
Submitted:
27 October 2023
Posted:
30 October 2023
You are already at the latest version
Abstract
(1) Background: P-cadherin (CDH3) is a transmembrane protein that plays a crucial role in maintaining the structural integrity of epithelial tissue and homeostasis. Its role in carcinogenesis remains a subject of debate, as its behavior can vary depending on the molecular context and the specific tumor cell model under study. In this study, we explored the role of P-cadherin in head and neck squamous cell carcinoma (HNSCC) and the mechanism underlying its function. (2) Methods: We analyzed P-cadherin expression in HNSCC patients using The Cancer Genome Atlas (TCGA) database. For in vitro functional analysis, we conducted proliferation, migration, invasion, and western blot assays after either suppressing or overexpressing P-cadherin. For in vivo functional analysis, we utilized mouse xenograft models. (3) Results: P-cadherin was significantly overex-pressed in tumor samples compared to normal samples in the TCGA-HNSCC cohort. P-cadherin knockdown resulted in decreased proliferation, migration, and invasion compared to control cells, while P-cadherin overexpression increased cell proliferation and migration in HNSCC cells. We discovered that c-Met functioned as an upstream regulator of P-cadherin. Surprisingly, we found that P-cadherin knockdown increased the phosphorylation of c-Met and STAT3. Combining P-cadherin siRNA with the c-Met inhibitor SU11274 resulted in a more effective reduction in HNSCC cell growth, both in vitro and in vivo, compared to either treatment alone. (4) Conclusions: Our study uncovered a previously unknown aspect of P-cadherin-mediated c-Met regulation. The enhanced activation of c-Met/STAT3 following P-cadherin inhibition could be responsible for the survival of resistant tumor cells. Therefore, dual inhibition of P-cadherin and c-Met may be a potentially effective approach for treating HNSCC.
Keywords:
P-cadherin
; c-Met
; cell proliferation
; cell migration
; Head and neck cancer
1. Introduction
Head and neck carcinoma (HNC) is the sixth most common cancer worldwide and affects > 500,000 people each year [1,2,3,4,5]. Squamous cell carcinoma (SCC), which presents as a highly heterogeneous disease, accounts for more than 90% of HNC cases [1,4,6,7]. Despite advancements in traditional therapies such as surgery, chemotherapy, and radiation therapy over the past three decades, the average 5-year survival rate post-diagnosis in developed countries remains between 42% and 64% [8,9]. Head and neck squamous cell carcinoma (HNSCC) is a multifactorial disease, with primary known risk factors including exposure to tobacco products, alcohol, and infection with high-risk human papillomavirus (HPV) strains [8,10,11,12]. The prognosis for HNSCC is generally poor, particularly when the disease has migrated regionally to cervical lymph nodes or metastasized to distant organs [13,14,15,16,17]. Therefore, a deeper understanding of this disease’s biological behavior could improve our ability to predict and guide HNSCC treatment.
Cadherins belong to a family of glycosylated Ca2+-dependent adhesion molecules that play a crucial role in maintaining tissue integrity and cellular localization [18,19]. The classic cadherin family is primarily divided into CDH1/E-cadherin (found in epithelial cells), CDH2/N-cadherin (found in neurons), CDH3/P-cadherin (found in the placenta), and CDH4/R-cadherin (found in the retina) [20]. These molecules mediate intercellular adhesion and facilitate the transduction of signals that impact several vital biological processes, such as proliferative activity, cellular motility, and apoptosis [21,22]. Among these classic cadherins, P-cadherin is a key epithelial molecule that is exclusively expressed in the basal layer of epithelial cells, similar to E-cadherin [23]. However, our understanding of its role in tissue integrity and homeostasis remains largely incomplete. Therefore, it is necessary to expand our understanding of the mechanisms underlying P-cadherin-mediated pathways in HNSCC.
The mesenchymal-epithelial transition factor (Met or c-Met), a receptor tyrosine kinase, is overexpressed in many cancers, including HNSCC, and plays a pivotal role in tumor development [4,24]. Numerous studies have reported that high c-Met expression was correlated with poor prognosis and tumor recurrence [25,26]. Additional research has demonstrated that c-Met promotes tumor progression through mechanisms such as cell proliferation, invasion, and angiogenesis [24,27,28]. However, data regarding the association between c-Met and P-cadherin, and the influence this has on signal transduction, remain scarce. In this study, we identified a correlation between c-Met and P-cadherin in HNSCC for the first time. We also discovered that suppressing P-cadherin amplifies the c-Met/STAT3 signaling pathway. Therefore, when P-cadherin is downregulated to achieve anticancer effects, it is crucial to consider the concurrent use of P-cadherin and c-Met inhibitors.
2. Materials and Methods
2.1. Cell lines and reagents
The human HNSCC cell lines SNU1076 (larynx), SNU1066 (larynx), SNU1041 (hypopharynx), FaDu (hypopharynx), SCC15 (oral), SCC25 (oral), and YD8 (oral) were obtained from the Korean Cell Line Bank (Seoul, South Korea). The primary human fibroblasts hFB were utilized as normal epithelial cells. The cell lines SNU1076, SNU1066, SNU1041, and YD8 were cultured in RPMI 1640 medium (WELGENE Inc., Gyeongsan, Korea). The SCC15 and SCC25 cell lines were maintained in DMEM/F12 (WELGENE Inc., Gyeongsan, Korea). The hFB and FaDu cell lines were cultured in high-glucose DMEM (WELGENE Inc., Gyeongsan, Korea). All cell lines were supplemented with 10% fetal bovine serum and 100 U/mL penicillin-streptomycin (HyClone, Logan, UT, USA). The cells were incubated at 37°C in a humidified atmosphere containing 5% CO2.
2.2. RNA isolation and quantitative real-time PCR (qRT-PCR)
Total cellular RNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). We then synthesized cDNA from 1 µg of the total RNA using TOPscriptTMRT DryMIX (Enzynomics Inc., Daejeon, Korea), following the manufacturer’s instructions. The amplification process was conducted using the SYBR Green qPCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA). The PCR reactions were executed over 40 cycles, each consisting of 95 °C for 15 seconds, 60 °C for 1 minute, and 72 °C for 1 minute. The primer sequences used were as follows: P-cadherin-F: 5′-ATG TGC CTG AGA ATG CAG TG-3′/P-cadherin-R: 5′- GTT TTT GGC CTC AAA ATC CA-3′, and GAPDH-F:5′-ACC CAG AAG ACT GTG GG-3′/GAPDH-R:5′-TTC TAG ACG GCA GGT CAG CT-3′. The Ct values provided by the real-time PCR instrument were imported into Excel. We then used the 2−ΔΔCt method to perform relative quantification of qRT-PCR fold changes.
2.3. Western blot analysis
Cells were lysed using a RIPA lysis buffer that contained 150 mM NaCl, 1.0% Nonidet P-40 (NP40), 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris (pH 8.0), and a protease inhibitor cocktail (Roche Applied Science, Vienna, Austria, pH 7.4). Electrophoresis was carried out as previously described [29,30]. For Western blot analysis, the following primary anti-human antibodies were utilized: anti-P-cadherin, anti-phospho-Met (Tyr1234/1235), anti-Met, anti-phospho-STAT3 (Tyr705), anti-STAT3, anti-phospho-FAK (Tyr397), anti-phospho-Src (Tyr416), anti-Phospho-Akt (Ser473), anti-Akt, anti-Phospho-ERK1/2 (Thr202/Tyr204), anti-ERK1/2, anti-ZEB1, anti-Slug, anti-β-Actin (1:1000; Cell Signaling Technology Inc, Danvers, MA, USA), anti-FAK, anti-Src, anti-E-cadherin, and anti-N-cadherin (1:1000; Santa Cruz Biotechnology, Dallas, TX, USA). The membranes were blocked with 5% skim milk for 1 hour at room temperature. After washing with Tris-buffered saline and 0.1% Tween 20 (TBST), the membranes were incubated with primary anti-human antibodies (Abs) at 4 °C. The membranes were then incubated with the corresponding horseradish peroxidase-conjugated secondary antibodies (1:1000; Cell Signaling Technology Inc, Danvers, MA, USA) for 1 hour at room temperature. Following four washes with TBST, the immunoreactive bands were visualized using enhanced chemiluminescence (ECL) detection.
2.4. Patients’ Samples and Ethics Statement
Samples from four patients diagnosed with HNSCC at CNUH were included in the study. Paired tumor and normal tissues were obtained from patients with HNSCC. All samples were gathered from patients after they provided informed consent according to the institutional guidelines of Chungnam National University Hospital. The protocol for this study was approved by the Institutional Review Board of CNUH (Reg. No. CNUH IRB No. 2017-07-005). Human tissues were homogenized in RIPA buffer and processed following the immunoblotting analysis protocol.
2.5. Small-interfering RNA (siRNA) and overexpression vector transfection
Cells were seeded at 2 × 105/well in six-well plates and subsequently cultured overnight to achieve a confluence of 60%-70%. The cells were then transfected with 50 nM of either P-cadherin siRNA-1 (sense: 5′-CUCUCUGGAAUGGAACCUU (dTdT)-3′; antisense: 5′-AAGGUUCCAUUCCAGAGAG (dTdT)-3′), P-cadherin siRNA-2 (sense: 5′-GACUGACCUACAGUGGACU (dTdT)-3′; antisense: AGUCCACUGUAGGUCAGUC (dTdT)-3′), or negative control siRNAs (#SN-1003, Bioneer, Daejeon, South Korea). These were dissolved in Opti-MEM medium (Gibco, Waltham, MA, USA) and transfected using Lipofectamine RNAiMAX (Thermo-Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturer’s protocol.
