
Submitted:
06 November 2023
Posted:
07 November 2023
You are already at the latest version
Abstract
Reactions of picolinamides with 1,3-propanesultone in methanol followed by the treatment with ketones led to a series of previously unknown chemical transformations, yielding first pyridinium salts (2a-f) with a protonated endocyclic nitrogen atom and then heterocyclic salts (3a-j) containing a 4-imidazoline ring. The structures of intermediate and final products were determined by IR and 1H, 13C NMR spectroscopy and X-ray study. The effects of the ketone and alcohol structures on the product yield were studied by quantum-chemical calculations. The stability of salts 3a-j towards hydrolysis and alcoholysis makes them excellent candidates for the search of new types of biologically active compounds.
Keywords:
4-imidazolidinones
; sulfobetaines
; NMR and FT-IR spectroscopy
; X-ray study
; quantum-chemical calculations
1. Introduction
Sulfobetaines, commonly prepared by the reactions of tertiary and aromatic amines with sultones, are objects of intense studies because of their unusual reactivity and broad spectrum of applications, including redox reagents [1], polymers, surfactants [2] and medical drugs [3,4,5,6,7,8].
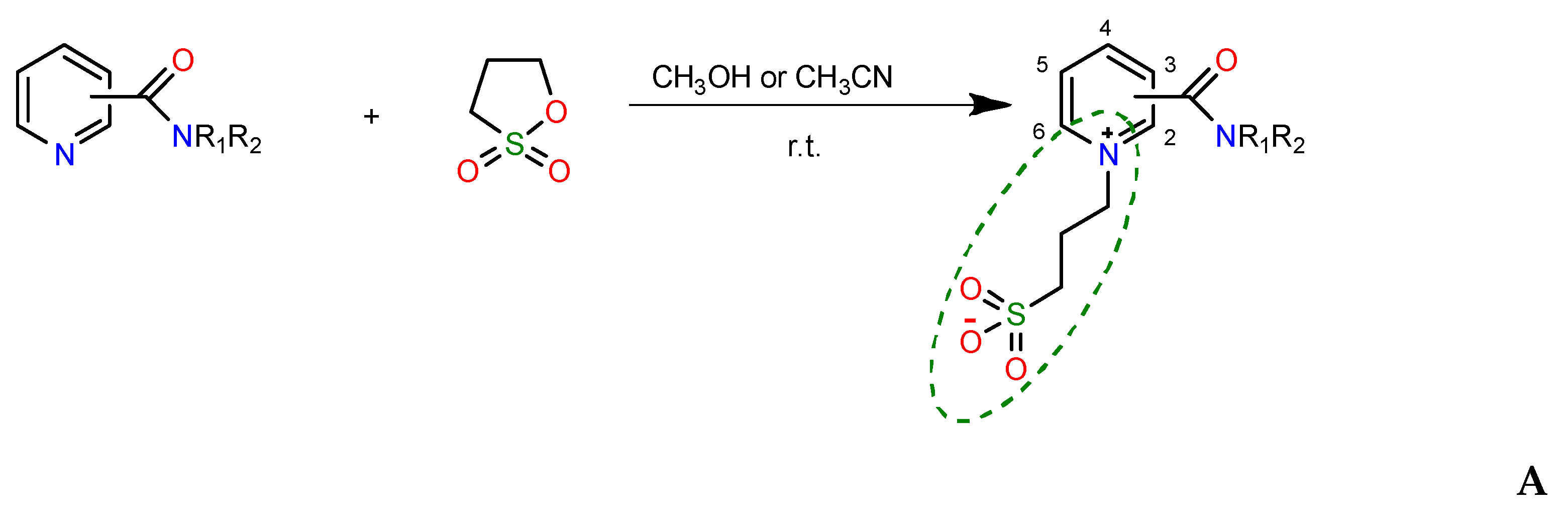
In our previous works, traditional synthetic approaches [9] were used for the preparation of novel pyridinecarboxamides (A) containing a homotaurin fragment [10]. Typically, the reactions proceeded through the opening of the sultone ring, as shown in Scheme 1.
For the reactions of 3- and 4-pyridinecarboxamides with sultones in boiling methanol, the position of the amido group has little effect on the yield of the final sulfobetaine A. In contrast, 2-pyridinecarboxamides (picolinamides) under similar conditions produce pyridinium salts B:
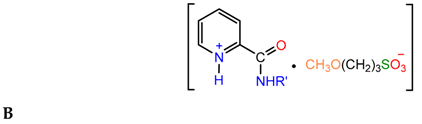
The low yield of product A in the case of picolinamides and the formation of salt B at high temperatures are likely to be a result of intramolecular bonding in the substrate involving the amido group and endocyclic nitrogen atom [10].
2. Results
2.1. Synthesis
The reaction between picolinamide 1a and 1,3-propanesultone in boiling methanol and subsequent treatment of the reaction mixture with acetone yielded a mixture of the expected salt B (2a) and another product, the derivative of 4-imidazoline 3a. By altering the reaction conditions, 3a could be isolated as individual compound and characterised by multinuclear (1H, 13C and 15N) and 2D NMR spectroscopy, IR spectroscopy and elemental analysis (Scheme 2).
To the best of our knowledge, compound 3a is the first example of 4-imidazolinium salts. According to literature, natural derivatives of 4-imidazoline, such as oxaline, neoxaline [11] and hetacillin [12], demonstrate a broad spectrum of antibacterial, antifungal and antitumour activity.
Biologically active synthetic 4-imidazolines include spiperone and mosapramine, which are potent dopamine receptor antagonists [13] and clinically important antipsychotic agents [14].
At present, 4-imidazolines are usually prepared by multistage synthetic processes that often require harsh conditions [15,16,17,18,19,20,21,22]. A typical route involves the synthesis of a linear α-aminoamide followed by cyclisation [23]:
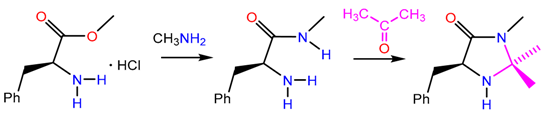
In some cases, a protective group must be used. The synthesis of N-substituted target compounds requires additional chemical transformations, such as condensations of α-acetaminoamides with aldehydes or ketones [24,25,26]. Other common approaches to N-substituted substrates include the Curtius rearrangement [27,28,29], Buchwald–Hartwig amination [30], reductive amination of carbonyl compounds [31] and Mitsunobu reaction [32].
Our attempt to prepare compound 3a by refluxing salt 2a in acetone for 3 h was only partially successful, as the yield was impractically low (12%). However, when a hot solution of 2a in methanol was treated with acetone, the yield increased to 80%. These results suggest that the formation of 3a in the second case could involve the reaction of 2a with a hemiketal intermediate.
Under similar conditions, the reactions of picolinamide 1a with various alcohols and ketones produced a broad range of pyridinium (2a-g) and 4-imidazolinium (3a-j) salts (Table 1 and Table 2). In the case of N-substituted amide 1b, the intermediate pyridinium salt 2g was found to be unreactive towards acetone, and the formation of the corresponding 4-imidazolinium derivative was not observed.
Most compounds 3a-j were obtained with high yields (75–94%, Table 2). The lower yields of compounds 3g (43%) and 3i (23%) could be caused by elimination reactions of alcohols used for the preparation of pyridinium salts 2c and 2e, respectively. The separation of 3i and 2e was very problematic, so the 1H NMR spectrum of the final mixture in D2O showed very broad signals of both compounds in 4:1 ratio, respectively. The IR spectrum of the mixture also showed the characteristic absorptions of both salts (see Experimental Part).
Our attempts to carry out the condensation of 2a with methyl tert-butyl ketone, anisole and benzaldehyde were unsuccessful – in all cases, only the original salt was isolated.
2.2. X-ray Study
According to the results of X-ray study, the values of all bond lengths and angles in salts 2a and 3a (Figure 1) fall within the ranges typical for pyridinium salts of alkylsulfonic acids. Crystallographic data for 2a and 3a are summarized in Table 3 (see Experimental Part). The parameters of hydrogen bonds are shown in Tables S6 and S7.
Salt 2a crystallised in monoclinic system and chiral space group P21. The unique part of unit cell contained four crystallographically independent cations and anions linked by strong hydrogen bonds between sulfo groups of anions and amido or pyridinium moieties of cations (Figure 2, left).
The supramolecular structure formed by hydrogen bonds can be described as a double layer, with ether groups of anions residing inside the layers while sulfo groups of anions and pyridinium moieties of cations forming the outer shell. In turn, the double layers are held together by weak interactions between sulfo groups and ipso-carbon atoms of pyridinium moieties.
In the crystal packing of 3a, cations and anions form dimers via hydrogen bonds between the sulfo group of the anion and the imidazolium ring of the cation. In turn, these dimers are assembled into a 3-D framework via weak C–H…O interactions (Figure 2, right).
2.3. Reactions of Hydrolysis and Alcoholysis of Compounds 3a, 3d and 3e
Many derivatives of 4-imidazoline are unstable in acidic and neutral aqueous environment. For example, the half-life of the antibacterial drug hetacillin in aqueous solutions at pH 3–8 is approximately 30 min [33]. The main hydrolysis product, ampicillin, is responsible for over 90% of the biological activity of hetacillin [34]. The N’-alkylation improves the hydrolytic stability of 4-imidazolinones both in human plasma and aqueous buffer with pH 7.4 [18].
In order to evaluate the applicability of salts 3a-j as potential drug candidates, we have studied the stability of compounds 3a, 3d and 3e towards water and alcohols.
In contrast to hetacillin, compound 3a was stable in water at room temperature (no hydrolysis was observed over a period of seven days). The reflux of compound 3a in water for 5 h led to the elimination of acetone and formation of salt 2a with a yield of 32% (Scheme 2). The hydrolysis of the same compound at moderate temperatures (70–80 °C) increased the yield of 2a to 63%.
The reaction of 3a with methanol produced a mixture of 2a and 3a, with 3a remaining the dominant component regardless of the reaction time (4–16 h). The IR spectra of the mixture showed two absorption bands of carbonyl groups in 2a and 3a (1707 and 1732 cm–1, respectively).
A similar behaviour was observed for compounds 3d and 3e. For example, the reaction of 3d with water at 70–80 °C for 5 h produced a mixture of 2a and 3d, with the IR spectrum showing two ν(C=O) bands at 1708 and 1727 cm–1, respectively.
2.4. Theoretical Study
Thermodynamic parameters of the chemical reactions shown in Scheme 2 were estimated using quantum-chemical calculations.
2.4.1. Thermodynamic Parameters of Formation for Compounds 2a-f
The first step of the reaction leading to salts 2a-f (Scheme 2) is the nucleophilic addition of an alcohol to 1,3-propanesultone, which produces 3-alkoxypropanesulfonic acids 1-IIa-f:
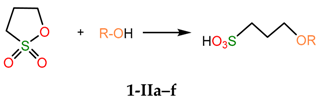
With the exception of R = Bu-i, the thermal effect of this step decreases when the size of the R substituent increases (Figure 3, Table S1).
Therefore, sterical hindrance is likely to be the major factor affecting the concentrations of 3-alkoxypropanesulfonic acids 1-IIa-f in the reaction mixture, which in turn affects the yields of respective salts 2a-f (Scheme 2).
2.4.2. Structures of the Cation–Anion Complexes
The actual thermodynamic parameters of chemical reactions shown in Scheme 2 depend on the mutual orientation of cations and anions under experimental conditions (in boiling methanol). Possible structures of salt 2a in the reaction medium could be predicted by analysing electrostatic potential (ESP) maps of the constituent ions (Figure 4A).
Red and blue dots in Figure 4A correspond to regions of ESP maps with low and high electron density, respectively. The lowest electron density for 1-Ia is observed near the nitrogen atom of the pyridinium ring while the highest electron density is concentrated around the sulfo group of the anion 1-IIa.
The most common mutual positions of ions 1-Ia and 1-IIa were identified by statistical analysis of molecular dynamics (MD) simulation for a system containing two ions of opposite charge and a large number of methanol molecules. This allowed to take into account the solvation of salt 2a by methanol and thus predict the most likely structure of the complex in solution (Figure 4B).
