
Submitted:
31 October 2023
Posted:
01 November 2023
You are already at the latest version
Abstract
The critical enzyme Dihydrofolate Reductase-Thymidylate Synthase in Leishmania major (LmDHFR-TS) serves a dual-purpose role and is essential for DNA synthesis, a cornerstone of the parasite's reproductive processes. Consequently, the development of inhibitors against LmDHFR-TS is crucial for the creation of novel anti-Leishmania chemotherapies. In this study, we employed an in-house database containing 314 secondary metabolites derived from cinnamic acid that occurred in the Asteraceae family. We conducted a combined ligand/structure-based virtual screening to identify potential inhibitors against LmDHFR-TS. By consensus analysis of both approaches, we identified three compounds, i.e., lithospermic acid (237), diarctigenin (306), and isolappaol A (308), that exhibited a high probability of being inhibitors according to both approaches and were consequently classified as promising hits. Subsequently, we expanded the binding mode examination of these compounds within the active site of the test enzyme through molecular dynamics simulations, revealing a high degree of structural stability and minimal fluctuations in its tertiary structure. The in-silico predictions were then validated through in-vitro assays to examine the inhibitory capacity of the top-ranked naturally-occurring compounds against LmDHFR-TS recombinant protein. The test compounds effectively inhibited the enzyme with IC50 values ranging from 6.1 to 10.1 μM. In contrast, other common cinnamic acid derivatives (i.e., flavonoid glycosides) from the Asteraceae family, such as hesperidin, isovitexin 4′-O-glucoside, and rutin, exhibited low activity against this target. The selective index (SI) for all tested compounds was determined using HsDHFR, with moderate inhibitory effect. Among these hits, lignans 306 and 308 demonstrated the highest selectivity, displaying superior SI values compared to methotrexate, the reference inhibitor of DHFR-TS. Therefore, continued research into the anti-leishmanial potential of these C6C3-hybrid butyrolactone lignans may offer a brighter outlook for combating this neglected tropical disease.
Keywords:
Leishmania
; Asteraceae
; DHFR-TS
; Lignans
; Flavonoids
; Natural products
; Machine learning.
1. Introduction
Leishmaniasis is a neglected tropical disease (NTD) caused by protozoan parasites of the genus Leishmania, which are transmitted by the bite of infected sandflies. This disease affects millions of people worldwide, particularly in developing countries with poor health infrastructure. The primary clinical forms of the disease are visceral, cutaneous, and mucocutaneous. According to the World Health Organization (WHO), the global burden of leishmaniasis is estimated to be around 700,000 to 1 million new cases each year, with 90% of the cases occurring in just six countries: Afghanistan, Algeria, Brazil, Colombia, Iran, and Syria [1,2]. The sandflies that transmit leishmaniasis are most active at night and breed in wet soil, organic matter, or animal burrows [3]. In Colombia, 10 out of the 20 species that can infect both humans and other living beings are present. The cutaneous leishmaniasis (CL) form is the most frequent (98-99%), with the population under five years old and immunocompromised individuals being the most affected [4,5]. The number of CL cases reported in Colombia in 2022 was 4,906, with the departments of Amazonas, Boyacá, Caquetá, Cesar, Córdoba, Cundinamarca, Putumayo, Santander, and Sucre being the most affected areas [6].
Since the late 1980s, Leishmania-HIV co-infection has been reported in 35 countries, and there have also been other cases of Leishmania-Malaria co-infection, which are associated with the worsening of the clinical condition of patients with leishmaniasis. This co-infection type has increased the disease's burden due to the greater difficulty of clinical treatment [7,8]. Currently, antimonial compounds are the primary treatment for leishmaniasis; however, they present high toxicity and resistance in some endemic regions. To address these challenges, alternative drugs have been developed, such as liposomal amphotericin B, which significantly reduces the side effects and treatment duration associated with free amphotericin B, but is expensive [9,10]. Other drugs, such as paromomycin and miltefosine, have been associated with high toxicity, resistance, and teratogenic and abortive effects, promoting the discovery and development of low-cost, highly effective drugs with low toxicity [11]. Furthermore, it is worth noting that while Leishmania is a parasitic disease mainly affecting humans, it also affects animals such as dogs and rodents, which can serve as reservoirs for the parasite and increase the risk of transmission to humans [12,13]. Therefore, efforts to develop effective treatments and control measures must be considered.
High-throughput screening (HTS) has been used since the early 1990s to test the activity of large numbers of molecules against different diseases and thereby identify potential hits for drug development [14]. However, the uncertainty of success, as well as the time and screening costs, limit the use of this technique [15]. In recent years, chemoinformatics tools (e.g., molecular docking, machine learning) have been utilized to conduct in-silico studies that can predict the interactions between a protein and a ligand, reducing the number of actual laboratory experiments and accelerating the drug discovery process more efficiently and cost-effectively [14, 16]. The different research conducted in this field has led to the development of increasingly efficient and better-classifying models, which take advantage of large compound databases, opening the possibility of studying diseases that mainly affect poorer populations (NTD), which are not attractive to large industries and big pharma [17].
Leishmaniasis is commonly treated with plants from the Asteraceae family in traditional medicine. Given the diversity of this family (32,913 species) and the wide range of phytochemicals they contain, including alkaloids, coumarins, flavonoids, benzofurans, sterols, and terpenoids, they are considered a promising source of new leishmanicidal compounds [18]. Some secondary metabolites studied in this family have been sesquiterpenoids [19,20], triterpenes [21], phytosterols [22], and kauranes [23]. However, although they have shown activity to inhibit the disease, their pIC50 is not large enough, and compounds that are effective at low concentrations and selective against the parasite are preferred. A group of compounds that have not yet been studied, with records reporting promising in-vitro activity, are derivatives of cinnamic acid belonging to the Asteraceae family [24-26].
Gouri et al. report some natural inhibitors against Leishmania amastigotes such as luteolin (IC50 = 3.12 μM), quercetin (IC50 = 10.5 μM), chrysin (IC50 = 13 μM), apigenin, myricetin, cinnamic acid (IC50 = 0.25 μM), licochalcone A (IC50 = 0.9 μM), which can play an important role in drug discovery [24]. Peixoto et al., on the other hand, evaluated the biological activity of 25 cinnamic acid derivatives against Leishmania braziliensis amastigotes, obtaining promising results and finding that aromatic rings with an oxygen as a heteroatom had a beneficial effect in terms of activity against Leishmania [25]. Considering that heterocyclic compounds have been of great importance for drug development in the pharmaceutical industry, derivatives of cinnamic acid, which is an aromatic carboxylic acid commonly substituted in trans position by an acrylic acid group, represent an interesting starting point for directing studies in the search for possible hits against different species of leishmaniasis [27]. Although some of these compounds have already been studied, many more remain to be analyzed.
In the present study, a computational approach was carried out to select potential inhibitors of the bifunctional enzyme dihydrofolate reductase-thymidylate synthase (DHFR-TS) of Leishmania major given that it plays a crucial role in the synthesis of DNA in trypanosomatids, which is essential for the parasite's reproduction [28]. In this regard, a custom-made, in-house library containing 314 specialized metabolites derived from cinnamic acid was virtually screened. Initially, a ligand-based predictive classification model was developed using experimental information on the IC50 values retrieved from in-vitro assays of reported compounds against Leishmania. In parallel, using a hybrid LmDHFR-TS model constructed based on its amino acid sequence [29], a structure-based ranking, through molecular docking calculations, was performed using the investigated specialized metabolite database. Through a consensus analysis, molecules with the highest probability of being inhibitors by both approaches were classified as possible hits. These secondary metabolites were evaluated through in-vitro assays using the recombinant LmDHFR-TS, and ADMET properties were calculated to determine their pharmacokinetic properties.
2. Results and Discussion
2.1. Combined Ligand-/Structure-Based Virtual Screening Approach using LmDHFR-TS
2.1.1. Ligand-Based Virtual Screening
Initially, a bank of compounds with inhibitory activity reported against LmDHFR-TS was created from the ChEMBL database. These molecules were classified as active or inactive based on their reported IC50 values, using a cut-off point of IC50 = 5.0. Duplicate molecules were removed to perform the data curation process and obtain a virtual screening model with high prediction efficiency. Additionally, molecules with an IC50 value within ±0.1 of the cut-off point were considered. Finally, a total of 790 molecules were selected for model training. Among these, 378 were inactive (47.8%) and 412 were active (52.2%).
In the ligand-based process, VolSurf+ (128) and AlvaDesc (more than 4000) molecular descriptors were calculated from the three-dimensional representation of each database compound. These descriptors were then used to create the Random Forest (RF) model in Knime software (KNIME 4.5.0, the Konstanz Information Miner, Copyright 2003–2014, www.knime.org), consisting of 200 decision trees. The Gini Index was employed as a split criterion in the RF model to reduce the number of false-positive results. The dataset underwent a five-fold cross-validation strategy, where it was divided into five subsets, each containing an 80% modeling set and a 20% validation set. The modeling set was exclusively used for model construction and further subdivided into multiple training and test sets, maintaining an 80%/20% split ratio. These procedures were conducted following the approach described by Fourches et al. [30].
Molecular descriptors play a crucial role in drug discovery and development, as they represent the molecular and chemical properties of the studied compounds. In this study, the employed descriptors were instrumental. VolSurf+ generates three-dimensional (3D) molecular descriptors based on the distribution of molecular electrostatic potentials and hydrophobicity, encompassing molecular surface properties such as size, shape, and electrostatic potential distribution [31,32]. On the other hand, AlvaDesc offers a wide range of descriptor types, including constitutional descriptors (the number and type of atoms, bonds, and functional groups in the molecule), topological descriptors (representing molecular shape, size, and complexity), electrostatic descriptors (conveying molecular polarity and charge distribution), and quantum mechanical descriptors (relating to the electronic structure and properties of the molecule) [33,34].
The performance of the RF model was evaluated to compare the effectiveness of the two types of descriptors. This evaluation involved calculating classification precision, recall, F1-score, and Matthew's correlation coefficient (MCC). Additionally, Receiver Operating Characteristic (ROC) curves were analyzed, and the Area Under the ROC Curve (AUC) was calculated (see Figure 1). These evaluation metrics are commonly used to measure the performance of binary classification models. ROC curves and their AUC are often employed to assess the performance of models that produce continuous output scores or probabilities. AUC serves as a scalar measure of the model's overall ability to distinguish between positive and negative cases [33,35]
According to the parameters displayed in Figure 1, it is evident that the MCC and AUC values for both the test sets and cross-validation are higher for AlvaDesc descriptors compared to those obtained for VolSurf descriptors. However, considering that a higher AUC value indicates a more remarkable classification ability of the model and that MCC is expressed in a range of -1 to 1 (where a high value close to 1 suggests a strong correlation between the predicted class and the true class), good values were obtained for both AlvaDesc (AUC: 0.863 and 0.906, MCC: 0.554 and 0.645) and VolSurf (AUC: 0.855 and 0.884, MCC: 0.539 and 0.598) descriptors.
Regarding precision, recall, and F1-score, good and similar values were obtained for both models, except for the recall for inactive compounds in the model created using VolSurf descriptors, which was low with a value of 0.69. Sensitivity and specificity measures were also calculated to assess the performance of the RF model. For AlvaDesc, the values were 0.807 and 0.752, while for VolSurf, the values were 0.843 and 0.690, respectively. These results indicate a tendency to have few false negatives, a higher value of true negatives, and a lower false-positive rate for both descriptors. The applicability domain was also determined, confirming that the regression model can provide reliable predictions.
