
Submitted:
21 November 2023
Posted:
23 November 2023
Read the latest preprint version here
Abstract
The evolutionary theory of aging, particularly antagonistic pleiotropy (AP), provides an account of the ultimate origins of aging. What remains unclear is the nature of the proximate mechanisms by which AP gives rise to diseases of aging, like cardiovascular disease and Alzheimer’s disease. Damage-centric theories focusing on loss of genetic and cellular function have been proposed, as well as programmatic theories focusing on unwanted gene and cellular function. The latter include the hyperfunction and developmental theory that view aging as the futile continuation, or run-on, of growth and developmental programmes into later life. Yet neither type of theory has performed well in explaining late-life disease aetiology, particularly with respect to disease onset, presentation and progression. What is proposed here in this review is a new programmatic theory of aging. We argue that the emergence of many specific diseases may involve quasi-programmes that are not the result of run-on, but rather are triggered by other factors in late life. Such triggers may be non-programmatic (e.g. infection, mechanical injury) or programmatic. Moreover, the consequent pre-pathological and pathological changes may in some cases trigger further changes, leading to futile and destructive cascades of quasi-programmes and pathology. The origins of triggered quasi-programmes can be traced to biological constraint i.e. the inability of organisms to optimise all functions at once. And, to some extent, the new theory presented here revises the understanding of AP. That is, because any gene can be triggered in an erroneous manner, every gene is potentially an AP gene that risks pathology, though level of risk varies according to constraint. To help validate the theory, we test it against several complex diseases of aging. The new model in this review attempts to provide a blueprint understanding that, to a certain extent, closes the gap in the causal chain of events between evolutionary causes of aging and the aetiology of age-related diseases. It also helps to explain why certain disorders mimic accelerated aging and how interventions, such as the suppression of IIS and mTOR retard many aspects of aging; notably, though, unlike prior programmatic theories, the new theory is not mTOR-centric. Finally, it provides new perspectives on possible treatment of aging.
Keywords:
ageing
; age-related disease
; antagonistic pleiotropy
; biological constraint
; hyperfunction
; hypofunction
; programmatic ageing
; trade-off
pAntagonistic pleiotropy “has so far failed to explain what is actually at the background of the ‘switching-over’ of the developmental effect of genes to the damaging one and at what stage” Vladimir Dilman(Dilman, 1986)
Introduction
Diseases of aging, ranging from osteoarthritis and chronic obstructive pulmonary disease (COPD) to type II diabetes (T2D) and cancer, are the main healthcare challenge in the world today. This is because their aetiologies are poorly understood, making them difficult to prevent and treat.
Why do diseases occur at all? One major cause of disease is disruption of the normal physiology, which is specified by our normal genome (i.e. the wild-type genotype). Disruptions include infectious pathogens (e.g. viruses, bacteria, protozoa and larger parasites), inherited and somatic mutations, mechanical injury, and diet-related disruptions (including malnutrition and over-eating). Modern medicine has developed great capability at preventing and repairing disruption of physiology and thereby restoring health - especially when that disruption occurs early in life.
When it comes to aging (or senescence, not to be confused with cellular senescence sensu Hayflick), there is something more than disruption at play. A clue to this “something more” lies in the large differences in the rate of aging and lifespans across even closely related species. For example, the maximum lifespan of our own species is around 110 years, while that of our closest relative, the common chimpanzee is only around 60 years. Similarly, mice only live up to around 3 years, while similar sized rodents like squirrels and the naked mole rat can live up to a quarter of a century. This implies that, disruptions aside, the underlying aging process and the rate at which it occurs are, like other aspects of function and physiology, somehow specified by the wild-type genome.
Based on the above discussion, we may deduce that the various causes of disease fall largely into one of the two categories defined: disruptions of wild-type function, and wild-type function itself (Dilman, 1994; Gems, 2022). Aetiologies of disruption are the predominant determinant of disease and mortality earlier in life. The question this review seeks to answer is how the wild-type genome causes aging and late-life disease (Figure 1A).
Why the wild-type genome should be a generator of disease and death is explained by the evolutionary theory of aging, particularly the antagonistic pleiotropy (AP) theory (Williams, 1957). Pleiotropic genes influence two or more phenotypic traits, and these traits can have opposing (beneficial and deleterious) effects on fitness, i.e. the pleiotropic effects on fitness are antagonistic. The action of specific AP genes generates trade-offs between beneficial and deleterious traits. A given AP gene variant is more likely to be favoured by natural selection if the cost presents later in life than the benefit because the force of natural selection declines with age (Hamilton, 1966; Medawar, 1952). To date, many genes have been identified in wild and laboratory animals that exhibit AP trade-offs, where early life fitness like reproduction is observed to come at the expense of later life condition and survival (Austad and Hoffman, 2018).
The AP theory provides an explanation in ultimate evolutionary terms for why the wild-type genome causes late-life disease: certain genes come with trade-offs. But it tells us little about how - about the actual proximate mechanisms of disease aetiology, i.e. what the costs of the trade-offs are. To answer this what is required is an explanatory account that spans several research disciplines, working from evolutionary biology, across biogerontology (the biology of aging) and through to medicine: a valid ultimate-proximate account. Such an account needs to explain the biological mechanisms (biochemical, cellular, physiological) through which AP genes exert their deleterious effects, i.e. the proximate mechanism through which AP genes cause diseases of old age.
The programmatic theory as proximate mechanism
Programmatic theories of aging argue that aging is driven by wild-type function, rather than loss of function. These theories stem from the general observation that inhibition of nutrient-sensitive pathways in laboratory animals increases their lifespan. To date, two quite similar programmatic theories have been proposed: the hyperfunction theory and the developmental theory (Blagosklonny, 2006; de Magalhães, 2005; Gems, 2022; Maklakov and Chapman, 2019). They argue that in early life nutrient-sensing pathways, like the mTOR and the linked IIS pathway, regulate growth and development to promote fitness, but due to attenuated selection in late-life, are not switched-off. As a consequence, their futile and uncontrolled continuation in late life causes disease; here it is an excess of function (or hyperfunction) that causes disease. The evolution of such run-on can be understood in terms of AP in genes governing these specific pathways and their associated growth and development programmes (Gems, 2022).
Notably, the later life deteriorative changes are specified by the wild-type genome, but are not adaptive. In other words, they are programmed in the mechanistic sense, but not in the beneficial sense. For this reason it is not accurate to say that they are programmed, yet they are programmatic.
In short, in the wild type nutrient-sensitive pathways like mTOR promotes growth and developmental programmes, which with age run on to become futile, hyper-functional and so senescence-promoting programmes, termed quasi-programmes (Blagosklonny, 2006) (Figure 1B).
Programmatic theories present a conceptual picture that differ from the predominant views of the biology of aging of the 1980s-2000s. A key difference is the abandonment of the hypothesis that molecular damage accumulation is the primary driver of aging (the damage/maintenance paradigm). Traditional hypotheses about proximate mechanisms of AP, such as the disposable soma theory (Kirkwood, 1977) (Figure 1B), view diseases of aging as originating from loss in basic cellular maintenance and function, due to oxidative and other molecular damage, that works upward to tissue, organ and system failure. However, work in animal models suggests that the observed late-life increases in oxidative damage are more consequence than cause of senescent pathology (Blagosklonny, 2006; Gems and Doonan, 2009; Shields et al., 2021). Moreover, for many age related diseases, like T2D and cardiovascular disease, the predominant role of molecular damage is far from clear (Blagosklonny, 2006; Blagosklonny, 2012; Gems and de la Guardia, 2013).
Limitations of the programmatic theory
For a theory of aging to be operational, it must explain age-related disease aetiology, including disease origins, onset, progression and presentation. Without such an understanding, attempting to intervene is shooting in the dark. To date hundreds of theories of aging have been proposed, but they all share the limitation of being unable to explain disease aetiology. And although conceptually very appealing, this limitation extends to current programmatic theories of aging (the hyperfunction theory and the developmental theory).
Focusing on disease origins, onset and progression: How exactly do useful biological programmes transition into destructive quasi-programmes? In human aging there is a large time span between sexual maturation and the appearance of senescent pathology. How is the length of this lag determined? Why do diseases take so long to appear? And what is the relationship between quasi-programmes and disease progression, if there is any?
Quasi-programmes are viewed as originating from programme run-on. Blagosklonny’s definition of a quasi-programme is “a purposeless continuation of a developmental programme that was not switched off after completion” (Blagosklonny, 2008). Here a slow cumulative effect throughout adulthood results in eventual pathology. There is one heavily-cited medical example: presbyopia (long-sightedness with age), the inability of the eye to focus on nearby objects. This is thought to result in part from the very gradual, continued growth of the eye lens during adulthood, which increases lens thickness thereby impairing ocular function (Figure 2A) (Blagosklonny, 2012; de Magalhães and Church, 2005; Gems, 2021; Strenk et al., 2005). Presbyopia provides an easy-to-grasp illustration of the general principle of quasi-programmes. But apart from it, no other clear examples are cited. Moreover, the chain of events involved in the emergence and progression of most age-related diseases appears to be more complex.
Moving now to predicting disease presentation, current programmatic theories do better than most theories of aging, though they too have limitations. According to current programmatic theories, run on of nutrient-sensitive pathways, like mTOR, causes disease not by damaging cells but rather by causing hyper-activity to a pathological level (Blagosklonny, 2006; Gems, 2022) (Figure 1C). Here a broad distinction may be made between cellular functions associated with basic maintenance, and those associated with cellular activity. For example, mechanisms such as DNA repair and maintenance of protein folding homeostasis are necessary for basic cellular maintenance and survival. Mechanisms involving cell growth, cell cycle regulation etc. on the other hand are more important for cell activity, and alterations in the level of these (increasing or decreasing) will, in the main, alter cellular behaviour rather than affect cell survival. By causing hyperfunction of cellular activity, quasi-programmes bring about changes that present at the tissue level as pathogenic hypertrophy, hyperplasia, dysplasia or hyper-secretion. These tissue level changes in turn will affect tissue, organ and organismal function (Blagosklonny, 2006; de Magalhaes and Church, 2005; Gems, 2022). And this is indeed what is seen in many age-related diseases, e.g. prostatic or endometrial hyperplasia, making this emphasis on cellular activity a key strength of the theory. But now the limitations. As just detailed, it is unclear how run-on of function (i.e. a continuation of function into late life) causes useful programmes to transition into pathological cellular activity. And, notably, many age-related diseases arise from pathological cellular activity in the form of a change in cellular activity - which can be as extreme as change in cell identity, i.e. pathological cellular transdifferentiation. Run on of function does not reconcile with a change in cellular activity.
For example, take vascular calcification that contributes to atherosclerosis. Here active extraskeletal ossification takes place as vascular cells undergo pathological transdifferentiation into to bone-like cells (Virchow, 1863). It occurs despite the presence of inhibitors and, once underway, recapitulates regulated osteogenesis (Sage et al., 2010). Also in atherosclerosis, another example of transdiferentiation involves mature medial smooth muscle cells undergoing clonal expansion and transdifferentiation into macrophage-like cells that contribute to unresolved inflammation and atherosclerotic lesions (Feil et al., 2014; Rong et al., 2003).
Triggered quasi-programmes
One possibility is that run on is not the only way in which futile, senescence-promoting genetic programmes can emerge. Consider the following example. Long-term exposure of the prostate gland to normal levels of the androgen dihydrotestosterone (DHT) promotes benign prostatic hyperplasia (Nacusi and Tindall, 2011; Waters et al., 2000) and increases risk of prostate cancer, a major cause of death in elderly men (Untergasser et al., 2005). Here pathology results from long-term signal exposure, but how exactly? One possibility is run on, with a very slow cumulative effect of DHT leading to pathology. Here the futile programme includes continued exposure to normal levels of DHT in later life until somehow a quasi-programme emerges.
But another possibility is that something changes in the prostate in later life, leading to a new, pathogenic response to DHT. In this case, that change marks the beginning of the futile programme (at least as far as promotion by DHT is concerned). One possible trigger for such a change in the prostate is a local, late-life increase in inflammation. This then disrupts the androgen-supported homeostasis between cell proliferation and cell death, allowing proliferative processes to predominate (Chughtai et al., 2016; Madersbacher et al., 2019; Robert et al., 2009). In this second case the emergence of the DHT-driven futile programme involves a trigger occurring during adulthood. Thus, one may distinguish futile genetic activity resulting from growth and developmental programme run on as originally proposed by the programmatic theory (Blagosklonny, 2006; de Magalhaes and Church, 2005; Gems, 2022) and triggered quasi-programmes that are activated in error (Figure 2A,B).
It is well-known that diseases of aging are typically multifactorial in origin, where a variety of factors (wild-type genome and disruption) influence risk, including cardiovascular disease, COPD, Alzheimer’s disease, osteoarthritis and osteoporosis (Al Anouti et al., 2019; Armstrong, 2013; Glocker et al., 2006; Higashi et al., 2019; Huertas and Palange, 2011). Triggered quasi-programmes is both consistent with this multifactorial view and explains one major form of interaction between different contributing aetiologies. As a different example of such aetiological interactions in aging, emergence of contained disruptions may be triggered by programmatic changes, as in the late-life recrudescence of Varicella zoster virus infection leading to herpes zoster (shingles) or recrudescence of Mycobacterium tuberculosis from granulomas. In principle, any number of causes could trigger quasi-programmes, both disruptions (e.g. infection, somatic mutation) and programmatic aetiologies, including other quasi-programmes.
