
Submitted:
27 October 2023
Posted:
27 October 2023
You are already at the latest version
Abstract
Cell migration crucial contributor to metastasis, a critical process associated with the mortality of cancer patients. The initiation of metastasis is triggered by epithelial-mesenchymal transition (EMT), along with the changes of the expression of EMT marker proteins. Inflammation plays a significant role in carcinogenesis and metastasis. Lipopolysaccharide (LPS), a typical inflammatory agent, promoted the generation of superoxide through the activation of p-Tyr42 RhoA, Rho-dependent kinase 2 (ROCK2) and the phosphorylation of p47phox. In addition, p-Tyr42 RhoA activated phospholipase D1 (PLD1), with PLD1 and phosphatidic acid (PA) being involved in superoxide production. PA also regulated the expression of EMT maker proteins. Consequently, we have identified MHY9 (Miosin IIA, NMIIA) as a PA-binding protein in response to LPS. MYH9 also contributed cell migration and the alteration of the expression of EMT marker proteins. P-Tyr42 RhoA, PLD1 and MYH9 associated to form a complex, which was distributed in both the cytosol and nucleus. In addition, we have found that p-Tyr42 RhoA and PLD1 associated with the ZEB1 promoter. Taken together, we propose that p-Tyr42 RhoA and PLD1, responsible for producing PA, and PA-bound MYH9 are involved in the regulation of ZEB1 expression, thereby promoting cell migration.

Keywords:
p-Tyr42 RhoA
; PLD1
; MYH9
; ZEB1
; EMT
1. Introduction
The migration of cells is vital in various biological phenomena, including embryogenesis, immune response, inflammation, morphogenesis, and wound healing. In particular, cancer cell migration is a critical step in tumor metastasis, which is the main cause of cancer-related deaths. Epithelial-mesenchymal transition (EMT) has been related to carcinogenesis and contributes to metastasis of cancer cells by promoting motility and invasion [1]. During EMT process, the expression of proteins for adhering adjacent cells, including E-cadherin, Occludin, and Claudin, decreased, while N-cadherin, Vimentin, and EMT transcription factors such as Zeb1, Snail 1/2, and Twist are upregulated [1]. Here, we focused on that inflammation is crucial for EMT process in cancer cells. Accordingly, a typical inflammatory agent, LPS was utilized for inducting inflammation in cancer cells.
Lipopolysaccharide (LPS), a distinctive constituent found in the cell wall of Gram-negative bacteria, serves as a well-known activator of the inflammasome. Within intestinal epithelial cells, the activation of the inflammasome is triggered by LPS through the initiation of Toll-like receptor 4 (TLR4)-mediated signal transduction pathways [2]. Numerous reports found that LPS stimulates inflammatory responses, causes epithelial cells to change into cancerous forms, and encourages carcinogenesis both in vitro and in vivo. For example, it has been shown that LPS promotes the invasion and migration of prostate cancer (PCa) cells [3] and promotes epithelial–mesenchymal transition (EMT) through TLR4 stimulated by LPS [4].
Typically, Rho GTPase is involved in regulating a range of cellular processes, including the modulation of cytoskeletal proteins, cellular morphology, migration, and cell proliferation. The activities of Rho GTPases are regulated by specific factors such as guanine nucleotide exchange factors (GEFs), GTPase activating proteins (GAPs), and the guanine nucleotide dissociation factor (GDI) [5]. The cell migration system encompasses extensively researched members of the Rho-GTPase family, including RhoA, Rac1, and Cdc42, which each play significant roles in regulating cell motility. RhoA specifically contributes to focal adhesion formation, actomyosin contraction, stress fiber formation, and retraction of the cell's tail, thereby facilitating cell migration [6]. Studies have indicated that LPS can trigger the disruption of tight junctions in brain microvascular endothelial cells (BMECs) through the activation of the RhoA/ROCK signaling pathway [7].
In mammals, intracellular enzymes phospholipase D (PLD) responds to external signals by hydrolyzing phosphatidylcholine (PC), the most abundant phospholipid in cell membranes, to generate the lipid second-messenger phosphatidic acid (PA). The resulting PA, produced by PLD, serves as a regulator for a wide range of cellular processes including vesicular trafficking, membrane fission and fusion, as well as cell growth, proliferation, and migration [8,9]. Two mammalian PLD, PLD1 and PLD2, have been identified both of which include splice variants [10]. Both in vitro and in vivo contexts, the activity of PLD1 is significantly boosted by protein kinase C (PKC) as well as small GTPases such as the ADP-ribosylation factor (Arf) and Rho families [11]. In particular, C-terminal domain of PLD1 interacts with the dominant active mutant RhoA G14V [12], and recently elucidated crystal structure of PLD1 provided activation mechanism by RhoA and PI(4,5)P2 [13]. Notably, G protein-coupled receptors (GPCRs) activates nuclear PLD1 in vascular smooth muscle cells in response to lysophosphatidic acid (LPA) [14]. It is also reported that PLD1, but not PLD2 is upregulated in non-small cell lung cancer tissue [15]. Our previous study revealed that p-Tyr42 RhoA contributes to amplifying superoxide production and induces cell migration of lung cancer A549 lung cancer cell line in response to PMA [16]. Based on these reports, we investigated the function of p-Tyr42 RhoA in PLD1 activation, and its downstream signaling pathway.
In this study, we observed that LPS promoted the p-Tyr42 RhoA levels, which activated PLD1 through the interaction between p-Tyr42 RhoA and PLD1 in A549 cells. LPS stimulated cell migration through the expression of EMT marker proteins, and particularly the promoter of which ZEB1were associated with p-Tyr42 RhoA and PLD1 in nucleus in response to LPS. Moreover, we found that Myosin IIA (MYH9) was a PA-binding proteins.
2. Materials and methods
2.1. Materials
The recombinant Tat-C3 fusion protein (consisting of Tat-peptide and C3 toxin) was isolated from E. coli using a His-Bind resin column (PMID: 16705092). Y27632 (SCM075) was obtained from Millipore Sigma (Burlington, USA). LPS (E. coli 055: B5), N-acetyl-L-cysteine (NAC, A7250), apocynin (A10809), Nonidet P-40 (NP-40), bovine serum albumin (BSA), and isopropyl β-D-thiogalactoside (IPTG) were procured from Sigma-Aldrich (St. Louis, USA). Fetal bovine serum (FBS, 12484010), Dulbecco's modified eagle's medium (DMEM, 11965092), and penicillin-streptomycin antibiotics (15140122) were obtained from GibcoBRL (New York, USA). Protease inhibitor cocktail was purchased from ApexBio (Boston, USA). Fetal bovine serum (FBS), penicillin, and streptomycin were purchased from Cambrex (Verviers, Belgium). LB Broth High Salt (MB-L4488) and skim milk powder (MB-S1667) were obtained from MBcell (SeoCho-Gu, Seoul, Korea). 4’6-diamidino-2- phenylindole (DAPI) was purchased from Invitrogen (Carlsbad, CA, USA). Alexa flour-568 and Alexa flour-594 reagents were obtained from Molecular Probes (Eugene, OR, USA). Protein A/G-agarose beads were from Amersham Biosciences (Piscataway, NJ, USA). The jetPRIME DNA/si-RNA transfection reagent was obtained from Polyplus-transfection (Seoul, Korea). si-PLD1 (sc-44000), si-RhoA (sc-29471) and control si-RNA (sc-37007) were procured from Santa Cruz Biotechnology.
The antibodies used in this study were obtained from various sources. An antibody against phosphorylated Tyr42 Rho was generated through immunization with a peptide corresponding to p-Tyr42 Rho (epitope peptide T37VFEN (phospho-) Y42VADIE47). This peptide was synthesized from the phospho-Tyr42 precursor obtained from Young-In Frontier, Seoul, Korea. Normal IgG (sc:2025,), actin (sc:58673,), PLD1 (sc-28314), Lamin B (sc:965962), Tubulin (sc:32293), ROCK I (sc-17794), ROCK II, (sc:398519), RhoA, (sc-418), N-cadherin (sc:393933), Vimentin (sc:6260), and Snail (sc:271977), were purchased from Santa Cruz Biotechnology (Texas, USA). MYH9 (14844-1-AP) was purchased from proteintech (Rosemont, USA). HA Tag (3724), p-Lamin A/C (cat; 2026) were purchased from Cell Signaling Technology. E-cadherin (ABP0083, Abbkine) and ZEB1 (ABP60963) were purchased from Abbkine (Wuhan, China). p-p47phox (#PA5-37806) was procured from Invitrogen (Waltham, USA). Goat anti-rabbit and goat anti-mouse IgG conjugated with HRP were obtained from Enzo Life Sciences (Farmingdale, NY, USA).
2.2. Cell culture
A549 cells, derived from human lung adenocarcinoma, were obtained from the Korean Cell Line Bank (Seoul, Korea). The cells were then cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% heat-inactivated fetal bovine serum (FBS) and antibiotics (100 U/ml of penicillin and 100 g/ml of streptomycin) in a humidified environment with 5% CO2 at 37 °C.
2.3. Superoxide measurement
The levels of superoxide were measured using the fluorescent probe 2′,7′-dichlorofluorescin diacetate (DCFDA) cellular ROS assay kit (ab113851). A549 cells (2x105) were stimulated with the specific treatment in serum-free media, followed by fixation using 4% formaldehyde for 15 minutes at room temperature. To induce fluorescence, the cells were treated with a solution of 5 μM DCFDA in 1x PBS (500 μl) for 30 minutes at 37°C in the dark. Fluorescence images were captured using a fluorescence microscope (Axiovert 200, Zeiss; Göttingen, Germany) with filters set an excitation wavelength of 485 nm and an emission wavelength of 530 nm
2.4. Transfection of Plasmid DNA and si-RNA
The A549 cells transfected with plasmid DNA and small interfering RNA (siRNA) using the Jetprime reagent (polyplus transfection, France). Plasmid constructs HA-RhoA WT, HA-RhoA Y42E, HA-RhoA-Y42F were utilized in this study. siRNAs targeting RhoA (sc-29471), PLD1 (sc-44000), and control siRNA (sc-37007) were obtained from Santa Cruz Biotechnology (Texas, USA). A549 cells were initially seeded to achieve 30%–40% confluences and then transfected with siRNAs using the jetprime transfection reagent as per the manufacturer's instructions. In six-well cell culture dishes, 2 µg plasmid DNA and 50nM-100 nM of si-RNAs were transfected. When performing both DNA and siRNA transfection in the same culture dishes, the siRNA was transfected first, followed by the transfection of DNA after 24 h using fresh media containing 5% serum. After 48 h for DNA transfection and 72 h for siRNA transfection, the cells were harvested, and protein expression levels were assessed through Western blot analysis.