Ligand-based virtual screening (VS) was then employed to predict the potential inhibitory activity of 314 compounds derived from cinnamic acid in the Asteraceae family, as documented in the literature. Figure 2 displays the structure and probability of the five best compounds classified using AlvaDesc descriptors. These compounds were (E)-2-hydroxy-3',6'-dimethoxychalcone (103) [36], apigenin 7-O-(6´´-caffeoyl)-glucoside (235) [37], montamine (63) [38], 3-O-p-coumaroyl-betulinic acid (150) [39], and cordoin (202) [40]. In addition, Figure 2 presents the top five compounds predicted using VolSurf descriptors: 6,8-di-C-β-glucopyranosylchrysin (242) [41], montamine (63) [38], dihydrocubebin (305) [42], prebalanophonin (312) [43], and 4-O-feruloyl 5-O-caffeoylquinic acid (96) [44].
Among all the tested compounds, 116 were classified as active using AlvaDesc molecular descriptors, with probability values ranging from 0.50 to 0.71. On the other hand, 93 compounds were considered active with VolSurf molecular descriptors, and their probability values ranged from 0.50 to 0.86. Some of these molecules were previously reported to exhibit various activities, such as analgesic activity (305), antimalarial activity (150), cytotoxic activity (63), acting as anticancer agents (202), and demonstrating antiproliferative properties (312) [38,40-42,45-47].
Regarding the best compounds, only one contains nitrogen in its structure (63). The rest have various oxygen atoms, forming heterocycles or containing carbonyl groups, ethers, and alcohols. Additionally, one of them is a steroid (150), and another is glycosylated (242).
2.1.2. Structure-Based Virtual Screening
The Structure-Based Virtual Screening (VS) was conducted utilizing a hybrid homology model of LmDHFR-TS [29], a bifunctional enzyme with a critical role in the metabolic pathway of Leishmania parasites, as well as several protozoa species. Leihsmania genus is autotrophic for folate and unconjugated pteridines, with the enzyme DHFR-TS playing a pivotal role in the reduction of dihydrofolate to tetrahydrofolate, a cofactor in the biosynthesis of thymine in nucleotide metabolism [48].
To evaluate the putative inhibitory capability of cinnamic acid derivatives against LmDHFR-TS, molecular docking calculations were performed using Molegro software. The results were validated by redocking the co-crystalized ligand, i.e., ethyl 4-(5-{[(2,4-diaminoquinazolin-6-yl)methyl]amino}-2-methoxyphenoxy)butanoate (DQ1) along with the reference inhibitor methotrexate (MTX) (Figure 3).
The compounds were ranked according to the predicted docking binding energy using the probability calculation shown below (Eq. 1), as previously reported by Herrera-Acevedo et al. [19, 20]. The ten compounds exhibiting the highest probability of being active are presented in Table 1. Ranked compounds that did not previously show high ligand-based probability values but appeared among the best-ranked derivatives through a structure-based approximation are represented in Figure 4, along with their respective structure-based probability (PSB) values.
Where is the structure-based probability; is the docking energy of compound , where ranges from 1 to 314 (cinnamic acid derivatives dataset); is the lowest energy value of the dataset; and is the ligand energy from co-crystalized inhibitor.
The results showed that energy-based scoring values were lower for the cinnamic acid derivatives than for the reference ligands, suggesting that the studied compounds have a better affinity with the LmDHFR-TS active site in the molecular recognition process. Additionally, the docking results showed that 24.5% of the 314 cinnamic acid derivatives dataset had values above 0.5, and 64 of these top-ranked compounds had a lower docking score than methotrexate, which obtained -114.15 kJ/mol.
Three of the top-ranked molecules predicted to have high ligand-based probability values based on the RF model also demonstrated high structure-based probability values. Specifically, compound 242, ranked fourth in the structure-based classification (Table 1), was the best classified in the ligand-based VS model with VolSurf descriptors. Compounds 235 and 63, positioned among the top ten compounds in structure-based VS with docking scores of -161.4 kJ/mol and -160.1 kJ/mol, respectively, also showed high ligand-based probabilities. Compound 235 was predicted to be the second-best structure with high potential for inhibition using the model built with AlvaDesc descriptors, while Compound 63 was classified in the top three for both RF models (AlvaDesc and VolSurf molecular descriptors).
The analysis of residues for the best poses in the top three compounds showed that the residues responsible for ligand binding (Val30, Val31, Ala32, Ile45, Trp47, Asp52, Met53, Phe56, Val87, Pro88, Fhe91, Leu94, Val156, Tyr162, and Thr180) have been previously reported in the literature as part of the active site [49]. Certain characteristics of these residues, such as accessibility and charge distribution, enable selective drug design against these protozoans without affecting human enzymes [49]. The interaction diagrams in Figure 5 illustrate that the compound with the highest docking score (compound 241, Figure 5C) possesses heterocyclic rings similar to the reference ligands, with oxygen atoms replacing the nitrogen atoms present in the reference ligands. However, due to the similar electronegativities of nitrogen and oxygen, these atoms favor nearly identical interactions with the enzyme's active site.
Compounds 164 and 21 lack heterocyclic rings but contain benzene rings, which participate in π–π and π–alkyl interactions. Additionally, these compounds exhibit a relevant number of oxygen-containing groups, such as esters, ethers, and carboxylic acids, facilitating interactions with both residues within the active site and other residues. Specifically, the carboxylic moiety facilitates Van Der Waals interactions, crucial as they occur with the amino groups in the reference ligands and appear to be important since they are present in the three top-ranked molecules. On the other hand, compounds 242 and 140, containing only hydroxyl groups, are less favorable in this binding mode. Although both compounds are isomeric, compound 164 has few favorable interactions (8 interactions), and compound 21 has more interactions (25 interactions), but it also has some unfavorable interactions (Ala32, Ala47, Ala119).
Furthermore, it was observed that all ligands adopted a U-shaped conformation like the reference ligands DQ1 and MTX (Figure 5F), and most of them have strong hydrogen bonding interactions with the enzyme (Val156, Val30, Lys95, Met53, Phe91, and Arg97), which are important determinants for binding [50]. To further explore this behavior, a topological polar surface area (TPSA) map was constructed for both the reference ligands and the best-ranked compounds (Figure 6).
The results of the TPSA maps confirmed a similar spatial distribution among the three top-ranked compounds with respect DQ1 and MTX. An electron-deficient region was identified at the top of the molecule (Figure 6, blue area), which is consistently present in all evaluated molecules, including the two reference ligands. This observation rationalized the similar binding behavior within the active site of LmDHFR-TS, particularly with Met53 as a common crucial contact for these test compounds.
2.1.3. Consensus Analysis of the Two VS Approaches
A combined approach was employed to ascertain the potential activity of cinnamic acids against the LmDHFR-TS enzyme and to mitigate the selection of false-positive compounds. This approach incorporated probability scores derived from both structure-based and ligand-based virtual screening (VS) methods in conjunction with the true-negative rate obtained from the RF model (Eq. 2) [19].
The design of this approach aimed to assign a higher weight to the ligand-based probability scores (considering their reliance on experimental pIC50 values), in contrast to the structure-based probability scores, which are founded on protein-ligand interactions. This weighting scheme significantly reduces the risk of incorrectly classifying inactive molecules as active (false positives) [23].
Where = combined-approach probability, = structure-based probability, TN = true-negative rate, and = ligand-based probability (AD=AlvaDesc descriptors and VS=VolSurf descriptors).
Table 2 presents the results of the best-ranked compounds calculated from the consensus analysis equation. The compounds ranked among the top five for each method are highlighted in bold. Except for 235, all compounds were classified as potentially active in all virtual screening approximations used in this study. The consensus analysis identified 110 compounds with combined-approach probability values greater than 0.5; however, only 47% of these compounds (52) were classified as active through the three in-silico models used in this study (Table S1). Compound 63 (montamine) was the top-ranked compound. Montamine is an indole alkaloid that has been isolated from Asteraceae species, such as Centaurea schischkinii and Centaurea montana. Previous studies have reported its anticancer properties [38,51], but its efficacy against Leishmania has not been investigated.
The second best-ranked compound is 6,8-di-C-β-glucopyranosylchrysin (242), a derivative of chrysin obtained from Lychnophora ericoides (Asteraceae). Compared to compound 69 (chrysin) and 231 (techtochrysin), classified as inactive, the glycosylated derivative 242 has more hydroxyl groups, enabling interactions with the enzyme's active site. In previous studies, chrysin was biofunctionalized with gold particles due to its low bioavailability, poor absorption, and rapid excretion issues, aiming to neutralize Leishmania parasites through its activity against the kinase-3 enzyme [52]. However, compound 242 could represent an alternative due to its hydrophilic character resulting from the glycosyl groups, potentially inhibiting Leishmania parasites by interacting with LmDHFR-TS.
The third- and fourth best-ranked compounds were 4-O-feruloyl-5-O-caffeoylquinic acid (96) and lucenin-2, 6,8-di-C-β-glucopyranosylluteolin (241), both extracted from the genus Lychnophora, specifically Lychnophora ericoides [41] and Lychnophora salicifolia [44], respectively. Additionally, apigenin 7-O-rutinoside (39), lithospermic acid (237), diarctigenin (306), and isolappaol A (308) – four cinnamic acid derivatives that previously exhibited moderate values in both RF models and the molecular docking calculations (all classified as active) – appeared among the top ten ranked compounds in the combined approach (Figure 7). Hence, these compounds emerge as interesting antileishmanial candidates, as they exhibit activity across all models and maintain consistency in their probability values. Notably, consensus scoring methods are known to enhance hit rates by diminishing the likelihood of false positives [53].
The compounds 4-(3,4-dihydroxybenzyl)-2-(3,4-dihydroxyphenyl)tetrahydrofuran-3-carboxy-O-β-D-glucopyranoside (306) and 7-(3,4-dihydroxyphenyl)-3′,4′-dihydroxy-7,8,7′,8′-tetrahydronaphtho[8,8′-c]furan-1(3H)-one (308) are two lignans found in certain species of Asteraceae. Notably, Hypochaeris radicata (native to Europe, northern Asia, and parts of North Africa) and Arctium lappa (native to Europe and Asia) have been reported as a natural sources of these compounds. However, A. lappa is widely disseminated in America and H. radicata has also become invasive in regions as far-flung as New Zealand and Chile [54]. Conversely, compound 237, lithospermic acid, is a common polycyclic phenolic carboxylic acid that has been isolated from species of multiple botanical families, including Lamiaceae and Asteraceae. It has demonstrated a wide range of beneficial properties, acting against cardiovascular diseases and hepatitis. It allows endothelium-dependent vasodilatation, lowers blood pressure, and produces antioxidant effects [55,56].
2.2. Molecular Dynamics Simulations
Molecular dynamics (MD) studies were conducted to assess the protein–ligand stabilities, considering factors such as solvent, ions, pressure, and temperature for compounds 237, 306, and 308. These three compounds were identified as potential inhibitors of LmDHFR-TS through the consensus analysis of the approaches outlined in this study. Methotrexate (MTX) was also used as the reference ligand.
The structural stability was evaluated through root-mean-square deviation (RMSD) measurements. During the simulated time (100 ns), all tested compounds showed similar behavior concerning the apoenzyme of LmDHFR-TS (apoLmDHFR-TS, the protein devoid of a ligand) and the LmDHFR-TS···MTX complex. After 20 ns, all ligands (including the reference ligand) displayed diminished disturbances, reaching values ranging from 0.23 to 0.27 nm. The derivative 237 exhibited a higher disturbance between 60 and 85 ns, with RMSD values varying close to 0.1 nm (Figure 8a).
Scheme 8. b), allowing us to examine residue flexibility in the presence of various ligands [23]. The RMSF plot revealed higher fluctuations in the N-terminus region, reaching approximately 0.35 nm, while the C-terminal residues exhibited lower fluctuations with values close to 0.20 nm. Regions characterized by well-defined tertiary structures, such as α-helices or β-sheets, consistently displayed RMSF values ranging from 0.10 to 0.20 nm.
In general terms, all evaluated compounds exhibited the same behavior; however, we identified some specific cases. The residues Glu218 and Thr410, located in the protein's loop regions, showed the highest fluctuations for the apoenzyme, with Glu218 having approximately twice the RMSF value compared to the complexes with MTX and the tested cinnamic acid derivatives. Among the selected compounds, compound 237 displayed higher fluctuations in the loop regions than the other derivatives and MTX, with Gly118, Arg254, and Arg380 being the most variable amino acids. Compounds 306 and 308 exhibited a similar behavior throughout the simulation, with reduced flexibility when complexed with LmDHFR-TS.
