
Submitted:
31 October 2023
Posted:
01 November 2023
You are already at the latest version
Abstract
Mesenchymal stromal cells (MSCs) are a subset of heterogeneous non-hematopoietic fibro-blast-like cells which play important roles in tissue repair, inflammation, and immune modula-tion. MSCs residing in the bone marrow microenvironment (BMME) functionally interact with hematopoietic stem progenitor cells regulating hematopoiesis. However, MSCs have also emerged in the last years as key regulator of tumor microenvironment. Indeed, they are now con-sidered as active players in the pathophysiology of hematologic malignancies rather than passive by-standers in hematopoietic microenvironment. Once the malignant event occurs, the BMME acquires cellular, molecular, and epigenetic abnormalities affecting tumor growth and progres-sion. In this context, MSC behavior is affected by signals coming from cancer cells. Furthermore, it has been showed that stromal cells themselves play a major role in several hematological malig-nancies’ pathogenesis. This bidirectional crosstalk creates a functional tumor niche unit wherein tumor cells acquire a selective advantage over their normal counterpart and are protected from drug treatment. It is therefore of critical importance to unveil the underlying mechanisms which activate a protumor phenotype of MSCs for defining unmasked vulnerabilities of hematological cancer cells which could be pharmacologically exploited to disrupt the tumor/MSCs coupling. The present review will focus on the current knowledge about MSC dysfunction mechanisms in the BMME of hematological cancers, sustaining tumor growth, immune escape, and cancer pro-gression.
Keywords:
MSCs
; tumor transformation
; hematological cancers
; senescence
; inflammation
1. Introduction
Mesenchymal stromal cells (MSCs) are a critical component of bone marrow (BMME) microenvironment whereby they provide newly formed osteoblasts and tightly regulate the homeostasis of hematopoietic stem and progenitor cells (HSPCs) [1]. In this context, MSCs are the major contributor of many key niche factors and maintain the dynamic balance between HSPCs self-renewal, quiescence, proliferation, and differentiation [2,3]. MSCs are located in the sites of hematopoiesis starting from embryonic developmental stages [4]. Importantly, MSCs and their progeny such as osteoblasts, chondrocytes and adipocytes, are structural components of both endosteal and perivascular niches [5]. Within these compartments, MSCs interact with both hematopoietic stem cells and more differentiated hematopoietic progenitors, thus regulating their quiescence, proliferation, and differentiation [6,7,8] (Figure 1).
Different MSC subtypes interact with HSPCs in specific regions of the niche [9]: CD271+ MSCs are bone-lining cells sustaining long-term HSPCs in low oxygen area, whereas CD146+ and CD271+CD146+ MSCs are located in BM sinusoids with activating and fast-proliferating HSPCs [10,11]. A plethora of supporting factors regulating HSPCs self-renewal and trafficking are provided by MSCs in the BM niche, such as CXCL12 and stem cells factor [12,13]. Notably, alteration of HSPC and BM-MSC interactions can alter normal hematopoiesis causing hematological malignancies [14,15,16,17].
MSC behavior is dynamically regulated both by intrinsic mechanisms and microenvironment factors, highlighting the high plasticity of these cells in adapting to tissue homeostasis and regenerative needs [18]. In this context, the therapeutic use of MSCs in the field of regenerative medicine relies on their ability to migrate to injured tissues and to promote endogenous regeneration sustaining the growth and differentiation of stem resident cells [19,20]. In details, MSC therapeutic potential for the treatment of immunological diseases result from their ability to suppress or control intensive immune activation by inhibiting immune cell proliferation and inducing immunosuppressive subsets though the secretion of anti-inflammatory factors or direct cell-to-cell contact [20,21,22]. However, MSCs also take part to tumorigenesis process contributing to BMME malignant transformation and maintenance, finally promoting tumor cell growth, survival, progression, and therapy resistance. Similar to HSPCs, the interactions between cancer cells and BM-MSCs can determinate tumor cell dormancy or proliferation. For example, leukemic stem cells (LSCs) co-localize with CXCL12-secreting MSCs in BM, inducing their quiescence [22]. Furthermore, accumulation of senescent MSCs in BM niche might promote the progression from premalignant to over hematological malignancy [23,24]. Senescence of MSCs is accompanied by several phenotypic changes, including enlarged cell morphology, decreased proliferative capacity and impaired differentiation ability [25]. Evidence suggests that the presence of increased numbers of senescent MSCs is a characteristic feature of several hematological cancers [26]. When functional and regenerative capacities of aging MSCs are diminished, they enter in a replicative senescence status which promotes BM inflammation and the dysregulation of hematopoiesis [27] (Figure 1). It is well known that senescent cells release pro-inflammatory factors, generally known as Senescence-Associated Secretory Phenotype (SASP) which contributes to tumor immunosuppressive microenvironment [28]. Furthermore, it has been shown that SASP factors such as IL-6, CXCL8 and GDF15 can alter HSPC homeostasis in vitro [29]. In detail, IL-6 secreted by aged BM-MSCs induces rapid HSPC expansion, thereby leading to depletion of HSPC pool and increased risk of genomic instability in these cells [23,30]. MSCs are subject to genetic alterations and chromosomal aberrations contributing to age-and disease-associated MSC dysfunctions [31,32]. Interestingly, it has been demonstrated that these alterations differ from ones observed in the hematopoietic tumor cells of the same patient, corroborating the idea that unstable MSCs might facilitate the expansion of malignant cells [33]. However, no recurring genetic mutations or cytogenetic aberrations have been found in MSCs from BMME of hematological cancers [33,34,35,36], unveiling that epigenetic modifications underlie the activation of their pro-tumor phenotype. In this context, cellular epigenetic architecture is modeled based on the environmental insults and physiological changes to maintain MSC functions, including their self-renewal, differentiation, and niche modulation abilities [37]. Notably, dysfunctions of MSC phenotype persist also after their expansion ex vivo, suggesting a heritable epigenetic dysregulation which persist despite the removal of disease associated BMME. In agreement, the methylome of MSC from hematological cancers was found to be distinct from healthy stromal cells [38,39,40,41].
Data from previous studies revealed that a cancer-associated fibroblast (CAF)-like phenotype is activated in MSCs from patients with hematological cancers [42,43,44]. Indeed, these cells express tumorigenic markers such as alpha smooth muscle actin (αSMA) and fibroblast activation protein (FAP) as consequence of the soluble factors produced by cancer cells [45,46,47]. In agreement, CAFs might derive from MSCs working as a subset of “specialized” stromal cells [48,49]. Paunescu and colleagues previously showed that MSCs and CAF have many similarities including their phenotype and the only difference is the secreted cytokines [50]. In their study, CAFs have been demonstrated to derive from a specialization process which converts MSCs inside the tumor structure for better serving tumor cells [50]. A mounting number of studies indicated that growth and survival of leukemic clone is promoted by inflammation-driven changes in the BM-MSCs [51]. In particular, naïve MSCs are able to exert bidirectional effect on tumor cells favoring or inhibiting its growth [52], while tumor “educated” MSCs promote tumor progression, in relation to the inflammatory microenvironment [53]. Compared to the healthy counterpart, MSCs from BM tumor milieu show a distinct transcriptional landscape characterized by cellular stress and upregulation of inflammatory molecules which sustain malignant over healthy clonal hematopoietic cell expansion [34,54,55]. The pro-leukemic role of MSCs can also be achieved indirectly by shaping the BMME immune infiltrate [56,57]. Indeed, the immunomodulatory effect of MSCs on innate and adaptive immunity is a major mechanism by which these cells can affect tumor initiation and progression [58,59,60,61]. This feature depends on the type and intensity of inflammatory signals into the BMME. A high inflammatory state causes MSCs to produce T cell suppression, while a low inflammatory state leads to MSC-induced T cell activation [62]. In hematological malignancies, senescent MSCs release more inflammatory signals feeding an inflammatory milieu able to scramble the delicate balance of pathways involved in tissue-specific regeneration and remodeling [63]. Although MSC immunomodulatory activity is primed by cytokines into BMME, it is also dependent on stimulation of toll like receptors (TLRs). In detail, MSCs can be polarized into two distinct phenotypes similar to macrophages, resulting in different immune-modulatory activity and secretome [64]. The TLR4-primed MSCs exhibit a proinflammatory phenotype (MSC1), while the TLR3-primed MSCs activate a more anti-inflammatory profile (MSC2). This concept of MSC polarization could explain the apparently contradictory roles of MSC in inflammation and immunomodulation [20]. Notably, the regulation of the functional profile of MSCs depends not only on the secretion of soluble factors but also on the communication and contact of MSCs with neighboring BM cells. MSCs can communicate with nearby cells through secretion of soluble factors, cell to cell contact, release of extracellular vesicles (EVs) and, as more recently evidenced, through tunneling nanotubes [65,66,67,68]. Evidence is arising that altered MSCs help leukemic cell growth and prompt drug resistance by providing nutrients, cytokines, pro-survivals signals and exchanging organelles [44,69,70]. Several recent studies have identified stroma-derived metabolites such as lactate, glutamine, and acetate to feed the tricarboxylic acid cycle (TCA) and lipid biosynthesis into hematopoietic cancer cells [71,72,73]. Of note, metabolism is adjusted during the development of drug resistance [73,74]. The complexity of this scenario is increased by a metabolic heterogeneity and dynamics of BMME which is mainly dependent on different access to oxygen and glucose and on different cell population co-existing in the BM milieu [75,76]. As result, cancer and stromal cells can compete and/or cooperate for nutrients. In recent years, the role of exosomes as mediators between cancer cells and tumor BMME ha gained increasing attention. For example, leukemia-derived exosomes induced a downregulation of HSPC-supporting factors in MSCs and reduced their capacity to support normal hematopoiesis [77]. Furthermore, while microvesicles (MVs) from healthy MSCs show anticancer action, MM-MSCs release MVs enriched in VLA-4, which facilitated MM cell uptake, enhance tumor cell phenotype and PCs growth [78,79].
In this review, we will highlight the role of MSCs in tumor microenvironment of hematological cancers, aiming to elucidate the mechanisms involved in the activation of their pro-tumor phenotype contributing to tumor growth and progression.
