
Submitted:
01 November 2023
Posted:
02 November 2023
Read the latest preprint version here
Abstract
Astrocytes regulate the shape and functions of the synaptic and vascular compartments through modulatory membrane-bound proteins or released factors. Their peculiar position further allows them to form a functional unit that “senses” the brain state and secrete factors in the bloodstream as a reflection of this state. Imbalanced astrocyte activity can therefore have a drastic negative impact on the formation of neuronal synaptic networks and lead to the development of severe brain pathologies. Clinical and pre-clinical studies show alterations in astrocyte cell number, morphology and molecular make-up in different affected brain regions in neurodevelopmental (ND) and neuropsychiatric (NP) disorders. The generation and differentiation/maturation of astrocytes (astrogenesis) occur during the critical period of early postnatal brain development. This is a time window of elevated astrocyte- and microglia-dependent synaptic phagocytosis (pruning), a process pivotal in refining neuronal networks. Therefore, any extrinsic and/or intrinsic cues altering this astrocyte-dependent synapse elimination during this critical period may result in altered synaptic remodeling and the onset of ND and NP disorders. Here, we collect recent advancements regarding astrogenesis and astrocyte-mediated regulation of neuronal network remodelling especially during early postnatal critical periods of brain development. We additionally propose alternative hypotheses to their putative involvement in the onset of ND and NP disorders. In light of the well-known differential prevalence of certain brain disorders between males and females, we also discuss sex-dependent effects on these neurodevelopmental processes.From a translational perspective, understanding age- and sex- dependent astrocyte-specific molecular and functional changes may help to identify biomarkers of distinct cellular (dys)functions in health and disease and may support the development of diagnostic tools or the selection of tailored treatment options for individual patients.
Keywords:
astrocytes
; critical period
; synapse phagocytosis
; sex differences
; neuropsychiatric and neurodevelopmental disorders
Main Text
Astrocytes are the most abundant subtype of glial cells that populate the brain and spinal cord [1]. At a microscopic level, they show a typical star-shaped morphology with few major processes extending from the soma. These processes become highly ramified at more distal locations [2]. Their numerous roles in the brain have been extensively described in the past years, ranging from their support of the formation and function of neuronal synapses to the development of a properly operating blood-brain barrier [3,4,5,6,7,8,9,10,11,12].
During embryogenesis, astrocytes are generated from radial glia (RG), which have the capability to self-renew and differentiate into neurons and macroglia cells, e.g. astrocytes, oligodendrocytes and Schwann cells [13,14]. Cell divisions of RGs are predominantly neurogenic at early/mid-gestation and acquire their gliogenic potential at late-gestation/early postnatal developmental stages [15]. The initial steps of gliogenesis produce astrocyte precursors, which then locally proliferate in the different brain areas to increase their numbers and give rise to mature astrocytes [14,16]. Several studies have pointed out the diversity of astrocyte morphologies and functions in various regions of the central nervous system (CNS); some specialized subtypes display very distinctive structural and functional properties such as the Bergmann and the Müller glia cells localized in the cerebellum and the retina, respectively [17,18]. In general, astrocytes show a great range of heterogeneity in the acquisition of various competencies selectively matching the function(s) of neighbouring cells [2,19,20,21]. To acquire such a high degree of intra- and inter-regional diversity, astrocytes must undergo positionally and temporally regulated developmental programmes, strictly modulated by the interaction between intrinsic and extrinsic factors, to ultimately guarantee their specification most aligned to the requirements of the local environment. Evidence that extrinsic signals play a role in the onset of astrogenesis came from co-culture experiments of mouse embryonic RG with cortical slices, which produced neurons when cultured on embryonic slices but shifted to a glial fate when cultured on postnatal slices [22]. Among such programmes, three pathways emerged as crucial players for astrogenesis: the Notch, the BMP, and the JAK-STAT signalling pathways [20,23,24]. These pathways, in combination with exogenously secreted molecules, induce chromatin changes, promoting astrocyte generation and differentiation from RGs [14,24]. As an example, the interaction between Notch and the JAK/STAT pathway induces RG differentiation into an astrocytic fate by demethylating and thereby activating astrocytic genes, such as glial fibrillary acidic protein (GFAP) or S100β. On the contrary, the activity of pro-neurogenic factors like Neurogenin1 (Ngn1), Ngn2, NeuroD and Mesh1 inhibits astrogenesis by either blocking the JAK/STAT pathway or favouring the expression of neurogenic genes [25,26,27]. Interestingly, the release of cytokines such as the gliogenic cardiotrophin-1 (CT-1) from newly generated neurons may further promote astrocyte differentiation and their depletion can severely impair astrogenesis [15,22,28]. Notably, a coordinated expression of cell-type-specific ligands and receptors is required to favour selected cell-cell interactions and support the appropriate activation of those intracellular pathways. Very recently, Voss and colleagues demonstrated that the synergistic and combinatorial activity of five ligand–receptor pairs, driven by the ligands TGFβ2, NLGN1, TSLP, DKK1 and BMP4, guides astrogenesis in both human cortical organoids and primary fetal tissue. In this work, they additionally identified a time frame of effective responsivity to gliogenic signals, which corresponds to the initial postnatal developmental stages [29] (Figure 1).
Thus, the tight regulation and coordination of all intracellular and extracellular stimuli along with cell-cell interactions implicated in controlling these events is critical for the establishment of a balanced ratio among all different cell types, their relative numbers, appropriate functional diversification, and correct formation of elaborated neuronal networks in each brain region [23].
Related to this, in different species from worms and insects to rodents and humans, the ratio glia to neurons increases with the complexity of the CNS [30], suggesting a correlation between the growing number of glial cells and advanced CNS functions. Therefore, the precise control of the mechanisms responsible for astrocyte proliferation and maturation, which go hand in hand with neuronal development, is essential in establishing the appropriate number of astrocytes with respect to neurons in any distinct brain region. This is critical for the proper acquisition and elaboration of increasingly more complex CNS functions, such as human mental and cognitive abilities [1,30].
Any disruption in these delicate processes might affect the formation and functions of astrocytic and neuronal networks, leading to the onset of severe brain disorders.
Astrocytes and the Critical Period of Early Postnatal Brain Development
All typical phases of astrogenesis (generation/proliferation, differentiation and maturation) take place within distinct time courses in various areas of the CNS during the so-called “critical periods” of brain development. These periods may vary both in length and cell types involved in different species, depending on the functions which must be refined [31,32,33,34]. Critical periods are also sometimes referred to as “sensitive” periods, although it is still debated whether critical and sensitive periods temporally overlap or should always be distinguished [34]. These are time frames during early postnatal life when brain plasticity and differentiation/maturation processes are strongly dependent on experience and environmental cues to customize neural circuits and connectivity to the needs of each individual. Therefore, the interaction between intrinsic molecular and biochemical programmes with extrinsic factors becomes crucial during early postnatal developmental periods in shaping neuronal circuits to respond with the most adapted behaviours in juvenile, adolescent and adult life [35,36] (Figure 1).
As described by Knudsen [34], during these developmental time windows structural changes such as axon elaboration, synapse elimination and synaptic consolidation are instructed by life experiences across brain areas in various species, further supporting the relevance of critical periods for neural circuit formation across animals.
In rodents, astrogenesis occurs during late-gestation and proceeds over the first three weeks of postnatal CNS development, concomitantly with synapse formation (synaptogenesis) and maturation, both considered key phases for the generation of synaptic networks [37]. In humans, it is still debated whether these periods are closed after the juvenile developmental stage (approximately corresponding to the first decade of life) or whether they might extend further into adolescence and early adulthood [32,38]. A deeper understanding of synaptic formation/elimination rates, the identification of the beginning and end of critical periods, and the isolation of main factors influencing their opening and closing time points, are essential to further investigate and study the establishment of cognitive abilities and duration of the time frame(s) for optimal acquisition of general mental abilities, as well as for the transformation of an individual personality. From a translational perspective, a better knowledge of the neurobiological, genetic and environmental determinants that affect the opening/closing of critical periods and the reactions upon exposure to particular detrimental chemico-physical or psychological stressors might be essential to identify time windows when therapeutic strategies are more effective in re-directing aberrant brain developmental trajectories.
In humans, it has been assumed that the closure of the critical period at the end of puberty marks the end for synaptic spine formation/elimination in the human cortex. However, this assumption mostly relies only on the first publications of Huttenlocher and colleagues [39,40], which contained only one brain specimen to support the claim. In contrast, more recent brain scan studies have suggested that several dynamic changes in gray matter density and developmental remodelling in the human cortex extend into the third decade of life [32,41,42]. Insofar as dendritic growth and branching in the cortex are mainly limited to early childhood stages, it has been postulated that changes in functional plasticity probably induce reorganization of neuronal circuitries via synaptic modifications, i.e. synapse formation/elimination processes, to favour the acquisition of higher brain functions typical of humans, such as modulation of emotions, cognitive flexibility, socialization skills and others.
Several hypotheses to explain late-onset neurological or neuropsychiatric (NP) disorders suggest defective pruning mechanisms of the initially supernumerary spine synapses [43,44,45,46]. It has been shown that the over-abundance of weak synaptic inputs negatively affects neuronal synchrony, ultimately disrupting functional connectivity of circuits in the limbic system [12,47,48]. A deeper understanding of these processes would support the development of therapeutic interventions tailored to the needs of individual patients.