The structural compactness and level of mobility of the protein-ligand complexes throughout the simulation period were evaluated using the radius of gyration (RoG) plot (Figure 8c) [23]. In the case of LmDHFR-TS, during the initial half of the 50 ns simulation, the complexes with cinnamic acid derivatives displayed RoG values that were indistinguishable from those of the control MTX and apoLmDHFR-TS (ranging from 2.64 nm to 2.70 nm). This finding suggests a high level of stability and low fluctuations in the tertiary structure. However, after 60 ns, compounds 237, 306, and 308 exhibited a similar behavior (varying between 2.64 nm to 2.70 nm) with increased perturbation compared to the DHFR-TS···MTX complex and the apoenzyme, which maintained a consistent mean value with fluctuations ranging from 2.62 to 2.64 nm.
After conducting molecular dynamic simulations, binding free energies for complexes involving compounds 237, 306, 308, and the control (MTX) with LmDHFR-TS were determined using the MM/PBSA method. The complexes of both benzylbutyrolactone-type lignans (306 and 308) and the polyphenolic acid (compound 237) with LmDHFR-TS showed binding free energies of -111.1 kJ/mol, -81.0 kJ/mol, and -91.6 kJ/mol, respectively. In all cases, the energy was higher than the -124.5 kJ/mol observed for the complex of MTX with LmDHFR-TS (Table 3).
All evaluated complexes, including the reference MTX, showed the same contribution pattern with negative energy values from van der Waals, electrostatic, and solvent-accessible surface area (SASA) parameters to the binding free energy. The van der Waals parameter presented the highest negative contribution, with values lower than -209 kJ/mol. This observation suggested that non-polar electrostatic interactions are the primary factor responsible for the molecular recognition of the LmDHFR-TS binding site by test compounds.
Regarding polar solvation, all compounds positively contributed to the total binding energy, with similar values for compounds 237, 308, and MTX. Conversely, diarctigenin (306) contributed less to this parameter. Moreover, electrostatic interactions negatively contributed to the binding free energies, being more relevant for MTX, with a negative contribution of -57.3 kJ/mol. Meanwhile, its influence was between 35% and 50% for the evaluated cinnamic acid derivatives compared to the reference ligand.
2.3. In-vitro Enzymatic Activity Inhibition for selected cinnamic acid derivatives (compounds 237, 306, and 308) against LmDHFR-TS and HsDHFR.
Five compounds from our in-house compound library were available to validate the results derived from the combined approach employing the two virtual screening (VS) methodologies through in-vitro enzymatic inhibition assays. Compounds 237, 306, and 308, classified as active in all approaches, along with hesperidin (140), an interesting flavonoid previously reported to exhibit antileishmanial activity through the induction of apoptosis and inhibition of sterol C-24 reductase [57], were included in this in-vitro exploration. Isovitexin 4′-O-glucoside and rutin, which displayed a moderate level of activity and were classified as inactive in one of the three approaches, were also evaluated against LmDHFR-TS, with methotrexate as the positive control.
The IC50 values were determined by analyzing concentration-response curves in the 0.1–128 μM range, using spectrophotometric monitoring of enzymatic activity under a standard DHFR assay. This analysis yielded a spectrum of values spanning from 6.1 to 53.2 μM, corresponding to pIC50 values ranging from 4.27 to 5.21. The compounds 237, 306, and 308 were the most active molecules against LmDHFR-TS. Hesperidin (IC50 = 21.6 μM) showed high activity against the target among the three tested flavonoids, with observed IC50 values of 53.2 μM and 41.7 μM for isovitexin 4′-O-glucoside and rutin, respectively (Table 4).
Structurally, we correlated the inhibitory activity against LmDHFR-TS with the relation between H-bond acceptors and donors (carbonyl and hydroxyl groups). Among the lignans 306 and 308, the presence of the γ-butyrolactone moiety showed that the most active compound (306) has a higher number of carbonyl groups compared to 308—a feature also observed in Lithospermic acid (237). Nevertheless, the low inhibitory activities of the glycosylated flavonoids (hesperidin, isovitexin 4′-O-glucoside, and rutin) indicate that the high presence of hydroxyl groups seems to have a negative effect on the inhibitory activity.
Subsequently, based on the obtained results from in-vitro tests using the recombinant protein of Homo sapiens (Hs) DHFR, the selectivity index (SI) was calculated. The IC50 values obtained against HsDHFR showed a different pattern, suggesting different mechanisms of action for these two proteins. Moderate SI values were revealed, with both benzylbutyrolactone-type lignans (compounds 306 and 308) showing the highest SI values at 4.6 and 4.4, respectively. Both lignans exhibited higher SI values than MTX, which was used as a positive control (Table 4).
2.4. Pharmacokinetic properties predictions.
Pharmacokinetic properties, i.e., absorption, distribution, metabolism, excretion, and toxicity (ADMET) of the compounds 237, 306, and 308, were predicted by using ADMETlab 2.0 and OSIRIS Datawarrior 5.5.0 [58,59]. Different approaches were used to assess the likelihood of oral bioavailability, and the results were mixed: on the one hand, all compounds were compliant with Lipinski's "rule of five" [60], but on the other hand, none of the above mentioned were accepted under the parameters established by Pfizer [61] and GSK [62]. Consequently, these compounds might not perform well regarding oral bioavailability (Table S2).
Concerning cytochrome P450 (CYP) and its isoenzymes, compound 237 was considerably likely to inhibit CYP2C9, and so were compounds 306 and 308 with CYP2C19, CYP2C9, and CYP3A4; this means that the metabolism of other drugs might be affected by the compounds evaluated herein. Meanwhile, compound 237 is likely to behave as a substrate for CYP2C9, and compounds 306 and 308 with CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4; thus, these compounds can be metabolized by the mentioned isoenzymes. Additionally, none of the studied compounds possess mutagenic, tumorigenic, reproductive, or irritant effects. Identifying potential hERG channel blockers has great importance when assessing the risk of cardiotoxicity [63]; for the three structures, the probabilities of hERG blocking were 0.212 at the most.
3. Materials and Methods
3.1. Cinnamic acid derivatives in-house dataset
A custom-made, in-house virtual library of 314 distinct cinnamic acid derivatives was built from 76 scientific articles using various search criteria, including keywords such as Asteraceae, Cinnamic Acid Derivatives, Lignans, Polyphenols, Flavonoids, and others. ChemAxon MarvinSketch [ChemAxon, version 21.18.0 (2021), a calculation module developed by ChemAxon, https://www.chemaxon.com/, accessed on 12 January 2023] was used to design all the structures.
The three-dimensional (3D) structures for the entire set were generated using Standardizer software [JChem, version 21.18.0 (2021), a calculation module developed by ChemAxon, https://www.chemaxon.com/, accessed on 12 January 2023]. This software standardized the structures, added hydrogens, performed aromatic form conversions, and refined molecular graphs in three dimensions. The final dataset was saved in Special Data File (.sdf) format.
3.2. Classificatory Machine Learning Models.
The analyses described below utilized Knime 4.5.0 software (KNIME 4.5.0, the Konstanz Information Miner, Copyright 2003–2014, www.knime.org) [64]. The process commenced with importing those descriptors generated by the Volsurf+ [31,32] and AlvaDesc [33,34] programs in CSV format.
Subsequently, these descriptors underwent segmentation via the 'Partitioning' node, implementing the stratified sampling option, with 80% of the initial dataset designated as the training set and the remaining 20% composing the test set. Random splits were also explored while maintaining consistent ratios for both training and test sets.
The model's creation processes entailed utilizing the modeling set and the RF algorithm, executed through a five-fold cross-validation procedure employing WEKA nodes. This approach provides a robust and efficient means to evaluate a model's performance by partitioning the data into five subsets for testing and training, facilitating model selection and generalization assessment [23].
RF models were configured with 200 trees, a random number of generator seed of 1, and employed the Gini Index as the split criterion for both training and cross-validation sets. Performance analysis of the selected models involved an assessment of their internal and external aspects, incorporating parameters such as sensitivity (true-positive rate), specificity (true-negative rate), and accuracy (overall predictability), derived from the confusion matrix.
To provide a more comprehensive understanding of the model's performance beyond accuracy, the ROC curve was employed, generated through a 'ROC curve' node, which relies on sensitivity and specificity. AUC values were obtained from this curve, where a value of 0.5 indicates an inability to distinguish between the two groups, and an AUC of 1 signifies perfect separation without overlap [65]. Furthermore, the MCC was calculated, with a value of 1 representing perfect prediction, 0 denoting random prediction, and -1 indicating complete disagreement between prediction and observation [66].
Additionally, a performance evaluation of the RF model using AlvaDesc and VolfSurf+ descriptors was conducted, incorporating precision, recall, and F1-score for both active and inactive sets.
3.3. Molecular docking calculations.
Molecular docking calculations involved the hybrid model of LmDHFR-TS bound to methotrexate (PDB ID: MTX) [29] and the three-dimensional structures of the cinnamic acid derivatives. We conducted these calculations using Molegro 6.0.1 software.
To ensure consistency, we removed all water molecules from both the enzyme and compound structures, and we prepared them to use the software's default settings. The MolDock scoring function was utilized, considering internal ES, internal H-bond, and Sp2-Sp2 torsions as criteria for evaluating the ligands.
The molecular docking process was executed through 10 runs utilizing the MolDock SE algorithm. It allowed for a maximum of 1500 interactions, maintained a population size of 50, included up to 300 steps, employed a neighbor distance factor of 1.00, and returned a maximum of 5 poses. To cover the enzyme's ligand-binding site, we established a grid with a 15 Å radius and 0.30 Å resolution [23,29].
Our results were categorized according to docking scores, reflecting the free energy or affinity of the interactions. Each calculation was repeated three times to ensure reliability. For comparison, we employed methotrexate (MTX) as a control.
Total Polar Surface Area (TPSA) maps were calculated using Spartan 14 for Windows Spartan'14 (Wavefunction Inc., Irvine, CA, USA) [67], and visualization of two-dimensional residual interaction diagrams was accomplished using Discovery Studio Visualizer v21.1.0.20298 (BIOVIA, Dassault Systèmes, San Diego, CA, USA) [23,29].
3.4. Molecular dynamics simulations.
Molecular dynamics (MD) simulations were conducted in YASARA Structure v. 19.12.14 [68], employing the AMBER14 force field to model the enzyme and ligand–enzyme systems. Before the simulations, each protein underwent hydrogen bond optimization, and chloride (Cl−) and (Na+) ions were added to the model systems through the transferable intermolecular potential 3-point (TIP3P) employing 0.997 g/L density for solvating the simulation cell. Acid dissociation constant values (pKa) were calculated for enzyme’s titratable amino acids with H-bonding network and the side-chain placement with a rotamer library (SCWRL) algorithm. Periodic boundary conditions were applied to facilitate the simulations, involving a cell size set 10 Å larger than the protein's size in all instances. An initial 5000-cycle energy minimization step was carried out using the steepest gradient approach. MD simulations used the particle-mesh Ewald (PME) method to account for long-range electrostatic interactions (8-Å cut-off distance). The simulations were performed under physiological conditions at 298 °K, pH 7.4, and 0.9% NaCl. Temperature control was maintained using a Berendsen thermostat while keeping the pressure constant. A multiple-time step algorithm with a time step of 2.00 fs was employed. Finally, MD simulations were run for 100 ns under constant pressure and the Berendsen thermostat, with snapshots saved at intervals of 100 p, using the YASARA macro (md_run.mcr) for all simulation phases. Subsequent analyses were also carried out using the default YASARA macro scripts. The molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) method was employed to calculate the binding free energies of apoenzyme and enzyme–ligand complexes from the resulting MD trajectories using the g_mmpbsa tool in Gromacs 5.0.5 (open source, http://www.gromacs.org) [69] on an Ubuntu 12.04 server, using NPT and periodic boundary conditions, as previously reported [29, 70].
