
Submitted:
19 November 2023
Posted:
21 November 2023
You are already at the latest version
Abstract
We have recently reported that a series of 1,3,4-oxadiazol-2-ylbenzenesulfonamides acts as potent and specific monoamine oxidase B (MAO-B) inhibitors with some compounds possessing potencies in the nanomolar range. While sulfonamide compounds (e.g., zonisamide) have been reported to inhibit the MAO enzymes, nanomolar MAO inhibition potencies for this class are not frequently observed. The present study expanded on these findings by replacing the 1,3,4-oxazole moiety with a 1,3-thiazole heterocycle. A series of 23 primary sulfonamides were synthesized by reaction of an α-haloketone with a substituted carbothioamide. The results of the MAO inhibition studies showed that the 1,3-thiazolylbenzenesulfonamides were specific inhibitors of human MAO-B. The most potent MAO-B inhibitor exhibited an in vitro IC50 value of 0.103 µM (3j). The derivatives were, however, weaker inhibitors of human MAO-A with the most potent compound exhibiting an IC50 value of 11.9 µM (3a). Examination of the structure-activity relationships (SARs) showed that the most potent MAO-B inhibition was obtained with substitution of the sulfonamide on the meta position of the phenyl rather than the para position. This study concludes that 1,3-thiazolylbenzenesulfonamides represent a class of potent and specific MAO-B inhibitors, which may serve as leads for the development of new treatments for disease states such as Parkinson’s disease.
Keywords:
inhibition
; monoamine oxidase
; MAO
; thiazole
; sulfonamide
; SAR
; synthesis
1. Introduction
The monoamine oxidase (MAO) enzymes are flavin adenine dinucleotide (FAD) dependent enzymes that are bound to the outer membranes of mitochondria (Youdim et al., 2006). The MAOs consist of two isoforms, MAO-A and MAO-B, which are products of different genes (Shih et al., 1999; Shih et al., 2018). The two MAO isoforms are similar in their three-dimensional structures and have approximately 70% homology of their amino acid sequences (Bach et al., 1988; Son et al., 2008; Binda et al., 2002). Among the 16 amino acid residues that comprise the active sites, only 6 differ between the MAO isoforms (Son et al., 2008). Due to these similarities, significant overlap in substrate and inhibitor specificities exist between MAO-A and MAO-B. For example, epinephrine, norepinephrine, dopamine, tyramine and tryptamine are metabolized efficiently by both MAO-A and MAO-B, while several clinically used inhibitors (e.g., tranylcypromine, phenelzine) inhibit both isoforms (Youdim et al., 2006). In spite of this, certain substrates (e.g., serotonin) are specific for MAO-A, while exogenous amines such as phenethylamine and benzylamine are specifically metabolized by MAO-B (Youdim et al., 2006). In addition, numerous clinically used and experimental inhibitors display specificity for a specific isoform. For example, harmine and safinamide are highly specific reversible inhibitors of MAO-A and MAO-B, respectively (Son et al., 2008; Binda et al., 2007). The molecular basis for the inhibitor specificities of MAO-A and MAO-B may be explained by the reported X-ray crystal structures of the MAOs (Son et al., 2008; Binda et al., 2002; Hubálek et al., 2005). The active site of MAO-A consists of a single cavity, while the active site of MAO-B consists of a larger substrate cavity where catalysis takes place, as well as a smaller entrance cavity that provides access from the protein surface to the site of catalysis. The aromatic residue Phe208 in MAO-A restricts the binding of larger inhibitors (e.g., safinamide) to MAO-A, while in MAO-B, the analogous residue is Ile199, the gating residue that normally separates the substrate and entrance cavities (Son et al., 2008; Binda et al., 2007). Since Ile199 is smaller in size, it can rotate into an alternate conformation to allow the MAO-B substrate and entrance cavities to fuse. When this occurs, larger cavity-spanning inhibitors bind to MAO-B while not being able to productively bind to the MAO-A active site. Similarly, Tyr326 in MAO-B would sterically prevent certain inhibitors (e.g., harmine) from binding to MAO-B, while the analogous residue in MAO-A (Ile335) is smaller and permits the binding of such inhibitors (Son et al., 2008; Binda et al., 2007).
The rapid degradation of neurotransmitter and dietary amines is critical for the regulation of synaptic neurotransmission and to prevent molecules from acting as false neurotransmitters. Imbalances of neurotransmitter levels has been associated with various neurogenic disorders (Kumar et al., 2017). For example, the monoamine theory of depression states that decreased concentrations of centrally available monoamines (e.g., serotonin, norepinephrine and dopamine) may precipitate depressive disorders (Mulinari, 2012). Although this may be an oversimplification of a complex mental disorder, it has been found that MAO-A levels in depression patients were elevated by approximately 34% in all regions of the brain compared to healthy individuals (Meyer et al., 2006). It may be concluded that elevated MAO-A density might be the primary monoamine lowering process during major depression, and that inhibitors of MAO-A would be effective antidepressant agents. Nonselective and selective MAO-A inhibitors have thus been used for the treatment of atypical depressions and phobic anxiety for several decades (Zisook, 1985; Lum & Stahl, 2012; Suchting et al., 2021).
