
Submitted:
07 November 2023
Posted:
08 November 2023
You are already at the latest version
Abstract
Due to the proliferation of genetic testing, pathogenic germline variants predisposing to hereditary hematological malignancy syndrome (HHMS) have been identified in an increasing number of genes. Consequently, the field of HHMS is gaining recognition among clinicians and scientists worldwide. Patients with germline genetic abnormalities often have poor outcomes and are candidates for allogeneic hematopoietic stem cell transplantation (HSCT). However, HSCT using blood from a related donor should be carefully considered because of the risk that the patient may inherit a pathogenic variant. At present, we now face the challenge of incorporating these advances into clinical practice for patients with myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) and optimizing the management and surveillance of patients and asymptomatic carriers, with the limitation that evidence-based guidelines are often inadequate. The 2016 revision of the WHO classification added a new section on myeloid malignant neoplasms, including MDS and AML with germline predisposition. The main syndromes can be classified into three groups. Those without pre-existing disease or organ dysfunction; DDX41, TP53, CEBPA, those with pre-existing platelet disorders; ANKRD26, ETV6, RUNX1, and those with other organ dysfunctions; SAMD9/SAMD9L, GATA2, and inherited bone marrow failure syndromes. In this review, we will outline the role of the genes involved in HHMS in order to clarify our understanding of HHMS.
Keywords:
HHMS
; AML
; MDS
; DDX41
; TP53
; SAMD9
; SAMD9L
; germline
; variant
1. Introduction
Most hematologic malignancies are thought to spontaneously arise due to acquired genetic lesions in hematopoietic stem and precursor cells (HSPC) [1]. However, in some cases of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS), a hereditary (mainly autosomal dominant) predisposition has been observed [2,3]. Typically, a family in which two or more first- or second-degree relatives have developed acute leukemia (AL), myeloid malignancies, characteristic cytopenias, or either MDS or AML, is defined as "familial MDS/AML" or, more broadly, hereditary hematologic malignancy syndrome (HHMS) [4,5,6]. The field of HHMS has gained increasing recognition among clinicians and scientists worldwide. Both myeloid and lymphoid malignancies may be present in individuals or families with these syndromes. Genetic predisposition should be considered in patients who present with bone marrow failure, MDS, or AML at a young age or who present with unexpected hematologic toxicity during treatment for malignancy at a young age [7,8]. Identifying characteristics of such patients include physical abnormalities, endocrine abnormalities, short stature, stunted growth, and immunodeficiency in patients with hematologic abnormalities such as cytopenia, unexplained macro-erythroblastosis, or overt malignancy. A genetic MDS/AML predisposition may also be indicated by a family history of first- or second-degree relatives with malignancy, cytopenia, congenital abnormalities, or excessive toxicity from chemotherapy or radiation therapy [9]. However, the absence of characteristic clinical features or a negative family history does not exclude the presence of a germline MDS/AML syndrome. Germline variants may occur de novo or result from parental gonadal mosaicism [10]. HHMS often shows marked inter- and intra-familial differences in latency, phenotype, expression, and penetrance. For example, some germline MDS syndromes lack obvious syndromic features or have variable penetrance or delayed expression. Cytogenetic clonal abnormalities common to certain inherited MDS disorders may warrant further investigation [11]. MDS with monosomy 7 frequently occurs in patients with germline variants in GATA-binding factor 2 (GATA2), sterile alpha motif domain containing 9 (SAMD9), sterile alpha motif domain containing 9 like (SAMD9L), or hereditary bone marrow failure syndrome [12]. Moreover, the involvement of hematopoietic transcription factor genes, such as CCAAT enhancer binding protein alpha (CEBPA), GATA2, runt-related transcription factor 1(RUNX1), ankyrin repeat domain containing 26 (ANKRD26), and ETS variant transcription factor 6 (ETV6), are traditionally associated with solid tumors such as MutS homolog 6 (MSH6) and breast cancer gene 1 (BRCA1); moreover, the recently identified genes DEAD-box helicase 41 (DDX41), SAMD9, SAMD9L are involved in leukemogenesis [13,14,15]. Many are found to be non-symptomatic and occur in various age groups. Studies suggest that about 10% of children and adults with MDS or AML may have heritable variants [5]. Importantly, these germline genetic abnormalities are not exclusive to the patient and may be shared by blood relatives, necessitating screening of blood relatives. As our diagnostic capabilities in HHMS improve, we now face the challenge of incorporating these advances into clinical practice with MDS/AML patients and how to optimize the management and surveillance of patients and asymptomatic carriers [16].
The discovery of novel syndromes combined with clinical, genetic, and epigenetic profiling of tumor samples has highlighted unique patterns of disease progression in HHMS. Despite these advances, causative lesions are identified in fewer than half of familial cases, and evidence-based guidelines are often inadequate. In the 2016 revision of the WHO classification, a new section was added for myeloid neoplasms with a germline predisposition, including cases of MDS, myeloproliferative neoplasms (MPN), and AL that develop on a background of predisposing germline variants [17]. As part of the diagnosis, specific underlying genetic abnormalities or predisposing syndromes should be considered. The major syndromes can be categorized into the following three groups: those without preexisting disease or organ dysfunction [e.g., DDX41, tumor protein p53 (TP53), CEBPA], those with pre-existing platelet disorders [e.g., ANKRD26, ETV6, RUNX1], and those with organ dysfunction [e.g., SAMD9/SAMD9L, GATA2, inherited bone marrow failure syndromes (IBMFSs)]. This review will outline the genes involved in the above HHMS.

Table 1.
Clinical characteristics, genetics, and prevalence of HHMS. Only the major genes discussed in this Review are included in this table.
Table 1.
Clinical characteristics, genetics, and prevalence of HHMS. Only the major genes discussed in this Review are included in this table.
| Gene | Chromosome location |
Disorder name | Penetrance and lifetime risk of HM |
Malignancy Types | Other manifestations |
| DDX41 | 5q35.3 | Familial MDS/AML with mutated DDX41 |
penetrance is incomplete | MDS, AML, t-MN, solid tumors, especially colon and prostate cancer and melanoma, but not yet definitively linked |
cytopenia, macrocytosis, autoimmune diseases |
| TP53 | 17p13.1 | Li-Fraumeni syndrome | lifetime risk of HM is about 6% | MDS, AML, lymphoma, ALL, t-MN, MM, osteosarcoma, breast cancer, brain tumors, soft tissue sarcoma, adrenocortica carcinoma and other solid tumors |
None |
| CEBPA | 19q13.1 | Familial AML with mutated CEBPA |
>80% lifetime risk of AML | AML | None |
| RUNX1 | 21q22.12 | Familial platelet disorder with propensity to myeloid malignancy |
unknown | MDS, AML, ALL, other lymphoid malignancies |
thrombocytopenia, platelet dysfunction, atopic and autoimmune disorders |
| ANKRD26 | 10p12.1 | Thrombocytopenia 2 | penetrance for thrombocytopenia is near complete, lifetime risk of HM is about 8% |
MDS, AML, CML, MPN, ALL, CLL, MM |
thrombocytopenia, leucocytosis, erythrocytosis, mild bleeding tendency |
| ETV6 | 12p13.2 | Thrombocytopenia 5 | Penetrance for thrombo- cytopenia is near complete |
ALL, MDS, AML, CMML, MM, GI cancers |
thrombocytopenia, macrocytosis, platelet dysfunction |
| SAMD9 | 7q21.2 | MIRAGE Syndrome | unknown | MDS, AML, CMML | bone marrow failure, cytopenia, infections, growth restriction, adrenal hypoplasia, enteropathy, genital abnormalities |
| SAMD9L | 7q21.2 | Ataxia Pancytopenia Syndrome |
unknown | MDS, AML, CMML | Systemic autoinflammatory disease, bone marrow failure, Ataxia |
| GATA2 | 3q21.3 | GATA2 deficiency syndrome |
penetrance is incomplete | MDS, AML, CMML, ALL | immunodeficiency, bone marrow failure, monocytopenia, lymphopenia, neutropenia, other cytopenia, infections, lymphedema, congenital deafness, pulmonary alveolar proteinosis, venous and arterial thrombosis |
| HM, Hematological malignancies; ALL, acute lymphoblastic leukemia; CML, chronic myeloid leukemia; CMML, chronic myelomonocytic leukemia; t-MN, therapy-related myeloid neoplasms; MM, multiple myeloma; MPN, myeloproliferative neoplasm; | |||||
2. Genes of Syndromes without Pre-Existing Disease or Organ Dysfunction
2.1. DDX41
RNA helicases are a series of enzymes that remodel RNA-RNA or RNA-protein interactions in an NTP-dependent manner. Humans have more than 70 helicases that are classified into superfamily (SF) 1 and SF2 based on differences in sequence motifs within the helicase core domain [18,19]. SF1 includes Upf1-like RNA helicases, while SF2 includes the DEAD-box, DEAH-box/RNA helicase A-like, Ski2-like and RIG-I-like families, with the DEAD-box family RNA helicases being the most numerous. While the DEAH-box RNA helicases are thought to translocate along the substrate RNA for remodeling, DEAD-box RNA helicases unwind substrate RNA locally; the mechanism of action of each is thus different, but they both play roles in virtually all processes that require RNA conformational changes, such as RNA transport, translation, RNA degradation, RNA splicing, and ribosome synthesis. As a single RNA helicase often exerts enzymatic activity in multiple cellular processes, it remains difficult to fully elucidate the pathogenesis of diseases due to abnormalities in RNA helicases.
In myeloid neoplasms, pathogenic variants in the gene encoding DDX41, a DEAD-box RNA helicase, are found in about 5% of cases [20]. It was recently shown that up to 13% of myeloid neoplasms have a genetic background [21], of which DDX41 variants account for about 80% of cases; MDS and AML occur in individuals with a heterozygous germline frameshift variant or a missense variant within the DEAD-box domain of DDX41 by later acquiring a somatic variant in the other allele, typically p.R525H (or p.G530D, etc. in a few cases) within the helicase domain [20,22,23] (Figure 1A). While many myeloid neoplasms with a genetic background develop at younger ages than those without a known genetic background, myeloid neoplasms with DDX41 variants are characterized by a late disease onset (mean age, 65 years) [24,25], which may have hindered identification of this gene as one of the genes responsible for genetic predisposition for myeloid leukemogenesis. In addition, the disease with a DDX41 variant is characterized by male dominancy, fewer proliferating tumor cells, hypoplastic bone marrow, and unique co-existing gene mutational patterns as compared to those in other myeloid neoplasms [26,27], with only DDX41 variants being often identified in many cases [20], suggesting a unique disease pathogenesis of myeloid neoplasms with DDX41 variants. In contrast, the disease phenotype may differ between cases with a single DDX41 variant and biallelic variants [28], and a report suggest that there is no clear difference in disease phenotype between cases with known pathogenic DDX41 variants and variants of unknown significance (VUS) [29]. Consequently, it is necessary to establish an validation system and database that can accurately interpret the significance of individual variant.