3.5. LmDHFR-TS and HsDHFR enzymatic inhibition assays.
Purification and kinetic characterization of the recombinant LmDHFR-TS protein were performed according to the previously reported procedures [29,71-72], while HsDHFR protein was obtained from the commercial assay kit (CS0340, Merck KGaA, Darmstadt, Germany). Thus, the in-vitro evaluation of top-ranked selected compounds (237, 306, 308, hesperidin, rutin, and isovitexin 4′-O-glucoside) for inhibitory activity against LmDHFR-TS and HsDHFR was conducted using a spectrophotometric assay under standard DHFR conditions. These tested compounds were available from our in-house compound library. Rutin, lithospermic acid, and rutin were commercially purchased (>98%, Merck KGaA, Darmstadt, Germany). Isolappaol and diarctigenin were isolated from a commercial A. lappa powdered root extract (Prescribed For Life, Fredericksburg, TX, USA) through successive column chromatography, whose spectroscopic data was identical to those of previous reports [73-74].
The assay consisted of LmDHFR-TS or HsDHFR (2.7 nM), bovine serum albumin (BSA, 1 mg/mL), N-[tris(hydroxymethyl)-methyl]-2-aminoethanesulfonic acid (TES) buffer (100 mM, pH 7.0, 150 mM β-mercaptoethanol, 2 mM ethylenediaminetetraacetic acid (EDTA)), and nicotinamide adenine dinucleotide phosphate (NADPH, 100 μM) with varying concentrations of the test compounds (0.1-128 μM). The reaction was initiated by adding the substrate (7,8-dihydrofolate (H2F), 20 μM) and was monitored for 360 seconds at 340 nm (i.e., oxidation of NADPH to NADP+) to determine the initial reaction rate (Vo) through linear regression analysis of the resulting absorbance profile. All measurements were performed in triplicate, and MTX was used as a positive control [29]. The resulting Vo values were used to calculate the % inhibition, as 100 - (Ri / Rc x 100), where Ri is the Vo in the presence of the inhibitor, and Rc is the Vo in the absence of inhibitors (1% DMSO v/v final concentration). The % inhibition was measured for at least five concentrations (0.1-128 μM) for each test compound (i.e., cinnamic acid derivatives and MTX), and concentration-response curves (% inhibition vs. Log[inhibitor]) were constructed using non-linear regression to determine the IC50 using GraphPad Prism 7.0 (GraphPad, San Diego, CA, USA) [29].
3.6. Pharmacokinetic properties predictions.
For the compounds 237, 306, and 308, the ADMET parameters were calculated using the ADMETlab 2.0, an integrated online platform for predictions of ADMET properties [58]. Drug toxicity prediction was performed using OSIRIS DataWarrior v.5.2.1, based on the following parameters: mutagenicity, tumorigenicity, reproductive effect, and irritability [59].
4. Conclusions
This study identified three cinnamic acid derivatives, Lithospermic acid (237), Diarctigenin (306), and Isolappaol A (308), as potential inhibitors of LmDHFR-TS using a combined Virtual Screening approach (structure/ligand-based). Two random forest models were built using different molecular descriptors. Sensitivity and specificity measures were obtained to evaluate the RF model performance. The models classified 116 (AlvaDesc) and 93 compounds (VolSurf) as active, showing a tendency to minimize false negatives.
Molecular docking revealed that 24.5% of the 314 cinnamic acid derivatives had values above 0.5, with 64 of them having a lower docking score than methotrexate, the reference ligand. A consensus analysis combining the RF models with molecular docking identified 110 compounds with combined-approach probability values greater than 0.5. From them, 47% were classified as active through the in-silico models, identifying some compounds with potential leishmanicidal activity that a single approach had not previously highlighted. Lithospermic acid (237), diarctigenin (306), and isolappaol A (308) were among the top-ranked compounds, and their binding mode was evaluated using molecular dynamics. Finally, in-vitro assays using recombinant LmDHFR-TS validated the computational results, with 237, 306, and 308 exhibiting significant activity against LmDHFR-TS. However, moderate selective indices (SIs) were observed when assays were performed using HsDHFR. Despite this finding, higher SI values than MTX were observed. Thus, these three tested compounds emerged as an interesting alternative as hits against LmDHFR-TS; however, specific assays against the parasitic forms of Leishmania major are required to extend a clearer prospect for fighting this neglected tropical disease.
Supplementary Materials
The following are available online. Table S1: Combined probabilities for the entire structure dataset obtained from random forest model and molecular docking calculations. Table S2: Prediction of pharmacokinetic properties.
Author Contributions
Conceptualization, M. T. S., E.C.-B., C.H.-A; methodology, formal analysis, investigation, M.C.M.-V., S.L.-H., A.S.-C.; software, M.T.S., L.S., E.C.-B., C.H.-A.; validation, C.H.-A.; resources, E.C.-B., C.H.-A.; data curation, and writing—original draft preparation, C.H.-A.; writing—review and editing, M.T.S., L.S., E.C.-B., C.H.-A.; supervision, project administration, C.H.-A.; funding acquisition, E.C.-B., M.T.S, C.H.-A All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by ECCI University through the project PIDi-13-2022, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), grant numbers 141341/2020-3 and 302469/2022-1 and Universidad Militar Nueva Granada (UMNG) through the project EXT-CIAS-3854.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The supplementary material data can be accessed directly from the authors upon request.
Acknowledgments
Authors thank Universidad ECCI, UMNG and CNPq for their financial support. CEINTECCI: Centro de Investigación e Innovación en Tecnología y Ciencia - Universidad ECCI. This study is a result of the Program + Woman + Science + Equity, an initiative of the Ministry of Science, Technology, and Innovation of Colombia in collaboration with the Organization of Ibero-American States for Education, Science, and Culture (OEI). Additionally, this study is a cooperative activity within the Research Network on Natural Products against Neglected Diseases (ResNet NPND, www.resnetnpnd.org, accessed on 29 September 2023).
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of all compounds are available from the authors.
References
- Leishmaniasis. https://www.who.int/health-topics/leishmaniasis (accessed 2023-06-30).
- Abadías-Granado, I.; Diago, A.; Cerro, P. A.; Palma-Ruiz, A. M.; Gilaberte, Y. Cutaneous and Mucocutaneous Leishmaniasis. Actas Dermosifiliogr (Engl Ed) 2021, 112, 601–618. [Google Scholar] [CrossRef]
- Wijerathna, T.; Gunathilaka, N. Diurnal Adult Resting Sites and Breeding Habitats of Phlebotomine Sand Flies in Cutaneous Leishmaniasis Endemic Areas of Kurunegala District, Sri Lanka. Parasit Vectors 2020, 13, 284. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Almario, J.; Hernández, C. A.; Ovalle, C. E. Geographical Distribution of Leishmania Species in Colombia, 1985-2017. Biomedica 2019, 39, 278–290. [Google Scholar] [CrossRef]
- Ovalle-Bracho, C.; Londoño-Barbosa, D.; Salgado-Almario, J.; González, C. Evaluating the Spatial Distribution of Leishmania Parasites in Colombia from Clinical Samples and Human Isolates (1999 to 2016). PLoS One 2019, 14, e0214124. [Google Scholar] [CrossRef] [PubMed]
- Boletín Epidemiológico. https://www.ins.gov.co/buscador-eventos/Paginas/Vista-Boletin-Epidemilogico.aspx (accessed 2023-05-28).
- Leishmaniasis - OPS/OMS | Organización Panamericana de la Salud. https://www.paho.org/es/temas/leishmaniasis (accessed 2023-06-05).
- Wilairatana, P.; Chanmol, W.; Rattaprasert, P.; Masangkay, F. R.; Milanez, G. D. J.; Kotepui, K. U.; Kotepui, M. Prevalence and Characteristics of Malaria Co-Infection among Individuals with Visceral Leishmaniasis in Africa and Asia: A Systematic Review and Meta-Analysis. Parasit Vectors 2021, 14, 545. [Google Scholar] [CrossRef]
- Herrera Acevedo, C.; Scotti, L.; Feitosa Alves, M.; Formiga Melo Diniz, M. D. F.; Scotti, M. T. Computer-Aided Drug Design Using Sesquiterpene Lactones as Sources of New Structures with Potential Activity against Infectious Neglected Diseases. Molecules 2017, 22, 79. [Google Scholar] [CrossRef]
- Kumari, S.; Kumar, V.; Tiwari, R. K.; Ravidas, V.; Pandey, K.; Kumar, A. Amphotericin B: A Drug of Choice for Visceral Leishmaniasis. Acta Trop 2022, 235, 106661. [Google Scholar] [CrossRef] [PubMed]
- Palić, S.; Beijnen, J. H.; Dorlo, T. P. C. An Update on the Clinical Pharmacology of Miltefosine in the Treatment of Leishmaniasis. Int J Antimicrob Agents 2022, 59, 106459. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, M.; Naveed, M.; Khan, Z. U.; Raza, A.; Awrejcewicz, J.; Soori, A. H.; Ul Haq, I.; Mohsin, M. Modeling the Spread of Leishmaniasis Disease via Delayed Analysis. Alex. Eng. J. 2022, 61, 11197–11209. [Google Scholar] [CrossRef]
- Sánchez-Suárez, J.; Bernal, F. A.; Coy-Barrera, E. Colombian Contributions Fighting Leishmaniasis: A Systematic Review on Antileishmanials Combined with Chemoinformatics Analysis. Molecules 2020, 25, 5704. [Google Scholar] [CrossRef]
- Hevener, K. E.; Pesavento, R.; Ren, J.; Lee, H.; Ratia, K.; Johnson, M. E. Chapter Twelve - Hit-to-Lead: Hit Validation and Assessment. In Methods in Enzymology; Lesburg, C. A., Ed.; Modern Approaches in Drug Discovery; Academic Press, 2018; Vol. 610, pp 265–309. [CrossRef]
- Bhattacharjee, A. K. 10 - Pharmacophore-Based Virtual Screening of Large Compound Databases Can Aid "Big Data" Problems in Drug Discovery. In Big Data Analytics in Chemoinformatics and Bioinformatics; Basak, S. C., Vračko, M., Eds.; Elsevier, 2023; pp 231–246. [CrossRef]
- Shreya; Shweta; Dagur, P.; Rakshit, G.; Ghosh, M. Chapter 7 - Virtual Screening of Phytochemicals for Drug Discovery. In Phytochemistry, Computational Tools and Databases in Drug Discovery; Egbuna, C., Rudrapal, M., Tijjani, H., Eds.; Drug Discovery Update; Elsevier, 2023; pp 149–179. [CrossRef]
- McGibbon, M.; Money-Kyrle, S.; Blay, V.; Houston, D. R. SCORCH: Improving Structure-Based Virtual Screening with Machine Learning Classifiers, Data Augmentation, and Uncertainty Estimation. J Adv Res 2022. [Google Scholar] [CrossRef]
