Submitted:
01 November 2023
Posted:
02 November 2023
You are already at the latest version
Abstract
The development of the ovarian antral follicle is a complex, highly regulated process. Oocytes orchestrate and coordinate the development of mammalian ovarian follicles, and the rate of follicular development is governed by a developmental program intrinsic to the oocyte. Characterizing oocyte signatures during this dynamic process is critical for understanding oocyte maturation and follicular development. Although the transcriptional signature of sheep oocytes matured in vitro and preovulatory oocytes have been previously described, the transcriptional changes of oocytes in antral follicles have not. Here, we used single-cell transcriptomics (SmartSeq2) to characterize sheep oocytes from small, medium, and large antral follicles. We characterized the transcriptomic landscape of sheep oocytes during antral follicle development, identifying unique features in the transcriptional atlas, stage-specific molecular signatures, oocyte-secreted factors, and transcription factor networks. Notably, we identified specific gene and signaling pathways in each antral follicle stage that may reflect oocyte quality and in vitro maturation competency. Additionally, we discovered key biological processes that drive the transition from small to large antral follicles, revealing hub genes involved in follicle recruitment and selection. Thus, our work provides a comprehensive characterization of the single-oocyte transcriptome, filling a gap in the mapping of the molecular landscape of sheep oogenesis. We also provide key insights into the transcriptional regulation of the critical stages of antral follicular development, which is essential for understanding how the oocyte orchestrates follicular development.
Keywords:
sheep oocytes
; antral follicle
; single-cell transcriptomics
; molecular signatures
; signaling pathways
1. Introduction
Ovarian folliculogenesis is the complex maturation of an ovarian follicle through the primordial, primary, secondary, antral, and preovulatory developmental stages [1]. This developmental complexity is reflected in the high level of synchronization among the different cell types within one follicle that together produce a competent mature oocyte [2]. The generation of healthy oocytes relies on the coordinated development of somatic and germ cells in the ovarian follicle, and the rate of follicular development is determined by a developmental program intrinsic to the oocyte [3]. Activated preantral follicles develop to the antral stage, where follicle growth increases rapidly. The granulosa cells facilitate the accumulation of follicular fluid into a central cavity known as an antrum, giving rise to growing antral follicles [4]. Antral follicles develop in a wave-like pattern during which one or more follicles, “selected” from a group of small follicles (2–3 mm), develop into dominant follicles ≥ 6 mm in diameter. However, in mono-ovulatory bovine and ovine species, only one follicle becomes dominant, and subordinate follicles regress or degenerate [5,6].
The late antral follicle stage, when the oocyte acquires developmental potential, is critical for oocyte maturation [7]. Thus, the quality of an oocyte depends on its follicle, and follicular size correlates with developmental outcomes [8]. Oocytes from larger follicles are more likely to reach the blastocyst stage than those from small follicles [9], but regulation of gene expression is important in determining oocyte quality [10]. Thus, the relationship between the follicle stage and the oocyte transcriptomic landscape should be determined and correlated with developmental competence. The oocyte depends on stored maternal transcripts that play a key role in the development of oocyte meiotic competence during the growth phase in preantral and early antral follicles. Oocytes at the antral follicle stage resume meiosis [11] and support early development [12]. In addition, transcripts involved in energy production and metabolism are present in oocytes [13]. Differences in transcripts in the antral follicle development stages could explain subsequent oocyte maturation and identify biomarkers of oocyte quality. Oocyte development requires continuous intercommunication using highly coordinated paracrine, autocrine, and endocrine regulators, as well as gap junctions [14]. These oocyte-specific factors (OSFs) also promote proliferation of follicular somatic cells and steroidogenesis. Transcription factors (TFs) that are translocated from the cytoplasm to the nucleus mediate signaling and regulate the cellular phenotype [15], and the formation of follicles may be coordinated by a TF expressed by the oocyte. Thus, it is important to 1) identify oocyte-secreted TFs and other proteins involved in oocyte development, 2) classify these proteins by antral follicle stage, and 3) describe the dynamic associations of key factors between the follicle different stages.
Single-cell RNA sequencing (scRNA-seq) is a key tool for characterizing tissue heterogeneity at a cellular level [16], and it identifies changes in gene transcripts at the single-cell level that are undetectable by bulk RNA sequencing. Advances in transcriptome technology have also been applied to characterizing oocytes [17], although most oocyte transcriptome studies use animal models [18,19]. The scRNA-seq transcriptomes of human oocytes at five follicular stages revealed candidate secretory biomarkers of ovarian reserve [20]. The scRNA-seq transcriptome in germinal vesicles (GVs) and in in vitro metaphase II (MII) human oocytes identified maturation differences in oocytes [21,22]. However, the developmental pattern and transcriptional regulatory networks in sheep oocytes during antral follicle development, which differs from human and mouse oocytes [23], is not well characterized.
Here, we analyzed the transcriptome of single oocytes from antral follicles of different stages to identify dynamic changes in gene expression throughout antral follicle growth. We identified unique features of the transcriptional machinery associated with developmental-stage-specific expression patterns. We also identified stage-specific gene signatures in sheep oocytes, including oocyte-specific genes, oocyte-secreted factors, and TFs, which could provide oocyte development-specific profiles. We found differences in transcripts associated with specific biological processes and key signaling pathways that were associated with antral follicle development, providing information on expression patterns of hub genes regulating oocyte maturation. Thus, our characterization of the transcriptional landscape of antral follicle oocytes provides insights into the regulation of follicle growth in sheep.
2. Results
2.1. Overview of single-oocyte sequence data
To identify changes in oocyte transcripts during the antral follicle stage, we collected oocytes from antral follicles at different developmental stages, including small (≤ 3 mm, SO1, SO2, SO3), medium (3–6 mm, MO1, MO2, MO3) and large (≥ 6 mm, LO1, LO2, LO3) antral follicles for single-oocyte RNA-seq using the Smart-seq2 protocol (Figure 1A–C and Table S1). Following library construction and deep sequencing of oocyte transcriptomes, we obtained a total of 372,027,250 bases from the nine sequence libraries, producing a total of 334,979,082 (90.06 %) clean reads (Figure S1A and Table S2). The mapping efficiency of the clean reads to the sheep genome was high, with an average of 97.55 % for the small antral oocytes (SO), 97.15 % for the medium antral oocytes (MO), and 96.68 % for the large antral oocytes (LO), and a unique mapped rate average of 91.22 % (Figure S1B). The quality of these mRNA libraries was high enough to use them for statistical analyses. We identified an average of 15,341 expressed genes per oocyte, with a total of 138,073 expressed genes; 14,512 were expressed in the SO group, 15,300 in the MO group, and 16,212 in the LO group; however, the LO had the most mRNAs. We found that RNA synthesis was still detectable in the period following oocyte growth arrest and germinal vesicle breakdown (GVBD), and transcription ceased only at GVBD. Therefore, it is possible to resolve differences in mRNA expression levels between stages and identify the factors involved in oocyte developmental competence.
Using principal component analysis (PCA) for the groups of oocytes, we observed three distinct subpopulations, which corresponded to the morphological classification of antral follicular development (Figure 1D, E). Thus, antral follicles at different developmental stages exhibited different gene expression dynamics, as evidenced by distinct transcriptomes.
2.2. Gene expression dynamics and transcriptional characteristics of oocytes during antral follicle development
To determine the global expression profiles during antral follicle development, we characterized the 9,400 differentially expressed genes (DEGs) for the stages of follicular development in oocytes (false discovery rate [FDR, q-value] < 0.05; Figure 2A, B; Figure S2A). To characterize gene expression dynamics between consecutive stages in antral follicle development, we compared DEGs and found 2,141 genes overrepresented in the SO stage, 2,058 genes overrepresented in the MO stage, and 4,486 genes overrepresented in the LO stage. Unsupervised cluster analysis identified nine clusters of genes according to their expression patterns. The genes in cluster 5 (2,122) and cluster 7 (1,304) were underexpressed in the SO stage and were upregulated during antral follicular development, with a peak expression in the LO stage, suggesting that they indicate oocyte maturation. However, the transcripts for genes in cluster 9 (1,137) and cluster 6 (1,004) accumulated before follicle selection and dominance, suggesting that they represent the resting follicle pool. The gene expression patterns of cluster 2 (586), cluster 8 (792), and cluster 4 (678) showed expression at the MO stage (Figure S2B). However, because oocyte mRNA increased substantially during antral follicular development, the putative DEGs might simply reflect changes in the relative abundance of transcripts, with only some of them corresponding to changes in transcriptional state.
To identify transcripts specifically enriched at each developmental stage from the DEG expression profiles, we considered those genes with a log2 transformed fold change (log2FC) > |2| and FDR < 0.05 as differentially expressed genes. This yielded 1,299 genes overrepresented in the LO, 246 genes overrepresented in the SO stage (Figure 2C), and 565 genes identified in a comparison between the MO and SO stage (Figure 2D). Compared to the SO stage, 476 genes were upregulated in the MO stage, which accounted for the majority of DEGs, and only 89 genes were downregulated. Using the same criteria, we screened 820 genes, including 195 upregulated genes and 625 downregulated genes in the MO vs. LO oocytes (Figure 2E). The top 10 upregulated or downregulated genes in the SO vs. LO, MO vs. SO, and MO vs. LO are listed in Table S3. Using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of DEGs in oocytes at different stages to characterize their function in antral follicle growth, we identified several significantly enriched biological processes associated with each stage of follicular development (Figure 2F, G; Figure S2 C, D).
2.3. Gene expression signatures of oocytes at different antral follicle stages
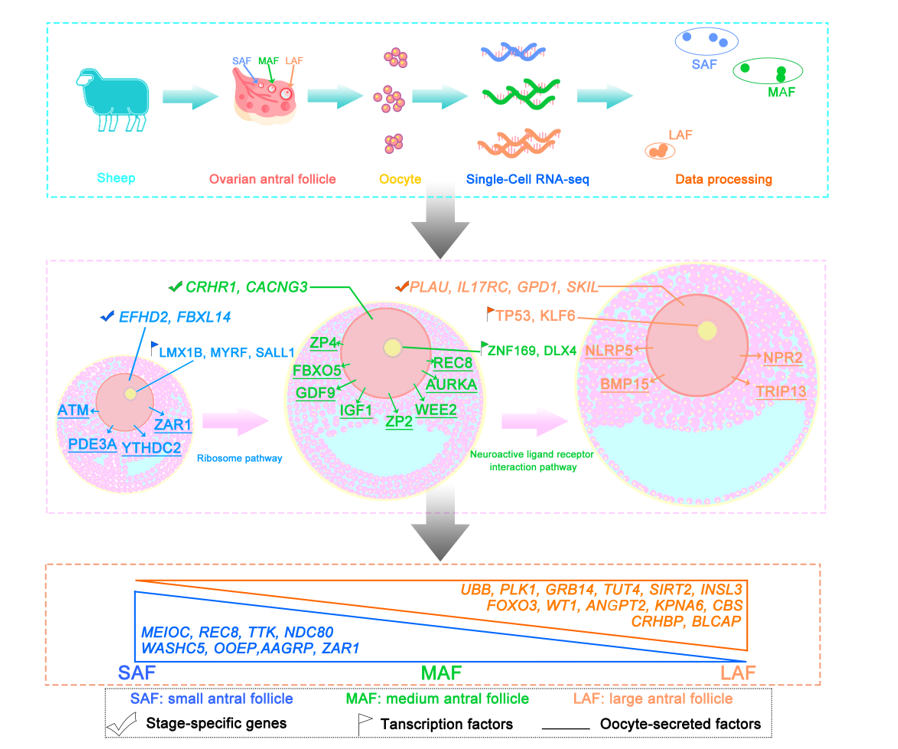
Using Venn diagrams, we identified 365 genes that overlapped the 625 genes (MO vs. LO) and 1,299 genes (SO vs. LO) in the LO stage (Figure 3A), and 45 genes overlapped in the MO stage, whereas 33 genes overlapped in the SO stage. Surprisingly, there was no overlap in the three groups for upregulated genes. We clustered the overlapping genes with DEGs (SO vs. MO vs. LO; 9,400) and considered those genes with FDR < 0.05 as the stage-specific genes. We identified 222 stage-specific genes, including genes expressed in the oocytes from each antral follicular stage (Figure 3B and Table S4). Most of these genes (208) were upregulated during antral follicular development, with the highest expression in the large antral follicle stage. In addition, eight and six genes were highly expressed in the MO and SO stages, respectively.
