
Submitted:
19 October 2023
Posted:
20 October 2023
You are already at the latest version
Abstract
Stable half-sandwich iron(II) dihalido complexes of the type [Fe(5-Cp’)X2] are extremely scarce, being limited to congeners containing the bulky C5H2-1,2,4-tBu3 ligand. We have extended this to homologues [Fe(5-Cp*)X2] (X = Cl – I) containing the particularly popular C5Me5 (Cp*) ligand. Corresponding ionic compounds ER4[Fe(5-Cp*)X2] are easily accessible from FeX2, MCp* (M = Li, K) and a suitable halide source R4EX (E = N, P) in THF. The new compounds NnPr4[Fe(5-Cp*)X2] (X = Cl, Br), NnPr4[Fe(5-Cp*)BrCl] and PPh4[Fe(5-Cp*)X2] (X = Cl, Br, I) have been structurally characterised by single-crystal X-ray diffraction. NnPr4[Fe(5-Cp*)Cl2] reacts readily with CO to afford [Fe(5-Cp*)Cl(CO)2], indicating the synthetic potential of ER4[Fe(5-Cp*)X2] in FeCp* half-sandwich chemistry.
Keywords:
crystal structures
; cyclopentadienyl complexes
; half-sandwich complexes
; halides
; iron
1. Introduction
Half-sandwich iron(II) complexes of the type [Fe(η5-Cp’)X] (Cp’ = C5H5 or substituted cyclopentadienyl, X = Cl – I) are useful as highly reactive cyclopentadienyliron(II) transfer reagents, which, due to their thermal lability, are usually generated in situ at low temperatures for immediate use [1]. Seminal work was published already in 1985 by Kölle, who described the generation of [Fe(η5-Cp*)Br] (Cp* = C5Me5) from LiCp* and [FeBr2(DME)] in THF at −80 °C [2]. The corresponding chlorido complex [Fe(η5-Cp*)Cl] is particularly popular as a Cp*Fe+ source [3,4,5,6,7,8,9,10]. In contrast to the thermal lability of this compound, its TMEDA chelate [Fe(η5-Cp*)Cl(TMEDA)] is perfectly stable at room temperature [4] and the same holds true for the closely related TMEDA complexes [Fe(η5-C5Me4Et)Cl(TMEDA)] [11] and [Fe(η5-Cp*)Br(TMEDA)] [12]. Similar to these TMEDA containing N,N-chelates, C,N-chelates [Fe(η5-Cp*)X(NHCN)] (X = Cl, I) containing N-heterocyclic carbenes functionalised with an N-donor moiety (NHCN) have also been described [14,15,16]; unchelated analogues [Fe(η5-Cp*)Cl(NHC)] proved sufficiently stable for isolation with the standard NHC IMes and the bulkier 1,3-diisopropyl-4,5-dimethylimidazolin-ylidene [17,18,19]. Stabilisation by external donors is not necessary for isolation when extremely bulky Cp’ ligands [20,21,22] are applied, leading to “self-stabilised” halido-bridged dimers [{Fe(η5-Cp’)(μ-X)}2] according to single-crystal X-ray diffraction (XRD) (Cp’ = C5iPr5, X = Br; Cp’ = C5HiPr4, X = Br, I; Cp’ = C5H2-1,2,4-tBu3, X = Br, I; Cp’ = C5(p-C6H4Et)5, X = Br) [23,24,25,26]. Manners and Walter independently found that [{Fe(η5-C5H2-1,2,4-tBu3)(μ-I)}2] undergoes heterolytic cleavage in toluene, affording [Fe(η5-C5H2-1,2,4-tBu3)(C7H8)]+ and [Fe(η5-C5H2-1,2,4-tBu3)I2]− [27,28]. In the same vein, deaggregation of [{Fe(η5-C5H2-1,2,4-tBu3)(μ-I)}2] was achieved by reaction with NR4I (R = Et, nBu), giving rise to the formation of NR4[Fe(η5-C5H2-1,2,4-tBu3)I2] [27,28]. The only other closely related compound is [Fe(η5-C5H2-1,2,4-tBu3)(μ-Br)2Na(DME)2], which Sitzmann had obtained by serendipity and in trace amounts only in the preparation of [{Fe(η5-C5H2-1,2,4-tBu3)(μ-Br)}2] from [FeBr2(DME)] and the corresponding sodium cyclopentadienide in DME [29]. This dinuclear complex might be viewed as contact ion pair [Na(DME)2][Fe(η5-C5H2-1,2,4-tBu3)Br2], thus exhibiting, cum grano salis, the [Fe(η5-C5H2-1,2,4-tBu3)Br2]− anion. In view of the mature state of half-sandwich iron(II) chemistry [1], the paucity of compounds containing simple anions of the type [Fe(η5-Cp’)X2]− is quite surprising. Together with the enormous popularity of the Cp* ligand [22], this prompted us to address the synthesis of compounds containing [Fe(η5-Cp*)X2]− (X = Cl – I).
2. Results and Discussion
The synthesis of our target compounds (Scheme 1) was inspired by the work of Manners and Walter mentioned above.
The addition of NnPr4Cl (1 equiv.) to [Fe(η5-Cp*)Cl], generated in situ from LiCp* and FeCl2 in THF at low temperatures, afforded a green solution. LiCl was precipitated by addition of toluene and subsequently removed by filtration. Storing of the filtrate at −40 °C afforded NnPr4[Fe(η5-Cp*)Cl2] as green crystals in 60% yield. The use of NnPr4Br instead of NnPr4Cl furnished NnPr4[Fe(η5-Cp*)BrCl] in 39% yield. Both compounds were structurally characterised by XRD. Their molecular structures are shown in Figure 1 and Figure 2 and pertinent metric parameters are collected in Table 1. Not surprisingly, the [Fe(η5-Cp*)BrCl]− anion exhibits a disorder of the halogen atoms.
Our attempts to prepare NnPr4[Fe(η5-Cp*)Br2] in an analogous way from Kölle’s compound [Fe(η5-Cp*)Br] and NnPr4Br furnished the product in 33% yield, but invariably afforded crystals whose structural investigation by XRD was fraught with problems due to severe cation disorder. Our best result is shown in Figure S1 in the Supporting Information. Although bond lengths and angles are given only for the heavy atoms in Table 1, these data should be treated with particular caution in the case of NnPr4[Fe(η5-Cp*)Br2], where they are not taken into consideration for our discussion. The problems encountered with the tetra-n-propylammonium cation prompted us to use the tetraphenylphosphonium cation instead. The preparation of PPh4[Fe(η5-Cp*)X2] (X = Cl, Br, I) by addition of PPh4X (1 equiv.) to [Fe(η5-Cp*)X] (prepared in situ from FeX2 and KCp*) turned out to be straightforward, although the isolated yields were unsatisfactorily poor (21% at most), probably due to the much lower solubility of PPh4X in comparison to NnPr4X. In contrast to the synthesis of NnPr4[Fe(η5-Cp*)X2], a trend towards even lower yields was observed when LiCp* was used instead of KCp*. The product was obtained as crystals suitable for XRD in each case and no disorder problems were encountered, as anticipated. The molecular structures of PPh4[Fe(η5-Cp*)X2] are shown in Figure 3 (X = Cl), Figure 4 (X = Br), and Figure 5 (X = I).
