
Submitted:
31 October 2023
Posted:
01 November 2023
You are already at the latest version
Abstract
With an estimated 10 million people infected, the deltaretrovirus human T-cell lymphotropic virus type 1 (HTLV-1) is the second most prevalent pathogenic retrovirus in humans after HIV-1. Like HIV-1, HTLV-1 overwhelmingly persists in a host via a reservoir of latently infected CD4+ T cells. Although most patients are asymptomatic, HTLV-1-associated pathologies are often debilitating and include childhood infective dermatitis and adult T-cell leukaemia/lymphoma (ATLL), which presents in mature adulthood and is associated with poor prognosis with short overall survival despite treatment. Curiously, the strongest indicator for the development of ATLL is the acquisition of HTLV-1 through breastfeeding. There are no therapeutic or preventative regimens for HTLV-1. However, antiretrovirals (ARVs), which target the essential retrovirus enzymes, have been developed for and transformed HIV therapy. Since the architectures of retroviral enzyme active sites are highly conserved, some HIV-specific compounds may be active against HTLV-1. Here, we expand on our work showing that integrase strand transfer inhibitors (INSTIs) and some nucleoside reverse transcriptase inhibitors (NRTIs) block HTLV-1 transmission in cell culture. Specifically, we find that dolutegravir, the INSTI currently recommended as the basis of all new combination antiretroviral therapy prescriptions, and the latest prodrug formulation of the NRTI tenofovir, tenofovir alafenamide, also potently inhibit HTLV-1 infection in cell culture. Our results, if replicated in a clinical setting, could see transmission rates of HTLV-1 and future caseloads of HTLV-1-associated pathologies like ATLL dramatically cut via the simple redistribution of already widely available HIV pills to HTLV-1 endemic areas. Considering our findings with the old medical saying ‘it’s better to prevent than cure’, we highly recommend the inclusion of INSTIs and tenofovir prodrugs in upcoming HTLV-1 clinical trials as potential prophylactics.
Keywords:
HTLV-1
; integrase
; INSTI
; dolutegravir
; reverse transcriptase
; TAF
; capsid
; lenacapavir
; vertical transmission
; PrEP
Introduction
There are few better indicators for the advances in medical care of the 20th century than our current abilities to treat and prevent infectious diseases1. Alongside improved sanitation and the advent of mass vaccination efforts, the availability of drugs to control infection has contributed enormously to improvements in humanity’s collective health. Nowadays, quick recovery with mild symptoms is the norm following diagnoses with some pathogens that would otherwise have meant certain death or physical disability less than a century ago. Human immunodeficiency virus (HIV) is one pathogen whose treatment has been radically transformed, exclusively, by the advent of synthetic antiretroviral (ARV) drugs. Specifically, the single-pill regimen for combination antiretroviral therapy (cART) provided a convenient option for patients, leading to improved adherence to therapy and, consequently, to sustained suppressed viraemia and a dramatic increase in life expectancy for those who promptly commenced treatment2.
Perhaps, owing to its endemic geographic distribution to non-western regions and long incubation period before the onset of fatality-associated disease, Human T-cell lymphotropic virus type 1 (HTLV-1), the first pathogenic human retrovirus to be identified, still has no approved therapeutic or preventative regimen 3. However, the mortality and morbidity stemming from the HTLV-1 associated diseases, acute T-cell leukaemia/lymphoma (ATLL) and HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP), necessitate the development of effective clinical interventions. ATLL is an aggressive lymphoproliferative CD4+ malignancy seen in 5% of HTLV-1 carriers, for which there are limited treatment options, with median survival following diagnosis remaining stagnant at less than a year since the 1990s. Another 1% of HTLV-1+ individuals develop HAM/TSP, a blanket term for a group of neurodegenerative disorders which can leave patients wheelchair dependent 4.
All retroviral genomes encode the enzymes reverse transcriptase (RT), integrase (IN) and protease5. RT converts single-stranded retroviral RNA genomes into double-stranded viral DNA (vDNA), which is then integrated into the host genome by IN to yield the provirus. Host-derived RNA polymerase and ribosomal machinery convert the provirus into viral polypeptides, which PR cleaves into mature, functional units. Of note, genetic and structural analysis indicates that the active site of each of these enzymes has a conserved architecture across all genera within the orthoretrovirinae subfamily 6–8. Therefore, orthosteric synthetics developed for a specific virus within the subfamily, like HIV-1, are likely to be effective against orthoretroviruses from an evolutionary distant genus. To this end, there already exists a small body of literature demonstrating that all approved INSTIs tested against HTLV-1 so far, and the NRTI tenofovir disoproxil fumarate (TDF), potently prevent cellular transmission of the deltaretrovirus 9–12.
In the present work, we set out to assess the activity of recently approved drugs, or their components, against HTLV-1 infection in cell culture. A single combination ARV pill comprised of the INSTI dolutegravir and the NRTIs emtricitabine and tenofovir alafenamide (DTG/FTC/TAF) developed by Viatris was approved by the US Food and Drug Administration as the first-line regimen in developing countries in 2018 13. More recently, the European Medicines Agency (EMA) and the US Federal Drugs Administration (FDA) has approved lenacapavir (LEN), a capsid inhibitor, as a first-in-class, twice-yearly option for persons with multi-drug resistant HIV-1 14,15. Here, we report that dolutegravir (DTG) and tenofovir alafenamide (TAF) both have potent activity against HTLV-1 in culture, with the former exhibiting an EC50 in the picomolar range whereas LEN, expectedly, does not prevent HTLV-1 transmission. This work further underscores the clinical potential of INSTIs and tenofovir prodrugs in preventing HTLV-1 transmission.
Materials and methods
Cell lines.
MT.2 and Jurkat E6.1 cells (obtained from ATCC) were grown and maintained at 37°C in RPMI medium (Gibco) supplemented with 10% foetal bovine serum (FCS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL Amphotericin B (Gibco). HEK 293T cells were maintained at 37°C in DMEM (Gibco) supplemented with 10% foetal calf serum (FCS), 1% non-essential amino acids (Gibco) and 1% penicillin/ streptomycin (P/S) (Gibco).
Structure visualisation and homology modelling.
All structural visualisation was performed in UCSF ChimeraX software 16. Predicted models of HTLV-1 RT and p24 monomers were generated using the AlphaFold 2 command tool on ChimeraX. Amino acid alignment and homology modelling of the best model HTLV-1 RT to HIV-1 p66 was previously executed exquisitely by Tardiota and colleagues17. Residue alignment of the best model of HTLV-1 p24 with HIV-1 p24 was determined after superimposition using rigid body alignment on ChimeraX.