2.6. Cell proliferation assay
Cells were seeded at 1 × 104/well in 96-well plates. After 24 hours, these cells were treated with either a control or P-cadherin siRNA for a duration of 48 hours. The viabilities of SNU1076 and SNU1041 cells were then assessed using the WST-1 cell proliferation reagent (Roche Diagnostics Corporation, Indianapolis, IN, USA), as previously described. The formazan product was quantitatively measured at 450 nm using an enzyme-linked immunosorbent assay reader.
2.7. Colony formation assay
Cells were seeded in 6-well plates at 1 × 103 cells/well in 2 mL of medium. The medium was replaced with new medium every three days, and the cells were allowed to grow for 12 days. The colonies were fixed in 4% formaldehyde, stained with crystal violet, and then imaged and counted.
2.8. Cell migration and invasion (Transwell) assay
Transwell chambers (24-well; Costar, Cambridge, MA, USA) with 6.5-mm diameter polycarbonate filters (8-µm pore size) were used to examine cell migration and invasion, as described previously. In brief, we coated the Transwell membranes with Matrigel (BD Biosciences) for 6 hours at 37 °C for the invasion assay, while we left the membranes uncoated for the migration assay. We then added 2 × 105 cells in 100 µL of serum-free medium to the upper chamber, and 700 µL of medium containing 10% fetal bovine serum to the lower chamber. We incubated the chamber for 48 hours in a 5% CO2 environment at 37 °C. After incubation, we removed non-migrated or non-invaded cells using a cotton swab. We stained the cells that had attached to the lower chamber (either migrating or invading cells) with crystal violet and counted them in four representative fields under a light microscope at ×200 magnification.
2.9. Bioinformatic Transcriptome Analysis
All genomic data for HNSCC (n=522) were sourced from The Cancer Genome Atlas (TCGA) via a specific portal (https://tcga-data.nci.nih.gov) and the UCSC Cancer Browser (https://genome-cancer.ucsc.edu). Ingenuity Pathways Analysis (IPA) was conducted to explore relevant pathways. To measure the dependence between gene expression signals, Spearman correlation coefficients were used.
2.10. Xenograft in vivo tumor model
Mice were maintained under specific pathogen-free conditions and utilized in accordance with the guidelines set by the Institutional Animal Care and Use Committee of Chungnam National University. This committee approved the animal research protocol (No. CNUH-020-A0036-1). We obtained 20 BALB/c-nude mice, each 6 weeks old, from Orient Bio Inc. (Seongnam, Republic of Korea). These mice were randomly divided into four groups: control, P-cadherin (100mg/kg) and SU11274 (20mg/kg), and P-cadherin siRNA+SU11274, with each group containing 5 mice. We inoculated luciferase-expressing FaDu (FaDu-luc) cells (1×106) subcutaneously into the lower right flanks of the BALB/c-nude mice. After 7 days, when the tumors had reached approximately 50 mm in diameter, we injected the tumors once every two days with 100 μL of each group’s respective treatment for 25 days. Following the final injection, we excised the tumors from the euthanized mice for subsequent western blot and histological analysis.
2.11. Immunohistochemistry
Tissue samples were preserved in a 4% formalin solution before being embedded in paraffin. The tissue sections were then deparaffinized in xylene, hydrated using graded alcohol solutions, and stained with hematoxylin and eosin (H&E). Following mounting, the samples were examined with an automatic digital slide scanner (Pannoramic MIDI; 3DHISTECH, Budapest, Hungary). For immunohistochemistry, the tissue sections underwent a similar process of deparaffinization in xylene and hydration in graded alcohol solutions. They were then heated at 100 °C for 15 minutes in Antigen Retrieval Citra Solution (pH 6.0) to retrieve antigens. In single immunostaining, endogenous peroxidase activity was inhibited using a 1% hydrogen peroxide solution (Sigma-Aldrich, St. Louis, MO, USA) in phosphate-buffered saline (PBS) with 0.3% Triton X-100. This process was carried out for 30 minutes at room temperature. The sections were then incubated with the specified antibodies at 4 °C overnight, followed by incubation with the corresponding horseradish peroxidase-conjugated secondary antibody. To detect these labeled antibodies, 3,3’-diaminobenzidine (DAB; Dako, Agilent, Santa Clara, CA, USA) was used, and the nucleus was stained with hematoxylin. After being rinsed with PBS, the samples were mounted and analyzed using the same automatic digital slide scanner (Pannoramic MIDI; 3DHISTECH, Budapest, Hungary).
2.12. Statistical analysis
All statistical analyses were conducted using SPSS for Windows version 20.0 (IBM Corp., Armonk, NY, USA). We employed the Pearson chi-square or Fisher exact test to examine the relationships between P-cadherin expression and clinicopathologic parameters. Survival curves were generated using the Kaplan–Meier method and compared with the log-rank test. All in vitro experiments were performed three times, and statistical significance was determined using the unpaired Student t-test. The data are represented as means ± standard deviation. A P-value of less than 0.05 was considered to indicate statistical significance (*P < 0.05; **P < 0.01; ***P < 0.001).
3. Results
3.1. P-cadherin is overexpressed in head and neck cancer
In a preliminary study, we identified significant differentially expressed genes (DEGs) from the RNA-seq data of the Chungnam National University Hospital (CNUH) HNSC cohort, which consisted of 9 tumors and 9 matched normal tissues [31]. P-cadherin was among the genes that exhibited high expression in tumor tissues compared to normal tissues. We first evaluated whether there was a disparity in P-cadherin expression between normal and tumor tissues from patients, using the TCGA-HNSCC database as a reference. We found that P-cadherin expression was elevated in tumor tissues compared to normal tissue (Figure 1A). We then examined the expression levels of P-cadherin in a normal cell line (hFB) and in seven HNSCC cell lines (FaDu, SNU1041, SNU1076, SNU1066, YD8, SCC15, SCC25). All HNSCC cell lines displayed significantly higher P-cadherin expression than the normal cell line, at both mRNA and protein levels (Figure 1B, C). We also investigated P-cadherin expression in four pairs of HNSCC patient tissues from CNUH. We found that P-cadherin expression levels were elevated in tumor tissues compared to their corresponding normal tissues (Figure 1D). In summary, these findings suggest that P-cadherin overexpression is associated with tumorigenesis, indicating that P-cadherin could potentially have an oncogenic role in HNSCC.
3.2. P-cadherin promotes cell proliferation, migration and invasion of HNSCC cells through EMT and FAK-Src signaling
To investigate the functional role of P-cadherin in HNSCC, we initially performed proliferation assays, cell counting, and colony formation assays after the knockdown and overexpression of P-cadherin in SNU1041 and SNU1076 cells. The cells were transiently transfected with either P-cadherin-specific siRNA or negative control siRNA. The knockdown of P-cadherin resulted in a decrease in both proliferative activity and colony formation capacity in both cell lines (Figure 2A-H). Conversely, to assess the impact of P-cadherin overexpression, we transiently transfected a P-cadherin overexpression vector into SNU1041 and SNU1076 cells. This overexpression of P-cadherin significantly increased cell proliferation and colony formation compared to the control (Figure 2I-N).
Next, we investigated the role of P-cadherin in HNSCC migration and invasion, which are recognized as critical steps in tumor metastasis. Cells transfected with either P-cadherin or negative control siRNA were allowed to migrate in Transwell chambers (cell migration) or chambers coated with Matrigel (cell invasion). The knockdown of P-cadherin significantly suppressed the migration and invasion of SNU1041 and SNU1076 cells (Figure 3A-D). In contrast, overexpression of P-cadherin resulted in an increase in migration and invasion of HNSCC cells (Figure 3G-J). These findings suggest that P-cadherin positively influences the proliferation, migration, and invasion of HNSCC cells.
To understand how P-cadherin regulates cell migration and invasion in HNSCC cells, we examined the epithelial-mesenchymal transition (EMT), as its activation is associated with cancer metastasis during cancer progression [32]. To determine whether P-cadherin regulates the EMT, we analyzed the epithelial marker (E-cadherin), mesenchymal marker (N-cadherin), and transcription factors (ZEB1, Slug, Snail) using western blot analysis. The knockdown of P-cadherin resulted in a significant decrease in N-cadherin, ZEB1, Slug, and Snail levels. Conversely, overexpression of P-cadherin had the opposite effect in SNU1041 and SNU1076 cells (Figure 3E, F, K, L).
Previous data have shown cross-talk between P-cadherin and integrin in breast cancer [33]. Moreover, numerous studies have found that the activated FAK-Src generates signals leading to tumor metastasis [34]. In our studies, we also examined whether P-cadherin affects the integrin-related FAK-Src signaling. The knockdown of P-cadherin resulted in a reduction of p-FAK and p-Src levels (Figure 3E, F, K, L). These findings suggest that P-cadherin promotes cancer progression by inducing EMT and the FAK-Src signaling pathway.