According to our calculations, salt 2a in solution is stabilised by hydrogen bonds involving one of the oxygen atoms in anion 1-IIa and hydrogen atoms at N1 and N2 in cation 1-Ia (Figure 4B). As expected, the calculated geometric parameters of the cation–anion complex in solution differ significantly from those in the solid state obtained by X-ray study (Figure 1). The most obvious difference is the mutual orientation of the acetamide fragment and pyridinium ring in 1-Ia. In solution (Figure 4B), the calculated value of the dihedral angle (φ) N1-C-C-N2 is close to zero while in the solid state (Figure 1) it approaches 180°. According to quantum-chemical calculations, the most stable conformation of non-protonated picolinamide 1a is achieved at φ ≈ 0° [10]. However, this is not the case for protonated picolinamide in 2a, where the conformation with φ ≈ 180° is by ca. 20 kJ/mol more stable (Figure 4C). The reason for this difference is the formation of hydrogen bonds: intramolecular in 1a (φ ≈ 0°) and interionic in the 2a complex (φ ≈ 180°). In the latter case, the AIM analysis reveals critical points of (3; –1) type (for more detail, see Table S2).
The activation energy for the proton migration from sulfonic acid to pyridine ring is relatively low (4.5 kJ/mol, M052X-D3/TZVP), which suggests that the formation of salt 2a might involve the following transition state:
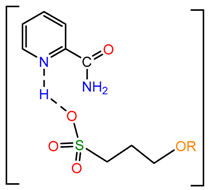
2.4.3. Thermodynamic Parameters of 2a–g Formation
Figure 5 shows a histogram where the yields of salts 2a–f are plotted together with relative differences of the reaction enthalpy and Gibbs free energy, with zero values for ΔΔrH° and ΔΔrG° corresponding to the lowest values of ΔrH° and ΔrG° from Table S2.
According to Figure 5, relative values ΔΔrH° and ΔΔrG° generally increase along with the size of the alkyl chain in the alcohol. The only exception is isopropanol, which also has the lowest values of ΔrH° and ΔrG° for the reaction with 1,3-propanesultone (Figure 5, Table S1). The least thermodynamically favourable reaction, the formation of salt 2e, proceeds with the lowest yield (25%). At the same time, similar ΔΔrH° and ΔΔrG° values are observed for salt 2f, which was obtained with the highest yield (89%). The formation of salt 2g is characterised by positive absolute values of ΔrH° and ΔrG° (Table S2).
2.4.4. Thermodynamic Parameters of 3a-j Formation
Analysis of thermodynamic parameters of salts 3a-j formation suggests that the observed chemical changes could be divided into three groups. The first group includes the formation of salts 3a-c by reactions of 2a with aliphatic ketones (Figure 6A).
In this group, an increase of the alkyl substituent size raises the ΔΔrH° and ΔΔrG° values and therefore lowers the reaction yield. (The absolute values of ΔrH° and ΔrG° are given in Table S4).
The second group includes the reactions of salt 2a with cyclic ketones. In this case, an increase in the Reactions of the third group include the formation of salts 3a and 3f-j. In these reactions, an increase in the size of the alkoxy substituent in the sulfonic acid raises the ΔΔrH° and ΔΔrG° values and therefore lowers the reaction yield (Figure 6B). These results correlate with the data shown in Figure 5. Ring size lowers the ΔΔrH° and ΔΔrG° values and therefore raises the reaction yield.
3. Materials and Methods
3.1. Chemistry
The purities of all compounds were assessed by elemental analysis and NMR and found to be ≥ 95%. NMR spectra were recorded on a Bruker Avance II 300 spectrometer at 300 MHz (1H) and 75 MHz (13C) in D2O in the pulse mode followed by Fourier transformation using Me4Si as internal standard. Spin multiplicities are designated as s (singlet), d (doublet), t (triplet), q (quartet) or m (multiplet). IR spectra in the solid phase were recorded on a Bruker Tensor-27 instrument with an attenuated total internal reflectance (ATR) module. Refraction parameters were measured using an IRF-454B2M refractometer. Melting points were determined using a Stuart SMP10 instrument. Elemental analyses were carried out at the Laboratory of Organic Microanalysis of INEOS RAS.
3.1.1. Synthesis
Compounds 1a, 1b, 1,3-propanesultone, ketones and alcohols were obtained as commercial reagents from Acros and Sigma–Aldrich and used without further purification.
General synthesis of compounds 2a-g and 3a-i. A mixture of 0.005 mmol of compound 1a or 1b and 0.006 mol of 1,3-propanesultone in an alcohol was refluxed for 4 h. The solvent was evaporated, the salt 2a-f obtained was treated with a ketone at reflux in methanol. The crystals of salt 3a-j formed were filtered out and dried.
Synthesis of 2-carbamoylpyridin-1-ium 3-methoxypropane-1-sulfonate (2a). According to the general protocol (MeOH, 4 mL), 1.52 g (65%) of 2a was obtained, m.p. 153–155 °C (benzene–acetonitrile, 1:2). IR spectrum (solid, ν/cm–1): 1707 s (C=O), 1602 (C=Cpyridine), 1179 s, 1124 s, 1035 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 1.88 (m, 2H, -H2CCH2CH2-), 2.81 (t, 2H, 3J=6.5, -CH2SO3), 3.46 (t, 2H, 3J=6.5, CH3OCH2CH2-), 3.23 (s, 3H, CH3OCH2), 8.35 (d, 1H, 3J=7.9, H3), 8.08 (t, 1H, 3J=7.9, H4), 8.58 (t, 1H, 3J=7.9, H5), 8.77 (d, 1H, 3J=7.9, H6); 13C-NMR (75.5 MHz, D2O, δ, ppm): 24.03, 47.88, 57.61, 70.47, 125.06, 129.57, 142.51, 143.21, 146.96, 162.39. Anal. calcd. for C10H16N2O5S: C, 43.47; H, 5.84; N, 10.14; S, 11.60; O, 28.95. Found: C, 42.85; H, 6.54; N, 9.09; S, 10.40; O, 31.13.
Synthesis of 2-carbamoylpyridine-1-ium-3-ethoxypropane-1-sulfonate (2b). A mixture of 0.61 g (0.005 mol) picolinamide 1a and 0.73 g (0.006 mol) 1,3-propanesultone in 4 mL of ethanol was refluxed for 3 h, the volatiles were removed in vacuum, and the residue was stirred with 10 mL of diethyl ether for 1 h. The crystals formed were isolated by filtration to yield 1.34 g (85%) of compound 2b ⋅ 1.5 H2O, m.p. 145–147 °C (CH3CN). IR spectrum (solid, ν/cm–1): 1707 s (C=O), 1602 w (C=Cpyridine), 1176 s, 1146 s, 1034 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 1.09 (t, 3H, 3J=7.0, CH3CH2-), 1.87 (m, 2H, -CH2CH2CH2-), 2.85 (t, 2H, 3J 7.2, -CH2SO3), 3.51 (m, 4H, -CH2OCH2-), 8.41 (d, 1H, 3J=8.1, H3), 8.13 (t, 1H, 3J=8.1, H4), 8.68 (t, 1H, 3J=8.1, H5), 8.84 (d, 1H, 3J=8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 14.14, 23.57, 47.99, 60.22, 72.29, 125.18, 129.68, 142.67, 143.38, 147.38, 162.58. Anal. calcd. for C11H21N2O6.5S: C, 41.63; H, 6.67; N, 8.83; S, 10.10. Found: C, 41.77; H, 6.35; N, 9.03; S, 9.61.
Synthesis of 2-carbamoylpyridine-1-ium-3-isopropoxypropane-1-sulfonate (2c). Similar to 2b, using isopropanol instead of ethanol, 1.43 g (59%) of compound 2c ⋅ HO(CH2)3SO3H ⋅ CH3CN was obtained, m.p. 180–181 °C (CH3CN). IR spectrum (solid, ν/cm–1): 1708 s (C=O), 1637 w (C=Cpyridine), 1176 s, 1146 s, 1036 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 1.05 (br. s, 6H, 2CH3), 3.60 (br. m, 1H, CH), 1.86 (m, 2H, -H2CCH2CH2-), 2.85 (br. m, 2H, 3J=7.1, -CH2SO3), 3.50 (br. t, 2H, 3J=7.1, OCH2-), 8.38 (d, 1H, 3J=8.1, H3), 8.11 (t, 1H, 3J=8.1, H4), 8.63 (t, 1H, 3J=8.1, H5), 8.79 (d, 1H, 3J=8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 21.15, 24.50, 48.01, 66.13, 72.28, 125.14, 129.66, 142.83, 143.54, 146.87, 162.79. Anal. calcd. for C17H31N3O9S2: C, 42.04; H, 6.43; N, 8.65; S, 13.21. Found (%): C, 41.91; H, 6.32; N, 8.85; S, 12.39.
Synthesis of 2-carbamoylpyridine-1-ium-3-isobutoxy propane-1-sulfonate (2d). Similar to 2b, using isobutanol at 78–80 °C instead of ethanol at reflux, 0.98 g (57%) of compound 2d ⋅ 1.5 H2O was obtained, m.p. 179–182 °C (CH3CN). IR spectrum (solid, ν/cm–1): 1708 s (C=O), 1601 w (C=Cpyridine), 1146 s, 1124 s, 1036 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 0.83 (d, 6H, 3J=7.1, 2CH3), 1.76 (m, 1H, CH), 1.96 (m, 2H, CH2CH2CH2), 2.91 (t, 2H, 3J=7.1, CH2SO3), 3.55 (t, 2H, 3J=7.1, OCH2), 3.23 (d, 2H, 3J=7.1, -CHCH2O), 8.42 (d, 1H, 3J=8.1, H3), 8.15 (t, 1H, 3J=8.1, H4), 8.65 (t, 1H, 3J=8.1, H5), 8.85 (d, 1H, 3J=8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 18.57, 24.30, 27.55, 48.06, 68.81, 77.48, 125.14, 129.66, 142.83, 143.54, 146.87, 162.77. Anal. calcd. for C13H25N2O6.5S: C, 45.20; H, 7.29; N, 8.10; S, 9.28. Found: C, 45.72; H, 6.69; N, 8.31; S, 8.71.
Synthesis of 2-carbamoylpyridine-1-ium-3-tert butoxy propane-1-sulfonate (2e). Similar to 2b, using tert-butanol instead of ethanol, 0.62 g (25%) of compound 2e ⋅ 1.5 HO(CH2)3SO3H ⋅ CH3CN was obtained, m.p. 149–150 °C (CH3CN). IR spectrum (solid, ν/cm–1): 1708 s (C=O), 1636 w, 1602 w (C=Cpyridine), 1147 s, 1036 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 1.15 (s, 9H, 3CH3), 1.87 (m, 2H, -H2CCH2CH2-), 2.91 (t, 2H, 3J=7.1, -CH2SO3), 3.49 (t, 2H, 3J=7.1, OCH2-), 8.42 (d, 1H, 3J=8.1, H3), 8.15 (t, 1H, 3J=8.1, H4), 8.64 (t, 1H, 3J=8.1, H5), 8.85 (d, 1H, 3J=8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 24.51, 26.61, 48.12, 60.24, 74.79, 125.18, 129.69, 142.77, 143.48, 146.96, 162.68. Anal. calcd. for C18H33N3O9S2: C, 43.27; H, 6.66; N, 8.41; S, 12.83. Found: C, 43.54; H, 6.41; N, 8.70; S, 12.35.
