
Submitted:
07 November 2023
Posted:
07 November 2023
You are already at the latest version
Abstract
Effective therapeutics for Alzheimer's disease (AD) are in great demand worldwide. In our previous work, we responded to this need by synthesizing novel drug candidates consisting of 4-amino-2,3-polymethylenequinolines conjugated with butylated hydroxytoluene via fixed-length alkylimine or alkylamine linkers (spacers) and studying their bioactivities pertaining to AD treatment. Here, we report significant extensions of these studies, including the use of variable-length spacers and more detailed biological characterizations. Conjugates were potent inhibitors of acetylcholinesterase (AChE, minimum IC50 15.1±0.2 nM) and butyrylcholinesterase (BChE, minimum IC50 5.96±0.58 nM), with weak inhibition of off-target carboxylesterase. Conjugates with alkylamine spacers were more effective cholinesterase inhibitors than alkylimine analogs. Optimal inhibition for AChE was exhibited by cyclohexaquinoline and for BChE by cycloheptaquinoline. Increasing spacer length elevated potency against both cholinesterases. Structure-activity relationships agreed with docking results. Mixed-type reversible AChE inhibition, dual docking to catalytic and peripheral anionic sites, and propidium iodide displacement suggested the potential of hybrids to block AChE-induced β-amyloid (Aβ) aggregation. Hybrids also exhibited inhibition of Aβ self-aggregation in the thioflavin test; those with a hexaquinoline ring and C8 spacer were the most active. Conjugates demonstrated high antioxidant activity in ABTS and FRAP assays as well as inhibition of luminol chemiluminescence and lipid peroxidation in mouse brain homogenates. Quantum-chemical calculations explained antioxidant results. Computed ADMET profiles indicated favorable blood-brain barrier permeability suggesting CNS activity potential. Thus, the conjugates could be considered promising multifunctional agents for potential treatment of AD.
Keywords:
Alzheimer's disease (AD)
; 4-amino-2
; 3-polymethylenequinolines
; butylated hydroxytoluene (BHT)
; acetylcholinesterase (AChE)
; butyrylcholinesterase (BChE)
; antioxidants
; ADMET
; β-amyloid
; molecular docking
; quantum-chemical calculations
1. Introduction
Considering the myriad possibilities of new products that could be produced by the judicious application of experimental and computational methodologies, we believe that one of the most pressing needs is to devise effective therapeutic agents for the prevention and/or treatment of Alzheimer's disease (AD).
AD is a devastating disorder that kills neurons in areas of the brain involved in retrieval and formation of memories, execution of other cognitive functions, and control of emotion and associated behaviors [1,2]. Apart from agents that provide mild cognition enhancement during the initial phases of the disease and palliative measures during the later severe stages, there are currently no effective preventions or treatments for AD. Moreover, given that the main risk factor for AD is advanced age, as the global elderly population increases, the negative impacts of the disease on afflicted individuals, their caregivers, and the economy are projected to intensify [3,4].
Intensive research has revealed pathological hallmarks of AD, but the cause and mechanism of the disease currently remain unknown. However, there is now general agreement that AD has multiple causes and that therapeutic agents will need to be designed to act on more than one target to be effective [5,6,7,8].
We have chosen to combine in single molecules the ability to mollify three processes known to participate in AD pathogenesis, as summarized below:
1. Inhibition of acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) to help restore decreased levels of acetylcholine. Three cholinesterase inhibitors are already in clinical use (Donepezil (Aricept), Galantamine (Reminyl), and Rivastigmine (Exelon)) [9,10,11]. However, these drugs produce a range of unpleasant side effects from cholinergic hyperstimulation in the peripheral and/or central nervous systems, indicating a need for refinement of the pharmacokinetic and pharmacodynamic profiles of new cholinesterase inhibitors intended as AD therapeutics [12,13].
2. Inhibition of oxidants and free radicals to limit oxidative stress that can both promote and be fueled by pathological states associated with neurodegeneration, such as the dysfunction of mitochondria, disruption of metal ion homeostasis, and formation and deposition of Aβ aggregates [14,15,16]. AD therapies employing antioxidants are currently under active investigation [17,18].
3. Inhibition of β-amyloid (Aβ) aggregation that can form pathogenic Aβ plaques in the brain [19,20]. Compounds that inhibit Aβ aggregation are thought to have an ameliorative disease-modifying effect [21,22].
Along with its main function to hydrolyze acetylcholine, AChE has proaggregant properties toward β-amyloid via involvement of its peripheral anionic site (PAS), which interacts with soluble β-amyloid peptides to promote their aggregation [23,24,25,26]. Moreover, dual-binding molecules that interact with both the catalytic active site (CAS) and the PAS of AChE can inhibit AChE activity and block its amyloidogenic properties as well. Such compounds could simultaneously improve cognitive function and exert positive disease-modifying properties [27,28,29].
BChE has also been shown to participate in one or more steps leading to Aβ aggregation [30,31]. Consequently, from the standpoints of cognition enhancement by elevating ACh levels as well as disease modification by blocking Aβ aggregation, it makes sense to search for compounds capable of inhibiting both AChE and BChE.
One of the promising paradigms in anti-AD drug development is the design of multi-target ligands, whereby two molecules with different pharmacological propensities are linked together through a spacer of varying length (throughout this article, we use the terms "spacer" and "linker" interchangeably). For example, a component exhibiting anticholinesterase activity could be joined with another moiety exerting antioxidant properties [32]. Tacrine is widely employed as an anticholinesterase pharmacophore to create multifunctional cholinesterase inhibitors possessing additional neuroprotective and disease-modifying properties [33,34,35,36,37,38]. In particular, hybrids of tacrine coupled with antioxidants have been a popular combination [38,39,40,41,42,43].
Recently, we applied the multifunctional approach to create new hybrid structures using the 4-amino-2,3-polymethylenequinoline scaffold, which included tacrine and its cyclic homologs, and the sterically hindered phenolic scaffold of butylated hydroxytoluene (BHT) as the antioxidant pharmacophore [44]. The present research described in this article represents our continuing development and characterization of multifunctional hybrids.
Here, we report significant extensions to our previously published work. The current investigation includes the expanded synthesis and more extensive biological activity evaluations of novel conjugates of 4-amino-2,3-polymethylenequinoline with various sizes of the aliphatic ring, which were connected to BHT by iminoalkyl or aminoalkyl spacers. Whereas our previous studies employed a fixed-length spacer, the present work involved spacers of increasing length. Given that these compounds were produced as potential multifunctional agents for the treatment of AD, our biological evaluations focused on endpoints relevant for this disease. First, we determined the esterase profile of the compounds, i.e., their inhibitory activities against AChE, BChE, and a structurally related off-target carboxylesterase (CES, EC 3.1.1.1). Second, we assessed the ability of the conjugates to inhibit Aβ self-aggregation and to displace propidium iodide from the PAS of AChE as a measure of their potential to block AChE-induced aggregation of Aβ. Third, we determined their primary antioxidant activity and their antioxidant activity in rat brain homogenate. Fourth, the experimentally observed effects were analyzed using detailed quantum-mechanical (QM) calculations and computational molecular modeling. Finally, we carried out computational predictions of ADMET properties of the conjugates.
2. Results and Discussion
2.1. Chemistry
As shown in Scheme 1, at the first synthetic stage, tricyclic 4-chloro derivatives of quinoline with different cycloalkyl fragments 5-7 were synthesized. They were obtained by condensation of anthranilic acid 1 with cyclic ketones: cyclopentanone 2, cyclohexanone 3 and cycloheptanone (suberone) 4. The condensation was carried out by boiling the starting components in phosphorus oxychloride (POCl3) [45]. At the second stage of the synthesis, alkyl spacers with different carbon chain lengths were added to the obtained tricyclic derivatives of 4-chloropyridine 5-7 by reaction with diaminoalkanes 8a-d.
The addition of diamines 8a-d was carried out by boiling the reaction mixture in pentanol [44], which afforded the aminoalkyl derivatives of 4-amino-2,3-polymethylene- quinoline 9–11a-d with a free amino group necessary for addition of the antioxidant phenolic fragment in the next step. At the second stage, conjugates of 4-amino-2,3-poly- methylenequinoline and an antioxidant were synthesized by adding a phenolic fragment to the free amino group of aminoalkyl derivatives of 4-amino-2,3-polymethylenequin- oline 9–11a-d. First, by boiling in a mixture of toluene-methanol (5:1), 3,5-di-tert-butyl- 4-hydroxybenzaldehyde 12 was added as an antioxidant fragment with the formation of conjugates in which two pharmacophores were connected via an imine bond 13–15a-d. The reduction of the obtained compounds with sodium borohydride in methanol led to the preparation of conjugates in which two pharmacophores were connected via an amine bond 16–18a-d.
2.2. Biological studies
2.2.1. Studies of AChE, BChE and CES Inhibition. Structure-Activity Relationships
We evaluated the esterase profile [46,47], of new potential anti-AD molecules that included the assessment of inhibition of cholinergic targets AChE and BChE, as well as off-target CES, which hydrolyzes numerous ester-containing drugs [47]. The inhibitory activities of the conjugates against the esterases were characterized as IC50 values or as the inhibition percent at an inhibitor concentration of 20 µM for low-activity compounds. Tacrine, an effective AChE and BChE inhibitor, and bis-4-nitrophenyl phosphate (BNPP), a selective CES inhibitor, were used as positive controls. The results are shown in Table 1.
The study of the esterase profile of the synthesized conjugates 13-18 showed that the compounds effectively inhibit cholinesterases with predominant BChE inhibition (Table 1) and rather weakly inhibit the off-target CES.
Effect of ring size. As can be seen from Table 1, compounds with a cyclohexaquinoline ring (m = 2, i.e., a tacrine pharmacophore) showed the maximum activity against AChE; and compounds with a cycloheptachinoline ring (m = 3) showed the maximum activity against BChE. This effect was observed for conjugates with different lengths (n) and types (immine or amine) of spacers.
Effect of spacer length. An increase in the length of the spacer led to a significant increase in anti-AChE activity, both for compounds with alkylimine (13-15) spacers (45- to 350-fold) and alkylamine (16-18) spacers (40- to 60-fold). Conjugates with spacer length n = 8 (14d, 15d; 17d, 18d) showed the maximum inhibitory activity against AChE, among which tacrine derivatives (m = 2) had the highest activity – 40 times higher than the activity of the basic pharmacophore tacrine.
An increase in the length of the spacer also led to an increase in anti-BChE activity. The most active BChE inhibitors with IC50 values in the nanomolar region were conjugates with spacer length n = 8 (14d, 15d; 17d, 18d). Considering the influence of the ring size, the cycloheptaquinoline (m = 3) conjugates 15d and 18d exhibited the maximum activity in this series with an IC50 of 6 nM.
Effect of spacer chemistry. The structure of the spacer also affected activity: in general, conjugates with an alkylamine spacer (16–18) were more effective at inhibiting AChE and BChE than their analogs with an alkylimine spacer (13–15), and the effect of replacing the imine spacer with an amine spacer was more pronounced for BChE inhibition.
2.2.2. Kinetic Studies of AChE and BChE Inhibition
The mechanism of inhibition of AChE and BChE by the conjugates was studied using compound 15d as an example. Graphical analysis of the kinetic data on AChE and BChE inhibition by the tested compound (Figure 1A,B) with Lineweaver-Burk plots demonstrated changes in both Km and Vmax values – a result consistent with a mixed type of inhibition. The values obtained for the competitive (Ki) and noncompetitive (αKi) components of the constants for AChE inhibition by compound 15d were 19.9±0.8 nM and 31.4±2.4 nM, respectively; and for BChE 4.91±0.42 nM and 5.68±0.06 nМ, respectively.

Table 2.
Inhibition constants of cholinesterases by conjugates 15a,c,d, 18c,d1.
| Compound | AChE | eqBChE | ||
|---|---|---|---|---|
| Ki, nM | αKi, nM | Ki, nM | αKi, nM | |
| 15a | 1580±100* | 4300±20* | 164±5* | 437±10* |
| 15c | 69.8±5.0 | 108±3 | 5.13±0.10 | 9.34±0.62 |
| 15d | 19.9±0.8 | 31.4±2.4 | 4.91±0.42 | 5.68±0.06 |
| 18c | 83.1±1.4 | 101±5 | 7.83±0.76 | 17.3±0.2 |
| 18d | 13.0±1.0 | 25.1±0.2 | 4.25±0.33 | 6.06±0.41 |
*Data from [44]. 1Values for Ki (competitive inhibition constant) and αKi (non-competitive inhibition constant) were determined from analyses of slopes of 1/V versus 1/S at various inhibitor concentrations. Values (means ± SEM) are from at least three separate experiments.
2.2.3. Molecular docking to AChE and BChE
Results of molecular docking were in good agreement with the experimentally observed effects of the ring size and linker length (Figure 2). According to the pKa estimations, the 4-amino-2,3-polymethylenequinoline fragment, secondary amine group of the linker would be protonated at the experimental conditions [38,48].
In contrast, for the imine group, calculated pKa values were close to 7. This indicates the possibility of co-existence of protonated and non-charged imino-groups at the experimental pH value; therefore, both possibilities were considered. Binding of alkylimine compounds with the protonated imine group was stronger than for non-protonated (Figure 2A,B). Due to their higher flexibility, binding of compounds with the alkylamino group was stronger than that of the protonated alkylimine derivatives (Figure 2C).
The increase of the length of the linker in all considered cases led to a better binding affinity due to increased occupancy of the PAS, which is typical of tacrine-based AChE inhibitors [37,38]. While the tacrine fragment interacted with the active site at the bottom of the gorge, the BHT fragment interacted with the PAS. For compounds with shorter linkers their hydroxyl groups formed a hydrogen bond with the Tyr341 main chain oxygen atom. In contrast, for compounds with the longest linker (n = 8), hydrogen bonding with polar atoms of Ser293 were possible (Figure 3A). The positively charged amine groups of the shorter linkers interacted with Asp70 and Tyr341 side chains. For the longest linkers, there were π-cation interactions with the Trp286 side chain. At the same time, the cyclohexaquinoline ring (m = 2) was optimal for AChE active site binding, while a further increase of the ring size led to displacement of the tacrine fragment (Figure 3B, [37,44]). This optimal structural binding was mirrored by minimal binding free energies (binding affinities) obtained from the molecular docking simulations. In the case of binding to BChE, which has a wider gorge than that of AChE, the best binding was achieved for the compound with the maximum spacer length (n = 8) and ring size (C-7, m = 3) (Figure 2D).
2.2.4. Displacement of Propidium from the PAS of EeAChE
The results of the study of propidium displacement from the AChE PAS by conjugates 13-18 are presented in Table 1.
As can be seen from Table 1, all studied conjugates with alkylimine (13-15) and alkylamine (16-18) spacers at a concentration of 20 μM reduced the fluorescence intensity by 12–20%. Accordingly, they displaced propidium from PAS of AChE almost at the level of the reference compound donepezil or exceeded it in efficiency. In addition, in the group of conjugates with an alkylimine spacer, the decrease in fluorescence intensity was somewhat stronger than for compounds with an alkylamine spacer and amounted to 14–20% and 12–17%, respectively.
These displacement results, along with the mixed type of AChE inhibition demonstrated by enzyme kinetics and the results of molecular docking, which showed binding of the conjugates to both the CAS and PAS of AChE, all support the ability of these compounds to block AChE-induced aggregation of Aβ42.
2.2.5. Inhibition of β-amyloid (1–42) (Aβ42) self-aggregation
The most active cholinesterase inhibitors with spacers of 6 and 8 methylene groups were studied (Table 3).
The results presented in Table 3 demonstrate that the studied conjugates exhibited inhibitory activity against Aβ42 self-aggregation in the range from 43.4% to 71.4%. Moreover, the degree of inhibition of Aβ42 self-aggregation depended on both the size of the aliphatic ring in the tacrine fragment (m = 1-3) and the length of the spacer (n = 6 or 8).
The most active compounds were the conjugates of BHT and 4-amino-2,3-poly- methylenequinoline containing a hexaquinoline ring (m = 2), combined with the alkylimine 14d and alkylamine spacer 17d (n = 8), which exhibited inhibitory activity against Aβ42 self-aggregation comparable to the level of the reference compound Myricetin (73.2±5.8%). Note that tacrine on its own exhibited minimal inhibition, and the inhibition by BHT alone was not detectable.
2.2.6. Molecular docking to Aβ42
Our previous studies demonstrated that the results of docking ligands to Aβ42 depend significantly on the conformation of the peptide [38,49]. Accordingly, as the docking target, we have used all 10 conformers of Aβ42 that are available in the NMR solution structure, PDB ID 1IYT. Among the different binding poses (Figure 4A), the majority was in the hydrophobic area of the turn segment and the C-terminal part. This region is crucial for the conformational transition at the initial phase of Aβ42 nucleation during fibril formation [50]. A long linker in the ligand ensures binding of one of the pharmacophores (BHT) to hydrophobic residues Lys16, Phe19, and Phe20 on one side (Figure 4B, compounds 16d, 17d), while the other pharmacophore (tacrine) binds with Val24, Ile31, and Leu34 at the turn segment and the C-terminal part of the peptide. The larger tacrine ring (m = 2 for 18d) did not fit well between residues Ile31 and Leu34, which changed the position of the compound relative to the peptide.
