Submitted:
23 October 2023
Posted:
24 October 2023
You are already at the latest version
Abstract
The results of the most recent investigation of triterpenoid-based antiviral agents effective namely in the HIV-1 and HSV-1 treatment were reviewed and summarized. Several key historical achievements are included to stress consequences and continuity in this research. Most of the agents studied belong into a series of compound derived from betulin or betulinic acid, and their synthetic derivative called bevirimat. A termination of clinical trials of bevirimat in the Phase IIb initiated a search for more successful compounds partly derived from bevirimat or designed independently of bevirimat structure. Surprisingly, a majority of bevirimat mimics are derivatives of betulinic acid, while other plant triterpenoids, such as ursolic acid, oleanolic acid, glycyrrhetinic acid or other miscellaneous triterpenoids, are relatively rarely involved in a search for novel antiviral agent. Therefore, this review article is divided into three parts based on the leading triterpenoid core structure.
Keywords:
plant triterpenoid
; structure modifier
; antiviral activity
; HIV-1
; HSV-1
; maturation inhibitor
1. Introduction
Currently, viral infections represent the main infectious disease worldwide [1,2], and they stand for more than 65% of total number of infectious diseases [3]. Viruses invading the human body can be divided into two categories: (a) Viruses being long-term parasites in the human body, e.g., chickenpox, rubella, herpes, measles, smallpox, polio, Japanese encephalitis, mumps, cytomegalovirus, hepatitis B/C virus, dengue virus, human influenza virus, human immunodeficiency virus or human papilloma virus, and (b) viruses being long-term parasites in animals usually living close to humans, e.g., chickens, dogs, pigs, horses or sheep. The viruses, capable of infecting humans across species, include avian influenza virus, rabies virus, hanta-virus, and others [4].
Due to the fact that viruses spread relatively easily, which effect may be documented by large-scale transmission of severe acute respiratory syndrome (SARS), Middle East respiratory syndrome (MERS), Ebola, and other viruses over the past decade, they have been considered to represent considerable threats to human health. The residual effects of viral infections often cause considerable deleterious effects, pain, and inconvenience to patients. The neurotoxicity induced by herpes zoster virus infection often causes ganglion inflammation and necrosis. After invading the sensory nerve endings of the skin, this virus can move along the nerve to the ganglion of the posterior root of the spinal cord, where it remains latent. At present, vaccines and screening antiviral drugs represent the main way used for the prevention and treatment of human viral infections [5]. However, it is difficult to develop an effective vaccine because the side effects that are often detected, are unpredictable [6], and vaccination is not effective in 100% of patients [7,8]. Presently, the main focus of antiviral drug research is limited to several types of viruses, such as HIV, herpes (HSV), influenza, hepatitis, and respiratory viruses [9]. Namely, anti-HIV-1 and anti-HSV-1 agents are focused in this review paper in more details.
The most effective anti-HIV-1 agents are compounds capable of inhibiting HIV-1 maturation [10]. Genetic and enzymatic investigation resulted in a finding that cleavage inhibition or even slowing cleavage at the CA-SP1 site is sufficient to disrupt the maturation process significantly, and to destroy virus infectivity quite efficiently. Therefore, maturation inhibitors interfering with CA-SP1 processing, are the most important candidates for augmenting the current ways of treatment of infection by HIV [11,12]. Biochemical and structural studies revealed that slow cleavage of CA-SP1 is due to structural separation of the proteolysis site [13]. Although the detailed mechanism of inhibition has not yet been determined, small-molecule maturation inhibitors, such as 3-O-(3′,3′-dimethylsuccinyl)-betulinic acid (bevirimat) and its analogs, are supposed to interfere with proteolysis by binding to the CA-SP1 junction and stabilizing the 6-helix bundle [14,15,16,17]. Maturation inhibitors do not interfere with substrate binding directly, they rather act indirectly by inhibiting the unfolding of the 6-helix bundle resulting in impeding access of the protease to its substrate. Despite being potent inhibitors of HIV infection under the laboratory conditions, maturation inhibitors have not yet been approved for clinical use. Bevirimat underwent Phase I and Phase II clinical trials, during which significant, dose-dependent viral load reductions in HIV-1-infected individuals were observed [18]. However, further studies revealed that in up to 50% of patients, bevirimat did not affect viral loads [19,20]. This resistance of bevirimat is clearly associated with naturally occurring viral sequence polymorphs.
Herpes simplex virus (HSV) is a double-stranded DNA virus [21]. It exists in two types of HSV strains: HSV-1 causing infection of the oral mucosa, and HSV-2 affecting the genital mucosa and causing neonatal infections [22]. If not treated effectively, the mortality rate of HSV-1 infections can reach 70% [23,24]. According to the latest report of the WHO, more than 65% of the world population being under the age of 50 are infected with HSV-1, and the incidence of herpes simplex virus encephalitis (HSE) caused by HSV-1 infection is increasing every year. Moreover, HSV-1 infections are associated with neurodegenerative diseases, e.g., Alzheimer disease [24,25]. Intracranial infection caused by HSV-1 was identified as an important factor in the pathogenesis of Alzheimer disease [25], and the brain changes in some patients with herpes simplex virus encephalitis were found to be similar to that of Alzheimer disease patients [25]. HSV-1 has also been identified for its association with certain types of cancers, e.g., cervical carcinoma or acute lymphocytic leukemia [26]. At present, anti-HSV-1 drugs mainly belong among nucleoside drugs (e.g., acyclovir, ganciclovir, valacyclovir etc.), capable of inhibiting viral replication by interfering with viral DNA polymerase. Based on the wide use of these drugs, drug-resistant strains of virus appeared within a relatively short time period [25]. In order to cope with the problem of drug resistance, investigation has been focused on developing novel anti-HSV agents targeting the viral DNA polymerase. The HSV-1 life cycle includes adsorption and entry into the host cell, intracellular transport to the nucleus, DNA replication, gene transcription, protein synthesis, nucleocapsid assembly and viral release [27]. Drugs theoretically affecting any stage in the life cycle could be inhibitory [27].
2. Triterpenoid-based agents for treating HIV-1 and HSV-1)
2.1. Plant triterpenoids of the lupane family
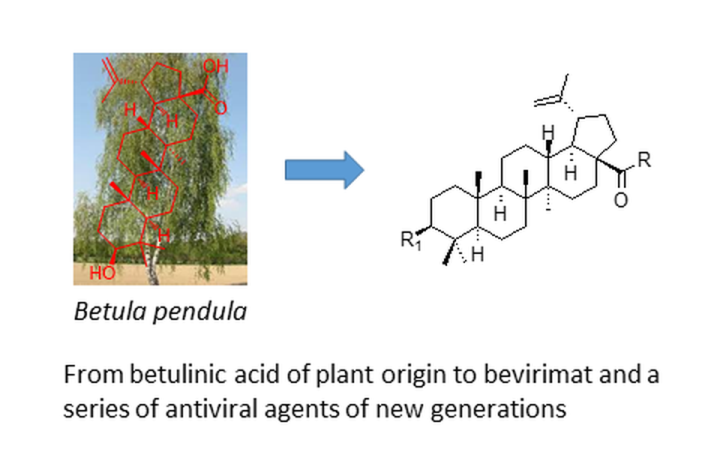
Betulin (1; Figure 1), a pentacyclic natural triterpenoid, represents one of the potent and for a long time known plant-derived product [28]. Mostly, it is found in the bark of various birch (Betula) species, and can be extracted therefrom. The biological effects of betulin (1) have been intensively investigated, and have resulted in a finding of its wide-ranging biological activity that involve antiviral, antibacterial, anticancer, and anti-inflammatory effects [29]. Antiviral properties of betulin (1) and its derivatives have been explored in the context of many different viruses [30,31,32]. Betulinic acid (2; Figure 1) represents another plant product extractable from the bark of birch and from other plant sources [33]. It can also be prepared synthetically by oxidation of betulin (1) [34]. The pharmacological characteristics of betulinic acid (2) are similar to betulin (1) [35,36,37]. 3-O-(3′,3′-Dimethylsuccinyl)betulinic acid (3; Figure 1), known as bevirimat, was one of the most promising derivatives of betulinic acid (2). It possesses potent anti-HIV activity with a novel mechanism of action [38]. Unfortunately, its further development was terminated at Phase IIb of clinical trials due to the reduced efficacy of the compound against certain HIV strains [39]. Nevertheless, discovery of bevirimat (3) initiated a subsequent investigation in the field of plant triterpenoids and their derivatives bearing various functional groups or structural modifiers, resulting in a synthesis of several early but successful bevirimat analogs (4–8; Figure 1) [40,41,42]. However, currently approved antiviral drugs have very diverse structures [28,43].
Bevirimat (3) was the first-in-class HIV-1 maturation inhibitor. It showed a low efficacy, essentially due to the natural polymorphism of its target, the CA-SP1 junction. Moreover, its low solubility in water and in physiological environment makes it difficult to study its interaction with the CA-SP1 junction. Therefore, designing new derivatives of bevirimat (3) was performed by introducing different hydrophilic substituents at the C-28 carboxyl group to improve solubility of the novel compounds in aqueous media. A synthesis of the novel derivatives, the effect of substituents at the C-28 carboxyl group, and their solubility in aqueous media were investigated intensively, and the ability of these molecules to inhibit viral infection and their cytotoxicity was carefully evaluated [43]. Compared to the well-known bevirimat (3), one of the prepared compounds (7) showed higher solubility in aqueous media associated with a 2.5-fold increase of activity, higher selectivity index, and a better antiviral profile (Table 1) [43]. Moreover, for the first time, a direct interaction between the prepared compound (7) and the domain CA-SP1 was shown by the NMR study [43]. Bevirimat (3) was launched by several pharmaceutical companies for further development and commercialization [44,45]. However, while bevirimat (3) succeeded in Phase IIa of the clinical trials, results obtained in Phase IIb of the clinical trials stopped the development on this new class of anti-HIV drug, principally because of the natural polymorphism of the CA-SP1 junction that lead to a natural resistance of the virus to maturation inhibitors. The prepared pioneer bevirimat-based compounds (4–8; Figure 1) represented novel, attractive and promising agents for future development of the next generation of HIV-1 maturation inhibitors [43]. Their anti-HIV-1 effects are summarized in Table 1.