3.3. P-cadherin is involved in the c-Met signaling pathway
To identify the upstream regulators of P-cadherin, we performed IPA using the TCGA-HNSCC database. Among the various potential upstream regulators of P-cadherin, HGFR (c-Met) has not yet been studied in relation to P-cadherin (Figure 4A). The HGF/c-Met signaling pathway is known to play a crucial role in tumorigenesis and tumor metastasis [35]. Based on our IPA results, we hypothesized that c-Met could be the primary upstream regulator of P-cadherin. To test this hypothesis, we evaluated the relationship between c-Met and P-cadherin using Spearman correlation analysis. In the TCGA-HNSCC database, we observed a significant positive correlation between the expression of c-Met and P-cadherin (Spearman coefficient=0.354, P=0.000) (Figure 4B). Next, we investigated whether the knockdown of c-Met would lead to a decrease in P-cadherin expression in HNSCC cells. We found that siRNA silencing of c-Met did indeed reduce P-cadherin expression, along with the phosphorylation of Akt, a well-known downstream target of c-Met in HNSCC cells (Figure 4C). In contrast, overexpression of c-Met led to an increase in both P-cadherin expression and Akt phosphorylation (Figure 4D). To determine whether the induction of P-cadherin was mediated by direct binding with c-Met, we evaluated the interaction between c-Met and P-cadherin using Co-IP. Our results showed that c-Met and P-cadherin did not bind directly, suggesting that c-Met modulates P-cadherin indirectly through some mechanisms (Supplementary Figure S1). Taken together, these data suggest that P-cadherin is involved in the c-Met signaling pathway.
3.4. P-cadherin blockade activates c-Met/STAT3 pathway
To elucidate the relationship between P-cadherin and c-Met, we examined the activation of c-Met following P-cadherin knockdown. Intriguingly, we discovered that the suppression of P-cadherin activates c-Met by enhancing the phosphorylation of c-Met and STAT3, a downstream component of c-Met, while Akt signaling was diminished in SNU1041 and SNU1076 cells (Figure 5A, B). To further investigate this novel mechanism, we studied the activation of c-Met after a time-dependent inhibition of P-cadherin. As depicted in figures 5C and D, no increase in c-Met phosphorylation was detected during the early hours of P-cadherin suppression. However, after 24 hours, P-cadherin suppression resulted in a significant increase in c-Met phosphorylation, while concurrently inducing Akt inhibition. Our findings suggest that although P-cadherin inhibition enhances the sensitivity of HNSCC, a subset of cells may survive, potentially due to the activation of other molecular pathways, including at least c-Met signaling.
3.5. Dual blockade of P-cadherin and c-Met inhibits HNSCC cell progression in vitro and in vivo
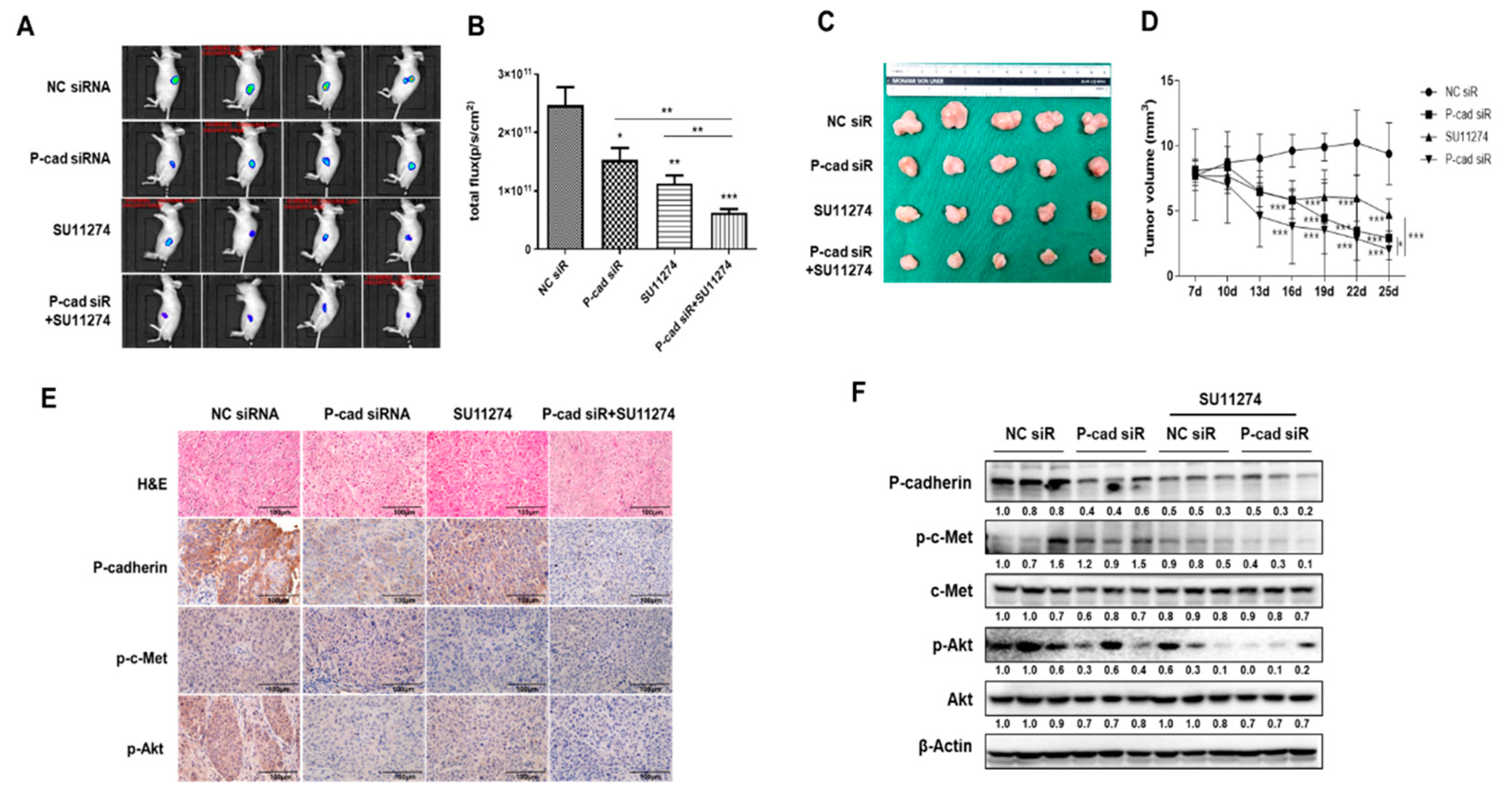
Since c-Met reactivation invariably follows P-cadherin inhibition, we assessed whether simultaneous inhibition of both P-cadherin and c-Met could yield a more potent anti-cancer effect in HNSCC. We treated SNU1041 and SNU1076 cells with the c-Met inhibitor SU11274 in conjunction with siRNA targeting P-cadherin, and compared the results to those of monotherapy, a c-Met inhibitor, or siRNA targeting P-cadherin alone. Intriguingly, the combined treatment of SU11274 and P-cadherin siRNA significantly diminished cell proliferation, migration, and invasion compared to cells treated solely with P-cadherin inhibition in both SNU1041 and SNU1076 cells (Figure 6A-J). Furthermore, to determine whether combined inhibition of P-cadherin and c-Met signaling could sensitize HNSCC in vivo, we established a xenograft mouse model from HNSCC. FaDu-Luc cells were subcutaneously injected into nude mice. Once tumors reached a volume of 300 mm3 post-inoculation, we administered P-cadherin siRNA and SU11274 either individually or in combination directly into the tumors. The region of interest and tumor volume of the groups that received the combined administration of P-cadherin and c-Met inhibitors were significantly reduced compared to the control or individually injected groups (Figure 7A-D). The expression levels of P-cadherin were notably lower in the group treated with the combination compared to the control group or those treated individually (Figure 7E, F). These results suggest that a combination treatment targeting P-cadherin and c-Met is more effective in curtailing the growth of head and neck cancer cells.
4. Discussion
In most cancers, recurrent or metastatic disease is typically associated with a dismal prognosis. There is a pressing need for the early detection of these diseases, precise prognosis prediction, and the development of biomarkers to guide appropriate treatment decisions. For HNSCC, several promising biomarkers have been identified. These have shown potential in the diagnosis, early detection, and prognosis of HNSCC. However, there are still limitations when it comes to treatment [36].
To identify genes involved in HNSCC carcinogenesis and to explore the mechanisms of progression, we utilized NGS technology to examine the transcriptomic profiles of non-tumor and tumor tissues from HNSCC patients. One of the validated findings was P-cadherin (CDH3), a protein previously associated with cancer progression and metastasis. However, the role of P-cadherin in cancer cells remains a topic of ongoing debate. P-cadherin has been observed to induce aggressive behavior in several types of cancer, including prostate, colon, breast, ovarian, gastric, pancreatic cancer, and glioblastoma [20,37,38,39]. In contrast, some reports suggest that P-cadherin functions as a tumor suppressor in other tumor models, such as melanoma, cholangiocarcinoma, hepatocarcinoma, and oral squamous cell carcinoma (OSCC) [37,40,41,42]. Notably, several studies have highlighted its potential tumor inhibitory effect in OSCC, a subtype of HNSCC. However, a recent study found no significant correlation between clinical parameters and P-cadherin expression in tongue squamous cell carcinoma [43]. P-cadherin has been reported to be associated with poor prognosis by promoting malignancy and chemoresistance in OSCC [44]. Our study discovered that high levels of P-cadherin enhance the proliferation, migration, and invasion of HNSCC cells, suggesting that P-cadherin may act as a key mediator of the aggressive tumor phenotype in all HNSCC cells, including OSCC (Supplementary Figure S2). Additionally, our research found that P-cadherin leads to increased activation of FAK and Src, as well as the EMT pathway, thereby promoting migration and invasion.