However, due to the limitations of human neuroimaging tools and the ethical issues associated with studies involving children and adolescents, understanding the cellular mechanisms behind these developmental periods and how perceived adverse events may become neurobiologically embedded in brain circuits and result in NP disorders, requires the continuous development of experimental models recapitulating the human conditions.
The use of animal models has been pivotal in revealing essential physiological underpinnings of critical periods and unravelling molecular pathways that respond to the simultaneous application of pharmacological treatments together with behavioural interventions, to ultimately remodel otherwise altered neuronal circuits [49,50,51]. These seminal studies examined changes in the plasticity of brain circuits in various brain regions, after the application of effectors aimed at reopening critical period-like states. Specifically in two of them, the chronic administration of the antidepressant fluoxetine showed remarkable effects on the reopening of critical period-like neuronal responses, further associated with phenotypical modifications. Interestingly, in a first study, fluoxetine-dependent changes helped to reinstate ocular dominance plasticity in adult mice and promoted the recovery of visual capabilities in amblyopic animals [50]. A further study showed that chronic treatment with fluoxetine induced the erasure of amygdala-dependent fear memories, when drug treatment was combined with behavioural extinction training to reduce fear symptoms [49]. In both studies, the critical role of brain-derived neurotrophic factor (BDNF) ligand and its receptor TrkB have been proposed to be essential for the fluoxetine-induced remodelling effects, with an enhanced BDNF expression measured after drug treatment in various brain regions. Interestingly, polymorphisms in the human Bdnf gene have been associated with impaired extinction, further sustaining its role as a memory modulator and possibly a selective target of fluoxetine to mediate its pharmacological and therapeutic effects in fear-based anxiety disorders [52].
More recently, an elegant study has highlighted the role of astrocytes to close the critical period for visual plasticity in the mouse [51]. Based on the knowledge that astrogenesis occurs during the early postnatal stages of brain development in rodents and that it is for the most part completed by the end of the critical period, Ribot, Breton and colleagues [51] postulated that the maturation of astrocytes and molecular factors involved in this process might be relevant for the closure of this period. In line with this, a previous study showed that transplanting immature astrocytes in the visual cortex of adult cats had a potent effect in reactivating a period of high brain flexibility, with an associated re-induction of ocular-dominance (OD) plasticity in adult animals [53]. In their study, Ribot, Breton and colleagues demonstrated that indeed a molecular switch from proliferative to mature astrocytes corresponded to the closure of this period of elevated neuronal network remodelling capability. A transcriptome analysis identified the astrocyte-specific gap-junction channel subunit connexin 30 (Cx30), an important modulator of hippocampal astrocyte maturation [54], as a pivotal regulator of the critical period closure for OD plasticity (Figure 1). Its lower expression in immature astrocytes and higher expression in mature astrocytes correlated with differential stages of neuronal network maturation and plasticity in the visual cortex. Interestingly, in a genetically modified mouse model lacking Cx30, ocular dominance plasticity continued to increase until postnatal (P) stage 50, indicating an impairment in the closure of the critical period, which usually happens around P28 [51].
It would be interesting to investigate whether and how immature astrocytes might characterize neurodevelopmental (ND) or NP disorders and potentially identify underlying molecular determinants, which might trigger the onset of astrocyte-dependent brain disorders. In humans, early-life adversity (ELA) has been associated with the appearance of severe neurological and mental symptoms later in life as well as with aberrant astrocyte functions [35,55]. Moreover, other chronic stress paradigms besides ELA are accompanied with a reduction in the numbers of GFAP-positive astrocytes and in the extent of GFAP-labeled processes that stem from their cell bodies in prefrontal cortical areas, the hippocampus or other brain regions [56,57,58,59]. Strikingly, a selectively lower expression of the astrocyte-specific protein Cx30 has been revealed in postmortem brain tissues of patients with major depressive disorder (MDD) [60], suggesting a link between stressful environmental triggers and the reinstatement of a critical period-like brain plasticity in brain disorders, plausibly characterized by aberrant astrocyte maturation and/or a complete lack of closure of critical developmental periods.
Several studies have pointed out the involvement of astrocytes in various brain pathologies other than MDD, such as schizophrenia (SCZ), bipolar disorder (BD), autism spectrum disorder (ASD) and epilepsy [61,62,63,64,65,66,67,68,69,70]. In MDD patients, especially younger subjects, the astrocyte pathology is characterized by a profound reduction in the numbers of GFAP- and S100β-positive astrocytes and an impaired astrocyte morphology in the prefrontal cortex, anterior cingulate cortex, and the hippocampus, all regions heavily affected in MDD [61,64,71,72,73,74]. Moreover, it is clear that antidepressants act on astrocytes and may be beneficial to reverse disease symptoms [62,75,76,77]. Conversely, in SCZ or BD, changes in astrocytes are less consistent, with different studies showing both increased and decreased numbers of astrocytes in postmortem tissue of SCZ patients [66,67]. Accordingly, the most reproduced findings show a dysfunction in the NMDA receptor-mediated neuronal transmission. Astrocytes regulate NMDA receptors’ functions through the release of gliotransmitters, such as D-serine, which acts as co-agonist of glutamate to enhance NMDA function [78]. The binding of D-serine to synaptic NMDARs additionally modulate the induction of long-term potentiation (LTP), the latter being the cellular correlate of memory formation [79]. Memory deficits are among the most common symptoms of various ND and NP disorders, thereby suggesting the involvement of gliotransmission in their etiopathogenesis. Indeed, recent clinical work has proposed D-serine as a promising therapeutic option for the treatment of ND disorders, most notably SCZ [80,81]. Using animal models, it has been further shown that mutations in Disc1, which reduce the stability of the D-serine synthesizing enzyme serine racemase, result in SCZ-like behavior [82]. Thus, in SCZ, not the number but rather astrocyte functionality seems to be mostly affected and influence disease onset and/or progression, principally driven by genetic factors. This might explain the inconsistent results from postmortem brain tissues, which did not show reproducible deficiencies in astrocyte counts [66,67]. Despite SCZ having been considered a disorder with a high genetic component, in reality the concordance rate among identical twins is only about 50% [83], with a substantial proportion of SCZ being of idiopathic origin. More interestingly, cortisol levels, an index of stress, measured in adolescents at-risk for SCZ were significantly elevated in subjects who transitioned to psychosis later in life, suggesting the negative impact of early life environmental factors such as stress on the worsening of NP disorders when encountered during critical periods of postnatal development [84,85]. Although the role of aberrant neurogenesis during the critical period has been described in SCZ and associated with its etiopathogenesis [86], only recent work has elucidated the putative role of astrocytes, with aberrant maturation impacting synaptic formation during critical periods as a possible cause of disrupted neuronal circuit formation and disease onset [68,87]. In ASD, astrocyte involvement in its etiology has been recently thoroughly investigated using patient-derived iPSCs [70,88]. Even in ASD, the essential role of the critical period has been proposed, with a shortened period of neuroplasticity suggested as a common cause of disease among various patients independently of their highly divergent genetic backgrounds. In particular, the language and social skill deficits observed in ASD have been led back to a premature closure of the critical period for their development, which should typically extend into adolescence. The hypothesis would be that a shorter critical period in ASD may possibly force the brain to rely on underdeveloped neuronal networks for learning language and social skills, among other mental abilities [89]. However, as of today no work has thouroughly investigated the possibility that an earlier closure of the critical period, marked by the premature differentiation of astrocytes, might be among the neurobiological underpinnings in the etiopathogenesis of ASD. In the same line, no work has ever considered a failure in the closure of this period among possible causes for other mental disorders such as depression (Figure 2).
Astrocytes and the Synaptic Compartments: Astrocyte-Mediated Phagocytosis for Neuronal Circuit Refinement
In the CNS, the peculiar position of astrocytes between synaptic and vascular compartments supports their formation of a functional unit (neurovascular, NVU) that “senses” the brain state and secrete factors in the bloodstream as a reflection of this state. In pathological conditions, such factors may serve as biomarkers of distinct cellular (dys)functions and help to improve diagnostic/treatment options specifically adapted to the needs of individual patients. Several excellent reviews have already described the role(s) of astrocyte in health and disease, in relation to synapse formation and a blood-brain barrier (BBB) breakdown [4,5,9,11,12,90,91,92,93]. We also validate and further characterized BBB deficits in an animal model of MDD [92,94] and identified the astrocyte-derived factor GDF15 as an essential modulator of simultaneous remodelling of disrupted astrocyte processes and loosened tight junctions between endothelial cells at the BBB upon treatment with the antidepressant fluoxetine [95]. Ideally, identifying cell-type specific molecular underpinnings of disease may support the discovery of disease biomarkers, which could improve diagnostic means. However, ND and NP disorders often share overlapping symptoms und underlying neurobiological alterations, making it difficult to distinguish disease-specific biomarkers for personalized medicine. Nevertheless, a combination of efforts and methodological approaches may offer valid guidance to reach this ambitious goal. Our contribution to those efforts includes the examination of less explored functions of astrocytes in health and disease. Among them, we focused on astrocyte-mediated phagocytosis in mood and depressive-like disorders.