- Joshi, T.; Sharma, P.; Joshi, T.; Mathpal, S.; Pandey, S. C.; Pandey, A.; Chandra, S. Chapter 4 - Recent Advances on Computational Approach towards Potential Drug Discovery against Leishmaniasis. In Pathogenesis, Treatment and Prevention of Leishmaniasis; Samant, M., Chandra Pandey, S., Eds.; Academic Press, 2021; pp 63–84. [CrossRef]
- Acevedo, C. H.; Scotti, L.; Scotti, M. T. In Silico Studies Designed to Select Sesquiterpene Lactones with Potential Antichagasic Activity from an In-House Asteraceae Database. ChemMedChem 2018, 13, 634–645. [Google Scholar] [CrossRef]
- Herrera-Acevedo, C.; Dos Santos Maia, M.; Cavalcanti, É. B. V. S.; Coy-Barrera, E.; Scotti, L.; Scotti, M. T. Selection of Antileishmanial Sesquiterpene Lactones from SistematX Database Using a Combined Ligand-/Structure-Based Virtual Screening Approach. Mol Divers 2021, 25, 2411–2427. [Google Scholar] [CrossRef] [PubMed]
- Ayeleso, T. B.; Matumba, M. G.; Mukwevho, E. Oleanolic Acid and Its Derivatives: Biological Activities and Therapeutic Potential in Chronic Diseases. Molecules 2017, 22, 1915. [Google Scholar] [CrossRef] [PubMed]
- Majid Shah, S.; Ullah, F.; Ayaz, M.; Sadiq, A.; Hussain, S.; Ali Shah, A.-U.-H.; Adnan Ali Shah, S.; Wadood, A.; Nadhman, A. β-Sitosterol from Ifloga Spicata (Forssk.) Sch. Bip. as Potential Anti-Leishmanial Agent against Leishmania Tropica: Docking and Molecular Insights. Steroids 2019, 148, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Acevedo, C.; Flores-Gaspar, A.; Scotti, L.; Mendonça-Junior, F. J. B.; Scotti, M. T.; Coy-Barrera, E. Identification of Kaurane-Type Diterpenes as Inhibitors of Leishmania Pteridine Reductase I. Molecules 2021, 26, 3076. [Google Scholar] [CrossRef] [PubMed]
- Gouri, V.; Upreti, S.; Samant, M. Evaluation of Target-Specific Natural Compounds for Drug Discovery against Leishmaniasis. Parasitol Int 2022, 91, 102622. [Google Scholar] [CrossRef]
- Rodrigues, M. P.; Tomaz, D. C.; Ângelo de Souza, L.; Onofre, T. S.; Aquiles de Menezes, W.; Almeida-Silva, J.; Suarez-Fontes, A. M.; Rogéria de Almeida, M.; Manoel da Silva, A.; Bressan, G. C.; Vannier-Santos, M. A.; Rangel Fietto, J. L.; Teixeira, R. R. Synthesis of Cinnamic Acid Derivatives and Leishmanicidal Activity against Leishmania Braziliensis. Eur J Med Chem 2019, 183, 111688. [Google Scholar] [CrossRef]
- Monzote, L.; Perera Córdova, W. H.; García, M.; Piñón, A.; Setzer, W. N. In-Vitro and in-Vivo Activities of Phenolic Compounds against Cutaneous Leishmaniasis. Rec. Nat. Prod. 2016, 10, 269–276. [Google Scholar]
- Kabir, E.; Uzzaman, M. A Review on Biological and Medicinal Impact of Heterocyclic Compounds. Results Chem. 2022, 4, 100606. [Google Scholar] [CrossRef]
- Sabt, A.; Eldehna, W. M.; Ibrahim, T. M.; Bekhit, A. A.; Batran, R. Z. New Antileishmanial Quinoline Linked Isatin Derivatives Targeting DHFR-TS and PTR1: Design, Synthesis, and Molecular Modeling Studies. Eur. J. Med. Chem. 2023, 246, 114959. [Google Scholar] [CrossRef]
- Herrera-Acevedo, C.; de Menezes, R. P. B.; de Sousa, N. F.; Scotti, L.; Scotti, M. T.; Coy-Barrera, E. Kaurane-Type Diterpenoids as Potential Inhibitors of Dihydrofolate Reductase-Thymidylate Synthase in New World Leishmania Species. Antibiotics 2023, 12, 663. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Pu, D.; Tassa, C.; Weissleder, R.; Shaw, S.Y.; Mumper, R.J.; Tropsha, A. Quantitative nanostructureactivity relationship modeling. ACS Nano 2010, 4, 5703–5712. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, G.; Pastor, M.; Guba, W. VolSurf: A New Tool for the Pharmacokinetic Optimization of Lead Compounds. Eur J Pharm Sci 2000, 11 (Suppl. 2), S29–S39. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, G.; Crivori, P.; Carrupt, P.-A.; Testa, B. Molecular Fields in Quantitative Structure–Permeation Relationships: The VolSurf Approach. J. Mol. Struct: THEOCHEM 2000, 503, 17–30. [Google Scholar] [CrossRef]
- Mauri, A. AlvaDesc: A Tool to Calculate and Analyze Molecular Descriptors and Fingerprints. In Ecotoxicological QSARs; Roy, K., Ed.; Methods in Pharmacology and Toxicology; Springer US: New York, NY, 2020; pp 801–820.
- Mauri, A.; Bertola, M. Alvascience: A New Software Suite for the QSAR Workflow Applied to the Blood–Brain Barrier Permeability. Int. J. Mol. Sci. 2022, 23, 12882. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Yan, F.; Liu, H. Screening Model of Candidate Drugs for Breast Cancer Based on Ensemble Learning Algorithm and Molecular Descriptor. Expert Syst. Appl. 2023, 213, 119185. [Google Scholar] [CrossRef]
- Ticha, L. A.; Klaasen, J. A.; Green, I. R.; Naidoo, S.; Baker, B.; Pietersen, R.-D. Phytochemical and Antimicrobial Screening of Flavanones and Chalcones from Galenia Africana and Dicerothamnus Rhinocerotis. Nat Prod Commun 2015, 10, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Repčák, M.; Krausová, T. Phenolic Glucosides in the Course of Ligulate Flower Development in Diploid and Tetraploid Matricaria Chamomilla. Food Chemistry 2009, 116, 19–22. [Google Scholar] [CrossRef]
- Shoeb, M.; MacManus, S. M.; Jaspars, M.; Trevidu, J.; Nahar, L.; Kong-Thoo-Lin, P.; Sarker, S. D. Montamine, a Unique Dimeric Indole Alkaloid, from the Seeds of Centaurea Montana (Asteraceae), and Its in Vitro Cytotoxic Activity against the CaCo2 Colon Cancer Cells. Tetrahedron 2006, 62, 11172–11177. [Google Scholar] [CrossRef]
- Zofou, D.; Kuete, V.; Titanji, V. P. K. 17 - Antimalarial and Other Antiprotozoal Products from African Medicinal Plants. In Medicinal Plant Research in Africa; Kuete, V., Ed.; Elsevier: Oxford, 2013; pp 661–709. [CrossRef]
- Genovese, S.; Epifano, F.; Curini, M.; Dudra-Jastrzebska, M.; Luszczki, J. J. Prenyloxyphenylpropanoids as a Novel Class of Anticonvulsive Agents. Bioorganic Med. Chem. Lett. 2009, 19, 5419–5422. [Google Scholar] [CrossRef]
- Gobbo-Neto, L.; Lopes, N. P. Online Identification of Chlorogenic Acids, Sesquiterpene Lactones, and Flavonoids in the Brazilian Arnica Lychnophora Ericoides Mart. (Asteraceae) Leaves by HPLC-DAD-MS and HPLC-DAD-MS/MS and a Validated HPLC-DAD Method for Their Simultaneous Analysis. J Agric Food Chem 2008, 56, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Borsato, M. L. C.; Grael, C. F. F.; Souza, G. E. P.; Lopes, N. P. Analgesic Activity of the Lignans from Lychnophora Ericoides. Phytochemistry 2000, 55, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Könye, R.; Tóth, G.; Sólyomváry, A.; Mervai, Z.; Zürn, M.; Baghy, K.; Kovalszky, I.; Horváth, P.; Molnár-Perl, I.; Noszál, B.; Béni, S.; Boldizsár, I. Chemodiversity of Cirsium Fruits: Antiproliferative Lignans, Neolignans and Sesquineolignans as Chemotaxonomic Markers. Fitoterapia 2018, 127, 413–419. [Google Scholar] [CrossRef]
- Athayde, A. E. de; Richetti, E.; Wolff, J.; Lusa, M. G.; Biavatti, M. W. "Arnicas" from Brazil: Comparative Analysis among Ten Species. Rev Bras Farmacogn 2019, 29, 401–424. [Google Scholar] [CrossRef]
- Amoa Onguéné, P.; Ntie-Kang, F.; Lifongo, L. L.; Ndom, J. C.; Sippl, W.; Mbaze, L. M. A. The potential of anti-malarial compounds derived from African medicinal plants. Part I: A pharmacological evaluation of alkaloids and terpenoids. Malar J 2013, 12, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Q. Z.; Jahan, N.; Ahmad, G. Nephroprotective effect of Kabab chini (Piper cubeba) in gentamycin-induced nephrotoxicity. Saudi J Kidney Dis Transpl 2012, 23, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Ostad, S. N.; Rajabi, A.; Khademi, R.; Farjadmand, F.; Eftekhari, M.; Hadjiakhoondi, A.; Khanavi, M. Cytotoxic potential of Centaurea bruguierana ssp. belangerana: The MTT assay. Acta Med Iran 2016, 583–589. [Google Scholar]
- Maganti, L.; Manoharan, P.; Ghoshal, N. Probing the Structure of Leishmania Donovani Chagasi DHFR-TS: Comparative Protein Modeling and Protein–Ligand Interaction Studies. J Mol Model 2010, 16, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Zuccotto, F.; Martin, A. C. R.; Laskowski, R. A.; Thornton, J. M.; Gilbert, I. H. Dihydrofolate Reductase: A Potential Drug Target in Trypanosomes and Leishmania. J Comput Aided Mol Des 1998, 12, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Maganti, L.; Manoharan, P.; Ghoshal, N. Probing the Structure of Leishmania Donovani Chagasi DHFR-TS: Comparative Protein Modeling and Protein–Ligand Interaction Studies. J Mol Model 2010, 16, 1539–1547. [Google Scholar] [CrossRef]
- Desam, N. R.; Al-Rajab, A. J. Chapter 19 - Herbal Biomolecules: Anticancer Agents. In Herbal Biomolecules in Healthcare Applications; Mandal, S. C., Nayak, A. K., Dhara, A. K., Eds.; Academic Press, 2022, 435–474. [CrossRef]
- Raj, S.; Sasidharan, S.; Tripathi, T.; Saudagar, P. Biofunctionalized Chrysin-Conjugated Gold Nanoparticles Neutralize Leishmania Parasites with High Efficacy. Int J Biol Macromol 2022, 205, 211–219. [Google Scholar] [CrossRef]
- Onawole, A. T.; Sulaiman, K. O.; Kolapo, T. U.; Akinde, F. O.; Adegoke, R. O. COVID-19: CADD to the rescue. Virus Res. 2020, 285, 198022. [Google Scholar] [CrossRef] [PubMed]
- Shulha, O.; Çiçek, S.S.; Wangensteen, H.; Kroes, J.; Mäder, M.; Girreser, U.; Sendker, J.; Jöhrer, K.; Greil, R.; Schühly, W.; Mangoni, A.; Grauso, L.; van Thriel, C.; Zidorn, C. Lignans and sesquiterpene lactones from Hypochaeris radicata subsp. neapolitana (Asteraceae, Cichorieae). Phytochem. 2019, 165, 112047. [Google Scholar] [CrossRef] [PubMed]
- Begum, T.; Prakash, R.; Duarah, G.; Gogoi, S. Utilization of Rh-carbenoid CH insertion reactions for the synthesis of bioactive natural products. Stud. Nat. Prod. Chem. 2020, 65, 349–380. [Google Scholar] [CrossRef]
- Liu, X., Chen, R., Shang, Y., Jiao, B., & Huang, C. Lithospermic acid as a novel xanthine oxidase inhibitor has anti-inflammatory and hypouricemic effects in rats. Chem. Biol. Interact. 2008, 176(2-3), 137-142. [CrossRef]
- Tabrez, S.; Rahman, F.; Ali, R.; Akand, S. K.; Alaidarous, M. A.; Banawas, S.; Bin Dukhyil, A.A.; Rub, A. Hesperidin targets Leishmania donovani sterol C-24 reductase to fight against leishmaniasis. ACS omega 2021, 6, 8112–8118. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization.
