
Submitted:
06 November 2023
Posted:
08 November 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The new coronavirus SARS-CoV-2 is the cause of the COVID-19 pandemic, which has created an incomparable global health problem. PLpro is a key protein involved in viral polyprotein processing and immune system evasion, making it a prime target for the development of antiviral drugs to combat COVID-19. The review begins by elucidating the functional and structural properties of SARS-CoV-2 PLpro, underscoring its significance in viral pathogenicity and replication. Employing homology modeling, a tertiary structure model of PLpro is constructed, laying the groundwork for subsequent investigations. To expedite the search for potential therapeutic candidates, the study harnesses computational methodologies. Molecular docking techniques are employed to explore binding sites for antiviral drugs within the catalytic region of PLpro. This computational approach not only aids in drug development but also provides crucial preliminary data before experimental research commences. The stability of drug-PLpro complexes is assessed through comprehensive all-atom molecular dynamics (MD) simulations, affording dynamic insights into drug-PLpro interactions. By evaluating binding energy estimates from MD simulations, stable drug-PLpro complexes with potential antiviral properties are identified. However, it is emphasized that experimental validation is imperative to confirm these computational findings, necessitating in vitro and in vivo tests to assess safety and efficacy. This pivotal stage bridges the computational realm with real-world clinical applications. The review emphasizes the imperative need for enhancements in the existing PLpro-targeting drugs currently on the market. Their actions are insufficient for clinical use and necessitate thorough characterization and improvement. The Supervised Molecular Dynamics (SuMD) simulation technique reveals critical information about drug unbinding pathways, shedding light on opportunities for refinement. In addition to optimizing existing drugs, the investigation of sub-structurally similar molecules emerges as a promising avenue for drug development. In conclusion, the review underscores the pivotal role of targeting SARS-CoV-2 PLpro in the ongoing battle against COVID-19. By integrating molecular dynamics simulations, structural modeling, and computational insights, a robust foundation is established for identifying promising antiviral drug candidates. The continual need for improvement in existing drugs and the exploration of novel compounds remain paramount in the global effort to combat COVID-19. The evolution and management of COVID-19 hinge on the symbiotic relationship between computational insights and experimental validation, illustrating the interdisciplinary synergy crucial to this endeavor.
Keywords:
SARS-CoV-2
; Papain-like protease
; COVID-19
; Molecular modelling
; Molecular dynamics simulations
; Drug discovery
; Antiviral drugs
; Inhibitors
; Therapeutic agents
; therapeutic targets
; Irreversible inhibitors
; Repurposing drugs
1. Introduction
A new coronavirus known as SARS-CoV-2, which exhibits structural similarities with the virus causing severe acute respiratory syndrome (SARS), emerged during the COVID-19 epidemic in December 2019 [1]. SARS-CoV-2 has resulted in a significant global burden of disease and mortality, with the Worldometer coronavirus monitoring tool providing daily updated status information on more than 696.4 million confirmed cases and 6.9 million fatalities. Following previous coronavirus illness outbreaks, such as SARS in 2002–2003 and Middle East respiratory syndrome (MERS) in 2012, this event presented serious obstaacles to the fields of public health, research, and medicine. SARS-CoV-2, the virus that causes COVID-19, started at a seafood market in Wuhan, China, and quickly spread to other parts of the world, sparking a pandemic with significant global implications [2,3].
Like other coronaviruses, the SARS-CoV-2 virus possesses a distinctive round or multishaped virion particle with a diameter of 120–160 nm that contains the triple Spike (S) protein, which is in charge of virus attachment and membrane fusion during infection [4]. In addition to the S protein, the viral genome codes for three other structural proteins that play distinct roles in the structure and replication of the virus: the Membrane (M) protein, the Envelope (E) protein, and the Nucleocapsid (N) protein [5].
SARS-CoV-2 is a member of the Betacoronavirus genus and possesses a large single-stranded RNA genome that codes for several different proteins, including accessory and structural proteins [6]. By means of a ribosomal frame-shifting mechanism, non-structural proteins (Nsps) are translated from big polyproteins, Pp1a and Pp1ab, during the replication process of SARS-CoV-2 [7]. The release and maturation of the 15 Nsps, which make up the replicase-transcriptase complex in charge of transcription and replication of the viral genome, depend on appropriate polyprotein processing [7].
Since it interacts to the angiotensin-converting enzyme 2 (ACE2) receptor on the surface of host cells, the spike (S) protein facilitates viral entry and is crucial to the SARS-CoV-2 infection process [8]. The two primary domains of the S protein are S1 and S2, where S1 mediates ACE2 binding and S2 facilitates viral membrane fusion with the host cell membrane [9]. The S1 domain’s receptor-binding domain (RBD) interacts with ACE2, and the entry of the virus into host cells depends on the cleavage of the S protein at specific areas [10].
The lipid membrane that envelops coronaviruses is produced from the host cell, and the spike (S) protein on their surface gives them their distinctive halo-like appearance [11]. Among all RNA viruses, the coronavirus genome is one of the biggest known [12]. It is a single-stranded RNA with positive polarity [13].
With multiple candidates in development and several vaccines in use globally by early 2022, the development of COVID-19 vaccines moved quickly after the SARS-CoV-2 genetic sequence was made public in January 2020 [14]. The virus has infected millions of people and significantly increased mortality rates despite vaccine efforts, particularly in low-income nations [15].
One of SARS-CoV-2’s non-structural proteins, papain-like protease (PLpro), has a variety of functions in the growth and replication of the virus [16]. It contributes to the cleavage of viral polyproteins and, through downregulating type I interferon production, impedes the host’s immune response to the virus [17]. The PLpro is a desirable target for therapeutic research because of its capacity to degrade ubiquitin-like interferon-stimulated gene 15 protein (ISG15) from interferon-responsive factor 3 (IRF3) [18].
Recent years have seen an increase in the use of the computational method known as molecular dynamics (MD) simulation to investigate peptides and peptide-like compounds that may be able to suppress SARS-CoV-2. SARS-CoV-2 PLpro in particular has drawn attention as a potential therapeutic target because of its vital function in both the host’s immune response regulation and viral replication [19]. To find potential drugs to treat SARS-CoV-2, computational techniques like homology modeling, molecular docking, and MD simulations have been used [20].
Our research study, "Unveiling the Inhibitory Potentials of Peptidomimetic Azanitriles and Pyridyl Esters towards SARS-CoV-2 Main Protease: A Molecular Modelling Investigation," describes how we evaluated new peptidomimetic nitriles that are azatripeptide and azatetrapeptide as possible inhibitors of the SARS-CoV-2 main protease. We used molecular docking, molecular dynamics (MD) simulations, and several post-MD analyses, such as percentage hydrogen occupancy, in this investigation. These methods were used to clarify the binding free energy potentials that the chosen compounds displayed in response to SARS-CoV-2. Additionally, we pinpointed the precise residues responsible for the drug-binding characteristics of the selected inhibitors with relation to SARS-CoV-2 Mpro. Furthermore, the focus of our current studies, "In Silico Analysis of Repurposed Antiviral Drugs as Prospective Therapeutic Agents for COVID-19: Molecular Docking and Dynamics Simulations Targeting the 3-Chymotryspine-Like Protease (3CLpro) and the Papain-Like Protease (PLPro)," is on how Ritonavir, Lopinavir, Ombitasvir, Paritaprevir, Isotretinoin, Ginkgolic Acids, and GRL0617 might reduce the activity of the 3CLpro enzyme. Targeting 3CLpro and PLpro makes sense because of its critical function in viral replication. In order to investigate these compounds’ potential as COVID-19 therapeutic agents, this study aims to shed light on the binding interactions of these compounds. This investigation entails examining their capacity to bind to and inhibit the activity of the 3CLpro and PLpro enzymes, essential targets in the development of coronavirus-targeting antiviral medications.
To sum up, the COVID-19 pandemic has increased the demand for efficient antiviral drugs, and there has been a lot of interest in the potential therapeutic role of SARS-CoV-2 PLpro [21]. In-depth analysis of the virus’s structural and functional features, vaccines developments, and the importance of PLpro targeting in the ongoing fight against COVID-19 are all covered in this review, which also emphasizes the use of computational methods like MD modeling in drug discovery.
2. Implications for Viral Evolution (Target Enzymes and Receptors)
The molecular interactions between the virus and host cells, particularly those involving viral enzymes and host receptors, have a complex connection to both the evolution of viruses and the control of COVID-19 [22]. In order to comprehend this relationship, we must go into the various phases of the SARS-CoV-2 life cycle and the potential targets for therapeutic treatments [6]. A crucial step in the life cycle of a virus is the attachment to host cells that occurs at the beginning of the infection [6]. Initially, the human angiotensin-converting enzyme 2 (hACE2) receptor is how SARS-CoV-2 binds to human cells [23]. One possible treatment approach to stop viral infection in its tracks is to interfere with this first virus-host contact [24]. Once within the host cell, the replication of SARS-CoV-2 is dependent on the action of particular viral enzymes [6]. In this setting, two crucial proteases are the 3-chymotrypsin-like protease (3CLpro) and the papain-like protease (PLpro) [25]. The cleavage of viral polyproteins by these proteases results in the production of non-structural proteins that are necessary for viral replication [26]. Thus, these proteases become excellent targets for the creation of anti-SARS-CoV-2 drugs that will successfully prevent viral replication, thereby lessening the severity and transmission of COVID-19 [27]. When comparing SARS-CoV-2 to its predecessor, SARS-CoV, which produced the SARS pandemic in 2002–2003, the importance of these proteases, particularly PLpro, in the context of SARS-CoV-2’s life cycle becomes even more evident [28]. Despite having a significant sequence identity with SARS-CoV, SARS-CoV-2 and SARS-CoV depend on different proteases, which is a crucial characteristic that distinguishes the two pathogenicities [29]. In particular, PLpro is essential for the cleavage of proteins in the host’s innate immune pathways, which permits the replication of SARS-CoV-2 [25]. In addition to viral proteases, RNA-dependent RNA polymerase (RdRp) is another essential enzyme involved in the viral life cycle [30]. This enzyme could be the target of a therapeutic intervention because it is essential for the synthesis of viral RNA [31]. While this role is well-recognized in the setting of SARS-CoV, it is equally significant in the case of SARS-CoV-2 [32].
Figure 1.
SARS-CoV-2 life cycle and the potential targets for therapeutic treatments as adapted from source.
Figure 1.
SARS-CoV-2 life cycle and the potential targets for therapeutic treatments as adapted from source.
