
Submitted:
04 November 2023
Posted:
06 November 2023
You are already at the latest version
Abstract
Poa crymophila is a perennial, cold-tolerant, native grass species, widely distributed to the Qing-hai-Tibet Plateau. However, the molecular mechanism behind the cold stress tolerance and role of key regulatory genes and pathways of P. crymophila are poorly understood yet. Therefore, the present study investigated the physiological and transcriptome responses of P. crymophila’s roots, stems, and leaves under cold stress to explore the molecular mechanism of cold tolerance. Results of the present study suggested that cold stress significantly changed the physiologic characteristics of roots, stems, and leaves of P. crymophila. In addition, the transcriptome results showed that 4434, 8793, and 14942 differentially expressed genes (DEGs) were identified in roots, stems, and leaves, respectively; however, 464 DEGs were commonly identified in these three tissues. The Gene On-tology (GO) results showed that a large numbers of DEGs were significantly enriched in the pho-tosynthetic related categories in leaves. In addition, the “response to stimulus” category was sig-nificantly enriched in roots and stems. The Kyoto Encyclopedia of Genes and Genomes (KEGG) results showed that DEGs involved in “phenylpropanoid biosynthesis” were significantly enriched in roots, “photosynthesis” and “circadian rhythm-plant” pathways significantly enriched in stems and leaves, starch and sucrose metabolism, and galactose metabolism were pathways significantly enriched in three tissues. Weighted gene coexpression network analysis (WGCNA) identified Hub genes involved in P. crymophila cold response. This study provides new insights into the molecular mechanisms underlying the cold tolerance of P. crymophila belowground and aboveground tissues. In addition, specific genes involved in Ca2+ signaling, ROS scavenging system, hormones, circadian clock, photosynthesis, and transcription factors (TFs) were identified in P. crymophila. The identi-fication of key genes may provide valuable resources for further functional genomic and breeding studies.
Keywords:
Poa crymophila
; cold stresses
; physiology
; transcriptome analysis
; weighted correlation network analysis (WGCNA)
; molecular mechanisms
1. Introduction
Global climate change imposes serious threats to crop growth and yields worldwide [1]. Cold is a common abiotic stress that has become a major environmental factor affecting plant life cycle and geographic distribution as well as crop yields by influencing plant growth and development [2]. The immense impact of cold stress on global agricultural productivity is undeniable, with cold damage costing approximately $2 billion annually which includes about 13 million hectares of rice each year [3,4]. Therefore, understanding the cold resistance mechanisms of plants to improve their cold tolerance is essential to maintain sustainable agricultural production.
Plants have evolved multiple methods to respond to cold stress during long-term environmental adaptations [5]. Cold stress signals are first sensed bythe outer parts of the plant through a highly organized signaling system that further generates a series of response mechanisms in vivo [6]. These responses mainly include modification of the membrane lipid composition, synthesis of osmoprotectants, activating the reactive oxygen species (ROS) scavenging system, signaling cascade, and changes in the expression profile of cold-responsive genes [5,7]. ICE (Inducer of CBF Expression) is a MYC-type bHLH transcription factor, including ICE1 and ICE2, which indces and positively regulates the expression of the CBFs, which enhances cold tolerance in Arabidopsis thaliana [8,9]. Additionally, CBFs/DREBs (C-repeat binding factors/dehydration-responsive element binding proteins), which include CBF1/DREB1B, CBF2/DREB1C, and CBF3/DREB1A, are transcription factors of the AP2/ERF (APETALA2/Ethylene-responsive factor) family that regulate the expression of CORs (cold-regulated genes) [2,10,11]. These genes encode osmoprotectants, protein kinases, lipids, hormones, and proteins, and directly participate in responding to cold stress to enhance plant tolerance [2,12]. Consequently, ICE, CBFs, and CORs constitute a crucial signaling pathway, the ICE-CBF-COR cascade which mitigates cold stress in plants [5,13].
In addition, plant responses to cold stress are regulated by other factors such as Ca2+ signaling, hormones, circadian rhythms, and photosynthesis [7,12]. The Ca2+ signaling pathway is considered as most common low-temperature response pathway in plants, including calmodulin (CaM), calmodulin-like protein (CML), calcineurin B-like protein (CBL), calcium-dependent protein kinase (CPK/CDPK), calcium-dependent protein kinase (CPK/CDPK), calmodulin-binding transcription activator (CAMTA), which interact and regulate the CBF/DREB1 genes in response to cold stress [5,12]. Moreover, Ca2+ is also associated with ROS signaling, triggering ROS and/or abscisic acid (ABA) to directly activate the mitogen-activated protein kinase (MAPK) cascade [2]. ROS induces the expression of TFs through the accumulation of plant hormones, and TFs alleviate the damage of ROS by regulating the expression of ROS-responsive genes [2,14]. Recent studies suggest that photosynthesis and circadian rhythms pathways are crucial pathways that play an important role in cold stress tolerance [15]. CBF genes are not only induced by cold stress but also regulated by the circadian clock and light signals [16].
Besides, in the recent years, transcriptome studies have provided a new perspective for research on the plant response to cold stress [3,7,17]. A previous study reported that the plant circadian pathway was crucial for the response of Helictotrichon virescens to cold stress; the expression of DEGs encoding LHY and HY5 were strongly up-regulated during cold stress [7]. Transcriptome analysis revealed that Ca2+, ROS, hormone signaling, the circadian clock, and photosynthetic antenna proteins may significantly contribute to cold tolerance in Vicia sativa L. through CBF-dependent or independent transcriptional pathways [17]. Transcriptome analysis of Triticum aestivum L. at 4°C low-temperature showed that starch and sucrose metabolism, glutathione metabolism, and plant hormone signal transduction pathways were significantly enriched, and many genes belonging to the bHLH, MYB, NAC, WRKY, and ERF TF families were highly expressed [18]. In addition, the WGCNA identified several key genes involved in the development of seedlings under cold stress [18]. Therefore, the previous studies suggested that transcriptome analysis can reveal the molecular mechanisms of plant stress tolerance and explore the potential functions of key genes.
The Qinghai-Tibet Plateau is known as the Roof of the World, the Third Pole and the Water Tower of Asia, which is an important shield for China’s ecological security, as well as an important animal husbandry production base [19]. However, the Qinghai-Tibet Plateau has a typical plateau continental climate, with a large temperature difference between day and night, low annual average temperature, and a short frost-free period which shows the average monthly temperature is between -20°C ~ -10°C [20,21]. The extreme climatic conditions of the Qinghai-Tibet Plateau impose selective pressures on the evolution of phenotypic and physiological characteristics. Many native species including Elymus s nutans, E. sibicicus, Poa pratensis and P. crymophila are widely distributed and utilized species on the Qinghai-Tibet Plateau due to their strong environmental adaptability and long-term natural domestication [20,22,23]. Of these native grasses, P. crymophila is widely distributed to humid grasslands, alpine grasslands, forest margins, hillsides, valleys, and tidal flats at an altitude of 2150 ~ 4800 m on the Qinghai-Tibet Plateau [20]. After more than 30 years of selection, cultivation, and domestication, P. crymophila has been cultivated as ecological grass variety widely used in ecological restoration of degraded grassland in the alpine region. It has excellent cold tolerance and can survive under about - 40°C [20]. However, the physiological and molecular mechanism behind the cold stress tolerance and role of key regulatory genes and pathways of P. crymophila are poorly understood yet.
In plants, both the aboveground and underground parts play a crucial role in perceiving environmental changes. Despite having different developmental trajectories, these two components are highly coordinated in their biological processes to respond to abiotic stress [17]. However, the specific and common molecular network and cold-responsive key genes in the aboveground and underground tissues of P. crymophila have not been reported yet. Therefore, the present study investigates the physiological and transcriptome responses of P. crymophila’s roots, stems, and leaves under cold stress to explore the molecular mechanism of cold tolerance. These results provide important clues for further studies on the molecular regulatory mechanisms between roots, stems, and leaves of P. crymophila in response to cold stress and further providing reference for studying the function of genes participating in cold response in P. crymophila.
2. Results
2.1. Physiological Responses of P. crymophila under Cold Stress
Forty-five days old P. crymophila seedlings were subjected to cold treatments for 3 and 6 days, respectively. Despite the 4°C treatment, no observable phenotypic differences were observed compared with control (Figure 1A). However, cold stress resulted in an increase in proline (excluding the roots treated for 3 days) and MDA content, as well as the activity of antioxidant enzymes (SOD, CAT, and POD) in P. crymophila (Figure 1B-D). The increase in proline content in each tissue was higher at 6 days (Figure 1B). Specifically, proline content of roots increased by 29.9%, and leaves by 10.6% compared with 3 days of cold stress. As compare to control, MDA content significantly increased under cold stress and reached a maximum at 6 days of cold treatment (an increase of at least 40%). Compared with roots, the increase in MDA content in stems and leaves was higher after 6 days of cold treatment (Figure 1B). In addition, the activities of antioxidants such as SOD, CAT, and POD in leaves were higher after 3 days of cold treatment when compared with roots (Figure 1B). Among them, the SOD, CAT, and POD activities in leaves after 3 days of cold stress were 1.92, 2.06, and 1.92 times higher than 6 days, respectively. For the roots, the SOD, CAT, and POD activities under 3 days of cold stress were 1.08, 1.02, and 1.29 times higher than those after 6 days of cold treatment, respectively.
2.2. Transcriptome Assembly and Annotation
A total of 27 samples were employed to construct the cDNA library. After trimming, 193.28 Gb databases were retrieved, or approximately 5.99 Gb per sample (three biological replicates) (Table S1). Furthermore, we obtained approximately 20.03 to 29.68 million clean reads per library, mapped at a ratio of 65.45%. The number of bases ranged from 5.99 billion to 8.85 billion. The Q3 base percentage was > 92.69%. The GC content was approximately 54.01% (Table S1). These results suggested that the sequencing output and high-quality reads were adequate for further analysis.
After de novo assembly of high-quality sequences by Trinity software, a total of 1,868,509 transcripts and 53,893 unigenes were generated with average lengths of 1108.27bp and 2243.7bp respectively, and the N50 lengths of 1681bp and 2941bp respectively (Table S2). The size-distribution analysis showed that the lengths of 41,330 (76.69%) unigenes were greater than 1000 bp (Table S2). These results demonstrated the effectiveness of Illumina sequencing in rapidly capturing a large portion of the transcriptome. Further, 46,542 (86.36%) of the unigenes were successfully annotated in nine databases (Table S3).
Currently, homologous species matched results in Nr database showed that the top 8 mostly annotated plants belong to Poaceae species, and Triticum aestivum (10,398, 23.26%) was the best match for P. crymophila, followed by Brachypodium distachyon (7,328, 16.39%) and Aegilops tauschii (6,662, 14.90%) (Figure 2A). In addition, the E-value distribution and similarity statistics of the annotated Unigenes showed that 89.36% of the unigenes had an E-value less than 1e-30 and had high homology (Figure 2B), unigenes with a similarity greater than 60% and greater than 80% accounted for 86.93% and 58.91%, respectively, indicating that the unigenes annotation information of P. crymophila has good reliability (Figure 2C).
2.3. Identification and Analysis of DEGs
A total of 22,233 DEGs were identified in all tissues under cold treatments. The number of DEGs in leaves was three times higher than in roots and 1.5 times higher than in stems (14942, 4434, and 8793 DEGs, respectively) (Figure 3A). Among them, 1975, 4892, and 9894 DEGs were found to be unique to roots, stems, and leaves, respectively. These results indicated that cold-induced transcriptome responses were largely tissue-specific. Notably, 464 genes were commonly expressed in three tissues (Figure 3A). The comparison of different treatment time points found more DEGs, 2888 and 7293 DEGs, were specifically expressed between R0 vs. R3 and L0 vs. L3, respectively (Figure 3A). Moreover, it was found that more up-regulated DEGs were obtained in roots (3012 DEGs) and leaves (5864 DEGs) under 3 days of cold treatment than under 6 days of cold treatment (Figure 3B). These results suggest that more transcripts responded to cold stress during 3 days than during 6 days. To obtain an overall picture of the impact of cold stress on transcriptional profiling, the expression patterns of all DEGs identified in the three tissues at the different time points were clustered together. Six expression patterns were identified in each tissue (P < 0.05) (Figure S1). The results showed that the expression of many more DEGs was up-regulated in leaves under cold stress, followed by stems and roots.
2.4. GO Enrichment Analysis of DEGs
To further determine the coordinated response mechanisms in the roots, stems, and leaves of P. crymophila seedlings under cold stress, GO category enrichment analysis was applied to determine the function of the DEGs expressed under cold stress. According to the P-value and number of DEGs associated with GO terms, the top 25 significantly enriched GO terms were selected and further analyzed. GO results showed that DEGs were most enriched in the “response to stimulus” category in roots and stems under cold stress, and most DEGs were up-regulated (Figure 4A, B; Table S4). In addition, categories associated with water response (“water transmembrane transporter activity”, “water channel activity”, and “water transport”) and photosynthesis (“chloroplast”, “plastid”, “obsolete chloroplast part”, “obsolete plastid part”, “chloroplast stroma”, “chloroplast envelope”, “plastid stroma”) were significantly enriched in roots and stems, respectively. For leaves, inversely, large numbers of DEGs were significantly enriched in photosynthesis-related categories (“chloroplast”, “obsolete chloroplast part”, “photosystem II”, “photosynthesis”, “plastid stroma”, “chloroplast stroma”, and “plastid”) under cold stress, and the DEGs involved with these categories were predominantly down-regulate (Figure 4C; Table S4).
