
Submitted:
01 November 2023
Posted:
03 November 2023
You are already at the latest version
Abstract
The TME, consisting of immune cells, fibroblasts, vessels, and the extracellular matrix, regulates tumor progression and therapy responses. TME-targeted therapies aim to transform this environment from supporting tumor growth to impeding it and fostering an effective immune response. This review examines the metabolic disparities between immune cells and cancer cells, their impact on immune function and therapeutic targeting, the TME components, and the complex interplay between cancer cells and non-tumoral cells. The success of TME-targeted therapies highlights their potential in achieving better cancer control or even a cure.
Keywords:
Tumor Microenvironment
; Immunotherapy
; Oncometabolites
1. Introduction
Malignant cells distinguish themselves from normal cells in a fundamental way: the genetic and metabolic programs of normal cells are dedicated to specialized functions, while malignant cells prioritize replication. Specialization and replication are mutually exclusive. In specialized tissues, regeneration is limited to tightly regulated stem cell niches with restricted differentiation abilities that have specific localizations within the tissue compartments [1]. These somatic stem cells are the origin of most malignant neoplasms. In contrast to their normal counterparts, transformed stem cells are no longer spatially confined but retain stem cell properties like the capacity for self-renewal, resiliency, pluripotency, and migration, but no longer respond to regulatory mechanisms and undergo rapid adaptation/evolution [2].
Over the years, tumor biologists have concentrated on unraveling the internal genetic and metabolic pathways that confer malignant cells a competitive growth and survival advantage. Consequently, therapies for unresectable malignancies have centered on exploiting the vulnerabilities of rapidly dividing cells, a strategy that has proven effective and remains the foundation of most chemotherapy and radiotherapy protocols, but that is associated with substantial toxicity to the stem cell compartments in different tissues. In a few gender-specific cancers, it was discovered that tumor growth and progression was influenced or driven by sex hormones, opening a path for hormone deprivation therapies [3]. Advances in cytogenetics led to the discovery that a small subset of malignancies is characterized by specific recurring translocations that produce predictable aberrant fusion or constitutively active proteins. In these neoplasms, targeting the function of those abnormal proteins induces maturation, suppression, or apoptosis of tumoral clones [2]. In recent years, more widespread access to advanced molecular-genetic technologies revealed that within the innumerable random chromosomal aberrations and mutations occurring in the unstable genomes of cancer cells, only a relatively limited subset of genetic alterations, termed "driver mutations," are pivotal for initiating and advancing malignancies [4]. In the 20th century, we came to understand the impact of environmental carcinogens through epidemiological research. In the 21st century, advances in epigenetics, encompassing DNA methylation, histone changes, chromatin modifications, and non-coding RNAs, began to unravel the mechanisms by which the environment contributes to tumor development [5].
Collectively, advances in cytogenetics, molecular genetics and epigenomics, have led to a new era of targeted oncologic therapies, which continues to evolve rapidly, but that has benefited only a small subset of patients. The idea of engaging the immune system to fight cancer cells began in the 19th century with the use of bacterial infections to induce tumor regression, and advanced further during the 20th century with advances in immunology and the realization that mutant proteins generated by malignant cells were immunogenic. Vaccines made from tumor lysates, conjugates of cancer-specific peptides with immune-boosting compounds, monoclonal antibodies, adoptive cell therapies and immunocytokines have demonstrated the immense potential of cancer immunotherapy, including the ability to offer durable benefit [6]. Despite the promise of cancer immunotherapies, only a small portion of patients will experience these long-lived benefits, giving birth to a new field of research focused on decoding how cancer cells are able to escape the immune response and induce immune tolerance through their interactions with the tumor microenvironment (TME) [7]. In this review we will delve into metabolic alterations of cancer cells that affect the TME causing an abnormal immune response, focusing on potential targets for therapeutic intervention.
2. Targeting the Tumor Microenvironment
The tumor microenvironment (TME) consists of key elements like immune cells, fibroblasts, vessels, and the extracellular matrix, which play a crucial role in regulating tumor progression and therapy responses. Immuno-oncology targets the local and systemic immune compartment and has revolutionized cancer treatment by using the immune system’s ability to detect and destroy cancer cells. Although the response to immunotherapy may be delayed compared to other treatment modalities, it can produce durable responses in advanced cancers for which until recently, only palliation could be offered [8]. Immuno-oncology currently employs four primary strategies: immune checkpoint inhibitors, adoptive cell therapy, immunocytokine and therapeutic cancer vaccines. Immune checkpoint inhibitors target proteins that normally inhibit immune cells from attacking cancer cells, including PD-1/PD-L1, CTLA4, and LAG-3 pathways (Figure 1). Adoptive cell therapy, on the other hand, enhances the natural capabilities of effector T-cells and NK-cells to combat cancer. Adoptive cell therapy encompasses therapies such as chimeric antigen receptor T cells (CAR-T), tumor-infiltrating lymphocytes (TIL), engineered T cell receptors (TCR), and NK-cell infusions [9]. Immunocytokines are endogenous small molecules that promote cellular interactions and modulate immune responses through regulation of proliferation and differentiation. While effective in promoting immune cell activation and anti-tumor activity, these treatments are often marred by significant toxicity and inconvenient administration [10]. Therapeutic vaccines have previously been employed in the fight against cancer though their efficacy has remained somewhat limited [11]. Advances in neoantigen target identification and novel delivery platforms have resulted in renewed optimism for this approach, though the full potential of these therapies has not yet been fully explored [12]. Additionally, the strategy of targeting tumoral angiogenesis with antiangiogenic agents has become a standard of care for certain malignancies. However, this approach has demonstrated limited success, primarily due to the development of tumor resistance and the occurrence of cardiovascular toxicity [13]. The success of these TME targeted therapies has reinforced the concept that this approach holds great potential for achieving cancer control or cure.