2. Materials and Methods
2.1. Role of MSCs in myelodysplastic syndromes
Myelodysplastic syndromes (MDS) is generally referred as a heterogenous group of clonal hematopoietic diseases characterized by ineffective hematopoiesis resulting in peripheral blood cytopenia potentially shifting to acute myeloid leukemia (AML) [80]. MDS patients display different degrees of cytopenia and dysplasia, therefore constituting the basis for Word health organization MDS classification criteria [81]. To date, no clinically effective treatment is available for preventing progression to AML. Half of patients show cytogenetic alteration, while nearly 90% of them harbor at least one somatic mutation affecting specific genes involved in spliceosome, transcription factors and epigenetics. Despite the clonal dominance, these mutations don’t provide a determined advantage for malignant cell growth, as suggested by their coexistence alongside normal HSPCs [82]. Therefore, MDS cells get an extrinsic support from BMME important for malignant cell cloning. Notably, support from BM milieu is essential to maintain MDS cells ex vivo. Concerning MSCs, MDS stromal cells are reprogramed to support uniquely MDS clones at the expense of normal HSPCs [35]. MDS-MSCs are characterized by a slower proliferation rate, which is independent by cell cycle distribution and apoptotic events [83]. Therefore, the reason characterizing this phenotype might involve cytogenetic aberrations, which have been difficult to characterize due to the lack of a specific isolation protocol allowing the comparison between different MSCs subpopulations. This goal has been achieved upon Aanei and colleagues published a robust immunoselection-based isolation protocol through two specific mesenchymal-associated markers, STRO-1 and CD73 [84]. Therefore, MDS-MSC cytological characterization highlighted genomic gains involving genes taking part in cell-cell adhesion processes and tumor development. In addition, MSCs isolated from patients harboring 5q- cytogenetic shared common traits including the overexpression of some genomic regions as 7p22.3, 19p13.3, and 19p13.11 [83]. Despite a cytogenetic signature characterizing MDS-MSCs is still missing, it is widely reported how these cells display all the typical marker related to cell senescence [85]. In this context, Fei et al. reported that isolated MDS-MSCs display a profound change in cytoskeletal architecture, in turn showing an increased size, longer podia and a disordered distribution of F-actin [86]. Moreover, MDS-MSCs also display an increased DNA damage level [87]. Coherently with this outcome, an hyperactivation of p53 signaling cascade has been detected in MDS-MSCs, therefore providing a further mechanism leading to MSC senescence [86]. Despite the efforts showing the essential role covered by BMME in MDS, the question asking who the first cell population is to impair the basal crosstalk, therefore triggering MDS pathogenesis, is still standing. A partial answer has been given by the studies describing that, in patients showing a complete hematological remission, the treatment was able to restore a MSC functionality comparable to healthy donors [88]. However, this study has the intrinsic assumption that the treatment has the HSC compartment as only target, excluding a direct effect on MSCs themselves. In this context, it has been recently demonstrated that the antileukemic activity of azacytidine in part depend on its direct effect on the hematopoietic supportive capacity of MDS-MSCs favoring expansion of healthy over malignant hematopoiesis [89]. These data highlighted the crucial role of an epigenetic treatment of dysfunctional MSCs. Other studies corroborate the hypothesis involving the crosstalk between stromal and hematopoietic compartments as driver of MDS pathogenesis, in turn rearranging the surrounding microenvironment to support the expansion of the malignant clone. Indeed, murine models depleted for Dicer or Sbds gene expression exclusively in stromal compartment have shown to develop an MDS-like phenotype characterized by ineffective hematopoiesis, marked dysplasia and leukemic progression, despite having no mutation in their HSPCs [90,91]. Medyouf and colleagues described a scenario in which MSCs are instructed by malignant HSCs to acquire MDS-MSC-like properties, eventually promoting the progression of the malignant clone over the healthy one [35]. This data introduced the “hematopoietic niche unit”, sustaining MDS progression also by the establishment of an altered secretome profile, where abundant levels of TNF-α, IFN-γ, IL-1α, IL-6, IL-17 and TGF-β have been detected [92]. These factors account for the establishment of an inflammatory BMME, in turn triggering genetic and epigenetics modifications on BM resident cell populations. Corroborating this, MDS-MSCs show several differentially methylated genes associated to alterations of their phenotype [36]. For instance, HHIP (Hh-interacting protein) gene is hypermethylated in MDS-MSCs [93]. Its downregulation accompanied by activation of the Hedgehog pathway in stromal cells sustain survival of MDS cells. Recently, our group have shown the relevance of an epigenetic-inflammatory interplay in MDS-MSCs supported by macroH2A1/TLR4 axis, prompting a replicative senescent phenotype, hypermethylation and metabolic rewiring which contribute to ineffective hematopoiesis [94]. In agreement, cellular stress and upregulation of inflammatory molecules with inhibitory effects on normal hematopoiesis has been described in MDS-MSCs [95]. In particular, activation of NFkB signaling in MSCs from patients with lower risk MDS (LR-MDS) attenuates normal hematopoiesis in accordance with cytopenia observed in these patients [96]. Moreover, overexpression of the alarmins S100A8/9 in stromal cell compartment has been shown to activate NFkB and a genotoxic stress in HSPCs associated to leukemic evolution in a subset of LR-MDS patients [91]. Supporting the crucial role played by the BM niche in MDS evolution, it has been proposed that the overexpression of CXCL12, in synergy with its receptor CXCR4, keep the myelodysplastic anchored inside the BM niche, in turn providing them with protection and support [97]. In this scenario an in vitro study highlighted the overproduction of IL-6, an interleukin possibly linked to the mechanism promoting MSCs senescence and chronic inflammation [98]. The crosstalk between MDS cells and MSCs is also orchestrated by a plethora of factors, as part of the two populations’ secretome. Releasing alarmins such as S100A9 and S100A8, tumor cells activate the inflammasome in stromal cells which results in higher secretion of pro-inflammatory cytokine [99]. Also, EVs secreted by MDS cells have been demonstrated to reduce the hematopoietic supportive capacity of MSCs inhibiting osteolineage differentiation of MSCs [100]. This perturbation of bone metabolism enables MDS clones to outcompete normal HSPCs. In turn, MDS-MSCs have been described to release EVs carrying miRNA, such as miR10a and miR15a, which increase the viability and clonogenicity of MDS cells [101]. Therefore, the multifaced aspects accounting for MSCs significancy need to be further dissected to provide more efficient strategies counteracting MDS progression.
2.2. Role of MSCs in acute leukemia
Acute leukemias are rapidly progressing malignant clonal disorders characterized by uncontrolled proliferation of immature and undifferentiated hematopoietic cells, associated to poor prognosis, and reduced overall survival. They are commonly divided, according to malignant cells lineage in acute myeloid or lymphoid leukemia (AML or ALL). Blast cells have been known to modify the BMME and disrupt non-malignant hematopoiesis [102,103]. The complex interactions within the tumor BMME significantly influence leukemia survival, disease progression and therapeutic response, with hematopoietic stem cell transplantation often being the only curative option for patients with refractory disease [104]. Several studies showed that the interaction of leukemic cells with MSCs resulted in a functionally disrupted niche specifically supporting tumor cells over healthy HSPCs and therefore establishing a self-reinforcing unit for the repopulation of leukemic cells [105,106]. In this context, blast cells might exploit physiological mechanisms regulating hematopoiesis as a strategy for gaining competitive advantages [107]. It has been recently described, using mouse models of leukemia, that both ALL and AML blasts express lymphotoxin α1β2 after colonizing the BM. Therefore, blasts trigger lymphotoxin beta receptor (LTβR) signaling in MSCs, turning off IL7 production and preventing non-malignant lymphopoiesis [107]. Among the changes in cytokine profile of AML-MSCs, the overproduction of CCL2 inhibits normal progenitors but not leukemic cells improving cancer survival [108,109]. Similarly, MSCs from T-ALL patients show reduced ability to support healthy HSCs blocking their differentiation in HPCs, without a direct leukemic MSC-induced damage [110]. This finding is consistent with the paradigm that, despite the exhaustion of HPCs in the leukemic milieu, HSCs remain functional upon relocation into non-leukemic BMME [110].
Hematopoietic insufficiency is the hallmark of AML with cytopenia-related complications such as bleeding and infections representing the major causes of death. In AML-MSCs, the downregulation of FOXM1, a member of the fork-head transcription factor family, impairs the hematopoietic MSCs support capacity [111]. Corroborating this, the silencing of this protein in healthy stromal cells affects the growth of CD34+ progenitor cells, mirroring the effects observed when using AML-derived MSCs [111]. Moreover, AML-MSCs displayed alterations in the expression of key hematopoiesis-regulating factors such as JAGGED1 and KITL corroborating that hematopoietic insufficiency in AML patients is at least in part mediated by BMME [112]. More recent studies provided evidence that, when acute leukemia occurs, blast cells remodel the resident MSCs establishing a physical connection and mediating a reprogrammed transcriptome [106,113]. Of note, healthy MSCs changed their gene expression profile after coculture with AML blasts displaying deregulation of genes matched with AML-MSCs [113]. This transcriptomic behavior, characterized by inflammatory factors and cytokine production pathways, correlate to AML suggesting a dynamic changes in MSCs occurring at leukemia onset as consequence of an instructive role of leukemic cells [112,113]. As a result, “reprogrammed” MSCs reset the niche crosstalk processes selectively suppressing normal hematopoiesis and favoring the clonal dominance of leukemic cells [106,114]. The heterogeneity of MSC subpopulation exhibit different BMME for leukemic cells contributing to the heterogeneous kinetics of leukemia relapse. In this context, Kim and colleagues evaluated whether differences in BM stromal cell partners at diagnosis can identify patients with a high risk of relapse [106]. They found that BMME of relapsed patients showed higher numbers of MSCs, osteoblasts and primitive nestin+ MSCs than AML patients who achieved complete remission (CR). Early relapsed patients have a greater primitive MSC content, while late relapsed ones exhibited more MSCs or osteoblasts than CR patients, corroborating a distinct BMME associated with early or late relapse [106]. This evidence suggests that leukemia-induced remodeling of BMME may be responsible for the heterogeneity of the AML clinical course. MSC lineage differentiated cells, including osteoblasts and adipocytes, are essential components of BMME contributing to hematopoiesis [115,116]. Increasing evidence suggests that the differentiation ability of AML-MSCs is altered [117,118]; however, the results are controversial. Indeed, some work report that AML cells induce an osteoblast-rich niche in the BM which in turn enhances AML expansion and favors disease relapse [106,119]. On the contrary, alterations of MSC osteoblastic plasticity resulted in selective promotion of leukemic cells in murine models [14,120]. Moreover, other researchers reported that leukemia educated MSCs are highly prone to adipocytes differentiation [118]. These conflicting results may be due to the heterogeneity of leukemia. For instance, AML cells of AML-M4 subtype induce MSCs towards adipogenic differentiation propensity [121]. The alterations of osteogenic differentiation capacity of AML-MSCs were also confirmed by specific methylation changes affecting genes regulating cell differentiation and skeletal development [112].
As MDS stromal cells, both AML and ALL-MSCs show accelerated cellular senescence contribute to their impairment in function associated with HSPC support and stemness properties [28,110]. MSCs exposed to leukemic blasts exhibit common characteristics to MSCs subjected to a physiological aging process, including overexpression of markers related to DNA damage and cell-cycle arrest [122]. Furthermore, leukemia-induced oxidative stress works as driver of pro-tumoral senescence in stromal cells [123]. Targeting senescent MSCs directly inhibits AML cell growth and improves survival of mice with leukemia, revealing the importance of a senescent milieu for the pathophysiology of leukemia [123]. Heterochromatin disorganization is a driver of MSC senescence [124]. AML-MSCs downregulate chromatin remodeling complex CHD1 (modulating chromatin condensation) whose reduction is associated with the decrease of HSPC supportive capacity [125]. Using an integrative approach of a multilevel molecular profiling combining genome-wide expression and DNA methylation high-throughput platforms, AML-MSCs exhibit selective transcriptional alterations associated to epigenetic ones, including adhesions molecules, endocytosis, and metabolic pathways [34]. In this context, accumulating evidence shows a complex metabolic coupling between leukemic cells and MSCs which allows tumors to respond to variations in nutrient availability to maximize cellular proliferation and to acquire survival advantages [126,127]. In leukemia patients, cancer stem cells or chemoresistant cells rely on mitochondrial oxidative phosphorylation (OXPHOS) [128,129]. MSCs directly provide the increased bioenergetic demand of AML cells, increasing OXPHOS and GSH-related ROS detoxifying tools which contribute to AML growth and chemoresistance [130]. Of note, MSCs supply mitochondria to leukemia cells [44,131,132,133], thus providing them with additional energy. In T-ALL, leukemic cells transfer their damaged mitochondria to MSCs through cell adhesion mechanisms, reducing intracellular ROS and promoting chemotherapy-induced apoptosis resistance [134]. Recently, it has been reported that AML-induced MSCs adipogenic differentiation propensity is associated to a switch from glycolysis to OXPHOS [121]. In this context, AML blasts modulate intracellular metabolism of adipocytes into a lipolytic state, resulting in the release of fatty acids (FAs) into the BMME [135]. Ultimately, free FAs are transferred to AML blasts fueling a FA oxidation signature beneficial to the leukemia counterpart [135]. Indeed, blocking lipolysis or inhibiting CPT1A (carnitine palmitoyltransferase 1a), essential for the transfer of FAs to the inner mitochondrial membrane and β-oxidation [136], reduced AML mitochondrial activity and survival [135]. Like AML blasts, ALL cells induce adipocytes to activate lipolysis to support their metabolism [137]. ALL blasts also release EVs which activate a metabolic switch from PXPHOS to aerobic glycolysis in MSCs leading to increased lactate into BMME which can be used by tumor cells [138].
