of ADCY9 is normally four times more controlled than the median kidney gene, we predict that expression manipulation of ADCY9 will totally jeopardize the PTB cells, have similar (high) negative effect on both normal and PTA cells, while stimulating the proliferation of CWM cells. Thus, at least for this patient, targeting ADCY9 could be both benefic and harmful.
The increased overall correlation of ADCY6 and AP2A1 with all other genes from the “Endocrine and other factor-regulated calcium reabsorption” pathway (Figure 3) in PTA but decrease in the other two cancer nodules indicate substantially different gene networks. Thus, according to Figure 5, in NOR, AP2A1 has 4 statistically significant synergistically (ADCY6, DNM2, FXYD2, PLCB1) and 4 antagonistically (ATP1A2, ATP2B3, ESR1, KLK2) expressed partners, while in PTA it has 11 synergistic (ADCY6, ATP1A2, ATP1A4, ATP2B3, CLTCL1, ESR1, GNAQ, GNAS, KLK2, PRKCB, VDR) and 5 antagonistic (ATP2M1, ATP1B3, CLTA, FXYD2) partners (bolded symbols indicate the genes that switched the expression correlation type in PTA with respect to NOR). The partnerships of AP2A1 was partially similar in PTB: synergism with ATP2B3, CLTB, CLTCL1, ESR1, and GNAQ, and antagonism with AP2S1, ATP1B1, ATP1B3, PLCB2, PRKCA, and SLC8A1 (underlined symbols indicate common partners in PTA and PTB).
Because ADCY6 is a promising target in cancer therapy [71], it is important to know how it relates to other excretory genes. Thus, in the normal tissue (NOR) it is synergistically expressed with AP2A1, ATP1B1, antagonistically expressed with ATP1A2, ATP2B3, ESR1, KLK2, and independently expressed with CLTB. In PTA, the synergism with AP2A1 is preserved but that with ATP1B1 is switched to antagonism. PTA adds synergism with GNAQ, GNAS and VDR and antagonism with AP2A2, ATP1B3. The overall coordination of ADCY6 with other genes from the pathway is diminished in PTB (synergism with ATP1A3 and antagonism with BDKRB2, CLTA and PLCB2) and CWM (synergism with ATP1A4 and KL, and antagonism with ATP1A3 and FXYD2). Therefore, for this patient, manipulating the expression of ADCY6 would have different effects on his cancer nodules.
The down-regulation of AQP3 (x = - 3.889) was reported recently [72] by performing meta-analysis of public datasets from the publicly accessible databases ONCOMINE [73] and UALCAN [74]. In the cited study, the authors compared the expressions of all aquaporin encoding genes (AQP1/2/3/4/5/6/7/8/9/10/11) in 533 tumor cases with 72 normal cases using the uniform fold-change threshold 1.5 and p-value < 0.01 to decide whether a gene is significantly regulated. Since no web resource specifies the exact locations in the kidney from which the samples have been collected, nor weather the tumors contained more cancer nodules, the authors implicitly assumed homogeneous gene expressions all over the tumor. In contrast, our results indicate that ccRCC tumors do not exhibit uniform but heterogeneous gene expressions and therefore the meta-analysis comparing tumor cases with normal cases have little biological relevance beyond the statistics exercise. Instead, the best reference for cancer nodules is the normal tissue still present in the tumor.
Figure 6 shows distinct statistically significant coordination partners of AVP, the neuropeptide hormone arginine vasopressin, a very important regulator of kidney salt and water homeostasis [75], with other excretory genes. Our analysis detailed also how AVP-dependent water reabsorption regulate the cAMP signaling pathway [76,77] through expression correlation with genes shared by the cAMP signaling pathway with the excretory pathways. Thus, in NOR, AVP is synergistically expressed with ATP1B2, CREB3L1, CREB3L4, and FXYD2, has no antagonistic partners, and is independently expressed with PIK3CB. In PTA, AVP is synergistically expressed with ATP1B2 and CREB3L3, antagonistically expressed with ATP1A1 and PRKACB, and independently expressed with ATP1A4, CREB3 and MAPK1. In PTB, AVP is synergistically expressed with CREB3L2 and independently expressed with ATP1A3, while in CWM it is synergistically expressed with ATP1A2 and antagonistically expressed with PIK3R1.
Interestingly, CREB3L4, known for its role in proliferating the prostate cancer cells [78], was significantly down-regulated in all kidney cancer nodules (x(PTA) = -1.40, x(PTB) = - 1.74, x(CWM) = -1.95). Out of 18 regulated excretory genes in PTA and 21 in PTB, only 5 were similarly regulated (ATP1B2, CREB3L4, DCTN2, PIK3R2, SLC4A4) and one (AQ3) was oppositely regulated.
Altogether, the differences among the transcriptomic alterations and the remodeling of the excretory pathways in the three cancer nodules indicate that the bio-assays used to identify the ccRCC presence by the regulation of certain gene biomarkers might not be always valid. For instance, CA9 (carbonic anhydrase IX) that should be expressed only in ccRCC [79,80] was found by us expressed not only in the cancer nodules but also in NOR and exhibited a significant regulation (x = -10.37) only in PTB.
5. Conclusions
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: Regulated genes in the KEGG-constructed functional pathway hsa04960 Aldosterone-regulated sodium reabsorption, Figure S2: Regulated genes in the KEGG-constructed functional pathway hsa04966 Collecting duct acid secretion, Figure S3: Regulated genes in the KEGG-constructed functional pathway hsa04961 Endocrine and other factor-regulated calcium reabsorption, Figure S4: Regulated genes in the KEGG-constructed functional pathway hsa04964 Proximal tubule bicarbonate reclamation.
Author Contributions
Conceptualization, D.A.I.; methodology, D.A.I. and S.I.; software, D.A.I.; validation, D.A.I. and S.I.; formal analysis, D.A.I.; investigation, S.I.; resources, D.A.I.; data curation, D.A.I.; writing—original draft preparation, D.A.I.; writing—review and editing, E.A.O.; visualization, D.A.I. and E.A.O.; supervision, D.A.I.; project administration, D.A.I.; funding acquisition, D.A.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The experimental dataset was retrieved from an investigation conducted according to the guidelines of the Declaration of Helsinki. At the time of the experiment, the study was part of D.A. Iacobas’ project approved by the Institutional Review Boards (IRB) of the New York Medical College’s (NYMC) and Westchester Medical Center (WMC) Committees for Protection of Human Subjects. The approved IRB (L11,376 from 2 October 2015) granted access to frozen cancer specimens from the WMC Pathology Archives and depersonalized pathology reports, waiving the patient’s informed consent.