Parkinson’s disease is another disorder where the symptoms are caused by abnormal neurotransmitter levels in the brain. In Parkinson’s disease, progressive degeneration of dopaminergic neurons in the nigrostriatal pathway results in diminished dopamine levels in this pathway. This affects the extrapyramidal system and ultimately gives rise to the characteristic motor symptoms (Przedborski, 2017; Chinta & Andersen, 2005). MAO-B inhibitors are of therapeutic value in Parkinson’s disease since they may prevent central dopamine degradation. Irreversible (e.g., selegiline and rasagiline) and reversible (e.g., safinamide) inhibitors of MAO-B are established therapy in Parkinson’s disease and although providing only moderate symptomatic benefit, these agents are used as monotherapy or in combination with levodopa in late-stage Parkinson’s disease while having few pharmacological adverse effects and safety concerns (Alborghetti et al., 2024; Murakami et al., 2023). MAO-mediated metabolism is also a prominent source of hydrogen peroxide production in the central nervous system, which may be converted to highly reactive hydroxyl free radicals if not detoxified (Pizzinat et al., 1999; Edmondson, 2014). The destructive processes associated with these reactive oxygen species have the potential to contribute to neurodegeneration in Parkinson’s disease. By decreasing the production of hydrogen peroxide, MAO-B inhibitors may have the potential to act as disease modifying agents in neurodegenerative disorders (Edmondson, 2014; Youdim & Bakhle, 2006). It is particularly relevant to note that the density of the MAO-B isoform increases in the brain with age, which provides a further rationale for MAO-B inhibitors as neuroprotective agents in aged-related neurodegenerative disorders (Fowler et al., 1997; Fowler et al., 1980). In contrast, the density of MAO-A in the brain remains unchanged with age.
Similar to the prooxidant role of MAO-B in the brain, MAO-A have been identified as a significant source of hydrogen peroxide in the heart, and the resultant oxidative damage to cardiac tissue might lead to cardiovascular diseases such as congestive heart failure (Kaludercic et al., 2014; Manni et al., 2016; Umbarkar et al., 2015). MAO-A inhibitors might therefore have a future role as cardioprotective agents. Interestingly MAO inhibitors are also under investigation for the treatment of other disease states such as prostate cancer and rheumatoid arthritis (Han et al., 2023; Bala, 2023).
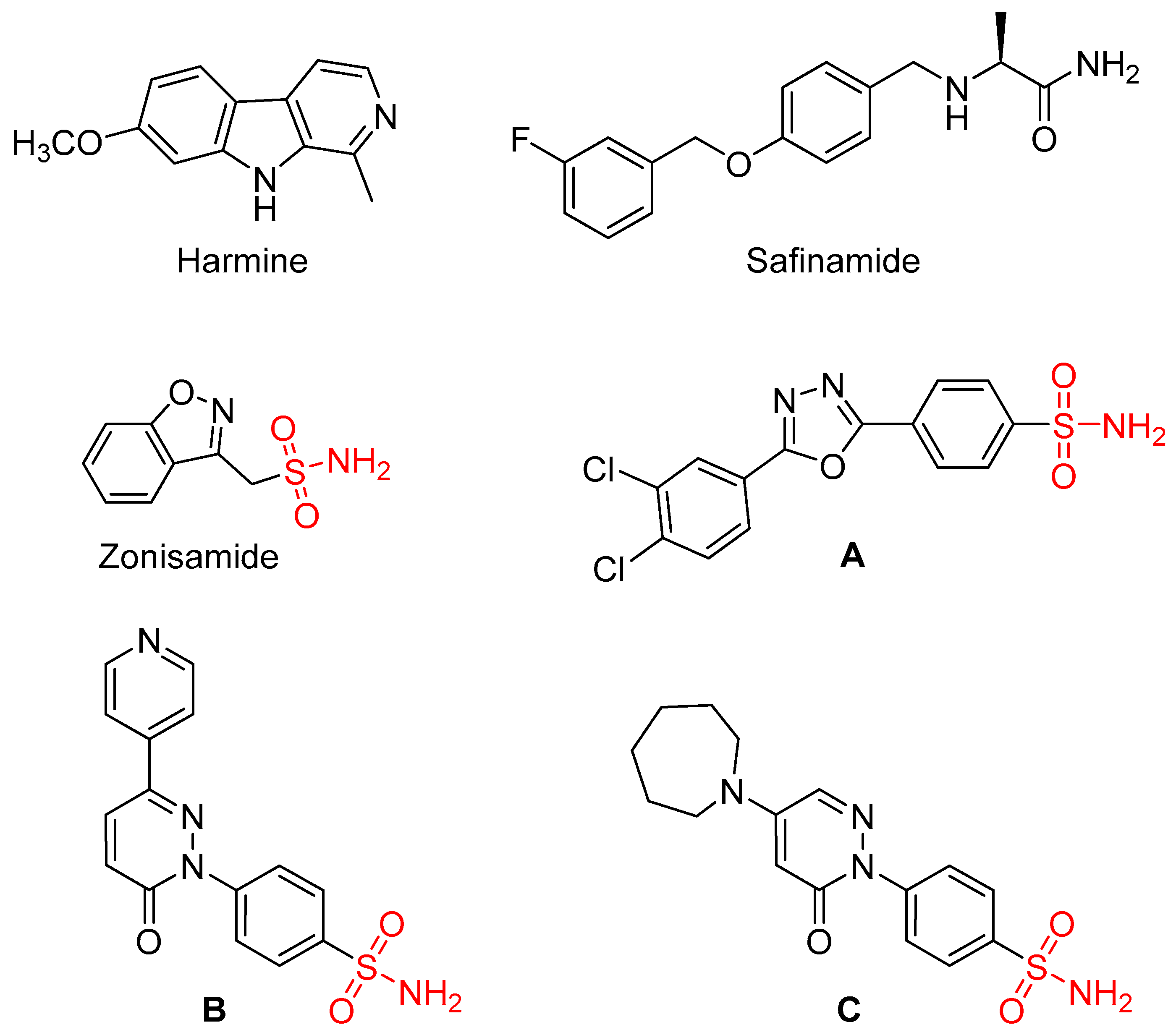
Since MAO has a significant role in the regulation of neurotransmitter levels and is a source of hydrogen peroxide and oxidative injury in certain tissues, inhibitors of these enzymes are of much scientific and medicinal interest. The discovery novel MAO inhibitors with favourable specificity and activity profiles are pursued by researchers in the academia and industry (Guglielmi et al., 2022). We have recently reported that a series of 1,3,4-oxadiazol-2-ylbenzenesulfonamides act as potent MAO-B inhibitors with a high degree of isoform specificity (Shetnev et al., 2019). For example, the most potent compound (A) presented with an IC50 value of 0.0027 µM. In a related study, we have discovered additional compounds with MAO inhibition properties among a series of pyridazinone-substituted benzenesulfonamides. Two compounds (B, C) exhibited specific inhibition of MAO-B with IC50 values of 2.90 and 4.36 μM, respectively (Petzer et al., 2023). An earlier study has shown that the sulfonamide compound, zonisamide, also inhibits MAO-B, although with moderate potency (IC50 = 24.8 µM) (Sonsalla et al., 2010). The crystal structure of zonisamide in complex with human MAO-B showed that zonisamide binds within the substrate cavity, leaving the entrance cavity unoccupied (Binda et al., 2011). While the sulfonamide functional group undergoes important interactions in the substrate cavity, the absence of interaction with the residues of the entrance cavity might explain the relatively lower MAO-B inhibition potency