
Submitted:
01 November 2023
Posted:
01 November 2023
You are already at the latest version
Abstract
Cellular locomotion is required for survival, fertility, proper embryonic development, regeneration, and wound healing. Cell migration is a major component of metastasis, which accounts for two-thirds of all solid tumor deaths. While many studies have demonstrated increased energy requirements, metabolic rates, and migration of cancer cells compared to normal cells, few have systematically compared normal and cancer cell migration as well as energy requirements side by side. Thus, we investigated how non-malignant and malignant cells migrate utilizing several cell lines from the breast and lung. Initial screening was done in an unbiased high-throughput manner for the ability to migrate/invade on collagen and/or Matrigel. We unexpectedly observed that all the non-malignant lung cells moved significantly faster than cells derived from lung tumors regardless of growth media used. Given the paradigm-shifting nature of our discovery, we pursued possible mechanisms responsible. Neither mass, cell doubling, nor volume, accounted for the individual speed and track length of the normal cells. Non-malignant cells had higher levels of ATP at premigratory-wound induction stages. Meanwhile, cancer cells also increased ATP at premigratory-wound induction – but not to the levels of the normal cells, indicating the possibility for further therapeutic investigation.
Keywords:
Cell Migration
; Cancer cell migration
; normal epithelial cell motility
1. Introduction
The complex process of cell migration usually initiates with cytoskeletal rearrangements and signaling cascades that trigger gene expression and shifting metabolic requirements. Numerous disease states including the metastatic spread of aggressive tumors have altered cell migration. Several studies have described various modes of movement within malignant and non-malignant cells as both individual cells and as collective sheets.
Despite the disease state or mode of cell migration the energy demands of cytoskeletal rearrangements, volume regulation, anoikis resistance and extracellular matrix remodeling are elevated in migrating cells compared to confluent/compressed cells [1,2,3,4,5,6,7]. The normal mammary epithelial cell line, MCF10A and the metastatic breast cancer cell line, MDA-MB-231, have been used concurrently and alone as exemplars of in vitro cell migration in hundreds of studies. Utilization of the MCF10A and MDA-MB-231 cell lines have led to a deeper understanding of the five W’s of cancer metastasis including: what is extracellular matrix [5], when growth factor (e.g TGFβ) signaling is unfavorable for tumorigenesis yet pro-metastatic[8,9]; how metastatic cells remodel distant organs [10,11]; and finally the debatable conclusion of why faster cancer cell migration is an indicator of metastatic potential [12,13,14,15].
Robust cell migration (quantified by speed, persistence, and track length) has been described as a hallmark of cancer metastasis[13,16]. Initially, we screened 15 cell lines: 12 lung lines and three mammary/breast lines derived from both non-malignant and malignant cells via scratch wound assay, to identify in an unbiased manner – fast and slow – migrators for further experimentation. Our data revealed an interesting result; non-malignant cells from the lung regardless of the media used for migration closed the wound faster than the lung cancer cells. This led us to modify our inquiry to ask, “Why are normal cells faster at wound closure than cancer cells if migration speed is a hallmark of cancer metastasis?”.
Cell migration/invasion necessitates a considerable expenditure of energy in the form of ATP[6,17]. We discovered nonmalignant cells (of both the lung and mammary) had greater basal ATP levels, than the cancer cells at wound induction. Additionally, all cell lines differentially increased ATP content at the onset of wound induction, indicating a critical demand for energy at wound induction during premigratory states regardless of disease state.
2. Materials and Methods
2.1. Cell Lines
Lung (Seven normal and six carcinoma) cell lines were screened for the ability to migrate on collagen and to invade Matrigel. All cancer cells were grown in RPMI 1640 supplemented with 5% FBS, maintained at 37 °C in an atmosphere of 5% CO2. All normal, non-tumorigenic cells were grown in, keratinocyte serum-free media kit (KSFM; Gibco, kit catalog no.17005042), except for MCF10A (grown in MEGM, Kit Catalog no. CC-4136) maintained at 37 °C in an atmosphere of 5% CO2. The HCC4017 cell line was additionally adapted to non-malignant cell media– KSFM, with and without the addition of 2% FBS. Lung cancer cells and human bronchial epithelial cells (HBEC), un-immortalized and immortalized were derived, deposited, and maintained by Hamon Cancer Center, UTSW Dallas TX. Human cell lines were authenticated to confirm origin, and all cell lines were pre-treated with Plasmocin until confirmed to be free of mycoplasma (Myco-Sniff-RapidTM Mycoplasma Luciferase Detection Kits, MP Biomedicals) before use.
2.2. Materials
RPMI 1640 and Trypsin-EDTA, were purchased from Sigma-Aldrich (St. Louis, MO). Keratinocyte SFM (1X), PBS pH7.4 (1X), and Gentamicin Reagent Solution were purchased from Thermo Fisher Scientific (Grand Island, NY). Cell culture plates were purchased from Corning Life Sciences (Corning, NY) and Thermo Fisher Scientific (Grand Island, NY). Plasmocin treatment reagent was purchased from InvivoGen (San Diego, CA). 96-well Wound Maker and IncuCyte CytoATP Lentivirus kit were purchased from Sartorius (Göttingen, Germany). ADP/ATP Ratio Assay Kit was from Abcam (Boston, MA). Myco-Sniff-RapidTM Mycoplasma Luciferase Detection Kits was purchased from MP Biomedicals (Santa Ana, CA). MEGMTM Mammary Epithelial Cell Growth Medium Bullet Kit was from Lonza (Basel, Switzerland).
2.3. In vitro scratch assay quantification
All cell lines tested (3.5-4 x 104 cells/well) were seeded onto a collagen-1-or Matrigel-coated 96-well ImageLock tissue culture plate (Sartorius) and incubated at 37 °C with 5% CO2 incubator for 24 hours. The wounds were made by the 96-well Wound Maker (Sartorius). The wounded cells were washed 3 times with 1XPBS plus gentamycin (Gibco) to remove the detached cells before imaging. Cancer and normal cells adapted to non-growth condition media were adapted for 48-72 hours prior to wounding and at wound induction.
2.4. Physical measurements of HBEC30UI, HBEC30KT, and HCC4017 on LIVECYTE
Quantification of individual cell: dry mass, doubling time, radius, sphericity, volume, track length, and speed were all performed on the Livecyte kinetic cytometer system. With the use of their quantitative phase imaging (QPI) technique known as Ptychography, to visualize cells with ultra-low powered laser to track proliferation, shape, dimensions and migration. Cells from each cell line were plated in duplicate, at density of 40,000 per/well. Raw data was analyzed using GraphPad-PRISM.