2.3. Role of MSCs in Myeloproliferative Neoplasms
Myeloproliferative Neoplasms (MPNs) are characterized by the clonal proliferation of one or more hematopoietic cell lineages, predominantly in the BMME. MPNs mainly include chronic myeloid leukemia (CML), polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF). CML is the BCR-ABL1 oncoprotein-positive MPN characterized by the Philadelphia (Ph) chromosome’s presence. Ph- MPNs include PV, ET and PMF, in which clonal proliferation is driven by somatically acquired driver mutations in JAK2, CALR and MPL genes [139]. Nevertheless, Ph- MPNs show a different clinical presentation and outcome [140]. PMF is defined by unique clinical features such as BM fibrosis, osteosclerosis, neo-angiogenesis and extramedullary hematopoiesis which characterize the natural history of PMF patients, significantly affecting quality of life and life expectancy [141]. Similar to MDS-MSCs, stromal cells from CML and higher fibrosis PMF patients display functional alterations, including low proliferative potential and precocious senescence [142,143]. Changes in MSC behavior are strongly associated with the dysfunction of T cells and the proliferation of Tregs in CML microenvironment [144]. Moreover, CML-MSCs directly orchestrate immunosuppression by also driving activation of myeloid cells in MDSCs [56]. Immune suppression of stromal cells can also be enhanced by CML cell secreted exosomes [145]. For instance, leukemic-derived exosome miR-130a/b has been demonstrated to promote the immunosuppressive properties of stromal cells through inhibition of connexin-43 [146]. In CML patients, CXCL12-expressing MSCs are crucial for maintaining quiescent leukemic stem cells and thus they represent a potential target to overcome drug resistance [147]. Indeed, analysis of gene expression profile reveal that abnormal alterations observed in CML-MSCs compared to normal counterpart persist in patients in deep molecular response after therapy with tyrosine kinase inhibitors [148], corroborating their role in leukemia relapse and drug resistance.
Neoplastic clone development in PMF is deeply influenced by alteration within BBME, highlighted by BM fibrosis, neo-angiogenesis and osteosclerosis [149,150]. In this context, it may be hypothesized that the progressive stromal cell alterations during myelofibrosis evolution affect the disease course [141]. These cells display an increased expression and deposition of fibronectin correlating with the fibrosis grades [151]. This outcome is further enhanced by megakaryocytes (Mks), aberrantly proliferating, and releasing several growth factors mitogenic for MSCs/fibroblasts and endothelial cells, such as TGF1 [152,153]. Corroborating this, in MPN biopsies, MSCs localize with Mks displaying an activated fibronectin-secretory phenotype [154]. This interaction is crucial for the priming of stromal cells in PMF. Compared to healthy or low fibrosis grade MSCs, stromal cells from high fibrosis grade PMF patients show higher capacity to support differentiation of Mks by fibronectin secretion [151], highlighting their key role in supporting Mks hyperproliferation observed in PMF BM biopsies [155]. In agreement with this, MSCs isolated from spleen of MF patients show higher expression of fibronectin to sustain extramedullary hematopoiesis and megakaryocytopoiesis [156]. In addition to this, other inflammatory molecules generated by malignant clones contribute to the microenvironment abnormalities of myelofibrosis niche. For instance, lipocalain-2 (LCN2) primes MSCs to differentiate into osteoblasts prompting matrix proteins deposition [157]. Moreover, Mk-derived PDGF activate MSCs and in particular, expression of its receptor strongly correlate with the intensity of MCS reaction and fibrosis grade [158]. We recently demonstrate the involvement of IGFBP6 (Insulin-like growth factor-binding protein 6) in the activation of a CAF-like phenotype of stromal cells controlling the fibrotic process through the activation of sonic hedgehog/TLR4 axis [159]. Using murine MPN models, Schneider and colleagues demonstrated a critical role for Gli1+ MSCs in the pathogenesis of BM fibrosis [160]. After their activation, dependent on Mk-produced Cxcl4, these cells are metabolically reprogrammed, particularly in fatty acid, and differentiate into matrix-producing myofibroblasts. The authors also demonstrated that genetic ablation of Gli1+ MSCs abolished BM fibrosis rescuing BM failure [160]. Of note, increased number of MSCs can be detected in the blood of PMF patients suggesting their involvement in the abnormal HSPC trafficking/homing leading to extramedullary hematopoiesis [161]. Analyzing the whole transcriptomic profile of MPN-MSCs, Martinaud and colleagues revealed a specific pro-fibrotic and inflammatory signature in PMF-MSCs, not observed in TE or PV patients, and characterized by increased osteogenic potential and endogenous production of TGFB1 [162]. Leimkuhler et found that MSCs transcriptionally downregulated niche support and decreased MSC multipotent progenitor status, but upregulated Mk-derived TGFB1 pathway and extracellular matrix proteins, specifically collagens [163]. MSCs from ET patients have also previously reported to decrease hematopoietic supportive capacity and increase ECM remodeling, suggesting an intrinsic defect of stromal cells already in pre-fibrotic MPN [154].
2.4. Role of MSCs in in Chronic lymphocytic leukemia
Chronic lymphocytic leukemia (CLL) is a lymphoproliferative disorder characterized by relentless accumulation of monoclonal mature B-lymphocytes in the peripheral blood, bone marrow and lymphoid tissue [164]. A plethora of molecular prognostic factors have been identified in CLL patients and among them, VLA-4, an exclusive member of the α4 integrin subfamily, represents a CLL negative prognostic marker [165]. VLA-4 plays a prominent role in the homing of high-risk CLL cells within BMME. Notably, MSC-CXCL12 triggers the activation of VLA-4, therefore highlighting a crucial role played by MSCs in CLL cells homing [166]. Furthermore, MSCs might also promote CLL B-cells resting by increasing their CD38 and CD71 expression therefore reflecting an activated phenotype that could be related to disease progression [167].
In agreement with this, CLL cells highly rely on the abundance of supporting stimuli generated by the neighboring cells in the microenvironment, including MSCs. In agreement with this, the resistance of CLL cells to normal apoptotic regulation has been reported to be mediated by direct contact with stromal cells, requiring the simultaneous engagement of β1 and β2 integrins [168,169]. Therefore, while CLL cells undergo rapid apoptosis when cultured alone, once cocultured with stromal cells they are easily propagated. This outcome is probably a consequence of MSC-EVs which have been recently reported to give to leukemic cells a survival advantage protecting them from spontaneous and drug-induced apoptosis [170]. In this scenario, CLL-derived exosomes establish a feedback loop by activating a CAF-like phenotype in MSCs, therefore improving the secretion of soluble factors promoting CLL cell survival [171]. Corroborating this scenario, CLL cells isolated from blood samples are non-dividing, despite their metabolism is still active [172]. In this context, Jitschin and colleagues reported that CLL cells acquire an increased glucose dependency upon contact with stromal cells [173], in turn promoting glucose uptake in CLL cells by decreasing mitochondrial stress and apoptosis [174]. However, this outcome is still debated. As recently reported, CLL cocultured with MSCs enhance their mitochondrial metabolism, sustaining ATP production along with a nucleotide pool without any change in their proliferation [175]. In agreement with this, it has been reported that CLL cells rely on OXPHOS, and this metabolic process has been associated with poor prognostic outcomes such as IGHV unmutated disease, ZAP70 positivity, increased Rai stage, and higher β2 microglobulin [175,176]. Therefore, as also described, is possible that leukemic cells modify MSCs metabolism to satisfy their energy demand. Recently, it has been reported that, after contact with CLL cells, MSCs switch their metabolism towards OXPHOS whit consequent lower glucose usage which might be an advantage for CLL survival [174].
As things stand, it might be speculated that MSCs in the CLL context have a crucial role in supporting the malignant clone. In agreement with this, Dig and colleagues demonstrated that platelet-derived growth factor (PDGF) secreted by CLL cells activate its receptor PDGFR on MSC membrane [177]. PDGF/PDGFR interaction enhances MSC proliferation, therefore enhancing the production of VEGF and promoting the neovascularization known to be related to disease progression [178,179].
2.5. Role of MSCs in in Multiple myeloma
Multiple myeloma (MM) is a hematological disease characterized by uncontrolled proliferation and expansion of monoclonal plasma cells (PCs) in the BMME that leads to the overproduction of abnormal monoclonal protein and immunoglobulin free light chain. MM evolves from an asymptomatic pre-malignant stage termed monoclonal gammopathy of undetermined clinical significance (MGUS), eventually progressing to an intermediate but more advanced pre-malignant stage defined as smoldering myeloma (SMM) and, finally, to overt myeloma [1]. Although the initiation of malignant transformation is based on genetic and epigenetic alterations occurring in MM cells, the BMME plays a key role in mediating survival, proliferation, drug resistance, and progression of the disease [2]. In particular, the interactions of the malignant PCs with other cells in the BM niche, including MSCs, adipocytes, endothelial cells, osteoclasts, osteoblasts and immune cells lead to a host of problems including hypercalcemia, anemia, kidney failure, or bone lesions (i.e., CRAB criteria) [3]. Specifically, mutual modulation of phenotype and functions are observed between PCs and MSCs as a consequence of their bidirectional cross-talking. Bone disease is one of the most prominent clinical symptoms of MM patients, affecting the 80% of MM patients, and seriously impact the quality of life of patients [4]. As MSCs are osteoblasts progenitors, MM-MSCs actively contribute to the pathogenesis of myeloma bone disease. The adhesion of myeloma PCs to the stroma promotes the tumor cell secretion of several proteins, such as DKK1, which prevents the differentiation of MSCs into osteoblasts [5,6]. Importantly, MSCs not only contribute to bone disease because of their reduced osteogenic potential, but also because they ultimately promote activation of osteoclasts. Interacting with tumor cells, MSCs upregulate RANKL and reduce its soluble receptor OPG, thus prompting osteoclastogenesis through activation of RANKL-RANK signaling in osteoclasts [7].
MM-MSCs exhibit a distinct gene expression profile when compared to MSCs from healthy donors [8,9,10,11]. Particularly, Fernando and colleagues showed that the main downregulated networks in MM-MSCs are related to cell cycle progression, immune activation and bone metabolism, which might contribute to MM physiopathology [10]. In addition, the expression of specific genes differentiate MGUS-, SMM- and MM-MSCs and, interestingly, gene expression profile of MSCs from patients with PCs dyscrasias have an independent prognostic impact on clinical outcome [9]. In detail, Schinke et al. identified a prognostic MSC three-gene score, including collagen type IV alpha 1 (COL4A1), natriuretic peptide receptor 3 (NPR3) and integrin beta like 1 (ITGBL1), which is able to predict progression-free survival in MM patients and progression of MGUS/SMM to MM [9]. Of note, as MSC from patients which underwent completed treatment show a transcriptome essentially identical to that of patients at diagnosis, a persistent printing could maintain a niche prone to relapse [12]. Single cell sequencing also confirmed that current antitumor therapy fails to counteract MSC inflammation, highlighting their role in disease persistence [13]. MM-MSCs have an early senescent profile characterized by higher cell size, β-galactosidase increased activity, a Senescence-Associated Secretory Phenotype (SASP) and reduced proliferation due to the accumulation of cells in S phase [193,194]. This phenotypic change is primed by tumor PCs because healthy MSCs showed a phenotype similar with MM-MSCs after exposure to tumor cells [195]. The senescence of MM-MSCs also impairs their differentiation potential and enhances their tumor-supporting capacity [194].