To identify a novel upstream regulator of P-cadherin, we explored various pathways implicated in tumorigenesis and/or metastasis. This was done using IPA to analyze a list of genes from the TCGA-HNSCC database. We discovered that HGFR (c-Met) acts as an upstream regulator of P-cadherin. Interestingly, no previous studies have reported a relationship between P-cadherin and c-Met. It is known that c-Met promotes cancer cell survival, proliferation, migration, and invasion, all of which are crucial for tumor progression [45,46]. Furthermore, HGF/c-Met signaling is involved in both cellular dissociation within the primary tumor and cellular re-association within the metastatic niche [47]. However, the mechanisms by which c-Met contributes to cell-to-cell dissociation remain unclear. Our analysis revealed a significant positive correlation between c-Met and P-cadherin expression, based on the TCGA-HNSCC database (Spearman coefficient=0.354, P=0.000). We then investigated whether c-Met could regulate P-cadherin in HNSCC cells. Our findings showed that c-Met knockdown by specific siRNA decreased P-cadherin expression, while overexpression of c-Met increased P-cadherin expression. However, this regulation did not result from direct binding between c-Met and P-cadherin.
Some reports have indicated that the inhibition of either MEK or ERK alone yields limited success due to its transient suppression of the MAPK pathway, insufficient pathway suppression, and probable pathway reactivation [48,49]. Furthermore, it has been proposed that targeting both MEK and ERK nodes simultaneously results in a deeper and longer-lasting suppression of MAPK signaling, which cannot be achieved with any dosage of a single agent, particularly in tumors where feedback reactivation occurs [49]. Unexpectedly, we found that the inhibition of P-cadherin induces c-Met activation through phosphorylation, suggesting a complementary enhancement of c-Met activation due to P-cadherin inhibition. We further explored whether a combination treatment targeting both P-cadherin and c-Met would be more effective in reducing HNSCC cell growth and migration. Our findings revealed that combination therapy using the c-Met inhibitor SU11274 and P-cadherin siRNA significantly reduced cell proliferation and migration more than each monotherapy in HNSCC cells and the HNSCC-xenograft mouse model.
P-cadherin is currently viewed as a promising target for solid-tumor treatment due to its overexpression in a variety of tumors. The humanized monoclonal antibody against P-cadherin, 90Y-FF-21101, has demonstrated high therapeutic efficacy for solid tumor treatment and is currently under evaluation in a first-in-human phase 1 study (NCT02454010) [50]. PCA062, another P-cadherin-specific antibody-drug conjugate, employs a clinically validated method to induce potent cytotoxicity in cell lines expressing high levels of P-cadherin. This includes breast, esophageal, and head and neck cell lines, and it is also being evaluated in a phase I trial (NCT02375958) [51]. Based on our finding, the complementary enhancement effect of c-Met activation due to P-cadherin inhibition should be considered when evaluating the therapeutic value of specific P-cadherin inhibitors such as 90Y-FF-21101 and PCA062 for HNSCC treatment.
In this study, we have, for the first time, demonstrated the association between c-Met and P-cadherin, highlighting the ability of c-Met to regulate P-cadherin expression. Furthermore, our findings suggest a potentially effective strategy involving the combined use of P-cadherin inhibitors and c-Met inhibitors. This approach could help overcome the issue of c-Met reactivation caused by P-cadherin inhibition. Considering c-Met inhibition in conjunction with P-cadherin suppression could serve as a crucial complementary strategy for therapy, potentially enhancing the sensitivity of HNSCC cells to therapeutics.
5. Conclusions
In this study, we identified a novel correlation between c-Met and P-cadherin in HNSCC. We also found that the suppression of P-cadherin enhances the c-Met/STAT3 signaling pathway. Therefore, when P-cadherin is downregulated in order to achieve anticancer effects, the combination of P-cadherin and c-Met inhibitors should be considered.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, L.L. and S.-N.J.; validation, Y.P. and Y.S.; investigation, C.O.; resources, Y.I.K.; data curation, S.O. and M.A.L; writing—original draft preparation, L.L. and S.-N. J.; writing—review and editing, B.S.K.; supervision, B.S.K., H.-R.W. and J.W.C.; project administration, L.L. and S.-N.J.; funding acquisition, B.S.K. All authors have read and agreed to the published version of the manuscript.”
Funding
This study was supported by a research fund grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), by the Ministry of Health and Welfare, Republic of Korea (grant number: HR22C1734), and by the National Research Foundation of Korea (NRF) (grant number: 2019R1A2C1084125, 2022R1C1C1008265, 2021R1C1C1014142, 2022R1I1A3071413, RS-2023-00241883), and by the Korea Medical Device Development Fund grant funded by the Korea government (the Ministry of Science and ICT, the Ministry of Trade, Industry and Energy, the Ministry of Health & Welfare, the Ministry of Food and Drug Safety) (Project Number: 1711138229, KMDF_PR_20200901_0124), and by research fund of Chungnam National University, and by the National Natural Science Foundation of China (NSCF, Grant number: 82304149).
Institutional Review Board Statement
The protocol for this study was approved by the Institutional Review Board of CNUH (Reg. No. CNUH IRB No. 2019-07-041-014). The animal study protocol was approved by the Institutional Animal Care and Use Committee of Chungnam National University (No. CNUH-020-A0036-1).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, D.; Su, L.; Huang, D.; Zhang, H.; Shin, D.M.; Chen, Z.G. Downregulation of E-Cadherin enhances proliferation of head and neck cancer through transcriptional regulation of EGFR. Mol. Cancer 2011, 10, 116. [Google Scholar] [CrossRef]
- Vigneswaran, N.; Williams, M.D. Epidemiologic trends in head and neck cancer and aids in diagnosis. Oral. Maxillofac. Surg. Clin. N. Am. 2014, 26, 123–141. [Google Scholar] [CrossRef]
- Larizadeh, M.H.; Damghani, M.A.; Shabani, M. Epidemiological characteristics of head and neck cancers in southeast of iran. Iran. J. Cancer Prev. 2014, 7, 80–86. [Google Scholar]
- Peltanova, B.; Raudenska, M.; Masarik, M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: a systematic review. Mol. Cancer 2019, 18, 63. [Google Scholar] [CrossRef]
- Liao, L.J.; Hsu, W.L.; Lo, W.C.; Cheng, P.W.; Shueng, P.W.; Hsieh, C.H. Health-related quality of life and utility in head and neck cancer survivors. BMC Cancer 2019, 19, 425. [Google Scholar] [CrossRef]
- Elkashty, O.A.; Ashry, R.; Tran, S.D. Head and neck cancer management and cancer stem cells implication. Saudi Dent. J. 2019, 31, 395–416. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Cognetti, D.M.; Weber, R.S.; Lai, S.Y. Head and neck cancer: an evolving treatment paradigm. Cancer 2008, 113, 1911–1932. [Google Scholar] [CrossRef]
- Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 2011, 61, 212–236. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef]
- Zabolotnyi, D.; Kizim, Y.; Zabolotna, D.; Sulaieva, O.; Kizim, V. Laryngopharyngeal Reflux Alters Macrophage Polarization in Human Papilloma Virus-Negative Squamous Cell Carcinoma of the Larynx in Males. Clin. Exp. Otorhinolaryngol. 2021, 14, 240–243. [Google Scholar] [CrossRef]
- Wong, T.S.; Gao, W.; Chan, J.Y. Interactions between E-cadherin and microRNA deregulation in head and neck cancers: the potential interplay. Biomed. Res. Int. 2014, 2014, 126038. [Google Scholar] [CrossRef]
- Pisani, P.; Airoldi, M.; Allais, A.; Aluffi Valletti, P.; Battista, M.; Benazzo, M.; Briatore, R.; Cacciola, S.; Cocuzza, S.; Colombo, A.; et al. Metastatic disease in head & neck oncology. Acta Otorhinolaryngol. Ital. 2020, 40, S1–s86. [Google Scholar] [CrossRef]
- Uz, U.; Eskiizmir, G. Association Between Interleukin-6 and Head and Neck Squamous Cell Carcinoma: A Systematic Review. Clin. Exp. Otorhinolaryngol. 2021, 14, 50–60. [Google Scholar] [CrossRef]
- Yin, C.Y.; Zhang, S.S.; Zhong, J.T.; Zhou, S.H. Pepsin and Laryngeal and Hypopharyngeal Carcinomas. Clin. Exp. Otorhinolaryngol. 2021, 14, 159–168. [Google Scholar] [CrossRef]
- Lee, D.Y.; Hah, J.H.; Jeong, W.J.; Chung, E.J.; Kwon, T.K.; Ahn, S.H.; Sung, M.W.; Kwon, S.K. The Expression of Defensin-Associated Genes May Be Correlated With Lymph Node Metastasis of Early-Stage Tongue Cancer. Clin. Exp. Otorhinolaryngol. 2022, 15, 372–379. [Google Scholar] [CrossRef]