of zonisamide. In contrast, modelling suggested that for the 1,3,4-oxadiazol-2-ylbenzenesulfonamides (e.g., A), the benzenesulfonamide group binds in the substrate cavity in proximity to the FAD, while the dichlorophenyl extends into the entrance cavity (Petzer et al., 2023). These compounds are thus cavity-spanning inhibitors and the additional interactions afforded by the residues of the entrance cavity may explain, in part, the high MAO-B inhibition potencies of these compounds. The present study aimed to expand on these findings by replacing the 1,3,4-oxazole moiety with a 1,3-thiazole heterocycle. This study thus synthesized a series of 1,3-thiazolylbenzenesulfonamides by reaction of an α-haloketone with a substituted carbothioamide, and the resulting compounds were evaluated in vitro as inhibitors of the human MAOs. The results showed that these compounds display specificity for the MAO-B isoform with some compounds exhibiting nanomolar potencies. Besides the 1,3,4-oxadiazol-2-ylbenzenesulfonamides reported earlier, this study is only the second example of sulfonamide compounds that exhibit such high potency MAO inhibition (Petzer et al., 2023).
Figure 1.
The structures of isoform-selected MAO inhibitors, 1,3,4-oxadiazol-2-ylbenzenesulfonamide A and pyridazinone-substituted benzenesulfonamides B and C.
Figure 1.
The structures of isoform-selected MAO inhibitors, 1,3,4-oxadiazol-2-ylbenzenesulfonamide A and pyridazinone-substituted benzenesulfonamides B and C.
2. Results and Discussion
2.1. Chemistry
For the synthesis of the 1,3-thiazolylbenzenesulfonamide derivatives, primary benzenesulfonamide fragments 2a,b and 4 were used building blocks (Figure 2). These are easily synthesized according to the methods described in literature (Scobie, 2018; Kaboudin, & Elhamifar, 2006). This strategy made it possible to avoid the use of dangerous and corrosive reagents, and reagents with low selectivity (e.g., chlorosulfonic acid and its derivatives) for the synthesis of the benzenesulfonamide derivatives.
The target 1,3-thiazolylbenzenesulfonamide derivatives were synthesized by reaction of thioamides with the corresponding α-haloketone, with the reactions being heated in isopropyl alcohol (i-PrOH) without any acceptor of the evolved hydrogen bromide. Remarkably, during the cyclization reaction to form 1,3-thiazole ring used (Figure 3 and Figure 4), the nucleophilic substitution reaction of the sulfur-containing nucleophile proceeded regioselectively without reaction at the second nucleophilic center, the primary sulfonamide moiety. By reacting a benzenesulfonamide-containing α-haloketone (2a,b) with a substituted carbothioamide derivative (1a–o), 1,3-thiazoles 3a–t were synthesized in yields of 40–91%. In a similar manner, benzenesulfonamide-containing carbothioamide (4) underwent a cyclocondensation reaction with a substituted α-haloketone (5, 7) to yield the corresponding 1,3-thiazoles 6a–b and 8 in yields of 44–72%. In most instances, the reaction products did not require additional purification and precipitated from the reaction medium after 24 h of boiling in i-PrOH.
2.2. In vitro biological evaluation – MAO inhibition
The 1,3-thiazolylbenzenesulfonamide derivatives (3, 6, 8) were evaluated as in vitro inhibitors of MAO by using the recombinant human MAO enzymes (Novaroli et al., 2005). Kynuramine was used as enzyme substrate for both MAO isoforms and is metabolized to give 4-hydroxyquioline. In this study the formation of 4-hydroxyquinoline was monitored by fluorescence spectrophotometry. MAO catalytic activity was thus measured in the presence of a range of concentrations (0.003–100 µM) of the test inhibitors and sigmoidal graphs of enzyme activity versus inhibitor concentration (Log[I]) were constructed from the activity measurements. These graphs were used to estimate IC50 values for the inhibition of MAO, which are reported as the mean ± standard deviation (SD) of triplicate measurements. The sigmoidal graphs for the inhibition of MAO-B by 3j, 3l and 3m are presented in Figure 5 as examples.