Figure 1.
Involvement of DDX41 variants in myeloid leukemogenesis.
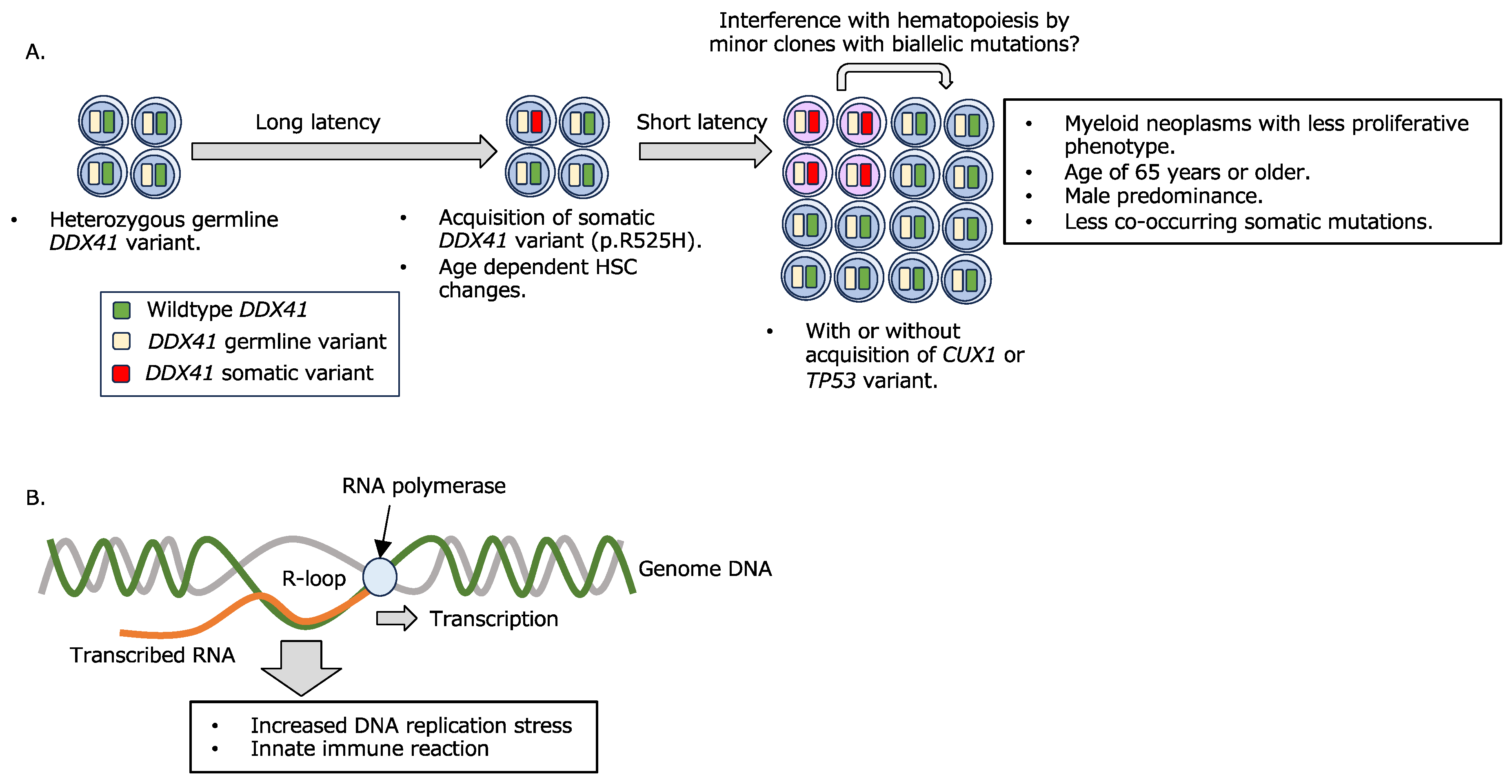
The prognosis of myeloid neoplasms with DDX41 variants is not necessarily worse than those without a known genetic background, regardless of the tendency to be categorized as high-risk. However, the development of disease at advanced ages often makes intensive treatment difficult. Several cases of donor-derived secondary leukemia in patients who received allogeneic hematopoietic stem cell transplantation (HSCT) have been reported [30,31,32,33], thus treatment decisions require careful consideration of genetic background. Recent reports describe the development of acute lymphocytic leukemia and solid cancers in individuals with DDX41 variants [34,35], but the extent to which DDX41 variants are involved in such diseases remains controversial [23].
DDX41 has been shown to be essential for hematopoiesis, with homozygous Ddx41 knockout mice being embryonic lethal, although heterozygous mice show no remarkable abnormalities [36,37]. Several mechanisms have been proposed for the actions of DDX41 variants in the development of myeloid neoplasms. It has been reported that R-loop, a nucleic acid structure on the genome consisting of a DNA:RNA hybrid and single-strand DNA, aberrantly accumulates in MDS with RNA splicing abnormalities, regardless of the type of responsible gene [38,39,40,41], and that R-loop accumulation causes DNA replication stress, DNA damage, and abnormal mitosis. Recently, DDX41 has also been shown to be involved in R-loop regulation [42,43,44], and it is suggested that R-loop accumulation due to dysfunction or decreased expression of DDX41 is involved in impaired hematopoiesis and aberrant innate immune responses (Figure 1B). One of the major functions of DDX41 is RNA splicing [45]. However, considering that DDX41 variants develop de novo AML in addition to MDS, DDX41 is thought to play different roles from those of typical RNA splicing factors associated with MDS development. Indeed, while SRSF2, SF3B1, and U2AF1 are all involved in the recognition of pre-mRNA 3’ splice sites with U2 snRNP [46], DDX41 has been shown to be incorporated into the spliceosome at the C complex stage, a late complex of the activated spliceosome [44,47]. Regarding the relationship between DDX41 and R-loops, there are reports showing that DDX41 can unwind R-loops on its own [43,48], while it has also been suggested that impaired DDX41 function leads to reduced efficiency of RNA splicing, thus resulting in conditions that facilitate R-loop formation [44]. The accumulation of R-loop has been shown to give rise to an excessive innate immune reaction mediated through the cGAS-STING signaling pathway, consequently inducing increased hematopoietic stem/progenitor cells [42]. However, the mechanisms by which R-loops activate the cGAS-STING pathway remain inconclusive. Recently, it was reported that DNA:RNA hybrids derived from R-loops are transported to the cytoplasm and thus trigger an innate immune response [49]. The relevance of this observation to impaired hematopoiesis caused by DDX41 variants is of interest.
DDX41 is also reported to promote the processing of small nucleolar RNA (snoRNA) from introns [37]. Some snoRNA are coded within introns of ribosomal protein genes and mature after being processed from the introns [50,51]. snoRNAs are classified into boxC/D type and boxH/ACA types depending on their sequences; the former catalyzes 2'-O-methylation and the latter is responsible for catalyzing pseudouridylation of uridine residues in ribosomal RNA, thereby promoting ribosomal biogenesis. Thus, loss of function or expression of DDX41 impairs ribosomal biogenesis [27,52]. Although the involvement of DDX41 in ribosomal biogenesis has been reported by other research groups, the process involving DDX41 may be different from those involving snoRNA processing.
Recently, myeloid neoplasms with germline DDX41 variants were shown to have a higher proportion of somatic CUX1 variants compared with those without a known germline background [20]. CUX1 is a transcription factor [53] that has also been shown to be directly involved in DNA damage repair by recruiting histone-modifying enzymes to damaged DNA regions [54]. Given that cells lacking sufficient CUX1 function can enter mitosis without completing DNA damage repair, the likelihood that loss of DDX41 function or expression causes DNA replication stress would be further increased. However, further studies are clearly needed to fully elucidate the mechanisms by which DDX41 variants lead to myeloid neoplasms.
- A.
- A combination of germline and somatic DDX41 variants confers myeloid disease development.
Hematopoietic cells with a germline DDX41 variant acquire a somatic DDX41 variant at an advanced age. Myeloid neoplasms are thought to develop shortly after biallelic DDX41 variant acquisition, with or without the addition of a limited number of somatic mutations in DNA repair-related genes, including CUX1 and TP53. It is also suggested that minor clones with biallelic DDX41 variants affect hematopoiesis by interfering with other cells [37].
- B.
- R-loop formation and its consequence.
R-loop accumulation due to impaired RNA splicing or other causes increases DNA replication stress and innate immune response, resulting in deficient hematopoiesis and leukemogenesis.
2.2. TP53
TP53 is one of the most frequently mutated genes, especially in adult-onset cancers. Genome sequencing of various human cancer cells has revealed that 42% of cases carry TP53 variants [55]. The p53 protein is a transcription factor that can activate the expression of multiple target genes, plays an important role in the regulation of the cell cycle, apoptosis, and genomic stability, and is widely known as “the guardian of the genome” [56,57]. The evidence accumulated to date suggest that p53 also regulates cell metabolism, ferroptosis, tumor microenvironment, and autophagy, which each contribute to tumor suppression [57]. Genomic instability caused by deletions and variants in TP53 may lead to the accumulation of more oncogenes and promote tumorigenesis, growth, metastasis, and drug resistance [58]. p53 variants confer metabolic plasticity to cancer cells, promoting adaptation to metabolic stress and increasing the possibility of proliferation and metastasis [59].
The major type of TP53 variant is a missense variant producing a single amino acid substitution, with the DNA-binding domain (DBD) being the most mutated region [60]. Structural variants can reduce the thermostability of the protein, resulting in protein misfolding at physiological temperatures and loss of its ability to bind DNA [61]. These variants not only bind wild-type p53 and cause dominant-negative (DN) effects, but may also be converted to oncogenic proteins via gain-of-function (GOF) [62,63]. p53 is mutated and inactivated in most malignancies, making it a very attractive target for the development of new anti-cancer drugs [64]. Until recently, however, p53 was considered an undruggable target, and the progress made in p53-targeted therapeutics has been limited.