Notably, triggers of quasi-programmes can originate within the affected tissue or beyond it. An example of the latter, in women later-life depletion of oocyte stocks triggers menopause, including a sustained decline in oestrogen levels. This leads to futile hyper-activation of osteoclasts and sustained bone resorption causing osteoporosis (Kern et al., 2023a).
Another example of an oestrogen-triggered quasi-programme in women is endometrial hyperplasia. This condition is a precursor to endometrial carcinoma, one of the most common gynaecological malignancies (Hannemann et al., 2007). The hyperplasia is caused by chronic oestrogen stimulation of endometrial cell growth, unopposed by progesterone that would otherwise cause cell shedding. Hyperplasia is triggered by factors that increase circulating oestrogen relative to progesterone, including high BMI, late age at menopause and nulliparity (Epplein et al., 2008; Hannemann et al., 2007) (Figure 2C).
Hyperfunction is usually viewed as a level of biological activity above that which is optimal to maintain healthy function. But triggered quasi-programmes can lead to both increased cellular activity and/or changes in cellular identity. In other words, triggered quasi-programmes can explain diseases involving pathological cellular trans-differentiation. For example, transdiferentiation of cells in vascular calcification that contributes to atherosclerosis is triggered by high calcium, high glucose and proinflammatory factors that initiate osteogenic gene expression (Abedin et al., 2004; Chen and Moe, 2012; Demer, 2002), suggesting an osteogenic triggered quasi-programme. Also, in atherosclerosis cholesterol loading of smooth muscle cells triggers phenotypic changes regulated at the mRNA level that result in the transdifferentiation to a macrophage-like state (Feil et al., 2014; Rong et al., 2003).
Costly programmes may be triggered later in life as quasi-programmes
Based on work with the roundworm Caenorhabditis elegans we recently defined a new class of programmatic mechanism of aging prevalent in this species. Evidence suggests that, as in Pacific salmon, C. elegans facilitates a large reproductive effort via repurposing its own biomass to the point of hastening its own death (reproductive death)(Kern et al., 2023b; Kern et al., 2021). In what we termed costly-programmes, a cost is incurred simultaneously with a benefit, forming the basis of cost-benefit trade-offs (Gems et al., 2020; Kern and Gems, 2022). Unlike run on quasi-programmes and triggered quasi-programmes, costly programmes are programmed in both the mechanistic and adaptive sense. The unusually large magnitude (in relative terms) of life-extension achievable in the aging research laboratory model C. elegans may therefore be attributable to suppression of reproductive death(Kern and Gems, 2022), similar to that seen when reproductive death in Pacific salmon is prevented (Robertson, 1961).
In species such as humans with multiple reproductive episodes and no reproductive death, costly programmes are typically transient and incur negligible cost (Kern and Gems, 2022; Kern and Gems, 2023b). As an example, bone loss at the time of lactation is a costly programme where calcium is translocated from bone to produce calcium-rich breast milk. This is at the expense of a transient increase in bone fracture risk, but in humans post-lactation bone regrowth fully restores lost bone (More et al., 2001).
At face value this suggests that costly-programmes are relatively unimportant in human aging and, for costly programmes occurring in their intended adaptive form, this is typically true (Gems et al., 2020; Kern and Gems, 2022; Kern and Gems, 2023b). The revelation of triggered quasi-programmes, however, suggests costly-programmes can also be initiated in a futile and uncontrolled pathogenic manner, in quasi-programmed forms, particularly in later life. And given “cost” is inherent of costly-programmes, futile triggering of costly-programmes risks a high level of damage.
An example of uncontrolled triggering of costly-programmes later in life is osteoporosis during the menopause. Bone misinterprets the sharp decline in oestrogen levels after menopause as the onset of lactation. This triggers a similar but more severe programme of bone resorption that contributes to the development of osteoporosis, in a futile lactational quasi-programme (Kern et al., 2023a).
Taking a step back, regarding the ultimate origins of aging and AP, this suggests that genes specifying fitness-promoting costly programmes may also result in two forms of pleiotropic programmatic cost: more modest and typically transient costs incurred by normal costly programmes, and greater costs caused by their later life quasi-programmed derivatives.
Another example of a costly-programme being triggered later in life as a quasi-programme is COPD. Notably, biomass repurposing is not the only way that costly-programmes can manifest. Another is in collateral damage from useful programmes, particularly when dealing with the crisis of infection. For instance, during infections of the lung, innate immune cells like neutrophils secrete elastases to enable them to burrow through tissue to sites of infection. In youth such programmes are highly accurate, with relatively high effectiveness of clearing infection and minimising collateral damage. Inflammaging in later life, however, causes aberrant neutrophil migration, likely via triggering hyper-activation of phosphoinositide 3-kinase (PI3K) in neutrophils. This translates to both poor immune clearance and severe tissue injury, to the point of contributing to COPD when infection is present (Sapey et al., 2014; Voynow and Shinbashi, 2021).
COPD also illustrates the complexity of many age-related diseases where multiple triggers may converge to induce pathology, some that prime the tissue (inflammaging) and some that then kick-start destruction once the tissue is primed (infection). As an analogy, for a fire you need both the spark, i.e. trigger, and fuel, i.e. priming of some form that increases susceptibility to the trigger.
Cascades of triggered quasi-programmes
In the epigram to this review, Vladimir Dilman raises a critical question: what determines how the good effects of an AP gene turn into the bad - and when? In the damage/maintenance paradigm the answer is slow accumulation of the negative effects with age (Kirkwood, 1977). The traditional programmatic theory suggests beneficial programmes gradually run on into quasi-programmes, in a slow process of developmental transition (Blagosklonny, 2006; de Magalhães, 2005; Gems, 2022; Maklakov and Chapman, 2019). But as argued, apart from presbyopia, it is not easy to find examples of quasi-programmes.
Here it is argued that, rather than run on quasi-programmes, diseases typically arise from triggered quasi-programmes that are triggered by disruptions or other, upstream, quasi-programmes. Thus, triggered quasi-programmes may act not only as primary drivers of pathogenesis, but also secondary and tertiary drivers.
This may be illustrated by the example of cancer development. Here primary causes include disruptions (particularly somatic mutations) and triggered quasi-programmes (e.g. causing hyperplasia that precedes somatic mutation). Progression to metastatic cancer requires angiogenesis, a quasi-programme triggered by earlier stages of cancer development.
If quasi-programmes act as triggers for other quasi-programmes, this suggests the presence of chains and cascades of quasi-programmes active in pathogenesis (Figure 3).
Two forms of triggered quasi-programme cascade may be distinguished. First, in multi-stage quasi-programme cascades a single quasi-programme drives pathology development, but multiple stages can be distinguished. Second, discrete stage quasi-programme cascade, where individual quasi-programmes are temporally or spatially separated from one another, and do not necessarily follow one to the next. To be precise, multi-stage and discrete stage quasi-programme cascades represent opposite ends of a continuum more so than discrete categories, with many cascades corresponding most closely to points in between. Some examples of multi-stage and discrete stage quasi-programme cascades follow.
Multi-stage quasi-programme cascades
The first disease to which this new theory of triggered-programmes was originally applied is rheumatoid arthritis (Kern, 2020); its relative simplicity makes concepts easy to grasp. It is also an example of a multi-stage triggered-programme cascade (Figure 4A). The disease is triggered by inflammation, typically increased sterile inflammation, induced by a range of causes. The arthritic disease process begins with immune cells attacking a healthy joint in a futile manner (stage 1). This causes the synovial tissue surrounding the joint to undergo vigorous proliferation and vascularisation and transform into a hypertrophied rheumatoid pannus, from the Latin pannus (table-cloth) reflecting the presence of a thickened layer of tissue (stage 2). The pannus then secretes proinflammatory cytokines that cause futile osteoclast-mediated bone resorption and loss (stage 3) (Distler et al., 2004; Karmakar et al., 2010).
In an additional stage, the hyperplastic synovium pointlessly invades cartilage and bone, further contributing to the destruction of the affected joints. The increased synovial fluid content in affected joints also increases intra-articular pressure. In addition to these elaborate self-destructive programmatic mechanisms, disruptions also pitch in and contribute to the demise of the joint. Here disruptions can act directly, or indirectly by acting as quasi-programme triggers, and/or making the tissue more susceptible to triggers and/or other disruptions (c.f. Figure 1A). Disruptions include mechanical injury, pathogen invasion and molecular damage (Figure 4A). Thus, in this complex disease a localized, tight succession of futile developmental processes leads to painful and disabling disease, in a multi-stage quasi-programme cascade. This is the core aetiology in a wider, multifactorial process.
Variations in the presentation and progression of the disease may be attributed to differences in the wild-type genome, and occurrence of disruptions (including triggers) (Figure 5).
Another multi-stage quasi-programme cascade is operative in the development of non-alcoholic fatty liver disease (NAFLD) and insulin resistance (Figure 4B). A major cause of the disorder is inflammation in adipose tissue, typically a consequence of obesity and/or aging. This induces adipocyte insulin resistance, which leads to increased export of free fatty acids (FFA) from the adipocytes and into the circulation. FFA are then taken up by other organs that are not adapted to store large amounts of fat, such as the liver (stage 1). This in turn leads to lipotoxicity (lipid-induced dysfunction) in those organs, which increases insulin resistance at those sites (stage 2) (Adams et al., 2005; Sears and Perry, 2015; Unger, 2003). It is here that the systemic consequences of insulin resistance really start to bite, as hepatic insulin resistance and lipid accumulation trigger a quasi-programme positive feedback loop, driving the development of the metabolic syndrome (Figure 4B).
Further pathological consequences of NAFLD include increased risk of hypertension and cardiovascular disease from excess FFA in the blood, and type II diabetes (T2D) as insulin resistance impairs control of blood glucose levels (Adams et al., 2005; Chen et al., 2017; Unger, 2003). Specifically, in the liver insulin resistance causes excess glucose secretion into the blood, and in muscle it reduces glucose uptake, which together send blood glucose levels soaring, and the development of T2D is on its way. Disruption-type aetiologies can exacerbate disease, e.g.infection and injury, to which the NAFLD liver is more susceptible (Adams et al., 2005). In conclusion, although NAFLD involves different organs (particularly adipose tissue and liver), the tight causal relationship between quasi-programmed changes define a multi-stage quasi-programme cascade.
Discrete stage quasi-programme cascades
In discrete stage quasi-programme cascades the relationship between primary and secondary quasi-programmes is looser and less inevitable. Here an example is insulin resistance leading to T2D (Unger, 2003), which can in turn sometimes cause diabetic cataracts. Diabetic cataracts develop due to excessive conversion of glucose into sorbitol (an example of hyperfunction), at a rate higher than sorbitol can be converted into fructose by the enzyme sorbitol dehydrogenase. The resulting build-up of sorbitol then generates increased osmotic stress in the lens fibres, causing them to swell and rupture (Pollreisz and Schmidt-Erfurth, 2010).
Returning to rheumatoid arthritis, a secondary triggered quasi-programme likely develops when calcium and phosphate released by bone degradation (stage 3 in Figure 4A) enter the bloodstream; excess blood calcium contributes to pathological arterial ossification, contributing to atherosclerosis as previously described. This is likely what causes the strong correlation between bone resorption diseases like osteoporosis and rheumatoid arthritis and arterial calcification (Wahlin, et al., 2016; Giles, et al., 2009) (Figure 4C).
Multi-stage cascades may also involve the action of multiple triggers, where preceding triggers prime tissue for subsequent pathology (c.f. a fire where a spark and fuel are required). This is particularly when the preceding trigger is a programmatic change, as in the example of COPD. As another example, osteoporosis, triggered by osteoclast hyperactivity, damages a joint and this in turn primes the joint for future triggers of osteoarthritis (Calvo et al., 2007; Horikawa et al., 2014). In osteoarthritis, chondrocyte hyperactivity is triggered by mechanical stress and induces a programme of new bone formation (osteogenesis), causing a thickening of subchondral bone, including the development of bone marrow pockets and a blood supply (echoing angiogenesis in cancer development), culminating in the formation of new bone outgrowths, called bone spurs (or osteophytes), which can restrict joint movement and cause pain (Glyn-Jones et al., 2015).
Returning also to NAFLD, another secondary quasi-programme can develop as lipotoxicity increases the susceptibility of the liver to injury, which can then trigger further inflammation (Adams et al., 2005). Such inflammation can then cause disease progression to non-alcoholic steatohepatitis (NASH), an aggressive form of fatty liver disease marked by liver inflammation, and advanced scarring and fibrosis (i.e. cirrhosis) as part of an exaggerated wound healing response. Here proinflammatory cytokines and hyperactivity of innate immune cells directly damage tissue. Concomitantly, these cytokines also trigger fibroblasts to secrete too much extracellular matrix including collagen, and the resulting fibrosis contributes to liver failure via disruption of the liver architecture and function (Figure 4D).