The potencies of MAO inhibition by the 1,3-thiazolylbenzenesulfonamide derivatives (3, 6, 8) are presented in Table 1. From the inhibition data it is evident that the derivatives are weak MAO-A inhibitors with most compounds exhibiting no inhibition at a maximal tested concentration of 100 µM. The most potent MAO-A inhibitors were 3a, 3f and 3m with IC50 values of 11.9, 18.7 and 17.2 µM, respectively. While clear structure-activity relationships (SARs) are not apparent, is may be noted that among the compounds with IC50 < 30 µM (e.g., 3a, 3b, 3f, 3j, 3l, 3m and 3t) the benzenesulfonamide group was substituted on C4 rather than C2 (6a,b) of the 1,3-thiazole moiety. Among these were compounds with substitution of the sulfonamide on both the meta and para positions of the phenyl.
In contrast, some of the 1,3-thiazolylbenzenesulfonamide derivatives were found to be potent inhibitors of MAO-B with the most potent inhibitors being 3j, 3l and 3m with IC50 values of 0.103, 0.193 and 0.172 µM, respectively. In fact, seven derivatives in total exhibited IC50 < 1 µM. SARs that were note included the following: (a) With the exception of 3j and 3q, all submicromolar inhibitors contained a halogen (F, Cl) on the phenyl ring; (b) For the submicromolar inhibitors, substitution of the benzenesulfonamide group occurred on either C4 or C2 (6b); (c) The most potent inhibitors (3j, l, m) all contained substitution of the sulfonamide on the meta position of the phenyl rather than the and para position. Most of the meta substituted compounds were more potent than the corresponding para substituted homologue (3j vs. 3b; 3l vs. 3f; 3m vs. 3d; 3n vs. 3k; 3q vs. 3c); (d) An aromatic substituent is a requirement for good potency MAO-B inhibition since derivatives with methyl substitution (3e, 3i, 8) were weak inhibitors.
2.3. Lineweaver-Burk graphs for the inhibition of MAO-B by 3j
This study has identified compound 3j as the most potent MAO-B inhibitor of the series. The mode of inhibition by this compound was further investigated by constructing sets of Lineweaver-Burk graphs. Six Lineweaver-Burk graphs were constructed at the following inhibitor concentrations: 0 × IC50, ¼ × IC50, ½ × IC50, ¾ × IC50, 1 × IC50 and 1¼ × IC50. For each graph, eight kynuramine concentrations (15 to 250 μM) were used to measure MAO-B activity. The Lineweaver-Burk graphs are shown in Figure 6 and suggest that compound 3j is a competitive inhibitor since the lines intersect on the y-axis. The enzyme-inhibitor dissociation constant (Ki) may be determined from a graph of the slopes of the Lineweaver-Burk graphs versus inhibitor concentration, and was estimated to be 0.21 µM.
2.4. Cytotoxicity in vitro
The effect of compounds 3j and 3m on human cell cultures were investigated with two cell lines, IMR-32 (neuroblastoma) and HS-5 (bone marrow stroma). In this respect, the cell viability after treatment with 0.5–100 µM of the test compounds were evaluated using the MTT assay (Gradiz et al., 2016; Mosmann, 1983)
The results of these experiments showed that compound 3j did not exhibit toxicity at the concentration range used for this study (Figure 7). Compound 3m, however, exhibited a TC50 value of approximately 4 µM for both cell lines evaluated.
2.5. Molecular docking
This investigation found that certain 1,3-thiazolylbenzenesulfonamide derivatives act as potent MAO-B inhibitors. A molecular docking study was carried out to determine potential binding orientations and interactions of a selected derivative (3m) with the MAO-B enzyme. Molecular docking was carried out according to the literature procedure (Mostert et al., 2015) using Discovery Studio 3.1 and the published X-ray crystal structure of MAO-B in complex with zonisamide (PDB code: 3PO7) (Binda et al., 2011). As anticipated, compound 3m binds with the benzenesulfonamide moiety in the substrate cavity, in proximity to the FAD where the sulfonamide group occupies the same space as the sulfonamide of zonisamide (Figure 8). The sulfonamide is involved in hydrogen bonding with Gln206 and a water molecule as well as a pi-sulfur interaction with the FAD. The dichlorophenyl substituent extends beyond Ile199 and Tyr326 into the entrance cavity where numerous van der Waals interactions occur with the surrounding amino acid residues. Interestingly, the thiazole ring forms a pi-pi interaction with Tyr326 and pi-sulfur interactions with Cys172 and Phe168. This analysis underscores the significant interactions of the sulfonamide and thiazole moieties with MAO-B, while further inhibitor stabilization is provided by interactions between the dichlorophenyl and the entrance cavity. A similar binding mode to MAO-B has been predicted for A (Shetnev et al., 2019).
3. Conclusion
The present study investigated the MAO inhibition properties of a series of 1,3-thiazolylbenzenesulfonamides. The compounds were found to be specific inhibitors of MAO-B, with three derivatives (3j, 3l and 3m) exhibiting IC50 values in the low nanomolar range. In contrast, the derivatives were much weaker MAO-A inhibitors with the most potent inhibition observed for 3a (IC50 = 11.9 µM). A molecular docking study predicts that both the sulfonamide and thiazole moieties productively interact with the MAO-B active site and thus contribute to inhibitor stabilization. This investigation is an extension of our previous study that showed 1,3,4-oxadiazol-2-ylbenzenesulfonamides to act as potent MAO-B inhibitors and is the second report of sulfonamide compounds with nonomolar inhibition potencies. Potent MAO-B inhibitors such as those identified here may be considered to be good leads for the development of new therapies for disorders such as Parkinson’s disease.