2.5. Western blotting
Western blot analysis was performed on A549 cells. The cells were rinsed 1x PBS and then lysed in RIPA buffer containing 20mM Tris- HCl pH7.4, 1mM MgCl2, 1% (v/v) Nonidet P40, and 125mM NaCl. Phosphatase/protease inhibitor cocktail was added to the lysates. The protein concentration in the supernatants was determined using a BCA protein quantification assay kit after centrifugation for 15 min at 13000xg at 4°C. The cell lysates were then separated using 10–14% SDS-PAGE and transferred to a PVDF membrane. Non-specific binding was suppressed by incubating the membrane with non-fat dried milk and 0.05% Tween-20 in Tris buffered saline solution. Specific antibodies were used to probe the target proteins, followed by washing and incubation with peroxidase-conjugated secondary antibodies. The bound antibodies were visualized using an ECL system.
2.6. Immunoprecipitation (IP)
Immunoprecipitation was performed on A549 cells. The cells were rinsed with 1× PBS and lysed in a buffer containing 20 mM Tris pH 7.4, 120 mM NaCl, 1 mM MgCl2, and 1% Nonidet P-40. A protease/phosphatase inhibitor mixture was added to the lysates. The lysates were clarified through centrifugation and then subjected to a preliminary clearance step using protein A/G-agarose beads for 1 hour. The cleared supernatant was incubated with an anti-PLD1 antibody (1:1000 dilutions) for overnight at 4 °C. Protein A/G-agarose beads (30 μl) were added to the lysate and the mixture was incubated with shaking. After the beads settled, mixtures were washed with a lysis buffer. The proteins attached to the beads were released and analyzed using immunoblotting with specific primary antibodies.
2.7. Cell fractionation
To separate cytosolic and nuclear fractions, NE-PER nuclear and cytoplasmic extraction reagents (CER: Thermo Scientific, 78833) were used. A549 cells were either stimulated with LPS or left unstimulated as a control. The cells were then collected in ice-cold 1 × PBS and centrifuged at 13,000×g for 15 minutes. A portion of the resulting cell pellet (20 μl) was mixed with ice-cold CER I (200 μl), CER II (11 μl), and protease inhibitors, and then vortexed and centrifuged obtain the cytoplasmic protein extract (supernatant). The remaining pellet, containing the nuclei, was re-suspended in ice-cold NER, vortexed, and centrifuged to obtain the nuclear extract. The fractions obtained were analyzed through immunoblotting using appropriate antibodies, with lamin B and tubulin proteins serving as markers for the nucleus and cytosol, respectively.
2.8. Monolayer Wound Healing Assay
A549 cells were seeded in 6-well plates at a density of 1.5 × 105 cells per well and incubated for 24 h. Then, the cells were subjected to different treatments, including plasmid DNA transfection, si-RNA transfection, or LPS stimulation. To prevent cell proliferation during migration, serum starvation was applied 12 h prior to creating the "wound" scratch. Once the cell monolayer reached confluence, a 200µl sterile plastic pipette tip was used to create scratches. The wounds were regular photographed (0, 24, and 48 h) and the area of cell-free wounds was measured using a microscope (Axiovert 200, Zeiss). The assay was performed in at least three independent experiments.
2.9. Chromatin immunoprecipitation (ChIP) and PCR
The ChIP assay was conducted based on a previously published paper (PMID: 19334278), with a slight modification. Briefly, A549 cells were exposed to LPS, and then formaldehyde was added as a crosslinker (final concentration: 0.75%) for 1 h. The crosslinking reaction was stopped by treating with 125 mM glycine for 20 minutes. After sonication, the fragmented chromatin-protein complex was incubated with p-Y42 RhoA and PLD1 antibodies, and then precipitated using protein A/G beads. Subsequently, the beads were washed, and the bound DNAs were eluted using elution buffer (containing 1% SDS and 100 mM NaHCO3). RNA and proteins were removed by incubation with RNAse and proteinase K. Finally, DNA was purified through phenol: chloroform extraction. PCR primers for ZEB1 (located on human chromosome 10, positions 31318016-31319215: forward primer: 5′- CAAACCTGCCCTTCCCCTCA-3’, reverse primer: 5′- CTCTACGGCCGGAACCTTGT-3’) and Snail1 (located on human chromosome 20, positions 48598513-48599512: forward primer: 5′- GAACGGGTGCTCTTGGCTAGCTG -3’, reverse primer: 5′- TCGAGCGAAGCGAGGCCTC -3’) were synthesized by Bioneer (Daejeon, Korea).
2.10. Immunofluorescent Staining
A549 cells were cultured and fixed with 4% paraformaldehyde for 10 minutes. To allow membrane permeabilization, a solution of 0.5% TX-100 detergents and PBS (1XPBS) was applied for 10 minutes, followed by rinsing with 1XPBS. The cells were then incubated overnight at 4°C with primary antibodies targeting p-Y42 RhoA and PLD1 (1:100 dilution). After incubation, the cells were washed with 1XPBS. To visualize the antibodies, an Alexa-546 conjugated anti-rabbit IgG (red color) was used at a dilution of 1:50 for 2 h at room temperature, while keeping the environment free from light. The nuclei were stained with DAPI (4’,6-diamidino-2-phenylindole), which was added 10 minutes before washing at the concentration of 1 μg/ml (PMID: 33388549). Fluorescence images were captured using a conventional fluorescence microscope (Axiovert 200, Zeiss, Oberkochen, Germany).
2.11. Statistical analysis
Data analysis and visualization were performed using GraphPad Prism 8 (GraphPad Software, San Diego, USA). All experiments were repeated at least three times. The data are presented as mean ± SE (standard error). Statistical comparisons were done using Student's t-test with the GraphPad Prism software, and differences between two groups were considered statistically significant if the P values were below the designated threshold (*P < 0.05, **P < 0.01, ***P < 0.001).
3. Results
3.1. LPS induces superoxide production through ROCK2 and p-p47phox.
In this study, we have observed that LPS induces superoxide generation in A549 lung cancer cell line in a concentration-dependent manner (Figure 1A). The LPS-mediated superoxide production was inhibited by Tat-C3, a Rho inhibitor, and Y27632, a ROCK inhibitor, highlighting the involvement of Rho and ROCK in regulating superoxide formation in response to LPS (Fig.1B). To distinguish ROCK types, we investigated the effects of ROCK1 or ROCK2 knockdown with siRNAs on superoxide production. Si-ROCK1 did not interfere with superoxide production, while si-ROCK2 prevented its generation in A549 cells, suggesting that ROCK2 primarily plays a role in superoxide production induced by LPS (Figure 1C). Consistent with these result, si-ROCK2, but not si-ROCK1, reduced the phosphorylation at Ser345 residue of p47phox (Figure 1D). Of particular interest, reagents reducing ROS level, namely apocynin, NAC and BHC, subsequently reduced p-p47phox level, suggesting that ROS induces the phosphorylation of p47phox and, consequently, NADPH oxidase activity (Figure 1E).
3.2. LPS upregulates p-Tyr42 RhoA phospholipase D1 (PLD1) levels with forming a protein complex.
LPS significantly increased both p-Tyr42 RhoA and PLD1 levels in concentration- and time-dependent manners (Figure 2A and 2B, respectively). Building on prior research indicating the interaction between that RhoA-GTP and PLD1, leading to PLD1 enzyme activation, we investigated whether p-Tyr42 RhoA interacts with PLD1. In response to LPS, PLD1 co-immunoprecipitated with p-Tyr42 RhoA, indicating an interaction between PLD1 and p-Tyr42 RhoA (Figure 2C). Remarkably, the knockdown of RhoA with siRNA led to a reduction of PLD1 enzyme activity, while reconstitution with RhoA WT and RhoA Y42E phosphomimetic form restored its activity. Notably, RhoA Y42F dephosphmimetic did not restore PLD1 activity, emphasizing a critical role of the p-Tyr42 residue in RhoA for PLD1 enzyme activity (Figure 2D). Next, we investigated whether the p-Tyr42 residue of RhoA is pivotal for superoxide generation. RhoA knockdown with siRNA and reconstitution with the RhoA Y42F dephospho-mimic form suppressed superoxide production, whereas reconstitution of RhoA WT and Y42E phospho-mimic form restored its generation (Figure 2E). Additionally, agents that mitigate ROS levels attenuated the levels of p-Tyr42 RhoA and PLD1 (Figure 2F). The noteworthy observation was that PLD1 co-immunoprecipitated with both RhoA WT and RhoA Y42E, a phospho-mimicking variant, while no interaction was detected with RhoA Y42F, a dephospho-mimicking variant. (Figure 2G). This implied that that the phosphorylation of Ty42 in RhoA plays a pivotal role in this interaction with PLD1.
3.3. p-Tyr42 RhoA is involved in EMT process and cell migration.
LPS significantly promoted cell migration (Figure 3A) and induced changes in the expression of EMT marker proteins, specifically a decrease in E-cadherin and an increase in N-cadherin, ZEB1 and Snail1 (Figure 3B). Furthermore, RhoA knockdown with siRNA led to a marked reduction in the expression of N-cadherin and ZEB1, coupled with an increase in E-cadherin levels (Figure 3C). Notably, the reconstitution of RhoA Y42F dephospho-mimetic form reversed the EMT process with an increase of E-cadherin expression and a decrease of ZEB1 and Snail1 levels, underscoring the critical role of the p-Tyr42 residue of RhoA in the EMT process triggered by LPS (Figure 3D). Consistent with these findings, si-RhoA and the reconstituted RhoA Y42F, dephospho-mimic form effectively prevented cell migration, whereas reconstitution with RhoA WT and RhoA Y42E, phospho-mimic form restored migratory behavior (Figure 3E).