Astrocytes derive from the embryonic ectodermal sheet and therefore share some developmental, genetic and functional similarities with other ectodermally-derived cells, such as neurons and oligodendrocytes, but less with other brain cells of mesodermal origin, such as microglia cells [96]. However, they might adopt analogous functions to microglia cells to cooperatively converge their efforts for the refinement of neuronal networks [97].
Together with microglia, astrocytes play a pivotal role in the remodelling of synaptic networks through the elimination of weak synapses (pruning) [98].
Early studies in the fly Drosophila melanogaster have shown that, during its metamorphosis from larvae to pupa stages, a high degree of tissue digestion occurs in its body to reshape its structures and refine its nervous system, allowing afterwards the extension of adult, mature, neuronal projections [99]. Furthermore, the sequence of events leading to such drastic changes occur in a developmentally regulated manner, activated by intracellular molecular mechanisms interacting with additional factors, such as the neuronal hormone ecdysone. In Drosophila, two genes, Draper and CED-6, were uncovered that are key for the engulfment of pruned axons by glia during metamorphosis [100]. Further studies showed that mutations affecting the genes encoding for Draper or Ced-6 and knockdown experiments to specifically reduce their expression in in glia cells by RNA interference, suppressed glia-mediated phagocytosis, resulting in the inhibition of axon pruning during metamorphosis. Genetic studies in Caenorhabditis elegans initially isolated the genes encoding for CED-1 and CED-6 as also essential to expressing the phagocytic molecules relevant for the clearance of apoptotic cells [101].
These studies opened the field of glia-mediated phagocytosis, which ended up in showing that such mechanisms are highly conserved across species, even when species-specific cellular and molecular mechanisms have diverged along evolution to adapt to the growing complexity of selected functions [30]. In mammals, the seminal work of Schafer and Stevens [102,103] and Chung [98] have been especially pivotal in identifying and describing the role of the classical complement cascade and MEGF10/MERTK, respectively, for the refinement of neuronal synaptic networks during postnatal developmental periods. Subsequently, they and others proposed a dysfunctional role of these signalling mechanisms in the etiopathogenesis of brain disorders [104,105]. Specifically, it has been postulated that so-called “eat-me” signals are key to determine which cells (or parts of them) should be excised/phagocytosed. Among such signals, the C1q has been described multiple times as a common tagging system shared between microglia cells and astrocytes to recognize the targets which need to be eliminated. This suggests that regulatory mechanisms controlling C1q expression/tagging and/or its binding partners may be responsible for sorting out which cell type between microglia cells and astrocytes should be recruited to execute the job. Because of the commonly shared target(s), it has also been proposed that a selective mechanism determining which cell type acts first may rely on the specific set of receptors localized either on astrocytes or on microglia cells, which might be activated alone or in combination with other molecules “on demand” [97]. Among them, MEGF10 has been identified in astrocytes and described to be highly relevant for synaptic remodelling during postnatal brain development and the maintenance of hippocampal homeostasis in adult brain [98,106]. MEGF10 can mediate its phagocytic functions on apoptotic material via the recognition of C1q bound to phosphatidylserine (PS) exposed on dying cells [46]. It is however still unknown whether this mechanism is also responsible for the astrocyte-mediated synaptic elimination and circuit refinement in mammalian brains. In contrast, the mechanisms of microglia-mediated synaptic pruning via the complement system with its ligand-receptor binding between C1q and the C3/CR4 receptor located on microglial processes have been thoroughly investigated and described both in health and disease [102,103,105,107] (Figure 3).
A reduced proliferation of astrocytes and/or disruption of astrocytic activity may either weaken mature synapses or prevent their maturation, thereby potentially hampering correct synaptic pruning. This highlights once more the underlying importance of the critical developmental periods, when astrocyte proliferation/maturation and synaptic formation take place simultaneously, in sculpting the developing and adult brain. Any disturbance in the sequence of these events may trigger the onset of brain disorders.
Sex-Dependent Differences in Astrogenesis and in Time Frames of Vulnerability to Perturbations and Disease Onset
Sex differences have been long recognized as a variable that affects many brain disorders in terms of predisposition, rates of incidence, age of onset, symptomatology and outcome, with skewed prevalence towards one sex in different pathologies [48,57,108,109,110]. However, research on sex-dependent pathologies and their neurobiological molecular and cellular causes is still scarce, thereby limiting the development of sex-specific diagnoses and treatments.
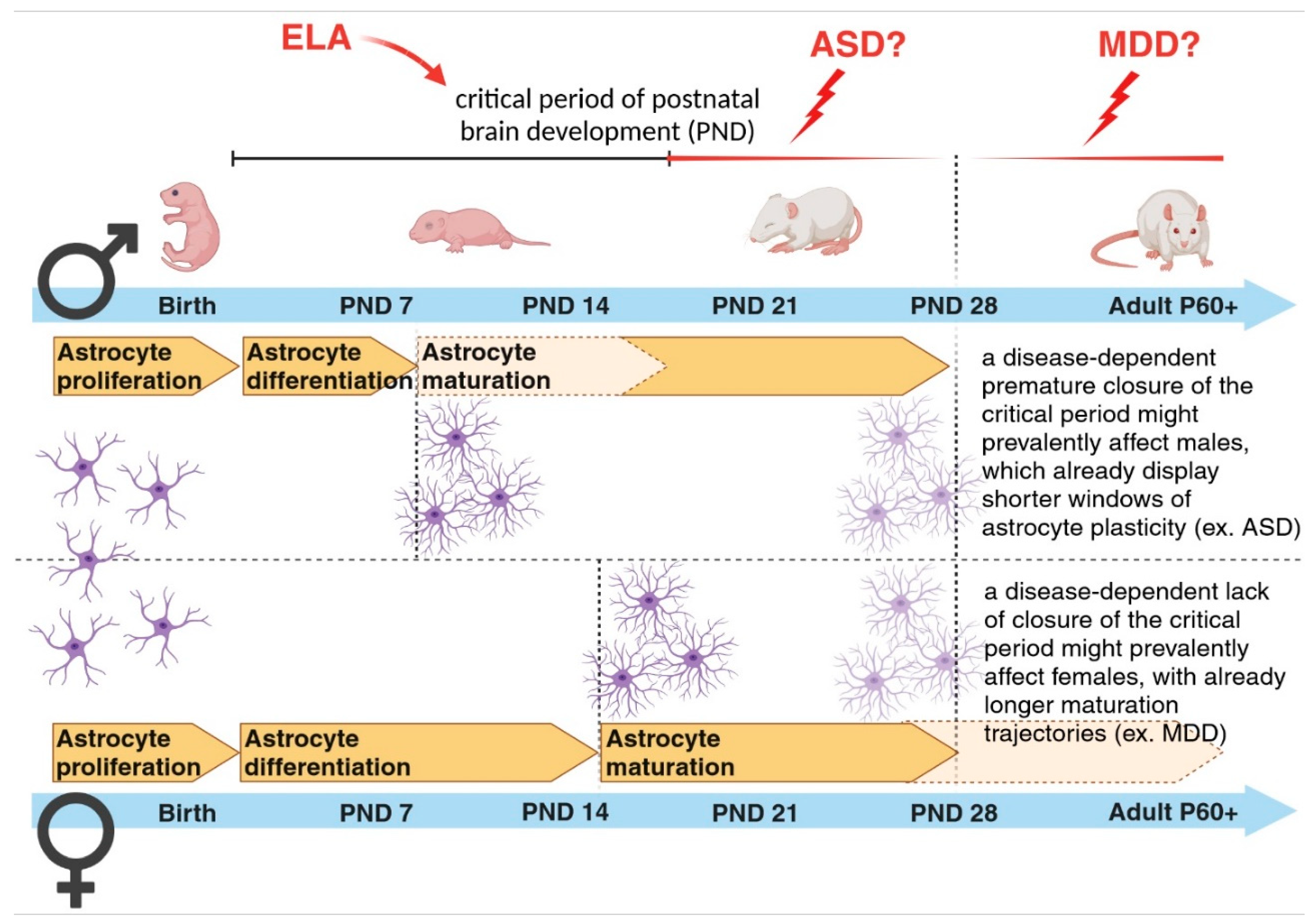
In the past, many human studies have often lacked sufficient sample availability to stratify results based on sex differences for any given disease category. Nowadays, the open access to huge genomic, transcriptomic and proteomic datasets help to discriminate the differential impact of sex on various biological parameters. However, our understanding of whether and how sex might affect astrogenesis, synaptogenesis and synapse elimination in humans and to which extent it might contribute to the development of distinct brain diseases is still in its early stages [111]. Currently, the overall idea is that mental conditions with early-onset neurodevelopmental origin, such as ASD, attention deficit/hyperactivity disorder and SCZ show a higher male prevalence. On the other hand, disorders with a higher emotional component, such as depression, anxiety disorder, and eating disorders, which usually start during puberty or later in life, show a higher female incidence [112]. This asymmetric development of psychopathologies has been analyzed across studies with the goal of elucidating how and which sex-specific developmental maturation trajectories might influence it. Interestingly, regional differences in volume and tissue density were found between sexes in areas implicated in sex-biased NP conditions, suggesting a few candidate regions to investigate the effect sex has on the developing brain [113]. Among those regions, the cortex has been implicated as one of the brain areas with a high sex-dependent diversification in cellular and synaptic densities [40,48,57,114]. Most of these differences have been attributed to remodelling processes occurring at the synaptic level, since the highest rate of dendritic growth is terminated during early childhood. An aberrant pruning of weaker synapses has been recently regarded as a mechanism relevant for the onset of brain disorders [47,48]. As previouslymentioned, most of these remodelling events take place during the critical periods of brain development, when astrogenesis, occurring in parallel with synaptogenesis, contributes to the formation and refinement of neuronal circuits [24,31,32,115]. Mechanisms controlling the opening and closure of these developmental time windows might be essential in building healthy neuronal networks and establishing appropriate behaviours in adult life. Recent work has highlighted sex differences in the maturation processes of astrocytes during these early postnatal developmental stages, showing that astrocytes in male mice reach a mature state earlier than those in female mice [116]. Sex-dependent maturational trajectories are influenced by the perinatal surge in testosterone aimed at masculinizing the brain after birth and establishing sexually dimorphic brain circuitries responsible for sex-differentiated behaviours and reproductive processes [117]. Astroglia respond to circulating gonadal hormones [118], which influence their relative sex-dependent maturation rates [116]. These may in turn correlate with sex-dependent closure times for temporal windows of high plasticity necessary for astrocyte-dependent modulation of neuronal circuit development. It would be highly interesting to investigate whether in ND diseases, for example, a further earlier closure of the critical period, accompanied by a putative higher premature differentiation of astrocytes, may account for the observed prevalence of disorders such as ASD in male individuals. Although this hypothesis has not yet been tested, we may speculate that a shorter time window for astrocyte maturation in males might make them more vulnerable to a disease state affecting astrocyte-dependent early-onset synaptic changes. This would explain some phenotypic effects seen in ASD. In such a case, there would be a narrower time frame for male individuals carrying those affected cells to recover from (or adapt to) detrimental environmental challenges occurring during this period and to rescue impaired neuronal circuits (Figure 4). Correspondingly, slower astrocyte maturation time frames in females might trigger the onset of female-biased disorders eventually characterized by an inappropriately longer immature brain state. Similar considerations can be true for other pathologies for which a sexual dimorphism has been already recognized, opening alternative lines for their investigations [119] (Figure 4).