2.5. ATP/ADP ratio assay kit
ATP/ADP ratios of unscratched and wound-induced cells were measured by ADP/ATP Ratio Assay Kit (#ab65313, abcam). Cell lysates were mixed with nucleotide releasing buffer and incubated for 5 min at room temperature with gentle shaking. Then, 100 μl prepared reaction mix was added and the background luminescence was read (Data A), then 50 μl sample was added and after 2 min the luminescence was read (Data B). To measure ADP levels in the cells, the samples were read again (Data C), then added 10 µL of 1× ADP converting enzyme and read the samples again after approximately 2 min (Data D). ATP/ADP ratio equation: ATP measurement / (ADP initial - ADP converted to ATP). Ratios were calculated from the ATP/ADP ratio of no scratch subtracted from ATP/ADP ratio of T=0 scratch.
2.6. Measurement of intracellular ATP dynamics
ATP levels are measured in live cells using an ATP-binding FRET indicator provided by the IncuCyte CytoATP Orange Lentivirus Reagent kit (catalog #4772; purchased from Sartorius). Analysis was done using the SX5 Metabolism Optical module and the ATP analysis software. Upon binding of cytoplasmic ATP conformational changes occurs in the ATP-binding sensor utilizing dual excitation (535X, mKOk orange; and 485X, cpmEGFP green) and fixed emission of 578M, increasing amounts of ATP lead to increased transmission and emission at 575M, which is used to evaluate dynamic changes when compared to a non-ATP binding sensor. Cancer, normal/immortalized and normal/primary cells were infected with the CytoATP binding, or non-binding lentivirus followed by puromycin selection. After wound initiation cells were visualized migrating into wound with the IncuCyte (model SX5).
2.7. Statistical Analysis
All graphs and statistical analyses were done using Graph Pad Prism 8 or 10. Statistical significance was assigned to data with the following criteria: P*<0.05 and N representing individual experiments greater than three, except for test of physical parameters done in isogenic cells. S.E.M., standard error of the mean. Data analyzed using unpaired t-test; one-way ANOVA, two-way ANOVA P*<0.05, **<0.01, ***<0.005, ****<0.0001.
3. Results
- Non-malignant cells migrate faster than cancer cells.
- Irrespective of media, normal cells outcompete cancer cells.
- Physical Differences do not account for faster migration speed.
- Increased ATP demands at wound induction.
- ATP levels oscillate throughout wound resolution in normal cells.
3.1. Non-malignant cells migrate faster than cancer cells.
Recent evidence has indicated that approved FDA drugs, including cisplatin, and paclitaxel to name a few, can promote metastasis and drug resistance in preclinical models[18,19,20]. Given these new revelations, we sought to develop a screen for migratory potential (Figure 1 and Figure 2). First, we screened malignant cell lines isolated from primary and metastatic tumors of lung and breast, together with their non-cancerous counterparts (as controls) for the ability to migrate and/or invade collagen/Matrigel (Figure 1 and Figure 2). Measuring the time for wound density to reach a defined percentage (Time for Density to reach 80% and 40%, – TD80 and TD40), gave us a quantifiable readout of migration potential (Figure 1b and Figure 2b). What we detected unexpectedly was the non-malignant cells of the lung had significantly higher migratory potential than tumor cells from their respective tissues (Figure 1b and 1c). Predictably, we observed the non-cancerous MCAF10A cell line was significantly slower than the breast tumor cell line, MDA-MB-231 (Figure 2b and 2c) as demonstrated in previous studies[13,14]. Based on these observations we redirected our investigation of “the differences between how non-malignant and malignant cells migrate” to “why are normal cells faster at wound closure than cancer cells”.
3.2. Irrespective of media, normal cells outcompete cancer cells.
Although, our initial screen was conducted in cancer cells grown in RPMI 1640 medium (with 5% fetal bovine serum, FBS) and normal non-malignant cells in (keratinocyte-serum free media, KSFM) we acknowledge that different medias have vastly different components. Experimentally, we chose to focus on the isogeneic lung cell lines HCC4017, isolated from tumor burdened lung, and HBEC30UI (30UI) and HBEC30KT (30KT), from normal contralateral lung of the same patient[21]. We adapted our lung cancer cells, HCC4017 to normal cell growth media, KSFM which contains no serum, and KSFM with 2% FBS (Figure 3a). Interestingly, we found normal cells adapted for migration experiments in KSFM with 2% FBS, or RPMI 1640 5% FBS were still able to close wounds (albeit, not statistically significant in RPMI 1640 with 5% FBS) faster (Figure 3b). Closer examination of wound closure dynamics indicated variations of both normal and cancer cells in RPMI 1640 5% FBS (Figure 3b-c).
3.3. Physical parameters do not account for faster migration speed.
To address our new query, we first investigated whether physical differences in cell doubling, mass, volume, or shape could account for the increased migration potential of the non-malignant cells (Figure 4). Between 1000-3200 individual cells were tracked, graphing only those cells that remained from the first to last frame (A1 Supplemental Video 1-3). We found the speed of the individual non-malignant cells though not statistically different from each other, were significantly faster than the malignant cells (Figure 4g). No gross differences in mass, shape (sphericity), or dimensions (radius, volume) between the normal (either 30KT or 30UI) and the cancer cells (Figure 4b-e). Statistically significant differences in mass, sphericity, and volume observed between 30KT and 30UI were attributed to the immortalization process (addition of constitutive expression hTERT and CDK4), as quantification of the same dimensions between 30KT and HCC4017, were not statistically significant. Taken together these data suggest the differences observed in the speed of malignant cells and non-malignant cells were not due to their physical parameters.
3.4. Increased ATP demands at wound induction.
A considerable expenditure of energy in the form of ATP is needed for cell migration. Differences between how normal and cancer cells meet this high demand for ATP during migration are currently not well understood. Intracellular ratios of ATP-to-ADP traditionally are used as a readout of cellular energy[22], where energy is derived from the hydrolysis of ATP to ADP[23]. We assessed energy changes during migration, beginning with premigratory-wound induction via two different ATP assays (Figure 5a). We measured the changes in ATP/ADP ratios in lysates from confluent/no scratched (NS) and premigratory-wound induced cells (T=0), there was an increase in ATP/ADP ratios from NS to T=0, (Figure 5b) indicating an acute energy demand. Next, we constructed cell lines stably expressing a lentiviral FRET-based ATP sensor in several non-malignant and malignant cells (Figure 5c). Like the first assay, most of the cells showed increased intracellular ATP levels going from NS to T=0. Subsequently, in the non-malignant cells of the breast and lung, there was also a higher fold change in the ATP/ADP ratios than in their respective cancer tissues (Figure 5d-e). Use of cells stably expressing the CytoATP sensor allowed for tracing intracellular ATP (iATP) levels throughout migration (Figure 5d-e). The isogeneic pair 30KT and HCC4017 revealed a spike in the iATP levels that occurred at the pre-migratory wound induction stage (T=0), which resolves and stabilizes over the course of wound closure (Figure 5e). The other cell lines likewise show the same results: there is an acute energy demand that accompanies wound induction in both normal and cancer cells (Figure 5d and 5e).