3. Divergent Metabolic Profiles of Cancer Cells and Immune Cells: Therapeutic Implications
3.1. Glucose Metabolism
3.1.1. Cancer cells
The metabolism of high-grade malignancies resembles that of embryonic stem cells, relying more on glycolysis than oxidative phosphorylation to support proliferation and self-renewal. This preferential use of aerobic glycolysis is known as the "Warburg effect" [15]. Although glycolysis yields a lower production of ATP than the citric acid cycle (CAC) and oxidative phosphorylation (OXPHOS), it allows higher levels of glucose uptake because it occurs in the cytoplasm and not the mitochondria, and it prevents the loss of carbon atoms as CO2, so that they can be reused in other biosynthetic pathways [15]. The NADH-dependent reduction of pyruvate to lactate by the lactate dehydrogenase recycles the NAD which is needed to sustain glycolysis. Pyruvate generated by glycolysis can enter the CAC through pyruvate dehydrogenase and carboxylase, yielding higher levels of energy and producing additional metabolic intermediates. Lactate released in the TME by the tumor cells reaches the circulation and can be converted back to pyruvate by normal cells and recaptured by tumor cells to further feed the CAC [16]. Metabolic reprogramming of tumor cells leads to increased expression of glucose transporters and glycolytic enzymes through activation of various oncogenes and signaling pathways common to several malignancies.
3.1.2. Immune Cells
Glucose utilization in immune cells is more regulated and more flexible. Resting cells rely on OXPHOS, which is more efficient in terms of ATP production. Activated immune cells can switch to glycolysis to meet increased energy demands, however malignant cells outcompete immune cells for glucose and oxygen uptake, and the increased production of lactate creates a hypoxic and acidotic TME that weakens the activation of immune cells and polarizes the cellular responses towards immune anergy [15]. Additionally, the presence of immune checkpoints in the TME have been associated with T cell metabolic dysfunction, including impaired mitochondrial ATP production limiting T cell self-renewal despite a glycolytic phenotype and promoting terminal differentiation [17]. The utilization of limited supplies of glucose within the TME is further complicated by presence of pro-tumor immune cells, including myeloid derived suppressor cells (MDSCs). Tumor associated monocytic MDSCs are characterized by their prominent utilization of glucose in the TME and generation of by-products that inhibit reactive oxygen species (ROS)-mediated apoptosis and promote polarization of early myeloid derived cells to immunosuppressive phenotypes [18]. Additionally, the effect of aberrant glucose metabolism in TME can be seen in the intrinsic regulation of tumor associated macrophages resulting in epigenetic remodeling and signal transduction that promotes an immunosuppressive phenotype [19]. Dendritic cells (DCs) play a crucial role in the activation of the adaptive immune response and their function is heavily influence by glucose metabolism. Restricted glycolysis coupled with increased lactate can inhibit DC activation resulting in diminished antigen presentation, cytokine production and T cell stimulation [20]. Moreover, limited availability of glucose may impair CCR7 oligomerization required for cytoskeletal remodeling and diminish DC trafficking to tumor-draining lymph nodes diminishing immune cell mobilization [21].
3.1.3. Therapeutic Targeting
The Warburg effect is the basis for using radiolabeled glucose analogs to stage, surveil and assess therapeutic response of malignancies through positron emission tomography (PET) scans (Figure 1). Numerous drugs that inhibit glycolysis, glucose, glutamine, and lactate metabolism are being investigated, however their efficacy has been limited by the numerous isoforms of metabolic enzymes, metabolic heterogeneity of tumor subpopulations, alternative pathways for energy production, and off-target side effects [22].
3.2. Amino Acid Metabolism
3.2.1. Cancer Cells
High-grade malignancies have increased amino acid requirements for their rapid proliferation. They often upregulate amino acid transporters to acquire them from the TME and use the pyruvate generated through glycolysis or the recirculation of lactate to support the CAC, which provides substrates for de novo amino acid synthesis [23]. Many tumors rely on glutaminolysis for energy and intermediate metabolite production, and some cancer cells lose their ability to synthesize specific non-essential amino acids, having to compensate with upregulation of specific transporters for their survival, creating dependencies that can be exploited through targeted therapies [23].
3.2.2. Immune Cells
Immune cells also require amino acids for protein synthesis, proliferation, and cytokine production. Their amino acid uptake is influenced by their activation state, however malignant cells outcompete immune cells causing depletion of amino acids in the TME, which contributes to a blunted immune response. Amino acid metabolism plays a crucial role in T cell biology including activation and clonal expansion through Slc7a5 interactions with mTORC1 and differentiation into effector and memory T cells through epigenetic modulation [24]. Deprivation of essential amino acids by MDSCs through arginase1 and nitric oxide synthase 1 and 2 activity also regulates T cell function, reduces the formation of memory T cells, and promotes release of peroxynitrite, which can induce T cell apoptosis [25,26,27,28]. The induction of indoleamine-pyrrole 2,3 dioxygenase (IDO-1) on both myeloid and tumors cells results in decreased levels of tryptophan and the presence of immunosuppressive catabolites such as kynurenine (Kyn) that promote Treg activity and diminish T cell effector function [29,30,31]. The broad impact of arginine and tryptophan catabolism and the formation of potent metabolites can also be seen on DCs, which are similarly impacted by the presence Kyn resulting in downstream activation of the aryl hydrocarbon receptor creating a positive feedback loop of IDO-1 activation [20,32,33]. The importance of IDO-1 as a modulator of immune cell activity resulted in the development of inhibitors across several solid tumors with numerous preclinical models and early phase drug testing demonstrating promising results [34]. Unfortunately, the clinical development of these drugs was marred by disappointing results in large clinical trials, marked by the failure of the phase 3 ECHO-301/KEYNOTE-252 trial to show a significant benefit with the addition of an IDO-1 inhibitor to pembrolizumab, a PD1 inhibitor, in the treatment of advanced melanoma [35]. A follow-up analysis evaluated IDO-1 expression via immunohistochemistry in primary and metastatic melanoma tumors and noted significant heterogeneity in IDO-1 expression across longitudinal samples [36].
3.2.3. Therapeutic Targeting
Targeting amino acid metabolism in cancer cells has shown success in a small subset of malignancies, for example the use of asparaginase in lymphoblastic leukemias and PEGylated arginine deaminase in hepatocellular carcinoma and mesothelioma. Treatment with glutamine inhibitors and recombinant methioninase have shown promising results in animal models or early clinical studies [37]. Deprivation therapies against non-essential amino acids should provide a competitive advantage to immune cells because they retain the capacity to synthesize these compounds and have shown synergistic effects with immunotherapy in animal models [23]. Despite the described setbacks in phase 3 clinical trials, ongoing studies continue to look at the role of IDO-1 inhibitors while honing predictive biomarkers, patient selection and combinatorial strategies to enhance efficacy [34] (Table 1).