- and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [CrossRef]
- Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Hughes, J. D.; Blagg, J.; Price, D. A.; Bailey, S.; DeCrescenzo, G. A.; Devraj, R. V. , et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorganic Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef]
- Gleeson, M. P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Xu, L.; Li, D.; Hou, T. Recent developments in computational prediction of HERG blockage. Curr. Top. Med. Chem. 2013, 13, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME-the Konstanz information miner: Version 2.0 and beyond. SIGKDD Explor. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J.; Ohlinger, W.A. Spartan ’14; Wavefunction Inc.: Irvine, CA, USA, 2014; pp. 429–507. ISBN 978-1-890661-42-2. [Google Scholar]
- Land, H.; Humble, M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. In Methods mol. biol.; Springer: New York, NY, USA, 2017; pp. 43–67. ISBN 1064-3745. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-2, 19–25. [Google Scholar] [CrossRef]
- Angarita-Rodríguez, A.; Quiroga, D.; Coy-Barrera, E. Indole-Containing Phytoalexin-Based Bioisosteres as Antifungals: In Vitro and In Silico Evaluation against Fusarium oxysporum. Molecules 2020, 25, 45. [Google Scholar] [CrossRef] [PubMed]
- Grumont, R.; Sirawaraporn, W.; Santi, D.V. Heterologous Expression of the Bifunctional Thymidylate Synthase-Dihydrofolate Reductase from Leishmania major. Biochemistry 1988, 27, 3776–3784. [Google Scholar] [CrossRef]
- Nare, B.; Hardy, L.W.; Beverley, S.M. The roles of pteridine reductase 1 and dihydrofolate reductase- thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J. Biol. Chem. 1997, 272, 13883–13891. [Google Scholar] [CrossRef]
- Park, S. Y.; Hong, S. S.; Han, X. H.; Hwang, J. S.; Lee, D.; Ro, J. S.; Hwang, B. Y. Lignans from Arctium lappa and Their Inhibition of LPS-Induced Nitric Oxide Production. Chem. Pharm. Bull. 2007, 55, 150–152. [Google Scholar] [CrossRef]
- Umehara, K.; Sugawa, A.; Kuroyanagi, M.; Ueno, A.; Taki, T. Studies on Differentiation-Inducers from Arctium Fructus. Chem. Pharm. Bull. 1993, 41, 1774–1779. [Google Scholar] [CrossRef]
Figure 1.
ROC curves comparison for the RF model using AlvaDesc and VolSurf descriptors for (a) test sets and (b) cross-validation. Performance evaluation of RF using (c) AlvaDesc and (d)VolSurf descriptors.
Figure 1.
ROC curves comparison for the RF model using AlvaDesc and VolSurf descriptors for (a) test sets and (b) cross-validation. Performance evaluation of RF using (c) AlvaDesc and (d)VolSurf descriptors.
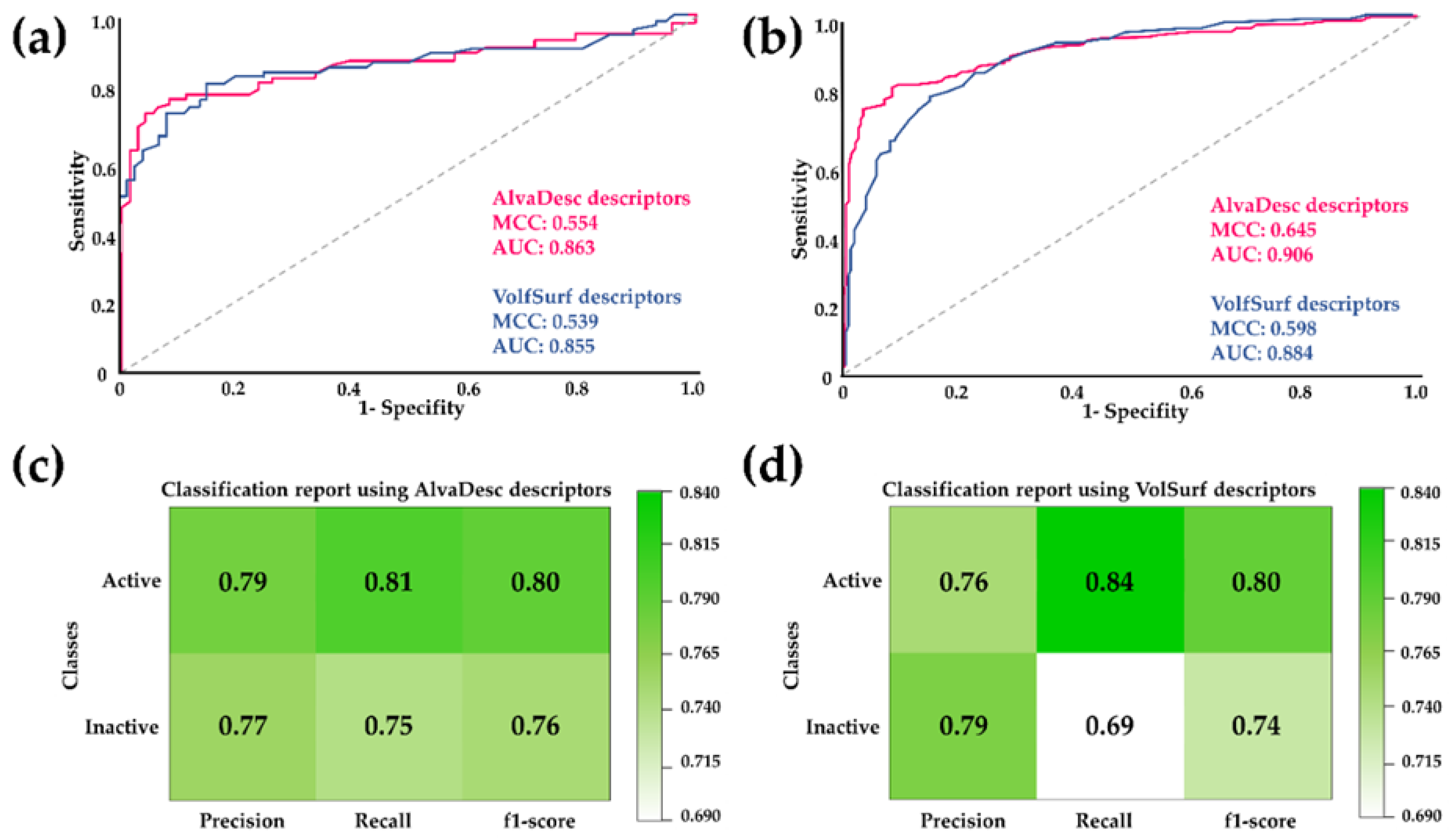
Figure 2.
Chemical Structures of the five top-ranked cinnamic acid derivatives using a ligand-based virtual screening (LB) with AlvaDesc and VolSurf+ descriptors; PLB =Active probability value.
Figure 2.
Chemical Structures of the five top-ranked cinnamic acid derivatives using a ligand-based virtual screening (LB) with AlvaDesc and VolSurf+ descriptors; PLB =Active probability value.
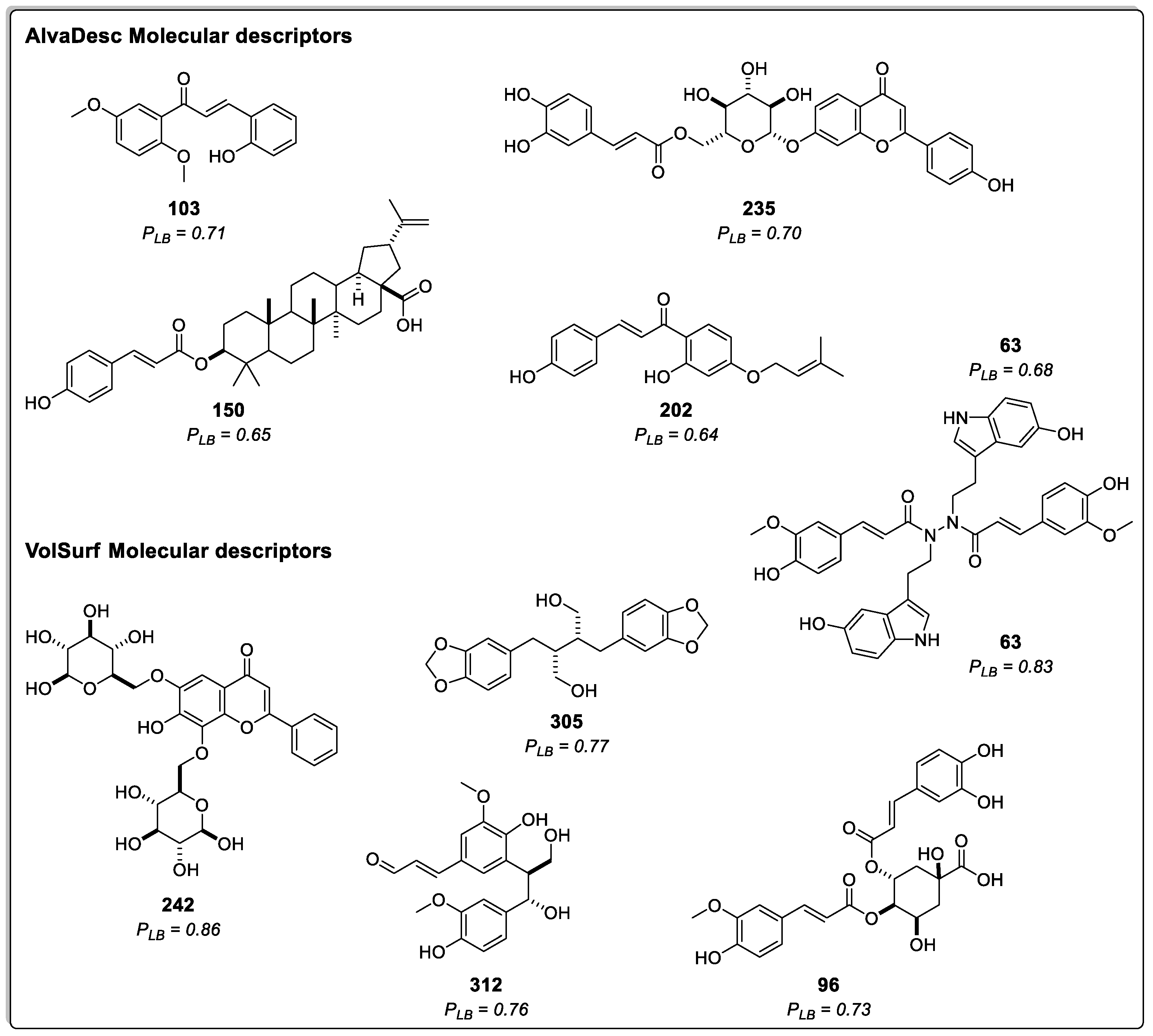
Figure 3.
Chemical structures of reference ligands: Methotrexate (MTX) and ethyl 4-(5-{[(2,4-diaminoquinazolin-6-yl)methyl]amino}-2-methoxyphenoxy)butanoate (DQ1).
Figure 3.
Chemical structures of reference ligands: Methotrexate (MTX) and ethyl 4-(5-{[(2,4-diaminoquinazolin-6-yl)methyl]amino}-2-methoxyphenoxy)butanoate (DQ1).