Cell treatment and Cell-to-cell infection of HTLV-1.
A detailed protocol outlining cell culture infection with authentic HTLV-1 can be found in our earlier works 9–11. Briefly, a day before infection, Jurkat cells were seeded in complete RPMI dosed with serial dilutions concentrations of TAF, raltegravir (RAL), dolutegravir (DTG) or LEN, and the drug vehicle (DMSO). On the day of infection, MT-2 cells, which are persistently infected with and transmit HTLV-1, were exposed to a sub-lethal dose of gamma-irradiation (400 Gray) and co-cultured with Jurkat cells at a 1:1 ratio in serum-free medium supplemented with the drugs mentioned above. After 18 hours, the co-culture was washed with PBS, resuspended in depletion buffer (0.1% FBS, 2 mM EDTA PBS), and gently tumbled (4 ◦C, 1-hour) with anti-CD25+ magnetic beads (DynaBeads, Thermo Fisher Scientific) to remove MT-2 cells. Unbound Jurkat cells were maintained and then expanded in drug-supplemented complete RPMI for 12-14 days, after which genomic DNA was harvested for downstream analysis. Cell viability assays were carried out alongside infection assays to determine drug cytotoxicity. Jurkat cells were treated with matched concentrations of drugs as infected samples and maintained for 12-14 days before cell viability was determined by alamarBlueTM (Thermo Fisher Scientific) following the manufacturer’s instructions.
Production and infection with HIV GFP reporter virus.
To generate pseudoparticles, 293T cells were co-transfected with three plasmids: an HIV (pCMV-d8.91) packaging construct, a GFP reporter plasmid (pCHGFPW) and an expression vector encoding vesicular stomatitis virus G protein (pCG-VSV-G) 21,22. Supernatants containing HIV-1 pseudoparticles were collected at 48 and 72 h post-transfection and filtered. To measure the early-stage anti-HIV-1 activity of LEN, Jurkat cells were seeded in complete RPMI dosed with serial dilutions concentrations of LEN a day before infection. The following day, Jurkat were transduced with HIV-1 pseudoparticles for 4 hours in serum free RPMI in the presence of LEN. After this, the cells were washed in PBS, reseeded in complete RPMI containing LEN and expanded for three days. Infection was determined by flow cytometry on either the LSR II or LSR Fortessa machines (Becton Dickinson), and data was analysed using FlowJo 10.9 software (FlowJo). Percentage infectivity was normalised to DMSO-treated samples. To measure effect on the drug at the late/egress stage of the viral life cycle, producer HEK 293T cells were maintained in DMEM containing LEN post-transfection. This pseudoparticle preparation was then used to transduce Jurkat cells for 4 hours, and the rest of the experiment proceeded as outlined above.
Result
The NRTI prodrug tenofovir alafenamide inhibits HTLV-1 transmission.
Retroviral RT is a multifunctional enzyme. It harbours both DNA- and RNA-dependent DNA polymerase and RNase H activities, which enables the removal of genomic RNA from newly synthesised vDNA 23,24. HIV RT is a heterodimer, with the active sites for both polymerase and RNase H located on the p66 subunit and the p51 subunit primarily serving a structural role. The NRTI tenofovir is an analogue of adenosine monophosphate. Mimicking nucleosides, NRTIs target the polymerase active site of RT, are incorporated into vDNA, and thus act as DNA chain terminators.
It is worth noting that although we lack a crystal structure of an active unit of HTLV-1 RT, both the alignment of HIV-1 and HTLV-1 RT amino acid sequences and the predicted model of a complete HTLV-1 RT monomer suggest that the active sites of the two enzymes are architecturally very similar (Figure 1A) 23. Our past observation that the first prodrug formulation of tenofovir, TDF, effectively inhibits HTLV-1 culture transmission (EC50 =17.78 ± 7.16 nM) supports this notion 9. Here, we assessed the anti-HTLV-1 activity of TAF (Figure 1B), which is purported to have a marginally better safety profile than TDF 25, by co-culturing persistently infected MT-2 cells with target Jurkat cells in varying prodrug concentrations. Following the depletion of MT-2 cells, de novo infection in Jurkat cells was quantified by determining the PVL following their expansion. TAF was observed to efficiently block HTLV-1 transmission without affecting cell viability in the range of concentration tested, achieving an EC50 of 15.10 ± 9.06 nM (Figure 1C, left panel). Although this figure is five times the average observed for a panel of 29 HIV-1 isolates26, it nonetheless indicates TAF possesses quite potent anti-HTLV-1 activity. Additionally, the highest concentration of TAF almost completely blocked HTLV-1 integration when quantifying for HTLV-1 integration products following Alu-PCR amplification (Figure 1C, right panel).
The 2nd generation INSTI dolutegravir potently inhibits HTLV-1 transmission.
Retroviral IN catalyses the requisite insertion of vDNA into host chromosomal DNA, a defining step that sets retroviruses apart from other viral families 6. IN engages the long terminal repeats (LTRs) at both termini of newly synthesised vDNA, forming a complex termed the intasome, and performs two enzymatic reactions. First, IN hydrolyses vDNA to remove two or three nucleotides following the invariant CA-dinucleotide, generating nucleophilic 3’-hydroxyl groups. Secondly, IN uses the reactive 3’-hydroxyl groups to cut both strands of chromosomal DNA and insert both 3’ ends of vDNA into the host genome simultaneously 27. The active site for both reactions is contained exclusively within the catalytic core domain (CCD) of the enzyme. This domain harbours a DDE catalytic triad, and these residues coordinate a pair of functionally critical Mg2+ cations. By binding this divalent cation, INSTIs displace the vDNA end from the integrase active site and, in doing so, deactivate the intasome 28,29.
Our previous work revealed approved INSTIs to be similarly potent in blocking HIV-1 and HTLV-1 cell culture transmission (see Table 1 in Barski et al. 9). In later structural work 10,30, we observed that INSTIs occupy the active sites of HIV-1 and STLV-1 intasomes in virtually identical conformations (Figure 2B), providing mechanistic insight for the high sensitivity of HTLV-1 to INSTIs, which are derivatives of metabolites isolated of their anti-HIV-1 IN activity 31–33.
