
Submitted:
27 October 2023
Posted:
31 October 2023
You are already at the latest version
Abstract
Valence tautomerism (VT) may occur if a molecule contains two chemically different redox-active units, which differ only slightly in their intrinsic redox potential. Herein, we present three new half-sandwich complexes [(η6arene)Cr(CO)2L]+ with a triarylmethylium substituent appended to the -coordinated arene and different coligands L (L = CO, P(OPh)3, PPh3, 1+ 3+) at the chromium atom. Ligand substitution purposefully lowers the half-wave potential for chromium oxidation and thereby the redox potential difference towards tritylium reduction. For the PPh3-substituted complex 3+, cyclic voltammetry measurements indicate that chromium oxidation and tritylium reduction occur at (almost) the same potential. This renders the diamagnetic Cr(0)C6H4CAr2+ form 3+ and its paramagnetic diradical Cr(I)+•C6H4CAr2• valence tautomer 3+•• energetically nearly degenerate. Temperature-dependent IR spectroscopy shows indeed two pairs of carbonyl bands assignable to a Cr(0) and a Cr(I) species, coexisting in a T-dependent equilibrium with almost equal quantities for both at 70 °C. The diradical form with one unpaired spin at the trityl unit engages in a monomer ⇌ dimer equilibrium, which was investigated by means of quantitative EPR spectroscopy. The diradical species 1+•• 3+•• were found to be highly reactive, leading to several identified reaction products, which presumably result from hydrogen atom abstraction by the trityl C atom, e.g. from the solvent.
Keywords:
valence tautomerism
; chromium half-sandwich complexes
; tritylium
; EPR spectroscopy
1. Introduction
Valence tautomerism (VT) is characterized by the coexistence of two (or more) redox isomers which differ in their charge and spin density distribution and hence the oxidation state assignment to the individual redox sites. VT may occur if a molecule contains two chemically different redox-active entities, which are oxidized, resp. reduced, at similar redox potentials [1]. Interconversion between individual valence tautomers occurs via an intramolecular electron transfer and can be induced by external triggers, e.g. irradiation with light [2,3,4,5] or X-ray [6,7], or variation of temperature [8,9,10,11,12]. This renders them stimuli-responsive.
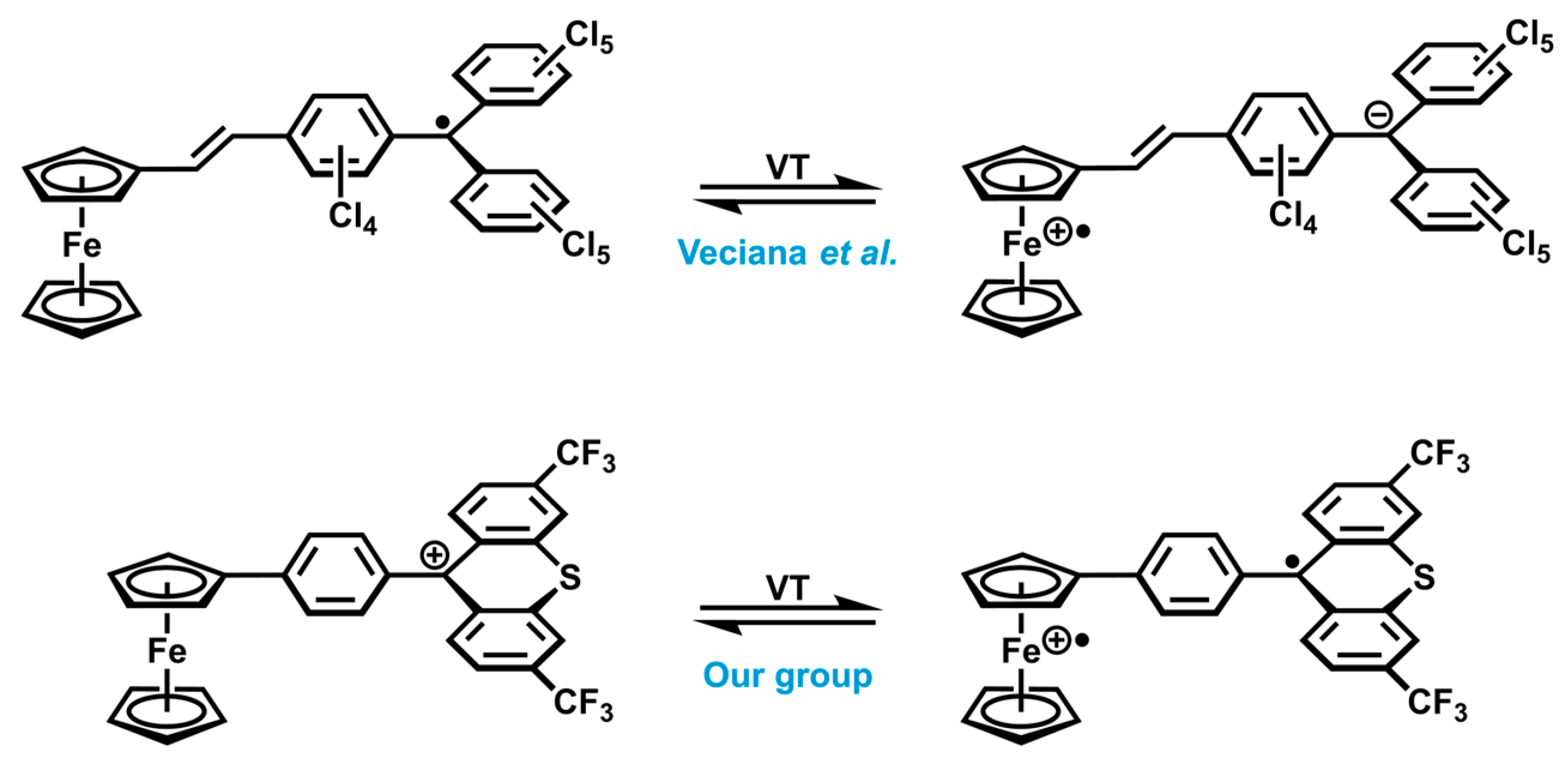
Several examples of metal complexes exhibiting VT are known to literature [1,13,14,15]. An early example of valence tautomerism occurring in sandwich complexes was provided by the group of Veciana in 2003 [12], when they presented a dyad comprising of a ferrocenyl building block and a perchlorotriphenylmethyl (PTM) radical as redox-active units, bridged by a vinylene linker. Thermally induced intramolecular electron transfer from the ferrocenyl unit to the PTM radical results in the zwitterionic ferrocenium-trityl complex shown at the top of Scheme 1. The valence tautomeric equilibrium was analyzed via T-dependent 57Fe-Mößbauer spectroscopy, showing both, Fe(II) and Fe(III) signals.
Trityl-type compounds CAr3n+ can not only exist as radicals CAr3• (n = 0) or anions CAr3− (n = -1), but also as the tritylium cations CAr3+ (n = +1) [17,18,19,20]. Attaching a suitable tritylium unit CAr3+ to ferrocene can therefore result in VT between a diamagnetic ferrocene-tritylium form and a paramagnetic ferrocenium-trityl diradical isomer (Scheme 1, bottom). This would render such complexes magnetochemical switches. While we have recently shown that VT can be purposefully implemented in ferrocene-tritylium complexes via tailoring the reduction potential of the tritylium unit [16,21], there are some inherent disadvantages of such dyads: (i) Altering the oxidation potential of the ferrocene unit (e.g. via methylation of the cyclopentadienide ligands) turned out to be synthetically challenging. (ii) Due to very fast electron spin relaxation, ferrocenium ions show extremely broad and low-intensity EPR signals, which are only observable at very low temperatures, usually at 10 K or below [22,23]. This made it very difficult for us to observe the EPR signatures of both types of radicals at the same time, since the diradical ferrocenium isomer is thermodynamically less stable [16]. We therefore sought for better alternatives for the ferrocenyl donor, which combines the treats of a facile adjustment of its oxidation potential and a more straightforward means to detect the actual oxidation state of the metal ion with the aid of molecular spectroscopy.
Examples of such candidates are chromium half-sandwich complexes [(η6-arene)Cr(CO)2L]. The simplest representative, [(η6-C6H6)Cr(CO)3], was first published by Fischer and Öfele in 1957 [24]. This was soon followed by a vast variety of other chromium half-sandwich complexes [25,26,27,28] and the development of more convenient synthetic approaches [29,30]. The herein proposed electron-rich [(η6-arene)Cr(CO)2L] complexes to be used as donors in such dyads have some advantages over the previously employed ferrocene. First, the oxidation potential of the chromium unit can be easily adjusted by substituting one CO ligand for a better σ-donor / weaker π-acceptor L [31]. This is demonstrated by the shift of the half-wave potential for chromium oxidation by -660 mV on substitution of one CO ligand in [(η6-C6H6)Cr(CO)3] for triphenylphosphine, PPh3 [32]. Secondly, CO ligands are excellent charge-sensitive labels for infrared (IR) spectroscopy. As the energies of the CO stretching vibrations of the Cr(0) and Cr(I) redox states differ by 100-150 cm-1 [31,32,33] and the CO band intensities vary only little with the chromium oxidation state [34], IR spectroscopy provides an easy handle for probing the valence tautomeric equilibrium. Thirdly, the chromium-centered radical is already EPR-active at 77 K [32,33,35], which is much more convenient as compared to the need for 10 K measurements for ferrocenium species.
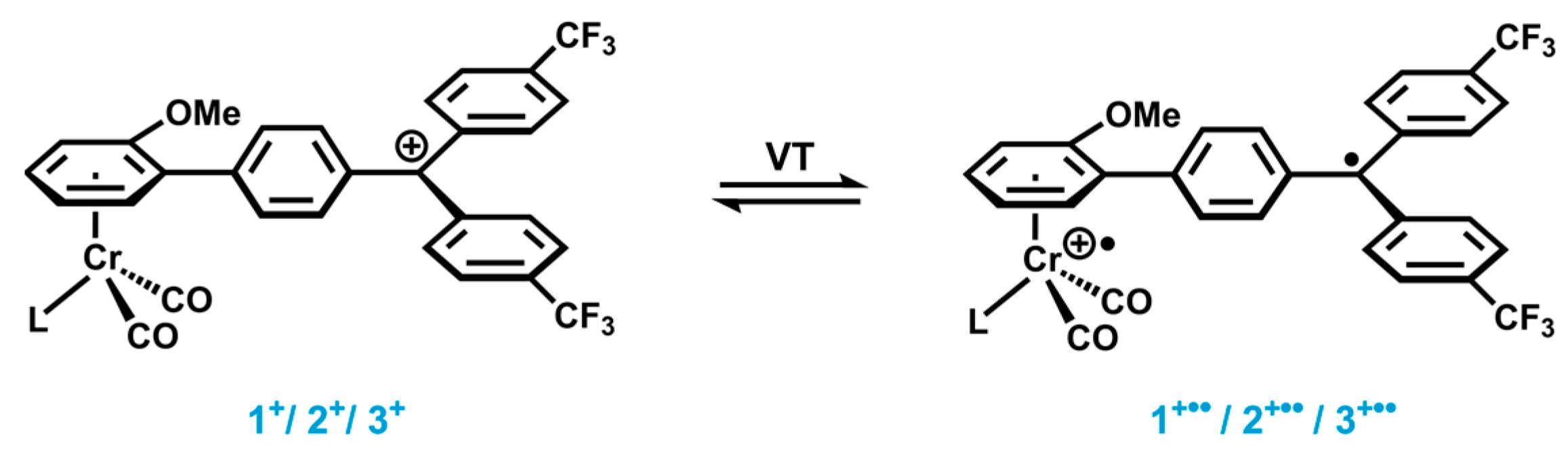
The main aim of this work is the purposeful implementation of VT into [(η6-arene)Cr(CO)2L]-triarylmethylium dyads via tuning the oxidation potential of the metal unit. The valence tautomeric equilibrium of the envisioned chromium half-sandwich complexes is designed to occur between a diamagnetic Cr(0)-C6H4-CAr2+ species and a paramagnetic Cr(I)+•-C6H4-CAr2• species (cf. Scheme 2).
2. Results and Discussion
2.1. Synthesis and Characterization
We prepared three chromium half-sandwich complexes with a different coligand L at the chromium atom, where a tritylium unit is attached to the π-coordinated anisole ring via a phenylene spacer. Variation in L is intended to adjust the redox potential difference between chromium oxidation and tritylium reduction and hence the free enthalpy difference ΔG between the two valence tautomers. In order to maintain the reduction potential of the tritylium unit in a viable range, we implemented electron-withdrawing trifluoromethyl groups in the para-positions of the remaining phenyl rings (see Scheme 2).
Complexes 1+ - 3+ were synthesized through the four- resp. five-step procedures shown in Scheme 3. [(η6-anisole)Cr(CO)3] was synthesized according to the literature [30] and converted to biaryl ester 1′ by Murahashi coupling with methyl-4-iodobenzoate [36]. Nucleophilic addition of two equivalents of 4-trifluromethylphenyl lithium to the ester functionality of 1′ resulted in the formation of the carbinol 1-OH. Photochemical ligand substitution [37,38] of one carbonyl ligand by either triphenylphosphite, P(OPh)3, or triphenylphosphine, PPh3, yielded precursors 2-OH and 3-OH. Due to the general light sensitivity of [(η6-arene)Cr(CO)3] complexes, the yields of the carbinol precursors were only moderate, ranging from 18 to 76%. The carbinols were characterized by NMR and IR spectroscopy as well as by mass spectrometry (MS). The corresponding spectra are provided as Figures S1–S15 in the Supplementary Materials. Subsequent protolytic dehydration via addition of Brookhart’s acid [H(OEt2)2]+ [B{C6H3(CF3)2-3,5}4]− [39], denoted henceforth as HBArF24, yielded the tritylium complexes 1+ - 3+. Although the very weakly nucleophilic [B{C6H3(CF3)2-3,5}4]– ([BArF24]−) counterion endowed ferrocene-tritylium complexes with sufficient stability to allow for the recording of their NMR spectra and their handling at r.t. [40,41,42], complexes 1+ - 3+ start to decompose already at -40 °C or even below. This precluded us from characterizing them by NMR spectroscopy and mass spectrometry. It was nevertheless possible to identify and investigate 1+ - 3+ by means of low-temperature IR and EPR spectroscopy as well as by cyclic voltammetry (CV). As will be shown later, their instability is mainly due to the high reactivity of the diradical redox isomers 1+•• - 3+••.