A search for new methods of antiviral therapy has ever been primarily focused on the use of substances of natural origin. In this context, lupane family triterpenoid compounds, betulin (1) and betulinic acid (2), proved to be good starting plant products for derivation [28]. Recently, a synthesis of a novel series of betulinic acid ester derivatives (9–16; Figure 2) was published [28]. The structures of the novel compounds were established, and the compounds were tested against DNA and RNA viruses, for antiviral activity against several types of viruses, including HSV-1 [28]. Virucidal and time-of-addition experiments confirmed research hypothesis and showed high antiviral effect of several derivatives against BEV, H1N1, and HSV-1. Compound 10c bearing a citraconic-piperidine moiety, exhibited 6-fold more potent activity against HSV-1 (EC50 = 17.2 µM) than the reference drug (acyclovir; EC50 = 111.1 µM) (Table 2). The phthalic-thiomorpholine derivative 13d possessed the highest selectivity index (13d, SI = 11.8; acyclovir, SI = 14.0). Overall, all active compounds showed high virus-specific activity, as none of them were active against more than one virus. Most of the active compounds were active at the later steps of the replication cycle, which suggested a mode of action during that step of nucleic acids/protein synthesis, assembly or maturation. The in silico study correlated well with in vitro data, and confirmed a high affinity of 10c to HSV-1 DNA polymerase. Additionally, all ester and amide derivatives were tested for the antiproliferative activity in A549 and MDCK cell lines (Table 3). Ester derivatives (9–16), glutaric acid amides (11b and 11c), and succinic acid amides (9a and 9b) showed strong cytotoxic effects. These findings provided valuable data for further investigation of the active compounds, as well as subsequent betulin derivation, in context of antiviral activity. In addition, the results indicated that natural resources have still been one of the most important sources of priority structures in a search for new drug candidates.
In a continuing search for novel HIV-1 maturation inhibitors, another series of promising compounds was designed and synthesized on a basis of a number of triterpenoid derivatives (17–17s; Figure 3), and namely the compound 18 (also known GSK3640254 or fipravirimat; Figure 3) [46]. Compound 18 exhibited significantly improved antiviral activity toward a range of clinically relevant polymorphic variants with reduced sensitivity toward the second generation maturation inhibitor (17s; also known as GSK3532795 or BMS-955176). The key structural difference between 18 and its earlier developed analogs (17–17s) is the replacement of the para-substituted benzoic acid moiety located at the C-3 position of the triterpenoid skeleton with a cyclohex-3-ene-1-carboxylic acid substituted with a CH2F moiety with the given absolute configuration (18; Figure 3). The sp3 carbon atom at this site of the molecule provided a new vector for structure-activity relationship exploration and resulted in the identification of compounds with improved polymorphic coverage while preserving the pharmacokinetic properties of the prototype. This structural element provided a new vector for exploring structure-activity relationships, and led to compounds with improved polymorphic coverage while preserving pharmacokinetic properties. The approach to the design of 18, the development of a synthetic route and its preclinical profile were clearly described in details in the original paper [46]. Compound 18 has completed the Phase IIa of the clinical trials, in which it demonstrated a dose-related reduction in plasma HIV-1 RNA over 7–10 days, and the compound is being advanced into the Phase IIb studies [46].
The investigation of the structure−activity relationships of a series of HIV-1 maturation inhibitors based on the compound 17s (Figure 3) continued by the subsequent incorporating novel C-17 amine substituents to reduce the overall basicity of the required compounds [47]. A replacement of the distal amine on the C-17 side chain present in 17s with a tertiary alcohol in combination with either a heterocyclic ring system or a cyclohexyl ring substituted with polar groups provided potent wild-type (WT) HIV-1 maturation inhibitors that also retained excellent potency against a T332S/V362I/prR41G variant, a laboratory strain that served as a surrogate to assess HIV-1 polymorphic virus coverage [47]. Compound 19 exhibited a broad-spectrum HIV-1 activity against an expanded panel of clinically relevant Gag polymorphic viruses and had the most desirable overall profile in this series of compounds. In pharmacokinetic studies, 19 had low clearance and exhibited 24 and 31% oral bioavailability in rats and dogs, respectively.
Compounds 18 and 19 had the most desirable overall profile in this series and was evaluated in rat and dog pharmacokinetic studies [47,48]. A comparison of basic anti-HIV-1 activity values is shown in Table 4. An overall summary of antiviral activity values of compounds 17–17s, 18 and 19 is presented in Table 5 [46,47,48].
Structurally similar compounds to those mentioned above [46,47,48] were reported recently as the second-generation maturation inhibitors (compounds 20–22 and 23a–23e; Figure 4), displaying effect higher than bevirimat (3) against HIV-1 subtype C [49]. In silico studies on interaction of with BVM and their analogs have been limited to HIV-1 subtype B (5I4T) due to the lack of an available 3D structure for HIV-1 subtype C virus. The authors [49] have developed a 3D model of HIV-1C Gag CA-SP1 region using protein homology modeling with HIV-1 subtype B (514T) as a template. The generated HIV-1 C homology model was extensively validated using several online tools and served as a template to perform molecular docking studies with eight well-characterized maturation inhibitors. The docked complex of HIV-1C and the studied maturation inhibitors were subjected to molecular dynamics simulation for 100 ns. Based on the obtained data, it was revealed that the investigation was probably a pioneering report on construction and validation of 3D model for the HIV-1C Gag CA-SP1, which could serve as a crucial tool in the structure-aided design of novel and broadly acting maturation inhibitors [49]. The docking studies confirmed that modifications at the C-28 positions in bevirimat analogs resulted in increased interactions with HIV-1C Gag CA-SP1 and higher binding energy as compared to the parental bevirimat (3), which may have conferred antiviral activity to these analogs [49]. The authors [49] presented no antiviral activity data, however, the in silico investigation brought a novel motivation in designing more effective antiviral agents of next generations. However, antiviral activity data of several compounds of the investigated series can be found in [46].
Phosphate and phosphonate derivatives of betulin (1), betulinic acid (2) and bevirimat (3) represent other types of antiviral compounds displaying better pharmacological characteristics than the parent compounds [1,50]. Several compounds of that series (24a–24h) are shown in Figure 5, and their antiviral activity values are summarized on Table 6. The inhibitory effect of compound 24a (Figure 5, Table 6) shows high value, as well as high therapeutic index (IC50 = 0.02 µM, TI = 1250) on viral replication, and it displayed high selectivity [1]. The capsid protein (CA) CTD-SP1 might be the target of compound 24a against HIV. Among additional phosphate and phosphonate derivatives of bevirimat (24b–24h), compound 24e showed antiviral activity comparable with that of 24a, however, with slightly worse therapeutic profile than displayed by 24a.
A novel compound (25; Figure 6), in principal also derived from bevirimat (3), bearing a pyrazolone system in the molecule, has been considered to represent HIV-1 maturation inhibitor of the third generation [51]. It displayed maturation inhibition effect in HIV-1 with the EC50 = 20.36 ± 2.85 nM [51]. The mechanism of action of 25 is identical to that of the first-generation antiviral maturation inhibitor bevirimat (3). However, the investigation showed that 25 displayed better antiviral potential than bevirimat (3) among the virus strains tested, regardless of the presence or absence of human serum [51]. Further designing and developing of suitable molecules resulted in a synthesis of a compound bearing selected, often called “privileged”, structural motifs (Figure 6) [52]. So far the most successful structure (26; Figure 6) showed high antiviral activity in HIV-1 NL4-3 (EC50 = 0.012 µM), which is higher than antiviral activity of 25. Within that series of novel piperazine-based compounds (27a–27g; Figure 6), none of them showed better antiviral effect and therapeutic profile than 26 (Table 7).
2.2. Peptide derivatives of triterpenoids of the lupane, ursane and oleanane family
To demonstrate variability in designing and developing effective structural modifications of bevirimat (3), specific series of peptide analogs of betulinic acid (2) should be mentioned. This part of the story started with triterpenoid saponins, natural products bearing glycoside units, which are a major group of active compounds of the natural origin with nonspecific antiviral activities. In turn, T20 peptide (enfuvirtide), containing a helix zone-binding domain, is a gp41-specific HIV-1 fusion inhibitor. One of the early approaches to the design, synthesis, and structure-activity relationship study of a group of hybrid molecules, in which bioactive triterpenoid sapogenins were covalently bound to the peptides containing the helix zone-binding domain, by the 1,3-dipolar cycloaddition, often known as click chemistry. Thus, a series of triterpenoid-peptide conjugates (28a–28n; Figure 7; Table 8) appeared as early as a decade ago [53]. The investigation resulted in a finding that either the triterpenoid or the peptide part separately showed only weak activity against HIV-1 Env-mediated cell-cell fusion, while the generated hybrid conjugates displayed strong cooperative effects [53]. Among them, P26−BApc (28k) exhibited anti-HIV-1 activity against both T20-sensitive and T20-resistant HIV-1 strains, and improved pharmacokinetic properties. The results proved that this scaffold design was a promising strategy for developing novel HIV-1 fusion inhibitors, and possibly, encouraging designing novel antiviral therapeutics against other viruses with class I fusion proteins (Table 9) [53].
2.3. Miscelaneous plant triterpenoids
Investigation of other plant triterpenoids than those of the lupane family can be found in the literature, nevertheless, it is less frequent than it could be expected, even if different plant triterpenoids have been reported to display antiviral effects [33].
Ursane-type triterpenoids and 28-nortriterpenoids (29a−29i; Figure 8) were isolated from Rhododendron latoucheae [54]. A hyphenated NMR technique (analytical HPLC with a DAD connected to MS, SPE, and NMR) has proven effective for the full structural analysis and identification of the isolated natural products in complex mixtures. Compounds 29a and 29i inhibited HSV-1 in Vero cells with IC50 values of 6.4 μM and 0.4 μM, respectively, while the compounds 29b–29h were less effective (Table 10) [54].
The chalcone derivatives of 20-oxo-lupanes have been synthesized and screened for several types of biological activity by Russian authors [55]. Investigating antiviral activity of the prepared series of compounds, two of them (30a and 30b; Figure 9) were evaluated as compounds displaying anti-HSV-1 activity (Table 11). The antiviral activity of 30a and 30b was evaluated against HCMV, and for compound 30b also against HSV-1 and HPV.
A large series of new pentacyclic triterpenoids, including oleanane-type, ursane-type and taraxerane-type, were isolated from the stems and branches of Enkianthus chinensis [56]. Their structures were elucidated by extensive spectroscopic analyses, X-ray crystallographic data and electronic circular dichroism (ECD) techniques. In the in vitro biological activity evaluation resulted in a finding that three compounds (31a–31c; Figure 10) showed antiviral activity. The most active compound of this small series of natural triterpenoids was 31c showing latent activity against HSV-1 with an IC50 value of 6.4 μM. Their structures and antiviral activity values (in comparison with those of acyclovir used as the positive reference agent) are summarized in Table 12.