- Bauer, K.; Dowejko, A.; Bosserhoff, A.K.; Reichert, T.E.; Bauer, R.J. P-cadherin induces an epithelial-like phenotype in oral squamous cell carcinoma by GSK-3beta-mediated Snail phosphorylation. Carcinogenesis 2009, 30, 1781–1788. [Google Scholar] [CrossRef]
- Niessen, C.M.; Leckband, D.; Yap, A.S. Tissue organization by cadherin adhesion molecules: dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol. Rev. 2011, 91, 691–731. [Google Scholar] [CrossRef]
- Xi, Y.; Zhang, X.; Yang, Z.; Zhang, X.; Guo, Q.; Zhang, Z.; Chen, S.; Zheng, H.; Hua, B. Prognositic significance of P-cadherin expression in breast cancer: Protocol for a meta-analysis. Medicine 2019, 98, e14924. [Google Scholar] [CrossRef]
- Hermiston, M.L.; Gordon, J.I. In vivo analysis of cadherin function in the mouse intestinal epithelium: essential roles in adhesion, maintenance of differentiation, and regulation of programmed cell death. J. Cell Biol. 1995, 129, 489–506. [Google Scholar] [CrossRef]
- Weber, G.F.; Bjerke, M.A.; DeSimone, D.W. Integrins and cadherins join forces to form adhesive networks. J. Cell Sci. 2011, 124, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Rübsam, M.; Mertz, A.F.; Kubo, A.; Marg, S.; Jüngst, C.; Goranci-Buzhala, G.; Schauss, A.C.; Horsley, V.; Dufresne, E.R.; Moser, M.; et al. E-cadherin integrates mechanotransduction and EGFR signaling to control junctional tissue polarization and tight junction positioning. Nat. Commun. 2017, 8, 1250. [Google Scholar] [CrossRef]
- Arnold, L.; Enders, J.; Thomas, S.M. Activated HGF-c-Met Axis in Head and Neck Cancer. Cancers (Basel) 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Akervall, J.; Nandalur, S.; Zhang, J.; Qian, C.N.; Goldstein, N.; Gyllerup, P.; Gardinger, Y.; Alm, J.; Lorenc, K.; Nilsson, K.; et al. A novel panel of biomarkers predicts radioresistance in patients with squamous cell carcinoma of the head and neck. Eur. J. Cancer 2014, 50, 570–581. [Google Scholar] [CrossRef]
- Baschnagel, A.M.; Williams, L.; Hanna, A.; Chen, P.Y.; Krauss, D.J.; Pruetz, B.L.; Akervall, J.; Wilson, G.D. c-Met expression is a marker of poor prognosis in patients with locally advanced head and neck squamous cell carcinoma treated with chemoradiation. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 701–707. [Google Scholar] [CrossRef]
- Xiang, C.; Chen, J.; Fu, P. HGF/Met Signaling in Cancer Invasion: The Impact on Cytoskeleton Remodeling. Cancers (Basel) 2017, 9. [Google Scholar] [CrossRef]
- Demkova, L.; Kucerova, L. Role of the HGF/c-MET tyrosine kinase inhibitors in metastasic melanoma. Mol. Cancer 2018, 17, 26. [Google Scholar] [CrossRef] [PubMed]
- Taniuchi, K.; Nakagawa, H.; Hosokawa, M.; Nakamura, T.; Eguchi, H.; Ohigashi, H.; Ishikawa, O.; Katagiri, T.; Nakamura, Y. Overexpressed P-cadherin/CDH3 promotes motility of pancreatic cancer cells by interacting with p120ctn and activating rho-family GTPases. Cancer Res. 2005, 65, 3092–3099. [Google Scholar] [CrossRef]
- Jung, S.N.; Shin, D.S.; Kim, H.N.; Jeon, Y.J.; Yun, J.; Lee, Y.J.; Kang, J.S.; Han, D.C.; Kwon, B.M. Sugiol inhibits STAT3 activity via regulation of transketolase and ROS-mediated ERK activation in DU145 prostate carcinoma cells. Biochem. Pharmacol. 2015, 97, 38–50. [Google Scholar] [CrossRef]
- Chang, J.W.; Seo, S.T.; Im, M.A.; Won, H.R.; Liu, L.; Oh, C.; Jin, Y.L.; Piao, Y.; Kim, H.J.; Kim, J.T.; et al. Claudin-1 mediates progression by regulating EMT through AMPK/TGF-β signaling in head and neck squamous cell carcinoma. Transl. Res. 2022, 247, 58–78. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes. Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.F.; Ribeiro, A.S.; Dionísio, M.R.; Sousa, B.; Nobre, A.R.; Albergaria, A.; Santiago-Gómez, A.; Mendes, N.; Gerhard, R.; Schmitt, F.; et al. P-cadherin signals through the laminin receptor α6β4 integrin to induce stem cell and invasive properties in basal-like breast cancer cells. Oncotarget 2014, 5, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Spina, A.; De Pasquale, V.; Cerulo, G.; Cocchiaro, P.; Della Morte, R.; Avallone, L.; Pavone, L.M. HGF/c-MET Axis in Tumor Microenvironment and Metastasis Formation. Biomedicines 2015, 3, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Economopoulou, P.; de Bree, R.; Kotsantis, I.; Psyrri, A. Diagnostic Tumor Markers in Head and Neck Squamous Cell Carcinoma (HNSCC) in the Clinical Setting. Front. Oncol. 2019, 9, 827. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.F.; Paredes, J. P-cadherin and the journey to cancer metastasis. Mol. Cancer 2015, 14, 178. [Google Scholar] [CrossRef]
- Turashvili, G.; McKinney, S.E.; Goktepe, O.; Leung, S.C.; Huntsman, D.G.; Gelmon, K.A.; Los, G.; Rejto, P.A.; Aparicio, S.A. P-cadherin expression as a prognostic biomarker in a 3992 case tissue microarray series of breast cancer. Mod Pathol 2011, 24, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Martins, E.P.; Gonçalves, C.S.; Pojo, M.; Carvalho, R.; Ribeiro, A.S.; Miranda-Gonçalves, V.; Taipa, R.; Pardal, F.; Pinto, A.A.; Custódia, C.; et al. Cadherin-3 is a novel oncogenic biomarker with prognostic value in glioblastoma. Mol. Oncol. 2022, 16, 2611–2631. [Google Scholar] [CrossRef]
- Bauer, R.; Valletta, D.; Bauer, K.; Thasler, W.E.; Hartmann, A.; Müller, M.; Reichert, T.E.; Hellerbrand, C. Downregulation of P-cadherin expression in hepatocellular carcinoma induces tumorigenicity. Int. J. Clin. Exp. Pathol. 2014, 7, 6125–6132. [Google Scholar] [CrossRef]
- Lo Muzio, L.; Pannone, G.; Mignogna, M.D.; Staibano, S.; Mariggiò, M.A.; Rubini, C.; Procaccini, M.; Dolci, M.; Bufo, P.; De Rosa, G.; et al. P-cadherin expression predicts clinical outcome in oral squamous cell carcinomas. Histol. Histopathol. 2004, 19, 1089–1099. [Google Scholar] [CrossRef]
- Jacobs, K.; Feys, L.; Vanhoecke, B.; Van Marck, V.; Bracke, M. P-cadherin expression reduces melanoma growth, invasion, and responsiveness to growth factors in nude mice. Eur. J. Cancer Prev. 2011, 20, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Seppälä, M.; Jauhiainen, L.; Tervo, S.; Al-Samadi, A.; Rautiainen, M.; Salo, T.; Lehti, K.; Monni, O.; Hautaniemi, S.; Tynninen, O.; et al. The expression and prognostic relevance of CDH3 in tongue squamous cell carcinoma. Apmis 2021, 129, 717–728. [Google Scholar] [CrossRef]
- Wu, T.; Xiao, Z.; Li, Y.; Jiao, Z.; Liang, X.; Zhang, Y.; Liu, H.; Yang, A. CDH3 is associated with poor prognosis by promoting the malignancy and chemoresistance in oral squamous cell carcinoma. Asian J. Surg. 2022, 45, 2651–2658. [Google Scholar] [CrossRef]
- Organ, S.L.; Tsao, M.S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–s19. [Google Scholar] [CrossRef]
- Wang, H.; Rao, B.; Lou, J.; Li, J.; Liu, Z.; Li, A.; Cui, G.; Ren, Z.; Yu, Z. The Function of the HGF/c-Met Axis in Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2020, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Delitto, D.; Vertes-George, E.; Hughes, S.J.; Behrns, K.E.; Trevino, J.G. c-Met signaling in the development of tumorigenesis and chemoresistance: potential applications in pancreatic cancer. World J. Gastroenterol. 2014, 20, 8458–8470. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Moffat, J.; Schaefer, G.; Chan, J.; Wang, X.; Orr, C.; Cheng, J.; Hunsaker, T.; Shao, L.; Wang, S.J.; et al. Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS One 2017, 12, e0185862. [Google Scholar] [CrossRef]
- Yang, S.; Liu, G. Targeting the Ras/Raf/MEK/ERK pathway in hepatocellular carcinoma. Oncol. Lett. 2017, 13, 1041–1047. [Google Scholar] [CrossRef]
- Subbiah, V.; Erwin, W.; Mawlawi, O.; McCoy, A.; Wages, D.; Wheeler, C.; Gonzalez-Lepera, C.; Liu, H.; Macapinlac, H.; Meric-Bernstam, F.; et al. Phase I Study of P-cadherin-targeted Radioimmunotherapy with (90)Y-FF-21101 Monoclonal Antibody in Solid Tumors. Clin. Cancer Res. 2020, 26, 5830–5842. [Google Scholar] [CrossRef]
- Sheng, Q.; D’Alessio, J.A.; Menezes, D.L.; Karim, C.; Tang, Y.; Tam, A.; Clark, S.; Ying, C.; Connor, A.; Mansfield, K.G.; et al. PCA062, a P-cadherin Targeting Antibody-Drug Conjugate, Displays Potent Antitumor Activity Against P-cadherin-expressing Malignancies. Mol. Cancer Ther. 2021, 20, 1270–1282. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
P-cadherin expression in HNSCC patient tissues and cell lines. (A) P-cadherin was expressed significantly more highly in tumor tissues than in normal tissues of HNC in The Cancer Genome Atlas (TCGA) database. Cell lysates were prepared from normal cell line (hFB) and seven HNSCC cell lines (FaDu, SNU1041, SNU1076, SNU1066, SCC15, SCC25, YD8) and examined the levels of P-cadherin by qRT-PCR (B) and western blot analysis (C). (D) P-cadherin expression was examined by western blot analysis using tissue samples obtained from head and neck cancer patients. Data were presented as mean ± SD of three independent experiments. Differences were considered relevant at p < 0.05 (* p < 0.05, ** p < 0.01, *** p < 0.001).