Single crystals of the biaryl precursor complex 1′ and of carbinol 3-OH suitable for X-ray crystallography were obtained by vapour diffusion of n-pentane into a solution of complex 1′ in dichloromethane or a diethylether solution of 3-OH. 1′ crystallizes in the triclinic space group P and 3-OH in the monoclinic space group P21/c as an ether disolvate, both as racemic mixtures of Sp and Rp enantiomers. Details to the data collection, structure solution and refinement are collected in Tables S2 and S3 of the Supplementary Materials. Table 1 compares the most important bond lengths and angles. A drawing of the molecular structure of complex 1′ can be found as Figure S16 of the Supplementary Materials. The molecular structure of the Sp enantiomer of carbinol 3-OH is displayed in Figure 1. The pseudo-tetrahedral [(η6-arene)Cr(CO)2(PPh3)] fragment in 3-OH adopts an eclipsed conformation, in which the PPh3 ligand is in anti-position with respect to the carbinol substituent. For 1′, the conformation is only nearly eclipsed. With a torsion angle of 17.4(4) (1′) and 5.6(4)° (3-OH), respectively, the methoxy substituent resides in the plane of the phenyl ring so as to improve its electron-donating capabilities towards the metal-coordinated arene ring, just as it was observed for other alkoxy-substituted chromium half-sandwich complexes [43,44,45,46,47,48]. The torsion angle between the planes of the coordinated anisole ring (PhCr) and the phenylene spacer (Ph1) is 53.63(17) and 52.55(14)°, respectively, similar to other [(biaryl)Cr(CO)3] complexes bearing an ortho-substituent [49]. Ring torsion between the coordinated π-perimeter and the linker is thus substantially larger than in [(η5-C5H5)Fe(η5-C5H4-C6H4)-C(OH)Ar2] complexes lacking an ortho-substituent at the biaryl connection, where they range from 12.3(3) to 24.19(12)° [42]. Enhanced ring torsion is expected to result in a more efficient decoupling of the two units, which itself is beneficial for achieving electronic bistability.
Both complexes show interesting packing motifs in the crystal lattice. As is shown in Figures S17b,c of the Supplementary Materials, the different enantiomers of 1′ (cf. Figure S17a) align in an antiparallel fashion as tail-to-tail dimers. They interact via pairwise C-H⋅⋅⋅O hydrogen bonds between methyl protons of the ester substituent and the O atom of a carbonyl ligand and the methoxy substituent of the coordinated benzene ring as well as a C-H⋅⋅⋅π interaction to the phenylene linker. The dimers pack in rows along the b axis of the unit cell with pairwise C-H⋅⋅⋅π interactions between η6-bonded anisole rings, while complexes with the same optical configuration align along the cell a axis via C-H⋅⋅⋅O contacts between a proton of the phenylene linker and the ester carbonyl.
In the case of 3-OH, the carbinol hydroxyl group forms an O-H⋅⋅⋅O hydrogen bond of 2.053(3) Å to one of the ether solvate molecules, whereas the other ether molecule occupies voids in the crystal lattice without forming any short contacts. In the crystal lattice, Rp and Sp enantiomers of 3-OH align alternately to chains that run along the b axis of the unit cell. These chains are held together by three short C-H⋅⋅⋅O hydrogen bonds of 2.528(2), 2.528(2) and 2.502(2) Å between one proton of the coordinated benzene ring and the PPh3 ligand each as well as a methoxy proton to the O atoms of the two CO ligands of each molecule (cf. Figure S18a in the Supplementary Materials). Along the c axis, pairs of Rp and Sp enantiomers associate via two weak C-H⋅⋅⋅π interactions between the phenylene spacer and the anisole ring as well as by five strong C-H⋅⋅⋅F and C-H⋅⋅⋅π hydrogen bonds that involve the methylene and methyl protons of an ethyl group of ether solvent molecules (cf. Figure S18b of the Supplementary Materials). As shown in Figure S18c, the molecular packing creates fluorous layers formed by the CF3 substituents along the bc plane of the unit cell.
2.2. Electrochemistry
The electrochemical properties of 1-OH - 3-OH were investigated in the very weakly nucleophilic electrolyte CH2Cl2/ 0.1 M [NBu4]+ [BArF24]−, which have not only the capability to stabilize the resulting tritylium complexes [40,41,42], but also increase the chemical reversibility of the oxidation in arene chromium half-sandwich complexes [32,50]. The cyclic voltammograms of the carbinol precursors 1-OH - 3-OH are displayed in Figure 2a and the respective data are listed in Table 2. All carbinols undergo a chemically (peak current ratios ip,a/ip,c ca. 1) and electrochemically (peak potential separations ΔEp = 68 - 81 mV) reversible one-electron oxidation of the chromium nucleus. The half-wave potential E1/2 of tricarbonyl complex 1-OH of 360 mV is nearly identical to that of [(η6-anisole)Cr(CO)3] (E1/2 = 350 mV) [51,52]. Substitution of one carbonyl ligand by P(OPh)3 lowers E1/2 by 250 mV, which matches with the data for [(estradiol)Cr(CO)2L] (L = CO, P(OPh)3) [32]. In keeping with the stronger donating properties and the weaker π-acidity of the PPh3 ligand, the half-wave potential is further lowered to -265 mV in 3-OH, consistent with the difference in Lever’s electrochemical ligand parameters EL [53]. The choice of ligand L thus varies E1/2 over a range of more than 600 mV.
For all further studies, including cyclic voltammetry, the cationic tritylium complexes were generated in situ from their carbinol precursors by treatment with an equimolar amount of Brookhart´s acid, which led to an instantaneous change of color from yellow to green or from bright to dark orange, respectively. As shown in Figure 2b, the cyclic voltammogram of 1+ exhibits two main redox waves, one for chromium oxidation at E1/2 = 750 mV, and one at E1/2 = 175 mV for tritylium reduction. Compared to the carbinol precursor, the oxidation potential is shifted by 390 mV to higher potential, which is due to the influence of the neighboring strongly electron-accepting triarylmethylium unit. The reduction potential of the tritylium unit resembles that of the uncoordinated ligand C6H4(2-OMe)-C6H4-C+{-C6H4(4-CF3)}2 (L+) (E1/2 = 135 mV). The half-wave potential difference ΔE1/2 between chromium oxidation and tritylium reduction thus amounts to 575 mV, which translates into a free-enthalpy difference ΔG of 55.5 kJ mol-1 for the two valence tautomers (cf. Scheme 2). In between these waves is located a third redox process, which is associated with smaller peak currents. Closer inspection of this wave shows that the peak current on the reverse scan, i.e. after traversing the oxidation wave of 1+, surmounts that of the forward (anodic) peak. This suggests that this wave belongs to a product arising from chemical decomposition which already ensues in the parent, cationic state, but occurs at an even faster rate after chromium oxidation. This particular wave could be assigned to the triphenylmethane-substituted complex 1-H (E1/2 = 435 mV) with a proton instead of the hydroxyl substituent of 1-OH (vide infra, Scheme 5).
The cyclic voltammogram of 2+ at room temperature shows two main redox processes which might be due to chromium oxidation and tritylium reduction of this complex (cf. Figure 2b). While the reduction wave is nearly unshifted with respect to 1+, the potential of the oxidation wave is lowered by ca. 300 mV, similar to the difference for carbinols 1-OH and 2-OH. Owing to the extreme thermosensitivity of this complex and the close similarity of the potentials of these waves to those of identified decomposition products 1-H and L+ (vide infra), the above assignment is however only tentative. An extreme tendency of complex 2+ to adsorb to any electrode surface (Au, Pt, glassy carbon) unfortunately prevented us from conducting CV measurements at low T.
Due to the inherent instability of 3+, we recorded its cyclic voltammograms at 195 K (cf. Figure 2b). To our surprise, we observed only one redox wave at -180 mV instead of the expected separate waves for metal oxidation and tritylium reduction. Intriguingly, the open circuit potential of 3+ (cf. Figure 2c, marked with an arrow) falls within the redox wave. This indicates a curious situation, where the half-wave potentials of both processes are so close that the two waves merge into a single, composite redox wave, corresponding to a half-wave potential difference of (almost) 0 mV. This means that the two possible valence tautomers are energetically degenerate or nearly so, which renders 3+ a particularly promising candidate for observing valence tautomerism.
2.3. IR Spectroscopy
Table 3 compiles the CO bands of all complexes of this study. For both, the carbinol complexes and their tritylium congeners, the increased electron density at the chromium atom on replacing one CO ligand by the weaker π-acceptors P(OPh)3 or PPh3 also manifest in their IR spectra (cf. Figure S15 of the Supplementary Materials) [31]. Hence, the average CO stretching frequency is red-shifted by 40 (2-OH) or 72 cm-1 (3-OH) with respect to 1-OH.
As expected from the large ΔE1/2 and ΔG, in situ generated 1+ shows only one pair of CO bands assignable to the diamagnetic Cr(0)-C6H4-CAr2+ valence tautomer within the accessible temperature range of -70 (Figure 3a) to -40 °C (cf. Figure S19 of the Supplementary Materials). The identical CO stretching energies of 1-OH and 1+ may look suspicious. We however note that ring substituents exert only minor effects on the CO bands in [(η6-arene)Cr(CO)3] complexes [55] and that cyclic voltammograms of 1+ did not show any remains of 1-OH under these conditions. Above -40 °C decomposition ensues as indicated by the emergence of new CO bands, which could later be assigned to specific follow products (vide infra). In the case of 2+, the IR spectrum shows a second set of carbonyl bands of considerably weaker intensity, which is characteristically shifted by 113 cm-1 to higher energies, as expected for a Cr(I) species (cf. Table 3 and Figure S20 of the Supplementary Materials). This hints at the presence of small amounts of the diradical valence tautomer. The rapid onset of decomposition already at -70 °C unfortunately precluded us from studying any temperature dependence of the equilibrium between the two isomers.
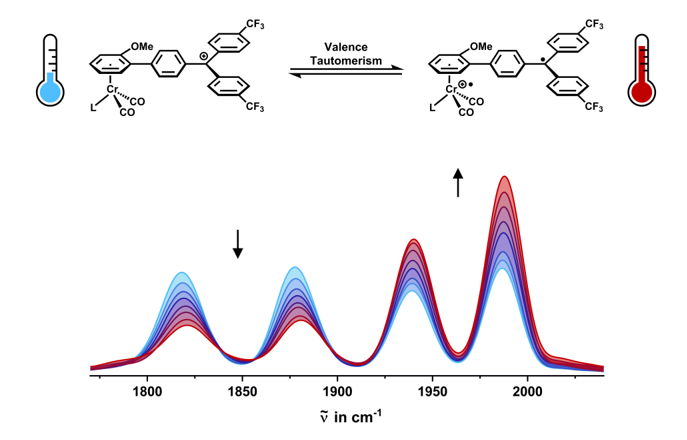
The IR spectroscopic data of 3+ are particularly interesting, as they show two pairs of CO bands immediately after addition of Brookhart’s acid to the carbinol precursor at -70 °C (Figure 3b). The energy difference of 117 cm-1 between the average CO values of the two pairs of bands strongly hints at the simultaneous presence of a Cr(0) and a Cr(I) species [31,32,33]. Increasing the temperature in steps of 5 °C, from -75 to -40 °C, resulted in an intensity decrease of the band pair at lower energies at the expense of the band pair at higher energies (Figure 3c). Upon re-cooling to -70 °C, the spectral changes were (almost) entirely reversible (cf. Figure S21 of the Supplementary Materials), in agreement with a T-dependent equilibrium. Therefore, the carbonyl bands at 1988 and 1940 cm-1 are tentatively assigned to the paramagnetic valence tautomer 3+•• with a positively charged Cr(I) unit, and the bands at 1878 and 1819 cm-1 to the diamagnetic valence tautomer with the positive charge at the triarylmethylium residue (cf. Scheme 2).
From the areas of the CO bands of each pair, we can even deduce the ratio of the quantities of the two valence tautomers of 3+ within the accessible temperature range as dictated from the limits of our cryostat and the onset of decomposition at T = -40 °C (cf. Figure S22 of the Supplementary Materials). In our analysis we assume that the oscillator strengths of the CO bands are identical for the Cr(I) and Cr(0) oxidation state, which is supported by literature [34]. Our finding that the total area over all carbonyl bands remained constant during the T-dependent measurements supports this notion and defies any significant decomposition during our measurements (cf. Figure S23 of the Supplementary Materials). The near degeneracy of the two isomers, which was already indicated by the coincident half-wave potentials for chromium oxidation and tritylium reduction, is thus duly reflected in the nearly identical intensities and integrals of both pairs of bands at -70 °C, corresponding to a ratio of the individual valence tautomers of ca. 1:1. The ratio of the isomers translates into the equilibrium constant K, from which ΔG can be calculated via the relationship ΔG = -RT⋅ln(K). When plotting our experimentally derived ΔG values as a function of T, we obtained an excellent linear fit (R2 = 0.9943), which allows us to determine ΔH = 15.8 ± 0.5 kJ mol-1 and ΔS = 77 ± 2 J (mol⋅K)-1 (cf. Figure S24 of the Supplementary Materials). We must however note that this analysis is compromised by partial dimerization of the diradical isomer 3+••, so that the thermodynamic parameters pertain to the combination of these two processes.