Even if triterpenoids and their natural saponin derivatives exhibit anti-HSV-1 activity, there has still been a lack of comprehensive information on the anti-HSV-1 activity of triterpenoids. Therefore, expanding information on the anti-HSV-1 activity of triterpenes and improving the efficiency of their exploration are urgently required. To improve the efficiency of the development of anti-HSV-1 active compounds, Japanese authors recently constructed a predictive model for the anti-HSV-1 activity of triterpenes by using the information obtained from previous studies [57]. They constructed a binary classification model (i.e., active or inactive) using a logistic regression algorithm. As a result, assay was performed on 20 triterpenes and triterpenoids, showing finally structure 32 (Figure 11) displaying potent anti-HSV-1 activity value (IC50 = 13.06 μM).
Several so far undescribed cycloartane triterpenoids, pseudolarnoids A−G, together with other known triterpenoids, were isolated from the seeds of Pseudolarix amabilis (J. Nelson) Rehder [58]. Their structures were elucidated on the basis of spectroscopic analysis, X-ray crystallography, and ECD data. Three of these natural products (33–35; Figure 12) proved their ability to display potent antiviral effects on HSV-1 in vitro (Table 13). Based on the therapeutic index values, structures 34 and 35 showed better therapeutic profile than 33 (Table 13). The structures of other less active or inactive cycloartane triterpenoids are not shown, they can be found in the original paper [58].
3. Conclusion
Even if the presented review paper covers a short period of investigation of antiviral agents, it shows a wide variety of triterpenoid-based compounds investigated as potential antiviral agents. Even if a majority of structures are—in principle—structurally derived from bevirimat (3), other types of triterpenoid structures were also included in this type of investigation. Because studies made with the lupane family of triterpenoids were always performed with high intensity, and possibly with high priority as well, the so far achieved results revealed that lupane-based agents seem to be the most successful structures among all triterpenoid-based ones. The review shows achievements in searching for novel structures and clearly shows that this intensive investigation is being resulted in designing perspective structures that have a great chance to pass over all phases of clinical trials to give a potent antiviral agent for application in human medicine.
Author Contributions
Conceptualization, Z.W.; methodology, U.B.; validation, M.W. and U.B.; writing—original draft preparation, Z.W., M.W. and U.B.; writing—review and editing, Z.W.; funding acquisition, Z.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Industry and Commerce (MPO), Czech Republic, grant number FV30300.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank MPO for financial support through the grant FV30300.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
References
- Liu, Y.; Yang, L.; Wang, H.; Xiong, Y. Recent Advances in Antiviral Activities of Triterpenoids. Pharmaceuticals 2022, 15, 1169. [Google Scholar] [CrossRef] [PubMed]
- Sander, W.J.; O’Neill, H.G.; Pohl, C.H. Prostaglandin E2 As a Modulator of Viral Infections. Front. Physiol. 2017, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Gao, Y.; Wen, Y.; Ke, X.; Ou, Z.; Li, Y.; He, H.; Chen, Q. Detection of Virus-Related Sequences Associated With Potential Etiologies of Hepatitis in Liver Tissue Samples From Rats, Mice, Shrews, and Bats. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Yang, X.W. Antiviral effect of glycyrrhizic acid. Mod. Chin. Med. 2020, 22, 533–541. [Google Scholar]
- Yang, J.; Yue, L.; Yang, Z.; Miao, Y.; Ouyang, R.; Hu, Y. Metal-Based Nanomaterials: Work as Drugs and Carriers against Viral Infections. Nanomaterials 2021, 11, 2129. [Google Scholar] [CrossRef]
- Finsterer, J. Neurological side effects of SARS-CoV-2 vaccinations. Acta Neurol. Scand. 2022, 145, 5–9. [Google Scholar] [CrossRef]
- Hacisuleyman, E.; Hale, C.; Saito, Y.; Blachere, N.E.; Bergh, M.; Conlon, E.G.; Schaefer-Babajew, D.J.; DaSilva, J.; Muecksch, F.; Gaebler, C.; Lifton, R. ; Nussenzweig. M. C.; Hatziioannou, T.; Bieniasz, P. D.; Darnell, R. B. Vaccine breakthrough infections with SARS-CoV-2 variants. N. Engl. J. Med. 2021, 384, 2212–2218. [Google Scholar]
- Yi, Y.; Li, J.; Lai, X.; Zhang, M.; Kuang, Y.; Bao, Y.-O.; Yu, R.; Hong, W.; Muturi, E.; Xue, H.; Wei, H.; Li, T.; Zhuang, H.; Qiao, X.; Xiang, K.; Yang, H.; Ye, M. Natural triterpenoids from licorice potently inhibit SARS-CoV-2 infection. J. Adv. Res. 2022, 36, 201–210. [Google Scholar] [CrossRef]
- Pu, J.-Y.; He, L.; Wu, S.-Y.; Zhang, P.; Huang, X. [Anti-virus research of triterpenoids in licorice]. . 2013, 29, 673–9. [Google Scholar]
- Pornillos, O.; Ganser-Pornillos, B.K. Maturation of retroviruses. Curr. Opin. Virol. 2019, 36, 47–55. [Google Scholar] [CrossRef]
- Kanamoto, T.; Kashiwada, Y.; Kanbara, K.; Gotoh, K.; Yoshimori, M.; Goto, T.; Sano, K.; Nakashima, H. Anti-Human Immunodeficiency Virus Activity of YK-FH312 (a Betulinic Acid Derivative), a Novel Compound Blocking Viral Maturation. Antimicrob. Agents Chemother. 2001, 45, 1225–1230. [Google Scholar] [CrossRef]
- Zhou, J.; Yuan, X.; Dismuke, D.; Forshey, B.M.; Lundquist, C.; Lee, K.-H.; Aiken, C.; Chen, C.H. Small-Molecule Inhibition of Human Immunodeficiency Virus Type 1 Replication by Specific Targeting of the Final Step of Virion Maturation. J. Virol. 2004, 78, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Adamson, C.S.; Ablan, S.D.; Boeras, I.; Goila-Gaur, R.; Soheilian, F.; Nagashima, K.; Li, F.; Salzwedel, K.; Sakalian, M.; Wild, C.T.; et al. In Vitro Resistance to the Human Immunodeficiency Virus Type 1 Maturation Inhibitor PA-457 (Bevirimat). J. Virol. 2006, 80, 10957–10971. [Google Scholar] [CrossRef] [PubMed]
- Pak, A.J.; Purdy, M.D.; Yeager, M.; Voth, G.A. Preservation of HIV-1 Gag Helical Bundle Symmetry by Bevirimat Is Central to Maturation Inhibition. J. Am. Chem. Soc. 2021, 143, 19137–19148. [Google Scholar] [CrossRef] [PubMed]
- Purdy, M.D.; Shi, D.; Christowicz, J.; Hattne, J.; Goner, T.; Yeager, M. MicroED structures of HIV-1GagCTD-SP1 reveal binding interactions with the maturation inhibitor bevirimat. Proc. Natl. Acad. Sci. USA 2018, 115, 13258–13263. [Google Scholar] [CrossRef] [PubMed]
- Adamson, C.S.; Salzwedel, K.; O Freed, E. Virus maturation as a new HIV-1 therapeutic target. Expert Opin. Ther. Targets 2009, 13, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, F.; Kashiwada, Y.; Cosentino, L.; Chen, C.-H.; Garrett, P.E.; Lee, K.-H. Anti-AIDS agents—XXVII. Synthesis and anti-HIV activity of betulinic acid and dihydrobetulinic acid derivatives. Bioorganic Med. Chem. 1997, 5, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.F.; Ogundele, A.; Forrest, A.; Wilton, J.; Salzwedel, K.; Doto, J.; Allaway, G.P.; Martin, D.E. Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-O-(3′,3′-dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob. Agents Chemother. 2007, 51, 3574–3581. [Google Scholar] [CrossRef]
- Van Baelen, K.; Salzwedel, K.; Rondelez, E.; Van Eygen, V.; De Vos, S.; Verheyen, A.; Steegen, K.; Verlinden, Y.; Allaway, G.P.; Stuyver, L.J. Susceptibility of Human Immunodeficiency Virus Type 1 to the Maturation Inhibitor Bevirimat Is Modulated by Baseline Polymorphisms in Gag Spacer Peptide 1. Antimicrob. Agents Chemother. 2009, 53, 2185–2188. [Google Scholar] [CrossRef]
- Adamson, C.S.; Sakalian, M.; Salzwedel, K.; O Freed, E. Polymorphisms in Gag spacer peptide 1 confer varying levels of resistance to the HIV- 1maturation inhibitor bevirimat. Retrovirology 2010, 7, 36–36. [Google Scholar] [CrossRef]
- Liu, X.; Main, D.; Ma, Y.; He, B. Herpes Simplex Virus 1 Inhibits TANK-Binding Kinase 1 through Formation of the Us11-Hsp90 Complex. J. Virol. 2018, 92, e00402–18. [Google Scholar] [CrossRef]
- Whitley, R.J. Herpes simplex encephalitis: Adolescents and adults. Antivir. Res. 2006, 71, 141–148. [Google Scholar] [CrossRef]
- Navid, M.H.; Laszczyk-Lauer, M.; Reichling, J.; Schnitzler, P. Pentacyclic triterpenes in birch bark extract inhibit early step of herpes simplex virus type 1 replication. Phytomedicine 2014, 21, 1273–1280. [Google Scholar] [CrossRef]
- Ye, J.; Wang, Z.; Jia, J.; Li, F.; Wang, Y.; Jiang, Y.; Wang, Y.; Ren, Z.; Pu, H. Lupeol impairs herpes simplex virus type 1 replication by inhibiting the promoter activity of the viral immediate early gene α0. Acta Virol. 2021, 65, 254–263. [Google Scholar] [CrossRef]
- Ramu, R.; Shirahatti, P.S.; Swamy, S.N.; Zameer, F.; Dhananjaya, B.L.; Prasad, M.N.N. Assessment of in vivo antidiabetic properties of umbelliferone and lupeol constituents of banana (Musa sp. var. Nanjangud Rasa Bale) flower in hyperglycaemic rodent model. Plos One 2016, 11, e0151135. [Google Scholar]
- Kolokotronis, A.; Doumas, S. Herpes simplex virus infection, with particular reference to the progression and complications of primary herpetic gingivostomatitis. Clin. Microbiol. Infect. 2006, 12, 202–211. [Google Scholar] [CrossRef]
- Coen, D.M.; Schaffer, P.A. Anti-herpesvirus drugs: a promising spectrum of new drugs and drug targets. Nat. Rev. Drug. Discov. 2003, 2, 278–288. [Google Scholar] [CrossRef]
- Pęcak, P.; Orzechowska, B.; Chrobak, E.; Boryczka, S. Novel betulin dicarboxylic acid ester derivatives as potent antiviral agents: Design, synthesis, biological evaluation, structure-activity relationship and in-silico study. Eur. J. Med. Chem. 2021, 225, 113738. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and its derivatives as novel compounds with different pharmacological effects. Biotechnol. Adv. 2020, 38. [Google Scholar] [CrossRef] [PubMed]
- Kazakova, O.B.; Medvedeva, N.I.; Baikova, I.P.; Tolstikov, G.A.; Lopatina, T.V.; Yunusov, M.S.; Zaprutko, L. Synthesis of triterpenoid acylates: Effective reproduction inhibitors of influenza A (H1N1) and papilloma viruses. Russ. J. Bioorganic Chem. 2010, 36, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.; Savinova, O.; Nikolaeva, S.; Boreko, E.; Flekhter, O. Antiviral activity of betulin, betulinic and betulonic acids against some enveloped and non-enveloped viruses. Fitoterapia 2003, 74, 489–492. [Google Scholar] [CrossRef]
- Sun, I.C.; Shen, J.K.; Wang, H.K.; Cosentino, L.M.; Lee, K.H. Anti-AIDS agents. 32. Synthesis and anti-HIV activity of betulin derivatives. Bioorg. Med. Chem. Lett. 1998, 1267–1272.