3.4. PLD1/PA effects on cell migration
Based on our earlier findings demonstrating that p-Tyr42 RhoA activates PLD1 activity (Figure 2D), we hypothesized that PA generated by activated PLD1 might play crucial physiological roles within cells. To explore this, we investigated the effect of PA on superoxide production. While LPS induced superoxide generation, knockdown of PLD1 with siRNA significantly suppressed its production (Figure 4A). Furthermore, our results consistently showed that PA stimulated superoxide generation (Figure 4B). Interestingly, PA also increased the phosphorylation of Tyr42 on RhoA (Figure 4C). To corroborate these observations, we assessed the impact of PLD1 knockdown, and found that siPLD1 significantly attenuated LPS-induced cell migration (Figure 4D). Additionally, given the strong connection between cell migration and EMT, we investigated the expression of EMT marker proteins. LPS induced a decrease of E-cadherin and an increase of N-cadherin, ZEB1 and Snail1. Conversely, PLD1 depletion with siPLD1 led to an increase of E-cadherin and a decrease of N-cadherin, ZEB1 and Snail1, underscoring the pivotal role of PLD1 in the EMT process triggered by LPS (Figure 4E). Notably, PA (5 μM) mimicked the effects of LPS on the regulation of EMT marker proteins, leading to a decrease of E-cadherin and increases of N-caherin, ZEB1 and Snail1 (Figure 4F). These results suggest that PA is involved in the regulation of EMT process and, indeed, we demonstrated that PA promoted cell migration (Figure 4G). Building on previous reports suggesting that p-Tyr42 RhoA translocates to nucleus [17,18], we examined the distribution of p-Tyr42 RhoA and PLD1 in response to LPS. Western blotting of nuclear and cytosolic fractions revealed that LPS induced an increase of nuclear p-Tyr42 RhoA and PLD1 compared to their cytosolic count-parts (Figure 4H). Furthermore, immunohistochemical images confirmed that LPS elevated the levels of p-Tyr42 RhoA and PLD1 within nucleus, with a portion of these proteins co-localizing (Figure 4I). Significantly, ChIP-PCR using antibodies against p-Tyr42 RhoA and PLD1 and ZEB1 promoter primers indicated that LPS increased the association of p-Tyr42 RhoA and PLD1 with ZEB1 promoter, but not that of Snail1 (Figure 4J and 4K, respectively).
3.5. p-Tyr42 RhoA and PLD1 bind to the promoter of ZEB1.
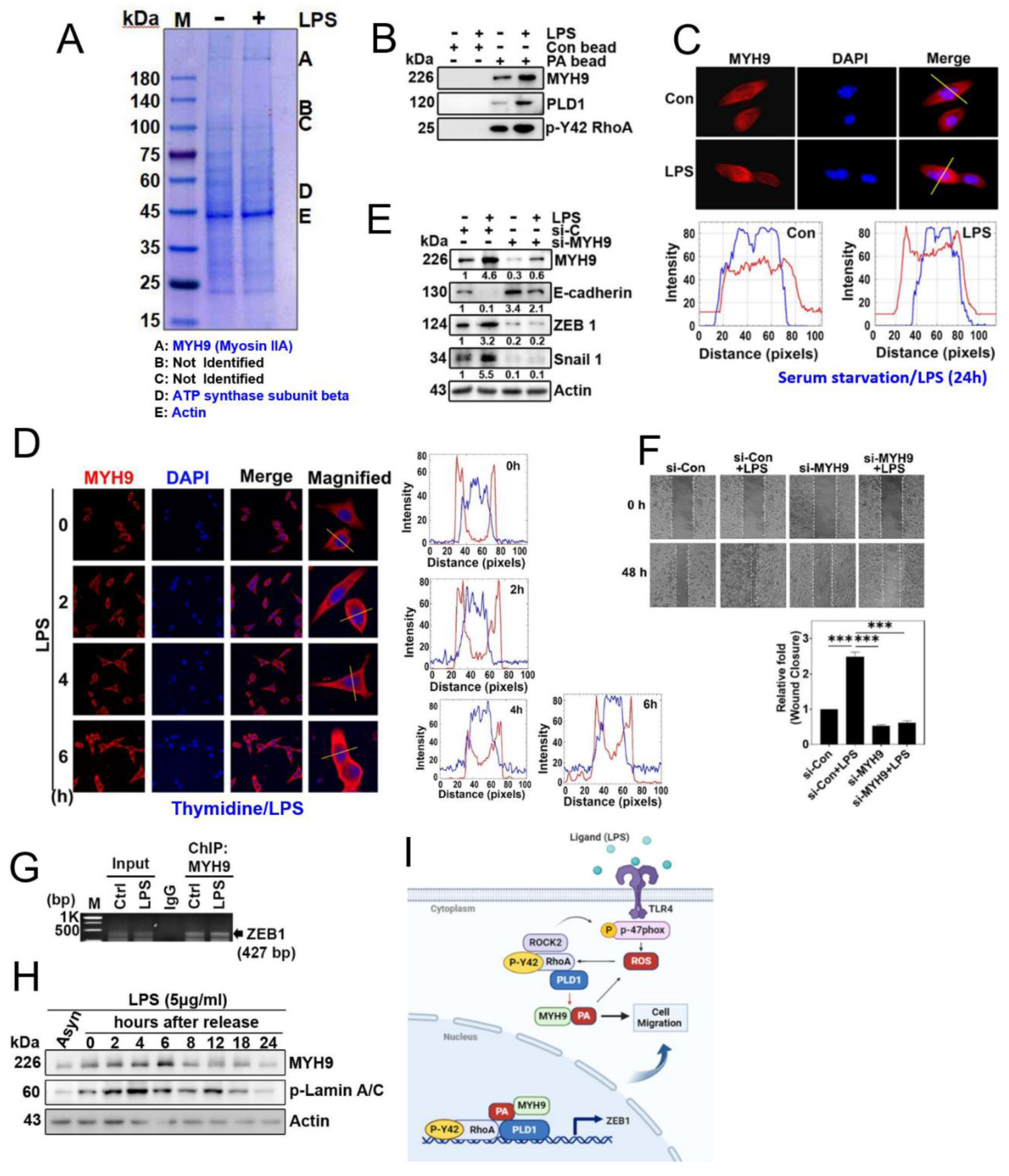
As a result, our objective was to investigate and identify the proteins that interact with phosphatidic acid (PA) in the downstream signaling pathway activated by LPS. We utilized PA-conjugated bead to capture proteins from lysates of the A549 cells stimulated with LPS. This approach led to the identification of MYH9, also referred to as Myosin IIA, non-muscle myosin IIA (NMIIA), and ATP synthase β as the protein bound to the PA-conjugated beads (Figure 5A). Furthermore, PA-conjugated beads were found to associate with MYH9, PLD1 and p-Tyr42 RhoA, indicating the formation of a PA/MYH9/PLD1/p-Tyr42 RhoA comp-lex (Figure 5B). Notably, MYH9 (Myosin IIA) was found to localize in both the cytosol and the nucleus, with a preference for the cytosol (Figure 5C). Myosin IIA has been previously linked to cell migration, metastasis, and EMT process [19]. Subsequently, we investigated the effect of MYH9 on cell migration, and found that knockdown of MYH9 with siRNA suppressed cell migration (Figure 5D). In line of these results, knockdown of MYH9 with siRNA lead to an increase in E-cadherin expression and a decrease in ZEB1 and Snail1 levels (Figure 5E). Significantly, we observed MYH9 binding to the ZEB1 promoter via ChIP-PCR, regardless of LPS stimulation (Figure 5F). Remarkably, we also noted that MYH9 expression exhibited regulation during cell cycle, coinciding with the phosphorylation of LaminA/C at Ser22 residue (Figure 5G).
4. Discussion
This study was initiated with the question of whether p-Tyr42 RhoA plays a regulatory role in PLD1 enzyme activity, given the well-established role of RhoA-GTP in activating PLD1. Recently, researchers elucidated the crystal structure of PLD1 in association with PIP2, PIP3 and RhoA-GTP. In this study, they discovered that C-terminal domain of PLD1 binds to the switch I domain of RhoA-GTP [13]. Notably, Tyr42 residue of RhoA is situated in the marginal region of switch I (see Supplementary Figure 2). Both RhoA and p-Tyr42 RhoA were found to interact with PLD1 (Figure 2C). However, it is worth mentioning that RhoA Y42F, which serves as a dephospho-mimic form, did not enhance PLD1 enzyme activity (Figure 2D). These findings support our hypothesis that p-Tyr42 residue of RhoA plays a critical role in the activation of PLD1.
Numerous proteins have been previously documented to bind to PA [20-23]. PA serves as a pivotal signaling molecule, contributing to a diverse range of regulatory functions within cellar processes. These functions encompass cell metabolism and growth, cell death, cytoskeletal remodeling, exocytosis, receptor endocytosis, membrane trafficking, and organelle dynamics [21]. Remarkably, alterations of PA levels have been linked to changes in actin filament dynamics. An increase of PA level is correlated with enhanced actin filament formation, while a decrease is associated with the disassembly of actin filaments. Notably, F-actin promotes PLD activity, whereas monomeric G-actin inhibits PLD activity [24]. To the best our knowledge, this study represents the first observation of the binding between PA and MYH9 (Myosin IIA). As a result, we postulate that PA plays a critical role in stimulating the interaction between F-actin and myosin, as PA, generated by PLD on F-actin, binds to and activates MYH9 (Myosin IIa), particularly during cell migration.