The compounds listed in Table 1 exhibit very similar iron–cyclopentadienyl ring centroid distances between 1.96 and 1.99 Å, which is much larger than the corresponding distances in the ferrocenes [Fe(η5-Cp*)2] (1.65 Å) [30] and [Fe(η5-C5H2-1,2,4-tBu3)2] (1.72 Å) [31] and marginally larger than those in the open-shell half-sandwich complexes [Fe(η5-Cp*){N(SiMe3)2}] (1.90 Å) [32], [Fe(η5-C5iPr5){N(SiMe3)2}] (1.92 Å) [13] and [{Fe(η5-C5H2-1,2,4-tBu3)(μ-X)}2] (1.92 and 1.93 Å for X = Br and I, respectively) [24,25]. The differences in the Fe–X bond lengths observed for X = Cl, Br and I are in accord with the different radii of the halogen atoms. A particularly good agreement is achieved with Pauling’s tetrahedral covalent radii, which reflect a convolution of covalent and dative bonding, the values being 0.99, 1.11 and 1.28 Å for Cl, Br and I, respectively [33]. Not surprisingly, the X–Fe–X angles of the Cp* complexes are wider (by ca. 5 °) than those of the congeners containing the bulkier C5H2-1,2,4-tBu3 ligand, whose comparatively less symmetric nature may be the reason for the significant difference of the two Fe–I bond lengths (Δd 0.09 Å) in the anion of NnBu4[Fe(η5-C5H2-1,2,4-tBu3)I2]. The tetraalkylammonium cations are engaged in CH···X contacts compatible with weak hydrogen bonds (indicated as dotted lines in Figure 1, not shown for the disordered species in Figure 2 and Figure 3).[34,35] The contacts of the two halogen atoms are almost equidistant in each case (CH···Cl 2.67 and 2.73 Å for NnPr4[Fe(η5-Cp*)Cl2], CH···I 3.10 and 3.14 Å for NnBu4[Fe(η5-C5H2-1,2,4-tBu3)I2]). The PPh4+ cations interact with the [Fe(η5-Cp*)X2]− anions through phenyl CH···X contacts (2.76–2.95, 2.96 and 3.09–3.15 Å for X = Cl, Br and I, respectively; not shown in Figure 3, Figure 4 and Figure 5). In addition, the para-H atom of a phenyl ring points towards the centre of the Cp* ligand (phenyl CH···C 2.53–2.74 Å, phenyl CH···Cp* ring centroid 2.28–2.38 Å shown as dotted lines in Figure 3, Figure 4 and Figure 5), indicating a CH···π interaction [36,37] similar to that in the T-shaped benzene dimer [38,39,40,41,42,43], for which a CH···C6H6 ring centroid distance of 2.25 Å was computed recently [44].
The electronic structure of the anion of NnBu4[Fe(η5-C5H2-1,2,4-tBu3)I2] has been scrutinised by SQUID magnetometry, EPR spectroscopy and ab initio Complete Active Space Self Consistent Field-Spin Orbit calculations, which revealed a high-spin d6 iron(II) centre with a strongly anisotropic S = 2 ground state [28]. This in-depth study by Manners makes an analogous investigation of our closely related compounds dispensable. The paramagnetic nature of their [Fe(η5-Cp*)X2]− anions is clearly evident from the NMR spectra. The Cp* ligand gives rise to a 1H NMR signal at δ ≈ 200 ppm. This may be compared with the data reported for the substituted cyclopentadienyl ligands of [{Fe(η5-C5iPr5)(μ-Br)}2] in C6D6 [δ(1H) = 95.7 (CHMe2), 11.3 (CHMe2), and −117.3 ppm (CHMe2)] [23] and of NnBu4[Fe(η5-C5H2-1,2,4-tBu3)I2] in THF-d8 [δ(1H) = −20.3 and −31.4 ppm (2 × tBu)] [28].
Kölle demonstrated the successful generation of the highly reactive compound [Fe(η5-Cp*)Br] by trapping reaction with carbon monoxide at −80 °C, which furnished the diamagnetic carbonyl complex [Fe(η5-Cp*)Br(CO)2] in 59% yield [2]. In the same vein, Walter obtained [Fe(η5-C5H2-1,2,4-tBu3)I(CO)2] by carbonylation of the “self-stabilised” halido-bridged dimer [{Fe(η5-C5H2-1,2,4-tBu3)(μ-I)}2] with CO at room temperature in 80% yield [25]. We have studied the carbonylation of our target compounds exemplarily with NnPr4[Fe(η5-Cp*)Cl2] and observed an essentially quantitative reaction with CO under the same mild conditions, affording the well-known carbonyl complex [Fe(η5-Cp*)Cl(CO)2] [45,46]. The crystal structure of this compound reported in 1988 had been determined at room temperature [46], which prompted us to redetermine the structure at 100 K (see the Supporting Information).
3. Materials and Methods
Experimental Details. All reactions were performed in an inert atmosphere (argon or dinitrogen) by using standard Schlenk techniques or a conventional glovebox. Solvents were dried with a commercial Solvent Purification System (M. Braun, MB SPS 7), degassed and stored over 3 Å molecular sieves under inert atmosphere. Starting materials were procured from standard commercial sources and used as received. LiCp* and KCp* were synthesised by deprotonation of pentamethylcyclopentadiene in n-hexane with n-butyllithium and potassium metal, respectively, and isolated by filtration or centrifugation. NMR spectra were recorded with a Varian MR-400 and Varian NMRS-500 spectrometers operating at 400 and 500 MHz, respectively, for 1H. Elemental analyses were carried out with a HEKAtech Euro EA-CHNS elemental analyser at the Institute of Chemistry, University of Kassel, Germany.
NnPr4[Fe(η5-Cp*)Cl2]: A Schlenk tube charged with LiCp* (176 mg, 1.24 mmol) and FeCl2 (156 mg, 1.23 mmol) was cooled to −60 °C. THF (3 mL) cooled to the same temperature was added. The stirred mixture was allowed to warm up to −20 °C. NnPr4Cl (275 mg, 1.24 mmol) was added. The stirred mixture was allowed to warm up to ambient temperature and was subsequently filtered through a Celite pad. Toluene (ca. 3 mL) was slowly added to the green filtrate until formation of an essentially colourless precipitate was observed. Insoluble material was removed by filtration through a Celite pad. Storing of the filtrate at −40 °C afforded the product as green crystals, which were separated from the yellow mother liquor, washed with n-hexane (5 mL) and dried under vacuum. Yield 327 mg (60%). Elemental Analysis for C22H43NCl2Fe (448.34 g/mol): Calculated (%): C 58.94, H 9.67, N 3.12. Found (%): 58.18, H 9.37, N 3.21. 1H NMR (400 MHz, THF-d8): δ 194.4 (15H, s, ν½ = 379 Hz, Cp*), 16.8 (8H, s, ν½ = 270 Hz, (CH2)2CH3), 9.9 (8H, s, ν½ = 217 Hz, (CH2)2CH3), 1.3 (12H, s, ν½ = 267 Hz, (CH2)2CH3).
NnPr4[Fe(η5-Cp*)BrCl]: This compound was obtained by a procedure analogous to that described above for NnPr4[Fe(η5-Cp*)Cl2] by using LiCp* (130 mg, 0.91 mmol), FeCl2 (116 mg, 0.92 mmol) and NnPr4Br (245 mg, 0.92 mmol) in THF (3 mL). Yield 175 mg (39%). Elemental Analysis for C22H43NBrClFe (492.79 g/mol): Calculated (%): C 53.62, H 8.80, N 2.84. Found (%): C 54.24, H 8.81, N 2.48. 1H NMR (400 MHz, THF-d8): δ 206.5 (15H, s, ν½ = 2860 Hz, Cp*), 25.8 (8H, s, ν½ = 501 Hz, (CH2)2CH3), 15.1 (8H, s, ν½ = 353 Hz, (CH2)2CH3), 2.7 (12H, s, ν½ = 649 Hz, (CH2)2CH3).
NnPr4[Fe(η5-Cp*)Br2]: This compound was obtained by a procedure analogous to that described above for NnPr4[Fe(η5-Cp*)Cl2] by using LiCp* (65 mg, 0.46 mmol), FeBr2 (99 mg, 0.46 mmol) and NnPr4Br (122 mg, 0.46 mmol) in THF (1.5 mL). Yield 81 mg (33%). An analytical sample was obtained by recrystallization from benzene. Elemental Analysis for C22H43NBr2Fe·½C6H6 (576.29 g/mol): Calculated (%): C 52.10, H 8.05, N 2.43. Found (%): C 52.18, H 8.24, N 1.76. 1H NMR (400 MHz, THF-d8): δ 203.5 (15H, s, ν½ = 561 Hz, Cp*), 16.7 (8H, s, ν½ = 312 Hz, (CH2)2CH3), 10.7 (8H, s, ν½ = 241 Hz, (CH2)2CH3), 2.23 (12H, s, ν½ = 194 Hz, (CH2)2CH3).