In conclusion, we propose astrocytes as critical novel targets for the development of efficacious medical treatments for NP and ND disorders. Moreover, specific attention to early postnatal brain developmental periods in combination with the study of sex-specific differential maturation patterns may be essential for improving our knowledge regarding physiological and pathological processes taking place during these time spans.
Acknowledgments
This work was supported by intramural funding from the Department of Psychiatry and Psychotherapy of the University of Regensburg and by the German Research Council (DFG-GRK2174 “Neurobiology of Emotion Dysfunction” (P1)) to BDB. The sponsors did not have any role in writing the report and in the decision to submit the article for publication. We would like to thank Dr. Nicholaus Meyers for proofreading the manuscript.
Conflict of Interest
The authors declare no conflict of interest in relation to the work described.
References
- Bandeira F, Lent R, Herculano-Houzel S. Changing numbers of neuronal and non-neuronal cells underlie postnatal brain growth in the rat. Proc Natl Acad Sci. 2009 Aug 18;106(33):14108–13. [CrossRef]
- Khakh BS, Sofroniew MV. Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci. 2015 Jul;18(7):942–52. [CrossRef]
- Magistretti PJ, Allaman I. Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci. 2018 Apr;19(4):235–49. [CrossRef]
- Allen NJ, Eroglu C. Cell Biology of Astrocyte-Synapse Interactions. Neuron. 2017 Nov;96(3):697–708. [CrossRef]
- Bosworth AP, Allen NJ. The diverse actions of astrocytes during synaptic development. Curr Opin Neurobiol. 2017 Dec;47:38–43. [CrossRef]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive Astrocytes Protect Tissue and Preserve Function after Spinal Cord Injury. J Neurosci. 2004 Mar 3;24(9):2143–55.
- Iram T, Frenkel D. Targeting the Role of Astrocytes in the Progression of Alzheimers Disease. Curr Signal Transduct Ther. 2012 Jan 1;7(1):20–7. [CrossRef]
- Molofsky AV, Kelley KW, Tsai HH, Redmond SA, Chang SM, Madireddy L, et al. Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature. 2014 May 8;509(7499):189–94. [CrossRef]
- Cabezas R, Ãvila M, Gonzalez J, El-Bachá RS, Báez E, Garcà a-Segura LM, et al. Astrocytic modulation of blood brain barrier: perspectives on Parkinsonâ€TMs disease. Front Cell Neurosci [Internet]. 2014 Aug 4 [cited 2022 Jan 12];8. Available from: http://journal.frontiersin.org/article/10.3389/fncel.2014.00211/abstract.
- Boulay AC, Saubaméa B, Adam N, Chasseigneaux S, Mazaré N, Gilbert A, et al. Translation in astrocyte distal processes sets molecular heterogeneity at the gliovascular interface. Cell Discov. 2017 Dec;3(1):17005. [CrossRef]
- Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. 2013 Dec;61(12):1939–58. [CrossRef]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010 Nov;468(7321):223–31. [CrossRef]
- Götz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005 Oct;6(10):777–88.
- Takouda J, Katada S, Nakashima K. Emerging mechanisms underlying astrogenesis in the developing mammalian brain. Proc Jpn Acad Ser B. 2017;93(6):386–98. [CrossRef]
- Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron. 2007 May 3;54(3):357–69. [CrossRef]
- Ge WP, Miyawaki A, Gage FH, Jan YN, Jan LY. Local generation of glia is a major astrocyte source in postnatal cortex. Nature. 2012 Apr;484(7394):376–80. [CrossRef]
- De Zeeuw CI, Hoogland TM. Reappraisal of Bergmann glial cells as modulators of cerebellar circuit function. Front Cell Neurosci [Internet]. 2015 Jul 2 [cited 2023 Jun 6];9. Available from: http://journal.frontiersin.org/Article/10.3389/fncel.2015.00246/abstract . [CrossRef]
- Güngör Kobat S. Importance of Müller Cells. Beyoglu Eye J [Internet]. 2020 [cited 2023 Jun 6]; Available from: http://beyoglueye.com/jvi.aspx?un=BEJ-28290&volume=.
- Farmer WT, Murai K. Resolving Astrocyte Heterogeneity in the CNS. Front Cell Neurosci. 2017 Sep 27;11:300. [CrossRef]
- Holt MG. Astrocyte heterogeneity and interactions with local neural circuits. Bolaños JP, editor. Essays Biochem. 2023 Mar 3;67(1):93–106. [CrossRef]
- Miller SJ. Astrocyte Heterogeneity in the Adult Central Nervous System. Front Cell Neurosci. 2018 Nov 15;12:401. [CrossRef]
- Morrow T, Song MR, Ghosh A. Sequential specification of neurons and glia by developmentally regulated extracellular factors. Dev Camb Engl. 2001 Sep;128(18):3585–94. [CrossRef]
- Oliveria JP, Li ZJ. critical role of astrogenesis and neurodevelopment in Fragile X Syndrome and Rett Syndrome. McMaster Univ Med J [Internet]. 2020 Dec 26 [cited 2023 May 31];17(1). Available from: https://journals.mcmaster.ca/mumj/article/view/2338 . [CrossRef]
- Kanski R, Van Strien ME, Van Tijn P, Hol EM. A star is born: new insights into the mechanism of astrogenesis. Cell Mol Life Sci. 2014 Feb;71(3):433–47.
- Sun Y, Nadal-Vicens M, Misono S, Lin MZ, Zubiaga A, Hua X, et al. Neurogenin Promotes Neurogenesis and Inhibits Glial Differentiation by Independent Mechanisms. Cell. 2001 Feb;104(3):365–76. [CrossRef]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, et al. Regulation of Gliogenesis in the Central Nervous System by the JAK-STAT Signaling Pathway. Science. 1997 Oct 17;278(5337):477–83. [CrossRef]
- Nieto M, Schuurmans C, Britz O, Guillemot F. Neural bHLH Genes Control the Neuronal versus Glial Fate Decision in Cortical Progenitors. Neuron. 2001 Feb;29(2):401–13. [CrossRef]
- Barnabé-Heider F, Wasylnka JA, Fernandes KJL, Porsche C, Sendtner M, Kaplan DR, et al. Evidence that Embryonic Neurons Regulate the Onset of Cortical Gliogenesis via Cardiotrophin-1. Neuron. 2005 Oct;48(2):253–65. [CrossRef]
- Voss AJ, Lanjewar SN, Sampson MM, King A, Hill EJ, Sing A, et al. Identification of ligand-receptor pairs that drive human astrocyte development. Nat Neurosci. 2023 Aug;26(8):1339–51. [CrossRef]
- Azevedo FAC, Carvalho LRB, Grinberg LT, Farfel JM, Ferretti REL, Leite REP, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol. 2009 Apr 10;513(5):532–41. [CrossRef]
- Berardi N, Pizzorusso T, Maffei L. Critical periods during sensory development. Curr Opin Neurobiol. 2000 Feb;10(1):138–45. [CrossRef]
- Chugani HT. A Critical Period of Brain Development: Studies of Cerebral Glucose Utilization with PET. Prev Med. 1998 Mar;27(2):184–8. [CrossRef]
- Sengpiel F. The critical period. Curr Biol. 2007 Sep;17(17):R742–3.