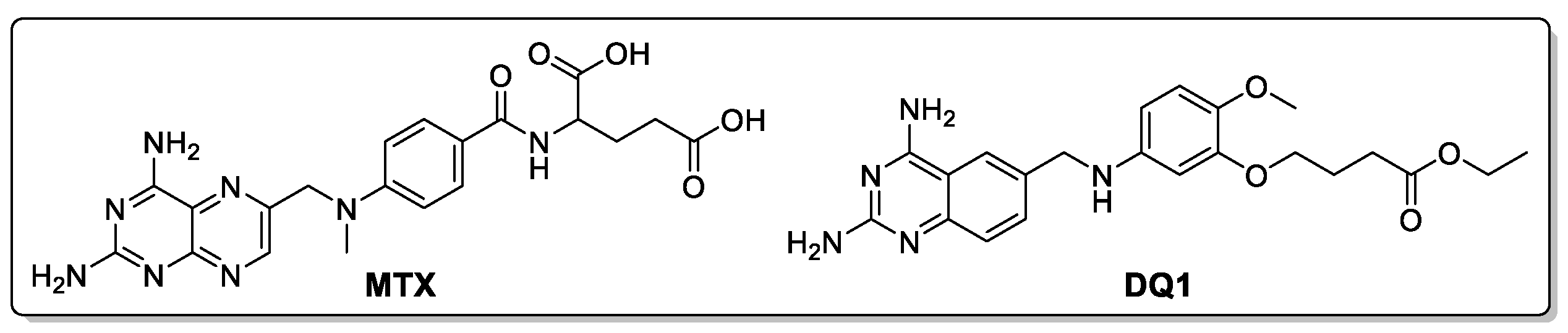
Figure 4.
Chemical Structure of six of the best-ranked cinnamic acid derivatives that appear as active in the structure-based virtual screening with their respective probability to be active. PSB=active probability value.
Figure 4.
Chemical Structure of six of the best-ranked cinnamic acid derivatives that appear as active in the structure-based virtual screening with their respective probability to be active. PSB=active probability value.
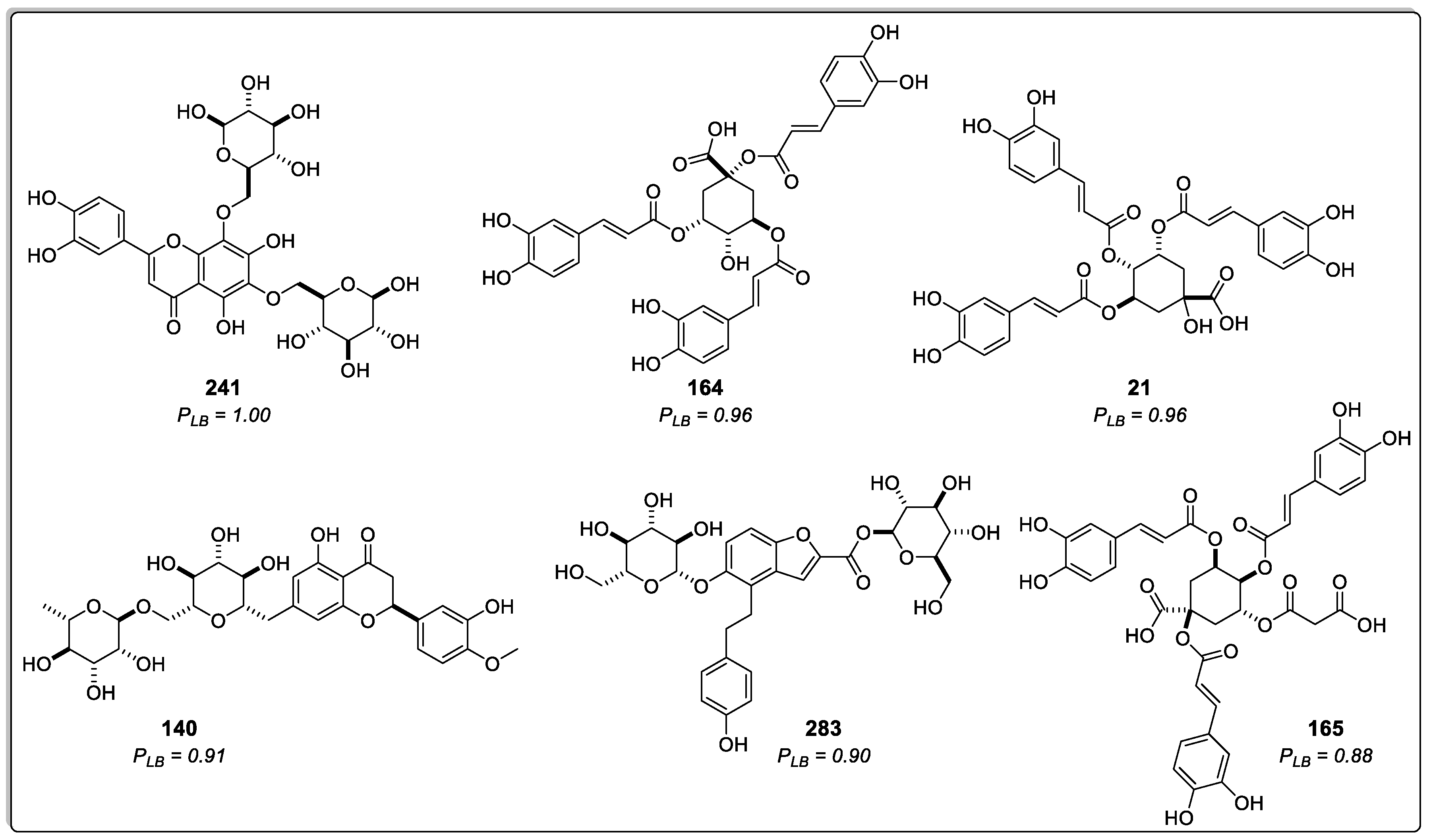
Figure 5.
Residual interaction diagrams of A) compound 241, B) compound 164, C) compound 21, D) DQ1, and E) methotrexate. Interacting residues are shown in colored circles depending on the type of interaction: H-bond (lime), Van der Waals (green), π-π (purple), π-alkyl (pink), unfavorable (red), interactions carbon H-bond (light green), π-anion (orange), π-sulfide (yellowish orange). F) structural conformations of the coupling between the LmDHFR-TS enzyme and the ligands: DQ1(red), compound 241 (green), compound 164 (pink), compound 21 (blue).
Figure 5.
Residual interaction diagrams of A) compound 241, B) compound 164, C) compound 21, D) DQ1, and E) methotrexate. Interacting residues are shown in colored circles depending on the type of interaction: H-bond (lime), Van der Waals (green), π-π (purple), π-alkyl (pink), unfavorable (red), interactions carbon H-bond (light green), π-anion (orange), π-sulfide (yellowish orange). F) structural conformations of the coupling between the LmDHFR-TS enzyme and the ligands: DQ1(red), compound 241 (green), compound 164 (pink), compound 21 (blue).
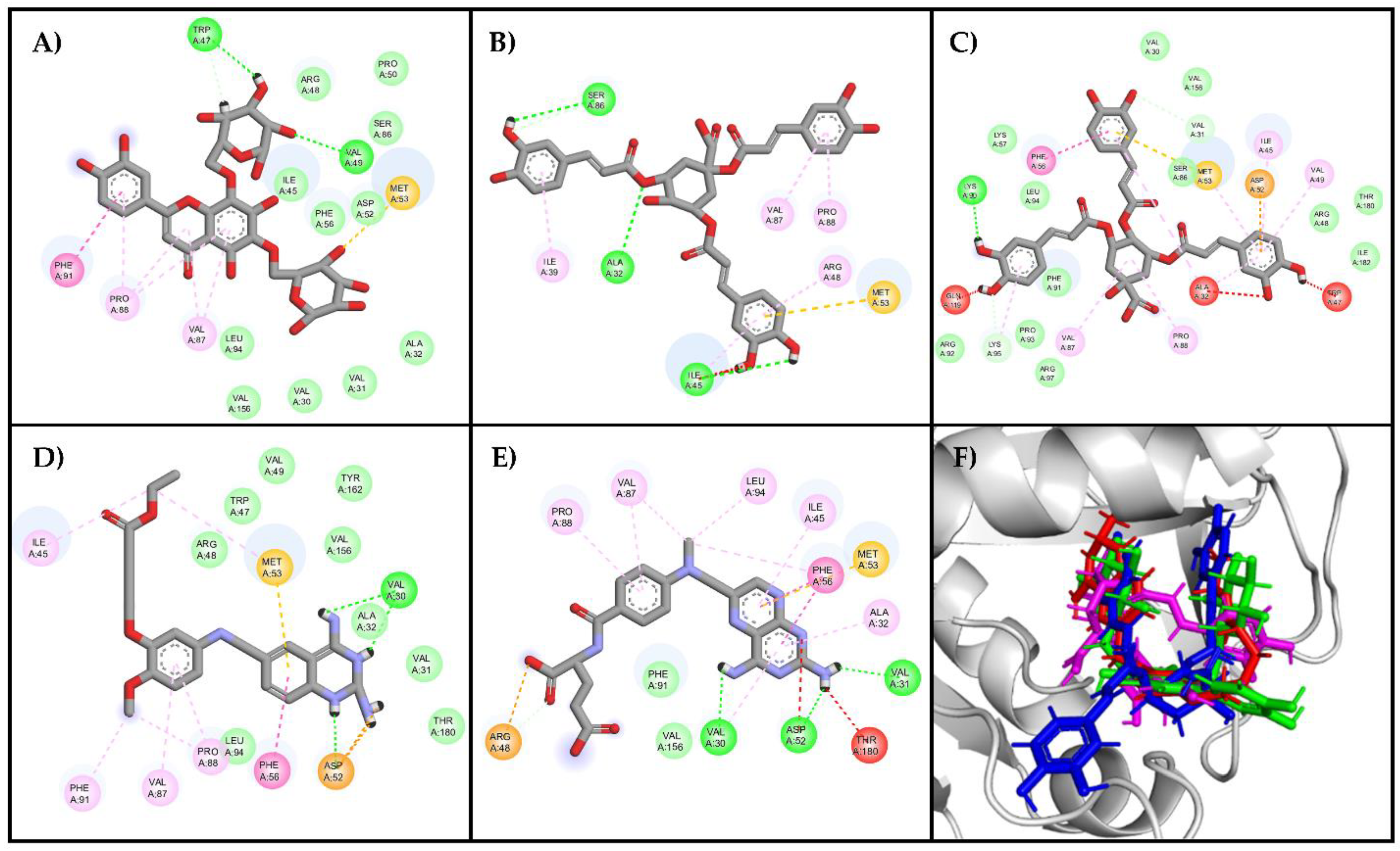
Figure 6.
Topological Polar Surface Area (TPSA) map of A) DQ1, B) methotrexate, C) compound 241, D) compound 164, and E) compound 21.
Figure 6.
Topological Polar Surface Area (TPSA) map of A) DQ1, B) methotrexate, C) compound 241, D) compound 164, and E) compound 21.
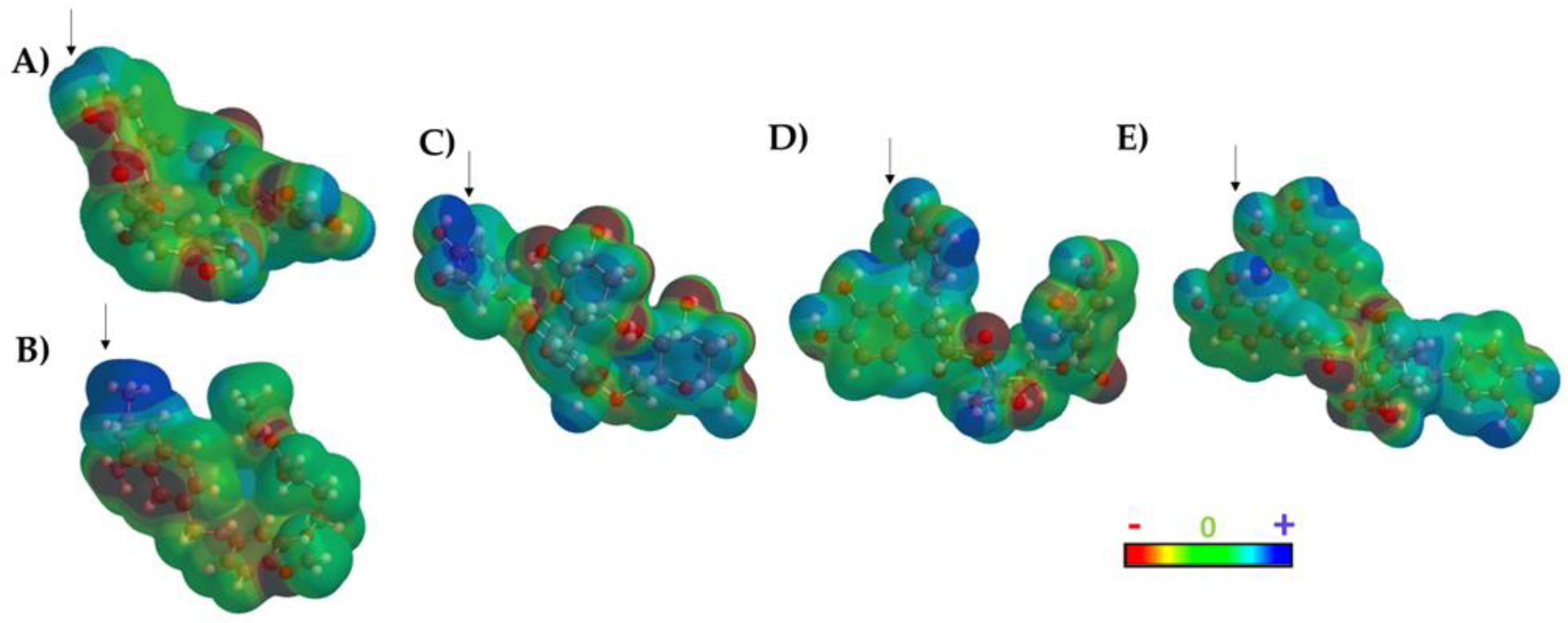
Figure 7.