We used GO analysis of genes specific to particular oocyte stages to determine the regulatory functions involved in antral follicle development (Figure 3C). Transcription coregulator binding (representative genes ESR1, JUND, MYC, NR5A2, and SMARCD3), glycolytic process (representative genes IER3, PFKL, PFKP, PGK1, and ENO1), vascular endothelial growth factor receptor signaling pathway (representative genes BCAR1, FGF10, FLT4, NRP1, PTK2B, VEGFA, and VEGFC), positive regulation of miRNA transcription (representative genes AR, JUN, MYC, and NGFR), response to hypoxia (DDIT4, EPO, HIF1A, LONP1, NOS2, P2RX2, PLAU, PLOD1, PLOD2, SOD3, and VEGFA), and cholesterol metabolic process (representative genes CH25H, CLN8, CYP11A1, LCAT, and SMPD1) were enriched in the LO stage, and GO enrichment items with FDR < 0.05 are shown (Figure 3D). Interestingly, regulation of calcium ion import (representative genes CACNG3), gap junction channel activity (representative genes GJB4), and steroid biosynthesis (representative genes HSD17B3) were enriched in the MO stage. We also identified candidate biomarkers uniquely expressed in the medium antral follicle stage: TENT5D, HGD, CRHR1, and CD86, which are involved in mRNA stabilization [24], tyrosine or L-phenylalanine metabolism [25], activation of G protein-coupled receptors [26], and adaptive immunity [27], respectively. In addition, ribosome biogenesis was enriched in the SO stage, with increased expression of ribosome-related genes (representative genes RPL23A and RPL24). GOLT1A plays a critical role in vesicle-mediated protein transport from the endoplasmic reticulum to the Golgi [28]. KBTBD6, as part of the Cul3 (KBTBD6/7)-RING ubiquitin ligase complex [29], is involved in the proteasome-mediated ubiquitin-dependent protein catabolic process and regulates RAC1 signal transduction and downstream biological processes, including the organization of the cytoskeleton, cell migration, and cell proliferation [30]. Members of the F-box protein family, such as FBXL14, are newly recognized E3 ligases that play a critical role in ubiquitylation [31]. EFHD2 is a conserved calcium-binding protein that plays a role in vitro in calcium signaling, apoptosis, actin cytoskeleton, and regulation of synapse formation [32]. Stage-specific gene expression profiles revealed a core hub of genes that regulate specific functions at each stage of oocyte development, including EFHD2 and FBXL14 in the SO stage, CRHR1 and CACNG3 in the MO stage, and PLAU, IL17RC, GPD1, and SKIL in the LO stage (Figure 3D). These signature genes may serve as potential cell-specific markers for each follicle stage. In addition, the subsets of 222 stage-specific signature genes in oocytes could serve as markers of oocyte developmental competency. Together, our results reveal the atlas of stage-specific transcriptional regulatory networks underlying antral follicle development in sheep.
2.4. Characterization of key pathways throughout antral follicle development
Gene Set Enrichment Analysis (GSEA) and KEGG analysis were used for pairwise comparisons of the follicular stages to identify changes in signaling pathways during the transition from the small stage to the medium stage during follicle recruitment (FDR<0.05). Ribosome and phosphatidylinositol signaling systems were upregulated in the SO stage. In contrast, 22 pathways were enriched in the MO stage, including ovarian steroidogenesis, fat digestion and absorption, glutathione metabolism, and neuroactive ligand-receptor interaction (Figure 4A). Notably, follicle selection is a critical step in oocyte maturation. We identified 25 functional pathways in the LO stage (FDR < 0.05), including glycolysis/gluconeogenesis, pentose phosphate pathway, biosynthesis of amino acids, apoptosis, and cellular senescence (Figure 4A). In addition, the nicotine addiction pathway and neuroactive ligand-receptor interaction were overrepresented in the MO stage. These intra-follicular metabolic signaling pathways likely mediate the transition from the medium to the large antral follicular stage, which involves the coordinated interaction of oocytes and granulosa cells for follicular development. Finally, the greatest increase in the number of signaling pathways occurred during the transition of oocytes from small to large antral follicles. Overrepresented pathways in the LO stage included ribosome, oxidative phosphorylation, aldosterone-regulated sodium reabsorption, and thermogenesis (Figure 4A).
To better understand whether these signaling pathways were enriched in sheep oocytes at specific antral follicle stages, we merged or overlapped the results described above to identify pathways that are specific or homogeneous during antral follicular development (Figure 4B). Ribosome genes were expressed predominantly in the SO stage with high expression of several ribosomal protein genes (representative genes RPL24, RPL27A, RPL36AL, and RPS12). Interestingly, the neuroactive ligand-receptor interaction signaling pathway (representative genes CRHR1 and OPRD1) was highly expressed in the MO stage. In addition, we identified 15 co-expressed signaling pathways at the LO stage (Figure 4C), and, among these, biosynthesis of amino acids and various types of N-glycan, arginine and proline metabolism, and protein processing in the endoplasmic reticulum play a key role in oocyte maturation. Because of the critical role of glycans in protein modification and the regulation of diverse cellular functions [33], we further analyzed the N-glycan biosynthesis pathway and found 12 highly expressed genes, including MGAT4B [34], ALG3 [35], and DDOST [36], suggesting that N-glycosylation was one of the key factors affecting sheep oocyte maturation.
2.5. Expression patterns of oocyte-specific genes at antral follicle developmental stages
We integrated our sheep oocyte transcriptomes with the human and mouse transcriptomes [37] to characterize the global expression profiles of oocyte-specific genes. To study the expression patterns of meiosis-related genes in oocytes, we obtained 38 genes involved in female meiosis I and nine genes involved in female meiosis II from the Human Protein Atlas (HPA) database and found 23 meiosis I genes and six meiosis II genes (FDR < 0.05) that were differentially expressed across different stages of oocyte maturation (Table S5). Based on the expression patterns, unsupervised cluster analysis identified 14 clusters of genes (Figure 5A). Most of these genes (including ESPL1, PSMC31P, PLK1, HSF2BP, MEIKIN, and UBB) showed upregulation as antral follicular development proceeds, with the highest expression at the large antral stage. Analysis of 44 genes from the Mouse Genome Informatics (MGI) and HPA databases involved in oocyte maturation revealed that some of these genes (including GRB14, TUT4, SIRT2, INSL3, FOXO3, and BMP15) increased gradually during oocyte development whereas others (such as REC8, FBXO5, WEE2, AURKA, and H3F3A) were expressed at high levels at the medium antral follicle stage (Figure 5B and Table S6). Among 62 maternal-effect genes, which play a critical role in mammalian development before zygote genome activation [38], 38 genes were expressed during antral follicle development (Table S7). The genes in cluster 5 (IHH, MBD2, LDOC11, and NLRP5), cluster 7 (ANGPT2 and KPNA6), cluster 8 (WT1), and cluster 11 (GET1, SDHD, and MELK) were upregulated during follicular maturation (Figure 5C). However, the genes in cluster 6 (HAVCR2 and PHLDA2), cluster 10 (PRDX3), cluster 12 (RGS2), and cluster 13 (BTG4) were expressed before follicle selection. Genes OOEP, AGRP, ZAR1, GRB10, UBE3A, LNPEP, NR2F2, BMPR2, and BICC1 were underexpressed in oocytes at all stages of follicular development. Among the maternal-to-zygotic transition genes, BTG4 and PABPN1L were relatively highly expressed, increasing in abundance during oocyte development (Table S8).
2.6. Identification of oocyte-secreted factors throughout antral follicle development
We identified 38 genes encoding sheep oocyte secretory proteins that were differentially expressed during antral follicular development (Table S9). The expression patterns of the genes from each cluster and their functions are shown in Figure 6A and 6B. Cluster 1 comprised nine genes expressed only in the LO stage, suggesting their involvement in the subsequent developmental competence of oocytes. Cluster 2 included nine genes expressed only in the MO stage, suggesting their involvement in the initiation of follicle selection. Cluster 3 consisted of nine genes that represented follicle recruitment. Protein-protein interaction (PPI) analysis of the oocyte-specific protein-coding genes showed that the PPI network was reconstructed from 18 gene symbols (Table S10). Among these, secretory factors GDF9, BMP15, NLRP5, ZP2, ZAR1, and Rec8 formed the core of the PPI network (Figure 6C, D; Table S11). These genes were related to oocyte development and were involved in progesterone-mediated oocyte maturation, the HIF-1 signaling pathway, and cytokine-cytokine receptor interactions. These oocyte-specific secretory factors reveal the diverse functions of oocytes in antral follicle development. Two important oocyte-secreted factorsare growth differentiation factor 9 (GDF9) and bone morphogenetic protein 15 (BMP15). They activate signaling pathways in cumulus cells (CCs) that regulate the differentiation and the distinctive phenotype of CCs. Also, the interaction of GDF9 and BMP15 with follicle-stimulating hormone (FSH) potentiates the expression of IGF-2 in human granulosa cells [39]. Nlrp5 is a maternal lethal effect gene in mammals, and loss of NLRP5 in mouse oocytes triggers premature activation of the mitochondrial pool, causing mitochondrial damage and apoptosis [40]. The zona pellucida surrounding all mammalian oocytes plays a vital role in fertilization and early development. Knockout of zona pellucida protein ZP2 in female mice produces oocytes without a ZP, resulting in fertilization failure and infertility [41]. Zygote arrest 1 (ZAR1) is the first oocyte-specific maternal-effect gene known to function in the oocyte-to-embryo transition [42]. Rec8-containing cohesin, bound to Smc3/Smc1a or Smc3/Smc1b, maintains bivalent cohesion in mammalian meiosis [43]. Sister chromatid cohesion mediated by the cohesin complex is essential for chromosome segregation in mitosis and meiosis [44]. Here, we found that the REC8 transcript decreased in oocytes from large antral follicles, which may indicate oocyte meiotic resumption. In addition, Wee1-like protein kinase 2 (WEE2), a key regulator of meiosis during prophase I, is involved in maintaining meiotic arrest [45]. Further evaluation of the expression levels of OSFs in the follicular fluid may identify novel biomarkers for assessing oocyte quality.
2.7. TF regulatory networks in the oocytes
To construct regulatory networks of TFs for the SO and LO stages, we obtained all 1,376 sheep TFs from the Animal Transcription Factor Database (TFDB v4.0), a comprehensive database for predicting and classifying genome-wide TFs. During the development from the small to large antral stages, we found 73 differentially expressed TF genes (Figure 7A); 63 TF genes were highly expressed in the LO stage, and the others were highly expressed in the SO stage. This suggests that these TFs play a critical role in the follicular cavity and oocyte maturation. During the transition from the small to the medium antral stage, 24 TF genes were upregulated, and five TF genes (NOTO, NR1I3, LMX1B, SOX2, and SNAPC4) were downregulated (Figure 7B). Genes for 37 TFs were overexpressed in the LO stage vs. the MO stage and, thus, are candidate regulators of the medium to large antral stage transition. Genes MIER3, PAX7, HELT, PPARD, and FOXJ2 were more highly expressed in the MO stage (Figure 7C). To identify the master regulators of sheep antral follicle development, we overlapped DEGs for TFs at different stages (Figure 7D). A large number of TFs were expressed only in the LO stage, indicating possible regulatory roles in the cytoplasmic and nuclear maturation of oocytes (Figure 7E). Searching for recurring instances of neighboring TFs with STRING provided a predicted PPI network for 50 TFs as hub genes for the LO stage. The cellular tumor antigen p53 (TP53) TF, along with Kruppel-like factor 6 (KLF6) and Jun proto-oncogene (JUN), are hub genes for the positive regulation of RNA polymerase genes (Figure 7F and Figure S3A; Table S12). Among 21 TFs that were co-expressed the MO and LO stages (Figure 7G and Figure S3B; Table S13), the gene for mothers against decapentaplegic homolog (SMAD4), the central mediator of TGF-β signaling, is indispensable for oocyte formation and development [46].
Next, we speculate that L3MBTL3, MYRF, SALL1, USF3, NOTO, NR1I3, LMX1B, and ZNF200 most likely initiate the transcription network in small antral follicles, whereas DLX4, ZNF169, and TBX15 could be the unique regulators of follicle selection. To determine the potential roles of TFs in regulating the expression of DEGs, a TF-gene regulatory network was established based on predicted stage-specific target genes. In the SO stage, key TFs (MYRF, SALL1, and LMX1B) showed a significant correlation with multiple DEGs (Figure 7H). Alkylglycerone phosphate synthase (AGPS), nuclear import carrier for Hsp70s (HIKESHI), and ectodermal-neural cortex 1(ENC1) encode proteins that promote oocyte stability through lipid metabolism [47], protein transport [48], and the cytoskeleton[49], respectively. In addition, we identified significant TF-target gene pairs ZNF169-ITGB3BP, ZNP169-SNRPD2, DLX4-ITGB3BP, and DLX4-SNRPD2 that may play a role in mRNA processing at the medium antral follicle stage (Figure 7I). Further characterization of these TFs will provide insights into the transcriptional control of oocyte-induced antral follicle development.