Another example of a discrete stage quasi-programme cascade involves immunosenescence and accumulation of cells that have undergone cellular senescence (cell cycle arrest, and entry into a hypertrophic, hypersecretory state) (Prata et al., 2018). Throughout life, fibroblasts undergo this type of differentiative changes to support wound healing (Demaria et al., 2014). Such “senescent” fibroblasts are subsequently cleared by the immune system, triggered by signals within the fibroblast senescence-associated secretory phenotype (SASP). In later life, efficiency of clearance of senescent fibroblasts is reduced due to immunosenescence. With the resulting accumulation of senescent fibroblasts, a programme for wound healing and tissue remodelling becomes a primary quasi-programme leading to disruption of cellular microenvironments which, in mice at least, promotes diverse diseases of aging (van Deursen, 2014). In a secondary quasi-programme, accumulating senescent cells can induce neighbouring cells to also undergo senescence (secondary senescence via autocrine and paracrine signals) (Admasu et al., 2021).
As a final example, general systemic inflammation, from causes such as those that give rise to NAFLD (Figure 4B), can then go on to further trigger neurodegeneration via microglia hyperaction. For instance, increasing evidence indicates that systemic inflammation might drive the initiation and progression of Alzheimer's disease as immune cues can pass into the brain through various routes. Examples include via the circumventricular organs, across the brain barriers, through activating vascular cells at the brain barriers and even neural routes like cytokines from the thoracic-abdominal cavity (e.g. Kupffer cells in the liver) directly activating the vagal nerve which signals to the brain (Xie et al., 2021). Neuroinflammation contributes to Alzheimer's disease (Heppner et al., 2015; Sala Frigerio et al., 2019), as it causes microglia hyperaction and exacerbates amyloid beta and tau pathologies that in turn cause neurodegeneration and cognitive decline (Ising et al., 2019). Likewise Parkinson’s disease is associated with neuroinflammation that systemic inflammation contributes to (Wang et al., 2015).
Moving away from a mTOR-centric model
Notably, the concept of triggered quasi-programmes moves away from the view that there is one central driving force behind the aging process, like damage accumulation, or mTOR (and other nutrient-sensing pathways that control growth and development) as proposed by existing damage-centric and programmatic theories, respectively. Instead, triggered quasi-programmes present a new primary pathophysiological principal. Think here of infectious diseases that differ greatly in their specific aetiologies, presentation, severity and so on, e.g. tuberculosis vs rabies, but can be understood as originating from the pathophysiological principal of infectious pathogens - of which there are many different kinds.
Regarding mTOR hyperactivity in particular, in all the examples of age-related disease described above, this is not a major driver. Take endometrial hyperplasia, for instance, where the immediate cause is an increase in the ratio of oestrogen to progesterone. Yes, mTOR function (hyperfunction) is a critical factor for such hyperplasia, but one that is downstream in the chain of events leading to pathology. To further emphasise this “downstream” role of mTOR, one can look at what happens when conversely there is too much progesterone vs oestrogen, as with the progesterone only birth control pill: atrophy of the endometrium. As other examples, in prostrate hyperplasia proximate drivers of pathology are DHT and inflammation. In the progression of fatty liver, proximate drivers are insulin resistance and lipotoxicity. In osteoporosis proximate drivers are disrupted signalling cues to osteoclasts, like disrupted oestrogen levels. In diabetic cataracts the proximate driver is too much glucose along with constraints on enzymatic conversion rates of its by-product sorbitol. In these examples, mTOR is a key player whose hyperfunction is integral to the development of pathological quasi-programmes. Yet mTOR hyperfunction is not the initial cause of quasi-programme development, but rather a mediator of upstream triggers.
Ironically, this account echoes previous criticisms of the oxidative damage theory. A major supporting argument for this theory was the increase in later life of oxidative damage. But it was then argued that this was a consequence of late-life pathology, whose origins lie in mTOR hyperfunction. Hence rapamycin can increase lifespan while antioxidants cannot. The proposition in this review is that dysregulated mTOR activity, which most typically is in the form of mTOR hyperfunction, is only a secondary cause of age-related disease. And this predicts that, like oxidative damage levels, mTOR hyperactivity in vivo will occur in the early stages of quasi-programme emergence, in mid to late life, rather than increasing gradually throughout life. Here it is argued that mTOR hyperfunction originates cell non autonomously, rather than cell autonomously.
Diseases prior to aging: triggered quasi-programmes in infectious disease
Medicine primarily views infectious disease from the angle of disruption to physiology; the key disruptor here being the infectious pathogen. Yet the wild-type genome also plays an integral role in the aetiology of many infectious diseases, primarily in the form of triggered quasi-programmes. This typically results from bystander effects as in autoimmune-driven pathology (an echo of late-life, chronic sterile inflammation). An example of this is Lyme disease, which results from infection with the tick-borne bacterium Borrelia burgdorferi. Here bystander T cell activation can alter synoviocytes (fibroblast-like cells) in articular joints. This can lead to joint pathology that includes the erosion of bone and cartilage, in a manner somewhat akin to the aetiology of rheumatoid arthritis (Whiteside et al., 2018) (Figure 4A). There are numerous other examples. For instance, in leishmaniasis (infection with the protozoan parasite Leishmania major), bystander activation can exacerbate disease, leading to skin lesions filled with copious numbers of monocytes, neutrophils and CD8+ T cells (Whiteside et al., 2018). Bystander activation after infection with the virus hepatitis C causes liver injury, closely resembling that of NAFLD and NASH (Adinolfi et al., 2016). Here in each case, triggered quasi-programme cellular behaviours arising from evolved, wild-type gene function contribute to pathology. And for infectious diseases where the pathogen that is the primary source of the disease proves difficult to treat, triggered quasi-programmes offer secondary potential sites of intervention. Notably, as with the age-related disease detailed above, the role of mTOR hyperfunction in these diseases is typically secondary.
AP is a feature of all wild-type genes, and constraint determines risk of disease
How well does the new theory work as an ultimate-proximate account of aging? If not run on of mTOR function, what other proximate mechanisms could account for the timing and appearance of triggered quasi-programmes? And: why have mechanisms to prevent the appearance of triggered quasi-programmes and cascades that cause age-related disease not evolved in animal species - especially where certain triggered quasi-programmes, such as those associated with infection, are occurring early in life.
One possible ultimate-proximate explanation for the cause of triggered quasi-programmes is biological constraint: any factor that affects either the production or presentation of a biological trait. Constraint is a consequence of the highly integrated nature of organismal biology, where, like with a machine, there is an inability to optimise all functions at once. And this leads to natural compromises in the form of trade-offs.
This idea of constraint differs from traditional programmatic theories, as the very concept of triggered quasi-programmes resulting from run on implies the absence of biological constraint (Blagosklonny, 2006; Blagosklonny, 2021; de Magalhães, 2005; Gems, 2022; Maklakov and Chapman, 2019).
Due to the presence of biological constraint, the function of many genes and proteins have the potential to provide a fitness benefit in one context and a cost in another. Here context can mean not only different ages, as in William’s account of AP in aging, but also in different tissues, different sexes, different genotypes and different environment - especially where the proximate mechanism of the cost is triggered quasi-programmes (see disease examples above).
Understanding AP and constraint in this way, one may reasonably argue that all genes in the human body exhibit AP. Consider for example, transformed breast epithelial tissues in metastatic breast cancer: here all the genes supporting cellular function have become pathogenic - all exhibit AP.
Whilst every gene has the potential to exhibit AP and be triggered as a quasi-programme, not all genes are equally likely to give rise to pathology. Specifically, the risk of a gene giving rise to pathology via quasi-programmes is a function of (i) the cost incurred if that gene is erroneously triggered, and (ii) the likelihood of the gene being erroneously triggered, formularised by a risk equation as follows.
Equation 1. Calculating risk of pathology from futile triggering of genes as quasi-programmes. Risk of pathology (Rp) is a function of the cost incurred when the gene is triggered in error (Ct), and the likelihood of the gene being triggered in error, i.e. the probability of the cost (pCt). Genes associated regulatory and signalling proteins like mTOR and IIS are at high risk of contributing to pathology. If triggered in a futile manner, such genes will have multiple knock on effects, making Ct high. And, as such genes typically have multiple upstream mediators, pCt is also high.
The probability of the gene being triggered (pCt) is determined by the probability of the occurrence of the trigger(s). Some triggers may be the product of biological constraint that exert a deleterious effect later in life, which is why you only see the pathology appearing later in life. For example, menopause is likely the result of constraint; it has been argued that the human menopause evolved, not because it promotes fitness in any way, but because as hominin longevity increased, it was simply not possible for reproductive lifespan to lengthen in hominin females (Austad, 1994; Marlowe, 2000). And the menopause triggers many pathologies, such as oestrogen decline inducing osteoclast hyperactivity and osteoporosis.
The level of cost is also determined by biological constraint, acting at multiple levels of organisation (for formularisation of cost using trade-off functions see (Kern and Gems, 2023b)) - i.e. ask yourself why evolution did not work to minimise cost even in the presence of the trigger.
As a simplification, AP effects are expected to be greater where more complex constraints are present, as where a given gene function plays a role in many different contexts. This is a particular issue in complex multicellular organisms where the same gene acting within diverse cell types and tissues may have different effects. For example, regulatory and signalling proteins may serve multiple functions which, similar to building the circuitry of a machine or software coding, result in signalling constraint that create risk of pathogenic off-target effects and cross-talk - and so deleterious quasi-programmes being triggered.
Syndromes of senescence: triggered quasi-programmes can act within wider aetiological webs
Complex though they are, triggered quasi-programme cascades are only part of a wider web of disease aetiology. The highly complex and interconnected nature of late-life disease can present a prospect of senescence as a hopelessly intractable condition. Yet in principle, it ought to be possible to map out the interconnected webs of causation, which include triggered quasi-programme cascades and other elements of multifactorial aging (Figure 6).
As an illustration of such an aetiological web, consider T2D triggered by menopause-related endocrine changes (Mauvais-Jarvis et al., 2017; Paschou and Papanas, 2019). Once developed, T2D can suppress the immune system via several different mechanisms involving further triggered quasi-programmes, and also molecular damage. As one mechanism, hyperglycaemia due to T2D leads to formation of advanced glycation end-products (AGEs). And this causes a shift in macrophage class frequency, with fewer M1 macrophages that encourage inflammation, and more M2 macrophages that favour tissue repair (He et al., 2020). In turn, this leads to a reduction in microbicidal capacity (Berbudi et al., 2020; Liu et al., 2012; Pavlou et al., 2018) which, along with reduced blood flow, increases risk of infection and impairs wound healing. Reduction in blood flow results from several causes, including increased blood viscosity, change in arterial wall tension, and hyperglycaemia which causes molecular damage like AGEs and triggered quasi-programme inflammation. This in turn causes vascular endothelial cell dysfunction (Cinar et al., 2001; Hadi and Suwaidi, 2007).
An example of a further diabetic complication is periodontitis (severe gum infection that can destroy teeth and the bone that supports them). In the mouth, the increased risk of infection from T2D is further compounded by hyposalivation (possibly due to impaired blood flow to the salivary glands) and an increase in salivary glucose levels (resulting from elevated plasma glucose) which promotes bacterial growth (Al-Maskari et al., 2011; Pérez-Ros et al., 2021). Hyposalivation is also bad because saliva contains antimicrobial proteins and peptides, and acts as a mechanical barrier preventing adhesion of microbes to the surface of the oral mucosa. Moreover, a lack of salvia production causes stagnation in flow; any stagnation in flow causes infection via bacterial build-up from a lack of mechanical washing (Iwabuchi et al., 2012). Bacterial build-up both directly destroys tissue, and has indirect effects such as lodging in between the gum and enamel thereby reducing adherence of gum to the enamel (leading to gum recession).
This complex example illustrates how diverging pathological outcomes from an aetiological web can reconverge to generate a further disease (here periodontitis). In other words, many diseases can converge to form one disease and one disease can proliferate to generate many more in a domino style effect.
Looking further back up the causal chain of aetiologies, it can be seen that menopause increases risk of periodontitis (Angeles-Albores et al., 2016; Deepa and Jain, 2016). This is due not only to increased risk of T2D, but also to other effects of oestrogen deficiency such as triggered quasi-programmed bone resorption contributing to loss of bone that supports teeth, and reduction in gingiva (gum) integrity (Bhardwaj and Bhardwaj, 2012; Kovacs, 2016). The latter arises from yet another triggered quasi-programme change arising from control by oestrogen of proliferation and differentiation of keratinocytes and fibroblasts in the gingiva (Bhardwaj and Bhardwaj, 2012).