Informed Consent Statement
The approved IRB granted access to frozen cancer specimens from the WMC Pathology Archives and depersonalized pathology reports, waiving the patient’s informed consent.
Data Availability Statement
Transcriptomic data are publicly available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE72304.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
A 3-gene cluster can be approximated with the superposition of three paired genes:
A 4-gene cluster can be approximated with four 3-gene clusters
that can be further approximated with six squared distributions of paired genes:
and so on, giving the recurrence relation:
From the normalization condition results that:
Therefore, one can neglect the contributions of clusters with more than 2 genes:
References
- Cancer Treatment options at Houston Methodist organization. Available on line at: https://www.houstonmethodist.org/cancer/treatment-options/. (accessed 05/03/2023).
- Pecoraro, A.; Campi, R.; Marchioni, M.; European Association of Urology Young Academic Urologists Renal Cancer Working Group. Techniques and outcomes of percutaneous tumour ablation for small renal masses. Curr Opin Urol. 2023; 33(5):360-366. [CrossRef]
- Lanza, C.; Carriero, S.; Ascenti, V.; Tintori, J.; Ricapito, F.; Lavorato, R.; Biondetti, P.; Angileri, S.A.; Piacentino, F.; Fontana F. et al. Percutaneous Application of High Power Microwave Ablation With 150 W for the Treatment of Tumors in Lung, Liver, and Kidney: A Preliminary Experience. Technol Cancer Res Treat. 2023 22:15330338231185277. [CrossRef]
- Key Statistics about Kidney Cancer. Available online: https://www.cancer.org/cancer/kidney-cancer.html ((accessed on Sept. 6th, 2023).
- Mieville, V.; Griffioen, A.W.; Benamran, D.; Nowak-Sliwinska, P. Advanced in vitro models for renal cell carcinoma therapy design. Biochim Biophys Acta Rev Cancer. 2023; 1878(5):188942. [CrossRef]
- Rotin, L.E.; Viswabandya, A.; Kumar, R.; Patriquin, C.J.; Kuo, K.H.M. A systematic review comparing allogeneic hematopoietic stem cell transplant to gene therapy in sickle cell disease. Hematology. 2023; 28(1):2163357. [CrossRef]
- Gong, Y.; Pang, H.; Yu, Z.; Wang, X.; Li, P.; Zhang, Q. Construction of inflammatory associated risk gene prognostic model of NSCLC and its correlation with chemotherapy sensitivity. Ann Med. 2023; 55(1):2200034. [CrossRef]
- Hara Y, Shiba N, Yoshida K, Yamato G, Kaburagi T, Shiraishi Y, Ohki K, Shiozawa Y, Kawamura M, Kawasaki H. et al. TP53 and RB1 alterations characterize poor prognostic subgroups in pediatric acute myeloid leukemia. Genes Chromosomes Cancer. 2023; 62(7):412-422. [CrossRef]
- Cancel-Tassin, G.; Koutros, S. Use of genomic markers to improve epidemiologic and clinical research in urology. Curr Opin Urol. 2023. [CrossRef]
- Wu, F.; Ning, H.; Sun, Y.; Wu, H.; Lyu, J. Integrative exploration of the mutual gene signatures and immune microenvironment between benign prostate hyperplasia and castration-resistant prostate cancer. Aging Male. 2023; 26(1):2183947. [CrossRef]
- Yerukala Sathipati, S.; Tsai, M.J.; Shukla, S.K.; Ho, S.Y. Artificial intelligence-driven pan-cancer analysis reveals miRNA signatures for cancer stage prediction. HGG Adv. 2023; 4(3):100190. [CrossRef]
- Yang, L.; Yang, M.; Cui, C.; Long, X.; Li, Y.; Dai, W.; Lang, T.; Zhou, Q. The myo-inositol biosynthesis rate-limiting enzyme ISYNA1 suppresses the stemness of ovarian cancer via Notch1 pathway. Cell Signal. 2023; 107:110688. [CrossRef]
- Huang, L.; Shao, J.; Xu, X.; Hong, W.; Yu, W.; Zheng, S.; Ge, X. WTAP regulates autophagy in colon cancer cells by inhibiting FLNA through N6-methyladenosine. Cell Adh Migr. 2023; 17(1):1-13. [CrossRef]
- Yang, R.Y.; Tan, J.Y.; Liu, Z.; Shen, X.L.; Hu, Y.J. Lappaol F regulates the cell cycle by activating CDKN1C/p57 in human colorectal cancer cells. Pharm Biol. 2023; 61(1):337-344. [CrossRef]
- Yavuz, M.; Takanlou, L.S.; Avcı, Ç.B.; Demircan, T. A selective androgen receptor modulator, S4, displays robust anti-cancer activity on hepatocellular cancer cells by negatively regulating PI3K/AKT/mTOR signalling pathway. Gene. 2023; 869:147390. [CrossRef]
- Ishiguro, M.; Fukushige, T.; Iwasaki, H. Establishment and Characterization of a TFE3-rearranged Renal Cell Carcinoma Cell Line (FU-UR-2) With the PRCC-TFE3 Fusion Transcript. Anticancer Res. 2023; 43(8):3463-3470. [CrossRef]
- NIH-National Cancer Institute Genomic Data Commons Data Portal. Available on line at: https://portal.gdc.cancer.gov/. Accessed 06/20/2023 .