2.5. KEGG Enrichment Analysis of DEGs
The DEGs were further annotated to the reference pathways in the KEGG database to explore the key biological pathways in response to cold stress in P. crymophila, and the top 20 pathways were screened as the most enriched pathways. The DEGs of the root were significantly enriched in the “phenylpropanoid biosynthesis” both in 3 days and 6 days of cold stress, and most of them were up-regulated (Figure 5A; Table S5). Furthermore, it was found that most of the DEGs were significantly enriched in the “MAPK signaling pathway-plant” (3 days), “plant hormone signal transduction” (6 days) pathways and were up-regulated in roots (Figure 5A; Table S5). The DEGs involved in the “Circadian rhythm-plant” (39 DEGs) and “photosynthesis” (41 DEGs) pathways were significantly enriched in stems, and were up-regulated (Figure 5B; Table S5). For leaves, “photosynthesis” and “starch and sucrose metabolism”, “galactose metabolism”, “circadian rhythm-plant”, “glutathione metabolism”, “arginine and proline metabolism”, “biosynthesis of amino acids” pathways were significantly enriched both at 3 and 6 days of cold stress (Figure 5C). Most of the DEGs involved in the “photosynthesis” pathway were down-regulated in leaves while in “circadian rhythm-plant” pathway were up-regulated (Table S5). Notably, all DEGs involved in “photosynthesis-antenna proteins” (33 DEGs) were significantly down-regulated after 3 days of cold stress (Table S5). In addition, “starch and sucrose metabolism” and “galactose metabolism” pathways were significantly enriched in roots (3 days), stems (6 days), and leaves (both at 3 and 6 days) (Figure 5).
2.6. Identification of DEGs Related to Transcription Factors (TFs)
Numerous transcription factor (TF) families, including bHLH, bZIP, AP2/ERF, WRKY, NAC, MYB, C2H2, HSF, and others, were identified in P. crymophila following exposure to cold treatment. The results indicated up-regulation of most of these families in roots (52 DEGs), stems (61 DEGs), and leaves (56 DEGs) during cold treatment (Table S6). Notably, most of WRKY (12 DEGs), AP2/ERF (23 DEGs), and NAC (6 DEGs) TFs were up-regulated in roots, particularly at 6 days, most of the bHLH (11 DEGs) and MYB (11 DEGs) TFs were up-regulated in the stems, most of the AP2/ERF (18 DEGs), bZIP (7 DEGs), and HSF (6 DEGs) TFs were up-regulated in the leaves (Table S6). All 23 DEGs belonged to 6 TF families (bHLH, AP2/ERF, WRKY, HSF, MYB, bZIP) were commonly expressed in roots, stems, and leaves under cold treatment, which were mainly up-regulated in roots and down-regulated in stems and leaves (Figure 6; Table S6). Furthermore, the largest group of TFs belonged to the AP2/ERF family; the 1, 1, and 5 AP2/ERF family members were up-regulated in roots, stems, and leaves under cold treatment for 3 and 6 days, and 4 and 8 AP2/ERF family members were down-regulated in stems and leaves under 3 and 6 days of cold stress (Table S6). The EREBP1 DEGs in roots (Unigene_197025) and stems (Unigene_316002) were specifically up-regulated after 6 days of cold treatment in roots and significantly up-regulated after 3 and 6 days in stems, respectively, while the EREBP1 DEGs in leaves (Unigene_071491) were down-regulated under 6 days of cold treatment (Table S6). In conclusion, most TFs were specifically expressed in the roots, stems, and leaves of P. crymophila.
2.7. Identification of Key DEGs Involved in Cold Responses
A total of 97 DEGs involved in the CBF signaling pathway were expressed in P. crymophila under cold treatment, divided into 14 categories (Table S7). Compared with roots and stems, leaves acquired more DEGs (61) involved in ICE-CBF-COR pathway, and most of them (31) were up-regulated, among which 23 DEGs were up-regulated after 3 and 6 days of cold treatment. Among the 37 DEGs identified in roots, 35 were up-regulated, 3 of which were up-regulated after 3 and 6 days of cold treatment, 25 of the 37 DEGs identified in stems were up-regulated, and 6 of them were up-regulated after 3 and 6 days of cold treatment (Table S7). Most of the DEGs showed tissue-specific expression patterns, among which 9, 13, and 9 were specifically up-regulated in roots, stems and leaves, respectively, especially ICE1, PAL, CSP, COR, CRPK, BLT14, and ZAT DEGs, and 1 ICE1 DEG was up-regulated in stems and leaves, 6 PAL DEGs were up-regulated in roots, 2 CSP and 3 COR DEGs were up-regulated in leaves, 2 each of CRPK and ZAT DEGs were up-regulated in stems, and3 BLT14 DEGs were specifically up-regulated in stems and leaves. In addition, 3, 4, and 6 DEGs were specifically down-regulated in roots, stems, and leaves, especially cold-responsive protein (1), PAL genes (3) and MADS-box protein (1). In P. crymophila, 1 CBF2/DREB1C, 2 CBF3/DREB1A DEGs were commonly expressed in roots, stems and leaves, and all of them were up-regulated in roots and leaves, down-regulated in stems (Figure 7A). Meanwhile, one each for COR410, COR413PM1, LEA1, and LEA14-A was significantly expressed in all tissues and up-regulated in stems and leaves, down-regulated in roots. One BLT14 and two ZAT DEGs were up-regulated in roots, and down-regulated in stems and leaves (Figure 7A).
In this study, 54, 103, and 120 DEGs related to Ca2+ signaling were identified in the roots, stems, and leaves of P. crymophila after cold treatment. Among them, 16 CML, 7 CPK/CDPK, 4 CAMTA, 12 CIPK, 2 ANN, 3 CNGC, and 2 CSC1 DEGs were up-regulated in roots (Table S8). 1 CML, 11 CPK/CDPK, 6 CAMTA, 23 CIPK, 3 CBL, 1 GLR, 5 ANN, 1 CNGC, and 3 CSC1 DEGs were up-regulated in the stems. 11 CML, 7 CPK/CDPK, 4 CAMTA, 30 CIPK, 4 CBL, 5 GLR, 4 CNGC, and 3 CSC1 DEGs were up-regulated in leaves (Table S8). In addition, 18co-induced DEGs were identified as associated with Ca2+ signaling, most of which were up-regulated in roots (especially 3 days) and down-regulated in stems and leaves (especially at 3 days) (Figure 7B, Table S8). Among them, 1 RLK, 1 CPK/CDPK, 1 CNGC, 7 CML and 4 CIPK DEGs were up-regulated in roots and down-regulated in stems and leaves (Figure 7B, Table S8). These results indicated that the Ca2+ signaling system plays an essential role in plant low-temperature signal transduction pathway in P. crymophila.
The physiological and transcriptome analysis results showed that ROS scavenging system of P. crymophila was activated under cold treatment. The activities of antioxidant enzymes (SOD, CAT, POD) in roots, stems, and leaves were significantly increased after cold treatment (Figure 1). According to the enrichment results of KEGG, the “MAPK signaling pathway-plant” was significantly enriched (Figure 5). In P. chillmophila, 68, 103, and 120 DEGs related to the ROS scavenging system were identified in roots, stems, and leaves, respectively. Among them, 1 SOD, 1 CAT, 23 POD, 11 MAPK, 9 GST, 1 MDAR, 1 GR, 1 GPX, 2 APX, 3 GDHs, 3 GSH, 4 RBOH, and 3 AOX DEGs were up-regulated in the roots, 2 CAT, 6 SOD, 9 POD, 12 MAPK, 12 GST, 3 MDAR, 5 GR, 4 GPX, 5 APX, 2 GDH, 2 GSH, 10 RBOH and 8 AOX DEGs were up-regulated in the stems, 1 CAT, 4 POD, 16 MAPK, 20 GST, 5 MDAR, 4 GR, 3 GPX, 2 APX, 3 GDH, 4 GSH, 12 RBOH and 6 AOX DEGs were up-regulated in the leaves (Table S9). In addition, 11 co-induced DEGs were identified to be related to ROS scavenging and most of them were down-regulated in roots and up-regulated in stems and leaves (especially at 3 days) (Figure 7C, Table S9). Among them, 2 POD, 5MAPK, 1 GST, and 1 GSH DEGs were up-regulated in roots and down-regulated in stems and leaves (Figure 8C).
Through transcriptome analysis, the “phytohormone signal transduction” was activated in P. crymophila under cold stress (Figure 5). In this study, 62, 84, and 136 DEGs related to hormone signaling were identified in the roots, stems, and leaves of P. crymophila, respectively. Among them, Abscisic acid (ABA)-related DEGs (PYLs, SnRK2s, PP2Cs) were mainly up-regulated in roots, stems, and leaves, auxin (IAA)-related DEGs (ARFs, IAAs, SAURs, GH3s) were mainly down-regulated in roots and leaves, and mainly up-regulated in stems, gibberellin (GA)-related (GA2OXs, GIDs, DELLAs) and ethylene (ETH)- related DEGs (EIN3s, EIN2s, ERFs, ETRs) showed different expression patterns in roots, stems, and leaves. Moreover, brassinosteroid (BR)-related DEGs (BRH1s, BRI1s) mainly up-regulated in roots, stems, and leaves and jasmonic acid (JA)-related DEGs (GH3s) mainly up-regulated in roots and stems (Table S10). In addition, 28 co-induced DEGs were identified to be related to hormone signaling, most of which were up-regulated in roots and down-regulated in stems and leaves (Figure 7D, Table S10). Among them, 2 CCR3s, 2 ERF4s, and 3 CIGR2s were up-regulated in roots and down-regulated in stems and leaves (especially at 3 days).
In P. crymophila, the “photosynthesis” and “circadian rhythm-plant” pathways were significantly enriched in stems and leaves under cold stress (Figure 5). Meanwhile, many DEGs involved in photosynthesis and plant circadian clock regulation of cold response were also identified, and they showed tissues-specific expression (Table S11). In this study, except that 4, 1, and 8 DEGs related to the plant circadian clock were down-regulated in roots, stems, and leaves, the rest were all up-regulated (Table S11). Among these up-regulated DEGs, 19 PRR, 5 LHY, 10 HY5, and 6 GI DEGs were identified in roots, stems, and leaves. Notably, 4 and 7 PRR DEGs were found to be up-regulated in stems and leaves at 3 and 6 days of cold treatment, respectively, and 6 GIs DEGs were up-regulated in stems and leaves at 3 and 6 days of cold treatment (Table S11). Moreover, only 3 co-induced DEGs in roots, stems, and leaves were found to be associated with plant circadian clock and photosynthesis. Among them, PRR DEG (Unigene_523659) was up-regulated in roots and down-regulated in stems and leaves, Adagio proteins (Unigene_074056) were down-regulated in roots and up-regulated in stems and leaves (Figure 7E). FDX3 (Unigene_183317) was associated with the electron transport chain in photosynthesis, which was up-regulated in roots and down-regulated in stems and leaves (Figure 7E). In addition, 8 DEGs related to the photosynthetic electron transport chain (1DEGs was up-regulated, 7 DEGs were down-regulated), 16 DEGs related to photosystem Ⅱ (10 DEGs was up-regulated, 6 DEGs were down-regulated) were identified in leaves. Notably, 33 DEGs related to photosynthesis-antenna were down-regulated, 23 DEGs related to porphyrin and chlorophyll metabolism were up-regulated (Table S11).
2.8. WGCNA and Identification of Key Genes
After screening the low-expressed genes, 7,439 DEGs were retained for the WGCNA. To ensure the network was scale-free and had more biological significance, a soft threshold power 16 was chosen when the correlation coefficient squared was 0.9 to define the adjacency matrix (Figure 8A). Then, based on the DynamicTreeCut algorithm, setting the minimum number of module genes as 30, and the maximum module distance as 0.25, the gene modules were generated, and the modules with high similarity were further merged (Figure 8B, C). As shown in Figure 8D, 17 gene modules were finally generated after merging. The results indicate a strong positive correlation (r > 0.9) between antioxidant enzyme activities (SOD, CAT, and POD) and the brown module, while a negative correlation (r < -0.6) was observed with the salmon module.
To analyze the brown and salmon modules, GO enrichment (Figure 9A) analysis and KEGG enrichment (Figure 9B) analysis were performed. Additionally, co-expression networks were observed between brown and salmon modules (Figure 9C). It was found that the response to stimulus (235) and endomembrane system (32) were the most significantly enriched GO categories of the brown module and the salmon module, respectively. “Circadian rhythm-plant” and “Phagosome” were the most significantly enriched KEGG pathways of the brown and salmon modules, respectively. The first six hub genes in the brown module were annotated: ethylene-responsive transcription factor-like protein (At4g13040), BEL1-like homeodomain protein (BLH1), ABC transporter C family member 2 (ABCC2), UDP-glycosyltransferase (UGT73C1), ATP-dependent zinc metalloprotease (FTSH 1) and Protein ACTIVITY OF BC1 COMPLEX KINASE 7 (ABC1K7). Serine/threonine-protein kinase CTR1 (CTR1), carbamoyl-phosphate synthase large chain, chloroplastic (CARB), and beta-glucuronosyltransferase GlCAT14A were the first three hub genes in the salmon module, and most of them were up-regulated (Figure 9C; Table S12). The nine hub genes with higher connectivity in the module are more consistent with the activity of antioxidant enzymes, which laid the foundation for the subsequent research on cold-responsive genes of P. crymophila.