The PX domain of p47phox features two distinct basic pockets on its membrane-binding surface, each designed for specific phospholipids, including PI(3,4)P2/PI(3)P and PA/PS (phosphatidyl serine). It is noteworthy that PA has been shown to enhance NADPH oxidase activity [25]. Similar findings were reported by another researcher, indicating that both PLD1 and PA stimulate ROS production, with PA binding and promoting NADPH oxidase activity [26]. Consequently, we can deduce that p-Tyr42 RhoA/PLD1/PA collectively play a critical role in superoxide generation. Furthermore, our previous research has unveiled reciprocal relationship between superoxide and Tyr42 phosphorylation in RhoA. Additionally, p-Tyr42 RhoA activates ROCK2, which, in turn, phosphorylates and activates p47phox [16]. In the light of this findings, we propose that superoxide and p-Tyr42 RhoA mutually regulate each other.
Our investigation unveiled that both p-Tyr42 RhoA and PLD1 bind to the promoter region of ZEB1 (Figure 4K). Moreover, MYH9 (Myosin IIA) was found to regulate the expression of EMT marker proteins (Figure 5E) and was observed to exist in nucleus (Figure 5C). MYH9 (Myosin IIA, NMIIA) is composed of 1960 amino acids in heavy chain (230 kDa) and two regulatory light chain (20kDa) that control the myosin activity, along with two essential light chain (17 kDa) that stabilize the heavy chain structure [27]. Phosphorylation of the C-terminal region of MYH9 by PKC, Casein kinase II (CKII), and transient receptor potential melastatin 7 (TRPM7), plays a pivotal role in the assembly-disassembly process [27]. Given that we identified the binding of MYH9 heavy chain, excluding the light chains, to PA-conjugated beads, we postulate that globular N-terminal region of MYH9 may contain the binding site for PA.
If PLD1 produces PA within the nucleus and binds to MYH9 (Myosin IIA), it raises the question of MYH9 (Myosin IIA)’s function within the nucleus. Additionally, it is well-established that myosin light chain kinase (MLCK) and ROCK phosphorylate regulatory subunit in MYH9 (Myosin II, NMIIA) to activate Mysosin IIA [27]. Building upon these findings, we speculate that PA-binding MYH9 (Myosin IIA), concurrently activated by p-Tyr42 RhoA/ROCK, may play a direct or indirect role may be in transcriptional regulation in EMT marker genes. Remarkably, the phosphorylation of Ser1916 in α-helical rod and Ser1943 in non-helical tail in MYH9 has been observed to increase during the TGF-β-induced EMT process [28]. Notably, PA can stimulate a variety of kinases, including Raf-1, PKCs, PKN, mTOR, mTORC2, Akt, PAK1, p70S6K1, Fer, GRK, LATS1 and KSR1 [29]. Based on these findings, we suggest that C-terminal phosphorylation of MYH9 may be implicated in transcriptional regulation of EMT marker proteins. Additionally, we propose that MYH9 may be involved in modulating the assembly and disassembly of the nuclear membrane throughout cell cycle via its interaction with F-actin. It is worth noting that previous studies have reported that exogenous PA and PLD can induce stress fibers and increased F-actin levels [30,31]. Furthermore, our observations show that actin binds to PA-conjugated beads irrespective of LPS stimulation (Figure 5A). Expanding upon these earlier findings, we postulate that PA plays a pivotal role in regulating cell migration and the transport of organelles or nuclear membranes, potentially through the collaborative actions of MYH9 and F-actin. This conjecture is supported by our observation of MYH9 alteration during cell proliferation processes (refer to Figure 5H). Nevertheless, several questions remain unanswered. The most significant among them is whether p-Tyr42 RhoA and PLD1 also regulate cytoskeletal dynamics within the nucleus. This aspect remains unexplored in this current paper and is a subject for future research endeavors.
5. Conclusion
The inflammatory agent LPS induces the generation of superoxide, leading to phosphorylation at the Tyrosine42 of RhoA. Subsequently, p-Tyr42 RhoA activates PDL1, resulting in the production of PA. PA then binds to MYH9 (myosin IIA), influencing ZEB1 expression and facilitating cell migration. Furthermore, the association of p-Tyr42 RhoA and PLD1 with the ZEB1 promoter increases its expression, further enhancing cell migration. This represents a novel mechanism for promoting cancer cell progress through inflammation.
Funding
This study was supported by National Research Foundation of Korea (RS-2023-00208724 and RS-2023-00217013 to JBP).
Conflicts of Interest
We declare there is no conflict for interest.
Abbreviations
DAPI: 4’,6-diamidino-2-phenylindole; DCFDA, fluorescent probe 2′,7′-dichlorofluorescin diacetate; EMT, Epithelial-mesenchymal transition; MYH9, Myosin 9 (Myosin IIA); LPS, Lipopolysaccharide; NMIIA, non-muscle myosin IIA; PA, phosphatidic acid; PLD1, phospholipase D1; ROCK2, Rho-dependent kinase 2; ROS, reactive oxygen species; Y42E, Tyr42Glu mutant; Y42F, Tyr42Phe mutant; ZEB1, zinc finger E-box binding homeobox 1
References
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu Rev Pathol 2018, 13, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Hornef, M.W.; Normark, B.H.; Vandewalle, A.; Normark, S. Intracellular recognition of lipopolysaccharide by toll-like receptor 4 in intestinal epithelial cells. J Exp Med 2003, 198, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.Y.; Zhang, Z.H.; Xu, S.; Hong, Q.; Tian, Q.X.; Ye, Q.L.; Wang, H.; Yu, D.X.; Xu, D.X.; Xie, D.D. Calcitriol inhibits lipopolysaccharide-induced proliferation, migration and invasion of prostate cancer cells through suppressing STAT3 signal activation. Int Immunopharmacol 2020, 82, 106346. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.Y.; Han, Z.P.; Sun, K.; Zhang, S.S.; Hou, J.; Liu, Y.; Li, R.; Gao, L.; Zhao, X.; Zhao, Q.D.; et al. Toll-like receptor 4 signaling promotes epithelial-mesenchymal transition in human hepatocellular carcinoma induced by lipopolysaccharide. BMC Med 2012, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Islam, R.; Cho, J.Y.; Jeong, H.; Cap, K.C.; Park, Y.; Hossain, A.J.; Park, J.B. Regulation of RhoA GTPase and various transcription factors in the RhoA pathway. J Cell Physiol 2018, 233, 6381–6392. [Google Scholar] [CrossRef] [PubMed]
- Narumiya, S.; Tanji, M.; Ishizaki, T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev 2009, 28, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Qin, J.; Liu, M.; Ruan, Q.; Li, Y.; Zhang, Z. Role of Rho kinase in lysophosphatidic acid-induced altering of blood-brain barrier permeability. Int J Mol Med 2014, 33, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Bruntz, R.C.; Lindsley, C.W.; Brown, H.A. Phospholipase D signaling pathways and phosphatidic acid as therapeutic targets in cancer. Pharmacol Rev 2014, 66, 1033–1079. [Google Scholar] [CrossRef] [PubMed]
- Tanguy, E.; Wang, Q.; Vitale, N. Role of Phospholipase D-Derived Phosphatidic Acid in Regulated Exocytosis and Neurological Disease. Handb Exp Pharmacol 2020, 259, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Steed, P.M.; Clark, K.L.; Boyar, W.C.; Lasala, D.J. Characterization of human PLD2 and the analysis of PLD isoform splice variants. FASEB J 1998, 12, 1309–1317. [Google Scholar] [CrossRef]
- Hammond, S.M.; Jenco, J.M.; Nakashima, S.; Cadwallader, K.; Gu, Q.; Cook, S.; Nozawa, Y.; Prestwich, G.D.; Frohman, M.A.; Morris, A.J. Characterization of two alternately spliced forms of phospholipase D1. Activation of the purified enzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, and Rho family monomeric GTP-binding proteins and protein kinase C-alpha. J Biol Chem 1997, 272, 3860–3868. [Google Scholar] [CrossRef] [PubMed]
- Mozzicato, S.; Joshi, B.V.; Jacobson, K.A.; Liang, B.T. Role of direct RhoA-phospholipase D1 interaction in mediating adenosine-induced protection from cardiac ischemia. FASEB J 2004, 18, 406–408. [Google Scholar] [CrossRef] [PubMed]