2.4. EPR Spectroscopy
Next, we analyzed 1+ and 3+ via EPR spectroscopy (complex 2+ proved too unstable to obtain reliable EPR data). The sheer existence of an EPR signal already hints at the presence of the diradical valence tautomer, since the Cr(0)-C6H4-CAr2+ isomer is EPR-silent. As is depicted in Figure S25a of the Supplementary Materials, the EPR spectrum of 1+, recorded at -70 °C, shows a weak isotropic signal with a g-value of 2.005, characteristic for a trityl radical [56,57], with resolved hyperfine splittings to the hydrogen and fluorine atoms in close proximity. Splitting constants and the g-value were obtained from a simulation using the EasySpin program [58]. The observability of the EPR signal of 1+•• demonstrates the superior sensitivity of this method as compared to IR spectroscopy, where no traces of this second valence tautomer were found. In agreement with the notion that the diradical isomer is entropically favored, the EPR signal intensity increases reversibly with increasing temperature (cf. Figure S25b-c of the Supplementary Materials), opposing the otherwise expected Boltzmann behavior [59]. Owing to the overall weak signal intensity, we were however only able to detect the organic, but not the metal-centered spin (cf. Figure S25d of the Supplementary Materials).

Table 4.
EPR data of the diradical valence tautomers 1+•• and 3+•• at different temperatures.
| T in K | Metal-centered spin | Organic spin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| g1 | g2 | g3 | A1 | A2 | A3 | giso | A | ||
| 1+ | 77 | -[a] | 2.003 | -[b] | |||||
| 203 | - | 2.005 | 5.4 (4H), 1.4 (2H), 2.7 (6F), 1.4 (1H) | ||||||
| 3+ | 77 | 2.095 | 2.031 | 1.991 | 34.6 (31P) | 33.9 (31P) | 27.8 (31P) | 2.003 | -[b] |
| 203 | - | 2.003 | 5.1 (4H), 1.4 (2H), 2.7 (6F), 1.4 (1H) | ||||||
Hyperfine splitting constants A are provided in Gauss [G] with the number of equivalent nuclei in parentheses as taken from simulations. [a] No metal-centered spin could be detected. [b] No hyperfine splitting resolved.
Investigations into the valence tautomeric equilibrium of 3+ at temperatures between -90 and -40 °C (fluid solution) and at 77 K (frozen solution) proved to be even more rewarding. Selected EPR spectra are depicted in Figure 4. At -70 °C, samples of 3+ provide an isotropic EPR signal with a g-value of 2.003, which can be ascribed to the organic spin of a trityl species (Figure 4a). Hyperfine couplings to hydrogen and fluorine atoms in close proximity are partially resolved and were modeled by digital simulation of the EPR spectrum at -70 °C. Upon gradually warming of the sample from -90 to -40 °C (Figure 4b), the signal intensity increases, which again opposes the expected Boltzmann behavior [59]. This finding agrees with a higher proportion of the paramagnetic form at higher temperatures. We however note that diradical 3+•• engages in a T-dependent monomer ⇌ dimer equilibrium, which also contributes to the intensity increase at higher T (vide infra). Changes induced by temperature variation were reversed on re-cooling the sample as long as the temperature was kept below -40 °C (cf. Figure S26 of the Supplementary Materials).
In frozen CH2Cl2, at 77 K, the EPR spectrum shows an additional rhombic signal with hyperfine coupling of all g-tensor components to one phosphorus atom (Figure 4c) side by side with that of the trityl-type radical. g-Values and hyperfine splitting constants are very similar to those reported for other oxidized [(η6-arene)Cr(CO)2(PPh3)]+ complexes [34,35]. The simultaneous presence of both kinds of EPR resonances in the same spectrum provides further compelling evidence for the coexisting diradical 3+••.
The high signal intensity already hints at substantial amounts of the diradical valence tautomer 3+••. In order to determine its amount, we conducted quantitative EPR measurements with spin counting at -70 °C. Scheme 4 illustrates the spin counting method as detailed in reference [42]. First, a sample of 3+ of known concentration was freshly prepared at -40 °C and rapidly cooled to -70 °C inside the cavity of the EPR spectrometer. An EPR spectrum with predefined parameters and instrument settings was recorded (cf. Experimental Section for details). The signal was then doubly integrated in order to relate to the spin concentration within the sample (step (1) in Scheme 4; note that the EPR signature is the first derivative of the pristine signal). The double integral (DI) of the sample was then compared to a regression line taken from EPR spectra of the sTable 2,2-diphenyl-1-picrylhydrazyl radical (dpph•) as a calibration standard [60], which were recorded at different concentrations, and at the same temperature as those of 3+ (step (2)). Assuming that dpph• is inert towards dimerization, even at low temperature, we can thereby determine the absolute spin concentration of the sample and compare it to the nominal concentration (step (3)). Our analysis yielded an amount of ca. 18% of the free diradical valence tautomer 3+•• in samples of 3+. The lower spin concentration as compared to the IR data at the same T, which indicated nearly equimolar amounts of both valence tautomers (vide supra), point to partial dimerization of the paramagnetic valence tautomer via the formation of a new C-C bond between the trityl C atoms. The same behavior was already documented for related ferrocene-tritylium complexes [42]. Dimerization results in the loss of the organic spin, while maintaining the oxidation state at the chromium center. Therefore, the pair of CO bands at 1988 and 1940 cm-1 are likely caused by both, the diradical valence tautomer 3+•• and its dimer 3+•-3+•.
2.5. Decomposition Pathways
Although the tritylium complexes were prepared and handled under strictly anaerobic and anhydrous conditions, decomposition commenced at temperatures above -40 °C (1+, 3+), or even -70 °C (2+). Following this process by means of IR and NMR spectroscopy allowed us to identify four decomposition products, some of them even analyzed by NMR and IR spectroscopy, ESI-MS, single crystal X-ray analysis and cyclic voltammetry. Relevant data are provided in Figures S27-S35 in the Supplementary Materials.
Scheme 5.
Identified decomposition products of tritylium complex 3+.
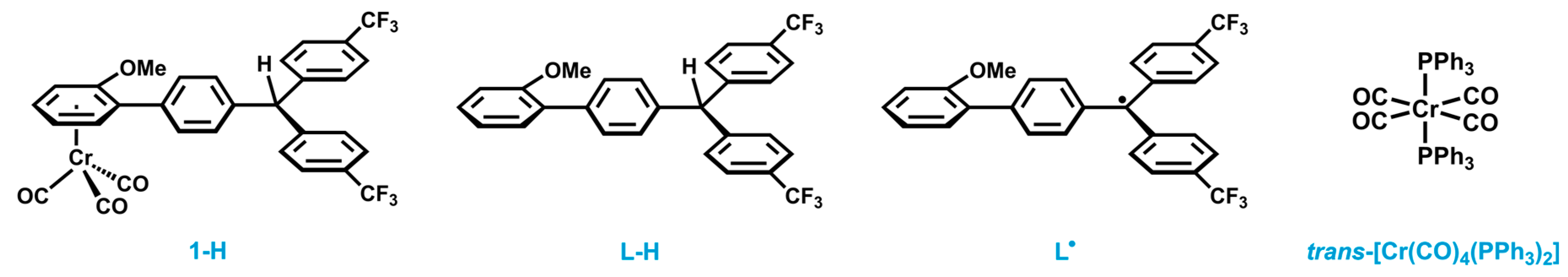
Tritylium complex 1+ proved to be the most stable out of the three in this study. We assume that this is due to the presence of only very minor amounts of the highly reactive diradical valence tautomer 1+••. Still, 1+ decomposes within several hours to chromium half-sandwich complex 1-H and minor amounts of the free ligand L-H, as revealed by the NMR spectra in Figure 5. The formation of 1-H was also confirmed by IR spectroscopy (cf. Figure S36 of the Supplementary Materials). The availability of an authentic sample of 1-H also allowed us to assign the additional redox wave observed in cyclic voltammograms of 1+ as arising from this species (vide supra). 1-H and the decoordinated, free ligand L-H putatively result from the diradical valence tautomer Cr(I)+•-C6H4-CAr2• (1+••) by hydrogen atom abstraction from the solvent. This would provide a Cr(I) species 1-H+•, which is gradually reduced. L-H then results from 1-H by loss of the arene ligand, a phenomenon, that we frequently observed during every synthesis of the chromium half-sandwich complexes in this work, or by hydrogen atom abstraction proceeding from L•.
Surprisingly, tritylium complexes 2+ and 3+ also form tricarbonyl complex 1-H and ligand L-H during their decomposition (cf. NMR and IR spectra depicted in Figures S36–S40 of the Supplementary Materials). This indicates that PR3 and CO ligands are scrambled between the metal fragments during this process. In this vein, we also isolated a single crystal of trans-[Cr(CO)4(PPh3)2] from a solution of decomposed 3+. Following the decomposition via IR spectroscopy (cf. Figure S38), hints at the intermediate formation of trans-[Cr(CO)4(PPh3)2]+ [61], which is gradually reduced to form trans-[Cr(CO)4(PPh3)2] [62].
Single crystals of 1-H and L-H as well as the crystal of trans-[Cr(CO)4(PPh3)2] were obtained by diffusion of n-pentane into a dichloromethane solution of 3+ at r.t. The molecular structures of 1-H and L-H are depicted in Figure 6, while Tables S4 and S5 of the Supplementary Materials provide the details of the data collection, structure solution and refinement. The Cr(CO)3 fragment in complex 1-H adopts an almost eclipsed conformation (cf. Figure 6, left), as it is typical of [(η6-anisole)Cr(CO)3] complexes [63]. As depicted in Figure S41 of the Supplementary Materials, Rp/Rp and Sp/Sp enantiomers interact via short C-H⋅⋅⋅O contacts of 2.672(8) and 2.798(7) Å between a proton of the coordinated anisole ring and the O atoms of two carbonyl ligands as well as pairwise C-H⋅⋅⋅O hydrogen bonds to carbonyl ligands involving the CH proton of the triarylmethane residue and a proton of the phenylene linker. The free ligand L-H (cf. Figure 6, right) exhibits a smaller torsion at the biphenyl linkage (Table 1). Individual molecules of L-H associate via several C-H⋅⋅⋅O and C-H⋅⋅⋅F hydrogen bonds in the crystal lattice (see Figure S42 of the Supplementary Materials).
4. Summary and Conclusions
We have reported on three new chromium half-sandwich triarylmethylium dyads [Cr(CO)2L(η6-C6H4(2-OMe)-C6H4-C+{-C6H4(4-CF3)}2] (1+ - 3+) with different coligands L (L = CO, P(OPh)3, PPh3) at the chromium atom. Cyclic voltammetry data of their carbinol precursors 1-OH - 3-OH indicate that variation in L shifts the chromium oxidation potential over a range > 600 mV. These compounds, symbolized as Cr(0)-C6H4-CAr2+, were investigated with respect to their propensity to coexist with their paramagnetic redox isomers (valence tautomers) Cr(I)+•-C6H4-CAr2•. The latter result from intramolecular electron transfer from the Cr(0) donor to the tritylium-type acceptor unit.
The potential difference of 575 mV between chromium-based oxidation and tritylium-based reduction in 1+, which corresponds to ΔG = 55.5 kJ mol-1, is too large to allow for significant quantities of the diradical valence tautomer. For complex 3+ however, the oxidation and reduction waves overlap, indicating that the two isomers are nearly degenerate. Indeed, the IR spectrum of in situ generated 3+ shows two pairs of carbonyl bands, separated by 117 cm-1, which is supportive of the simultaneous presence of a Cr(0)-C6H4-CAr2+ and a Cr(I)+•-C6H4-CAr2• isomer. As was shown by IR spectroscopy, the equilibrium between the two valence tautomers is T-dependent with higher proportions of the thermodynamically less stable, but entropically favored diradical isomer present at higher T. The spectral changes are reversible as long as T is kept below -40 °C, where decomposition sets in. Quantitative EPR spectroscopy suggests that, at -70 °C, ca. two out of three molecules of the paramagnetic isomer are trapped in a dimer 3+•-3+•. In frozen solution, the typical EPR signature of the half-sandwich Cr(I) entity was observed together with the isotropic signal of the trityl-type radical, confirming the simultaneous presence of both types of paramagnetic centers.
While the near degeneracy of the diamagnetic and diradical isomer in 3+ constitutes an ideal situation for magnetoswitching, i.e. the thermoresponsive and reversible alteration of magnetic properties, its low thermal stability poses a serious obstacle to practical applications. 3+ and its congeners 1+ and 2+ decompose even at low temperatures and under inert gas conditions. We were able to identify the triarylmethane-appended complex 1-H and the respective free ligand L-H as the main products generated during this process. This hints at the decisive role of the diradical valence tautomers 1+•• - 3+•• from which 1-H is putatively formed by H atom abstraction and subsequent decoordination of the ligand L-H. This concurs with the observation that 1+, which generates the smallest quantities of the paramagnetic isomer, is the most stable complex of the three. Since 1-H and L-H were also identified as arising from decomposition of complexes 2+ and 3+, their decomposition must ensue with exchange of CO and PR3 ligands.
Our results nevertheless show that valence tautomerism can be purposefully implemented into chromium half-sandwich triarylmethylium complexes by tailoring the oxidation potential of the metal unit via ligand substitution. The presence of CO ligands at the metal ion and the detectability of the EPR signal of the paramagnetic metal entity under well-accessible conditions provide highly convenient handles for an in-depth analysis of such systems. In particular, they alleviate studies of the valence tautomeric equilibria when compared to related ferrocenyl systems. Further improvements might be achieved by steric shielding of the trityl unit in order to thwart dimerization and taming the reactive trityl site through enhanced spin density delocalization. This will be the subject of further forays in our laboratories.
5. Materials and Methods
Experimental Methods and Materials. All syntheses were carried out under a nitrogen atmosphere using standard Schlenk and Glovebox techniques. Solvents were taken from a solvent purification system (SPS). All reagents were purchased from commercial sources and used without further purification. [NBu4]+ [B{C6H3(CF3)2-3,5}4]− [64,65], Brookhart’s acid [39] and complex 1′ [36] were prepared according to published procedures. Ligand substitution was conducted photochemically by UV irradiation using a high-pressure mercury lamp and a Duran glass irradiation apparatus [38,66].