Interestingly, the mechanism behind the establishment of such a phenotype is still unkown. Dicer1, an RNAse III endonuclease essential for miRNA biogenesis, has been demonstrated to be one of the key promoters of cellular senescence in MSCs [17,194]. Specifically, upregulation of Dicer1 in MM-MSCs reversed cellular senescence and promoted cell differentiation [194]. More recently, Cao et al. provided evidence for a link between MSC senescence and MM progression investigating genes coexpressed by tumor PCs and MM-MSCs [63]. The authors identified a set of signatures of fourteen genes linked to MSC senescence which is essential in predicting MM progression [63].
Immunosuppression is a common feature of MM associated with disease evolution [196]. Concerning this, our group previously demonstrated that MM-MSCs promote immunosuppressive abilities of surrounding myeloid cells by promoting the expansion of granulocyte-like myeloid derived suppressor cells (G-MDSCs) [197] and immunosuppressive neutrophils [61], leading to cancer cell immune evasion. As the immunological dysfunction of MSCs was observed already in SMM stromal cells but not in MGUS ones, the activation of a MSC-induced immunosuppressive microenvironment might contribute to the transition from MGUS to MM as an evolutionary advantage acquired during the multistep development of MM. Of note, MSC from relapsed patients have an increased immunosuppressive ability compared to those from patients in remission [191]. The support of malignant clone proliferation by MM-MSCs is mediated by stromal activation of PD1/PDL-1 axis disrupting T cell immune response [198,199]. Similarly, MM-MSCs are able to induce NK cell exhaustion by activation of CD155/TIGT signaling [200]. Furthermore, the tumorigenic behavior of MM-MSCs is directly mediated by tumor PCs through the activation of a TLR4-primed inflammatory phenotype [61]. Using a single-cell transcriptomic approach, De Jong et al. identified specific inflammatory MSCs in MM milieu [192]. As successful antimyeloma therapy is unable to revert MSC inflammatory status, not even in patients whose are undetectable by flow cytometry [192], inflammatory-primed MSCs could be also epigenetically reprogrammed maintaining their dysfunction also in absence of tumor cells. In agreement, epigenetic alterations of stromal cells have been recently associated to the impairment of bone formation in MM patients [39,201]. Furthermore, members of the Homeobox family, known as key drivers of osteogenic differentiation, result epigenetically and transcriptionally deregulated in MM-MSCs [39]. Of note, epigenetic alterations in stromal compartment already occur in the asymptomatic phases of myeloma and most of these changes are specific to each stage [39]. This phenomenon could be associated to the expansion of MSCs subpopulations which promote tumor progression just as in MM cells [202].
The activation of an immunosuppressive and pro-inflammatory phenotype has been associated to a metabolic rewire of MSCs towards a more glycolytic metabolism, in turn required to sustain the secretion of immunosuppressive factors [203]. In agreement, we recently showed that MM-MSCs are more glycolytic than the normal counterpart [70]. Their relatively independence on mitochondrial metabolism impact MM cell energy making MM-MSCs inclined to transfer more mitochondria to tumor cells [70]. The uptake of functional mitochondria from MM-MSCs occur through several mechanisms, including tunneling nanotubes and CD38 [204], EVs as well as cell-to-cell contact and CXCL12/CXCR4 axis [70]. This mitochondrial trafficking supports the oxidative metabolism of tumor PCs favoring cancer growth and drug resistance [70,205,206,207,208].
3.1. Concluding Remarks and future perspectives
MSCs are key component of the BMME, wherein they exert multiple functions for supporting hematopoietic niche, tissue homeostasis, and immune system modulation. The interest in dissecting the role of MSCs in hematopoietic malignancies has vastly grown over the past years. As we discussed, the BM milieu leukemic transformation causes profound modifications of MSC phenotype, including their morphology and functions with the acquisition of the SASP, which strongly contributes to the development of a proinflammatory microenvironment. Evidence suggests that SASP-related secretome of MSCs might contribute to the progression from benign states to over malignancies [24]. Indeed, the progression of hematological cancers towards a more aggressive phenotype does not solely rely on intrinsic leukemic cell factors but are independently impacted by the biology of the surrounding microenvironment, including MSCs (Figure 2).
Reprogrammed stromal cells provide a nurturing niche that sustain tumor growth, clonal evolution, and drug resistance. Although it has been reported that MSCs from AML patients at time of disease remission recover healthy activities [113], inheritance of epigenetic alterations associated to MSC imprinting could lead to an autonomous status of stromal cells from neoplastic clone. In “absence/decrease” of clonal cells after targeted therapies, the persistence of this pathologic inflamed phenotype of MSCs might be a key component partially explaining disease relapse [209]. Moreover, the importance of BMME is highlighted by the prolonged time to stabilize engraftment after autologous HSPC transplantation. In this case, the prerequisite for transplant success is the rebuilding of the interplay between BMME and HSPCs.
For this reason, targeting the BM niche might represent a valuable novel strategy counteracting blood malignancy. Among the emerging targets the CXCL12/CXCR4 axis disrupts leukemic cell adhesion to MSCs mobilizing tumor cells into circulation and increasing drug-induced apoptosis [210,211,212]. In our own previous research, the inhibition of this axis also affects tumor/MSC metabolic coupling inhibiting mitochondria trafficking [70]. In this context, it is becoming increasingly clear the importance of the metabolic interplay between stromal and leukemic cells for promoting disease establishment and progression. The mitochondrial transfer support leukemic cell bioenergetics and antioxidant defenses, sparing them from high energetic cost of mitochondrial biogenesis. To understand which metabolic vulnerabilities can be targeted in the leukemic BMME might open new avenues to improve cancer therapy. Recently, niche-calcium homeostasis has been found to be involved in the reprogramming of MSCs into leukemic niche [113]. Compounds blocking the inward movement of calcium modify the transcriptomic and secretome profile of AML-MSCs restoring healthy functions [113]. Furthermore, the current focus has also been on age-related changes of MSCs which characterize the development of hematological cancers. For this reason, pharmacological approaches to eliminate senescent cells have been investigated [213]. Concerning MSCs, targeting senescent MSCs it has been demonstrated a possible strategy to recover hematopoietic supportive capacity of stromal cells improving metabolic fitness of HSPCs [214]. Therefore, the utility of senolytic agents as a potential intervention in the context of hematological cancer might be a promising new strategy to both inhibit pro-tumorigenic effects of inflamed MSCs and improve their hematopoietic supportive capacity.
In the framework of BMME, the complex interplay between leukemic cells and MSCs include dynamic cell–cell interactions and organization, release of soluble factors and EVs, immunoregulatory properties which hide unrecognized leukemogenic events with innovative treatment opportunities. Therefore, extended investigations of relationship occurring in the leukemic niche may revolutionize treatment strategies to disadvantage cancer cells using niche-directed therapies.
Author Contributions
Conceptualization: S.G., A.D., C.G., D.T. and GAP; review design: S.G., G.C., I.D., T.Z., A.S., ELS; interpretation: S.G., G.S., FDR, A.D., C.G., A.R., D.T. and GAP.; figures and artworks: D.T., C.G.; writing—original draft: S.G., A.D., FDR, A.S., C.G., D.T. and GAP. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wei, Q. and P.S. Frenette, Niches for Hematopoietic Stem Cells and Their Progeny. Immunity, 2018. 48(4): p. 632-648. [CrossRef]
- Ding, L. and S.J. Morrison, Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature, 2013. 495(7440): p. 231-5. [CrossRef]
- Cordeiro Gomes, A., et al., Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity, 2016. 45(6): p. 1219-1231. [CrossRef]
- Mendes, S.C., C. Robin, and E. Dzierzak, Mesenchymal progenitor cells localize within hematopoietic sites throughout ontogeny. Development, 2005. 132(5): p. 1127-36. [CrossRef]
- Fallati, A., et al., Mesenchymal Stromal Cells (MSCs): An Ally of B-Cell Acute Lymphoblastic Leukemia (B-ALL) Cells in Disease Maintenance and Progression within the Bone Marrow Hematopoietic Niche. Cancers (Basel), 2022. 14(14). [CrossRef]
- Kim, J.A., et al., Identification of a stroma-mediated Wnt/beta-catenin signal promoting self-renewal of hematopoietic stem cells in the stem cell niche. Stem Cells, 2009. 27(6): p. 1318-29. [CrossRef]
- Stier, S., et al., Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med, 2005. 201(11): p. 1781-91. [CrossRef]
- Nakamura, Y., et al., Isolation and characterization of endosteal niche cell populations that regulate hematopoietic stem cells. Blood, 2010. 116(9): p. 1422-32. [CrossRef]
- Crippa, S. and M.E. Bernardo, Mesenchymal Stromal Cells: Role in the BM Niche and in the Support of Hematopoietic Stem Cell Transplantation. Hemasphere, 2018. 2(6): p. e151. [CrossRef]
- Tormin, A., et al., CD146 expression on primary nonhematopoietic bone marrow stem cells is correlated with in situ localization. Blood, 2011. 117(19): p. 5067-77. [CrossRef]
- Sacchetti, B., et al., Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell, 2007. 131(2): p. 324-36. [CrossRef]
- Sugiyama, T., et al., Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity, 2006. 25(6): p. 977-88. [CrossRef]
- Jung, Y., et al., Regulation of SDF-1 (CXCL12) production by osteoblasts; a possible mechanism for stem cell homing. Bone, 2006. 38(4): p. 497-508. [CrossRef]
- Krevvata, M., et al., Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood, 2014. 124(18): p. 2834-46. [CrossRef]
- Bowers, M., et al., Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood, 2015. 125(17): p. 2678-88. [CrossRef]
- Kode, A., et al., Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature, 2014. 506(7487): p. 240-4. [CrossRef]
- Zhao, Y., et al., Down-regulation of Dicer1 promotes cellular senescence and decreases the differentiation and stem cell-supporting capacities of mesenchymal stromal cells in patients with myelodysplastic syndrome. Haematologica, 2015. 100(2): p. 194-204. [CrossRef]