- Bowling, F.Z.; Salazar, C.M.; Bell, J.A.; Huq, T.S.; Frohman, M.A.; Airola, M.V. Crystal structure of human PLD1 provides insight into activation by PI(4,5)P(2) and RhoA. Nat Chem Biol 2020, 16, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M. Nuclear phospholipase D1 in vascular smooth muscle: specific activation by G protein-coupled receptors. Circ Res 2006, 99, 116–118. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.J.; Park, S.Y.; Kim, W.K.; Cho, J.H.; Chang, B.J.; Kim, D.J.; Ahn, J.S.; Park, K.; Han, J.S. A Single Nucleotide Polymorphism in the Phospholipase D1 Gene is Associated with Risk of Non-Small Cell Lung Cancer. Int J Biomed Sci 2012, 8, 121–128. [Google Scholar] [PubMed]
- Cap, K.C.; Kim, J.G.; Hamza, A.; Park, J.B. P-Tyr42 RhoA GTPase amplifies superoxide formation through p47phox, phosphorylated by ROCK. Biochem Biophys Res Commun 2020, 523, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Cap, K.C.; Jung, Y.-J.; Choi, B.Y.; Hyeon, S.J.; Kim, J.-G.; Min, J.-K.; Islam, R.; Hossain, A.J.; Chung, W.-S.; Suh, S.W. Distinct dual roles of p-Tyr42 RhoA GTPase in tau phosphorylation and ATP citrate lyase activation upon different Aβ concentrations. Redox biology 2020, 32, 101446. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Mahmud, S.; Min, J.K.; Lee, Y.B.; Kim, H.; Kang, D.C.; Park, H.S.; Seong, J.; Park, J.B. RhoA GTPase phosphorylated at tyrosine 42 by src kinase binds to beta-catenin and contributes transcriptional regulation of vimentin upon Wnt3A. Redox Biol 2021, 40, 101842. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Qi, X.; Liu, J.; Zhou, R.; Lin, C.; Shangguan, J.; Zhang, Z.; Zhao, L.; Li, G. MYH9 Promotes Growth and Metastasis via Activation of MAPK/AKT Signaling in Colorectal Cancer. J Cancer 2019, 10, 874–884. [Google Scholar] [CrossRef]
- Stace, C.L.; Ktistakis, N.T. Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim Biophys Acta 2006, 1761, 913–926. [Google Scholar] [CrossRef]
- Zhou, H.; Huo, Y.; Yang, N.; Wei, T. Phosphatidic acid: from biophysical properties to diverse functions. FEBS J 2023. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, F.; He, J.; Wu, P.; Tay, L.W.R.; Cai, M.; Nian, W.; Weng, Y.; Qin, L.; Chang, J.T.; et al. Binding of PLD2-Generated Phosphatidic Acid to KIF5B Promotes MT1-MMP Surface Trafficking and Lung Metastasis of Mouse Breast Cancer Cells. Dev Cell 2017, 43, 186–197 e187. [Google Scholar] [CrossRef] [PubMed]
- Kattan, R.E.; Han, H.; Seo, G.; Yang, B.; Lin, Y.; Dotson, M.; Pham, S.; Menely, Y.; Wang, W. Interactome Analysis of Human Phospholipase D and Phosphatidic Acid-Associated Protein Network. Mol Cell Proteomics 2022, 21, 100195. [Google Scholar] [CrossRef] [PubMed]
- Pleskot, R.; Li, J.; Zarsky, V.; Potocky, M.; Staiger, C.J. Regulation of cytoskeletal dynamics by phospholipase D and phosphatidic acid. Trends Plant Sci 2013, 18, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Karathanassis, D.; Stahelin, R.V.; Bravo, J.; Perisic, O.; Pacold, C.M.; Cho, W.; Williams, R.L. Binding of the PX domain of p47(phox) to phosphatidylinositol 3,4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J 2002, 21, 5057–5068. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, H.; Zhang, Q.; Li, M.; Yan, M.; Wang, R.; Wang, L.; Welti, R.; Zhang, W.; Wang, X. Phospholipase dalpha1 and phosphatidic acid regulate NADPH oxidase activity and production of reactive oxygen species in ABA-mediated stomatal closure in Arabidopsis. Plant Cell 2009, 21, 2357–2377. [Google Scholar] [CrossRef] [PubMed]
- Pecci, A.; Ma, X.; Savoia, A.; Adelstein, R.S. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene 2018, 664, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Beach, J.R.; Hussey, G.S.; Miller, T.E.; Chaudhury, A.; Patel, P.; Monslow, J.; Zheng, Q.; Keri, R.A.; Reizes, O.; Bresnick, A.R.; et al. Myosin II isoform switching mediates invasiveness after TGF-beta-induced epithelial-mesenchymal transition. Proc Natl Acad Sci U S A 2011, 108, 17991–17996. [Google Scholar] [CrossRef] [PubMed]
- Sakane, F.; Hoshino, F.; Murakami, C. New Era of Diacylglycerol Kinase, Phosphatidic Acid and Phosphatidic Acid-Binding Protein. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Ha, K.S.; Exton, J.H. Activation of actin polymerization by phosphatidic acid derived from phosphatidylcholine in IIC9 fibroblasts. J Cell Biol 1993, 123, 1789–1796. [Google Scholar] [CrossRef]
- Lee, S.; Park, J.; Lee, Y. Phosphatidic acid induces actin polymerization by activating protein kinases in soybean cells. Mol Cells 2003, 15, 313–319. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
LPS increases superoxide generation via ROCK2 and p-p47phox. (A) Representative image of the measurement of cellular ROS/superoxide production using the fluorescent probe 2′,7′-dichlorofluorescin diacetate (DCFDA) in A549 cell by LPS treatment at different concentration (0, 1, 2, 5, 10 μg/ml) for 24 h. Produced superoxide was visualized with 10 μM (DCFDA) for 20 min, and detected using a fluorescence microscope (Axiovert 200, Carl Zeiss). (B) A549 cells underwent an 8-hour period of serum starvation, followed by pretreatment with Tat-C3 (1 μg/ml) and Y27632 (10 μM) for 1 hour. Subsequently, the cells were stimulated with LPS (5 μg/ml) for 24 hours, and the measurement of reactive oxygen species (ROS) was conducted using DCFDA. (C) Similarly, superoxide was measured after the knockdown of ROCK1 and ROCK2 by si-RNA for 48 h, followed by the stimulation of LPS (5 μg/ml) for 24 h in A549 cells. (D) p-p47phox protein levels were measured by western blotting in A549 cells stimulated with LPS for 24 h after administration of si-RNA mediated knockdown as ROCK1 and ROCK2 together. (E) A549 cells were pretreated with apocynin (1 μM), NAC (10 mM) and BHA (10 μM) for 1 h, then stimulated with LPS (5 μg/ml) for 24 h and the indicated proteins were detected by western blotting. Each western blot representative images were shown and band intensity was measured by Adobe Photoshop (version 8) software. The data are mean ± SD of three independent experiments (*, p<0.05; **, p<0.01, and ***, p<0.001) in case without particular remark. Western blot data are representative of at least three independent experiments.
Figure 1.
LPS increases superoxide generation via ROCK2 and p-p47phox. (A) Representative image of the measurement of cellular ROS/superoxide production using the fluorescent probe 2′,7′-dichlorofluorescin diacetate (DCFDA) in A549 cell by LPS treatment at different concentration (0, 1, 2, 5, 10 μg/ml) for 24 h. Produced superoxide was visualized with 10 μM (DCFDA) for 20 min, and detected using a fluorescence microscope (Axiovert 200, Carl Zeiss). (B) A549 cells underwent an 8-hour period of serum starvation, followed by pretreatment with Tat-C3 (1 μg/ml) and Y27632 (10 μM) for 1 hour. Subsequently, the cells were stimulated with LPS (5 μg/ml) for 24 hours, and the measurement of reactive oxygen species (ROS) was conducted using DCFDA. (C) Similarly, superoxide was measured after the knockdown of ROCK1 and ROCK2 by si-RNA for 48 h, followed by the stimulation of LPS (5 μg/ml) for 24 h in A549 cells. (D) p-p47phox protein levels were measured by western blotting in A549 cells stimulated with LPS for 24 h after administration of si-RNA mediated knockdown as ROCK1 and ROCK2 together. (E) A549 cells were pretreated with apocynin (1 μM), NAC (10 mM) and BHA (10 μM) for 1 h, then stimulated with LPS (5 μg/ml) for 24 h and the indicated proteins were detected by western blotting. Each western blot representative images were shown and band intensity was measured by Adobe Photoshop (version 8) software. The data are mean ± SD of three independent experiments (*, p<0.05; **, p<0.01, and ***, p<0.001) in case without particular remark. Western blot data are representative of at least three independent experiments.
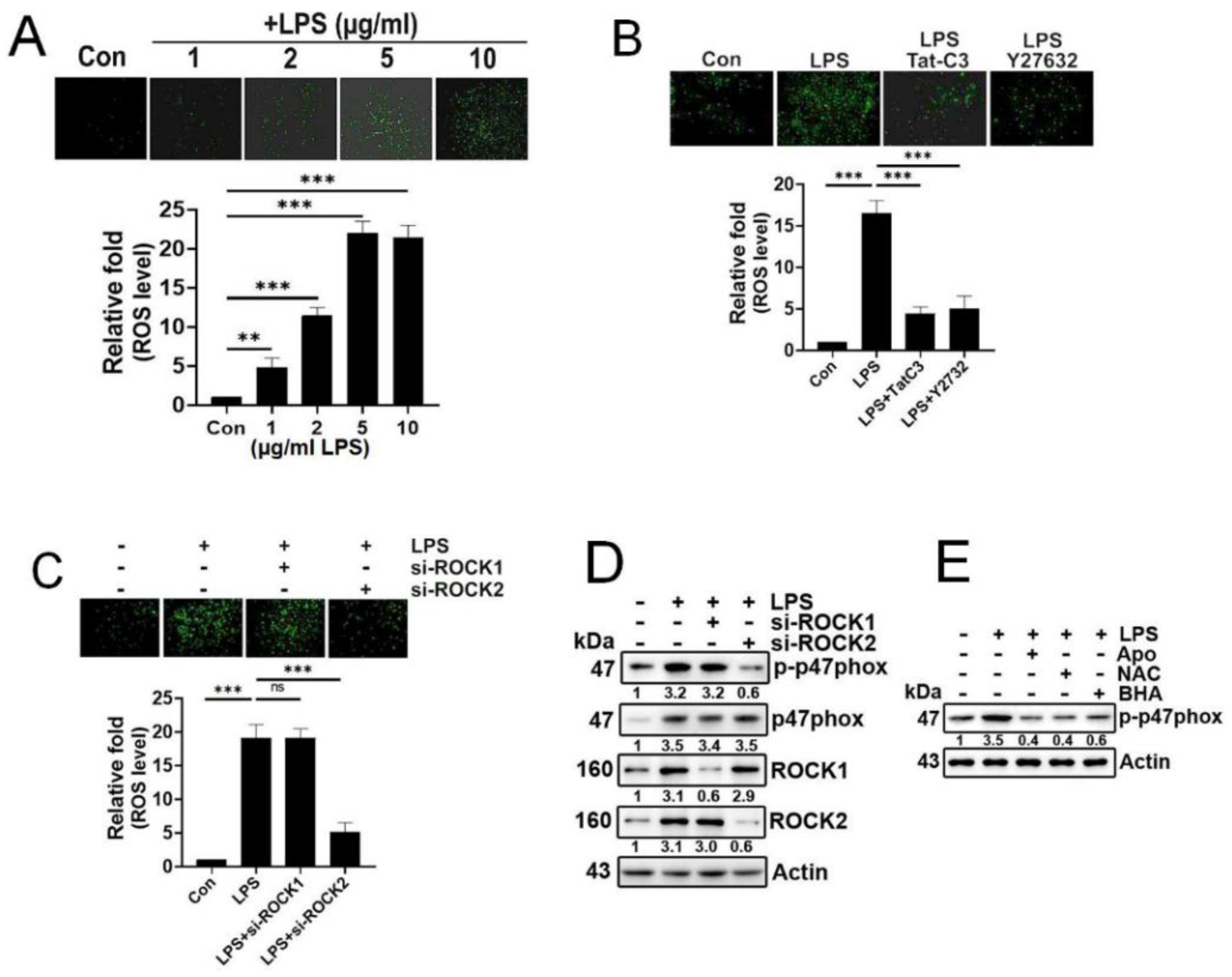
Figure 2.