- Knudsen EI. Sensitive Periods in the Development of the Brain and Behavior. J Cogn Neurosci. 2004 Oct 1;16(8):1412–25. [CrossRef]
- Nelson CA, Gabard-Durnam LJ. Early Adversity and Critical Periods: Neurodevelopmental Consequences of Violating the Expectable Environment. Trends Neurosci. 2020 Mar;43(3):133–43. [CrossRef]
- Virolainen SJ, VonHandorf A, Viel KCMF, Weirauch MT, Kottyan LC. Gene–environment interactions and their impact on human health. Genes Immun. 2022 Dec 30;24(1):1–11. [CrossRef]
- Milbocker KA, Campbell TS, Collins N, Kim S, Smith IF, Roth TL, et al. Glia-Driven Brain Circuit Refinement Is Altered by Early-Life Adversity: Behavioral Outcomes. Front Behav Neurosci. 2021 Dec 2;15:786234. [CrossRef]
- Juraska JM, Willing J. Pubertal onset as a critical transition for neural development and cognition. Brain Res. 2017 Jan;1654:87–94. [CrossRef]
- Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997 Oct 20;387(2):167–78.
- Peter R. H. Synaptic density in human frontal cortex — Developmental changes and effects of aging. Brain Res. 1979 Mar;163(2):195–205.
- Dosenbach NUF, Nardos B, Cohen AL, Fair DA, Power JD, Church JA, et al. Prediction of Individual Brain Maturity Using fMRI. Science. 2010 Sep 10;329(5997):1358–61. [CrossRef]
- Petanjek Z, Judaš M, Šimić G, Rašin MR, Uylings HBM, Rakic P, et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci. 2011 Aug 9;108(32):13281–6. [CrossRef]
- Eltokhi A, Janmaat IE, Genedi M, Haarman BCM, Sommer IEC. Dysregulation of synaptic pruning as a possible link between intestinal microbiota dysbiosis and neuropsychiatric disorders. J Neurosci Res. 2020 Jul;98(7):1335–69.
- Cardozo PL, De Lima IBQ, Maciel EMA, Silva NC, Dobransky T, Ribeiro FM. Synaptic Elimination in Neurological Disorders. Curr Neuropharmacol. 2019 Oct 2;17(11):1071–95. [CrossRef]
- Zhuang Y, Xu X, Li H, Niu F, Yang M, Ge Q, et al. Megf10-related engulfment of excitatory postsynapses by astrocytes following severe brain injury. CNS Neurosci Ther. 2023 Apr 20;cns.14223. [CrossRef]
- Iram T, Ramirez-Ortiz Z, Byrne MH, Coleman UA, Kingery ND, Means TK, et al. Megf10 Is a Receptor for C1Q That Mediates Clearance of Apoptotic Cells by Astrocytes. J Neurosci. 2016 May 11;36(19):5185–92. [CrossRef]
- Pattwell SS, Liston C, Jing D, Ninan I, Yang RR, Witztum J, et al. Dynamic changes in neural circuitry during adolescence are associated with persistent attenuation of fear memories. Nat Commun. 2016 May 24;7(1):11475. [CrossRef]
- Honeycutt JA, Demaestri C, Peterzell S, Silveri MM, Cai X, Kulkarni P, et al. Altered corticolimbic connectivity reveals sex-specific adolescent outcomes in a rat model of early life adversity. eLife. 2020 Jan 20;9:e52651.
- Karpova NN, Pickenhagen A, Lindholm J, Tiraboschi E, Kulesskaya N, Ágústsdóttir A, et al. Fear Erasure in Mice Requires Synergy Between Antidepressant Drugs and Extinction Training. Science. 2011 Dec 23;334(6063):1731–4. [CrossRef]
- Vetencourt JFM, Sale A, Viegi A, Baroncelli L, De Pasquale R, F. O’Leary O, et al. The Antidepressant Fluoxetine Restores Plasticity in the Adult Visual Cortex. Science. 2008 Apr 18;320(5874):385–8. [CrossRef]
- Ribot J, Breton R, Calvo CF, Moulard J, Ezan P, Zapata J, et al. Astrocytes close the mouse critical period for visual plasticity. Science. 2021 Jul 2;373(6550):77–81. [CrossRef]
- Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS, et al. A Genetic Variant BDNF Polymorphism Alters Extinction Learning in Both Mouse and Human. Science. 2010 Feb 12;327(5967):863–6. [CrossRef]
- Müller CM, Best J. Ocular dominance plasticity in adult cat visual cortex after transplantation of cultured astrocytes. Nature. 1989 Nov;342(6248):427–30. [CrossRef]
- Ghézali G, Calvo CF, Pillet LE, Llense F, Ezan P, Pannasch U, et al. Connexin 30 controls astroglial polarization during postnatal brain development. Development. 2018 Feb 15;145(4):dev155275. [CrossRef]
- Abbink MR, Deijk AF, Heine VM, Verheijen MH, Korosi A. The involvement of astrocytes in early-life adversity induced programming of the brain. Glia. 2019 Apr 30;glia.23625. [CrossRef]
- Codeluppi SA, Chatterjee D, Prevot TD, Bansal Y, Misquitta KA, Sibille E, et al. Chronic Stress Alters Astrocyte Morphology in Mouse Prefrontal Cortex. Int J Neuropsychopharmacol. 2021 Oct 23;24(10):842–53. [CrossRef]
- Woodburn SC, Bollinger JL, Wohleb ES. Synaptic and behavioral effects of chronic stress are linked to dynamic and sex-specific changes in microglia function and astrocyte dystrophy. Neurobiol Stress. 2021 May;14:100312. [CrossRef]
- Dolotov OV, Inozemtseva LS, Myasoedov NF, Grivennikov IA. Stress-Induced Depression and Alzheimer’s Disease: Focus on Astrocytes. Int J Mol Sci. 2022 Apr 30;23(9):4999. [CrossRef]
- Naskar S, Chattarji S. Stress Elicits Contrasting Effects on the Structure and Number of Astrocytes in the Amygdala versus Hippocampus. eNeuro. 2019;6(1):ENEURO.0338-18.2019. [CrossRef]
- Miguel-Hidalgo JJ, Moulana M, Deloach PH, Rajkowska G. Chronic Unpredictable Stress Reduces Immunostaining for Connexins 43 and 30 and Myelin Basic Protein in the Rat Prelimbic and Orbitofrontal Cortices. Chronic Stress. 2018 Jan;2:247054701881418. [CrossRef]
- Rajkowska G, Miguel-Hidalgo JJ. Glial Pathology in Major Depressive Disorder: An Approach to Investigate the Coverage of Blood Vessels by Astrocyte Endfeet in Human Postmortem Brain. In: Di Benedetto B, editor. Astrocytes [Internet]. New York, NY: Springer New York; 2019 [cited 2023 May 9]. p. 247–54. (Methods in Molecular Biology; vol. 1938). Available from: http://link.springer.com/10.1007/978-1-4939-9068-9_17.
- Benedetto B, Rupprecht R. Targeting Glia Cells: Novel Perspectives for the Treatment of Neuropsychiatric Diseases. Curr Neuropharmacol. 2013 Mar 1;11(2):171–85. [CrossRef]
- Roman C, Egert L, Di Benedetto B. Astrocytic-neuronal crosstalk gets jammed: Alternative perspectives on the onset of neuropsychiatric disorders. Eur J Neurosci. 2021 Sep;54(5):5717–29. [CrossRef]
- Nagy C, Suderman M, Yang J, Szyf M, Mechawar N, Ernst C, et al. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol Psychiatry. 2015 Mar;20(3):320–8. [CrossRef]
- Martins-Macedo J, Salgado AJ, Gomes ED, Pinto L. Adult brain cytogenesis in the context of mood disorders: From neurogenesis to the emergent role of gliogenesis. Neurosci Biobehav Rev. 2021 Dec;131:411–28. [CrossRef]
- Feresten AH, Barakauskas V, Ypsilanti A, Barr AM, Beasley CL. Increased expression of glial fibrillary acidic protein in prefrontal cortex in psychotic illness. Schizophr Res. 2013 Oct;150(1):252–7. [CrossRef]
- Tarasov VV, Svistunov AA, Chubarev VN, Sologova SS, Mukhortova P, Levushkin D, et al. Alterations of Astrocytes in the Context of Schizophrenic Dementia. Front Pharmacol. 2020 Feb 7;10:1612.