- Sarka,r O.S.; Donninger, H.; Al Rayyan, N.; Chew, L.C.; Stamp, B.; Zhang, X.; Whitt, A.; Li, C.; Hall, M.; Mitchell, R.A. et al. Monocytic MDSCs exhibit superior immune suppression via adenosine and depletion of adenosine improves efficacy of immunotherapy. Sci Adv. 2023; 9(26):eadg3736. [CrossRef]
- Liu, C.L.; Huang, W.C.; Cheng, S.P.; Chen, M.J.; Lin, C.H.; Chang, S.C.; Chang, Y.C. Characterization of Mammary Tumors Arising from MMTV-PyVT Transgenic Mice. Curr Issues Mol Biol. 2023; 45(6):4518-4528. [CrossRef]
- Montico, F.; Lamas, C.A.; Rossetto, I.M.U.; Baseggio, A.M.; Cagnon, V.H.A.. Lobe-specific responses of TRAMP mice dorsolateral prostate following celecoxib and nintedanib therapy. J Mol Histol. 2023. [CrossRef]
- Ding, W.Y.; Kuzmuk, V.; Hunter, S.; Lay, A.; Hayes, B.; Beesley, M.; Rollason, R.; Hurcombe, JA.; Barrington, F.; Masson, C. et al. Adeno-associated virus gene therapy prevents progression of kidney disease in genetic models of nephrotic syndrome. Sci Transl Med. 2023; 15(708):eabc8226. [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Urban-Maldonado, M.; Spray, D.C. Sensitivity of the brain transcriptome to connexin ablation, Biochim Biofis Acta. 2005; 1711: 183-196. Review. [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Li, W.E.; Zoidl, G.; Dermietzel, R.; Spray, D.C. Genes controlling multiple functional pathways are transcriptionally regulated in connexin43 null mouse heart. Physiol Genomics 2005; 20: 211-223. [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Spray, D.C. Connexin-dependent transcellular transcriptomic networks in mouse brain. Prog Biophys Mol Biol. 2007; 94(1-2):168-184. Review. [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Urban-Maldonado, M.; Scemes, E.; Spray, D.C Similar transcriptomic alterations in Cx43 knock-down and knock-out astrocytes. Cell Commun. Adhes. 2008; 15:1, 195-206. [CrossRef]
- Iacobas, S.; Iacobas, D.A.; Spray, D.C.; Scemes, E. The connexin43 transcriptome during brain development: importance of genetic background. Brain Research. 2012; 1487: 131-139. [CrossRef]
- Tolkach, Y.; Kristiansen, G. The Heterogeneity of Prostate Cancer: A Practical Approach. Pathobiology 2018, 85, 108–116. [CrossRef]
- Tu, S.-M.; Zhang, M.; Wood, C.G.; Pisters, L.L. Stem Cell Theory of Cancer: Origin of Tumor Heterogeneity and Plasticity. Cancers, 2021, 13, 4006. [CrossRef]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstråhle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.;
- Larsson, L.; et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. [CrossRef]
- Brady, L.; Kriner, M.; Coleman, I.; Morrissey, C.; Roudier, M.; True, L.D.; Gulati, R.; Plymate, S.R.; Zhou, Z.; Birditt, B.; et al. Inter and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat. Commun. 2021, 12, 1426. [CrossRef]
- Iacobas, S.; Iacobas, D.A. Personalized 3-Gene Panel for Prostate Cancer Target Therapy. Curr. Issues Mol. Biol. 2022, 44, 360-382. [CrossRef]
- Zhang, G.; Feng, Z.; Zeng, Q.; Huang, P. Exploring Cancer Dependency Map genes and immune subtypes in colon cancer, in which TIGD1 contributes to colon cancer progression. Aging (Albany NY). 2023; 15. [CrossRef]
- Gui, Z.; Du, J.; Wu, N.; Shen, N.; Yang, Z.; Yang, H.; Wang, X.; Zhao, N.; Zeng, Z.; Wei R. et al. Immune regulation and prognosis indicating ability of a newly constructed multi-genes containing signature in clear cell renal cell carcinoma. BMC Cancer. 2023; 23(1):649. [CrossRef]
- Qiagen Ingenuity Pathway Analysis. Available online: https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ (accessed on July 12th 2023).
- DAVID Functional Annotation Bioinformatics Microarray Analysis. Available online: https://david.ncifcrf.gov (accessed on July 12th 2023).
- Kyoto Encyclopedia of Genes and Genomes. Available online: https://www.kegg.jp/kegg/pathway.html (accessed on July 12th 2023).
- Iacobas, D.A.; Mgbemena, V.; Iacobas, S.; Menezes, K.M.; Wang, H.; Saganti, P.B. Genomic fabric remodeling in metastatic clear cell renal cell carcinoma (ccRCC): A new paradigm and proposal for a personalized gene therapy approach. Cancers 2020, 12(12), 3678; [CrossRef]
- Iacobas, D.A.; Tuli, N.; Iacobas, S.; Rasamny, J.K.; Moscatello, A.; Geliebter, J.; Tiwari, R.M. Gene master regulators of papillary and anaplastic thyroid cancer phenotypes. Oncotarget 2018, 9(2), 2410-2424. [CrossRef]
- Iacobas, S.; Iacobas, D.A. A Personalized Genomics Approach of the Prostate Cancer. Cells 2021, 10, 1644. https://www.mdpi.com/2073-4409/10/7/1644.
- Remodeling of major genomic fabrics and their interplay in metastatic clear cell renal carcinoma. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE72304.
- Iacobas, D.A.; Iacobas, S.; Spray, D.C. Connexin43 and the brain transcriptome of newborn mice. Genomics. 2007, 89(1), 113-123. [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Lee, P.R.; Cohen, J.E.; Fields, R.D. Coordinated Activity of Transcriptional Networks Responding to the Pattern of Action Potential Firing in Neurons. Genes 2019, 10, 754. [CrossRef]
- Ebbing D, Gammon SD. General Chemistry – Standalone book. Student Edition. Cengage Learning: Boston, MA, U.S.A. 2015; pp. 88-100.
- Hansen J-P.; McDonald I.R. Chapter 7 – Time-dependent Correlation and Response Functions. In Theory of Simple Liquids, 4th ed.; Academic Press: London, UK, 2013. pp. 265-310.
- Eisen, M.; Spellman, P.; Brown, P.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [CrossRef]
- Butte, A.J.; Tamayo, P.; Slonim, D.; Golub, T.R.; Kohane, I.S. Discovering functional relationships between RNA expression and chemotherapeutic susceptibility using relevance networks. Proc. Natl. Acad. Sci. USA, 2000, 97, 12182–12186. [CrossRef]
- Horvath, S.; Dong, J. Geometric Interpretation of Gene Coexpression Network Analysis. PLoS Comput. Biol. 2008, 4, e1000117. [CrossRef]
- Oldham, M.C.; Langfelder, P.; Horvath, S. Network methods for describing sample relationships in genomic datasets: Application to Huntington’s disease. BMC Syst. Biol. 2012, 6, 63. [CrossRef]
- Marbach, D.; Costello, J.C.; Küner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Aderhold, A.; Bonneau, R.; Chen, Y.; et al. Wisdom of the crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [CrossRef]
- P Value from Pearson (R) Calculator. Available on line at: https://www.socscistatistics.com/pvalues/pearsondistribution.aspx (accessed 09/01/2023).
- Mathew, R.; Iacobas, S.; Huang, J.; Iacobas, D.A. Metabolic Deregulation in Pulmonary Hypertension. Curr Issues Mol Biol. 2023; 45(6):4850-4874. [CrossRef]
- Iacobas, S.; Ede, N.; Iacobas, D.A. The Gene Master Regulators (GMR) Approach Provides Legitimate Targets for Personalized, Time-Sensitive Cancer Gene Therapy. Genes 2019, 10, 560. [CrossRef]
- Iacobas DA. Biomarkers, Master Regulators and Genomic Fabric Remodeling in a Case of Papillary Thyroid Carcinoma. Genes 2020. 11(9):E1030. [CrossRef]
- Iacobas DA. Powerful Quantifiers for Cancer Transcriptomics. World J Clinical Oncology 2020, 11(9):679-704. https://www.wjgnet.com/2218-4333/full/v11/i9/679.htm.