4. Materials and Methods
4.1. Chemicals and instrumentation
All reagents and solvents were obtained from commercial sources (Aldrich, Fluka, Merck) and were used without further purification. Reactions were monitored with silica gel TLC using a mixture of toluene, acetone and petroleum ether (60:100:100) as eluent. Melting points were measured with a Stuart SMP30 apparatus in capillary tubes and are uncorrected. Electrospray ionization (ESI) mass spectra were obtained on a Bruker maXis spectrometer equipped with an ESI source. The instrument was operated in positive ion mode using an m/z range of 50–1200. The flow rate of the nebulizer gas was set to 1.0 bar, and that of the drying gas was set to 4.0 L/min. NMR spectra were recorded on a Bruker Avance 400 at ambient temperature, and were referenced to the residual solvent signal. 1H NMR spectra were recorded at 400 MHz while 13C NMR spectra were recorded at 100 MHz. Chemical shifts are reported in parts per million (ppm).
Recombinant human MAO-A and MAO-B, kynuramine and reference MAO inhibitors used for the biological experiments were obtained from Sigma-Aldrich. A SpectraMax iD3 multi-mode microplate reader (Molecular Devices) was used to record the fluorescence measurements of the enzymatic reactions.
4.2. General procedure for the preparation of thiazoles (3) (GP1).
Thioamide 1 (0.9 mmol, 1 equiv.) was dissolved in i-PrOH (2.5 mL) and sulfonamide 2 (0.9 mmol, 1 equiv.) was added. The reaction mixture was stirred at room temperature for 1 h and then stirred overnight at 82.5 °C. The solvent was evaporated under reduced pressure and the reaction mixture was diluted with cold water (5 mL). The resulting precipitate was collected by filtration, washed with water (15 mL) and air-dried at 50 °C.
4-(2-(Thiophen-2-yl)thiazol-4-yl)benzenesulfonamide (3a). The reaction of 2-thiophenecarbothioamide (0.9 mmol, 0.13 g) and 4-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.24 g (83%) of 3a isolated as the beige solid. m.p. 248-250 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (s, 1H), 8.17 (d, J = 8.5 Hz, 2H), 7.92 (d, J = 8.6 Hz, 2H), 7.78 (d, J = 5.0 Hz, 1H), 7.76 (dd, J = 3.7, 1.0 Hz, 1H), 7.42 (s, 2H), 7.21 (dd, J = 5.0, 3.7 Hz, 1H). 13C NMR (101 MHz, DMSO- d6) δ 161.98, 153.75, 144.13, 137.18, 136.84, 129.94, 129.18, 128.49, 127.04, 126.98, 116.99. HRMS (ESI), m/z: [M+H]+ calcd. for C13H10N2O2S3 322.9977; found 322.9978.
4-(2-Phenylthiazol-4-yl)benzenesulfonamide (3b). The reaction of thiobenzamide (1.8 mmol, 0.26 g) and 4-(2-bromoacetyl)benzenesulfonamide (1.8 mmol, 0.25 g) according to general procedure GP1 afforded 0.51 g (89%) of 3b isolated as the beige solid. m.p. 215-217 °C. 1H NMR (400 MHz, DMSO- d6) δ 8.37 (s, 1H), 8.24 (d, J = 8.4 Hz, 2H), 8.06 (d, J = 2.2 Hz, 1H), 8.04 (d, J = 1.7 Hz, 1H), 7.93 (d, J = 8.4 Hz, 2H), 7.56 (s, 1H), 7.54 (t, J = 2.2 Hz, 2H), 7.41 (s, 2H). 13C NMR (101 MHz, DMSO- d6) δ 168.12, 154.35, 144.10, 137.53, 133.45, 131.25, 129.97, 127.09, 126.97 (2С), 117.62. HRMS (ESI), m/z: [M+H]+ calcd. for C15H12N2O2S2 317.0413; found 317.0410.
4-(2-(4-(Dimethylamino)phenyl)thiazol-4-yl)benzenesulfonamide (3c). The reaction of 4-(dimethylamino)benzenecarbothioamide (0.9 mmol, 0.162 g) and 4-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.29 g (91%) of 3c isolated as the grey solid. m.p. 254-256 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, J = 8.4 Hz, 2H), 8.15 (s, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.85 (d, J = 8.8 Hz, 2H), 7.40 (s, 2H), 6.81 (d, J = 8.9 Hz, 2H), 3.01 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 168.96, 153.77, 152.34, 143.81, 137.85, 128.13, 126.95, 126.89, 121.19, 115.07, 112.51, 39.58. HRMS (ESI), m/z: [M+H]+ calcd. for C17H17N3O2S2 360.0835; found 360.0839.
4-(2-(3,4-Dichlorophenyl)thiazol-4-yl)benzenesulfonamide (3d). The reaction of 3,4-dichlorobenzenecarbothioamide (0.9 mmol, 0.19 g) and 4-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.30 g (86%) of 3d isolated as the beige solid. m.p. 189-191 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.45 (s, 1H), 8.29 (d, J = 2.0 Hz, 1H), 8.26 (d, J = 8.4 Hz, 2H), 8.02 (dd, J = 8.4, 2.0 Hz, 1H), 7.93 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 8.4 Hz, 1H), 7.41 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.26, 154.55, 144.23, 137.18, 133.79, 133.61, 132.83, 132.18, 128.26, 127.18, 127.09, 126.97, 118.83. HRMS (ESI), m/z: [M+H]+ calcd. for C15H10Cl2N2O2S2 384.9634; found 384.9632.