LPS upregulates p-Tyr42 RhoA, phospholipase D1 (PLD1) levels via generating a protein complex. (A, B) The levels of RhoA, p-Y42 RhoA, and PLD1 were quantified using western blotting in A549 treatments with varying concentrations of LPS for 24 h (A) and concentration of 5 μg/ml for different time periods (B). (C) Immunoprecipitation of PLD1 was performed in LPS-stimulated A549 cells to evaluate the interaction with p-Y42 RhoA, and PLD1 using specific antibodies. (D, E) A549 cells transfected with si-RhoA (50 nM) were further transfected with RhoA WT, Y42E, and Y42F plasmid DNA (2 μg) and subsequently stimulated with 5 μg/ml LPS for 24 h. The activity of PLD1 was determined using a colorimetric Phospholipase D activity assay kit at an optical density of 570 nm (D), while superoxide production was measured (E). (F) Prior to LPS stimulation (5 μg/ml) for 24 h, A549 cells were pretreated with apocynin (1 μM), NAC (10 mM) and BHA (10 μM) for 1 h. Western blotting was employed to examine changes in the levels of PDL1 and p-Y42 RhoA. (G) A549 cells were transfected with RhoA wild type as well as Y42E and Y42F both phospho mimic and de-phospho mimic form (2 μg/ml each) for 24 hours. Then cells were cultured in a serum depleted media for 8 h and stimulated by LPS. Then, PLD1 interaction was checking by immunoprecipitation with HA probe and RhoA, PLD1 and HA protein levels were detected by western blotting. The data are mean ± SD of three independent experiments (*, p<0.05; **, p<0.01, and ***, p<0.001) in case without particular remark. Western blot data are representative of at least three independent experiments.
Figure 2.
LPS upregulates p-Tyr42 RhoA, phospholipase D1 (PLD1) levels via generating a protein complex. (A, B) The levels of RhoA, p-Y42 RhoA, and PLD1 were quantified using western blotting in A549 treatments with varying concentrations of LPS for 24 h (A) and concentration of 5 μg/ml for different time periods (B). (C) Immunoprecipitation of PLD1 was performed in LPS-stimulated A549 cells to evaluate the interaction with p-Y42 RhoA, and PLD1 using specific antibodies. (D, E) A549 cells transfected with si-RhoA (50 nM) were further transfected with RhoA WT, Y42E, and Y42F plasmid DNA (2 μg) and subsequently stimulated with 5 μg/ml LPS for 24 h. The activity of PLD1 was determined using a colorimetric Phospholipase D activity assay kit at an optical density of 570 nm (D), while superoxide production was measured (E). (F) Prior to LPS stimulation (5 μg/ml) for 24 h, A549 cells were pretreated with apocynin (1 μM), NAC (10 mM) and BHA (10 μM) for 1 h. Western blotting was employed to examine changes in the levels of PDL1 and p-Y42 RhoA. (G) A549 cells were transfected with RhoA wild type as well as Y42E and Y42F both phospho mimic and de-phospho mimic form (2 μg/ml each) for 24 hours. Then cells were cultured in a serum depleted media for 8 h and stimulated by LPS. Then, PLD1 interaction was checking by immunoprecipitation with HA probe and RhoA, PLD1 and HA protein levels were detected by western blotting. The data are mean ± SD of three independent experiments (*, p<0.05; **, p<0.01, and ***, p<0.001) in case without particular remark. Western blot data are representative of at least three independent experiments.
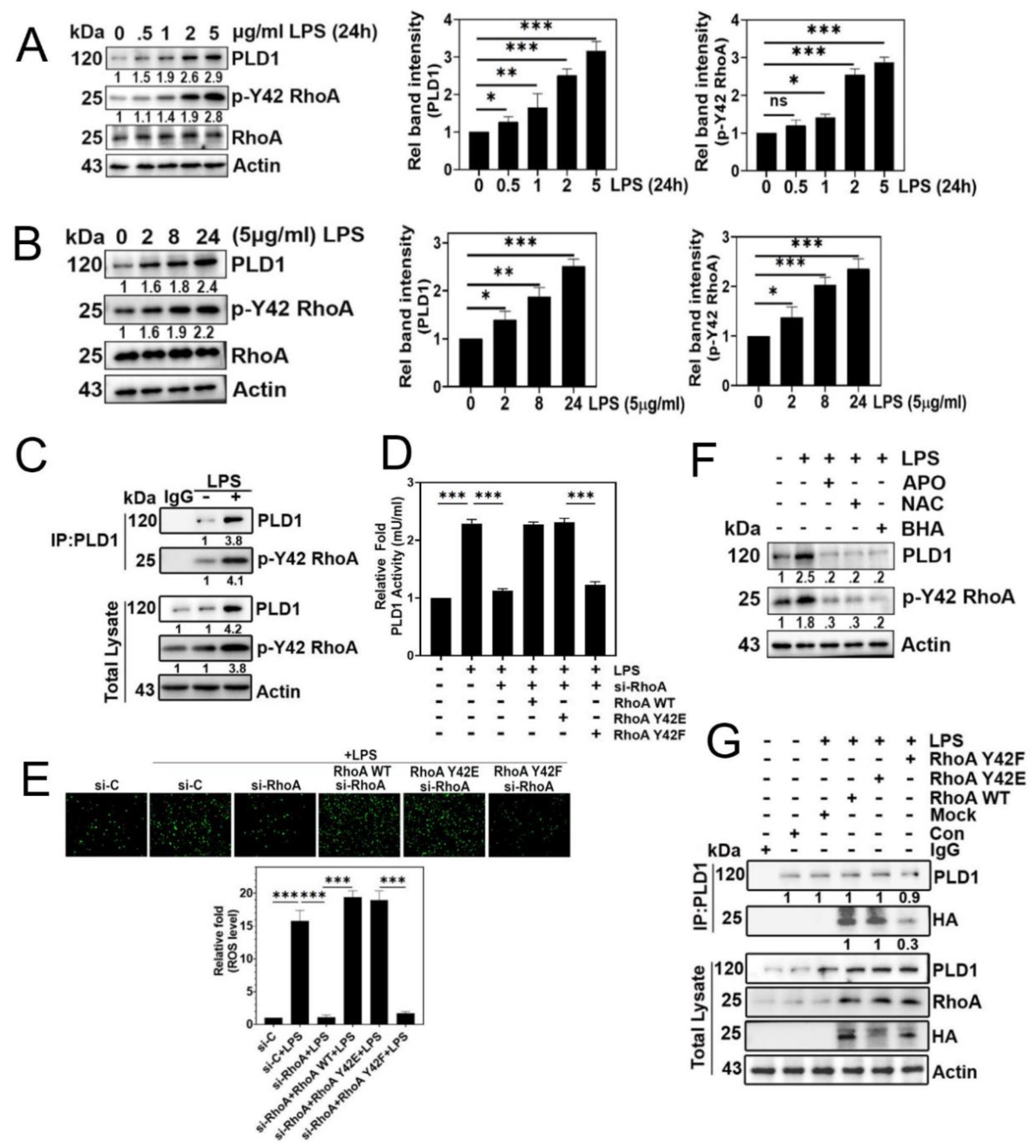
Figure 3.
EMT and cell migration are facilitated by p-Tyr42 RhoA. (A) A549 cells were activated different concentrations of LPS for 24 h. Cell migration was assessed at 0 and 48 h by observing under a microscope after scratching with a plastic pipette tip. (B) After treating A549 cells with LPS at varying concentrations for 24 h, changes in expression of E-cadherin, N-cadherin, ZEB1, and Snail were assessed using immunoblotting. (C) A549 cells were transfected with si-Con (20 nM) and si-RhoA (50 nM) were transfected into for 48 h, followed by a 12 h starvation period before stimulation with LPS (5 μg/ml) for 24 h. Immunoblotting was used to detect the expression of E-cadherin, N-cadherin, ZEB1, RhoA, and p-Y42 RhoA which were then plotted in a bar diagram. (D-E) After transfection with si-con and si-RhoA for 48 h, A549 cells were further transfected with Mock, RhoA WT, Y42E, and Y42F plasmids DNA (2 μg) for 24 h. Subsequently, the cells were stimulated with 5 μg/ml LPS for 24 h. (D) Western blot analysis was used to determine the levels of E-cadherin, N-cadherin, ZEB1, PLD1, Snail 1, HA, RhoA, and Actin as a loading control, (E) while cell migration was evaluated according to the methods described in fig 3(A). The data are mean ± SD of three independent experiments (*, p<0.05; **, p<0.01, and ***, p<0.001) in case without particular remark. Western blot data are representative of at least three independent experiments. .
Figure 3.
EMT and cell migration are facilitated by p-Tyr42 RhoA. (A) A549 cells were activated different concentrations of LPS for 24 h. Cell migration was assessed at 0 and 48 h by observing under a microscope after scratching with a plastic pipette tip. (B) After treating A549 cells with LPS at varying concentrations for 24 h, changes in expression of E-cadherin, N-cadherin, ZEB1, and Snail were assessed using immunoblotting. (C) A549 cells were transfected with si-Con (20 nM) and si-RhoA (50 nM) were transfected into for 48 h, followed by a 12 h starvation period before stimulation with LPS (5 μg/ml) for 24 h. Immunoblotting was used to detect the expression of E-cadherin, N-cadherin, ZEB1, RhoA, and p-Y42 RhoA which were then plotted in a bar diagram. (D-E) After transfection with si-con and si-RhoA for 48 h, A549 cells were further transfected with Mock, RhoA WT, Y42E, and Y42F plasmids DNA (2 μg) for 24 h. Subsequently, the cells were stimulated with 5 μg/ml LPS for 24 h. (D) Western blot analysis was used to determine the levels of E-cadherin, N-cadherin, ZEB1, PLD1, Snail 1, HA, RhoA, and Actin as a loading control, (E) while cell migration was evaluated according to the methods described in fig 3(A). The data are mean ± SD of three independent experiments (*, p<0.05; **, p<0.01, and ***, p<0.001) in case without particular remark. Western blot data are representative of at least three independent experiments. .