- arek, J.; Kvasnica, M.; Vlk, M.; Urban, M.; Džubák, P.; Hajdúch, M. The potential of triterpenoids in the treatment of melanoma. In Research on Melanoma—A Glimpse into Current Directions and Future Trends; Murph, M., Ed.; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- Kim, D.S.H.L.; Chen, Z.; Nguyen, v.T.; Pezzuto, J.M.; Qiu, S.; Lu, Z.-Z. A Concise Semi-Synthetic Approach to Betulinic Acid from Betulin. Synth. Commun. 1997, 27, 1607–1612. [Google Scholar] [CrossRef]
- Alakurtti, S.; Mäkelä, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar] [CrossRef]
- Bildziukevich, U.; Özdemir, Z.; Wimmer, Z. Recent Achievements in Medicinal and Supramolecular Chemistry of Betulinic Acid and Its Derivatives ‡. Molecules 2019, 24, 3546. [Google Scholar] [CrossRef]
- zdemir, Z.; Wimmer, Z. Selected plant triterpenoids and their amide derivatives in cancer treatment: A review. Phytochemistry 2022, 203, 113340. [Google Scholar] [CrossRef]
- E Martin, D.; Salzwedel, K.; Allaway, G.P. Bevirimat: A Novel Maturation Inhibitor for the Treatment of HIV-1 Infection. Antivir. Chem. Chemother. 2008, 19, 107–113. [Google Scholar] [CrossRef]
- Martin, D.E.; Blum, R.; Doto, J.; Galbraith, H.; Ballow, C. Multiple-dose pharmacokinetics and safety of bevirimat, a novel inhibitor of HIV maturation, in healthy volunteers. Clin. Pharmacokinet. 2007, 46, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Jones, S.A.; Jeffrey, J.L.; Miranda, S.R.; Galardi, C.M.; Irlbeck, D.M.; Brown, K.W.; McDanal, C.B.; Johns, B.A. Discovery of a novel and potent class of anti-HIV-1 maturation inhibitors with improved virology profile against gag polymorphisms. Bioorganic Med. Chem. Lett. 2017, 27, 2689–2694. [Google Scholar] [CrossRef]
- Marciniec, K.; Chrobak, E.; Dąbrowska, A.; Bębenek, E.; Kadela-Tomanek, M.; Pęcak, P.; Boryczka, S. Phosphate Derivatives of 3-Carboxyacylbetulin: SynThesis, In Vitro Anti-HIV and Molecular Docking Study. Biomolecules 2020, 10, 1148. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Zadrozny, K.K.; Zadorozhnyi, R.; Russell, R.W.; Quinn, C.M.; Kleinpeter, A.; Ablan, S.; Meshkin, H.; Perilla, J.R.; Freed, E.O.; et al. Structural basis of HIV-1 maturation inhibitor binding and activity. Nat. Commun. 2023, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Coric, P.; Turcaud, S.; Souquet, F.; Briant, L.; Gay, B.; Royer, J.; Chazal, N.; Bouaziz, S. Synthesis and biological evaluation of a new derivative of bevirimat that targets the Gag CA-SP1 cleavage site. Eur. J. Med. Chem. 2013, 62, 453–465. [Google Scholar] [CrossRef]
- Martin, D.E.; Blum, R.; Wilton, J.; Doto, J.; Galbraith, H.; Burgess, G.L.; Smith, P.C.; Ballow, C. Safety and Pharmacokinetics of Bevirimat (PA-457), a Novel Inhibitor of Human Immunodeficiency Virus Maturation, in Healthy Volunteers. Antimicrob. Agents Chemother. 2007, 51, 3063–3066. [Google Scholar] [CrossRef]
- Smith, P.F.; Ogundele, A.; Forrest, A.; Wilton, J.; Salzwedel, K.; Doto, J.; Allaway, G.P.; Martin, D.E. Phase I and II study of the safety, virological effect, and pharmacokinetics/pharmacodynamics of single-dose 3-O-(3′,3′-dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob. Agents Chemother. 2007, 51, 3574–3581. [Google Scholar] [CrossRef]
- Regueiro-Ren, A.; Sit, S.-Y.; Chen, Y. , Chen, J. ; Swidorski, J.J.; Liu, Z.; Venables, B.L.; Sin. N.; Hartz, R.A.; Protack, T.; Lin, Z.; Zhang, S.; Li, Z.; Wu, D.-R.; Li, P.; Kempson, J.; Hou, X.; Gupta, A.; Rampulla, R.; Mathur, A.; Park, H.; Sarjeant, A.; Benitex, Y.; Rahematpura, S.; Parker, D. Phillips, T.; Haskell, R.; Jenkins, S.; Santone, K.S.; Cockett, M.; Hanumegowda, U.; Dicker, I.; Meanwell, N.A.; Krystal, M. The discovery of GSK3640254, a next-generation inhibitor of HIV-1 maturation. J. Med. Chem. 2022, 65, 11927–11948. [Google Scholar]
- Hartz, R.A.; Xu, L.; Sit, S.-Y.; Chen, J.; Venables, B.L.; Lin, Z.; Zhang, S.; Li, Z.; Parker, D.; Simmons, T.S.; Jenkins, S.; Hanumegowda, U.M.; Dicker, I.; Krystal, M.; Meanwell, N.A.; Regueiro-Ren, A. Synthesis, structure−activity relationships, and in vivo evaluation of novel C-17 amine derivatives based on GSK3640254 as HIV-1 maturation inhibitors with broad spectrum activity. J. Med. Chem. 2022, 65, 15935–15966. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Sub-stoichiometric Modulation of Viral Targets─Potent Antiviral Agents That Exploit Target Vulnerability. ACS Med. Chem. Lett. 2023, 14, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- C, Y.K.; Pal, S.; Nitz, T.J.; Wild, C.; Gaur, R. Construction of a HIV-1 subtype C 3D model using homology modeling and in-silico docking, molecular dynamics simulation, and MM-GBSA calculation of second-generation HIV-1 maturation inhibitor(s). J. Biomol. Struct. Dyn. 2023, 1–10. [Google Scholar] [CrossRef]
- Chrobak, E.; Marciniec, K.; Dąbrowska, A.; Pęcak, P.; Bębenek, E.; Kadela-Tomanek, M.; Bak, A.; Jastrzębska, M.; Boryczka, S. New Phosphorus Analogs of Bevirimat: Synthesis, Evaluation of Anti-HIV-1 Activity and Molecular Docking Study. Int. J. Mol. Sci. 2019, 20, 5209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; He, H.H.; Ou-Yang, T.; Liu, D.F.; Jiang, C.H.; Yang, H.P.; Wang, P.; Xie, N.; Yan, S.S. Pre-clinical pharmacological profile of QF-036, a potent HIV-1 maturation inhibitor. Basic Clin. Pharm. Toxicol. 2021, 128, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, C.-H.; Morris-Natschke, S.L.; Lee, K.-H. Design, synthesis, and structure activity relationship analysis of new betulinic acid derivatives as potent HIV inhibitors. Eur. J. Med. Chem. 2021, 215, 113287–113287. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lu, L.; Na, H.; Li, X.; Wang, Q.; Jiang, X.; Xu, X.; Yu, F.; Zhang, T.; Li, J.; et al. Conjugation of a Nonspecific Antiviral Sapogenin with a Specific HIV Fusion Inhibitor: A Promising Strategy for Discovering New Antiviral Therapeutics. J. Med. Chem. 2014, 57, 7342–7354. [Google Scholar] [CrossRef]
- Liu, F.; Wang, Y.-N.; Li, Y.; Ma, S.-G.; Qu, J.; Liu, Y.-B.; Niu, C.-S.; Tang, Z.-H.; Li, Y.-H.; Li, L.; et al. Minor Nortriterpenoids from the Twigs and Leaves of Rhododendron latoucheae. J. Nat. Prod. 2018, 81, 1721–1733. [Google Scholar] [CrossRef]
- Khusnutdinova, E.; Galimova, Z.; Lobov, A.; Baikova, I.; Kazakova, O.; Thu, H.N.T.; Van Tuyen, N.; Gatilov, Y.; Csuk, R.; Serbian, I.; et al. Synthesis of messagenin and platanic acid chalcone derivatives and their biological potential. Nat. Prod. Res. 2021, 36, 5189–5198. [Google Scholar] [CrossRef]
- Wang, H.-Q.; Ma, S.-G.; Zhang, D.; Li, Y.-H.; Qu, J.; Li, Y.; Liu, Y.-B.; Yu, S.-S. Oxygenated pentacyclic triterpenoids from the stems and branches of Enkianthus chinensis. Bioorganic Chem. 2021, 111, 104866. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Nakamura, S.; Oguri, H.; Ryu, K.; Yoneda, T.; Hosoki, R. Effective Search of Triterpenes with Anti-HSV-1 Activity Using a Classification Model by Logistic Regression. Front. Chem. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-T.; Yu, M.-H.; Su, S.-Y.; Shi, X.-L.; Lei, C.; Hou, A.-J. Cycloartane triterpenoids from Pseudolarix amabilis and their antiviral activity. Phytochemistry 2020, 171, 112229. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Betulin (1), betulinic acid (2), bevirimat (3) and its early stage analogs (4–8).