Triggered quasi-programme cascades are amenable to medical intervention
A good understanding of disease aetiology is important for devising means of disease prevention and treatment. In the case of triggered quasi-programme cascades and the wider aetiological webs in which they operate, such understanding requires careful enumeration of the diverse links in the aetiological causal chains involved. There are several possible reasons why such chains and networks are not a stronger focus of efforts by doctors and scientists. First, as detailed in the introduction, medical research has tended to look to disruptions of normal function for explanations of disease aetiology, whereas the main cause of senescence is wild-type function (Figure 1A). Second, much past research on aging was guided by the damage/maintenance paradigm, looking to accumulation of molecular damage and loss of cellular function for the causes of this or that disease of aging (Figure 1C). Third, complex chains of disease causation involve different steps at different times in different organs and tissues, creating a complex picture. But, by understanding the aetiological webs that drive senescence, opportunities for interventions to improve health come into view. For example, the observation that hormone replacement therapy in post-menopausal women reduces risk of periodontitis is potentially explicable in terms of the cascade of triggered quasi-programmes described above (Angeles-Albores et al., 2016). Triggered quasi-programme cascade maps are a potential means for rational design of such long-distance intervention approaches (i.e. preventing a cause far upstream in the cascade of the proximate cause of a disease).
Specifically, one approach for intervention is to identify and target the initiating triggers of quasi-programmes. This may be particularly effective where trigger amplification has occurred, as in the triggering of metabolic syndrome by inflammaging (Figure 4B). Yet for many diseases of senescence, triggers of quasi-programme may be hard to pinpoint, variable, and numerous making intervention difficult.
An alternative approach is to try and eliminate as many secondary systemic triggered quasi-programmes as possible - given the new theory reveals much of age-related disease is a secondary consequence of initial triggers due to quasi-programme cascades and arises in a cell non autonomous manner. One way to do this is via readjusting cell signalling cues in blood; likely why parabiosis (linking of the blood supplies) of young and old mice works to alleviate some age-related pathology, at least as far as clonal animals are concerned.
Rapamycin as an anti-aging drug
What about means to target all triggered quasi-programmes that emerge in later life at once? Is say the drug rapamycin (sirolimus), an mTOR inhibitor (Blagosklonny, 2019; Harrison et al., 2009; Wilkinson et al., 2012), a possible candidate. The beneficial effects of IIS/mTOR inhibition on the lifespan of laboratory models led to the development of the programmatic theory in the first place (Blagosklonny, 2006; de Magalhaes and Church, 2005; Gems, 2022). The theory argues that IIS/mTOR suppression increases lifespan by preventing developmental programmes, particularly governed by mTOR, from running-on into quasi-programmes in later life (Figure 1B and Figure 2A). But how does all this reconcile with triggered quasi-programmes, and is general mTOR/IIS inhibition still a good approach?
There are two points to note here. First, simply because something works in laboratory animals, does not make it a good intervention for humans; mechanisms driving aging in model organisms are not necessarily relevant to human aging (Kern and Gems, 2022).
Second, while mTOR inhibition is a therapy against triggered quasi-programmes, it is not necessarily the best therapy. Rapamycin against aging can be likened to chemotherapy against cancer. Chemotherapy works by damaging genes associated with cellular proliferation, with the consequence that all highly proliferative cells are most affected. Treatment with chemotherapy works because the vital cells in us are slow dividing (highly proliferative cells like hair and skin are less essential) and so a balance is struck between killing the cancer cells (in order to cure or control the disease) and sparing vital cells, but at an expense (many unpleasant side effects). You wouldn’t want to be on chemotherapy 24/7. General suppression of IIS/mTOR with a drug like rapamycin to treat aging is likely to be beneficial in the same way as chemotherapy: a balance will be struck between preventing gross pathological change by dampening all cellular activity, whilst trying to maintain vital functions. But there will be costs like immunosuppression, and reduced wound healing (from mTOR hypofunction). As a specific example, rapamycin dampens bone remodelling and so delays fracture healing (Holstein et al., 2008), but multiple papers also suggest it is beneficial against osteoporosis. But to be clear this is not an argument against the potential efficacy of mTOR inhibitors to treat late-life disease when no other intervention is available.
Predictions of the new theory
An operative theory of aging must be able to explain age-relate disease aetiology, as the new blueprint-theory does for many diseases. A further means to validate it is to see if it makes testable predictions, some of which may involve explaining past observations. Below are a number of such predictions.
Prediction 1: If triggered quasi-programmes is what is giving rise to many age-related diseases, the same pathologies should present earlier in life if the trigger is present earlier. This is indeed what is seen. Numerous disorders exhibit features that resemble acceleration of some aspects of aging, so-called progeroid conditions (Martin, 1978). And in many cases this reflects earlier occurrence of triggered quasi-programme cascades. For example, oophorectomy (removal of the ovaries) prior to menopause leads to a sharp decline in circulating oestrogen which causes osteoporosis (Hibler et al., 2016), in a premature menopausal triggered qausi-programme cascade (Kern et al., 2023a).
Obesity causes premature onset of several aging-related diseases such as cardiovascular disease, NAFLD, T2D and stroke, and may therefore be viewed as a progeroid condition (Horvath et al., 2014; Salvestrini et al., 2019). A contributor to obesity-triggered progeria is chronic sterile inflammation, or premature inflammaging (Mancuso and Bouchard, 2019). This develops because adipocytes reach their limit of expansion, causing hypoxia which triggers proinflammatory pathways and systemic insulin resistance (Sears and Perry, 2015). This in turn can lead to conditions such as NAFLD and metabolic syndrome as described above (Figure 4B) much earlier in life.
This could be why interventions that are anti-diabetic and promote weight loss, like metformin and ozempic (semaglutide), exhibit some features of “anti-aging”. Rather than being anti-aging per se, such drugs are likely beneficial via preventing accelerated aging. For examples involving progeroid congenital syndromes, see Appendix.
Prediction 2: The evolutionary conservation across animal species of cellular signalling and physiology (and hence biological constraint) predicts that cascade will also show some evolutionary conservation. This prediction is consistent with the presence of similar diseases of aging in many mammalian species. As one example, canine rheumatoid arthritis shows cascades and stages that are very similar to those seen in the human disease (Figure 4A) (Bennett, 1987; Innes & Clegg, 2010).
Prediction 3: According to the new theory, the more different contexts a given gene acts in, the more it will be subject to constraint and, therefore, pathology-generation. A prediction that follows is that cell and tissue types that show greater plasticity of function (such as those with active tissue remodelling during adulthood) proffer more biological contexts for gene function, and therefore are expected to create more biological constraint and AP. In line with this, the liver, skin and colon for example, which exhibit high levels of cell replenishment from stem cells and division of differentiated cells, are relatively highly prone to cancer and autoimmune disease (Cooper et al., 2009; NCI, 2023). By contract, myocardial, neurological and germline tissues that are structurally more static are less prone to such pathologies (Cooper et al., 2009; NCI, 2023). Operation of this risky plasticity principle can also be seen in sexual dimorphism, with greater susceptibility to some diseases of aging in the more “plastic” sex, i.e. females that undergoe greater change in tissue status for reproduction, both in laboratory model animals and humans. For details of this and cellular plasticity correlating with aging disease (i.e. mesenchymal stem cell lineages and risk of triggered quasi-programmes) see appendix.
Prediction 4: Senescence should be commonly observed in a wild animal’s populations. Prior programmatic theories argue aging is the result of run-on of nutrient-sensing pathways like mTOR (Blagosklonny, 2006; de Magalhães, 2005; Gems, 2022; Maklakov and Chapman, 2019), and rely on the absence of “off-switches” to be a part of the evolution of aging - a consequence of most animals not surviving to late ages where the benefits of such off-switches can be realised, due to high extrinsic mortality (i.e. death from external causes like predation) (Blagosklonny, 2006). This predicts that senescence should be restricted to late ages that the majority of the population do not reach in the wild, i.e. they predict senescence should not be commonly observed in wild animal populations. In contrast to run-on, the new model predicts features of aging should be common in wild animal populations due to biological constraint and triggered quasi-programmes. And field studies ranging from baboons and monkeys to sheep, bears, whales, swallows and a multitude of other animals are in line with this prediction (Nussey et al., 2013). For details, see appendix.
Prediction 5: Predicting results of prior tests of evolutionary theories. The antagonistic pleiotropy (AP) theory is one of two early theories proposed for the evolution of aging due to late-life selection shadows. The other is Peter Medawar’s mutational accumulation (MA) theory (Medawar, 1952). This argued that wholly deleterious mutations may accumulate in populations if their effects are exerted only later in life. While AP and MA are not mutually exclusive, the importance of MA in the evolution of aging remains uncertain and is much debated. This is partly because it is difficult to test directly for the existence of MA (Hughes, 2010; Rose and Charlesworth, 1980). There also seem to be few clear examples of MA. Even the Huntington’s disease Htt mutation, long quoted as an example of MA (Haldane, 1941), has been reported to increase number of children (Shokeir, 1975; Walker et al., 1983) and resistance to cancer (McNulty et al., 2018; Sorenson et al., 1999), i.e. to exhibit AP.
This being said, strong support for MA has been derived indirectly by testing predictions of MA that differ from those of AP. However, the existence of triggered quasi-programme cascades changes many of the previous predictions about AP, and therefore affects interpretations of prior attempts to distinguish AP and MA - and provides an explanation for the outcome of a range of such studies, ranging from quantitative trait locus (QTL) mapping to human genome wide association studies (GWAS), that were previously used to support MA over AP. For details see appendix.
Conclusions
This article is one of a trio, that together attempt to build a much needed unified theory of aging - stretching from understanding effects of ecology on life-history strategy, down to presentation of age-related disease, and finally effects on population survival curves and other aspects of biodemography. The first article (Kern and Gems, 2023b) integrates life history theory with evolutionary theory and so offers an explanation for differences in aging rate and presentation between species. It also provides a means to account for how variation in levels of constraint influence presentation of proximate mechanisms of aging. For a list of the types of constraint associated with AP and aging and associated examples, see (Gems and Kern, 2022). The second article focuses on explaining effects of interventions into the aging process on population survival curves, challenging current theory on biodemography (Kern and Gems, 2023a). Finally, this review presents a new programmatic theory of aging, and in doing so integrates the evolutionary theory of aging to the aetiology of age-related disease.
In recent decades aging research has largely focused on the accumulation of molecular damage and loss of information and cell function as the primary cause of aging and late-life disease. This view has predominated in biogerontology, evolutionary biology (e.g. the disposable soma theory, Figure 1B) and, to some extent, in studies of individual diseases of aging (Kirkwood, 2005; Sohal and Weindruch, 1996). This focus has been relatively fruitless in terms of a general account of the origins of actual diseases. Molecular damage accumulation and deficiency in cellular maintenance seem insufficient to explain the wide spectrum of late-life afflictions, such as rheumatoid arthritis, osteoporosis and T2D. For most diseases of aging (though not cancer), molecular damage appears to play only a secondary role: it is a consequence rather than a cause of senescence (Figure 3). Programmatic theories of aging provide an alternative ultimate-proximate mechanism to the previous damage-centric ones, but to date they have shared the same limitation: not being able to explain age-related disease aetiology.
In this article what is described is a a new blueprint-style account of the principles of aetiology governing much of senescent disease. Biological organisms can be likened to machines, where all functions cannot be optimised at once, resulting in desperate trade-offs - rather than being blind, the watchmaker is forced to compromise. But here the real danger is domino style system collapse through triggered quasi-programmes and quasi-programme cascades.
Glossary
Antagonistic pleiotropy (AP): Where action of a given gene is both beneficial and detrimental to fitness. If the latter occurs later in life and is therefore subject to weaker selection, such a gene may be favoured by natural selection, and promote aging (Williams, 1957).
Biological constraint: A property of organisms and/or their ecology that prevents the evolution of traits that would increase fitness.
Damage/maintenance paradigm: Theory that aging is largely caused by accumulation of molecular damage, which can be prevented by somatic maintenance functions. Various theories of aging are based on this broad assumption.
Discrete stage quasi-programme cascade (New term): A series of quasi-programmes triggered in a causal chain where quasi-programme are temporally or spatially separated from one another (c.f. multi-stage quasi-programme cascade).
Disposable soma theory: Theory proposing that natural selection favors investment of limited resources into reproduction rather than somatic maintenance, accelerating damage accumulation and, therefore, senescence (Kirkwood, 1977).
Hyperfunction: Where wild-type gene function actively leads to senescent pathology, as opposed to passive random damage or wear and tear (Blagosklonny, 2006).
Multi-stage quasi-programme cascade (New term): Where a single quasi-programme driving pathology progression involves a cascade of distinguishable stages (c.f. discrete stage quasi-programme cascade).
Programmed aging: Where complex biological processes contributes to senescence, but not necessarily to fitness (cf. quasi-programmes and triggered quasi-programmes).
Quasi-programmed aging: Senescence caused by futile gene action. This can arise from a futile run-on of wild-type programmes that promote fitness earlier in life (Blagosklonny, 2006), or (new addition), as argued in this review, may be triggered by other factors (programmatic or non-programmatic) like infection or injury. Tissue may undergoe changes with age that prime it and make it more susceptible to triggers.
Run on: Futile continuation of gene function or processes in later life, leading to pathology (cf. quasi-programme).
Signaling constraint (new term): Where a given signaling molecule (e.g. hormone or growth factor, receptor, signaling kinase, transcription factor) acts in diverse contexts (cell types, tissues, organs), such that optimization of function in all contexts is not possible.
Selection shadow: Decrease in selection with increasing age, leading to weaker selection against genes with deleterious effects on fitness and health the later in life those effects are expressed. Environmental factors that increase mortality rate (e.g. predators, infectious pathogens, starvation) can deepen the selection shadow.