A concerning link between the 2nd generation INSTI DTG and neural-tube defects in Botswanan newborns was reported in 2018; consequently, we excluded this drug from bygone cell culture evaluation34. However, recent pharmacovigilance analyses did not uphold the link, dispelling safety concerns 35. We thus set out to determine whether DTG is as potent as other INSTIs against HTLV-1 transmission in the present work. We first reconfirmed the sensitivity of HTLV-1 to the first-in-class INSTI RAL in cell culture to ensure comparisons between the new and past data sets are appropriate. As before, RAL strongly inhibited HTLV-1 transmission and showed no signs of cytotoxicity at the range of concentration tested (Figure 2 C, left panel). An EC50 of 5.63 ± 3.91 nM was calculated from the dose-response curve, consistent with the previous value of 6.42 ± 4.24 nM. As observed for other 2nd gen INSTIs, bictegravir and cabotegravir, DTG more potently prevented HTLV-1 transmission than RAL (Figure 2 D, left panel). Fittingly, the EC50 of 0.24 ± 0.42 nM is in the same ballpark as those calculated for bictegravir and cabotegravir 9–11. Furthermore, we were unable to amplify any HTLV-1 integration products from Jurkat cells challenged with the highest concentration of RAL or DTG during co-culture with MT-2 cells, indicating complete blockade of integration and thus infection (Figures 2C and 2D, right panels).
The HIV-1 capsid inhibitor lenacapavir is inactive against HTLV-1.
Targeting the trifecta of retroviral enzymes has transformed the lives of HIV-1/2+ positive patients the world over. However, the emergence of multidrug-resistant strains as contributors to cART failure and the necessity of daily administration for current therapy impelled pharmaceutical companies to search for new amenable targets on the HIV polypeptide 36. For Gilead Sciences, this quest culminated in the development of lenacapavir LEN (Figure 3A) 37. This drug functions by tightly stabilising adjoining capsid/p24 subunits, thereby preventing the disassembly of the curve capsid lattice, and interfering with capsid association to essential HIV-1 cellular cofactors38.
Striking findings from phase I clinical trials (NCT037339866C) suggest that six-month dosing intervals with LEN could effectively suppress HIV-1. Preexposure prophylaxis (PrEP) that can be administered twice yearly is poised to be clinically more attractive than that requiring daily administration. To evaluate whether LEN can prevent HTLV-1 transmission, we first generated a model of HTLV-1 p24 using the AlphaFold protein prediction tool on ChimeraX since its crystal structure is yet to be solved. While superimposition implied the four alpha helices composing the N terminal domain of the best model of HTLV-1 p24 overlapped significantly with HIV-1 p24 (1E6J), structure-guided alignment revealed poor residue conservation in the helices binding LEN (Figure 3B). Consistent with this, results from our cell culture experiments dismissed all prospects for adopting LEN as an HTLV-1 preventative intervention. Since LEN has been shown to interfere with both the early and late stages of HIV-1 replication, we preincubated either the target Jurkat cells or HTLV-1 producer MT-2 cells with LEN before co-culture and witnessed no discernible antiviral activity in both conditions (Figure 3C). Equivalent experiments with GFP reporter HIV-1 pseudoparticles confirmed this batch of LEN was active (Figure 3D).
Discussion
A recent meta-analysis of epidemiological studies has suggested that HTLV-1+ status may be linked to at least a dozen diseases, including seborrheic dermatitis and Sjogren’s syndrome, in addition to the previously identified fatality- and myelopathy-associated pathologies 39. Even asymptomatic patients have reported higher incidences of malaise, discomfort, and depression compared to the general population 40. The reduced quality of life, the risk of developing currently untreatable ATLL and HAM/TSP, and the likely underestimation of the population living with HTLV-1 all highlight the urgent need for prophylactics and therapeutics in the clinic. Most prophylactic research efforts have understandably been directed towards developing vaccine candidates for HTLV-1, but none have advanced beyond small animal trials. Likewise, curative or genuinely transformative therapeutics have yet to be discovered for either of the main HTLV-1 pathologies.
Due to the excessive disease burden resulting from the two conditions, early HTLV-1 clinical trials with ARVs predominantly examined whether the drugs could improve the prognosis of patients with ATLL or alleviate debilitation imposed by HAM/TSP 41. In the nineties, several studies indicated a combination of the NRTI azidothymidine (AZT) and interferon-alpha (IFNα) extended the median survival of ATL patients, although not beyond that observed following chemotherapy 42–44. Despite their poor efficacies and high relapse rates after remission, AZT/IFNα combination therapy, chemotherapy and stem-cell transplantation are the only regimens available to ATLL patients. Regarding HAM/TSP, AZT and tenofovir failed to alleviate patient illness in clinical trials. Indeed, a consortium of international experts recently concluded there is insufficient evidence for the use of ARVs in managing HTLV-1-dependent myeloneuropathy 45. The proviral load of HTLV-1 carriers was not noticeably reduced following 6-month treatment with RAL 46. Notably, HTLV-1+ CD4+ T cell numbers are maintained by mitotic spread and not infectious spread (where ARVs antagonise replicating virus), as required for HIV-1. This rationalises the poor clinical efficacy of ARVs against ATLL and HAM/TSP since these conditions develop in adulthood, where the contribution of de novo infection to preserving the HTLV-1+ T cell population is negligible.
Preventing the establishment of infection is the main objective of HIV PrEP. In this work, we demonstrate the anti-HTLV-1 activities of TAF and DTG in cell culture, confirming all approved INSTIs and tenofovir prodrugs can potently inhibit HTLV-1 transmission. Tenofovir prodrugs and the second-generation INSTI cabotegravir are already approved by EMA and FDA as HIV PrEP. TDF-based PrEP entered the clinic a decade ago, leading to a dramatic fall in HIV incidence rates among vulnerable groups because strict adherence to the recommended daily dose is 99% effective at preventing infection. TAF-based PrEP is as effective as TDF, but TAF possesses a better safety profile and has longer bioavailability 47,48. The most transformative advance in PrEP is probably the advent of long-acting cabotegravir, which reduces the need for daily pill dosing to as few as six injections a year. There is little biological indication that current PrEP regimens would fail to protect persons at risk from contracting HTLV-1 49. Ultimately, the target cells required for optimal drug function and the transmission via bodily fluids are shared between HIV and HTLV-1. Indeed, the genomic stability of HTLV-1 relative to HIV-1 means there’s a reduced risk for the emergence of drug-resistant strains following the theoretical widespread adoption of ARVs as HTLV-1 PrEP 50.