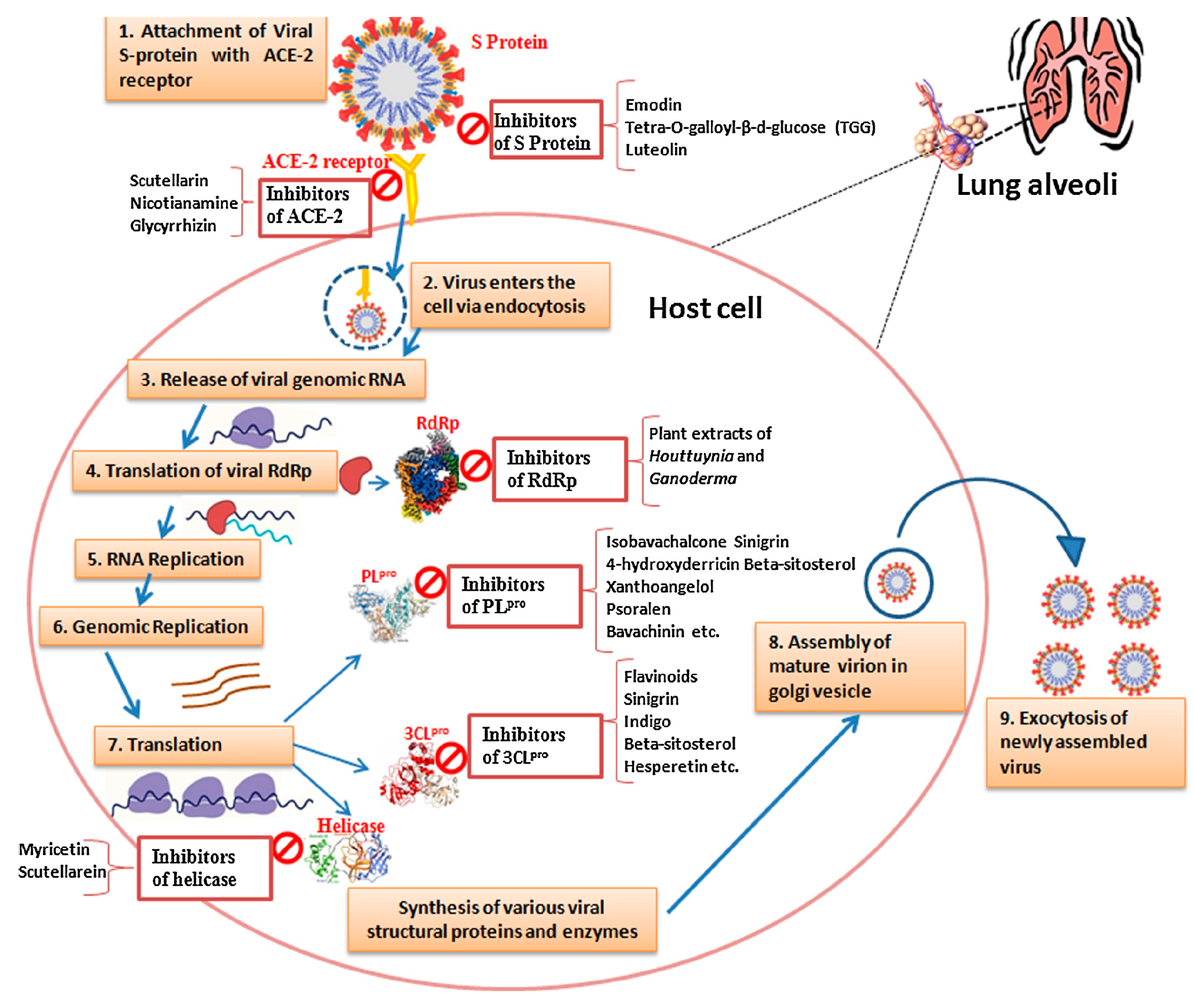
One key factor in the development of COVID-19 is the interaction between the virus and host receptors, particularly ACE2 receptor [33]. How the virus impacts distinct organs is largely dependent on the expression of ACE2 in the lungs, heart, kidneys, and intestines, among other host tissues [34]. It is noteworthy that ACE2 seems to have two roles in infections with SARS-CoV-2 [35]. It protects the lungs from acute respiratory lung injury, including damage brought on by the SARS-spike protein, but it also increases viral susceptibility [36]. Apart from ACE2, it has been shown that SARS-CoV-2 entrance and pathogenesis are mediated by other receptors and proteases, including TMPRSS2, DPP4, and cathepsin B and L [37]. Presently discovery of the exact processes by which these receptors and proteases aid in viral entrance are still studied [38]. However, these results highlight their potential as therapeutic intervention targets.
The spike protein and the activation of viral entry by cellular proteases are other examples of the interaction between the virus and host [39]. Inhibitors that target TMPRSS2, in particular, have demonstrated promise in reducing viral infection. TMPRSS2 is essential in priming the spike protein for viral entry [40,41].
Proteomics and genomics of SARS-CoV-2 are equally intriguing. Proteases such as 3CLpro are essential in breaking down polyproteins into useful proteins [42]. The virus codes for a variety of structural and non-structural proteins. The Replicase/Transcriptase Complex (RTC), which is necessary for viral transcription, replication, and maturation, is formed in part by this process [26,43]. Enzymes like nsp3, nsp4, nsp5, nsp12, nsp13, and nsp14 are among the many components of RTC that work together to coordinate various stages of viral replication [44]. These enzymes reveal themselves as excellent possibilities for the creation of antiviral drugs since they lack near homologues in hosts [45].
A crucial step in the viral entry process is the interaction of SARS-CoV-2 with a variety of proteases and receptors, such as TMPRSS2, CatB/L, and furin proteases [46]. Finding inhibitors for these proteases is a promising approach to stop the spread of viruses [47]. Effective antiviral drugs require a thorough understanding of the host receptors and viral enzymes involved in SARS-CoV-2 infection [9,48]. We might discover novel approaches to stop viral replication, lessen the severity of COVID-19, and possibly contain this worldwide epidemic by focusing on particular proteins and receptors [49].

Table 1.
Summary the various enzymes, receptors, and their functions in the context of SARS-CoV-2.
| Enzyme/Receptor | Function/Role | Target for Therapeutic Development | Virus-Host Interaction | Implication in Viral Evolution and SARS-CoV-2 Management | References |
|---|---|---|---|---|---|
| ACE2 | Viral Entry, Lung Protection | Therapeutic Target | Facilitates viral entry and cross-talk with host cells | Dual role in COVID-19 infections; potential protection from acute lung injury and ARDS; increased susceptibility due to high ACE2 expression | [50,51] |
| TMPRSS2 | Viral Entry, Priming | Potential Inhibitor | Mediates viral entry and spike protein cleavage | Important for viral entry; inhibitors may prevent infection and reduce viral spread | [52] |
| DPP4 | Possible Receptor | Role Uncertain | Possible binding to SARS-CoV-2 | Role in virus-host interaction needs further investigation; may be involved in viral entry | [53] |
| PLpro (Protease) | Viral Replication, Polyprotein Cleavage | Drug Target for Inhibition | Essential for viral replication | Critical for cleaving polyproteins; potential drug target for inhibiting viral replication | [54] |
| 3CLpro (Mpro) | Polyprotein Cleavage, Replication | Drug Target for Inhibition | Essential for viral replication | Key for cleaving polyproteins; promising target for antiviral strategies | [55,56] |
| RdRp (RNA Polymerase) | RNA Synthesis, Replication | Potential Drug Target | Crucial for viral genome replication | Required for replication; promising drug target for antiviral strategies | [44] |
| Helicase (NSP13) | RNA Unwinding, Replication | Potential Target | Facilitates RNA unwinding and replication | Important for viral genome replication; potential therapeutic target for anti-COVID-19 strategies | [57] |
| Cathepsin B/L | Viral Entry | Target for Inhibiting Entry | Involved in viral entry | Blocking cathepsin activity can prevent viral entry | [58,59,60] |
| Furin | S Protein Cleavage | Potential Drug Target | Cleavage of S protein and virus entry | Promising target for inhibiting viral entry and spread | [60,61] |
3. Structure and Function of SARS-CoV-2 PLpro
A key player in the genomic RNA replication of SARS-CoV, and also important for the closely related SARS-CoV-2, is the papain-like cysteine protease (PLpro) [18]. Three nonstructural proteins (NSPs-1, 2, and 3) are produced when polyproteins (PPs) have their N-terminal regions cut by PLpro. PLpro’s catalytic core domain, which includes the consensus sequence LXGG, is a crucial component. Since this sequence is essential for cleaving the replicase substrate, PLpro is an excellent candidate for the creation of antiviral drugs that specifically target SARS-CoV-2 [62]. Three domains have been identified in the crystal structure of SAR-CoV-2 PLpro, which highlights the catalytic cysteine cleavage domain, a potential labile Zn-binding domain, and a ubiquitin domain [63]. The labile ZnII ion is essential for preserving the structural integrity of SAR CoV-2 PLpro, while the catalytic site, is defined by the amino acid triad Cys111-His272-Asp286 [64]. All of these domains are viable targets for therapy because the ubiquitin domain is connected to the host’s innate immune responses [65].
Playing a dual purpose is one of the primary roles of the conserved cysteine residue in viral PLpro [62]. First and foremost, it binds to the ZnII ion, which is necessary to preserve correct protein folding and stability of the local geometry [66]. Secondly, it attacks the substrate in the capacity of a nucleophile, changing PLpro’s structure and functionality [67]. Potential antiviral methods to interfere with PLpro activity and hinder viral replication include inhibiting the catalytic cysteine at the active site or focusing on the elimination of the labile ZnII ion [68,69].
Figure 2.
Redrawn SARS-CoV-2 papain-like protease structure as adapted from source].
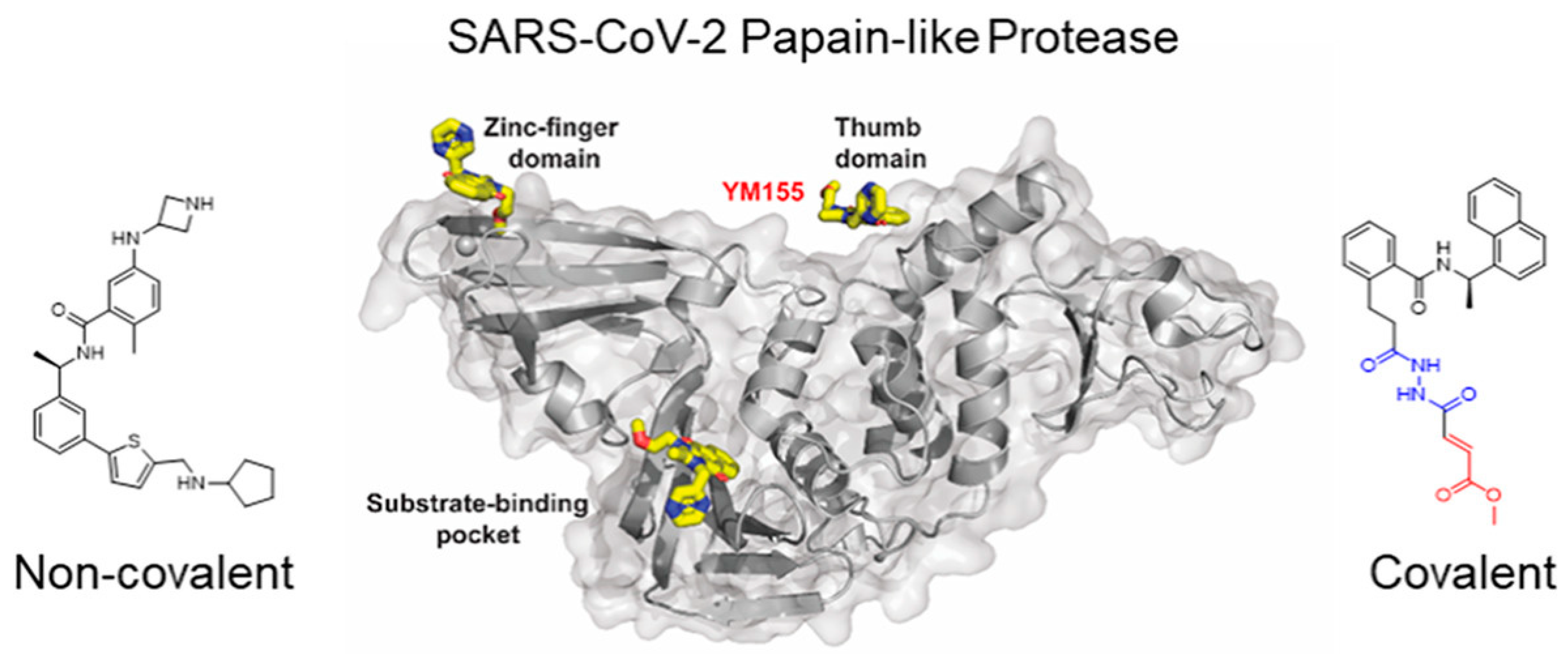
SARS-CoV-2 drug development interests seem to be drawn to PLpro, although other subdomains such as nsp3, nsp10, and nsp13 also have putative labile Zn sites, which broadens the pool of possible targets. As a result of their weaker selectivity, Zn-ejector drugs present a problem in the development process because they could affect different Zn-containing proteins in the human body [70]. Substance-based inhibitors, on the other hand, exhibit promise in stopping viral replication and hence reducing infection since they specifically bind the catalytic cysteine at the active site of SARS-CoV-2 PLpro. Peptide-based inhibitors offer potential for increased antiviral efficacy and allow for more rational design. As a result, thiol-targeting inhibitors are a potentially effective treatment for SARS-CoV-2.