- Novoseletskaya, E., et al., Mesenchymal Stromal Cell-Produced Components of Extracellular Matrix Potentiate Multipotent Stem Cell Response to Differentiation Stimuli. Front Cell Dev Biol, 2020. 8: p. 555378. [CrossRef]
- Aithal, A.P., L.K. Bairy, and R.N. Seetharam, Safety and therapeutic potential of human bone marrow-derived mesenchymal stromal cells in regenerative medicine. Stem Cell Investig, 2021. 8: p. 10. [CrossRef]
- Bernardo, M.E. and W.E. Fibbe, Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell, 2013. 13(4): p. 392-402. [CrossRef]
- Nauta, A.J., et al., Mesenchymal stem cells inhibit generation and function of both CD34+-derived and monocyte-derived dendritic cells. J Immunol, 2006. 177(4): p. 2080-7. [CrossRef]
- Tyndall, A. and A. Gratwohl, Adult stem cell transplantation in autoimmune disease. Curr Opin Hematol, 2009. 16(4): p. 285-91. [CrossRef]
- Plakhova, N., et al., Mesenchymal stromal cell senescence in haematological malignancies. Cancer Metastasis Rev, 2023. 42(1): p. 277-296. [CrossRef]
- Ghobrial, I.M., et al., The bone-marrow niche in MDS and MGUS: implications for AML and MM. Nat Rev Clin Oncol, 2018. 15(4): p. 219-233. [CrossRef]
- Weng, Z., et al., Mesenchymal Stem/Stromal Cell Senescence: Hallmarks, Mechanisms, and Combating Strategies. Stem Cells Transl Med, 2022. 11(4): p. 356-371. [CrossRef]
- Hu, D., et al., Cellular senescence and hematological malignancies: From pathogenesis to therapeutics. Pharmacol Ther, 2021. 223: p. 107817. [CrossRef]
- Chen, X., et al., Senescent Mesenchymal Stem Cells in Myelodysplastic Syndrome: Functional Alterations, Molecular Mechanisms, and Therapeutic Strategies. Front Cell Dev Biol, 2020. 8: p. 617466. [CrossRef]
- Vanegas, N.P., et al., Leukemia-Induced Cellular Senescence and Stemness Alterations in Mesenchymal Stem Cells Are Reversible upon Withdrawal of B-Cell Acute Lymphoblastic Leukemia Cells. Int J Mol Sci, 2021. 22(15). [CrossRef]
- O’Hagan-Wong, K., et al., Increased IL-6 secretion by aged human mesenchymal stromal cells disrupts hematopoietic stem and progenitor cells’ homeostasis. Oncotarget, 2016. 7(12): p. 13285-96. [CrossRef]
- Adams, P.D., H. Jasper, and K.L. Rudolph, Aging-Induced Stem Cell Mutations as Drivers for Disease and Cancer. Cell Stem Cell, 2015. 16(6): p. 601-12. [CrossRef]
- Blau, O., et al., Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp Hematol, 2007. 35(2): p. 221-9. [CrossRef]
- Huang, J.C., et al., Mesenchymal stromal cells derived from acute myeloid leukemia bone marrow exhibit aberrant cytogenetics and cytokine elaboration. Blood Cancer J, 2015. 5(4): p. e302. [CrossRef]
- Blau, O., et al., Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood, 2011. 118(20): p. 5583-92. [CrossRef]
- von der Heide, E.K., et al., Molecular alterations in bone marrow mesenchymal stromal cells derived from acute myeloid leukemia patients. Leukemia, 2017. 31(5): p. 1069-1078. [CrossRef]
- Medyouf, H., et al., Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell, 2014. 14(6): p. 824-37. [CrossRef]
- Geyh, S., et al., Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia, 2013. 27(9): p. 1841-51. [CrossRef]
- Sui, B.D., et al., Epigenetic Regulation of Mesenchymal Stem Cell Homeostasis. Trends Cell Biol, 2020. 30(2): p. 97-116. [CrossRef]
- Bhagat, T.D., et al., Epigenetically Aberrant Stroma in MDS Propagates Disease via Wnt/beta-Catenin Activation. Cancer Res, 2017. 77(18): p. 4846-4857. [CrossRef]
- Garcia-Gomez, A., et al., Targeting aberrant DNA methylation in mesenchymal stromal cells as a treatment for myeloma bone disease. Nat Commun, 2021. 12(1): p. 421. [CrossRef]
- Poon, Z., et al., Correction: Bone marrow MSCs in MDS: contribution towards dysfunctional hematopoiesis and potential targets for disease response to hypomethylating therapy. Leukemia, 2019. 33(6): p. 1542. [CrossRef]
- Huang, J., et al., Use of methylation profiling to identify significant differentially methylated genes in bone marrow mesenchymal stromal cells from acute myeloid leukemia. Int J Mol Med, 2018. 41(2): p. 679-686. [CrossRef]
- Frassanito, M.A., et al., Halting pro-survival autophagy by TGFbeta inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia, 2016. 30(3): p. 640-8. [CrossRef]
- Zhai, Y., et al., Growth differentiation factor 15 contributes to cancer-associated fibroblasts-mediated chemo-protection of AML cells. J Exp Clin Cancer Res, 2016. 35(1): p. 147. [CrossRef]
- Burt, R., et al., Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood, 2019. 134(17): p. 1415-1429. [CrossRef]
- Zi, F.M., et al., Fibroblast activation protein protects bortezomib-induced apoptosis in multiple myeloma cells through beta-catenin signaling pathway. Cancer Biol Ther, 2014. 15(10): p. 1413-22. [CrossRef]
- Duan, C.W., et al., Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell, 2014. 25(6): p. 778-93. [CrossRef]
- Pan, C., et al., Mesenchymal Stem Cells With Cancer-Associated Fibroblast-Like Phenotype Stimulate SDF-1/CXCR4 Axis to Enhance the Growth and Invasion of B-Cell Acute Lymphoblastic Leukemia Cells Through Cell-to-Cell Communication. Front Cell Dev Biol, 2021. 9: p. 708513. [CrossRef]
- Miyazaki, Y., et al., Adipose-derived mesenchymal stem cells differentiate into pancreatic cancer-associated fibroblasts in vitro. FEBS Open Bio, 2020. 10(11): p. 2268-2281. [CrossRef]
- Rubinstein-Achiasaf, L., et al., Persistent Inflammatory Stimulation Drives the Conversion of MSCs to Inflammatory CAFs That Promote Pro-Metastatic Characteristics in Breast Cancer Cells. Cancers (Basel), 2021. 13(6). [CrossRef]
- Paunescu, V., et al., Tumour-associated fibroblasts and mesenchymal stem cells: more similarities than differences. J Cell Mol Med, 2011. 15(3): p. 635-46. [CrossRef]
- Longhitano, L., et al., The Role of Inflammation and Inflammasome in Myeloproliferative Disease. J Clin Med, 2020. 9(8). [CrossRef]
- Zhu, Y., et al., Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia, 2009. 23(5): p. 925-33. [CrossRef]
- Sun, Z., S. Wang, and R.C. Zhao, The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J Hematol Oncol, 2014. 7: p. 14. [CrossRef]
- Zhang, L., et al., Bone marrow-derived mesenchymal stem/stromal cells in patients with acute myeloid leukemia reveal transcriptome alterations and deficiency in cellular vitality. Stem Cell Res Ther, 2021. 12(1): p. 365. [CrossRef]
- Garcia-Gomez, A., et al., Transcriptomic profile induced in bone marrow mesenchymal stromal cells after interaction with multiple myeloma cells: implications in myeloma progression and myeloma bone disease. Oncotarget, 2014. 5(18): p. 8284-305. [CrossRef]
- Giallongo, C., et al., Mesenchymal Stem Cells (MSC) Regulate Activation of Granulocyte-Like Myeloid Derived Suppressor Cells (G-MDSC) in Chronic Myeloid Leukemia Patients. PLoS One, 2016. 11(7): p. e0158392. [CrossRef]
- Poggi, A. and M. Giuliani, Mesenchymal Stromal Cells Can Regulate the Immune Response in the Tumor Microenvironment. Vaccines (Basel), 2016. 4(4). [CrossRef]
- Azevedo, R.I., et al., Mesenchymal stromal cells induce regulatory T cells via epigenetic conversion of human conventional CD4 T cells in vitro. Stem Cells, 2020. 38(8): p. 1007-1019. [CrossRef]
- Zheng, L., et al., The immunological role of mesenchymal stromal cells in patients with myelodysplastic syndrome. Front Immunol, 2022. 13: p. 1078421. [CrossRef]
- Holthof, L.C., et al., Bone Marrow Mesenchymal Stromal Cells Can Render Multiple Myeloma Cells Resistant to Cytotoxic Machinery of CAR T Cells through Inhibition of Apoptosis. Clin Cancer Res, 2021. 27(13): p. 3793-3803. [CrossRef]
- Giallongo, C., et al., TLR4 signaling drives mesenchymal stromal cells commitment to promote tumor microenvironment transformation in multiple myeloma. Cell Death Dis, 2019. 10(10): p. 704. [CrossRef]
- Li, W., et al., Mesenchymal stem cells: a double-edged sword in regulating immune responses. Cell Death Differ, 2012. 19(9): p. 1505-13. [CrossRef]
- Cao, Y.J., et al., MSC Senescence-Related Genes Are Associated with Myeloma Prognosis and Lipid Metabolism-Mediated Resistance to Proteasome Inhibitors. J Oncol, 2022. 2022: p. 4705654. [CrossRef]
- Waterman, R.S., et al., A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS One, 2010. 5(4): p. e10088. [CrossRef]
- Jackson, M.V., et al., Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells, 2016. 34(8): p. 2210-23. [CrossRef]
- Wang, L., et al., Regulation of Inflammatory Cytokine Storms by Mesenchymal Stem Cells. Front Immunol, 2021. 12: p. 726909. [CrossRef]
- Loussouarn, C., et al., Mesenchymal Stromal Cell-Derived Extracellular Vesicles Regulate the Mitochondrial Metabolism via Transfer of miRNAs. Front Immunol, 2021. 12: p. 623973. [CrossRef]
- Thomas, M.A., et al., Human mesenchymal stromal cells release functional mitochondria in extracellular vesicles. Front Bioeng Biotechnol, 2022. 10: p. 870193. [CrossRef]
- Phetfong, J., et al., Bone marrow-mesenchymal stem cell-derived extracellular vesicles affect proliferation and apoptosis of leukemia cells in vitro. FEBS Open Bio, 2022. 12(2): p. 470-479. [CrossRef]
- Giallongo, C., et al., CXCL12/CXCR4 axis supports mitochondrial trafficking in tumor myeloma microenvironment. Oncogenesis, 2022. 11(1): p. 6. [CrossRef]
- Vilaplana-Lopera, N., et al., Crosstalk between AML and stromal cells triggers acetate secretion through the metabolic rewiring of stromal cells. Elife, 2022. 11. [CrossRef]
- Matamala Montoya, M., et al., Metabolic changes underlying drug resistance in the multiple myeloma tumor microenvironment. Front Oncol, 2023. 13: p. 1155621. [CrossRef]
- Chiu, M., et al., Myeloma Cells Deplete Bone Marrow Glutamine and Inhibit Osteoblast Differentiation Limiting Asparagine Availability. Cancers (Basel), 2020. 12(11). [CrossRef]
- Giallongo, C., et al., Inhibition of TLR4 Signaling Affects Mitochondrial Fitness and Overcomes Bortezomib Resistance in Myeloma Plasma Cells. Cancers (Basel), 2020. 12(8). [CrossRef]
- Nemkov, T., A. D’Alessandro, and J.A. Reisz, Metabolic underpinnings of leukemia pathology and treatment. Cancer Rep (Hoboken), 2019. 2(2): p. e1139. [CrossRef]
- Kim, J. and R.J. DeBerardinis, Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab, 2019. 30(3): p. 434-446. [CrossRef]
- Kumar, B., et al., Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia, 2018. 32(3): p. 575-587. [CrossRef]
- Dabbah, M., et al., Multiple myeloma BM-MSCs increase the tumorigenicity of MM cells via transfer of VLA4-enriched microvesicles. Carcinogenesis, 2020. 41(1): p. 100-110. [CrossRef]
- Dabbah, M., et al., Microvesicles derived from normal and multiple myeloma bone marrow mesenchymal stem cells differentially modulate myeloma cells’ phenotype and translation initiation. Carcinogenesis, 2017. 38(7): p. 708-716. [CrossRef]
- Dotson, J.L. and Y. Lebowicz, Myelodysplastic Syndrome, in StatPearls. 2023: Treasure Island (FL) ineligible companies. Disclosure: Yehuda Lebowicz declares no relevant financial relationships with ineligible companies.