Li-Fraumeni syndrome (LFS) is caused by a germline variant in the TP53 gene and is characterized by an increased risk of developing various solid tumors and hematologic malignancies at a young age [65,66]. LFS is inherited in an autosomal dominant manner, although de novo instances occur in 7–20% of cases. The tumor spectrum includes soft-tissue sarcomas, premenopausal breast cancer, central nervous system tumors, adrenocortical carcinomas, and pancreatic tumors, as well as MDS and lymphoid and myeloid malignancies. Germline TP53 variants are found in approximately 50% of pediatric patients with hypoploid acute lymphoblastic leukemia (ALL) and are associated with poor outcomes [67,68]. In the Le-Fraumeni lineage, leukemia is relatively uncommon, with only approximately 4% of children and adolescents presenting with hypodiploid ALL, treatment-related, or de novo MDS/AML [69].
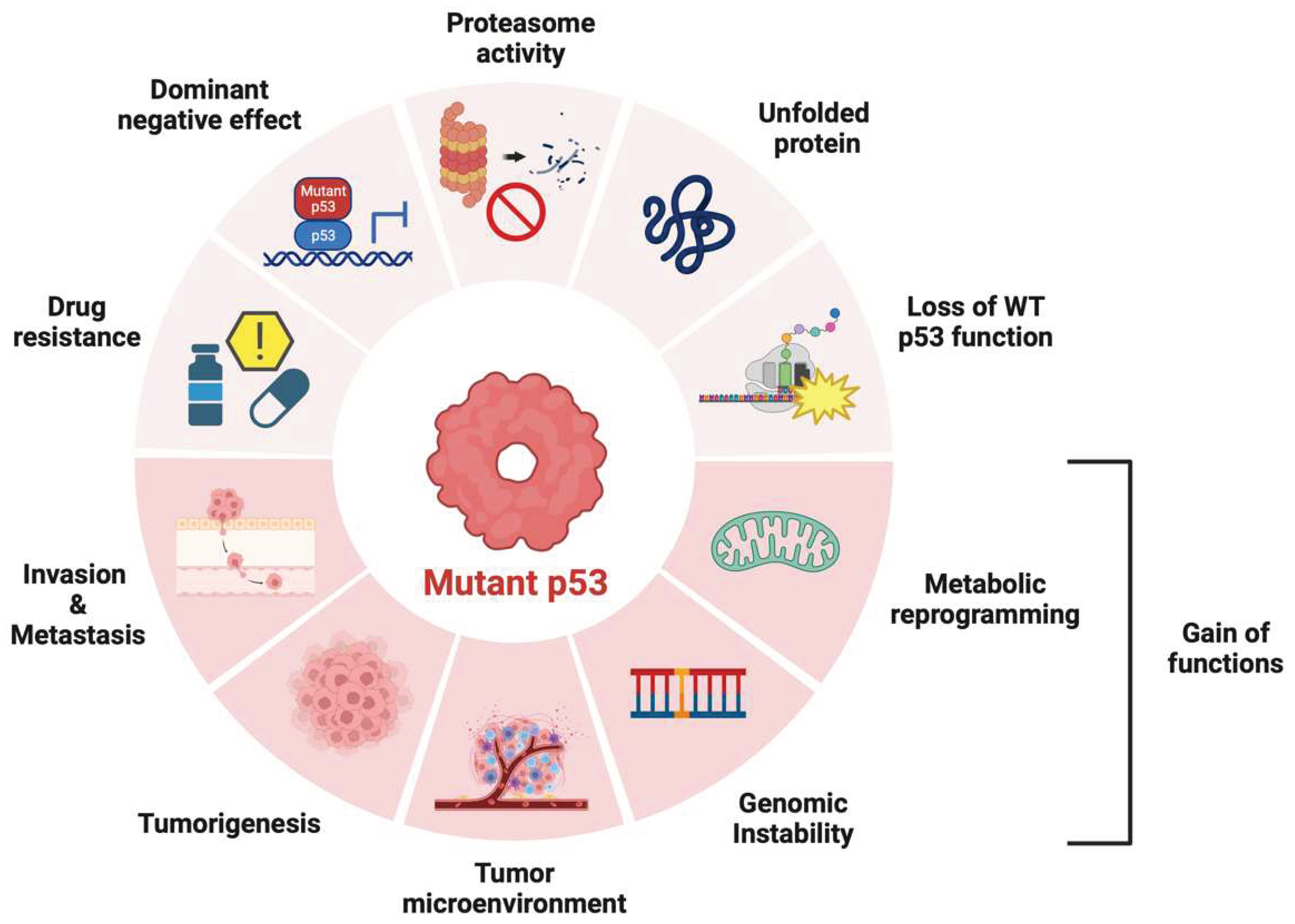
Figure 2.
Role of p53 variants in cancer. p53 variants produce drug resistance, dominant negative effects on wild-type p53, proteasome repression, and loss of function of wild-type p53. In cases of gain of function (GOF), it promotes various cellular responses such as carcinogenesis, cancer cell proliferation, invasion, metastasis, tumor microenvironment establishment, genomic instability, and metabolic reprogramming.
Figure 2.
Role of p53 variants in cancer. p53 variants produce drug resistance, dominant negative effects on wild-type p53, proteasome repression, and loss of function of wild-type p53. In cases of gain of function (GOF), it promotes various cellular responses such as carcinogenesis, cancer cell proliferation, invasion, metastasis, tumor microenvironment establishment, genomic instability, and metabolic reprogramming.
2.3. CEBPA
The CEBPA gene is located on chromosome 19q13.1 and gene variants are a common genetic alteration in AML. Patients present with de novo AML [French American-British (FAB) classification; AML M1, M2, and M4 subtypes] and a group of differentiation abnormalities [70]. These germline variants are generally frameshift or nonsense variants near the amino terminus of the encoded protein; somatic variants in CEBPA often occur in the other allele, leading to a biallelic variant in CEBPA. This triggers the development of AML [71]. CEBPA-associated familial AML is defined as the presence of heterozygous germline CEBPA pathogenic variants in AML patients and/or in families with one or more AML patient. In contrast, sporadic CEBPA-associated AML is defined as AML in which the CEBPA pathogenic variant is identified in leukemic cells and not in non-leukemic cells [72]. AML with germline CEBPA variants generally occurs in an autosomal-dominant inheritance without preceding abnormal blood cell counts or myelodysplasia [73]. Approximately 10% of CEBPA-associated AMLs have been shown to carry germline CEBPA variants [2]. In contrast to the incomplete penetrance observed in other HHMS, germline CEBPA variants cause AML with almost complete penetrance (lifetime risk estimated to be >80%) [74]. In the majority of CEBPA-associated familial AML, the age of onset appears to be earlier than in sporadic CEBPA-associated AML [72]. Onset usually occurs in the 20th or 30th year of life, and many patients develop AML before 50 years of age; the median age of onset for AML is 25 years [75]. The prognosis of CEBPA-associated familial AML appears to be better than that of sporadic CEBPA-associated AML [76,77]. Patients with CEBPA-associated familial AML with a cured initial presentation are at high risk of developing additional independent leukemic episodes in addition to the risk of relapse from a pre-existing clone; the clinical observation that AML patients with CEBPA variants are more likely to develop a secondary leukemia despite their favorable prognosis is likely due to this pattern of progression [78]. Lifelong surveillance is recommended in patients with familial AML because of the high risk of late leukemia relapse [16]. It is important to avoid the use of allogeneic or consanguineous donors for HSCT without prior evaluation of the donor's germline CEBPA pathogenic variant [79].
3. Genes of Syndromes Associated with Preexisting Platelet Disorders
Most predisposition syndromes are associated with specific hematopoietic cell lineage abnormalities and each exhibits a different tumor profile. For example, germline variants in RUNX1, ANKRD26, and ETV6 all predispose to thrombocytopenia and hematologic malignancies [80]. However, there are marked differences in cancer predisposition: the ANKRD26 variant predisposes to myeloid malignancies, ETV6 predominantly predisposes to B-cell ALL, and RUNX1 is associated with myeloid malignancies, and to a lesser extent, predisposed to T-cell ALL [81]. Three different types of germ cell lineage predisposition are associated with highly variable penetrance in both myeloid and lymphoid systems. In both myeloid and lymphoid leukemias, the disease phenotype is likely influenced by both intrinsic and extrinsic cellular factors [80].
3.1. RUNX1
RUNX1 encodes a heterodimeric transcription factor essential for hematopoiesis, megakaryopoiesis, and platelet function [82]. It functions as a transcriptional activator for some genes and a transcriptional repressor for others. Somatic variants in RUNX1 are among the most common variants in adults and children with ALL, AML, or MDS, including recurrent fusions in B-ALL (ETV6-RUNX1) and AML (RUNX1-RUNX1T1) [83]. RUNX1 was identified as a gene located at a truncation site on chromosome 21 in t (8;21), which is found in AML [84]. Somatic variants in the RUNX1 gene are one of the most frequently identified variants and have been identified in patients with various myeloid malignancies, including MDS, MPN, and AML [85]. In most cases, these RUNX1 variants are considered "subclonal variants" [86]. A high frequency of RUNX1 variants (30-50%) has been reported in treatment-related and radiation-related MDS and AML [87,88]. It is generally believed that RUNX1 variants lead to the loss of RUNX1 function [89]. In contrast, germline variants in the RUNX1 gene cause familial myeloid malignant platelet disorders (FPD/AML) with autosomal dominant inheritance, typically presenting with quantitative/qualitative platelet defects and a predisposition to myeloid malignancies like MDS and AML [90]. In this case, heterozygous inherited RUNX1 variants play a fundamental role in the etiology of FPD/AML [91]. However, these inherited RUNX1 variants are not sufficient to cause leukemia. It is thought that the accumulation of various variants, such as the CDC25C biallelic RUNX1 variant, and the TET2 variant, progresses to preleukemic clones and eventually develops hematologic malignancies [92,93].