2.2.7. Antioxidant activity
Primary antioxidant activity of the conjugates was determined using two spectrophotometric tests: the ABTS radical-scavenging assay and the Fe3+ reducing antioxidant power (FRAP) assay.
2.2.7.1. ABTS assay
All synthesized conjugates exhibited high ABTS•+-scavenging activity, close to or exceeding the activity of the standard antioxidant Trolox and the basic antioxidant pharmacophore BHT (TEAC = 0.78 - 1.5) (Table 4). The structure of the spacer influenced the ABTS•+-binding activity. Thus, the replacement of an alkylimine spacer (conjugates 13-15) with an alkylamine one (conjugates 16-18) generally led to an increase in the radical-scavenging activity with a maximum difference of 1.5-fold. Most alkylamino analogs (16-18) demonstrated a radical-scavenging activity noticeably higher than that of Trolox (TEAC = 1.2 - 1.5).
For an adequate assessment of the antiradical activity of tested compounds and for revealing structure-activity relationships, it is also important to consider the initial reaction rate of ABTS radical scavenging [51,52]. Our experiments showed that the lead conjugates with alkylamine spacers (16c, 17b, 17c, 17d, 18b, 18d) had a rather high initial rate of ABTS radical scavenging. For these compounds, the time to reach the activity level of Trolox when used in a concentration equal to its IC50 (20 μM) was less than 1 min, whereas for the other alkylamine derivatives this interval increased to 3 to 5 min.
Alkylimine analogs (13, 14, 15) exhibited antiradical activity at the level of Trolox (TEAC = 0.78 - 1.13). However, they demonstrated a lower initial rate of binding of the ABTS radical: the time to reach the degree of radical binding at the Trolox level ranged from 5 to 60 min.
An increase in the size of the aliphatic ring of the 4-amino-2,3-polymethylene- quinoline ("tacrine") fragment, as well as a change in the length of the spacer, did not substantially affect the antiradical activity of conjugates with either alkylimine or alkylamine spacers.
Thus, the results showed that the synthesized conjugates of cyclic homologs of tacrine and BHT exhibited high radical-scavenging activity in the ABTS test. Conjugates with an alkylamine spacer were more active compared to their alkylimine analogs. Maximum activity, exceeding the activity of the standard antioxidant Trolox, was demonstrated by conjugates 16c, 17b, 17c, 17d, 18b, 18d.
2.2.7.2. FRAP assay
As can be seen from Table 4, conjugates 13-15 and 16-18 had a high iron-reducing ability, which, however, was somewhat lower than that of Trolox and the basic pharmacophore BHT. In general, conjugates with alkylimine spacer were somewhat more active than their alkylamine analogs.
The size of the aliphatic “tacrine” ring had practically no effect on the ability of the conjugates to reduce Fe3+. The activity somewhat increased with elongation of the spacer, which was more evident in the case of conjugates with an alkylimine spacer.
2.2.8. Antioxidant activity of conjugates in a biological system
To obtain information about the antioxidant activity of conjugates in biological systems, we estimated their free radical scavenging activity in mouse brain homogenate by a chemiluminescence (CL) method and studied their ability to suppress spontaneous lipid peroxidation (LP) by the TBARS assay. Three pairs of conjugates with an imine and amine fragment in the spacer and tacrine moiety ring 6 (m = 2) and 7 (m =3 ) were selected for study. For conjugates with a 7-membered ring in the tacrine moiety, compounds with spacer lengths (CH2)6 and (CH2)8 were studied. The results are presented in Table 6.
Table 5.
Antioxidant activity of conjugates in biological system.
| No | m | n | Radical scavenging capacity, luminol chemiluminescence assay | Inhibition of spontaneous lipid peroxidation in mouse brain homogenate, TBARS assay |
|---|---|---|---|---|
| IC50, µM | IC50, μM | |||
| 14d | 2 | 8 | 1.9±0.1 | 20.4±2.3 |
| 15d | 3 | 8 | 3.0±0.1 | 29.1±2.4 |
| 15c | 3 | 6 | 2.3±0.3 | 27.6±1.7 |
| 17d | 2 | 8 | 11.3±1.5 | 17.4±2.1 |
| 18c | 3 | 6 | 14.6±0.8 | 24.6±2.8 |
| 18d | 3 | 8 | 9.2±0.6 | 20.2±3.3 |
| Tacrine | n.a. | n.a. | ||
| BHT | 70.4±4.1 | 6.9±0.3 | ||
m, n - see footnote for Table 1. n.a. = not active.
2.2.8.1. Radical scavenging activity in mouse brain homogenate. Luminol CL assay.
In this method, luminol is used as a CL enhancer. The CL assay of the radical-scavenging capacity of the compounds was based on assessing the reduction in the luminol CL mediated by its interaction with free radicals whose formation in the mouse brain homogenate was initiated by tert-butyl hydroperoxide (TBHP) [53]. Luminol allows detecting hydrogen peroxide (H2O2), hydroxyl radicals (OH), hypochlorite (ClO–), peroxynitrite (ONOO–), and lipid peroxyl radicals.
The radical scavenging capacity of the tested compounds was characterized by IC50 values. As can be seen from Table 5, the conjugates demonstrated a high radical scavenging capacity, which was markedly higher than that of the basic pharmacophore BHT. In addition, radical-scavenging capacity was significantly higher for compounds with an alkylimine rather than an alkylamine spacer.
2.2.8.2. Inhibition of spontaneous LP in mouse brain homogenate. TBARS assay
The reaction of malondialdehyde (MDA) with thiobarbituric acid (TBA) has been widely used as a sensitive method for LP assay in animal tissues [54]. We studied the impact of three pairs of conjugates on the formation of TBARS by following the reaction of oxidized lipids with TBA under the conditions of spontaneous LP in mouse brain homogenate. Inhibition of LP was characterized by IC50 values (Table 5).
As can be seen from Table 5, conjugates effectively inhibited the process of spontaneous LP, and compounds with an amine-containing spacer were more effective than those with an imine-containing spacer. However, their activity was lower than that of the basic BHT pharmacophore.
2.2.9. Quantum-Chemical Calculations
Quantum chemical methods were used to assess the primary antioxidant activity (AOA) of conjugates with imine- and amine-containing spacers. For this purpose, two compounds, 14b (imine) and 17b (amine), were chosen as examples.
Analysis of all experimental conditions and quantum chemical calculations, taking into account the pKa predictions described in section 3.2.3, shows that in all tests for assessing AOA (ABTS test with pH ≈ 4.5, FRAP test with pH = 3.5, CL and LP with pH = 7.4) both imines and amines would have been doubly protonated in the tacrine moiety and the spacer (for details, see the Supplementary Materials). Below, the state of protonation in the tacrine moiety and the spacer is denoted by the subscripts t and s, respectively.
The compounds under consideration demonstrate different results in different antioxidant tests. In the ABTS test, 14b has a lower AOA than 17b (1.0 for 14b vs. 1.4 for 17b, see Table 4). Whereas in the FRAP test, the AOA of 14b is higher than that of 17b (0.72 for 14b vs. 0.46 for 17b, see Table 4). In the CL and LP tests, 14b and 17b were not investigated. However, the results for other compounds from corresponding series indicate that CL radical-scavenging capacity of imines was much higher (3-6 times) than that of amines, and both imines and amines demonstrate markedly higher CL radical scavenging capacity than the basic pharmacophore BHT (see Table 5). In the LP test, amines are more effective than imines. However, their LP activity is lower than that of the basic BHT pharmacophore (see Table 5).
Such different results of antioxidant tests can be explained by different mechanisms of AOA. To further characterize the AOA of the studied compounds on a theoretical basis using quantum-chemical calculations, the bond dissociation enthalpy (BDE), vertical ionization potential (IP), vertical proton affinity (EAv) and proton dissociation enthalpy (PDE) were computed as is commonly done [55,56,57] and displayed in Table 6. The solvation enthalpies of the proton and electron in water and ethanol were taken from the literature [58].
Table 6.
Calculated energetic characteristics of BHT, 14bts and 17bts in water, kcal/mol.
| Compound | BDE1 | IP | EA | PDE1 | PDE2 |
|---|---|---|---|---|---|
| BHT | 75.1 | 105.3 | -12.7 | 41.0 | n.a |
| 14bts | 83.7 | 113.9 | 38.1 | 26.7 | 31.4 |
| 17bts | 79.2 | 114.0 | 27.3 | 37.0 | 32.9 |
1For the OH bond in the BHT moiety; 2For the NH bond in the spacer.
The lowest BDE value is given for each compound, which is for the OH bond in the BHT moiety. PDE values were also calculated for the OH bond in the BHT moiety (PDE1) and for NH bond in the spacer (PDE2). The mechanisms of AOA of the studied compounds in the CL, LP, ABTS and FRAP tests, as well as the main energetic characteristics describing these mechanisms, are discussed below.
2.2.9.1. Luminol chemiluminescence
Superoxide anion O2• − plays a key role in the CL assay [59]. It does not react directly with luminol. Instead, it reacts with the luminol radical anion (L• −) formed in the reactions of luminol with various radicals (e.g., HO• and CO3• −). The reaction between the luminol radical and superoxide results in the formation of 3-aminophyhalane in the excited state (see Figure 5). The luminescence of the latter is detected in the experiment [60]. Thus, in the CL test, the luminol luminescence can be reduced by quenching superoxide, which would be present in mouse brain homogenates [61]. The fundamentally different results obtained in the CL and LP tests (see Table 5) suggest that it is the superoxide radical quenching that is responsible for the decrease in luminol luminescence in the CL assay.
Analysis of free energy changes in possible reactions for the superoxide quenching [62,63,64] (for details see Supplemental Materials) shows that the superoxide quenching proceeds through proton donation by the antioxidant , which is similar to the dismutation of the superoxide radical (O2•-) into hydrogen peroxide (H2O2) and oxygen (O2) in biological systems as depicted in reaction (1) [65]:
where AH is the antioxidant molecule and A- is the deprotonated antioxidant. The energy benefit in reaction (1) is determined by the PDE. As the PDE decreases, the ease of proton donation by the antioxidant molecule increases. BHT and 14bts can donate a proton from the OH bond in the BHT moiety with PDE values of 41.0 and 26.7 kcal/mol, respectively; compound 17bts donates a proton from the NH bond in the spacer with a PDE value of 32.9 kcal/mol. Thus, the AOA in the CL test is determined by the PDE; a difference in PDE values of 6 kcal/mol decreases the IC50 value in the CL test about 6-fold.
2O2•- + 2AH → H2O2 + O2 + 2A-
2.2.9.2. Inhibition of spontaneous lipid peroxidation
In spontaneous lipid peroxidation (LP) many different radicals are formed in brain homogenate. We estimated the ability to inhibit LP by the interaction of the considered antioxidants with the most active hydroxyl radical HO•. The direct pathway for hydroxyl radical quenching is the H-atom transfer from the antioxidant to the radical [66,67,68]:
AOH + HO• → AO• + H2O
The energy benefit in reaction (2) is determined by the BDE. The lower the BDE, the easier it is for the antioxidant molecule to donate an H-atom. The BDE value of 17bts is lower than that of 14bts, resulting in a higher AOA of 17bts than that of 14bts. The BDE value of BHT is lower than that of either 14bts or 17bts, which correlates with the higher activity of BHT in the LP test.
In LP, a mixture of many different radicals is formed, the quenching mechanism of which may differ from the hydroxyl radical. Despite this, the BDE can be considered a rough but adequate characterization of AOA [69].
2.2.9.3. ABTS and FRAP tests
In both the ABTS and FRAP tests, specially pre-generated cation-radicals/cations (ABTS•+ in the ABTS test and Fe3+[TPTZ]2 in the FRAP test) are reduced to their parent form by acquiring an electron because of the interaction with an antioxidant molecule. Regardless of the antioxidant mechanism, the pre-generated cation acquires an electron because of the antioxidant reaction. ABTS•+ and Fe3+[TPTZ]2 cations serve as markers. The decrease in their concentration characterizes the antioxidant capacity of the tested compounds. The decrease in the concentration of ABTS•+ and the increase in the concentration of Fe2+[TPTZ]2 are measured by the intensity of the characteristic bands in the UV-vis spectrum (734 and 593 nm in the ABTS and FRAP tests, respectively) [70].
The electron transfer (ET) from one molecule (donor) to another molecule (acceptor) proceeds via formation of a reaction complex [71]. In antioxidant reactions, the antioxidant and cation molecules in the reaction complex are linked either by hydrogen bonding [72] or by Coulomb interaction. This is the case with the outer-sphere ET mechanism.
The ET process is illustrated in Figure 6 for a neutral donor (D) and acceptor (A). The process occurs in three stages. At the first stage, the precursor complex [D, A] is formed during thermal diffusion.
At the second rate-limiting stage, the intra-complex ET results in the successor complex [D+, A-] formation. The electron passes from the donor HOMO to the acceptor LUMO. The ET occurs only if the donor HOMO lies energetically above the acceptor LUMO. The efficiency of this stage depends on the energy characteristics of the donor and acceptor. The greater the difference between the energies of the donor HOMO and the acceptor LUMO, the faster the ET.
The third stage of the ET is a diffusive separation of the [D+, A-] successor complex.
Thus, ET efficiency is generally determined by two factors. The first is the energy benefit of the ET from a donor to an acceptor. For the same acceptor, ET is more efficient for a donor with a higher HOMO energy, i.e., a lower IP.
The second factor is the efficiency of the precursor complex formation during thermal diffusion. The probability of precursor complex formation depends on the geometric structure (mutual orientation of molecules suitable for ET) and diffusion characteristics (probability of contact) of the donor and acceptor. Obviously, imines and amines have different diffusion characteristics due to different spacer rigidity resulting in different probabilities of precursor complex formation. (The same applies to the marker cations ABTS•+ and Fe3+[TPTZ]2.) The donor and acceptor in the complex should be oriented so that the electron can move from the donor HOMO to the acceptor LUMO. For the sterically hindered propeller-shaped Fe3+[TPTZ]2, complex formation is more difficult than for the quasi-one-dimensional ABTS•+.
In addition, the presence of other species, such as cations and anions (buffer acetate anion CH3COO- in the FRAP test, ammonium cation NH4+ and sulfate anion SO42- in the ABTS test and some others), should influence the diffusion due to their interaction with both charged (protonated) antioxidants and the marker cations ABTS•+ and Fe2+[TPTZ]2.
Thus, the same order of AOA of imines and amines in the ABTS and FRAP tests are explained by their nearly equivalent IP values (113.9 and 114.0 kcal/mol for 14bt and 17bt, respectively, see Table 6) characterizing the ability to donate an electron. Some difference in AOA is associated with different thermal diffusion behavior due to the different spacer rigidity of both the antioxidant molecules and the marker cations ABTS•+ and Fe3+[TPTZ] and their interaction with other cations and anions in a reaction solution, which leads to different efficiencies of precursor complex formation.
2.3. Predicted ADMET Profiles and PAINS Analysis
The computational estimates of selected ADMET and physicochemical properties for compounds 13–18 are shown in Table 8. All the compounds had high predicted values for intestinal absorption, enabling their oral administration. Moreover, we could expect reasonable CNS activity in view of rather high predicted blood–brain barrier permeability (brain concentration is about 1.7- to 8-fold greater than the plasma concentration). The cardiac toxicity risk parameters (hERG pKi and pIC50) varied from 5.7 to 7.4 log units for all the analyzed compounds, which falls within the middle range of their possible range (3–9 log units).
According to the commonly accepted drug-likeness guidelines, the molecular weights of the compounds, as well as the predicted lipophilicities and aqueous solubilities, were within or close to their desirable ranges for potential drug compounds. However, the LogP values exceeded the original Rule-of-5 limits and the solubilities were in the micromolar or nanomolar range. Nevertheless, given that some of the compounds were outside of the model applicability domain, the predicted values were not fully reliable. The integral quantitative estimates of drug-likeness (QED) are in the 0.2–0.4 range (based on the data for oral drugs, QED > 0.2 is desirable). The Pan Assay INterference compoundS (PAINS) filter check did not identify any alerts.
Overall, the predicted ADMET, physicochemical, and PAINS properties of the compounds seem acceptable for potential lead compounds in the discovery phase. Nevertheless, additional studies and structure optimization would be desirable to improve the pharmacokinetic profile and maximize safety.