Nsp3, which contains the PLpro domain, stands out as a crucial element in the extensive genome of SARS-CoV-2. Between a nucleic acid-binding domain (NAB) and the SARS unique domain (SUD/HVR), in Nsp3, lies a large multidomain protein that includes PLpro. It is often found in two copies, known as PL1pro and PL2pro, and is highly conserved across coronaviruses [71]. The cleavage of peptide bonds at particular places by these cysteine proteases releases Nsp1, Nsp2, and Nsp3, which are essential for viral replication [72]. The identification and cleavage of PLpro depend on the LXGG motif present in Pp1a/Pp1ab. Beyond its involvement in viral replication, PLpro performs a variety of other tasks that affect host immunological responses, such as deubiquitinating and deISG15ylating [71]. It is a prime candidate for treatment since it affects many host cellular pathways, suggesting that it plays a crucial role in the growth and replication of viruses [73].
Previous research on SARS-CoV-2 antiviral drug development has mostly concentrated on particular Nsp proteins, such as Nsp3 PLpro, Nsp5 Mpro, and Nsp12 RNA-dependent RNA polymerase, which were found from investigations on other coronaviruses [73]. The argument for focusing on the SARS-CoV-2 Nsp3 domain, PLpro, is discussed in this context. This enzyme’s potential as a therapeutic target is highlighted by its conservation across coronaviruses and low sequence similarity to human enzymes [48]. Many coronaviruses have structural characterizations of PLpro available, which are a great resource for structure-based drug development [74]. Nevertheless, obstacles have arisen in the development of antivirals against SARS-CoV and SARS-CoV-2, requiring new approaches [75].
Despite having less research done on it than Mpro, PLpro is a multifunctional protein that functions as a deubiquitinase and a cysteine protease, which opens the door for the development of powerful PLpro inhibitors [64]. This special combination of roles affects host immune responses in addition to viral protein processing [64]. Thus, PLpro is an appealing target for antivirals since blocking its functions may prevent viral replication and interfere with host immune systems [76]. The expertise obtained from investigating SARS-CoV PLpro can be quickly used to the investigation of SARS-CoV-2 PLpro, hence accelerating the development of new antivirals and retargeting drug methods for SARS-CoV-2 [74]. Developing effective inhibitors will need a full understanding of the substrate specificity, structure, and processes of SARS-CoV-2 PLpro [25]. This presents a promising path for the creation of antiviral medications to battle the COVID-19 pandemic.
SARS-CoV-2, the agent responsible for causing COVID-19, is a positive-sense genomic RNA that encodes ten open reading frames (ORFs) [77]. Of these, the majority of the genome is made up of ORF1ab. ORF1ab translates into two big polypeptide products, pp1a and pp1ab, in the host cell [42]. Proteolytic cleavage of these polypeptides is necessary to produce the crucial nonstructural proteins (nsps) required for viral replication [26]. Notably, two cysteine proteases, PLpro (Nsp3) and MPro (Nsp5), catalyze the autocatalytic cleavage process [78]. Nsp1, Nsp2, and Nsp3, which are essential for host regulation and viral replication, are released when PLpro recognizes and cleaves LXGG patterns between viral proteins [71]. Furthermore, PLpro is a desirable target for the development of antiviral drugs due to its deubiquitinating and deISGylating actions that influence the host’s innate immune responses [18,79].
Since PLpro is a difficult pharmacological target, there hasn’t been much research done on its inhibitors [80]. When contrast to MPro, its active site pocket is flatter [81]. Finding initial hits with potential PLpro inhibitors for additional optimization can be facilitated by screening for cysteine protease and deubiquitinase inhibitors, given its dual activity as a cysteine protease and deubiquitinase [82].
Viral protein processing is greatly aided by PLpro, which is distinguished by its Cys-His-Asp catalytic triad and is essential for immune evasion [21]. Not only does inhibition of PLpro prevent viral replication, but it also interferes with different host immunological responses, underscoring its potential as a crucial target for drug development [49].
The insights and expertise obtained by studying SARS-CoV PLpro can be applied immediately to improve the development of antivirals targeting SARS-CoV-2 PLpro [48]. A thorough comprehension of the substrate specificity, structure, and mechanism of SARS-CoV-2 PLpro will be essential for designing potent PLpro inhibitors and opening the door for the creation of antiviral drugs to treat COVID-19 [74].
4. Multifaceted Approach with MD Simulations Targeting SARS-CoV-2 Papain-Like Protease (PLpro)
In the midst of the ongoing SARS-CoV-2 pandemic, researchers have taken a variety of approaches to finding efficient ways to fight the virus [83]. Targeting Papain-like Protease (PLpro), the virus’s main protease enzyme, is essential to this mission [74]. To tackle the problems caused by this multifunctional protein, scientists are using thiol-reacting inhibitors, substrate-based peptides, FDA-approved drugs repurposing, and molecular dynamics (MD) simulations [84].
Finding thiol-reacting inhibitors is a potential path, especially if they can covalently bind to the conserved cysteine residues in SARS-CoV-2-PLpro [85]. By forming a Cys-inhibitor complex through this covalent connection, the PLpro is rendered inactive and viral replication is impeded [86]. The capacity of Zn-ejector compounds to remove zinc ions (ZnII) from the PLpro active site has drawn significant interest [87]. Disulfiram (DSF) and other disulfide-based thiol-reacting inhibitors are notable among them [87]. The potential of DSF, an FDA-approved drug for treating persistent alcohol abuse, as a Zn-ejector has been thoroughly investigated [88]. DSF has potential as a Zn-ejector drug against CoV-2-PLpro because of the close genetic and structural similarities between SARS-CoV-2-PLpro and SARS-CoV-PLpro [89]. The multifunctionality of DSF has been revealed by recent investigations [90]. In addition to acting as a Zn-ejector, it also modifies cysteines, causing damage to SARS-CoV-2 PLpro’s structural and functional integrity [91]. Due to their combined effects, which limit viral replication, additional FDA-approved sulfur-based drugs are being investigated for their potential to be effective against COVID-19 [92]. Tests have been conducted on compounds such as diethyldithiocarbamate (DDC), captopril, 2,2′-dithiodipyridine, 2,2′-dithiobis(benzothiazole), and 6-thioguanine (6-TG) [89]. Notably, in cell cultures, 6-TG has demonstrated a significant inhibition of SARS-CoV-2 PLpro [21]. Moreover, even at low micromolar concentrations, the FDA-approved drug auranofin used to treat rheumatoid arthritis has shown a strong ability to hinder SARS-CoV-2 replication in human cells [93].
As mentioned above, SARS-CoV-2 PLpro, which recognizes and cleaves peptide bonds at LXGG patterns between nonstructural proteins (nsps), is a crucial component in the virus’s replication mechanism [71]. These motifs are the target of small peptides that are developed to function as substrate-based inhibitors [87]. The creation of two highly selective tetrapeptides that effectively inhibit SARS-CoV-2 PLpro, VIR250 and VIR251, is the result of recent breakthroughs [74]. The purpose of these peptides is to bind to the catalytic Cys111 residue via thioether linkage, so blocking the enzyme’s ability to cleave polyproteins and stopping the spread of the virus [94]. Despite its greater potency, VIR251 displays less selectivity for binding SARS-CoV-2 PLpro because of variations in the catalytic site’s amino acid orientation [30].
Repurposing FDA-approved drugs is an alternative therapy approach being studied for COVID-19 [95]. The FDA has approved Remdesivir, an RNA-dependent RNA polymerase inhibitor, as the only antiviral drug for treating COVID-19 [96]. Its efficacy in patients is, however, rather moderate [97]. Ritonavir has been used to block SARS-CoV-2 PLpro which it is well-known anti-HIV-1 properties [48]. This drug affects the coordination of signals within infected cells by targeting polyproteins produced by viral proteases [49].
Understanding the stability and interactions between small molecules and proteins is made possible through the use of molecular dynamics simulations [98]. This computational method sheds light on the kinetics of binding and interactions between drugs and proteins.
Table 2.