NMR Spectroscopy. 1H-NMR (400/600/800 MHz), 19F{1H}-NMR (376/752 MHz), 31P{1H}-NMR (162 MHz) and 13C{1H}-NMR (101/151/202 MHz) spectra were recorded in CD2Cl2 at 300 K on a Bruker Avance III 400, Bruker Avance III 600 or Bruker Avance Neo 800 spectrometer. NMR spectra were referenced to residual solvent signals (1H) resp. the solvent signal itself (13C). 19F- and 31P-NMR spectra were referenced to the external reference of the spectrometer (δ(19F/31P) = 0 ppm for CFCl3, or H3PO4, respectively). Assignment of the signals is based on 2D spectra. Mass Spectrometry. Mass spectra were recorded in the positive mode on an ESI-calibrated LTQ Orbitrap Velos Spectrometer with direct injection of dichloromethane solutions. IR Spectroscopy. Room temperature IR spectra were recorded in dichloromethane on a Bruker Tensor II FT-IR spectrometer. Low-temperature IR spectra of the tritylium complexes were recorded inside a nitrogen filled glovebox on a Nicolet iS10 spectrometer with the aid of a Remspec fiber-optic transmission IR probe. The custom-build setup was already described in an earlier publication [16]. It is composed of two cups with 5 mL capacity each, made of thermally conducting Al2O3 (one for the reference, one for the sample), which can be switched automatically. Cooling was done with a cryostat (-40 °C) with thermal connection to a cascading Peltier cooler inside the glovebox, which allows for cooling to -75 °C (the temperature is measured directly at the Al vials). The IR spectra were baseline-corrected. Thermodynamic Analysis. The pairs of carbonyl bands in T-dependent IR spectra of 3+ were integrated separately for both valence tautomers. Assuming that the band intensities are invariant to the chromium oxidation state allowed us to calculate the equilibrium constant K from the ratio of the peak areas for the paramagnetic and the diamagnetic valence tautomers. This was done for every temperature in the range of -70 to -40 °C. Next, the corresponding ΔG values were calculated according to the equation ΔG = -RT⋅ln(K) for every data point. The resulting plot of ΔG against T is depicted in Figure S24 in the Supplementary Materials. ΔH was taken from the intercept, and ΔS from the slope. X-Ray Crystallography. A STOE IPDS-II image plate diffractometer equipped with a Mo-Kα or Cu-Kα radiation source was used. Data acquisition was conducted at 100 K. The program package X-Area was used for data processing. Depending on the structure, either semiempirical or spherical absorption corrections were performed. The structure was solved and refined with SHELXT [67,68] and OLEX2 [69]. All non-hydrogen atoms were refined anisotropically. EPR Spectroscopy. EPR samples of the cationic complexes were freshly prepared inside a nitrogen-filled glovebox, using glassware and dichloromethane that were precooled to -40 °C. Prior to filling, the EPR tube was inserted into suitable boring in an Al block that was precooled to -40°C to serve as cold reservoir in order to prevent decomposition. T-dependent EPR spectra were recorded in dichloromethane on a X-band benchtop spectrometer MiniScope MS5000. Quantitative EPR measurements of the complexes and the dpph• standard were recorded with defined measurement parameters (B = 330-345 mT, modulation = 0.3 mT, power = 6.3096 mW, sweep time 60 s) to ensure comparability [42]. EPR spectra in frozen solution were conducted at 77 K on a X-band ELEXSYS E580 spectrometer. Simulation of the EPR spectra was performed with the Matlab software package EasySpin using the ‘garlic’ resp. ‘pepper’ functions [58]. Cyclic Voltammetry. Cyclic voltammetry measurements were conducted under argon atmosphere at room temperature (1-OH - 3-OH, 1+, 2+) or at -78 °C (3+). The cyclic voltammograms were recorded in dry dichloromethane using the very weakly nucleophilic electrolyte [NBu4]+ [B{C6H3(CF3)2-3,5}4]−. A platinum counter electrode, an Ag/AgCl (pseudo)reference electrode, and a glassy carbon working electrode were used. The working electrode was polished with first 1 μm and then 0.25 μm diamond pastes prior to every measurement. Data acquisition was conducted with a computer-controlled BASi potentiostat. After completion of the measurements, ferrocene resp. decamethylferrocene was added as an internal reference and the measurements were repeated in the presence of the standard. The cyclic voltammograms were then referenced to the Cp2Fe0/+ redox couple with Cp*2Fe0/+ = -540 mV.
Synthesis of 1-OH. To a solution of 4-bromo-1-trifluoromethylbenzene (0.23 mL, 1.65 mmol) in 20 mL THF was slowly added n-butyllithium (2.5 M in hexane, 0.73 mL, 1.82 mmol) at -78 °C. The solution was stirred for 2 h at -78 °C and [(biaryl ester)Cr(CO)3] 1′ (250 mg, 0.66 mmol) dissolved in 10 mL THF was added dropwise. The reaction mixture was allowed to warm to room temperature overnight and subsequently quenched by addition of water (10 mL). The phases were separated and the aqueous phase was extracted with diethylether (3 x 20 mL). The combined organic phases were washed with water (20 mL) and brine (20 mL), dried over Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified via column chromatography (silica gel, n-pentane/ethyl acetate 3:1 - 2:1) to give yellow 1-OH in 76% yield (321 mg, 0.50 mmol). For atom numbering, see Figure S1 of the Supplementary Materials. 1H-NMR (400 MHz, CD2Cl2) δ [ppm] = 7.64 (d, 3JHH = 8.4 Hz, 4H, H-15), 7.54 (d, 3JHH = 8.7 Hz, 2H, H-9), 7.51 (d, 3JHH = 8.4 Hz, 4H, H-14), 7.26 (d, 3JHH = 8.7 Hz, 2H, H-10), 5.82 (dd, 3JHH = 6.3 Hz, 4JHH = 1.4 Hz, 1H, H-6), 5.69 (ddd, 3JHH = 6.8, 6.3 Hz, 4JHH = 1.4 Hz, 1H, H-4), 5.20 (dd, 3JHH = 6.8 Hz, 4JHH = 0.8 Hz, 1H, H-3), 5.03 (vt, 3JHH = 6.3 Hz, 4JHH = 0.8 Hz, 1H, H-5), 3.75 (s, 3H, H-1), 3.01 (s, 1H, OH). 19F{1H}-NMR (376 MHz, CD2Cl2) δ [ppm] = -62.8 (s, CF3).13C{1H}-NMR (101 MHz, CD2Cl2) δ [ppm] = 233.6 (CO), 150.3 (C-13), 146.0 (C-11), 142.3 (C-2), 134.7 (C-8), 130.7 (C-9), 130.0 (q, 2JCF = 33 Hz, C-16), 128.6 (C-14), 127.9 (C-10), 125.6 (q, 3JCF = 4 Hz, C-15), 124.6 (q, 1JCF = 272 Hz, C-17), 101.1 (C-7), 99.2 (C-6), 95.6 (C-4), 85.4 (C-5), 81.7 (C-12), 74.3 (C-3), 56.3 (C-1). IR (CH2Cl2) [cm-1] = 1965, 1886. ESI-MS calculated for C31H20CrF6O5+: m/z = 485.1335 (M+ -CrCO3 -OH), 621.0587 (M+ -OH), 638.0615 (M+), found: 485.1345, 621.0592, 638.0591 (measured in CH2Cl2, direct injection, calibrated).
Ligand Substitution (2-OH). Complex 1-OH (50 mg, 0.08 mmol) was dissolved in tetrahydrofuran (30 mL) and irradiated under IR monitoring. Subsequently, triphenylphosphite (24 mg, 0.08 mmol), dissolved in 5 mL tetrahydrofuran, was added and the reaction mixture was stirred for 30 min. The solution was evaporated to dryness and the mixture was separated via column chromatography (silica gel, n-pentane/diethylether 2:1) giving 2-OH as light orange solid in 18% yield (13 mg, 14 μmol). For atom numbering, see Figure S5 of the Supplementary Materials. 1H-NMR (400 MHz, CD2Cl2) δ [ppm] = 7.62 (d, 3JHH = 8.3 Hz, 4H, H-15), 7.50 (m, 6H, H-9+H-14), 7.40-7.30 (m, 14H, H-19+H-20), 7.21-7.14 (m, 5H, H-10+H-21), 5.40 (d, 3JHH = 6.1 Hz, 1H, H-6), 5.23 (dt, 3JHH = 6.7, 6.1 Hz, 1H, H-4), 3.87 (d, 3JHH = 6.7 Hz, 1H, H-3), 3.52 (vt, 3JHH = 6.1 Hz, 1H, H-5), 3.25 (s, 3H, H-1), 3.03 (s, 1H, OH). 13C{1H}-NMR (101 MHz, CD2Cl2) δ [ppm] = 236.5 (d, 2JCP = 34 Hz, CO-a), 236.3 (d, 2JCP = 31 Hz, CO-b), 152.8 (d, 2JCP = 6 Hz, C-18), 150.3 (C-13), 145.4 (C-11), 140.6 (C-2), 135.8 (C-8), 130.7 (C-9), 130.0 (C-20), 129.9 (q, 2JCF = 32 Hz, C-16), 128.6 (C-14), 127.7 (C-10), 125.5 (q, 3JCF = 4 Hz, C-15), 124.7 (C-21), 124.6 (q, 1JCF = 272 Hz, C-17), 122.2 (d, 3JCP = 4 Hz, C-19), 96.9 (C-6), 96.1 (C-7), 92.6 (C-4), 82.7 (C-5), 81.7 (C-12), 71.4 (C-3), 55.6 (C-1). 19F{1H}-NMR (376 MHz, CD2Cl2) δ [ppm] = -62.9 (s, CF3). 31P{1H}-NMR (162 MHz, CD2Cl2) δ [ppm] = 202.1 (s, P(OPh)3). IR (CH2Cl2) [cm-1] = 1910, 1853. ESI-MS calculated for C48H35CrF6O7P+: m/z = 920.1424 (M+), found: 920.1385 (measured in CH2Cl2, direct injection, calibrated).
Ligand Substitution (3-OH). Complex 1-OH (110 mg, 0.17 mmol) and triphenylphosphine (68 mg, 0.26 mmol) were dissolved in toluene (30 mL) and irradiated under IR monitoring. Subsequently, the solution was evaporated to dryness and the mixture was separated via column chromatography (silica gel, n-pentane/diethylether 2:1) to give 3-OH as dark orange solid in 69% yield (103 mg, 0.12 mmol). A crystal suitable for single crystal X-ray diffraction was obtained via vapor diffusion of n-pentane into a diethylether solution. X-ray crystallographic data is provided in the Supplementary Materials. For atom numbering, see Figure S10 of the Supplementary Materials. 1H-NMR (400 MHz, CD2Cl2) δ [ppm] = 7.64 (d, 3JHH = 8.3 Hz, 4H, H-15), 7.63 (d, 3JHH = 8.5 Hz, 2H, H-9), 7.54 (d, 3JHH = 8.3 Hz, 4H, H-14), 7.52-7.47 (m, 6H, H-19), 7.36-7.33 (m, 9H, H-20+H-21), 7.23 (d, 3JHH = 8.5 Hz, 2H, H-10), 5.38 (d, 3JHH = 6.0 Hz, 1H, H-6), 4.76 (vq, 3JHH = 6.3 Hz, 1H, H-4), 4.58 (vt, 3JHH = 6.0 Hz, 1H, H-5), 3.93 (d, 3JHH = 6.6 Hz, 1H, H-3), 3.51 (s, 3H, H-1), 3.02 (s, 1H, OH). 19F{1H}-NMR (376 MHz, CD2Cl2) δ [ppm] = -62.8 (s, CF3). 31P{1H}-NMR (162 MHz, CD2Cl2) δ [ppm] = 90.9 (s, PPh3).13C{1H}-NMR (101 MHz, CD2Cl2) δ [ppm] = 241.9 (d, 2JCP = 21 Hz, CO-a), 240.4 (d, 2JCP = 20 Hz, CO-b), 150.4 (C-13), 145.1 (C-11), 139.8 (d, 1JCP = 34 Hz, C-18), 139.6 (C-2), 136.8 (C-8), 133.3 (d, 2JCP = 11 Hz, C-19), 130.8 (C-9), 129.9 (q, 2JCF = 33 Hz, C-16), 129.4 (d, 4JCP = 2 Hz, C-21), 128.7 (C-14), 128.3 (d, 3JCP = 9 Hz, C-20), 127.7 (C-10), 125.5 (q, 3JCF = 4 Hz, C-15), 124.6 (q, 1JCF = 272 Hz, C-17), 94.1 (C-4), 93.9 (C-6), 93.2 (C-7), 84.9 (C-5), 81.7 (C-12), 73.6 (C-3), 55.8 (C-1). IR (CH2Cl2) [cm-1] = 1880, 1821. ESI-MS calculated for C48H35CrF6O4P+: m/z = 872.1577 (M+), found: 872.1608 (measured in CH2Cl2, direct injection, calibrated).
Synthesis of L-OH. Irradiation (λ = 465 nm) of 1-OH (60 mg, 94 μmol) in dichloromethane (10 mL) and subsequent purification via column chromatography (silica gel, n-pentane/dichloromethane 1:1) provided L-OH as white solid in 75% yield (35 mg, 69 μmol). 1H-NMR (400 MHz, CD2Cl2) δ [ppm] = 7.63 (d, 3JHH = 8.4 Hz, 4H, H-15), 7.53 (d, 3JHH = 8.4 Hz, 4H, H-14), 7.52 (d, 3JHH = 8.6 Hz, 2H, H-9), 7.36-7.30 (m, 2H, H-4+H-6), 7.26 (d, 3JHH = 8.6 Hz, 2H, H-10), 7.03 (vt, 3JHH = 7.5 Hz, 1H, H-5), 7.01 (d, 3JHH = 8.3 Hz, 1H, H-3), 3.80 (s, 3H, H-1), 3.02 (s, 1H, OH). 19F{1H}-NMR (376 MHz, CD2Cl2) δ [ppm] = -62.8 (s, CF3). 13C{1H}-NMR (101 MHz, CD2Cl2) δ [ppm] = 156.9 (C-2), 150.5 (C-13), 144.4 (C-11), 138.9 (C-8), 131.1 (C-6), 130.0 (C-7), 129.9 (C-4), 129.9 (q, 2JCF = 33 Hz, C-16), 129.4 (C-9), 128.7 (C-14), 127.8 (C-10), 125.5 (q, 3JCF = 4 Hz, C-15), 124.6 (q, 1JCF = 272 Hz, C-17), 121.2 (C-5), 111.7 (C-3), 81.8 (C-12), 55.8 (C-1). ESI-MS calculated for C28H20F6O2: m/z = 485.1335 (M+ -OH), found: 485.1331 (measured in CH2Cl2, direct injection, calibrated). Representations of the spectra can be found as Figures S43 to S46 of the Supplementary Materials.