3. Discussion
Understanding the mechanisms underlying oocyte maturation is essential for understanding reproductive biology. From the GV stage, oocytes reduce transcription and rely on stored mRNA and the translational machinery to control gene expression [50]. Thus, to understand oocyte maturation, it is important to determine the transcriptional profile of genes that regulate this process. In this study, we removed the oocytes from single follicles and generated scRNA-seq data from oocytes at the small, medium, and large antral follicle stages for a comprehensive assessment of gene expression. We identified DEGs by pairwise comparisons of the three stages and found that the LO stage had more specifically expressed genes than the SO and MO stages. The oocyte is transcriptionally active when the antral follicle first forms, but stored transcripts are critical for the initial stages of embryonic development until transcription is activated. A total of 565 DEGs were found in the MO stage vs. the SO stage, representing protein transport, glycolytic processes, and regulation of mRNA stability. Circulating FSH supports the growth of a group of antral follicles until the largest follicle produces estradiol, which occurs in sheep when the largest follicle is ~6 mm. GO analysis revealed that the biological processes that were enriched in the LO vs. the MO stage included regulation of transcription, positive regulation of fibroblast proliferation, and response to hypoxia, which may indicate intrinsic differences in these follicle stages. These signature genes may serve as cell-specific markers for each follicle stage. Also, EFHD2 and FBXL14 genes in oocytes from the SO stage, CRHR1 and CACNG3 in oocytes from the MO stage, and the top 15 DEGs in oocytes from the LO stage could serve as markers of oocyte maturation and developmental competency and may provide essential genetic tools for stage-specific labeling.
Although the transition from the small to the large antral is a key step in oocyte development, the molecular mechanisms and signaling pathways that drive this transition are unknown. Here, we identified signaling pathways associated with oocyte development and maturation based on the expression patterns of maternal transcripts and genes for key pathways. We found that ribosome biogenesis was regulated in the small antral follicle stage, producing sufficient ribosomes to support early embryo development until the initiation of zygotic transcription [51]. Thus, the oocyte translation program plays a critical role in the preparation for oocyte development. Oocyte-granulosa cell communication, mediated by several ligand-receptor pairs, is essential for oocyte development [52], and ligand-receptor pairs transmit signals between granulosa cells and oocytes to coordinate oocyte-granulosa cell functions (e.g., regulation of transcription, translation, apoptosis, cell differentiation, and transport). We found that signaling between the oocyte and CCs is regulated by the neuroactive ligand-receptor interactions pathway. Moreover, the key active pathways in large antral follicles are primarily in protein synthesis and glucose metabolism, although we also found that the thyroid hormone signaling pathway was upregulated in the oocytes from large antral follicles. Interestingly, thyroid hormones may directly affect oocytes, as specific binding sites for thyroxin are found in mouse and human oocytes. Thyroid hormone status may also affect the estrogenic sensitivity of the ovary [53], which is required for follicular dominance. Our results indicate that regulation of mRNA translation is important in follicle recruitment and that the neuroactive ligand-receptor interaction pathway is critical for the activation of oocyte maturation.
Oocyte quality determines the ability of the oocyte to overcome meiotic arrest and progress to the MII stage [54]. Thus, we identified the gene expression signatures for meiosis-related and oocyte maturation-associated genes and found that MLH1 and AURKA, which are associated with spindle building and chromosome segregation [55], were among the genes highly expressed in medium antral follicles. Lodde et al. [56] demonstrated a temporal relationship between chromatin remodeling and the main morpho-functional events that characterize the final growth phase in bovine oocytes. MEIOC, together with YTHDC2, promotes a meiotic cell cycle program via post-transcriptional control [57]. In addition, changes in the expression of maternal-effect genes revealed different hub genes at each oocyte stage. The maternal-effect genes, which are the mRNAs produced and stored in the cytoplasm of growing oocytes [58], are the sole substrates for protein synthesis after transcription is silenced in oocytes. The transcript levels of ANGPT2, KPNA6, CBS, CRHBP, and BLCAP decreased sharply with antral follicle development. CBS is required for the acetylation of α-tubulin for proper spindle assembly during meiotic maturation, which affects oocyte quality [59]. Here, we provide evidence that meiotic and developmental competence is acquired gradually during the antral follicular process. This involves sequential events regulated by maternal transcripts within the oocyte, including transcriptional regulation, spindle assembly, and chromosomal recombination. Our high-quality data reveal extensive changes in gene expression associated with oocyte development at the single-cell level.
OSFs are critical for the regulation of oocyte maturation and follicular development, and the regulation of the ovarian follicular microenvironment through OSFs may be important for oocyte quality. Two oocyte-specific growth factors, growth differentiation factor 9 (GDF9) and bone morphogenetic protein 15 (BMP15), are critical players in the follicular development of sheep [60]. Interestingly, we found expression of the gene for natriuretic peptide receptor (NPR2) in the MO stage. NPR2 in sheep, unlike in mice and pigs, is not only in CCs but also in oocyte membranes, indicating that C-type natriuretic peptide (CNP) may activate intra-oocyte cGMP production via the membrane NPR2 in addition to the CC-mediated pathway [61]. In meiosis, Aurora kinase A (AURKA) regulates spindle building and chromosome segregation in oocytes [62], whereas thyroid hormone receptor interactor 13 (TRIP13), which is involved in recombination, is required early in meiotic recombination [63]. We showed here that meiotic resumption is activated at the medium antral follicle stage, whereas chromosome alignment and segregation occur at the large antral follicle stage, suggesting that oocytes from large antral follicles are competent to resume meiosis. These secretory protein-coding genes might serve as biomarkers for certain outcomes of interest.
TFs, which regulate mammalian development, bind to the regulatory regions of genes, forming a complex system that guides the expression of the genome [64]. We found expression of key TFs during antral follicle development, including Homeobox, helix-loop-helix (bHLH), and zf-C2H2, which are essential for transcriptional regulation of developmental processes [65,66]. We also determined that LMX1B, MYRF, and SALL1 play critical roles in the small antral follicle stage, whereas ZNF169 and DLX4 are critical in regulating follicle selection. TP53 and KLF6 serve as regulatory hubs for many TFs in large antral follicles. We found that the regulation of TFs is strongly associated with the follicle development stages, and we characterized the interactions between TFs and their target genes to identify the transcriptional regulators of DEGs. The functions of these TFs in oocyte development are only partially known; thus, understanding the role of these TF-DEG pairs in developmental regulation and the underlying molecular mechanisms requires further oocyte studies.
Our work revealed unique features in the transcriptional machinery and gene signatures of sheep oocytes at different antral follicular stages. These oocyte-specific genes and TFs may provide valuable clues for future functional studies. First, we identified the gene expression profiles for three antral follicle stages of oocytes and the functions of stage-specific transcribed genes. Second, we identified key regulatory networks and signaling pathways associated with oocyte development, including those for ribosome biogenesis and a neuroactive ligand receptor. Third, we identified oocyte-specific changes in gene expression that elucidated the regulation of oocyte maturation by maternal transcripts at different stages. Fourth, we identified subsets of oocyte-specific secretory factors that might serve as biomarkers for specific outcomes of interest. Fifth, we identified stage-specific transcriptional regulators and constructed a regulatory network of core transcriptional regulators and their target genes for the stages of antral follicle development. Together, these discoveries provide key information on oocyte development and broaden our understanding of sheep folliculogenesis.
4. Materials and Methods
4.1. Ovarian stimulation and oocyte collection
A total of 12 healthy adult Chinese Hu sheep (1.5–2.5-year-old), procured in April 2023 from the Inner Mongolia JinLai Animal Husbandry Technology Co., Ltd. (Hohhot, Inner Mongolia), were subjected to hormonal treatment as described by Bragança [67]. The animals received a progestogen-releasing device for 6 d before the initiation of superovulation by FSH soon after normal ovulation, i.e., during the emergence of the first follicular wave. We synchronized estrus in all ewes using the intravaginal progesterone device (CIDR, Pharmacia & Upjohn, Hartwell, Australia) for 6 days. One day prior to the removal of the CIDR, intramuscular injections of 300 IU eCG (Sansheng, Ningbo, China) and 0.12 mg cloprostenol sodium (Sansheng, Ningbo, China) were administered, and then 36 h after removing the CIDR 0.025 mg lecirelin (Sansheng, Ningbo, China) was administered. On the day of ovarian stimulation initiation (D0), each ewe was treated with a new intravaginal progesterone device (CIDR). Thereafter, the ewes received 360 IU of FSH (Sansheng, Ningbo, China) in six decreasing doses (75, 75, 60, 60, 45, and 45 IU) every 12 h (Figure 1A).
Oocyte retrieval was performed as described previously [68]. Briefly, cumulus-oocyte complexes (COCs) were recovered from each donor and classified according to antral follicle size: small (< 3 mm; representing early antral follicle stage), medium (3–6 mm, indicative of the follicle selection stage), and large (˃ 6 mm; indicating the follicular dominance stage). The oocytes and GCs were mechanically stripped by pipet several times. Subsequently, the naked oocytes were collected from small, medium, and large follicles and were labeled as small oocytes (SO), medium oocytes (MO), and large oocytes (LO). The oocytes from each stage were separated into different samples (three samples for each stage, SO1, SO2, SO3; MO1, MO2; MO3; LO1, LO2, LO3; 10–20 oocytes/each sample) and total RNA was extracted and purified as described previously [69].
4.2. Single-cell RNA sequencing and data analysis
Single-cell full-length cDNA libraries using Smart-seq2 were prepared as previously described [70] using an Illumina NovaseqTM 6000 sequencing platform (Illumina, San Diego, CA, USA). Paired-end reads were processed using an RNA-seq pipeline. The pipeline first performed quality control on the raw reads using Cutadapt (v1.9) and FastQC (v0.11.9), then aligned the reads to the ovine genome sequence (Ovisaries.Oarv3.1) using the HISAT2 (v2.21) package. An average of 92.5% of the reads per sample were mapped to the reference genome. Next, the mapped reads of each sample were assembled by StringTie (v2.1.6) using the default parameters. All the transcriptomes from all samples were merged to reconstruct a comprehensive transcriptome using gffcompare software (v0.9.8). For the final transcriptome, StringTie and Ballgown were used to estimate the expression levels of all transcripts, and the abundance of the mRNAs was calculated from the fragment per kilobase mapped reads (FPKM).
4.3. PCA and identification of DEGs
We applied the Seurat method to analyze the single-cell data to create the whole clustering profile [71]. Only highly variable marker genes (coefficient of variation > 0.5) were used as inputs for PCA. The marker genes in the PCA plots were plotted by the FeaturePlot function in the Seurat package (v4.0.3). For better accuracy, we complemented the PCA oocyte clustering separately using the FactoMineR (v2.8) package in R (v4.1.0). The multiple t-test in the DESeq2 software was used to obtain the statistical significance of DEGs in each stage (one stage versus all other stages). Only genes with significant p-values, an FDR < 0.05, and a log2FC transformed FPKM (fragments per kilobase of exon per million fragments mapped) larger than |2| were considered to be DEGs. The Enhanced Volcano R package was used to visualize the significance of each gene based on its log2FC, and the top-ranked 20 markers for each development stage were plotted.
To identify unique gene expression functions and regulatory pathways of oocytes during antral follicle development, we performed GO and KEGG enrichment analysis using the DEGs generated by the R package ClusterProfiler [72]. Gene numbers for terms in GO and KEGG were calculated, and a hyper-geometric test was used to find significantly enriched GO terms (biological process) and KEGG pathways. We defined a term or pathway with an FDR < 0.05 as significantly enriched.
4.4. Identification of stage-specific DEGs
Only genes with (log2FC) greater than |2|, an FDR < 0.05, and expressed in both groups were considered as stage-specific DEGs. GO analysis of stage-specific DEGs was performed with ClusterProfiler. Next, we analyzed stage-specific processes. GO enrichment items for the three samples were obtained based on GSEA (v4.1.0). We selected GSEA terms with p-values < 0.05 and listed the top-ranked 10 terms based on the normalized p-value enrichment score. We used GSEA and the KEGG pathway enrichment analysis to identify the significantly enriched pathways in oocytes in the transition from medium to large antral stages. The gene sets with FDR < 0.05 were considered to be enriched.
4.5. Identifying expression patterns of oocyte-specific genes from DEGs
To characterize the expression patterns of oocyte-specific genes and identify candidate markers for enhancing oocyte developmental competence in vitro, we focused on oocyte-specific functions. The meiosis-related genes and maternal-effect genes were downloaded from the HPA database (https://www.proteinatlas.org/). The genes involved in oocyte maturation were downloaded from the MGI database (https://www.informatics.jax.org/) and incorporated into the analysis. Sheep oocyte DEGs were overlapped with genes expressed in human or mouse oocytes to find sheep oocyte candidate genes. The average FPKM value of genes at each time point was imported into STEM software (v1.3.13; School of Computer Science, Carnegie Mellon University) to generate gene expression clusters.