3.3. Nucleotide Metabolism
3.3.1. Cancer Cells
High-grade malignant cells have very high nucleotide requirements. Nucleotides are essential for nucleic acid synthesis, enzyme regulation and metabolism. Cancer cells rely more on de novo synthesis than external sources, a process that requires large amounts of energy, and that leads to depletion of nucleotide precursors in the TME. The de novo synthesis by cancer cells involves upregulation of the numerous enzymatic pathways required for the synthesis, modification, and assembly of precursors such as ribose for purines, deoxyribose for pyrimidines, CO2, amino acids, and tetrahydrofolate [38]. Under normal circumstances the synthesis of pyrimidines is simpler than that of purines; purines exert an inhibitory effect over the enzymes participating in purine synthesis, but activate enzymes needed for pyrimidine formation and vice versa [39]. However, in cancer cells these processes are upregulated and dysregulated, usually due to the abnormal function of oncogenes (e.g., K-RAS, c-MYC), tumor suppressor genes (e.g., p53, RB1) or genes that have dual oncogenic and tumor suppressor functions (e.g., ATF3) leading to genomic instability and tumor progression [40].
3.3.2. Immune Cells
Immune cells also require nucleotides for replication, but in contrast to malignant cells they can switch between de novo synthesis and salvage pathways. In the salvage pathway bases and nucleosides resulting from the degradation of nucleic acids are recycled. In general, immune cells are more efficient, use less energy and are less dependent on precursors from the TME, making immune cells less vulnerable to antimetabolite therapies. However, dysregulated nucleotide metabolism affects the immune response through Toll-like receptors, RIG-like receptors, NOD-like receptors, purinergic receptors, and adenosine receptors. In many tumors the net effect is inhibitory through recruitment and expansion of suppressor Tregs [41]. The adenosine pathway and its implication on TME and immune cell composition has been an area of interest for therapeutic development. The accumulation of adenosine in the TME, produced by CD39 and CD73 and driven by the downregulation of intracellular transport in the setting of hypoxia, creates a pro-angiogenic effect with modulation of the local immune system [42]. The adenosine receptors, A1, A2A, A2B, and A3, are ubiquitously expressed on myeloid cells and lymphocytes and appear to primarily attenuate the immune response via inactivation of TNF-α production, augmentation of IL-10 production, suppression of IL-2 secretion, and upregulation of immune checkpoints such as CTLA4 and PD1 [43].
3.3.3. Therapeutic Targeting
Nucleoside analogs and folate antagonists comprise a large proportion of the standard chemotherapy regimens and have been part of the oncologic armamentarium for a long time; while effective they are associated with significant toxicity to stem cells of tissues with high turnover rates. Newer therapies targeting nucleotide metabolism include highly specific inhibitors of specific enzymes downstream from driver mutations in different types of cancer. Most of these agents are being tested in phase I/II clinical trials. Several of these agents have shown synergistic effects when combined with immunotherapy in animal models [44]. Targeting nucleotide metabolism should also produce imbalances in the purine/pyrimidine ratios, leading to increased tumor mutational burden (TMB), and thus augment the effectiveness of immunotherapy [45]. Given the potential synergy of adenosine directed therapies with immunotherapy, several studies have sought to evaluate the role of combination therapy in the treatment of cancer as summarized in Table 2.
3.4. Fatty Acid Metabolism
3.4.1. Cancer Cells
High-grade cancer cells have an increased demand of fatty acids for energy, membrane synthesis, and for generation of signaling intermediates such as eicosanoids, fatty acid carnitines, thioesters, and N-acyl ethanolamines [46]. Fatty acids also modulate the function of proteins involved in cell growth, proliferation, motility, survival through the PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways [47]. High-grade cancer cells favor de novo synthesis from non-lipid substrates, a process in which fatty acids are generated from simple precursors predominantly in the cytosol, and to a lesser extent in the mitochondria [48]. Additionally, they also upregulate fatty acid transporters to capture fatty acids from extracellular sources either from the circulation or the TME through passive diffusion or transport proteins leading to their depletion in the TME [49]. Lipid droplets are common in many types of tumors and serve as energy reserves through lipolysis and lipophagy. Metabolic shift towards fatty acid oxidation appears to be a common mechanism of therapy resistance.
3.4.2. Immune Cells
Immune cells also require fatty acids for similar purposes. However, the uptake of these fatty acids is regulated and influenced by their activation state. Activated immune cells may shift towards increased fatty acid oxidation to meet their energy needs. Insufficient fatty acids in the TME can impair an effective anti-tumor response. De novo fatty acid synthesis remains an integral part to T cell activation coupled with the shift towards the glycolytic pathway. T cell differentiation appears to be highly coupled to intracellular programs of lipid metabolism and extracellular lipid exposure [50,51]. Similarly, MDSC and DC differentiation and function are completed with lipid metabolisms with the accumulation of oxidized fatty acids leading to augmented suppressive functions of polymorphonuclear, MDSCs and DC dysfunction through impaired antigen presentation and immune tolerance [52,53,54].
3.4.3. Therapeutic Targeting
Inhibitors of enzymes involved in fatty acid synthesis or fatty acid transporters are currently under investigation as potential cancer treatments. However, research in this area is still in its early stages. Enzymes such as fatty acid synthase (FASN), acyl-CoA thioesterases (ACOTs), and acetyl CoA carboxylase (ACC), which play a crucial role in the de novo synthesis of fatty acids, have been targeted resulting in restricted growth, proliferation, and metastasis in both cancer cell lines and animal models. [55,56]
3.5. Oxygen Dependency
3.5.1. Cancer Cells
High-grade cancer cells frequently exhibit elevated hypoxia tolerance. They achieve this by promoting the development of abnormal vascular networks (tumoral neoangiogenesis), which reduce blood flow inducing hypoxia, impede the delivery of intravenous chemotherapy drugs, and enhance the entry of tumor cells into the bloodstream [57]. Cancer stem cells and their offspring capable of adapting to low-oxygen environments through the activation of hypoxia-inducible factors (HIF-1α, HIF-2α, HIF-1β) exhibit more aggressive phenotypes compared to those that cannot adapt to hypoxia and undergo cell death. Hypoxia-inducible factors (HIF) proteins regulate a multitude of genes and intricate RNA networks, encompassing those responsible for redirecting metabolism toward anaerobic pathways such as glycolysis or fatty acid oxidation for energy production, as discussed earlier. Additionally, HIF proteins govern pathways associated with angiogenesis, cell survival, and invasive behavior [58]. Hypoxia promotes a stem-like phenotype in tumor cells through activation of genes such as OCT4, SOX2, c-MYC, CD44, CD133, WNT, Notch and NANOG leading to dedifferentiated and undifferentiated tumor phenotypes associated with more aggressive biology [57]. Hypoxia has recently been recognized for its ability to induce epithelial-mesenchymal-transition (EMT) in epithelial cells. This process is driven by the activation of genes such as SNAIL, ZEB1, TWIST, TCF3, and NF-κβ, mediated by HIF-1α and other mechanisms. EMT entails the activation of signaling pathways that enable epithelial cells to adopt characteristics similar to stromal cells involved in reparative/regenerative processes. These characteristics include rapid growth, migration, angiogenesis induction, tissue remodeling, and the capacity to trigger an inflammatory response. EMT is linked to sarcomatoid carcinoma phenotypes, heightened local tumor aggressiveness, and increased metastatic potential [59] (Figure 1).