3.5. ATP levels oscillate throughout wound resolution in normal cells.
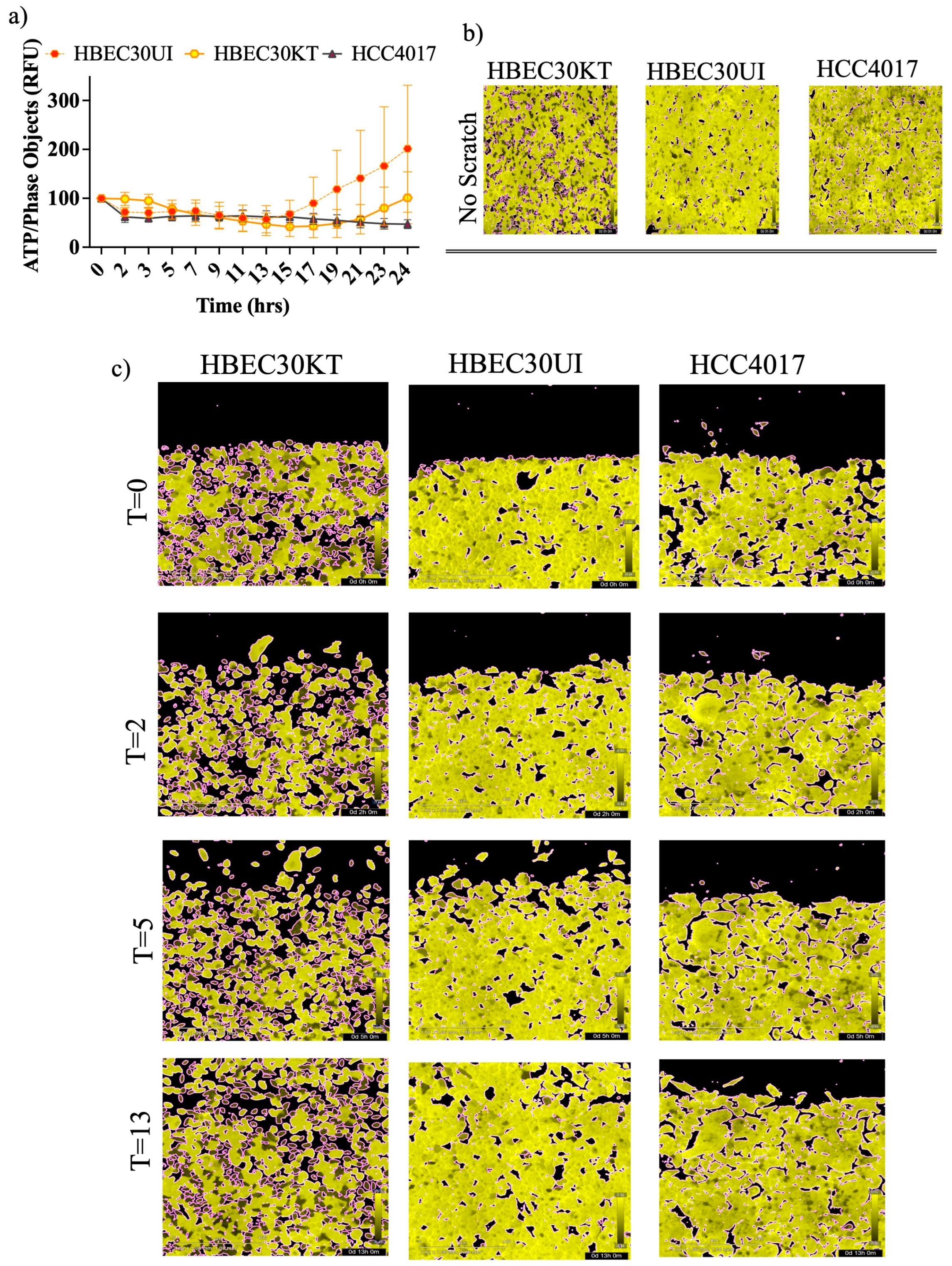
Various modes and morphologies of cancer and normal epithelial cell migration have been described ranging from single cell migration (e.g., amoeboid or mesenchymal state) to collective migration (e.g., mesenchymal chains, clusters, and multicellular sheets undergoing biophysical unjamming)[24,25,26,27,28]. Recently hybrid states of collective migration have also been reported describing the ability of invading cells to adopt leader-follower hierarchies [24,25,26,29,30]. Mechanisms responsible for plasticity of migratory modes ranges from variations in genetic/epigenetic signals, cell state of premalignant lesions, and more recently cellular energetics[31,32,33]. Previous literature suggests a need to maintain certain thresholds of ATP to maintain leader positions during collective migration[33]. While, our results indicate an initial burst of iATP at wound induction (Figure 5b and Figure 5c) we also find iATP levels are stabilized throughout migration until wound resolution where there is an increase in iATP in the normal cells upon wound closure that correlated with speed of the cells (Figure 6a).
Variations in iATP levels in singular cells were detected in the immortalized (30KT) cells compared to the primary unimmortalized cells (30UI) in confluence (Figure 6b) that lessened throughout migration (Figure 6c). Additional dissimilarities in migration modes were also observed whereby the 30UI cells moved primarily as collective sheets led by discrete “leader” cells, while the 30KT cells exhibited single and pockets of collective migration like the HCC4017 cancer cells. We attributed the migration mode differences in the normal cells to the immortalization process. In our model, we observed no gross increase in iATP dynamics specifically at the migratory edge of the wound (Figure 6c).
4. Discussion
Previous reports have highlighted the vast discrepancies between plasticity, proliferation rates, and metabolic needs of malignant cells compared to non-malignant cells. Few studies, however, have systematically compared these same parameters side-by-side during migration; resulting in a lack of understanding of what are the basic requirements for cancer and normal cell locomotion that may be exploited therapeutically[5,15,28,34,35,36,37]. We conducted wound-healing/migration screens of normal and cancer cells isolated from breast and lung and found that normal lung cells closed wounds faster than lung cancer cells. Our working hypothesis for potential differences led us to investigate ATP. The function of ATP as a switch for increased cell migration has been well documented primarily in brain and immune system[38,39,40,41,42,43,44]. These pleiotropic effects of ATP range from paracrine – indicator of injury, stimulator of immune response and repair, to autocrine – sensor and regulator of intracellular metabolic demand. Intracellular and extracellular ATP sensing carried out via the purinergic receptor family, which in addition to their modulation of calcium release act in autocrine manner to increase subsequently more ATP increases migration via cytoskeletal and matrix remodeling.