Germline variants in RUNX1 are among the most frequently detected variants in the pathogenesis of HHMS [93]; the RUNX1 gene encodes a DNA-binding subunit that contains a highly conserved runt-homology domain (RHD) for sequence-specific DNA binding [94]. Truncation lesions occur throughout the gene, but missense variants within the RHD are the most common. Others include nonsense, frameshifts, duplications, partial or total gene deletions, and gene rearrangements; many RUNX1 variants cause haploinsufficiency [89]. RUNX1 variants cause defects in hematopoietic differentiation, resulting in decreased hematopoietic progenitor cell numbers and abnormal megakaryocyte differentiation. Tumorigenesis is most commonly caused by the somatic second hit of RUNX1; typical clinical features of FPD/AML are gradual thrombocytopenia, aspirin-like qualitative platelet abnormalities, and a tendency to develop hematologic tumors [95]. Approximately 20-60% of FPD/AML families develop hematologic neoplasms during their lifetime [95]. The latency period to transformation is relatively long, with the average age at diagnosis reported to be 33 years (maximum 76 years) [83]. Similar to what is observed in sporadic hematologic malignancies, additional acquired genetic events cooperate with the hereditary RUNX1 variant to progress the manifestation of the malignant phase. Although most cases develop MDS or AML, other phenotypes have also been reported, including secondary leukemia, T-cell acute lymphoblastic leukemia( T-ALL) and non Hodgkin lymphoma (NHL) [95]. Interestingly, the location of variants within the RUNX1 gene does not seem to affect disease phenotype among individuals, and phenotypic heterogeneity is often observed even within families with lesions of the same germ lineage [93].
3.2. ANKRD26
ANKRD26 is a gene located at 10p12.1 that regulates megakaryocyte development and thrombocytopenia [96]. RUNX1 and FLi1 co-regulate ANKRD26 by binding to the ANKRD26 promoter and repressing gene activity [97]. ANKRD26-related thrombocytopenia (ANKRD26 RT) is an autosomal dominant thrombocytopenia caused by a single nucleotide substitution in the ANKRD26 gene, characterized by quantitative and qualitative platelet disorders and an increased risk of MDS and AML [98]. ANKRD26 encodes a protein with an ankyrin repeat domain at its N-terminus and is thought to function in protein-protein interactions; while the function of the ANKRD26 protein is unknown, expression profiling has demonstrated its presence in megakaryocytes [98]. Germline variants in ANRK26 are usually point mutations located in the 5' untranslated region (UTR) of the gene, although deletions and point mutations within the coding region have also been reported [99]. Variants in the 5’UTR affect binding of repressive transcription factors such as RUNX1 and FLi1 to this regulatory region, abnormally increasing the expression of ANKRD26 and impairing platelet production [80]. The age of diagnosis generally ranges from the early 20s to 70s. The incidence of myeloid malignancies is high in these patients, with an estimated 5% for AML, 2.2% for MDS, and 1.3% for chronic myeloid leukemia, with an estimated risk of these malignancies of 23, 12, and 21 times that of the general population, respectively [14].
3.3. ETV6
Patients with thrombocytopenia 5, an autosomal dominant disorder of thrombocytopenia with bleeding tendency, usually present in childhood and have been found to have germline variants in ETV6 [100]. Clinical features include thrombocytopenia, abnormal platelet function, and increased bleeding tendency [101]. Leukemia is estimated to occur in about 30% of carriers, most commonly in ALL, but more than 30 translocation partners of ETV6 have been reported in AML, MDS, MPN, and T-cell lymphomas. ETV6 is one of the most commonly translocated genes in human AL and MDS [102]. ETV6 is located on chromosome 12p13.2 and encodes a transcriptional repressor important for hematopoiesis, megakaryopoiesis, and embryogenesis, and is involved in angiogenesis, cell growth and differentiation [103]. The gene encodes an N-terminal or C-terminal zinc finger but the majority of variants are clustered within the DNA-binding ETS domain. Somatic rearrangements (most commonly with RUNX1), deletions, and sequence variants are observed in ALL. Second-hit variants (especially deletions) in ETV6 are common in ETV6-RUNX1 rearranged leukemias [104]. In addition, somatic rearrangements with RUNX1 are observed in a quarter of ALL patients [105]. Studies using umbilical cord blood from healthy newborns have shown that ETV6-RUNX1 translocations can occur in more than 1% of the healthy population [106].
4. Genes of Syndromes Associated with Other Organ Dysfunction
4.1. SAMD9/SAMD9L
SAMD9 and SAMD9L are a homologous gene pair at the head and tail of 7q21 and are interferon-inducible genes that are widely expressed in human tissues [107,108]. Both negatively regulate cell proliferation and function as tumor suppressors. Genetic variants in SAMD9/SAMD9L were initially shown to cause multisystem syndromes characterized by various neurological and/or endocrine abnormalities, as well as MDS with monosomy 7 and del7q [107,109]. Little is known about the biochemical activity of the SAMD9 and SAMD9L proteins and their domain structures, but they cluster in the latter half of the protein, in or near the putative P-loop [110]. The SAMD9 and SAMD9L proteins appear to be involved in endocytosis and cytokine signaling [111,112]; moreover, they have been reported to play a role in antiviral responses, similar to DDX41. Specifically, SAMD9 and SAMD9L are known to be host-restricted factors in poxvirus infection [113,114].
Germline variants in these genes are strongly associated with monogenic and familial pediatric MDS and potential full or partial deletions of adult chromosome 7 [115]. Germline variants in SAMD9 or SAMD9L are heterozygous gain-of-function missense variants, leading to proliferative arrest when expressed exogenously in the cell [108]. Carriers are at high risk for MDS and AML with cytopenia and monosomy 7/del7q. Many other patients who do not develop monosomy 7 acquire somatic variants in SAMD9 or SAMD9L resulting in the loss of function of the mutant protein [116]. Overexpression of SAMD9 or SAMD9L results in decreased proliferation and increased apoptosis, ultimately leading to the hypocellular phenotype observed in patients. The effects on ribosome biology, DNA damage, and the resulting genomic instability are thought to promote the observed apoptotic phenotype [117,118] and ultimately lead to reduced bone marrow cellularity. Unrepaired DNA defects in hematopoietic cells cause significant long-term functional disruption and are a major driving force for the accumulation of further variants, thus promoting clonal expansion and malignant transformation [119,120,121].
Germline variants in SAMD9 cause a syndrome known by the acronym MIRAGE; MIRAGE syndrome is an autosomal-dominant multisystem disorder characterized by six core features [122,123,124,125,126]. The features include bone marrow failure, progression to MDS and AML, infection, intrauterine dysplasia, adrenal hypoplasia, genital abnormalities, and enteropathy (chronic diarrhea with colonic dilatation). Germline variants in SAMD9L cause ataxia-pancytopenia syndrome, an autosomal dominant disorder with early onset gait and balance disturbances, nystagmus, mild pyramidal signs, and marked cerebellar atrophy [127,128,129,130]. Hematologic abnormalities include pancytopenia, bone marrow failure, and progression to MDS and AML. Germline variants in these two genes are found in 8-17% of pediatric MDS cases [107].
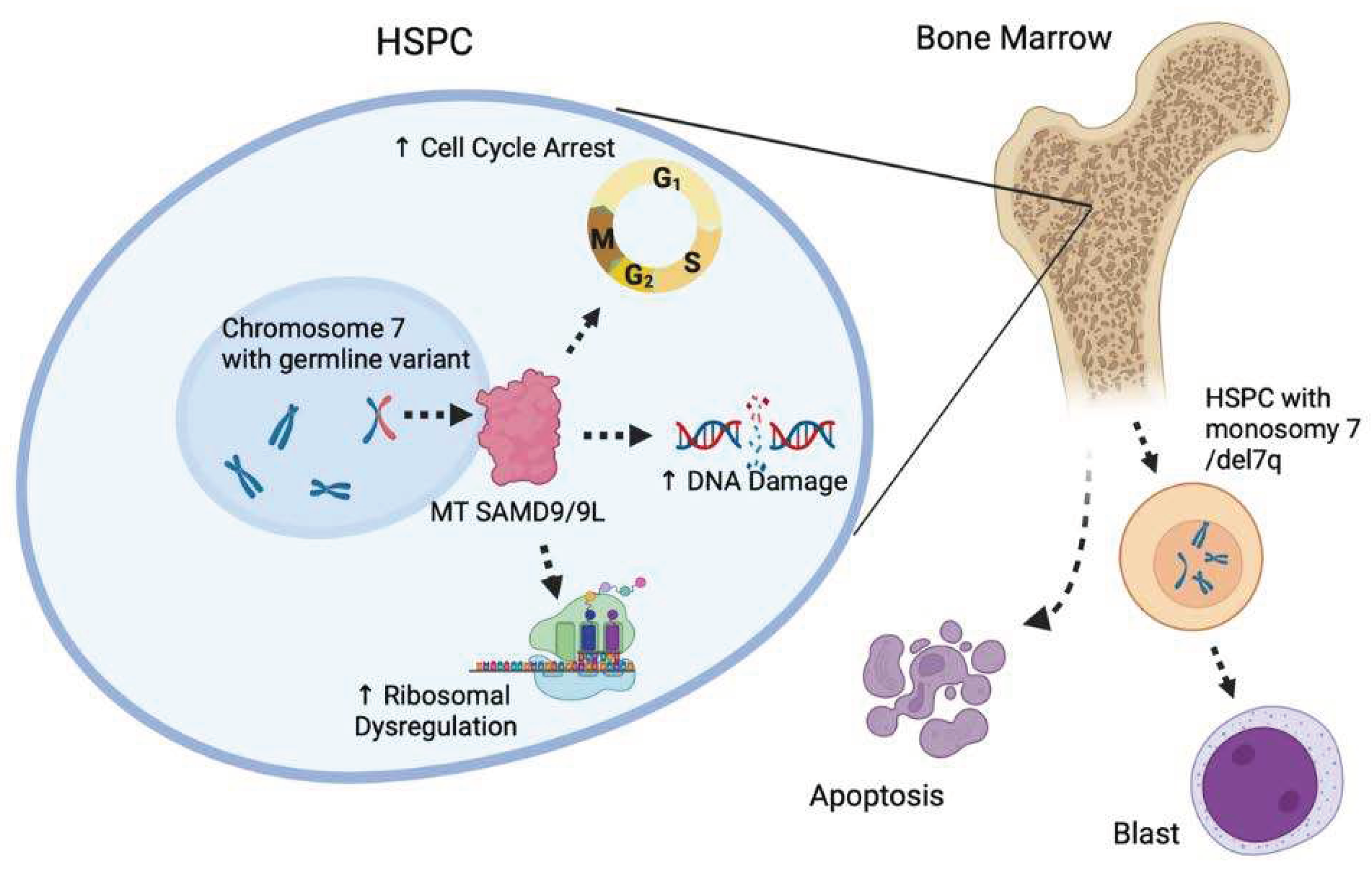
Figure 3.