PPh4[Fe(η5-Cp*)Cl2]: A Schlenk tube charged with KCp* (40 mg, 0.23 mmol) and FeCl2 (29 mg, 0.23 mmol) was cooled to −60 °C. THF (0.5 mL) cooled to the same temperature was added. The stirred mixture was allowed to warm up to −20 °C. PPh4Cl (86 mg, 0.23 mmol) was added. The stirred mixture was allowed to warm up to ambient temperature and was subsequently filtered through a Celite pad. The yellow filtrate was carefully layered with n-hexane, resulting in the slow formation of yellow crystals, which were separated from the mother liquor, washed with n-hexane (2 mL) and dried under vacuum. Yield 8 mg (6%). In view of the unsatisfactorily low yield, elemental analysis was not performed for this compound. 1H NMR (500 MHz, THF-d8): δ 188.2 (15H, s, ν½ = 311 Hz, Cp*), 13.2 (8H, ν½ = 95 Hz, Ph), 10.7 (8H, ν½ = 98 Hz, Ph), 10.2 (4H, ν½ = 80 Hz, Ph).
PPh4[Fe(η5-Cp*)Br2]: This compound was obtained by a procedure analogous to that described above for PPh4[Fe(η5-Cp*)Cl2] by using KCp* (40 mg, 0.23 mmol), FeBr2 (50 mg, 0.23 mmol) and PPh4Br (96 mg, 0.23 mmol) in THF (0.5 mL). Yield 12 mg (8%). In view of the unsatisfactorily low yield, elemental analysis was not performed for this compound. 1H NMR (500 MHz, THF-d8): δ 193.7 (15H, s, ν½ = 647 Hz, Cp*), 11.3 (8H, ν½ = 162 Hz, Ph), 9.2 (8H, ν½ = 194 Hz, Ph), 8.6 (4H, ν½ = 188 Hz, Ph).
PPh4[Fe(η5-Cp*)I2]: This compound was obtained by a procedure analogous to that described above for PPh4[Fe(η5-Cp*)Cl2] by using KCp* (40 mg, 0.23 mmol), FeI2 (71 mg, 0.23 mmol) and PPh4I (107 mg, 0.23 mmol) in THF (0.5 mL). Yield 38 mg (21%). In view of the unsatisfactorily low yield, elemental analysis was not performed for this compound. 1H NMR (500 MHz, THF-d8): δ 209.9 (15H, s, ν½ = 5.34 Hz, Cp*), 10.1 (8H, ν½ = 55 Hz, Ph), 9.1 (8H, ν½ = 60 Hz, Ph), 8.7 (4H, ν½ = 55 Hz, Ph).
[Fe(η5-Cp*)Cl(CO)2]: A solution of NnPr4[Fe(η5-Cp*)Cl2] (40 mg, 0.09 mmol) in THF (2 mL) was subjected to an atmospheric pressure of CO, which led to an immediate colour change from green to red. The solution was stirred for 10 min. Volatile components were removed under vacuum. Benzene (0.7 mL) was added to the residue. Insoluble material was removed by filtration through a Celite pad. Slow evaporation of the filtrate afforded the product as red crystals. Yield 23 mg (92%). Spectroscopic data were found to be in good agreement with published values [45,46].
X-Ray Crystallography: For all data collections a single crystal was mounted on a micro-mount and all geometric and intensity data were taken from this sample by ω-scans at 100(2) K. Data collections were carried out either on a Stoe StadiVari diffractometer equipped with a 4-circle goniometer and a DECTRIS Pilatus 200K detector (for NnPr4[Fe(η5-Cp*)Cl2], NnPr4[Fe(η5-Cp*)Br2] and PPh4[Fe(η5-Cp*)Cl2]) or on a Stoe IPDS2 diffractometer equipped with a 2-circle goniometer and an area detector (for NnPr4[Fe(η5-Cp*)BrCl], PPh4[Fe(η5-Cp*)Br2], PPh4[Fe(η5-Cp*)I2] and [Fe(η5-Cp*)Cl(CO)2]). The data sets were corrected for absorption (by multi scans), Lorentz and polarisation effects. The structures were solved by direct methods (SHELXT 2014/7) [47] and refined using alternating cycles of least-squares refinements against F2 (SHELXL2014/7) [47]. H atoms were included to the models in calculated positions with the 1.2 fold isotropic displacement parameter of their bonding partner. Experimental details for each diffraction experiment are given in Table S1 (Supplementary Materials). CCDC 2300615–2300621 contain supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre, www.ccdc.cam.uk/structures.
4. Conclusions
Thermally stable half-sandwich iron(II) dihalido complexes of the type [Fe(η5-Cp’)X2]− reported in the literature are so far limited to a small number of salts containing the anion [Fe(η5-C5H2-1,2,4-tBu3)I2]−. We have extended this to homologues [Fe(η5-Cp*)X2]− (X = Cl – I) containing the widely used Cp* ligand. Corresponding ionic compounds ER4[Fe(η5-Cp*)X2] are easily accessible from FeX2, MCp* (M = Li, K) and a suitable halide source R4EX (E = N, P). While yields of up to 60% could be achieved with ER4 = NnPr4, unsatisfactorily low yields were obtained with ER4 = PPh4, which, however, turned out to be superior to NnPr4 in terms of the quality of crystals needed for XRD. The high-yield synthesis of [Fe(η5-Cp*)Cl(CO)2] from NnPr4[Fe(η5-Cp*)Cl2] and CO under mild conditions exemplarily demonstrates that such anions are amenable to halido ligand substitution reactions and may thus provide facile access to a range of pentamethylcyclopentadienyliron half-sandwich complexes.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: X-ray crystallographic details; Figure S1: Molecular structure of NnPr4[Fe(η5-Cp*)Br2] in the crystal; Figure S2: 1H NMR spectrum of NnPr4[Fe(η5-Cp*)Cl2]; Figure S3: 1H NMR spectrum of NnPr4[Fe(η5-Cp*)BrCl]; Figure S4: 1H NMR spectrum of NnPr4[Fe(η5-Cp*)Br2]; Figure S5: 1H NMR spectrum of PPh4[Fe(η5-Cp*)Cl2]; Figure S6: 1H NMR spectrum of PPh4[Fe(η5-Cp*)Br2]; Figure S7: 1H NMR spectrum of PPh4[Fe(η5-Cp*)I2]; Figure S8: 1H NMR spectrum of [Fe(η5-Cp*)Cl(CO)2]; Figure S9: 13C NMR spectrum of [Fe(η5-Cp*)Cl(CO)2].