3. Materials and Methods
3.1. Chemistry
All solvents, chemicals, and reagents were obtained commercially and used without purification. 1H-NMR (200 MHz) spectra were recorded on a DPX-200 NMR spectrometer (Bruker, Karlsruhe, Germany) using tetramethylsilane as an internal standard. Chemical shifts, δ, are given in parts per million (ppm), and spin multiplicities are given as s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet) or m (multiplet). Coupling constants, J, are expressed in hertz (Hz). Melting points were recorded on the Stuart SMP10 Melting Point Apparatus (Stuart, Staffordshire, UK) and are uncorrected. Yields refer to isolated pure products and were not maximized. CHN analysis was performed on the ER-20 analyzer (Carlo-Erba, Val-de-Reuil, France). All compounds exhibited analytical and spectroscopic data that strongly agreed with their expected structures.
3.1.1. General procedure for the preparation of derivatives 13–15a-d and 16–18a-d
A mixture of aminoquinoline 9–11a-d (1.0 mmol) and 3,5-di-tert-butyl-4-hydroxy-benzaldehyde 12 (234 mg, 1.0 mmol) in toluene (10 mL) was stirred for 3 h at boiling point. Then, the solution was evaporated under vacuum and the residue was washed with ether, yielding the target conjugates 13–15a-d.
To a solution of compounds 13–15a-d (1.0 mmol) in 5 mL of methanol, 57 mg (1.0 mmol) of sodium borohydride was added and the mixture was stirred for 1 h at room temperature. Methanol was evaporated, 15 mL of methylene chloride was added and washed with water (2×10 mL). The organic layer was dried over anhydrous sodium sulfate. The drying agent was filtered, the filtrate was evaporated, and the residue was recrystallized in a benzene-methanol (5:1) mixture to give target conjugates 16–18a-d.
3.1.2. Synthesis of compounds
The compounds 13a, 14a, 15a, 16a and 18a were synthesized and described earlier in [44].
2,6-Di-tert-butyl-4-{[4-(2,3-dihydro-1H-cyclopenta[b]quinolin-9-ylamino)-butylimino]-methyl}-phenol (13b). Yellow solid; Yield 67%, m.p. 91–93 °C. 1H-NMR (CDCl3), δ: 1.45 (s, 18H, 6×CH3), 1.66-1.86 (m, 4H, 2×CH2), 2.02-2.23 (m, 2H, CH2), 3.08 (t, 2H, J = 7.4 Hz, CH2), 3.19 (t, 2H, J = 7.4 Hz, CH2), 3.50-3.82 (m, 4H, 2×CH2), 5.11 (br.s, 1Н, ОН), 7.12-7.42 (m, 2H, 2×Har), 7.26 (s, 2×1H, 2×Har), 7.77 (d, 1H, J = 8.6 Hz, Har), 7.94 (d, 1H, J = 8.6 Hz, Har), 8.19 (s, 1Н, =CН). Anal. Calcd. for C31H41N3O: C, 78.94; H, 8.76; N, 8.91. Found: C, 78.82; H, 8.84; N, 8.82.
2,6-Di-tert-butyl-4-{[6-(2,3-dihydro-1H-cyclopenta[b]quinolin-9-ylamino)-hexylimino]-methyl}-phenol (13c). Light brown solid; Yield 72%, m.p. 92–94°C. 1H-NMR (CDCl3), δ: 1.15-1.30 (m, 2Н, CH2), 1.44 (s, 18H, 6×CH3), 1.56-1.82 (m, 6H, 3×CH2), 2.12 (pent, 2H, J = 7.2 Hz, CH2), 3.07 (t, 2H, J = 7.6 Hz, CH2), 3.19 (t, 2H, J = 7.1 Hz, CH2), 3.41-3.72 (m, 4H, 2×CH2), 5.08 (br.s, 1Н, ОН), 7.28-7.43 (m, 1H, Har), 7.43-7.65 (m, 3H, 3×Har), 7.79 (d, 1H, J = 8.1 Hz, Har), 7.92 (d, 1H, J = 8.1 Hz, Har), 8.07 (s, 1Н, =CН). 13C-NMR (CDCl3), δ: 22.43, 23.20, 26.13, 26.56, 26.97, 30.12 (6), 30.93, 31.14, 34.36, 34.77, 45.61, 113.93, 118.75, 119.75, 123.96, 125.34, 125.42, 125.77, 127.61, 128.30, 128.73, 136.72, 146.65, 147.78, 168.19. Anal. Calcd. for C33H45N3O: C, 79.31; H, 9.08; N, 8.41. Found: C, 79.43; H, 9.00; N, 8.50.
2,6-Di-tert-butyl-4-{[8-(2,3-dihydro-1H-cyclopenta[b]quinolin-9-ylamino)-octylimino]-methyl}-phenol (13d). Light brown solid; Yield 71%, m.p. 75–78°C. 1H-NMR (CDCl3), δ: 1.29-1.40 (m, 6Н, 3×CH2), 1.45 (s, 18H, 6×CH3), 1.53-1.78 (m, 6H, 3×CH2), 2.13 (pent, 2H, J = 7.2 Hz, CH2), 3.07 (t, 2H, J = 7.5 Hz, CH2), 3.21 (t, 2H, J = 7.2 Hz, CH2), 3.40-3.76 (m, 4H, 2×CH2), 4.72 (br.s, 1Н, NН), 5.45 (br.s, 1Н, ОН), 7.36 (s, 2×1H, 2×Har), 7.45-7.63 (m, 2H, 2×Har), 7.74 (d, 1H, J = 8.1 Hz, Har), 7.92 (d, 1H, J = 8.1 Hz, Har), 8.11 (s, 1Н, =CН). 13C-NMR (CDCl3), δ: 22.44, 23.23, 26.13, 26.72, 27.30, 29.46, 30.28 (6), 30.81, 31.19, 34.26, 35.00, 45.72, 54.48, 114.02, 118.82, 119.56, 123.82, 124.81, 125.04, 128.10, 129.17, 131.07, 135.77, 146.28, 148.35, 152.68, 168.63. Anal. Calcd. for C35H49N3O: C, 79.65; H, 9.36; N, 7.96. Found: C, 79.53; H, 9.27; N, 8.05.
2,6-Di-tert-butyl-4-{[4-(1,2,3,4-tetrahydro-acridin-9-ylamino)-butylimino]-methyl}-phenol (14b). Yellow solid; Yield 67%, m.p. 90–93°C. 1H-NMR (CDCl3), δ: 1.45 (s, 18H, 6×CH3), 1.64-1.83 (m, 4H, 2×CH2), 1.83-2.07 (m, 4H, 2×CH2), 2.56-2.80 (m, 2H, CH2), 2.91-3.18 (m, 2H, CH2), 3.36-3.75 (m, 4H, 2×CH2), 4.11 (br.s, 1H, NH), 5.51 (br.s, 1Н, ОН), 7.21-7.32 (m, 1H, Har), 7.41-7.70 (m, 1H, Har), 7.55 (s, 2×1H, 2×Har), 7.90 (d, 1H, J = 8.8 Hz, Har), 7.96 (d, 1H, J = 8.8 Hz, Har), 8.19 (s, 1Н, =CН). 13C-NMR (CDCl3), δ: 22.77, 23.04, 24.86, 28.47, 29.51, 30.16 (6), 34.04, 34.33 (2), 49.25, 61.15, 115.81, 120.19, 122.82, 123.53, 125.25 (2), 127.64, 128.20, 128.71, 136.14 (2), 147.45, 150.68, 156.27, 158.41, 161.73. Anal. Calcd. for C32H43N3O: C, 79.13; H, 8.92; N, 8.65. Found: C, 79.28; H, 8.84; N, 8.53.
2,6-Di-tert-butyl-4-{[6-(1,2,3,4-tetrahydro-acridin-9-ylamino)-hexylimino]-methyl}-phenol (14с). Yellow solid; Yield 65%, m.p. 79–81°C. 1H-NMR (CDCl3), δ: 1.30-1.40 (m, 4H, 2×CH2), 1.46 (s, 18H, 6×CH3), 1.66-1.87 (m, 4H, 2×CH2), 1.83-2.10 (m, 4H, 2×CH2), 2.54-2.82 (m, 2H, CH2), 2.96-3.16 (m, 2H, CH2), 3.37-3.78 (m, 4H, 2×CH2), 4.11 (br.s, 1H, NH), 5.51 (br.s, 1Н, ОН), 7.23-7.32 (m, 1H, Har), 7.44-7.75 (m, 1H, Har), 7.58 (s, 2×1H, 2×Har), 7.91 (d, 1H, J = 8.8 Hz, Har), 7.98 (d, 1H, J = 8.8 Hz, Har), 8.18 (s, 1Н, =CН). 13C-NMR (CDCl3), δ: 21.76, 22.94, 24.26, 27.11, 27.67, 28.57, 29.78, 30.11 (6), 33.93, 34.26 (2), 48.12, 60.12, 119.28, 120.51, 123.13, 124.37, 125.66, 127.62, 128.16, 128.55, 128.22, 137.13 (2), 146.81, 151.13, 160.02, 163.75. Anal. Calcd. for C34H47N3O: C, 79.49; H, 9.22; N, 8.18. Found: C, 79.38; H, 9.30; N, 8.27.
2,6-Di-tert-butyl-4-{[8-(1,2,3,4-tetrahydro-acridin-9-ylamino)-octylimino]-methyl}-phenol (14d). Yellow solid; Yield 65%, m.p. 68-69°C. 1H-NMR (CDCl3), δ: 1.20-1.37 (m, 8H, 4×CH2), 1.44 (s, 18H, 6×CH3), 1.56-1.74 (m, 4H, 2×CH2), 1.82-2.02 (m, 4H, 2×CH2), 2.59-2.80 (m, 2H, CH2), 2.98-3.15 (m, 2H, CH2), 3.38-3.68 (m, 4H, 2×CH2), 4.11 (br.s, 1H, NH), 5.45 (br.s, 1Н, ОН), 7.36 (s, 2×1H, 2×Har), 7.44-7.69 (m, 3H, =CН, 2×Har), 7.94 (t, 2H, J = 8.2 Hz, 2×Har). 13C-NMR (CDCl3), δ: 22.43, 22.69, 22.99, 24.61, 24.71, 26.85, 27.11, 29.30, 30.10 (6), 31.02, 31.73, 33.80, 34.38, 49.45, 115.59, 120.03, 122.87, 123.58, 125.30, 125.49, 127.61, 128.38, 136.69, 147.13, 150.94, 158.12, 161.45. Anal. Calcd. for C36H51N3O: C, 79.80; H, 9.49; N, 7.76. Found: C, 79.68; H, 9.39; N, 7.85.
2,6-Di-tert-butyl-4-{[4-(7,8,9,10-tetrahydro-6H-cyclohepta[b]quinolin-11-ylamino)-butylimino]-methyl}-phenol (15b). Light yellow solid; Yield 68%. m.p. 138-140 °C. 1H-NMR (CDCl3), δ: 1.45 (s, 18H, 6×CH3), 1.59-1.96 (m, 10H, 5×CH2), 2.79-3.00 (m, 2H, CH2), 3.08-3.23 (m, 2H, CH2), 3.35 (t, 2H, J = 6.6 Hz, CH2), 3.60 (t, 2H, J = 5.6 Hz, CH2), 5.50 (br.s, 1Н, ОН), 7.30-7.45 (m, 1H, Har), 7.54 (s, 2×1H, 2×Har), 7.49-7.66 (m, 1H, Har), 7.93 (t, 2H, J = 9.2 Hz, 2×Har), 8.16 (s, 1Н, =CН). 13C-NMR (CDCl3), δ: 26.85, 27.68, 28.26, 28.58, 29.22, 30.14 (6), 31.97, 34.34, 39.88, 50.47, 65.82, 121.94, 120.00, 123.75, 124.70, 124.83, 125.29, 128.27, 128.83, 129.20, 136.31, 146.46, 149.88, 161.56, 165.16. Anal. Calcd. for C33H45N3O: C, 79.31; H, 9.08; N, 8.41. Found: C, 79.45; H, 9.15; N, 8.31.
2,6-Di-tert-butyl-4-{[6-(7,8,9,10-tetrahydro-6H-cyclohepta[b]quinolin-11-ylamino)-hexylimino]-methyl}-phenol (15c). Light yellow solid; Yield 68%, m.p. 62-65°C. 1H-NMR (CDCl3), δ: 1.21-1.33 (m, 2H, CH2), 1.44 (s, 18H, 6×CH3), 1.58-1.91 (m, 12H, 6×CH2), 2.84-2.97 (m, 2H, CH2), 3.17-3.28 (m, 2H, CH2), 3.29-3.41 (m, 2H, CH2), 3.54 (t, 2H, J = 7.1 Hz, CH2), 4.27 (br.s, 1Н, NН), 5.47 (br.s, 1Н, ОН), 7.41 (t, 1H, J = 7.5 Hz, Har), 7.51 (s, 2×1H, 2×Har), 7.59 (t, 1H, J = 7.5 Hz, Har), 7.93 (d, 1H, J = 8.4 Hz, Har), 8.05 (d, 1H, J = 8.4 Hz, Har), 8.07 (s, 1Н, =CН). 13C-NMR (CDCl3), δ: 27.19, 27.25, 27.46, 28.03, 28.52, 30.58 (6), 31.39, 31.83, 32.33, 34.85, 39.55, 50.91, 60.87, 121.87, 122.50, 123.36, 125.36, 125.95, 127.61, 128.10, 128.35, 129.22, 137.23, 145.84, 151.03, 161.04, 164.85. Anal. Calcd. for C35H49N3O: C, 79.65; H, 9.36; N, 7.96. Found: C, 79.52; H, 9.45; N, 7.88.
2,6-Di-tert-butyl-4-{[8-(7,8,9,10-tetrahydro-6H-cyclohepta[b]quinolin-11-ylamino)-octylimino]-methyl}-phenol (15d). Light yellow solid; Yield 66%, m.p. 77-78°C. 1H-NMR (CDCl3), δ: 1.18-1.39 (m, 6H, 3×CH2), 1.44 (s, 18H, 6×CH3), 1.55-1.97 (m, 12H, 6×CH2), 2.85-3.00 (m, 2H, CH2), 3.13-3.34 (m, 4H, 2×CH2), 3.52 (t, 2H, J = 7.1 Hz, CH2), 4.07 (br.s, 1Н, NН), 5.49 (br.s, 1Н, ОН), 7.32-7.65 (m, 4H, 4×Har), 7.83-8.15 (m, 3H, 2×Har, =CН). 13C-NMR (CDCl3), δ: 26.84, 26.93, 27.13, 27.64, 28.23, 29.14, 29.27, 29.31, 30.11 (6), 31.02, 31.45, 31.95, 34.37, 39.85, 50.66, 121.87, 123.71, 124.70, 125.37, 128.30, 128.80, 146.45, 149.96, 160.32, 165.11. Anal. Calcd. for C37H53N3O: C, 79.95; H, 9.61; N, 7.56. Found: C, 79.85; H, 9.54; N, 7.68.
2,6-Di-tert-butyl-4-{[4-(2,3-dihydro-1H-cyclopenta[b]quinolin-9-ylamino)-butylamino]-methyl}-phenol (16b). Light yellow solid; Yield 65%, m.p. 65–67°C. 1H-NMR (CDCl3), δ: 1.44 (s, 18H, 6×CH3), 1.62-1.82 (m, 4H, 2×CH2), 2.13 (pent, 2H, J = 7.1 Hz, CH2), 2.66 (t, 2H, J = 7.2 Hz, CH2), 3.05 (t, 2H, J = 6.8 Hz, CH2), 3.28 (t, 2H, J = 6.8 Hz, CH2), 3.62 (кв, 2H, J = 6.3 Hz, CH2), 3.68 (s, 2H, CH2), 4.64 (br.s, 1H, NH), 5.15 (br.s, 1Н, ОН), 7.10 (s, 2×1H, 2×Har), 7.55 (t, 1H, J = 7.2 Hz, Har), 7.55 (t, 1H, J = 7.2 Hz, Har), 7.75 (t, 1H, J = 8.1 Hz, Har), 7.90 (d, 1H, J = 8.0 Hz, Har). Anal. Calcd. for C31H43N3O: C, 78.60; H, 9.15; N, 8.87. Found: C, 78.72; H, 9.06; N, 8.78.
2,6-Di-tert-butyl-4-{[6-(2,3-dihydro-1H-cyclopenta[b]quinolin-9-ylamino)-hexylamino]-methyl}-phenol (16c). Light yellow solid; Yield 75%, m.p. 62-65°C. 1H-NMR (CDCl3), δ: 1.16-1.33 (m, 4Н, CH2), 1.42 (s, 18H, 6×CH3), 1.51-1.75 (m, 6H, 3×CH2), 2.11 (pent, 2H, J = 7.2 Hz, CH2), 2.65 (t, 2H, J = 5.9 Hz, CH2), 3.03 (t, 2H, J = 7.2 Hz, CH2), 3.18 (t, 2H, J = 6.7 Hz, CH2), 3.47-3.62 (m, 2H, CH2), 3.66 (s, 2Н, CH2), 4.62 (br.s, 1Н, NН), 5.11 (br.s, 1Н, ОН), 7.08 (s, 2×1H, 2×Har), 7.38 (t, 1H, J = 7.5 Hz, Har), 7.57 (t, 1H, J = 7.4 Hz, Har), 7.70 (d, 1H, J = 8.2 Hz, Har), 7.88 (d, 1H, J = 8.2 Hz, Har). 13C-NMR (CDCl3), δ: 22.42, 23.21, 26.10, 26.71, 27.13, 30.27 (6), 30.79, 34.25, 34.97, 45.66, 49.60, 54.48, 114.05, 118.84, 119.56, 123.82, 124.76, 128.08, 129.17, 131.04, 135.82, 146.26, 148.36, 152.69, 168.61. Anal. Calcd. for C33H47N3O: C, 79.00; H, 9.44; N, 8.37. Found: C, 79.13; H, 9.08; N, 8.45.