MD simulation studies for SARS-CoV-2 PLpro inhibitors.
| Inhibitor/Drug Candidate | Simulation Method | Key/Type of Interaction | Simulation Length | Force Field Used | Binding Free Energy | Mechanism of Action | Ref. |
|---|---|---|---|---|---|---|---|
| GRL-0617 | Molecular Dynamics Simulation | Non-covalent binding | 100 ns | AMBER | -21.5 kcal/mol | Non-covalent inhibition of PLpro. | [99] |
| VIR250 and VIR251 | Molecular Dynamics Simulation | Irreversible binding | 50 ns | OPLS-AA | Not available | Irreversible inhibitors of PLpro. | [100,101] |
| Neobavaisoflavone | Molecular Dynamics Simulation | Low energy binding | 75 ns | CHARMM36 | Not available | Binding to the catalytic triad of PLpro. | [102] |
| Ritonavir | Molecular Dynamics Simulation | Binding analysis | 50 ns | GROMOS | -8.2 kcal/mol | Investigated for potential PLpro inhibition. | [20] |
| Dasabuvir (A17) | Molecular Dynamics Simulation | Stable binding | 100 ns | CHARMM27 | -11.7 kcal/mol | Stable binding with PLpro. | [103] |
| Methisazone (A34) | Molecular Dynamics Simulation | Stable binding | 75 ns | AMBER | -12.3 kcal/mol | Exhibits stable dynamic behavior in complex. | [103] |
| Vaniprevir (A53) | Molecular Dynamics Simulation | High binding affinity | 100 ns | OPLS-AA | Not available | Shows high binding affinity to PLpro. | [103] |
| Baicalein | Molecular Dynamics Simulation | Binding to active site | 50 ns | CHARMM36 | -12.8 kcal/mol | Binds to the active site of PLpro. | [104] |
| Disulfiram | Molecular Dynamics Simulation | Inhibition analysis | 75 ns | GROMOS | Not available | Repurposed for potential PLpro inhibition. | [105] |
| Carmofur | Molecular Dynamics Simulation | Binding to PLpro | 100 ns | AMBER | -10.5 kcal/mol | Demonstrates binding to PLpro. | [106] |
| Ebselen | Molecular Dynamics Simulation | Antiviral activity | 75 ns | CHARMM27 | Not available | Investigated for its antiviral activity. | [107] |
| Tideglusib | Molecular Dynamics Simulation | Potential inhibitor | 50 ns | CHARMM36 | Not available | Explored for its potential as an inhibitor. | [106] |
| Shikonin | Molecular Dynamics Simulation | Active site binding | 100 ns | AMBER | -15.6 kcal/mol | Binds to the active site of PLpro. | [108] |
| PX-12 (Belinostat) | Molecular Dynamics Simulation | Inhibition potential | 75 ns | GROMOS | Not available | Investigated for its inhibition potential. | [49] |
| Sub-structurally Similar Compounds with Ritonavir | Molecular Dynamics Simulation | Antiviral drug potential | 50 ns | OPLS-AA | Not available | Explored for their antiviral potential. | [109] |
Since they are commercially available and have a track record of safety, FDA-approved drugs make perfect repurposing candidates [110]. One important goal is to identify the most effective options among these drugs based on their robust binding ability to SARS-CoV-2 PLpro [74]. To assess the stability, binding affinity, and interactions of drug-PLpro binding, molecular mechanics/generalized Born surface area (MM/GBSA) binding energy estimates are used after molecular dynamics simulations [111].
Molecular dynamics simulations are utilized to study the stability and interactions of small molecules bonded to proteins in more detail [112]. Since they are readily available on the market, have a track record of safety, and may be used directly without requiring further preclinical or clinical testing, FDA-approved drugs are the main alternatives for treatment in emergency situations [113]. The ultimate goal of this research is to repurpose drugs for the inhibition of SARS-CoV-2 by identifying the best candidates among FDA-approved drugs with substantial binding potential for SARS-CoV-2 PLpro [48]. Using molecular dynamics simulations over 50 ns, the most promising drugs based on the anticipated docking scores for COVID-19 PLpro will be examined in further detail [114]. Throughout the MD simulated time, drug-PLpro binding energies (ΔGbinding) will be assessed using a molecular mechanics/generalized Born surface area (MM/GBSA) technique [115]. Estimates will also be made of the drug-PLpro binding’s stability, affinity, and interactions [116].
Three antioxidants and cell protectants (NAD+, quercitrin, and oxiglutatione), three antivirals (ritonavir, moroxydine, and zanamivir), two antimicrobials (doripenem and sulfaguanidine), two anticancer drugs, three benzimidazole anthelmintics, one antacid (famotidine), three anti-hypertensive ACE receptor blockers (candesartan, losartan, and valsartan), and other various systemically or topically acting drugs were among the top results expected to bind with SARS-CoV-2 PLpro strongly [117]. These drugs binding patterns were superior to those of 6-mercaptopurine (6-MP), the previously discovered SARS CoV PLpro inhibitor, indicating the possibility of using these drugs as an alternate way to treat COVID-19 [118].
One drug that has been repurposed to inhibit PLpro function in the replication process is ritonavir, which is an inhibitor of protease that works against the Human Immunodeficiency Virus Type 1 (HIV-1) [119]. Ritonavir provides a multifaceted approach to stop the growth of viruses by interfering with the cleavage of the polyproteins produced by viral proteases, which is necessary for the transcription and replication of viral RNA [120].
The use of SuperNatural Database conducted a high throughput virtual screening (HTVS) program with the aim of finding inhibitors that target Papain-Like Proteases (PLpro) [121]. This later, revealed that two substances, SN00334175 and SN00162745, showed docking scores of -10.58 kcal/mol and -9.93 kcal/mol, respectively, according to the XP docking results [122]. Furthermore, Van der Waal energy and hydrophobic energy components were identified as significant contributors to the overall binding free energy in the PRIME MMGB-SA investigations [123]. SN00334175/7JN2 and SN00162745/7JN2 were shown to be stabilized by ligand binding, which formed interacts with Gly266, Asn267, Tyr268, Tyr273, Thr301, and Asp302, Lys157, Leu162, Asp164, Arg166, Glu167, Pro248, and Tyr264 [123]. This information was obtained from a 100 ns molecular dynamics simulation of these complexes [123].
A study screened 21 antiviral, antifungal, and anticancer compounds using an in silico molecular docking technique to find potential inhibitors of the SARS-CoV-2 papain-like protease [124]. This study showed that Neobavaisoflavone is the one with the highest binding energy to the papain-like SARS-CoV-2 protease among them [120]. These compounds may bind close to the ISGylation, ubiquitination, and papain-like protease critical catalytic triad of SARS-CoV-2: Trp106, Asn109, Cys111, Met208, Lys232, Pro247, Tyr268, Gln269, His272, Asp286 and Thr301 [125]. These compounds may be useful options for therapeutic intervention against COVID-19 since inhibiting the papain-like protease is a crucial tactic in the battle against viruses [125].
To sum up, the multifunctional technique utilizing MD simulations to target SARS-CoV-2-PLpro is a comprehensive approach to suppress viral replication and control COVID-19 [126]. In the fight against the current pandemic, thiol-reacting inhibitors, substrate-based peptides, FDA-approved drug repurposing, and molecular dynamics simulations are combined to provide information on potential candidates and modes of action.
5. Clinical and Preclinical Studies: Integrating MD Simulation Insights
Targeting the SARS-CoV-2 Papain-like Protease (PLpro) has been the focus of extensive studies due to the urgent need to discover new therapeutic techniques to attack COVID-19 [25]. We examine preclinical and clinical research on PLpro inhibition as well as the knowledge obtained from molecular dynamics (MD) simulations in this thorough review.
Targeting SARS-CoV-2 PLpro, GRL-0617, a non-covalent inhibitor, demonstrated potential which primary its intent purpose was for SARS-CoV PLpro [127]. Because of the structural similarities between the two viral proteases and the compound’s efficacy against SARS-CoV-2 PLpro, there is potential to repurpose existing PLpro inhibitors for SARS-CoV-2 [25,76,128]. The conserved Tyr268 in SARS-CoV-2 PLpro and GRL-0617 share a same binding mechanism, which was further supported by computational simulations [85].
Although vaccines like those from Moderna, Johnson & Johnson, and Pfizer/BioNTech provide promise in controlling the pandemic, their efficacy against novel virus strains remains uncertain because they mostly target the Spike protein [129]. Moreover, vaccinations act as prevention measures, which makes the creation of antiviral drugs necessary for the treatment of COVID-19 necessary [130].
Recent research has shown irreversible inhibitors with great selectivity for SARS-CoV-2 PLpro, such as VIR250 and VIR251 [74]. These structural discoveries provide the groundwork for logical drug design approaches that efficiently target SARS-CoV-2 PLpro [131].
Molecular docking and molecular dynamics simulations are two examples of computational techniques that have been very useful in determining the binding affinities and stability of different drugs to PLpro [114]. These techniques are essential for drug development since they have revealed compounds that may have antiviral qualities [132]. Furthermore, structurally related substances from many sources, including marine species and medicinal plants, have been also the focus of exploration [133]. Compounds that may inhibit SARS-CoV-2 PLpro have been effectively found through computational screening, expanding the pool of possible antiviral options [76]. Using Supervised Molecular Dynamics (SuMD) simulations, the unbinding mechanisms of PLpro inhibitors have been explained [134]. The significance of the BL2 loop and its function in inhibitor unbinding were brought to light by these simulations [135]. This information can help optimize inhibitors to stop natural fluctuations in the loop and improve binding efficacy [136].
To sum up, this analysis highlights the diverse strategy employed to specifically target SARS-CoV-2 PLpro in the COVID-19 pandemic [128]. It includes the repurposing of well-known inhibitors, the investigation of structurally related molecules, and the application of simulations and computational techniques to drug discovery [137]. This comprehensive strategy continues to be essential for creating potent antiviral therapies as the pandemic progresses [137].
6. Challenges and Future Directions: Guided by MD Simulations
There have been numerous challenges in the way of finding efficient coronavirus protease inhibitors, especially those that target the papain-like protease (PLpro) and major protease (Mpro) of SARS-CoV-2 [25]. The main causes of these obstacles are the inhibitory efficacy and pharmacokinetic properties of potential drugs [138]. In order to be deemed a promising drugs or drug candidate, a molecule needs to accomplish two things: it needs to successfully reach target in the body and trigger the desired biological reactions [139,140]. These are important factors to take into account while developing antiviral drugs, particularly in light of the critical need to eradicate COVID-19 [141,142].
Baicalein, Disulfiram, Carmofur, Ebselen, Tideglusib, Shikonin, and PX-12 are only a few of the compounds that have proven to exhibit drug-like qualities and fit the requirements to be taken into consideration for drug development [143]. Nevertheless, these substances still struggle to attain the appropriate level of potency and selectivity against coronavirus proteases [144]. This paradox is also shown in the case of SARS-CoV-2 PLpro inhibitors, where it is still difficult to strike a precise balance between drug-like qualities and the capacity to elicit strong and targeted protease inhibition [145]. It is critical to address these problems because they have an immediate influence on the viability of creating antiviral drugs [145].
Studies on the structure-activity relationship (SAR) have provided important new information on the properties of protease inhibitors [146]. Covalent and peptididomimetic inhibitors have demonstrated the ability to selectively and potently inhibit these enzymes’ active sites [147]. But when it comes to pharmacokinetic factors, these inhibitors often come up short, even when they show promise [147]. They are not acceptable as drug candidates because of their poor metabolism, excretion, absorption, distribution, and toxicological properties [147]. On the other hand, low-molecular-weight or non-peptidomimetic compounds show potential in meeting drug- like criteria [142]; yet, they encounter difficulties in obtaining the required potency and selectivity against coronavirus proteases [148]. In order to close this gap, it is imperative to concentrate on lead optimization of low-molecular-weight, non-peptidomimetic drugs [149].
To address these issues in the creation of protease inhibitors, fragment-based drug design (FBDD) appears to be a potential strategy [150]. It entails identifying low-molecular-weight compounds as potential protease inhibitors, using a combination of computational and experimental methods to select advantageous fragments from peptidomimetic compounds, and incorporating these fragments into the lead optimization process of low-molecular-weight compounds [151]. The last phase is carefully optimizing hybrid compounds to achieve desirable pharmacokinetic and pharmacodynamic characteristics [152]. This approach may offer a viable way to create protease inhibitors that are more potent in the future.