2.9. Experimental Validation
To validate the RNA sequencing (RNA-seq) data of the present study, the expression profiles of a subset of 12 DEGs involved in oxidation-reduction, membrane components, and hormone signal transduction were selected for qRT-PCR assays. The variation trends and errors of these 12 DEGs at different treatment points showed a high degree of consistency with the change tendency of the transcript FPKM values (Figure 10). The coefficients of determination obtained by linear regression analysis between the qRT-PCR and transcriptome data of the roots, stem, and leaves were R2 = 0.7, R2 = 0.78, and R2 = 0.85, respectively, and the correlations were positive (Figure S2). The high congruence between the RNA-seq and qRT-PCR results indicated the reliability of the gene expression values in our experiment.
3. Discussion
3.1. Physiological Changes in P. crymophila Responding to Cold Stress
When plants encounter cold stress, their physiology or cells will be disturbed, such as cell membrane permeability, membrane lipid peroxidation, plant protection enzyme activity and photosynthesis [10]. Plants can accumulate more compatible substances to mitigate damage caused by cold stress, such as proline, which acts as an osmoprotectant and allows plants to tolerate cold stress [23,24]. Cold stress leads to overproduction of ROS in the cells that damages various biological molecules and causes membrane lipid peroxidation [8,24]. To maintain ROS homeostasis plants have evolved defensive mechanisms composed of enzymatic and non-enzymatic components such as SOD, CAT, and POD [3,11,12]. In P. crymophila, the proline content, MDA content, and antioxidant enzyme activity increased in all three tissues under cold stress. Furthermore, the MDA content and antioxidant enzyme activity in stems and leaves exceeded those of roots during each stress period (Figure 1). Thus, it can be seen that the aboveground tissues of P. crymophila were more responsive to cold signals and better adapted to handle cold stress quickly.
3.2. Tissue-specific RNA-seq Strategy Provides More Comprehensive Information
In a previous study, the mean length and N50 of assembled unigenes generated by RNA-seq of cold-stress P. crymophila leaves were 616 nt and 804 nt, respectively [25]. A limited study has been reported as compared to the present study. Besides, the annotation rate of total unigenes in our study is up to 86.36%, which is much higher than previously reported in P. crymophila (66.94%) [25]. These comparisons further proved the great advantage of transcriptome sequencing in different tissues in improving assembly integrity and reference annotation efficiency, which facilitate the identification of the regulatory networks and comprehensive resistance mechanisms in P. crymophila under cold stress. Our research shows that the expression changes and function of enriched DEGs showed the highly coordinated and dynamic changes in responses to cold stress in P. crymophila roots, stems and leaves.
3.3. TFs involved in the cold-stress response
TFs are important regulators of various biological and molecular functions in plants that activate the downstream gene transcripts to show responses against cold stresses [18,26,27]. For instance, OsERF096 modified IAA accumulation and signaling in regulating cold tolerance of rice [28], the japonica version of bZIP73 interacts with bZIP71 to regulate ABA levels and ROS homeostasis for cold acclimation in japonica rice [29]. In addition, differences in the types and levels of TFs expression can significantly affect the ability of plants to adapt to cold stress. It has been shown that E. nutans specifically expresses bHLH, bZIP, C2H2, WRKY, and MYB TFs at the early stage of cold stress [22], and that the temporal specificity in the expression levels of the TFs enhances the cold tolerance of the plant [30]. In present study, the main differentially expressed TFs in P. crymophila at 3 days of cold treatment were AP2/ERF, bHLH, WRKY, MYB, bZIP, C2H2, and HSF TFs. Additionally, MADS-box, C2C2-Dof, and E2FA TFs were up-regulated only in stems or leaves (Table S6). Thus, specific types of TFs in different tissues and different expression levels under different cold stress conditions may be the key to improving cold tolerance.
3.4. Role of ICE-CBF-COR in the Cold Response
The ICE-CBF-COR signaling pathway has been the best characterized cold stress signaling pathway [3,8]. In Arabidopsis, the cold-induced AtCBF activates the expression of CORs, which in turn initiates a variety of mechanisms such as membrane stabilization and scavenging of ROS to mitigate cold damage [5,31]. LEAs are antioxidants and stabilizers of membranes and proteins, and the expression of AtCBF3/AtDREB1A improves cold tolerance in Arabidopsis by increasing the levels of LEAs [31,32]. CSPs were highly expressed in the cold-resistant plants, and it has also been reported that CSPs were up-regulated by the CBFs in response to cold stress in Arabidopsis [33]. In this study, the ICE-CBF-COR signaling pathway also plays an important role in the response to cold stress in P. crymophila (Table S7). LEA1, LEA14-A, COR413PM, COR413IM2, COR410, and BLT14 were expressed in stems and leaves to enhance cold tolerance of P. crymophila by scavenging ROS to attenuate the cellular damage caused by cold stress (Table S7). In addition, ICE1 and CSP showed higher transcriptional abundance in stems and leaves than in roots, which enables aboveground tissues to more effectively activate downstream defense reponses and further improve tolerance to cold stress.
3.5. Role of Ca2+ Signaling, Hormone Signaling, and ROS Scavenging System in the Cold Response
Ca2+, hormones and ROS act as signaling molecules that are involve in response to various stresses [2,5]. In P. crymophila, many genes encoding calcium-related proteins responding to cold stress, including Ca2+ channel proteins CNGC and GLR, Ca2+ sensors CML, CBL, CPK/CDPK, CIPK, Ca2+-binding transcription factor CAMTA (Table S8). To ensure the normal transmission of cold stress signals with the fluctuation of Ca2+concentration, P. crymophila has a higher expression level of the calcium channel proteins [20]. Ca2+ sensor protein senses temperature changes through monitoring intracellular calcium levels, interprets this signal as stress and passes it downstream, acting as a master switch to regulate various stress genes that confer cold tolerance [5,20]. CAMTA has CaM-binding activity and contributes to the cold induction of CBF genes, when the CAMTA3 binds to the CBF2 promoter, CBF2 is activated, which increases the cold tolerance of plants [2]. Therefore, it was speculated that Ca2+ signaling pathway components are expressed earlier in leaves and stems than in roots, making P. crymophila more responsive to cold signals and getting ready for cold sooner.
IAA, GA and BR are growth-promoting hormones that promote cell elongation and division, ABAs, ETHs and JAs often act stress signaling and play an important role in regulating plant tolerance to abiotic stresses [34,35,36]. GH3 proteins are usually involved in regulating the synthesis and accumulation of JA and IAA [37,38]. In rice, overexpression of OsGH3-2 inhibited the accumulation of free IAA, which leads to the enhancement of ROS scavenging capacity and ultimately promotes cold tolerance [37]. Both GA metabolism and signaling can affect the cold stress responses of plants, and studies have shown that CBF upregulates multiple genes involved in GA metabolism (GA2ox6, GA2ox9) and signaling (RGL3) under cold stress, thereby reducing bioactive GA levels [34]. The activity of DELLA is central to GA signaling, DELLA proteins interact with the GA receptors, to alleviate DELLA repression on GA signaling [35]. Moreover, BR was able to enhance the expression of AtCBF1 and AtCOR47, which ultimately improved cold tolerance in Arabidopsis [39]. In response to cold, reduced expression of brassinosteroid response 1 (BRH1) and increased expression of brassinosteroid LRR receptor kinase to activate BR signaling in Magnolia wufengensis [40]. Therefore, it could suggest that P. crymophila reduced the level of IAA, GA and BR, while low expression of GH3 reduced the decomposition of IAA, and increased the level of JA in roots and stems to make it more cold-tolerant.
Studies have shown that ABA is a core regulator of cold stress signaling and plays an important role in CBF-dependent pathway [34,41]. Abscisic acid insensitive 1 (ABI1), abscisic acid insensitive 2 (ABI2) and Hypersensitive to Abscisic acid 1 (HAB1) have been shown to interact with ABA receptors PYR/PYLs (Pyrabactin-resistant/PYR1-like proteins) to regulate SNF1-Related Protein Kinase 2 (SnRK2s) and Protein phosphatase 2C (PP2Cs) [34,35]. In this study, P. crymophila may promote PP2C and SnRK2 DEGs expression through the downregulation of PYR/PYL DEGs in the ABA regulatory pathway, thereby conferring cold tolerance (Table S10). Furthermore, ethylene-insensitive 3 (EIN3) acts as an antagonist in the ETH signaling pathway, controlling cold acclimation by inhibiting CBF [12,35]. Under cold stress, ABA, and JA hormones can induce the expression of ERF genes, and ERFs bind to GCC box and DRE elements, conferring cold stress tolerance in A. thaliana and T. aestivum [12]. Therefore, these DEGs will be crucial for cold stress response in P. crymophila, and down-regulated EIN3 can weaken the CBF pathway's inhibition, thus enhancing P.crymophila's cold tolerance.
ROS can function as a cold stress marker that impacts downstream gene expression. It may be due to protein denaturation, nucleic acid mutations, and cellular damage [42]. It has been found that cold stress induces SOD, POD, and CAT activities to scavenge ROS, thereby enhancing cold tolerance in plants [3,12,43]. In this study, we found that cold stress induced up-regulation of SOD, POD, and CAT synthesis-related DEGs (Table S9), resulting in a significant increase in their activities (Figure 1B, C, and D). In addition, APX, GSH and GPX, as the most important peroxidases in H2O2 detoxification, play an important role in scavenging ROS to enhance cold tolerance in plants [8,42]. In winter wheat Dn1and Capsicum annuum L., the higher activities of RBOH, DHAR and MDHAR enzymes have a significant effect on alleviating the symptoms of cold stress [26,44]. In summary, these enzymes of the ROS scavenging system protect cells from damage and ensure normal life under stress, which is also the strategy of P. crymophila cold adaptation.
The MAPK cascade is a universal signal transduction module, including MAPK, MAP kinase kinase (MAPKK/MKK/MMK) and MAP kinase kinase kinase (MAPKKK/MEKK), lie downstream of ROS, Ca2+ and hormones, and play vital roles in plant responses to cold stress [45,46]. The overexpression of OsMKK6 activated the OsMPK3 and enhanced the cold resistance of rice [47]. Zhao et al. [48] have reported that the MAP kinase pathway (MEKK1-MKK1/2-MPK4) increased the CBFs expression and freezing tolerance by inhibiting the MKK4/5-MPK3/6 cascade. Collectively, the enhanced expression of transcripts for Ca2+, hormones, and ROS in P. crymophila activated a complicated signaling cascade, regulating various downstream genes that respond to cold tolerance. These DEGs could be intriguing candidates to investigate during P. crymophila seedling cold stress responses.
3.6. Role of the Circadian Clock and Photosynthesis in the Cold Response
Studies have shown that core components of the circadian clock, circadian clock-associated 1 (CCA1) and late-elongating hypocotyl (LHY), play an active role in CBF expression, while the nocturnal component, pseudoresponse regulator (PRR), acts as a repressor of CBF expression [16,49,50]. A cold-induced clock component GIGANTEA (GI) can also regulate cold responses in a CBF-independent manner,positively regulates cold stress in Arabidopsis [24,49,51]. In P. crymophila, PRRs, LHYs, and GIs DEGs were enhanced under cold, which indicated the circadian clock that may be an efficient way to increase cold resistance in P. crymophila.
Photosystem II (PSII) is one of the most sensitive components of photosynthesis [52,53]. The decline in photosynthesis under cold stress is mainly due to cold stress-induced inhibition of the activity of key enzymes associated with photosynthesis [7,40]. The light-harvesting complex (LHC) of photosynthetic antenna proteins can rapidly capture light energy to promote photosynthesis in plants [14,16]. In P. crymophila, we found that most of photosynthesis-related DEGs were down-regulated (especially in leaves) (Table S12). These results confirm that P. crymophila may adapt to low temperature by reducing leaf photosynthesis and reducing energy loss.
3.7. Cold Tolerance Hub genes Selected by WGCNA
ABCC3, a member of the ABCC subfamily of the ABC (ATP-binding cassette) transporter family, was found to be involved in the MAPK signaling pathway to enhance freezing tolerance in alfalfa [54]. This study indicated that ABCC2 was significantly up-regulated when exposed to cold stress (Table S12), potentially impacting MAPK signaling. The activity of the Protein ACTIVITY OF BC1 COMPLEX KINASE 7 (ABC1K7) protein increased under cold stress in P. crymophila (Table S12). It has been suggested that ABC1K7 and ABC1K8 contribute to cross-talk between ABA and ROS signaling, indicating a potential involvement of PcABC1K7 in the regulation of ABA and ROS signaling [55]. Furthermore, this investigation uncovered significant up-regulation of genes associated with chlorophyll synthesis, namely PcBLH1 and PcFTSH1 [56,57], arginine biosynthesis-related gene CARB [58], and regulator of the ETH signaling pathway CTR1 gene [34,59], indicating their important role in responding to cold stress in P. crymophila. In short, these genes may serve as potential targets for future research aimed at improving plant cold tolerance.
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
The seeds of P. crymophila were provided by Qinghai Modern Grass Industry Development Co., Ltd. Seeds are vernalized at 4°C for 3 days, and sown in 0.5-L pots (three plants per pot) filled with a mixture of black soil and peat in a 2:1 (v:v) ratio. Germinated seedlings were grown in greenhouse (25°C/20°C under16-h-light/8-h-dark cycle, light intensity approximately 500 μmol m−2 s−1, 60% relative humidity). To avoid nutrient deficiencies, the 1/2 Hoagland nutrient solution was watered every two days during the tillering and stem elongation periods.
After 45 days, uniform seedlings were selected and used for treatments. The seedlings were randomly divided into three treatments: (1) control (0 d), (2) cold treatment for 3 days (3 d), and (3) cold treatment for 6 days (6 d). The plants were watered with Hoagland nutrient solution every 3 days. The leaves (L), stems (S), and roots in each treatment were collected after 0 d, 3 d, and 6 d, respectively. For transcriptome experiments, a total 27 samples were uniformly harvested at the end of the treatment and immediately frozen in liquid nitrogen and stored at -80 °C. The samples were collected in triplicate at each time point.