2,6-Di-tert-butyl-4-{[8-(2,3-dihydro-1H-cyclopenta[b]quinolin-9-ylamino)-octylamino]-methyl}-phenol (16d). Light grey solid; Yield 76%, m.p. 60-63°C. 1H-NMR (CDCl3), δ: 1.27-1.38 (m, 8Н, 4×CH2), 1.44 (s, 18H, 6×CH3), 1.53-1.77 (m, 4H, 2×CH2), 2.13 (pent, 2H, J = 7.1 Hz, CH2), 2.65 (t, 2H, J = 7.4 Hz, CH2), 3.05 (t, 2H, J = 7.5 Hz, CH2), 3.21 (t, 2H, J = 7.1 Hz, CH2), 3.63 (кв, 2H, J = 6.2 Hz, CH2), 3.75 (s, 2Н, CH2), 4.63 (br.s, 1Н, NН), 5.14 (br.s, 1Н, ОН), 7.10 (s, 2×1H, 2×Har), 7.36 (t, 1H, J = 7.5 Hz, Har), 7.55 (t, 1H, J = 7.5 Hz, Har), 7.75 (d, 1H, J = 8.2 Hz, Har), 7.90 (d, 1H, J = 8.1 Hz, Har). Anal. Calcd. for C35H51N3O: C, 79.35; H, 9.70; N, 7.93. Found: C, 79.23; H, 9.78; N, 8.02.
2,6-Di-tert-butyl-4-{[2-(1,2,3,4-tetrahydro-acridin-9-ylamino)-ethylamino]-methyl}-phenol (17a). Yellow solid; Yield 76%. m.p. 66-68°C. 1H-NMR (CDCl3) δ: 1.45 (s, 18H, 6×CH3), 1.83-2.02 (m, 4H, 2×CH2), 2.70-2.84 (m, 2H, CH2), 2.86-2.99 (m, 2H, CH2), 3.00-3.13 (m, 2H, CH2), 3.53-3.66 (m, 2H, CH2), 3.74 (s, 2Н, CH2), 5.18 (br.s, 1Н, ОН), 7.16 (s, 2H, 2×Har), 7.32 (t, 1H, J = 7.6 Hz, Har), 7.54 (t, 1H, J = 7.6 Hz, Har), 7.89 (d, 1H, J = 8.4 Hz, Har), 8.03 (d, 1H, J = 8.4 Hz, Har). 13C-NMR (CDCl3), δ: 22.82, 23.10, 24.82, 30.27 (6), 33.99, 34.28, 48.15, 49.39, 53.89, 116.02, 120.32, 122.89, 123.42, 124.76, 128.11, 128.64, 130.69, 135.92, 147.44, 150.96, 152.85, 158.38. Anal. Calcd. for C30H41N3O: C, 78.39; H, 8.99; N, 9.14. Found: C, 78.28; H, 8.91; N, 9.03.
2,6-Di-tert-butyl-4-{[4-(1,2,3,4-tetrahydro-acridin-9-ylamino)-butylamino]-methyl}-phenol (17b). Light grey solid; Yield 67%, m.p. 59–62°C. 1H-NMR (CDCl3), δ: 1.42 (s, 18H, 6×CH3), 1.55-1.80 (m, 4H, 2×CH2), 1.81-2.02 (m, 4H, 2×CH2), 2.49-2.85 (m, 4H, 2×CH2), 2.90-3.19 (m, 2H, CH2), 3.34-3.60 (m, 2H, CH2), 3.67 (c, 2H, CH2), 4.02 (br.s, 1H, NH), 5.14 (br.s, 1Н, ОН), 7.08 (s, 2×1H, 2×Har), 7.31 (t, 1H, J = 7.8 Hz, Har), 7.54 (t, 1H, J = 7.8 Hz, Har), 7.77-8.03 (m, 2H, 2×Har). 13C-NMR (CDCl3), δ: 22.79, 23.06, 24.87, 27.58, 29.62, 30.28 (6), 34.09, 34.28 (2), 49.28, 49.44, 54.46, 115.91, 120.23, 122.78, 123.54, 124.77 (2), 128.19, 128.78 (2), 130.89, 135.82, 147.51, 150.65, 152.75, 158.46. Anal. Calcd. for C32H45N3O: C, 78.80; H, 9.30; N, 8.62. Found: C, 78.68; H, 9.39; N, 8.54.
2,6-Di-tert-butyl-4-{[6-(1,2,3,4-tetrahydro-acridin-9-ylamino)-hexylamino]-methyl}-phenol (17c). Light grey solid; Yield 68%, m.p. 68–70°C. 1H-NMR (CDCl3), δ: 1.20-1.30 (m, 8H, 4×CH2), 1.41 (s, 18H, 6×CH3), 1.50-1.70 (m, 4H, 2×CH2), 1.84-2.05 (m, 4H, 2×CH2), 2.47-2.86 (m, 4H, 2×CH2), 2.97-3.19 (m, 2H, CH2), 3.44-3.57 (m, 2H, CH2), 3.71 (s, 2H, CH2), 4.10 (br.s, 1H, NH), 5.12 (br.s, 1Н, ОН), 7.13 (s, 2×1H, 2×Har), 7.36 (t, H, J = 8.2 Hz, Har), 7.54 (t, H, J = 6.8 Hz, Har), 7.95 (t, 2H, J = 8.3 Hz, 2×Har). 13C-NMR (CDCl3), δ: 22.39, 23.26, 24.81, 27.19, 27.55, 28.51, 29.88, 30.27 (6), 34.19, 34.22 (2), 49.21, 49.54, 55.48, 118.92, 121.21, 122.76, 124.55, 123.71 (2), 128.34, 126.72 (2), 131.19, 136.12, 146.53, 151.05, 161.24, 165.05. Anal. Calcd. for C34H49N3O: C, 79.18; H, 9.58; N, 8.15. Found: C, 79.07; H, 9.49; N, 8.24.
2,6-Di-tert-butyl-4-{[8-(1,2,3,4-tetrahydro-acridin-9-ylamino)-octylamino]-methyl}-phenol (17d). Yellow solid; Yield 75%, m.p. 71-73°C. 1H-NMR (CDCl3), δ: 1.23-1.37 (m, 8H, 4×CH2), 1.43 (s, 18H, 6×CH3), 1.53-1.75 (m, 4H, 2×CH2), 1.82-2.04 (m, 4H, 2×CH2), 2.49-2.89 (m, 4H, 2×CH2), 2.99-3.17 (m, 2H, CH2), 3.41-3.58 (m, 2H, CH2), 3.70 (s, 2H, CH2), 4.12 (br.s, 1H, NH), 5.14 (br.s, 1Н, ОН), 7.12 (s, 2×1H, 2×Har), 7.34 (t, H, J = 8.1 Hz, Har), 7.55 (t, H, J = 6.9 Hz, Har), 7.95 (t, 2H, J = 8.3 Hz, 2×Har). 13C-NMR (CDCl3), δ: 22.40, 22.57, 22.91, 24.63, 26.79, 27.18, 29.21, 29.34, 29.62, 30.23 (6), 30.85, 31.64, 33.55, 34.24, 49.38, 115.35, 119.81, 122.90, 123.59, 124.99, 128.06, 128.48, 130.66, 135.33, 135.80, 146.74, 151.06, 157.82. Anal. Calcd. for C36H53N3O: C, 79.51; H, 9.82; N, 7.73. Found: C, 79.64; H, 9.90; N, 7.83.
2,6-Di-tert-butyl-4-{[4-(7,8,9,10-tetrahydro-6H-cyclohepta[b]quinolin-11-ylamino)-butylamino]-methyl}-phenol (18b). Light grey solid; Yield 64%, m.p. 65–67°C. 1H-NMR (CDCl3), δ: 1.43 (s, 18H, 6×CH3), 1.60-1.89 (m, 10H, 5×CH2), 2.71 (t, 2H, J = 6.3 Hz, CH2), 2.82-2.99 (m, 2H, CH2), 3.10-3.23 (m, 2H, CH2), 3.30 (t, 2H, J = 6.8 Hz, CH2), 3.69 (c, 2H, CH2), 5.14 (br.s, 1Н, ОН), 7.10 (s, 2×1H, 2×Har), 7.33-7.46 (m, 1H, Har), 7.57 (t, 1H, J = 7.8 Hz, Har), 7.92 (t, 2H, J = 8.7 Hz, 2×Har). 13C-NMR (CDCl3), δ: 26.90, 27.70 (2), 28.31, 29.33, 30.29 (6), 32.00, 34.28, 40.13 (2), 49.33, 50.68, 54.47, 121.86, 124.02, 124.67, 124.81 (2), 125.04, 128.17, 129.12 (2), 130.87, 135.84, 146.79, 149.72, 152.77, 165.38. Anal. Calcd. for C33H47N3O: C, 79.00; H, 9.44; N, 8.37. Found: C, 79.14; H, 9.35; N, 8.43.
2,6-Di-tert-butyl-4-{[6-(7,8,9,10-tetrahydro-6H-cyclohepta[b]quinolin-11-ylamino)-hexylamino]-methyl}-phenol (18c). White powder; Yield 68%, m.p. 71-73°C. 1H-NMR (CDCl3), δ: 1.12-1.32 (m, 4H, 2×CH2), 1.43 (s, 18H, 6×CH3), 1.55-1.98 (m, 10H, 5×CH2), 2.66 (t, 2H, J = 6.7 Hz, Har), 2.84-3.01 (m, 2H, CH2), 3.11-3.36 (m, 4H, 2×CH2), 3.69 (s, 2H, CH2), 5.17 (br.s, 1Н, ОН), 7.11 (s, 2×1H, 2×Har), 7.41 (t, 1H, J = 7.7 Hz, Har), 7.58 (t, 1H, J = 7.2 Hz, Har), 7.89 (d, 1H, J = 8.4 Hz, Har), 7.95 (d, 1H, J = 8.4 Hz, Har). 13C-NMR (CDCl3), δ: 26.86, 27.14, 27.64, 28.25, 29.66, 30.26 (6), 31.40, 31.97, 34.27, 40.00, 49.33, 50.66, 54.22, 60.36, 121.85, 122.05, 123.97, 124.68, 124.98, 128.21, 128.98, 135.36, 135.82, 146.63, 149.75, 152.84, 165.26. Anal. Calcd. for C35H51N3O: C, 79.35; H, 9.70; N, 7.93. Found: C, 79.49; H, 9.59; N, 7.86.
2,6-Di-tert-butyl-4-{[8-(7,8,9,10-tetrahydro-6H-cyclohepta[b]quinolin-11-ylamino)-octylamino]-methyl}-phenol (18d). White powder; Yield 76%, m.p. 63-65°C. 1H-NMR (CDCl3), δ: 1.16-1.38 (m, 8H, 4×CH2), 1.44 (s, 18H, 6×CH3), 1.57-1.99 (m, 10H, 5×CH2), 2.66 (t, 2H, J = 7.2 Hz, CH2), 2.85-2.99 (m, 2H, CH2), 3.12-3.33 (m, 4H, 2×CH2), 3.69 (s, 2H, CH2), 5.13 (br.s, 1Н, ОН), 7.15 (s, 2×1H, 2×Har), 7.41(t, 1H, J = 7.5 Hz, Har), 7.57 (t, 1H, J = 7.5 Hz, Har), 7.92 (t, 2H, J = 9.2 Hz, 2×Har),). 13C-NMR (CDCl3), δ: 26.85, 27.62, 28.26, 29.30, 29.40, 29.55, 30.33 (6), 30.84, 31.42, 31.95, 34.20, 40.08, 49.57, 50.70, 54.32, 58.32, 121.82, 123.96, 124.59, 124.84, 125.00, 128.10, 129.07, 130.71, 135.36, 135.80, 146.76, 149.69, 165.30. Anal. Calcd. for C37H55N3O: C, 79.66; H, 9.94; N, 7.53. Found: C, 79.78; H, 9.84; N, 7.62.
3.2. Biological testing
3.2.1. In vitro AChE, BChE and CES inhibition
All experiments were carried out in accordance with the standard protocols approved by IPAС RAS.
Human erythrocyte AChE and equine serum BChE were purchased from Milamed (Perm, Russia). Porcine liver CES, substrates, and reference compounds were from Sigma-Aldrich (St. Louis, MO, USA). The activity of enzymes was measured spectrophotometrically, as described in detail in [73] using ATCh iodide, BTCh iodide, and 4-NPA as substrates for AChE, BChE, and CES, respectively. Experimental conditions: K,Na-phosphate buffer (100 mM), 25 °С, pH 7.5 for AChE and BChE and pH 8.0 for CES assay. Measurements were carried out on a FLUOStar Optima microplate reader (BMG Labtech, Ortenberg, Germany).
Test compounds were dissolved in DMSO; final concentration of solvent in the incubation mixture was 2% (v/v). Initial assessment of inhibitory activity was carried out by determining the degree of enzyme inhibition at a compound concentration of 20 µM. For active compounds (inhibition ≥35%), IC50 values were determined.
Mechanism of AChE and BChE inhibition was assessed by a detailed analysis of enzyme kinetics with three increasing concentrations of inhibitor and six substrate concentrations as described in detail in [73].
3.2.2. Propidium displacement from EeAChE PAS
The ability of the test compounds to competitively displace propidium, a selective ligand of the PAS of AChE, was evaluated by the fluorescence method [74,75] as described in detail in [44]. Propidium iodide, donepezil, and Electric eel AChE (EeAChE, type VI-S, lyophilized powder) were purchased from Sigma-Aldrich (Saint Louis, MO, USA).
EeAChE (7 μM) was incubated with the test compound at a concentration of 20 μM in 1 mM Tris-HCl buffer pH 8.0, 25 °C, for 15 min. After that, propidium iodide was added at final concentration of 8 μM. The samples were incubated for 15 min and the fluorescence spectrum (530 nm (excitation) and 600 nm (emission)) was taken. Donepezil and tacrine were used as reference compounds. The measurements were carried out in triplicate on a FLUOStar Optima microplate reader (BMG LabTech, Ortenberg, Germany).
3.2.3. Effect on β-Amyloid self-aggregation
The inhibitory effect of the test compounds toward Aβ42 self-aggregation was determined using the thioflavin T (ThT) fluorescence method [24,27,76] with minor modifications as described in detail in [38]. Lyophilized HFIP-pretreated Aβ42 from BACHEM (Bubendorf, Switzerland) was used.
For the measurement of Aβ42 self-aggregation and assessment of inhibition of amyloid fibril formation by the tested compounds, aliquots of 500 μM Aβ42 stock solution in DMSO were diluted in 215 mM Na-phosphate buffer pH 8.0 to a final concentration of 50 μM Aβ42 and incubated for 24 h at 37 °C in the absence or presence of the tested compounds at a concentration of 100 µM. After that, the samples were incubated with 5 μM ThT in 50 mM glycine-NaOH buffer pH 8.5 for 10 min and the fluorescence was measured at 440 nm (excitation) and 485 nm (emission). Myricetin and propidium iodide were used as reference compounds (positive controls). Analyses were performed with a FLUOStar Optima microplate reader (BMG LabTech, Ortenberg, Germany).
3.2.4. Antioxidant Activity
3.2.4.1. ABTS radical cation scavenging activity assay
Radical scavenging activity of the compounds was evaluated using the ABTS radical cation (2,2ʹ-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid, ABTS•+) decolorization assay [77] with minor modifications described in detail in [78]. The reduction in absorbance was measured spectrophotometrically at 734 nm using a xMark UV/VIS microplate spectrophotometer (Bio-Rad, Hercules, CA, USA) for 1 h with an interval of 1-10 min compared to a standard synthetic antioxidant, Trolox (6-hydroxy-2,5,7,8- tetramethyl- chroman-2-carboxylic acid).
The antioxidant activity of the compounds was reported as Trolox equivalent antioxidant capacity (TEAC values) – the ratio of the slopes of the concentration−response curves, test compound/Trolox. The IC50 values for the test compounds (compound concentration required for 50% reduction of the ABTS radical), were also determined.