4.6. Identifying OSFs at sequential antral follicle developmental stages
To identify candidate biomarkers for predicting oocyte development, we focused on 103 genes encoding oocyte-derived secretory proteins that we downloaded from the HPA database and selected those that overlapped the 9,400 sheep oocyte-secreted protein-coding DEGs. These candidate genes were analyzed using the online Search Tool for Recurring Instances of Neighboring Genes (STRING, https://cn.string-db.org/) to construct a PPI network. We set the cut-off as a combined score > 0.4 based on the number of associations with other genes in the PPI network and visualized the results using CytoScape software (v3.9.1).
4.7. Construction of transcriptional regulatory factor network in oocytes
We identified SO vs. LO stage DEGs (log2(FC) > |2| and FDR < 0.05) and selected those that overlapped with 1,376 sheep TFs (AnimalTFDB, v4.0) as candidates TF to construct the network of transcriptional regulators and TF families. To identify the TFs associated with specific biological processes, we used STRING to visualize PPI interaction networks for key TFs. The active interaction sources for our search criteria were limited to Experiments, Databases, Co-expression, Neighborhood, Gene Fusion, and Co-occurrence, with a minimum interaction score set to the medium confidence value of 0.400. We also characterized the TF regulatory network and their differential target genes at different antral follicle stages. We used the Gene Transcription Regulation Database (GTRD) [73] and the Human Transcription Factor Targets Database (hTFtarget) [74] to find possible targets (within the 9,400 DEGs) that could bind the stage-specific TFs. Finally, the TF-target regulatory network was visualized using CytoScape software.
4.8. Statistical analysis
The experimental data were analyzed statistically using a two-tailed t-test to compare differences between different stages with PRISM software (GraphPad 6 Software). An FDR < 0.05 was considered statistically significant. The boxplot represents the median, the first quartile, and the third quartile values.
5. Conclusions
In summary, our results highlight the complexity and dynamic nature of oocyte development. We identified stage-specific genes, OSFs, and transcriptional regulators that contribute to the functional and morphological changes that occur during oocyte maturation. Overall, these findings provide insights into the molecular mechanisms that regulate oocyte maturation, with important implications for improving assisted reproductive technologies in domestic animals.
Supplementary Materials
The following are available online at www.mdpi.com/xxx/s1.
Author Contributions
Conceptualization, Jiaxin Zhang and Yukun Song; methodology, Jiaxin Zhang and Qi Lv; formal analysis, Yukun Song and Nan Zhang.; data curation, Yukun Song, Nan Zhang, Yu Zhang, and Junlan Wang; writing—original draft preparation, Yukun Song; writing—review and editing, Jiaxin Zhang and Qi Lv; visualization, Nan Zhang and Yu Zhang; supervision, Jiaxin Zhang and Qi Lv; project administration, Jiaxin Zhang; funding acquisition, Jiaxin Zhang. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants from the National Key R&D Program of China (grant numbers 2021YFD1300901), the Natural Science Foundation of Inner Mongolia Autonomous Region (grant numbers 2023MS03001), and the 12th "Grassland Talents" High-level Talent Training Project.
Institutional Review Board Statement
The protocols using animals were approved by the Ethics Committee of Experimental Animals of Inner Mongolia Agricultural University, Inner Mongolia Autonomous Region, China (No. XMXK20180016).
Acknowledgments
The authors are grateful to the staff of Inner Mongolia Jinlai Animal Husbandry Technology Co., Ltd for providing assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gong, X.; Zhang, Y.; Ai, J.; Li, K. , Application of Single-Cell RNA Sequencing in Ovarian Development. Biomolecules 2022, 13, (1), 47. [Google Scholar] [CrossRef] [PubMed]
- Jozkowiak, M.; Hutchings, G.; Jankowski, M.; Kulcenty, K.; Mozdziak, P.; Kempisty, B.; Spaczynski, R. Z.; Piotrowska-Kempisty, H. , The Stemness of Human Ovarian Granulosa Cells and the Role of Resveratrol in the Differentiation of MSCs-A Review Based on Cellular and Molecular Knowledge. Cells 2020, 9, (6), 1418. [Google Scholar] [CrossRef]
- Eppig, J. J.; Wigglesworth, K.; Pendola, F. L. , The mammalian oocyte orchestrates the rate of ovarian follicular development. Proc Natl Acad Sci U S A 2002, 99, (5), 2890–2894. [Google Scholar] [CrossRef]
- Wang, S.; Gong, Y.; Wang, Z.; Greenbaum, J.; Xiao, H. M.; Deng, H. W. , Cell-specific network analysis of human folliculogenesis reveals network rewiring in antral stage oocytes. J Cell Mol Med 2021, 25, (6), 2851–2860. [Google Scholar] [CrossRef]
- Evans, A. C. , Ovarian follicle growth and consequences for fertility in sheep. Anim Reprod Sci 2003, 78, (3–4), 289-306. [Google Scholar] [CrossRef] [PubMed]
- Bartlewski, P. M.; Beard, A. P.; Rawlings, N. C. , An ultrasound-aided study of temporal relationships between the patterns of LH/FSH secretion, development of ovulatory-sized antral follicles and formation of corpora lutea in ewes. Theriogenology 2000, 54, (2), 229–245. [Google Scholar] [CrossRef]
- Keefe, D.; Kumar, M.; Kalmbach, K. , Oocyte competency is the key to embryo potential. Fertil Steril 2015, 103, (2), 317–322. [Google Scholar] [CrossRef]
- Leroy, J. L.; Valckx, S. D.; Jordaens, L.; De Bie, J.; Desmet, K. L.; Van Hoeck, V.; Britt, J. H.; Marei, W. F.; Bols, P. E. , Nutrition and maternal metabolic health in relation to oocyte and embryo quality: critical views on what we learned from the dairy cow model. Reprod Fertil Dev 2015, 27, (4), 693–703. [Google Scholar] [CrossRef]
- Lequarre, A. S.; Vigneron, C.; Ribaucour, F.; Holm, P.; Donnay, I.; Dalbiès-Tran, R.; Callesen, H.; Mermillod, P. , Influence of antral follicle size on oocyte characteristics and embryo development in the bovine. Theriogenology 2005, 63, (3), 841–859. [Google Scholar] [CrossRef]
- Ntostis, P.; Iles, D.; Kokkali, G.; Vaxevanoglou, T.; Kanavakis, E.; Pantou, A.; Huntriss, J.; Pantos, K.; Picton, H. M. , The impact of maternal age on gene expression during the GV to MII transition in euploid human oocytes. Hum Reprod 2021, 37, (1), 80–92. [Google Scholar] [CrossRef]
- Sorensen, R. A.; Wassarman, P. M. , Relationship between growth and meiotic maturation of the mouse oocyte. Dev Biol 1976, 50, (2), 531–536. [Google Scholar] [CrossRef]
- Eppig, J. J.; Schultz, R. M.; O'Brien, M.; Chesnel, F. , Relationship between the developmental programs controlling nuclear and cytoplasmic maturation of mouse oocytes. Dev Biol 1994, 164, (1), 1–9. [Google Scholar] [CrossRef]
- Kirillova, A.; Smitz, J. E. J.; Sukhikh, G. T.; Mazunin, I. , The Role of Mitochondria in Oocyte Maturation. Cells 2021, 10, (9), 2484. [Google Scholar] [CrossRef]
- Bernabé, B. P.; Woodruff, T.; Broadbelt, L. J.; Shea, L. D. , Ligands, Receptors, and Transcription Factors that Mediate Inter-Cellular and Intra-Cellular Communication during Ovarian Follicle Development. Reprod Sci 2020, 27, (2), 690–703. [Google Scholar] [CrossRef]
- Guo, Y.; Cai, L.; Liu, X.; Ma, L.; Zhang, H.; Wang, B.; Qi, Y.; Liu, J.; Diao, F.; Sha, J.; Guo, X. , Single-Cell Quantitative Proteomic Analysis of Human Oocyte Maturation Revealed High Heterogeneity in In Vitro-Matured Oocytes. Mol Cell Proteomics 2022, 21, (8), 100267. [Google Scholar] [CrossRef]
- Svensson, V.; Vento-Tormo, R.; Teichmann, S. A. , Exponential scaling of single-cell RNA-seq in the past decade. Nat Protoc 2018, 13, (4), 599–604. [Google Scholar] [CrossRef]
- Freour, T.; Vassena, R. , Transcriptomics analysis and human preimplantation development. J Proteomics 2017, 162, 135–140. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, Q.; Dai, J.; Cui, Y.; Zhang, C.; Wen, X.; Li, J.; Xiao, Y.; Peng, X.; Liu, M.; Shen, B.; Sha, J.; Hu, Z.; Li, J.; Shu, W. , Single-cell RNA-Seq reveals a highly coordinated transcriptional program in mouse germ cells during primordial follicle formation. Aging Cell 2021, 20, (7), e13424. [Google Scholar] [CrossRef]
- Wang, J. J.; Ge, W.; Zhai, Q. Y.; Liu, J. C.; Sun, X. W.; Liu, W. X.; Li, L.; Lei, C. Z.; Dyce, P. W.; De Felici, M.; Shen, W. , Single-cell transcriptome landscape of ovarian cells during primordial follicle assembly in mice. PLoS Biol 2020, 18, (12), e3001025. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, Z.; Qin, Q.; Nisenblat, V.; Chang, H. M.; Yu, Y.; Wang, T.; Lu, C.; Yang, M.; Yang, S.; Yao, Y.; Zhu, X.; Xia, X.; Dang, Y.; Ren, Y.; Yuan, P.; Li, R.; Liu, P.; Guo, H.; Han, J.; He, H.; Zhang, K.; Wang, Y.; Wu, Y.; Li, M.; Qiao, J.; Yan, J.; Yan, L. , Transcriptome Landscape of Human Folliculogenesis Reveals Oocyte and Granulosa Cell Interactions. Mol Cell 2018, 72, (6), 1021–1034.e4. [Google Scholar] [CrossRef]
- Llonch, S.; Barragán, M.; Nieto, P.; Mallol, A.; Elosua-Bayes, M.; Lorden, P.; Ruiz, S.; Zambelli, F.; Heyn, H.; Vassena, R.; Payer, B. , Single human oocyte transcriptome analysis reveals distinct maturation stage-dependent pathways impacted by age. Aging Cell 2021, 20, (5), e13360. [Google Scholar] [CrossRef]
- Hu, W.; Zeng, H.; Shi, Y.; Zhou, C.; Huang, J.; Jia, L.; Xu, S.; Feng, X.; Zeng, Y.; Xiong, T.; Huang, W.; Sun, P.; Chang, Y.; Li, T.; Fang, C.; Wu, K.; Cai, L.; Ni, W.; Li, Y.; Yang, Z.; Zhang, Q. C.; Chian, R.; Chen, Z.; Liang, X.; Kee, K. , Single-cell transcriptome and translatome dual-omics reveals potential mechanisms of human oocyte maturation. Nat Commun 2022, 13, (1), 5114. [Google Scholar] [CrossRef]
- Maside, C.; Sánchez-Ajofrín, I.; Medina-Chávez, D.; Alves, B.; Garde, J. J.; Soler, A. J. , Oocyte Morphometric Assessment and Gene Expression Profiling of Oocytes and Cumulus Cells as Biomarkers of Oocyte Competence in Sheep. Animals (Basel) 2021, 11, (10),2818. [Google Scholar] [CrossRef] [PubMed]
- Mroczek, S.; Chlebowska, J.; Kuliński, T. M.; Gewartowska, O.; Gruchota, J.; Cysewski, D.; Liudkovska, V.; Borsuk, E.; Nowis, D.; Dziembowski, A. , The non-canonical poly(A) polymerase FAM46C acts as an onco-suppressor in multiple myeloma. Nat Commun 2017, 8, (1), 619. [Google Scholar] [CrossRef]
- Nemethova, M.; Radvanszky, J.; Kadasi, L.; Ascher, D. B.; Pires, D. E.; Blundell, T. L.; Porfirio, B.; Mannoni, A.; Santucci, A.; Milucci, L.; Sestini, S.; Biolcati, G.; Sorge, F.; Aurizi, C.; Aquaron, R.; Alsbou, M.; Lourenço, C. M.; Ramadevi, K.; Ranganath, L. R.; Gallagher, J. A.; van Kan, C.; Hall, A. K.; Olsson, B.; Sireau, N.; Ayoob, H.; Timmis, O. G.; Sang, K. H.; Genovese, F.; Imrich, R.; Rovensky, J.; Srinivasaraghavan, R.; Bharadwaj, S. K.; Spiegel, R.; Zatkova, A. , Twelve novel HGD gene variants identified in 99 alkaptonuria patients: focus on 'black bone disease' in Italy. Eur J Hum Genet 2016, 24, (1), 66–72. [Google Scholar] [CrossRef]
- Kwon, M.; Lee, J. H.; Yoon, Y.; Pleasure, S. J.; Yoon, K. , The CRHR1/CREB/REST signaling cascade regulates mammalian embryonic neural stem cell properties. EMBO Rep 2023, 24, (2), e55313. [Google Scholar] [CrossRef]
- Kennedy, A.; Waters, E.; Rowshanravan, B.; Hinze, C.; Williams, C.; Janman, D.; Fox, T. A.; Booth, C.; Pesenacker, A. M.; Halliday, N.; Soskic, B.; Kaur, S.; Qureshi, O. S.; Morris, E. C.; Ikemizu, S.; Paluch, C.; Huo, J.; Davis, S. J.; Boucrot, E.; Walker, L. S. K.; Sansom, D. M. , Differences in CD80 and CD86 transendocytosis reveal CD86 as a key target for CTLA-4 immune regulation. Nat Immunol 2022, 23, (9), 1365–1378. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, R.; Cheng, Y.; Wu, X.; Xi, S.; Sun, Y.; Jiang, H. , Lidocaine inhibits the proliferation of lung cancer by regulating the expression of GOLT1A. Cell Prolif. 2017, 50, (5), e12364. [Google Scholar] [CrossRef]