Drug development efforts are greatly aided by in silico studies, however it is crucial to validate these results with in vivo research [153]. These in vivo investigations demonstrate the therapeutic potential of these possible drug candidates in addition to confirming their method of action [154]. Therefore, a multidisciplinary strategy that incorporates in vivo research, pharmacokinetics, pharmacodynamics, and computational drug design is crucial [155]. When combined, these tactics offer the best chance of surmounting current obstacles and advancing the creation of potent antiviral drugs for COVID-19 treatment [156]. Without a doubt, the entire effort will support the ongoing fight against the worldwide epidemic and aid in the creation of vital drug compounds.
7. Conclusions and Author Insights on Targeting SARS-CoV-2 Papain-Like Protease
We have examined the crucial role of focusing on the SARS-CoV-2 Papain-like Protease (PLpro) in the development and control of COVID-19 in this review. The discovery of prospective antiviral drugs and our understanding of this important enzyme have both benefited greatly from the application of innovative computational techniques [157,158,159]. A tertiary structure model for SARS-CoV-2 PLpro was created by homology modelling, and the catalytic region of the PLpro protein was found to have binding sites for antiviral drugs through the use of molecular docking techniques [160]. In the process of creating new drugs, this computational method that combines modelling and docking is a useful first step that lays the groundwork for further experimental studies [161].
Moreover, all-atom molecular dynamics (MD) simulations were performed to guarantee the stability of the drug-PLpro complexes in a water solvent environment [162]. Important insights into the dynamics and interactions between the drug candidates and the PLpro protein were provided by MD simulations [76]. The most stable complexes were found using the binding energy estimations from MD simulations, which is crucial in the search for effective antiviral drugs that inhibit SARS-CoV-2 by inhibiting PLpro [163]. This versatile computational method has shown to be an effective method in the search for new antiviral drugs [164].
Although the promise of certain therapeutic candidates targeting SARS-CoV-2 PLpro has been revealed by computational approaches, it is crucial to stress that these results need to undergo thorough in vitro and in vivo examinations [165]. To assess these recommended drugs therapeutic worth, real-world experimental settings must validate their safety and effectiveness [166]. These crucial next steps need to be completed in accordance with acceptable scientific methods in order to guarantee that encouraging computational results materialize in real progress against COVID-19. [167,168]
This review makes it clear that the existing range of drugs intended to suppress PLpro is insufficient. Their weak actions against this enzyme necessitate careful definition and refinement before they may be considered clinically useful. Therefore, in order to fully utilize these drugs in the therapeutic context, researchers must set out to optimize and improve their performance. Unbinding events of PLpro inhibitors, like GRL0617 and its derivatives, have been recorded through a thorough understanding of the unbinding pathways attained via the Supervised Molecular Dynamics (SuMD) simulation method, providing important insights into drug behaviour and opportunities for improvement [136].
Apart from optimizing existing drugs, investigating sub-structurally similar molecules is a potentially fruitful path [169]. A variety of computational techniques have been used to evaluate the possibility of compounds similar to Ritonavir that were obtained from the PubChem database as antiviral drugs for SARS-CoV-2 [170]. Once again, the use of homology modeling, molecular docking, and MD studies together proved to be quite helpful in identifying new drug candidates that may be effective in treating SARS-CoV-2 by specifically targeting PLpro.
The comprehensive review emphasizes how important it is to focus on SARS-CoV-2 PLpro in the fight against COVID-19. We have made significant progress in locating and analyzing possible antiviral drugs by utilizing computational methods including homology modeling, molecular docking, and MD simulations. It is crucial to understand that these results are preliminary and need to be rigorously validated by experiments. Moreover, the continuous battle against this worldwide pandemic would require a concentrated effort to improve currently available drugs and investigate new chemical compounds. This multidisciplinary strategy, which combines experimental validation with computational insights, is critical to the development and control of COVID-19.
Author Contributions
Conceptualization, N.N.M, A.G-A.M., S.C.U and H.K.; writing—original draft preparation, A.G-A.M. and N.N.M: writing—review and editing, A.G-A.M, N.A.M and S.C.U; supervision, R.B.K and H.M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge the College of Health Sciences of the University of Kwazulu-Natal for the support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): an update. Cureus 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: a brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respiratory research 2020, 21, 1–14. [Google Scholar] [CrossRef]
- Acter, T.; Uddin, N.; Das, J.; Akhter, A.; Choudhury, T.R.; Kim, S. Evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) as coronavirus disease 2019 (COVID-19) pandemic: A global health emergency. Science of the Total Environment 2020, 730, 138996. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, G.; Cai, X.-p.; Deng, J.-w.; Zheng, L.; Zhu, H.-h.; Zheng, M.; Yang, B.; Chen, Z. An overview of COVID-19. Journal of Zhejiang University. Science. B 2020, 21, 343. [Google Scholar] [CrossRef]
- Satarker, S.; Nampoothiri, M. Structural proteins in severe acute respiratory syndrome coronavirus-2. Archives of medical research 2020, 51, 482–491. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nature Reviews Microbiology 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Astuti, I. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes & Metabolic Syndrome: Clinical Research & Reviews 2020, 14, 407–412. [Google Scholar]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Critical Care 2020, 24, 1–10. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.-f.; Xu, W.; Liu, S.-w. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacologica Sinica 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Applied microbiology and biotechnology 2017, 101, 3103–3119. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: evolving the largest RNA virus genome. Virus research 2006, 117, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, Z.; Lai, M.; Shu, S.; Du, Y.; Zhou, Z.H.; Sun, R. In situ structures of the genome and genome-delivery apparatus in a single-stranded RNA virus. Nature 2017, 541, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Alturki, S.O.; Alturki, S.O.; Connors, J.; Cusimano, G.; Kutzler, M.A.; Izmirly, A.M.; Haddad, E.K. The 2020 pandemic: current SARS-CoV-2 vaccine development. Frontiers in immunology 2020, 11, 1880. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; Plotkin, S.A. Impact of vaccines; health, economic and social perspectives. Frontiers in microbiology 2020, 11, 1526. [Google Scholar] [CrossRef] [PubMed]
- Gusev, E.; Sarapultsev, A.; Solomatina, L.; Chereshnev, V. SARS-CoV-2-specific immune response and the pathogenesis of COVID-19. International journal of molecular sciences 2022, 23, 1716. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Onoguchi, K.; Yoneyama, M. Regulation of RIG-I-like receptor-mediated signaling: interaction between host and viral factors. Cellular & molecular immunology 2021, 18, 539–555. [Google Scholar]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan, S.; Selvaraj, C.; Singh, S.K.; Dubey, V.K.; Kumar, S.; Fialho, A.M.; Saudagar, P. Bacterial protein azurin and derived peptides as potential anti-SARS-CoV-2 agents: insights from molecular docking and molecular dynamics simulations. Journal of Biomolecular Structure and Dynamics 2021, 39, 5706–5721. [Google Scholar] [CrossRef]
- Muralidharan, N.; Sakthivel, R.; Velmurugan, D.; Gromiha, M.M. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. Journal of Biomolecular Structure and Dynamics 2021, 39, 2673–2678. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M.; Shahabi, D.; Chapman, M.E.; Mesecar, A.D. Drug development and medicinal chemistry efforts toward SARS-coronavirus and Covid-19 therapeutics. ChemMedChem 2020, 15, 907–932. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef] [PubMed]
- Beyerstedt, S.; Casaro, E.B.; Rangel, É.B. COVID-19: angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. European journal of clinical microbiology & infectious diseases 2021, 40, 905–919. [Google Scholar]
- Bost, P.; Giladi, A.; Liu, Y.; Bendjelal, Y.; Xu, G.; David, E.; Blecher-Gonen, R.; Cohen, M.; Medaglia, C.; Li, H. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell 2020, 181, 1475–1488.e1412. [Google Scholar] [CrossRef] [PubMed]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. The EMBO journal 2020, 39, e106275. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. Journal of medicinal chemistry 2020, 65, 2716–2746. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.K.; Singh, A.; Prakash, S.; Kumar, M.; Singh, A.K. Race to arsenal COVID-19 therapeutics: Current alarming status and future directions. Chemico-Biological Interactions 2020, 332, 109298. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nature reviews Molecular cell biology 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Jiang, H.; Yang, P.; Zhang, J. Potential inhibitors targeting papain-like protease of SARS-CoV-2: two birds with one stone. Frontiers in chemistry 2022, 10, 822785. [Google Scholar] [CrossRef]
- Huang, J.; Song, W.; Huang, H.; Sun, Q. Pharmacological therapeutics targeting RNA-dependent RNA polymerase, proteinase and spike protein: from mechanistic studies to clinical trials for COVID-19. Journal of clinical medicine 2020, 9, 1131. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Diabetes, obesity, metabolism, and SARS-CoV-2 infection: the end of the beginning. Cell metabolism 2021, 33, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Kai, H.; Kai, M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors—lessons from available evidence and insights into COVID-19. Hypertension Research 2020, 43, 648–654. [Google Scholar] [CrossRef] [PubMed]