Role of SAMD9 and SAMD9L in HSPC function. The SAMD9 and SAMD9L genes regulate proteins involved in the cell cycle, DNA damage repair, and ribosome regulation. Mutant SAMD9 and SAMD9L proteins significantly enhance these functions, which cause decreased hematopoietic potential and apoptosis in the bone marrow, promoting monosomy 7/del 7 HSPC production. Hematopoietic stem and progenitor cell (HSPC), myelodysplastic syndrome (MDS), mutant type (MT).
Figure 3.
Role of SAMD9 and SAMD9L in HSPC function. The SAMD9 and SAMD9L genes regulate proteins involved in the cell cycle, DNA damage repair, and ribosome regulation. Mutant SAMD9 and SAMD9L proteins significantly enhance these functions, which cause decreased hematopoietic potential and apoptosis in the bone marrow, promoting monosomy 7/del 7 HSPC production. Hematopoietic stem and progenitor cell (HSPC), myelodysplastic syndrome (MDS), mutant type (MT).
4.2. GATA2
GATA2 is a zinc finger transcription factor that plays important roles in hematopoiesis, homeostasis of hematopoietic stem cells (HSC), and lymphocyte development, specifically interacting with RUNX1 to control HSC survival [131]. GATA2 haploinsufficiency is caused by a missense variant or deletion in the GATA2 located on chromosome 3q21.3 [132]. Other causative variants have been detected throughout the gene, including nonsense, frameshift, splice site, and synonymous variants that cause splice abnormalities, as well as variants that target enhancers deep within introns [133]. GATA2 haploinsufficiency is an autosomal dominant inherited bone marrow failure and immunodeficiency syndrome predisposing to MDS and AML. The syndrome results from loss-of-function variants or deletions in the GATA2 gene [134]. Notably, GATA2 deficiency syndromes show marked heterogeneity in inter- and intra-familial phenotypes, all within the spectrum of the single condition GATA2 deficiency syndrome [13,135].
Phenotypes range from isolated chronic neutropenia to MDS/AML, bone marrow failure, severe immunodeficiency, and alveolar proteinosis. Patients may present with isolated neutropenia and bone marrow failure without syndromic features or family history [136]. Atypical mycobacterial infections, viral and fungal infections are common, often overlapping with prolonged neutropenia, monocytopenia, B-cell deficiency, NK-cell deficiency, monocytopenia with Mycobacterium avium complex (MonoMAC) syndrome, or dendritic cell-monocyte-B-NK lymphocyte (DCML) deficiency [137,138]. Other symptoms include sensorineural hearing loss and lymphoedema (Emberger syndrome) [139,140]. Of particular note is that MDS/AML may present with one or more of these features, either years before the onset of MDS/AML or in isolation with MDS/AML. MDS with germline GATA2 variants is often associated with monosomy 7/del7q(-7) or trisomy 8, especially in children and young adults [138,141]. A study of 426 pediatric MDS cases identified germline GATA2 variants in 37% of patients with primary MDS with -7 and 16% of MDS cases with trisomy 8 [142]. In contrast, no germline GATA2 variants were found in treatment-related MDS.
4.3. IBMFS
Inherited bone marrow failure syndrome (IBMFS) is an inherited disease associated with decreased bone marrow cell production [143,144,145]. It is associated with a specific clinical phenotype and variable risk of developing MDS or AML. Traditionally, the distinction has been made based on the presence or absence of classical physical manifestations [146] such as abnormal nails, reticulate pigmentation of the skin, and oral leukoplakia in congenital dyskeratosis. Fanconi anemia (FA) [147,148,149], Diamond-Blackfan anemia (DBA) [150,151,152], dyskeratosis congenita (DC) [153,154,155] or telomere biology disorders (TBDs) [156], and Schwachman-Diamond syndrome (SDS) [157] are well-known predisposing factors for MDS/AML and exhibit characteristic physical symptoms and signs.
FA is an X-linked or autosomal recessive disorder characterized by genomic instability, hypersensitivity to DNA cross-linking agents, bone marrow failure, and predisposition to hematologic malignancies and solid tumors [143,144,145]. Hematologic abnormalities vary and include cytopenia, erythrocytosis, hypocellular bone marrow with mild dysplasia, and bone marrow failure with increased risk of MDS or AML. The incidence of leukemia is even higher in the FANCD1/BRCA2 subtype of FA, with most cases occurring at less than 5 years of age [158]. This clinically and genetically diverse syndrome is caused by germline mutations in any of at least 23 FA genes (FANCA- FANCW) that function cooperatively in DNA repair. The risk of progression to MDS or AML is very high (cumulative incidence of AML at age 50 years is 10% and MDS at age 50 years is 40%) [159]. Unlike other MDS that are cured by HSCT, these patients have higher post-transplant morbidity and a higher risk of solid tumors compared to non-transplant patients.
DBA usually presents in infancy with macrocytic anemia and reticulocytopenia. Bone marrow histology usually shows aplasia of erythrocytes in normocytic bone marrow. Major causes of morbidity and mortality are associated with side effects of treatment and long-term risk of malignancy [150,151,152]. X-linked variants in GATA1, which encodes a transcription factor important for erythropoiesis, are also a cause of DBA [160]. Disease mechanisms include p53-mediated apoptosis induced by ribosomal stress, increased cell death due to excess free heme with delayed globin production, increased autophagy, and translational changes in selective erythroid-specific transcripts such as GATA1 [161].
DC/TBDs encompass genetically heterogeneous disorders associated with impaired telomere maintenance [153,154,155,156]. They are often associated with hematologic complications such as bone marrow failure, MDS, and AML. The cumulative incidence of MDS in DC/TBDs is estimated to be 2% by age 50 [162]. DC/TBD is associated with many non-hematologic complications, particularly pulmonary fibrosis, liver function abnormalities, and vascular abnormalities. Screening for TBD involves assessing the telomere length of lymphocytes; further genetic testing for specific gene mutations is diagnostically useful. because telomere shortening can also be seen in other diseases [163]. Telomeres shorten as the DNA replication cycle progresses. Critical shortening of telomere length leads to senescence and cell death [164].
SDS is characterized by pancreatic exocrine dysfunction and other physical findings. The most common nonhematologic abnormality is neurologic decompensation, which may be mild or severe, transient or persistent [157]. Other hematologic complications include bone marrow failure, MDS, and AML. In a French cohort of 102 SDS patients, the cumulative incidence of MDS/AML was 18.8% at age 20 and 36.1% at age 30 [165]; SDS is most often caused by an autosomal recessive mutation in the eponymous SBDS gene, resulting in low levels of SDS protein. SDS is involved in the binding of the large and small ribosomal subunits and functions as an elongation factor-like cofactor that removes the anti-binding factor eukaryotic initiation factor 6 (eIF6) from the large subunit [166]. SDS is also involved in the stabilization of mitotic spindles. The spectrum of SBDS variants, including missense, splice site, nonsense, frameshift, and partial or total gene deletions, has been confirmed; AML has been reported in patients with variants in the autosomal recessive gene at DnaJ Heat Shock Protein Family Member C21 genes (DNAJC21) and various clinical features of SDS [167].
5. Conclusions & Perspectives
HHMS exhibits a variety of phenotypes and most HHMS-related genes have clearly defined functions that contribute to hematopoietic regulation. However, the precise nature of this association requires further investigation. Multi-cancer gene panel testing, is beginning to reveal germline abnormalities in genes associated with solid tumor predisposition. For example, variants in breast cancer gene type 1/2 (BRCA1/2), partner and localizer of BRCA2 (PALB2), and TP53 occur in primary or treatment-related hematological malignancies, including AML, ALL, and MDS, narrowing the apparent distinction between solid tumors and hematologic cancer predisposition [168,169,170]. Future development of a hematologic cancer testing panel that is also useful in detecting refractory cytopenia and the risk of relapse refractoriness after leukemia-directed therapy is warranted.
There is a growing need for expert consultation and clinical surveillance of patients with germline predisposition to hematologic malignancies [171]. Troublingly, prognosis and disease progression are slow. Therefore, consultation and treatment strategies must be tailored to the individual patient. Patients and family members with suspected HHMS should be advised of the indications for genetic testing, the limitations of genetic testing, and genetic counseling. This is because curative therapy influences the outcome of allogeneic HSCT, regardless of the phenotypic spectrum or clinical presentation of HHMS [172]. The outcome in these patients is often poor, making them candidates for allogeneic HSCT. Compatible blood stem cell donors should be carefully considered, and donors with known germline variants or unknown retention status should be avoided. There are reports of cases of leukemia after allogeneic transplantation from blood donors [30]. DDX41, CEBPA, GATA2, and others have been reported to be present in 1~2% of allogeneic post-transplant relapses [173] with a median time of recurrence of 5.2 years [174]; there are also reports of onset 10 years after transplantation [31]. The clinical significance of germline predisposition remains unclear, and further case accumulation is desirable. There are many problems characteristic of hematopoietic tumors, such as donor selection, and the establishment of a follow-up system, including genetic counseling and confirmatory testing, is an important area for future research.
Author Contributions
H.A., H.M., S.C. and Y.M. were responsible for the preparation and writing of the manuscript; Y.U., S.M. and N.A. gave academic advice related to the theme of the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This paper was supported by a National Cancer Research and Development expenses grant (2021-A-11), funded by the National Cancer Center, Japan.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Corces-Zimmerman, M.R. and R. Majeti, Pre-leukemic evolution of hematopoietic stem cells: the importance of early mutations in leukemogenesis. Leukemia, 2014. 28(12): p. 2276-82. [CrossRef]
- Godley, L.A., Germline mutations in MDS/AML predisposition disorders. Curr Opin Hematol, 2021. 28(2): p. 86-93. [CrossRef]
- Fenwarth, L., et al., Hereditary Predisposition to Acute Myeloid Leukemia in Older Adults. Hemasphere, 2021. 5(4): p. e552. [CrossRef]
- Guijarro, F., et al., Germ line variants in patients with acute myeloid leukemia without a suspicion of hereditary hematologic malignancy syndrome. Blood Adv, 2023. [CrossRef]
- Churpek, J.E., Familial myelodysplastic syndrome/acute myeloid leukemia. Best Pract Res Clin Haematol, 2017. 30(4): p. 287-289. [CrossRef]
- Hamidi, A., et al., Clinical guideline variability in the diagnosis of hereditary hematopoietic malignancy syndromes. Leuk Lymphoma, 2023. 64(9): p. 1562-1565. [CrossRef]
- Stieglitz, E. and M.L. Loh, Genetic predispositions to childhood leukemia. Ther Adv Hematol, 2013. 4(4): p. 270-90. [CrossRef]
- Babushok, D.V. and M. Bessler, Genetic predisposition syndromes: when should they be considered in the work-up of MDS? Best Pract Res Clin Haematol, 2015. 28(1): p. 55-68. [CrossRef]
- Kotmayer, L., K. Kallay, and C. Bodor, [Hereditary haematological malignancies]. Magy Onkol, 2020. 64(1): p. 43-55.