Despite DTG being the backbone of most modern cART, no pill containing this INSTI is currently slated for use as HIV PrEP since the options mentioned above are already highly effective, and pharmaceutical efforts are shifting to developing more long-acting agents. To this end, a recently developed intramuscularly administered long-acting form of DTG showed early promise in animal experiments, with pharmacokinetics suggesting that three-month dosing intervals with the prodrug could be possible 51. Irrespective of the form of first-in-class HTLV-1 PrEP, daily orals or long-lasting injections, dosing adjustments would be required for their potential use in breastfeeding infants 52. Breastmilk is the major route for vertical (mother-to-child) transmission of HTLV-1 53. As already discussed, ARVs are likely ineffective in controlling chronic HTLV-1 because the virus principally persists via mitotic spread. Therefore, whereas cART taken by HIV+ mothers protects infants during breastfeeding, PrEP may have to be administered to infants to guarantee protection from HTLV-1.
Breastfeeding is also a leading indicator for ATLL development. As such, interventions directed at the first year of life, including withholding breastmilk in favour of formula as implemented in Japan, may reduce the future incidence rates for the most mortal of HTLV-1-associated diseases. Screening mothers for HTLV-1 and feeding infants on formula are strategies that are currently exclusive to wealthy countries and would not be economically viable, culturally acceptable or safe (when no access to clean water can be guaranteed) in other regions where HTLV-1 is endemic. As such, in the absence of a vaccine, PrEP provides the only cost-effective option for stemming childhood infections and future caseloads of ATLL in lower-middle-income countries. If rolled out alongside campaigns to increase HTLV-1 awareness, it’s likely clinically successful PrEP would drastically reduce sexual transmission rates, offering the first effective transmission barrier to condomless sex, through which 70% of new HTLV-1 cases occur 49,54. Moreover, since sexual transmission is associated with myeloneuropathy prognosis, PrEP could prove an efficient measure in quelling HAM/TSP cases.
It’s paramount to be prepared for the unexpected. Therefore, some HTLV-1-specific compounds must be developed for the unlikely scenario where INSTIs and tenofovir prodrugs fail as HTLV-1 prophylactics in vivo. Recent cutting-edge HIV-1 drugs to reach clinical trials do not target enzymatic active sites. Instead, allosteric inhibitors of integrase (ALLINIs) target an allosteric surface on the HIV-1 intasome, inducing IN multimerization and aberrant virion formation, whereas LEN tightly binds adjacent p24 monomers to prevent capsid disassembly 38,55,56. Structures of deltaretroviral intasomes are now available, and an allosteric surface corresponding to ALLINIs interface may exist but is yet to be determined. The crystal structure of HTLV-1 p24, let alone the capsid lattice, is unresolved, so developing a capsid inhibitor via structure-guided methods is currently impossible. All in all, HTLV-1-specific structural antivirals are a long way from reaching clinical trials.
While the field awaits the discovery of HTLV-1 antivirals, a feat entirely dependent on sufficient funding, we may not need to reinvent the wheel, as the saying goes. This work culminates a series of studies showing INSTIs and tenofovir prodrugs to be equally efficacious at inhibiting cell culture transmission of HIV and HTLV-1. We have provided the structural and mechanistic specifics underpinning conserved functionality for these drugs toward the distant orthoretroviruses, which should give confidence as to their potential in preventing new cases of HTLV-1. Poor HTLV-1 awareness and a dearth of treatment options for the two major HTLV-1-associated pathologies necessitate a prophylactic measure. Our cumulative findings suggest this prophylactic may already exist in the form of ARVs. Considering the wide availability and implementation of cART and PrEP, it is pertinent for INSTIs and tenofovir prodrugs to be trialled as HTLV-1 PrEP at pre-clinical and clinical stages.
Author Contributions
M.D.K. – Investigation, Formal analysis, Data curation, Visualisation & Writing. G.N.M. – Conceptualisation, Methodology, Supervision, Review and editing & Funding acquisition.
Acknowledgements
This work was supported by Blood Cancer UK (ref. 22002). We thank Ian Taylor for providing us with a generous aliquot of lenacapavir. We also thank all members of the Maertens lab for valuable discussions and critiques.
Conflicts of Interest
Both authors declare there was no conflict of interests.
References
- Hutchings, M., Truman, A. & Wilkinson, B. Antibiotics: past, present and future. Curr Opin Microbiol 51, 72–80 (2019). [CrossRef]
- Choudhary, M. C. & Mellors, J. W. The transformation of HIV therapy: One pill once a day. 27, (2022). [CrossRef]
- Futsch, N., Mahieux, R. & Dutartre, H. HTLV-1, the Other Pathogenic Yet Neglected Human Retrovirus: From Transmission to Therapeutic Treatment. Viruses 2018, Vol. 10, Page 1 10, 1 (2017). [CrossRef]
- Bangham, C. R. M., Araujo, A., Yamano, Y. & Taylor, G. P. HTLV-1-associated myelopathy/tropical spastic paraparesis. Nature Reviews Disease Primers 2015 1:1 1, 1–17 (2015). [CrossRef]
- Marie Skalka, A. The Retroviral Enzymes. Article in Annual Review of Biochemistry (1994). [CrossRef]
- Maertens, G. N., Engelman, A. N. & Cherepanov, P. Structure and function of retroviral integrase. Nat Rev Microbiol 20, 20–34 (2022). [CrossRef]
- Herschhorn, A. & Hizi, A. Retroviral reverse transcriptases. Cellular and Molecular Life Sciences 2010 67:16 67, 2717–2747 (2010). [CrossRef]