- South, A.M.; Diz, D.I.; Chappell, M.C. COVID-19, ACE2, and the cardiovascular consequences. American Journal of Physiology-Heart and Circulatory Physiology 2020. [Google Scholar]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. European journal of internal medicine 2020, 76, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-Y.; Zheng, B.; Zhang, Y.; Li, J.-P. Role and mechanism of angiotensin-converting enzyme 2 in acute lung injury in coronavirus disease 2019. Chronic Diseases and Translational Medicine 2020, 6, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W. SARS-CoV-2 receptor ACE 2 and TMPRSS 2 are primarily expressed in bronchial transient secretory cells. The EMBO journal 2020, 39, e105114. [Google Scholar] [CrossRef] [PubMed]
- Mohamadian, M.; Chiti, H.; Shoghli, A.; Biglari, S.; Parsamanesh, N.; Esmaeilzadeh, A. COVID-19: Virology, biology and novel laboratory diagnosis. The journal of gene medicine 2021, 23, e3303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, J.; Liu, X.; Xu, D.; Shi, L.; Liu, J.; Zeng, Q.; Wang, X. 5-Aminolaevulinic acid photodynamic therapy amplifies intense inflammatory response in the treatment of acne vulgaris via CXCL8. Experimental Dermatology 2021, 30, 923–931. [Google Scholar] [CrossRef]
- Shapira, T.; Monreal, I.A.; Dion, S.P.; Buchholz, D.W.; Imbiakha, B.; Olmstead, A.D.; Jager, M.; Désilets, A.; Gao, G.; Martins, M. A TMPRSS2 inhibitor acts as a pan-SARS-CoV-2 prophylactic and therapeutic. Nature 2022, 605, 340–348. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; Rohde, C. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life science alliance 2020, 3. [Google Scholar] [CrossRef]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Rohaim, M.A.; El Naggar, R.F.; Clayton, E.; Munir, M. Structural and functional insights into non-structural proteins of coronaviruses. Microbial pathogenesis 2021, 150, 104641. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A structural view of SARS-CoV-2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef]
- Ma, Y.; Frutos-Beltrán, E.; Kang, D.; Pannecouque, C.; De Clercq, E.; Menéndez-Arias, L.; Liu, X.; Zhan, P. Medicinal chemistry strategies for discovering antivirals effective against drug-resistant viruses. Chemical Society Reviews 2021, 50, 4514–4540. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Khan, S.A.; Zia, K.; Ashraf, S.; Uddin, R.; Ul-Haq, Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. Journal of Biomolecular Structure and Dynamics 2021, 39, 2607–2616. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.Á.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E. COVID-19: drug targets and potential treatments. Journal of medicinal chemistry 2020, 63, 12359–12386. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). The Journal of pathology 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Davidson, A.M.; Wysocki, J.; Batlle, D. Interaction of SARS-CoV-2 and other coronavirus with ACE (angiotensin-converting enzyme)-2 as their main receptor: therapeutic implications. Hypertension 2020, 76, 1339–1349. [Google Scholar] [CrossRef]
- Sarker, J.; Das, P.; Sarker, S.; Roy, A.K.; Momen, A.R. A review on expression, pathological roles, and inhibition of TMPRSS2, the serine protease responsible for SARS-CoV-2 spike protein activation. Scientifica 2021, 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cameron, K.; Rozano, L.; Falasca, M.; Mancera, R.L. Does the SARS-CoV-2 spike protein receptor binding domain interact effectively with the DPP4 (CD26) receptor? A molecular docking study. International Journal of Molecular Sciences 2021, 22, 7001. [Google Scholar] [CrossRef] [PubMed]
- Mouffouk, C.; Mouffouk, S.; Mouffouk, S.; Hambaba, L.; Haba, H. Flavonols as potential antiviral drugs targeting SARS-CoV-2 proteases (3CLpro and PLpro), spike protein, RNA-dependent RNA polymerase (RdRp) and angiotensin-converting enzyme II receptor (ACE2). European journal of pharmacology 2021, 891, 173759. [Google Scholar] [CrossRef] [PubMed]
- Chitranshi, N.; Gupta, V.K.; Rajput, R.; Godinez, A.; Pushpitha, K.; Shen, T.; Mirzaei, M.; You, Y.; Basavarajappa, D.; Gupta, V. Evolving geographic diversity in SARS-CoV2 and in silico analysis of replicating enzyme 3CL pro targeting repurposed drug candidates. Journal of Translational Medicine 2020, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Mitra, D.; Paul, M.; Chaudhary, P.; Kamboj, A.; Thatoi, H.; Janmeda, P.; Jain, D.; Panneerselvam, P.; Shrivastav, R. Potential inhibitors of SARS-CoV-2 (COVID 19) proteases PLpro and Mpro/3CLpro: molecular docking and simulation studies of three pertinent medicinal plant natural components. Current Research in Pharmacology and Drug Discovery 2021, 2, 100038. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y. Host cellular RNA helicases regulate SARS-CoV-2 infection. Journal of Virology 2022, 96, e00002–00022. [Google Scholar] [CrossRef]
- Padmanabhan, P.; Desikan, R.; Dixit, N.M. Targeting TMPRSS2 and Cathepsin B/L together may be synergistic against SARS-CoV-2 infection. PLoS computational biology 2020, 16, e1008461. [Google Scholar] [CrossRef]
- Pišlar, A.; Mitrović, A.; Sabotič, J.; Pečar Fonović, U.; Perišić Nanut, M.; Jakoš, T.; Senjor, E.; Kos, J. The role of cysteine peptidases in coronavirus cell entry and replication: The therapeutic potential of cathepsin inhibitors. PLoS Pathogens 2020, 16, e1009013. [Google Scholar] [CrossRef]
- Essalmani, R.; Jain, J.; Susan-Resiga, D.; Andréo, U.; Evagelidis, A.; Derbali, R.M.; Huynh, D.N.; Dallaire, F.; Laporte, M.; Delpal, A. Distinctive roles of furin and TMPRSS2 in SARS-CoV-2 infectivity. Journal of virology 2022, 96, e00128–00122. [Google Scholar] [CrossRef]
- Cheng, Y.-W.; Chao, T.-L.; Li, C.-L.; Chiu, M.-F.; Kao, H.-C.; Wang, S.-H.; Pang, Y.-H.; Lin, C.-H.; Tsai, Y.-M.; Lee, W.-H. Furin inhibitors block SARS-CoV-2 spike protein cleavage to suppress virus production and cytopathic effects. Cell reports 2020, 33. [Google Scholar] [CrossRef]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharmaceutica Sinica B 2021, 11, 237–245. [Google Scholar] [CrossRef]
- Gupta, A.; Pradhan, A.; Maurya, V.K.; Kumar, S.; Theengh, A.; Puri, B.; Saxena, S.K. Therapeutic approaches for SARS-CoV-2 infection. Methods 2021, 195, 29–43. [Google Scholar] [CrossRef]
- Dubey, R.; Dubey, K. SARS-CoV-2: Potential drug targets and its virtual screening. Modeling, control and drug development for COVID-19 outbreak prevention 2022, 203–244. [Google Scholar]
- Liu, J.; Cheng, Y.; Zheng, M.; Yuan, B.; Wang, Z.; Li, X.; Yin, J.; Ye, M.; Song, Y. Targeting the ubiquitination/deubiquitination process to regulate immune checkpoint pathways. Signal Transduction and Targeted Therapy 2021, 6, 28. [Google Scholar] [CrossRef]
- Hill, D.J.; Mio, M.J.; Prince, R.B.; Hughes, T.S.; Moore, J.S. A field guide to foldamers. Chemical Reviews 2001, 101, 3893–4012. [Google Scholar] [CrossRef]
- Sanders, B.C.; Pokhrel, S.; Labbe, A.D.; Mathews, I.I.; Cooper, C.J.; Davidson, R.B.; Phillips, G.; Weiss, K.L.; Zhang, Q.; O’Neill, H. Potent and selective covalent inhibition of the papain-like protease from SARS-CoV-2. Nature Communications 2023, 14, 1733. [Google Scholar] [CrossRef]
- de Paiva, R.E.; Neto, A.M.; Santos, I.A.; Jardim, A.C.; Corbi, P.P.; Bergamini, F.R. What is holding back the development of antiviral metallodrugs? A literature overview and implications for SARS-CoV-2 therapeutics and future viral outbreaks. Dalton Transactions 2020, 49, 16004–16033. [Google Scholar] [CrossRef]
- Gupta, Y.; Maciorowski, D.; Zak, S.E.; Jones, K.A.; Kathayat, R.S.; Azizi, S.-A.; Mathur, R.; Pearce, C.M.; Ilc, D.J.; Husein, H. Bisindolylmaleimide IX: A novel anti-SARS-CoV2 agent targeting viral main protease 3CLpro demonstrated by virtual screening pipeline and in-vitro validation assays. Methods 2021, 195, 57–71. [Google Scholar] [CrossRef]
- Li, G.; Hilgenfeld, R.; Whitley, R.; De Clercq, E. Therapeutic strategies for COVID-19: progress and lessons learned. Nature Reviews Drug Discovery 2023, 1–27. [Google Scholar] [CrossRef]
- Osipiuk, J.; Azizi, S.-A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nature communications 2021, 12, 743. [Google Scholar] [CrossRef]
- Arya, R.; Kumari, S.; Pandey, B.; Mistry, H.; Bihani, S.C.; Das, A.; Prashar, V.; Gupta, G.D.; Panicker, L.; Kumar, M. Structural insights into SARS-CoV-2 proteins. Journal of molecular biology 2021, 433, 166725. [Google Scholar] [CrossRef]
- Murgolo, N.; Therien, A.G.; Howell, B.; Klein, D.; Koeplinger, K.; Lieberman, L.A.; Adam, G.C.; Flynn, J.; McKenna, P.; Swaminathan, G. SARS-CoV-2 tropism, entry, replication, and propagation: Considerations for drug discovery and development. PLoS pathogens 2021, 17, e1009225. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti–COVID-19 drug design. Science advances 2020, 6, eabd4596. [Google Scholar] [CrossRef]
- Nikolich-Zugich, J.; Knox, K.S.; Rios, C.T.; Natt, B.; Bhattacharya, D.; Fain, M.J. SARS-CoV-2 and COVID-19 in older adults: what we may expect regarding pathogenesis, immune responses, and outcomes. Geroscience 2020, 42, 505–514. [Google Scholar] [CrossRef]
- Prajapati, J.; Patel, R.; Rao, P.; Saraf, M.; Rawal, R.; Goswami, D. Perceiving SARS-CoV-2 Mpro and PLpro dual inhibitors from pool of recognized antiviral compounds of endophytic microbes: an in silico simulation study. Structural Chemistry 2022, 33, 1619–1643. [Google Scholar] [CrossRef]
- Malik, Y.A. Properties of coronavirus and SARS-CoV-2. The Malaysian journal of pathology 2020, 42, 3–11. [Google Scholar]
- Cao, W.; Cho, C.-C.D.; Geng, Z.Z.; Shaabani, N.; Ma, X.R.; Vatansever, E.C.; Alugubelli, Y.R.; Ma, Y.; Chaki, S.P.; Ellenburg, W.H. Evaluation of SARS-CoV-2 main protease inhibitors using a novel cell-based assay. ACS central science 2022, 8, 192–204. [Google Scholar] [CrossRef]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and noncovalent inhibition of the deubiquitinase and deISGylase activity of SARS-CoV-2 papain-like protease. ACS infectious diseases 2020, 6, 2099–2109. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Y.; Liu, Y.; Yang, Q.; Wu, X.; Huang, X.; Liu, H.; Cai, W.; Ma, G. Medication therapy strategies for the coronavirus disease 2019 (COVID-19): recent progress and challenges. Expert Review of Clinical Pharmacology 2020, 13, 957–975. [Google Scholar] [CrossRef]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Communications Biology 2022, 5, 169. [Google Scholar] [CrossRef]
- Steuten, K.; Kim, H.; Widen, J.C.; Babin, B.M.; Onguka, O.; Lovell, S.; Bolgi, O.; Cerikan, B.; Neufeldt, C.J.; Cortese, M. Challenges for targeting SARS-CoV-2 proteases as a therapeutic strategy for COVID-19. ACS infectious diseases 2021, 7, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.; Carriere, M.; Fusco, L.; Capua, I.; Regla-Nava, J.A.; Pasquali, M.; Scott, J.A.; Vitale, F.; Unal, M.A.; Mattevi, C. Toward nanotechnology-enabled approaches against the COVID-19 pandemic. ACS nano 2020, 14, 6383–6406. [Google Scholar] [CrossRef]