4.2. Determination of MDA, Proline Contents, and Antioxidant Enzymatic Activities
All samples were collected from non-stressed and cold stressed plants. Malondialdehyde (MDA) content was measured according to the thiobarbituric acid-reactive-substances (TBARS) assay [60]. Free proline content was determined by the sulfosalicylic acid method as previously described [1]. For the estimation of antioxidant enzyme activities, the superoxide dismutase (SOD), catalase (CAT), and peroxidase (POD) were measured according to the instructions of the kit manufacturer instructions (Shanghai Enzyme-linked Biotechnology Co., Ltd. China).
4.3. Total RNA Extraction and Sequencing
To obtain the transcriptome of leaves, stems, and roots of P. crymophila, total RNA was extracted from each tissues using a UNIQ-10 Column TRIzol Total RNA Extraction Kit (Sangon Biotech, Shanghai) as previously described [61]. The purity, concentration, and integrity of RNA samples were detected by Nanodrop, Qubit 2.0, and Agilent 2100 methods, respectively. A total of 27 cDNA libraries were sequenced with a read length of 150 bp using the Illumina HiSeq 2000 platform with a paired-end module at Biomarker CoLtd. (BMKcloud, Beijing, China). Sequencing libraries were generated using NEBNext®Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer’s instructions and index codes were added to attribute sequences to each sample. Each library was sequenced on an Illumina NovaSeq 6000 (Illumina, San Diego, CA) in 150-bp paired-end format according to the Illumina Paired-End Sequencing protocol. The raw reads were further processed using the bioinformatics pipeline tool, BMKCloud (www.biocloud.net) online platform.
4.4. Sequence Data Processing, De novo Assembly, and Annotation
Raw data were obtained after transcriptome sequencing of the library constructed by P. crymophila. After filtering raw data, the joint sequences and primer sequences in the raw data were removed, empty sequences and low-quality sequences were screened out, and clean data were obtained for assembly. Unigenes were obtained by de novo transcriptome assembly by Trinity-v2.5.1 (https://github.com/trinityrnaseq/trinityrnaseq/wiki).
The obtained functional gene sequences were aligned by Blastx to protein databases such as NR (NCBI non-redundant protein sequences); Pfam (Protein family); KOG/COG/eggNOG (Clusters of Orthologous Groups of proteins); Swiss-Prot (A manually annotated and reviewed protein sequence database); KEGG (Kyoto Encyclopedia of Genes and Genomes); GO (Gene Ontology) (E-value≤1e-5). Using KOBAS-v3.0 software (http://kobas.cbi.pku.edu.cn/kobas3) to obtain the results of KEGG Orthology of unigene in KEGG, InterProScan (https://www.ebi.ac.uk/interpro/download/) uses the database integrated by InterPro to analyze the results of GO Orthology of new genes [62]. HMMER-v3.1b2 software (http://hmmer.org/) was used for comparison with the Pfam database [63] (E-value≤1e-10).
4.5. Analysis of Differentially Expressed Genes
The fragments per kilobase of gene per million mapped reads (FPKM) method was used in estimating the expression levels of each transcript. Differential expression analysis between the control/cold conditions was conducted using the DESeq R package (1.10.1) [64]. Genes were identified by DESeq with false discovery rate (FDR) <0.01, adjusted P-value < 0.05, and |fold change (FC)| ≥2 defined as DEGs [17]. The differential expression analysis between the control/cold conditions in the P. crymophila were compared as; control root (R0) vs. stressed at 4°C for 3 d (R3), R0 vs. stressed at 4°C for 6 d (R6), R3 vs. R6, control shoot (S0) vs. stressed at 4°C for 3 d (S3), S0 vs. stressed at 4°C for 6 d (S6), S3 vs. S6, control leaf (L0) vs. stressed at 4°C for 3 d (L3), L0 vs. stressed at 4°C for 6 d (L6) and L3 vs. L6.
4.6. Weighted Gene Co-Expression Network Analysis (WGCNA)
All the DEGs were used for the construction of weighted gene co-expression networks. Filtering the DEGs to remove low-expression genes, DEGs with FPKM > 1 in at least 27 samples were retained. A total of 7,439 DEGs were obtained for WGCNA. The WGCNA assumed that the genetic network followed scale-free networks. The similarity matrix of gene co-expression and gene network formed the adjacency function. The similarity matrix was transformed into the adjacency matrix and then, the expression correlation coefficient was calculated to construct the gene’s hierarchical clustering tree. Modules were divided according to the clustering relationship between genes, and modules with similar expression patterns were merged. Finally, the correlation between phenotypic traits of modules and samples was analyzed, and the hub genes in the network were found by the R language package [65].
4.7. Validation Analysis of Transcriptome Data by qRT-PCR
To verify the reliability and accuracy of the RNA-seq data, a random selection of 12 genes from P. crymophila were selected for validation by qRT-PCR (Table S13). The same RNA and cDNA were used as for the RNA sequencing experiments. Total RNA was extracted from the 27 samples as described previously and followed the manufacturer’s protocol to generate cDNA (TaKaRa, Japan). The qRT-PCR was performed using a ChamQ Universal SYBR qPCR Master Mix kit (Vazyme, Nanjing, China) on a Roche LightCycler480 quantitative PCR. The relative level of gene expression was calculated using the 2-ΔΔct formula [66]. The qRT-PCR analysis included three independent technical repeats with three biological replicates. The beta-actin gene (PcACTIN) was used as the internal control gene [25]. Gene-specific primers were designed with DNAMAN software (Lynnon BioSoft, Van-dreuil, Quebec, Canada).
5. Conclusions
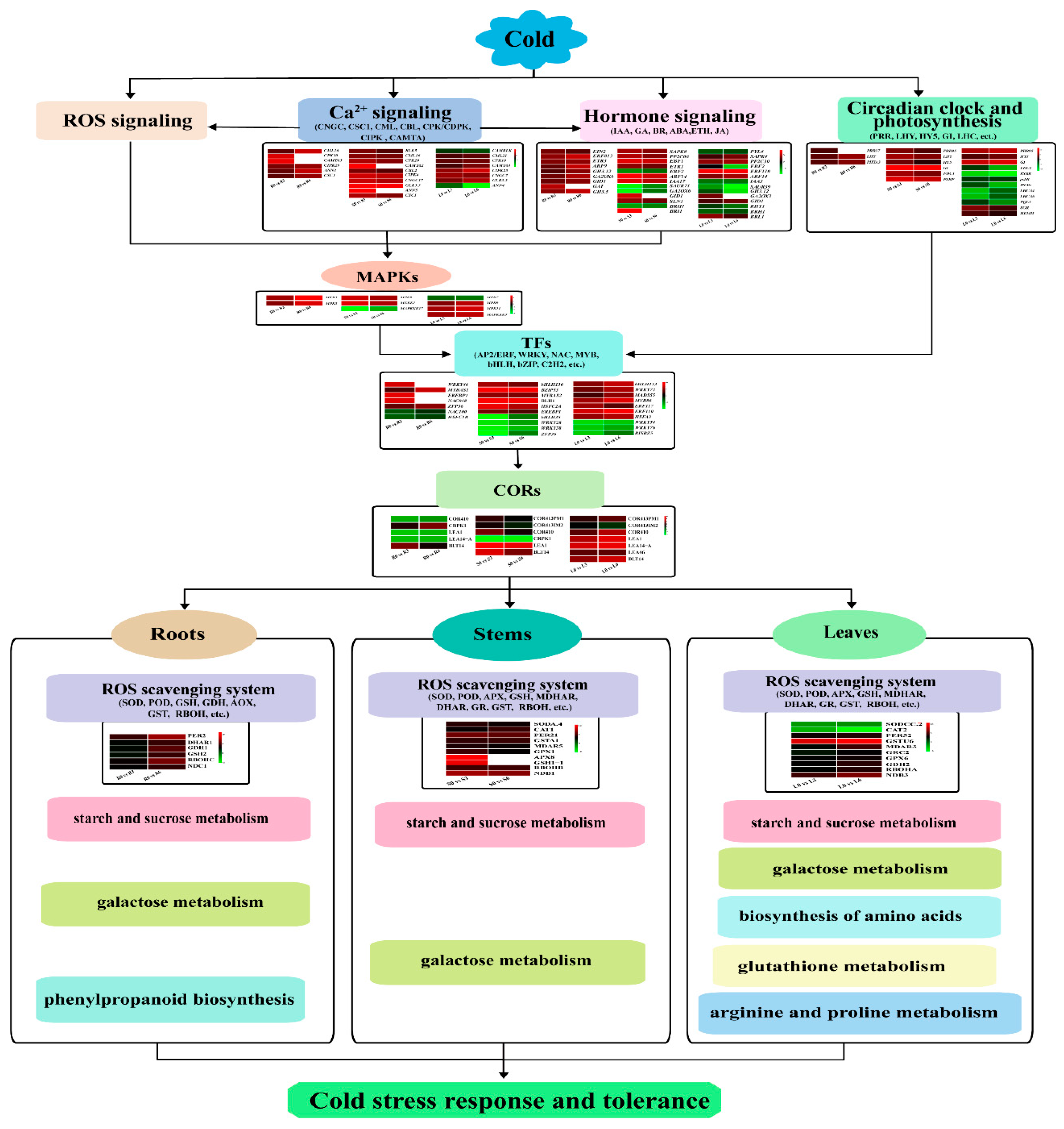
The present study concluded that physiological and transcriptome analyses identified key genes and molecular pathways responsible for cold tolerance in P. crymophila. The Ca2+ signaling, ROS signaling, hormone signaling, circadian clock, and photosynthesis related DEGs are coupled to TFs and play important roles in response to cold stress through trigger a cascade of downstream CORs, CORs activations further modulates each tissue's cellular metabolic homeostasis and enhanced tolerance (Figure 11), and most of the DEGs were induced in the early stage of cold stress (3 d). Compared with roots, DEGs in the stems and leaves of P. crymophila were more active under cold stress. In addition, WGCNA identified nine key genes involved in P. crymophila responses to cold stress. Altogether, our results provide important insights into the future understanding of the biochemical and molecular mechanisms underlying the response of P. crymophila to cold stress. Furthermore, the functional genes involved have the potential to be used in the development of new forage grass varieties for enhanced productivity and stress resistance.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Differentially expressed gene expression patterns in roots, stems and leaves across three time; Figure S2: Correlation analysis; Table S1: Summary of Illumina sequencing data and assembled results; Table S2: Statistics of assembly results; Table: S3 BLAST analysis of transcripts against public databases; Table S4: The top 25 significantly enriched GO terms of DEGs in roots; stems, and leaves under cold stress; Table S5: The top 20 significantly enriched KEGG pathway of DEGs in roots, stems, and leaves under cold stress; Table S6: Differentially expressed genes involved in the TFs of roots, stems and leaves under cold stress; Table S7: Differently expressed genes related to CBF pathway; Table S8: Differently expressed genes related to Ca2+ signaling of roots, stems and leaves under cold stress; Table S9: Differently expressed genes related to ROS scavenging system of roots, stems and leaves under cold stress; Table S10: Differently expressed genes related to hormone signaling of roots, stems and leaves under cold stress; Table S11: Differently expressed genes related to the circadian clocks and photosynthesis of roots, stems and leaves under cold stress; Table S12: The hub genes were selected in the brown module and the salmon module; Table S13 Primers used for qRT-PCR analysis.