- Lockbaum, G. J. et al. Inhibiting HTLV-1 Protease: A Viable Antiviral Target. ACS Chem Biol 16, 529–538 (2021). [CrossRef]
- Barski, M. S., Minnell, J. J. & Maertens, G. N. Inhibition of HTLV-1 infection by HIV-1 first-and second-generation integrase strand transfer inhibitors. Front Microbiol 10, 475549 (2019). [CrossRef]
- Barski, M. S. et al. Structural basis for the inhibition of HTLV-1 integration inferred from cryo-EM deltaretroviral intasome structures. Nature Communications 2021 12:1 12, 1–10 (2021). [CrossRef]
- Schneiderman, B. S., Barski, M. S. & Maertens, G. N. Cabotegravir, the Long-Acting Integrase Strand Transfer Inhibitor, Potently Inhibits Human T-Cell Lymphotropic Virus Type 1 Transmission in vitro. Front Med (Lausanne) 9, 889621 (2022). [CrossRef]
- Seegulam, M. E. & Ratner, L. Integrase inhibitors effective against human T-cell leukemia virus type 1. Antimicrob Agents Chemother 55, 2011–2017 (2011). [CrossRef]
- Lockman, S. et al. Efficacy and safety of dolutegravir with emtricitabine and tenofovir alafenamide fumarate or tenofovir disoproxil fumarate, and efavirenz, emtricitabine, and tenofovir disoproxil fumarate HIV antiretroviral therapy regimens started in pregnancy (IMPAACT 2010/VESTED): a multicentre, open-label, randomised, controlled, phase 3 trial. The Lancet 397, 1276–1292 (2021). [CrossRef]
- Paik, J. Lenacapavir: First Approval. Drugs 82, 1499–1504 (2022). [CrossRef]
- Prather, C., Lee, A. & Yen, C. Lenacapavir: A first-in-class capsid inhibitor for the treatment of highly treatment-resistant HIV. American Journal of Health-System Pharmacy (2023). [CrossRef]
- Pettersen, E. F. et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004). [CrossRef]
- Tardiota, N., Jaberolansar, N., Lackenby, J. A., Chappell, K. J. & O’donnell, J. S. Title: HTLV-1 reverse transcriptase homology model provides structural basis for sensitivity to 1 existing nucleoside/nucleotide reverse transcriptase inhibitors 2 3 4. biorxiv (2023). [CrossRef]
- Rowan, A. G. et al. T Cell Receptor Vβ Staining Identifies the Malignant Clone in Adult T cell Leukemia and Reveals Killing of Leukemia Cells by Autologous CD8 + T cells. (2016). [CrossRef]
- Lairmore, M. et al. Absence of Human T-Cell Lymphotropic Virus Type I Coinfection in Human Immunodeficiency Virus-Infected Hemophilic Men. Blood 74, 2596–2599 (1989). [CrossRef]
- Alais, S., Mahieux, R. & Dutartre, H. Viral Source-Independent High Susceptibility of Dendritic Cells to Human T-Cell Leukemia Virus Type 1 Infection Compared to That of T Lymphocytes. J Virol 89, 10580–10590 (2015). [CrossRef]
- Van Maele, B., De Rijck, J., De Clercq, E. & Debyser, Z. Impact of the Central Polypurine Tract on the Kinetics of Human Immunodeficiency Virus Type 1 Vector Transduction. J Virol 77, 4685–4694 (2003). [CrossRef]
- Ulm, J. W., Perron, M., Sodroski, J. & C. Mulligan, R. Complex determinants within the Moloney murine leukemia virus capsid modulate susceptibility of the virus to Fv1 and Ref1-mediated restriction. Virology 363, 245–255 (2007). [CrossRef]
- Tuske, S. et al. Structures of HIV-1 RT–DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nature Structural & Molecular Biology 2004 11:5 11, 469–474 (2004). [CrossRef]
- Coffin, J. M. & Fan, H. The Discovery of Reverse Transcriptase. 3, 29–51 (2016). [CrossRef]
- Wang, H., Lu, X., Yang, X. & Xu, N. The efficacy and safety of tenofovir alafenamide versus tenofovir disoproxil fumarate in antiretroviral regimens for HIV-1 therapy: Meta-analysis. Medicine (United States) 95, (2016). [CrossRef]
- Callebaut, C., Stepan, G., Tian, Y. & Miller, M. D. In Vitro Virology Profile of Tenofovir Alafenamide, a Novel Oral Prodrug of Tenofovir with Improved Antiviral Activity Compared to That of Tenofovir Disoproxil Fumarate. Antimicrob Agents Chemother 59, 5909 (2015). [CrossRef]
- Cherepanov, P., Maertens, G. N. & Hare, S. Structural insights into the retroviral DNA integration apparatus. Curr Opin Struct Biol 21, 249–256 (2011). [CrossRef]
- Hare, S., Gupta, S. S., Valkov, E., Engelman, A. & Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010 464:7286 464, 232–236 (2010). [CrossRef]
- Passos, D. O., Li, M., Craigie, R. & Lyumkis, D. Retroviral integrase: Structure, mechanism, and inhibition. Enzymes (Essen) 50, 249–300 (2021). [CrossRef]
- Passos, D. O. et al. Structural basis for strand-transfer inhibitor binding to HIV intasomes. Science (1979) 367, 810–814 (2020). [CrossRef]
- Hazuda, D. et al. Isolation and Characterization of Novel Human Immunodeficiency Virus Integrase Inhibitors from Fungal Metabolites. 10, 63–70 (1999). [CrossRef]
- Espeseth, A. S. et al. HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc Natl Acad Sci U S A 97, 11244–11249 (2000). [CrossRef]
- Summa, V. et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem 51, 5843–5855 (2008). [CrossRef]
- Zash, R., Makhema, J. & Shapiro, R. L. Neural-Tube Defects with Dolutegravir Treatment from the Time of Conception. New England Journal of Medicine 379, 979–981 (2018). [CrossRef]
- Chouchana, L., Pariente, A., Pannier, E., Tsatsaris, V. & Treluyer, J.-M. Dolutegravir and neural tube defects: a new insight. Lancet Infect Dis 20, 405–406 (2020). [CrossRef]
- Temereanca, A. & Ruta, S. Strategies to overcome HIV drug resistance-current and future perspectives. Frontiers in Microbiology vol. 14 Preprint at https://doi.org/10.3389/fmicb.2023.1133407 (2023). [CrossRef]
- Link, J. O. et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 584, 614–618 (2020). [CrossRef]
- Bester, S. M. et al. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science (1979) 370, 360–364 (2020). [CrossRef]
- Schierhout, G. et al. Association between HTLV-1 infection and adverse health outcomes: a systematic review and meta-analysis of epidemiological studies. Lancet Infect Dis 20, 133–143 (2020). [CrossRef]