3.5.2. Immune cells
Effector immune cells require oxygen for the generation of reactive oxygen species (ROS) and the production of cytokines for mounting a robust immune response. ROS are particularly important for innate immune responses such as respiratory burst and inflammasome activation [60]. In a hypoxic TME there is an accumulation of lactic acid and adenosine due to the Warburg effect. The previously described A2A receptor (A2AR) can hinder the function and proliferation of effector cells by interacting with high levels of adenosine in the TME, initiating a series of intracellular events that diminish the responsiveness of effector cells to IL-2 through mTOR pathway blockade [61]. Furthermore, within hypoxic TMEs, there is an elevation in the levels of chemokines CCL28 and CCL2 produced by tumor cells. These chemokines play a role in recruiting Tregs and macrophages. Tregs promote angiogenesis and continue to suppress effector immune cells and antigen-presenting cells through both contact-dependent mechanisms and independent mechanisms, such as the secretion of IL-10 and TGF-β. This contributes to immune evasion within the TME [62,63].
The combination of a hypoxic and acidotic TME along with an expanded population of Tregs has been linked to the reprogramming of macrophages within the TME, skewing tumor associated macrophages toward the M2 phenotype. M2 macrophages release cytokines such as IL-10, IL-1β, TGF-β, and VEGF, which exhibit anti-inflammatory, immunosuppressive, and pro-angiogenic properties. They also secrete matrix metalloproteins that contribute to the remodeling of the tumor stroma, further hindering the recruitment of effector cells and the efficient diffusion of chemotherapeutic agents. This combined effect of Tregs and M2 macrophages leads to the inhibition of CD4+ helper cells, CD8+ cytotoxic T cells, increased tumoral angiogenesis, stromal activation and remodeling and is considered a critical factor in tumor immune evasion and progression [64,65].
3.5.3. Therapeutic Targeting
Therapeutic strategies aimed at modifying the hypoxic TME include antiangiogenic agents (e.g., bevacizumab, ramucirumab, trebananib), HIF inhibitors (belzutifan, 6RK73), hypoxia-activated/bioreductive prodrugs (e.g., tirapazamine, evofosfamide, apaziquone) and less commonly, hyperbaric medicine. These agents are currently undergoing preclinical and clinical development, with some having received approval for specific clinical scenarios, typically in combination with traditional chemotherapy and immunotherapy regimens [65] (Table 2).
4. Conclusions
The realization of the importance of non-tumoral TME for the survival and propagation of cancer cells has been one of the most important breakthroughs in the understanding of tumor biology. The stem-cell-like properties of aggressive cancer cells allow them to reprogram non-tumoral cells in the TME to align them with their goal of favoring replication in detriment of specialization. Oncologic therapies have traditionally focused on exploiting vulnerabilities of tumor cells and remain the mainstay of oncologic treatment. The recent development of therapies targeting the non-tumoral TME have yielded some of the most significant advances in recent years. In our review we described some of the advances in the understanding of the incredibly complex crosstalk between cancer cells and non-tumoral cells in the TME, focusing on areas that appear promising for enhancing immunotherapies.
Author Contributions
Conceptualization, all authors; writing—original draft preparation, RY, HM.; writing—review and editing, RM, JK; supervision, RM, HM; project administration, HM. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable, review article.
Acknowledgments
The Department of pathology and Laboratory Medicine of Indiana University School of Medicine, Indianapolis IN, provided support for this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauß A, Blaudszun AR, Yevsa T, Fricke S, Kossatz-Boehlert U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front Immunol. 2020; 11:1280. [CrossRef]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144 (5): 646-74.