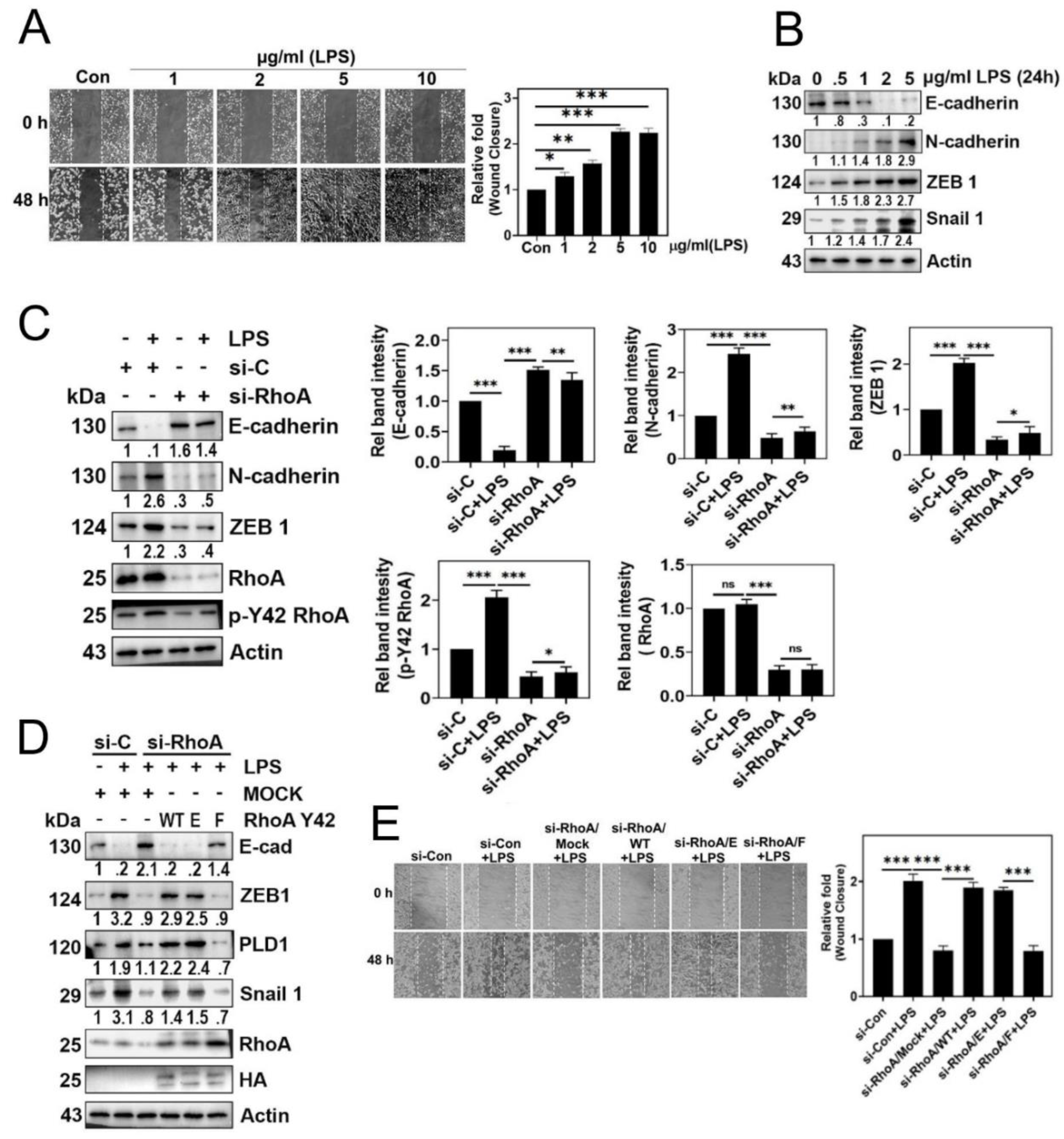
Figure 4.
Effects of PLD1/PA on cell migration. (A) A549 cells were transfected with si-Control (20 nM) and si-RhoA (50 nM) for 48 h. Following 12 h of starvation, cells were stimulated with 5 μg/ml LPS for 24 h to assess ROS generation using DCFDA, as depicted in Fig 1 (A). (B, C). A549 cells were activated with various concentrations of phosphatidic acid (PA) for 24 h. (B) ROS levels were then quantified as mentioned previously in Fig 1 (A) and (C) western blot analysis performed to determine the expression levels of RhoA and p-Y42 RhoA. (D, E). A549 cells were transfected with si-Con (20 nM) and si-RhoA (50 nM) for 48 h. After 12 h of starvation, cells were stimulated with 5 (μg/ml) LPS for 24 h. (D) Cell migration rate was assessed according to Fig 3(A). (E) Immunoblotting was conducted to determine the expression levels of E-cadherin, N-cadherin, ZEB1, PLD1, Snail 1, and actin as a loading control. (F, G) A549 cells stimulated with various concentrations of phosphatidic acid (PA) for 24 h. (F) Immunoblotting was utilized to measure the expression levels of E-cadherin, N-cadherin, ZEB1, Snail 1, and actin as a loading control. (G) Cell migration was measured as depicted in Fig 3(A). (H) A549 cells were treated 5 μg/ml LPS for 24 h, following which cytosolic and nuclear fractions were isolated. Western blotting was performed to identify PLD1 and p-Y42 RhoA, with lamin B and tubulin expression levels serving as loading controls for the cytosolic and nuclear fractions respectively. (I) HEK 293 T cells were seeded for 24h and after 8 h serum starvation, cells were stimulated with 5 μg/ml LPS for 24 h. Then cells were treated with 5 μg/ml p-Y 42 RhoA (Rabbit antibody) and 5 μg/ml PLD1 (Mouse antibody) and kept overnight at 4°C. Confocal microscopy was used to determine the co-localization of PLD1 (green) and p-Y 42 RhoA (red) with the nucleus labeled using 5 μg/ml DAPI (blue). (J) Confocal microscopy was used to determine the co-localization of p-Y42 RhoA (red) and PLD1 (green) in A549 cells as described in fig 4(I). (K, L) ChIP-PCR experiments were conducted on A549 cells exposed to 5 μg/ml of LPS for 24 h, using primers specific for Zeb1 (K) and snail1 (L), aiming to investigate the binding of p-Y42 RhoA and PLD1. The data are mean ± SD of three independent experiments (*, p<0.05; **, p<0.01; ***, p<0.001; and ****, p<0.0001) in case without particular remark. Western blot data are representative of at least three independent experiments.
Figure 4.
Effects of PLD1/PA on cell migration. (A) A549 cells were transfected with si-Control (20 nM) and si-RhoA (50 nM) for 48 h. Following 12 h of starvation, cells were stimulated with 5 μg/ml LPS for 24 h to assess ROS generation using DCFDA, as depicted in Fig 1 (A). (B, C). A549 cells were activated with various concentrations of phosphatidic acid (PA) for 24 h. (B) ROS levels were then quantified as mentioned previously in Fig 1 (A) and (C) western blot analysis performed to determine the expression levels of RhoA and p-Y42 RhoA. (D, E). A549 cells were transfected with si-Con (20 nM) and si-RhoA (50 nM) for 48 h. After 12 h of starvation, cells were stimulated with 5 (μg/ml) LPS for 24 h. (D) Cell migration rate was assessed according to Fig 3(A). (E) Immunoblotting was conducted to determine the expression levels of E-cadherin, N-cadherin, ZEB1, PLD1, Snail 1, and actin as a loading control. (F, G) A549 cells stimulated with various concentrations of phosphatidic acid (PA) for 24 h. (F) Immunoblotting was utilized to measure the expression levels of E-cadherin, N-cadherin, ZEB1, Snail 1, and actin as a loading control. (G) Cell migration was measured as depicted in Fig 3(A). (H) A549 cells were treated 5 μg/ml LPS for 24 h, following which cytosolic and nuclear fractions were isolated. Western blotting was performed to identify PLD1 and p-Y42 RhoA, with lamin B and tubulin expression levels serving as loading controls for the cytosolic and nuclear fractions respectively. (I) HEK 293 T cells were seeded for 24h and after 8 h serum starvation, cells were stimulated with 5 μg/ml LPS for 24 h. Then cells were treated with 5 μg/ml p-Y 42 RhoA (Rabbit antibody) and 5 μg/ml PLD1 (Mouse antibody) and kept overnight at 4°C. Confocal microscopy was used to determine the co-localization of PLD1 (green) and p-Y 42 RhoA (red) with the nucleus labeled using 5 μg/ml DAPI (blue). (J) Confocal microscopy was used to determine the co-localization of p-Y42 RhoA (red) and PLD1 (green) in A549 cells as described in fig 4(I). (K, L) ChIP-PCR experiments were conducted on A549 cells exposed to 5 μg/ml of LPS for 24 h, using primers specific for Zeb1 (K) and snail1 (L), aiming to investigate the binding of p-Y42 RhoA and PLD1. The data are mean ± SD of three independent experiments (*, p<0.05; **, p<0.01; ***, p<0.001; and ****, p<0.0001) in case without particular remark. Western blot data are representative of at least three independent experiments.