Senescence: The overall process of deterioration with age or the resulting pathological condition (not to be confused with cellular senescence (sensu Hayflick), which is a particular form of cell cycle arrest affecting some vertebrate cell types). Although aging has several meanings, in the biological context it is usually synonymous with senescence.
Quasi-programme cascade (New term): A causal chain of quasi-programmes, in which each triggers the next in the chain (c.f. multi-stage quasi-programme cascade and discrete quasi-programme cascade).
Ultimate-proximate theories of aging: These combine explanations of the evolutionary (ultimate) and mechanistic (proximate) causes of aging into a single integrated account.
Wild-type: Genes maintained in the population, i.e. non atypical mutant.
References
- Abedin, M.; Tintut, Y. and Demer, L.L. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. 2004, 24, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Angulo, P. and Lindor, K.D. Nonalcoholic fatty liver disease. Canad. Med. Assoc. J 2005, 172, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, L.E.; Rinaldi, L.; Guerrera, B.; Restivo, L.; Marrone, A.; Giordano, M. and Zampino, R.; NAFLD and NASH in HCV Infection: Prevalence and Significance in Hepatic and Extrahepatic Manifestations. Int J Mol Sci 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Admasu, T.D.; Rae, M. and Stolzing, A. Dissecting primary and secondary senescence to enable new senotherapeutic strategies. Ageing Res Rev 2021, 70, 10. [Google Scholar] [CrossRef]
- Al-Maskari, A.Y.; Al-Maskari, M.Y. and Al-Sudairy, S. Oral Manifestations and Complications of Diabetes Mellitus: A review. Sult. Qab. Uni. med. J 2021, 11, 179–186. [Google Scholar]
- Al Anouti, F.; Taha, Z.; Shamim, S.; Khalaf, K.; Al Kaabi, L. and Alsafar, H. An insight into the paradigms of osteoporosis: From genetics to biomechanics. Bone Rep 2019, 11, 100216. [Google Scholar] [CrossRef] [PubMed]
- Angeles-Albores, D.; Lee, R.; Chan, J. and Sternberg, P. Tissue enrichment analysis for C. elegans genomics. BMC Bioinformatics 2016, 17, 366. [Google Scholar]
- Armstrong, R.A. What causes Alzheimer's disease? Folia Neuropathol. 2013, 51, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Austad, S. and Hoffman, J. Is antagonistic pleiotropy ubiquitous in aging biology? Evol Med Public Health 2018, 287–294. [Google Scholar] [CrossRef]
- Austad, S.N. Menopause: An evolutionary perspective. Exp Gerontol 1994, 29, 255–263. [Google Scholar] [CrossRef]
- Berbudi, A.; Rahmadika, N.; Tjahjadi, A.I. and Ruslami, R. Type 2 diabetes and its impact on the immune system. Curr. Diabetes Rev 2020, 16, 442–449. [Google Scholar] [PubMed]
- Bhardwaj, A. and Bhardwaj, S.V. Effect of menopause on women's periodontium. J. Midliife Health 2012, 3, 5–9. [Google Scholar]
- Blagosklonny, M.V. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle 2006, 5, 2087–2102. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Aging: ROS or TOR. Cell Cycle 2008, 7, 3344–3354. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Answering the ultimate question "what is the proximal cause of aging? ". Aging (Albany NY) 2012, 4, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Rapamycin for longevity: opinion article. Aging (Albany NY) 2019, 11, 8048–8067. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. The hyperfunction theory of aging: three common misconceptions. Oncoscience 2021, 8, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Castañeda, S.; Largo, R.; Fernández-Valle, M.E.; Rodríguez-Salvanés, F. and Herrero-Beaumont, G. Osteoporosis increases the severity of cartilage damage in an experimental model of osteoarthritis in rabbits. Osteoarthritis and Cartilage 2007, 15, 69–77. [Google Scholar] [CrossRef]
- Chen, N.X. and Moe, S.M. Vascular calcification: pathophysiology and risk factors. Curr. Hypertens. Rep 2012, 14, 228–237. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, R.; Xiong, Y.; Du, F. and Zhu, S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids in Health and Disease 2017, 16, 203. [Google Scholar] [CrossRef]
- Chughtai, B.; Forde, J.C.; Thomas, D.D.; Laor, L.; Hossack, T.; Woo, H.H.; Te, A.E. and Kaplan, S.A. Benign prostatic hyperplasia. Nat Rev Dis Primers 2016, 2, 16031. [Google Scholar] [CrossRef] [PubMed]
- Cinar, Y.; Senyol, A.M. and Duman, K. Blood viscosity and blood pressure: role of temperature and hyperglycemia. Am J Hypertens 2001, 14, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.S.; Bynum, M.L. and Somers, E.C. Recent insights in the epidemiology of autoimmune diseases: improved prevalence estimates and understanding of clustering of diseases. J Autoimmun 2009, 33, 197–207. [Google Scholar] [CrossRef] [PubMed]
- de Magalhaes, J. and Church, G. Genomes optimize reproduction: aging as a consequence of the developmental program. Physiology 2005, 20, 252–259. [Google Scholar] [CrossRef] [PubMed]
- de Magalhães, J.P. Open-minded scepticism: inferring the causal mechanisms of human ageing from genetic perturbations. Ageing Res. Rev 2005, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- de Magalhães, J.P. and Church, G.M. Genomes optimize reproduction: aging as a consequence of the developmental program. Physiology 2005, 20, 252–259. [Google Scholar] [CrossRef]
- Deepa, D. and Jain, G. Assessment of periodontal health status in postmenopausal women visiting dental hospital from in and around Meerut city: Cross-sectional observational study. J Midlife Health 2016, 7, 175–179. [Google Scholar] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.; Rodier, F.; Toussaint, W.; Mitchell, J.; Laberge, R.; Vijg, J.; Van Steeg, H.; Dollé, M.; Hoeijmakers, J.; de Bruin, A.; Hara, E. and Campisi, J. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Demer, L.L. Vascular calcification and osteoporosis: inflammatory responses to oxidized lipids. International Journal of Epidemiology 2002, 31, 737–741. [Google Scholar] [CrossRef]
- Dilman, V.M. Ontogenetic model of ageing and disease formation and mechanisms of natural selection. J Theor Biol 1986, 118, 73–81. [Google Scholar] [CrossRef]
- Dilman, V.M. Development, Aging and Disease: A New Rationale for an Intervention Strategy Harwood Academic Publishers: 1994.
- Distler, J.H.W.; Wenger, R.H.; Gassmann, M.; Kurowska, M.; Hirth, A.; Gay, S. and Distler, O. Physiologic responses to hypoxia and implications for hypoxia-inducible factors in the pathogenesis of rheumatoid arthritis. Arthritis & Rheumatism 2004, 50, 10–23. [Google Scholar]
- Epplein, M.; Reed, S.D.; Voigt, L.F.; Newton, K.M.; Holt, V.L. and Weiss, N.S. Risk of Complex and Atypical Endometrial Hyperplasia in Relation to Anthropometric Measures and Reproductive History. Am. J Epidem 2008, 168, 563–570. [Google Scholar] [CrossRef]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M. and Feil, R. Transdifferentiation of Vascular Smooth Muscle Cells to Macrophage-Like Cells During Atherogenesis. Circulation Research 2014, 115, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Gems, D. Understanding hyperfunction: an emerging paradigm for the biology of aging. Preprints. 2021. [Google Scholar] [CrossRef]
- Gems, D. The hyperfunction theory: an emerging paradigm for the biology of aging. Ageing. Res. Rev 2022, 74, 101557. [Google Scholar] [CrossRef] [PubMed]
- Gems, D. and de la Guardia, Y. Alternative perspectives on aging in C. elegans: reactive oxygen species or hyperfunction? Antioxid Redox Signal 2013, 19, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Gems, D. and Doonan, R. Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong? Cell Cycle 2009, 8, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Gems, D. and Kern, C. C. Biological constraint as a cause of aging. In preparation. 2022.
- Gems, D.; Kern, C.C.; Nour, J. and Ezcurra, M. Semelparity and reproductive death in Caenorhabditis elegans. Preprints 2020, 2020110019. [Google Scholar]
- Glocker, M.O.; Guthke, R.; Kekow, J. and Thiesen, H.J. Rheumatoid arthritis, a complex multifactorial disease: on the way toward individualized medicine. Med Res Rev 2006, 26, 63–87. [Google Scholar] [CrossRef]
- Glyn-Jones, S.; Palmer, A.J.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H. and Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Hadi, H.A.R. and Suwaidi, J.A. Endothelial dysfunction in diabetes mellitus. Vasc. Health Risk Manag 2007, 3, 853–876. [Google Scholar] [PubMed]
- Haldane, J.B.S. New Paths in Genetics Allen and Unwin, London. 1941. [Google Scholar]
- Hamilton, W.D. The moulding of senescence by natural selection. J. Theor. Biol 1966, 12, 12–45. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, M.M.; Alexander, H.M.; Cope, N.J. and Acheson, N. Endometrial hyperplasia: a clinician's review. Obst. Gyn. & Reproduct. Med 2007, 17, 169–172. [Google Scholar]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; Pahor, M.; Javors, M.A.; Fernandez, E. and Miller, R.A. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Hu, Q.; Xu, X.; Niu, Y.; Chen, Y.; Lu, Y.; Su, Q. and Qin, L. Advanced glycation end products enhance M1 macrophage polarization by activating the MAPK pathway. Biochem Biophys Res Commun 2020, 525, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M. and Becher, B. Immune attack: the role of inflammation in Alzheimer disease. Nature Reviews Neuroscience 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Hibler, E.A.; Kauderer, J.; Greene, M.H.; Rodriguez, G.C. and Alberts, D. S. Bone loss after oophorectomy among high-risk women: an NRG oncology/gynecologic oncology group study. Menopause (New York, N.Y.) 2016, 23, 1228–1232. [Google Scholar]
- Higashi, Y.; Gautam, S.; Delafontaine, P. and Sukhanov, S.; 2019. IGF-1 and cardiovascular disease. Growth Horm. IGF Res 2021, 45, 6–16. [Google Scholar]
- Holstein, J.H.; Klein, M.; Garcia, P.; Histing, T.; Culemann, U.; Pizanis, A.; Laschke, M.W.; Scheuer, C.; Meier, C.; Schorr, H.; Pohlemann, T. and Menger, M.D. Rapamycin affects early fracture healing in mice. British journal of pharmacology 2008, 154, 1055–1062. [Google Scholar] [CrossRef]
- Horikawa, A.; Miyakoshi, N.; Shimada, Y. and Kodama, H. The Relationship between Osteoporosis and Osteoarthritis of the Knee: A Report of 2 Cases with Suspected Osteonecrosis. Case Rep Orthop 2014, 2014, 514058. [Google Scholar] [PubMed]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; von Schoenfels, W.; Ahrens, M.; Heits, N.; Bell, J.T.; Tsai, P.C.; Spector, T.D.; Deloukas, P.; Siebert, R.; Sipos, B.; Becker, T.; Roecken, C.; Schafmayer, C. and Hampe, J. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci U S A 2014, 111, 15538–15543. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A. and Palange, P. COPD: a multifactorial systemic disease. Ther. Adv. Respir. Dis 2011, 5, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.A. Mutation and the evolution of ageing: from biometrics to system genetics. Phil. Trans. R. Soc. B 2010, 365, 1273–1279. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M. and Tejera, D. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Iwabuchi, H.; Fujibayashi, T.; Yamane, G.; Imai, H. and Nakao, H. Relationship between Hyposalivation and Acute Respiratory Infection in Dental Outpatients. Gerontology 2012, 58, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Kay, J. and Gravallese, E.M. Bone damage in rheumatoid arthritis: mechanistic insights and approaches to prevention. Rheum. Dis. Clin. North Am 2010, 36, 385–404. [Google Scholar] [CrossRef] [PubMed]
- Kern, C. and Gems, D. How biological mechanisms shape population dynamics and lifespan through ageing. In preperation.. 2023. [Google Scholar]
- Kern, C.C. Ageing through reproductive death in Caenorhabditis elegans. (UCL) University College London:. 2020. [Google Scholar]
- Kern, C.C. and Gems, D. Semelparous Death as one Element of Iteroparous Aging Gone Large. Front Genet 2022, 13, 880343. [Google Scholar] [CrossRef]
- Kern, C.C. and Gems, D. Unifying theory: Life history strategy, trade-offs and constraints shape ageing mechanisms and pathology. In preparation. 2023b.
- Kern, C.C.; Morley, H. and Gems, D. 2023a. Lactation reactivation syndrome. In preparation. 2023b.