Cinnamic acid derivatives as potential inhibitors of LmDHFR-TS were identified using an approach that combines ligand-based and structure-based virtual screening (VS). CALm represents the combined probability value.
Figure 7.
Cinnamic acid derivatives as potential inhibitors of LmDHFR-TS were identified using an approach that combines ligand-based and structure-based virtual screening (VS). CALm represents the combined probability value.
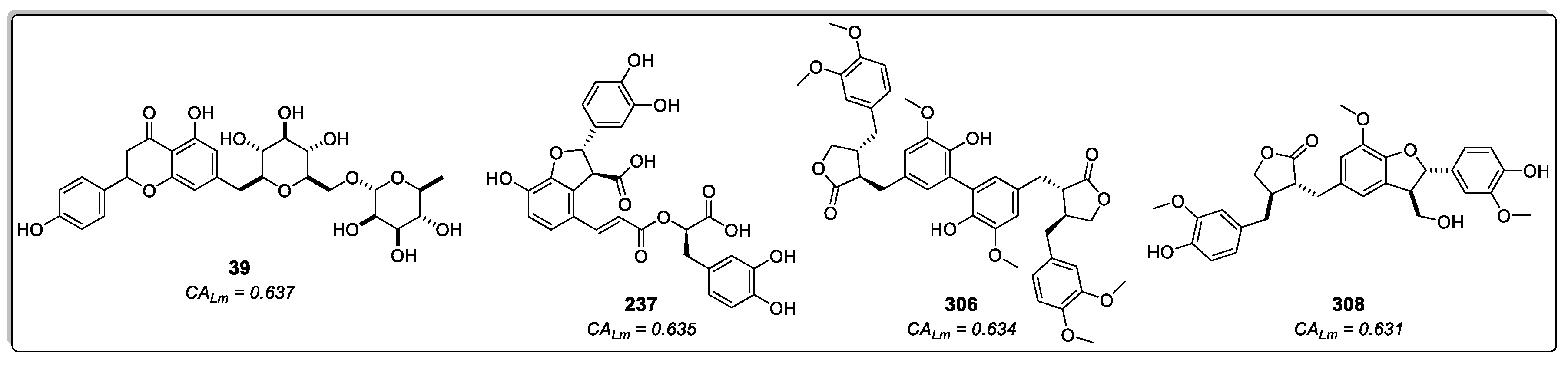
Figure 8.
(a) Root-mean-square deviation (RMSD), (b) root-mean-square fluctuation (RMSF), and (c) radius of gyration (RoG) values within the LmDHFR-TS binding site, obtained after molecular dynamics simulations. Apoenzyme (blue); DHFR-TS···MTX complex (cyan); DHFR-TS···237 complex (light green); DHFR-TS···306 complex (yellow) and DHFR-TS···308 complex (pink).
Figure 8.
(a) Root-mean-square deviation (RMSD), (b) root-mean-square fluctuation (RMSF), and (c) radius of gyration (RoG) values within the LmDHFR-TS binding site, obtained after molecular dynamics simulations. Apoenzyme (blue); DHFR-TS···MTX complex (cyan); DHFR-TS···237 complex (light green); DHFR-TS···306 complex (yellow) and DHFR-TS···308 complex (pink).
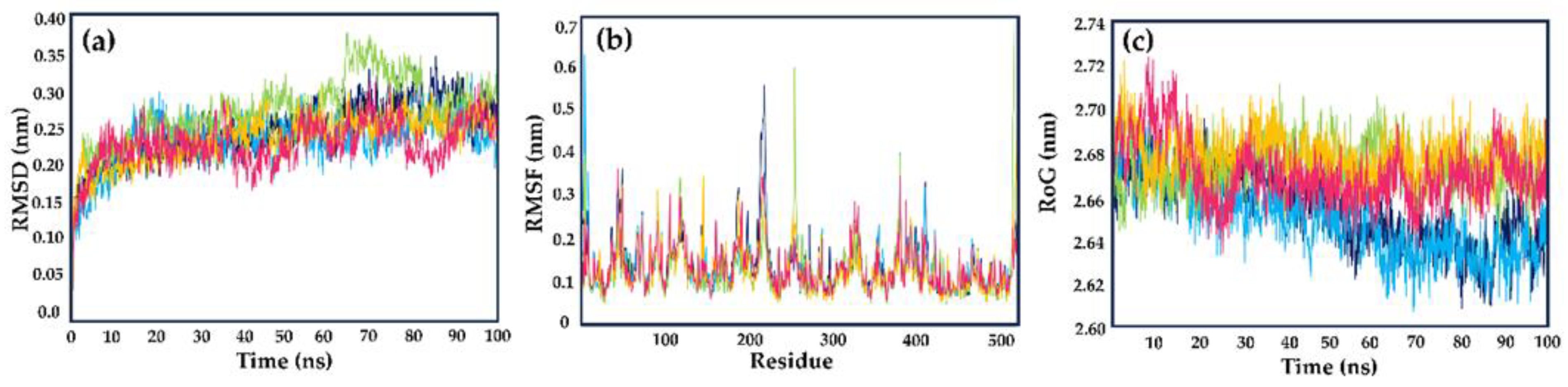

Table 1.
Chemical Structure of six of the best-ranked cinnamic acid derivatives that appear as active in the structure-based virtual screening with their respective probability to be active. PSB=active probability value.
Table 1.
Chemical Structure of six of the best-ranked cinnamic acid derivatives that appear as active in the structure-based virtual screening with their respective probability to be active. PSB=active probability value.
| Rank | Ligand | Docking Score (kJ/mol) | SD | RMSD |
|---|---|---|---|---|
| 1 | 241 | -182.8 | 5.4 | 1.0 |
| 2 | 164 | -175.6 | 7.1 | 1.8 |
| 3 | 21 | -175.5 | 11.2 | 1.0 |
| 4 | 242 | -169.6 | 1.9 | 1.2 |
| 5 | 140 | -167.0 | 3.3 | 0.4 |
| 6 | 283 | -165.4 | 4.8 | 1.7 |
| 7 | 165 | -161.8 | 7.4 | 1.2 |
| 8 | 235 | -161.4 | 5.9 | 0.9 |
| 9 | 285 | -160.9 | 8.8 | 1.2 |
| 10 | 63 | -160.1 | 5.2 | 1.1 |
| Ref | methotrexate | -114.2 | 2.2 | 0.3 |
| Ref | DQ1 | -134.4 | 2.5 | 0.3 |
Table 2.
Cinnamic acid derivatives are classified as active by combining ligand-based and structure-based VS. The numbers in italics represent those compounds classified as active in all three in-silico models, but they were not previously identified as the best-ranked compounds in any approach.
Table 2.
Cinnamic acid derivatives are classified as active by combining ligand-based and structure-based VS. The numbers in italics represent those compounds classified as active in all three in-silico models, but they were not previously identified as the best-ranked compounds in any approach.
| Rank | Ligand | PLB(AD) | PLB(VS) | Pdocking | CALm |
|---|---|---|---|---|---|
| 1 | 63 | 0.68 | 0.83 | 0.88 | 0.78 |
| 2 | 242 | 0.52 | 0.86 | 0.93 | 0.74 |
| 3 | 96 | 0.55 | 0.73 | 0.77 | 0.67 |
| 4 | 241 | 0.53 | 0.55 | 1.00 | 0.64 |
| 5 | 39 | 0.57 | 0.64 | 0.77 | 0.64 |
| 6 | 237 | 0.61 | 0.55 | 0.84 | 0.64 |
| 7 | 306 | 0.63 | 0.53 | 0.83 | 0.63 |
| 8 | 165 | 0.53 | 0.60 | 0.88 | 0.63 |
| 9 | 140 | 0.59 | 0.51 | 0.91 | 0.63 |
| 10 | 308 | 0.57 | 0.59 | 0.81 | 0.63 |
Table 3.
Binding free energies (kJ/mol) from the MM/PBSA calculations for compounds 237, 306, and 308 in the active site of LmDHFR-TS; MTX was used as the reference ligand.
Table 3.
Binding free energies (kJ/mol) from the MM/PBSA calculations for compounds 237, 306, and 308 in the active site of LmDHFR-TS; MTX was used as the reference ligand.
| 237 | 306 | 308 | MTX | |||||
|---|---|---|---|---|---|---|---|---|
| Energy contribution | kJ/mol | SD | kJ/mol | SD | kJ/mol | SD | kJ/mol | SD |
| van der Waals | -218.3 | 6.2 | -209.7 | 4.6 | -217.6 | 6.2 | -239.5 | 8.2 |
| Electrostatic | -31.3 | 4.1 | -38.0 | 3.9 | -29.0 | 4.6 | -57.3 | 4.3 |
| Polar solvation | 181.5 | 6.5 | 157.6 | 6.3 | 185.6 | 6.5 | 194.6 | 8.5 |
| SASA | -23.6 | 1.8 | -21.0 | 1.9 | -20.0 | 1.2 | -22.4 | 2.2 |
| Binding energy | -91.6 | 4.7 | -111.1 | 4.2 | -81 | 4.6 | -124.5 | 5.8 |
Table 4.
Results of enzymatic activity against LmDHFR-TS and HsDHFR for selected cinnamic acid derivatives. CI = Confidence Interval (95%). SI = Selectivity Index.
Table 4.
Results of enzymatic activity against LmDHFR-TS and HsDHFR for selected cinnamic acid derivatives. CI = Confidence Interval (95%). SI = Selectivity Index.
| Compound | LmDHFR-TS | HsDHFR | SI | ||
| IC50 (µM) | CI (95%) | IC50 (µM) | CI (95%) | ||
| hesperidin | 21.6 | 20.2-23.1 | 86.5 | 82.3-87.2 | 4.0 |
| lithospermic acid (237) | 7.5 | 6.8-7.9 | 22.6 | 21.3-24.7 | 3.0 |
| diarctigenin (306) | 6.1 | 5.7-6.4 | 27.9 | 26.8-28.6 | 4.6 |
| isolappaol A (308) | 10.1 | 9.7-10.3 | 44.8 | 42.4-45.9 | 4.4 |
| isovitexin 4′-O-glucoside | 53.2 | 51.1-54.1 | 125.7 | 122.8-127.8 | 2.4 |
| rutin | 41.7 | 40.3-43.1 | 188.9 | 186.2-190.6 | 4.5 |
| MTX | 1.4 | 1.1-1.5 | 4.9 | 4.7-5.1 | 3.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