Synthesis of Tritylium Complexes 1+ - 3+, L+. To the respective carbinol (1-OH - 3-OH, L-OH) in dichloromethane was added an equimolar amount of Brookhart’s acid at -70 °C (IR, for 1-OH - 3-OH), -78 °C (CV, for 3-OH) resp. r.t. (CV, for 1-OH, 2-OH, L-OH) or -40 °C (EPR, only for 1+ and 3+).
1-H and L-H. Decomposition products 1-H and L-H were isolated from the decomposition of 3+ in CD2Cl2 at room temperature via column chromatography (silica gel, n-pentane/ethyl acetate 4:1). 1-H: For atom numbering, see Figure S27 of the Supplementary Materials. 1H-NMR (600 MHz, CD2Cl2) δ [ppm] = 7.60 (d, 3JHH = 8.0 Hz, 4H, H-15), 7.51 (d, 3JHH = 8.0 Hz, 2H, H-9), 7.30 (d, 3JHH = 8.0 Hz, 4H, H-14), 7.12 (d, 3JHH = 8.0 Hz, 2H, H-10), 5.81 (d, 3JHH = 6.1 Hz, 1H, H-6), 5.71 (s, 1H, H-12), 5.68 (vt, 3JHH = 6.5 Hz, 1H, H-4), 5.20 (d, 3JHH = 6.7 Hz, 1H, H-3), 5.03 (vt, 3JHH = 6.1 Hz, 1H, H-5), 3.75 (s, 3H, H-1). 19F{1H}-NMR (376 MHz, CD2Cl2) δ [ppm] = -62.8 (s, CF3). 13C{1H}-NMR (151 MHz, CD2Cl2) δ [ppm] = 233.7 (CO), 147.3 (C-13), 142.7 (C-11), 142.2 (C-2), 133.6 (C-8), 131.0 (C-9), 130.2 (C-14), 129.3 (C-10), 129.3 (q, 2JCF = 32 Hz, C-16), 125.9 (q, 3JCF = 4 Hz, C-15), 124.7 (q, 1JCF = 270 Hz, C-17), 101.5 (C-7), 99.2 (C-6), 95.6 (C-4), 85.4 (C-5), 74.3 (C-3), 56.5 (C-12), 56.3 (C-1). IR (CH2Cl2) [cm-1] = 1880, 1821. ESI-MS calculated for C31H20CrF6O4+: m/z = 468.1413 (M+ -CrCO3), 623.0744 (M+) found: 486.1412, 623.0697 (measured in CH2Cl2, direct injection, calibrated). X-ray crystallographic data is provided in the Supplementary Materials. L-H: For atom numbering, see Figure S32 of the Supplementary Materials. 1H-NMR (800 MHz, CD2Cl2) δ [ppm] = 7.59 (d, 3JHH = 8.3 Hz, 4H, H-15), 7.49 (d, 3JHH = 8.3 Hz, 2H, H-9), 7.34-7.28 (m, 5H, H-4+H-14), 7.30 (d, 3JHH = 7.5 Hz, 1H, H-6), 7.13 (d, 3JHH = 8.3 Hz, 2H, H-10), 7.02 (vt, 3JHH = 7.5 Hz, 1H, H-5), 7.00 (d, 3JHH = 8.3 Hz, 1H, H-3), 5.71 (s, 1H, H-12), 3.80 (s, 3H, H-1). 19F{1H}-NMR (752 MHz, CD2Cl2) δ [ppm] = -62.7 (s, CF3). 13C{1H}-NMR (202 MHz, CD2Cl2) δ [ppm] = 156.9 (C-2), 147.7 (C-13), 141.0 (C-11), 137.8 (C-8), 131.0 (C-6), 130.2 (C-7+C-14+C-9), 129.3 (C-10), 129.2 (C-4), 129.1 (q, 2JCF = 32 Hz, C-16), 125.8 (q, 3JCF = 4 Hz, C-15), 124.7 (q, 3JCF = 272 Hz, C-17), 121.2 (C-5), 111.6 (C-3), 56.5 (C-12), 55.8 (C-1). ESI-MS calculated for C28H20F6O2+: m/z = 486.1413 (M+), found: 486.1410 (measured in CH2Cl2, direct injection, calibrated). X-ray crystallographic data is provided in the Supplementary Materials.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org: (to be inserted) characterization data (NMR, IR and mass spectra), crystal data for 1′, 3-OH, 1-H, L-H, additional NMR, IR and EPR spectra for cations 1+ - 3+. CCDC 2303541 (1-H), 2303542 (L-H), 2303543 (1′) and 2303544 (3-OH) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
Author Contributions
Conceptualization, A.R. and R.F.W.; methodology, A.R., M.L., M.A.; crystal structure analysis, M.L.; investigation, A.R., M.L. and M.A.; resources, R.F.W. and M.D.; data curation, A.R. and M.L.; writing – original draft preparation, A.R. and R.F.W.; writing – review and editing, A.R. and R.F.W.; visualization, A.R.; supervision, R.F.W., M.L.; project administration, R.F.W.; funding acquisition, R.F.W., M.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Data are contained within the article or in the Supplementary Materials.
Acknowledgments
We thank Malin Bein for ESI-MS and Anke Friemel from the NMR core facility for NMR spectroscopic measurements and Kai Jellinek for his aid with the synthesis of carbinols 1-OH and 3-OH.
Conflicts of Interest
The authors declare no conflict of interest.
Dedication
This work is dedicated to Prof. Otto J. Scherer, a full-blood, enthusiastic chemist and inspiring mentor, at the occasion of his 90th birthday.
References
- Pierpont, C.G. Studies on charge distribution and valence tautomerism in transition metal complexes of catecholate and semiquinonate ligands. Coord. Chem. Rev. 2001, 216, 99–125. [Google Scholar] [CrossRef]
- Adams, D.M.; Li, B.; Simon, J.D.; Hendrickson, D.N. Photoinduced Valence Tautomerism in Cobalt Complexes Containing Semiquinone Anion as Ligand: Dynamics of the High-Spin [CoII(3,5-dtbsq)2] to Low-Spin [CoIII(3,5-dtbsq)(3,5-dtbcat)] Interconversion. Angew. Chem. Int. Ed. 1995, 34, 1481–1483. [Google Scholar] [CrossRef]
- Adams, D.M.; Hendrickson, D.N. Pulsed Laser Photolysis and Thermodynamics Studies of Intramolecular Electron Transfer in Valence Tautomeric Cobalt o-Quinone Complexes. J. Am. Chem. Soc. 1996, 118, 11515–11528. [Google Scholar] [CrossRef]
- Sato, O.; Cui, A.; Matsuda, R.; Tao, J.; Hayami, S. Photo-induced Valence Tautomerism in Co Complexes. Acc. Chem. Res. 2007, 40, 361–369. [Google Scholar] [CrossRef]
- Tao, J.; Maruyama, H.; Sato, O. Valence Tautomeric Transitions with Thermal Hysteresis around Room Temperature and Photoinduced Effects Observed in a Cobalt−Tetraoxolene Complex. J. Am. Chem. Soc. 2006, 128, 1790–1791. [Google Scholar] [CrossRef] [PubMed]
- Francisco, T.M.; Gee, W.J.; Shepherd, H.J.; Warren, M.R.; Shultz, D.A.; Raithby, P.R.; Pinheiro, C.B. Hard X-ray-Induced Valence Tautomeric Interconversion in Cobalt-o-Dioxolene Complexes. J. Phys. Chem. Lett. 2017, 8, 4774–4778. [Google Scholar] [CrossRef]
- Poneti, G.; Mannini, M.; Sorace, L.; Sainctavit, P.; Arrio, M.-A.; Otero, E.; Criginski Cezar, J.; Dei, A. Soft-X-ray-Induced Redox Isomerism in a Cobalt Dioxolene Complex. Angew. Chem. Int. Ed. 2010, 49, 1954–1957. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, R.M.; Pierpont, C.G. Tautomeric Catecholate-Semiquinone Interconversion via Metal-Ligand Electron Transfer. Structural, Spectral, and Magnetic Properties of (3,5-Di-tert-butylcatecholato)-(3,5-di-tert-butylsemiquinone)(bipyridyl)cobalt(III), a Complex Containing Mixed-Valence Organic Ligands. J. Am. Chem. Soc. 1980, 102, 4951–4957. [Google Scholar] [CrossRef]
- Caneschi, A.; Dei, A.; Fabrizi de Biani, F.; Gütlich, P.; Ksenofontov, V.; Levchenko, G.; Hoefer, A.; Renz, F. Pressure- and Temperature-Induced Valence Tautomeric Interconversion in a o-Dioxolene Adduct of a Cobalt-Tetraazamacrocycle Complex. Chem. Eur. J. 2001, 7, 3926–3930. [Google Scholar] [CrossRef]
- Carbonera, C.; Dei, A.; Létard, J.-F.; Sangregorio, C.; Sorace, L. Thermally and Light-Induced Valence Tautomeric Transition in a Dinuclear Cobalt–Tetraoxolene Complex. Angew. Chem. Int. Ed. 2004, 43, 3136–3138. [Google Scholar] [CrossRef]
- Leroy, L.; Francisco, T.M.; Shepherd, H.J.; Warren, M.R.; Saunders, L.K.; Shultz, D.A.; Raithby, P.R.; Pinheiro, C.B. Controlled Light and Temperature Induced Valence Tautomerism in a Cobalt-o-Dioxolene Complex. Inorg. Chem. 2021, 60, 8665–8671. [Google Scholar] [CrossRef]
- Ratera, I.; Ruiz-Molina, D.; Renz, F.; Ensling, J.; Wurst, K.; Rovira, C.; Gütlich, P.; Veciana, J. A New Valence Tautomerism Example in an Electroactive Ferrocene Substituted Triphenylmethyl Radical. J. Am. Chem. Soc. 2003, 125, 1462–1463. [Google Scholar] [CrossRef]
- Evangelio, E.; Ruiz-Molina, D. Valence Tautomerism: New Challenges for Electroactive Ligands. Eur. J. Inorg. Chem. 2005, 2005, 2957–2971. [Google Scholar] [CrossRef]
- Evangelio, E.; Ruiz-Molina, D. Valence tautomerism: More actors than just electroactive ligands and metal ions. C. R. Chim. 2008, 11, 1137–1154. [Google Scholar] [CrossRef]
- Tezgerevska, T.; Alley, K.G.; Boskovic, C. Valence tautomerism in metal complexes: Stimulated and reversible intramolecular electron transfer between metal centers and organic ligands. Coord. Chem. Rev. 2014, 268, 23–40. [Google Scholar] [CrossRef]
- Casper, L.A.; Linseis, M.; Demeshko, S.; Azarkh, M.; Drescher, M.; Winter, R.F. Tailoring Valence Tautomerism by Using Redox Potentials: Studies on Ferrocene-Based Triarylmethylium Dyes with Electron-Poor Fluorenylium and Thioxanthylium Acceptors. Chem. Eur. J. 2021, 27, 10854–10868. [Google Scholar] [CrossRef] [PubMed]
- Arnett, E.M.; Flowers, R.A.; Ludwig, R.T.; Meekhof, A.E.; Walek, S.A. Triarylmethanes and 9-arylxanthenes as prototypes amphihydric compounds for relating the stabilities of cations, anions and radicals by C-H bond cleavage and electron transfer. J. Phys. Org. Chem. 1997, 10, 499–513. [Google Scholar] [CrossRef]
- Cheng, J.; Handoo, K.L.; Parker, V.D. Hydride affinities of carbenium ions in acetonitrile and dimethyl sulfoxide solution. J. Am. Chem. Soc. 1993, 115, 2655–2660. [Google Scholar] [CrossRef]
- Breslow, R.; Mazur, S. Electrochemical determination of pKR+ for some antiaromatic cyclopentadienyl cations. J. Am. Chem. Soc. 1973, 95, 584–585. [Google Scholar] [CrossRef]
- Strohbusch, F. Polarographische Untersuchungen der Substituenteneffekte in Triarylmethylkationen. Ber. Bunsenges. Phys. Chem. 1972, 76, 622–628. [Google Scholar] [CrossRef]
- Nau, M.; Casper, L.A.; Haug, G.; Linseis, M.; Demeshko, S.; Winter, R.F. Linker permethylation as a means to foster valence tautomerism and thwart dimerization in ferrocenyl-triarylmethylium cations. Dalton Trans. 2023, 52, 4674–4677. [Google Scholar] [CrossRef] [PubMed]
- Prins, R. Electronic structure of the ferricenium cation. Mol. Phys. 1970, 19, 603–620. [Google Scholar] [CrossRef]
- Prins, R.; Reinders, F.J. Electron spin resonance of the cation of ferrocene. J. Am. Chem. Soc. 1969, 91, 4929–4931. [Google Scholar] [CrossRef]
- Fischer, E.O.; Öfele, K. Über Aromatenkomplexe von Metallen, XIII Benzol-Chrom-Tricarbonyl. Chem. Ber. 1957, 90, 2532–2535. [Google Scholar] [CrossRef]
- Berger, A.; Djukic, J.-P.; Michon, C. Metalated (η6-arene)tricarbonylchromium complexes in organometallic chemistry. Coord. Chem. Rev. 2002, 225, 215–238. [Google Scholar] [CrossRef]
- Rosillo, M.; Domínguez, G.; Pérez-Castells, J. Chromium arene complexes in organic synthesis. Chem. Soc. Rev. 2007, 36, 1589–1604. [Google Scholar] [CrossRef] [PubMed]
- Semmelhack, M.F. Transition Metal Arene Complexes: Nucleophilic Addition. In Comprehensive Organometallic Chemistry II; Elsevier, 1995; pp. 979–1015. ISBN 9780080465197. [Google Scholar]
- Nicholls, B.; Whiting, M.C. The Organic Chemistry of the Transition Elements. Part I. Tricarbonylchromium Derivatives of Aromatic Compounds. J. Chem. Soc. 1959, 551–556. [Google Scholar] [CrossRef]
- Strohmeier, W. Eine verbesserte Darstellung von Aromaten- und Cycloheptatrien-Chromtricarbonylen. Chem. Ber. 1961, 94, 2490–2493. [Google Scholar] [CrossRef]
- Mahaffy, C.A.L.; Pauson, P.L.; Rausch, M.D.; Lee, W. (η6-Arene)Tricarbonylchromium Complexes. In Inorganic Syntheses; John Wiley & Sons, Ltd, 1979; pp. 154–158. ISBN 9780470132500. [Google Scholar]