- Quon H, Loblaw DA. Androgen deprivation therapy for prostate cancer-review of indications in 2010. Curr Oncol. 2010; 17 Suppl 2: S38-44. [CrossRef]
- Raimondi D, Passemiers A, Fariselli P, et al. Current cancer driver variant predictors learn to recognize driver genes instead of functional variants. BMC Biol. 2021; 19(3). [CrossRef]
- Lodewijk I, Nunes SP, Henrique R, et al. Tackling tumor microenvironment through epigenetic tools to improve cancer immunotherapy. Clin Epigenet.2021; 13(1): 63. [CrossRef]
- Dobosz P, Dzieciątkowski T. The Intriguing History of Cancer Immunotherapy. Front Immunol. 2019; 10: 2965. [CrossRef]
- Zhu S, Zhang T, Zheng L, et al. Combination strategies to maximize the benefits of cancer immunotherapy. J Hematol Oncol. 2021; 14, 156. [CrossRef]
- Li Z, Song W, Rubinstein M, Liu D. Recent updates in cancer immunotherapy: a comprehensive review and perspective of the 2018 China Cancer Immunotherapy Workshop in Beijing. J Hematol Oncol. 2018; 11(1): 142. [CrossRef]
- Yang L, Ning Q, Tang SS. Recent Advances and Next Breakthrough in Immunotherapy for Cancer Treatment. J Immunol Res. 2022; 2022:8052212. [CrossRef]
- Pabani A, Gainor JF. Facts and Hopes: Immunocytokines for Cancer Immunotherapy. Clin Cancer Res. 2023; 29(19): 3841-3849. [CrossRef]
- Gardner TA, Elzey BD, Hahn NM. Sipuleucel-T (Provenge) autologous vaccine approved for treatment of men with asymptomatic or minimally symptomatic castrate-resistant metastatic prostate cancer. Hum Vaccin Immunother. 2012; 8(4): 534-9. [CrossRef]
- Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer. 2021; 21(6): 360-378. [CrossRef]
- Oguntade AS, Al-Amodi F, Alrumayh A, Alobaida M, Bwalya M. Anti-angiogenesis in cancer therapeutics: the magic bullet. J Egypt Natl Canc Inst. 2021; 33(1): 15. [CrossRef]
- Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020; 368 (6487): eaaw5473. [CrossRef]
- Lin X, Xiao Z, Chen T, Liang SH, Guo H. Glucose Metabolism on Tumor Plasticity, Diagnosis, and Treatment. Front Oncol. 2020; 10: 317. [CrossRef]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Zhan, Yanxiang Guo J, White E, Rabinowitz JD. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017; 551 (7678): 115-118. [CrossRef]
- Vardhana SA, Hwee MA, Berisa M, Wells DK, Yost KE, King B, Smith M, Herrera PS, Chang HY, Satpathy AT, van den Brink MRM, Cross JR, Thompson CB. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat Immunol. 2020; 21(9): 1022-1033. [CrossRef]
- Boulos JC, Omer EA, Rigano D, Formisano C, Chatterjee M, Leich E, Klauck SM, Shan LT, Efferth T. Cynaropicrin disrupts tubulin and c-Myc-related signaling and induces parthanatos-type cell death in multiple myeloma. Acta Pharmacol Sin. 2023. Epub ahead of print. [CrossRef]
- Liu J, Peng Y, Wei W. Cell cycle on the crossroad of tumorigenesis and cancer therapy. Trends Cell Biol. 2022; 32(1): 30-44. [CrossRef]
- Peng X, He Y, Huang J, Tao Y, Liu S. Metabolism of Dendritic Cells in Tumor Microenvironment: For Immunotherapy. Front Immunol. 2021; 12:613492. [CrossRef]
- Guak H, Al Habyan S, Ma EH, Aldossary H, Al-Masri M, Won SY, Ying T, Fixman ED, Jones RG, McCaffrey LM, Krawczyk CM. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat Commun. 2018; 9(1): 2463. [CrossRef]
- Zhang Y, Li Q, Huang Z, Li B, Nice EC, Huang C, Wei L, Zou, B. Targeting Glucose Metabolism Enzymes in Cancer Treatment: Current and Emerging Strategies. Cancers 2022, 14, 4568. [CrossRef]
- Endicott M, Jones M, Hull J. Amino acid metabolism as a therapeutic target in cancer: a review. Amino Acids. 2021; 53 (8): 1169-1179. [CrossRef]
- Han C, Ge M, Ho PC, Zhang L. Fueling T-cell Antitumor Immunity: Amino Acid Metabolism Revisited. Cancer Immunol Res. 2021; 9(12): 1373-1382. [CrossRef]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009; 9(3): 162-74. [CrossRef]
- Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008; 111(8): 4233-44. [CrossRef]
- Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, Pisarev V, Sherman S, Sporn MB, Gabrilovich D. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. 2011; 121(10): 4015-29. [CrossRef]
- De Sanctis F, Sandri S, Ferrarini G, Pagliarello I, Sartoris S, Ugel S, Marigo I, Molon B, Bronte V. The emerging immunological role of post-translational modifications by reactive nitrogen species in cancer microenvironment. Front Immunol. 2014; 5:69. [CrossRef]
- Campesato LF, Budhu S, Tchaicha J, Weng CH, Gigoux M, Cohen IJ, Redmond D, Mangarin L, Pourpe S, Liu C, Zappasodi R, Zamarin D, Cavanaugh J, Castro AC, Manfredi MG, McGovern K, Merghoub T, Wolchok JD. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat Commun. 2020; 11(1): 4011. [CrossRef]
- Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, Fiskesund R, Xie J, Liu J, Yin X, Jin X, Chen D, Tang K, Ma J, Zhang H, Yu J, Yan J, Liang H, Mo S, Cheng F, Zhou Y, Zhang H, Wang J, Li J, Chen Y, Cui B, Hu ZW, Cao X, Xiao-Feng Qin F, Huang B. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell. 2018; 33(3): 480-494.e7. [CrossRef]
- Halaby MJ, McGaha TL. Amino Acid Transport and Metabolism in Myeloid Function. Front Immunol. 2021; 12: 695238. [CrossRef]
- Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011; 478(7368): 197-203. [CrossRef]
- Mondanelli G, Bianchi R, Pallotta MT, Orabona C, Albini E, Iacono A, Belladonna ML, Vacca C, Fallarino F, Macchiarulo A, Ugel S, Bronte V, Gevi F, Zolla L, Verhaar A, Peppelenbosch M, Mazza EMC, Bicciato S, Laouar Y, Santambrogio L, Puccetti P, Volpi C, Grohmann U. A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity. 2017; 46(2): 233-244. [CrossRef]
- Fujiwara Y, Kato S, Nesline MK, Conroy JM, DePietro P, Pabla S, Kurzrock R. Indoleamine 2,3-dioxygenase (IDO) inhibitors and cancer immunotherapy. Cancer Treat Rev. 2022; 110: 102461. [CrossRef]
- Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, Arance A, Carlino MS, Grob JJ, Kim TM, Demidov L, Robert C, Larkin J, Anderson JR, Maleski J, Jones M, Diede SJ, Mitchell TC. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019; 20(8): 1083-1097. [CrossRef]
- Gide TN, Allanson BM, Menzies AM, Ferguson PM, Madore J, Saw RPM, Thompson JF, Long GV, Wilmott JS, Scolyer RA. Inter- and intrapatient heterogeneity of indoleamine 2,3-dioxygenase expression in primary and metastatic melanoma cells and the tumour microenvironment. Histopathology. 2019; 74(6):817-828. [CrossRef]
- Zehui Li, Grace Yang, Shuang Zhou, Xin Wang, Xiyan Li; Abstract C065: Dietary deprivation of non-essential amino acids improves anti-PD-1 immunotherapy in murine colon cancer. Mol Cancer Ther. 2019; 18 (12_Supplement): C065. [CrossRef]
- Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005; 8 (1): 7–12.
- Lieberman, M. Lieberman, M., and A. Peet, eds. Marks' Basic Medical Biochemistry: A Clinical Approach, 5th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2018, Chapter 27: Pentose Phosphate Pathway, Chapter 39: Purine and Pyrimidine Synthesis.
- Burhans WC, Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007; 35(22): 7545–56.
- Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol. 2014; 5: 304.
- Mazziotta C, Rotondo JC, Lanzillotti C, Campione G, Martini F, Tognon M. Cancer biology and molecular genetics of A3 adenosine receptor. Oncogene. 2022; 41(3):301-308. [CrossRef]
- Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008; 7(9): 759-70. [CrossRef]
- Wu HL, Gong Y, Ji P, Xie YF, Jiang YZ, Liu GY. Targeting nucleotide metabolism: a promising approach to enhance cancer immunotherapy. J Hematol Oncol. 2022; 15 (1): 45. [CrossRef]
- Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019; 51(2): 202–6.
- Nagarajan SR, Butler LM, Hoy AJ. The diversity and breadth of cancer cell fatty acid metabolism. Cancer Metab. 2021; 9 (1):2. [CrossRef]
- Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell. 2017; 170(4): 605–35.
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007; 7(10): 763–77.
- Munir R, Lisec J, Swinnen JV, Zaidi N. Lipid metabolism in cancer cells under metabolic stress. Br J Cancer. 2019; 120 (12): 1090–8.
- Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015; 36(2): 81-91. [CrossRef]
- Lim SA, Su W, Chapman NM, Chi H. Lipid metabolism in T cell signaling and function. Nat Chem Biol. 2022; 18(5): 470-481. [CrossRef]
- Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, To TKJ, Schug Z, Basu S, Wang F, Ricciotti E, DiRusso C, Murphy ME, Vonderheide RH, Lieberman PM, Mulligan C, Nam B, Hockstein N, Masters G, Guarino M, Lin C, Nefedova Y, Black P, Kagan VE, Gabrilovich DI. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019; 569(7754): 73-78. [CrossRef]
- Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018; 19(2): 108-119. [CrossRef]
- O'Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016; 213(1): 15-23. [CrossRef]
- Oren Y, Tsabar M, Cuoco MS, Amir-Zilberstein L, Cabanos HF, Hütter JC, Hu B, Thakore PI, Tabaka M, Fulco CP, Colgan W, Cuevas BM, Hurvitz SA, Slamon DJ, Deik A, Pierce KA, Clish C, Hata AN, Zaganjor E, Lahav G, Politi K, Brugge JS, Regev A. Cycling cancer persister cells arise from lineages with distinct programs. Nature. 2021; 596 (7873): 576-582. [CrossRef]
- Jung SH, Lee HC, Hwang HJ, Park HA, Moon YA, Kim BC, et al. Acyl-CoA thioesterase 7 is involved in cell cycle progression via regulation of PKCzeta-p53-p21 signaling pathway. Cell Death Dis. 2017; 8(5): e2793.
- Quail DF, Taylor MJ, Postovit LM. Microenvironmental regulation of cancer stem cell phenotypes. Curr Stem Cell Res Ther. 2012; 7(3): 197-216. [CrossRef]
- Emami Nejad A, Najafgholian S, Rostami A, Sistani A, Shojaeifar S, Esparvarinha M, Nedaeinia R, Haghjooy Javanmard S, Taherian M, Ahmadlou M, Salehi R, Sadeghi B, Manian M. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: a novel approach to developing treatment. Cancer Cell Int. 2021; 21(1): 62. [CrossRef]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119(6): 1420-8. Erratum in: J Clin Invest. 2010; 120(5): 1786. [CrossRef]
- Shekhova E. Mitochondrial reactive oxygen species as major effectors of antimicrobial immunity. PLoS Pathog. 2020; 16(5): e1008470. [CrossRef]
- Sasidharan Nair V, Saleh R, Toor SM, Cyprian FS, Elkord E. Metabolic reprogramming of T regulatory cells in the hypoxic tumor microenvironment. Cancer Immunol Immunother. 2021; 70(8): 2103-2121. [CrossRef]
- Emami Nejad A, Najafgholian S, Rostami A, Sistani A, Shojaeifar S, Esparvarinha M, Nedaeinia R, Haghjooy Javanmard S, Taherian M, Ahmadlou M, Salehi R, Sadeghi B, Manian M. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: a novel approach to developing treatment. Cancer Cell Int. 2021; 21(1): 62. [CrossRef]
- Jin J, Lin J, Xu A, Lou J, Qian C, Li X, Wang Y, Yu W, Tao H. CCL2: An Important Mediator Between Tumor Cells and Host Cells in Tumor Microenvironment. Front Oncol. 2021 Jul 27; 11:722916. [CrossRef]
- Liu J, Geng X, Hou J, Wu G. New insights into M1/M2 macrophages: key modulators in cancer progression. Cancer Cell Int. 2021; 21(1): 389. [CrossRef]
- Chen G, Wu K, Li H, Xia D, He T. Role of hypoxia in the tumor microenvironment and targeted therapy. Front Oncol. 2022; 12: 961637. [CrossRef]
- Zhang RS, Li ZK, Liu J, Deng YT, Jiang Y. WZB117 enhanced the anti-tumor effect of apatinib against melanoma via blocking STAT3/PKM2 axis. Front Pharmacol. 2022; 13:976117. [CrossRef]
- Pliszka M, Szablewski L. Glucose Transporters as a Target for Anticancer Therapy. Cancers (Basel). 2021; 13(16): 4184. [CrossRef]
- Linke C, Wösle M, Harder A. Anti-cancer agent 3-bromopyruvate reduces growth of MPNST and inhibits metabolic pathways in a representative in-vitro model. BMC Cancer. 2020; 20(1): 896. [CrossRef]
- Gebbia V, Borsellino N, Testa A, Latteri MA, Milia V, Valdesi M, Giotta F, Gebbia N, Colucci G. Cisplatin and epirubicin plus oral lonidamine as first-line treatment for metastatic breast cancer: a phase II study of the Southern Italy Oncology Group (GOIM). Anticancer Drugs. 1997; 8(10): 943-8. [CrossRef]
- Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, Rocha Lima CM, Schlesselman JJ, Tolba K, Langmuir VK, Kroll S, Jung DT, Kurtoglu M, Rosenblatt J, Lampidis TJ. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013; 71(2): 523-30. [CrossRef]
- ClinicalTrials.gov ID: NCT03163667: CB-839 With Everolimus vs. Placebo with Everolimus in Participants With Renal Cell Carcinoma (RCC) (ENTRATA). https://clinicaltrials.gov/study/NCT03163667.