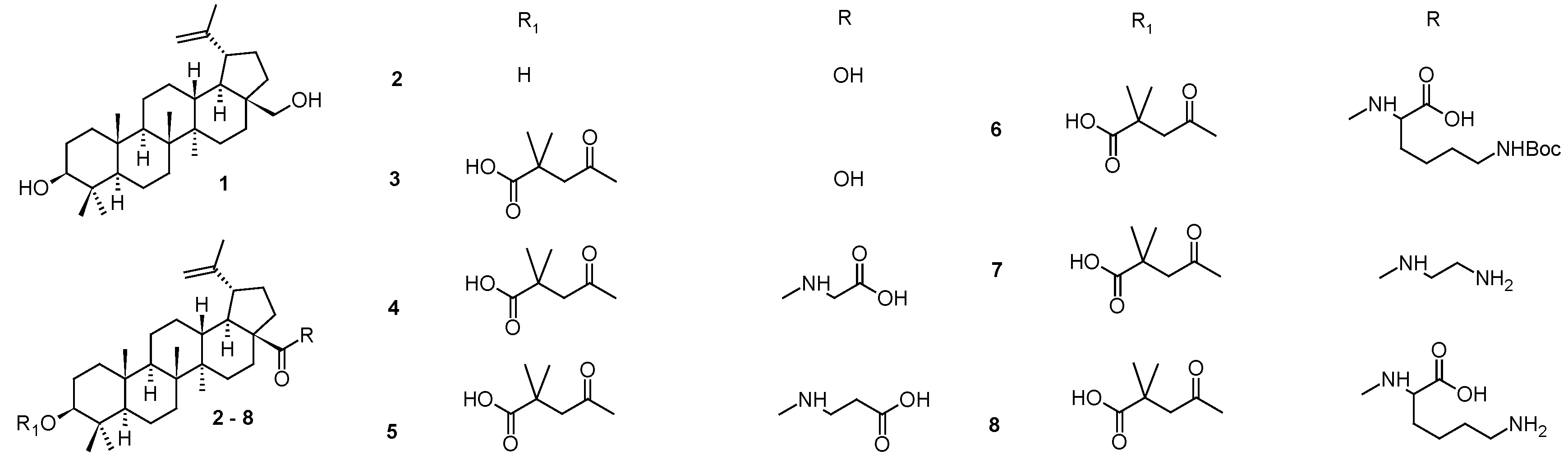
Figure 2.
Structures of compounds 9–16.
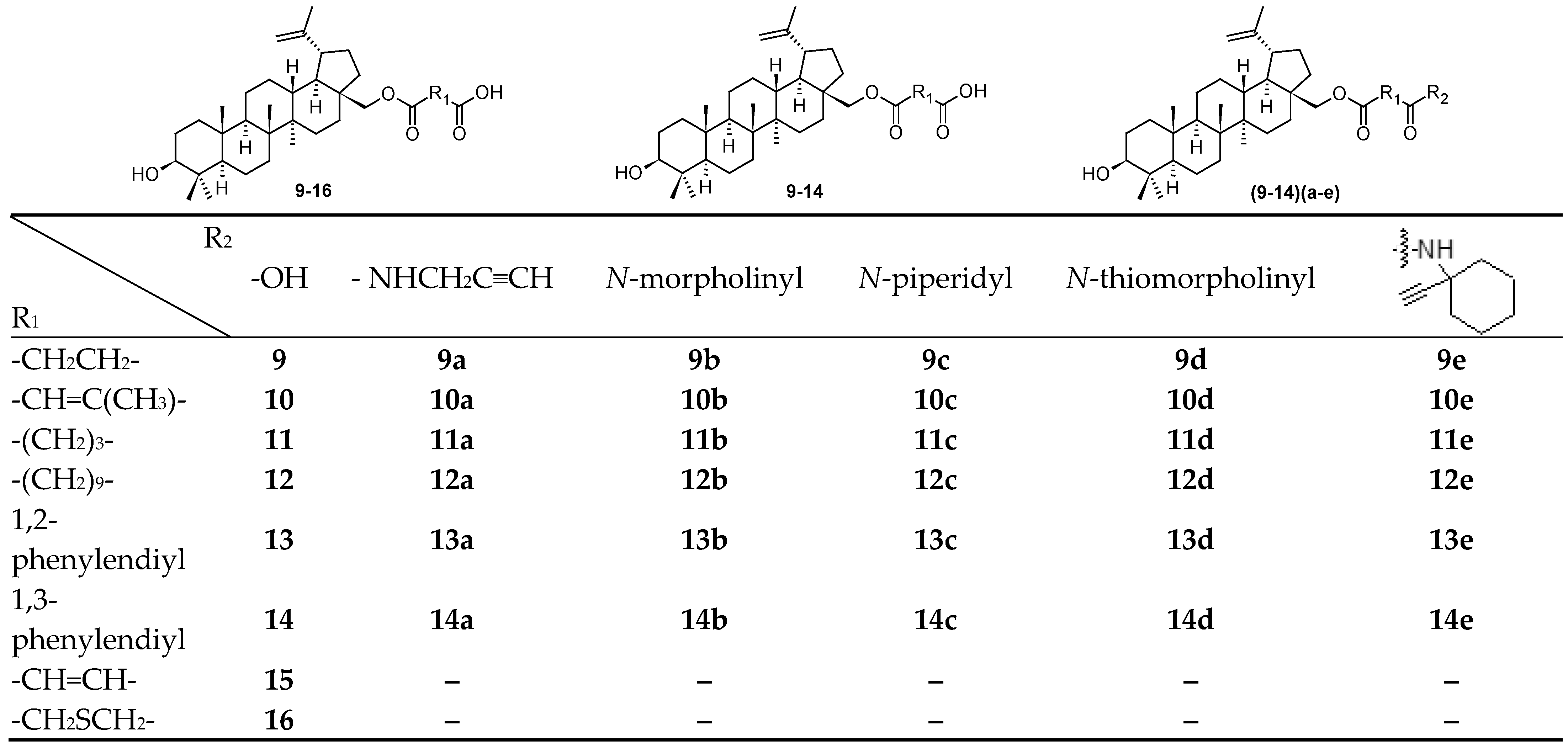
Figure 3.
Structures of the compounds 17–17s, 18 and 19.
Figure 4.
Structures of compounds 20–22 and 23a–23e.

Figure 5.
Structures of compounds 24a–24h.
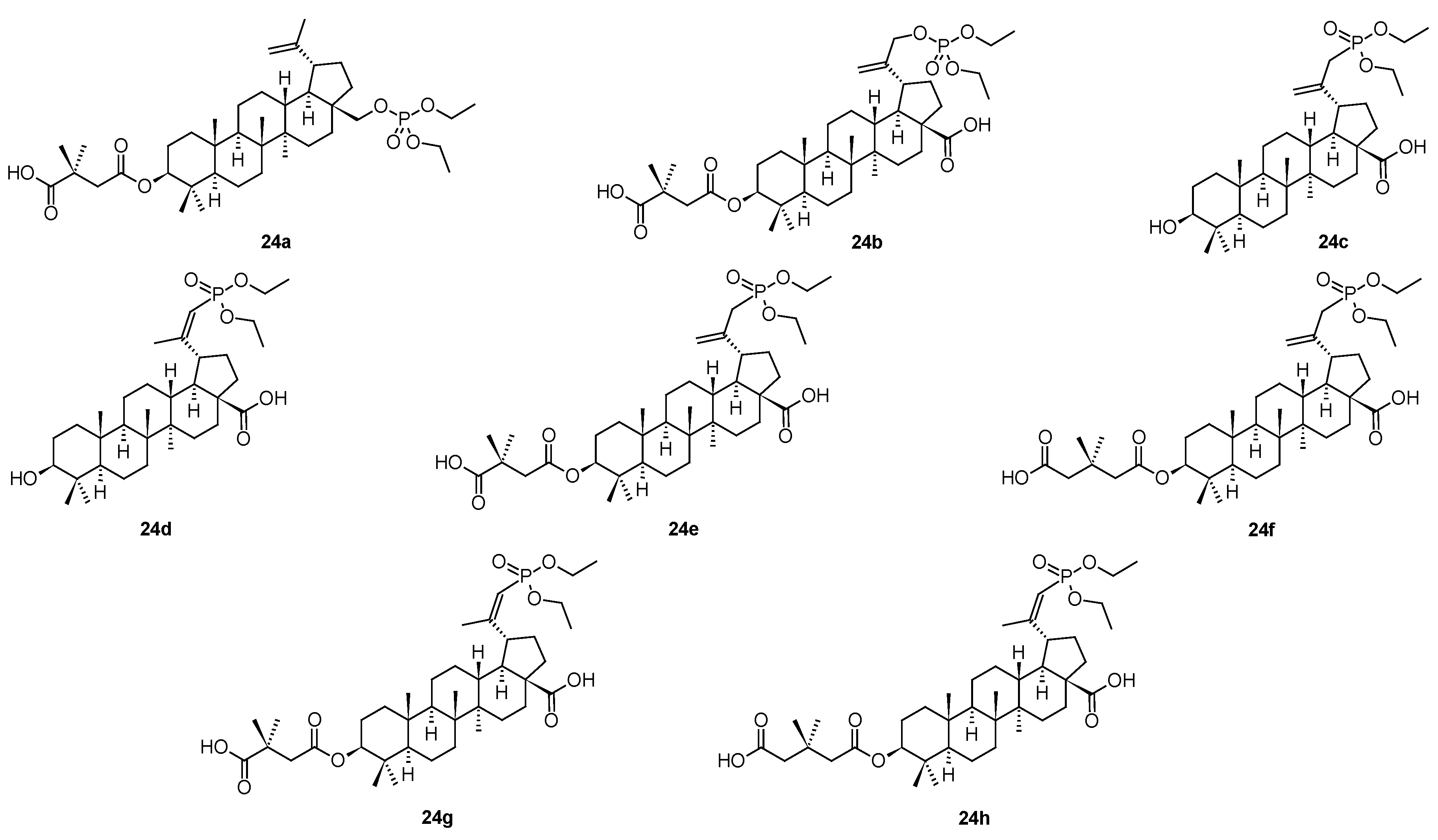
Figure 6.