Author Contributions
Conceptualization, Ulrich Siemeling; Formal analysis, Clemens Bruhn; Investigation, Julian Zinke and Clemens Bruhn; Methodology, Julian Zinke; Project administration, Ulrich Siemeling; Supervision, Ulrich Siemeling; Writing—original draft, Ulrich Siemeling; Writing—review & editing, Ulrich Siemeling.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available in the supporting information.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Münster, K.; Walter, M.D. Monocyclopentadienyl and Other Half-Sandwich Complexes of Iron. In Comprehensive Organometallic Chemistry, 4th ed.; Parkin, G., Meyer, K., O’Hare, D., Eds.; Elsevier: Kidlington, UK, 2022; Volume 7, pp. 46–184. [Google Scholar] [CrossRef]
- Kölle, U.; Fuss, B.; Khouzami, F.; Gersdorf, J. Pentamethylcyclopentadienyl-Übergangsmetall-Komplexe: X. Halbsandwichkomplexe des Fe und Ni als reaktive Zwischenprodukte. J. Organomet. Chem. 1985, 290, 77–83. [Google Scholar] [CrossRef]
- Sun, R.; Deng, W.-H.; Yu, B.; Lu, Y.; Zhai, X.; Liao, R.-Z.; Tung, C.-H.; Wang, W. Hydroboration of the (C5Me5)Fe(1,2-Ph2PC6H4) System To Derive Hydridoborate and Hydridosilicate Complexes. Organometallics 2022, 41, 2504–2512. [Google Scholar] [CrossRef]
- Sun, T.; Xu, S.; Yang, D.; Su, L.; Wang, B.; Qu, J. Catalytic Disproportionation of Hydrazine Promoted by Biomimetic Diiron Complexes with Benzene-1,2-Dithiolate Bridge Modified by Different Substituents. Eur. J. Inorg. Chem. 2020, 4263–4269. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Yang, D.; Li, Y.; Sun, P.; Wang, B.; Qu, J. Synthesis and Reactivity of Thioether-Dithiolate-Bridged Multi-iron Complexes. Organometallics 2015, 34, 1661–1667. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Wang, B.; Luo, Y.; Yang, D.; Tong, P.; Zhao, J.; Luo, L.; Zhou, Y.; Chen, S.; Cheng, F.; Qu, J. Ammonia formation by a thiolate-bridged diiron amide complex as a nitrogenase mimic. Nat. Chem. 2013, 5, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, S.; Ogura, S.-i.; Yo, H.; Hosokoshi, Y.; Kamikawa, K.; Matsuzaka, H. Diiron Amido−Imido Complex [(Cp*Fe)2(μ2-NHPh)(μ2-NPh)]: Synthesis and a Net Hydrogen Atom Abstraction Reaction To Form a Bis(imido) Complex. Inorg. Chem. 2006, 45, 4871–4873. [Google Scholar] [CrossRef] [PubMed]
- Shintani,R. ; Fu, G.C. Copper-Catalyzed Enantioselective Conjugate Addition of Diethylzinc to Acyclic Enones in the Presence of Planar-Chiral Phosphaferrocene-Oxazoline Ligands. Org. Lett. 2002, 4, 3699–3702. [Google Scholar] [CrossRef]
- Hettrich, R.; Kaschke, M.; Wadepohl, H.; Weinmann, W.; Stephan, M.; Pritzkow, H.; Siebert, W.; Hyla-Kryspin, I.; Gleiter, R. Electron-poor 2,3-Dihydro-1,3-diborolyl Complexes of Iron and Ruthenium: Synthesis, Reactivity, and Crystal and Electronic Structures of an Iron Sandwich Complex. Chem. Eur. J. 1996, 2, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Stephan, M.; Müller, P.; Zenneck, U.; Pritzkow, H.; Siebert, W.; Grimes, R.N. Organotransition-Metal Metallacarboranes. 37. Paramagnetic Iron−Cobalt and Dicobalt Triple-Decker Sandwich Complexes. Inorg. Chem. 1995, 34, 2058–2067. [Google Scholar] [CrossRef]
- Jonas, K.; Klusmann, P.; Goddard, R. Pentamethylcyclopentadienylbis(ethen)eisen − ein 17e-Halbsandwichkomplex mit leicht verdrängbaren Ethenliganden, Z. Naturforsch. B 1995, 50, 394–404. [Google Scholar] [CrossRef]
- Schneider, J.J.; Spickermann, D.; Lehmann, C.W.; Magull, J.; Krüger, H.-J.; Ensling, J.; Gütlich, P. Decacyclene as Complexation Manifold: Synthesis, Structure and Properties of Its Fe2 and Fe4 Slipped Triple-Decker Complexes. Chem. Eur. J. 2006, 12, 1427–1435. [Google Scholar] [CrossRef]
- Groß, O.A.; Lauk, S.; Müller, C.; Gidt, W.; Sun, Y.; Demeshko, S.; Meyer, F.; Sitzmann, H. Iron(II) High-Spin and Low-Spin Complexes from Pentaisopropylcyclopentadienyliron(II) Bis(trimethylsilyl)amide. Eur. J. Inorg. Chem. 2017, 3635–3643. [Google Scholar] [CrossRef]
- Liang, Q.; Song, D. Syntheses and Reactivity of Piano-Stool Iron Complexes of Picolyl-Functionalized N-Heterocyclic Carbene Ligands. Organometallics 2021, 40, 3943–3951. [Google Scholar] [CrossRef]
- Takahashi, H.; Watanabe, T.; Tobita, H. Bifunctional Iron-Amino Complexes: Highly Efficient Catalysts for Dehydrogenation of Ammonia-Borane. Chem. Lett. 2018, 47, 296–299. [Google Scholar] [CrossRef]
- Liang, Q.; Osten, K.M.; Song, D. Iron-Catalyzed gem-Specific Dimerization of Terminal Alkynes. Angew. Chem. Int. Ed. 2017, 56, 6317–6320. [Google Scholar] [CrossRef]
- Ohki, Y.; Hatanaka, T.; Tatsumi, K. C−H Bond Activation of Heteroarenes Mediated by a Half-Sandwich Iron Complex of N-Heterocyclic Carbene. J. Am. Chem. Soc. 2008, 130, 17174–17186. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Suárez, A.; Nelson, D.J.; Nolan, S.P. Quantifying and understanding the steric properties of N-heterocyclic carbenes. Chem. Commun. 2017, 53, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Clavier, H.; Nolan, S.P. Percent buried volume for phosphine and N-heterocyclic carbene ligands: steric properties in organometallic chemistry. Chem. Commun. 2010, 46, 841–861. [Google Scholar] [CrossRef] [PubMed]
- Lauk, S.; Schäfer, A. Pentaisopropyl Cyclopentadienyl: An Overview across the Periodic Table. Eur. J. Inorg. Chem. 2021, 5026–5036. [Google Scholar] [CrossRef]
- Field, L.D.; Lindall, C.M.; Masters, A.F.; Clentsmith, G.K.B. Penta-arylcyclopentadienyl complexes, Coord. Chem. Rev. 2011, 255, 1733–1790. [Google Scholar] [CrossRef]
- Janiak, C.; Schumann, H. Bulky or Supracyclopentadienyl Derivatives in Organometallic Chemistry. Adv. Organomet. Chem. 1991, 33, 291–393. [Google Scholar] [CrossRef]
- Bauer, H.; Weismann, D.; Wolmershäuser, G.; Sun, Y.; Sitzmann, H. Iron-Mediated Coupling of Two Ethyl Anions to Form a 2-Butyne Ligand. Eur. J. Inorg. Chem. 2014, 3072–3084. [Google Scholar] [CrossRef]