Synthesis of 2-carbamoylpyridine-1-ium-3-cyclohexane hydroxypropane-1-sulfonate (2f). Similar to 2b, using cyclohexanol instead of ethanol, 1.78 g (89%) of compound 2f ⋅ CH3CN ⋅ H2O was obtained, m.p. 173–175 °C (CH3CN). IR spectrum (solid, ν/cm–1): 1707 s (C=O), 1636 w, 1602 w (C=Cpyridine), 1178 s, 1147 s, 1037 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 1.91 (m, 2H, -H2CCH2CH2-), 2.93 (t, 2H, 3J=7.0, -CH2SO3), 1.17–1.68 (m, 10H, C6H10), 3.39 (m, 1H, OCH-), 3.63 (t, 2H, 3J=7.0, OCH2-), 8.43 (d, 1H, 3J=8.1, H3), 8.16 (t, 1H, 3J=8.1, H4), 8.65 (t, 1H, 3J=8.1, H5), 8.85 (d, 1H, 3J=8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 23.84, 24.78, 25.24, 31.73, 48.02, 65.92, 78.28, 125.13, 129.66, 142.86, 143.58, 146.82, 162.78. Anal. calcd. for C17H29N3O6S: C, 50.60; H, 7.24; N, 10.41; S, 7.94. Found: C, 51.01; H, 7.00; N, 10.12; S, 7.41.
Synthesis of 2-(methylcarbamoyl)pyridin-1-ium 3-methoxypropane-1-sulfonate (2g). Reaction was performed in 5 mL of methanol. Mixture was heated under reflux for 4 h. Obtained 2.00 g (81% yield) oily complex, m.p. 60–64 °C. IR (solid, ν/cm–1): 1679 s (C=O), 1604 (C=Cpyridine), 1212, 1108, 1032, (SO3). 1H-NMR (300.1 MHz, D2O, ppm, J/Hz): δ 1.92 (m, 2H, C-CH2-C), 2.85 (t, 2H, 3J = 7.1 CH2SO3), 3.50 (t, 2H, 3J = 7.3, CH3O-CH2), 3.26 (s, 3H, CH3O-CH2), 2.95 (s, 3H, HNCH3), 8.12 (t, 1H, 3J = 7.0, H5), 8.81 (d, 1H, 3J = 8.1, H6), 8.34 (d, 1H, 3J = 6.1, H4), 8.63 (t, 1H, 3J = 7.0, H5); 13C-NMR (75.5 MHz, D2O, ppm): δ 24.03, 26.90, 47.78, 57.61, 70.47, 125.06, 129.57, 143.21, 146.96, 162.39 [10].
Synthesis of 3,3-dimethyl-1-oxo-2,3-dihydro-1H-imidazo [1,5-a]pyridin-4-ium 3-methoxypropane-1-sulfonate (3a).
1) A mixture of 0.61 g (0.005 mol) of 1a and 0.73 g (0.006 mol) of 1,3-propanesultone in 5 mL of methanol was refluxed for 3 h, then 4 mL of acetone was added to the hot solution, and the mixture was refluxed for further 1.5 h. Next day, the volatiles were removed in vacuum, the residue was stirred with diethyl ether for 2 h, and the crystals formed were filtered and dried to afford 1.27 g (80%) of compound 3a, m.p. 174–177 °C (methanol–acetone, 1:20). IR spectrum (solid, ν/cm–1): 1729 s (C=O), 1637 w (C=Cpyridine), 1208 s, 1160 s, 1033 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 1.84 (m, 2H, -H2CCH2CH2-), 1.89 (s, 6H, 2CH3), 2.84 (t, 2H, 3J 7.2, -CH2SO3), 3.25 (s, 3H, 3J=7.2, CH3O), 3.47 (t, 2H, 3J=7.2, OCH2-), 8.42 (d, 1H, 3J=8.1, H3), 8.31 (t, 1H, 3J=8.1, H4), 8.76 (t, 1H, 3J=8.1, H5), 9.31 (d, 1H, 3J=8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 24.10, 26.48, 47.65, 57.69, 70.57, 84.15, 123.80, 130.93, 138.48, 141.63, 148.03, 158.99. Anal. calcd. for C13H23.5N2O6.75S (3a ⋅1.75H2O): C, 44.87; H, 6.80; N, 8.05; S, 9.21. Found: C, 44.89; H, 6.18; N, 8.28; S, 10.53.
2) 0.82 g (0.003 mol) of salt 2a was refluxed for 3 h in 10 mL of acetone. Next day, the volatiles were removed in vacuum, the residue was stirred with diethyl ether for 2 h, and the crystals formed were filtered and dried to afford 0.51 g (62%) of unreacted compound 2a are obtained. By evaporation, 0.11 g (11.7%) of compound 3a was isolated, m.p. 170–174 °C (methanol–acetone, 1:20). IR spectrum (solid, ν, cm–1): 1738 s (C=O), 1633 w (C=Cpyridine), 1194 s, 1148 s, 1032 s (SO3).
Synthesis of 3-ethyl-3-methyl-1-oxo-2,3-dihydro-1H-imidazo[1,5-a]pyridin-4-ium 3-methoxypropane-1-sulfonate (3b). Similar to 3a, using butanone instead of acetone, 1.58 g (91%) of 3b ⋅ 0.5 H2O was obtained, m.p. 162–165 °C (acetonitrile–acetone, 1:30). IR spectrum (solid, ν/cm–1): 1738 s (C=O), 1633 w (C=Cpyridine), 1194 s, 1148 s, 1032 s (SO3). 1H NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 0.67 (t, 3H, 3J=7.2, CH3CH2-), 1.95 (m, 2H, -H2CCH2CH2-), 2.41 (q, 2H, 3J=7.2, -CH2CH3), 2.89 (t, 2H, 3J=7.2, -CH2SO3), 3.31 (s, 3H, CH3O), 3.53 (t, 2H, 3J=7.2, -OCH2), 8.53 (d, 1H, 3J=8.1, H3), 8.42 (t, 1H, 3J=8.1, H4), 8.85 (t, 1H, 3J=8.1, H5), 9.31 (d, 1H, 3J=8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 5.99, 22.97, 26.43, 36.69, 48.03, 57.75, 70.64, 86.95, 123.94, 131.01, 138.54, 141.79, 148.27, 159.44. Anal. calcd. for C14H23N2O5.5S: C, 49.54; H, 6.82; N, 8.25; S, 9.44. Found: C, 49.69; H, 6.37; N, 8.27; S, 9.98.
Synthesis of 1-oxo-3,3-dipropyl-2,3-dihydro-1H-imidazo[1,5-a]pyridin-4-ium 3-methoxypropane-1-sulfonate (3c). 1.23 g (0.0044 mol) of 2a was refluxed in a mixture of 4 mL of methanol and 4 mL of dipropyl ketone for 15 h. The volatiles were removed in vacuum, the residue was stirred with diethyl ether for 2 h, and the crystals formed were filtered and dried to afford 2.28 (75%) of 3c ⋅ 2 HO(CH2)3SO3H ⋅ CH3CN, m.p. 88–91 °C (CH3CN). IR spectrum (solid, ν/cm–1): 1733 s (C=O), 1630 w (C=Cpyridine), 1147 s, 1115 s, 1031 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 0.79 (t, 6H, 3J=7.2, 2 CH3CH2CH2-), 1.27 (m, 4H, 2 CH3CH2CH2-), 1.94 (m, 2H, -CH2CH2CH2-), 2.48 (m, 4H, 2 CH3CH2CH2-), 2.91 (t, 2H, 3J 7.2, CH2SO3), 3.33 (s, 3H, CH3O), 3.55 (t, 2H, 3J 7.2, OCH2), 8.50 (d, 1H, 3J 8.1, H3), 8.40 (t, 1H, 3J 8.1, H4), 8.80 (t, 1H, 3J 8.1, H5), 9.24 (d, 1H, 3J 8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 12.59, 15.10, 24.17, 40.23, 47.94, 57.75, 70.65, 89.11, 124.10, 131.38, 138.58, 148.43, 159.10. Anal. calcd. for C25H47N3O13S3: C, 43.27; H, 6.82; N, 6.05; S, 13.86. Found: C, 42.54; H, 6.64; N, 6.46; S, 13.20.
Synthesis of 1’-oxo-1’,2’-dihydrospiro[cyclopentane-1,3’-imidazo[1,5-a]pyridin]-4’-ium 3-methoxypropane-1-sulfonate (3d). Similar to 3a, using cyclopentanone instead of acetone, 1.60 g (85%) of 3d ⋅ 2 H2O was obtained, m.p. 177–179 °C (acetonitrile–acetone, 1:3). IR spectrum (solid, ν/cm–1): 1726 s (C=O), 1637 w (C=Cpyridine), 1169 s, 1112 s, 1039 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 1.88–2.55 (m, 8H cycle; 2H, -H2CCH2CH2-), 2.89 (t, 2H, 3J=7.2, CH2SO3), 3.29 (s, 3H, CH3O), 3.49 (t, 2H, 3J=7.2, -OCH2), 8.45 (d, 1H, 3J=8.1, H3), 8.35 (t, 1H, 3J=8.1, H4), 8.78 (t, 1H, 3J=8.1, H5), 9.30 (d, 1H, 3J=8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 22.80, 24.16, 40.33, 47.93, 57.75, 70.64, 92.40, 123.29, 131.00, 138.51, 142.18, 148.11, 159.20. Anal. calcd. for C15H26N2O7S. Calculated: C, 47.61; H, 6.92; N, 7.40; S, 8.47. Found: C, 47.12; H, 6.65; N, 7.87; S, 8.79.
Synthesis of 1’-oxo-1’,2’-dihydrospiro[cyclohexane-1,3’-imidazo[1,5-a]pyridin]-4’-ium 3-methoxypropane-1-sulfonate (3e). Similar to 3a, using cyclohexanone instead of acetone, 1.71 g (94%) of compound 3e ⋅ 0.5 H2O was obtained, m.p. 213–216 °C (CH3CN). IR spectrum (solid, ν/cm–1): 1735 s (C=O), 1638 w (C=Cpyridine), 1169 s, 1114 s, 1036 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 1.4–2.3 (m, 10H cycle; 2H -H2CCH2CH2-), 2.90 (t, 2H, 3J=7.2, -CH2SO3), 3.29 (s, 3H, CH3O), 3.53 (t, 2H, 3J=7.2, OCH2-), 8.48 (d, 1H, 3J 8.1, H3), 8.34 (t, 1H, 3J 8.1, H4), 8.81 (t, 1H, 3J 8.1, H5), 9.33 (d, 1H, 3J 8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 22.57, 22.98, 23.17, 24.17, 47.94, 57.75, 70.65, 86.88, 123.87, 130.84, 138.67, 141.48, 148.11, 159.64. Anal. calcd. for C16H25N2O5.5S: C, 52.58; H, 6.89; N, 7.66; S, 8.72. Found: C, 52.49; H, 6.65; N, 8.02; S, 8.96.
Synthesis of 3,3-dimethyl-1-oxo-2,3-dihydro-1H-imidazo[1,5-a]pyridin-4-ium 3-ethoxypropane-1-sulfonate (3f). Similar to 3a, using ethanol instead of methanol, 2.05 g (82%) of 3f ⋅ HO(CH2)3SO3H ⋅ CH3CN was obtained, m.p. 118–121 °C (CH3CN). IR spectrum (solid, ν/cm–1): 1732 s (C=O), 1635 w (C=Cpyridine), 1156 s, 1123 s, 1034 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 1.10 (t, 3H, 3J=6.9, CH3CH2), 1.89 (s, 6H, 2CH3), 1.94 (m, 2H, -H2CCH2CH2-), 2.91 (t, 2H, 3J=7.0, -CH2SO3), 3.49 (t, 2H, 3J=7.0, OCH2-), 3.63 (q, 2H, 3J=6.9, CH3CH2-), 8.45 (d, 1H, 3J=8.1, H3), 8.39 (t, 1H, 3J=8.1, H4), 8.79 (t, 1H, 3J=8.1, H5), 9.32 (d, 1H, 3J=8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 14.18, 21.17, 24.35, 26.61, 48.02, 68.52, 84.21, 122.81, 131.02, 138.54, 141.64, 148.12, 159.01. Anal. calcd. for C19H33N3O9S2. Calculated: C, 44.60; H, 6.50; N, 8.21; S, 12.53. Found: C, 44.09; H, 6.46; N, 8.38; S, 11.79.