- Notter T. Astrocytes in schizophrenia. Brain Neurosci Adv. 2021 Jan;5:239821282110091. [CrossRef]
- Vakilzadeh G, Martinez-Cerdeño V. Pathology and Astrocytes in Autism. Neuropsychiatr Dis Treat. 2023;19:841–50. [CrossRef]
- Allen M, Huang BS, Notaras MJ, Lodhi A, Barrio-Alonso E, Lituma PJ, et al. Astrocytes derived from ASD individuals alter behavior and destabilize neuronal activity through aberrant Ca2+ signaling. Mol Psychiatry. 2022 May;27(5):2470–84. [CrossRef]
- Rajkowska G, Miguel-Hidalgo J. Gliogenesis and Glial Pathology in Depression. CNS Neurol Disord - Drug Targets. 2007 Jun 1;6(3):219–33. [CrossRef]
- Belleau EL, Treadway MT, Pizzagalli DA. The Impact of Stress and Major Depressive Disorder on Hippocampal and Medial Prefrontal Cortex Morphology. Biol Psychiatry. 2019 Mar;85(6):443–53. [CrossRef]
- Malykhin NV, Carter R, Seres P, Coupland NJ. Structural changes in the hippocampus in major depressive disorder: contributions of disease and treatment. J Psychiatry Neurosci. 2010 Sep;35(5):337–43. [CrossRef]
- Geng R, Huang X. Identification of major depressive disorder disease-related genes and functional pathways based on system dynamic changes of network connectivity. BMC Med Genomics. 2021 Dec;14(1):55. [CrossRef]
- Czéh B, Simon M, Schmelting B, Hiemke C, Fuchs E. Astroglial Plasticity in the Hippocampus is Affected by Chronic Psychosocial Stress and Concomitant Fluoxetine Treatment. Neuropsychopharmacology. 2006 Aug;31(8):1616–26. [CrossRef]
- Czéh B, Di Benedetto B. Antidepressants act directly on astrocytes: Evidences and functional consequences. Eur Neuropsychopharmacol. 2013 Mar;23(3):171–85.
- Czéh B, Nagy SA. Clinical Findings Documenting Cellular and Molecular Abnormalities of Glia in Depressive Disorders. Front Mol Neurosci. 2018 Feb 27;11:56. [CrossRef]
- Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, et al. Synaptic and Extrasynaptic NMDA Receptors Are Gated by Different Endogenous Coagonists. Cell. 2012 Aug;150(3):633–46. [CrossRef]
- Henneberger C, Papouin T, Oliet SHR, Rusakov DA. Long-term potentiation depends on release of d-serine from astrocytes. Nature. 2010 Jan 14;463(7278):232–6. [CrossRef]
- Heresco-Levy U, Javitt DC, Ebstein R, Vass A, Lichtenberg P, Bar G, et al. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol Psychiatry. 2005 Mar;57(6):577–85. [CrossRef]
- Kantrowitz JT, Malhotra AK, Cornblatt B, Silipo G, Balla A, Suckow RF, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res. 2010 Aug;121(1–3):125–30. [CrossRef]
- Ma TM, Abazyan S, Abazyan B, Nomura J, Yang C, Seshadri S, et al. Pathogenic disruption of DISC1-serine racemase binding elicits schizophrenia-like behavior via D-serine depletion. Mol Psychiatry. 2013 May;18(5):557–67. [CrossRef]
- Cardno AG, Gottesman II. Twin studies of schizophrenia: From bow-and-arrow concordances to Star Wars Mx and functional genomics. Am J Med Genet. 2000;97(1):12–7.
- Walker EF, Trotman HD, Pearce BD, Addington J, Cadenhead KS, Cornblatt BA, et al. Cortisol Levels and Risk for Psychosis: Initial Findings from the North American Prodrome Longitudinal Study. Biol Psychiatry. 2013 Sep;74(6):410–7. [CrossRef]
- Selemon LD, Zecevic N. Schizophrenia: a tale of two critical periods for prefrontal cortical development. Transl Psychiatry. 2015 Aug 18;5(8):e623–e623. [CrossRef]
- Sheu JR, Hsieh CY, Jayakumar T, Tseng MF, Lee HN, Huang SW, et al. A Critical Period for the Development of Schizophrenia-Like Pathology by Aberrant Postnatal Neurogenesis. Front Neurosci. 2019 Jun 18;13:635.
- De Oliveira Figueiredo EC, Calì C, Petrelli F, Bezzi P. Emerging evidence for astrocyte dysfunction in schizophrenia. Glia. 2022 Sep;70(9):1585–604. [CrossRef]
- Russo FB, Freitas BC, Pignatari GC, Fernandes IR, Sebat J, Muotri AR, et al. Modeling the Interplay Between Neurons and Astrocytes in Autism Using Human Induced Pluripotent Stem Cells. Biol Psychiatry. 2018 Apr;83(7):569–78. [CrossRef]
- Berger JM, Rohn TT, Oxford JT. Autism as the Early Closure of a Neuroplastic Critical Period Normally Seen in Adolescence. Biol Syst Open Access [Internet]. 2012 [cited 2023 Jul 23];02(03). Available from: https://www.omicsgroup.org/journals/autism-as-the-early-closure-of-a-neuroplastic-critical-period-normally-seen-in-adolescence-2329-6577-1000118.php?aid=43859.
- Hashimoto Y, Greene C, Munnich A, Campbell M. The CLDN5 gene at the blood-brain barrier in health and disease. Fluids Barriers CNS. 2023 Mar 28;20(1):22. [CrossRef]
- Igarashi Y, Utsumi H, Chiba H, Yamada-Sasamori Y, Tobioka H, Kamimura Y, et al. Glial Cell Line-Derived Neurotrophic Factor Induces Barrier Function of Endothelial Cells Forming the Blood–Brain Barrier. Biochem Biophys Res Commun. 1999 Jul;261(1):108–12.
- Rajkowska G, Hughes J, Stockmeier CA, Javier Miguel-Hidalgo J, Maciag D. Coverage of Blood Vessels by Astrocytic Endfeet Is Reduced in Major Depressive Disorder. Biol Psychiatry. 2013 Apr;73(7):613–21. [CrossRef]
- Lee E, Chung WS. Glial Control of Synapse Number in Healthy and Diseased Brain. Front Cell Neurosci. 2019 Feb 13;13:42. [CrossRef]
- Di Benedetto B, Malik VA, Begum S, Jablonowski L, Gómez-González GB, Neumann ID, et al. Fluoxetine Requires the Endfeet Protein Aquaporin-4 to Enhance Plasticity of Astrocyte Processes. Front Cell Neurosci [Internet]. 2016 Feb 2 [cited 2022 Jan 13];10. Available from: http://journal.frontiersin.org/Article/10.3389/fncel.2016.00008/abstract . [CrossRef]
- Malik VA, Zajicek F, Mittmann LA, Klaus J, Unterseer S, Rajkumar S, et al. GDF15 promotes simultaneous astrocyte remodeling and tight junction strengthening at the blood–brain barrier. J Neurosci Res. 2020 Jul;98(7):1433–56. [CrossRef]
- Williams BP, Price J. Evidence for multiple precursor cell types in the embryonic rat cerebral cortex. Neuron. 1995 Jun;14(6):1181–8. [CrossRef]
- Konishi H, Koizumi S, Kiyama H. Phagocytic astrocytes: Emerging from the shadows of microglia. Glia. 2022 Jun;70(6):1009–26. [CrossRef]
- Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013 Dec 19;504(7480):394–400. [CrossRef]
- Logan MA, Freeman MR. The scoop on the fly brain: glial engulfment functions in Drosophila. Neuron Glia Biol. 2007 Feb;3(1):63–74. [CrossRef]
- Freeman MR, Delrow J, Kim J, Johnson E, Doe CQ. Unwrapping Glial Biology. Neuron. 2003 May;38(4):567–80. [CrossRef]
- Reddien PW, Horvitz HR. THE ENGULFMENT PROCESS OF PROGRAMMED CELL DEATH IN CAENORHABDITIS ELEGANS. Annu Rev Cell Dev Biol. 2004 Nov 1;20(1):193–221.
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell. 2007 Dec;131(6):1164–78. [CrossRef]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron. 2012 May;74(4):691–705. [CrossRef]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science. 2011 Sep 9;333(6048):1456–8. [CrossRef]
- Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci. 2010 Apr 27;107(17):7975–80. [CrossRef]
- Lee JH, Kim J young, Noh S, Lee H, Lee SY, Mun JY, et al. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature. 2021 Feb 25;590(7847):612–7. [CrossRef]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016 May 6;352(6286):712–6. [CrossRef]
- Dion-Albert L, Bandeira Binder L, Daigle B, Hong-Minh A, Lebel M, Menard C. Sex differences in the blood-brain barrier: implications for mental health. Front Neuroendocrinol. 2022 Mar;100989. [CrossRef]
- Blokland GAM, Grove J, Chen CY, Cotsapas C, Tobet S, Handa R, et al. Sex-Dependent Shared and Nonshared Genetic Architecture Across Mood and Psychotic Disorders. Biol Psychiatry. 2022 Jan;91(1):102–17. [CrossRef]
- Riecher-Rössler A. Sex and gender differences in mental disorders. Lancet Psychiatry. 2017 Jan;4(1):8–9. [CrossRef]
- 111. Hyer MM, Phillips LL, Neigh GN. Sex Differences in Synaptic Plasticity: Hormones and Beyond. Front Mol Neurosci. 2018 Jul 31;11:266. [CrossRef]
- Rutter M, Caspi A, Moffitt TE. Using sex differences in psychopathology to study causal mechanisms: unifying issues and research strategies: Using sex differences in psychopathology to study causal mechanisms. J Child Psychol Psychiatry. 2003 Nov;44(8):1092–115.