- Genau, H. M.; Huber, J.; Baschieri, F.; Akutsu, M.; Dötsch, V.; Farhan, H.; Rogov, V.; Behrends, C. , CUL3-KBTBD6/KBTBD7 ubiquitin ligase cooperates with GABARAP proteins to spatially restrict TIAM1-RAC1 signaling. Mol Cell 2015, 57, (6), 995–1010. [Google Scholar] [CrossRef]
- Liu, Y. T.; Liu, F.; Cao, L.; Xue, L.; Gu, W. T.; Zheng, Y. Z.; Tang, H.; Wang, Y.; Yao, H.; Zhang, Y.; Xie, W. Q.; Ren, B. H.; Xiao, Z. H.; Nie, Y. J.; Hu, R.; Wu, Z. B. , The KBTBD6/7-DRD2 axis regulates pituitary adenoma sensitivity to dopamine agonist treatment. Acta Neuropathol 2020, 140, (3), 377–396. [Google Scholar] [CrossRef]
- Cui, Y. H.; Kim, H.; Lee, M.; Yi, J. M.; Kim, R. K.; Uddin, N.; Yoo, K. C.; Kang, J. H.; Choi, M. Y.; Cha, H. J.; Kwon, O. S.; Bae, I. H.; Kim, M. J.; Kaushik, N.; Lee, S. J. , FBXL14 abolishes breast cancer progression by targeting CDCP1 for proteasomal degradation. Oncogene 2018, 37, (43), 5794–5809. [Google Scholar] [CrossRef]
- Vega, I. E. , EFhd2, a Protein Linked to Alzheimer's Disease and Other Neurological Disorders. Front. Neurosci. 2016, 10, (2), 150. [Google Scholar] [CrossRef]
- Li, H.; You, L.; Tian, Y.; Guo, J.; Fang, X.; Zhou, C.; Shi, L.; Su, Y. Q. , DPAGT1-Mediated Protein N-Glycosylation Is Indispensable for Oocyte and Follicle Development in Mice. Adv Sci (Weinh) 2020, 7, (14), 2000531. [Google Scholar] [CrossRef]
- Tan, Z.; Lu, W.; Li, X.; Yang, G.; Guo, J.; Yu, H.; Li, Z.; Guan, F. , Altered N-Glycan expression profile in epithelial-to-mesenchymal transition of NMuMG cells revealed by an integrated strategy using mass spectrometry and glycogene and lectin microarray analysis. J Proteome Res 2014, 13, (6), 2783–2795. [Google Scholar] [CrossRef]
- Anyaogu, D. C.; Hansen, A. H.; Hoof, J. B.; Majewska, N. I.; Contesini, F. J.; Paul, J. T.; Nielsen, K. F.; Hobley, T. J.; Yang, S.; Zhang, H.; Betenbaugh, M.; Mortensen, U. H. , Glycoengineering of Aspergillus nidulans to produce precursors for humanized N-glycan structures. Metab Eng 2021, 67, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Elsharkawi, I.; Wongkittichote, P.; Daniel, E. J. P.; Starosta, R. T.; Ueda, K.; Ng, B. G.; Freeze, H. H.; He, M.; Shinawi, M. , DDOST-CDG: Clinical and molecular characterization of a third patient with a milder and a predominantly movement disorder phenotype. J Inherit Metab Dis 2023, 46, (1), 92–100. [Google Scholar] [CrossRef]
- Li, R.; Albertini, D. F. , The road to maturation: somatic cell interaction and self-organization of the mammalian oocyte. Nat Rev Mol Cell Biol 2013, 14, (3), 141–152. [Google Scholar] [CrossRef]
- Innocenti, F.; Fiorentino, G.; Cimadomo, D.; Soscia, D.; Garagna, S.; Rienzi, L.; Ubaldi, F. M.; Zuccotti, M. , Maternal effect factors that contribute to oocytes developmental competence: an update. J Assist Reprod Genet 2022, 39, (4), 861–871. [Google Scholar] [CrossRef]
- Hobeika, E.; Armouti, M.; Kala, H.; Fierro, M. A.; Winston, N. J.; Scoccia, B.; Zamah, A. M.; Stocco, C. , Oocyte-Secreted Factors Synergize With FSH to Promote Aromatase Expression in Primary Human Cumulus Cells. J Clin Endocrinol Metab 2019, 104, (5), 1667–1676. [Google Scholar] [CrossRef]
- Fernandes, R.; Tsuda, C.; Perumalsamy, A. L.; Naranian, T.; Chong, J.; Acton, B. M.; Tong, Z. B.; Nelson, L. M.; Jurisicova, A. , NLRP5 mediates mitochondrial function in mouse oocytes and embryos. Biol Reprod 2012, 86, (5), 138, 1–10. [Google Scholar] [CrossRef]
- Rankin, T. L.; O'Brien, M.; Lee, E.; Wigglesworth, K.; Eppig, J.; Dean, J. , Defective zonae pellucidae in Zp2-null mice disrupt folliculogenesis, fertility and development. Development 2001, 128, (7), 1119–1126. [Google Scholar] [CrossRef]
- Wu, X.; Viveiros, M. M.; Eppig, J. J.; Bai, Y.; Fitzpatrick, S. L.; Matzuk, M. M. , Zygote arrest 1 (Zar1) is a novel maternal-effect gene critical for the oocyte-to-embryo transition. Nat Genet 2003, 33, (2), 187–191. [Google Scholar] [CrossRef]
- Tachibana-Konwalski, K.; Godwin, J.; van der Weyden, L.; Champion, L.; Kudo, N. R.; Adams, D. J.; Nasmyth, K. , Rec8-containing cohesin maintains bivalents without turnover during the growing phase of mouse oocytes. Genes Dev 2010, 24, (22), 2505–2516. [Google Scholar] [CrossRef]
- Nasmyth, K.; Haering, C. H. , Cohesin: its roles and mechanisms. Annu Rev Genet 2009, 43, 525–558. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Chen, T.; Yu, M.; Bian, Y.; Cao, Y.; Ning, Y.; Su, S.; Zhang, J.; Zhao, S. , Novel WEE2 gene variants identified in patients with fertilization failure and female infertility. Fertil Steril 2019, 111, (3), 519–526. [Google Scholar] [CrossRef]
- Sun, R.; Li, M.; He, N.; Wen, X.; Zhang, J. , Molecular Characterization, Expression Profiles of SMAD4, SMAD5 and SMAD7 Genes and Lack of Association with Litter Size in Tibetan Sheep. Animals (Basel) 2022, 12, (17), 2232. [Google Scholar] [CrossRef]
- Wang, S.; Yang, H.; Fu, Y.; Teng, X.; Wang, C.; Xu, W. , The Key Role of Peroxisomes in Follicular Growth, Oocyte Maturation, Ovulation, and Steroid Biosynthesis. Oxid. Med. Cell. Longev. 2022, 2022, (4), 7982344. [Google Scholar] [CrossRef]
- Yang, C. X.; Song, Z. Q.; Pei, S.; Yu, X. X.; Miao, J. K.; Liang, H.; Miao, Y. L.; Du, Z. Q. , Single cell RNA-seq reveals molecular pathways altered by 7, 12-dimethylbenz[a]anthracene treatment on pig oocytes. Theriogenology 2020, 157, (1), 449–457. [Google Scholar] [CrossRef]
- Kim, S. G.; Jang, S. J.; Soh, J.; Lee, K.; Park, J. K.; Chang, W. K.; Park, E. W.; Chun, S. Y. , Expression of ectodermal neural cortex 1 and its association with actin during the ovulatory process in the rat. Endocrinology 2009, 150, (8), 3800–3806. [Google Scholar] [CrossRef]
- Conti, M.; Franciosi, F. , Acquisition of oocyte competence to develop as an embryo: integrated nuclear and cytoplasmic events. Hum Reprod Update 2018, 24, (3), 245–266. [Google Scholar] [CrossRef]
- Mercer, M.; Jang, S.; Ni, C.; Buszczak, M. , The Dynamic Regulation of mRNA Translation and Ribosome Biogenesis During Germ Cell Development and Reproductive Aging. Front Cell Dev Biol 2021, 9, 710186. [Google Scholar] [CrossRef] [PubMed]
- Biase, F. H.; Kimble, K. M. , Functional signaling and gene regulatory networks between the oocyte and the surrounding cumulus cells. BMC Genomics 2018, 19, (1), 351. [Google Scholar] [CrossRef] [PubMed]
- Rae, M. T.; Gubbay, O.; Kostogiannou, A.; Price, D.; Critchley, H. O.; Hillier, S. G. , Thyroid hormone signaling in human ovarian surface epithelial cells. J Clin Endocrinol Metab 2007, 92, (1), 322–327. [Google Scholar] [CrossRef]
- Sánchez, F.; Romero, S.; De Vos, M.; Verheyen, G.; Smitz, J. , Human cumulus-enclosed germinal vesicle oocytes from early antral follicles reveal heterogeneous cellular and molecular features associated with in vitro maturation capacity. Hum Reprod 2015, 30, (6), 1396–1409. [Google Scholar] [CrossRef]
- Singh, P.; Fragoza, R.; Blengini, C. S.; Tran, T. N.; Pannafino, G.; Al-Sweel, N.; Schimenti, K. J.; Schindler, K.; Alani, E. A.; Yu, H.; Schimenti, J. C. , Human MLH1/3 variants causing aneuploidy, pregnancy loss, and premature reproductive aging. Nat Commun 2021, 12, (1), 5005. [Google Scholar] [CrossRef]
- Lodde, V.; Modina, S.; Maddox-Hyttel, P.; Franciosi, F.; Lauria, A.; Luciano, A. M. , Oocyte morphology and transcriptional silencing in relation to chromatin remodeling during the final phases of bovine oocyte growth. Mol Reprod Dev 2008, 75, (5), 915–924. [Google Scholar] [CrossRef]
- Soh, Y. Q. S.; Mikedis, M. M.; Kojima, M.; Godfrey, A. K.; de Rooij, D. G.; Page, D. C. , Meioc maintains an extended meiotic prophase I in mice. PLoS Genet 2017, 13, (4), e1006704. [Google Scholar] [CrossRef] [PubMed]
- Tora, L.; Vincent, S. D. , What defines the maternal transcriptome? Biochem Soc Trans 2021, 49, (5), 2051–2062. [Google Scholar] [CrossRef]
- Cao, Y.; Zhu, X.; Zhen, P.; Tian, Y.; Ji, D.; Xue, K.; Yan, W.; Chai, J.; Liu, H.; Wang, W. , Cystathionine β-synthase is required for oocyte quality by ensuring proper meiotic spindle assembly. Cell Prolif 2022, 55, (12), e13322. [Google Scholar] [CrossRef]
- Kona, S. S.; Praveen Chakravarthi, V.; Siva Kumar, A. V.; Srividya, D.; Padmaja, K.; Rao, V. H. , Quantitative expression patterns of GDF9 and BMP15 genes in sheep ovarian follicles grown in vivo or cultured in vitro. Theriogenology 2016, 85, (2), 315–322. [Google Scholar] [CrossRef]
- Xi, G.; An, L.; Jia, Z.; Tan, K.; Zhang, J.; Wang, Z.; Zhang, C.; Miao, K.; Wu, Z.; Tian, J. , Natriuretic peptide receptor 2 (NPR2) localized in bovine oocyte underlies a unique mechanism for C-type natriuretic peptide (CNP)-induced meiotic arrest. Theriogenology 2018, 106, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Blengini, C. S.; Ibrahimian, P.; Vaskovicova, M.; Drutovic, D.; Solc, P.; Schindler, K. , Aurora kinase A is essential for meiosis in mouse oocytes. PLoS Genet 2021, 17, (4), e1009327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, B.; Fu, J.; Li, R.; Diao, F.; Li, C.; Chen, B.; Du, J.; Zhou, Z.; Mu, J.; Yan, Z.; Wu, L.; Liu, S.; Wang, W.; Zhao, L.; Dong, J.; He, L.; Liang, X.; Kuang, Y.; Sun, X.; Sang, Q.; Wang, L. , Bi-allelic Missense Pathogenic Variants in TRIP13 Cause Female Infertility Characterized by Oocyte Maturation Arrest. Am J Hum Genet 2020, 107, (1), 15–23. [Google Scholar] [CrossRef]
- Lambert, S. A.; Jolma, A.; Campitelli, L. F.; Das, P. K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T. R.; Weirauch, M. T. , The Human Transcription Factors. Cell 2018, 172, (4), 650–665. [Google Scholar] [CrossRef]
- Wu, S.; Tong, X.; Li, C.; Lu, K.; Tan, D.; Hu, H.; Liu, H.; Dai, F. , Genome-wide identification and expression profiling of the C2H2-type zinc finger protein genes in the silkworm Bombyx mori. PeerJ 2019, 7, e7222. [Google Scholar] [CrossRef] [PubMed]
- Torres-Machorro, A. L. , Homodimeric and Heterodimeric Interactions among Vertebrate Basic Helix-Loop-Helix Transcription Factors. Int. J. Mol. Sci. 2021, 22, (23), 12855. [Google Scholar] [CrossRef]
- Bragança, G. M.; Souza-Fabjan, J. M. G.; Ribeiro, L. S.; Brair, V. L.; Côrtes, L. R.; Souza, C. V.; Batista, R.; Fonseca, J. F.; Menchaca, A.; Brandão, F. Z. , Exogenous progestogen does not affect first-wave follicle populations and oocyte quality during ovarian stimulation with FSH in sheep. Domest Anim Endocrinol 2020, 72, 106369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Fan, X.; Li, R.; Zhang, C.; Zhang, J. , Effects of pre-incubation with C-type natriuretic peptide on nuclear maturation, mitochondrial behavior, and developmental competence of sheep oocytes. Biochem Biophys Res Commun 2018, 497, (1), 200–206. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, P.; Ge, W.; Feng, Y.; Li, L.; Sun, Z.; Zhang, H.; Shen, W. , Alginate oligosaccharides improve germ cell development and testicular microenvironment to rescue busulfan disrupted spermatogenesis. Theranostics 2020, 10, (7), 3308–3324. [Google Scholar] [CrossRef]
- Picelli, S.; Björklund Å, K.; Faridani, O. R.; Sagasser, S.; Winberg, G.; Sandberg, R. , Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 2013, 10, (11), 1096–1098. [Google Scholar] [CrossRef]
- Macosko, E. Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A. R.; Kamitaki, N.; Martersteck, E. M.; Trombetta, J. J.; Weitz, D. A.; Sanes, J. R.; Shalek, A. K.; Regev, A.; McCarroll, S. A. , Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, (5), 1202–1214. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; Fu, X.; Liu, S.; Bo, X.; Yu, G. , clusterProfiler 4. 0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2021, 2, (3), 100141. [Google Scholar]
- Yevshin, I.; Sharipov, R.; Kolmykov, S.; Kondrakhin, Y.; Kolpakov, F. , GTRD: a database on gene transcription regulation-2019 update. Nucleic Acids Res 2019, 47, (D1), D100–D105. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, W.; Zhang, H. M.; Xie, G. Y.; Miao, Y. R.; Xia, M.; Guo, A. Y. , hTFtarget: A Comprehensive Database for Regulations of Human Transcription Factors and Their Targets. Genomics Proteomics Bioinformatics 2020, 18, (2), 120–128. [Google Scholar] [CrossRef]
Figure 1.