3.2.4.2. Ferric reducing antioxidant power (FRAP) assay
The ferric reducing antioxidant power (FRAP) assay proposed by Benzie and Strain [79,80] modified to be performed in 96-well microplates as described in detail in [44] was used. 10 µL (0.5 mM) of the tested compound or reference compound were mixed with 240 µL of FRAP reagent and the absorbance of the mixture was measured spectrophotometrically (λ = 593 nm) with a FLUOStar OPTIMA microplate reader (BMG LabTech, Ortenberg, Germany) at 600 nm after a 1 h incubation at 37 ºC against a blank. Trolox was used as a reference compound. The results were expressed as Trolox equivalents (TE) – the ratio of the concentrations of Trolox and the test compound resulting in the same effect on ferric reducing activity.
3.2.4.3. Tissue preparation
The work was carried out in accordance with the EU Directive 2010/63/EU. Hybrid BDF1 mice (about 6 months old) were sacrificed by decapitation. Each brain was rapidly excised, frozen in liquid nitrogen, and stored at –80°C until use. The brains were thawed and homogenized using a Wisd WiseTis HG-15D homogenizer (Daihan Scientific, Wonju, South Korea)for 2 min in a buffer (0.01 M PBS, pH 7.4 or 0.1 M Tris-HCl, pH 7.4). Protein concentrations were determined by the Lowry method [81].
3.2.4.4. Luminol chemiluminescence assay of radical-scavenging activity of conjugates in mouse brain homogenate
Luminol-dependent chemiluminescence produced by mouse brain homogenates was measured using the Luminometer 1250 (LKB Wallac, Turku, Finland) according to the assay by Di Meo et al. [53] with minor modifications as described in detail in [44]. The reaction mixture consisted of mouse brain homogenate (protein concentration 0.1 mg/mL) in 0.1 M Tris-HCl (Sigma-Aldrich), pH 7.4, luminol (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) (0.05 mmol/L), tert-butyl hydroperoxide (TBHP) (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) (0.073 mol/L) and tested compounds in a concentration range of 0.01-100 μМ. Compounds were dissolved in DMSO (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany). IC50 values represent the concentration that caused 50% reduction of luminescence.
3.2.4.5. TBARS Assay of the effect of compounds on spontaneous LP
The procedure was performed in accordance with the method of Ohkawa et al. [54] with minor modifications as described in detail in [44].
Briefly, mouse brain homogenate (protein concentration 1 mg/mL) in PBS (0.1 M, pH 7.4) was incubated for 30 min at 37°C with tested conjugates, and the reaction was terminated by addition of 0.4 mL of 17% (w/v) trichloroacetic acid. Following centrifugation for 20 min at 1300×g, 0.5 mL of 0.8% (w/v) of TBA (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) was added to 1 mL of supernatant, heated for 30 min at 95°C and then cooled to RT.
The optical density of the TBARS, which corresponds to the produced MDA, was measured at 532 nm against a blank using an Agilent Cary 60 UV-Vis spectrophotometer (Agilent, Santa Clara, United States). Trolox and BHT were used as reference antioxidants. Compounds were dissolved in DMSO (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) and tested in a concentration range 0.01-100 μМ. IC50 values represent the concentration that caused 50% inhibition of LP.
3.3. Molecular Modeling Studies
3.3.1. Molecular docking
The Calculator Plugins of MarvinSketch 21.14.0, ChemAxon (https://www.chemaxon.com, accessed on 27 January 2023) and MolGpKa [82] (https://xundrug.cn/molgpka, accessed on 27 January 2023) were used to estimate the pKa values of the ligands. The ligand compounds were optimized using a DFT quantum chemistry method (B3LYP/6-31G*, GAMESS-US [83] software). For molecular docking, the optimized structures of the ligands were employed, with partial atomic charges obtained from QM results based on the Löwdin scheme [84].
The protein targets used for docking included X-ray structures of human AChE co-crystallized with donepezil (PDB: 4EY7, chain A) [85], and an optimized X-ray structure of human BChE (PDB: 1P0I) [86,87]. For modeling of conformational flexibility, all conformers in the NMR structure of the soluble α-helical form of Aβ42 PDB ID 1IYT [88] were used.
AutoDock 4.2.6 software [89] was applied to perform molecular docking. The docking grid box was set to cover the entire active site gorge of AChE (22.5 Å×22.5 Å × 22.5 Å) and BChE (15 Å × 20.25 Å × 18 Å), as well as the entire Aβ42 molecule for all conformers (43.5 Å × 28.5 Å × 54.75 Å), with a grid spacing of 0.375 Å used in all cases. The Lamarckian Genetic Algorithm (LGA) [90] was used with 256 runs, 25×106 evaluations, 27×104 generations, and a population size of 3000. Figures were created using PyMol (www.pymol.org, accessed on 21 July 2016).
3.3.2. QM calculation of antioxidant activity
The quantum chemical calculations were performed by a density-functional theory (DFT) method using Gaussian 16 [91] and Priroda 19 [92,93] packages. The Priroda 19 package was used for a preliminary conformational search in the gas phase with the PBE0 functional [94] and TZVP basis set [95]. The lowest-energy conformations were used as initial geometries for optimization in solvent (water or ethanol) by the Gaussian 16 package. The optimization was done using the B3LYP functional [96,97] and 6-31++G(d,p) basis set [98] with the empirical Grimme correction DFT-D3BJ [99]. The solvent effects were taken into account using the SMD continuum solvation model [100].
3.3.3. Prediction of ADMET, Physicochemical, and PAINS Profiles
Lipophilicity (LogPow) and aqueous solubility (pSaq) were estimated by the ALogPS 3.0 neural network model implemented in the OCHEM platform [101]. Human intestinal absorption (HIA) [102], blood–brain barrier distribution/permeability (LogBB) [103,104], and hERG-mediated cardiac toxicity risk (channel affinity pKi and inhibitory activity pIC50) [105] were estimated using the integrated online service for the prediction of ADMET properties [106]This service implements predictive QSAR models based on accurate and representative training sets, fragmental descriptors, and artificial neural networks. The quantitative estimate of drug-likeness (QED) values [107] were calculated and the Pan Assay INterference compoundS (PAINS) alerts were checked using RDKit version 2021.09.2 software[108]
3.4. Statistical Analyses
All tests were performed at least in triplicate in three independent experiments. Results are presented as mean ± SEM calculated using GraphPad Prism version 6.05 for Windows (San Diego CA, USA). Plots, linear regressions, and IC50 values were determined using Origin 6.1 for Windows, OriginLab (Northampton, MA, USA).
4. Conclusions
In summary, first, we synthesized two series of new conjugates of 4-amino-2,3-polymethylenequinolines and butylated hydroxytoluene that were linked together with alkylimine and alkylamine spacers. In contrast to our previous studies [44], the present work employed spacers of variable length. Next, we assessed biological activities of the new compounds with respect to their potential as multi-target agents for AD treatment. These assessments included new or more detailed biological characterizations than were conducted in our previous investigations and revealed some substantial improvements in the anticholinesterase properties of the conjugates.
All new conjugates were potent inhibitors of AChE and BChE with selectivity toward BChE. Conjugates very weakly inhibited the off-target CES indicating the likely absence of certain unwanted drug-drug interactions in clinical applications.
Conjugates with an alkylamine spacer (16–18) were more effective at inhibiting AChE and BChE than their alkylimine analogs (13–15). Maximum inhibition for AChE was achieved by compounds with a cyclohexaquinoline ring and for BChE by compounds with cycloheptaquinoline ring. An increase in the length of the spacer resulted in a significant increase in both anti-AChE and anti-BChE activity.
Consequently, conjugates 14d and 17d showed the maximum inhibitory activity against AChE (IC50=0.0171±0.0016 and 0.0151±0.002 μM), being 40 times more active than tacrine. Conjugates 15d and 18d exhibited the maximum anti-BChE activity with IC50 values of about 6 nM, 5 times more active than tacrine. It is also noteworthy that anticholinesterase potencies achieved by compounds in the present study were markedly improved over those from the previous investigation. For example, with respect to AChE inhibition, conjugate 14d from the current work was 350 times more potent than compound 14a (corresponding to compound 7b from the previous study).
Patterns of structure-activity relationships were in full agreement with the results of molecular docking.
Kinetics revealed mixed-type reversible inhibition of AChE and BChE by representative conjugates and molecular docking results indicated dual binding to the CAS and PAS of AChE. These results, along with experimental data on propidium iodide displacement, suggest their potential to block AChE-induced β-amyloid aggregation.
Conjugates also demonstrated the ability to block β-amyloid self-aggregation; compounds 14d and 17d with a hexaquinoline ring (m = 2) and spacer n = 8 were the most active, which agrees with the results of molecular docking to Aβ42 for conjugate 17d.
High radical-scavenging activity was exhibited by the compounds in the ABTS test, Conjugates with an alkylamine spacer were somewhat more active than the alkylimine analogs. Maximum activity (TEAC=1.2-1.5) was demonstrated by conjugates 16c, 17b, 17d, 18b, 18d. In the FRAP assay, conjugates 13-15 and 16-18 had a high iron-reducing ability, which, however, was somewhat lower (TE=0.4-0.78) than the activity of BHT. In general, conjugates with an alkylimine spacer were somewhat more active than their alkylamine analogs.
In a biological system consisting of a mouse brain homogenate, the conjugates demonstrated high antioxidant activity. Conjugates with imine spacers were 3-6 times more active than their amine analogs and much more active than BHT when assessed by the CL assay. In contrast, in the spontaneous LP assay, the AOA of imines was somewhat lower than that of amines, and much lower than BHT activity.
Quantum-chemical calculations served to explain the variety of results obtained for the conjugates in various systems for assessing antioxidant activity.
Computed ADMET profiles of the conjugates showed high predicted values for intestinal absorption, enabling their oral administration and favorable blood–brain barrier permeability suggesting the potential for CNS activity. Thus, pending further development and optimization, the conjugates could be considered promising multifunctional CNS agents for potential treatment of AD.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1: Figures S1-S33: 1H and 13C spectra of compounds 13b-d, 14b-d, 15b-d, 16b-d, 17a-d, 18b-d; Quantum-Chemical Calculations of AOA.
Author Contributions
All authors contributed to the study conception and design. Conceptualization, G.F.M.; S.O.B.; methodology, G.F.M., I.V.S., S.V.L., T.Y.A.; synthesis, A.N.P., I.V.S.; validation, G.F.M., V.A.P., and R.J.R.; formal analysis, investigation, N.V.K., E.V.Ru., N.P.B., S.V.L., T.Y.A., E.N.T., Y.V.S., D.A.P., V.A.M., E.V.R.; writing—original draft preparation, N.V.K., E.V.Ru., N.P.B., S.V.L., I.I.F., T.Y.A., A.N.P., I.V.S., E.V.R., A.A.T.; writing—review and editing, G.F.M., N.V.K., S.V.L., N.P.B., E.V.Ru., T.Y.A., V.A.P., R.J.R.; supervision, G.F.M., R.J.R., S.O.B.; project administration, G.F.M., S.O.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Russian Ministry of Science and Higher Education grant #075-15-2020-777.
Institutional Review Board Statement
All animal procedures for preparing rat brain homogenate were approved by the Bioethics Committee of IPAC RAS (Approval No. 41, 26 November 2019).
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank the Centre for Collective Use of IPAC RAS (IPAC research topic FFSN-2021-0005) and the Medicinal Chemistry Research and Education Center of RAS (research topic AAAAA19-119071890015-6) for using the equipment cited in the Methods. E.N.T. and T.Y.A. thank the Joint Supercomputer Center of RAS (JSCC) for supercomputer time (IBCP RAS research topic 122041400110-4).
Conflicts of Interest
RJR currently serves as a member of the advisory board of NeuroX1, a startup biotech company that is developing a software platform for the discovery and development of drugs for neurodegenerative diseases. All other authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available.
References
- Huang, Y.; Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef]
- Tahami Monfared, A.A.; Byrnes, M.J.; White, L.A.; Zhang, Q. Alzheimer's Disease: Epidemiology and Clinical Progression. Neurol Ther 2022, 11, 553–569. [Google Scholar] [CrossRef]
- 2020 Alzheimer's disease facts and figures. Alzheimers Dement. 2020, 16, 391–460. [CrossRef]
- Weller, J.; Budson, A. Current understanding of Alzheimer's disease diagnosis and treatment. F1000Res 2018, 7. [Google Scholar] [CrossRef]
- Carreiras, M.C.; Mendes, E.; Perry, M.J.; Francisco, A.P.; Marco-Contelles, J. The multifactorial nature of Alzheimer's disease for developing potential therapeutics. Curr. Top. Med. Chem. 2013, 13, 1745–1770. [Google Scholar] [CrossRef]
- Rossi, M.; Freschi, M.; de Camargo Nascente, L.; Salerno, A.; de Melo Viana Teixeira, S.; Nachon, F.; Chantegreil, F.; Soukup, O.; Prchal, L.; Malaguti, M.; et al. Sustainable Drug Discovery of Multi-Target-Directed Ligands for Alzheimer's Disease. J. Med. Chem. 2021, 64, 4972–4990. [Google Scholar] [CrossRef]
- Blaikie, L.; Kay, G.; Kong Thoo Lin, P. Current and emerging therapeutic targets of alzheimer's disease for the design of multi-target directed ligands. MedChemComm 2019, 10, 2052–2072. [Google Scholar] [CrossRef]
- Blaszczyk, J.W. Pathogenesis of Dementia. Int. J. Mol. Sci. 2022, 24. [Google Scholar] [CrossRef]
- Ballard, C.; Greig, N.; Guillozet-Bongaarts, A.; Enz, A.; Darvesh, S. Cholinesterases: Roles in the Brain During Health and Disease. Curr. Alzheimer Res. 2005, 2, 307–318. [Google Scholar] [CrossRef]
- Agatonovic-Kustrin, S.; Kettle, C.; Morton, D.W. A molecular approach in drug development for Alzheimer's disease. Biomed. Pharmacother. 2018, 106, 553–565. [Google Scholar] [CrossRef]
- Moreta, M.P.; Burgos-Alonso, N.; Torrecilla, M.; Marco-Contelles, J.; Bruzos-Cidón, C. Efficacy of Acetylcholinesterase Inhibitors on Cognitive Function in Alzheimer's Disease. Review of Reviews. Biomedicines 2021, 9, 1689. [Google Scholar] [CrossRef]
- McGleenon, B.M.; Dynan, K.B.; Passmore, A.P. Acetylcholinesterase inhibitors in Alzheimer's disease. Br. J. Clin. Pharmacol. 1999, 48, 471–480. [Google Scholar] [CrossRef]
- Ruangritchankul, S.; Chantharit, P.; Srisuma, S.; Gray, L.C. Adverse Drug Reactions of Acetylcholinesterase Inhibitors in Older People Living with Dementia: A Comprehensive Literature Review. Ther. Clin. Risk. Manag. 2021, 17, 927–949. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of multifunctional molecules as potential therapeutic candidates for Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis in the last decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef]
- Pohanka, M. Oxidative stress in Alzheimer disease as a target for therapy. Bratisl. Lek. Listy 2018, 119, 535–543. [Google Scholar] [CrossRef]
- Cassidy, L.; Fernandez, F.; Johnson, J.B.; Naiker, M.; Owoola, A.G.; Broszczak, D.A. Oxidative stress in alzheimer’s disease: A review on emergent natural polyphenolic therapeutics. Complement Ther. Med. 2020, 49, 102294. [Google Scholar] [CrossRef]
- Moosmann, B.; Behl, C. Antioxidants as treatment for neurodegenerative disorders. Expert Opin Investig Drugs 2002, 11, 1407–1435. [Google Scholar] [CrossRef]