- Alaggio, R., et al., The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia, 2022. 36(7): p. 1720-1748. [CrossRef]
- Calvi, L.M., A.J. Li, and M.W. Becker, What is the role of the microenvironment in MDS? Best Pract Res Clin Haematol, 2019. 32(4): p. 101113. [CrossRef]
- Lopez-Villar, O., et al., Both expanded and uncultured mesenchymal stem cells from MDS patients are genomically abnormal, showing a specific genetic profile for the 5q- syndrome. Leukemia, 2009. 23(4): p. 664-72. [CrossRef]
- Aanei, C.M., et al., Intrinsic growth deficiencies of mesenchymal stromal cells in myelodysplastic syndromes. Stem Cells Dev, 2012. 21(10): p. 1604-15. [CrossRef]
- Farr, J.N., et al., Identification of Senescent Cells in the Bone Microenvironment. J Bone Miner Res, 2016. 31(11): p. 1920-1929. [CrossRef]
- Fei, C., et al., Senescence of bone marrow mesenchymal stromal cells is accompanied by activation of p53/p21 pathway in myelodysplastic syndromes. Eur J Haematol, 2014. 93(6): p. 476-86. [CrossRef]
- Jann, J.C., et al., Bone marrow derived stromal cells from myelodysplastic syndromes are altered but not clonally mutated in vivo. Nat Commun, 2021. 12(1): p. 6170. [CrossRef]
- Maurizi, G., et al., DNA demethylating therapy reverts mesenchymal stromal cells derived from high risk myelodysplastic patients to a normal phenotype. Br J Haematol, 2017. 177(5): p. 818-822. [CrossRef]
- Wenk, C., et al., Direct modulation of the bone marrow mesenchymal stromal cell compartment by azacitidine enhances healthy hematopoiesis. Blood Adv, 2018. 2(23): p. 3447-3461. [CrossRef]
- Raaijmakers, M.H., et al., Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature, 2010. 464(7290): p. 852-7. [CrossRef]
- Zambetti, N.A., et al., Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell, 2016. 19(5): p. 613-627. [CrossRef]
- Lambert, C., Y. Wu, and C. Aanei, Bone Marrow Immunity and Myelodysplasia. Front Oncol, 2016. 6: p. 172. [CrossRef]
- Kobune, M., et al., Stromal cells expressing hedgehog-interacting protein regulate the proliferation of myeloid neoplasms. Blood Cancer J, 2012. 2(9): p. e87. [CrossRef]
- Giallongo, C., et al., MacroH2A1.1 as a crossroad between epigenetics, inflammation and metabolism of mesenchymal stromal cells in myelodysplastic syndromes. Cell Death Dis, 2023. 14(10): p. 686. [CrossRef]
- Chen, S., et al., Massive parallel RNA sequencing of highly purified mesenchymal elements in low-risk MDS reveals tissue-context-dependent activation of inflammatory programs. Leukemia, 2016. 30(9): p. 1938-42. [CrossRef]
- Ping, Z., et al., Activation of NF-kappaB driven inflammatory programs in mesenchymal elements attenuates hematopoiesis in low-risk myelodysplastic syndromes. Leukemia, 2019. 33(2): p. 536-541. [CrossRef]
- Pleyer, L., P. Valent, and R. Greil, Mesenchymal Stem and Progenitor Cells in Normal and Dysplastic Hematopoiesis-Masters of Survival and Clonality? Int J Mol Sci, 2016. 17(7). [CrossRef]
- Calkoen, F.G., et al., Despite differential gene expression profiles pediatric MDS derived mesenchymal stromal cells display functionality in vitro. Stem Cell Res, 2015. 14(2): p. 198-210. [CrossRef]
- Kim, M., et al., Increased expression of interferon signaling genes in the bone marrow microenvironment of myelodysplastic syndromes. PLoS One, 2015. 10(3): p. e0120602. [CrossRef]
- Hayashi, Y., et al., MDS cells impair osteolineage differentiation of MSCs via extracellular vesicles to suppress normal hematopoiesis. Cell Rep, 2022. 39(6): p. 110805. [CrossRef]
- Muntion, S., et al., Microvesicles from Mesenchymal Stromal Cells Are Involved in HPC-Microenvironment Crosstalk in Myelodysplastic Patients. PLoS One, 2016. 11(2): p. e0146722. [CrossRef]
- Jager, P., et al., Acute myeloid leukemia-induced functional inhibition of healthy CD34+ hematopoietic stem and progenitor cells. Stem Cells, 2021. 39(9): p. 1270-1284. [CrossRef]
- Pan, C., et al., Bone marrow mesenchymal stem cells in microenvironment transform into cancer-associated fibroblasts to promote the progression of B-cell acute lymphoblastic leukemia. Biomed Pharmacother, 2020. 130: p. 110610. [CrossRef]
- Leotta, S., et al., Prevention and Treatment of Acute Myeloid Leukemia Relapse after Hematopoietic Stem Cell Transplantation: The State of the Art and Future Perspectives. J Clin Med, 2022. 11(1). [CrossRef]
- Colmone, A., et al., Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science, 2008. 322(5909): p. 1861-5. [CrossRef]
- Kim, J.A., et al., Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res, 2015. 75(11): p. 2222-31. [CrossRef]
- Feng, X., et al., Cell circuits between leukemic cells and mesenchymal stem cells block lymphopoiesis by activating lymphotoxin beta receptor signaling. Elife, 2023. 12. [CrossRef]
- Mazur, G., et al., Increased monocyte chemoattractant protein 1 (MCP-1/CCL-2) serum level in acute myeloid leukemia. Neoplasma, 2007. 54(4): p. 285-9.
- Binato, R., et al., The molecular signature of AML mesenchymal stromal cells reveals candidate genes related to the leukemogenic process. Cancer Lett, 2015. 369(1): p. 134-43. [CrossRef]
- Lim, M., et al., Altered mesenchymal niche cells impede generation of normal hematopoietic progenitor cells in leukemic bone marrow. Leukemia, 2016. 30(1): p. 154-62. [CrossRef]
- Falconi, G., et al., Impairment of FOXM1 expression in mesenchymal cells from patients with myeloid neoplasms, de novo and therapy-related, may compromise their ability to support hematopoiesis. Sci Rep, 2022. 12(1): p. 21231. [CrossRef]
- Geyh, S., et al., Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia, 2016. 30(3): p. 683-91. [CrossRef]
- Borella, G., et al., Targeting the plasticity of mesenchymal stromal cells to reroute the course of acute myeloid leukemia. Blood, 2021. 138(7): p. 557-570. [CrossRef]
- Glait-Santar, C., et al., Functional Niche Competition Between Normal Hematopoietic Stem and Progenitor Cells and Myeloid Leukemia Cells. Stem Cells, 2015. 33(12): p. 3635-42. [CrossRef]
- Huang, X., S. Cho, and G.J. Spangrude, Hematopoietic stem cells: generation and self-renewal. Cell Death Differ, 2007. 14(11): p. 1851-9. [CrossRef]
- Beerman, I., et al., The evolving view of the hematopoietic stem cell niche. Exp Hematol, 2017. 50: p. 22-26. [CrossRef]
- Le, Y., et al., Adipogenic Mesenchymal Stromal Cells from Bone Marrow and Their Hematopoietic Supportive Role: Towards Understanding the Permissive Marrow Microenvironment in Acute Myeloid Leukemia. Stem Cell Rev Rep, 2016. 12(2): p. 235-44. [CrossRef]
- Azadniv, M., et al., Bone marrow mesenchymal stromal cells from acute myelogenous leukemia patients demonstrate adipogenic differentiation propensity with implications for leukemia cell support. Leukemia, 2020. 34(2): p. 391-403. [CrossRef]
- Battula, V.L., et al., AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight, 2017. 2(13). [CrossRef]
- Hanoun, M., et al., Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell, 2014. 15(3): p. 365-375. [CrossRef]
- Zhang, L., et al., Acute Myeloid Leukemia Cells Educate Mesenchymal Stromal Cells toward an Adipogenic Differentiation Propensity with Leukemia Promotion Capabilities. Adv Sci (Weinh), 2022. 9(16): p. 2105811. [CrossRef]
- Ruiz-Aparicio, P.F. and J.P. Vernot, Bone Marrow Aging and the Leukaemia-Induced Senescence of Mesenchymal Stem/Stromal Cells: Exploring Similarities. J Pers Med, 2022. 12(5). [CrossRef]
- Abdul-Aziz, A.M., et al., Acute myeloid leukemia induces protumoral p16INK4a-driven senescence in the bone marrow microenvironment. Blood, 2019. 133(5): p. 446-456. [CrossRef]
- Collado, M., M.A. Blasco, and M. Serrano, Cellular senescence in cancer and aging. Cell, 2007. 130(2): p. 223-33. [CrossRef]
- Lee, H.R., et al., The Chromatin Remodeling Complex CHD1 Regulates the Primitive State of Mesenchymal Stromal Cells to Control Their Stem Cell Supporting Activity. Stem Cells Dev, 2021. 30(7): p. 363-373. [CrossRef]
- Samudio, I., et al., The warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res, 2008. 68(13): p. 5198-205. [CrossRef]
- Wilde, L., et al., Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin Oncol, 2017. 44(3): p. 198-203. [CrossRef]
- Baccelli, I., et al., Mubritinib Targets the Electron Transport Chain Complex I and Reveals the Landscape of OXPHOS Dependency in Acute Myeloid Leukemia. Cancer Cell, 2019. 36(1): p. 84-99 e8. [CrossRef]
- Farge, T., et al., Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov, 2017. 7(7): p. 716-735. [CrossRef]
- Forte, D., et al., Bone Marrow Mesenchymal Stem Cells Support Acute Myeloid Leukemia Bioenergetics and Enhance Antioxidant Defense and Escape from Chemotherapy. Cell Metab, 2020. 32(5): p. 829-843 e9. [CrossRef]
- Marlein, C.R., et al., NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood, 2017. 130(14): p. 1649-1660. [CrossRef]
- Moschoi, R., et al., Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood, 2016. 128(2): p. 253-64. [CrossRef]
- Mistry, J.J., et al., Daratumumab inhibits acute myeloid leukaemia metabolic capacity by blocking mitochondrial transfer from mesenchymal stromal cells. Haematologica, 2021. 106(2): p. 589-592. [CrossRef]
- Wang, J., et al., Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J Hematol Oncol, 2018. 11(1): p. 11. [CrossRef]
- Shafat, M.S., et al., Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood, 2017. 129(10): p. 1320-1332. [CrossRef]
- Lee, K., J. Kerner, and C.L. Hoppel, Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J Biol Chem, 2011. 286(29): p. 25655-62. [CrossRef]
- Tucci, J., et al., Adipocytes Provide Fatty Acids to Acute Lymphoblastic Leukemia Cells. Front Oncol, 2021. 11: p. 665763. [CrossRef]
- Johnson, S.M., et al., Metabolic reprogramming of bone marrow stromal cells by leukemic extracellular vesicles in acute lymphoblastic leukemia. Blood, 2016. 128(3): p. 453-6. [CrossRef]
- Barbui, T., et al., The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J, 2018. 8(2): p. 15. [CrossRef]
- Nasillo, V., et al., Inflammatory Microenvironment and Specific T Cells in Myeloproliferative Neoplasms: Immunopathogenesis and Novel Immunotherapies. Int J Mol Sci, 2021. 22(4). [CrossRef]