Figure 1.
P-cadherin expression in HNSCC patient tissues and cell lines. (A) P-cadherin was expressed significantly more highly in tumor tissues than in normal tissues of HNC in The Cancer Genome Atlas (TCGA) database. Cell lysates were prepared from normal cell line (hFB) and seven HNSCC cell lines (FaDu, SNU1041, SNU1076, SNU1066, SCC15, SCC25, YD8) and examined the levels of P-cadherin by qRT-PCR (B) and western blot analysis (C). (D) P-cadherin expression was examined by western blot analysis using tissue samples obtained from head and neck cancer patients. Data were presented as mean ± SD of three independent experiments. Differences were considered relevant at p < 0.05 (* p < 0.05, ** p < 0.01, *** p < 0.001).
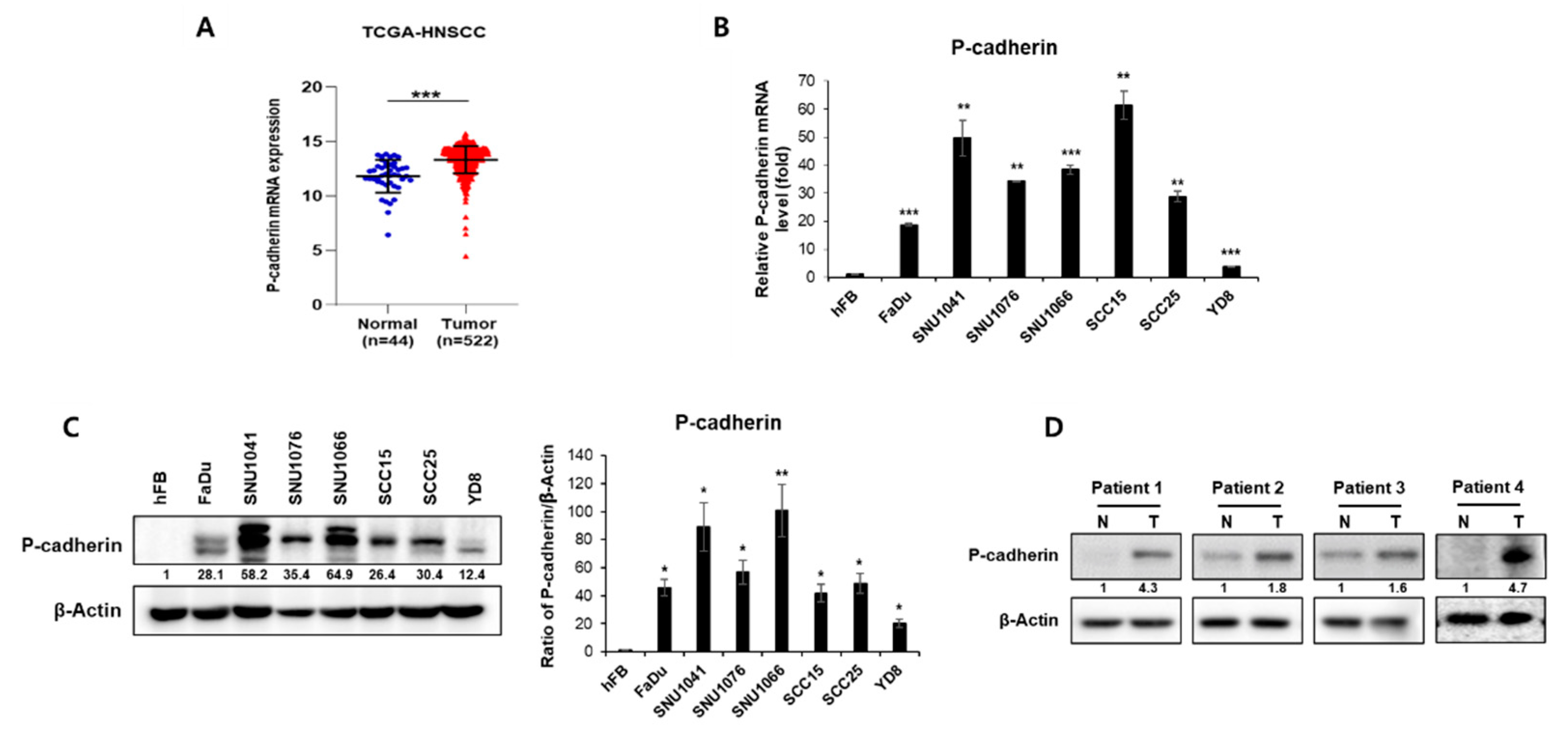
Figure 2.
P-cadherin promotes proliferation of HNSCC cells. SNU1041 and SNU1076 cells were transfected with P-cadherin siRNAs or negative control siRNA for 48h. (A, B) P-cadherin protein levels were assessed by western blot analysis. (C, D) The proliferation of SNU1041 and SNU1076 cells was analyzed by WST-1 assay. (E, F) Cell counting assessed using a hemocytometer. (G, H) Colony formation assays were conducted. (I, J) P-cadherin protein levels were assessed by western blot analysis after P-cadherin overexpression vector was transiently transfected into SNU1041 and SNU1076 cells. After P-cadherin overexpression, the proliferation of SNU1041 and SNU1076 cells analyzed by WST-1 assay (K, L) and colony formation assays were conducted (M, N). Data were presented as mean ± SD of three independent experiments. Differences were considered relevant at p < 0.05 (* p < 0.05, ** p < 0.01, *** p < 0.001).
Figure 2.
P-cadherin promotes proliferation of HNSCC cells. SNU1041 and SNU1076 cells were transfected with P-cadherin siRNAs or negative control siRNA for 48h. (A, B) P-cadherin protein levels were assessed by western blot analysis. (C, D) The proliferation of SNU1041 and SNU1076 cells was analyzed by WST-1 assay. (E, F) Cell counting assessed using a hemocytometer. (G, H) Colony formation assays were conducted. (I, J) P-cadherin protein levels were assessed by western blot analysis after P-cadherin overexpression vector was transiently transfected into SNU1041 and SNU1076 cells. After P-cadherin overexpression, the proliferation of SNU1041 and SNU1076 cells analyzed by WST-1 assay (K, L) and colony formation assays were conducted (M, N). Data were presented as mean ± SD of three independent experiments. Differences were considered relevant at p < 0.05 (* p < 0.05, ** p < 0.01, *** p < 0.001).
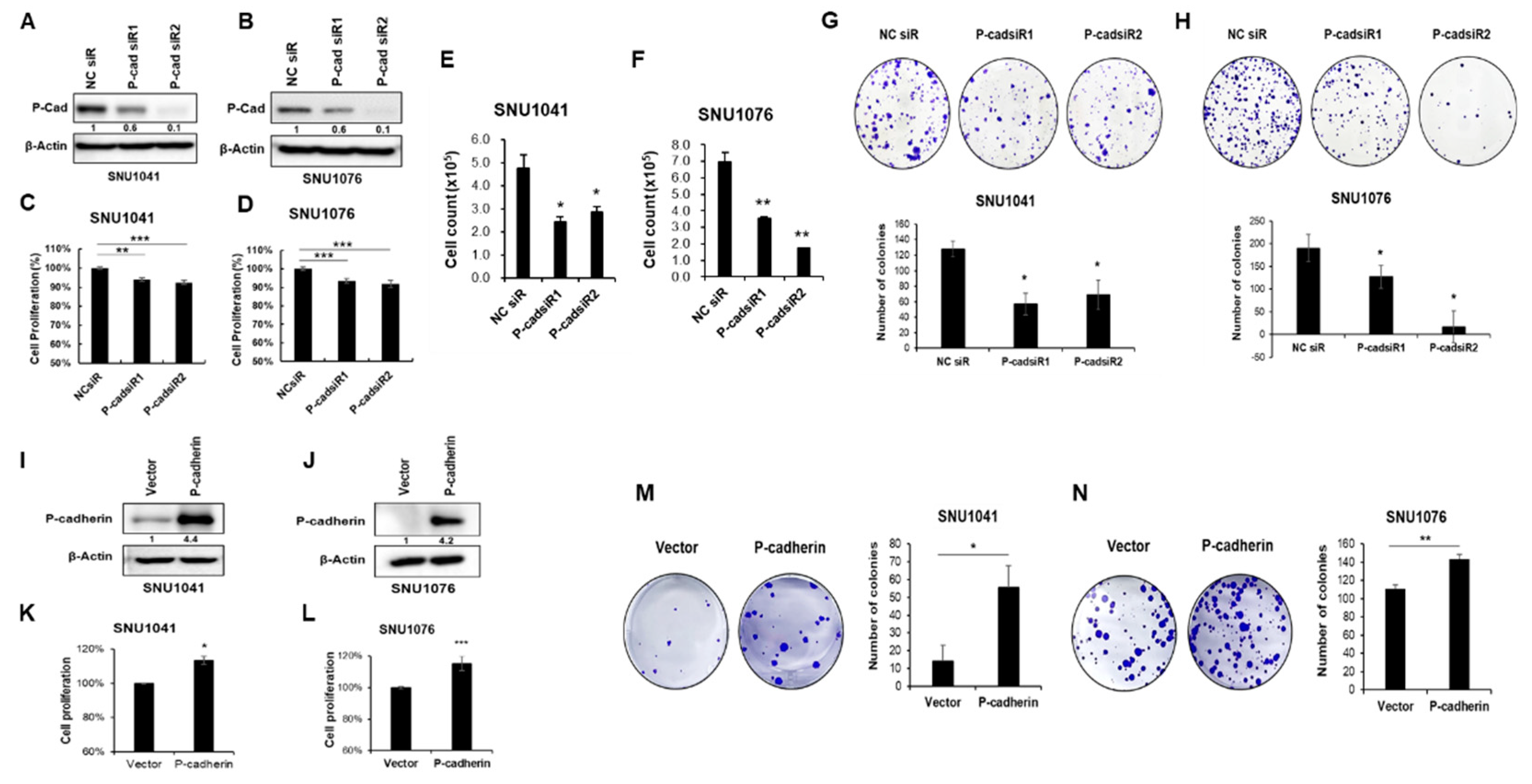
Figure 3.