- Wallasch, M.; Wolmershäuser, G.; Sitzmann, H. Phenolate Complexes of Iron(II) in Different Spin States. Angew. Chem. Int. Ed. 2005, 44, 2597–2599. [Google Scholar] [CrossRef]
- Walter, M.D.; White, P.S. [Cp′FeI]2 as convenient entry into iron-modified pincer complexes: bimetallic η6,κ1-POCOP-pincer iron iridium compounds. New J. Chem. 2011, 35, 1842–1854. [Google Scholar] [CrossRef]
- Chakraborty, U.; Modl, M.; Mühldorf, B.; Bodensteiner, M.; Demeshko, S.; van Velzen, N.J.C.; Scheer, M.; Harder, S.; Wolf, R. Pentaarylcyclopentadienyl Iron, Cobalt, and Nickel Halides. Inorg. Chem. 2016, 55, 3065–3074. [Google Scholar] [CrossRef]
- Reiners, M.; Maekawa, M.; Baabe, D.; Zaretzke, M.-K.; Schweyen, P.; Daniliuc, C.G.; Freytag, M.; Raeder, J.; Hohenberger, J.; Sutter, J.; Meyer, K.; Walter, M.D. Monomeric Fe(III) half-sandwich complexes [Cp′FeX2] – synthesis, properties and electronic structure. Dalton Trans. 2018, 47, 10517–10526. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.; Chilton, N.F.; Kumar, A.; Colebatch, A.L.; Whittell, G.R.; Sparkes, H.A.; Weller, A.S.; Manners, I. Iron Precatalysts with Bulky Tri(tert-butyl)cyclopentadienyl Ligands for the Dehydrocoupling of Dimethylamine-Borane. Chem. Eur. J. 2018, 24, 14127–14136. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, G.Y.; Wallasch, M.W.; Saurenz, D.; Eger, T.R.; Bauer, H.; Wollmershäuser, G.; Prosenc, M.H.; Sitzmann, H. Benzylidyne Bridges from Diphenylacetylene and a Methylidyne Bridge from Methylmagnesium Chloride. Organometallics 2015, 34, 644–652. [Google Scholar] [CrossRef]
- Malischewski, M.; Seppelt, K.; Sutter, J.; Munz, D.; Meyer, K. A Ferrocene-Based Dicationic Iron(IV) Carbonyl Complex. Angew. Chem. Int. Ed. 2018, 57, 14597–14601. [Google Scholar] [CrossRef]
- Goodwin, C.A.P.; Giansiracusa, M.J.; Greer, S.M.; Nicholas, H.M.; Evans, P.; Vonci, M.; Hill, S.; Chilton, N.F.; Mills, D.P. Isolation and electronic structures of derivatized manganocene, ferrocene and cobaltocene anions. Nat. Chem. 2021, 13, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Siemeling, U.; Vorfeld, U.; Neumann, B.; Stammler, H.-G. Bis(trimethylsilyl)amido](η5-pentamethylcyclopentadienyl)-iron(II): A Diamagnetic 14-Electron Complex with a “Pogo-Stick” Structure. Organometallics 1998, 17, 483–484. [Google Scholar] [CrossRef]
- Pauling, L.; Huggins, M.L. Covalent Radii of Atoms and Interatomic Distances in Crystals containing Electron-Pair Bonds. Z. Kristallogr. 1934, 87, 205–238. [Google Scholar] [CrossRef]
- van den Berg, J.-A.; Seddon, K.R. Critical Evaluation of C−H···X Hydrogen Bonding in the Crystalline State. Cryst. Growth Des. 2003, 3, 643–661. [Google Scholar] [CrossRef]
- Thallapally, P.K.; Nangia, A. A Cambridge Structural Database analysis of the C–H⋯Cl interaction: C–H⋯Cl− and C–H⋯Cl–M often behave as hydrogen bonds but C–H⋯Cl–C is generally a van der Waals interaction. CrystEngComm 2001, 3, 114–119. [Google Scholar] [CrossRef]
- Hwang, J.w.; Li, P.; Shimizu, K.D. Synergy between experimental and computational studies of aromatic stacking interactions. Org. Biomol. Chem. 2017, 15, 1554–1564. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with Aromatic Rings in Chemical and Biological Recognition. Angew. Chem. Int. Ed. 2003, 42, 1210–1250. [Google Scholar] [CrossRef] [PubMed]
- Tummanapelli, A.K.; Vasudevan, S. Response to “Comment on ‘Communication: Benzene dimer—The free energy landscape’”. J. Chem. Phys. 2014, 140, 227102. [Google Scholar] [CrossRef]
- van der Avoird, A.; Podeszwa, R.; Ensing, B.; Szalewicz, K. Comment on “Communication: Benzene dimer—The free energy landscape”. J. Chem. Phys. 2014, 140, 227101. [Google Scholar] [CrossRef]
- Tummanapelli, A.K.; Vasudevan, S. Benzene dimer—The free energy landscape. J. Chem. Phys. 2013, 139, 201102. [Google Scholar] [CrossRef]
- Schnell, M.; Erlekam, U.; Bunker, P.R.; von Helden, G.; Grabow, J.-U.; Meijer, G.; van der Avoird, A. Structure of the Benzene Dimer—Governed by Dynamics. Angew. Chem. Int. Ed. 2013, 52, 5180–5183. [Google Scholar] [CrossRef]
- Sinnokrot, M.O.; Sherrill, C.D. High-Accuracy Quantum Mechanical Studies of π−π Interactions in Benzene Dimers. J. Phys. Chem. A 2006, 110, 10656–10668. [Google Scholar] [CrossRef] [PubMed]
- Podeszwa, R.; Bukowski, R.; Szalewicz, K. Potential Energy Surface for the Benzene Dimer and Perturbational Analysis of π−π Interactions. J. Phys. Chem. A 2006, 110, 10345–10354. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.K.; Venkatnarayan, R. Substituents’ influence on the C–H···π interaction in the T-shaped benzene dimer. Theor. Chem. Acc. 2018, 137, 72. [Google Scholar] [CrossRef]
- Jutzi, P.; Mix, A. Oxidative Addition von Halogenpentamethylcyclopentadienen an Metallcarbonyle: ein einfaches Verfahren zur Synthese von (η5-Pentamethylcyclopentadienyl)metallhalogeniden. Chem. Ber. 1990, 123, 1043–1045. [Google Scholar] [CrossRef]
- Clegg, W.; Compton, N.A.; Errington, R.J.; Norman, N.C. Synthesis and Structural Characterisation of some Cyclopentadienyliron–Bismuth Complexes. J. Chem. Soc. Dalton Trans. 1988, 1671–1678. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of the target compounds (X = Cl, Br, I; M = Li, K; ER4 = NnPr4, PPh4).
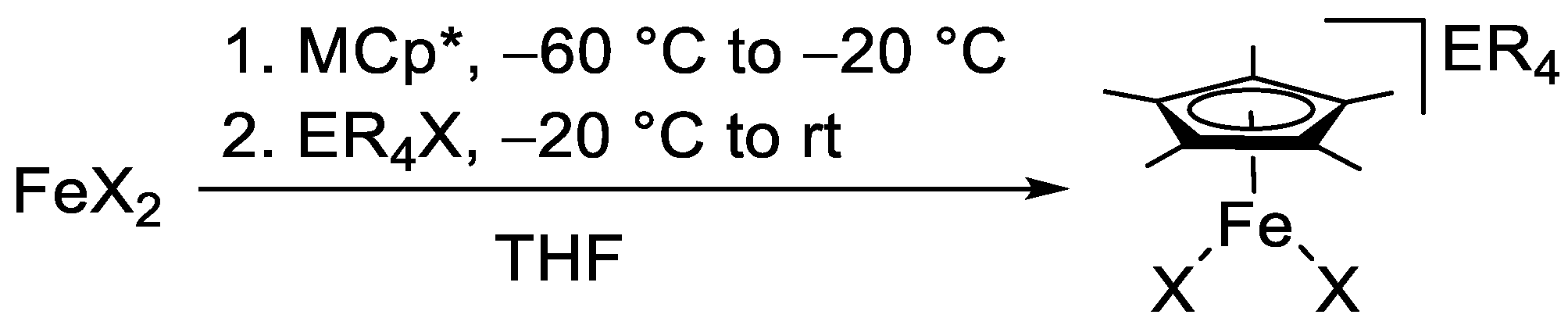
Figure 1.
Molecular structure of NnPr4[Fe(η5-Cp*)Cl2] in the crystal (ORTEP with 50% probability ellipsoids). The anion exhibits CH···Cl contacts compatible with weak hydrogen bonds (indicated by dotted lines) to two tetra-n-propylammonium cations, which are both shown.
Figure 1.
Molecular structure of NnPr4[Fe(η5-Cp*)Cl2] in the crystal (ORTEP with 50% probability ellipsoids). The anion exhibits CH···Cl contacts compatible with weak hydrogen bonds (indicated by dotted lines) to two tetra-n-propylammonium cations, which are both shown.