Structures of compounds 25, 26 and 27a–27g.
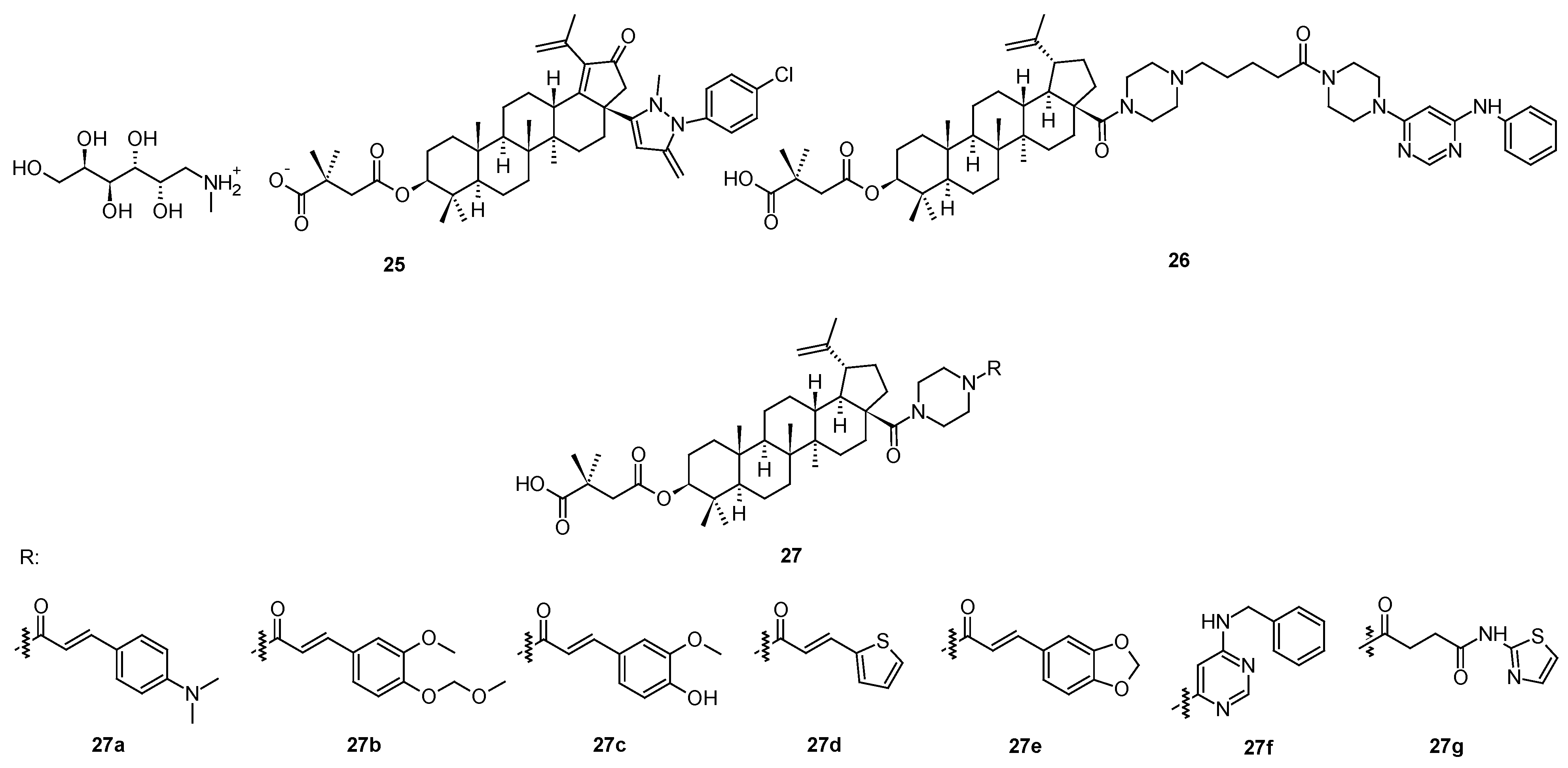
Figure 7.
Triterpenoid-peptide hybrid conjugates. For explanation of the substituent R, see Table 8.
Figure 7.
Triterpenoid-peptide hybrid conjugates. For explanation of the substituent R, see Table 8.
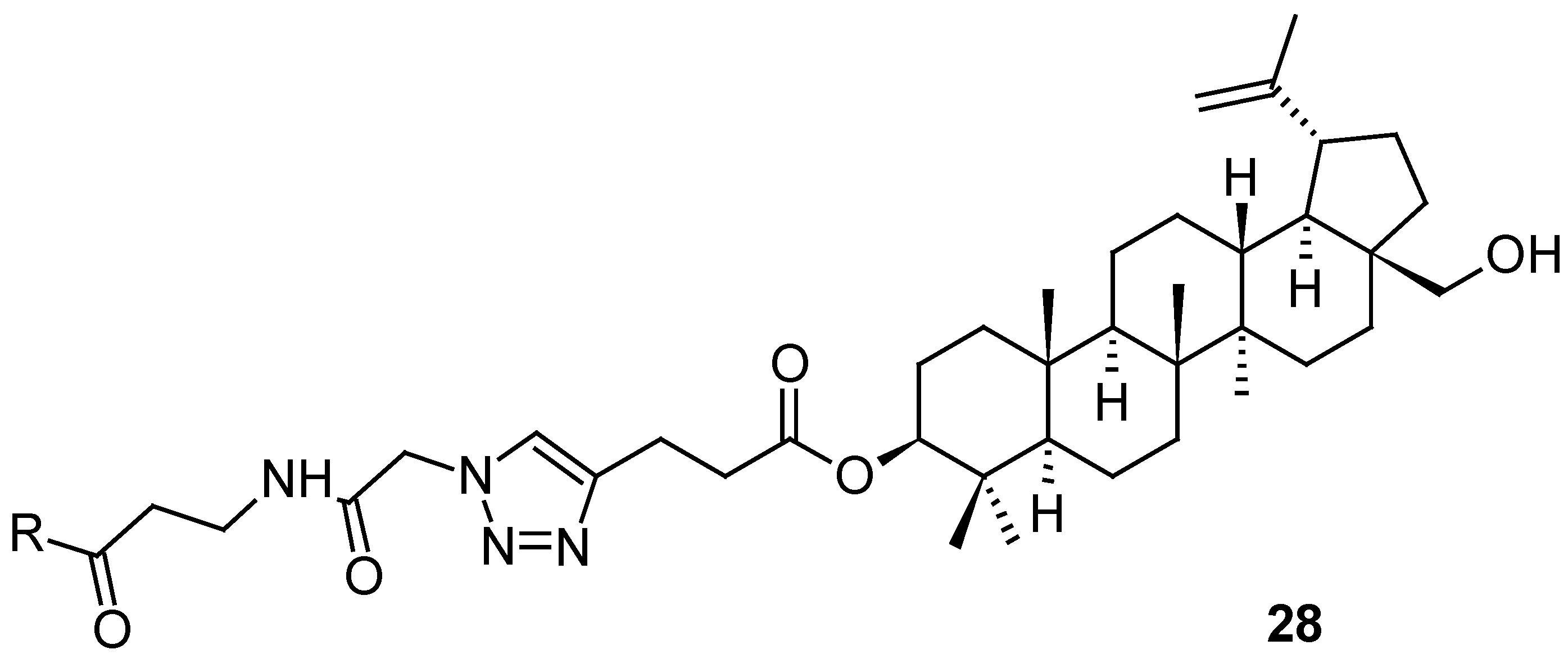
Figure 8.
Structures of the compounds 29a–29i.
Figure 9.
Structures of the compounds 30a and 30b.
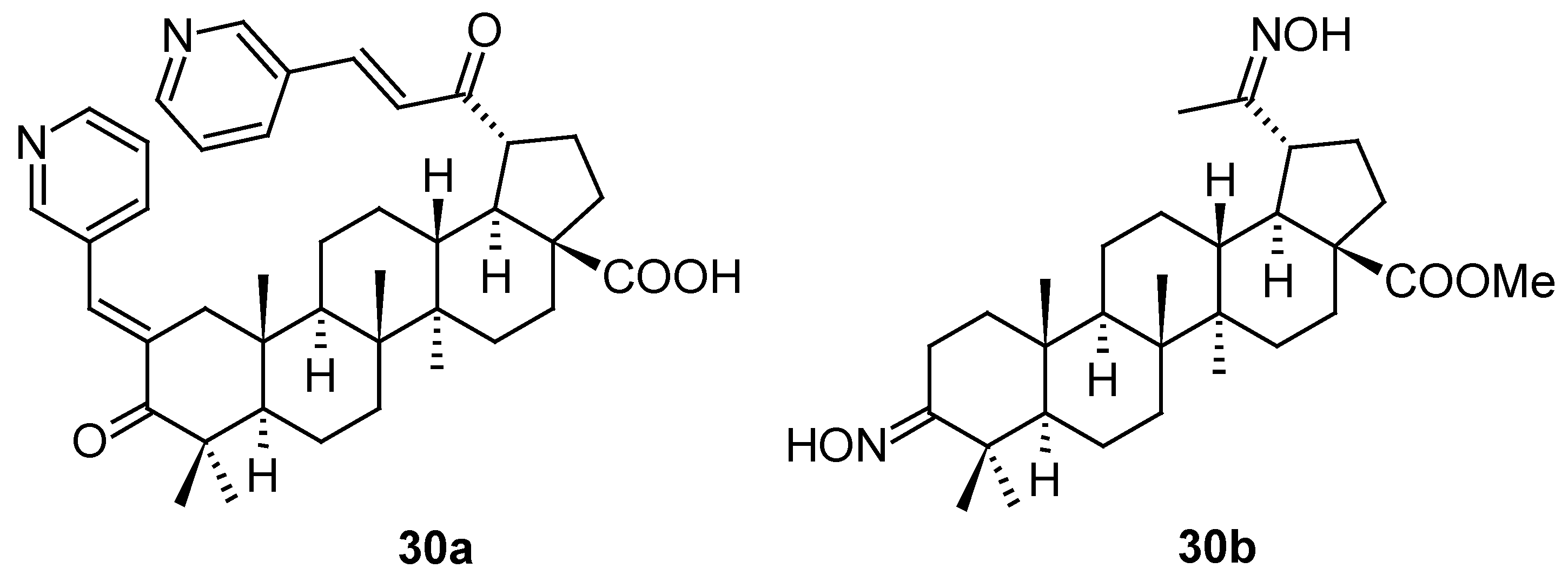
Figure 10.