- Furutani, E. and A. Shimamura, Germline Genetic Predisposition to Hematologic Malignancy. J Clin Oncol, 2017. 35(9): p. 1018-1028. [CrossRef]
- Zahid, M.F., et al., Cytogenetic Abnormalities in Myelodysplastic Syndromes: An Overview. Int J Hematol Oncol Stem Cell Res, 2017. 11(3): p. 231-239. [CrossRef]
- Yoshida, M., et al., Prevalence of germline GATA2 and SAMD9/9L variants in paediatric haematological disorders with monosomy 7. Br J Haematol, 2020. 191(5): p. 835-843. [CrossRef]
- Sahoo, S.S., E.J. Kozyra, and M.W. Wlodarski, Germline predisposition in myeloid neoplasms: Unique genetic and clinical features of GATA2 deficiency and SAMD9/SAMD9L syndromes. Best Pract Res Clin Haematol, 2020. 33(3): p. 101197. [CrossRef]
- Rafei, H. and C.D. DiNardo, Hereditary myeloid malignancies. Best Pract Res Clin Haematol, 2019. 32(2): p. 163-176. [CrossRef]
- Zhang, M.Y., et al., Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet, 2015. 47(2): p. 180-5. [CrossRef]
- Tawana, K., A.L. Brown, and J.E. Churpek, Integrating germline variant assessment into routine clinical practice for myelodysplastic syndrome and acute myeloid leukaemia: current strategies and challenges. Br J Haematol, 2022. 196(6): p. 1293-1310. [CrossRef]
- Arber, D.A., et al., The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood, 2016. 127(20): p. 2391-405. [CrossRef]
- Bohnsack, K.E., et al., Cellular functions of eukaryotic RNA helicases and their links to human diseases. Nat Rev Mol Cell Biol, 2023. [CrossRef]
- Fairman-Williams, M.E., U.P. Guenther, and E. Jankowsky, SF1 and SF2 helicases: family matters. Curr Opin Struct Biol, 2010. 20(3): p. 313-24. [CrossRef]
- Makishima, H., et al., Germ line DDX41 mutations define a unique subtype of myeloid neoplasms. Blood, 2023. 141(5): p. 534-549. [CrossRef]
- Yang, F., et al., Identification and prioritization of myeloid malignancy germline variants in a large cohort of adult patients with AML. Blood, 2022. 139(8): p. 1208-1221. [CrossRef]
- Li, P., et al., The genetic landscape of germline DDX41 variants predisposing to myeloid neoplasms. Blood, 2022. 140(7): p. 716-755. [CrossRef]
- Cheloor Kovilakam, S., et al., Prevalence and significance of DDX41 gene variants in the general population. Blood, 2023. [CrossRef]
- Makishima, H., T.V. Bowman, and L.A. Godley, DDX41-associated susceptibility to myeloid neoplasms. Blood, 2023. 141(13): p. 1544-1552. [CrossRef]
- Sébert, M., et al., Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood, 2019. 134(17): p. 1441-1444. [CrossRef]
- Molteni, E., et al., Prevalence and clinical expression of germline predisposition to myeloid neoplasms in adults with marrow hypocellularity. Blood, 2023.
- Kadono, M., et al., Biological implications of somatic DDX41 p.R525H mutation in acute myeloid leukemia. Exp Hematol, 2016. 44(8): p. 745-754.e4. [CrossRef]
- Tierens, A., et al., Biallelic disruption of DDX41 activity is associated with distinct genomic and immunophenotypic hallmarks in acute leukemia. Front Oncol, 2023. 13: p. 1153082. [CrossRef]
- Badar, T., et al., Clinical and molecular correlates of somatic and germline DDX41 variants in patients and families with myeloid neoplasms. Haematologica, 2023. [CrossRef]
- Kobayashi, S., et al., Donor cell leukemia arising from preleukemic clones with a novel germline DDX41 mutation after allogenic hematopoietic stem cell transplantation. Leukemia, 2017. 31(4): p. 1020-1022. [CrossRef]
- Rolles, B., et al., DDX41 germline variants causing donor cell leukemia indicate a need for further genetic workup in the context of hematopoietic stem cell transplantation. Blood Cancer J, 2023. 13(1): p. 73. [CrossRef]
- Berger, G., et al., Re-emergence of acute myeloid leukemia in donor cells following allogeneic transplantation in a family with a germline DDX41 mutation. Leukemia, 2017. 31(2): p. 520-522. [CrossRef]
- Hirsch, P., et al., Successive relapses from donor and host cells in a patient with DEAD-box helicase 41 (DDX41)-associated myelodysplastic syndrome: The lessons to be learned. Br J Haematol, 2022. 199(4): p. 623-626. [CrossRef]
- Huo, L., et al., Causative germline variant p.Y259C of DDX41 recurrently identified in acute lymphoblastic leukaemia. Br J Haematol, 2023. 202(1): p. 199-203.
- Jelloul, F.Z., et al., DDX41 mutations in patients with non-myeloid hematologic neoplasms. Am J Hematol, 2023. 98(8): p. E193-e196.
- Ma, J., et al., DDX41 is needed for pre- and postnatal hematopoietic stem cell differentiation in mice. Stem Cell Reports, 2022. 17(4): p. 879-893. [CrossRef]
- Chlon, T.M., et al., Germline DDX41 mutations cause ineffective hematopoiesis and myelodysplasia. Cell Stem Cell, 2021. 28(11): p. 1966-1981.e6. [CrossRef]
- Chen, L., et al., The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol Cell, 2018. 69(3): p. 412-425.e6. [CrossRef]
- Nguyen, H.D., et al., Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Res, 2018. 78(18): p. 5363-5374. [CrossRef]
- Singh, S., et al., SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia, 2020. 34(9): p. 2525-2530. [CrossRef]
- Cusan, M., et al., SF3B1 mutation and ATM deletion co-drive leukemogenesis via centromeric R-loop dysregulation. J Clin Invest, 2023.
- Weinreb, J.T., et al., Excessive R-loops trigger an inflammatory cascade leading to increased HSPC production. Dev Cell, 2021. 56(5): p. 627-640.e5. [CrossRef]
- Mosler, T., et al., R-loop proximity proteomics identifies a role of DDX41 in transcription-associated genomic instability. Nat Commun, 2021. 12(1): p. 7314. [CrossRef]
- Shinriki, S., et al., DDX41 coordinates RNA splicing and transcriptional elongation to prevent DNA replication stress in hematopoietic cells. Leukemia, 2022. 36(11): p. 2605-2620. [CrossRef]
- Polprasert, C., et al., Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell, 2015. 27(5): p. 658-70. [CrossRef]
- Yoshida, K., et al., Frequent pathway mutations of splicing machinery in myelodysplasia. Nature, 2011. 478(7367): p. 64-9. [CrossRef]
- Cvitkovic, I. and M.S. Jurica, Spliceosome database: a tool for tracking components of the spliceosome. Nucleic Acids Res, 2013. 41(Database issue): p. D132-41. [CrossRef]
- Singh, R.S., et al., DDX41 is required for cGAS-STING activation against DNA virus infection. Cell Rep, 2022. 39(8): p. 110856.
- Crossley, M.P., et al., R-loop-derived cytoplasmic RNA-DNA hybrids activate an immune response. Nature, 2023. 613(7942): p. 187-194. [CrossRef]
- Challakkara, M.F. and R. Chhabra, snoRNAs in hematopoiesis and blood malignancies: A comprehensive review. J Cell Physiol, 2023. 238(6): p. 1207-1225. [CrossRef]
- Dong, J., et al., Small but strong: Pivotal roles and potential applications of snoRNAs in hematopoietic malignancies. Front Oncol, 2022. 12: p. 939465. [CrossRef]
- Tungalag, S., et al., Ribosome profiling analysis reveals the roles of DDX41 in translational regulation. Int J Hematol, 2023. 117(6): p. 876-888. [CrossRef]
- Ramdzan, Z.M. and A. Nepveu, CUX1, a haploinsufficient tumour suppressor gene overexpressed in advanced cancers. Nat Rev Cancer, 2014. 14(10): p. 673-82. [CrossRef]
- Imgruet, M.K., et al., Loss of a 7q gene, CUX1, disrupts epigenetically driven DNA repair and drives therapy-related myeloid neoplasms. Blood, 2021. 138(9): p. 790-805. [CrossRef]
- Leroy, B., M. Anderson, and T. Soussi, TP53 mutations in human cancer: database reassessment and prospects for the next decade. Hum Mutat, 2014. 35(6): p. 672-88. [CrossRef]
- Hernandez Borrero, L.J. and W.S. El-Deiry, Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim Biophys Acta Rev Cancer, 2021. 1876(1): p. 188556.
- Eisenstein, M., p53: an anticancer protein's chequered past and promising future. Nature, 2022. 603(7899): p. S1.
- Usman, R.M., et al., Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia Pac J Clin Oncol, 2021. 17(3): p. 193-208. [CrossRef]
- Mantovani, F., L. Collavin, and G. Del Sal, Mutant p53 as a guardian of the cancer cell. Cell Death Differ, 2019. 26(2): p. 199-212. [CrossRef]
- Lapke, N., et al., Missense mutations in the TP53 DNA-binding domain predict outcomes in patients with advanced oral cavity squamous cell carcinoma. Oncotarget, 2016. 7(28): p. 44194-44210. [CrossRef]
- Hansen, S., T.R. Hupp, and D.P. Lane, Allosteric regulation of the thermostability and DNA binding activity of human p53 by specific interacting proteins. CRC Cell Transformation Group. J Biol Chem, 1996. 271(7): p. 3917-24. [CrossRef]
- Alvarado-Ortiz, E., et al., Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front Cell Dev Biol, 2020. 8: p. 607670. [CrossRef]
- Gencel-Augusto, J. and G. Lozano, p53 tetramerization: at the center of the dominant-negative effect of mutant p53. Genes Dev, 2020. 34(17-18): p. 1128-1146. [CrossRef]
- Zhu, G., et al., Mutant p53 in Cancer Progression and Targeted Therapies. Front Oncol, 2020. 10: p. 595187. [CrossRef]
- Keymling, M., et al., [Li-Fraumeni syndrome]. Radiologie (Heidelb), 2022. 62(12): p. 1026-1032.