- Kern, C.C.; Srivastava, S.; Ezcurra, M.; Hsiung, K.C.; Hui, N.; Townsend, S.; Maczik, D.; Zhang, B.; Tse, V.; Konstantellos, V.; Bähler, J. and Gems, D. C. elegans ageing is accelerated by a self-destructive reproductive programme. Nature Communications. 2023, 14, 4381. [Google Scholar] [PubMed]
- Kern, C.C.; Townsend, S.; Salzmann, A.; Rendell, N.; Taylor, G.; Comisel, R.M.; Foukas, L.; Bähler, J. and Gems, D. C. elegans feed yolk to their young in a form of primitive lactation. Nature Communications 2021, 12, 5801. [Google Scholar] [CrossRef]
- Kirkwood, T.B. Understanding the odd science of aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B.L. Evolution of ageing. Nature 1977, 270, 301–304. [Google Scholar] [CrossRef]
- Kovacs, C.S. Maternal mineral and bone metabolism during pregnancy, lactation, and post-weaning recovery. Physiological reviews 2016. [Google Scholar]
- Liu, H.F.; Zhang, H.J.; Hu, Q.X.; Liu, X.Y.; Wang, Z.Q.; Fan, J.Y.; Zhan, M. and Chen, F.L. Altered polarization, morphology, and impaired innate immunity germane to resident peritoneal macrophages in mice with long-term type 2 diabetes. J Biomed Biotechnol 2012, 2012, 867023. [Google Scholar] [CrossRef]
- Madersbacher, S.; Sampson, N. and Culig, Z. Pathophysiology of benign prostatic hyperplasia and benign prostatic enlargement: A mini-review. Gerontology 2019, 65, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Maklakov, A.A. and Chapman, T. Evolution of ageing as a tangle of trade-offs: energy versus function. Proc Biol Sci 2019, 286, 20191604. [Google Scholar]
- Mancuso, P. and Bouchard, B. The impact of aging on adipose function and adipokine synthesis. Front. Endocrinol 2019, 10, 137–137. [Google Scholar] [CrossRef]
- Marlowe, F. The patriarch hypothesis: An alternative explanation of menopause. Human Nature 2000, 11, 27–42. [Google Scholar] [CrossRef]
- Martin, G. Genetic syndromes in man with potential relevance to the pathobiology of aging. Birth Defects Orig Artic Ser 1978, 14, 5–39. [Google Scholar]
- Mauvais-Jarvis, F.; Manson, J.E.; Stevenson, J.C. and Fonseca, V.A. Menopausal Hormone Therapy and Type 2 Diabetes Prevention: Evidence, Mechanisms, and Clinical Implications. Endo. revs 2017, 38, 173–188. [Google Scholar] [CrossRef]
- McNulty, P.; Pilcher, R.; Ramesh, R.; Necuiniate, R.; Hughes, A.; Farewell, D.; Holmans, P.; Jones, L. and Network, R.I.o.t.E.H.s.D. Reduced cancer incidence in Huntington's disease: analysis in the registry study. J. Huntingtons Dis 2018, 7, 209–222. [Google Scholar]
- Medawar, P.B. An Unsolved Problem Of Biology H.K. Lewis, London. 1952. [Google Scholar]
- More, C.; Bettembuk, P.; Bhattoa, H.P. and Balogh, A. The effects of pregnancy and lactation on bone mineral density. Osteoporos Int 2001, 12, 732–737. [Google Scholar] [CrossRef]
- Nacusi, L.P. and Tindall, D.J. Targeting 5alpha-reductase for prostate cancer prevention and treatment. Nat Rev Urol 2011, 8, 378–384. [Google Scholar] [CrossRef]
- NCI. Surveillance, Epidemiology, and End Results Program http://seer.cancer. 2023. Available online: http://seer.cancer.gov/.
- Nussey, D.H.; Froy, H.; Lemaitre, J.F.; Gaillard, J.M. and Austad, S. N. Senescence in natural populations of animals: widespread evidence and its implications for bio-gerontology. Ageing Res Rev 2013, 12, 214–225. [Google Scholar]
- Paschou, S.A. and Papanas, N. Type 2 diabetes mellitus and menopausal hormone therapy: an update. Diabetes Ther 2019, 10, 2313–2320. [Google Scholar] [CrossRef]
- Pavlou, S.; Lindsay, J.; Ingram, R.; Xu, H. and Chen, M. Sustained high glucose exposure sensitizes macrophage responses to cytokine stimuli but reduces their phagocytic activity. BMC Immunol 2018, 19, 24. [Google Scholar] [CrossRef]
- Pérez-Ros, P.; Navarro-Flores, E.; Julián-Rochina, I.; Martínez-Arnau, F.M. and Cauli, O. Changes in Salivary Amylase and Glucose in Diabetes: A Scoping Review. Diagnostics (Basel).
- Pollreisz, A. and Schmidt-Erfurth, U. Diabetic cataract-pathogenesis, epidemiology and treatment. J Ophthalmol 2010, 2010, 608751. [Google Scholar]
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T. and Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Seminars in immunology 2018, 40, 101275–101275. [Google Scholar] [CrossRef]
- Robert, G.; Descazeaud, A.; Nicolaïew, N.; Terry, S.; Sirab, N.; Vacherot, F.; Maillé, P.; Allory, Y. and de la Taille, A. Inflammation in benign prostatic hyperplasia: a 282 patients' immunohistochemical analysis. Prostate 2009, 69, 1774–1780. [Google Scholar]
- Robertson, O.H. Prolongation of the life span of kokanee salmon (Oncorhynchus nerka kennerlyi) by castration before the beginning of gonad development. Proc. Natl. Acad. Sci. USA 1961, 47, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Shapiro, M.; Trogan, E. and Fisher, E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [PubMed]
- Rose, M. and Charlesworth, B. A test of evolutionary theories of senescence. Nature 1980, 287, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Tintut, Y. and Demer, L.L. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol 2010, 7, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.; Woodbury, M. and Srivastava, G. The major risk factors for Alzheimer’s disease: age, sex and genes modulate the microglia response to Aβ plaques. Cell Rep 2019, 27, 1293–1306. [Google Scholar]
- Salvestrini, V.; Sell, C. and Lorenzini, A. Obesity may accelerate the aging process. Front. Endocrinol 2019, 10, 266. [Google Scholar] [CrossRef] [PubMed]
- Sapey, E.; Greenwood, H.; Walton, G.; Mann, E.; Love, A.; Aaronson, N.; Insall, R.H.; Stockley, R.A. and Lord, J.M. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood 2014, 123, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Sears, B. and Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis 2015, 14, 121–121. [Google Scholar] [CrossRef]
- Shields, H.J.; Traa, A. and Van Raamsdonk, J.M. Beneficial and detrimental effects of reactive oxygen species on lifespan: a comprehensive review of comparative and experimental studies. Front. Cell Dev. Biol 2021, 9, 628157. [Google Scholar] [CrossRef] [PubMed]
- Shokeir, M. Investigation on Huntington’s disease in the Canadian Prairies. II. Fecundity and fitness. Clin. Genet 1975, 7, 349–353. [Google Scholar]
- Sohal, R.S. and Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar] [CrossRef]
- Sorenson, S.A.; Fenger, K. and Olsen, J. Significantly lower incidence of cancer among patients with Huntington’s disease. Cancer 1999, 6, 1342–1346. [Google Scholar] [CrossRef]
- Strenk, S.A.; Strenk, L.M. and Koretz, J.F. The mechanism of presbyopia. Prog. Retin. Eye Res 2005, 24, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 2003, 144, 5159–5165. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, G.; Madersbacher, S. and Berger, P. Benign prostatic hyperplasia: age-related tissue-remodeling. Exp Gerontol 2005, 40, 121–128. [Google Scholar] [CrossRef] [PubMed]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Cellular Pathology, as Based upon Physiological and Pathological Histology Dover Publications, Inc, New York, NY. 1863. [Google Scholar]
- Voynow, J.A. and Shinbashi, M. Neutrophil Elastase and Chronic Lung Disease. Neutrophil Elastase and Chronic Lung Disease. Biomolecules 2021, 11. [Google Scholar]
- Walker, D.A.; Harper, P.S.; Newcombe, R.G. and Davies, K. Huntington's chorea in South Wales: mutation, fertility, and genetic fitness. J. Med. Genet 1983, 20, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y. and Zhou, J. Neuroinflammation in Parkinson's disease and its potential as therapeutic target. Transl Neurodegener 2015, 4, 19. [Google Scholar] [CrossRef]
- Waters, D.J.; Shen, S. and Glickman, L.T. Life expectancy, antagonistic pleiotropy, and the testis of dogs and men. The Prostate 2000, 43, 272–277. [Google Scholar] [CrossRef]
- Whiteside, S.K.; Snook, J.P.; Williams, M.A. and Weis, J.J. Bystander T Cells: A Balancing Act of Friends and Foes. Trends Immunol 2018, 39, 1021–1035. [Google Scholar] [CrossRef]
- Wilkinson, J.E.; Burmeister, L.; Brooks, S.V.; Chan, C.C.; Friedline, S.; Harrison, D.E.; Hejtmancik, J.F.; Nadon, N.; Strong, R.; Wood, L.K.; Woodward, M.A. and Miller, R.A. Rapamycin slows aging in mice. Aging Cell 2012, 11, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.C. Pleiotropy, natural selection and the evolution of senescence. Evolution 1957, 11, 398–411. [Google Scholar] [CrossRef]
- Xie, J.; Van Hoecke, L. and Vandenbroucke, R.E. The Impact of Systemic Inflammation on Alzheimer's Disease Pathology. Front Immunol 2021, 12, 796867. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Theories of aging and the origins of late-life disease. A, A simplified, two cause scheme for understanding aging as a multifactorial process, drawing on the programmatic theory: disruption of wild-type function (left), and wild-type function itself (due to the evolution of aging, particularly effects of AP) (right) (Dilman, 1994; Gems, 2022). B, Two ultimate-proximate theories of aging (simplified schemes). Top, the force of natural selection declines with advancing age (selection shadow). Middle, the disposable soma theory. Here senescence is caused by stochastic molecular damage accumulation, and the inadequacy of somatic maintenance mechanisms that could prevent it (Kirkwood, 1977). Constraint results from limited resource availability to optimize both reproduction and somatic maintenance. Bottom, Current programmatic theories (hyperfunction and developmental theories). Here senescence results from non-adaptive continuation or activation of wild-type biological programmes (Blagosklonny, 2006; de Magalhães, 2005; Gems, 2022; Maklakov and Chapman, 2019). C, Different views of the role of molecular damage accumulation in aging. Left, traditional view: molecular damage accumulation is the major, primary cause of aging. Right, alternative programmatic theory view: the major cause of aging is programmatic (i.e. futile gene and cell action). The observed increases in molecular damage with advancing age are largely a consequence of late-stage pathology.
Figure 1.
Theories of aging and the origins of late-life disease. A, A simplified, two cause scheme for understanding aging as a multifactorial process, drawing on the programmatic theory: disruption of wild-type function (left), and wild-type function itself (due to the evolution of aging, particularly effects of AP) (right) (Dilman, 1994; Gems, 2022). B, Two ultimate-proximate theories of aging (simplified schemes). Top, the force of natural selection declines with advancing age (selection shadow). Middle, the disposable soma theory. Here senescence is caused by stochastic molecular damage accumulation, and the inadequacy of somatic maintenance mechanisms that could prevent it (Kirkwood, 1977). Constraint results from limited resource availability to optimize both reproduction and somatic maintenance. Bottom, Current programmatic theories (hyperfunction and developmental theories). Here senescence results from non-adaptive continuation or activation of wild-type biological programmes (Blagosklonny, 2006; de Magalhães, 2005; Gems, 2022; Maklakov and Chapman, 2019). C, Different views of the role of molecular damage accumulation in aging. Left, traditional view: molecular damage accumulation is the major, primary cause of aging. Right, alternative programmatic theory view: the major cause of aging is programmatic (i.e. futile gene and cell action). The observed increases in molecular damage with advancing age are largely a consequence of late-stage pathology.