- Selvaraj, C.; Dinesh, D.C.; Panwar, U.; Abhirami, R.; Boura, E.; Singh, S.K. Structure-based virtual screening and molecular dynamics simulation of SARS-CoV-2 Guanine-N7 methyltransferase (nsp14) for identifying antiviral inhibitors against COVID-19. Journal of Biomolecular Structure and Dynamics 2021, 39, 4582–4593. [Google Scholar] [CrossRef] [PubMed]
- Frances-Monerris, A.; Hognon, C.; Miclot, T.; Garcia-Iriepa, C.; Iriepa, I.; Terenzi, A.; Grandemange, S.; Barone, G.; Marazzi, M.; Monari, A. Molecular basis of SARS-CoV-2 infection and rational design of potential antiviral agents: modeling and simulation approaches. Journal of Proteome Research 2020, 19, 4291–4315. [Google Scholar] [CrossRef] [PubMed]
- Báez-Santos, Y.M.; John, S.E.S.; Mesecar, A.D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral research 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Maiti, B.K. Can Papain-like Protease Inhibitors Halt SARS-CoV-2 Replication? ACS Pharmacol Transl Sci 2020, 3, 1017–1019. [Google Scholar] [CrossRef] [PubMed]
- Lanz, J.; Biniaz-Harris, N.; Kuvaldina, M.; Jain, S.; Lewis, K.; Fallon, B.A. Disulfiram: Mechanisms, Applications, and Challenges. Antibiotics (Basel) 2023, 12, 524. [Google Scholar] [CrossRef]
- Maiti, B.K. Can papain-like protease inhibitors halt SARS-CoV-2 replication? ACS pharmacology & translational science 2020, 3, 1017–1019. [Google Scholar]
- Akram, M.W.; Hasannuzaman, M.; Cuce, E.; Cuce, P.M. Global technological advancement and challenges of glazed window, facade system and vertical greenery-based energy savings in buildings: A comprehensive review. Energy and Built Environment 2023, 4, 206–226. [Google Scholar] [CrossRef]
- Andreini, C.; Arnesano, F.; Rosato, A. The zinc proteome of SARS-CoV-2. Metallomics 2022, 14. [Google Scholar] [CrossRef]
- Debnath, S.K.; Debnath, M.; Srivastava, R.; Omri, A. Drugs repurposing for SARS-CoV-2: new insight of COVID-19 druggability. Expert Review of Anti-infective Therapy 2022, 20, 1187–1204. [Google Scholar] [CrossRef]
- Owji, H.; Negahdaripour, M.; Hajighahramani, N. Immunotherapeutic approaches to curtail COVID-19. International immunopharmacology 2020, 88, 106924. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: open the door for novel therapies. Signal transduction and targeted therapy 2022, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug repurposing approach to fight COVID-19. Pharmacological Reports 2020, 72, 1479–1508. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life sciences 2020, 253, 117592. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Cao, Z.; Han, M.; Wang, Z.; Chen, J.; Sun, W.; Wu, Y.; Xiao, W.; Liu, S.; Chen, E. Hydroxychloroquine in patients with mainly mild to moderate coronavirus disease 2019: open label, randomised controlled trial. bmj 2020, 369. [Google Scholar] [CrossRef] [PubMed]
- Martinotti, C.; Ruiz-Perez, L.; Deplazes, E.; Mancera, R.L. Molecular dynamics simulation of small molecules interacting with biological membranes. ChemPhysChem 2020, 21, 1486–1514. [Google Scholar] [CrossRef] [PubMed]
- Parmar, P.; Rao, P.; Sharma, A.; Shukla, A.; Rawal, R.M.; Saraf, M.; Patel, B.V.; Goswami, D. Meticulous assessment of natural compounds from NPASS database for identifying analogue of GRL0617, the only known inhibitor for SARS-CoV2 papain-like protease (PLpro) using rigorous computational workflow. Molecular diversity 2022, 26, 389–407. [Google Scholar] [CrossRef]
- Sanachai, K.; Mahalapbutr, P.; Sanghiran Lee, V.; Rungrotmongkol, T.; Hannongbua, S. In silico elucidation of potent inhibitors and rational drug design against SARS-CoV-2 papain-like protease. The Journal of Physical Chemistry B 2021, 125, 13644–13656. [Google Scholar] [CrossRef]
- Naidoo, D.; Kar, P.; Roy, A.; Mutanda, T.; Bwapwa, J.; Sen, A.; Anandraj, A. Structural insight into the binding of cyanovirin-n with the spike glycoprotein, mpro and PLpro of SARS-CoV-2: Protein–protein interactions, dynamics simulations and free energy calculations. Molecules 2021, 26, 5114. [Google Scholar] [CrossRef]
- Selvaraj, V.; Rathinavel, T.; Ammashi, S.; Nasir Iqbal, M. Polyphenolic phytochemicals exhibit promising SARS-COV-2 papain like protease (PLpro) inhibition validated through a computational approach. Polycyclic Aromatic Compounds 2023, 43, 5545–5566. [Google Scholar] [CrossRef]
- Bera, K.; Reeda, V.J.; Babila, P.; Dinesh, D.C.; Hritz, J.; Karthick, T. An in silico molecular dynamics simulation study on the inhibitors of SARS-CoV-2 proteases (3CLpro and PLpro) to combat COVID-19. Molecular Simulation 2021, 47, 1168–1184. [Google Scholar] [CrossRef]
- Yan, F.; Gao, F. An overview of potential inhibitors targeting non-structural proteins 3 (PLpro and Mac1) and 5 (3CLpro/Mpro) of SARS-CoV-2. Computational and structural biotechnology journal 2021, 19, 4868–4883. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Tong, J.; Wu, Y.; Zhao, S.; Lin, B.-L. A computational evaluation of targeted oxidation strategy (TOS) for potential inhibition of SARS-CoV-2 by disulfiram and analogues. Biophysical Chemistry 2021, 276, 106610. [Google Scholar] [CrossRef]
- Ma, C.; Hu, Y.; Townsend, J.A.; Lagarias, P.I.; Marty, M.T.; Kolocouris, A.; Wang, J. Ebselen, disulfiram, carmofur, PX-12, tideglusib, and shikonin are nonspecific promiscuous SARS-CoV-2 main protease inhibitors. ACS pharmacology & translational science 2020, 3, 1265–1277. [Google Scholar]
- Zmudzinski, M.; Rut, W.; Olech, K.; Granda, J.; Giurg, M.; Burda-Grabowska, M.; Zhang, L.; Sun, X.; Lv, Z.; Nayak, D. Ebselen derivatives are very potent dual inhibitors of SARS-CoV-2 proteases-PLpro and Mpro in in vitro studies. BioRxiv 2020. 2020.2008. 2030.273979. [Google Scholar]
- Wang, Z.; Yang, L.; Zhao, X.-E. Co-crystallization and structure determination: An effective direction for anti-SARS-CoV-2 drug discovery. Computational and Structural Biotechnology Journal 2021, 19, 4684–4701. [Google Scholar] [CrossRef]
- Arwansyah, A.; Arif, A.; Ramli, I.; Kurniawan, I.; Sukarti, S.; Nur Alam, M.; Illing, I.; Farid Lewa, A.; Manguntungi, B. Molecular modelling on SARS-CoV-2 papain-like protease: an integrated study with homology modelling, molecular docking, and molecular dynamics simulations. SAR and QSAR in Environmental Research 2021, 32, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A.; Bellera, C.L. Challenges and opportunities with drug repurposing: finding strategies to find alternative uses of therapeutics. Expert Opinion on Drug Discovery 2020, 15, 397–401. [Google Scholar] [CrossRef]
- Pang, J.; Gao, S.; Sun, Z.; Yang, G. Discovery of small molecule PLpro inhibitor against COVID-19 using structure-based virtual screening, molecular dynamics simulation, and molecular mechanics/Generalized Born surface area (MM/GBSA) calculation. Struct Chem 2021, 32, 879–886. [Google Scholar] [CrossRef]
- Hu, X.; Zeng, Z.; Zhang, J.; Wu, D.; Li, H.; Geng, F. Molecular dynamics simulation of the interaction of food proteins with small molecules. Food Chemistry 2023, 405, 134824. [Google Scholar] [CrossRef] [PubMed]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated research and development investment needed to bring a new medicine to market, 2009-2018. Jama 2020, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Zrieq, R.; Ahmad, I.; Snoussi, M.; Noumi, E.; Iriti, M.; Algahtani, F.D.; Patel, H.; Saeed, M.; Tasleem, M.; Sulaiman, S. Tomatidine and patchouli alcohol as inhibitors of SARS-CoV-2 enzymes (3CLpro, PLpro and NSP15) by molecular docking and molecular dynamics simulations. International Journal of Molecular Sciences 2021, 22, 10693. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, N.; Albratty, M. Benchmarked molecular docking integrated molecular dynamics stability analysis for prediction of SARS-CoV-2 papain-like protease inhibition by olive secoiridoids. J King Saud Univ Sci 2023, 35, 102402. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Athar, M.; Jha, P.C. Computational investigation of binding of chloroquinone and hydroxychloroquinone against PLPro of SARS-CoV-2. J Biomol Struct Dyn 2022, 40, 3071–3081. [Google Scholar] [CrossRef]
- Kandeel, M.; Abdelrahman, A.; Oh-Hashi, K.; Ibrahim, A.; Venugopala, K.; Morsy, M.; Ibrahim, M. Repurposing of FDA-approved antivirals, antibiotics, anthelmintics, antioxidants, and cell protectives against SARS-CoV-2 papain-like protease. Journal of Biomolecular Structure and Dynamics 2020, 39, 1–8. [Google Scholar] [CrossRef]
- Joshi, S.; Joshi, M.; Degani, M.S. Tackling SARS-CoV-2: proposed targets and repurposed drugs. Future medicinal chemistry 2020, 12, 1579–1601. [Google Scholar] [CrossRef]
- Zhou, Y.-W.; Xie, Y.; Tang, L.-S.; Pu, D.; Zhu, Y.-J.; Liu, J.-Y.; Ma, X.-L. Therapeutic targets and interventional strategies in COVID-19: mechanisms and clinical studies. Signal transduction and targeted therapy 2021, 6, 317. [Google Scholar] [CrossRef]
- Russo, M.; Moccia, S.; Spagnuolo, C.; Tedesco, I.; Russo, G.L. Roles of flavonoids against coronavirus infection. Chemico-biological interactions 2020, 328, 109211. [Google Scholar] [CrossRef]
- Kumar, B.K.; Faheem, n.; Sekhar, K.V.G.C.; Ojha, R.; Prajapati, V.K.; Pai, A.; Murugesan, S. Pharmacophore based virtual screening, molecular docking, molecular dynamics and MM-GBSA approach for identification of prospective SARS-CoV-2 inhibitor from natural product databases. Journal of Biomolecular Structure and Dynamics 2022, 40, 1363–1386. [Google Scholar] [CrossRef]
- Kumar, B.K.; Faheem, *!!! REPLACE !!!*; Sekhar, K.; Ojha, R.; Prajapati, V.K.; Pai, A.; Murugesan, S. Pharmacophore based virtual screening, molecular docking, molecular dynamics and MM-GBSA approach for identification of prospective SARS-CoV-2 inhibitor from natural product databases. J Biomol Struct Dyn 2022, 40, 1363–1386. [Google Scholar] [CrossRef] [PubMed]
- Jupudi, S.; Rajagopal, K.; Murugesan, S.; Kumar, B.K.; Raman, K.; Byran, G.; Chennaiah, J.; pillai Muthiah, V.; Sankaran, S. Identification of Papain-Like Protease inhibitors of SARS CoV-2 through HTVS, Molecular docking, MMGBSA and Molecular dynamics approach. South African Journal of Botany 2022, 151, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Vardhan, S.; Sahoo, S.K. In silico ADMET and molecular docking study on searching potential inhibitors from limonoids and triterpenoids for COVID-19. Computers in biology and medicine 2020, 124, 103936. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Luan, J.; Zhang, L. Molecular docking of potential SARS-CoV-2 papain-like protease inhibitors. Biochemical and Biophysical Research Communications 2021, 538, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Abdelrahman, A.H.; Oh-Hashi, K.; Ibrahim, A.; Venugopala, K.N.; Morsy, M.A.; Ibrahim, M.A. Repurposing of FDA-approved antivirals, antibiotics, anthelmintics, antioxidants, and cell protectives against SARS-CoV-2 papain-like protease. Journal of Biomolecular Structure and Dynamics 2021, 39, 5129–5136. [Google Scholar] [CrossRef] [PubMed]