- Iacobas, D.A.; Xi, L. Theory and Applications of the (Cardio) Genomic Fabric Approach to Post-Ischemic and Hypoxia-Induced Heart Failure. J Pers Med. 2022; 12(8):1246. [CrossRef]
- Aldosterone-regulated sodium reabsorption. Available on line at: https://www.genome.jp/pathway/hsa04960 (accessed 09/01/2023.
- Collecting duct acid secretion. Available on line at: https://www.genome.jp/pathway/hsa04966 (accessed 09/01/2023).
- Endocrine and other factor-regulated calcium reabsorption. Available on line at: https://www.genome.jp/pathway/hsa04961 (accessed on Sept 1, 2023).
- Proximal tubule bicarbonate reclamation. Available online at: https://www.genome.jp/kegg-bin/show_pathway?hsa04964 (accessed 01/09/2023).
- Vasopressin-regulated water reabsorption. Available online at: https://www.genome.jp/kegg-bin/show_pathway?hsa04962 (accessed 09/01/2023).
- Thomas, W.; Harvey, B.J. Estrogen-induced signalling and the renal contribution to salt and water homeostasis. Steroids. 2023; 199:109299. [CrossRef]
- Li, L.; Tan, H.; Zhou, J.; Hu, F. Predicting response of immunotherapy and targeted therapy and prognosis characteristics for renal clear cell carcinoma based on m1A methylation regulators. Sci Rep. 2023; 13(1):12645. [CrossRef]
- Wang, X.X.; Myakala, K.; Libby, A.E.; Krawczyk, E.; Panov, J.; Jones, B.A.; Bhasin, K.; Shults, N.; Qi, Y.; Krausz, K.W. et al. Estrogen-Related Receptor Agonism Reverses Mitochondrial Dysfunction and Inflammation in the Aging Kidney. Am J Pathol. 2023 17:S0002-9440(23)00321-8. [CrossRef]
- Adamopoulos, P.G.; Kontos, C.K.; Diamantopoulos, M.A.; Scorilas A. Molecular cloning of novel transcripts of the adaptor-related protein complex 2 alpha 1 subunit (AP2A1) gene, using Next-Generation Sequencing. Gene. 2018, 678:55-64. [CrossRef]
- Zhang, G.; Xi, M.; Li, Y.; Wang, L.; Gao, L.; Zhang, L.; Yang, Z.; Shi, H. The ADCY9 genetic variants are associated with glioma susceptibility and patient prognosis. Genomics. 2021; 113(2):706-716. [CrossRef]
- Tang, Y.; Wang, T.; Zhang, A.; Zhu, J.; Zhou, T.; Zhou, Y.L.; Shi, J. ADCY9 functions as a novel cancer suppressor gene in lung adenocarcinoma. J Thorac Dis. 2023; 15(3):1018-1035. [CrossRef]
- Chao, X.; Jia, Y.; Feng, X.; Wang, G.; Wang, X.; Shi, H.; Zhao, F.; Jiang, C. A Case-Control Study of ADCY9 Gene Polymorphisms and the Risk of Hepatocellular Carcinoma in the Chinese Han Population. Front Oncol. 2020; 10:1450. [CrossRef]
- Li, H.; Liu, Y.; Liu, J.; Sun, Y.; Wu, J.; Xiong, Z.; Zhang, Y.; Li, B.; Jin, T. Assessment of ADCY9 polymorphisms and colorectal cancer risk in the Chinese Han population. J Gene Med. 2021; 23(2):e3298. [CrossRef]
- Chen, Q.; Cai, L.; Liang, J. Construction of prognosis model of bladder cancer based on transcriptome. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2022; 51(1):79-86. English. [CrossRef]
- Lee, Y.H.; Gyu Song, G. Genome-wide pathway analysis in pancreatic cancer. J BUON. 2015; 20(6):1565-75. PMID: 26854454.
- Guo, R.; Liu, T.; Shasaltaneh, M.D.; Wang, X.; Imani, S.; Wen, Q. Targeting Adenylate Cyclase Family: New Concept of Targeted Cancer Therapy. Front Oncol. 2022; 12:829212. [CrossRef]
- Wang, H.; Zhang, W.; Ding, Z.; Xu, T.; Zhang, X.; Xu, K. Comprehensive exploration of the expression and prognostic value of AQPs in clear cell renal cell carcinoma. Medicine (Baltimore). 2022; 101(41):e29344. [CrossRef]
- Oncomine Solutions For Next-Generation Sequencing, available online at: http://www.oncomine.org. Accessed 10/01/2024.
- The University of ALabama at Birmingham CANcer data analysis Portal, available online at: http://ualcan.path.uab.edu. Accessed 10/01/2023.
- Sinha, S.; Dwivedi, N.; Tao, S.; Jamadar, A.; Kakade, V.R.; Neil, M.O.; Weiss, R.H.; Enders, J.; Calvet, J.P.; Thomas, S.M.; et al. Targeting the vasopressin type-2 receptor for renal cell carcinoma therapy. Oncogene. 2020; 39(6):1231-1245. [CrossRef]
- Baltzer, S.; Bulatov, T.; Schmied, C.; Krämer, A.; Berger, B.T.; Oder, A.; Walker-Gray, R.; Kuschke, C.; Zühlke, K.; Eichhorst, J.; et al. Aurora Kinase A Is Involved in Controlling the Localization of Aquaporin-2 in Renal Principal Cells. Int J Mol Sci. 2022; 23(2):763. [CrossRef]
- KEGG-constructed cAMP signaling pathway. Available on line at https://www.genome.jp/pathway/hsa04024+109 (accessed 10/10/2023).
- Kim, T.H.; Park, J.M.; Kim, M.Y.; Ahn, Y.H. The role of CREB3L4 in the proliferation of prostate cancer cells. Sci Rep. 2017; 7:45300. [CrossRef]
- Giménez-Bachs, J.M.; Salinas-Sánchez, A.S.; Serrano-Oviedo, L.; Nam-Cha, S.H.; Rubio-Del, Campo, A.; Sánchez-Prieto, R. Carbonic anhydrase IX as a specific biomarker for clear cell renal cell carcinoma: comparative study of Western blot and immunohistochemistry and implications for diagnosis. Scand J Urol Nephrol. 2012; 46(5):358-64. [CrossRef]
- Tostain, J.; Li, G.; Gentil-Perret, A.; Gigante, M. Carbonic anhydrase 9 in clear cell renal cell carcinoma: a marker for diagnosis, prognosis and treatment. Eur J Cancer. 2010; 46(18):3141-8. [CrossRef]
Figure 1.