Single-cell RNA-seq transcriptome patterns in sheep oocytes:(A) Estrus synchronization and superovulation treatment schedule. On day 0, an intravaginal progesterone device was inserted, and ovarian stimulation was performed using multiple decreasing doses of FSH. PGF2α, prostaglandin F2α; eCG, equine chorionic gonadotropin; GnRH, gonadotrophin-releasing hormone; FSH, follicle-stimulating hormone; LOPU, laparoscopic ovum pick-up; (B) Flowchart for sheep oocyte scRNA-seq. Cumulus-oocyte complexes (COCs) were recovered from each sheep and classified according to antral follicle size: small (≤ 3 mm), medium (3–6 mm), and large (≥ 6 mm). The oocytes and granulosa cells (GCs) were stripped mechanically, and oocyte transcriptomes were compared by single-cell RNA sequencing analysis (Smart-seq2); (C)Representative image of sheep ovaries (a) with or (b) without stimulation treatment. Small, medium, and large antral follicles are depicted in yellow, blue, green, and circles, respectively, whereas red circles highlight the corpus luteum; (D)Heatmap with color highlighting the distance matrix (Pearson correlation). The color gradient ranges from dark red (minimum distance) to light red. Horizontal bars represent the antral follicle phase. LO, oocyte from large antral follicle; MO, oocyte from medium antral follicle; SO, oocyte from small antral follicle; (E) Principal component analysis (PCA) of the transcriptomes from all oocytes. Purple indicates SO, blue indicates MO, and green indicates LO.
Figure 1.
Single-cell RNA-seq transcriptome patterns in sheep oocytes:(A) Estrus synchronization and superovulation treatment schedule. On day 0, an intravaginal progesterone device was inserted, and ovarian stimulation was performed using multiple decreasing doses of FSH. PGF2α, prostaglandin F2α; eCG, equine chorionic gonadotropin; GnRH, gonadotrophin-releasing hormone; FSH, follicle-stimulating hormone; LOPU, laparoscopic ovum pick-up; (B) Flowchart for sheep oocyte scRNA-seq. Cumulus-oocyte complexes (COCs) were recovered from each sheep and classified according to antral follicle size: small (≤ 3 mm), medium (3–6 mm), and large (≥ 6 mm). The oocytes and granulosa cells (GCs) were stripped mechanically, and oocyte transcriptomes were compared by single-cell RNA sequencing analysis (Smart-seq2); (C)Representative image of sheep ovaries (a) with or (b) without stimulation treatment. Small, medium, and large antral follicles are depicted in yellow, blue, green, and circles, respectively, whereas red circles highlight the corpus luteum; (D)Heatmap with color highlighting the distance matrix (Pearson correlation). The color gradient ranges from dark red (minimum distance) to light red. Horizontal bars represent the antral follicle phase. LO, oocyte from large antral follicle; MO, oocyte from medium antral follicle; SO, oocyte from small antral follicle; (E) Principal component analysis (PCA) of the transcriptomes from all oocytes. Purple indicates SO, blue indicates MO, and green indicates LO.
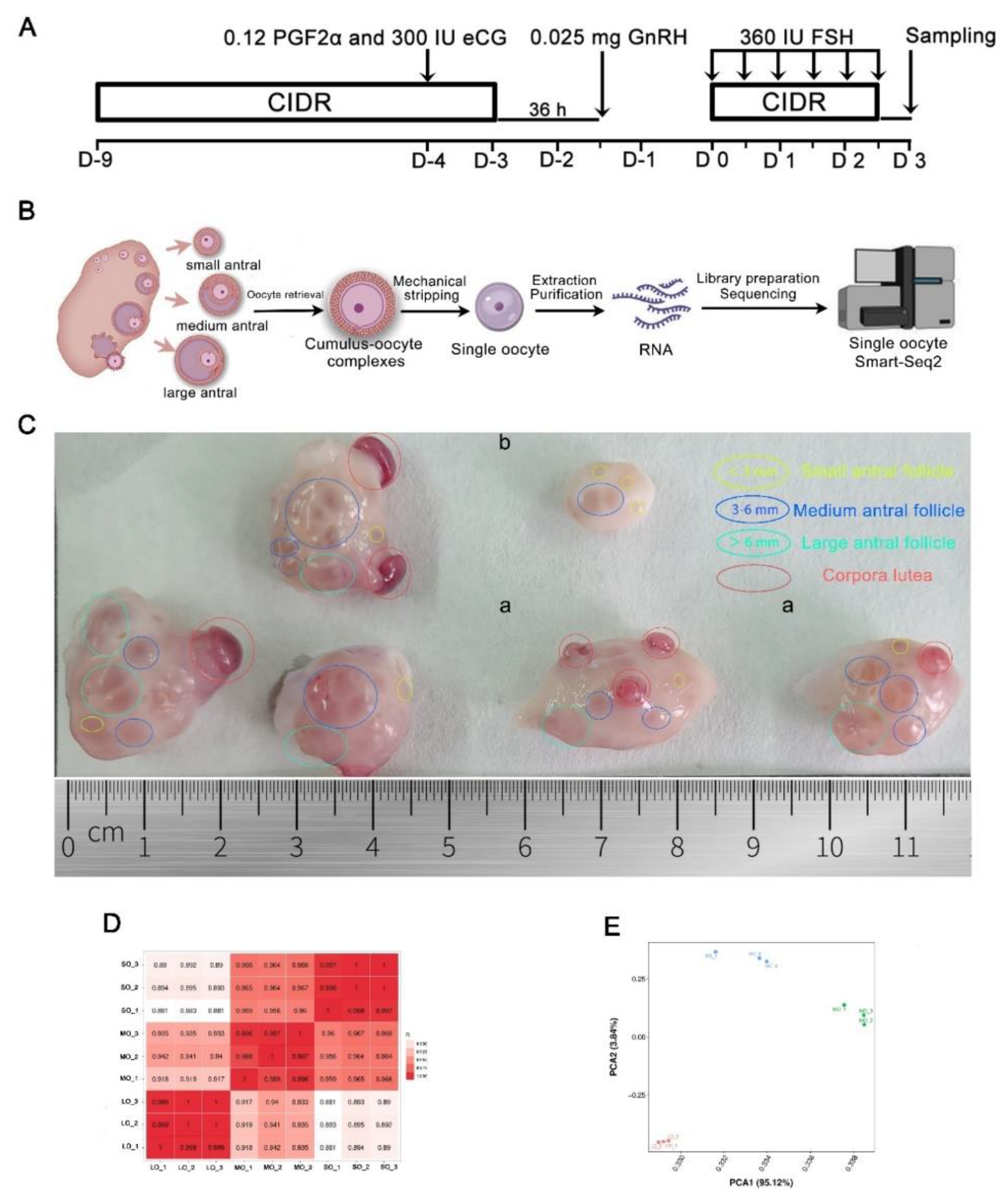
Figure 2.
Gene expression dynamics and transcriptional characteristics of oocytes: (A)Heatmap of 9,400 differentially expressed genes (DEGs) in oocytes at three stages of antral follicle development. The number of DEGs is indicated on the y-axis and the stages of follicular development on the x-axis. The color key indicates the relative gene expression from blue (high) to red (low); (B) Above: Clustering analysis of significant DEGs. DEGs were classified into 16 profiles by hierarchical cluster analysis. Each box represents a model expression profile. Below: profiles 12 and profile 3 represent upregulated and downregulated expression during antral follicle development, respectively; (C, D, and E) Volcano plots showing DEGs for oocytes in different groups: red, upregulated DEGs; blue, downregulated DEGs (log2 (FC)> |2| and FDR < 0.05); (F) Significantly enriched GO terms (biological processes) for DEGs in the oocytes at three antral follicle stages. The top 10 terms for each stage are based on the normalized enrichment score (NES). FDR< 0.05; (G) Significantly enriched KEGG pathways for DEGs in oocytes at three antral follicle stages.
Figure 2.
Gene expression dynamics and transcriptional characteristics of oocytes: (A)Heatmap of 9,400 differentially expressed genes (DEGs) in oocytes at three stages of antral follicle development. The number of DEGs is indicated on the y-axis and the stages of follicular development on the x-axis. The color key indicates the relative gene expression from blue (high) to red (low); (B) Above: Clustering analysis of significant DEGs. DEGs were classified into 16 profiles by hierarchical cluster analysis. Each box represents a model expression profile. Below: profiles 12 and profile 3 represent upregulated and downregulated expression during antral follicle development, respectively; (C, D, and E) Volcano plots showing DEGs for oocytes in different groups: red, upregulated DEGs; blue, downregulated DEGs (log2 (FC)> |2| and FDR < 0.05); (F) Significantly enriched GO terms (biological processes) for DEGs in the oocytes at three antral follicle stages. The top 10 terms for each stage are based on the normalized enrichment score (NES). FDR< 0.05; (G) Significantly enriched KEGG pathways for DEGs in oocytes at three antral follicle stages.

Figure 3.
Dynamic gene expression signatures of oocytes at three antral follicle stages: (A) Upset plot showing the number of overlapping upregulated and downregulated DEGs in each group. The histogram represents the number of co-expressed genes; (B) Heatmap showing gene expression levels in different stages. The number of DEGs is indicated on the y-axis and the stages of follicular development on the x-axis. Right: representative stage-specific DEGs are shown. The three clusters of genes represent their functions; (C) A boxplot of expression levels (log2 [FPKM]) of key genes in SO, MO, and LO stages. The two-tailed t-test p-value is indicated; (D) Representative GO terms enriched for SO, MO, and LO stages. The top 15 terms for each stage are based on the normalized enrichment score (NES). FDR < 0.05.