- Olufunmilayo, E.O.; Gerke-Duncan, M.B.; Holsinger, R.M.D. Oxidative Stress and Antioxidants in Neurodegenerative Disorders. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Hardy, J.; Bogdanovic, N.; Winblad, B.; Portelius, E.; Andreasen, N.; Cedazo-Minguez, A.; Zetterberg, H. Pathways to Alzheimer's disease. J. Intern. Med. 2014, 275, 296–303. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Nie, Q.; Du, X.G.; Geng, M.Y. Small molecule inhibitors of amyloid beta peptide aggregation as a potential therapeutic strategy for Alzheimer's disease. Acta Pharmacol. Sin. 2011, 32, 545–551. [Google Scholar] [CrossRef]
- Jeremic, D.; Jimenez-Diaz, L.; Navarro-Lopez, J.D. Past, present and future of therapeutic strategies against amyloid-beta peptides in Alzheimer's disease: a systematic review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Canales, M.A.; Shin, I.; Weiner, L.M.; Silman, I.; Inestrosa, N.C. A structural motif of acetylcholinesterase that promotes amyloid beta-peptide fibril formation. Biochemistry 2001, 40, 10447–10457. [Google Scholar] [CrossRef]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. β-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions. Implications for Alzheimer's disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef]
- Lushchekina, S.V.; Kots, E.D.; Novichkova, D.A.; Petrov, K.A.; Masson, P. Role of Acetylcholinesterase in β-Amyloid Aggregation Studied by Accelerated Molecular Dynamics. BioNanoScience 2017, 7, 396–402. [Google Scholar] [CrossRef]
- Muñoz-Ruiz, P.; Rubio, L.; García-Palomero, E.; Dorronsoro, I.; del Monte-Millán, M.; Valenzuela, R.; Usán, P.; de Austria, C.; Bartolini, M.; Andrisano, V.; et al. Design, Synthesis, and Biological Evaluation of Dual Binding Site Acetylcholinesterase Inhibitors: New Disease-Modifying Agents for Alzheimer's Disease. J. Med. Chem. 2005, 48, 7223–7233. [Google Scholar] [CrossRef]
- Camps, P.; Formosa, X.; Galdeano, C.; Gomez, T.; Munoz-Torrero, D.; Ramirez, L.; Viayna, E.; Gomez, E.; Isambert, N.; Lavilla, R.; et al. Tacrine-based dual binding site acetylcholinesterase inhibitors as potential disease-modifying anti-Alzheimer drug candidates. Chem. Biol. Interact. 2010, 187, 411–415. [Google Scholar] [CrossRef]
- Zueva, I.; Dias, J.; Lushchekina, S.; Semenov, V.; Mukhamedyarov, M.; Pashirova, T.; Babaev, V.; Nachon, F.; Petrova, N.; Nurullin, L.; et al. New evidence for dual binding site inhibitors of acetylcholinesterase as improved drugs for treatment of Alzheimer's disease. Neuropharmacology 2019, 155, 131–141. [Google Scholar] [CrossRef]
- Ramanan, V.K.; Risacher, S.L.; Nho, K.; Kim, S.; Swaminathan, S.; Shen, L.; Foroud, T.M.; Hakonarson, H.; Huentelman, M.J.; Aisen, P.S.; et al. APOE and BCHE as modulators of cerebral amyloid deposition: a florbetapir PET genome-wide association study. Mol. Psychiatry 2014, 19, 351–357. [Google Scholar] [CrossRef]
- Darvesh, S. Butyrylcholinesterase as a diagnostic and therapeutic target for Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 1173–1177. [Google Scholar] [CrossRef]
- Przybylowska, M.; Dzierzbicka, K.; Kowalski, S.; Chmielewska, K.; Inkielewicz-Stepniak, I. Therapeutic Potential of Multifunctional Derivatives of Cholinesterase Inhibitors. Curr. Neuropharmacol. 2021, 19, 1323–1344. [Google Scholar] [CrossRef]
- Spilovska, K.; Korabecny, J.; Nepovimova, E.; Dolezal, R.; Mezeiova, E.; Soukup, O.; Kuca, K. Multitarget tacrine hybrids with neuroprotective properties to confront Alzheimer's disease. Curr. Top. Med. Chem. 2017, 17, 1006–1026. [Google Scholar] [CrossRef]
- Sameem, B.; Saeedi, M.; Mahdavi, M.; Shafiee, A. A review on tacrine-based scaffolds as multi-target drugs (MTDLs) for Alzheimer's disease. Eur. J. Med. Chem. 2017, 128, 332–345. [Google Scholar] [CrossRef]
- Przybylowska, M.; Kowalski, S.; Dzierzbicka, K.; Inkielewicz-Stepniak, I. Therapeutic Potential of Multifunctional Tacrine Analogues. Curr. Neuropharmacol. 2019, 17, 472–490. [Google Scholar] [CrossRef]
- Bubley, A.; Erofeev, A.; Gorelkin, P.; Beloglazkina, E.; Majouga, A.; Krasnovskaya, O. Tacrine-Based Hybrids: Past, Present, and Future. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Boltneva, N.P.; Lushchekina, S.V.; Astakhova, T.Y.; Rudakova, E.V.; Proshin, A.N.; Serkov, I.V.; Radchenko, E.V.; Palyulin, V.A.; et al. New Hybrids of 4-Amino-2,3-polymethylene-quinoline and p-Tolylsulfonamide as Dual Inhibitors of Acetyl- and Butyrylcholinesterase and Potential Multifunctional Agents for Alzheimer's Disease Treatment. Molecules 2020, 25, 3915. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Rudakova, E.V.; Boltneva, N.P.; Grishchenko, M.V.; Lushchekina, S.V.; Astakhova, T.Y.; Serebryakova, O.G.; Timokhina, E.N.; Zhilina, E.F.; et al. Conjugates of Tacrine and Salicylic Acid Derivatives as New Promising Multitarget Agents for Alzheimer's Disease. Int. J. Mol. Sci. 2023, 24, 2285. [Google Scholar] [CrossRef]
- Pi, R.; Mao, X.; Chao, X.; Cheng, Z.; Liu, M.; Duan, X.; Ye, M.; Chen, X.; Mei, Z.; Liu, P.; et al. Tacrine-6-ferulic acid, a novel multifunctional dimer, inhibits amyloid-beta-mediated Alzheimer's disease-associated pathogenesis in vitro and in vivo. PLoS One 2012, 7, e31921. [Google Scholar] [CrossRef]
- Nepovimova, E.; Korabecny, J.; Dolezal, R.; Babkova, K.; Ondrejicek, A.; Jun, D.; Sepsova, V.; Horova, A.; Hrabinova, M.; Soukup, O.; et al. Tacrine-Trolox Hybrids: A Novel Class of Centrally Active, Nonhepatotoxic Multi-Target-Directed Ligands Exerting Anticholinesterase and Antioxidant Activities with Low In Vivo Toxicity. J. Med. Chem. 2015, 58, 8985–9003. [Google Scholar] [CrossRef] [PubMed]
- Scipioni, M.; Kay, G.; Megson, I.L.; Kong Thoo Lin, P. Synthesis of novel vanillin derivatives: novel multi-targeted scaffold ligands against Alzheimer's disease. MedChemComm 2019, 10, 764–777. [Google Scholar] [CrossRef]
- Roldan-Pena, J.M.; Romero-Real, V.; Hicke, J.; Maya, I.; Franconetti, A.; Lagunes, I.; Padron, J.M.; Petralla, S.; Poeta, E.; Naldi, M.; et al. Tacrine-O-protected phenolics heterodimers as multitarget-directed ligands against Alzheimer's disease: Selective subnanomolar BuChE inhibitors. Eur. J. Med. Chem. 2019, 181, 111550. [Google Scholar] [CrossRef]
- Serkov, I.V.; Proshin, A.N.; Kovaleva, N.V.; Boltneva, N.P.; Rudakova, E.V.; Makhaeva, G.F.; Bachurin, S.O. Synthesis and properties of new derivatives of 4-amino-2,3-polymethylenequinolines with antioxidant function. Russ. Chem. Bull. 2023, 72, 802–806. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Rudakova, E.V.; Boltneva, N.P.; Lushchekina, S.V.; Faingold, II; Poletaeva, D.A.; Soldatova, Y.V.; Kotelnikova, R.A.; Serkov, I.V.; et al. New Multifunctional Agents Based on Conjugates of 4-Amino-2,3-polymethylenequinoline and Butylated Hydroxytoluene for Alzheimer's Disease Treatment. Molecules 2020, 25, 5891. [Google Scholar] [CrossRef]
- Carlier, P.R.; Han, Y.F.; Chow, E.S.; Li, C.P.; Wang, H.; Lieu, T.X.; Wong, H.S.; Pang, Y.P. Evaluation of short-tether bis-THA AChE inhibitors. A further test of the dual binding site hypothesis. Bioorg. Med. Chem. 1999, 7, 351–357. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Rudakova, E.V.; Serebryakova, O.G.; Aksinenko, A.Y.; Lushchekina, S.V.; Bachurin, S.O.; Richardson, R.J. Esterase profiles of organophosphorus compounds in vitro predict their behavior in vivo. Chem. Biol. Interact. 2016, 259, 332–342. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Rudakova, E.V.; Kovaleva, N.V.; Lushchekina, S.V.; Boltneva, N.P.; Proshin, A.N.; Shchegolkov, E.V.; Burgart, Y.V.; Saloutin, V.I. Cholinesterase and carboxylesterase inhibitors as pharmacological agents. Russ. Chem. Bull. 2019, 68, 967–984. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Boltneva, N.P.; Lushchekina, S.V.; Rudakova, E.V.; Stupina, T.S.; Terentiev, A.A.; Serkov, I.V.; Proshin, A.N.; Radchenko, E.V.; et al. Conjugates of tacrine and 1,2,4-thiadiazole derivatives as new potential multifunctional agents for Alzheimer's disease treatment: Synthesis, quantum-chemical characterization, molecular docking, and biological evaluation. Bioorg. Chem. 2020, 94, 103387. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Proshin, A.N.; Kovaleva, N.V.; Rudakova, E.V.; Boltneva, N.P.; Lushchekina, S.V.; Astakhova, T.Y.; Serkov, I.V.; Kalashnikova, I.P.; Bachurin, S.O. Synthesis and study of the biological activity of thiourea-containing amiridine derivatives as potential multi-target drugs for the treatment of Alzheimer’s disease. Russ. Chem. Bull. 2022, 71, 2404–2415. [Google Scholar] [CrossRef]
- Jokar, S.; Erfani, M.; Bavi, O.; Khazaei, S.; Sharifzadeh, M.; Hajiramezanali, M.; Beiki, D.; Shamloo, A. Design of peptide-based inhibitor agent against amyloid-beta aggregation: Molecular docking, synthesis and in vitro evaluation. Bioorg. Chem. 2020, 102, 104050. [Google Scholar] [CrossRef]
- Schaich, K.M.; Tian, X.; Xie, J. Hurdles and pitfalls in measuring antioxidant efficacy: A critical evaluation of ABTS, DPPH, and ORAC assays. J. Funct. Foods 2015, 14, 111–125. [Google Scholar] [CrossRef]
- Tian, X.; Schaich, K.M. Effects of molecular structure on kinetics and dynamics of the trolox equivalent antioxidant capacity assay with ABTS(+*). J. Agric. Food. Chem. 2013, 61, 5511–5519. [Google Scholar] [CrossRef]
- Di Meo, S.; Venditti, P.; Piro, M.C.; De Leo, T. Enhanced luminescence study of liver homogenate response to oxidative stress. Arch. Physiol. Biochem. 1995, 103, 187–195. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Baj, A.; Cedrowski, J.; Olchowik-Grabarek, E.; Ratkiewicz, A.; Witkowski, S. Synthesis, DFT Calculations, and In Vitro Antioxidant Study on Novel Carba-Analogs of Vitamin E. Antioxidants (Basel) 2019, 8. [Google Scholar] [CrossRef]
- Xue, Y.; Zheng, Y.; An, L.; Dou, Y.; Liu, Y. Density functional theory study of the structure-antioxidant activity of polyphenolic deoxybenzoins. Food Chem. 2014, 151, 198–206. [Google Scholar] [CrossRef]
- Hossen, J.; Pal, T.K.; Hasan, T. Theoretical investigations on the antioxidant potential of 2,4,5-trihydroxybutyrophenone in different solvents: A DFT approach. Results Chem. 2022, 4. [Google Scholar] [CrossRef]
- Marković, Z.; Tošović, J.; Milenković, D.; Marković, S. Revisiting the solvation enthalpies and free energies of the proton and electron in various solvents. Comput. Theor. Chem. 2016, 1077, 11–17. [Google Scholar] [CrossRef]
- Bedouhène, S.; Moulti-Mati, F.; Hurtado-Nedelec, M.; My-Chan Dang, P.; El-Benna, J. Luminol-amplified chemiluminescence detects mainly superoxide anion produced by human neutrophils. Am. J. Blood Res. 2017, 7, 41–48. [Google Scholar]
- Villaverde, A.; Netherton, J.; Baker, M.A. From Past to Present: The Link Between Reactive Oxygen Species in Sperm and Male Infertility. Antioxidants (Basel) 2019, 8. [Google Scholar] [CrossRef]
- Kudin, A.P.; Malinska, D.; Kunz, W.S. Sites of generation of reactive oxygen species in homogenates of brain tissue determined with the use of respiratory substrates and inhibitors. Biochim. Biophys. Acta 2008, 1777, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Raghuvanshi, R.S. Superoxide Anion Radical, A Multipotent Reagent: A Review. Nat. Volatiles & Essent. Oils 2020, 7, 75–82. [Google Scholar]
- Galano, A.; Vargas, R.; Martinez, A. Carotenoids can act as antioxidants by oxidizing the superoxide radical anion. Phys. Chem. Chem. Phys. 2010, 12, 193–200. [Google Scholar] [CrossRef]
- Ahmed, S.; Shakeel, F. Antioxidant activity coefficient, mechanism, and kinetics of different derivatives of flavones and flavanones towards superoxide radical. Czech J. Food Sci. 2012, 30, 153–163. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, R.; Yan, X.; Fan, K. Superoxide dismutase nanozymes: an emerging star for anti-oxidation. J. Mater. Chem. B 2021, 9, 6939–6957. [Google Scholar] [CrossRef]
- Tahan, A.; Shiroudi, A. Oxidation reaction mechanism and kinetics between OH radicals and alkyl-substituted aliphatic thiols: H-abstraction pathways. Prog. React. Kinet. Mech. 2020, 45, 1–21. [Google Scholar] [CrossRef]
- Yadav, A.; Mishra, P.C. Modeling the activity of glutathione as a hydroxyl radical scavenger considering its neutral non-zwitterionic form. J. Mol. Model. 2013, 19, 767–777. [Google Scholar] [CrossRef]
- Mitroka, S.; Zimmeck, S.; Troya, D.; Tanko, J.M. How solvent modulates hydroxyl radical reactivity in hydrogen atom abstractions. J. Am. Chem. Soc. 2010, 132, 2907–2913. [Google Scholar] [CrossRef]
- Valgimigli, L. Lipid Peroxidation and Antioxidant Protection. Biomolecules 2023, 13, 1291. [Google Scholar] [CrossRef]
- Cacique, A.P.; Barbosa, É.S.; de Pinho, G.P.; Silvério, F.O. Miniaturized Methodologies for Determining the Total Phenol and Flavonoid Concentrations and the Antioxidant Activity. Food Anal. Methods 2021, 14, 1110–1120. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Kochi, J.K. Continuum of outer- and inner-sphere mechanisms for organic electron transfer. Steric modulation of the precursor complex in paramagnetic (ion-radical) self-exchanges. J. Am. Chem. Soc. 2007, 129, 3683–3697. [Google Scholar] [CrossRef]
- Cheng, T.; Shen, D.X.; Meng, M.; Mallick, S.; Cao, L.; Patmore, N.J.; Zhang, H.L.; Zou, S.F.; Chen, H.W.; Qin, Y.; et al. Efficient electron transfer across hydrogen bond interfaces by proton-coupled and -uncoupled pathways. Nat. Commun. 2019, 10, 1531. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Lushchekina, S.V.; Boltneva, N.P.; Serebryakova, O.G.; Rudakova, E.V.; Ustyugov, A.A.; Bachurin, S.O.; Shchepochkin, A.V.; Chupakhin, O.N.; Charushin, V.N.; et al. 9-Substituted acridine derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors possessing antioxidant activity for Alzheimer's disease treatment. Bioorg. Med. Chem. 2017, 25, 5981–5994. [Google Scholar] [CrossRef]
- Taylor, P.; Lappi, S. Interaction of fluorescence probes with acetylcholinesterase. Site and specificity of propidium binding. Biochemistry 1975, 14, 1989–1997. [Google Scholar] [CrossRef]
- Taylor, P.; Lwebuga-Mukasa, J.; Lappi, S.; Rademacher, J. Propidium—a fluorescence probe for a peripheral anionic site on acetylcholinesterase. Mol. Pharmacol. 1974, 10, 703–708. [Google Scholar]
- LeVine, H. 3rd. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999, 309, 274–284. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Elkina, N.A.; Shchegolkov, E.V.; Boltneva, N.P.; Lushchekina, S.V.; Serebryakova, O.G.; Rudakova, E.V.; Kovaleva, N.V.; Radchenko, E.V.; Palyulin, V.A.; et al. Synthesis, molecular docking, and biological evaluation of 3-oxo-2-tolylhydrazinylidene-4,4,4-trifluorobutanoates bearing higher and natural alcohol moieties as new selective carboxylesterase inhibitors. Bioorg. Chem. 2019, 91, 103097. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of "antioxidant power": the FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. Ferric reducing/antioxidant power assay: direct measure of total antioxidant activity of biological fluids and modified version for simultaneous measurement of total antioxidant power and ascorbic acid concentration. Methods Enzymol. 1999, 299, 15–27. [Google Scholar] [CrossRef]
- Waterborg, J.H. The Lowry Method for Protein Quantitation. in Walker J.M. (eds) The Protein Protocols Handbook. Springer Protocols Handbooks. Humana Press. 2002, pp, 7-10. [CrossRef]
- Pan, X.; Wang, H.; Li, C.; Zhang, J.Z.H.; Ji, C. MolGpka: A Web Server for Small Molecule pKa Prediction Using a Graph-Convolutional Neural Network. J. Chem. Inf. Model. 2021, 61, 3159–3165. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comp. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Löwdin, P.-O. On the nonorthogonality problem. In Advances in Quantum Chemistry, Per-Olov, L., Ed. Academic Press: New York, London, 1970; Vol. Volume 5, pp. 185-199.