- Rambaldi, B., et al., Heterogeneity of the bone marrow niche in patients with myeloproliferative neoplasms: ActivinA secretion by mesenchymal stromal cells correlates with the degree of marrow fibrosis. Ann Hematol, 2021. 100(1): p. 105-116. [CrossRef]
- Xie, J., et al., Bone mesenchymal stromal cells exhibit functional inhibition but no chromosomal aberrations in chronic myelogenous leukemia. Oncol Lett, 2019. 17(1): p. 999-1007. [CrossRef]
- Avanzini, M.A., et al., Functional and genetic aberrations of in vitro-cultured marrow-derived mesenchymal stromal cells of patients with classical Philadelphia-negative myeloproliferative neoplasms. Leukemia, 2014. 28(8): p. 1742-5. [CrossRef]
- Zhao, Z.G., et al., The characteristics and immunoregulatory functions of regulatory dendritic cells induced by mesenchymal stem cells derived from bone marrow of patient with chronic myeloid leukaemia. Eur J Cancer, 2012. 48(12): p. 1884-95. [CrossRef]
- Jafarzadeh, N., et al., Alteration of cellular and immune-related properties of bone marrow mesenchymal stem cells and macrophages by K562 chronic myeloid leukemia cell derived exosomes. J Cell Physiol, 2019. 234(4): p. 3697-3710. [CrossRef]
- Chai, C., et al., BCR-ABL1-driven exosome-miR130b-3p-mediated gap-junction Cx43 MSC intercellular communications imply therapies of leukemic subclonal evolution. Theranostics, 2023. 13(12): p. 3943-3963. [CrossRef]
- Agarwal, P., et al., Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell, 2019. 24(5): p. 769-784 e6. [CrossRef]
- Aggoune, D., et al., Bone marrow mesenchymal stromal cell (MSC) gene profiling in chronic myeloid leukemia (CML) patients at diagnosis and in deep molecular response induced by tyrosine kinase inhibitors (TKIs). Leuk Res, 2017. 60: p. 94-102. [CrossRef]
- Schepers, K., et al., Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell, 2013. 13(3): p. 285-99. [CrossRef]
- La Spina, E., et al., Mesenchymal stromal cells in tumor microenvironment remodeling of BCR-ABL negative myeloproliferative diseases. Front Oncol, 2023. 13: p. 1141610. [CrossRef]
- Abbonante, V., et al., Altered fibronectin expression and deposition by myeloproliferative neoplasm-derived mesenchymal stromal cells. Br J Haematol, 2016. 172(1): p. 140-4. [CrossRef]
- Ponce, C.C., et al., The relationship of the active and latent forms of TGF-beta1 with marrow fibrosis in essential thrombocythemia and primary myelofibrosis. Med Oncol, 2012. 29(4): p. 2337-44. [CrossRef]
- Badalucco, S., et al., Involvement of TGFbeta1 in autocrine regulation of proplatelet formation in healthy subjects and patients with primary myelofibrosis. Haematologica, 2013. 98(4): p. 514-7. [CrossRef]
- Schneider, R.K., et al., Activated fibronectin-secretory phenotype of mesenchymal stromal cells in pre-fibrotic myeloproliferative neoplasms. J Hematol Oncol, 2014. 7: p. 92. [CrossRef]
- Ciurea, S.O., et al., Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood, 2007. 110(3): p. 986-93. [CrossRef]
- Avanzini, M.A., et al., The spleen of patients with myelofibrosis harbors defective mesenchymal stromal cells. Am J Hematol, 2018. 93(5): p. 615-622. [CrossRef]
- Lu, M., et al., Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood, 2015. 126(8): p. 972-82. [CrossRef]
- Bedekovics, J., et al., Platelet derived growth factor receptor-beta (PDGFRbeta) expression is limited to activated stromal cells in the bone marrow and shows a strong correlation with the grade of myelofibrosis. Virchows Arch, 2013. 463(1): p. 57-65. [CrossRef]
- Longhitano, L., et al., IGFBP-6/sonic hedgehog/TLR4 signalling axis drives bone marrow fibrotic transformation in primary myelofibrosis. Aging (Albany NY), 2021. 13(23): p. 25055-25071. [CrossRef]
- Schneider, R.K., et al., Gli1(+) Mesenchymal Stromal Cells Are a Key Driver of Bone Marrow Fibrosis and an Important Cellular Therapeutic Target. Cell Stem Cell, 2017. 20(6): p. 785-800 e8. [CrossRef]
- Lataillade, J.J., et al., Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood, 2008. 112(8): p. 3026-35. [CrossRef]
- Martinaud, C., et al., Osteogenic Potential of Mesenchymal Stromal Cells Contributes to Primary Myelofibrosis. Cancer Res, 2015. 75(22): p. 4753-65. [CrossRef]
- Leimkuhler, N.B., et al., Heterogeneous bone-marrow stromal progenitors drive myelofibrosis via a druggable alarmin axis. Cell Stem Cell, 2021. 28(4): p. 637-652 e8. [CrossRef]
- Nasnas, P., et al., How I Manage Chronic Lymphocytic Leukemia. Hematol Rep, 2023. 15(3): p. 454-464. [CrossRef]
- Gattei, V., et al., Relevance of CD49d protein expression as overall survival and progressive disease prognosticator in chronic lymphocytic leukemia. Blood, 2008. 111(2): p. 865-73. [CrossRef]
- Hartmann, T.N., et al., Circulating B-cell chronic lymphocytic leukemia cells display impaired migration to lymph nodes and bone marrow. Cancer Res, 2009. 69(7): p. 3121-30. [CrossRef]
- Ding, W., et al., Bi-directional activation between mesenchymal stem cells and CLL B-cells: implication for CLL disease progression. Br J Haematol, 2009. 147(4): p. 471-83. [CrossRef]
- Lagneaux, L., et al., Adhesion to bone marrow stroma inhibits apoptosis of chronic lymphocytic leukemia cells. Leuk Lymphoma, 1999. 35(5-6): p. 445-53. [CrossRef]
- Lee, S., et al., Adhesion of B-Cell Chronic Lymphocytic Leukemia Cells to Marrow Stromal Cells is Mediated by alpha4beta1 but not beta2alphaL Integrin: MSC also Prevent Apoptosis of B-CLL Cells. Hematology, 2001. 5(6): p. 463-73. [CrossRef]
- Crompot, E., et al., Extracellular vesicles of bone marrow stromal cells rescue chronic lymphocytic leukemia B cells from apoptosis, enhance their migration and induce gene expression modifications. Haematologica, 2017. 102(9): p. 1594-1604. [CrossRef]
- Paggetti, J., et al., Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood, 2015. 126(9): p. 1106-17. [CrossRef]
- Vangapandu, H.V., et al., B-cell Receptor Signaling Regulates Metabolism in Chronic Lymphocytic Leukemia. Mol Cancer Res, 2017. 15(12): p. 1692-1703. [CrossRef]
- Jitschin, R., et al., Stromal cell-mediated glycolytic switch in CLL cells involves Notch-c-Myc signaling. Blood, 2015. 125(22): p. 3432-6. [CrossRef]
- von Heydebrand, F., et al., Protein kinase C-beta-dependent changes in the glucose metabolism of bone marrow stromal cells of chronic lymphocytic leukemia. Stem Cells, 2021. 39(6): p. 819-830. [CrossRef]
- Vangapandu, H.V., et al., The Stromal Microenvironment Modulates Mitochondrial Oxidative Phosphorylation in Chronic Lymphocytic Leukemia Cells. Neoplasia, 2017. 19(10): p. 762-771. [CrossRef]
- Rassenti, L.Z., et al., ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med, 2004. 351(9): p. 893-901. [CrossRef]
- Ding, W., et al., Platelet-derived growth factor (PDGF)-PDGF receptor interaction activates bone marrow-derived mesenchymal stromal cells derived from chronic lymphocytic leukemia: implications for an angiogenic switch. Blood, 2010. 116(16): p. 2984-93. [CrossRef]
- Kay, N.E., D.F. Jelinek, and L. Peterson, Angiogenesis in B-chronic lymphocytic leukemia. Leuk Res, 2001. 25(8): p. 709-10. [CrossRef]
- Kini, A.R., N.E. Kay, and L.C. Peterson, Increased bone marrow angiogenesis in B cell chronic lymphocytic leukemia. Leukemia, 2000. 14(8): p. 1414-8. [CrossRef]
- Zhang, W., et al., Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol, 2012. 14(3): p. 276-86. [CrossRef]
- Korde, N., S.Y. Kristinsson, and O. Landgren, Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): novel biological insights and development of early treatment strategies. Blood, 2011. 117(21): p. 5573-81. [CrossRef]
- Kawano, Y., et al., Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev, 2015. 263(1): p. 160-72. [CrossRef]
- Brigle, K. and B. Rogers, Pathobiology and Diagnosis of Multiple Myeloma. Semin Oncol Nurs, 2017. 33(3): p. 225-236. [CrossRef]
- Rasch, S., et al., Multiple Myeloma Associated Bone Disease. Cancers (Basel), 2020. 12(8). [CrossRef]
- Gunn, W.G., et al., A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK1 and interleukin-6: a potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells, 2006. 24(4): p. 986-91. [CrossRef]
- Fowler, J.A., et al., Bone marrow stromal cells create a permissive microenvironment for myeloma development: a new stromal role for Wnt inhibitor Dkk1. Cancer Res, 2012. 72(9): p. 2183-9. [CrossRef]
- Garcia-Gomez, A., et al., Multiple myeloma mesenchymal stromal cells: Contribution to myeloma bone disease and therapeutics. World J Stem Cells, 2014. 6(3): p. 322-43. [CrossRef]
- Todoerti, K., et al., Distinct transcriptional profiles characterize bone microenvironment mesenchymal cells rather than osteoblasts in relationship with multiple myeloma bone disease. Exp Hematol, 2010. 38(2): p. 141-53. [CrossRef]
- Schinke, C., et al., The Pattern of Mesenchymal Stem Cell Expression Is an Independent Marker of Outcome in Multiple Myeloma. Clin Cancer Res, 2018. 24(12): p. 2913-2919. [CrossRef]
- Fernando, R.C., et al., Transcriptome Analysis of Mesenchymal Stem Cells from Multiple Myeloma Patients Reveals Downregulation of Genes Involved in Cell Cycle Progression, Immune Response, and Bone Metabolism. Sci Rep, 2019. 9(1): p. 1056. [CrossRef]
- Lemaitre, L., et al., Imprinting of Mesenchymal Stromal Cell Transcriptome Persists even after Treatment in Patients with Multiple Myeloma. Int J Mol Sci, 2020. 21(11). [CrossRef]
- de Jong, M.M.E., et al., The multiple myeloma microenvironment is defined by an inflammatory stromal cell landscape. Nat Immunol, 2021. 22(6): p. 769-780. [CrossRef]
- Andre, T., et al., Evidences of early senescence in multiple myeloma bone marrow mesenchymal stromal cells. PLoS One, 2013. 8(3): p. e59756. [CrossRef]
- Guo, J., et al., Dicer1 downregulation by multiple myeloma cells promotes the senescence and tumor-supporting capacity and decreases the differentiation potential of mesenchymal stem cells. Cell Death Dis, 2018. 9(5): p. 512. [CrossRef]
- Yaccoby, S., et al., Inhibitory effects of osteoblasts and increased bone formation on myeloma in novel culture systems and a myelomatous mouse model. Haematologica, 2006. 91(2): p. 192-9.