- Yang JC, Sequist LV, Geater SL, Tsai CM, Mok TS, Schuler M, Yamamoto N, Yu CJ, Ou SH, Zhou C, Massey D, Zazulina V, Wu YL. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015; 16(7): 830-8. [CrossRef]
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/020896s037lbl.pdf.
- https://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021790s000_Dacogen_Approv.pdf.
- Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev. 2017; 276(1): 121-144. [CrossRef]
- Kurago Z, Guo G, Shi H, Bollag RJ, Groves MW, Byrd JK, Cui Y. Inhibitors of the CD73-adenosinergic checkpoint as promising combinatory agents for conventional and advanced cancer immunotherapy. Front Immunol. 2023; 14: 1212209. [CrossRef]
- Mozolewska P, Duzowska K, Pakiet A, Mika A, ŚledziŃski T. Inhibitors of Fatty Acid Synthesis and Oxidation as Potential Anticancer Agents in Colorectal Cancer Treatment. Anticancer Res. 2020; 40(9): 4843-4856. [CrossRef]
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125477s002lbl.pdf.
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/125085s0169lbl.pdf.
- ClinicalTrials.gov ID NCT00479817: Phase 2 AMG 386 in Comb. Paclitaxel for Subjects with Advanced Recurrent Epithelial Ovarian or Primary Peritoneal Cancer. https://clinicaltrials.gov/study/NCT00479817.
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215383s000lbl.pdf.
- Mondal M, Conole D, Nautiyal J, Tate EW. UCHL1 as a novel target in breast cancer: emerging insights from cell and chemical biology. Br J Cancer. 2022; 126(1): 24-33. [CrossRef]
- ClinicalTrials.gov ID NCT03145558: TATE Versus TACE in Intermediate Stage HCC (TATE). https://clinicaltrials.gov/study/NCT03145558.
- ClinicalTrials.gov ID NCT04895748: DFF332 as a Single Agent and in Combination with Everolimus & Immuno-Oncology Agents in Advanced/ Relapsed Renal Cancer & Other Malignancies. https://www.clinicaltrials.gov/study/NCT04895748.
- ClinicalTrials.gov ID NCT02564614: A Study of Hypoxia-inducible Factor 1a (HIF1A) Messenger Ribonucleic Acid (mRNA) Antagonist (RO7070179), to Demonstrate Proof-of-mechanism in Adult Participants With Hepatocellular Carcinoma (HCC). https://clinicaltrials.gov/study/NCT02564614.
- ClinicalTrials.gov ID NCT05935748: NKT2152 With Palbociclib & Sasanlimab in Subjects With Advanced Clear Cell Renal Cell Carcinoma (ccRcc). https://clinicaltrials.gov/study/NCT05935748.
- Brenner AJ, Floyd J, Fichtel L, Michalek J, Kanakia KP, Huang S, Reardon D, Wen PY, Lee EQ. Phase 2 trial of hypoxia activated evofosfamide (TH302) for treatment of recurrent bevacizumab-refractory glioblastoma. Sci Rep. 2021; 11(1): 2306. [CrossRef]
- ClinicalTrials.gov ID NCT00598806: Single Dose Intravesical Apaziquone Postoperative in Patients Undergoing TURBT for Noninvasive Bladder Cancer (SPI-612). https://clinicaltrials.gov/study/NCT00598806.
Figure 1.
Figure 1. Tumor Microenvironment. A. Warburg Effect: Positron emission tomography visualizes a tongue base carcinoma (upper arrowhead) and neck lymph node metastasis (lower arrowhead) using radioactive glucose analogs. B. Cancer-Associated Inflammation: The image shows multinucleated giant cells (yellow right-pointing arrowhead), atypical cancer cell mitosis (yellow left-pointing arrowhead), mononuclear leukocytes (red right-pointing arrowhead), and granulocyte (red left-pointing arrowhead).(H&E, 40x magnification) C. PD-L1 Expression (brown): The immunostain shows gradient expression of this immune checkpoint protein in mucosal cancer, negative in non-invasive component (top) and highest at the deepest invasion front (bottom) (20x magnification). D. Epithelial-Mesenchymal-Transition: Carcinoma with classic epithelioid cells (left) undergoes sarcomatoid/mesenchymal transformation with spindle morphology (middle) and loss of keratin expression by immunohistochemistry (right). (H&E, 40x magnification) E. Tumor-Immune Cell Crosstalk: Tumor cells (bottom-left) activate osteoclasts (arrowhead) to induce bone resorption (upper-right) in a lytic bone lesion. (H&E, 40x magnification).
Figure 1.
Figure 1. Tumor Microenvironment. A. Warburg Effect: Positron emission tomography visualizes a tongue base carcinoma (upper arrowhead) and neck lymph node metastasis (lower arrowhead) using radioactive glucose analogs. B. Cancer-Associated Inflammation: The image shows multinucleated giant cells (yellow right-pointing arrowhead), atypical cancer cell mitosis (yellow left-pointing arrowhead), mononuclear leukocytes (red right-pointing arrowhead), and granulocyte (red left-pointing arrowhead).(H&E, 40x magnification) C. PD-L1 Expression (brown): The immunostain shows gradient expression of this immune checkpoint protein in mucosal cancer, negative in non-invasive component (top) and highest at the deepest invasion front (bottom) (20x magnification). D. Epithelial-Mesenchymal-Transition: Carcinoma with classic epithelioid cells (left) undergoes sarcomatoid/mesenchymal transformation with spindle morphology (middle) and loss of keratin expression by immunohistochemistry (right). (H&E, 40x magnification) E. Tumor-Immune Cell Crosstalk: Tumor cells (bottom-left) activate osteoclasts (arrowhead) to induce bone resorption (upper-right) in a lytic bone lesion. (H&E, 40x magnification).