Author Contributions
W.X. conceived and designed the study; L.T. performed the experiment; L.T, H.W and Y.Z collected and analyzed the data; L.T and I. K wrote the manuscript; W.X. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by CARS (CARS-34), Chinese National Natural Science Foundation (31971751), the Leading Scientist Project of Qinghai Province (2023- NK -147), Gansu Provincial Science and Technology Major Projects (22ZD6NA007), and the Fundamental Research Fund for the Central Universities (Izujbky -2021-ct21).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
We thank the College of Pastoral Agriculture Science and Technology of Lanzhou University for their support and contribution to this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, L.; Jian, Z.; Wang, P.; Zhao, L.; Chen, K.; Chaves, M. Combined physiological responses and differential expression of drought-responsive genes preliminarily explain the drought resistance mechanism of Lotus corniculatus. Functional Plant Biology 2022, 50, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Yang, S. Surviving and thriving: How plants perceive and respond to temperature stress. Developmental Cell 2022, 57, 947–958. [Google Scholar] [CrossRef]
- Dong, R.; Luo, B.; Tang, L.; Wang, Q.-x.; Lu, Z.-J.; Chen, C.; Yang, F.; Wang, S.; He, J. A comparative transcriptomic analysis reveals a coordinated mechanism activated in response to cold acclimation in common vetch (Vicia sativa L.). BMC Genomics 2022, 23, 814. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Sun, M.; Su, R.; Wang, X.; Lu, X.; Xiao, Y.; Deng, H.; Liu, X.; Tang, W.; Zhang, G. Transcriptomic profiling of cold stress-induced differentially expressed genes in seedling stage of Indica rice. Plants 2023, 12, 2675. [Google Scholar] [CrossRef]
- Gusain, S.; Joshi, S.; Joshi, R. Sensing, signalling, and regulatory mechanism of cold-stress tolerance in plants. Plant Physiology and Biochemistry 2023, 197, 107646. [Google Scholar] [CrossRef] [PubMed]
- Nouri, M.-Z.; Komatsu, S. Subcellular protein overexpression to develop abiotic stress tolerant plants. Frontiers in Plant Science 2013, 4, 2. [Google Scholar] [CrossRef]
- Cheng, M.; Pan, Z.; Cui, K.; Zheng, J.; Luo, X.; Chen, Y.; Yang, T.; Wang, H.; Li, X.; Zhou, Y.; Lei, X.; Li, Y.; Zhang, R.; Iqbal, M.Z.; He, R. RNA sequencing and weighted gene co-expression network analysis uncover the hub genes controlling cold tolerance in Helictotrichon virescens seedlings. Frontiers in Plant Science 2022, 13, 938859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, J.; Gong, Z.; Zhu, J.-K. Abiotic stress responses in plants. Nature Reviews Genetics 2021, 23, 104–119. [Google Scholar] [CrossRef]
- Fursova, O.V.; Pogorelko, G.V.; Tarasov, V.A. Identification of ICE2, a gene involved in cold acclimation which determines freezing tolerance in Arabidopsis thaliana. Gene 2009, 429, 98–103. [Google Scholar] [CrossRef]
- Shu, Y.; Li, W.; Zhao, J.; Zhang, S.; Xu, H.; Liu, Y.; Guo, C. Transcriptome sequencing analysis of alfalfa reveals CBF genes potentially playing important roles in response to freezing stress. Genetics and Molecular Biology 2017, 40, 824–833. [Google Scholar] [CrossRef]
- Zhuo, C.; Liang, L.; Zhao, Y.; Guo, Z.; Lu, S. A cold responsive ethylene responsive factor from Medicago falcata confers cold tolerance by up-regulation of polyamine turnover, antioxidant protection, and proline accumulation. Plant, Cell & Environment 2018, 41, 2021–2032. [Google Scholar]
- Ritonga, F.N.; Chen, S. Physiological and molecular mechanism involved in cold Stress tolerance in plants. Plants 2020, 9, 560. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Cui, Y.; Dong, S.; Luo, D.; Fang, L.; Shi, Z.; Liu, W.; Wang, Z.; Nan, Z.; Liu, Z. Integrative physiological, transcriptome, and metabolome analyses reveal the associated genes and metabolites involved in cold stress response in common vetch (Vicia sativa L.). Food and Energy Security 2023, 12, e484. [Google Scholar] [CrossRef]
- Zhu, J.-K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, C.; Ren, J.; Dong, J.; Shi, X.; Zhao, X.; Wang, X.; Wang, J.; Zhong, C.; Zhao, S.; Liu, X.; Gao, S.; Yu, H. An advanced lipid metabolism system revealed by transcriptomic and lipidomic analyses plays a central role in peanut cold tolerance. Frontiers in Plant Science 2020, 11, 1110. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Shi, Y.; Terzaghi, W.; Yang, S.; Li, J. Integration of light and temperature signaling pathways in plants. Journal of Integrative Plant Biology 2022, 64, 393–411. [Google Scholar] [CrossRef]
- Min, X.; Liu, Z.; Wang, Y.; Liu, W. Comparative transcriptomic analysis provides insights into the coordinated mechanisms of leaves and roots response to cold stress in Common Vetch. Industrial Crops and Products 2020, 158, 112949. [Google Scholar] [CrossRef]
- Li, L.; Han, C.; Yang, J.; Tian, Z.; Jiang, R.; Yang, F.; Jiao, K.; Qi, M.; Liu, L.; Zhang, B.; Niu, J.; Jiang, Y.; Li, Y.; Yin, J. comprehensive transcriptome analysis of responses during cold Stress in Wheat (Triticum aestivum L.). Genes 2023, 14, 844. [Google Scholar] [CrossRef]
- Zou, F.; Hu, Q.; Li, H.; Lin, J.; Liu, Y.; Sun, F. Dynamic disturbance analysis of grasslands using nural networks and spatiotemporal indices fusion on the Qinghai-Tibet Plateau. Frontiers in Plant Science 2022, 12, 760551. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Hou, X.; Chen, Y.; Li, C.; Ma, X. Flexible response and rapid recovery strategies of the plateau forage Poa crymophila to cold and drought. Frontiers in Plant Science 2022, 13, 970496. [Google Scholar] [CrossRef]
- Hua, Q.; Yu, Y.; Dong, S.; Li, S.; Shen, H.; Han, Y.; Zhang, J.; Xiao, J.; Liu, S.; Dong, Q.; Zhou, H.; Wessell, K. Leaf spectral responses of Poa crymophila to nitrogen deposition and climate change on Qinghai-Tibetan Plateau. Agriculture, Ecosystems & Environment 2019, 284, 106598. [Google Scholar]
- Fu, J.; Miao, Y.; Shao, L.; Hu, T.; Yang, P. De novo transcriptome sequencing and gene expression profiling of Elymus nutans under cold stress. BMC Genomics 2016, 17, 870. [Google Scholar] [CrossRef]
- Dong, W.; Ma, X.; Jiang, H.; Zhao, C.; Ma, H. Physiological and transcriptome analysis of Poa pratensis var. anceps cv. Qinghai in response to cold stress. BMC Plant Biology 2020, 20, 362. [Google Scholar] [CrossRef]
- Qi, W.; Wang, F.; Ma, L.; Qi, Z.; Liu, S.; Chen, C.; Wu, J.; Wang, P.; Yang, C.; Wu, Y.; Sun, W. Physiological and biochemical mechanisms and cytology of cold tolerance in Brassica napus. Frontiers in Plant Science 2020, 11, 1241. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.-Y.; Li, C.-X.; He, Y.; Hou, X.-Y.; Ma, X.-R. The regulation of adaptation to cold and drought stresses in Poa crymophila Keng revealed by integrative transcriptomics and metabolomics analysis. Frontiers in Plant Science 2021, 12, 631117. [Google Scholar] [CrossRef]
- Tian, Y.; Peng, K.; Lou, G.; Ren, Z.; Sun, X.; Wang, Z.; Xing, J.; Song, C.; Cang, J. Transcriptome analysis of the winter wheat Dn1 in response to cold stress. BMC Plant Biology 2022, 22, 277. [Google Scholar] [CrossRef]
- Waititu, J.K.; Cai, Q.; Sun, Y.; Sun, Y.; Li, C.; Zhang, C.; Liu, J.; Wang, H. Transcriptome profiling of maize (Zea mays L.) leaves reveals key cold-responsive genes, transcription factors, and metabolic pathways regulating cold stress tolerance at the seedling stage. Genes 2021, 12, 1638. [Google Scholar] [CrossRef]
- Cai, X.; Chen, Y.; Wang, Y.; Shen, Y.; Yang, J.; Jia, B.; Sun, X.; Sun, M. A comprehensive investigation of the regulatory roles of OsERF096, an AP2/ERF transcription factor, in rice cold stress response. Plant Cell Reports 2023. Advance online publication. [Google Scholar] [CrossRef]
- Liu, C.; Ou, S.; Mao, B.; Tang, J.; Wang, W.; Wang, H.; Cao, S.; Schläppi, M.R.; Zhao, B.; Xiao, G.; Wang, X.; Chu, C. Early selection of bZIP73 facilitated adaptation of japonica rice to cold climates. Nature Communications 2018, 9, 3302. [Google Scholar] [CrossRef]
- Cheng, M.; Cui, K.; Zheng, M.; Yang, T.; Zheng, J.; Li, X.; Luo, X.; Zhou, Y.; Zhang, R.; Yan, D.; Yao, M.; Iqbal, M.Z.; Zhou, Q.; He, R. Physiological attributes and transcriptomics analyses reveal the mechanism response of Helictotrichon virescens to low temperature stress. BMC Genomics 2022, 23, 280. [Google Scholar] [CrossRef]
- Novillo, F.; Medina, J.; Salinas, J. Arabidopsis CBF1 and CBF3 Have a Different Function than CBF2 in Cold Acclimation and Define Different Gene Classes in the CBF Regulon. Proceedings of the National Academy of Sciences of the United States of America 2007, 104, 21002–21007. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, L.; Baral, R.; Paudel, D.; Min, D.; Makaju, S.O.; Poudel, H.P.; Acharya, J.P.; Missaoui, A.M. Cold stress in plants: Strategies to improve cold tolerance in forage species. Plant Stress 2022, 4, 100081. [Google Scholar] [CrossRef]
- Shah, F.A.; Ni, J.; Yao, Y.; Hu, H.; Wei, R.; Wu, L. Overexpression of karrikins receptor gene sapium sebiferum KAI2 promotes the cold stress tolerance via regulating the redox homeostasis in Arabidopsis thaliana. Frontiers in Plant Science 2021, 12, 657960. [Google Scholar] [CrossRef] [PubMed]
- Waadt, R.; Seller, C.A.; Hsu, P.-K.; Takahashi, Y.; Munemasa, S.; Schroeder, J.I. Plant hormone regulation of abiotic stress responses. Nature Reviews Molecular Cell Biology 2022, 23, 680–694. [Google Scholar] [CrossRef] [PubMed]
- Eremina, M.; Rozhon, W.; Poppenberger, B. Hormonal control of cold stress responses in plants. Cellular and Molecular Life Sciences 2015, 73, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Li, H.; Chen, X.; Xiang, X.; Guo, Z.; Yu, J.; Zhou, Y. The role of calcium-dependent protein kinase in hydrogen peroxide, nitric oxide and ABA-dependent cold acclimation. Journal of Experimental Botany 2018, 69, 4127–4139. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Wu, N.; Fu, J.; Wang, S.; Li, X.; Xiao, J.; Xiong, L. A GH3 family member, OsGH3-2, modulates auxin and abscisic acid levels and differentially affects drought and cold tolerance in rice. Journal of Experimental Botany 2012, 63, 6467–6480. [Google Scholar] [CrossRef]
- Shi, Q.; Zhang, Y.; To, V.-T.; Shi, J.; Zhang, D.; Cai, W. Genome-wide characterization and expression analyses of the auxin/indole-3-acetic acid (Aux/IAA) gene family in barley (Hordeum vulgare L.). Scientific Reports 2020, 10, 10242. [Google Scholar] [CrossRef] [PubMed]
- Kagale, S.; Divi, U.K.; Krochko, J.E.; Keller, W.A.; Krishna, P. Brassinosteroid confers tolerance in Arabidopsis thaliana and Brassica napus to a range of abiotic stresses. Planta 2006, 225, 353–364. [Google Scholar] [CrossRef]
- Deng, S.; Ma, J.; Zhang, L.; Chen, F.; Sang, Z.; Jia, Z.; Ma, L. De novo transcriptome sequencing and gene expression profiling of Magnolia wufengensis in response to cold stress. BMC Plant Biology 2019, 19, 321. [Google Scholar] [CrossRef]
- Xiang, N.; Hu, J.-g.; Yan, S.; Guo, X. Plant hormones and volatiles response to temperature stress in sweet corn (Zea mays L.) seedlings. Journal of Agricultural and Food Chemistry 2021, 69, 6779–6790. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Jiang, C.; Chen, L.; Paul, A.; Chatterjee, A.; Shen, G. Achieving abiotic stress tolerance in plants through antioxidative defense mechanisms. Frontiers in Plant Science 2023, 14, 1110622. [Google Scholar] [CrossRef]
- Mittler, R.; Zandalinas, S.I.; Fichman, Y.; Van Breusegem, F. Reactive oxygen species signalling in plant stress responses. Nature Reviews Molecular Cell Biology 2022, 23, 663–679. [Google Scholar] [CrossRef]
- Kantakhoo, J.; Imahori, Y. Antioxidative responses to pre-storage hot water treatment of red sweet pepper (Capsicum annuum L.) fruit during cold storage. Foods 2021, 10, 3031. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wu, T.; Li, S.; He, Q.; Yang, Z.; Zhang, W.; Gan, Y.; Sun, P.; Xiang, G.; Zhang, H.; Deng, H. The methylation patterns and transcriptional responses to chilling stress at the seedling stage in rice. International Journal of Molecular Sciences 2019, 20, 5089. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zhang, L.; Wang, H.; Lu, J.; Wei, H.; Yu, S. Transcriptomic profiling of young cotyledons response to chilling stress in two contrasting cotton (Gossypium hirsutum L.) genotypes at the seedling stage. International Journal of Molecular Sciences 2020, 21, 5095. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Kato, H.; Imai, R. Biochemical identification of the OsMKK6–OsMPK3 signalling pathway for chilling stress tolerance in rice. Biochemical Journal 2012, 443, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, P.; Si, T.; Hsu, C.-C.; Wang, L.; Zayed, O.; Yu, Z.; Zhu, Y.; Dong, J.; Tao, W.A.; Zhu, J.-K. MAP kinase cascades regulate the cold response by modulating ICE1 protein stability. Developmental Cell 2017, 43, 618–629.e5. [Google Scholar] [CrossRef] [PubMed]
- Hung, F.-Y.; Chen, F.-F.; Li, C.; Chen, C.; Chen, J.-H.; Cui, Y.; Wu, K. The LDL1/2-HDA6 histone modification complex interacts with TOC1 and regulates the core circadian clock components in Arabidopsis. Frontiers in Plant Science 2019, 10, 233. [Google Scholar] [CrossRef]
- Hong, W.-J.; Jiang, X.; Ahn, H.R.; Choi, J.; Kim, S.-R.; Jung, K.-H. Systematic analysis of cold Stress response and diurnal rhythm using transcriptome data in rice reveals the molecular networks related to various biological processes. International Journal of Molecular Sciences 2020, 21, 6872. [Google Scholar] [CrossRef]
- Li, Y.; Shi, Y.; Li, M.; Fu, D.; Wu, S.; Li, J.; Gong, Z.; Liu, H.; Yang, S. The CRY2–COP1–HY5–BBX7/8 module regulates blue light-dependent cold acclimation in Arabidopsis. The Plant Cell 2021, 33, 3555–3573. [Google Scholar] [CrossRef]
- Wu, H.; Wu, Z.; Wang, Y.; Ding, J.; Zheng, Y.; Tang, H.; Yang, L. Transcriptome and metabolome analysis revealed the freezing resistance mechanism in 60-year-old overwintering Camellia sinensis. Biology 2021, 10, 996. [Google Scholar] [CrossRef]
- Ren, C.; Fan, P.; Li, S.; Liang, Z. Advances in understanding cold tolerance in grapevine. Plant Physiology 2023, 192, 1733–1746. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kang, W.; Wu, F.; Miao, J.; Shi, S. Comparative transcriptome analysis reveals new insight of alfalfa (Medicago sativa L.) cultivars in response to abrupt freezing stress. Frontiers in Plant Science 2022, 13, 798118. [Google Scholar] [CrossRef]
- Manara, A.; DalCorso, G.; Furini, A. The role of the atypical kinases ABC1K7 and ABC1K8 in abscisic acid responses. Frontiers in Plant Science 2016, 7, 366. [Google Scholar] [CrossRef]
- Wu, Q.; Han, T.; Yang, L.; Wang, Q.; Zhao, Y.; Jiang, D.; Ruan, X. The essential roles of OsFtsH2 in developing the chloroplast of rice. BMC Plant Biology 2021, 21, 445. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Fan, Z.; Zhang, Q.; Wang, C.; Gao, Y.; Deng, Y.; Zhu, B.; Zhu, H.; Chen, J.; Shan, W.; Yin, X.; Zhong, S.; Grierson, D.; Jiang, C.Z.; Luo, Y.; Fu, D.Q. BEL1-LIKE HOMEODOMAIN 11 regulates chloroplast development and chlorophyll synthesis in tomato fruit. The Plant Journal 2018, 94, 1126–1140. [Google Scholar] [CrossRef] [PubMed]
- Ke, N.J.; Kumka, J.E.; Fang, M.X.; Weaver, B.; Burstyn, J.N.; Bauer, C.E. RedB, a member of the CRP/FNR family, functions as a transcriptional redox brake. Microbiology spectrum 2022, 10, e0235322. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, X.; Bürger, M.; Chory, J.; Wang, X. The role of ethylene in plant temperature stress response. Trends in Plant Science 2023, 28, 808–824. [Google Scholar] [CrossRef]
- Wang, X.; Yu, C.; Liu, Y.; Yang, L.; Li, Y.; Yao, W.; Cai, Y.; Yan, X.; Li, S.; Cai, Y.; Li, S.; Peng, X. GmFAD3A, a ω-3 fatty acid desaturase gene, enhances cold tolerance and seed germination rate under low temperature in rice. International Journal of Molecular Sciences 2019, 20, 3796. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, N.; Zhang, Z.; Liu, W.; Xie, W. Identification of flowering regulatory networks and Hub genes expressed in the leaves of Elymus sibiricus L. using comparative transcriptome analysis. Frontiers in Plant Science 2022, 13, 877908. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.Z.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; Pesseat, S.; Quinn, A.F.; Sangrador-Vegas, A.; Scheremetjew, M.; Yong, S.Y.; Lopez, R.; Hunter, S. InterProScan 5: genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–63. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome biology 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef]
- Adnan, M.; Morton, G.; Hadi, S. Analysis of rpoS and bolA gene expression under various stress-induced environments in planktonic and biofilm phase using 2(-ΔΔCT) method. Molecular and cellular biochemistry 2011, 357, 275–282. [Google Scholar] [CrossRef]
Figure 1.