- Rosadas Id, C. et al. Health state utility values in people living with HTLV-1 and in patients with HAM/TSP: The impact of a neglected disease on the quality of life. (2020). [CrossRef]
- Petruzziello, A. et al. Distribution pattern of hepatitis C Virus genotypes and correlation with viral load and risk factors in chronic positive patients. Intervirology (2014). [CrossRef]
- Arkash, P. et al. Treatment of Adult T-Cell Leukemia–Lymphoma with a Combination of Interferon Alfa and Zidovudine. 332, 1744–1748 (1995). [CrossRef]
- Livier Ermine, O. H. et al. Treatment of Adult T-Cell Leukemia-Lymphoma with Zidovudine and Interferon Alfa. 332, 1749–1751 (1995). [CrossRef]
- Marino-Merlo, F. et al. Antiretroviral Therapy in HTLV-1 Infection: An Updated Overview. Pathogens 9, (2020). [CrossRef]
- Araujo, A. et al. Management of HAM/TSP. Neurol Clin Pract 11, 49–56 (2021). [CrossRef]
- Treviño, A. et al. Antiviral effect of raltegravir on HTLV-1 carriers. Journal of Antimicrobial Chemotherapy 67, 218–221 (2012). [CrossRef]
- Kearney, B. P., Flaherty, J. F. & Shah, J. Tenofovir disoproxil fumarate: Clinical pharmacology and pharmacokinetics. Clin Pharmacokinet 43, 595–612 (2004). [CrossRef]
- Ray, A. S., Fordyce, M. W. & Hitchcock, M. J. M. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of Human Immunodeficiency Virus. Antiviral Res 125, 63–70 (2016). [CrossRef]
- Bradshaw, D. & Taylor, G. P. HTLV-1 Transmission and HIV Pre-exposure Prophylaxis: A Scoping Review. Frontiers in Medicine vol. 9 Preprint at https://doi.org/10.3389/fmed.2022.881547 (2022). [CrossRef]
- Afonso, P. V., Cassar, O. & Gessain, A. Molecular epidemiology, genetic variability and evolution of HTLV-1 with special emphasis on African genotypes. Retrovirology vol. 16 Preprint at https://doi.org/10.1186/s12977-019-0504-z (2019). [CrossRef]
- Deodhar, S. et al. Transformation of dolutegravir into an ultra-long-acting parenteral prodrug formulation. Nature Communications 2022 13:1 13, 1–15 (2022). [CrossRef]
- Bevers, L. A. H. et al. Pharmacokinetic Data of Dolutegravir in Second-line Treatment of Children With Human Immunodeficiency Virus: Results From the CHAPAS4 Trial. Clinical Infectious Diseases (2023). [CrossRef]
- Carneiro-Proietti, A. B. F. et al. Mother-to-Child Transmission of Human T-Cell Lymphotropic Viruses-1/2: What We Know, and What Are the Gaps in Understanding and Preventing This Route of Infection. J Pediatric Infect Dis Soc 3, S24–S29 (2014). [CrossRef]
- Soltani, A. et al. Molecular targeting for treatment of human T-lymphotropic virus type 1 infection. (2018). [CrossRef]
- Jurado, K. A. et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. 110, (2013). [CrossRef]
- Singer, M. R. et al. The Drug-Induced Interface That Drives HIV-1 Integrase Hypermultimerization and Loss of Function. mBio 14, (2023). [CrossRef]
Figure 1.
The activity of tenofovir alafenamide (TAF) against HTLV-1. (A) Structures and active site sequence of HIV-1 and HTLV-1 reverse transcriptase (RT). Top panel: Surface structure of the HIV RT p66 monomer (PDB: 1T05) and the predicted structure of an HTLV-1 monomer generated using the AlphaFold protein prediction tool on ChimeraX. The active sites are coloured in tangerine according to HIV-1 RT p66. Bottom panel: Alignment of residues in the active sites of HIV- and HTLV-1 RT, boxes filled in tangerine indicate residues identified to bind TAF in the 1T05 crystal structure. (B) Chemical structure of the prodrug TAF. (C) TAF blocks HTLV-1 transmission to Jurkat cells. Left panel – Jurkat cells were pre-treated with a dilution of TAF (10 concentrations starting from 8 μM two-fold down to 2 μM, then ten-fold down to 0.2 pM) and infected by co-culture with MT-2 cells. Infection is represented as % of PVL relative to DMSO-treated cells. Cell viability is also represented relative to DMSO-treated cells. Right panel – The percentage of integrated provirus of 8 µM, 200 nM and DMSO-treated Jurkat cells was determined relative to DMSO-treated samples. Data in C are from three independent experiments & performed in triplicate. Error bars represent standard deviation. Asterisks denote statistical significance, following ordinary ANOVA using Dunnett’s multiple comparison test with the DMSO-treated samples as the control (****p<0.0001).
Figure 1.
The activity of tenofovir alafenamide (TAF) against HTLV-1. (A) Structures and active site sequence of HIV-1 and HTLV-1 reverse transcriptase (RT). Top panel: Surface structure of the HIV RT p66 monomer (PDB: 1T05) and the predicted structure of an HTLV-1 monomer generated using the AlphaFold protein prediction tool on ChimeraX. The active sites are coloured in tangerine according to HIV-1 RT p66. Bottom panel: Alignment of residues in the active sites of HIV- and HTLV-1 RT, boxes filled in tangerine indicate residues identified to bind TAF in the 1T05 crystal structure. (B) Chemical structure of the prodrug TAF. (C) TAF blocks HTLV-1 transmission to Jurkat cells. Left panel – Jurkat cells were pre-treated with a dilution of TAF (10 concentrations starting from 8 μM two-fold down to 2 μM, then ten-fold down to 0.2 pM) and infected by co-culture with MT-2 cells. Infection is represented as % of PVL relative to DMSO-treated cells. Cell viability is also represented relative to DMSO-treated cells. Right panel – The percentage of integrated provirus of 8 µM, 200 nM and DMSO-treated Jurkat cells was determined relative to DMSO-treated samples. Data in C are from three independent experiments & performed in triplicate. Error bars represent standard deviation. Asterisks denote statistical significance, following ordinary ANOVA using Dunnett’s multiple comparison test with the DMSO-treated samples as the control (****p<0.0001).