Independence of: (a) AVE, (b) REV and (c) COR with ESR1 characteristics for 37 genes involved in the KEGG-constructed pathway “Endocrine and other factor-regulated calcium absorption”. The CORESR1,ESR1 = 1 values in all conditions validates the coordination analysis. Observe the differences in all three gene characteristics between the two equally graded and located close to each-other nodules PTA and PTB.
Figure 1.
Independence of: (a) AVE, (b) REV and (c) COR with ESR1 characteristics for 37 genes involved in the KEGG-constructed pathway “Endocrine and other factor-regulated calcium absorption”. The CORESR1,ESR1 = 1 values in all conditions validates the coordination analysis. Observe the differences in all three gene characteristics between the two equally graded and located close to each-other nodules PTA and PTB.
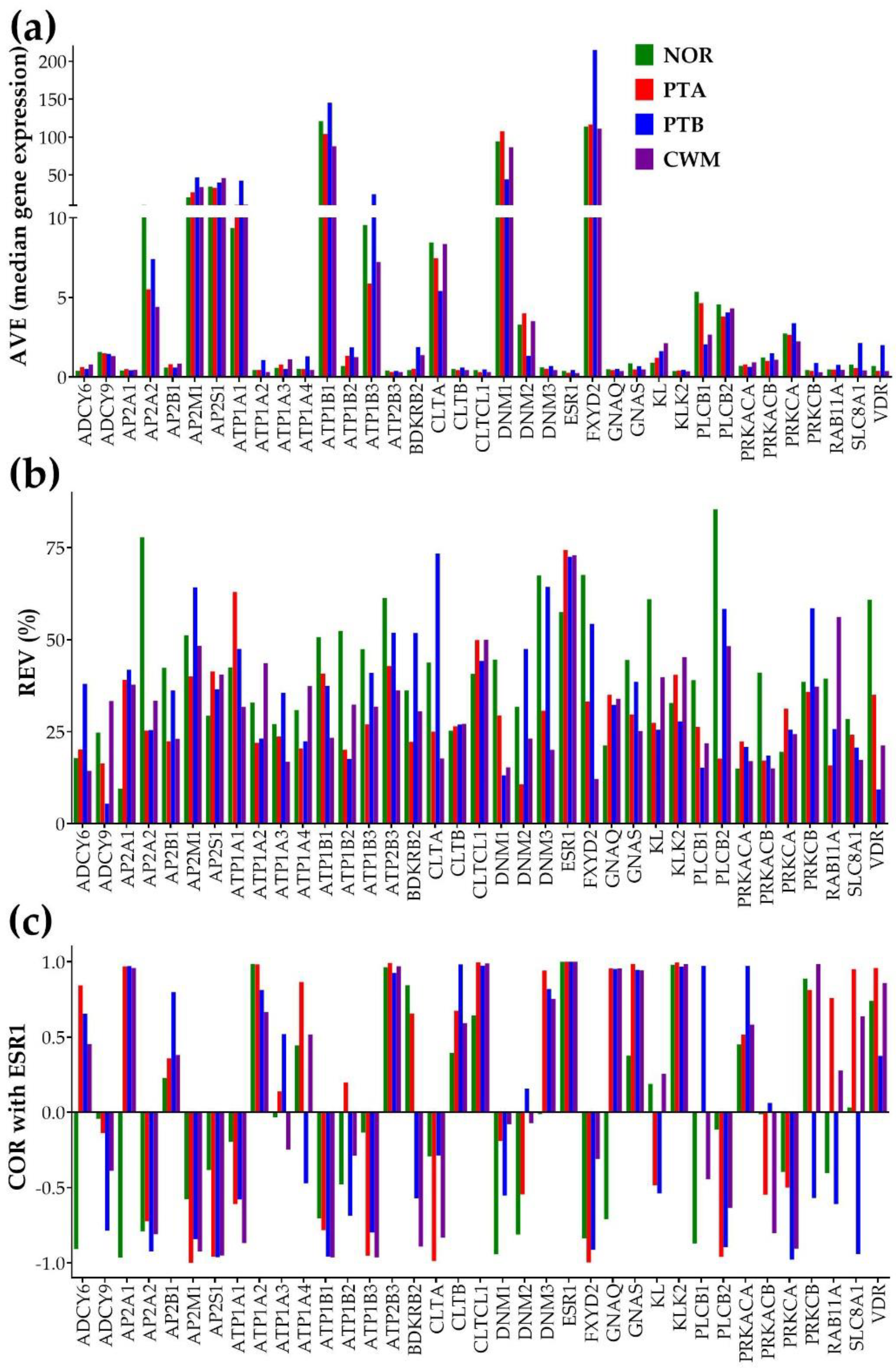
Figure 2.
Gene Commanding Height (GCH) scores of the genes involved in the five KEGG-constructed excretory pathways: (a) Aldosterone-regulated sodium reabsorption, (b) Collecting duct acid secretion, (c) Proximal tubule bicarbonate reclamation, (d) Endocrine and other factor-regulated calcium reabsorption, (e) Vasopressin-regulated water reabsorption.
Figure 2.
Gene Commanding Height (GCH) scores of the genes involved in the five KEGG-constructed excretory pathways: (a) Aldosterone-regulated sodium reabsorption, (b) Collecting duct acid secretion, (c) Proximal tubule bicarbonate reclamation, (d) Endocrine and other factor-regulated calcium reabsorption, (e) Vasopressin-regulated water reabsorption.

Figure 3.
Six measures of individual gene regulation in the KEGG-constructed pathway hsa04961 “Endocrine and other factor-regulated calcium reabsorption”. (a) Uniform (+1/-1 for significant up-/down regulation). (b) Expression ratio x (negative for down-regulation). (c) Weighted individual (gene) regulation (WIR, negative for down-regulation). (d) Regulation of transcript abundance control mechanisms (negative for decreased control). (e) Regulation of expression coordination (with respect to every other gene of the pathway, negative for reduced correlation); (f) Transcriptomic distance of individual (gene) (here with respect to all its partners within the pathway). Observe that all but the uniform measure takes into account the contributions of every single gene.
Figure 3.