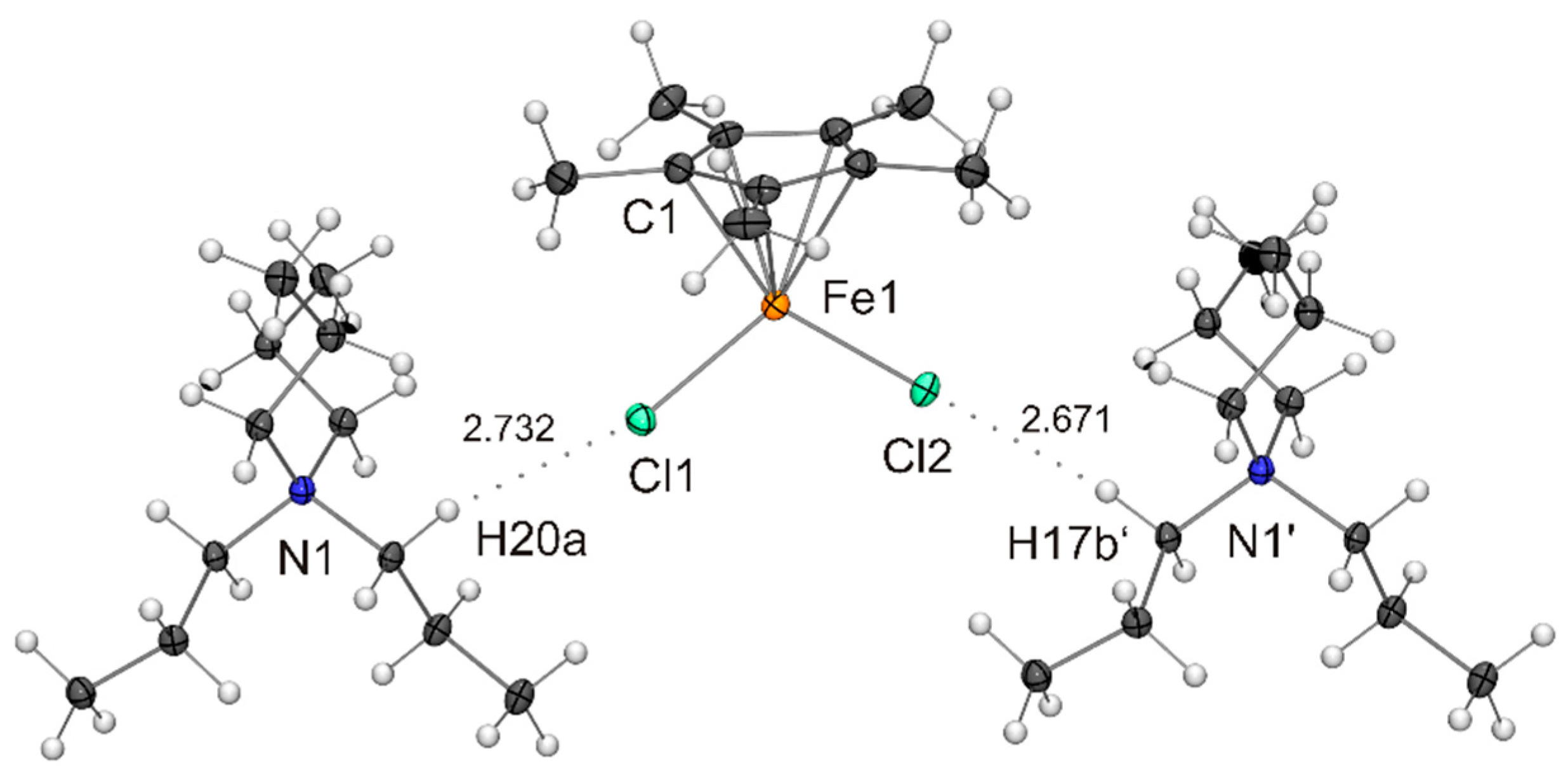
Figure 2.
Molecular structure of NnPr4[Fe(η5-Cp*)BrCl] in the crystal (ORTEP with 50% probability ellipsoids). The atom sites with the higher occupancy (58%) of the disordered halogen atoms are shown. The anion is engaged in CH···X contacts (X = Cl, Br) with neighbouring cations (not shown).
Figure 2.
Molecular structure of NnPr4[Fe(η5-Cp*)BrCl] in the crystal (ORTEP with 50% probability ellipsoids). The atom sites with the higher occupancy (58%) of the disordered halogen atoms are shown. The anion is engaged in CH···X contacts (X = Cl, Br) with neighbouring cations (not shown).
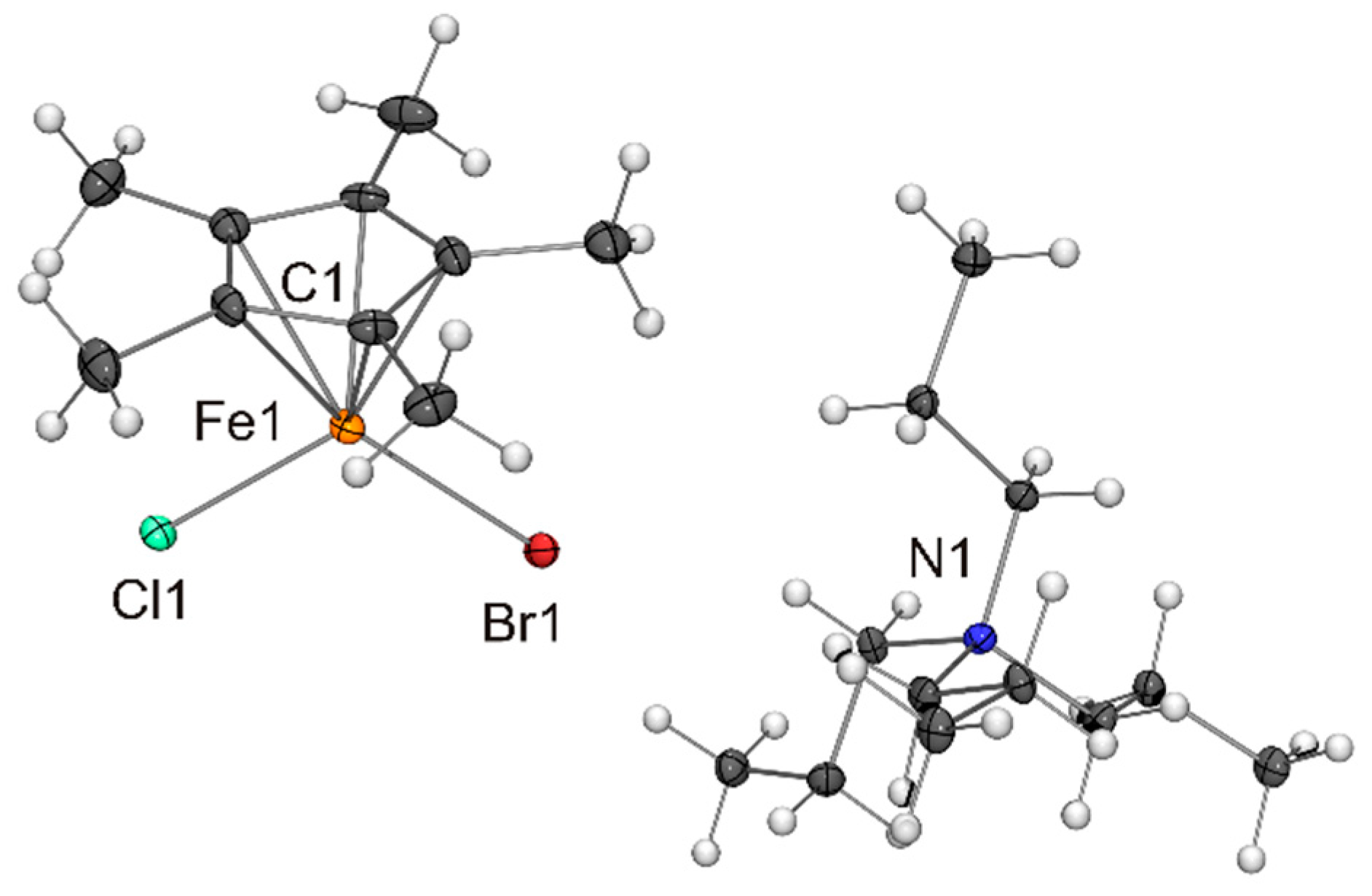
Figure 3.