Figure 3.
Dynamic gene expression signatures of oocytes at three antral follicle stages: (A) Upset plot showing the number of overlapping upregulated and downregulated DEGs in each group. The histogram represents the number of co-expressed genes; (B) Heatmap showing gene expression levels in different stages. The number of DEGs is indicated on the y-axis and the stages of follicular development on the x-axis. Right: representative stage-specific DEGs are shown. The three clusters of genes represent their functions; (C) A boxplot of expression levels (log2 [FPKM]) of key genes in SO, MO, and LO stages. The two-tailed t-test p-value is indicated; (D) Representative GO terms enriched for SO, MO, and LO stages. The top 15 terms for each stage are based on the normalized enrichment score (NES). FDR < 0.05.
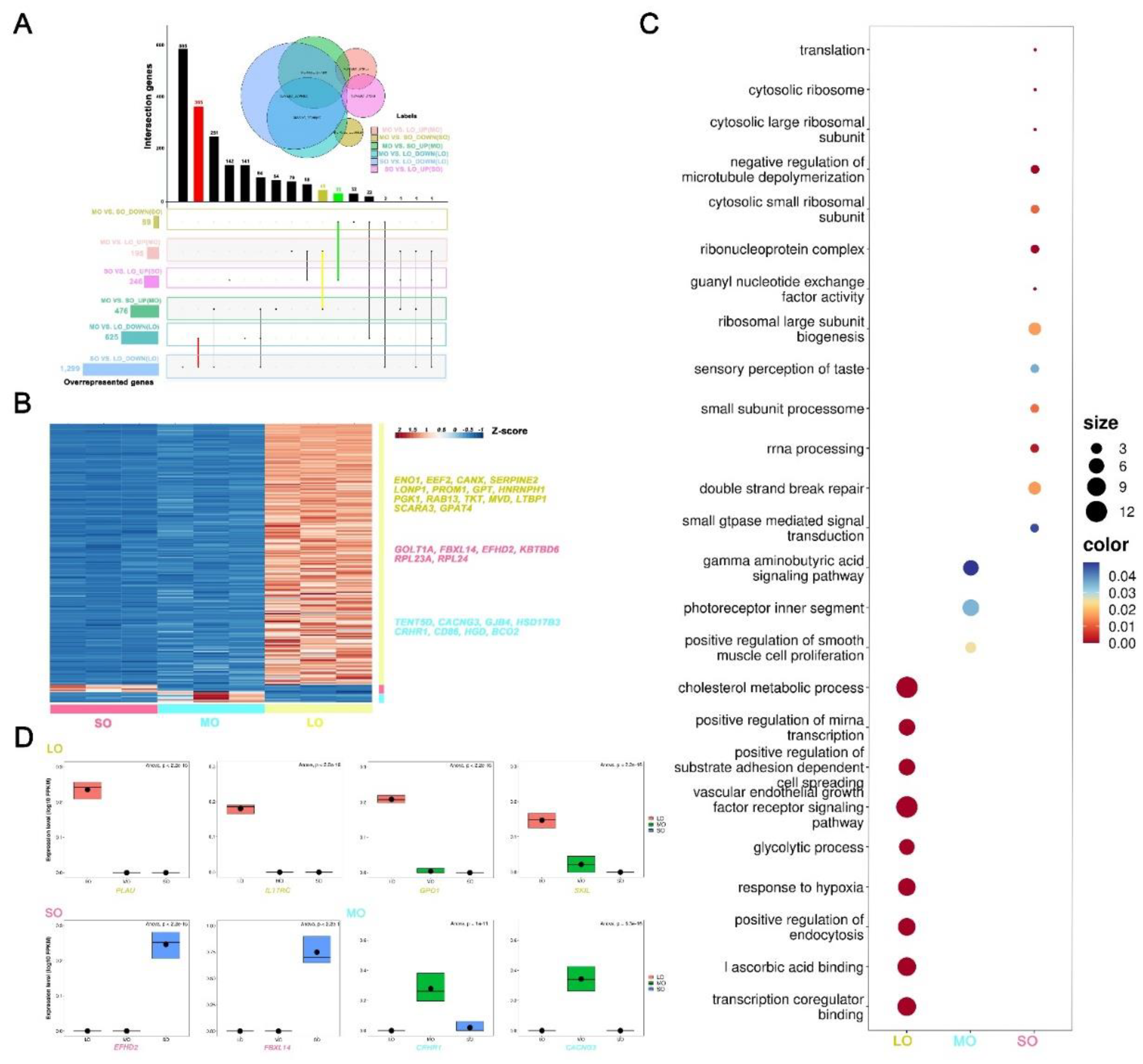
Figure 4.
Signaling pathways enriched at each antral follicle stage: (A) Representative KEGG terms for upregulated enrichment at each oocyte stage. KEGG terms for each stage are based on the normalized enrichment score (NES). FDR < 0.05; (B) Left: GSEA enrichment plots of KEGG signaling pathways. Right: boxplots of key component genes in oocytes from the medium and small antral follicle; (C) Left: GSEA enrichment plots of KEGG signaling pathways. Right: boxplots of key component genes in oocytes from large antral follicles.
Figure 4.
Signaling pathways enriched at each antral follicle stage: (A) Representative KEGG terms for upregulated enrichment at each oocyte stage. KEGG terms for each stage are based on the normalized enrichment score (NES). FDR < 0.05; (B) Left: GSEA enrichment plots of KEGG signaling pathways. Right: boxplots of key component genes in oocytes from the medium and small antral follicle; (C) Left: GSEA enrichment plots of KEGG signaling pathways. Right: boxplots of key component genes in oocytes from large antral follicles.
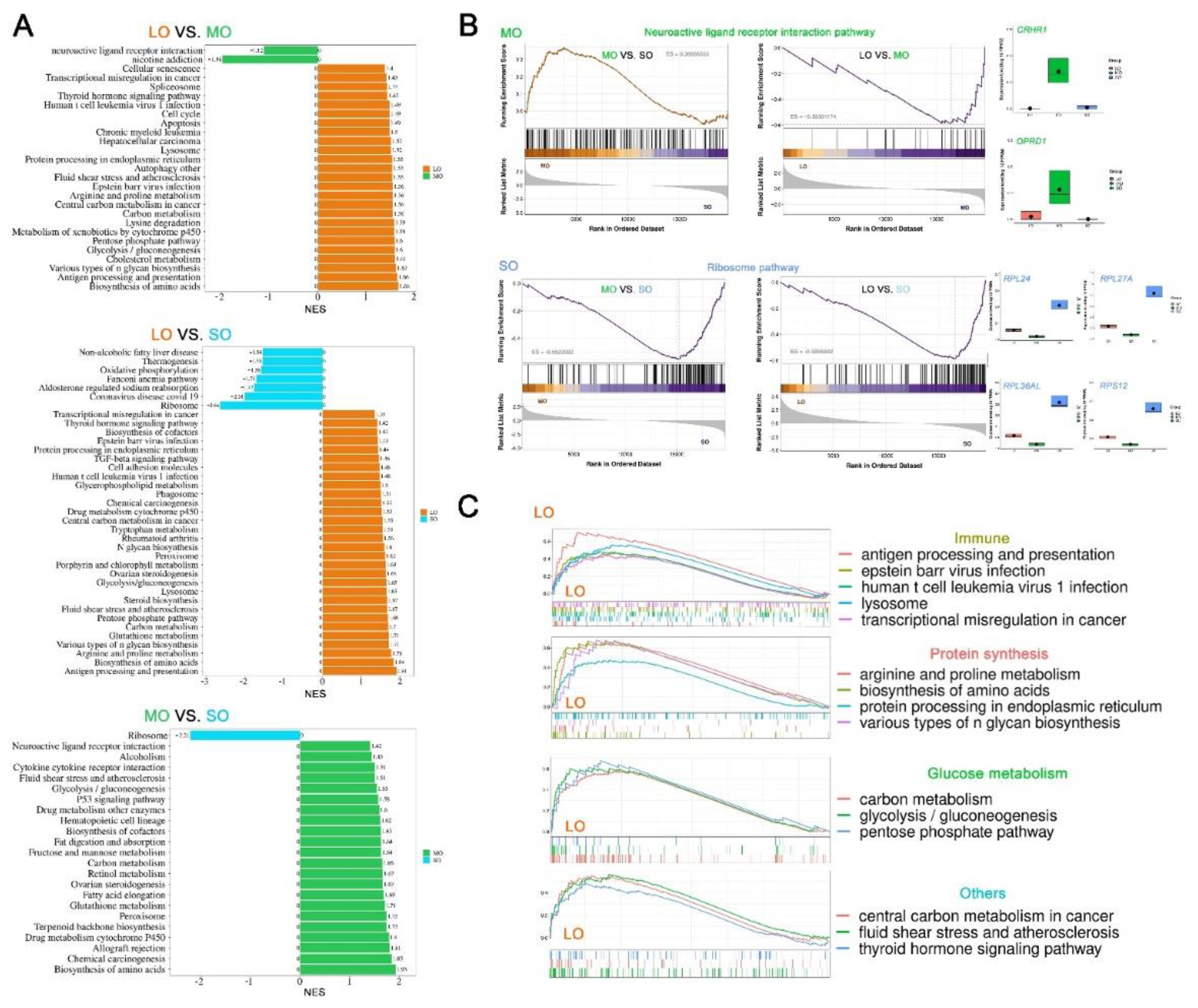
Figure 5.
Expression patterns of oocyte-specific genes: (A) Classification of meiosis-related gene expression clusters. The stages of antral follicular development are presented on the x-axis. The y-axis shows the relative expression for each cluster; (B) Classification of oocyte maturation gene expression clusters; (C) Classification of maternal-effect gene expression clusters.
Figure 5.
Expression patterns of oocyte-specific genes: (A) Classification of meiosis-related gene expression clusters. The stages of antral follicular development are presented on the x-axis. The y-axis shows the relative expression for each cluster; (B) Classification of oocyte maturation gene expression clusters; (C) Classification of maternal-effect gene expression clusters.
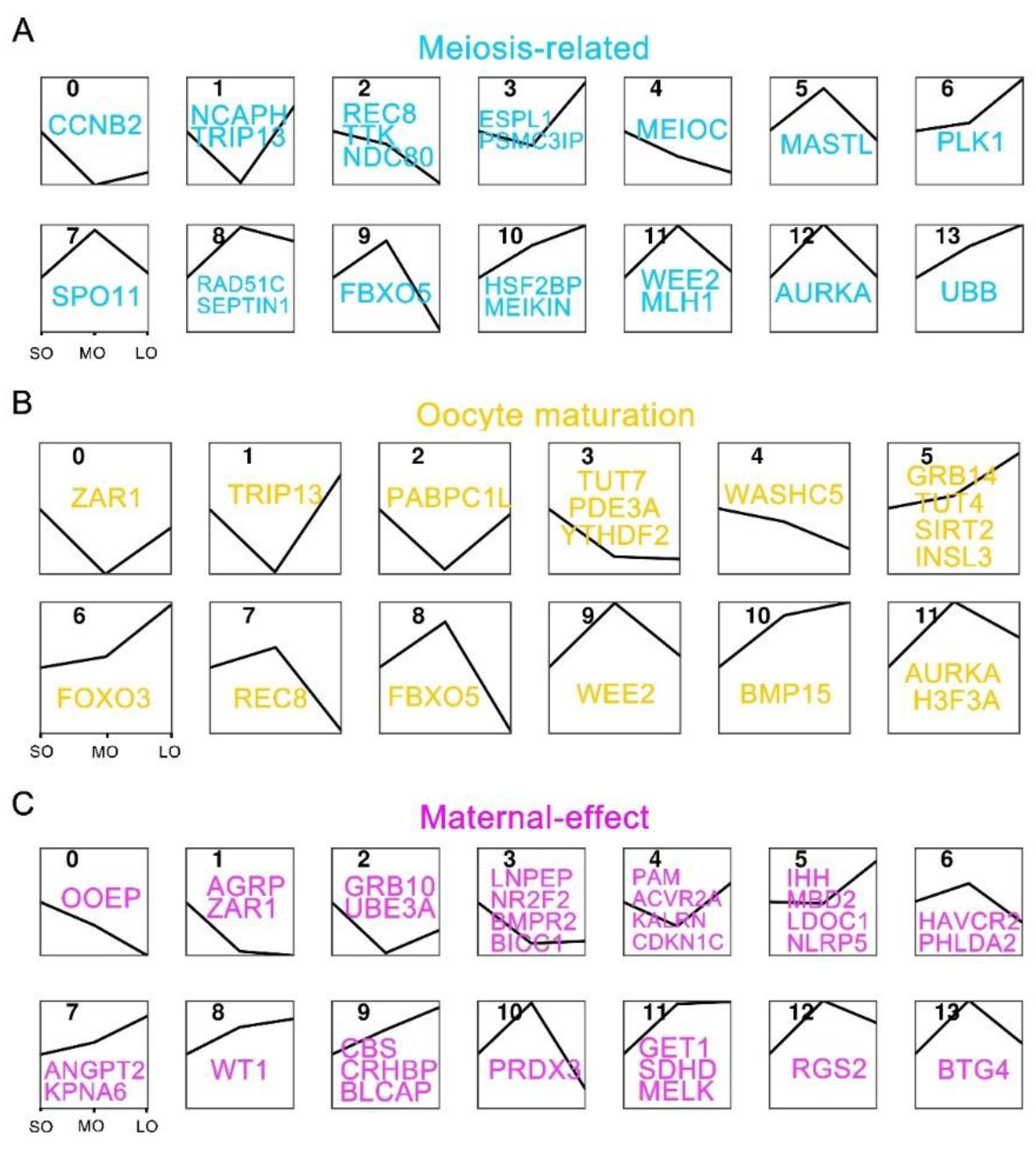
Figure 6.