P-cadherin promotes migration and invasion of HNSCC cells.SNU1041 and SNU1076 cells were transfected with P-cadherin siRNAs or negative control siRNA for 48h. Cells were allowed to migrate for 48h in transwell chambers (Cell Migration) (A, B) or for 72 h in chambers coated with Matrigel (Cell invasion) (C, D). (E, F) The expression levels of FAK, Src, Akt, ERK, and the EMT-related proteins were evaluated by western blot analysis. After P-cadherin overexpression, Cells were allowed to migrate for 48h in transwell chambers (Cell Migration) (G, H) or for 72 h in chambers coated with Matrigel (Cell invasion) (I, J). (K, L) After P-cadherin overexpression, the expression levels of FAK, Src, Akt, ERK, and the EMT-related proteins were evaluated by western blot analysis. Data were presented as mean ± SD of three independent experiments. Differences were considered relevant at p < 0.05 (* p < 0.05, ** p < 0.01).
Figure 3.
P-cadherin promotes migration and invasion of HNSCC cells.SNU1041 and SNU1076 cells were transfected with P-cadherin siRNAs or negative control siRNA for 48h. Cells were allowed to migrate for 48h in transwell chambers (Cell Migration) (A, B) or for 72 h in chambers coated with Matrigel (Cell invasion) (C, D). (E, F) The expression levels of FAK, Src, Akt, ERK, and the EMT-related proteins were evaluated by western blot analysis. After P-cadherin overexpression, Cells were allowed to migrate for 48h in transwell chambers (Cell Migration) (G, H) or for 72 h in chambers coated with Matrigel (Cell invasion) (I, J). (K, L) After P-cadherin overexpression, the expression levels of FAK, Src, Akt, ERK, and the EMT-related proteins were evaluated by western blot analysis. Data were presented as mean ± SD of three independent experiments. Differences were considered relevant at p < 0.05 (* p < 0.05, ** p < 0.01).
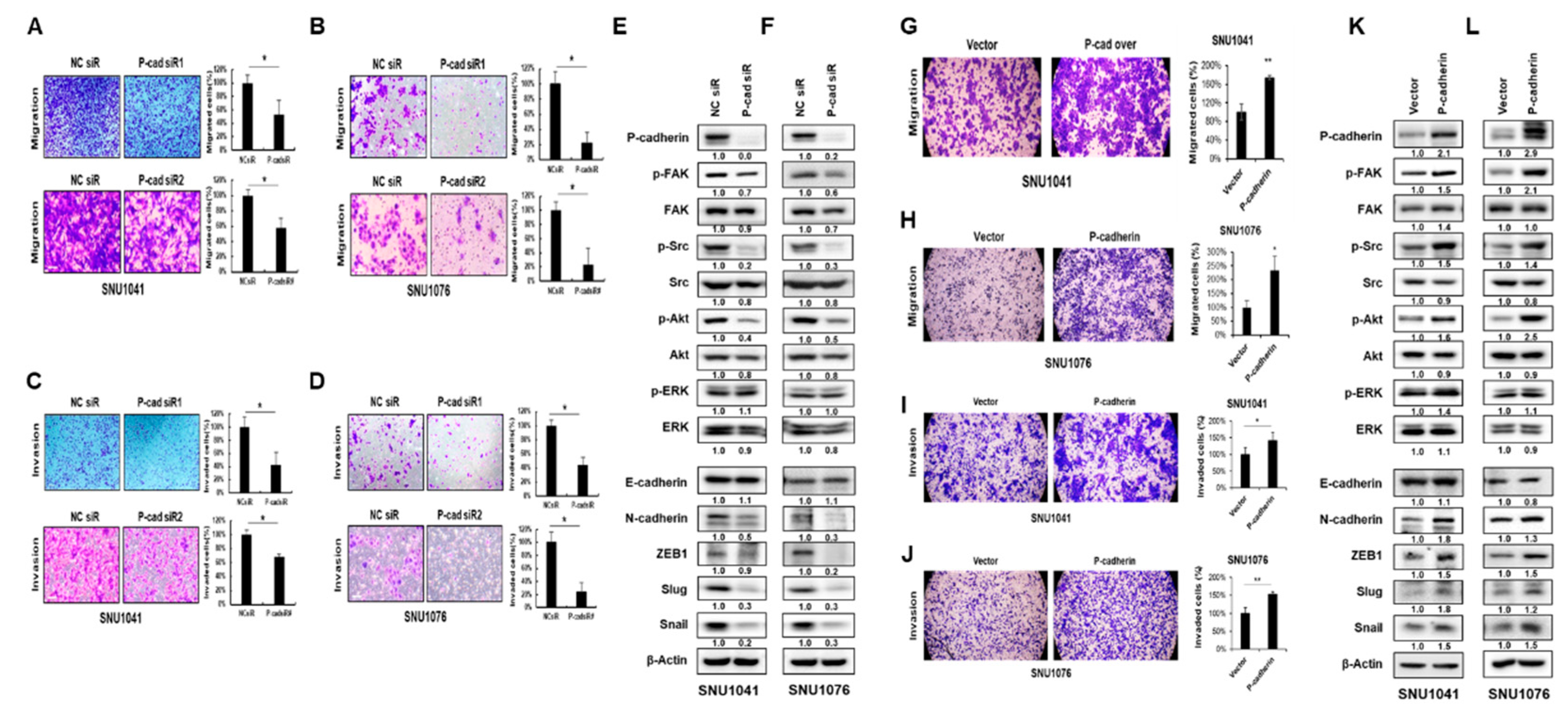
Figure 4.
P-cadherin was involved in the c-Met signaling pathway. (A) Upstream regulators of P-cadherin by IPA analysis. (B) Scatter plots showing the relationship between the expression of P-cadherin and c-Met using the Spearman’s correlation co-efficient. (C, D) SNU1041 and SNU1076 cells were transfected with c-Met siRNAs and c-Met overexpression vector or negative control for 48h. p-c-Met, total c-Met, P-cadherin, p-Akt, total Akt levels were examined by western blot analysis. Data were presented as mean ± SD of three independent experiments.
Figure 4.
P-cadherin was involved in the c-Met signaling pathway. (A) Upstream regulators of P-cadherin by IPA analysis. (B) Scatter plots showing the relationship between the expression of P-cadherin and c-Met using the Spearman’s correlation co-efficient. (C, D) SNU1041 and SNU1076 cells were transfected with c-Met siRNAs and c-Met overexpression vector or negative control for 48h. p-c-Met, total c-Met, P-cadherin, p-Akt, total Akt levels were examined by western blot analysis. Data were presented as mean ± SD of three independent experiments.

Figure 5.
P-cadherin blockade activates c-Met/STAT3 pathway. (A, B) SNU1041 and SNU1076 cells were transfected with P-cadherin siRNAs or negative control siRNA for 48h. The levels of phospho-c-Met, total c-Met, P-cadherin, phospho-STAT3, total STAT3, phospho-Akt, total Akt, phospho-ERK were examined by Western blot analysis. (C, D) SNU1041 and SNU1076 cells were transfected with P-cadherin siRNAs or negative control siRNA in time-dependent manner. To examine the levels of various proteins, western blot analysis was conducted. Data were presented as mean ± SD of three independent experiments. .
Figure 5.
P-cadherin blockade activates c-Met/STAT3 pathway. (A, B) SNU1041 and SNU1076 cells were transfected with P-cadherin siRNAs or negative control siRNA for 48h. The levels of phospho-c-Met, total c-Met, P-cadherin, phospho-STAT3, total STAT3, phospho-Akt, total Akt, phospho-ERK were examined by Western blot analysis. (C, D) SNU1041 and SNU1076 cells were transfected with P-cadherin siRNAs or negative control siRNA in time-dependent manner. To examine the levels of various proteins, western blot analysis was conducted. Data were presented as mean ± SD of three independent experiments. .