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Lushchekina, S.; Schopfer, L.M.; Lockridge, O. Effects of viscosity and osmotic stress on the reaction of human butyrylcholinesterase with cresyl saligenin phosphate, a toxicant related to aerotoxic syndrome: kinetic and molecular dynamics studies. Biochem. J. 2013, 454, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D'Ursi, A.M.; Temussi, P.A.; Picone, D. Solution structure of the Alzheimer amyloid beta-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur. J. Biochem. 2002, 269, 5642–5648. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comp. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comp. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16 Revision C. 01. 2016, Gaussian Inc.: Wallingford, CT, USA: 2016.
- Laikov, D.N.; Ustynyuk, Y.A. PRIRODA-04: a quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing. Russ. Chem. Bull. 2005, 54, 820–826. [Google Scholar] [CrossRef]
- Laikov, D.N. PRIRODA. Electronic Structure Code, 19; 2020.
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 2002, 98, 11623–11627. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comp. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- ACS Chem NeurosciActa NeuropatholMarenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Korner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y.; et al. Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information. J. Comput. Aided Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef] [PubMed]
- Radchenko, E.V.; Dyabina, A.S.; Palyulin, V.A.; Zefirov, N.S. Prediction of human intestinal absorption of drug compounds. Russ. Chem. Bull. 2016, 65, 576–580. [Google Scholar] [CrossRef]
- Dyabina, A.S.; Radchenko, E.V.; Palyulin, V.A.; Zefirov, N.S. Prediction of blood-brain barrier permeability of organic compounds. Dokl. Biochem. Biophys. 2016, 470, 371–374. [Google Scholar] [CrossRef]
- Radchenko, E.V.; Dyabina, A.S.; Palyulin, V.A. Towards Deep Neural Network Models for the Prediction of the Blood-Brain Barrier Permeability for Diverse Organic Compounds. Molecules 2020, 25, 5901. [Google Scholar] [CrossRef]
- Radchenko, E.V.; Rulev, Y.A.; Safanyaev, A.Y.; Palyulin, V.A.; Zefirov, N.S. Computer-aided estimation of the hERG-mediated cardiotoxicity risk of potential drug components. Dokl. Biochem. Biophys. 2017, 473, 128–131. [Google Scholar] [CrossRef] [PubMed]
- ADMET Prediction Service. http://qsar.chem.msu.ru/admet/ (accessed on 15 June 2022).
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- RDKit: open-source cheminformatics software. Available online: http://www.rdkit.org (accessed on 15 June 2022).
Scheme 1.
Synthesis of conjugates 13-15 and 16-18 showing variations in both ring size and spacer length.
Scheme 1.
Synthesis of conjugates 13-15 and 16-18 showing variations in both ring size and spacer length.
Figure 1.
Steady state inhibition of AChE (A) and BChE (B) by compound 15d.
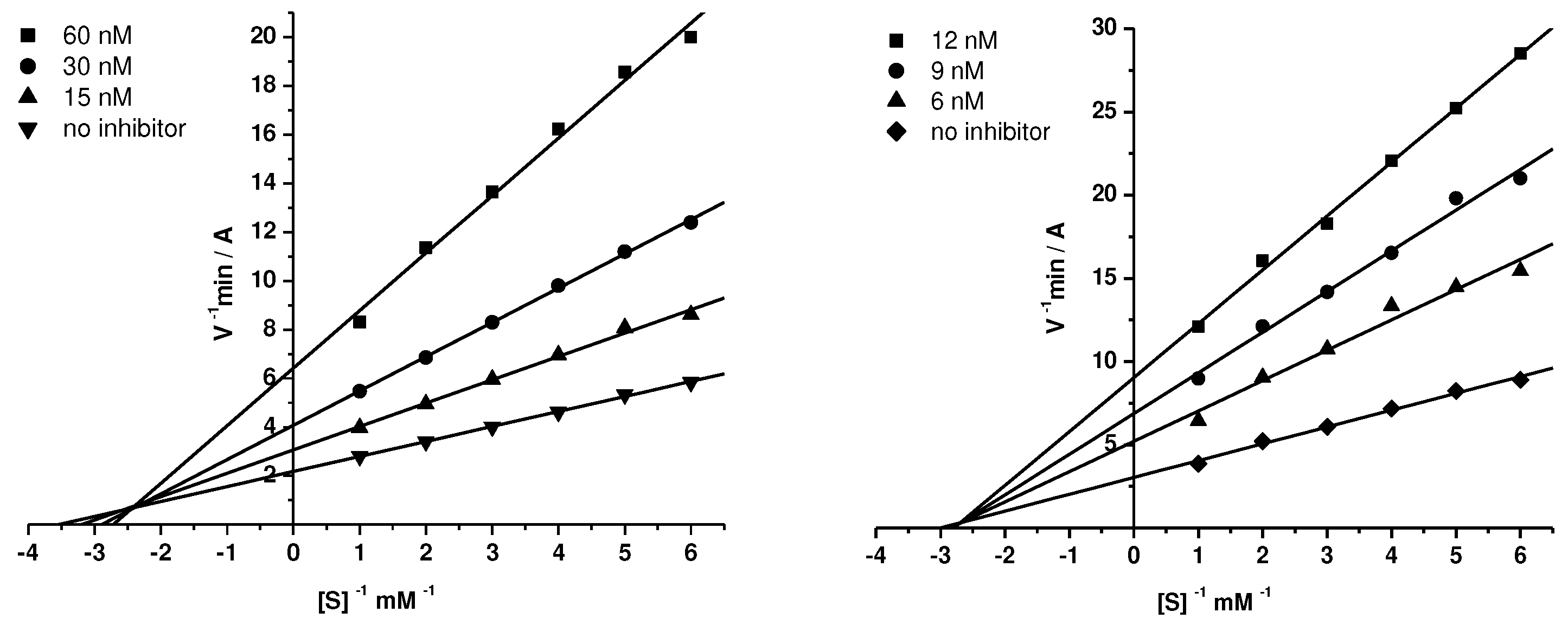
Figure 2.
Estimated binding affinities from molecular docking to AChE of alkylimine (13-15) compounds with protonated tacrine group (A), and both tacrine and imine groups protonated (B) in comparison with results for alkylamine (16-18) derivatives with both tacrine and the secondary amine groups protonated docked to AChE (C) and BChE (D). Plots A-C have the same energy scale, which differ from the energy scale for BChE in panel D.
Figure 2.
Estimated binding affinities from molecular docking to AChE of alkylimine (13-15) compounds with protonated tacrine group (A), and both tacrine and imine groups protonated (B) in comparison with results for alkylamine (16-18) derivatives with both tacrine and the secondary amine groups protonated docked to AChE (C) and BChE (D). Plots A-C have the same energy scale, which differ from the energy scale for BChE in panel D.
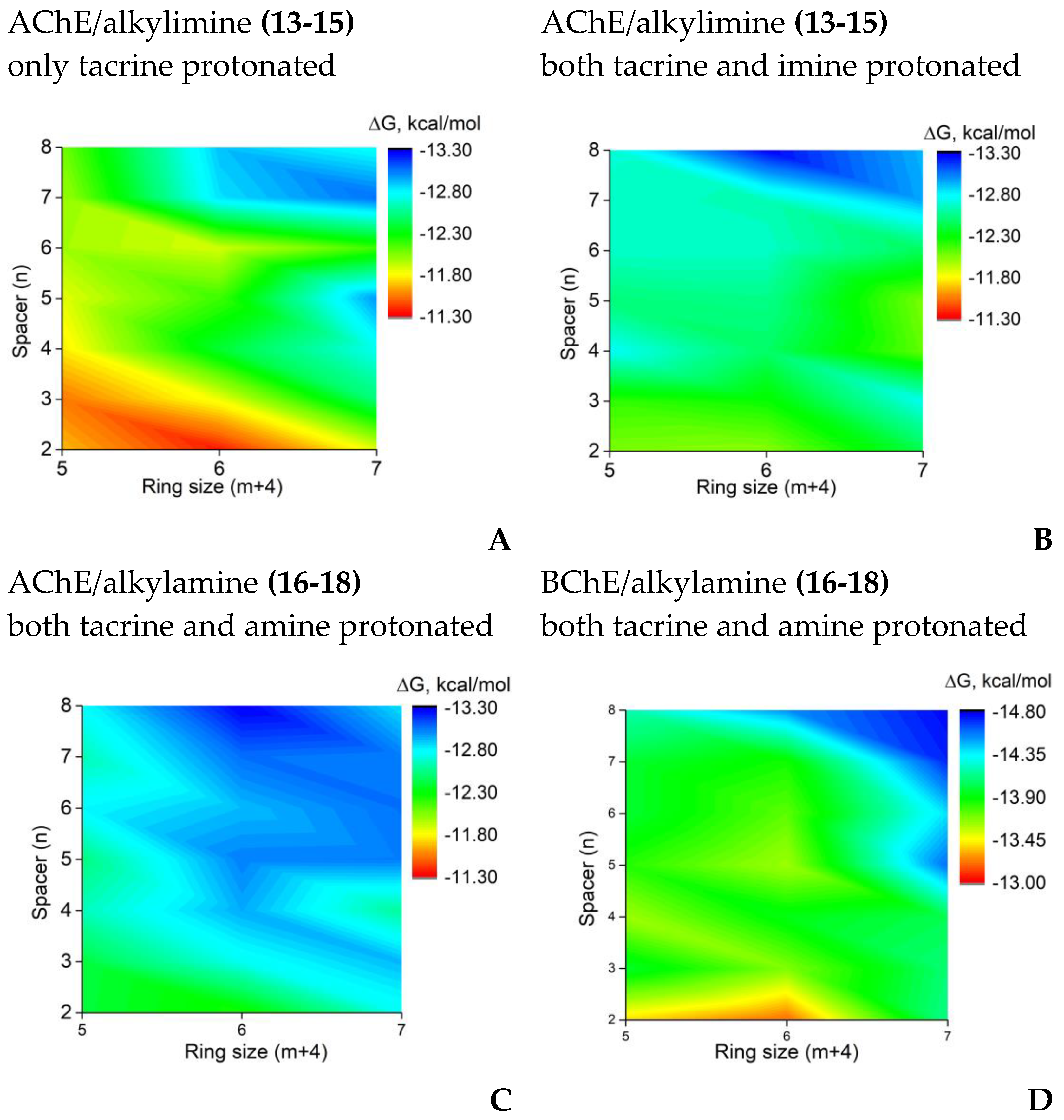
Figure 3.
Binding poses inside AChE for alkylamine derivatives with (A) increasing linker length (17a-17d, carbon atoms are colored with different shades of green from the lightest for n = 2 to the darkest for n = 8) and (B) increasing ring size (16d, carbon atoms are shown violet, 17d – purple, 18d – deep purple).
Figure 3.
Binding poses inside AChE for alkylamine derivatives with (A) increasing linker length (17a-17d, carbon atoms are colored with different shades of green from the lightest for n = 2 to the darkest for n = 8) and (B) increasing ring size (16d, carbon atoms are shown violet, 17d – purple, 18d – deep purple).
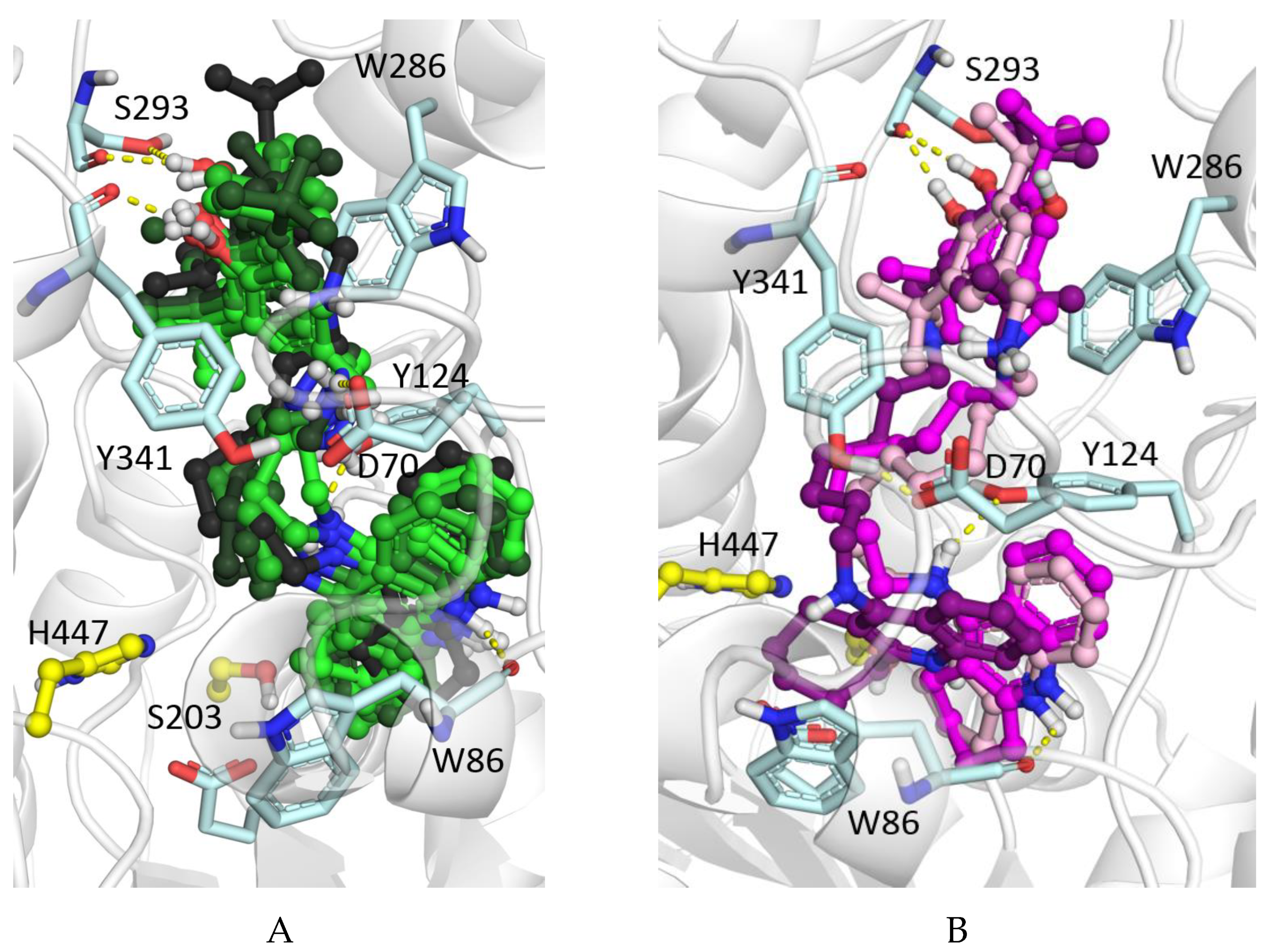
Figure 4.
Molecular docking to Aβ42. (A) all conformers of PDB ID 1IYT and compound 17d (the color gradually changes from red through white to blue from the first to the tenth Aβ42 conformer); (B) Aβ42 conformer #10 with the best energy of binding of compound 17d (carbon atoms colored in magenta) in comparison with positions of compounds 16d (carbon atoms colored in cyan), and 18d (carbon atoms colored in yellow).
Figure 4.
Molecular docking to Aβ42. (A) all conformers of PDB ID 1IYT and compound 17d (the color gradually changes from red through white to blue from the first to the tenth Aβ42 conformer); (B) Aβ42 conformer #10 with the best energy of binding of compound 17d (carbon atoms colored in magenta) in comparison with positions of compounds 16d (carbon atoms colored in cyan), and 18d (carbon atoms colored in yellow).