- Romano, A., et al., Immunological dysregulation in multiple myeloma microenvironment. Biomed Res Int, 2014. 2014: p. 198539. [CrossRef]
- Giallongo, C., et al., Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget, 2016. 7(52): p. 85764-85775. [CrossRef]
- Chen, D., et al., Bone marrow-derived mesenchymal stem cells promote cell proliferation of multiple myeloma through inhibiting T cell immune responses via PD-1/PD-L1 pathway. Cell Cycle, 2018. 17(7): p. 858-867. [CrossRef]
- Liu, Z., et al., Bone marrow-derived mesenchymal stem cells inhibit CD8(+) T cell immune responses via PD-1/PD-L1 pathway in multiple myeloma. Clin Exp Immunol, 2021. 205(1): p. 53-62. [CrossRef]
- Liu, Z.Y., et al., CD155/TIGIT signalling plays a vital role in the regulation of bone marrow mesenchymal stem cell-induced natural killer-cell exhaustion in multiple myeloma. Clin Transl Med, 2022. 12(7): p. e861. [CrossRef]
- Giuliani, N., et al., Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood, 2005. 106(7): p. 2472-83. [CrossRef]
- van Nieuwenhuijzen, N., et al., From MGUS to Multiple Myeloma, a Paradigm for Clonal Evolution of Premalignant Cells. Cancer Res, 2018. 78(10): p. 2449-2456. [CrossRef]
- Liu, Y., et al., Commitment to Aerobic Glycolysis Sustains Immunosuppression of Human Mesenchymal Stem Cells. Stem Cells Transl Med, 2019. 8(1): p. 93-106. [CrossRef]
- Marlein, C.R., et al., CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res, 2019. 79(9): p. 2285-2297. [CrossRef]
- Matula, Z., et al., Stromal Cells Serve Drug Resistance for Multiple Myeloma via Mitochondrial Transfer: A Study on Primary Myeloma and Stromal Cells. Cancers (Basel), 2021. 13(14). [CrossRef]
- Matula, Z., et al., The Effect of Belantamab Mafodotin on Primary Myeloma-Stroma Co-Cultures: Asymmetrical Mitochondrial Transfer between Myeloma Cells and Autologous Bone Marrow Stromal Cells. Int J Mol Sci, 2023. 24(6). [CrossRef]
- Barbato, A., et al., Lactate trafficking inhibition restores sensitivity to proteasome inhibitors and orchestrates immuno-microenvironment in multiple myeloma. Cell Prolif, 2023. 56(4): p. e13388. [CrossRef]
- Barbato, A., et al., Mitochondrial Bioenergetics at the Onset of Drug Resistance in Hematological Malignancies: An Overview. Front Oncol, 2020. 10: p. 604143. [CrossRef]
- Desterke, C., et al., Inflammation as a Keystone of Bone Marrow Stroma Alterations in Primary Myelofibrosis. Mediators Inflamm, 2015. 2015: p. 415024. [CrossRef]
- Azab, A.K., et al., CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood, 2009. 113(18): p. 4341-51. [CrossRef]
- Cancilla, D., M.P. Rettig, and J.F. DiPersio, Targeting CXCR4 in AML and ALL. Front Oncol, 2020. 10: p. 1672. [CrossRef]
- Giannoni, P., et al., An interaction between hepatocyte growth factor and its receptor (c-MET) prolongs the survival of chronic lymphocytic leukemic cells through STAT3 phosphorylation: a potential role of mesenchymal cells in the disease. Haematologica, 2011. 96(7): p. 1015-23. [CrossRef]
- Pan, J., et al., Inhibition of Bcl-2/xl With ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int J Radiat Oncol Biol Phys, 2017. 99(2): p. 353-361. [CrossRef]
- Hellmich, C., et al., p16INK4A-dependent senescence in the bone marrow niche drives age-related metabolic changes of hematopoietic progenitors. Blood Adv, 2023. 7(2): p. 256-268. [CrossRef]
Figure 1.
Schematic representation showing dynamic activity of HSPC and MSC when BMME is at homeostasis. The continuous interplay between HSCPs and stromal cells is essential for ensuring normal hematopoiesis. In the hematological cancer scenario, this complex interplay is deeply dysregulated favoring trafficking and infiltration of cancer cells into a protective BM niche.
Figure 1.
Schematic representation showing dynamic activity of HSPC and MSC when BMME is at homeostasis. The continuous interplay between HSCPs and stromal cells is essential for ensuring normal hematopoiesis. In the hematological cancer scenario, this complex interplay is deeply dysregulated favoring trafficking and infiltration of cancer cells into a protective BM niche.
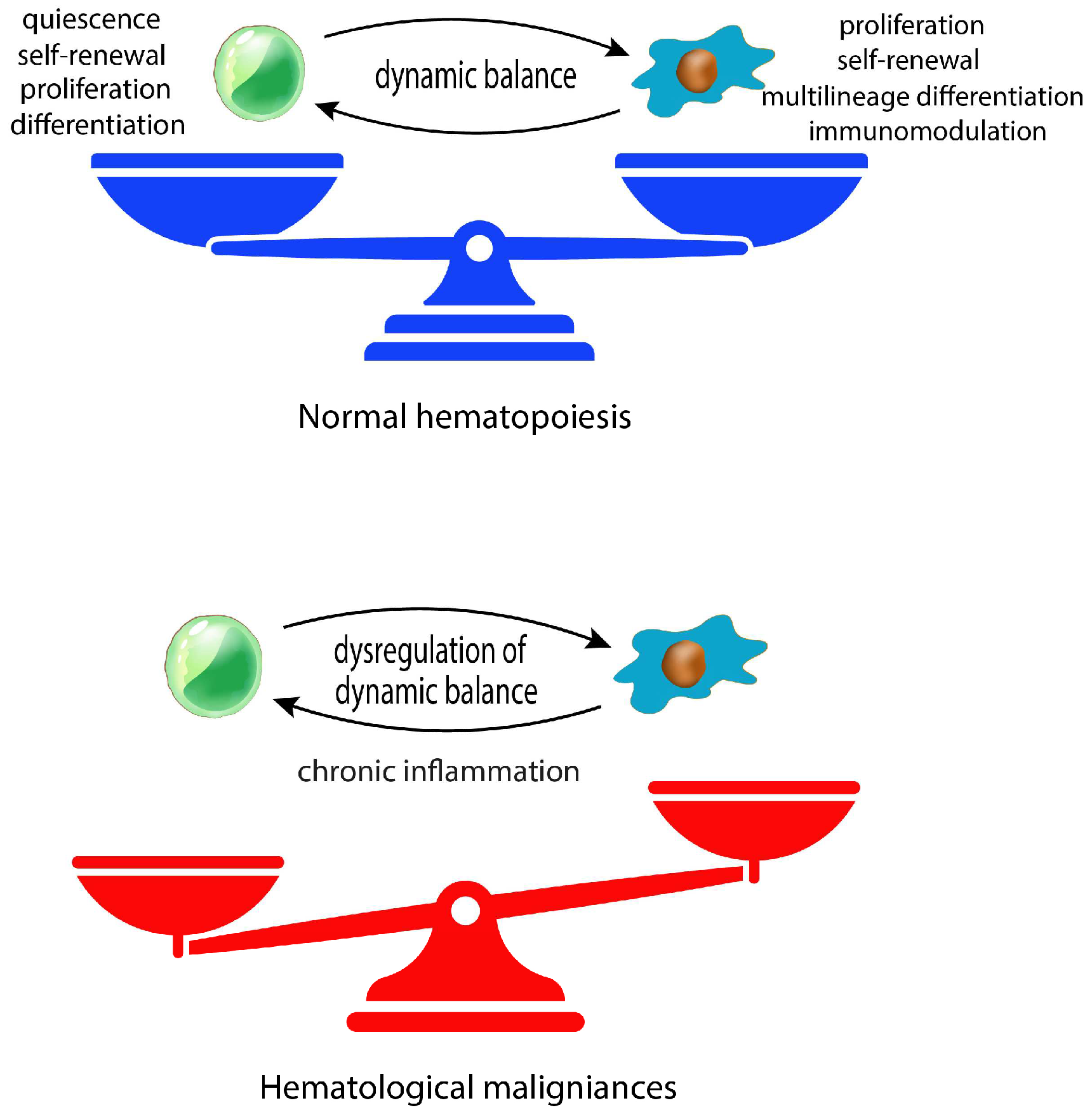
Figure 2.
The role of MSCs in different hematological malignancies. The illustration shows how dynamic interactions between malignant cells and MSCs eventually inducing transcriptomic and epigenetic alterations affect stromal secretome and multipotency. A common hallmark of MSCs in hematological cancer is the acquisition of a senescent phenotype associated to a pro-inflammatory profile. Moreover, the interplay between cancer and stromal cells supports a complex metabolic coupling sustaining immune suppressive activity, tumor growth and drug resistance.
Figure 2.
The role of MSCs in different hematological malignancies. The illustration shows how dynamic interactions between malignant cells and MSCs eventually inducing transcriptomic and epigenetic alterations affect stromal secretome and multipotency. A common hallmark of MSCs in hematological cancer is the acquisition of a senescent phenotype associated to a pro-inflammatory profile. Moreover, the interplay between cancer and stromal cells supports a complex metabolic coupling sustaining immune suppressive activity, tumor growth and drug resistance.
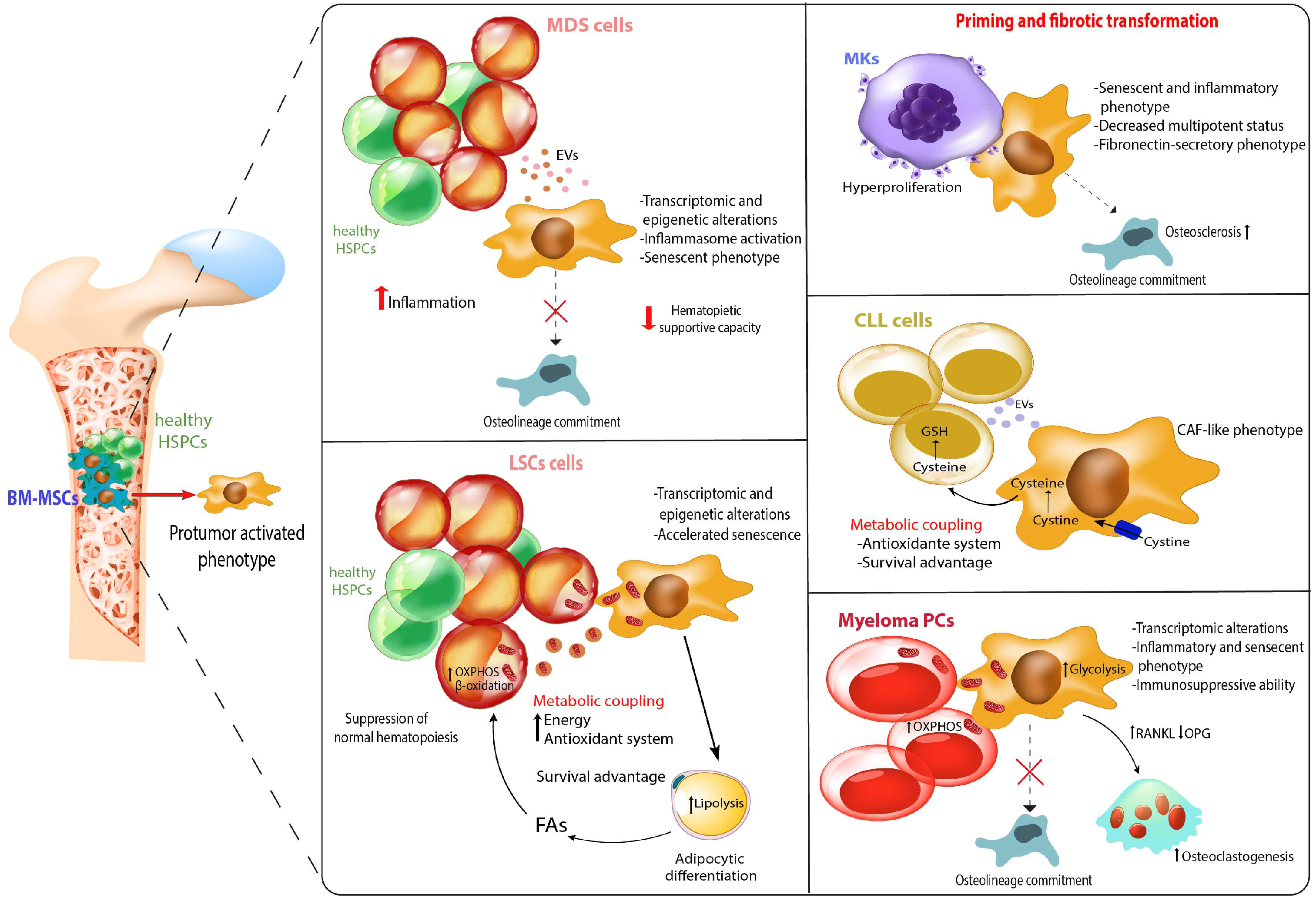
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