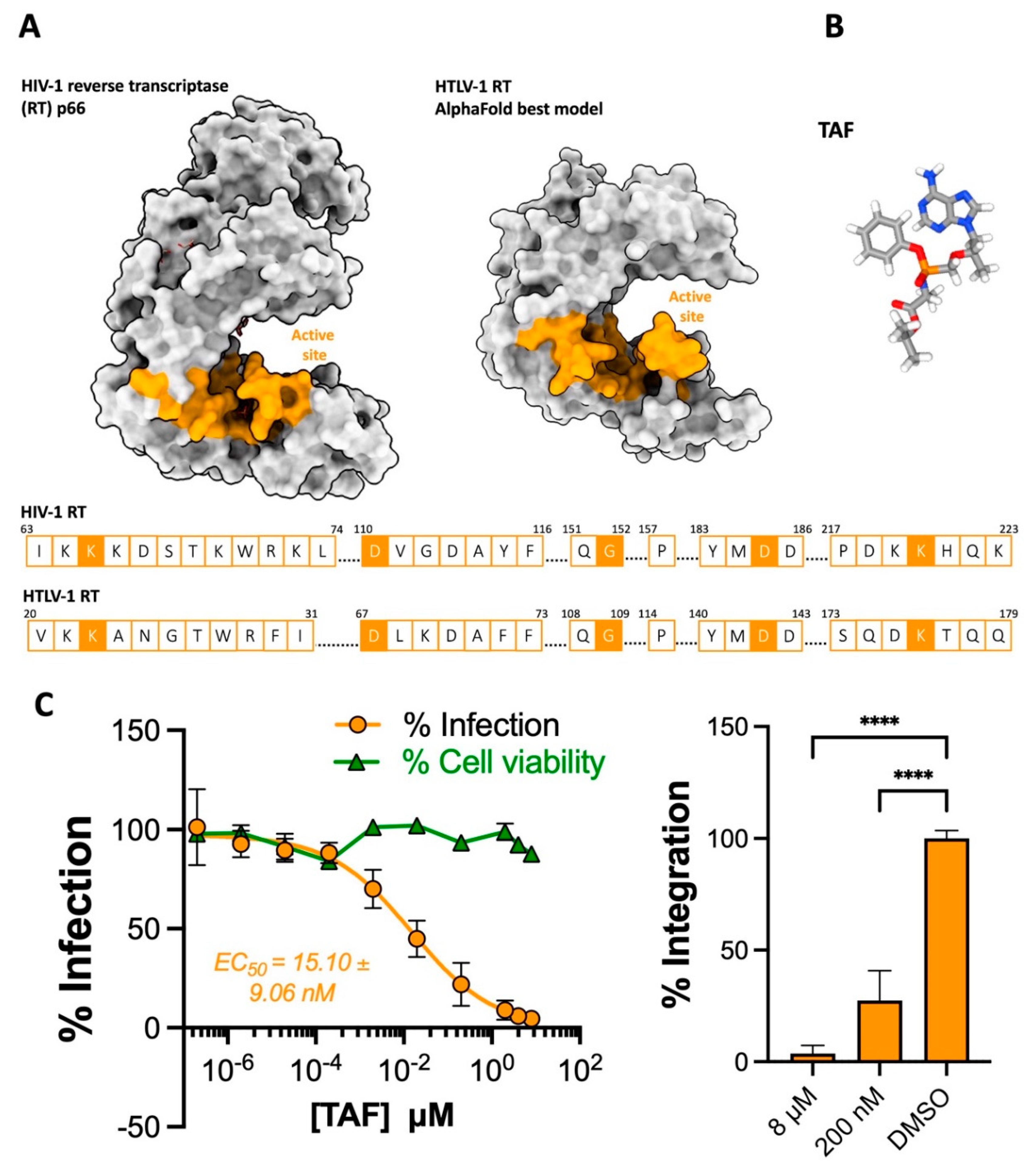
Figure 2.
The activity of dolutegravir against HTLV-1. (A) Chemical structures of the 1st generation INSTI raltegravir (RAL) and the 2nd generation INTSIs bictegravir and dolutegravir (DTG). (B) View of the HIV-1 (PDB: 6PUW) and STLV-1 (7OUF) intasome active sites bound to bictegravir, vDNA chains are coloured in cyan and pink. Residues comprising the DDE catalytic triad are highlighted in blue, with their sidechains displayed as sticks. (C) RAL blocks HTLV-1 transmission to Jurkat cells. Left panel – Jurkat cells were pre-treated with a serial dilution of RAL (10 concentrations starting from 20 μM log-diluted down to 20 fM) and infected by co-culture with MT-2 cells. Infection is represented as % of PVL relative to DMSO-treated cells. Cell viability is also represented relative to DMSO-treated cells. Right panel – The percentage of integrated provirus of 20 µM, 2 nM and DMSO-treated Jurkat cells was calculated relative to DMSO-treated samples. (D) DTG potently blocks HTLV-1 transmission to Jurkat cells. Left panel – Jurkat cells were pre-treated with a serial dilution of DTG (10 concentrations starting from 800 nM log-diluted down to 0.8 fM) and infected by co-culture with MT-2 cells. Infection is represented as % of PVL relative to DMSO-treated cells. Cell viability is also represented relative to DMSO-treated cells. Right panel – The percentage of integrated provirus of 800 nM, 80 pM and DMSO-treated Jurkat cells was calculated relative to DMSO-treated samples. Error bars represent standard deviation. Asterisks denote statistical significance, following ordinary ANOVA using Dunnett’s multiple comparison test with the DMSO-treated samples as the control (****p<0.0001).
Figure 2.
The activity of dolutegravir against HTLV-1. (A) Chemical structures of the 1st generation INSTI raltegravir (RAL) and the 2nd generation INTSIs bictegravir and dolutegravir (DTG). (B) View of the HIV-1 (PDB: 6PUW) and STLV-1 (7OUF) intasome active sites bound to bictegravir, vDNA chains are coloured in cyan and pink. Residues comprising the DDE catalytic triad are highlighted in blue, with their sidechains displayed as sticks. (C) RAL blocks HTLV-1 transmission to Jurkat cells. Left panel – Jurkat cells were pre-treated with a serial dilution of RAL (10 concentrations starting from 20 μM log-diluted down to 20 fM) and infected by co-culture with MT-2 cells. Infection is represented as % of PVL relative to DMSO-treated cells. Cell viability is also represented relative to DMSO-treated cells. Right panel – The percentage of integrated provirus of 20 µM, 2 nM and DMSO-treated Jurkat cells was calculated relative to DMSO-treated samples. (D) DTG potently blocks HTLV-1 transmission to Jurkat cells. Left panel – Jurkat cells were pre-treated with a serial dilution of DTG (10 concentrations starting from 800 nM log-diluted down to 0.8 fM) and infected by co-culture with MT-2 cells. Infection is represented as % of PVL relative to DMSO-treated cells. Cell viability is also represented relative to DMSO-treated cells. Right panel – The percentage of integrated provirus of 800 nM, 80 pM and DMSO-treated Jurkat cells was calculated relative to DMSO-treated samples. Error bars represent standard deviation. Asterisks denote statistical significance, following ordinary ANOVA using Dunnett’s multiple comparison test with the DMSO-treated samples as the control (****p<0.0001).