- Connelly, N.G.; Demidowicz, Z.; Kelly, R.L. Reactions of tricarbonyl(η-hexamethylbenzene)chromium derivatives with nitrosonium and benzenediazonium ions: reversible oxidation versus nitrosyl- or areneazo-complex formation. J. Chem. Soc., Dalton Trans. 1975, 2335–2340. [Google Scholar] [CrossRef]
- Ohrenberg, N.C.; Paradee, L.M.; DeWitte, R.J.; Chong, D.; Geiger, W.E. Spectra and Synthetic-Time-Scale Substitution Reactions of Electrochemically Produced [Cr(CO)3(η6-arene)]+ Complexes. Organometallics 2010, 29, 3179–3186. [Google Scholar] [CrossRef]
- van Order Jr, N.; Geiger, W.E.; Bitterwolf, T.E.; Rheingold, A.L. Mixed-valent cations of dinuclear chromium arene complexes: electrochemical, spectroscopic, and structural considerations. J. Am. Chem. Soc. 1987, 109, 5680–5690. [Google Scholar] [CrossRef]
- Pierce, D.T.; Geiger, W.E. Mixed-valent interactions in rigid dinuclear systems: electrochemical and spectroscopic studies of CrICr0 ions with controlled torsion of the biphenyl bridge. Inorg. Chem. 1994, 33, 373–381. [Google Scholar] [CrossRef]
- Castellani, M.P.; Connelly, N.G.; Pike, R.D.; Rieger, A.L.; Rieger, P.H. EPR Spectra of [Cr(CO)2L(η-C6Me6)]+ (L= PEt3, PPh3, P(OEt)3, P(OPh)3): Analysis of Line Widths and Determination of Ground State Configuration from Interpretation of 31P Couplings. Organometallics 1997, 16, 4369–4376. [Google Scholar] [CrossRef]
- Momoi, Y.; Okano, K.; Tokuyama, H. Generation of Aryl Grignard Reagents from Arene Chromium Tricarbonyl Complexes by Mg(TMP)2·2LiCl and Their Application to Murahashi Coupling. Synlett 2014, 25, 2503–2507. [Google Scholar] [CrossRef]
- Strohmeier, W.; Hellmann, H. Photochemisch hergestellte Derivate von Aromatenchromtricarbonylen und ihre Stabilität als Funktion der Substituenten am Benzolring. Chem. Ber. 1963, 96, 2859–2866. [Google Scholar] [CrossRef]
- Cais, M.; Kaftory, M.; Kohn, D.H.; Tatarsky, D. Structural and Catalytic Activity Studies on Phosphine- and Phosphite-Dicarbonylchromium Complexes of Phenanthrene and Naphthalene. J. Organomet. Chem. 1980, 184, 103–112. [Google Scholar] [CrossRef]
- Brookhart, M.; Grant, B.; Volpe Jr., A.F. [(3,5-(CF3)2C6H3)4B]-[H(OEt2)2]+: A Convenient Reagent for Generation and Stabilization of Cationic, Highly Electrophilic Organometallic Complexes. Organometallics 1992, 11, 3920–3922. [Google Scholar] [CrossRef]
- Oßwald, S.; Casper, L.A.; Anders, P.; Schiebel, E.; Demeshko, S.; Winter, R.F. Electrochemical, Spectroelectrochemical, Mößbauer, and EPR Spectroscopic Studies on Ferrocenyl-Substituted Tritylium Dyes. Chem. Eur. J. 2018, 24, 12524–12538. [Google Scholar] [CrossRef]
- Casper, L.A.; Oßwald, S.; Anders, P.; Rosenbaum, L.-C.; Winter, R.F. Extremely Electron-Poor Bis(diarylmethylium)-Substituted Ferrocenes and the First Peroxoferrocenophane. Z. Anorg. Allg. Chem. 2020, 646, 712–725. [Google Scholar] [CrossRef]
- Casper, L.A.; Wursthorn, L.; Geppert, M.; Roser, P.; Linseis, M.; Drescher, M.; Winter, R.F. 4-Ferrocenylphenyl-Substituted Tritylium Dyes with Open and Interlinked C+Ar2 Entities: Redox Behavior, Electrochromism, and a Quantitative Study of the Dimerization of Their Neutral Radicals. Organometallics 2020, 39, 3275–3289. [Google Scholar] [CrossRef]
- Tan, Y.-L.; White, A.J.P.; Widdowson, D.A.; Wilhelm, R.; Williams, D.J. Dilithiation of arenetricarbonylchromium(0) complexes with enantioselective quench: application to chiral biaryl synthesis. J. Chem. Soc., Perkin Trans. 2001, 3269–3280. [Google Scholar] [CrossRef]
- Uemura, M.; Nishikawa, N.; Take, K.; Ohnishi, M.; Hirotsu, K.; Higuchi, T.; Hayashi, Y. Arene-metal complex in organic synthesis: directed regioselective lithiation of (π-substituted benzene)chromium tricarbonyl complexes. J. Org. Chem. 1983, 48, 2349–2356. [Google Scholar] [CrossRef]
- Carter, O.L.; McPhail, A.T.; Sim, G.A. Metal–carbonyl and metal–nitrosyl complexes. Part II. Crystal and molecular structure of the tricarbonylchromiumanisole–1,3,5-trinitrobenzene complex. J. Chem. Soc. A 1966, 822–838. [Google Scholar] [CrossRef]
- Alberico, E.; Braun, W.; Calmuschi-Cula, B.; Englert, U.; Salzer, A.; Totev, D. Expanding the Range of “Daniphos”-Type P∩P- and P∩N-Ligands: Synthesis and Structural Characterisation of New [(η6-arene)Cr(CO)3] Complexes. Eur. J. Inorg. Chem. 2007, 2007, 4923–4945. [Google Scholar] [CrossRef]
- Cambie, R.C.; Clark, G.R.; Gourdie, A.C.; Rutledge, P.S.; Woodgate, P.D. Synthesis and crystal structures of the α and β stereoisomers of tricarbonyl[(8,9,11,12,13,14-η)-methyl 12-methoxypodocarpa-8,11,13-trien-19-oate]chromium. J. Organomet. Chem. 1985, 297, 177–184. [Google Scholar] [CrossRef]
- Gilday, J.P.; Widdowson, D.A. Lithiation of 2-, 3-, and 4-fluoroanisole(tricarbonyl)chromium(0) complexes: a reversal of normal regiocontrol. J. Chem. Soc., Chem. Commun. 1986, 1235–1237. [Google Scholar] [CrossRef]
- Batuecas, M.; Luo, J.; Gergelitsová, I.; Krämer, K.; Whitaker, D.; Vitorica-Yrezabal, I.J.; Larrosa, I. Catalytic Asymmetric C-H Arylation of (η6-Arene)Chromium Complexes: Facile Access to Planar-Chiral Phosphines. ACS Catal. 2019, 9, 5268–5278. [Google Scholar] [CrossRef]
- Camire, N.; Nafady, A.; Geiger, W.E. Characterization and Reactions of Previously Elusive 17-Electron Cations: Electrochemical Oxidations of (C6H6)Cr(CO)3 and (C5H5)Co(CO)2 in the Presence of [B(C6F5)4]-. J. Am. Chem. Soc. 2002, 124, 7260–7261. [Google Scholar] [CrossRef]
- Allen, D. Hunter; Vivian Mozol; Stanislaus D. Tsai. Nonlinear Substituent Interactions and the Electron Richness of Substituted (η6-Arene)Cr(CO)3 Complexes as Measured by IR and 13C NMR Spectroscopy and Cyclic Voltammetry: Role of π-Donor and π-Acceptor Interactions. Organometallics 1992, 11, 2251–2262. [Google Scholar]
- Connelly, N.G.; Geiger, W.E. Chemical redox agents for organometallic chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef]
- Lever, A.B.P. Electrochemical Parametrization of Metal Complex Redox Potentials, Using the Ruthenium(III)/Ruthenium(II) Couple to Generate a Ligand Electrochemical Series. Inorg. Chem. 1990, 29, 1271–1285. [Google Scholar] [CrossRef]
- Richardson, D.E.; Taube, H. Determination of E20-E10 in multistep charge transfer by stationary-electrode pulse and cyclic voltammetry: application to binuclear ruthenium ammines. Inorg. Chem. 1981, 20, 1278–1285. [Google Scholar] [CrossRef]
- Mahaffy, C.A.L.; Pauson, P.L.; Rausch, M.D.; Lee, W. (η6-Arene)Tricarbonylchromium Complexes. In Reagents for transition metal complex and organometallic syntheses; Angelici, R.J., Basolo, F., Eds.; Wiley: New York, 1990; pp. 136–140. ISBN 9780470132593. [Google Scholar]
- Trapp, C.; Wang, C.-S.; Filler, R. Electron Spin Resonance Absorption in the Tris(pentafluorophenyl)methyl Radical. J. Chem. Phys. 2004, 45, 3472–3474. [Google Scholar] [CrossRef]
- Hoffmann, K.F.; Battke, D.; Golz, P.; Rupf, S.M.; Malischewski, M.; Riedel, S. The Tris(pentafluorophenyl)methylium Cation: Isolation and Reactivity. Angew. Chem. Int. Ed. 2022, 61, e202203777. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S.S.; Eaton, G.R. Signal Area Measurements in EPR. Bull. Magn. Reson. 1980, 1, 130–138. [Google Scholar]
- Holden, A.N.; Kittel, C.; Merritt, F.R.; Yager, W.A. Determination of g-Values in Paramagnetic Organic Compounds by Microwave Resonance. Phys. Rev. 1950, 77, 147–148. [Google Scholar] [CrossRef]
- Bagchi, R.N.; Bond, A.M.; Brain, G.; Colton, R.; Henderson, T.L.E.; Kevekordes, J.E. Electrochemical, chemical and spectroscopic characterization of the trans-[tetracarbonylbis(triphenylphosphine)chromium]+/0 redox couple. Organometallics 1984, 3, 4–9. [Google Scholar] [CrossRef]
- Bond, A.M.; Colton, R.; Kevekordes, J.E. Redox reactions of chromium tetracarbonyl and tricarbonyl complexes: thermodynamic, kinetic, and catalytic aspects of isomerization in the fac/mer-tricarbonyltris(trimethyl phosphite)chromium(1+/0) system. Inorg. Chem. 1986, 25, 749–756. [Google Scholar] [CrossRef]
- Hunter, A.D.; Shilliday, L.; Furey, W.S.; Zaworotko, M.J. Substituent interactions in η6-arene complexes. 1. Systematic x-ray crystallographic study of the structural manifestations of π-donor and π-acceptor substituent effects in substituted chromium (η6-arene)Cr(CO)3 complexes. Organometallics 1992, 11, 1550–1560. [Google Scholar] [CrossRef]
- Reger, D.L.; Wright, T.D.; Little, C.A.; Lamba, J.J.S.; Smith, M.D. Control of the Stereochemical Impact of the Lone Pair in Lead(II) Tris(pyrazolyl)methane Complexes. Improved Preparation of Na{B [3,5-(CF3)2C6H3]4}. Inorg. Chem. 2001, 40, 3810–3814. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Josowicz, M.; Tolbert, L.M. Diferrocenyl Molecular Wires. The Role of Heteroatom Linkers. J. Am. Chem. Soc. 2010, 132, 10374–10382. [Google Scholar] [CrossRef] [PubMed]
- Hunter, G.; Weakley, T.J.R.; Weissensteiner, W. The Triphenylphosphine Cone Angle and Restricted Rotation about the Chromium–Phosphorus Bond in Dicarbonyl(η6-hexa-alkylbenzene)(triphenylphosphine)chromium(0) Complexes. Crystal and Molecular Structure of Dicarbonyl(η6-hexa-n-propylbenzene)(triphenylphosphine)chromium(0). J. Chem. Soc., Dalton Trans. 1987, 1545–1550. [Google Scholar] [CrossRef]
- Kratzert, D.; Holstein, J.J.; Krossing, I. DSR: enhanced modelling and refinement of disordered structures with SHELXL. J. Appl. Crystallogr. 2015, 48, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
Scheme 1.
Valence tautomerism (VT) in literature known ferrocene-trityl(ium) complexes relevant to this work [12,16].
Scheme 2.
Anticipated valence tautomeric equilibrium between diamagnetic [(η6-arene)Cr(CO)2L]-triarylmethylium complexes 1+ - 3+ (left) and their paramagnetic valence tautomers 1+•• - 3+•• (right).
Scheme 2.
Anticipated valence tautomeric equilibrium between diamagnetic [(η6-arene)Cr(CO)2L]-triarylmethylium complexes 1+ - 3+ (left) and their paramagnetic valence tautomers 1+•• - 3+•• (right).
Scheme 3.
Synthesis of carbinol precursors 1-OH - 3-OH and subsequent conversion to tritylium complexes 1+ - 3+: (i) 4-trifluoromethylphenyl lithium, THF, -78 °C - r.t., 16 h; (ii) corresponding ligand L, THF for 2-OH, toluene for 3-OH, hν, r.t., 5-10 min; (iii) HBArF24, CH2Cl2, -70 °C, 5 min.
Scheme 3.
Synthesis of carbinol precursors 1-OH - 3-OH and subsequent conversion to tritylium complexes 1+ - 3+: (i) 4-trifluoromethylphenyl lithium, THF, -78 °C - r.t., 16 h; (ii) corresponding ligand L, THF for 2-OH, toluene for 3-OH, hν, r.t., 5-10 min; (iii) HBArF24, CH2Cl2, -70 °C, 5 min.