Six measures of individual gene regulation in the KEGG-constructed pathway hsa04961 “Endocrine and other factor-regulated calcium reabsorption”. (a) Uniform (+1/-1 for significant up-/down regulation). (b) Expression ratio x (negative for down-regulation). (c) Weighted individual (gene) regulation (WIR, negative for down-regulation). (d) Regulation of transcript abundance control mechanisms (negative for decreased control). (e) Regulation of expression coordination (with respect to every other gene of the pathway, negative for reduced correlation); (f) Transcriptomic distance of individual (gene) (here with respect to all its partners within the pathway). Observe that all but the uniform measure takes into account the contributions of every single gene.

Figure 4.
The regulated genes from the KEGG-constructed pathway hsa04962 “Vasopressin-regulated water reabsorption” in the three cancer nodules with respect to the surrounding normal (NOR) tissue in the right kidney: (a) PTA, (b) PTB, (c) CWM. Red/green background of the gene symbol indicates significant up-/down-regulation, yellow background indicates not statistically significant regulation, while blank background indicates that that gene was not quantified. Significantly regulated genes: ADCY6 (adenylate cyclase 6), AQP3, CREB3 (cAMP responsive element binding protein 3), CREB3L2/3/4 (cAMP responsive element binding protein 3-like 2/3/4), DCTN1/2 (dynactin 1/2), DYNC2LI1 (dynein, cytoplasmic 2, light intermediate chain 1), GNAS (GNAS complex locus), and VAMP2 (vesicle-associated membrane protein 2 (synaptobrevin 2)). Note the differences among the three nodules including that AQP3 is down-regulated in PTA and CWM but up-regulated in PTB.
Figure 4.