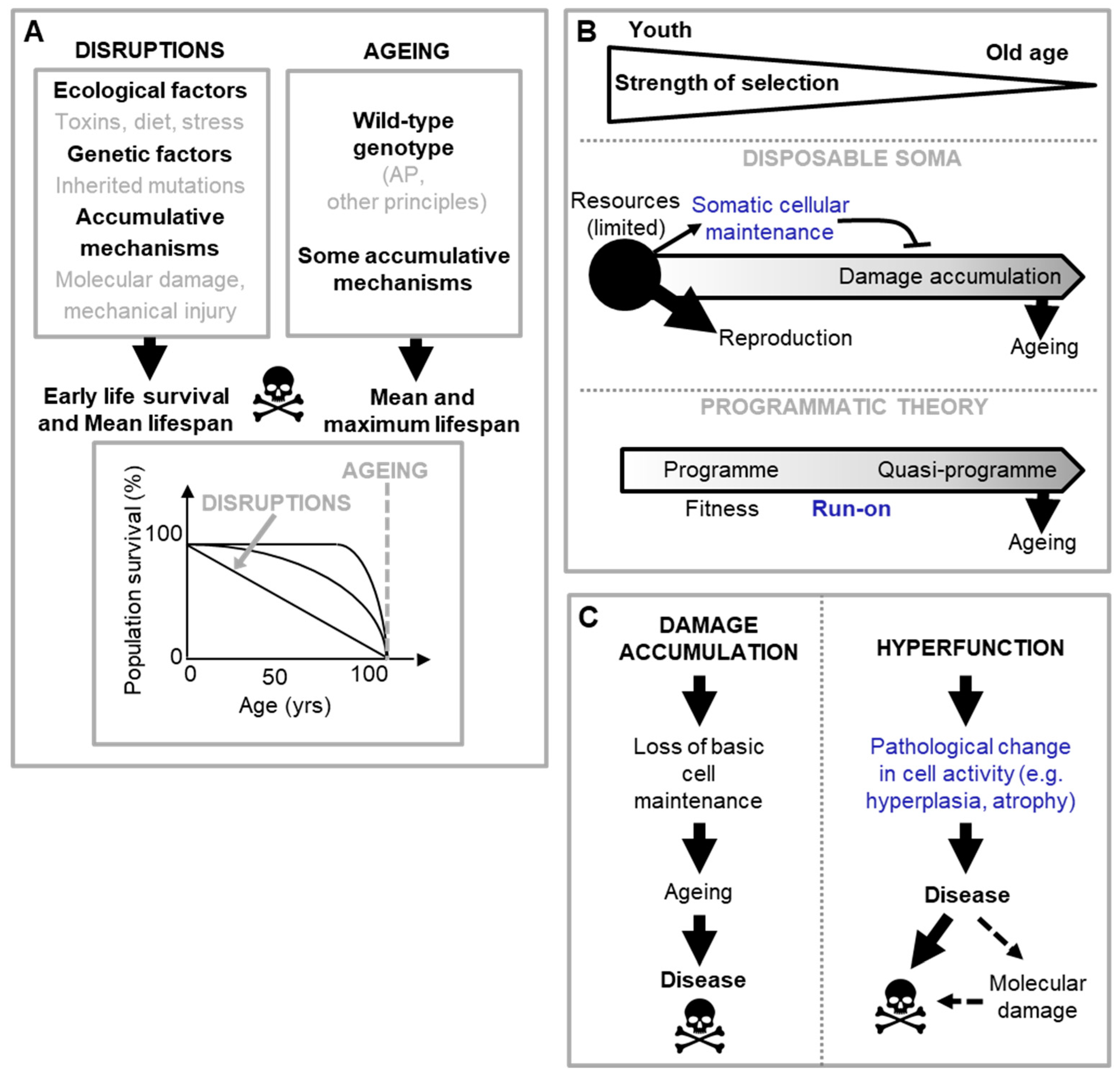
Figure 2.
Different origins of pathological programmatic change. A, Quasi-programmes resulting from developmental run on, e.g. presbyopia. B, Futile quasi-programmes can originate from developmental run on, or be triggered in later life. C, An example of a triggered quasi-programme. Listed triggers: elevate oestrogen to progesterone ratio causing endometrial hyperplasia, increasing risk of endometrial carcinoma. Inflammatory changes also increase this likelihood.
Figure 2.
Different origins of pathological programmatic change. A, Quasi-programmes resulting from developmental run on, e.g. presbyopia. B, Futile quasi-programmes can originate from developmental run on, or be triggered in later life. C, An example of a triggered quasi-programme. Listed triggers: elevate oestrogen to progesterone ratio causing endometrial hyperplasia, increasing risk of endometrial carcinoma. Inflammatory changes also increase this likelihood.
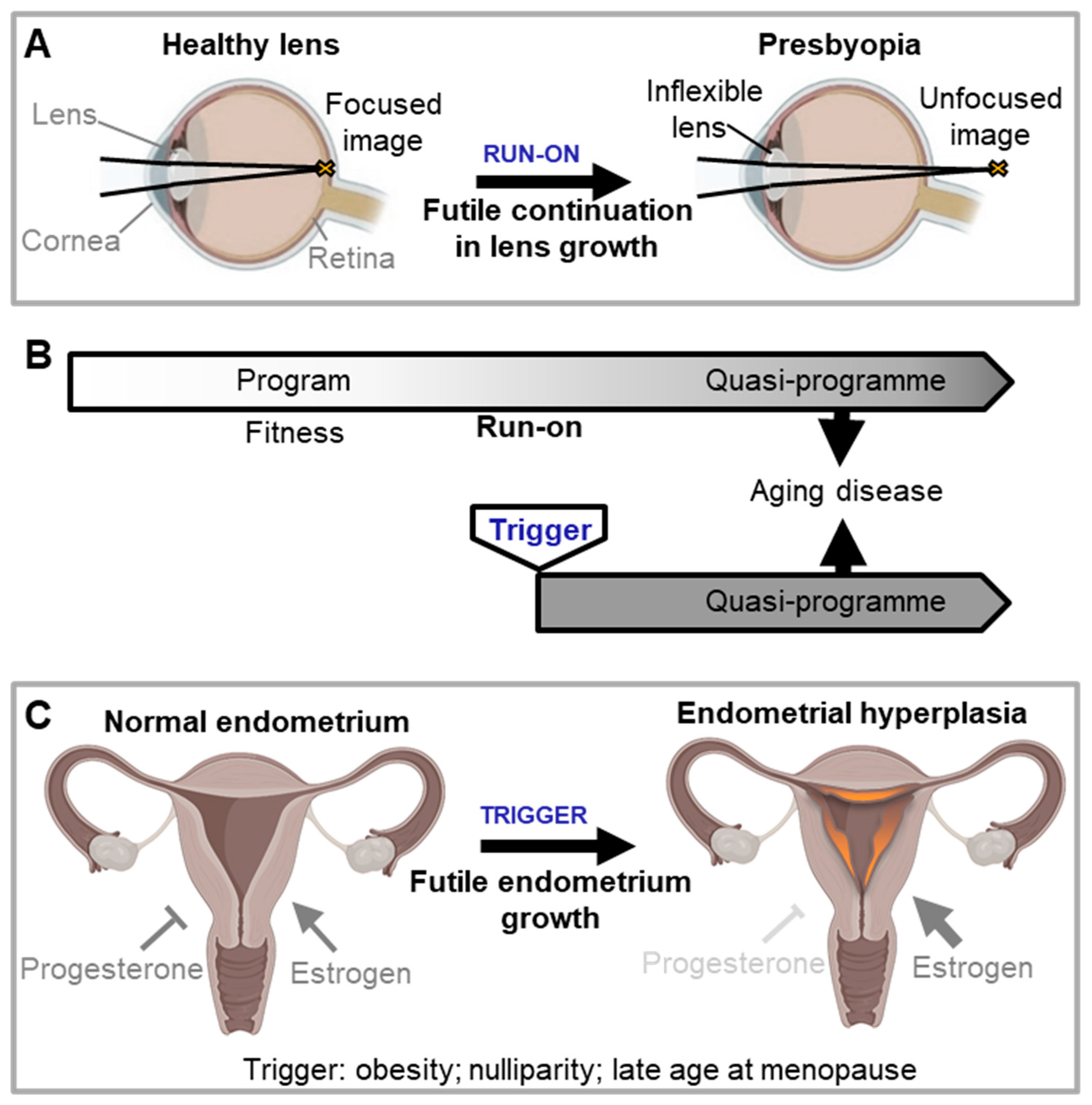
Figure 3.
Cascades of primary and secondary quasi-programmes promote organismal senescence (hypothetical scheme). Other forms of AP that may contribute to aging directly, or indirectly by increasing tissue susceptibility to triggered quasi-programmes, include costly programmes. As an example of the latter, high fever protects against infection but can cause hyperthermic injury as a costly programme. Disruptions can directly contribute to pathology as well as increase tissue susceptibility to triggers.
Figure 3.
Cascades of primary and secondary quasi-programmes promote organismal senescence (hypothetical scheme). Other forms of AP that may contribute to aging directly, or indirectly by increasing tissue susceptibility to triggered quasi-programmes, include costly programmes. As an example of the latter, high fever protects against infection but can cause hyperthermic injury as a costly programme. Disruptions can directly contribute to pathology as well as increase tissue susceptibility to triggers.
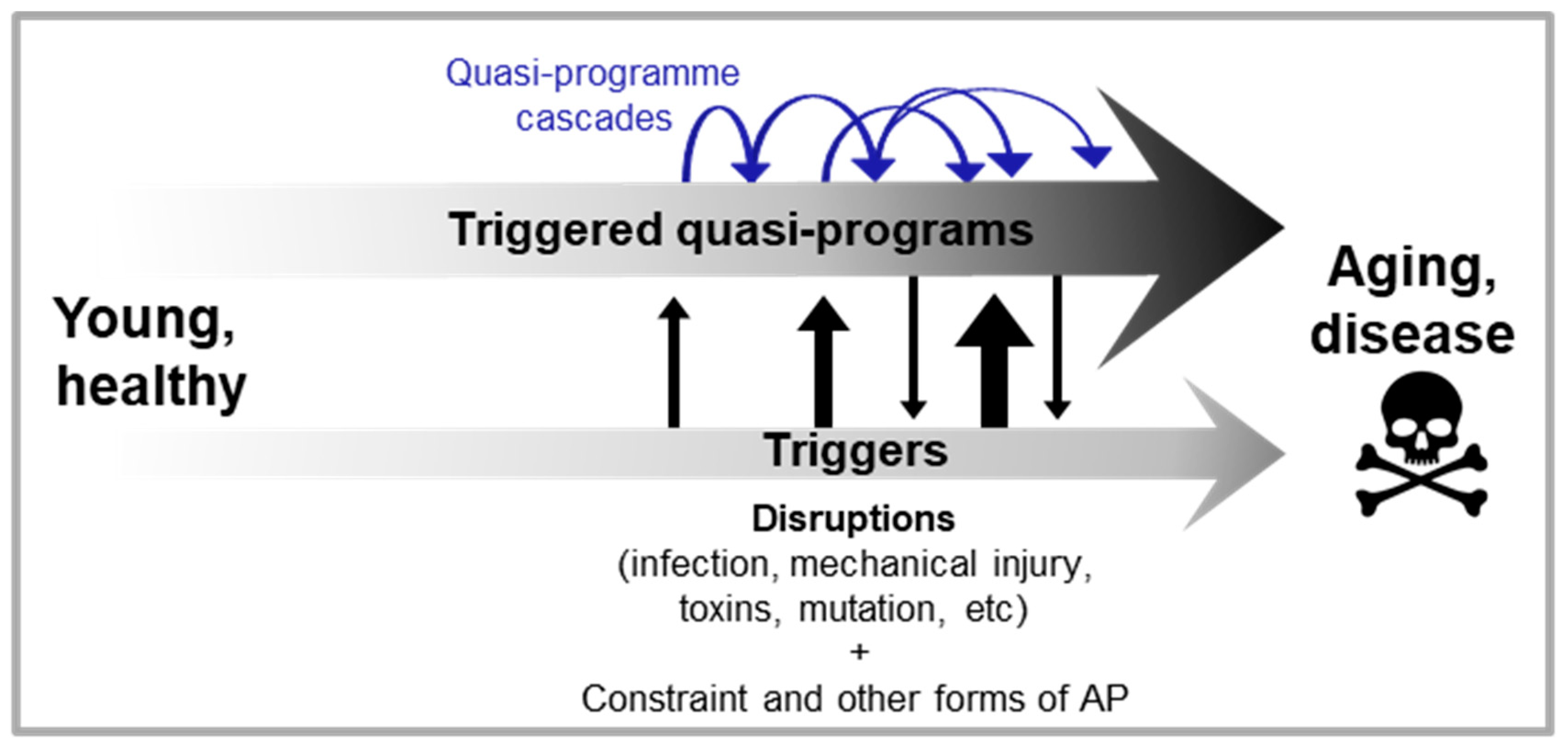
Figure 4.
Multi-stage and discrete stage tripped-programme cascades. Both programmatic and disruption aetiologies are included (cf. Figure 1A). A, B, Multi-stage tripped-programme cascades. A, Rheumatoid arthritis. B, Non-alcoholic fatty liver disease. Hepatic insulin resistance and continued free fatty acid (FFA) uptake generates an imbalance between factors that favour hepatic lipid accumulation (e.g. lipid influx and de novo lipid synthesis) and ones that reduce lipid build-up (e.g. lipid export or oxidation). C, D, Discrete stage tripped-programme cascades. C, Calcium and phosphate movement into blood leads to vascular calcification. D, Lipotoxicity in NAFLD increases susceptibility to injury, resulting in further inflammation, scarring, and non-alcoholic steatohepatitis (NASH).
Figure 4.
Multi-stage and discrete stage tripped-programme cascades. Both programmatic and disruption aetiologies are included (cf. Figure 1A). A, B, Multi-stage tripped-programme cascades. A, Rheumatoid arthritis. B, Non-alcoholic fatty liver disease. Hepatic insulin resistance and continued free fatty acid (FFA) uptake generates an imbalance between factors that favour hepatic lipid accumulation (e.g. lipid influx and de novo lipid synthesis) and ones that reduce lipid build-up (e.g. lipid export or oxidation). C, D, Discrete stage tripped-programme cascades. C, Calcium and phosphate movement into blood leads to vascular calcification. D, Lipotoxicity in NAFLD increases susceptibility to injury, resulting in further inflammation, scarring, and non-alcoholic steatohepatitis (NASH).