Molecular structure of PPh4[Fe(η5-Cp*)Cl2] in the crystal (ORTEP with 50% probability ellipsoids). The CH···π interaction between cation and anion is indicated by a dotted line. The anion is engaged in CH···Cl contacts with neighbouring cations (not shown).
Figure 3.
Molecular structure of PPh4[Fe(η5-Cp*)Cl2] in the crystal (ORTEP with 50% probability ellipsoids). The CH···π interaction between cation and anion is indicated by a dotted line. The anion is engaged in CH···Cl contacts with neighbouring cations (not shown).
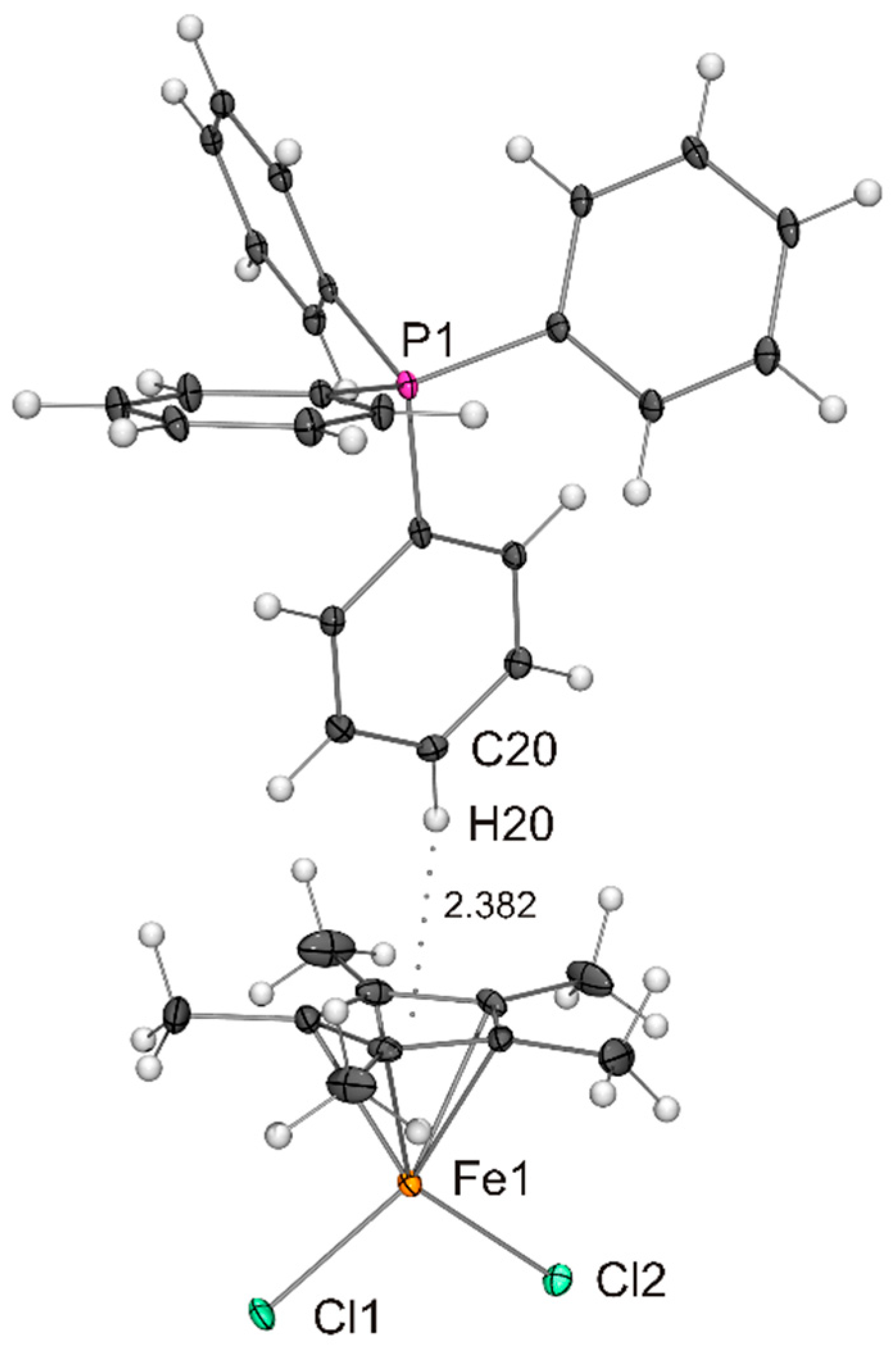
Figure 4.
Molecular structure of PPh4[Fe(η5-Cp*)Br2] in the crystal (ORTEP with 50% probability ellipsoids). The CH···π interaction between cation and anion is indicated by a dotted line. The anion is engaged in CH···Br contacts with neighbouring cations (not shown).
Figure 4.
Molecular structure of PPh4[Fe(η5-Cp*)Br2] in the crystal (ORTEP with 50% probability ellipsoids). The CH···π interaction between cation and anion is indicated by a dotted line. The anion is engaged in CH···Br contacts with neighbouring cations (not shown).
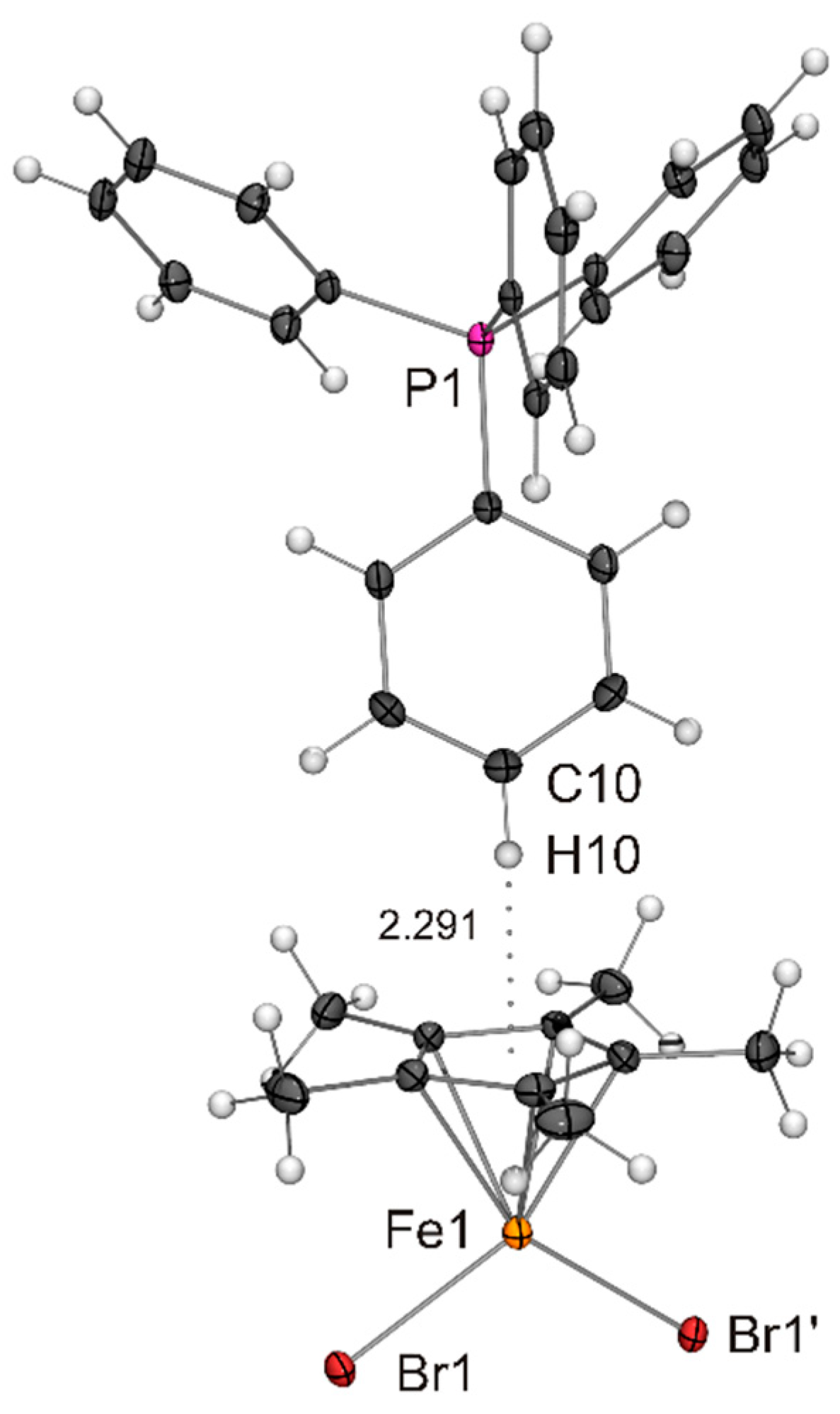
Figure 5.