Oocyte-secreted factors at three antral follicle stages: (A) Heatmap of DEGs for oocyte-secreted factors (OSFs). The genes are shown on the y-axis according to the three clusters based on the similarity of expression patterns, and the stages of antral follicular development are shown on the x-axis. The color key indicates relative gene expression from green (low) to purple (high); (B) Screening and enrichment analysis of oocyte-specific secretory factors. FDR < 0.05; (C) Analysis of the protein-protein interaction (PPI) network of OSFs for each follicle stage; (D) Visualization of the gene network of OSFs using CytoScape. The 16 genes with the highest connectivity within the network are charted, and hub genes within the network are shown by the size of the dots. The nodes represent proteins, and the edges represent the interaction.
Figure 6.
Oocyte-secreted factors at three antral follicle stages: (A) Heatmap of DEGs for oocyte-secreted factors (OSFs). The genes are shown on the y-axis according to the three clusters based on the similarity of expression patterns, and the stages of antral follicular development are shown on the x-axis. The color key indicates relative gene expression from green (low) to purple (high); (B) Screening and enrichment analysis of oocyte-specific secretory factors. FDR < 0.05; (C) Analysis of the protein-protein interaction (PPI) network of OSFs for each follicle stage; (D) Visualization of the gene network of OSFs using CytoScape. The 16 genes with the highest connectivity within the network are charted, and hub genes within the network are shown by the size of the dots. The nodes represent proteins, and the edges represent the interaction.
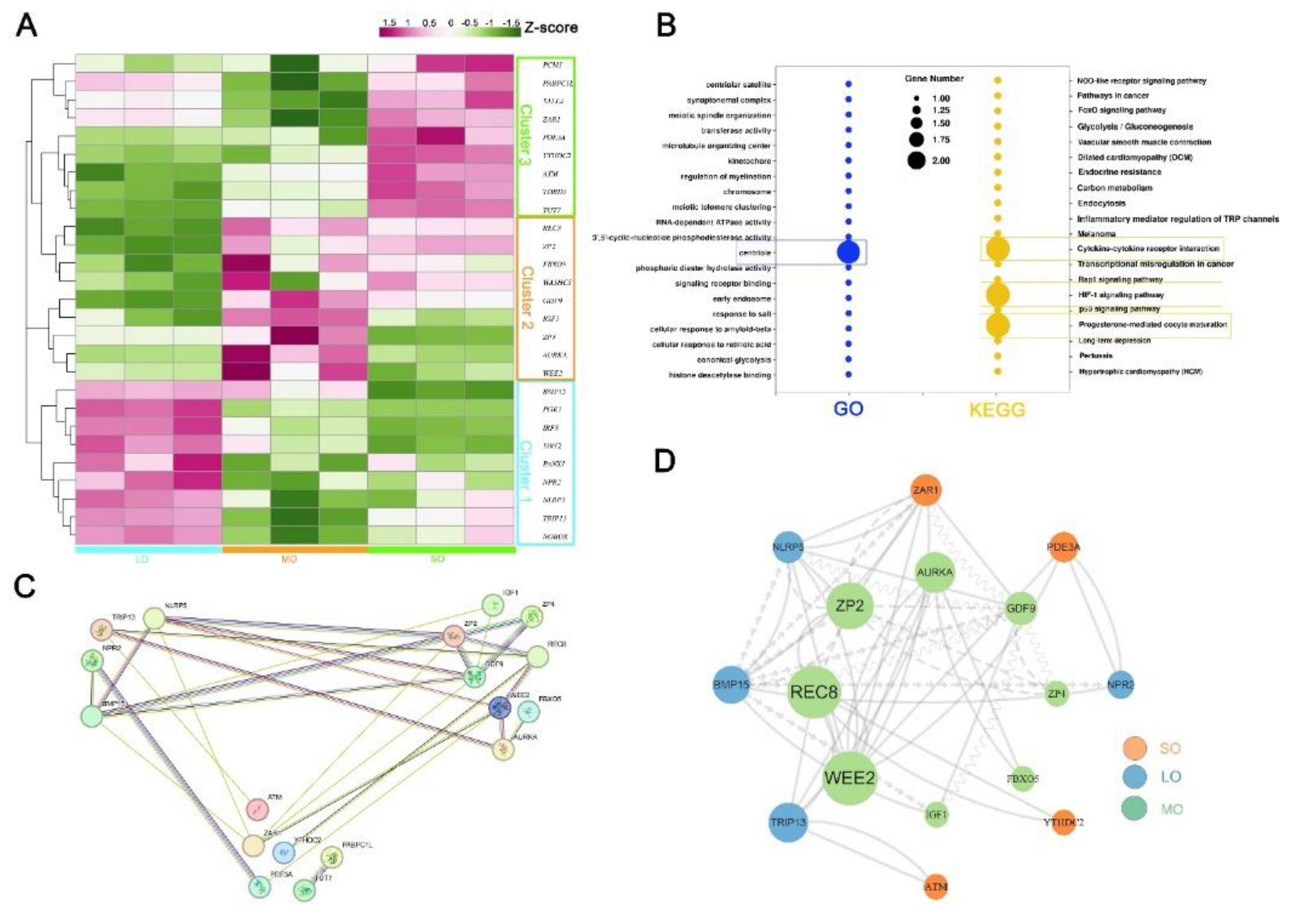
Figure 7.
Mapping TF regulatory networks in oocytes from three antral follicle stages: (A, B, and C) Visualization by CytoScape of the TFs and TF family regulatory networks at the transition of each stage of antral follicle development. The inner circle shows the TF family, whereas the traits indicate the number of TFs contained. An upward triangle indicates a significantly upregulated TF and an inverted triangle indicates a significantly downregulated TF; (D) Venn diagram showing key TFs among pairwise comparisons of the three antral follicle stages; (E) Visualization by CytoScape of TF regulatory networks in oocytes from the large antral stage. The inner circle shows TF families, whereas the different shapes represent key transcription families, and the outer circle (triangles) shows TFs; (F) The PPI network of the 18 TFs in oocytes from the large antral follicle was constructed using the STRING database. The nodes represent proteins, and the edges represent the interactions. Outer circles represent hub TFs, and different sizes indicate criticality; (G) The PPI network of the seven TFs co-expressed in small and medium antral follicles was constructed using the STRING database. The nodes represent proteins, and the edges represent the interactions; (H) CytoScape plots of the TF-DEG regulatory network in small antral follicles. Yellow circles represent TFs, and triangles and diamonds represent co-regulated target genes; (I) CytoScape plots of the TF-DEG regulatory networks in medium antral follicles. Yellow circles represent TFs, and triangles and diamonds represent co-regulated target genes.
Figure 7.
Mapping TF regulatory networks in oocytes from three antral follicle stages: (A, B, and C) Visualization by CytoScape of the TFs and TF family regulatory networks at the transition of each stage of antral follicle development. The inner circle shows the TF family, whereas the traits indicate the number of TFs contained. An upward triangle indicates a significantly upregulated TF and an inverted triangle indicates a significantly downregulated TF; (D) Venn diagram showing key TFs among pairwise comparisons of the three antral follicle stages; (E) Visualization by CytoScape of TF regulatory networks in oocytes from the large antral stage. The inner circle shows TF families, whereas the different shapes represent key transcription families, and the outer circle (triangles) shows TFs; (F) The PPI network of the 18 TFs in oocytes from the large antral follicle was constructed using the STRING database. The nodes represent proteins, and the edges represent the interactions. Outer circles represent hub TFs, and different sizes indicate criticality; (G) The PPI network of the seven TFs co-expressed in small and medium antral follicles was constructed using the STRING database. The nodes represent proteins, and the edges represent the interactions; (H) CytoScape plots of the TF-DEG regulatory network in small antral follicles. Yellow circles represent TFs, and triangles and diamonds represent co-regulated target genes; (I) CytoScape plots of the TF-DEG regulatory networks in medium antral follicles. Yellow circles represent TFs, and triangles and diamonds represent co-regulated target genes.
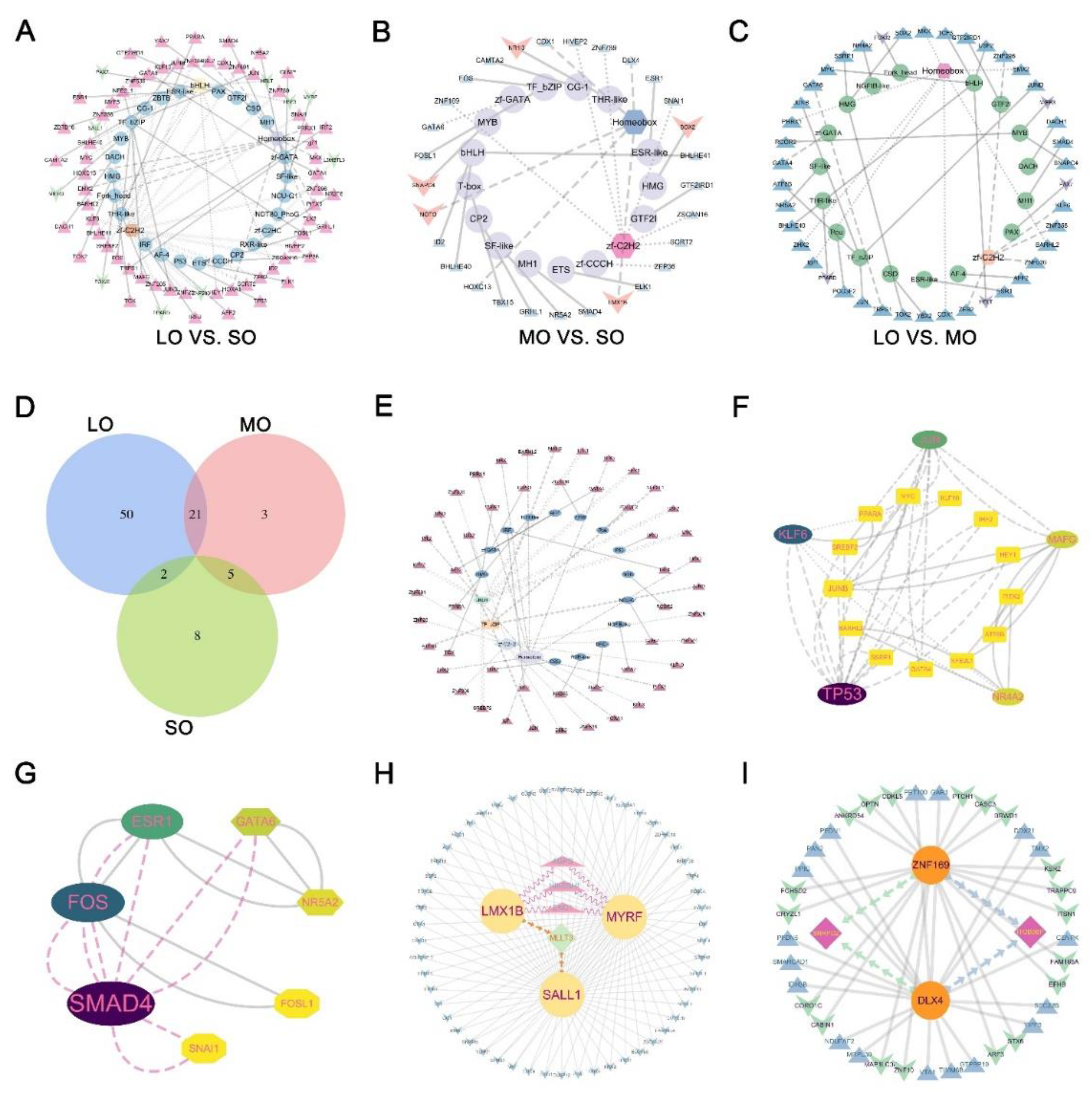
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