- Sejben, A., et al., [Li-Fraumeni syndrome]. Orv Hetil, 2019. 160(6): p. 228-234.
- Qian, M., et al., TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J Clin Oncol, 2018. 36(6): p. 591-599. [CrossRef]
- Comeaux, E.Q. and C.G. Mullighan, TP53 Mutations in Hypodiploid Acute Lymphoblastic Leukemia. Cold Spring Harb Perspect Med, 2017. 7(3). [CrossRef]
- Swaminathan, M., et al., Hematologic malignancies and Li-Fraumeni syndrome. Cold Spring Harb Mol Case Stud, 2019. 5(1). [CrossRef]
- Preudhomme, C., et al., Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: a study from the Acute Leukemia French Association (ALFA). Blood, 2002. 100(8): p. 2717-23. [CrossRef]
- Pathak, A., et al., Whole exome sequencing reveals a C-terminal germline variant in CEBPA-associated acute myeloid leukemia: 45-year follow up of a large family. Haematologica, 2016. 101(7): p. 846-52. [CrossRef]
- Tawana, K. and J. Fitzgibbon, CEBPA-Associated Familial Acute Myeloid Leukemia (AML), in GeneReviews((R)), M.P. Adam, et al., Editors. 1993: Seattle (WA).
- West, A.H., L.A. Godley, and J.E. Churpek, Familial myelodysplastic syndrome/acute leukemia syndromes: a review and utility for translational investigations. Ann N Y Acad Sci, 2014. 1310(1): p. 111-8. [CrossRef]
- Godley, L.A., Inherited predisposition to acute myeloid leukemia. Semin Hematol, 2014. 51(4): p. 306-21. [CrossRef]
- Harrigan, A.M. and A.M. Trottier, Hereditary acute myeloid leukemia associated with C-terminal CEBPA germline variants. Fam Cancer, 2023. 22(3): p. 331-339. [CrossRef]
- Pabst, T., et al., Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet, 2001. 27(3): p. 263-70.
- Frohling, S., et al., CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations. J Clin Oncol, 2004. 22(4): p. 624-33. [CrossRef]
- Brown, A.L., C.N. Hahn, and H.S. Scott, Secondary leukemia in patients with germline transcription factor mutations (RUNX1, GATA2, CEBPA). Blood, 2020. 136(1): p. 24-35. [CrossRef]
- Xiao, H., et al., First report of multiple CEBPA mutations contributing to donor origin of leukemia relapse after allogeneic hematopoietic stem cell transplantation. Blood, 2011. 117(19): p. 5257-60. [CrossRef]
- Homan, C.C., H.S. Scott, and A.L. Brown, Hereditary platelet disorders associated with germ line variants in RUNX1, ETV6, and ANKRD26. Blood, 2023. 141(13): p. 1533-1543.
- Galera, P., A. Dulau-Florea, and K.R. Calvo, Inherited thrombocytopenia and platelet disorders with germline predisposition to myeloid neoplasia. Int J Lab Hematol, 2019. 41 Suppl 1: p. 131-141. [CrossRef]
- Asou, N., The role of a Runt domain transcription factor AML1/RUNX1 in leukemogenesis and its clinical implications. Crit Rev Oncol Hematol, 2003. 45(2): p. 129-50.
- Forster, A., et al., Beyond Pathogenic RUNX1 Germline Variants: The Spectrum of Somatic Alterations in RUNX1-Familial Platelet Disorder with Predisposition to Hematologic Malignancies. Cancers (Basel), 2022. 14(14). [CrossRef]
- Okumura, A.J., et al., t(8;21)(q22;q22) Fusion proteins preferentially bind to duplicated AML1/RUNX1 DNA-binding sequences to differentially regulate gene expression. Blood, 2008. 112(4): p. 1392-401.
- Homan, C.C., et al., The RUNX1 database (RUNX1db): establishment of an expert curated RUNX1 registry and genomics database as a public resource for familial platelet disorder with myeloid malignancy. Haematologica, 2021. 106(11): p. 3004-3007. [CrossRef]
- Kamath-Loeb, A.S., et al., Accurate detection of subclonal variants in paired diagnosis-relapse acute myeloid leukemia samples by next generation Duplex Sequencing. Leuk Res, 2022. 115: p. 106822. [CrossRef]
- Zharlyganova, D., et al., High frequency of AML1/RUNX1 point mutations in radiation-associated myelodysplastic syndrome around Semipalatinsk nuclear test site. J Radiat Res, 2008. 49(5): p. 549-55.
- Sendker, S., et al., RUNX1 mutation has no prognostic significance in paediatric AML: a retrospective study of the AML-BFM study group. Leukemia, 2023. 37(7): p. 1435-1443. [CrossRef]
- Sood, R., Y. Kamikubo, and P. Liu, Role of RUNX1 in hematological malignancies. Blood, 2017. 129(15): p. 2070-2082. [CrossRef]
- Hayashi, Y., et al., Myeloid neoplasms with germ line RUNX1 mutation. Int J Hematol, 2017. 106(2): p. 183-188. [CrossRef]
- Bellissimo, D.C. and N.A. Speck, RUNX1 Mutations in Inherited and Sporadic Leukemia. Front Cell Dev Biol, 2017. 5: p. 111. [CrossRef]
- Ng, I.K., et al., Preleukemic and second-hit mutational events in an acute myeloid leukemia patient with a novel germline RUNX1 mutation. Biomark Res, 2018. 6: p. 16. [CrossRef]
- Brown, A.L., et al., RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv, 2020. 4(6): p. 1131-1144. [CrossRef]
- Hong, D., et al., RUNX1-dependent mechanisms in biological control and dysregulation in cancer. J Cell Physiol, 2019. 234(6): p. 8597-8609. [CrossRef]
- Deuitch, N., et al., RUNX1 Familial Platelet Disorder with Associated Myeloid Malignancies, in GeneReviews((R)), M.P. Adam, et al., Editors. 1993: Seattle (WA).
- Vyas, H., et al., Prevalence and natural history of variants in the ANKRD26 gene: a short review and update of reported cases. Platelets, 2022. 33(8): p. 1107-1112. [CrossRef]
- Ferrari, S., et al., A novel RUNX1 mutation with ANKRD26 dysregulation is related to thrombocytopenia in a sporadic form of myelodysplastic syndrome. Aging Clin Exp Res, 2021. 33(7): p. 1987-1992. [CrossRef]
- Sullivan, M.J., E.L. Palmer, and J.P. Botero, ANKRD26-Related Thrombocytopenia and Predisposition to Myeloid Neoplasms. Curr Hematol Malig Rep, 2022. 17(5): p. 105-112. [CrossRef]
- Kennedy, A.L. and A. Shimamura, Genetic predisposition to MDS: clinical features and clonal evolution. Blood, 2019. 133(10): p. 1071-1085. [CrossRef]
- Melazzini, F., et al., Clinical and pathogenic features of ETV6-related thrombocytopenia with predisposition to acute lymphoblastic leukemia. Haematologica, 2016. 101(11): p. 1333-1342. [CrossRef]
- Noetzli, L., et al., Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet, 2015. 47(5): p. 535-538. [CrossRef]
- Wang, Q., et al., ETV6 mutation in a cohort of 970 patients with hematologic malignancies. Haematologica, 2014. 99(10): p. e176-8. [CrossRef]
- Di Paola, J. and C.C. Porter, ETV6-related thrombocytopenia and leukemia predisposition. Blood, 2019. 134(8): p. 663-667.
- Rodriguez-Hernandez, G., et al., The Second Oncogenic Hit Determines the Cell Fate of ETV6-RUNX1 Positive Leukemia. Front Cell Dev Biol, 2021. 9: p. 704591. [CrossRef]
- Filipiuk, A., et al., Genetic Disorders with Predisposition to Paediatric Haematopoietic Malignancies-A Review. Cancers (Basel), 2022. 14(15). [CrossRef]
- Schafer, D., et al., Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood, 2018. 131(7): p. 821-826. [CrossRef]
- Wong, J.C., et al., Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight, 2018. 3(14). [CrossRef]
- Tesi, B., et al., Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood, 2017. 129(16): p. 2266-2279. [CrossRef]
- Pastor, V.B., et al., Constitutional SAMD9L mutations cause familial myelodysplastic syndrome and transient monosomy 7. Haematologica, 2018. 103(3): p. 427-437. [CrossRef]
- Mekhedov, S.L., K.S. Makarova, and E.V. Koonin, The complex domain architecture of SAMD9 family proteins, predicted STAND-like NTPases, suggests new links to inflammation and apoptosis. Biol Direct, 2017. 12(1): p. 13. [CrossRef]
- Nagamachi, A., et al., Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell, 2013. 24(3): p. 305-17. [CrossRef]
- Nagamachi, A., et al., Multiorgan failure with abnormal receptor metabolism in mice mimicking Samd9/9L syndromes. J Clin Invest, 2021. 131(4).
- Meng, X. and Y. Xiang, RNA granules associated with SAMD9-mediated poxvirus restriction are similar to antiviral granules in composition but do not require TIA1 for poxvirus restriction. Virology, 2019. 529: p. 16-22. [CrossRef]
- Zhang, F., et al., Human SAMD9 is a poxvirus-activatable anticodon nuclease inhibiting codon-specific protein synthesis. Sci Adv, 2023. 9(23): p. eadh8502. [CrossRef]
- Davidsson, J., et al., SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia, 2018. 32(5): p. 1106-1115. [CrossRef]
- Nagata, Y., et al., Germline loss-of-function SAMD9 and SAMD9L alterations in adult myelodysplastic syndromes. Blood, 2018. 132(21): p. 2309-2313. [CrossRef]
- Yahata, T., et al., Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood, 2011. 118(11): p. 2941-50. [CrossRef]
- Zhou, T., et al., Myelodysplastic syndrome: an inability to appropriately respond to damaged DNA? Exp Hematol, 2013. 41(8): p. 665-74.