Structures of the compounds 31a–31c.
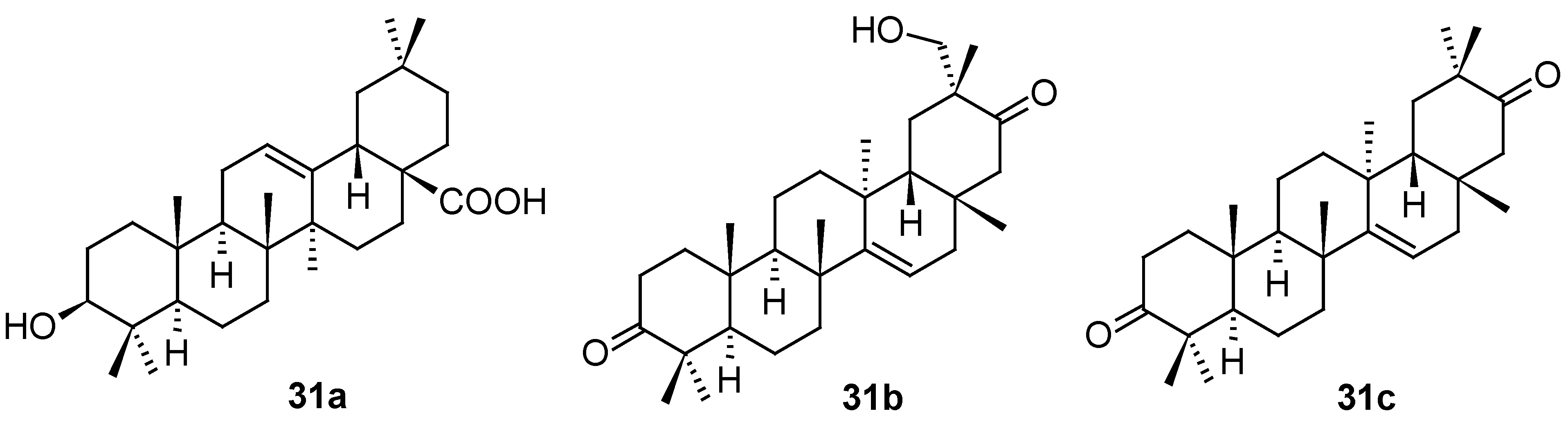
Figure 11.
Structure of the natural compound 32.
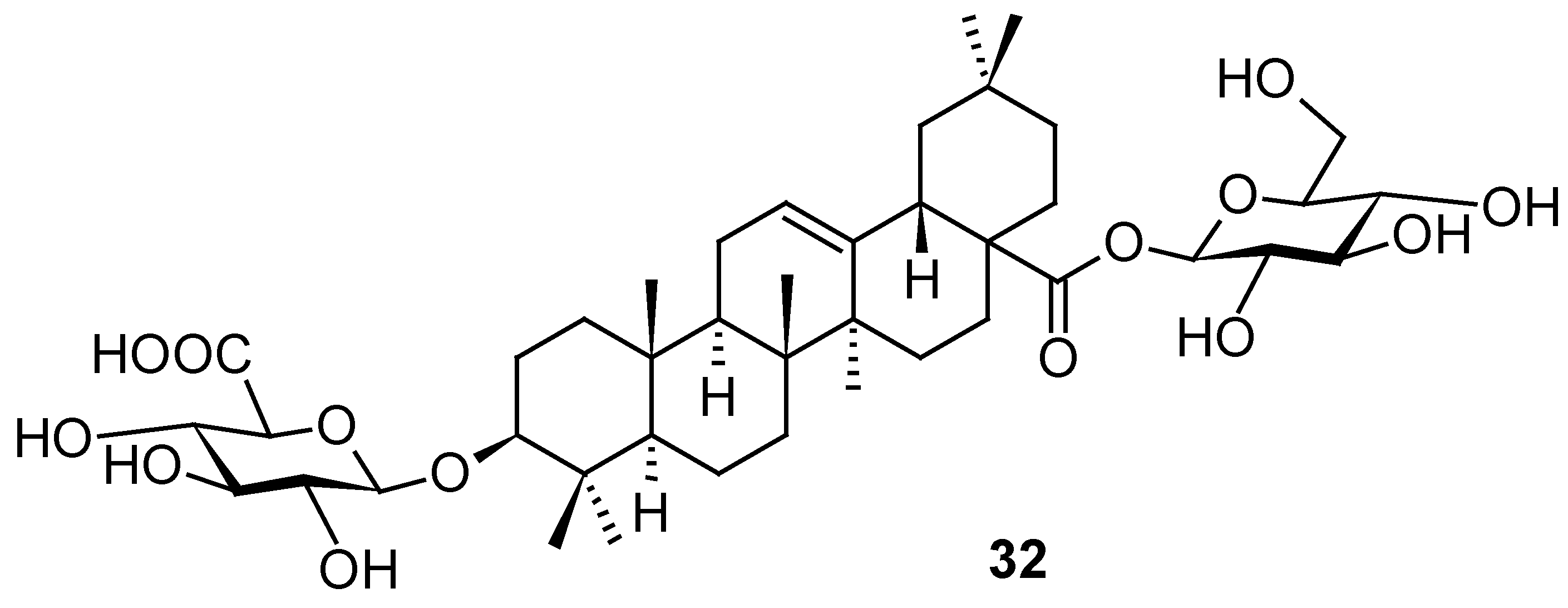
Figure 12.
Structures of the natural compounds 33–35.

Table 1.
Efficiency of HIV-1 infection inhibition by betulinic acid derivatives.
| Compound | IC50 [µM] | CC50 [µM] | SIa |
| 2 | 5.315 | 4.52 | 0.85 |
| 3 | 0.040 | 31.00 | 775.00 |
| 4 | 0.160 | 49.50 | 309.37 |
| 5 | 0.118 | 48.50 | 411.01 |
| 6 | 0.170 | 33.90 | 199.41 |
| 7 | 0.016 | 33.90 | 2118.75 |
| 8 | 4.330 | 48.70 | 11.24 |
a The selectivity index (SI) represented the CC50/IC50 ratio.
Table 2.
Anti-HSV-1 activity of the studied compounds in the mode III post-treatment assay. Compounds that reduced virus titers by at least 2 logarithms were considered active and are presented here. Presented values are medians from three independent experiments.
Table 2.
Anti-HSV-1 activity of the studied compounds in the mode III post-treatment assay. Compounds that reduced virus titers by at least 2 logarithms were considered active and are presented here. Presented values are medians from three independent experiments.
| Compd. | CC50 [μM] | EC50 [μM] | SI | Compd. | CC50 [μM] | EC50 [μM] | SI |
| 10c | 68.7 | 17.2 | 4.0 | 13b | 143.5 | 35.9 | 4.0 |
| 11 | 80.8 | 22.4 | 3.6 | 13c | 65.4 | 32.7 | 2.0 |
| 11b | 150.8 | 18.9 | 8.0 | 13d | 349.6 | 29.6 | 11.8 |
| 12b | 164.1 | 34.8 | 4.7 | Acyclovir | 1555.6 | 111.1 | 14.0 |
Table 3.
In vitro cytotoxicity of tested compounds 9–16 and (9–14)(a–e) on human lung cancer cell line (A549) and normal canine kidney cell line (MDCK).
Table 3.
In vitro cytotoxicity of tested compounds 9–16 and (9–14)(a–e) on human lung cancer cell line (A549) and normal canine kidney cell line (MDCK).
| Compd. | CC20 [µM]a,b | Compd. | CC20 [µM]a,b | ||
| A549 | MDCK | A549 | MDCK | ||
| 1 | 28.22 | 28.22 | 12a | 38.46 | 153.85 |
| 9 | 11.51 | 11.51 | 12b | 34.77 | 139.08 |
| 9a | 10.78 | 5.34 | 12c | 127.23 | 127.84 |
| 9b | 9.63 | 4.78 | 12d | 114.81 | 1141.81 |
| 9c | 69.83 | 17.46 | 12e | 102.46 | 102.46 |
| 9d | 69.42 | 124.84 | 13 | 21.15 | 42.30 |
| 9e | 110.38 | 0.77 | 13a | 39.81 | 159.24 |
| 10 | 11.26 | 5.63 | 13b | 35.87 | 143.47 |
| 10a | 10.56 | 84.46 | 13c | 65.45 | 32.72 |
| 10b | 37.82 | 37.82 | 13d | 58.89 | 7.30 |
| 10c | 34.34 | 4.26 | 13e | 104.82 | 1.57 |
| 10d | 61.50 | 61.50 | 14 | 21.15 | 42.30 |
| 10e | 54.47 | 54.47 | 14a | 159.24 | 159.24 |
| 11 | 22.44 | 11.22 | 14b | 71.74 | 143.47 |
| 11a | 42.09 | 1.18 | 14c | 130.89 | 130.89 |
| 11b | 18.85 | 18.85 | 14d | 117.79 | 117.79 |
| 11c | 17.12 | 4.25 | 14e | 104.82 | 104.82 |
| 11d | 122.70 | 61.35 | 15 | 11.55 | 5.73 |
| 11e | 108.70 | 108.70 | 16 | 21.08 | 21.08 |
| 12 | 5.09 | 10.20 | Acyclovir | 196.08 | NTc |
a Presented values are medians from three independent experiments; b CC20, concentrations required to reduce A549 and MDCK cells viability by 20%; c NT = not tested.
Table 4.