Here, we report that normal lung epithelial cells have higher basal and wound-induced intracellular ATP levels than lung tumor cells. These findings have broad reaching implications for aggressive carcinomas from other non-breast tissues such as colon and pancreas. Our data also rouses further important questions concerning ATP generation and it’s use in response to varied fuel sources or other metabolic needs during normal wound healing and throughout the metastatic cascade. Recent studies have both described heightened and suppressed Warburg effects during cancer cell migration, thus additional investigations are critically needed to elucidate differential mechanisms in normal cell migration as well[6,17,33,45]. The therapeutic connections to various nonmalignant lung diseases are also intriguing and range from tissue regeneration post-respiratory infection, autoimmune infiltration and wound repair linked to asthma, pulmonary fibrosis, and acute or COPD (Chronic Obstructive Pulmonary Disease) to name a few.
5. Conclusions
Unbiased screening for the migratory potential of cells from both normal and cancer tissue of the lung revealed that normal bronchial epithelial cells (both immortalized and unimmortalized/primary) migrated faster than: 1) established lung cancer cell lines 2) the isogeneic tumor tissue established from the same patient. We were able to repeat data from countless other studies demonstrating the augmented migratory potential of metastatic breast cancer cells of the MDA-MB-231 compared to the normal immortalized MCF10A cell line, validifying of our experimental set-up. Further examination indicated higher basal levels of intracellular ATP in normal cells of both breast and lung, that increased upon wound-induction.
Supplementary Materials
Supplemental video, 1- HBEC30KT; 2-HEC30UI; 3-HCC4017.
Author Contributions
Conceptualization, J.K.O., A.L., and B.K.; Methodology, B.K. and A.L. Writing—original draft preparation, J.K.O.; writing—review and editing, J.K.O., B.K., I.T.; Investigations performed by B.K., A.L., M.M., M.O.; Validation, resources, I.T.; Resources provided by B.G. and Hamon Cancer Center; Supervision, J.K.O.; Funding acquisition, J.K.O. All authors have read and agreed to the published version of the manuscript.
Funding
Awarded to JKO: CPRIT (Cancer Prevention Research Institute of Texas) grant number RR210016; V Foundation V2021-028, Awarded to B.K. Cancer Prevention and Research Institute of Texas (CPRIT) Training Grant (RP210041).
Acknowledgments
We would sincerely like to thank Elma Zaganjor, John D. Minna and Svetlana Earnest for critical review of work and manuscript. We would also like to thank Dionne Ware for admin support.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Appendix A
The appendix is an optional section that can contain details and data supplemental to the main text—for example, explanations of experimental details that would disrupt the flow of the main text but nonetheless remain crucial to understanding and reproducing the research shown; figures of replicates for experiments of which representative data is shown in the main text can be added here if brief, or as Supplementary data. Mathematical proofs of results not central to the paper can be added as an appendix.
References
- Bressan, C. et al. The dynamic interplay between ATP/ADP levels and autophagy sustain neuronal migration in vivo. Elife 9, (2020). [CrossRef]
- Bressan, C. & Saghatelyan, A. AMPK-induced autophagy as a key regulator of cell migration. Autophagy 17, 828-829, (2021). [CrossRef]
- LeBleu, V. S. et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 16, 992-1003, 1001-1015, (2014). [CrossRef]
- Pacheco-Velazquez, S. C. et al. Energy Metabolism Drugs Block Triple Negative Breast Metastatic Cancer Cell Phenotype. Mol Pharm 15, 2151-2164, (2018). [CrossRef]
- Schafer, Z. T. et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109-113, (2009). [CrossRef]
- Zanotelli, M. R. et al. Regulation of ATP utilization during metastatic cell migration by collagen architecture. Mol Biol Cell 29, 1-9, (2018). [CrossRef]
- Cunniff, B., McKenzie, A. J., Heintz, N. H. & Howe, A. K. AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol Biol Cell 27, 2662-2674, (2016). [CrossRef]
- Iavarone, A. & Massague, J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-beta in cells lacking the CDK inhibitor p15. Nature 387, 417-422, (1997). [CrossRef]
- Shi, Y., Hata, A., Lo, R. S., Massague, J. & Pavletich, N. P. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature 388, 87-93, (1997). [CrossRef]
- Kang, Y. et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537-549, (2003). [CrossRef]
- Minn, A. J. et al. Genes that mediate breast cancer metastasis to lung. Nature 436, 518-524, (2005). [CrossRef]
- Clark, A. G. & Vignjevic, D. M. Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol 36, 13-22, (2015). [CrossRef]
- Desai, S. P., Bhatia, S. N., Toner, M. & Irimia, D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J 104, 2077-2088, (2013). [CrossRef]
- Kashani, A. S. & Packirisamy, M. Cancer cells optimize elasticity for efficient migration. R Soc Open Sci 7, 200747, (2020). [CrossRef]
- Simpson, K. J. et al. Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat Cell Biol 10, 1027-1038, (2008). [CrossRef]
- Welch, D. R. & Hurst, D. R. Defining the Hallmarks of Metastasis. Cancer Res 79, 3011-3027, (2019). [CrossRef]
- Zanotelli, M. R. et al. Energetic costs regulated by cell mechanics and confinement are predictive of migration path during decision-making. Nat Commun 10, 4185, (2019). [CrossRef]
- Sanchez-Laorden, B. et al. BRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signal 7, ra30, (2014). [CrossRef]
- Steeg, P. S. Targeting metastasis. Nat Rev Cancer 16, 201-218, (2016). [CrossRef]
- Volk-Draper, L. et al. Paclitaxel therapy promotes breast cancer metastasis in a TLR4-dependent manner. Cancer Res 74, 5421-5434, (2014). [CrossRef]
- Ramirez, R. D. et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res 64, 9027-9034, (2004). [CrossRef]