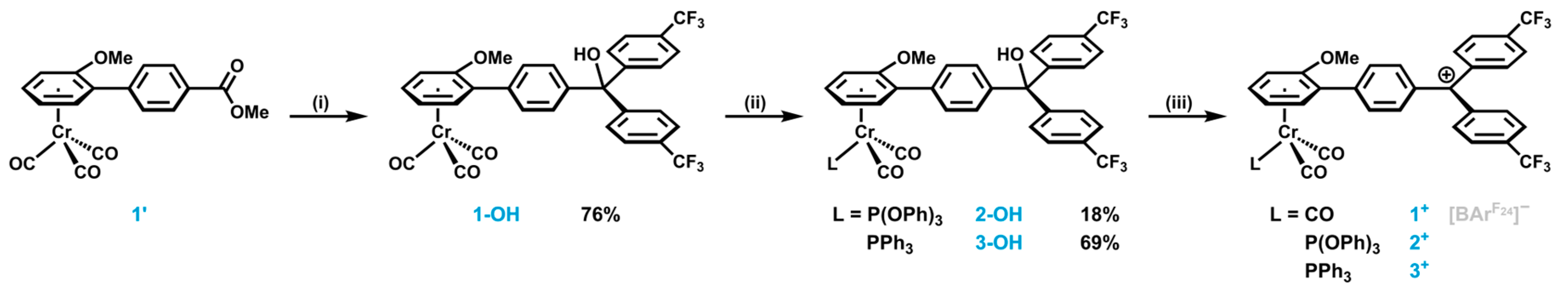
Figure 1.
ORTEP of the Sp enantiomer of carbinol 3-OH with the atomic numbering and the hydrogen-bonded ether molecule. Ellipsoids are displayed at the 50% probability level. The hydrogen atoms of the triphenylphosphine ligand, a second ether solvent molecule, and disorders of the CF3 groups are omitted for clarity reasons.
Figure 1.
ORTEP of the Sp enantiomer of carbinol 3-OH with the atomic numbering and the hydrogen-bonded ether molecule. Ellipsoids are displayed at the 50% probability level. The hydrogen atoms of the triphenylphosphine ligand, a second ether solvent molecule, and disorders of the CF3 groups are omitted for clarity reasons.
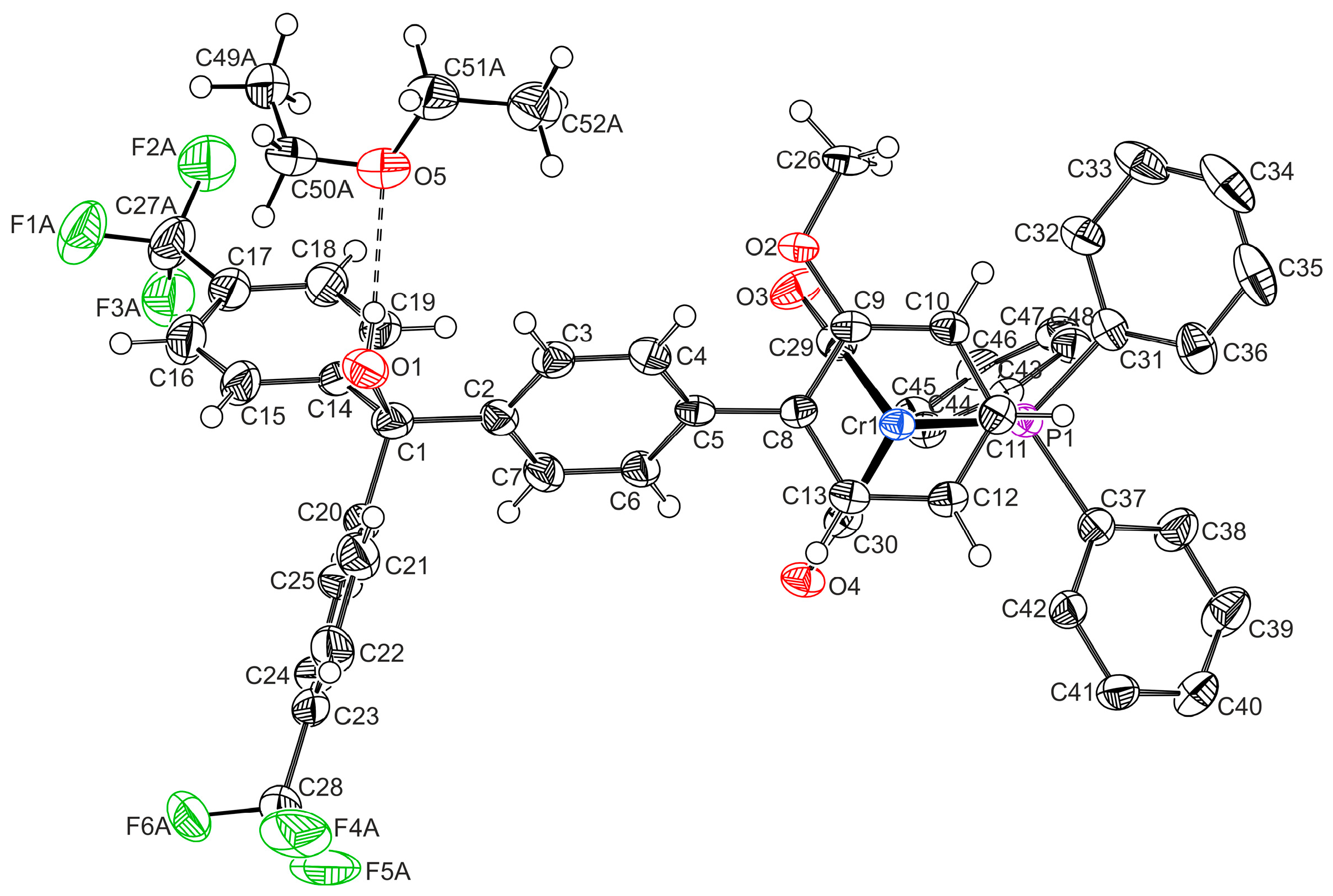
Figure 2.
(a) Cyclic voltammograms of carbinols 1-OH - 3-OH (293±3 K). (b) Cyclic voltammograms of cations 1+ - 3+ (293±3, resp. 195±3 K for 3+) and of identified decomposition products 1-H and L+. Vertical lines in grey and blue indicate the half-wave potentials of the decomposition products. (c) Three-segment cyclic voltammogram of 3+ (195±3 K), starting at the open circuit potential marked by the arrow. Supporting electrolyte: CH2Cl2/ 0.1 M [NBu4]+ [BArF24]−, scan rate v = 100 mV s-1.
Figure 2.
(a) Cyclic voltammograms of carbinols 1-OH - 3-OH (293±3 K). (b) Cyclic voltammograms of cations 1+ - 3+ (293±3, resp. 195±3 K for 3+) and of identified decomposition products 1-H and L+. Vertical lines in grey and blue indicate the half-wave potentials of the decomposition products. (c) Three-segment cyclic voltammogram of 3+ (195±3 K), starting at the open circuit potential marked by the arrow. Supporting electrolyte: CH2Cl2/ 0.1 M [NBu4]+ [BArF24]−, scan rate v = 100 mV s-1.
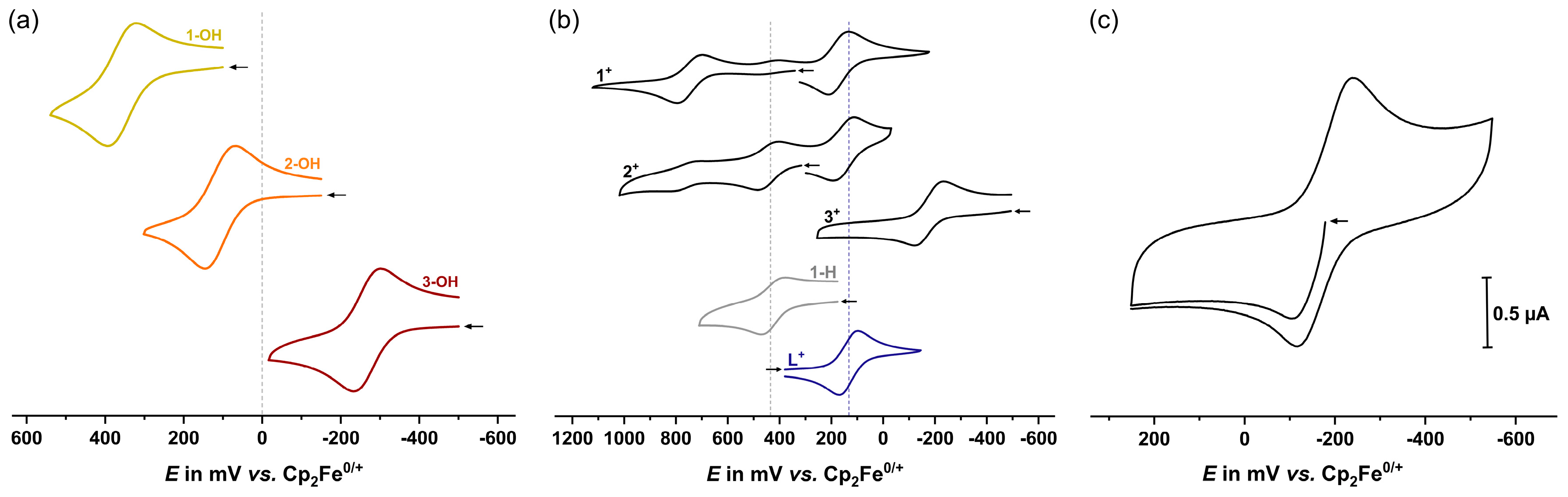
Figure 3.
(a) IR spectra of 1-OH and 1+ in CH2Cl2 at -70 °C, (b) IR spectra of 3-OH and 3+ in CH2Cl2 at -70 °C, (c) baseline-corrected IR spectra of 3+ in the temperature range from -75 to -40 °C.
Figure 3.
(a) IR spectra of 1-OH and 1+ in CH2Cl2 at -70 °C, (b) IR spectra of 3-OH and 3+ in CH2Cl2 at -70 °C, (c) baseline-corrected IR spectra of 3+ in the temperature range from -75 to -40 °C.
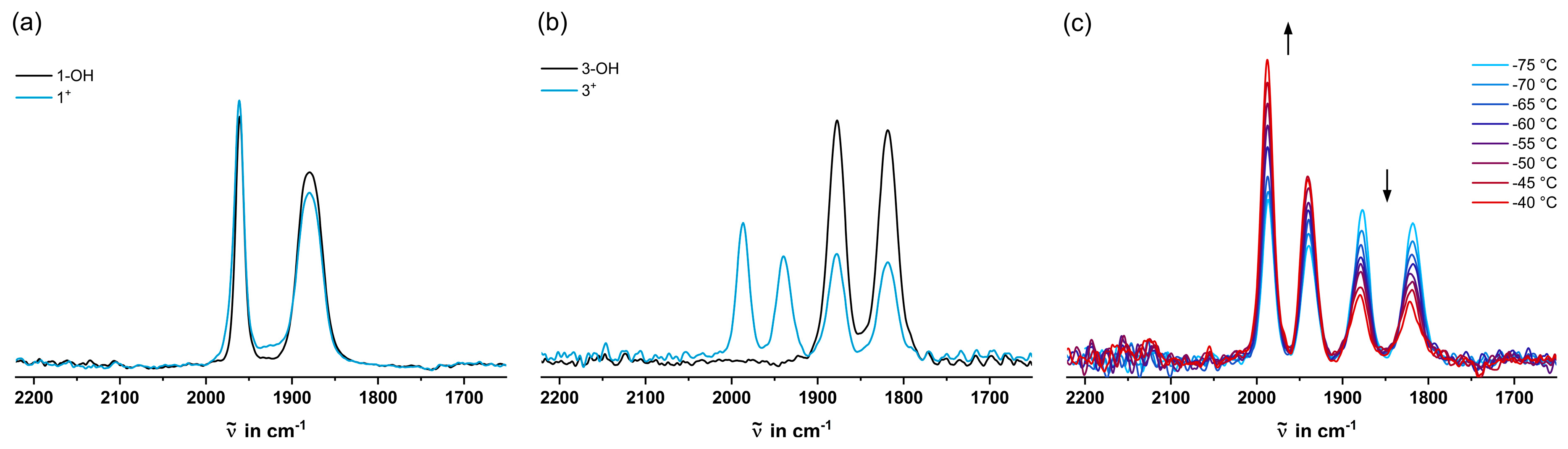
Figure 4.
Experimental and simulated EPR spectra of diradical 3+•• at different temperatures. (a) Spectra in fluid solution at -70 °C; (b) in the temperature range from -90 to -40 °C; (c) in frozen CH2Cl2 at 77 K.
Figure 4.
Experimental and simulated EPR spectra of diradical 3+•• at different temperatures. (a) Spectra in fluid solution at -70 °C; (b) in the temperature range from -90 to -40 °C; (c) in frozen CH2Cl2 at 77 K.