Synthesis of 3,3-dimethyl-1-oxo-2,3-dihydro-1H-imidazo[1,5-a]pyridin-4-ium 3-isopropoxypropane-1-sulfonate (3g). Similar to 3a, using isopropanol instead of methanol, 1.12 g (43%) of 3g ⋅ HO(CH2)3SO3H ⋅ CH3CN was obtained, m.p. 139–140 °C (CH3CN). IR spectrum (solid, ν/cm–1): 1734 s (C=O), 1638 w (C=Cpyridine), 1156 s, 1123 s, 1033 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 0.86 (d, 6H, 3J=7.0, 2CH3), 1.93 (m, 1H, CHO), 1.90 (m, 2H, -H2CCH2CH2-), 1.94 (s, 6H, 2CH3), 2.89 (t, 2H, 3J=7.0, -CH2SO3), 3.54 (t, 2H, 3J=7.0, OCH2-), 8.48 (d, 1H, 3J=8.1, H3), 8.37 (t, 1H, 3J=8.1, H4), 8.79 (t, 1H, 3J=8.1, H5), 9.34 (d, 1H, 3J=8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 18.63, 24.34, 26.66, 27.59, 48.06, 68.85, 77.51, 84.24, 122.83, 131.06, 138.57, 141.63, 148.16, 158.99. Anal. calcd. for C20H35N3O9S2. Calculated: C, 45.69; H, 6.71; N, 7.99; S, 12.20. Found: C, 46.35; H, 6.35; N, 7.75; S, 12.75.
Synthesis of 3,3-dimethyl-1-oxo-2,3-dihydro-1H-imidazo[1,5-a]pyridin-4-ium 3-isobutoxixipropane-1-sulfonate (3h). Similar to 3a, using isobutanol instead of methanol, 1.96 g (73%) of 3h ⋅ HO(CH2)3SO3H ⋅ CH3CN was obtained, m.p. 147–150 °C (CH3CN). IR spectrum (solid, ν/cm–1): 1740 s (C=O), 1632 w (C=Cpyridine), 1149 s, 1031 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 0.85 (d, 6H, 3J 7.0, (CH3)2CH-), 2.02 (m, 6H, 2CH3), 1.87 (m, 1H, (CH3)2CH-)), 1.98 (s, 6H, 2CH3), 2.94 (t, 2H, 3J=7.0, CH2SO3), 3.15 (d, 2H, 3J=7.0, CHCH2O), 3.64 (t, 2H, 3J=7.0, OCH2-), 8.52 (d, 1H, 3J=8.1, H3), 8.43 (t, 1H, 3J=8.1, H4), 8.85 (t, 1H, 3J=8.1, H5), 9.38 (d, 1H, 3J=8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 18.63, 24.34, 26.66, 27.59, 48.06, 60.27, 68.85, 77.51, 84.24, 122.83, 131.06, 138.57, 141.63, 148.16, 158.99. Anal. calcd. for C21H37N3O9S2. Calculated (%): C, 46.74; H, 6.91; N, 7.79; S, 11.88. Found: C, 46.25; H, 6.81; N, 8.01; S, 11.29.
Synthesis of 3,3-dimethyl-1-oxo-2,3-dihydro-1H-imidazo[1,5-a]pyridin-4-ium 3-tretbutoxipropane-1-sulfonate (3i). Similar to 3a, using tert-butanol instead of methanol, 0.45 g (23%) of 3i ⋅ 2 H2O was obtained, m.p. 118–121 °C. IR spectrum (solid, ν/cm–1): 1735 s (C=O), 1638 w (C=Cpyridine), 1152 s, 1033 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 1.14 (br. s, 9H, 3CH3), 1.86-2.13 (m, 2H, -H2CCH2CH2-; s, 6H, 2CH3), 2.93 (br. m, 2H, -CH2SO3), 3.52 (br. m, 2H, OCH2-), 8.53 (br. m, 1H, H3), 8.44 (br. m, H4), 8.89 (br. m, 1H, H5), 9.42 (br. m, 1H, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 15.63, 23.19, 24.54, 26.64, 31.75, 48.14, 69.83, 74.80, 85.00, 123.91, 131.04, 138.55, 142.77, 148.14, 162.28. Anal. calcd. for C16H30N2O7S. Calculated: C, 48.71; H, 7.66; N, 7.10; S, 8.12. Found: C, 48.53; H, 7.30; N, 7.50; S, 8.81.
Synthesis of 3,3-dimethyl-1-oxo-2,3-dihydro-1H-imidazo[1,5-a]pyridin-4-ium 3-cyclohexaneoxipropane-1-sulfonate (3j). Similar to 3a, using cyclohexanol at 78–80 °C instead of methanol at reflux, 1.68 g (80%) of 3j ⋅ 2 H2O was obtained, m.p. 167–170 °C (CH3CN). IR spectrum (solid, ν/cm–1): 1735 s (C=O), 1632 w (C=Cpyridine), 1158 s, 1037 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 1.91 (m, 2H, -CH2CH2CH2-), 1.95 (s, 6H, 2CH3), 2.93 (t, 2H, 3J=7.0, -CH2SO3), 1.17–1.68 (m, 10H, C6H10), 3.39 (m, 1H, OCH), 3.63 (t, 2H, 3J=7.0, OCH2-), 8.51 (d, 1H, 3J=7.2, H3), 8.43 (t, 1H, 3J=7.2, H4), 8.85 (t, 1H, 3J=7.2, H5), 9.38 (d, 1H, 3J=7.2, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 23.87, 24.79, 25.26, 26.62, 31.75, 48.14, 65.94, 78.32, 84.23, 122.54, 123.90, 131.03, 138.54, 148.14, 158.50. Anal. calcd. for C18H32N2O7S. Calculated: C, 51.41; H, 7.67; N, 6.66; S, 7.62. Found: C, 51.02; H, 7.42; N, 7.03; S, 8.10.
Reactions of Hydrolysis and Alcoholysis of Compounds 3a,d,e
Synthesis of 2-carbamoylpyridine-1-ium-3-methoxypropane-1-sulfonate (3a).
- a)
- 0.11 g (0.3 mmol) of 3a in 3 mL of water was stirred for 7 days at room temperature. After evaporation, 0.10 g (91%) of 3a was obtained.
- b)
- 0.36 g (1.1 mmol) of 3a in 4 mL of water was refluxed for 4 h. After evaporation and recrystallisation of the residue from CH3CN, 0.10 g (32%) of 2a was obtained.
- c)
- 0.31 g (0.97 mmol) of 3a in 4 mL of water was stirred at 70–80 °C for 5 h. After evaporation, 0.17 g (63%) of 2a was obtained.
- d)
- 0.11 g (0.3 mmol) of 3d in 4 mL of water was stirred at 70–80 °C for 5 h. After evaporation, 0.08 g of a mixture of 3a and 3d was obtained.
- e)
- 0.16 g (0.4 mmol) of 3e in 4 mL of methanol was refluxed for 16 h. After evaporation, 0.10 g of a mixture of 2a and 3e was obtained.
- f)
- 0.11 g (0.3 mmol) of 3e in 4 mL of water was stirred at 70–80 °C for 5 h. After evaporation, 0.08 g (97%) of 3a was obtained.
3.2. Calculation Details
Quantum-chemical calculations were carried out using Gaussian software, ver. 09 rev. C01 [35] and visualised using ChemCraft software, ver. 1.8 [36]. All geometric and energy-related values were obtained using the M052X hybrid functional [37] with empirical dispersion [38] and the TZVP basis set [39]. Chemical shifts were calculated using the continuous set of gauge transformations (CSGT) method [39,40,41]. The correlation coefficients for experimental and calculated chemical shifts in NMR spectra were around 99% (Table S4).
The above method was used for the full optimisation of the structures of reactants and products (S1 and S2, Supporting information). The calculations were carried out in the approximation of isolated molecules. The solvent effects were taken into account using the integral equation formalism variant of the polarisable continuum model (IEFPCM).
The correspondence of the calculated structures to minima on the potential energy surface was assessed by the absence of negative elements in the diagonalised Hessian matrix. The transition states were identified by the presence of a single negative element in the matrix.
Thermal effects of reactions and activation enthalpies were calculated as the difference between the absolute enthalpies of the final (or transition) and initial states of the process. Absolute enthalpies were calculated as the sum of total energy, zero-point energy and thermal correction for the enthalpy change from zero to 298 K. The latter values were obtained by frequency calculations using common equations of statistical thermodynamics.
The mutual arrangement of the protonated picolinamide cation and the sulfonate anion was determined using electrostatic potential (ESP) maps, which were calculated using MultiWFN software, ver. 3.8 [42] and visualised using VMD software [43].
The structure of the pre-reaction complex was determined using molecular dynamics (MD) modelling of a system containing a protonated picolinamide cation, a sulfonate anion, a molecule of acetone and 2000 solvent (methanol) molecules (Figure S1). MD modelling was performed for a cube-shaped system with periodic boundary conditions (PBC) using the OPLS4 force field [44].
The simulation of the isobaric-isothermal process was carried out using the NPT molecular ensemble. The dynamics simulation was recorded for 10 ns at 337 K (the boiling point of methanol). A total of 5000 frames were used for the statistical analysis. The analysis of the MD trajectory and the construction of volumetric maps for the PBC space were carried out using VMD software [43].
3.3. X-ray Crystallographic Studies
Single-crystal X-ray studies of compounds 2a and 3a were carried out in the Center for Molecule Composition Studies of INEOS RAS using APEX3 software [45]. The data obtained were then integrated with SAINT. SADABS was used for scaling, empirical absorption corrections and generation of data files for structure solution and refinement.
The structures were solved by dual-space algorithm and refined in anisotropic approximation for non-hydrogen atoms against F2(hkl). The positions of hydrogen atoms in methyl, methylene and aromatic fragments were calculated for idealised geometry and refined with constraints applied to C–H and N–H bond lengths and equivalent displacement parameters [Ueq(H) = 1.2Ueq(X) for XH2 groups and Ueq(H) = 1.5Ueq(Y) for YH3 groups]. All structures were solved using ShelXT [46] and refined using ShelXL software [47]. Molecular graphics were drawn using OLEX2 software [48]. Structure 2a was refined as a two-component non-merohedral twin using PLATON software [49]. The scale factors for the twin components were 0.880(4) and 0.120(4). Structure 3a was refined as a two-component non-merohedral twin using TWINABS program implemented in APEX3 software [45].
The supplementary crystallographic data for 2a and 3a (2287812 and 2287813) are available free of charge from the Cambridge Crystallographic Data Centre at https://www.ccdc.cam.ac.uk/structures.
4. Conclusions
In contrast to meta- and para-pyridinecarboxamides, their ortho-analogues (picolinamides) react with 1,3-propanesultone in hot methanol to give pyridinium salts with a protonated endocyclic nitrogen atom (10). In this work, the scope of this reaction has been extended to other alcohols. In the presence of ketones, the reaction products 2a–f form new type of heterocyclic salts 3a–j containing a 4-imidazoline ring. According to X-ray study, the main structural parameters of compounds 2a and 3a are typical for pyridinium salts of alkylsulfonic acids. The calculated thermodynamic parameters of reactions leading to the formation of 2a and 3a correlate with the size of alkyl substituents in substrates and reaction yields.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Molecular dynamics box with a system containing ortho-pyridinecarboxamide1,2 (black), sulfonate (blue), acetone (red), methanol (blue); Table S1: The enthalpy (ΔrH0) and free Gibbs energy (ΔrG0), Figure S2: Proton migration transition state, S1, XYZ coordinates of the stationary points of 2a-f products; calculation by the M052X-D3/ TZVP+IEEPCM approximation; Table S2: The enthalpy (ΔrH0) and free Gibbs energy (ΔrG0) of the formation reaction of salts 2a-f; S2, XYZ coordinates of the stationary points of 3a-j products; calculation by the M052X-D3/ TZVP+IEEPCM approximation; Table S3: The enthalpy (ΔrH0) and free Gibbs energy (ΔrG0) of the formation reaction of salts 3a-j; Table S4: 13C NMR chemical shifts (theoretical calculations), 1H, 13C NMR experimental data; X-Ray diffraction data.