- Ruigrok ANV, Salimi-Khorshidi G, Lai MC, Baron-Cohen S, Lombardo MV, Tait RJ, et al. A meta-analysis of sex differences in human brain structure. Neurosci Biobehav Rev. 2014 Feb;39(100):34–50. [CrossRef]
- Knickmeyer RC, Styner M, Short SJ, Lubach GR, Kang C, Hamer R, et al. Maturational Trajectories of Cortical Brain Development through the Pubertal Transition: Unique Species and Sex Differences in the Monkey Revealed through Structural Magnetic Resonance Imaging. Cereb Cortex. 2010 May;20(5):1053–63. [CrossRef]
- Dehorter N, Del Pino I. Shifting Developmental Trajectories During Critical Periods of Brain Formation. Front Cell Neurosci. 2020;14:283. [CrossRef]
- Rurak GM, Simard S, Freitas-Andrade M, Lacoste B, Charih F, Van Geel A, et al. Sex differences in developmental patterns of neocortical astroglia: A mouse translatome database. Cell Rep. 2022 Feb;38(5):110310. [CrossRef]
- Clarkson J, Herbison AE. Hypothalamic control of the male neonatal testosterone surge. Philos Trans R Soc Lond B Biol Sci. 2016 Feb 19;371(1688):20150115. [CrossRef]
- Acaz-Fonseca E, Avila-Rodriguez M, Garcia-Segura LM, Barreto GE. Regulation of astroglia by gonadal steroid hormones under physiological and pathological conditions. Prog Neurobiol. 2016 Sep;144:5–26. [CrossRef]
- Rurak GM, Woodside B, Aguilar-Valles A, Salmaso N. Astroglial cells as neuroendocrine targets in forebrain development: Implications for sex differences in psychiatric disease. Front Neuroendocrinol. 2021 Jan;60:100897. [CrossRef]
Figure 1.
The critical period for astrogenesis and synaptic circuit formation during postnatal brain development in rodents and humans. The timelimes show the major processes occurring from pre- and postnatal early stages through adulthood in rodents and humans to develop neuronal synaptic networks. A few examples of intracellular signalling molecules which determine the switch between neurogenic to gliogenic cell fates are depicted, as well as specific proteins relevant for astrocyte-specific differentiation/maturation processes (e.g. Cx30 and TGFβ/NLGN1/TLSP/DKK1/BMP4 ligands). Cx30, connexin 30. Figure created with BioRender.com.
Figure 1.
The critical period for astrogenesis and synaptic circuit formation during postnatal brain development in rodents and humans. The timelimes show the major processes occurring from pre- and postnatal early stages through adulthood in rodents and humans to develop neuronal synaptic networks. A few examples of intracellular signalling molecules which determine the switch between neurogenic to gliogenic cell fates are depicted, as well as specific proteins relevant for astrocyte-specific differentiation/maturation processes (e.g. Cx30 and TGFβ/NLGN1/TLSP/DKK1/BMP4 ligands). Cx30, connexin 30. Figure created with BioRender.com.
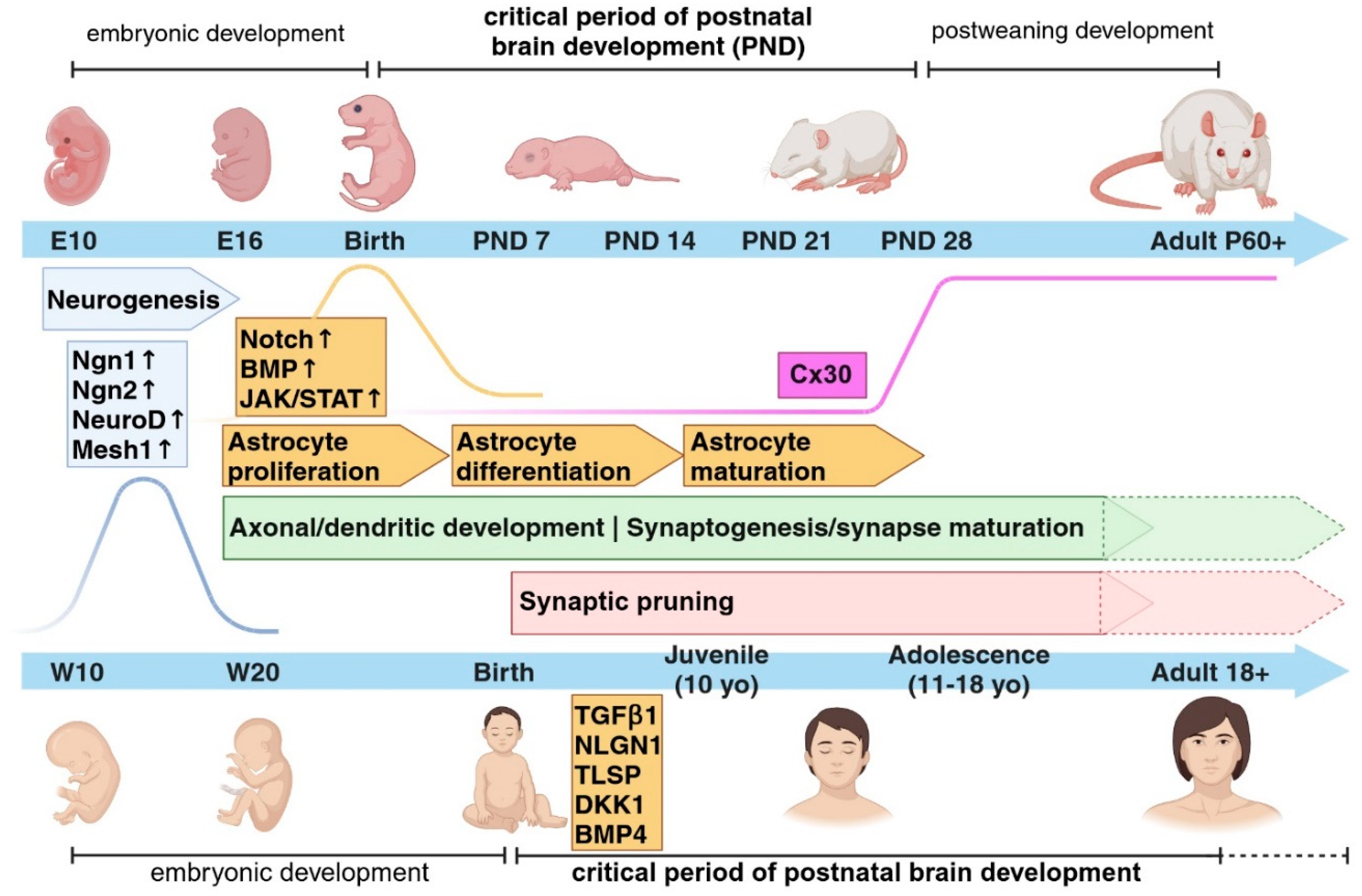
Figure 2.
The critical period in neurodevelopmental and neuropsychiatric disorders. Extrinsic and intrinsic cues may interact with each other during the critical period of brain development to drive the proper formation of neuronal circuits. Any alterations in the sequences of events occurring during these time frames might lead to the onset of neurodevelopmental (ND) disorders such as Autism Spectrum Disorder (ASD) or neuropsychiatric (NP) disorders such as Schizophrenia (SCZ) or Major depressive disorder (MDD). Early-life adversity (ELA) such as stress experienced during the critical period show a higher impact on the development of ND and NP disorders later in life. Molecules like Cx30 may play a role in the closure of the critical period of astrocyte development, which is necessary for the proper formation of brain circuits. Figure created with BioRender.com.
Figure 2.
The critical period in neurodevelopmental and neuropsychiatric disorders. Extrinsic and intrinsic cues may interact with each other during the critical period of brain development to drive the proper formation of neuronal circuits. Any alterations in the sequences of events occurring during these time frames might lead to the onset of neurodevelopmental (ND) disorders such as Autism Spectrum Disorder (ASD) or neuropsychiatric (NP) disorders such as Schizophrenia (SCZ) or Major depressive disorder (MDD). Early-life adversity (ELA) such as stress experienced during the critical period show a higher impact on the development of ND and NP disorders later in life. Molecules like Cx30 may play a role in the closure of the critical period of astrocyte development, which is necessary for the proper formation of brain circuits. Figure created with BioRender.com.