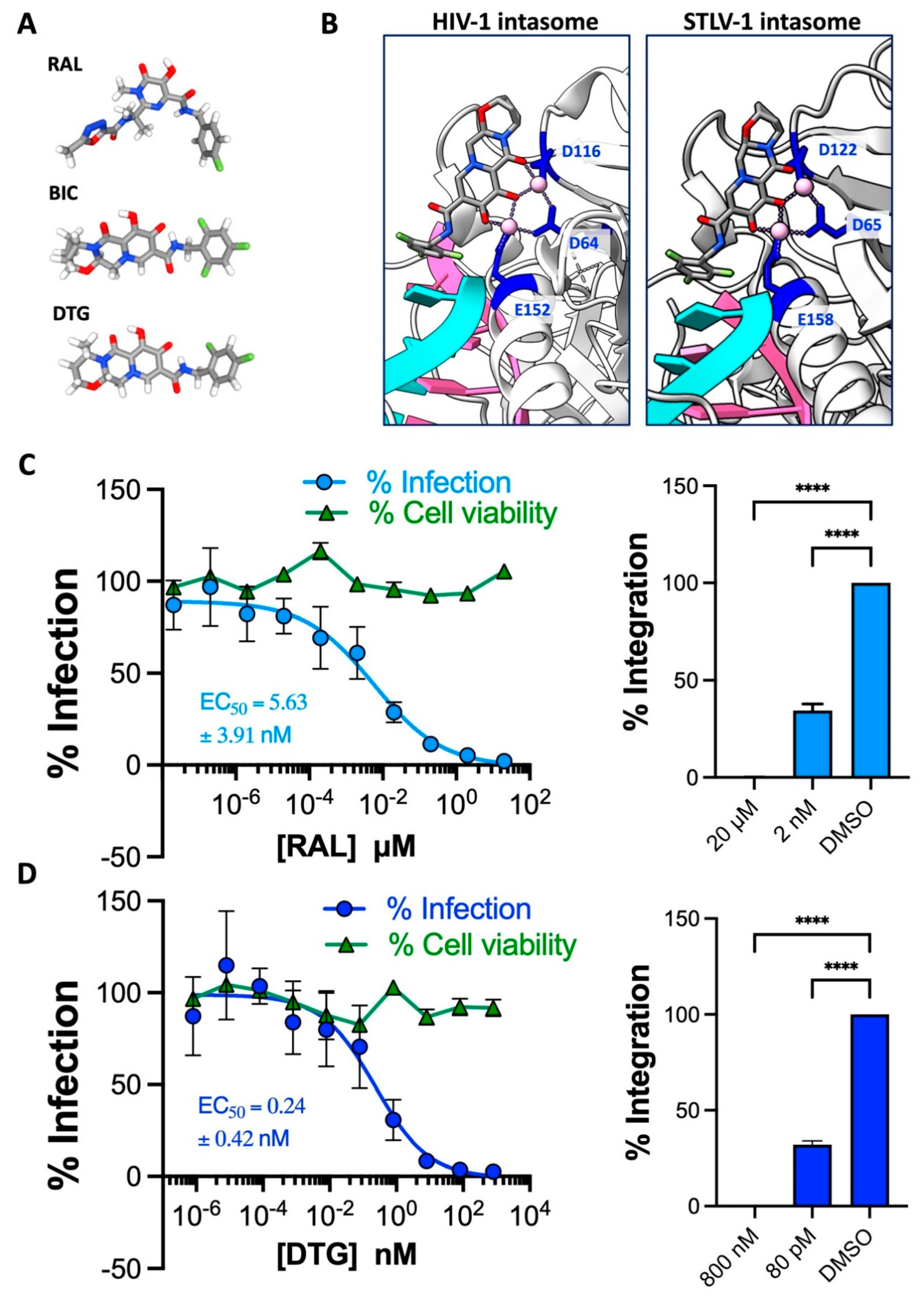
Figure 3.
The activity of lenacapavir against HTLV-1. (A) Chemical structure of the HIV-1 capsid targeting lenacapavir (LEN). (B) The predicted structure of an HTLV-1 p24 coloured in blue generated on AlphaFold superimposed onto HIV-1 p24 coloured in salmon (PDB: 1E6J) using ChimeraX visualisation software. The residues involved in HIV-1 p24-LEN binding are displayed in the boxes with salmon outlines. Filled boxes in salmon indicate residues that bind LEN. Corresponding residues in HTLV-1 p24 following superimposition or alignment are displayed in the boxes with blue outlines. (C) LEN is ineffective at blocking HTLV-1 transmission to Jurkat cells. Salmon points: Jurkat cells were pre-treated with a serial dilution of LEN (7 concentrations from 100 nM diluted 3-fold down to 140 pM) and infected by co-culture with MT-2 cells to measure anti-HTLV-1 activity in the early stages of viral replication. Blue points: MT-2 cells were pre-treated with a serial dilution of LEN before co-culture with Jurkat cells to measure anti-HTLV-1 activity in the late stages of viral replication. Infection is represented as % of PVL relative to DMSO-treated cells. (D) LEN potently blocks HIV-1 cell-free (VSV-G) pseudotyped infection of Jurkat cells. LEN is active against HIV-1 when pre-treated with both producer cells and target cells, as previously demonstrated. Data in C and D are from two independent experiments performed in triplicate. Error bars represent standard deviation.
Figure 3.
The activity of lenacapavir against HTLV-1. (A) Chemical structure of the HIV-1 capsid targeting lenacapavir (LEN). (B) The predicted structure of an HTLV-1 p24 coloured in blue generated on AlphaFold superimposed onto HIV-1 p24 coloured in salmon (PDB: 1E6J) using ChimeraX visualisation software. The residues involved in HIV-1 p24-LEN binding are displayed in the boxes with salmon outlines. Filled boxes in salmon indicate residues that bind LEN. Corresponding residues in HTLV-1 p24 following superimposition or alignment are displayed in the boxes with blue outlines. (C) LEN is ineffective at blocking HTLV-1 transmission to Jurkat cells. Salmon points: Jurkat cells were pre-treated with a serial dilution of LEN (7 concentrations from 100 nM diluted 3-fold down to 140 pM) and infected by co-culture with MT-2 cells to measure anti-HTLV-1 activity in the early stages of viral replication. Blue points: MT-2 cells were pre-treated with a serial dilution of LEN before co-culture with Jurkat cells to measure anti-HTLV-1 activity in the late stages of viral replication. Infection is represented as % of PVL relative to DMSO-treated cells. (D) LEN potently blocks HIV-1 cell-free (VSV-G) pseudotyped infection of Jurkat cells. LEN is active against HIV-1 when pre-treated with both producer cells and target cells, as previously demonstrated. Data in C and D are from two independent experiments performed in triplicate. Error bars represent standard deviation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