Author Contributions
Conceptualization, V.V.N. and Y.I.B.; methodology, E.P.K., A.A.K., A.R.R., D.V.T., D.L., S.S.B. and T.A.S.; software, S.S.B., E.M.K. and A.R.Y.; formal analysis, S.Y.B.; writing—review and editing, S.Y.B. and V.V.N.; funding acquisition, V.V.N. and Y.I.B.; supervision, V.V.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation, grant number 21-73-20250.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
All spectra and XRD data are available from the authors.
Acknowledgments
The authors are grateful to the theoretical group “Quanta and Dynamics”: https://monrel.ru/.
Conflicts of Interest
The authors declare no conflict of interest.
References
- DeBruler, C.; Hu, B.; Moss, J.; Liu, X.; Luo, J.; Sun, Y.; and Liu, T.L. Designer Two-Electron Storage Viologen Anolyte Materials for Neutral Aqueous Organic Redox Flow Batteries. Chem. 2017, 3, 961–978. [Google Scholar] [CrossRef]
- Ichikawa, T.; Kato, T.; and Ohno, H. (2012) 3D Continuous Water Nanosheet as a Gyroid Minimal Surface Formed by Bicontinuous Cubic Liquid-Crystalline Zwitterions. J. of the Amer. Chem. Soc. 2012, 134, 11354–11357. [Google Scholar] [CrossRef] [PubMed]
- Fadda, A.A.; El-Mekawy, R.E.-D.; and AbdelAal, M.T. Synthesis and antimicrobial evaluation of some new N-pyridinium, quinolinium, and isoquinolinium sulfonate derivatives. Phosphorus, Sulfur, and Silicon and the Related Elements 2016, 191, 1148–1154. [Google Scholar] [CrossRef]
- Frederickson, M.; Vuillard, L.; and Abell, C. Novel selenium containing non-detergent sulphobetaines. Tetrahedron Lett. 2003, 44, 7925–7928. [Google Scholar] [CrossRef]
- Bondock, S.; Khalifa, W.; Fadda, A.A. Synthesis and antimicrobial evaluation of some new thiazole, thiazolidinone and thiazoline derivatives starting from 1-chloro-3,4-dihydronaphthalene-2-carboxaldehyde. Eur. J.l of Med. Chem. 2007, 42, 948–954. [Google Scholar] [CrossRef]
- Mostafa S. Amany, Bialy A. El Serry, Bayoumi A. Waleed, Ueda Youki, Ikeda Masanori, Nobuyuki, K.; Ali, A.M. Synthesis and In Vitro Activity of Pyrrolo [3,4-d]pyrimidine-2,5-diones as Potential Non-nucleoside HCV Inhibitors. Curr. Enz. Inhib. 2016, 12, 170–176. [CrossRef]
- Tian, X.; Liu, T.; Fang, B.; Wang, A.; Zhang, M.; Hussain, S.; Luo, L.; Zhang, R.; Zhang, Q.; Wu, J.; Battaglia, G.; Li, L.; Zhang, Z.; and Tian, Y. NeuN-Specific Fluorescent Probe Revealing Neuronal Nuclei Protein and Nuclear Acids Association in Living Neurons under STED Nanoscopy. ACS Appl. Mater. & Interf. 2018, 10, 31959–31964. [Google Scholar]
- Yan, P.; Acker, C.D.; and Loew, L.M. Tethered Bichromophoric Fluorophore Quencher Voltage Sensitive Dyes. ACS Sens. 2018, 3, 2621–2628. [Google Scholar] [CrossRef]
- Willner, I.; Ford, W.E. Preparation of zwitterionic electron acceptors and donors. J. of Heteroc. Chem. 1983, 20, 1113–1114. [Google Scholar] [CrossRef]
- Kramarova, E.P.; Borisevich, S.S.; Khamitov, E.M.; Korlyukov, A.A.; Dorovatovskii, P.V.; Shagina, A.D.; Mineev, K.S.; Tarasenko, D.V.; Novikov, R.A.; Lagunin, A.A.; Boldyrev, I.; Ezdoglian, A.A.; Karpechenko, N.Y.; Shmigol, T.A.; Baukov, Y.I.; and Negrebetsky, V.V. Pyridine Carboxamides Based on Sulfobetaines: Design, Reactivity, and Biological Activity. Molecules 2022, 27, 7542. [Google Scholar] [CrossRef]
- Koizumi, Y.; Arai, M.; Tomoda, H.; Omura, S. Oxaline, a fungal alkaloid, arrests the cell cycle in M phase by inhibition of tubulin polymerization. Biochim Biophys Acta 2004, 1693, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Gottstein, W.J.; Kim, C.U.; Shih, K.M.; McGregor, D.N. Hetacillin (R)- and (S)-sulfoxides. Synthesis and structure-activity relationships. J. Med. Chem. 1978, 21, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Burns, H.D.; Dannals, R.F.; Langström, B.; Ravert, H.T.; Zemyan, S.E.; Duelfer, T.; Wong, D.F.; Frost, J.J.; Kuhar, M.J.; and Wagner, H.N. (3-N-[11C]methyl)spiperone, a ligand binding to dopamine receptors: radiochemical synthesis and biodistribution studies in mice. J. Nuc.l Med. 1984, 25, 1222–1227. [Google Scholar]
- Barrow, J.C.; Rittle, K.E.; Ngo, P.L.; Selnick, H.G.; Graham, S.L.; Pitzenberger, S.M.; McGaughey, G.B.; Colussi, D.; Lai, M.T.; Huang, Q.; Tugusheva, K.; Espeseth, A.S.; Simon, A.J.; Munshi, S.K.; Vacca, J.P. Design and synthesis of 2,3,5-substituted imidazolidin-4-one inhibitors of BACE-1. Chem. Med. Chem. 2007, 2, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Augelli-Szafran, C.E.; Roth, B.D.; Essenburg, A.; Hamelehle, K.L.; Krause, B.R.; Stanfield, R.L. Imidazolidinones and pyrazolones as novel ACAT inhibitors: Chemistry and biological activity. Bioorg. Med. Chem. Lett. 1994, 4, 1095–1100. [Google Scholar] [CrossRef]
- Rinnová, M.; Nefzi, A.; Houghten, R.A. Opioid activity of 4-imidazolidinone positional analogues of Leu-Enkephalin. Bioorg. Med. Chem. Lett. 2002, 12, 3175–3178. [Google Scholar]
- Liu, J.; Cui, G.; Zhao, M.; Cui, C.; Ju, J.; and Peng, S. Dual-acting agents that possess reversing resistance and anticancer activities: Design, synthesis, MES-SA/Dx5 cell assay, and SAR of Benzyl 1,2,3,5,11,11a-hexahydro-3,3-dimethyl-1-oxo-6H-imidazo [3’,4’:1,2]pyridin [3,4-b]indol-2-substitutedacetates. Bioorg. Med. Chem. 2007, 15, 7773–7788. [Google Scholar] [CrossRef]
- Vale, N.; Matos, J.; Gut, J.; Nogueira, F.; do Rosário, V.; Rosenthal, P.J.; Moreira, R.; Gomes, P. Imidazolidin-4-one peptidomimetic derivatives of primaquine: synthesis and antimalarial activity. Bioorg. Med. Chem. Lett. 2008, 18, 4150–4153. [Google Scholar]
- Vale, N.; Prudêncio, M.; Marques, C.A.; Collins, M.S.; Gut, J.; Nogueira, F.; Matos, J.; Rosenthal, P.J.; Cushion, M.T.; do Rosário, V.E.; Mota, M.M.; Moreira, R.; Gomes, P. Imidazoquines as antimalarial and antipneumocystis agents. J. Med. Chem. 2009, 52, 7800–7807. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, M.; Qian, K.; Zhang, X.; Lee, K.H.; Wu, J.; Liu, Y.N.; Peng, S. Benzyl 1,2,3,5,11,11a-hexahydro-3,3-dimethyl-1-oxo-6H-imidazo [3’,4’:1,2]pyridin [3,4-b]indole-2-substituted acetates: One-pot-preparation, anti-tumor activity, docking toward DNA and 3D QSAR analysis. Bioorg. Med. Chem. 2010, 18, 1910–1917. [Google Scholar] [CrossRef]
- Mostardeiro, M.A.; Ilha, V.; Dahmer, J.; Caro, M.S.; Dalcol, II, da Silva, U.F.; Morel, A.F. Cyclopeptide alkaloids: stereochemistry and synthesis of the precursors of discarines C and D and myrianthine A. J. Nat. Prod. 2013, 76, 1343–1350. [CrossRef]
- Ross, T.M.; Battista, K.; Bignan, G.C.; Brenneman, D.E.; Connolly, P.J.; Liu, J.; Middleton, S.A.; Orsini, M.; Reitz, A.B.; Rosenthal, D.I.; Scott, M.K.; Vaidya, A.H. A selective small molecule NOP (ORL-1 receptor) partial agonist for the treatment of anxiety. Bioorg. Med. Chem. Lett. 2015, 25, 602–606. [Google Scholar] [CrossRef]
- Morgan, B.J.; Kozlowski, M.C.; Song, A.; Wang, W. (2016) (5S)-2,2,3-Trimethyl-5-(phenylmethyl)-4-imidazolidinone. In Encyclopedia of Reagents for Organic Synthesis (EROS), 2016, 1-5.
- Bedos, P.; Amblard, M.; Subra, G.; Dodey, P.; Luccarini, J.M.; Paquet, J.L.; Pruneau, D.; Aumelas, A.; Martinez, J. A rational approach to the design and synthesis of a new bradykinin B(1) receptor antagonist. J. Med. Chem. 2000, 43, 2387–2394. [Google Scholar] [CrossRef]
- Vale, N.; Nogueira, F.; do Rosário, V.E.; Gomes, P.; Moreira, R. Primaquine dipeptide derivatives bearing an imidazolidin-4-one moiety at the N-terminus as potential antimalarial prodrugs. Eur. J. Med. Chem. 2009, 44, 2506–2516. [Google Scholar] [CrossRef]
- Blackmore, T.R.; and Thompson, P.E. (2012) ChemInform Abstract: Imidazolidin-4-ones: Their Syntheses and Applications. Heterocycl. 2011, 83, 1953–1975. [Google Scholar] [CrossRef]
- Curtius, T. Hydrazide und Azide organischer Säuren I. Abhandlung. Journal für Praktische Chem. 1894, 50, 275–294. [Google Scholar] [CrossRef]
- Mailyan, A.K.; Eickhoff, J.A.; Minakova, A.S.; Gu, Z.; Lu, P.; Zakarian, A. Cutting-Edge and Time-Honored Strategies for Stereoselective Construction of C–N Bonds in Total Synthesis. Chem. Rev. 2016, 116, 4441–4557. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Brindisi, M.; Sarkar, A. The Curtius Rearrangement: Applications in Modern Drug Discovery and Medicinal Chemistry. Chem. Med. Chem. 2018, 13, 2351–2373. [Google Scholar] [CrossRef]
- Bariwal, J.; Van der Eycken, E. C–N bond forming cross-coupling reactions: an overview. Chem. Soc. Rev. 2013, 42, 9283–9303. [Google Scholar] [CrossRef]
- Gomez, S.; Peters, J.A.; Maschmeyer, T. (2002) The Reductive Amination of Aldehydes and Ketones and the Hydrogenation of Nitriles: Mechanistic Aspects and Selectivity Control. Adv. Synt. & Cat. 2002, 344, 1037–1057. [Google Scholar]
- Swamy, K.C.; Kumar, N.N.; Balaraman, E.; Kumar, K.V. Mitsunobu and related reactions: advances and applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar]
- Magni, L.; Örtengren, B.; Sjöberg, B.; Wahlqvist, S. (1967) Stability, Absorption and Excretion Studies with Hetacillin. Scand. J. Clin. and Lab. Invest. 1967, 20, 195–201. [Google Scholar] [CrossRef]
- Brown, D.M.; Hannan, D.P.; Langley, P.F. Biotransformation of hetacillin to ampicillin in man. Tox. Appl. Pharm. 1969, 15, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A.; Bloino, J.; Janesko, B.G.; Gomperts, R.; Mennucci, B.; Hratchian, H.P.; Ortiz, J.V.; Izmaylov, A.F.; Sonnenberg, .; J.L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V.G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Jr. Montgomery, J.A.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd J. J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; J.; Millam, M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Farkas, O.; Foresman, J.B.; Fox, D.J. (2016) Gaussian 09, Revision C. Wallingford CT.