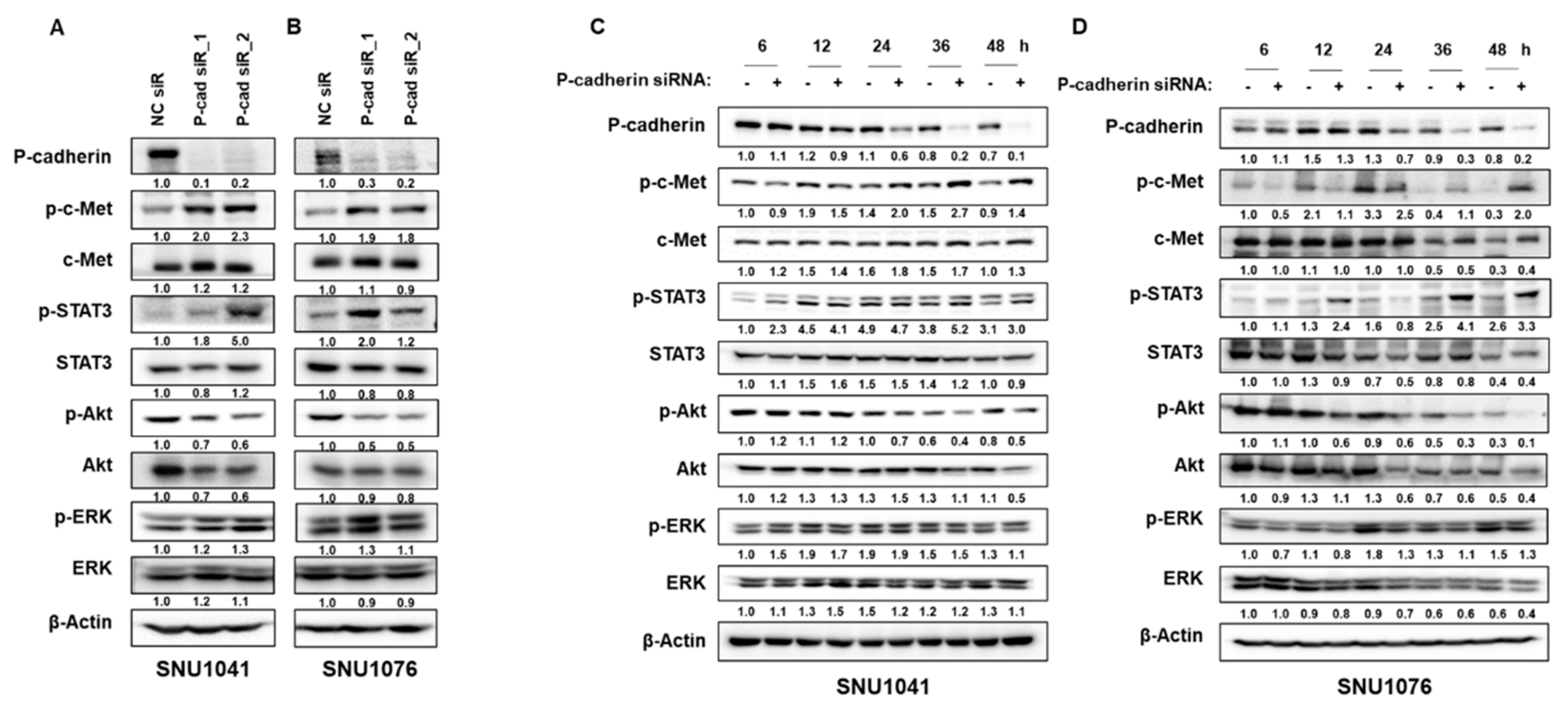
Figure 6.
Combination treatment of P-cadherin siRNA and c-Met inhibitor reduces head and neck cancer cell growth and migration. SNU1041 and SNU1076 cells were treated with siRNA targeting P-cadherin, SU11274 (5µM) or a combination of siRNA targeting P-cadherin and SU11274. (A, B) The expression levels of P-cadherin, phospho-c-Met, and c-Met were evaluated by western blot analysis. Cell proliferation assays (WST-1 assay) (C, D) and colony formation assays (E, F) were conducted. Cell migration (G, H) and invasion (I, J) assays were conducted. Data were presented as mean ± SD of three independent experiments. Differences were considered relevant at p < 0.05 (* p < 0.05, ** p < 0.01, *** p < 0.001).
Figure 6.
Combination treatment of P-cadherin siRNA and c-Met inhibitor reduces head and neck cancer cell growth and migration. SNU1041 and SNU1076 cells were treated with siRNA targeting P-cadherin, SU11274 (5µM) or a combination of siRNA targeting P-cadherin and SU11274. (A, B) The expression levels of P-cadherin, phospho-c-Met, and c-Met were evaluated by western blot analysis. Cell proliferation assays (WST-1 assay) (C, D) and colony formation assays (E, F) were conducted. Cell migration (G, H) and invasion (I, J) assays were conducted. Data were presented as mean ± SD of three independent experiments. Differences were considered relevant at p < 0.05 (* p < 0.05, ** p < 0.01, *** p < 0.001).
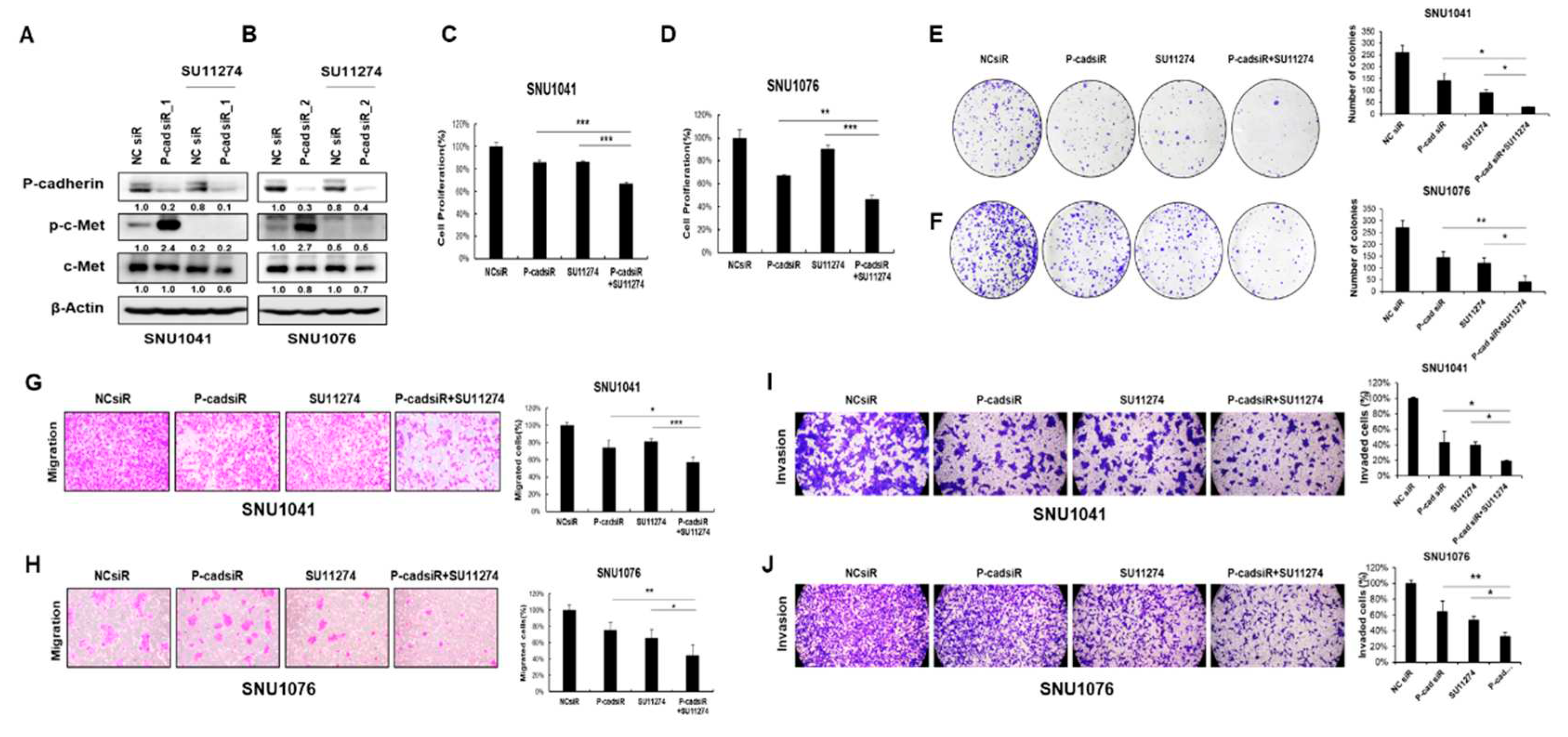
Figure 7.
Dual blockade of P-cadherin and c-Met inhibits cancer growth in a head and neck cancer xenograft model in vivo.Luciferase-expressing FaDu (FaDu-Luc) HNSCC cells were injected subcutaneously into BALB/c-nude mice and treated with siRNA targeting P-cadherin (100mg/kg), SU11274 (20mg/kg) or a combination of siRNA targeting P-cadherin and SU11274. (A) Final tumor images of cancer cells tracked with the IVIS imaging system following the injection of mice with FaDu-Luc cells. (B) The luminescence radiance of the ROI of the tumor was determined. (C) Final tumor images (D) Tumor volume was measured every 3 days for 18 days. (E) Representative images of H&E and immunohistochemical staining of P-cadherin. (F) The protein expression levels of P-cadherin, p-c-Met, and, c-Met in the xenograft tissues of each group were evaluated by western blot analysis. Differences were considered relevant at p < 0.05 (* p < 0.05, ** p < 0.01, *** p < 0.001).
Figure 7.
Dual blockade of P-cadherin and c-Met inhibits cancer growth in a head and neck cancer xenograft model in vivo.Luciferase-expressing FaDu (FaDu-Luc) HNSCC cells were injected subcutaneously into BALB/c-nude mice and treated with siRNA targeting P-cadherin (100mg/kg), SU11274 (20mg/kg) or a combination of siRNA targeting P-cadherin and SU11274. (A) Final tumor images of cancer cells tracked with the IVIS imaging system following the injection of mice with FaDu-Luc cells. (B) The luminescence radiance of the ROI of the tumor was determined. (C) Final tumor images (D) Tumor volume was measured every 3 days for 18 days. (E) Representative images of H&E and immunohistochemical staining of P-cadherin. (F) The protein expression levels of P-cadherin, p-c-Met, and, c-Met in the xenograft tissues of each group were evaluated by western blot analysis. Differences were considered relevant at p < 0.05 (* p < 0.05, ** p < 0.01, *** p < 0.001).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