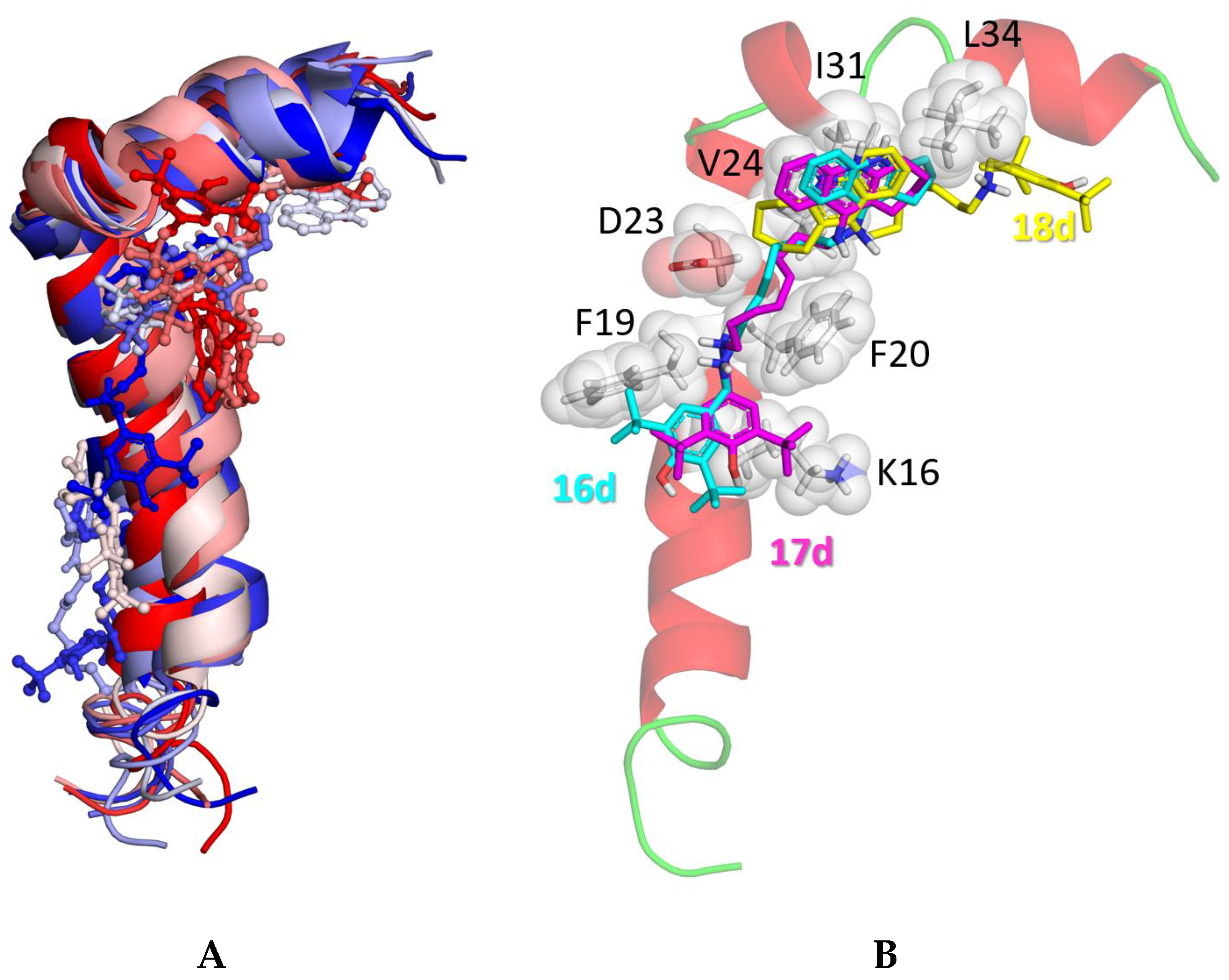
Figure 5.
Main stages that control light emission in the CL test.

Figure 6.
Three stages of the outer-sphere ET mechanism.
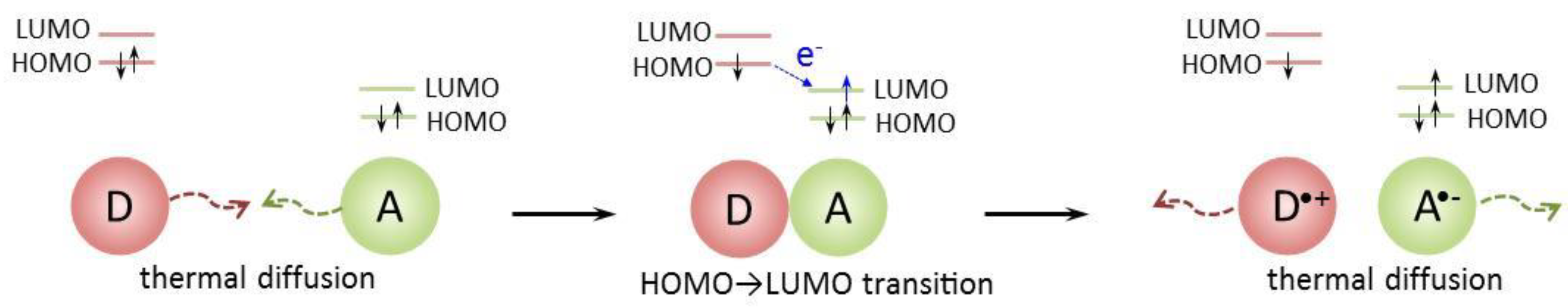
Table 1.
Esterase profiles of the compounds and their ability to displace propidium from the peripheral anionic site of Electrophorus electricus AChE (EeAChE).
Table 1.
Esterase profiles of the compounds and their ability to displace propidium from the peripheral anionic site of Electrophorus electricus AChE (EeAChE).
| No | m | n | Inhibitory activity against AChE, BChE and CES IC50, μM or % inhibition at 20 μM |
Propidium displacement, (%) | ||
|---|---|---|---|---|---|---|
| AChE | BChE | CES | ||||
| 13a | 1 | 2 | 4.86±0.01* | 1.92±0.11* | 26.1±0.7%* | 18.1±1.6* |
| 13b | 1 | 4 | 1.30±0.07 | 0.351±0.001 | 32.7±1.9% | 20.0±1.6 |
| 13c | 1 | 6 | 0.210±0.010 | 0.172±0.017 | 31.3±2.1% | 16.5±1.3 |
| 13d | 1 | 8 | 0.107±0.009 | 0.0417±0.0003 | 26.3±1.7% | 15.1±0.9 |
| 14a | 2 | 2 | 5.98±0.13* | 1.61±0.04* | 28.6±1.6%* | 18.2±1.6* |
| 14b | 2 | 4 | 0.424±0.022 | 0.385±0.031 | 26.6±2.2% | 17.6±1.4 |
| 14c | 2 | 6 | 0.0712±0.0012 | 0.055±0.005 | 25.1±1.9% | 19.7±1.5 |
| 14d | 2 | 8 | 0.0171±0.0016 | 0.00939±0.00042 | 28.8±2.0% | 14.8±1.0 |
| 15a | 3 | 2 | 4.03±0.03* | 0.419±0.040* | 30.1 ± 2.5%* | 16.3±1.3* |
| 15b | 3 | 4 | 0.524±0.020 | 0.131±0.004 | 26.3±0.4% | 18.4±1.4 |
| 15c | 3 | 6 | 0.151±0.013 | 0.0106±0.0002 | 25.4±0.9% | 16.8±1.2 |
| 15d | 3 | 8 | 0.0260±0.0024 | 0.00624±0.00054 | 25.5±1.1% | 14.9±1.2 |
| 16a | 1 | 2 | 3.50±0.33* | 0.652±0.05* | 19.6±1.3%* | 16.4±1.4* |
| 16b | 1 | 4 | 0.912±0.016 | 0.177±0.017 | 17.2±3.3% | 17.5±1.5 |
| 16c | 1 | 6 | 0.205±0.012 | 0.0488±0.0005 | 18.7±0.7% | 15.8±1.4 |
| 16d | 1 | 8 | 0.094±0.006 | 0.0170±0.0016 | 17.2±0.2% | 13.9±1.1 |
| 17a | 2 | 2 | 2.88±0.19 | 0.464±0.041 | 18.4±1.1% | 13.4±1.1 |
| 17b | 2 | 4 | 0.279±0.022 | 0.111±0.009 | 15.8±1.4% | 15.4±1.3 |
| 17c | 2 | 6 | 0.0702±0.0011 | 0.0361±0.023 | 18.5±1.6% | 16.1±1.1 |
| 17d | 2 | 8 | 0.0151±0.002 | 0.00756±0.00042 | 23.6±2.1% | 13.4±1.2 |
| 18a | 3 | 2 | 1.90±0.26* | 0.0838±0.0082* | 26.0±3.9%* | 13.6±1.2* |
| 18b | 3 | 4 | 0.436±0.016 | 0.0678±0.0061 | 21.4±1.8% | 15.7±1.2 |
| 18c | 3 | 6 | 0.103±0.004 | 0.0149±0.0003 | 16.8±0.5% | 12.5±0.9 |
| 18d | 3 | 8 | 0.0308±0.0002 | 0.00596±0.00058 | 13.9±1.2% | 12.3±0.8 |
| Tacrine | 0.601±0.047 | 0.0295±0.0002 | n.a. | 4.4 ± 0.6 | ||
| BHT | 6.0±1.5% | 18.9±1.7% | 5.6±0.2% | n.d. | ||
| BNPP | n.a. | n.a. | 99.1±0.9%1 | n.d. | ||
| Donepezil | n.d. | n.d. | n.d. | 11.9 ± 0.9 | ||
*Data from [44]. 1BNPP IC50 CES = 1.80±0.11 µM. Data are presented as mean ± SEM, n = 3; m+4 = number of methylene units in the cycloalkyl ring; n = number of methylene units in the spacer; n.a. – not active; n.d. – not determined.
Table 3.
Inhibition of Aβ42 self-aggregation by selected conjugates.
| No | m | n | Inhibition of Aβ42 Self- aggregation, % 1 |
|
|---|---|---|---|---|
| 13d | 1 | 8 | 49.4±4.3 | |
| 14c | 2 | 6 | 62.4±4.9 | |
| 14d | 2 | 8 | 70.4±5.6 | |
| 15c | 3 | 6 | 43.4±3.9 | |
| 15d | 3 | 8 | 63.5±5.0 | |
| 16d | 1 | 8 | 54.6±3.9 | |
| 17c | 2 | 6 | 64.1±5.7 | |
| 17d | 2 | 8 | 71.4±4.9 | |
| 18c | 3 | 6 | 47.8±3.8 | |
| 18d | 3 | 8 | 59.6±4.7 | |
| Tacrine | 5.9±0.5 | |||
| Myricetin | 73.2±5.8 | |||
| Propidium iodide | 89.3±7.1 | |||
1Inhibition of Aβ42 (50 µM) self-aggregation by the tested compound at 100 µM concentration. m, n - see footnote for Table 1.
Table 4.
Primary antioxidant activity of conjugates 13-18.
| No | m | n | ABTS•+-scavenging activity |
Ferric reducing antioxidant power |
|
|---|---|---|---|---|---|
| TEAC | IC50, µM | TE | |||
| 13a | 1 | 2 | 0.92±0.03* | 22.3±1.5 | 0.51±0.03* |
| 13b | 1 | 4 | 0.78±0.04 | 25.4±1.6 | 0.60±0.02 |
| 13c | 1 | 6 | 0.98±0.04 | 19.4±0.9 | 0.59±0.01 |
| 13d | 1 | 8 | 0.85±0.03 | 22.7±1.2 | 0.71±0.03 |
| 14a | 2 | 2 | 1.13±0.05* | 17.8±1.5 | 0.58±0.03* |
| 14b | 2 | 4 | 1.00±0.05 | 19.7±0.9 | 0.72±0.03 |
| 14c | 2 | 6 | 1.10±0.05 | 18.6±0.9 | 0.71±0.02 |
| 14d | 2 | 8 | 1.06±0.03 | 18.2±0.7 | 0.70±0.03 |
| 15a | 3 | 2 | 1.11±0.04* | 18.8±0.8 | 0.52±0.01* |
| 15b | 3 | 4 | 0.89±0.04 | 22.5±1.4 | 0.46±0.01 |
| 15c | 3 | 6 | 0.90±0.03 | 22.3±1.1 | 0.52±0.02 |
| 15d | 3 | 8 | 1.00±0.03 | 19.6±0.8 | 0.73±0.02 |
| 16a | 1 | 2 | 1.39±0.05* | 14.6±0.8 | 0.57±0.02 |
| 16b | 1 | 4 | 0.90 ±0.04 | 23.6±1.3 | 0.44±0.02 |
| 16c | 1 | 6 | 1.50±0.06 | 13.4±0.7 | 0.51±0.02 |
| 16d | 1 | 8 | 1.00±0.03 | 21.3±1.2 | 0.61±0.01 |
| 17a | 2 | 2 | 1.20±0.05 | 16.7±0.8 | 0.52±0.02 |
| 17b | 2 | 4 | 1.40±0.06 | 14.3±0.6 | 0.46±0.01 |
| 17c | 2 | 6 | 1.32±0.08 | 15.7±0.6 | 0.44±0.02 |
| 17d | 2 | 8 | 1.27±0.05 | 15.2±0.7 | 0.57±0.01 |
| 18a | 3 | 2 | 1.35±0.06* | 15.3±0.6 | 0.44±0.06 |
| 18b | 3 | 4 | 1.36±0.06 | 14.6±0.5 | 0.38±0.01 |
| 18c | 3 | 6 | 1.00±0.03 | 21.6±1.1 | 0.43±0.02 |
| 18d | 3 | 8 | 1.20±0.05 | 15.8±0.6 | 0.45±0.01 |
| BHT | 0.98 ± 0.03 | 22.4±1.4 | 0.96±0.02 | ||
| Trolox | 1.0 | 20.1±1.2 | 1.0 | ||
Table 8.
Predicted ADMET and physicochemical profiles of compounds 13-18.
| Compound | m | n | MW | LogPow | pSaq | LogBB | HIA, % | hERG pKi | hERG pIC50 | QED |
|---|---|---|---|---|---|---|---|---|---|---|
| 13a | 1 | 2 | 443.63 | 5.82 | 6.82 | 0.24 | 100 | 6.22 | 6.17 | 0.35 |
| 13b | 1 | 4 | 471.69 | 6.35 | 7.39 | 0.48 | 100 | 6.18 | 6.55 | 0.28 |
| 13c | 1 | 6 | 499.74 | 6.99 | 7.96 | 0.40 | 100 | 6.09 | 6.95 | 0.23 |
| 13d | 1 | 8 | 527.79 | 7.38 | 8.24 | 0.77 | 100 | 6.45 | 7.39 | 0.19 |
| 14a | 2 | 2 | 457.66 | 6.14 | 7.19 | 0.27 | 100 | 6.27 | 6.06 | 0.32 |
| 14b | 2 | 4 | 485.71 | 6.64 | 7.65 | 0.51 | 100 | 6.22 | 6.43 | 0.27 |
| 14c | 2 | 6 | 513.77 | 7.31 | 8.10 | 0.43 | 100 | 6.13 | 6.82 | 0.22 |
| 14d | 2 | 8 | 541.82 | 7.55 | 8.56 | 0.79 | 100 | 6.49 | 7.26 | 0.19 |
| 15a | 3 | 2 | 471.69 | 6.40 | 7.44 | 0.30 | 100 | 6.27 | 6.17 | 0.23 |
| 15b | 3 | 4 | 499.74 | 6.98 | 7.96 | 0.54 | 100 | 6.22 | 6.55 | 0.20 |
| 15c | 3 | 6 | 527.79 | 7.36 | 8.32 | 0.46 | 100 | 6.13 | 6.95 | 0.17 |
| 15d | 3 | 8 | 555.85 | 7.70 | 8.72 | 0.82 | 100 | 6.49 | 7.39 | 0.14 |
| 16a | 1 | 2 | 445.65 | 5.33 | 5.68 | 0.22 | 100 | 5.87 | 5.83 | 0.39 |
| 16b | 1 | 4 | 473.70 | 5.89 | 6.23 | 0.49 | 100 | 6.43 | 6.29 | 0.31 |
| 16c | 1 | 6 | 501.76 | 6.42 | 6.80 | 0.48 | 100 | 6.23 | 6.43 | 0.25 |
| 16d | 1 | 8 | 529.81 | 6.88 | 7.30 | 0.84 | 100 | 6.60 | 6.81 | 0.21 |
| 17a | 2 | 2 | 459.68 | 5.65 | 5.96 | 0.25 | 100 | 5.92 | 5.74 | 0.36 |
| 17b | 2 | 4 | 487.73 | 6.18 | 6.51 | 0.52 | 100 | 6.47 | 6.19 | 0.29 |
| 17c | 2 | 6 | 515.78 | 6.67 | 7.06 | 0.51 | 100 | 6.27 | 6.33 | 0.24 |
| 17d | 2 | 8 | 543.84 | 7.11 | 7.50 | 0.87 | 100 | 6.64 | 6.71 | 0.20 |
| 18a | 3 | 2 | 473.70 | 5.92 | 6.28 | 0.28 | 100 | 5.93 | 5.82 | 0.27 |
| 18b | 3 | 4 | 501.76 | 6.40 | 6.81 | 0.55 | 100 | 6.47 | 6.28 | 0.22 |
| 18c | 3 | 6 | 529.81 | 6.85 | 7.28 | 0.54 | 100 | 6.28 | 6.43 | 0.18 |
| 18d | 3 | 8 | 557.86 | 7.39 | 7.68 | 0.90 | 100 | 6.64 | 6.82 | 0.15 |
MW—molecular weight, LogPow—octanol-water partition coefficient, pSaq—aqueous solubility [−log(M)], LogBB—blood–brain barrier distribution, HIA—human intestinal absorption [%], hERG pKi—hERG potassium channel affinity [−log(M)], hERG pIC50—hERG potassium channel inhibitory activity [−log(M)], QED—quantitative estimate of drug-likeness.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