- Tantama, M., Martinez-Francois, J. R., Mongeon, R. & Yellen, G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun 4, 2550, (2013. [CrossRef]
- Vander Heiden, M. G. & DeBerardinis, R. J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657-669, (2017). [CrossRef]
- Friedl, P. Prespecification and plasticity: shifting mechanisms of cell migration. Curr Opin Cell Biol 16, 14-23, (2004). [CrossRef]
- Friedl, P. & Alexander, S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992-1009, (2011). [CrossRef]
- Friedl, P., Locker, J., Sahai, E. & Segall, J. E. Classifying collective cancer cell invasion. Nat Cell Biol 14, 777-783, (2012). [CrossRef]
- Park, J. A., Atia, L., Mitchel, J. A., Fredberg, J. J. & Butler, J. P. Collective migration and cell jamming in asthma, cancer and development. J Cell Sci 129, 3375-3383, (2016). [CrossRef]
- Park, J. A. & Fredberg, J. J. Cell Jamming in the Airway Epithelium. Ann Am Thorac Soc 13 Suppl 1, S64-67, (2016). [CrossRef]
- George, S., Martin, J. A. J., Graziani, V. & Sanz-Moreno, V. Amoeboid migration in health and disease: Immune responses versus cancer dissemination. Front Cell Dev Biol 10, 1091801, (2022). [CrossRef]
- Graziani, V., Rodriguez-Hernandez, I., Maiques, O. & Sanz-Moreno, V. The amoeboid state as part of the epithelial-to-mesenchymal transition programme. Trends Cell Biol 32, 228-242, (2022). [CrossRef]
- Vilchez Mercedes, S. A. et al. Decoding leader cells in collective cancer invasion. Nat Rev Cancer 21, 592-604, (2021). [CrossRef]
- Westcott, J. M. et al. An epigenetically distinct breast cancer cell subpopulation promotes collective invasion. J Clin Invest 125, 1927-1943, (2015). [CrossRef]
- Zhang, J. et al. Energetic regulation of coordinated leader-follower dynamics during collective invasion of breast cancer cells. Proc Natl Acad Sci U S A 116, 7867-7872, (2019). [CrossRef]
- De Marzio, M. et al. Genomic signatures of the unjamming transition in compressed human bronchial epithelial cells. Sci Adv 7, (2021). [CrossRef]
- DeCamp, S. J. et al. Epithelial layer unjamming shifts energy metabolism toward glycolysis. Sci Rep 10, 18302, (2020). [CrossRef]
- Delgado, O. et al. Multipotent capacity of immortalized human bronchial epithelial cells. PLoS One 6, e22023, (2011). [CrossRef]
- Larsen, J. E. et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J Clin Invest 126, 3219-3235, (2016). [CrossRef]
- Idzko, M., Ferrari, D. & Eltzschig, H. K. Nucleotide signalling during inflammation. Nature 509, 310-317, (2014). [CrossRef]
- Idzko, M., Ferrari, D., Riegel, A. K. & Eltzschig, H. K. Extracellular nucleotide and nucleoside signaling in vascular and blood disease. Blood 124, 1029-1037, (2014). [CrossRef]
- Liu, X., Hashimoto-Torii, K., Torii, M., Haydar, T. F. & Rakic, P. The role of ATP signaling in the migration of intermediate neuronal progenitors to the neocortical subventricular zone. Proc Natl Acad Sci U S A 105, 11802-11807, (2008). [CrossRef]
- Liu, X. H. et al. Rapid inhibition of ATP-induced currents by corticosterone in rat dorsal root ganglion neurons. Pharmacology 82, 164-170, (2008). [CrossRef]
- Maeda, T. et al. ATP increases the migration of microglia across the brain endothelial cell monolayer. Biosci Rep 36, (2016). [CrossRef]
- Saez, P. J. et al. ATP promotes the fast migration of dendritic cells through the activity of pannexin 1 channels and P2X(7) receptors. Sci Signal 10, (2017). [CrossRef]
- Zeng, J. W. et al. Inhibition of ATP-induced glutamate release by MRS2179 in cultured dorsal spinal cord astrocytes. Pharmacology 82, 257-263, (2008). [CrossRef]
- Zanotelli, M. R., Zhang, J. & Reinhart-King, C. A. Mechanoresponsive metabolism in cancer cell migration and metastasis. Cell Metab 33, 1307-1321, (2021). [CrossRef]
Figure 1.
Non-malignant lung cells close wounds faster than cancer cells a) Table of lung cell lines tested for the ability to migrate on collagen and invade Matrigel. KT- CDK4/hTERT overexpression; UI un-immortalized/primary a Isogeneic Cell lines from same patient AA Black/African descent. b) Normal and cancer cells were plated in duplicate in a 96-well plate at a density between 35,000 to 50,000/ per well. Heatmaps for panel (b) represent the quantification of time to closure. Graph of panel for b) quantitate the time for wounds to reach a density of 80% (TD80) in lung. Wounds were initiated using the 96-well Incucyte® Wound Maker. c) Images were collected every 1-2 hours on the SX5 Incucyte ®Live cell imaging instrument and analyzed using the Sartorius 96-well cell migration software application. Yellow lines in images indicate the initial wounded area; red lines represent cells moving into the wound. Data analyzed using unpaired t-test ** P<0.01, Error bars represent +/- S.E.M N is quantitation of individual experiments. N=3 for normal cells: HBEC3KT,14UI, 14KT, 30KT; for cancer cells N=5 for HCC2352; N=4 for HCC2814; N=5 for HCC4017.
Figure 1.
Non-malignant lung cells close wounds faster than cancer cells a) Table of lung cell lines tested for the ability to migrate on collagen and invade Matrigel. KT- CDK4/hTERT overexpression; UI un-immortalized/primary a Isogeneic Cell lines from same patient AA Black/African descent. b) Normal and cancer cells were plated in duplicate in a 96-well plate at a density between 35,000 to 50,000/ per well. Heatmaps for panel (b) represent the quantification of time to closure. Graph of panel for b) quantitate the time for wounds to reach a density of 80% (TD80) in lung. Wounds were initiated using the 96-well Incucyte® Wound Maker. c) Images were collected every 1-2 hours on the SX5 Incucyte ®Live cell imaging instrument and analyzed using the Sartorius 96-well cell migration software application. Yellow lines in images indicate the initial wounded area; red lines represent cells moving into the wound. Data analyzed using unpaired t-test ** P<0.01, Error bars represent +/- S.E.M N is quantitation of individual experiments. N=3 for normal cells: HBEC3KT,14UI, 14KT, 30KT; for cancer cells N=5 for HCC2352; N=4 for HCC2814; N=5 for HCC4017.