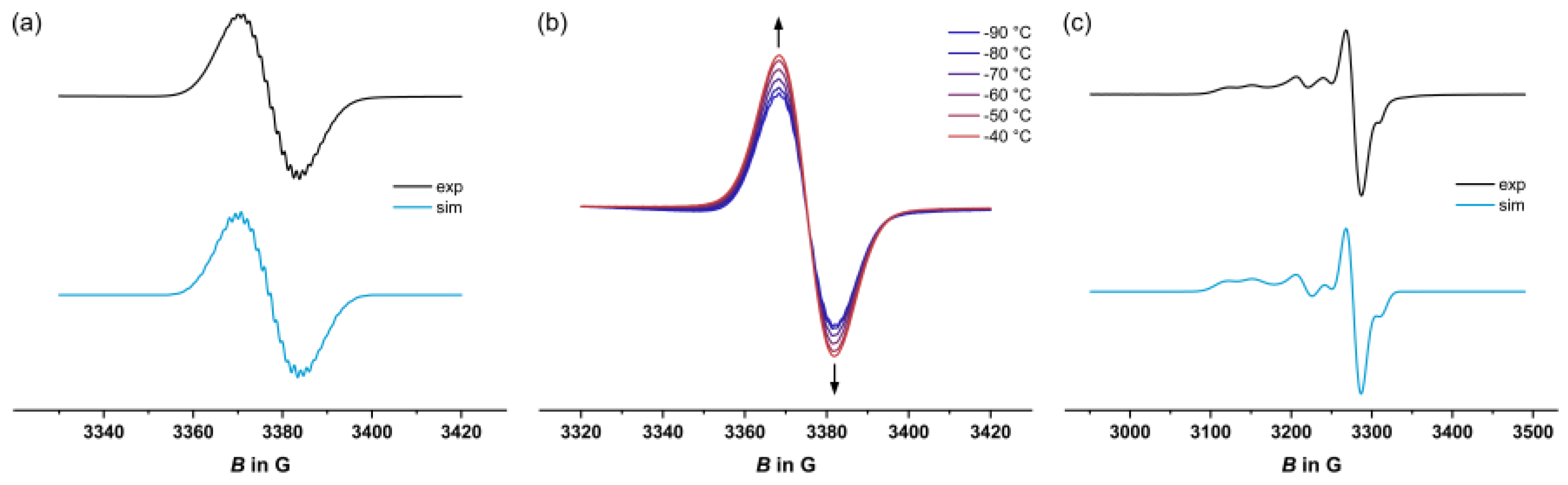
Scheme 4.
Schematic representation of the procedure for quantitative EPR spectroscopy of 3+ and the dimerization of the paramagnetic valence tautomer 3+•• to 3+•-3+•. (1) Double integration of the EPR signal (DI), (2) comparison of the DI to a regression line recorded from the sTable 2,2-diphenyl-1-picrylhydrazyl radical (dpph•) to yield the effective spin concentration, and (3) comparison of the spin concentration to the nominal concentration [42].
Scheme 4.
Schematic representation of the procedure for quantitative EPR spectroscopy of 3+ and the dimerization of the paramagnetic valence tautomer 3+•• to 3+•-3+•. (1) Double integration of the EPR signal (DI), (2) comparison of the DI to a regression line recorded from the sTable 2,2-diphenyl-1-picrylhydrazyl radical (dpph•) to yield the effective spin concentration, and (3) comparison of the spin concentration to the nominal concentration [42].
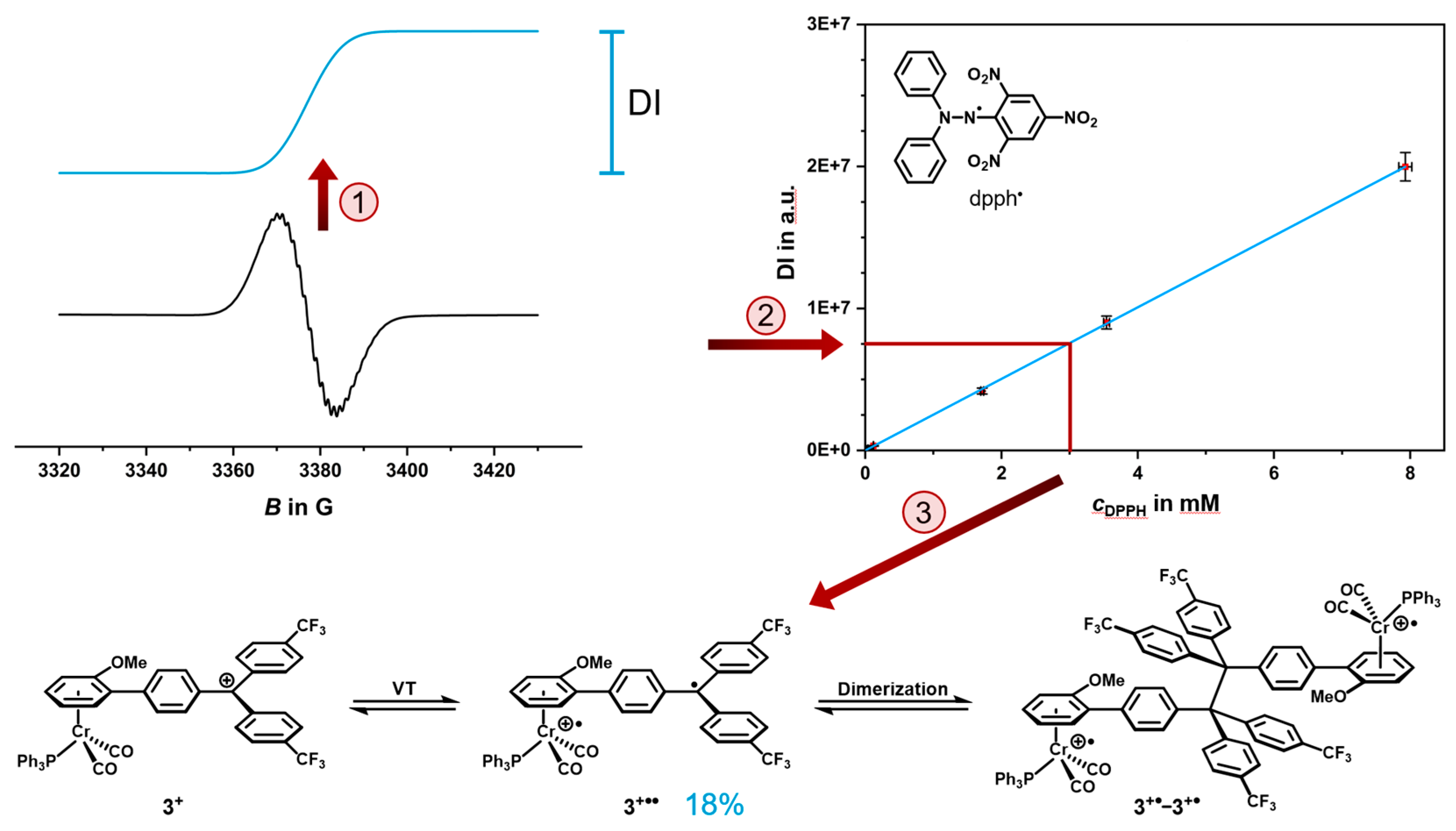
Figure 5.
1H-NMR spectra of 1+ in CD2Cl2 after 12 h at r.t. (top), and of the purified decomposition products 1-H (middle) and L-H (bottom). [BArF24]− signals are marked with asterisks.
Figure 5.
1H-NMR spectra of 1+ in CD2Cl2 after 12 h at r.t. (top), and of the purified decomposition products 1-H (middle) and L-H (bottom). [BArF24]− signals are marked with asterisks.
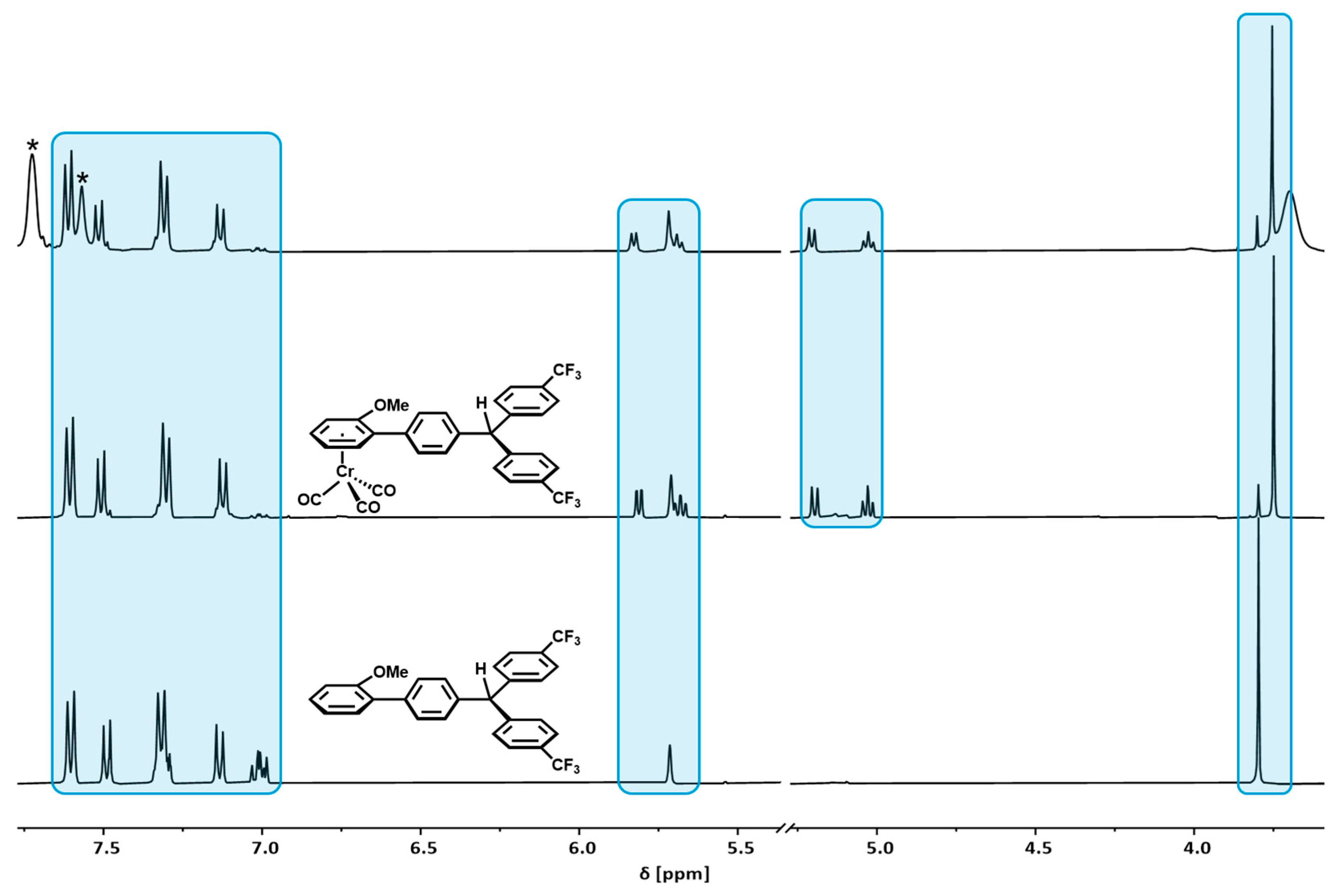
Figure 6.
ORTEPs of the Rp enantiomer of 1-H (left) and L-H (right) with the atomic numbering. Ellipsoids are displayed at the 50% probability level. Disorders are omitted for clarity reasons.
Figure 6.
ORTEPs of the Rp enantiomer of 1-H (left) and L-H (right) with the atomic numbering. Ellipsoids are displayed at the 50% probability level. Disorders are omitted for clarity reasons.
Table 1.
Comparison of important bond lengths and angles of complexes 1′, 3-OH, 1-H, and L-H.
| 1′ | 3-OH | 1-H | L-H | |
|---|---|---|---|---|
| Crystal system | triclinic | monoclinic | monoclinic | orthorhombic |
| Space group | P21/c | P21/n | Pbca | |
| ∠PhCr-Ph1 | 53.63(17)° | 52.55(14)° | 55.3(5)° | 47.09(8)° |
| ∠Ph1-Ph2 | n.a. | 75.00(16)° | 78.1(5)° | 77.00(8)° |
| ∠Ph1-Ph3 | n.a. | 76.50(18)° | 84.1(5)° | 82.95(8)° |
| ∠Ph1-C-OH | n.a. | 110.1(3)° | n.a. | n.a. |
| ∠Ph2-C-OH | n.a. | 105.9(3)° | n.a. | n.a. |
| ∠Ph3-C-OH | n.a. | 106.8(3)° | n.a. | n.a. |
| d C-OH | n.a. | 1.433(4) | n.a. | n.a. |
| d Cr-PhCr | 1.7151(5) | 1.703(5) | 1.720(5) | n.a. |
| d Cr-CO1 | 1.837(4) | 1.821(3) | 1.819(12) | n.a. |
| d Cr-CO2 | 1.837(3) | 1.810(3) | 1.786(12) | n.a. |
| d Cr-CO3/P | 1.844(5) | 2.303(1) | 1.801(12) | n.a. |
Bond lengths d in Å.
Table 2.
Electrochemical data for carbinol precursors, cations and decomposition products.
| Oxidation | Reduction | Potential Difference |
|||
|---|---|---|---|---|---|
| E1/2 | ΔEp | E1/2 | ΔEp | ΔE1/2 | |
| 1-OH | 360 | 71 | - | - | - |
| 2-OH | 110 | 81 | - | - | - |
| 3-OH | -265 | 68 | - | - | - |
| 1+[a] | 750 | 96 | 175 | 78 | 575 |
| 2+[b] | 445 | 77 | 155 | 79 | 290 |
| 3+[c] | -180 | 110 | -180 | 110 | < 50 |
| 1-H | 435 | 83 | - | - | - |
| L+ | - | - | 135 | 72 | - |
All potentials in mV (±5 mV) relative to the Cp2Fe0/+ redox couple. Measured in CH2Cl2/ 0.1 M [NBu4]+ [BArF24]− at 293±3 K at a scan rate v of 100 mV s-1. [a] Additional redox wave at E1/2 = 435 mV can be assigned to 1-H. [b] Tentative assignment, see text. [c] Measured at 195±3 K; ΔE1/2 estimated from wave broadening of the convoluted redox wave [54].
Table 3.
CO stretching vibrations for all complexes.
| [cm-1] | [cm-1] [a] | |
|---|---|---|
| 1-OH | 1965, 1886 | 1912 |
| 2-OH | 1910, 1853 | 1872 |
| 3-OH | 1880, 1821 | 1840 |
| 1+ | 1965, 1886 | 1912 |
| 2+ |
2012, 1964[b] 1904, 1848 |
1980 1867 |
| 3+ |
1988, 1940[b] 1878, 1819 |
1956 1839 |
| 1-H | 1965, 1888 | 1914 |
Measured in CH2Cl2 at 293±3 K (neutral carbinols), resp. 203±3 K (cationic complexes). [a] Weighted average counting the doubly degenerate asymmetric vibration twice. [b] IR data for the diradical valence tautomer.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