The regulated genes from the KEGG-constructed pathway hsa04962 “Vasopressin-regulated water reabsorption” in the three cancer nodules with respect to the surrounding normal (NOR) tissue in the right kidney: (a) PTA, (b) PTB, (c) CWM. Red/green background of the gene symbol indicates significant up-/down-regulation, yellow background indicates not statistically significant regulation, while blank background indicates that that gene was not quantified. Significantly regulated genes: ADCY6 (adenylate cyclase 6), AQP3, CREB3 (cAMP responsive element binding protein 3), CREB3L2/3/4 (cAMP responsive element binding protein 3-like 2/3/4), DCTN1/2 (dynactin 1/2), DYNC2LI1 (dynein, cytoplasmic 2, light intermediate chain 1), GNAS (GNAS complex locus), and VAMP2 (vesicle-associated membrane protein 2 (synaptobrevin 2)). Note the differences among the three nodules including that AQP3 is down-regulated in PTA and CWM but up-regulated in PTB.
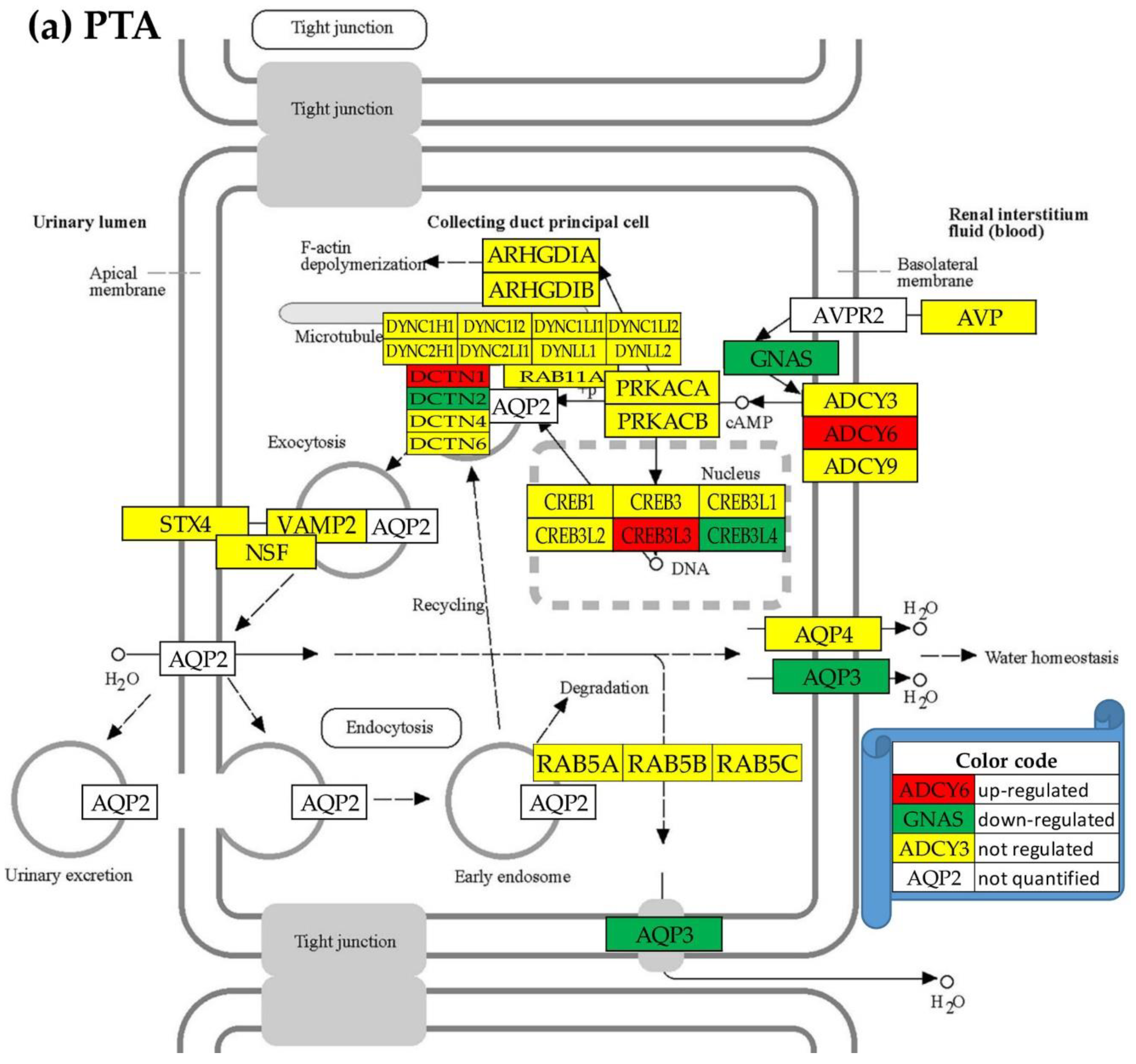

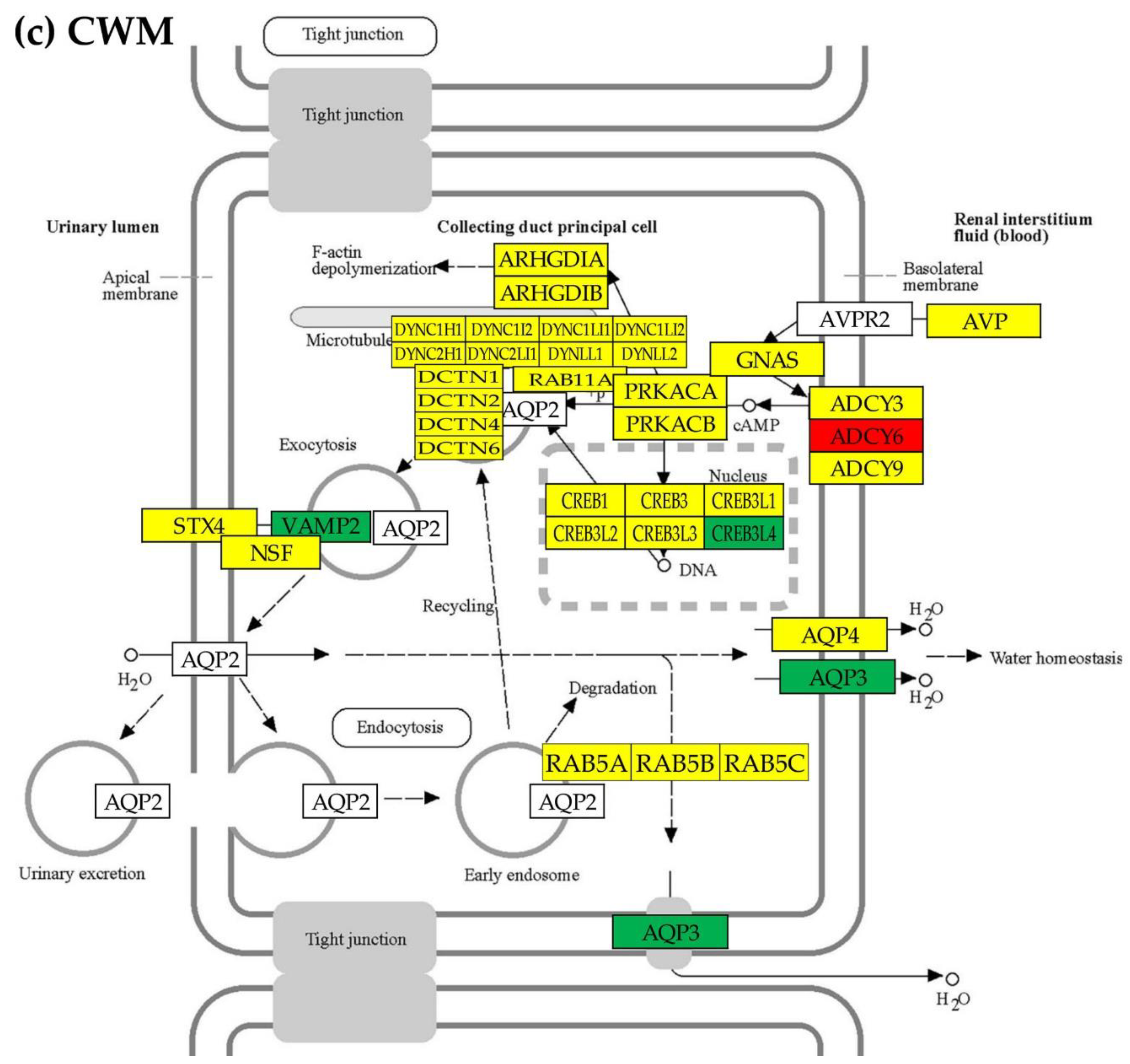
Figure 5.
(p < 0.05) significant synergism, antagonism and independence among the genes responsible for the Endocrine and other factor-regulated calcium reabsorption [56]. A red/blue/yellow square indicates significant synergism/antagonism/independence of the genes labeling the intersecting raw and column, while a blank square means lack of statistical significance of the expression correlation.
Figure 5.
(p < 0.05) significant synergism, antagonism and independence among the genes responsible for the Endocrine and other factor-regulated calcium reabsorption [56]. A red/blue/yellow square indicates significant synergism/antagonism/independence of the genes labeling the intersecting raw and column, while a blank square means lack of statistical significance of the expression correlation.
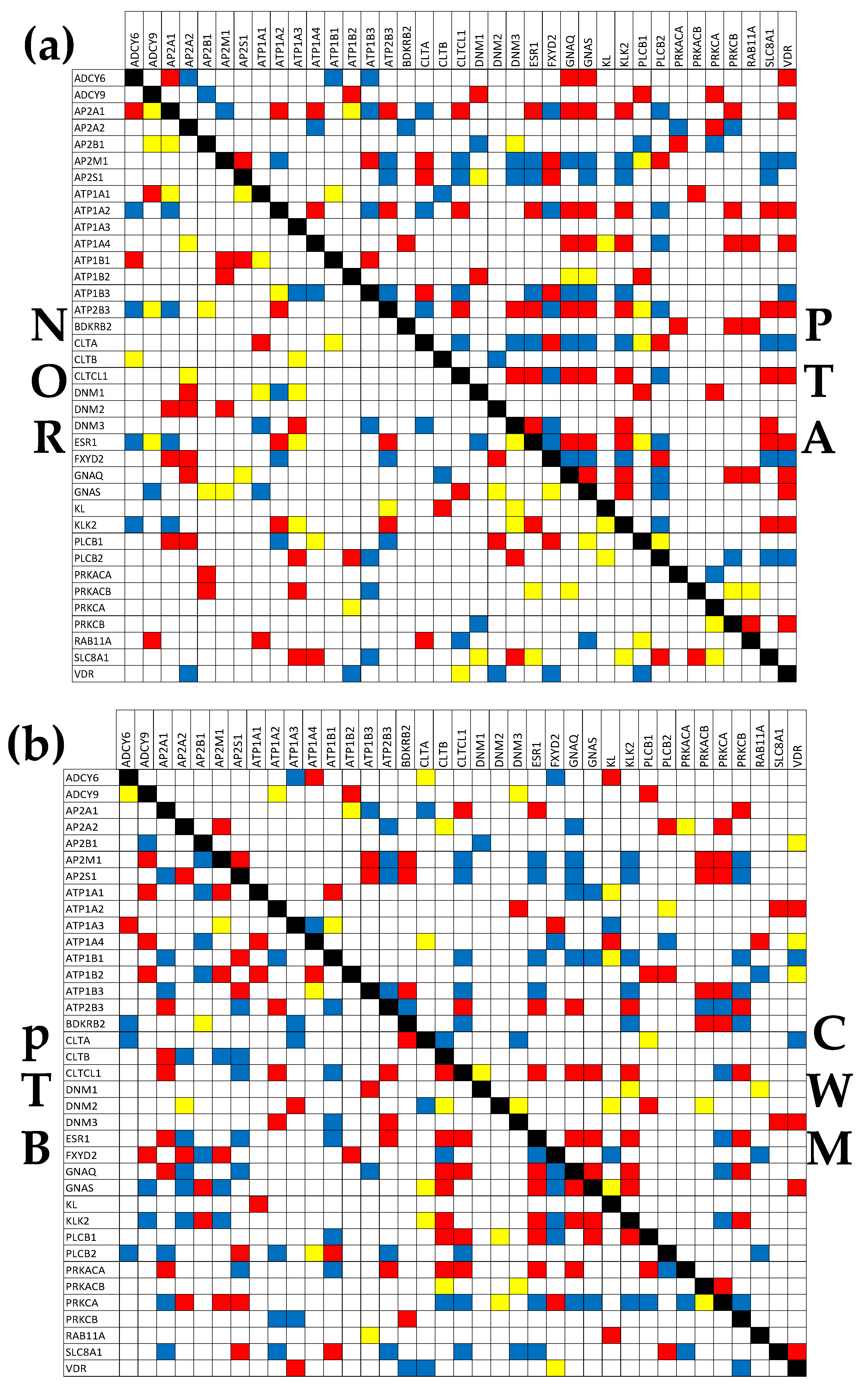
Figure 6.