4-(2-Methylthiazol-4-yl)benzenesulfonamide (3e). The reaction of thioacetamide (1.9 mmol, 0.15 g) and 4-(2-bromoacetyl)benzenesulfonamide (1.9 mmol, 0.5 g) according to general procedure GP1 afforded 0.41 g (89%) of 3e isolated as the white solid. m.p. 250-252 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (d, J = 3.4 Hz, 2H), 8.11 (s, 1H), 7.87 (d, J = 8.6 Hz, 2H), 7.37 (s, 2H), 2.74 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.81, 152.99, 143.75, 137.74, 126.91, 126.86, 116.83, 19.59. HRMS (ESI), m/z: [M+H]+ calcd. for C10H10N2O2S2 255.0256; found 255.0257.
4-(2-(4-Fluorophenyl)thiazol-4-yl)benzenesulfonamide (3f). The reaction of 4-fluorothiobenzamide (1.8 mmol, 0.29 g) and 4-(2-bromoacetyl)benzenesulfonamide (1.8 mmol, 0.5 g) according to general procedure GP1 afforded 0.46 g (77%) of 3f isolated as the beige solid. m.p. 203-205 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.36 (s, 1H), 8.23 (d, J = 8.3 Hz, 2H), 8.11 (d, J = 5.4 Hz, 1H), 8.09 (d, J = 5.6 Hz, 1H), 7.92 (d, J = 8.4 Hz, 2H), 7.41 (s, 2H), 7.38 (d, J = 8.6 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.90, 164.01, 162.78, 154.26, 144.06, 137.41, 130.08, 129.34, 129.25, 127.06, 126.94, 117.71, 117.10, 116.88. HRMS (ESI), m/z: [M+H]+ calcd. for C15H11FN2O2S2 335.0319; found 335.0317.
4-(2-(5-Methylthiophen-2-yl)thiazol-4-yl)benzenesulfonamide (3g). The reaction of 5-methyl-2-thiophenecarbothioamide (0.9 mmol, 0.14 g) and 4-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.24 g) according to general procedure GP1 afforded 0.23 g (79%) of 3g isolated as the beige solid. m.p. 241-243 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.24 (s, 1H), 8.15 (d, J = 8.4 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H), 7.55 (d, J = 3.6 Hz, 1H), 7.40 (s, 2H), 6.91 (d, J = 3.0 Hz, 1H), 2.51 (s, 3H). 13C NMR (101 MHz, DMSO- d6) δ 162.07, 153.53, 144.03, 143.77, 137.22, 134.44, 128.51, 127.60, 126.96 (2С), 116.42, 15.79. HRMS (ESI), m/z: [M+H]+ calcd. for C14H12N2O2S3 337.0134; found 337.0131.
4-(2-(4-(Trifluoromethoxy)phenyl)thiazol-4-yl)benzenesulfonamide (3h). The reaction of 4-(trifluoromethoxy)benzenecarbothioamide (0.9 mmol, 0.2 g) and 4-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.22 g (61%) of 3h isolated as the beige solid. m.p. 194-196 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (s, 1H), 8.24 (d, J = 8.5 Hz, 2H), 8.18 (d, J = 8.8 Hz, 2H), 7.93 (d, J = 8.4 Hz, 2H), 7.55 (d, J = 8.1 Hz, 2H), 7.42 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.46, 154.52, 150.29, 144.20, 137.36, 132.57, 129.02, 127.11, 126.98, 122.37, 118.28. HRMS (ESI), m/z: [M+H]+ calcd. for C16H11F3N2O3S2 401.0236; found 401.0233.
3-(2-Methylthiazol-4-yl)benzenesulfonamide (3i). The reaction of thioacetamide (0.9 mmol, 0.07 g) and 3-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.17 g (74%) of 3i isolated as the beige solid. m.p. 168-170 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.43 (s, 1H), 8.14 (d, J = 7.6 Hz, 1H), 8.07 (s, 1H), 7.78 (d, J = 7.4 Hz, 1H), 7.63 (t, J = 7.4 Hz, 1H), 7.41 (s, 2H), 2.75 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.81, 152.95, 145.46, 135.43, 130.13, 129.51, 125.49, 123.78, 115.97, 19.57. HRMS (ESI), m/z: [M+H]+ calcd. for C10H10N2O2S2 255.0256; found 255.0257.
3-(2-Phenylthiazol-4-yl)benzenesulfonamide (3j). The reaction of thiobenzamide (0.9 mmol, 0.12 g) and 3-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.18 g (64%) of 3j isolated as the brown solid. m.p. 180-181 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.55 (s, 1H), 8.33 (s, 1H), 8.25 (d, J = 7.7 Hz, 1H), 8.07 (d, J = 6.7 Hz, 2H), 7.84 (d, J = 7.8 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.56 (d, J = 5.9 Hz, 3H), 7.47 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.14, 154.35, 145.59, 135.28, 133.49, 131.24, 130.24, 129.99, 129.79, 126.95, 125.86, 123.94, 116.85. HRMS (ESI), m/z: [M+H]+ calcd. for C15H12N2O2S2 317.0413; found 317.0410.
4-(2-(4-Isopropylphenyl)thiazol-4-yl)benzenesulfonamide (3k). The reaction of 4-isopropylthiobenzamide (0.9 mmol, 0.16 g) and 4-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.25 g (78%) of 3k isolated as the beige solid. m.p. 177-179 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.33 (d, J = 2.6 Hz, 1H), 8.23 (d, J = 8.4 Hz, 2H), 7.96 (d, J = 8.2 Hz, 2H), 7.92 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 3.2 Hz, 2H), 3.02 – 2.91 (m, 1H), 1.30 – 1.20 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 168.23, 154.25, 151.87, 144.07, 137.60, 131.28, 127.89, 127.07 (2С), 126.96, 117.17, 34.02, 24.28. HRMS (ESI), m/z: [M+H]+ calcd. for C18H18N2O2S2 359.0882; found 359.0883.