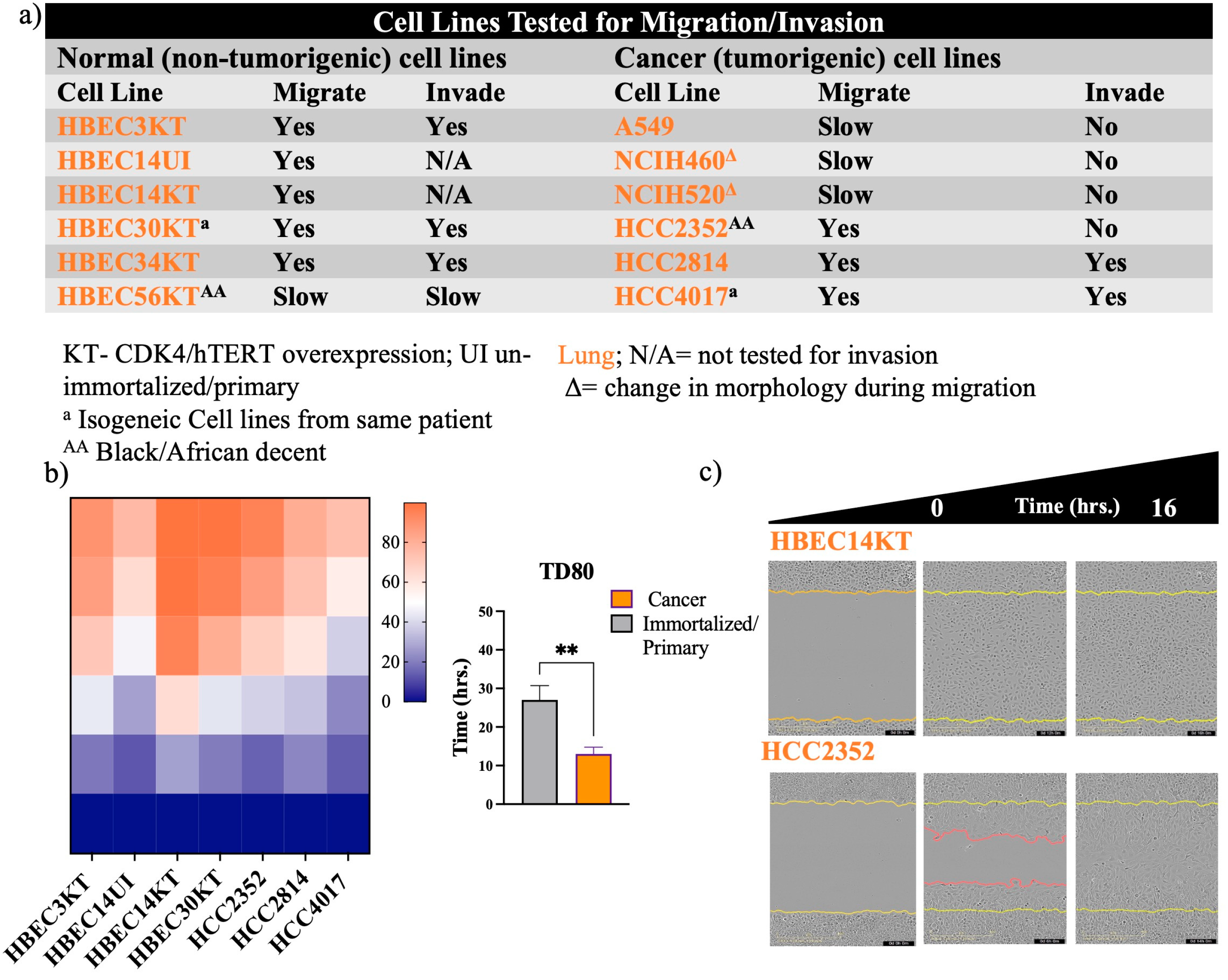
Figure 2.
MDA-MB-231 is at faster wound closure than MCF7 and MCF10A a) Table of breast/mammary cell lines tested for the ability to migrate on collagen and invade Matrigel. Cells were plated in duplicate in a 96-well plate at a density between 35,000 to 50,000/ per well. Heatmaps for panel (b) represents quantification of time to closure. Graph of panel for b) quantitate the time for wounds to reach a density of 40% (TD40). Error bars represent S.E.M. N is quantitation of individual experiments. N=3. Data was analyzed using one-way ANOVA, P*<0.05 for MCF10A compared to MDA-MB-231.
Figure 2.
MDA-MB-231 is at faster wound closure than MCF7 and MCF10A a) Table of breast/mammary cell lines tested for the ability to migrate on collagen and invade Matrigel. Cells were plated in duplicate in a 96-well plate at a density between 35,000 to 50,000/ per well. Heatmaps for panel (b) represents quantification of time to closure. Graph of panel for b) quantitate the time for wounds to reach a density of 40% (TD40). Error bars represent S.E.M. N is quantitation of individual experiments. N=3. Data was analyzed using one-way ANOVA, P*<0.05 for MCF10A compared to MDA-MB-231.
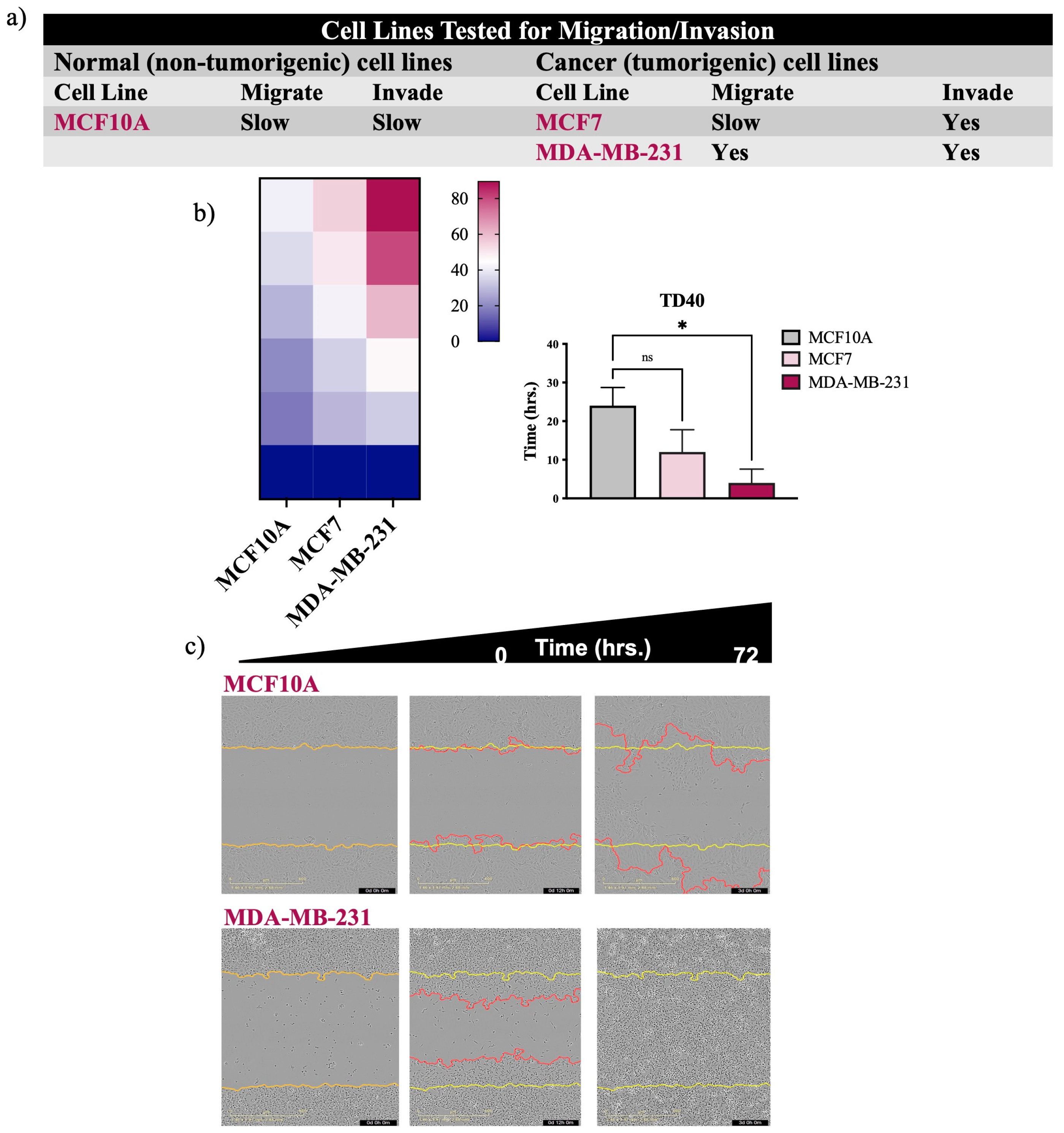
Figure 3.