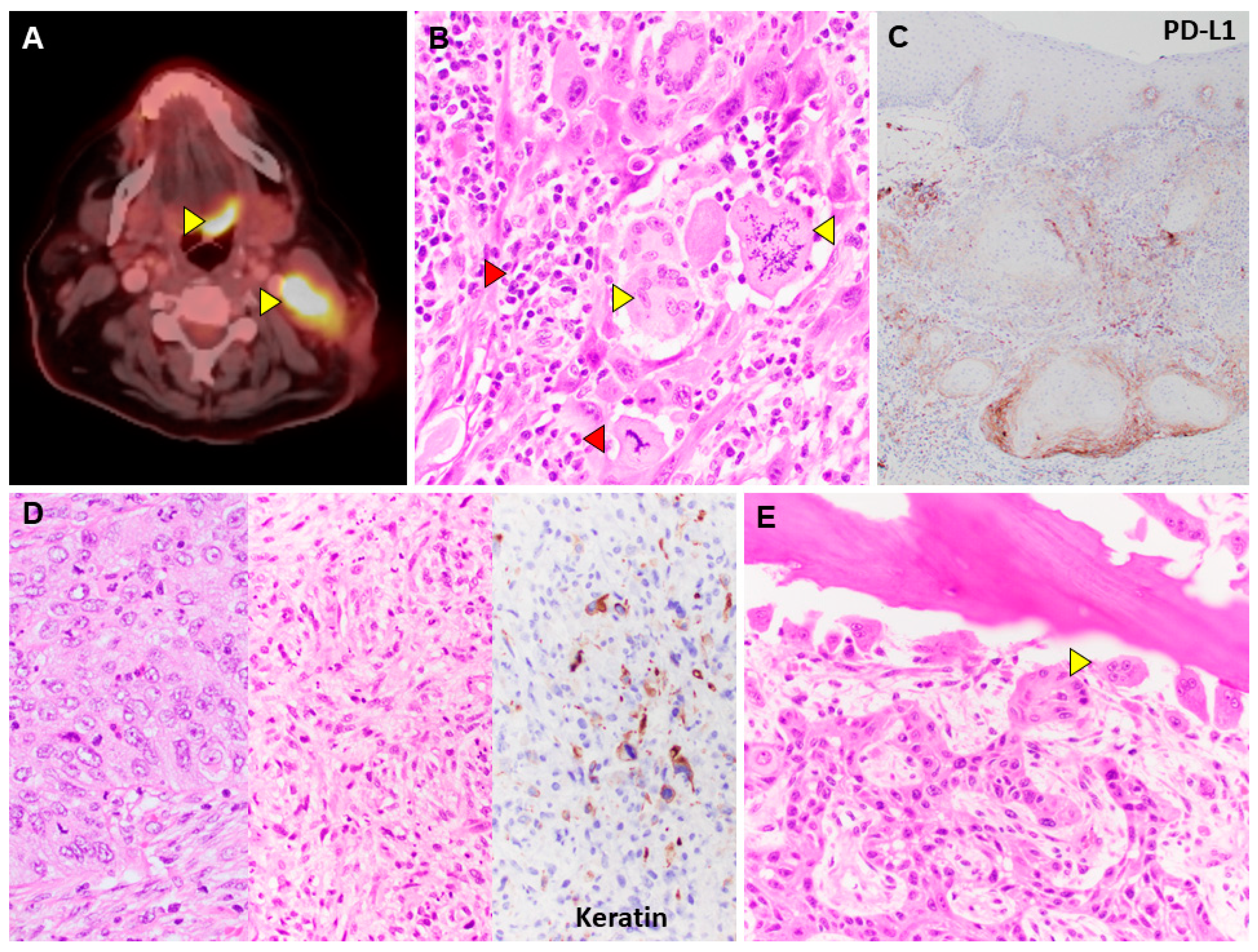

Table 1.
Critical Disparities in Metabolic Processes Between Normal and Tumor Cells.
| Metabolite | Immune Cells | Malignant Cells | Net Effect |
|---|---|---|---|
| Glucose | Regulated uptake Resting cells: Oxidative phosphorylation Activated cells: glycolysis |
Unregulated uptake Glycolysis -> Lactate, hypoxia, acidosis |
Malignant cells outcompete immune cells; acidotic environment leads to impaired immune function |
| Amino acids |
Regulated, influenced by activation state |
Dysregulated, upregulation of specific pathways, e.g., glutamine-> tricarboxylic acid cycle |
Tumor induced amino acid depletion leads to impaired immune function |
| Nucleotides | Synthesis is tightly regulated, switch between de novo and salvage/recycling pathways |
Upregulation of de novo synthesis pathways over recycling leads to depletion of local resources | Nucleotide depletion leads to impaired immune activation and proliferation |
| Fatty acids | Fatty acid transporters are tightly regulated, skewed towards lipolysis | Upregulation of fatty acid transporters and de novo synthesis for energy and membrane assembly |
Depletion of fatty acids leads to impaired immune activation and proliferation |
| Oxygen | Highly dependent on oxygen for generation of reactive oxygen species and production of cytokines | High resistance to hypoxia through activation of hypoxia-inducible factors and glycolysis | Hypoxia hinders the movement and function of immune cells – Favors function of regulatory over effector T-cells |
Table 2.
Pharmacological agents targeting the TME.
| Metabolic pathway |
Mechanism of action | Pharmacological Agent | Stage of development |
| Glucose | GLUT, glucose transporter inhibitor Hexokinase inhibitor |
WZB117 BAY-876 3-Bromopyruvic acid (3-BrPA) Lonidamine (LND) 2-Deoxy-d-glucose (2-DG) |
Preclinical 66 Preclinical 67 Preclinical 68 Phase II 69 Phase I 70 |
| Amino acid | Selective glutaminase inhibitor BCAT1 inhibitor |
Telaglenastat Gabapentin |
Phase II 71 Preclinical 23 |
| Nucleotide | Targeting purine or pyrimidine pathways DNA synthesis blockage Adenosine pathway inhibitors |
CAD blockage: Afatinib Capecitabine Decitabine Oleclumab Dalutrafusp alfa HLX23 Anti CD73 antibodies |
FDA approved 72 FDA approved 73 FDA approved 74 Phase II 75 ~50 phase I/II trials 76 |
| Fatty acid | Acetyl-CoA carboxylase (ACC) inhibitor ATP-citrate lyase (ACLY) inhibitors |
5-tetradecyloxy-2-furancarboxylic acid (TOFA) GSK165 |
Preclinical 77 Preclinical 78 |
| Hypoxia | Antiangiogenic agents HIF inhibitors Hypoxia-activated/bioreductive prodrugs |
Ramucirumab Bevacizumab Trebananib Belzutifan 6RK73 DFF332 RO7070179 NKT2152 Tirapazamine, Evofosfamide Apaziquone |
FDA approved 79 FDA approved 80 Phase II 81 FDA approved 82 Preclinical 83 Phase I 84 Phase I 85 Phase II 86 Phase II 87 Phase II 88 Phase III 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