3-(2-(4-Fluorophenyl)thiazol-4-yl)benzenesulfonamide (3l). The reaction of 4-fluorothiobenzamide (0.9 mmol, 0.14 g) and 3-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.26 g (87%) of 3l isolated as the beige solid. m.p. 189-191 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.31 (s, 1H), 8.23 (d, J = 7.7 Hz, 1H), 8.10 (dd, J = 8.6, 5.5 Hz, 2H), 7.82 (d, J = 7.8 Hz, 1H), 7.68 (t, J = 7.8 Hz, 1H), 7.45 (s, 2H), 7.39 (t, J = 8.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.94, 164.04 (d, J = 248.4 Hz), 154.31, 145.57, 135.18, 130.00 (d, J = 46.0 Hz), 129.28 (d, J = 8.5 Hz), 125.86, 123.92, 117.12, 116.91. HRMS (ESI), m/z: [M+H]+ calcd. for C15H11FN2O2S2 335.0319; found 335.0322.
3-(2-(3,4-Dichlorophenyl)thiazol-4-yl)benzenesulfonamide (3m). The reaction of 3,4-dichlorobenzenecarbothioamide (0.9 mmol, 0.19 g) and 4-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.3 g (86%) of 3m isolated as the beige solid. m.p. 177-179 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.40 (s, 1H), 8.26 (d, J = 2.0 Hz, 1H), 8.23 (s, 1H), 8.02 (d, J = 2.0 Hz, 1H), 8.00 (d, J = 1.4 Hz, 1H), 7.82 (t, J = 9.5 Hz, 1H), 7.68 (t, J = 7.8 Hz, 1H), 7.47 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.28, 154.59, 145.61, 134.96, 133.84, 133.64, 132.87, 132.21, 130.27, 129.87, 128.23, 127.05, 126.00, 123.95, 118.09. HRMS (ESI), m/z: [M+H]+ calcd. for C15H10Cl2N2O2S2 384.9634; found 384.9637.
3-(2-(4-Isopropylphenyl)thiazol-4-yl)benzenesulfonamide (3n). The reaction of 4-isopropylthiobenzamide (0.9 mmol, 0.16 g) and 3-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.17 g (65%) of 3n isolated as the beige solid. m.p. 140-142 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.28 (s, 1H), 8.24 (d, J = 7.7 Hz, 1H), 7.97 (d, J = 8.1 Hz, 2H), 7.83 (d, J = 7.7 Hz, 1H), 7.68 (t, J = 7.8 Hz, 1H), 7.46 (s, 2H), 7.42 (d, J = 8.2 Hz, 2H), 2.97 (dt, J = 13.8, 6.9 Hz, 1H), 1.25 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 168.24, 154.25, 151.88, 145.57, 135.33, 131.30, 130.23, 129.77, 127.90, 127.05, 125.81, 123.91, 116.40, 34.02, 24.28. HRMS (ESI), m/z: [M+H]+ calcd. for C18H18N2O2S2 359.0882; found 359.0880.
3-(2-(3,4-Dimethoxyphenyl)thiazol-4-yl)benzenesulfonamide (3o). The reaction of 3,4-dimethoxybenzenecarbothioamide (0.9 mmol, 0.18 g) and 3-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.29 g (85%) of 3o isolated as the beige solid. m.p. 139-141 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.51 (s, 1H), 8.24 (d, J = 10.7 Hz, 2H), 7.82 (d, J = 7.8 Hz, 1H), 7.68 (t, J = 7.8 Hz, 1H), 7.59 (d, J = 8.3 Hz, 1H), 7.56 (d, J = 1.8 Hz, 1H), 7.47 (s, 2H), 7.11 (d, J = 8.4 Hz, 1H), 3.89 (s, 3H), 3.84 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.23, 154.06, 151.65, 149.80, 145.53, 135.36, 130.19, 129.81, 126.44, 125.73, 123.84, 120.28, 115.91, 112.74, 110.07, 56.40 (2C). HRMS (ESI), m/z: [M+H]+ calcd. for C17H16N2O4S2 377.0624; found 377.0621.
4-[2-(3,4-Dimethoxyphenyl)-1,3-thiazol-4-yl]benzenesulfonamide (3p). The reaction of 3,4-dimethoxybenzenecarbothioamide (0.9 mmol, 0.18 g) and 4-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.25 g (74%) of 3p isolated as the beige solid. m.p. 185-187 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.26 (s, 1H), 8.21 (d, J = 8.3 Hz, 2H), 7.89 (d, J = 8.3 Hz, 2H), 7.55 (d, J = 6.3 Hz, 2H), 7.39 (s, 2H), 7.07 (d, J = 8.8 Hz, 1H), 3.84 (d, J = 20.4 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 168.19, 154.04, 151.63, 149.79, 144.02, 137.64, 127.07 (2C), 126.92 (2C), 126.40, 120.29, 116.69, 112.68, 109.98, 56.38 (2C). HRMS (ESI), m/z: [M+H]+ calcd. for C17H16N2O4S2 377.0624; found 377.0628.