- Thomas, M.E., 3rd, et al., Pediatric MDS and bone marrow failure-associated germline mutations in SAMD9 and SAMD9L impair multiple pathways in primary hematopoietic cells. Leukemia, 2021. 35(11): p. 3232-3244.
- Milyavsky, M., et al., A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell, 2010. 7(2): p. 186-97. [CrossRef]
- Parker, J.E., et al., The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood, 2000. 96(12): p. 3932-8.
- Tanase-Nakao, K., T.S. Olson, and S. Narumi, MIRAGE Syndrome, in GeneReviews((R)), M.P. Adam, et al., Editors. 1993: Seattle (WA).
- Yoshizaki, K., et al., MIRAGE syndrome with recurrent pneumonia probably associated with gastroesophageal reflux and achalasia: A case report. Clin Pediatr Endocrinol, 2019. 28(4): p. 147-153. [CrossRef]
- Viaene, A.N. and B.N. Harding, The Neuropathology of MIRAGE Syndrome. J Neuropathol Exp Neurol, 2020. 79(4): p. 458-462. [CrossRef]
- Basilious, A., et al., Lacrimal Gland Hypoplasia and Corneal Anesthesia in MIRAGE Syndrome: A Case Report and Literature Review. Cornea, 2022. 41(8): p. 1041-1044.
- Janjua, D., et al., MIRAGE Syndrome Enteropathy Responding to Pancrelipase Despite Normal Pancreatic Fecal Elastase: A Case Report. Am J Case Rep, 2022. 23: p. e937057. [CrossRef]
- Gorcenco, S., et al., Ataxia-pancytopenia syndrome with SAMD9L mutations. Neurol Genet, 2017. 3(5): p. e183. [CrossRef]
- Vaughan, D., et al., Ataxia pancytopenia syndrome due to SAMD9L mutation presenting as demyelinating neuropathy. J Peripher Nerv Syst, 2020. 25(4): p. 433-437. [CrossRef]
- King-Robson, J., et al., Ataxia-Pancytopenia Syndrome due to a de Novo SAMD9L Mutation. Neurol Genet, 2021. 7(3): p. e580. [CrossRef]
- Raskind, W.H., D.H. Chen, and T. Bird, SAMD9L Ataxia-Pancytopenia Syndrome, in GeneReviews((R)), M.P. Adam, et al., Editors. 1993: Seattle (WA).
- de Pater, E., et al., Gata2 is required for HSC generation and survival. J Exp Med, 2013. 210(13): p. 2843-50.
- Santiago, M., et al., The Clinical Spectrum, Diagnosis, and Management of GATA2 Deficiency. Cancers (Basel), 2023. 15(5).
- Wehr, C., et al., A novel disease-causing synonymous exonic mutation in GATA2 affecting RNA splicing. Blood, 2018. 132(11): p. 1211-1215. [CrossRef]
- Oleaga-Quintas, C., et al., Inherited GATA2 Deficiency Is Dominant by Haploinsufficiency and Displays Incomplete Clinical Penetrance. J Clin Immunol, 2021. 41(3): p. 639-657. [CrossRef]
- Calvo, K.R. and D.D. Hickstein, The spectrum of GATA2 deficiency syndrome. Blood, 2023. 141(13): p. 1524-1532. [CrossRef]
- McReynolds, L.J., K.R. Calvo, and S.M. Holland, Germline GATA2 Mutation and Bone Marrow Failure. Hematol Oncol Clin North Am, 2018. 32(4): p. 713-728. [CrossRef]
- Mir, M.A., et al., Spectrum of myeloid neoplasms and immune deficiency associated with germline GATA2 mutations. Cancer Med, 2015. 4(4): p. 490-9. [CrossRef]
- Wlodarski, M.W., M. Collin, and M.S. Horwitz, GATA2 deficiency and related myeloid neoplasms. Semin Hematol, 2017. 54(2): p. 81-86. [CrossRef]
- Spinner, M.A., et al., GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood, 2014. 123(6): p. 809-21. [CrossRef]
- Hsu, A.P., L.J. McReynolds, and S.M. Holland, GATA2 deficiency. Curr Opin Allergy Clin Immunol, 2015. 15(1): p. 104-9.
- Shimamura, A., Aplastic anemia and clonal evolution: germ line and somatic genetics. Hematology Am Soc Hematol Educ Program, 2016. 2016(1): p. 74-82. [CrossRef]
- Wlodarski, M.W., et al., Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood, 2016. 127(11): p. 1387-97; quiz 1518. [CrossRef]
- Park, M., Overview of inherited bone marrow failure syndromes. Blood Res, 2022. 57(S1): p. 49-54. [CrossRef]
- Li, J. and J.R. Bledsoe, Inherited bone marrow failure syndromes and germline predisposition to myeloid neoplasia: A practical approach for the pathologist. Semin Diagn Pathol, 2023. [CrossRef]
- Deng, J. and L.J. McReynolds, Inherited bone marrow failure syndromes: a review of current practices and potential future research directions. Curr Opin Pediatr, 2023. 35(1): p. 75-83. [CrossRef]
- Dokal, I. and T. Vulliamy, Inherited bone marrow failure syndromes. Haematologica, 2010. 95(8): p. 1236-40.
- Bhandari, J., P.K. Thada, and Y. Puckett, Fanconi Anemia, in StatPearls. 2023: Treasure Island (FL) ineligible companies. Disclosure: Pawan Thada declares no relevant financial relationships with ineligible companies. Disclosure: Yana Puckett declares no relevant financial relationships with ineligible companies.
- Dufour, C. and F. Pierri, Modern management of Fanconi anemia. Hematology Am Soc Hematol Educ Program, 2022. 2022(1): p. 649-657.
- Thakur, B. and K.M. Hiwale, Fanconi Anemia: A Rare Genetic Disorder. Cureus, 2023. 15(5): p. e38899. [CrossRef]
- Da Costa, L., T. Leblanc, and N. Mohandas, Diamond-Blackfan anemia. Blood, 2020. 136(11): p. 1262-1273.
- Da Costa, L.M., I. Marie, and T.M. Leblanc, Diamond-Blackfan anemia. Hematology Am Soc Hematol Educ Program, 2021. 2021(1): p. 353-360.
- Gadhiya, K. and C. Wills, Diamond Blackfan Anemia, in StatPearls. 2023: Treasure Island (FL) ineligible companies. Disclosure: Christina Wills declares no relevant financial relationships with ineligible companies.
- AlSabbagh, M.M., Dyskeratosis congenita: a literature review. J Dtsch Dermatol Ges, 2020. 18(9): p. 943-967. [CrossRef]
- Gitto, L., et al., Dyskeratosis congenita. Autops Case Rep, 2020. 10(3): p. e2020203.
- Garofola, C., A. Nassereddin, and G.P. Gross, Dyskeratosis Congenita, in StatPearls. 2023: Treasure Island (FL) ineligible companies. Disclosure: Ali Nassereddin declares no relevant financial relationships with ineligible companies. Disclosure: Gary Gross declares no relevant financial relationships with ineligible companies.
- Nelson, N., et al., Functional genomics for curation of variants in telomere biology disorder associated genes: A systematic review. Genet Med, 2023. 25(3): p. 100354. [CrossRef]
- Cordell, V. and L. Osoba, Pregnancy in a patient with Schwachman-Diamond syndrome. BMJ Case Rep, 2015. 2015. [CrossRef]
- Woodward, E.R. and S. Meyer, Fanconi Anaemia, Childhood Cancer and the BRCA Genes. Genes (Basel), 2021. 12(10).
- Alter, B.P., et al., Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol, 2010. 150(2): p. 179-88. [CrossRef]
- van Dooijeweert, B., et al., GATA-1 Defects in Diamond-Blackfan Anemia: Phenotypic Characterization Points to a Specific Subset of Disease. Genes (Basel), 2022. 13(3).
- Mello, F.V., et al., Maturation-associated gene expression profiles during normal human bone marrow erythropoiesis. Cell Death Discov, 2019. 5: p. 69. [CrossRef]
- Savage, S.A. and C. Dufour, Classical inherited bone marrow failure syndromes with high risk for myelodysplastic syndrome and acute myelogenous leukemia. Semin Hematol, 2017. 54(2): p. 105-114. [CrossRef]
- Feurstein, S., et al., Telomere biology disorder prevalence and phenotypes in adults with familial hematologic and/or pulmonary presentations. Blood Adv, 2020. 4(19): p. 4873-4886. [CrossRef]
- Victorelli, S. and J.F. Passos, Telomeres and Cell Senescence - Size Matters Not. EBioMedicine, 2017. 21: p. 14-20. [CrossRef]
- Myers, K.C., S.M. Davies, and A. Shimamura, Clinical and molecular pathophysiology of Shwachman-Diamond syndrome: an update. Hematol Oncol Clin North Am, 2013. 27(1): p. 117-28, ix. [CrossRef]
- Tan, S., et al., EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome. Blood, 2019. 134(3): p. 277-290. [CrossRef]
- Godley, L.A., DNAJC21: the new kid on the SDS block. Blood, 2017. 129(11): p. 1413-1414. [CrossRef]
- Tawana, K., M.W. Drazer, and J.E. Churpek, Universal genetic testing for inherited susceptibility in children and adults with myelodysplastic syndrome and acute myeloid leukemia: are we there yet? Leukemia, 2018. 32(7): p. 1482-1492. [CrossRef]
- Roloff, G.W. and E.A. Griffiths, When to obtain genomic data in acute myeloid leukemia (AML) and which mutations matter. Blood Adv, 2018. 2(21): p. 3070-3080. [CrossRef]
- Padella, A., et al., Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J Hematol Oncol, 2022. 15(1): p. 10. [CrossRef]
- Godley, L.A. and A. Shimamura, Genetic predisposition to hematologic malignancies: management and surveillance. Blood, 2017. 130(4): p. 424-432. [CrossRef]
- Saygin, C., et al., Allogeneic hematopoietic stem cell transplant outcomes in adults with inherited myeloid malignancies. Blood Adv, 2023. 7(4): p. 549-554. [CrossRef]
- Williams, L., et al., Genetics of donor cell leukemia in acute myelogenous leukemia and myelodysplastic syndrome. Bone Marrow Transplant, 2021. 56(7): p. 1535-1549. [CrossRef]
- Gibson, C.J., et al., Donor Clonal Hematopoiesis and Recipient Outcomes After Transplantation. J Clin Oncol, 2022. 40(2): p. 189-201. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