Statistically significant synergism, antagonism and independence of excretory genes with arginine vasopressin (AVP) in all four profiled regions. A continuous red/blue line indicates synergism/antagonism, while a dashed black line indicates significant independence. Letters A, C, E, P, V indicate the pathways affiliations of the genes: A = Aldosterone-regulated sodium reabsorption [54], C = Collecting duct acid secretion [55], E = Endocrine and other factor-regulated calcium reabsorption [56], P = Proximal tubule bicarbonate reclamation [57], and V = Vasopressin-regulated water reabsorption [58]. Note: only the genes with significant correlations in at least one region were included in the figure.
Figure 6.
Statistically significant synergism, antagonism and independence of excretory genes with arginine vasopressin (AVP) in all four profiled regions. A continuous red/blue line indicates synergism/antagonism, while a dashed black line indicates significant independence. Letters A, C, E, P, V indicate the pathways affiliations of the genes: A = Aldosterone-regulated sodium reabsorption [54], C = Collecting duct acid secretion [55], E = Endocrine and other factor-regulated calcium reabsorption [56], P = Proximal tubule bicarbonate reclamation [57], and V = Vasopressin-regulated water reabsorption [58]. Note: only the genes with significant correlations in at least one region were included in the figure.


Table 1.
Numbers of mutated genes in 14 primary sites (data from [17]). Note that the number of mutated genes in the listed individual sites represents from 86.94% (prostate) to 95.07% (bone marrow) out of the 22,588 mutated genes reported in all 88,991 cancer cases located in all 68 primary sites.
Table 1.
Numbers of mutated genes in 14 primary sites (data from [17]). Note that the number of mutated genes in the listed individual sites represents from 86.94% (prostate) to 95.07% (bone marrow) out of the 22,588 mutated genes reported in all 88,991 cancer cases located in all 68 primary sites.
| Primary site | # of cases | # of genes | Protein coding | # of mutations | Primary site | # of cases | # of genes | Protein coding | # of mutations | |
|---|---|---|---|---|---|---|---|---|---|---|
| Bladder | 1,725 | 20,183 | 19,692 | 114,662 | Lung | 12,262 | 21,318 | 19,790 | 443,974 | |
| Bone marrow | 11,027 | 21,474 | 19,705 | 163,756 | Ovary | 3,381 | 20,266 | 19,673 | 64,142 | |
| Brain | 1,452 | 20,343 | 19,729 | 93,128 | Pancreas | 2,776 | 19,874 | 19,502 | 36,676 | |
| Breast | 9,121 | 20,454 | 19,727 | 113,777 | Prostate | 2,387 | 19,638 | 19,402 | 27,468 | |
| Colorectal | 8,140 | 21,060 | 19,794 | 337,634 | Skin | 2,893 | 20,739 | 19,770 | 353,213 | |
| Head & neck | 2,792 | 20,535 | 19,712 | 116,274 | Stomach | 1,631 | 20,336 | 19,739 | 182,493 | |
| Kidney | 3,501 | 20,129 | 19,631 | 65,471 | Uterus | 2,803 | 21,471 | 19,781 | 769,622 |
Table 2.
Overall regulation of the genes from the KEGG-constructed Excretory System pathways: hsa04960 (Aldosterone-regulated sodium reabsorption), hsa04966 (Collecting duct acid secretion), hsa04961 (Endocrine and other factor-regulated calcium reabsorption), hsa04964 (Proximal tubule bicarbonate reclamation), and hsa04962 (Vasopressin-regulated water reabsorption) in the three cancer nodules. Note the differences between the equally ranking nodules PTA and PTB. .
Table 2.
Overall regulation of the genes from the KEGG-constructed Excretory System pathways: hsa04960 (Aldosterone-regulated sodium reabsorption), hsa04966 (Collecting duct acid secretion), hsa04961 (Endocrine and other factor-regulated calcium reabsorption), hsa04964 (Proximal tubule bicarbonate reclamation), and hsa04962 (Vasopressin-regulated water reabsorption) in the three cancer nodules. Note the differences between the equally ranking nodules PTA and PTB. .
| KEGG pathway | PTA | PTB | CWM | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Code | Genes | % up | % down | WPR | % up | % down | WPR | % up | % down | WPR |
| hsa04960 | 26 | 11.54 | 11.54 | 1.12 | 30.77 | 0.00 | 8.19 | 11.54 | 11.54 | 2.32 |
| hsa04966 | 16 | 6.25 | 12.50 | 5.20 | 12.50 | 0.00 | 16.36 | 12.50 | 0.00 | 9.69 |
| hsa04961 | 37 | 5.41 | 2.70 | 0.88 | 18.92 | 5.41 | 7.68 | 8.11 | 5.41 | 2.15 |
| hsa04964 | 18 | 16.67 | 0.00 | 0.96 | 38.89 | 0.00 | 9.04 | 11.11 | 0.00 | 2.12 |
| hsa04962 | 36 | 8.33 | 11.11 | 0.69 | 11.11 | 5.56 | 0.93 | 2.78 | 8.33 | 0.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