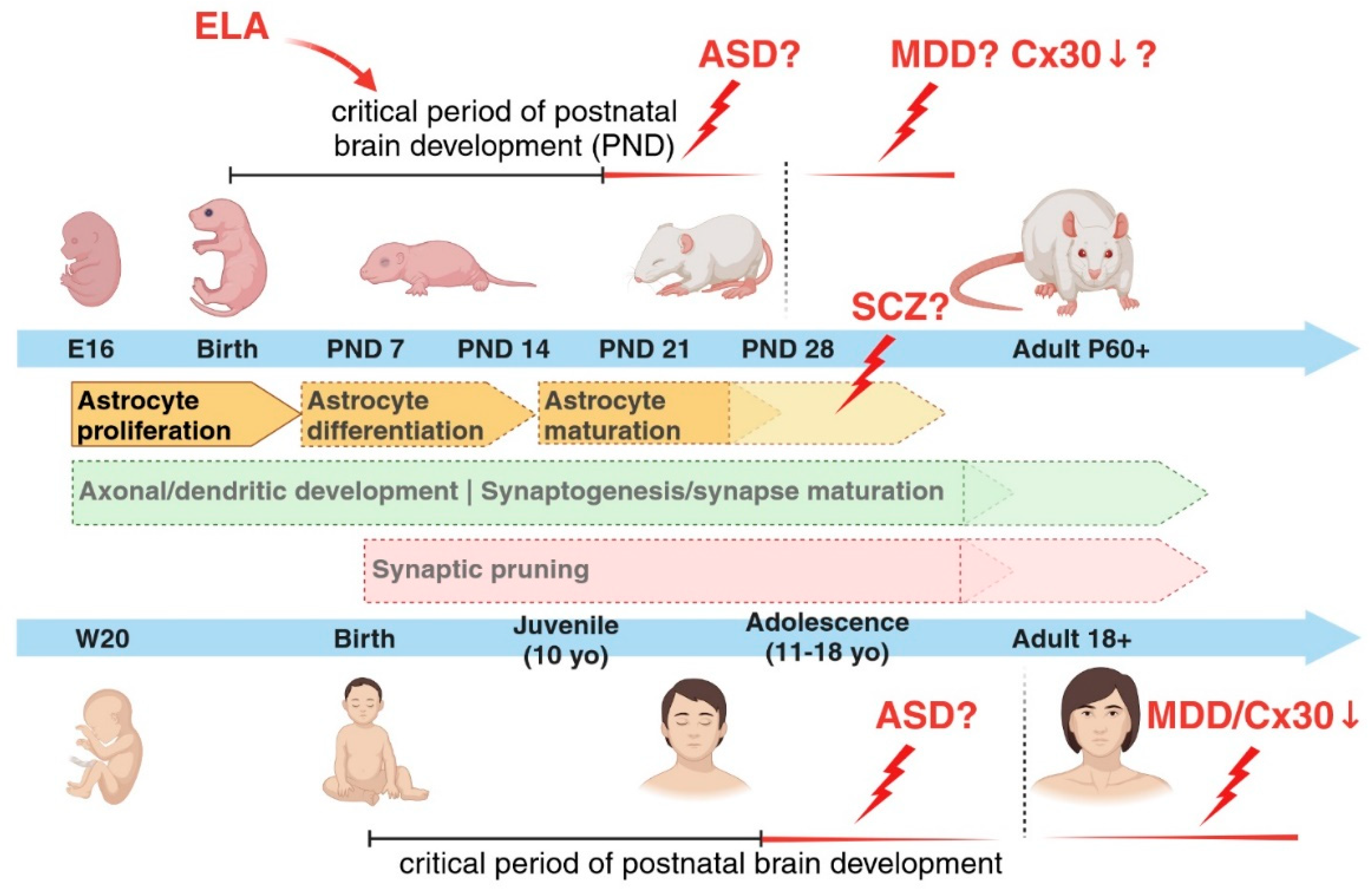
Figure 3.
Glia-mediated synaptic pruning. Astrocytes and microglia cells can both regulate the refinement of synaptic neuronal networks through the elimination of weak synapses during early postnatal developmental stages. Signalling molecules called “eat me” signals, such as C1q and phosphatidylserine (PS), recruited during these events have been identified, as well as binding partners located on the respective cells, like MEGF10 receptor on astrocytes and the receptors of the complement cascade on microglia cells. It is however still unclear whether PS and C1q are the molecules responsible for an astrocyte-mediated synaptic elimination. Figure created with BioRender.com.
Figure 3.
Glia-mediated synaptic pruning. Astrocytes and microglia cells can both regulate the refinement of synaptic neuronal networks through the elimination of weak synapses during early postnatal developmental stages. Signalling molecules called “eat me” signals, such as C1q and phosphatidylserine (PS), recruited during these events have been identified, as well as binding partners located on the respective cells, like MEGF10 receptor on astrocytes and the receptors of the complement cascade on microglia cells. It is however still unclear whether PS and C1q are the molecules responsible for an astrocyte-mediated synaptic elimination. Figure created with BioRender.com.
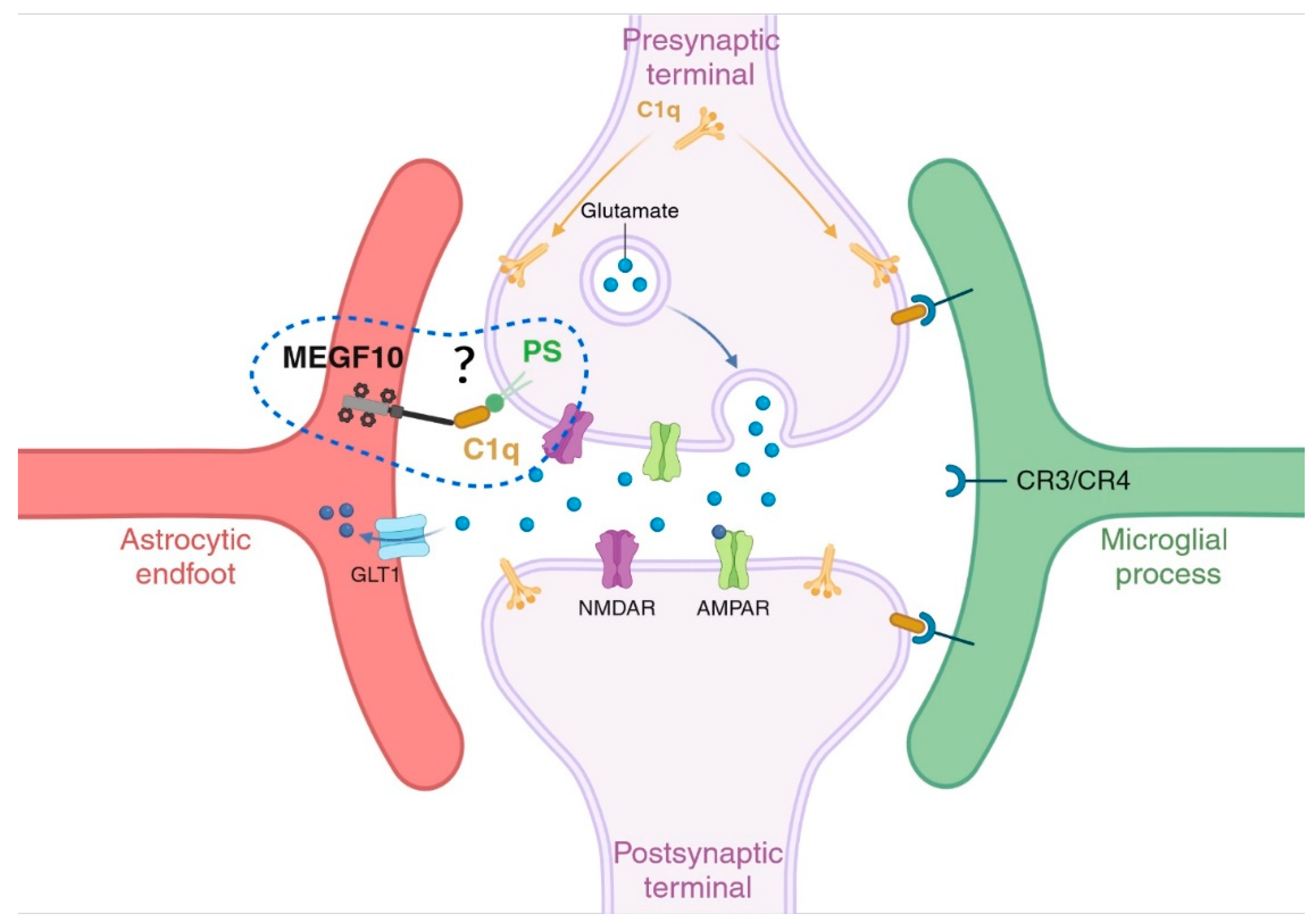
Figure 4.
Sex-dependent differences in astrogenesis and vulnerability to disease onset. A sex-dependent predisposition to brain disorders has been long recognized for many brain pathologies. The differentially time-shifted maturation trajectories for astrocytes and astrocyte-mediated processes between males and females may account for such sex-dependent biased windows of vulnerability to disease onset. Figure created with BioRender.com.
Figure 4.
Sex-dependent differences in astrogenesis and vulnerability to disease onset. A sex-dependent predisposition to brain disorders has been long recognized for many brain pathologies. The differentially time-shifted maturation trajectories for astrocytes and astrocyte-mediated processes between males and females may account for such sex-dependent biased windows of vulnerability to disease onset. Figure created with BioRender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