- Zhurko, G.A. ChemCraft. Version 1.6 (build 332).
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theor. Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 15410. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A.; Bader, R.F.W. Calculation of magnetic response properties using atoms in molecules. Chem. Phys. Lett. 1992, 194, 1–8. [Google Scholar] [CrossRef]
- Keith, T.A.; Bader, R.F.W. Calculation of magnetic response properties using a continuous set of gauge transformations. Chemical Physics Lett. 1993, 210, 223–231. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; Abel, R.; Friesner, R.A.; Harder, E.D. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Bruker. (2016) APEX3, RLATT, CELL_NOW, TWINABS, SAINT-Plus and SADABS.
- Sheldrick, G. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. Sec. 2015, A 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sec. 2015, C 71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; and Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
Scheme 1.
Representative synthetic routes for compounds A.
Scheme 2.
Representative synthetic routes for compounds 2a-g and 3a-j.
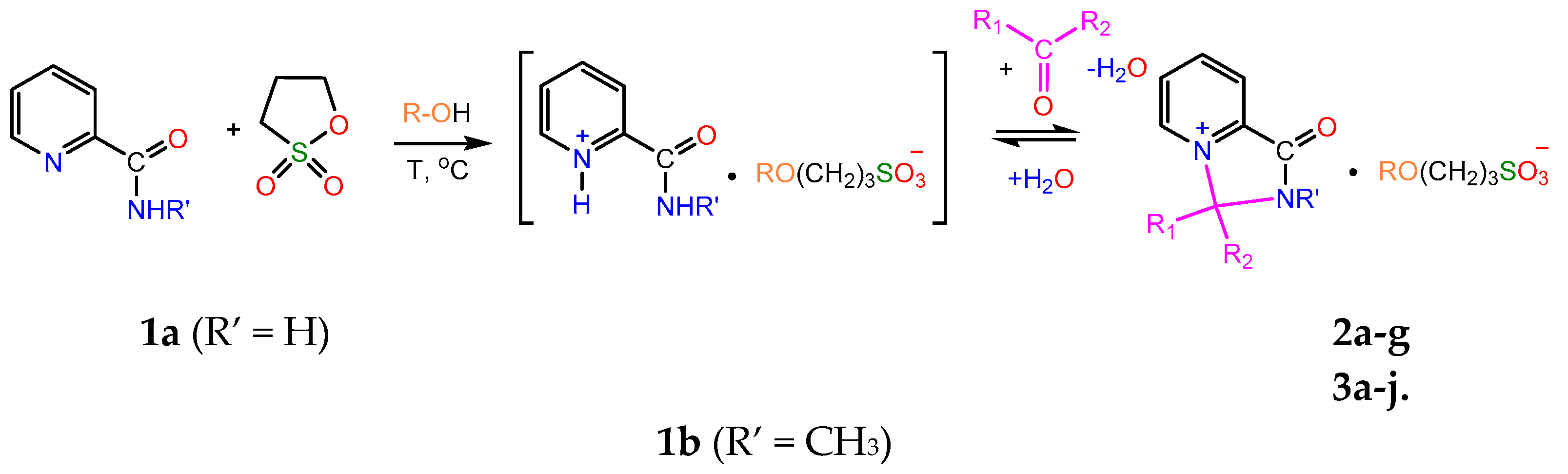
Figure 1.
Molecular structures of 2a (left) and 3a (right) showing thermal ellipsoids at the 50% probability level.
Figure 1.
Molecular structures of 2a (left) and 3a (right) showing thermal ellipsoids at the 50% probability level.
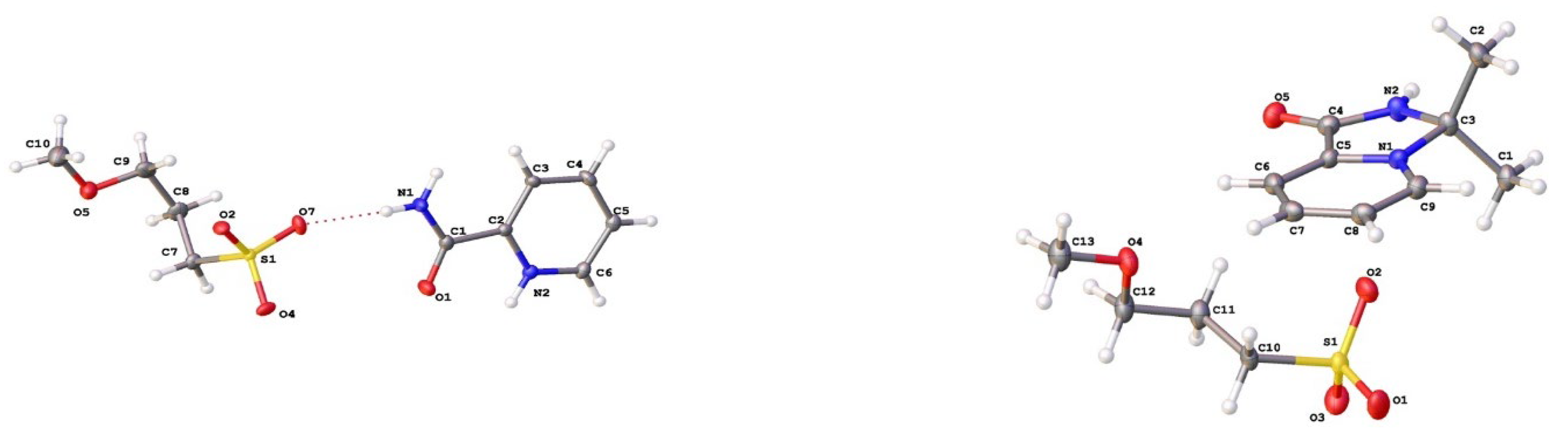
Figure 2.
Crystal packing of 2a (left) and 3a (right).
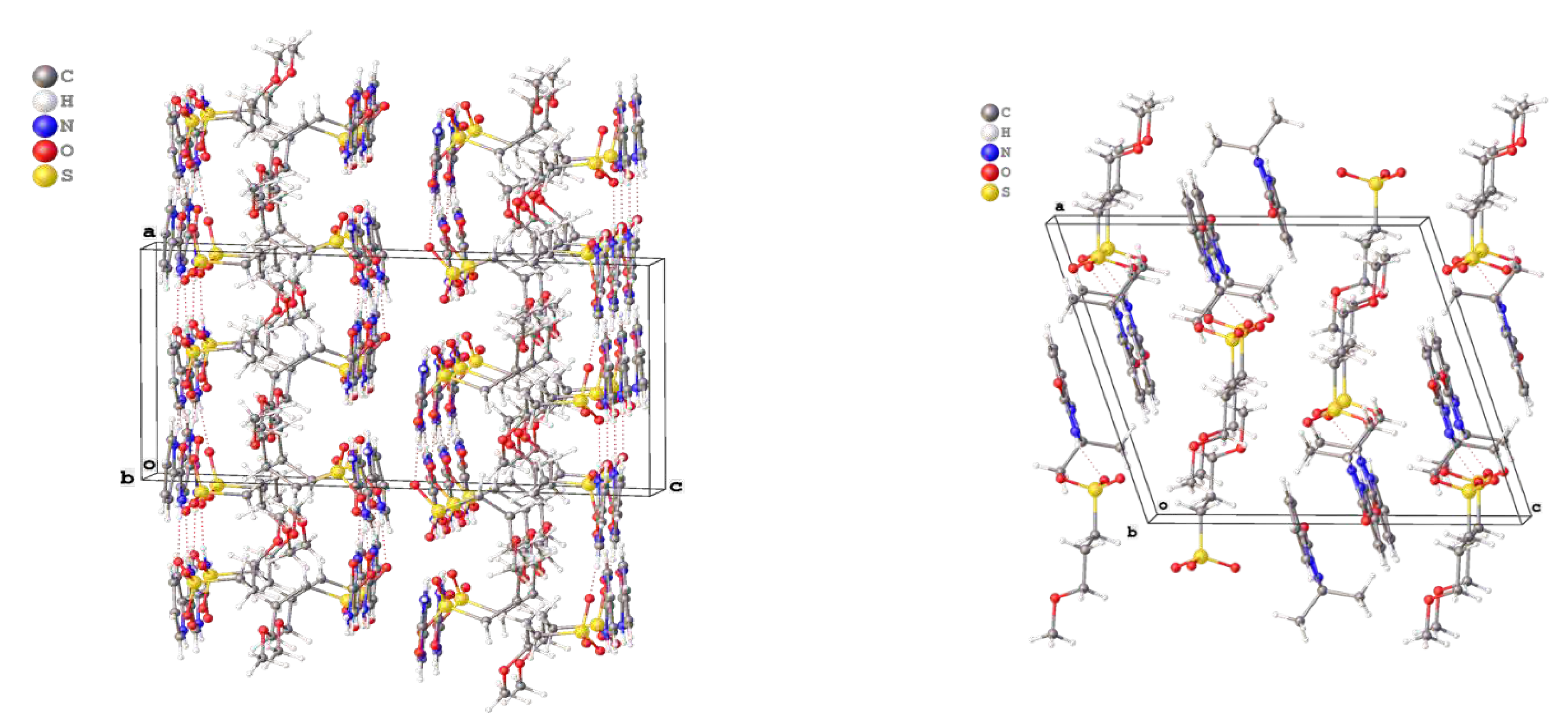
Figure 3.
Thermodynamic parameters (M052X/TZVP) of the formation of 3-alkoxypropanesulfonic acids.
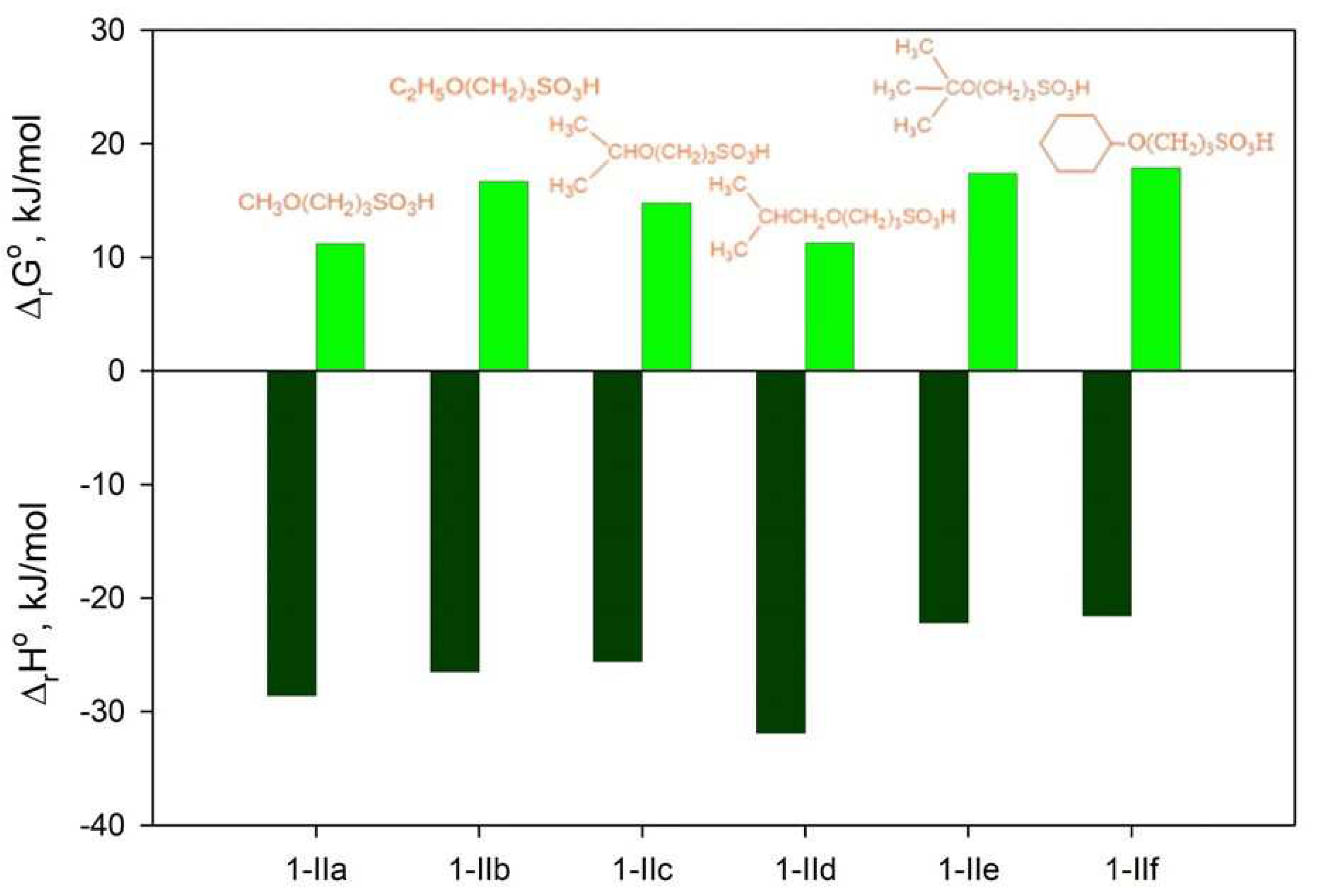
Figure 4.