Phenotype and physiological responses of P. crymophila under cold stress. (A) Phenotypic status of P. crymophila under different cold treatment time. Analysis of dynamic physiological effects on (B) roots, (C) stems and (D) leaves of P. crymophila under cold treatment. The data in (B-D) are means ± SE (n=5). The different letters indicate significant differences (P < 0.05, Tukey’s test).
Figure 1.
Phenotype and physiological responses of P. crymophila under cold stress. (A) Phenotypic status of P. crymophila under different cold treatment time. Analysis of dynamic physiological effects on (B) roots, (C) stems and (D) leaves of P. crymophila under cold treatment. The data in (B-D) are means ± SE (n=5). The different letters indicate significant differences (P < 0.05, Tukey’s test).
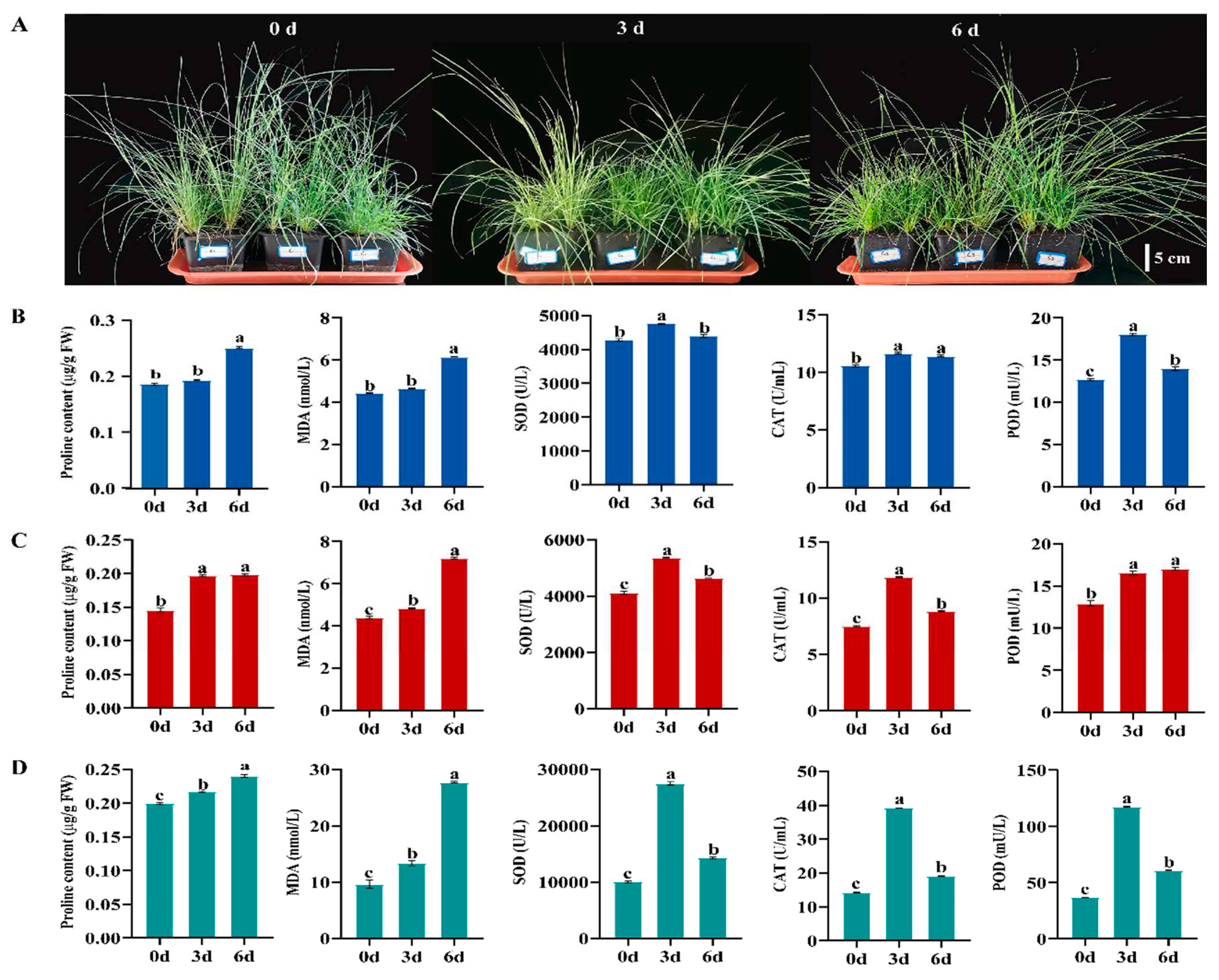
Figure 2.
The E-value distribution and similarity statistics of the annotated unigenes. (A)The species distribution of homologous based on Nr. (B)The E-value distribution of unigenes blasted to the Nr database. (C)The similarity distribution of unigenes blasted to the Nr database.
Figure 2.
The E-value distribution and similarity statistics of the annotated unigenes. (A)The species distribution of homologous based on Nr. (B)The E-value distribution of unigenes blasted to the Nr database. (C)The similarity distribution of unigenes blasted to the Nr database.
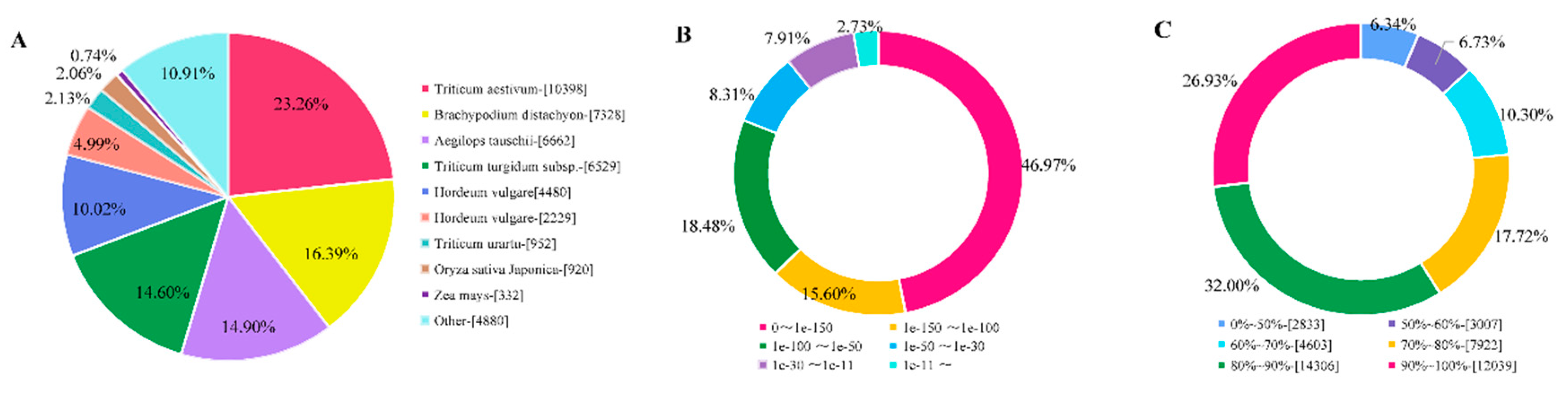
Figure 3.
Summary of differentially expressed genes (DEGs). (A) Summary of the numbers of roots, stems, and leaves DEGs under cold treatments. (B) The number of genes whose expression is differentially regulated under cold treatments.
Figure 3.
Summary of differentially expressed genes (DEGs). (A) Summary of the numbers of roots, stems, and leaves DEGs under cold treatments. (B) The number of genes whose expression is differentially regulated under cold treatments.
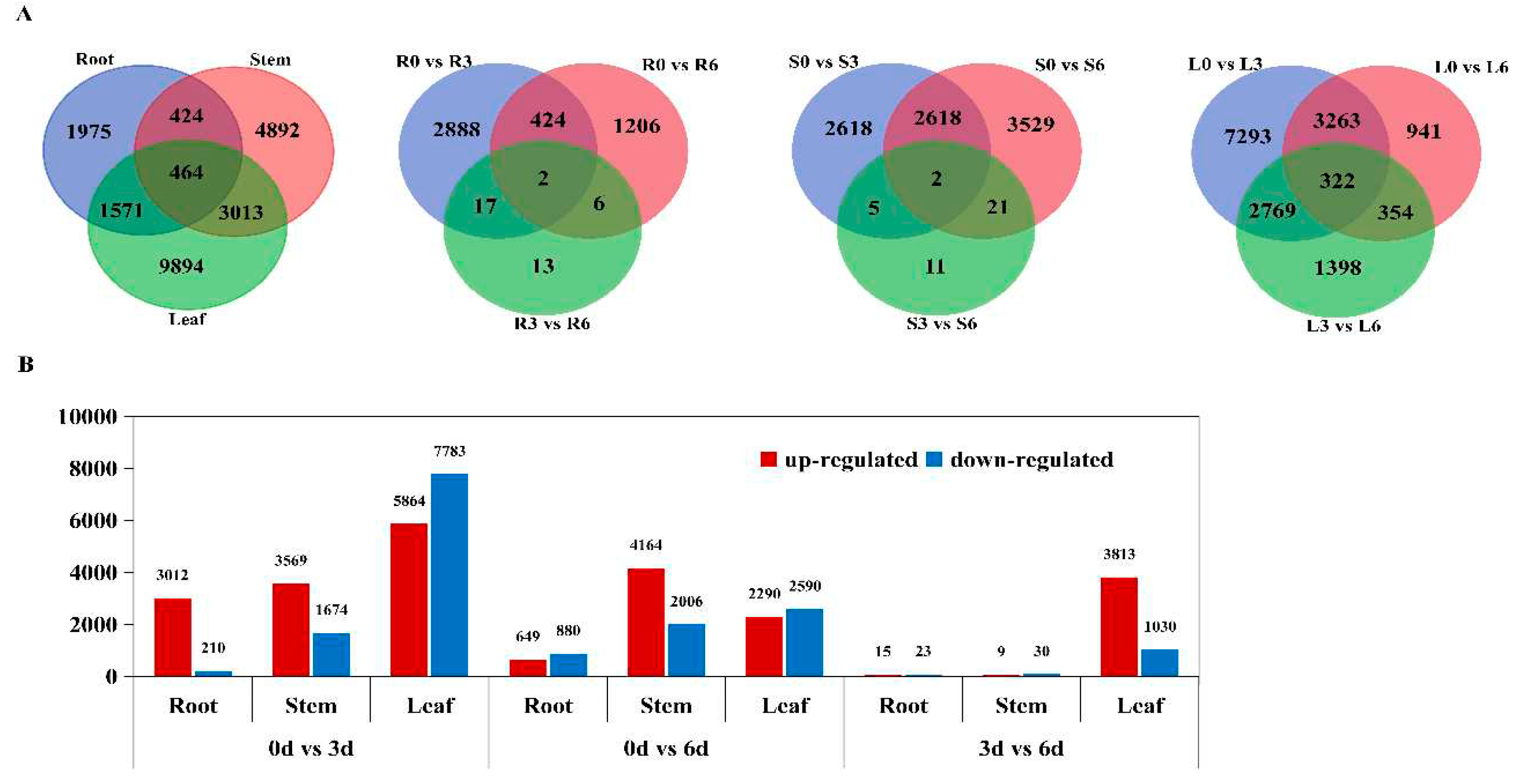
Figure 4.