Irrespective of media, normal cells outcompete cancer cells. a) Graph of isogeneic lung cells quantitating the time for wounds to reach a density of 50% (TD50) in various media. b-d) Measurements of wound width (in μm) of each cell line in different media. Error bars in a) represent individual experiments of N=3 for each cell line +/-S.E.M. P*<0.05 achieved using two-way ANOVA. Wound measurements are representative of one experiment from (a).
Figure 3.
Irrespective of media, normal cells outcompete cancer cells. a) Graph of isogeneic lung cells quantitating the time for wounds to reach a density of 50% (TD50) in various media. b-d) Measurements of wound width (in μm) of each cell line in different media. Error bars in a) represent individual experiments of N=3 for each cell line +/-S.E.M. P*<0.05 achieved using two-way ANOVA. Wound measurements are representative of one experiment from (a).
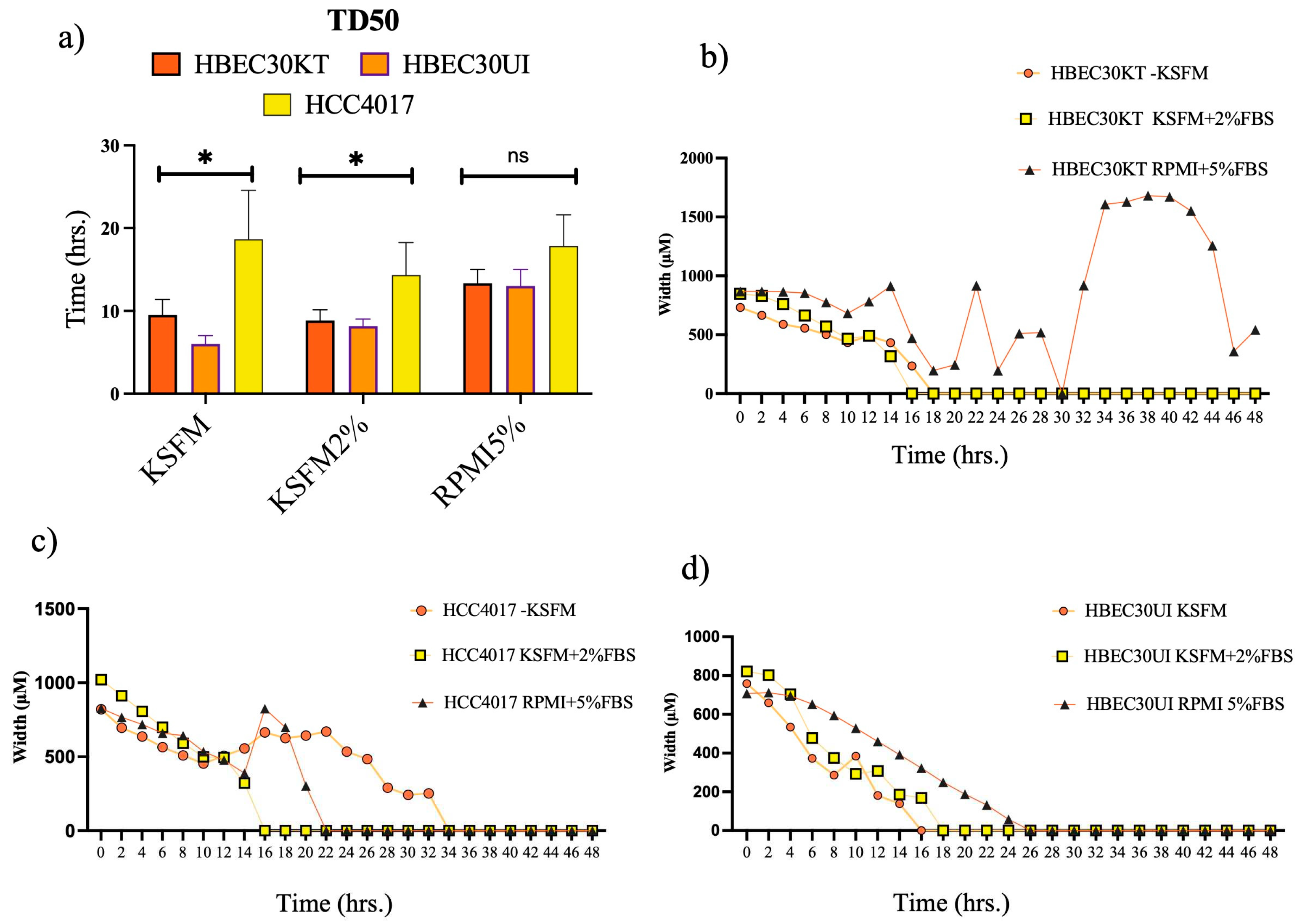
Figure 4.
Physical Parameters do not account for faster migration speed. Each cell line was plated at a density of 40,000/ per well. Wounds were initiated using the 96-well Incucyte® Wound Maker. Images were collected every 30 minutes on the LiveCyte imager. A total of 45 frames were collected. A combination of 1000 (HBEC30UI) cells, 2500 (HCC4017), 3200 (HBEC30KT) cells were initially tracked and analyzed. The first 50 individual cells that were visible from frame 1 to frame 45 were graphed. Graphs represent 2 individual experiments run simultaneously with 3 replicas per cell line. Statistics of a) cell counts, b) dry mass, c) radius, d) sphericity, e) volume, f) track length, and g) speed were all achieved using one-way ANOVA, P*<0.05, **<0.01, ***<0.005, ****<0.0001.
Figure 4.
Physical Parameters do not account for faster migration speed. Each cell line was plated at a density of 40,000/ per well. Wounds were initiated using the 96-well Incucyte® Wound Maker. Images were collected every 30 minutes on the LiveCyte imager. A total of 45 frames were collected. A combination of 1000 (HBEC30UI) cells, 2500 (HCC4017), 3200 (HBEC30KT) cells were initially tracked and analyzed. The first 50 individual cells that were visible from frame 1 to frame 45 were graphed. Graphs represent 2 individual experiments run simultaneously with 3 replicas per cell line. Statistics of a) cell counts, b) dry mass, c) radius, d) sphericity, e) volume, f) track length, and g) speed were all achieved using one-way ANOVA, P*<0.05, **<0.01, ***<0.005, ****<0.0001.
Figure 5.