Structures and coordination of ions: (A) – ESP maps (kJ/mol) of 1-Ia and 1-IIa; (B) – calculated structure of 2a in solution and actual structure of solid 2a determined by X-ray study; (C) – possible conformations of 1-Ia. Bond lengths are given in Å; hydrogen bond energies (bold italic) are given in kJ/mol.
Figure 4.
Structures and coordination of ions: (A) – ESP maps (kJ/mol) of 1-Ia and 1-IIa; (B) – calculated structure of 2a in solution and actual structure of solid 2a determined by X-ray study; (C) – possible conformations of 1-Ia. Bond lengths are given in Å; hydrogen bond energies (bold italic) are given in kJ/mol.
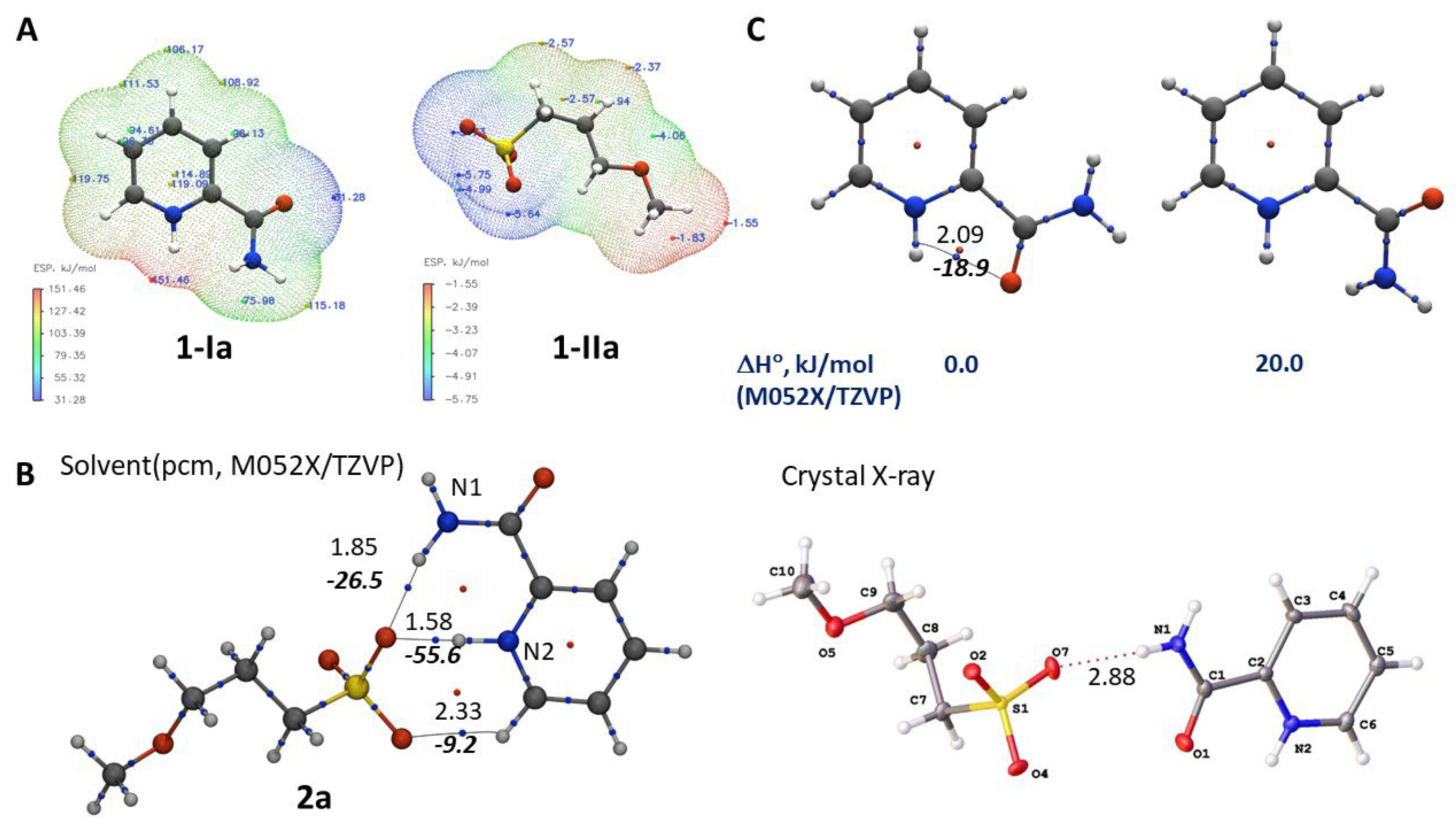
Figure 5.
Thermodynamic parameters of salts 2a–f formation reactions.
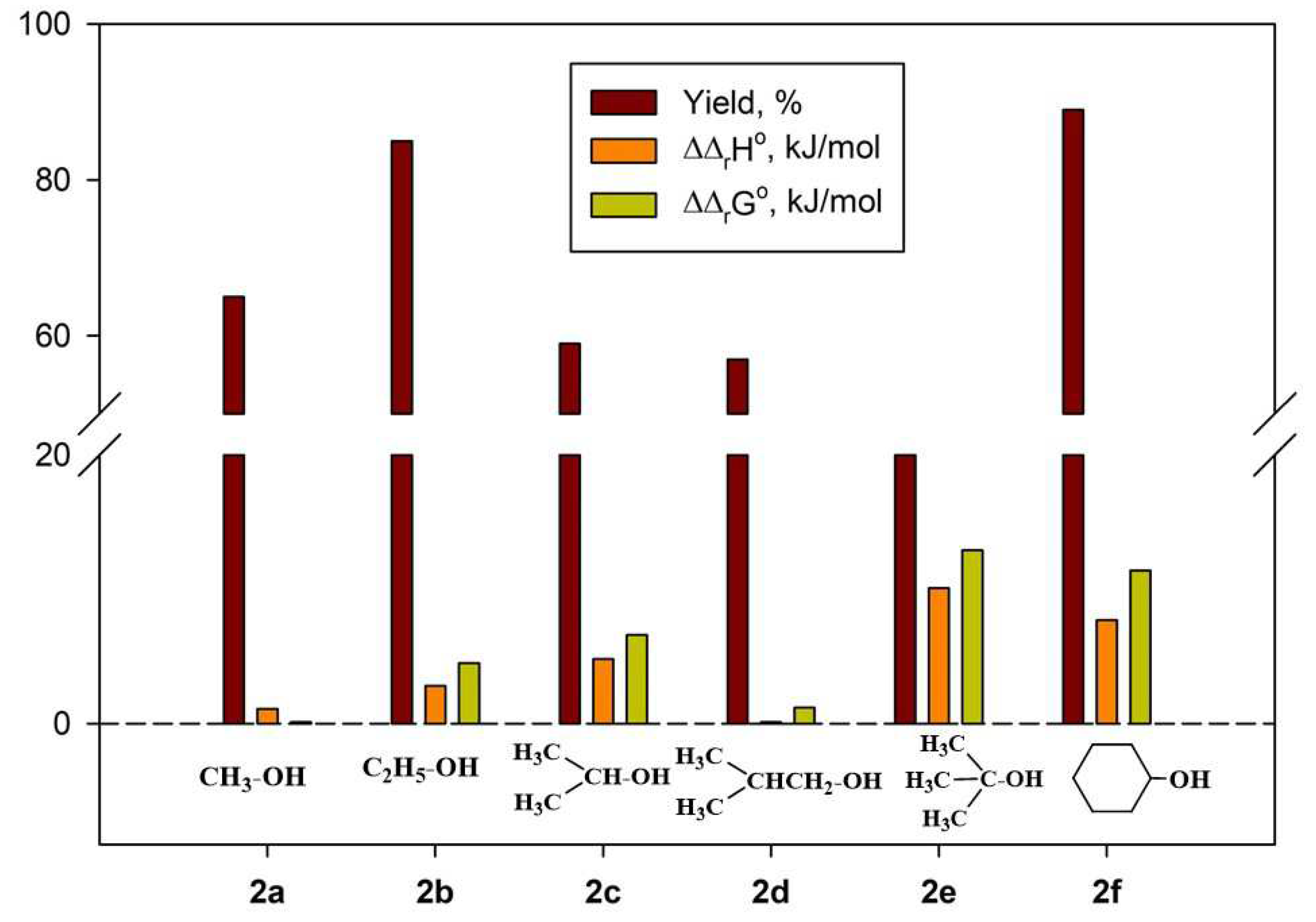
Figure 6.
Thermodynamic parameters of salts 3a–j formation reactions.