- Çalışkaner, Z.O. Determination of Binding Potential of HCV Protease Inhibitors Against to SARS-CoV-2 Papain-like Protease wtih Computational Docking. Letters in Drug Design & Discovery 2021, 18, 949–960. [Google Scholar]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nature communications 2021, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Tregoning, J.S.; Flight, K.E.; Higham, S.L.; Wang, Z.; Pierce, B.F. Progress of the COVID-19 vaccine effort: viruses, vaccines and variants versus efficacy, effectiveness and escape. Nature reviews immunology 2021, 21, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Dhama, K.; Sharun, K.; Tiwari, R.; Dadar, M.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W. COVID-19, an emerging coronavirus infection: advances and prospects in designing and developing vaccines, immunotherapeutics, and therapeutics. Human vaccines & immunotherapeutics 2020, 16, 1232–1238. [Google Scholar]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. An updated review of computer-aided drug design and its application to COVID-19. BioMed research international 2021, 2021. [Google Scholar] [CrossRef]
- Asif, M.; Saleem, M.; Saadullah, M.; Yaseen, H.S.; Al Zarzour, R. COVID-19 and therapy with essential oils having antiviral, anti-inflammatory, and immunomodulatory properties. Inflammopharmacology 2020, 28, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Süntar, I. Importance of ethnopharmacological studies in drug discovery: role of medicinal plants. Phytochemistry Reviews 2020, 19, 1199–1209. [Google Scholar] [CrossRef]
- Kaliamurthi, S.; Selvaraj, G.; Selvaraj, C.; Singh, S.K.; Wei, D.-Q.; Peslherbe, G.H. Structure-based virtual screening reveals ibrutinib and zanubrutinib as potential repurposed drugs against COVID-19. International Journal of Molecular Sciences 2021, 22, 7071. [Google Scholar] [CrossRef]
- Freire, M.C.; Noske, G.D.; Bitencourt, N.V.; Sanches, P.R.; Santos-Filho, N.A.; Gawriljuk, V.O.; de Souza, E.P.; Nogueira, V.H.; de Godoy, M.O.; Nakamura, A.M. Non-toxic dimeric peptides derived from the bothropstoxin-I are potent SARS-CoV-2 and papain-like protease inhibitors. Molecules 2021, 26, 4896. [Google Scholar] [CrossRef]
- Sohraby, F.; Aryapour, H. Unraveling the unbinding pathways of SARS-CoV-2 Papain-like proteinase known inhibitors by Supervised Molecular Dynamics simulation. PLoS ONE 2021, 16, e0251910. [Google Scholar] [CrossRef] [PubMed]
- Rudrapal, M.; Khairnar, S.J.; Jadhav, A.G. Drug repurposing (DR): an emerging approach in drug discovery. Drug repurposing-hypothesis, molecular aspects and therapeutic applications 2020, 10. [Google Scholar]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal transduction and targeted therapy 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Fu, W.; Jiang, D.; Sun, H.; Wang, J.; Zhang, X.; Weng, G.; Liu, H.; Tao, P.; Hou, T. VAD-MM/GBSA: A Variable Atomic Dielectric MM/GBSA Model for Improved Accuracy in Protein–Ligand Binding Free Energy Calculations. Journal of Chemical Information and Modeling 2021, 61, 2844–2856. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nature reviews drug discovery 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Consortium, W.S.T. Repurposed antiviral drugs for Covid-19—interim WHO solidarity trial results. New England journal of medicine 2021, 384, 497–511. [Google Scholar] [CrossRef]
- Abd El-Aziz, T.M.; Stockand, J.D. Recent progress and challenges in drug development against COVID-19 coronavirus (SARS-CoV-2)-an update on the status. Infection, Genetics and Evolution 2020, 83, 104327. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 Mpro: A potential target for peptidomimetics and small-molecule inhibitors. Biomolecules 2021, 11, 607. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative inhibitors of SARS-CoV-2 main protease from a library of marine natural products: a virtual screening and molecular modeling study. Marine drugs 2020, 18, 225. [Google Scholar] [CrossRef] [PubMed]
- Minetti, C.A.; Remeta, D.P. Forces driving a magic bullet to its target: revisiting the role of thermodynamics in drug design, development, and optimization. Life 2022, 12, 1438. [Google Scholar] [CrossRef] [PubMed]
- Badavath, V.N.; Kumar, A.; Samanta, P.K.; Maji, S.; Das, A.; Blum, G.; Jha, A.; Sen, A. Determination of potential inhibitors based on isatin derivatives against SARS-CoV-2 main protease (mpro): a molecular docking, molecular dynamics and structure-activity relationship studies. Journal of Biomolecular Structure and Dynamics 2022, 40, 3110–3128. [Google Scholar] [CrossRef] [PubMed]
- Boike, L.; Henning, N.J.; Nomura, D.K. Advances in covalent drug discovery. Nature Reviews Drug Discovery 2022, 21, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Banerjee, S.; Ghosh, K.; Gayen, S.; Jha, T. Protease targeted COVID-19 drug discovery and its challenges: Insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorganic & medicinal chemistry 2021, 29, 115860. [Google Scholar]
- Al-Karmalawy, A.A.; El-Gamil, D.S.; El-Shesheny, R.; Sharaky, M.; Alnajjar, R.; Kutkat, O.; Moatasim, Y.; Elagawany, M.; Al-Rashood, S.T.; Binjubair, F.A. Design and statistical optimisation of emulsomal nanoparticles for improved anti-SARS-CoV-2 activity of N-(5-nitrothiazol-2-yl)-carboxamido candidates: in vitro and in silico studies. Journal of Enzyme Inhibition and Medicinal Chemistry 2023, 38, 2202357. [Google Scholar] [CrossRef] [PubMed]
- de Souza Neto, L.R.; Moreira-Filho, J.T.; Neves, B.J.; Maidana, R.L.B.R.; Guimarães, A.C.R.; Furnham, N.; Andrade, C.H.; Silva, F.P., Jr. In silico strategies to support fragment-to-lead optimization in drug discovery. Frontiers in chemistry 2020, 8, 93. [Google Scholar] [CrossRef]
- Trisciuzzi, D.; Villoutreix, B.O.; Siragusa, L.; Baroni, M.; Cruciani, G.; Nicolotti, O. Targeting protein-protein interactions with low molecular weight and short peptide modulators: insights on disease pathways and starting points for drug discovery. Expert Opinion on Drug Discovery 2023, 1–16. [Google Scholar] [CrossRef]
- Kumar, H.M.S.; Herrmann, L.; Tsogoeva, S.B. Structural hybridization as a facile approach to new drug candidates. Bioorganic & Medicinal Chemistry Letters 2020, 30, 127514. [Google Scholar]
- Brogi, S.; Ramalho, T.C.; Kuca, K.; Medina-Franco, J.L.; Valko, M. In silico methods for drug design and discovery. 2020, 8, 612. [Google Scholar] [PubMed]
- Salehi, B.; Venditti, A.; Sharifi-Rad, M.; Kręgiel, D.; Sharifi-Rad, J.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B.; Novellino, E. The therapeutic potential of apigenin. International journal of molecular sciences 2019, 20, 1305. [Google Scholar] [CrossRef] [PubMed]
- Pantaleão, S.Q.; Fernandes, P.O.; Gonçalves, J.E.; Maltarollo, V.G.; Honorio, K.M. Recent advances in the prediction of pharmacokinetics properties in drug design studies: a review. ChemMedChem 2022, 17, e202100542. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, X.; Shu, Y.; Guo, M.; Zhang, H.; Tao, W. Insights from nanotechnology in COVID-19 treatment. Nano today 2021, 36, 101019. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Lin, X. A review on applications of computational methods in drug screening and design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef] [PubMed]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. European Journal of Medicinal Chemistry 2021, 224, 113705. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Pannecouque, C.; De Clercq, E.; Liu, X. Anti-HIV drug discovery and development: current innovations and future trends: miniperspective. Journal of medicinal chemistry 2016, 59, 2849–2878. [Google Scholar] [CrossRef] [PubMed]
- Rakib, A.; Nain, Z.; Sami, S.A.; Mahmud, S.; Islam, A.; Ahmed, S.; Siddiqui, A.B.F.; Babu, S.O.F.; Hossain, P.; Shahriar, A. A molecular modelling approach for identifying antiviral selenium-containing heterocyclic compounds that inhibit the main protease of SARS-CoV-2: An in silico investigation. Briefings in bioinformatics 2021, 22, 1476–1498. [Google Scholar] [CrossRef]
- Carracedo-Reboredo, P.; Liñares-Blanco, J.; Rodríguez-Fernández, N.; Cedrón, F.; Novoa, F.J.; Carballal, A.; Maojo, V.; Pazos, A.; Fernandez-Lozano, C. A review on machine learning approaches and trends in drug discovery. Computational and structural biotechnology journal 2021, 19, 4538–4558. [Google Scholar] [CrossRef]
- Huynh, T.; Cornell, W.; Luan, B. In silico Exploration of Inhibitors for SARS-CoV-2’s Papain-Like Protease. Front Chem 2020, 8, 624163. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors–an in silico docking and molecular dynamics simulation study. Journal of Biomolecular Structure and Dynamics 2021, 39, 4362–4374. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. Journal of chemical information and modeling 2020, 60, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Yosri, N.; Abd El-Wahed, A.A.; Ghonaim, R.; Khattab, O.M.; Sabry, A.; Ibrahim, M.A.; Moustafa, M.F.; Guo, Z.; Zou, X.; Algethami, A.F. Anti-viral and immunomodulatory properties of propolis: Chemical diversity, pharmacological properties, preclinical and clinical applications, and in silico potential against SARS-CoV-2. Foods 2021, 10, 1776. [Google Scholar] [CrossRef] [PubMed]
- Chekroud, A.M.; Bondar, J.; Delgadillo, J.; Doherty, G.; Wasil, A.; Fokkema, M.; Cohen, Z.; Belgrave, D.; DeRubeis, R.; Iniesta, R. The promise of machine learning in predicting treatment outcomes in psychiatry. World Psychiatry 2021, 20, 154–170. [Google Scholar] [CrossRef] [PubMed]
- Pearce, J.M. A review of open source ventilators for COVID-19 and future pandemics. F1000Research 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, G.; Biagi, F.; Costa, P.; Karpiński, Z.; Mazza, J. The likely impact of COVID-19 on education: Reflections based on the existing literature and recent international datasets; Publications Office of the European Union Luxembourg, 2020; Volume 30275. [Google Scholar]
- Alanine, A.; Nettekoven, M.; Roberts, E.; Thomas, A.W. Lead generation-enhancing the success of drug discovery by investing in the hit to lead process. Combinatorial chemistry & high throughput screening 2003, 6, 51–66. [Google Scholar]
- Aftab, S.O.; Ghouri, M.Z.; Masood, M.U.; Haider, Z.; Khan, Z.; Ahmad, A.; Munawar, N. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. Journal of translational medicine 2020, 18, 1–15. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