Molecular structure of PPh4[Fe(η5-Cp*)I2] in the crystal (ORTEP with 50% probability ellipsoids). The CH···π interaction between cation and anion is indicated by a dotted line. The anion is engaged in CH···I contacts with neighbouring cations (not shown).
Figure 5.
Molecular structure of PPh4[Fe(η5-Cp*)I2] in the crystal (ORTEP with 50% probability ellipsoids). The CH···π interaction between cation and anion is indicated by a dotted line. The anion is engaged in CH···I contacts with neighbouring cations (not shown).
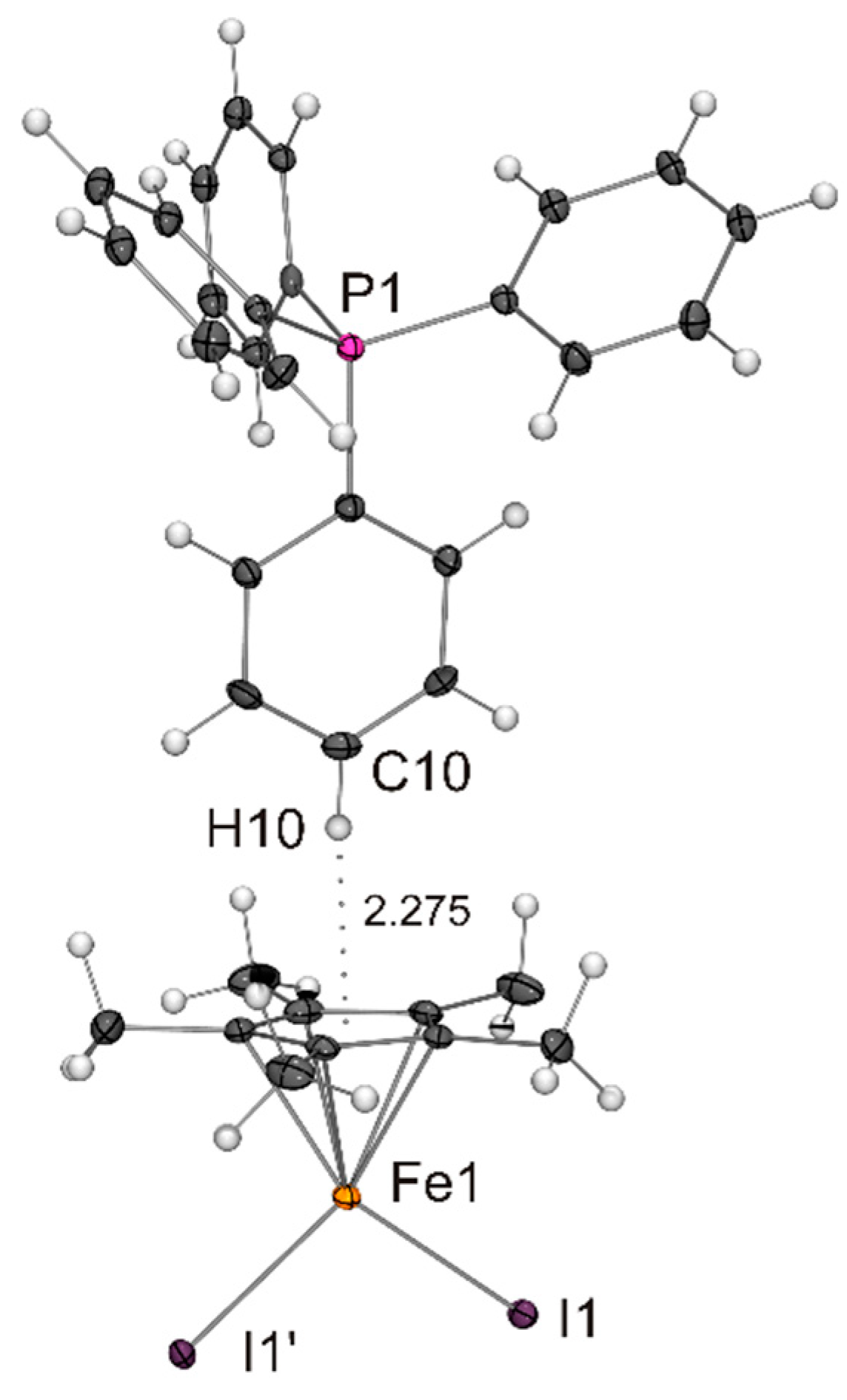

Table 1.
Selected metric parameters (distances in Å, angles in deg) of the compounds of this study and, for comparison, of previously reported closely related compounds.
Table 1.
Selected metric parameters (distances in Å, angles in deg) of the compounds of this study and, for comparison, of previously reported closely related compounds.
| Fe–Cp*centroid | Fe–X | X–Fe–X | |
|---|---|---|---|
| NnPr4[Fe(η5-Cp*)Cl2] | 1.975 | 2.2953(8) 2.2814(8) |
106.27(3) |
| NnPr4[Fe(η5-Cp*)BrCl] 1 | 1.970 | 2.27(2) 2 2.357(9) 3 |
109.0(6) |
| NnPr4[Fe(η5-Cp*)Br2] 4, 5 | 1.958 1.961 1.999 1.995 |
2.432(5) 2.406(5) 2.404(5) 2.446(5) 2.415(5) 2.431(5) 2.405(5) 2.415(5) |
103.8(2) 103.8(2) 104.8(2 103.6(2) |
| PPh4[Fe(η5-Cp*)Cl2] | 1.988 | 2.288(2) 2.284(2) |
107.07(7) |
| PPh4[Fe(η5-Cp*)Br2] | 1.972 | 2.4278(8) | 107.86(5) |
| PPh4[Fe(η5-Cp*)I2] | 1.958 | 2.6201(5) | 106.56(3) |
| NnBu4[Fe(η5-C5H2-1,2,4-tBu3)I2] 6 | 1.989 | 2.7003(6) 2.6144(6) |
102.20(2) |
| [Na(DME)2][Fe(η5-C5H2-1,2,4-tBu3)Br2] 7 | 1.967 | 2.4633(7) 2.4316(7) |
102.36(2) |
1 Disorder of the halogen atoms, the atom sites with the higher occupancy (58%) were chosen. 2 X = Cl. 3 X = Br. 4 Four cations and anions are each present in the asymmetric unit. 5 Caution: The structure solution lacks quality because the arrangement and disorder of the tetra-n-propylammonium cations imposes non-crystallographic symmetry. 6 Ref. [28]. 7 Ref. [29].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