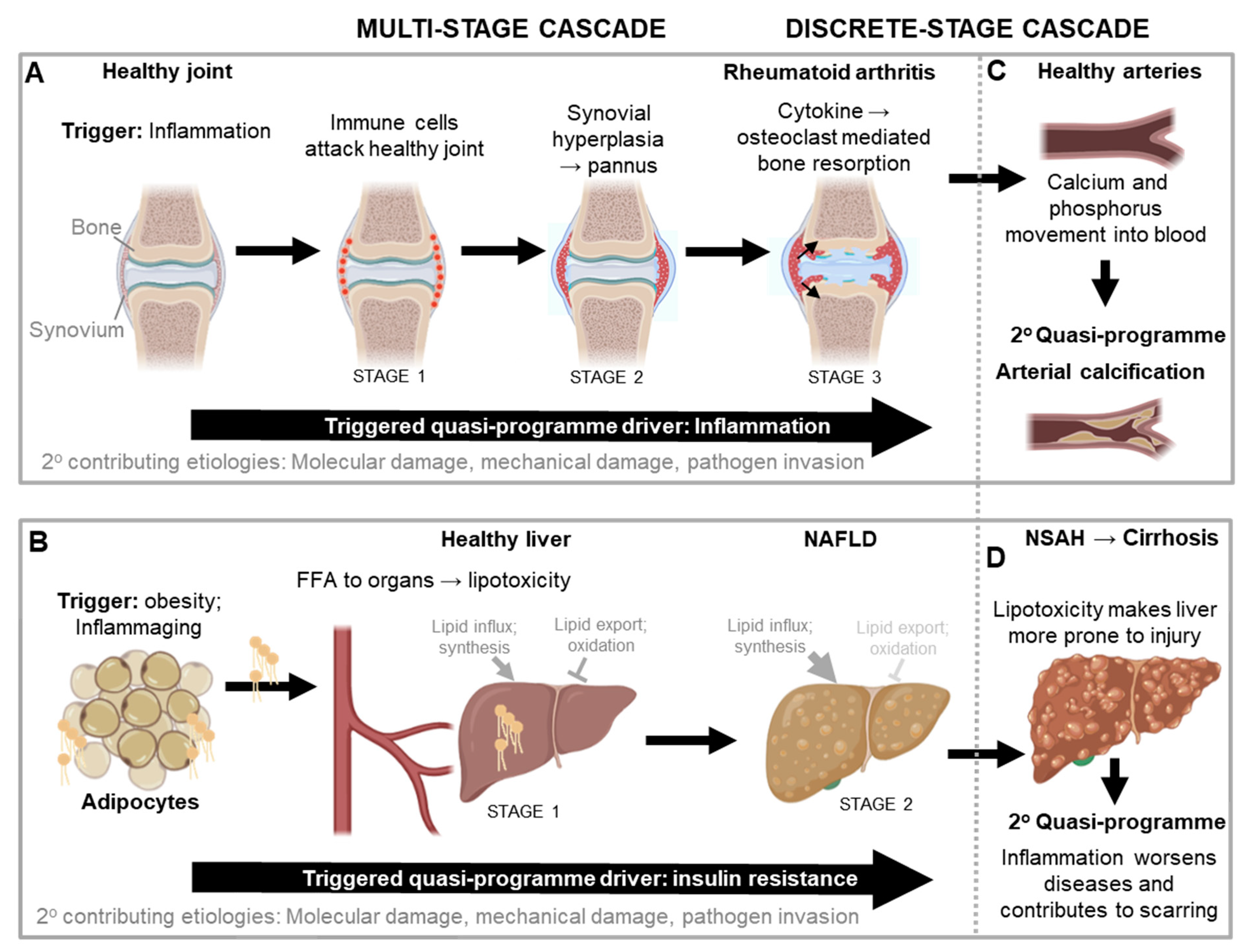
Figure 5.
A blueprint model for the causes of aging and late-life disease (hypothetical scheme). The main cause of programmatic aging is the evolutionary process, in which biological constraint plays a major role. Due to the presence of constraint and late-life selection shadows, the compromises made by the evolutionary process generates late-life cost which manifest as diseases of aging. Broadly speaking, two main elements combine to cause aging and its attendant diseases: disruptions and programmatic changes specified by the wild-type genome. Diseases of aging typically arise from combinations of disruptions and programmatic changes, but can also arise from the latter alone. Programmatic changes include costly programmes, and triggered quasi-programmes that may result from run on or, arguably more often, by being triggered in later life.
Figure 5.
A blueprint model for the causes of aging and late-life disease (hypothetical scheme). The main cause of programmatic aging is the evolutionary process, in which biological constraint plays a major role. Due to the presence of constraint and late-life selection shadows, the compromises made by the evolutionary process generates late-life cost which manifest as diseases of aging. Broadly speaking, two main elements combine to cause aging and its attendant diseases: disruptions and programmatic changes specified by the wild-type genome. Diseases of aging typically arise from combinations of disruptions and programmatic changes, but can also arise from the latter alone. Programmatic changes include costly programmes, and triggered quasi-programmes that may result from run on or, arguably more often, by being triggered in later life.
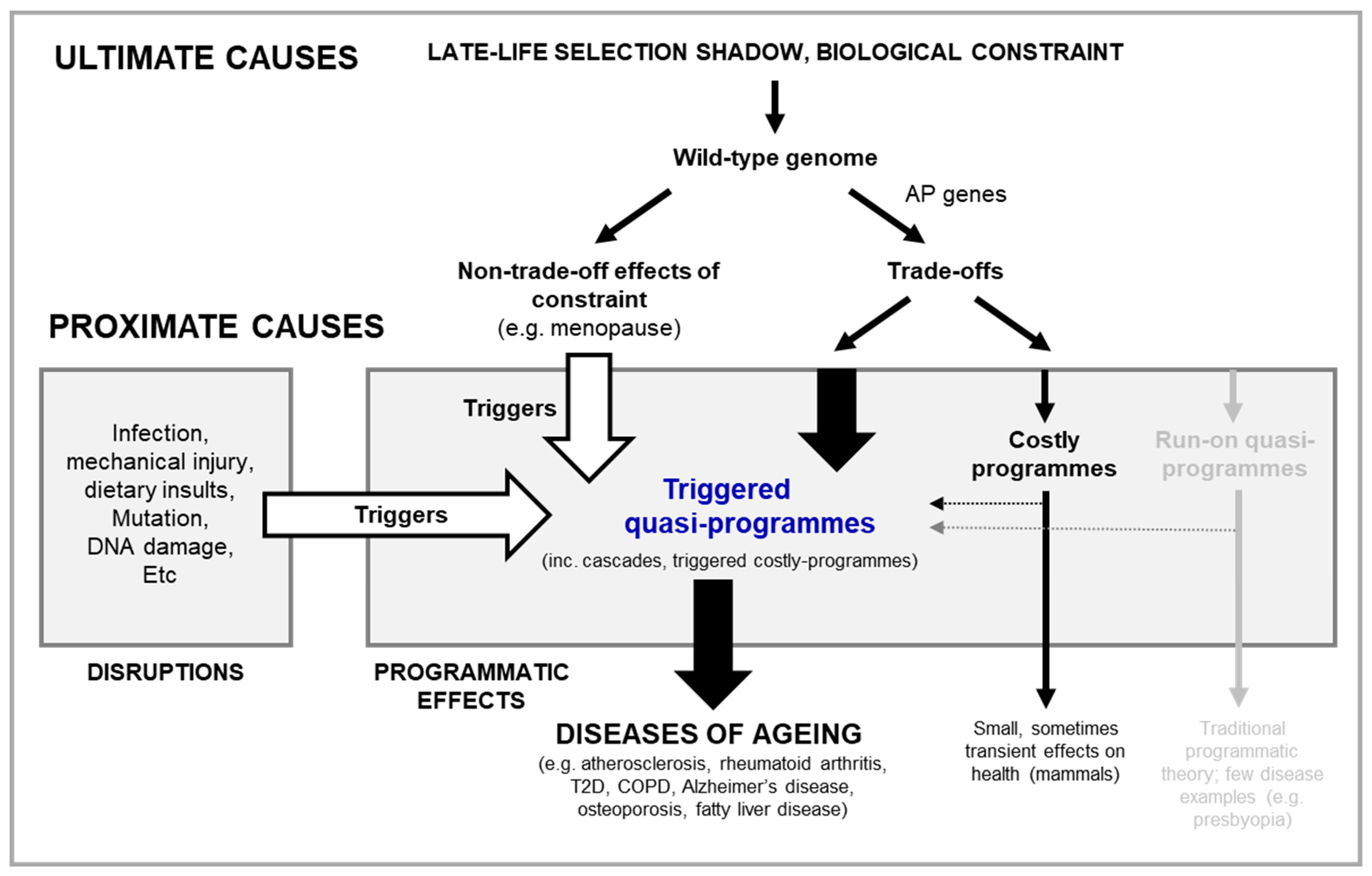
Figure 6.
Representation of a hypothetical web of disease aetiologies, largely triggered quasi-programme driven. One disease can give rise to many different diseases via a knock-on effect, and many different conditions can converge to give rise to another multi-factorial disease. TP: triggered quasi-programme.
Figure 6.
Representation of a hypothetical web of disease aetiologies, largely triggered quasi-programme driven. One disease can give rise to many different diseases via a knock-on effect, and many different conditions can converge to give rise to another multi-factorial disease. TP: triggered quasi-programme.
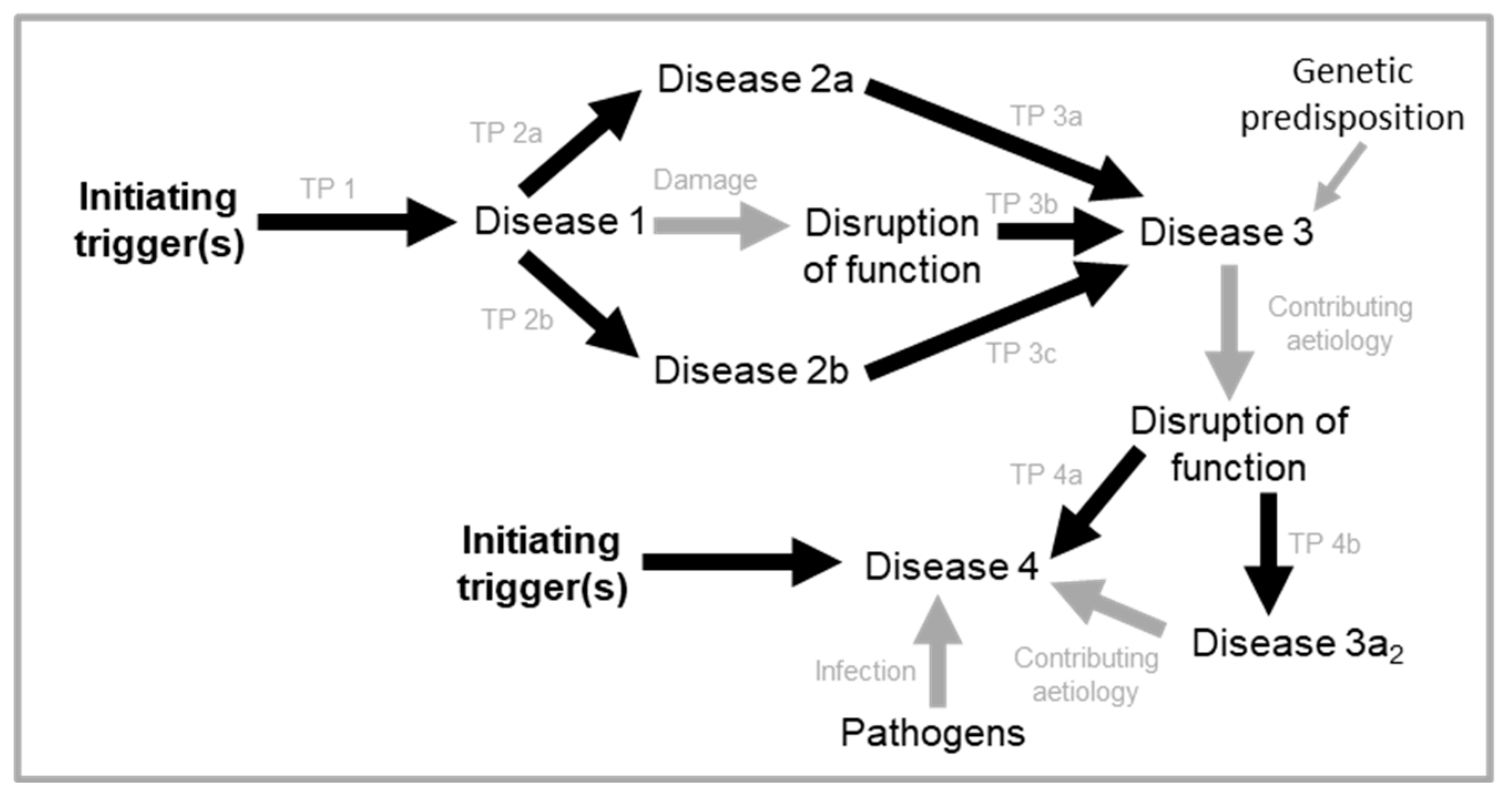
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