GO enrichment analysis of DEGs in (A) roots, (B) stems, and (C) leaves under 3 and 6 days of cold stress. For more detailed information about GO term, please refer to Table S4.
Figure 4.
GO enrichment analysis of DEGs in (A) roots, (B) stems, and (C) leaves under 3 and 6 days of cold stress. For more detailed information about GO term, please refer to Table S4.
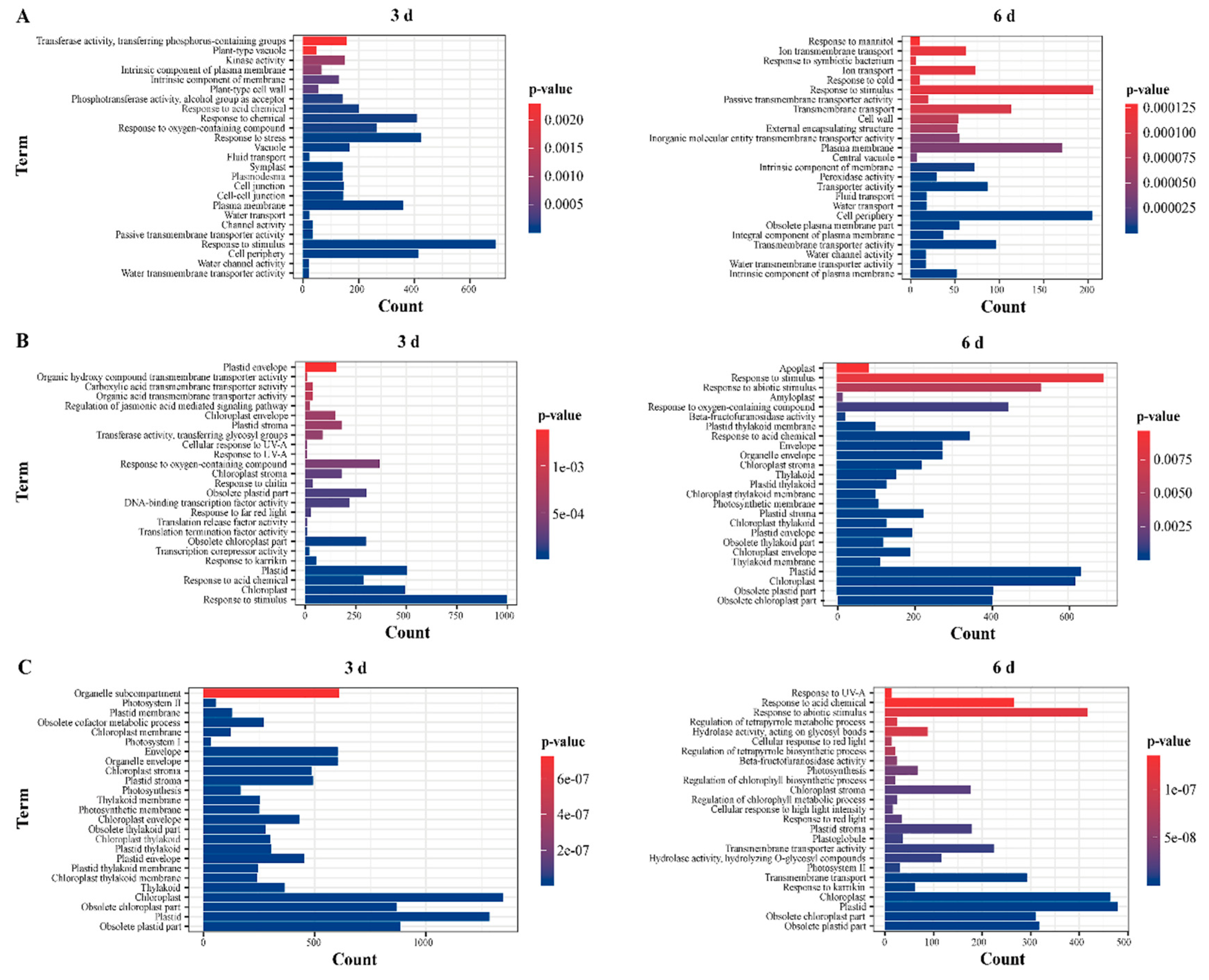
Figure 5.
KEGG pathway enrichment analysis of DEGs in (A) roots, (B) stems, and (C) leaves under 3 and 6 days of cold stress. For more detailed information about KEGG pathway, please refer to Table S5.
Figure 5.
KEGG pathway enrichment analysis of DEGs in (A) roots, (B) stems, and (C) leaves under 3 and 6 days of cold stress. For more detailed information about KEGG pathway, please refer to Table S5.
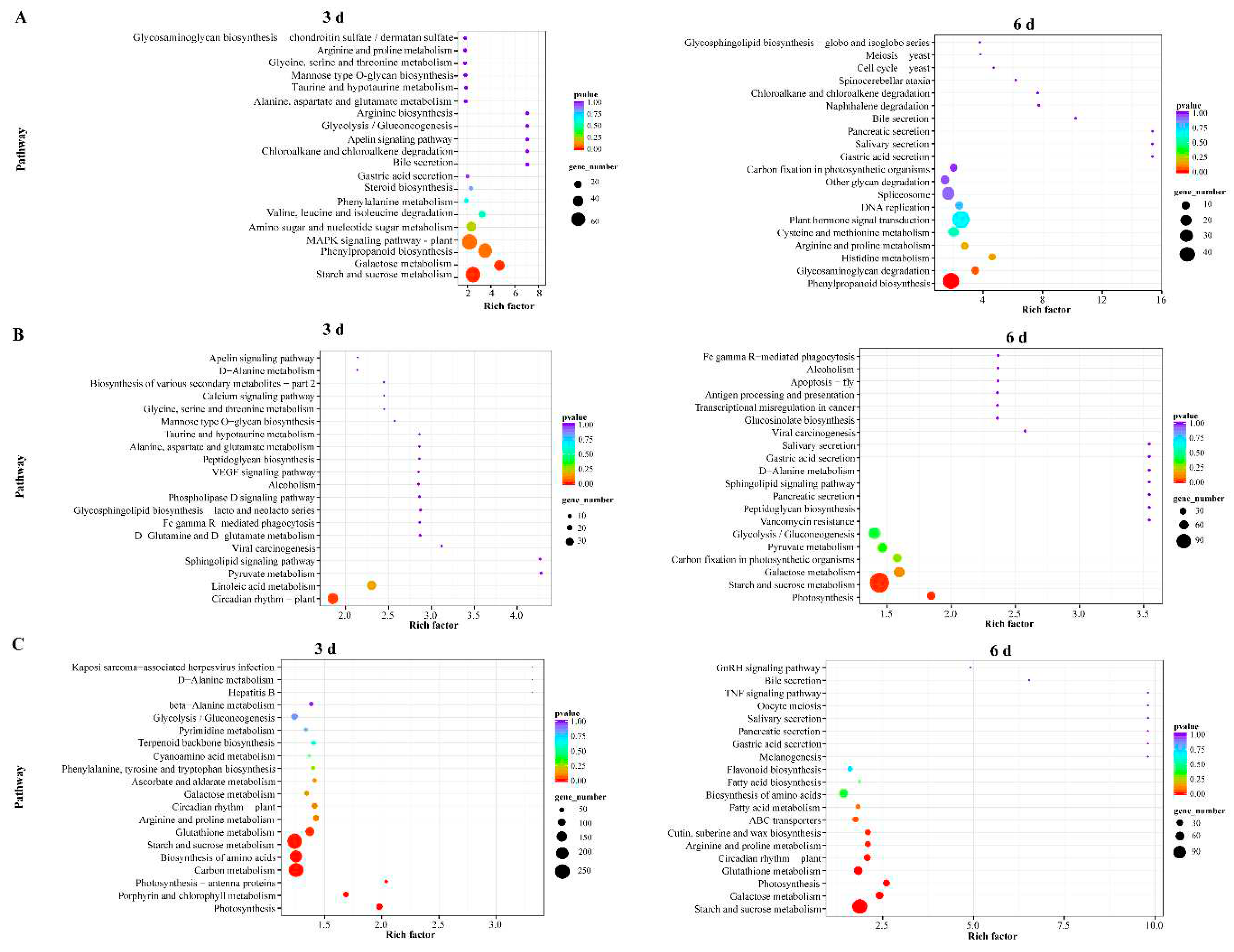
Figure 6.
Heat maps of DEGs involving transcription factors were co-induced in roots, stems, and leaves. Red and green indicate that the expression pattern of the gene is up-regulated and down-regulated respectively.
Figure 6.
Heat maps of DEGs involving transcription factors were co-induced in roots, stems, and leaves. Red and green indicate that the expression pattern of the gene is up-regulated and down-regulated respectively.
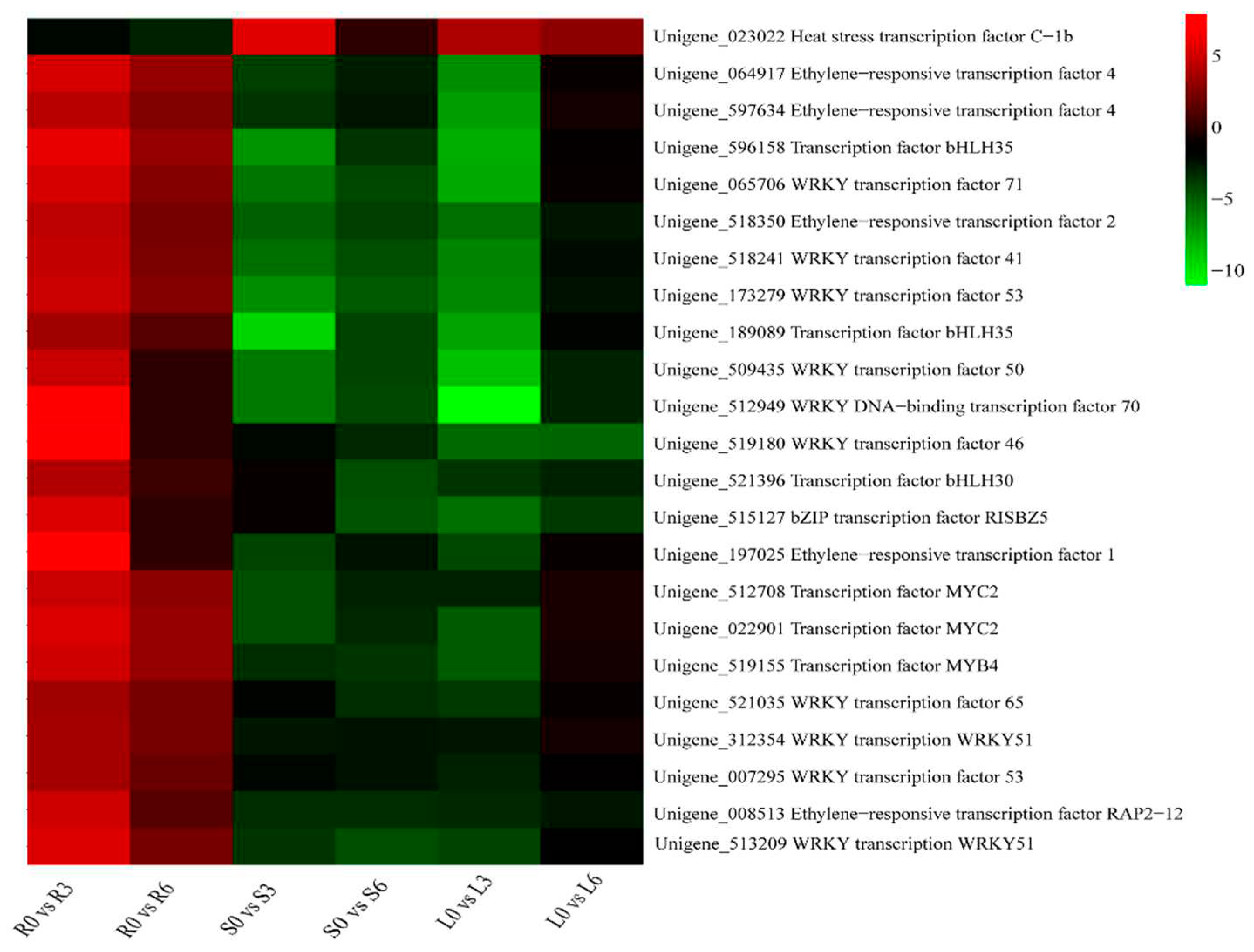
Figure 7.