A comparison of anti-HIV-1 activity values of bevirimat (3) and the compounds 17–19.
| 3 | 17 | 18 | 19 |
|---|---|---|---|
| WT, EC50 = 10.0 nM | WT, EC50 = 5.0 nM | WT, EC50 = 3.0 nM | WT, EC50 = 1.6 nM |
| 40% HS, EC50 = 970.0 nM (97 x) | 40% HS, EC50 = 10.0 nM (2 x) | 40% HS, EC50 = 14.0 nM (4.6 x) | 40% HS, EC50 = 9.3 nM (5.8 x) |
| V370A, EC50 = 552.0 nM | V370A, EC50 = 6.0 nM | V370A, EC50 = 2.0 nM | V370A, EC50 = 3.0 nM |
| ΔV370, EC50 = > 10,000 nM | ΔV370, EC50 = 6.0 nM | ΔV370, EC50 = 3.6 nM | ΔV370, EC50 = 5.1 nM |
| T332S/V362I/prR41G, EC50 = 704 nM | T332S/V362I/prR41G, EC50 = 7.0 nM | T332S/V362I/prR41G, EC50 = 6.4 nM |
Table 5.
Anti-HIV-1 activity of compounds 17–17s, 18 and 19.
| Compd. | EC50 [nM] | EC50 V370A [nM] | EC50 ΔV370 [nM] | Compd. | EC50 [nM] | EC50 V370A [nM] | EC50 ΔV370 [nM] |
| 17 | 16 | 233 | > 3000 | 17k | 7 | 10 | 359 |
| 17a | 59 | 67 | – | 17l | 7 | 12 | 77 |
| 17b | 369 | NT | – | 17m | 42 | 15 | 61 |
| 17c | 106 | > 2000 | – | 17n | 10 | 51 | 160 |
| 17d | 37 | 427 | > 4000 | 17o | 3 | 9 | 64 |
| 17e | 6 | 32 | 2000 | 17p | 2 | 6 | 60 |
| 17f | 7 | 41 | > 2000 | 17q | 1 | 2 | 13 |
| 17g | 17 | 150 | – | 17r | 1 | 2 | 13 |
| 17h | 5 | 24 | 361 | 17s | 2 | 3 | 13 |
| 17i | 3 | 8 | 31 | 18 | 3 | 2 | 3.6 |
| 17j | 47 | 101 | 1600 | 19 | 1.6 | 3.0 | 5.1 |
Table 6.
Anti-HIV-1 activity (EC50 [µM]) of compounds 24a–24h.
| Compound | 24a | 24b | 24c | 24d | 24e | 24f | 24g | 24h |
| EC50 [µM] | 0.02a | 1 | > 10 | > 10 | 0.02 | 0.9 | 4 | 0.6 |
a Therapeutic index, TI = 1250.
Table 7.
Antiviral activity of the piperazine-type compounds 27a–27g, compared to 25 and 26 in HIV-1 NL4-3.
Table 7.
Antiviral activity of the piperazine-type compounds 27a–27g, compared to 25 and 26 in HIV-1 NL4-3.
| Compound | 25 | 26 | 27a | 27b | 27c | 27d | 27e | 27f | 27g |
| EC50 [µM] | 0.021 | 0.012 | 0.027 | 0.025 | 0.018 | 0.032 | 0.022 | 0.037 | 0.040 |
Table 8.
Inhibitory activities of sapogenin−peptide conjugates 28a–28n on HIV-1 Env-mediated cell-cell fusion.
Table 8.
Inhibitory activities of sapogenin−peptide conjugates 28a–28n on HIV-1 Env-mediated cell-cell fusion.
| Compd. | Compd. codea | Sequenceb | EC50 [nM]c |
| 28a | BAo−P26 | BAo-a-NNYTSLIHSLIEESQNQQEKNEQELL | 89.4 ± 2.4 |
| 28b | UAo−P26 | UAo-a-NNYTSLIHSLIEESQNQQEKNEQELL | 145 ± 17 |
| 28c | OAo−P26 | OAo-a-NNYTSLIHSLIEESQNQQEKNEQELL | 176 ± 45 |
| 28d | BAc−P26 | BAc-a-NNYTSLIHSLIEESQNQQEKNEQELL | 15.1 ± 2.5 |
| 28e | UAc−P26 | UAc-a-NNYTSLIHSLIEESQNQQEKNEQELL | 51.5 ± 25 |
| 28f | OAc−P26 | OAc-a-NNYTSLIHSLIEESQNQQEKNEQELL | 28.6 ± 5.1 |
| 28g | BApc−P26 | BApc-a-NNYTSLIHSLIEESQNQQEKNEQELL | 197 ± 55 |
| 28h | BApo−P26 | BApo-a-NNYTSLIHSLIEESQNQQEKNEQELL | 327 ± 21 |
| 28i | P26−BAo | NNYTSLIHSLIEESQNQQEKNEQELL-a-K(BAo) | 19.6 ± 5.0 |
| 28j | P26−BAc | NNYTSLIHSLIEESQNQQEKNEQELL-a-K(BAc) | 44.2 ± 10 |
| 28k | P26−BApc | NNYTSLIHSLIEESQNQQEKNEQELL-a-K(BApc) | 3.94 ± 0.3 |
| 28l | P26−BApo | NNYTSLIHSLIEESQNQQEKNEQELL-a-K(BApo) | 7.94 ± 1.5 |
| 28m | P26−UApc | NNYTSLIHSLIEESQNQQEKNEQELL-a-K(UApc) | 3.35 ± 1.1 |
| 28n | P26−OApc | NNYTSLIHSLIEESQNQQEKNEQELL-a-K(OApc) | 3.31 ± 1.0 |
| 2 | BA | Betulinic acid | > 1000000 |
| – | UA | Ursolic acid | > 1000000 |
| – | OA | Oleanolic acid | > 1000000 |
| – | P26 | NNYTSLIHSLIEESQNQQEKNEQELL | 3240 ± 560 |
| – | Ptrz−P26 | Ptrz-a-NNYTSLIHSLIEESQNQQEKNEQELL | 3580 ± 156 |
| – | P26−Ptrz | NNYTSLIHSLIEESQNQQEKNEQELL-a-K(Ptrz) | 2183 ± 786 |
| – | P26 + BAo | NNYTSLIHSLIEESQNQQEKNEQELL + BAo | 2390 ± 612 |
| – | T20 | YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF | 10.1 ± 1.4 |
a When a non-peptide moiety is conjugated to the N-terminus of P26, the hybrid has carboxyamide at the C-terminus. When a non-peptide moiety is attached to the C-terminus of P26, the conjugate has an acetyl group at the N-terminus and carboxyamide at the C-terminus. P26 and T20 have an acetyl group at the N-terminus and carboxyamide at the C-terminus; b a = β-alanine; Ptrz = 4-propyl-1H-1,2,3-triazol; c Compounds were tested in triplicate, and the data are presented as the mean ± standard deviation (SD).
Table 9.
Anti-HIV-1 Activities and Cytotoxicities of the Conjugates 28a–28n.
| Compound | Compd. code | EC50 [nM] for inhibitinga | |||
| HIV-1IIIB replication |
HIV-1BaL replication |
CC50 [μM] | SIb | ||
| 28a | BAo−P26 | 475 ± 87 | 456 ± 72 | > 25 | > 52 |
| 28b | UAo−P26 | 565 ± 130 | 288 ± 32 | > 25 | > 44 |
| 28c | OAo−P26 | 369 ± 2.0 | 498 ± 87 | > 25 | > 68 |
| 28d | BAc−P26 | 133 ± 53 | 98.0 ± 26 | > 25 | > 187 |
| 28e | UAc−P26 | 387 ± 252 | 113 ± 55 | > 25 | > 64 |
| 28f | OAc−P26 | 94.0 ± 15 | 150 ± 21 | > 25 | > 266 |
| 28g | BApc−P26 | 242 ± 11 | 519 ± 98 | > 25 | > 103 |
| 28h | BApo−P26 | 501 ± 122 | 99.0 ± 12 | > 25 | > 52 |
| 28i | P26−BAo | 154 ± 9.0 | 135 ± 15 | > 25 | > 50 |
| 28j | P26−BAc | 61.6 ± 16 | 83.1 ± 8.3 | > 25 | > 406 |
| 28k | P26−BApc | 4.28 ± 0.7 | 6.90 ± 0.1 | 14.3 ± 1.0 | 3348 |
| 28l | P26−BApo | 475 ± 87 | 456 ± 72 | > 25 | > 52 |
| 28m | P26−UApc | 565 ± 130 | 288 ± 32 | > 25 | > 44 |
| 28n | P26−OApc | 369 ± 2.0 | 498 ± 87 | > 25 | > 68 |
a Compounds were tested in triplicate, and the data are presented as the mean ± standard deviation; b SI (selectivity index) = CC50/EC50 for inhibiting HIV-1IIIB infection.
Table 10.
Antiviral activity against HSV-1 and cytotoxicity for compounds 29a–29i in Vero cellsa.
| Compound | CC50 [µM]b | IC50 [µM] | SIc |
| 29a | 23.11 | 6.41 | 3.6 |
| 29b | 57.74 | > 11.11 | - |
| 29c | 33.33 | > 11.11 | - |
| 29d | 69.34 | > 33.33 | - |
| 29e | 3.70 | > 1.23 | - |
| 29f | 11.11 | >3.70 | - |
| 29g | 5.34 | > 1.23 | - |
| 29h | 1.78 | > 0.41 | - |
| 29i | 1.78 | 0.41 | 4.3 |
| Acyclovir | > 100 | 0.41 | > 243.9 |
a Data represent mean values for three independent determinations; b Cytotoxic concentration required to inhibit Vero cell growth by 50%; c Selectivity index value equaled CC50/IC50.
Table 11.
Antiviral activity values found for 30a and 30b.
| Compd. | EC50 [μM] | Compd. | EC50 [μM] |
| 30a | > 0.24 (HCMV) | 30b | 1.20 (HSV-1) |
| 3.47 (HPV) |
Table 12.
Antiviral activities of 31a–31c against HSV-1 in Vero cells.
| Compound | CC50 [μM] | IC50 [μM] | SIa |
| 31a | 33.3 | 11.1 | 3.0 |
| 31b | 57.7 | 14.3 | 4.0 |
| 31c | 57.7 | 6.4 | 9.0 |
| Acyclovir | > 100 | 0.3 | > 370.4 |
a Selectivity index value calculated as a ratio CC50/IC50.
Table 13.
Antiviral activity values found for 33–35 against HSV-1.
| Compound | IC50 [μM] | SIa |
| 33 | 15.3 ± 1.9 | 2.4 ± 0.3 |
| 34 | 1.1 ± 0.2 | 6.8 ± 0.9 |
| 35 | 4.3 ± 0.4 | 7.8 ± 0.7 |
| Acyclovir | 11.9 ± 1.4 | > 50 |
a Selectivity index value calculated as a ratio CC50/IC50; CC50 values were calculated in silico.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