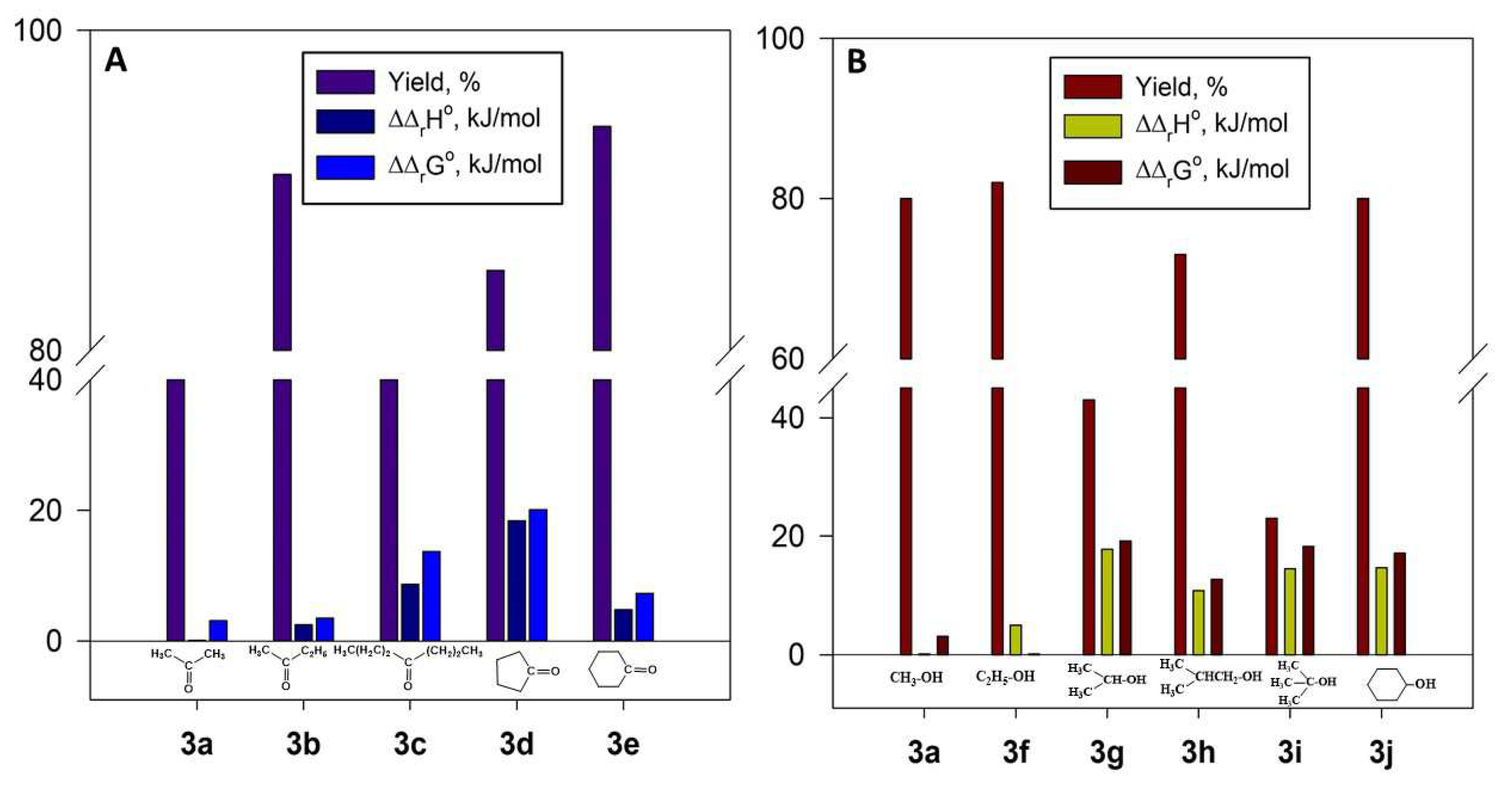

Table 1.
Structure and yields of salts 2a-g.
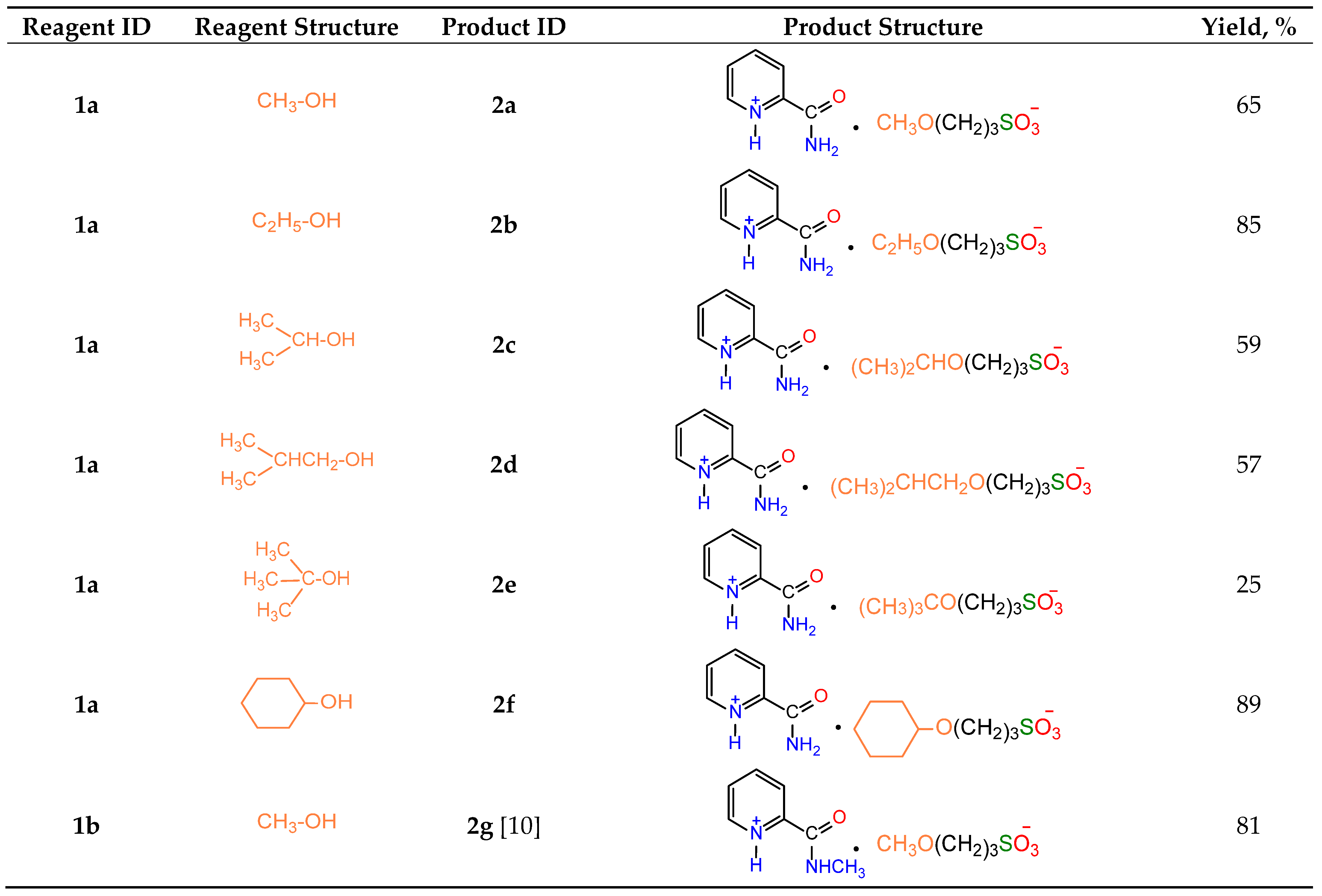 |
Table 2.
Structure and yields of salts 3a-j.
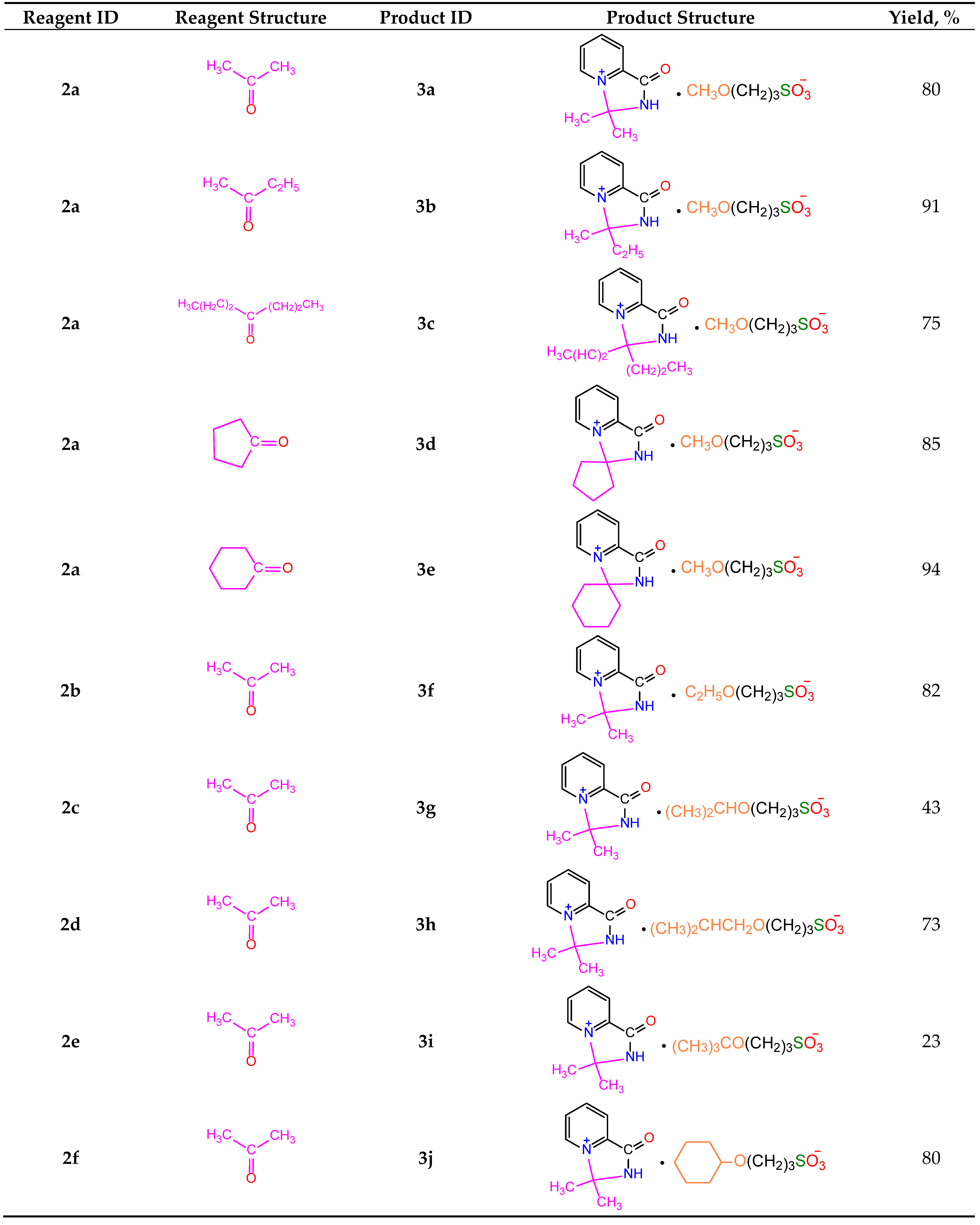 |
Table 3.
Crystallographic data for 2a and 3a.
| Datablock | 2a | 3a |
|---|---|---|
| Formula moiety | C4H9O4S, C6H7N2O | 2(C4H9O4S), 2(C9H11N2O) |
| Brutto formula | C10H16N2O5S | C26H40N4O10S2 |
| Formula weight | 276.31 | 632.74 |
| Diffractometer | Bruker QUEST | Bruker QUEST |
| Scan mode | ω and ϕ scans | ω and ϕ scans |
| Anode [Wavelength, Å] | MoKα [0.71073] microfocus sealed X-ray tube | MoKα [0.71073] microfocus sealed X-ray tube |
| Crystal Dimensions, mm | 0.04 × 0.07 × 0.14 | 0.03 × 0.05 × 0.1 |
| Crystal colour | colourless | colourless |
| Crystal system | monoclinic | monoclinic |
| a, Å | 9.9079(5) | 12.422(2) |
| b, Å | 12.6799(6) | 8.7237(15) |
| c, Å | 19.8194(10) | 14.699(2) |
| α, ° | 90 | 90 |
| β, ° | 92.227(3) | 109.106(6) |
| γ, ° | 90 | 90 |
| Volume, Å3 | 2488.1(2) | 1505.1(4) |
| Density, g cm–3 | 1.475 | 1.396 |
| Temperature, K | 100 | 100 |
| Tmin/Tmax | 0.497553/0.746069 | 0.5954/0.7461 |
| μ, mm⁻¹ | 0.276 | 0.238 |
| Space group | P1211 | P121/n1 |
| Z | 8 | 2 |
| F(000) | 1168 | 672 |
| Reflections collected | 29400 | 13353 |
| Independent reflections | 29400 | 3544 |
| Reflections (I > 2σ(I)) | 27603 | 3030 |
| Parameters | 654 | 193 |
| Rint | 0.00 | 0.0598 |
| 2θmin – 2θmax, ° | 3.814 – 55.750 | 5.226 – 55.752 |
| wR2 (all reflections) | 0.1531 | 0.2011 |
| R1(I > σ(I)) | 0.0572 | 0.0730 |
| GOF | 1.055 | 0.972 |
| ρmin/ρmax, e Å–3 | –0.748/1.719 | –0.445/0.676 |
| Restraints | 1 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