Heat map of DEGs involving (A) the CBF pathway, (B) Ca2+ signaling, (C) hormone signaling, (D) ROS scavenging, and (E) circadian clock and photosynthesis were co-induced in roots, stems, and leaves. Red and green indicate that the expression pattern of the gene is up-regulated and down-regulated respectively.
Figure 7.
Heat map of DEGs involving (A) the CBF pathway, (B) Ca2+ signaling, (C) hormone signaling, (D) ROS scavenging, and (E) circadian clock and photosynthesis were co-induced in roots, stems, and leaves. Red and green indicate that the expression pattern of the gene is up-regulated and down-regulated respectively.
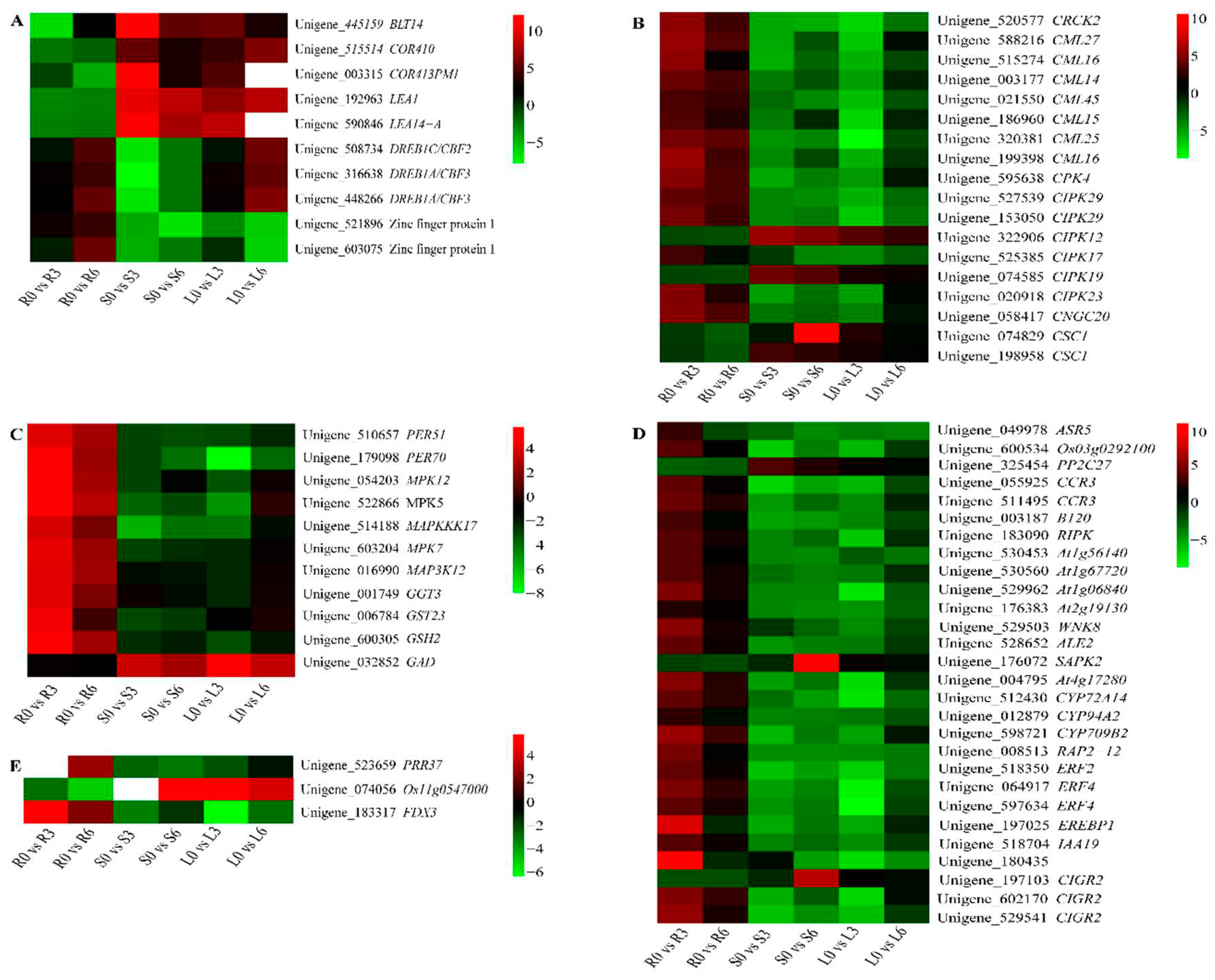
Figure 8.
WGCNA associated with cold response. (A) Scale independence and mean connectivity. (B) Gene cluster tree. (C) Module diagram. (D) Correlation heatmap between modules and traits.
Figure 8.
WGCNA associated with cold response. (A) Scale independence and mean connectivity. (B) Gene cluster tree. (C) Module diagram. (D) Correlation heatmap between modules and traits.
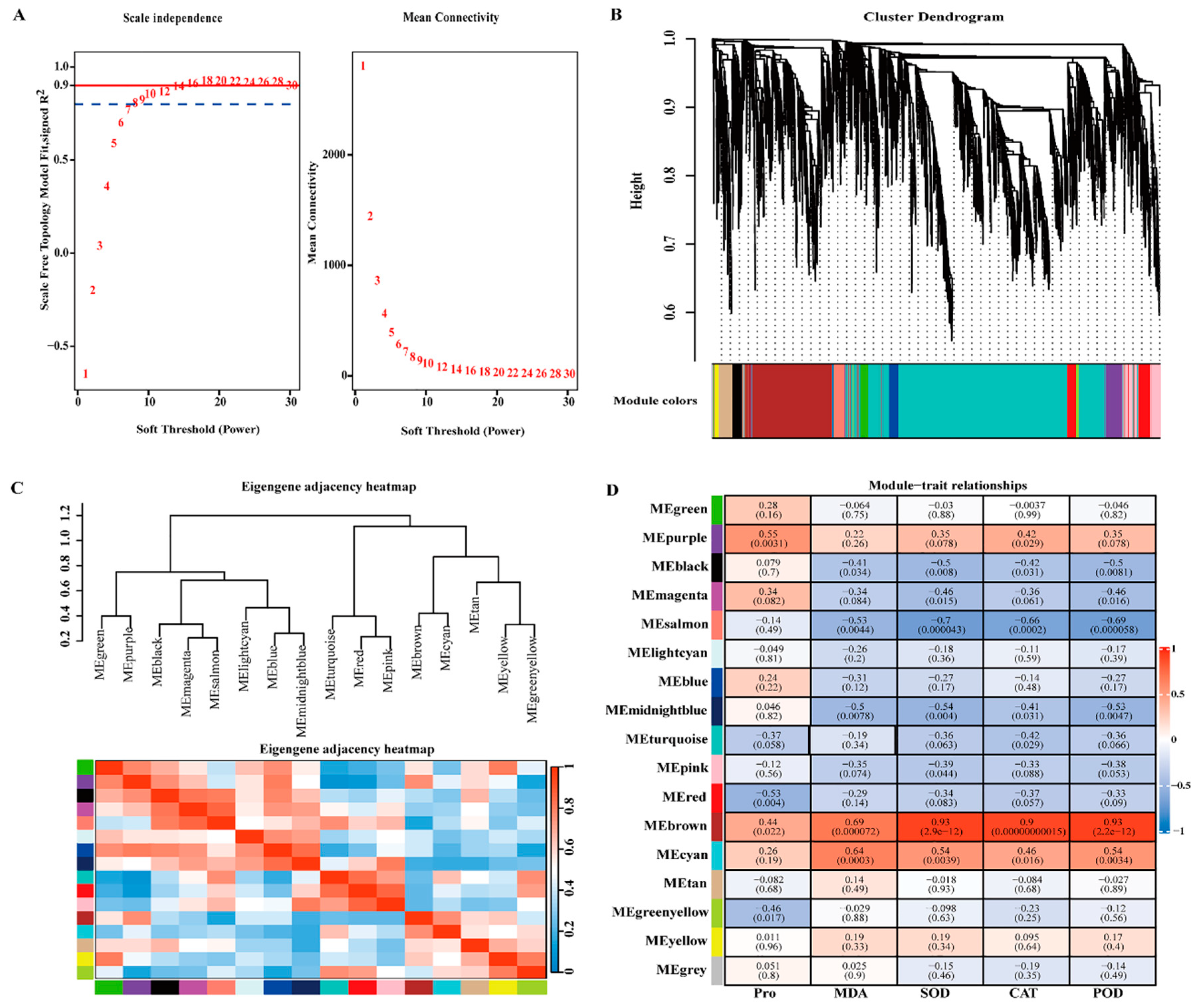
Figure 9.
Analysis of DEGs for brown and salmon modules. (A) GO enrichment analysis and (B) KEGG pathway enrichment analysis of DEGs in brown module and salmon module. (C) Co-expression network visualization of brown and salmon module.
Figure 9.
Analysis of DEGs for brown and salmon modules. (A) GO enrichment analysis and (B) KEGG pathway enrichment analysis of DEGs in brown module and salmon module. (C) Co-expression network visualization of brown and salmon module.
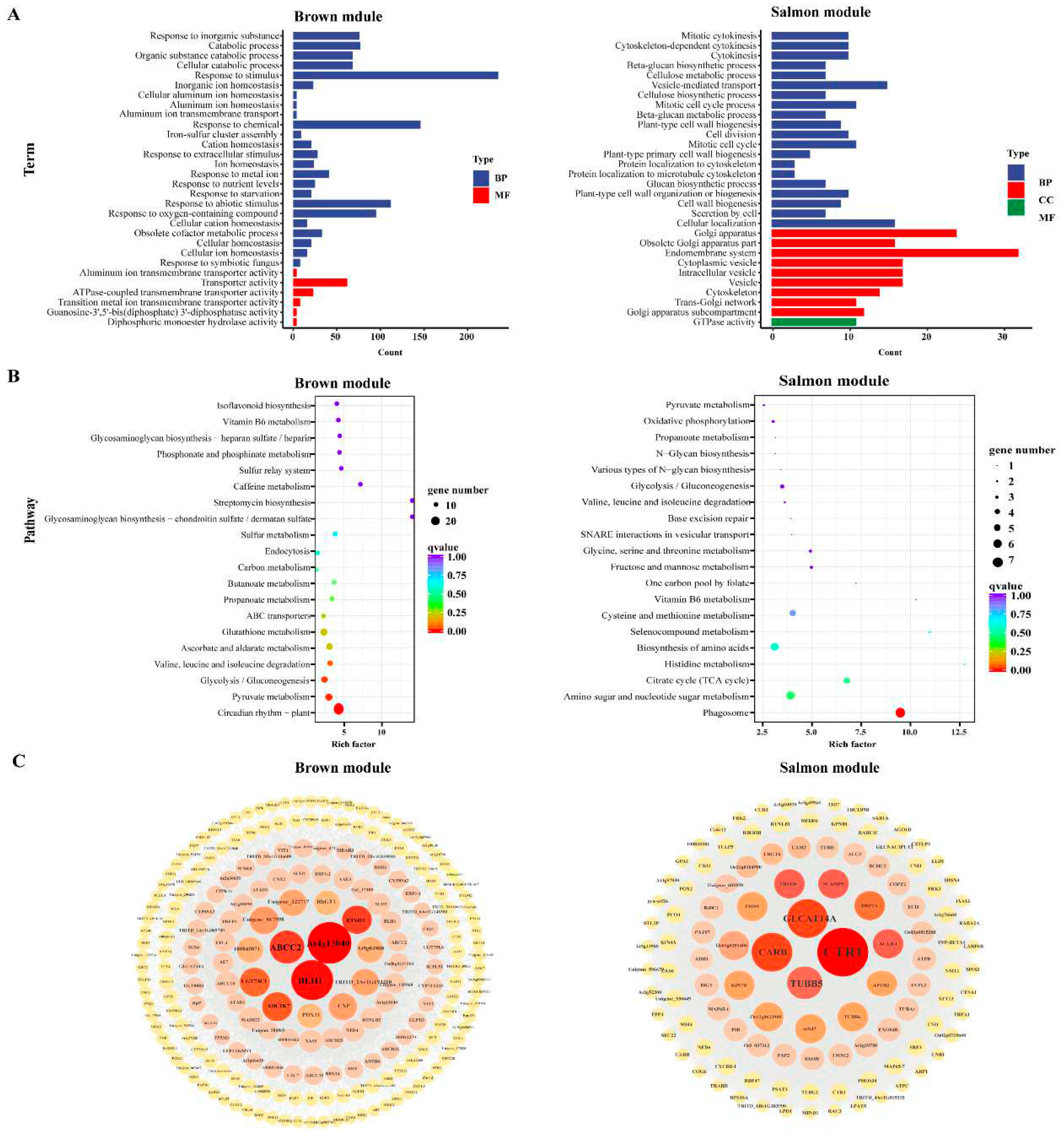
Figure 10.
Validation of the expression by qRT-PCR. Bar charts indicate values of qRT-PCR. Line plots indicate values of FPKM value.
Figure 10.
Validation of the expression by qRT-PCR. Bar charts indicate values of qRT-PCR. Line plots indicate values of FPKM value.

Figure 11.
Schematic diagram of the signaling pathways involved in the cold response mechanism of P. crymophila. The model was constructed based on the major cold response components identified in this report and previously described plant abiotic stress pathway schemes.
Figure 11.
Schematic diagram of the signaling pathways involved in the cold response mechanism of P. crymophila. The model was constructed based on the major cold response components identified in this report and previously described plant abiotic stress pathway schemes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