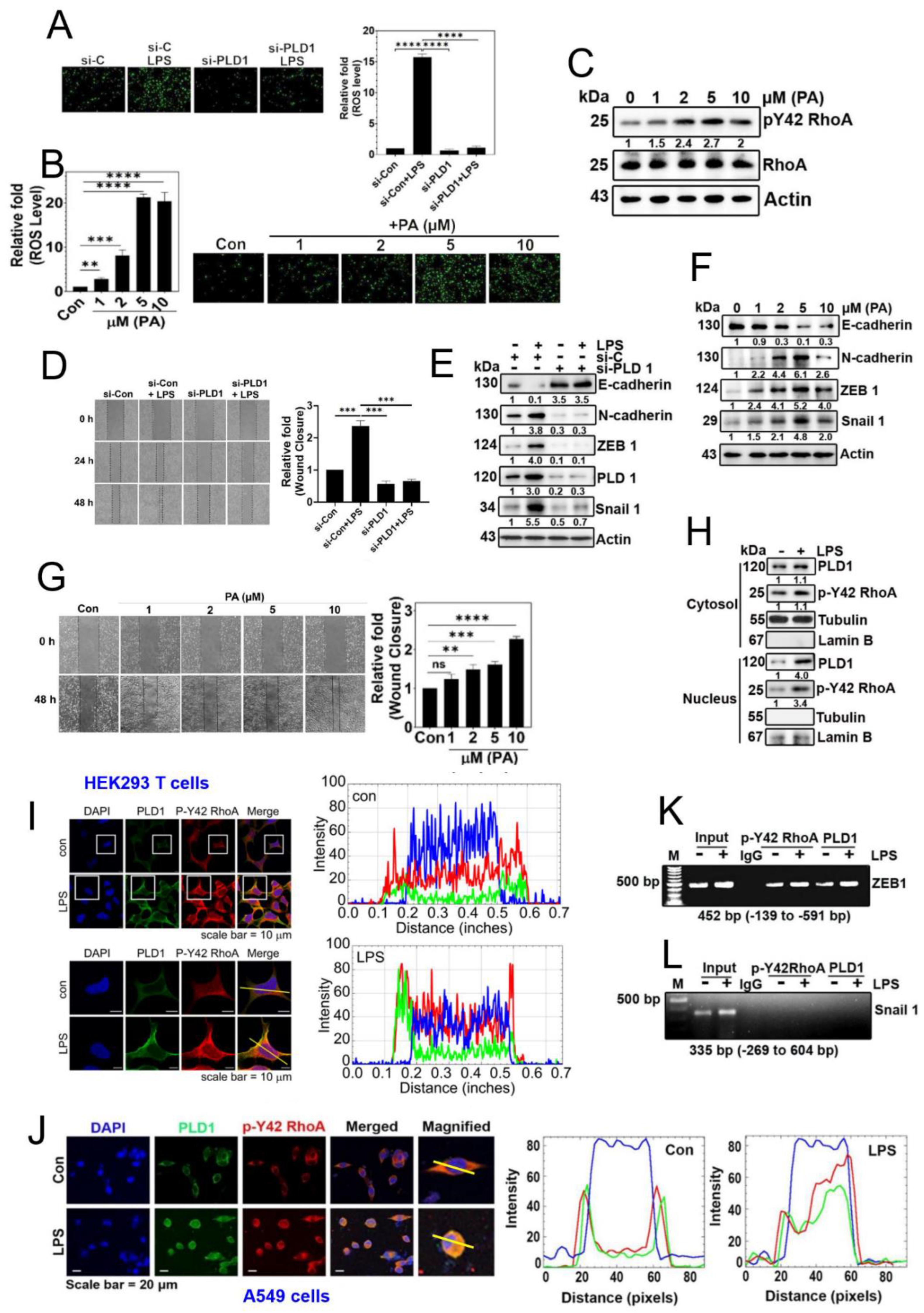
Figure 5.
MYH9 is a PA-binding protein. (A) A549 cells were incubated with LPS (5 μg/ml) for 24 h to induce stimulation. After that, the cell lysate was incubated with phosphatidic acid (PA) conjugated beads overnight, followed by separation of proteins using SDS-PAGE and determination of target protein species using MALDI-TOF analysis. (B) A549 cells were stimulated with LPS at a concentration of 5 μg/ml for a duration of 24 hours. Subsequently, the cell lysate underwent incubation overnight with both control beads and beads linked to phosphatidic acid (PA). Following this, protein separation was achieved through SDS-PAGE, and immunoblotting was employed to assess the protein expression levels of MYH9, PLD1, and p-Y42 RhoA. (C) A549 cells were cultured for 24 h and after serum starvation for 8 h cells were treated with LPS (5 µg/ml) for 24 h. Localization of MYH9 was assessed by immunofluorescence staining (red) and the nucleus was stained with DAPI (blue), and the images were captured with immunofluorescence microscopy. (D) After 16 h later of seeding of A549 cells were treated with thymidine (2 mM) for 16h for first block of cell cycle arrest. Subsequently, the cells were rinsed with PBS and exposed to fresh media. A second cycle arrest was initiated by treating the cells with thymidine (2 mM) for an additional 16 hours. Upon completing the second arrest, the cells were then treated with 5 μg/ml of LPS, and the localization of MYH9 (in red) was observed at various time points (0, 2, 4, 6 hours) using confocal microscopy. (E) A549 cells underwent transfection with si-control (20 nM) and si-MYH9 (50 nM) for duration of 48 hours. Following an 8-hour period of starvation, the cells were exposed to 5 μg/ml of LPS. Subsequently, alterations in the protein expression levels of MYH9, E-cadherin, ZEB1, Snail 1, and actin were evaluated through immunoblotting. (F) A549 cells underwent transfection with si-Con (20 nM) and si-MYH9 (50 nM) over a 48-hour period. Following a 8-h starvation of the cells, they were then exposed to 5 μg/ml of LPS for an additional 48 hours. The evaluation of the cell migration rate aligns with the details presented in Figure 3A. (G) Representative ChiP data of ZEB1 which was performed with the primer pair covering the promotor region of ZEB1. A549 cells were cultured without serum for 8 h and then stimulated with LPS (5 µg/ml) for 24 h. MYH9 antibody was used for the immunoprecipitation. Normal IgG was used as a negative control for the IP. (H) A549 cells were initially seeded and then synchronized using the double thymidine block method. In the first block, the cells were exposed to 2 mM thymidine for 16 hours. Subsequently, after rinsing with PBS, fresh media containing 5 μg/ml LPS was applied and maintained for 12 hours. Following this, a second round of synchronization was carried out using 2 mM thymidine for another 16 hours (secondary block). After completing the secondary block, 5 μg/ml LPS was administered, and the cells were released from arrest. At various time points, cells were harvested and subjected to immunoblotting to examine the levels of MYH9 and phosphorylated Lamin A/C. (I) A proposed diagram illustrating the function of the PLD1/p-Tyr42 RhoA/PA/MYH9 complex in the migration of A549 lung cancer cells. The data are mean ± SD of three independent experiments (*, p<0.05; **, p<0.01, and ***, p<0.001) in case without particular remark. Western blot data are representative of at least three independent experiments.
Figure 5.
MYH9 is a PA-binding protein. (A) A549 cells were incubated with LPS (5 μg/ml) for 24 h to induce stimulation. After that, the cell lysate was incubated with phosphatidic acid (PA) conjugated beads overnight, followed by separation of proteins using SDS-PAGE and determination of target protein species using MALDI-TOF analysis. (B) A549 cells were stimulated with LPS at a concentration of 5 μg/ml for a duration of 24 hours. Subsequently, the cell lysate underwent incubation overnight with both control beads and beads linked to phosphatidic acid (PA). Following this, protein separation was achieved through SDS-PAGE, and immunoblotting was employed to assess the protein expression levels of MYH9, PLD1, and p-Y42 RhoA. (C) A549 cells were cultured for 24 h and after serum starvation for 8 h cells were treated with LPS (5 µg/ml) for 24 h. Localization of MYH9 was assessed by immunofluorescence staining (red) and the nucleus was stained with DAPI (blue), and the images were captured with immunofluorescence microscopy. (D) After 16 h later of seeding of A549 cells were treated with thymidine (2 mM) for 16h for first block of cell cycle arrest. Subsequently, the cells were rinsed with PBS and exposed to fresh media. A second cycle arrest was initiated by treating the cells with thymidine (2 mM) for an additional 16 hours. Upon completing the second arrest, the cells were then treated with 5 μg/ml of LPS, and the localization of MYH9 (in red) was observed at various time points (0, 2, 4, 6 hours) using confocal microscopy. (E) A549 cells underwent transfection with si-control (20 nM) and si-MYH9 (50 nM) for duration of 48 hours. Following an 8-hour period of starvation, the cells were exposed to 5 μg/ml of LPS. Subsequently, alterations in the protein expression levels of MYH9, E-cadherin, ZEB1, Snail 1, and actin were evaluated through immunoblotting. (F) A549 cells underwent transfection with si-Con (20 nM) and si-MYH9 (50 nM) over a 48-hour period. Following a 8-h starvation of the cells, they were then exposed to 5 μg/ml of LPS for an additional 48 hours. The evaluation of the cell migration rate aligns with the details presented in Figure 3A. (G) Representative ChiP data of ZEB1 which was performed with the primer pair covering the promotor region of ZEB1. A549 cells were cultured without serum for 8 h and then stimulated with LPS (5 µg/ml) for 24 h. MYH9 antibody was used for the immunoprecipitation. Normal IgG was used as a negative control for the IP. (H) A549 cells were initially seeded and then synchronized using the double thymidine block method. In the first block, the cells were exposed to 2 mM thymidine for 16 hours. Subsequently, after rinsing with PBS, fresh media containing 5 μg/ml LPS was applied and maintained for 12 hours. Following this, a second round of synchronization was carried out using 2 mM thymidine for another 16 hours (secondary block). After completing the secondary block, 5 μg/ml LPS was administered, and the cells were released from arrest. At various time points, cells were harvested and subjected to immunoblotting to examine the levels of MYH9 and phosphorylated Lamin A/C. (I) A proposed diagram illustrating the function of the PLD1/p-Tyr42 RhoA/PA/MYH9 complex in the migration of A549 lung cancer cells. The data are mean ± SD of three independent experiments (*, p<0.05; **, p<0.01, and ***, p<0.001) in case without particular remark. Western blot data are representative of at least three independent experiments.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