Increased ATP demands at wound induction a) Experimental scheme for ATP assessment. For b-i) Wounds were initiated using the 96-well Incucyte® Wound Maker. Images were collected every 2 hours on the SX5 Incucyte ®Live cell imaging instrument and analyzed using the Sartorius (96-well) SX5 Metabolism Optical module and the ATP analysis software. b) Cells were plated to confluency, lysates extracted from non-scratch control cells (NS) and at wound induction (T=0). ATP/ADP ratios of wound induction were subtracted from non-scratch controls (ΔATP/ADP). Error bars are representative of 2 out of 4 experiments. c-d) Intracellular ATP measurements of cell lines c) Non-malignant, and malignant cancer cells analyzed only for NS and T=0. Error bars on c) and d) are of 3 independent experiments. Intracellular ATP measurements of e) normal and cancer cells of the lung. Cells for (c-i) were all plated at a density of 40,000/ per well. Error bars graphs in e) are N=4 for 30KT and N=3 for HCC4017; heatmaps f-i) represent standard deviation of N=3 independent experiments. P*<0.05 Statistical significance was assessed using a two-way ANOVA for d) and e). Upper* indicates statistical difference between HBEC30KT and HCC4017 at wound induction and lower* indicates a statistical difference before wound induction.
Figure 5.
Increased ATP demands at wound induction a) Experimental scheme for ATP assessment. For b-i) Wounds were initiated using the 96-well Incucyte® Wound Maker. Images were collected every 2 hours on the SX5 Incucyte ®Live cell imaging instrument and analyzed using the Sartorius (96-well) SX5 Metabolism Optical module and the ATP analysis software. b) Cells were plated to confluency, lysates extracted from non-scratch control cells (NS) and at wound induction (T=0). ATP/ADP ratios of wound induction were subtracted from non-scratch controls (ΔATP/ADP). Error bars are representative of 2 out of 4 experiments. c-d) Intracellular ATP measurements of cell lines c) Non-malignant, and malignant cancer cells analyzed only for NS and T=0. Error bars on c) and d) are of 3 independent experiments. Intracellular ATP measurements of e) normal and cancer cells of the lung. Cells for (c-i) were all plated at a density of 40,000/ per well. Error bars graphs in e) are N=4 for 30KT and N=3 for HCC4017; heatmaps f-i) represent standard deviation of N=3 independent experiments. P*<0.05 Statistical significance was assessed using a two-way ANOVA for d) and e). Upper* indicates statistical difference between HBEC30KT and HCC4017 at wound induction and lower* indicates a statistical difference before wound induction.
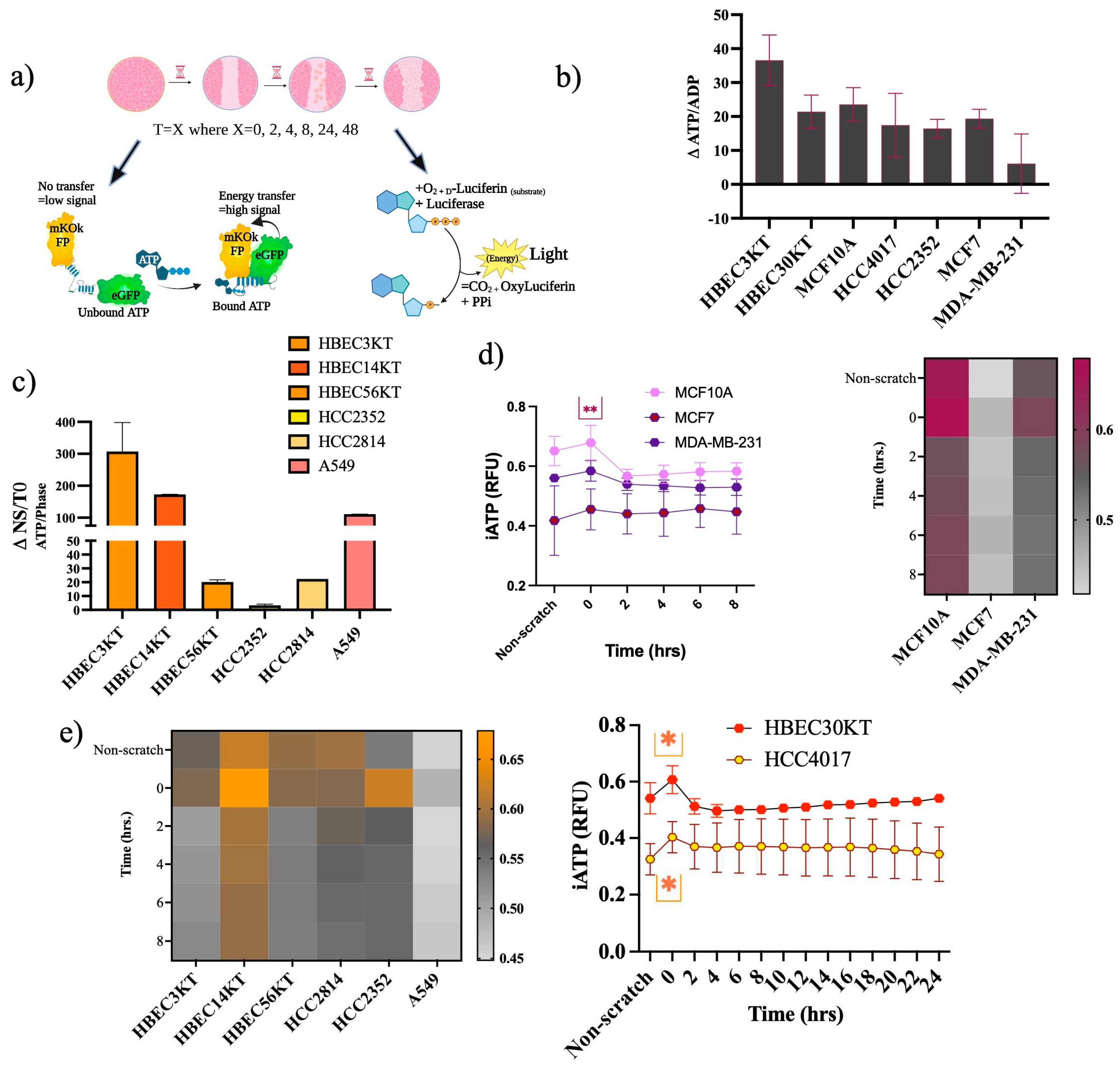
Figure 6.
Increased ATP demands at wound induction Isogeneic cells stably expressing the iATP construct were seeded at a density of 35-40,000/ per well. a) Images of the FRET channel (fixed emission of 578M, Yellow) were used to quantitate iATP per individual cells migrating into the wound (phase).
Figure 6.
Increased ATP demands at wound induction Isogeneic cells stably expressing the iATP construct were seeded at a density of 35-40,000/ per well. a) Images of the FRET channel (fixed emission of 578M, Yellow) were used to quantitate iATP per individual cells migrating into the wound (phase).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