3-{2-[4-(Dimethylamino)phenyl]-1,3-thiazol-4-yl}benzenesulfonamide (3q). The reaction of 4-(dimethylamino)benzenecarbothioamide (0.9 mmol, 0.16 g) and 3-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.25 g (78%) of 3q isolated as the green solid. m.p. 134-136 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.51 (s, 1H), 8.21 (d, J = 7.9 Hz, 1H), 8.10 (s, 1H), 7.85 (d, J = 8.7 Hz, 2H), 7.80 (d, J = 7.7 Hz, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.46 (s, 2H), 6.81 (d, J = 8.8 Hz, 2H), 3.00 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 168.89, 153.78, 152.12, 145.49, 135.55, 130.12, 129.67, 128.13, 125.58, 123.86, 121.64, 114.37, 112.85, 39.82. HRMS (ESI), m/z: [M+H]+ calcd. for C17H17N3O2S2 360.0835; found 360.0836.
3-(2-(4-(Trifluoromethoxy)phenyl)thiazol-4-yl)benzenesulfonamide (3r). The reaction of 4-(trifluoromethoxy)benzenecarbothioamide (0.9 mmol, 0.2 g) and 4-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.14 g (40%) of 3r isolated as the beige solid. m.p. 146-148 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.54 (s, 1H), 8.38 (s, 1H), 8.25 (d, J = 7.9 Hz, 1H), 8.18 (d, J = 8.8 Hz, 2H), 7.84 (d, J = 7.8 Hz, 1H), 7.69 (t, J = 7.8 Hz, 1H), 7.56 (d, J = 8.1 Hz, 2H), 7.46 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.49, 154.53, 150.31, 145.61, 135.10, 132.61, 130.28, 129.82, 129.01, 125.96, 123.94, 122.41, 121.97, 119.41, 117.57. HRMS (ESI), m/z: [M+H]+ calcd. for C16H11F3N2O3S2 401.0236; found 401.0233.
4-(2-(3-(Trifluoromethyl)phenyl)thiazol-4-yl)benzenesulfonamide (3s). The reaction of 3-(trifluoromethyl)benzenecarbothioamide (0.9 mmol, 0.18 g) and 4-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.3 g (87%) of 3s isolated as the white solid. m.p. 158-160 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.54 (s, 1H), 8.37 (s, 1H), 8.25 (d, J = 8.1 Hz, 1H), 8.18 (d, J = 8.9 Hz, 2H), 7.84 (d, J = 8.4 Hz, 1H), 7.69 (t, J = 7.7 Hz, 1H), 7.56 (d, J = 8.1 Hz, 2H), 7.47 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.48, 154.52, 150.30, 145.59, 135.09, 132.61, 130.29, 129.81, 129.01, 125.95, 123.92, 122.43, 117.59. HRMS (ESI), m/z: [M+H]+ calcd. for C16H11F3N2O3S2 401.0236; found 401.0237.
3-(2-(4-Methoxyphenyl)thiazol-4-yl)benzenesulfonamide (3t). The reaction of 4-methoxybenzothioamide (0.9 mmol, 0.15 g) and 3-(2-bromoacetyl)benzenesulfonamide (0.9 mmol, 0.25 g) according to general procedure GP1 afforded 0.27 g (87%) of 3u isolated as the yellow solid. m.p. 160-162 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.23 (d, J = 4.5 Hz, 2H), 7.99 (d, J = 8.6 Hz, 2H), 7.81 (d, J = 7.8 Hz, 1H), 7.67 (t, J = 7.8 Hz, 1H), 7.46 (s, 2H), 7.11 (d, J = 8.6 Hz, 2H), 3.85 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.03, 161.79, 154.05, 145.53, 135.37, 130.19, 129.74, 128.56, 126.32, 125.73, 123.90, 115.82, 115.34, 56.13. HRMS (ESI), m/z: [M+H]+ calcd. for C16H14N2O3S2 347.0519; found 347.0515.
4.3. Preparation and characterization of isomeric thiazoles (6)
General procedure for the preparation of isomeric thiazoles (6) (GP2). 4-(Aminosulfonyl)benzenecarbothioamide 4 (1.2 mmol, 1 equiv.) was dissolved in i-PrOH (2.5 mL) and bromide 5 (1.2 mmol, 1 equiv.) was added. The reaction mixture was stirred at room temperature for 1 h and then stirred overnight at 82.5 °C. The solvent was evaporated under reduced pressure and the reaction mixture was diluted with cold water (5 mL). The resulting precipitate was collected by filtration, washed with water (15 mL) and air-dried at 50 °C.
4-(4-(Benzo[d][1,3]dioxol-5-yl)thiazol-2-yl)benzenesulfonamide (6a). The reaction of 1-(1,3-benzodioxol-5-yl)-2-bromoethanone (0.9 mmol, 0.22 g) and 4-(aminosulfonyl)benzenecarbothioamide (0.9 mmol, 0.19 g) according to general procedure GP2 afforded 0.14 g (44%) of 6a isolated as the white solid. m.p. 181-183 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, J = 8.3 Hz, 1H), 8.15 (s, 1H), 7.95 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 6.8 Hz, 2H), 7.50 (s, 2H), 7.02 (d, J = 8.6 Hz, 2H), 6.08 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.69, 156.00, 148.50, 148.10, 145.87, 136.32, 128.79, 127.30 (2C), 120.92, 115.10, 109.24, 107.17, 101.94. HRMS (ESI), m/z: [M+H]+ calcd. for C16H12N2O4S2 361.0311; found 361.0310.
4-(4-(4-Chlorophenyl)thiazol-2-yl)benzenesulfonamide (6b). The reaction of 2′-bromo-4-chloroacetophenone (0.9 mmol, 0.21 g) and 4-(aminosulfonyl)benzenecarbothioamide (0.9 mmol, 0.19 g) according to general procedure GP2 afforded 0.18 g (60%) of 6b isolated as the white solid. m.p. 158-160 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.35 (s, 1H), 8.22 (d, J = 8.2 Hz, 2H), 8.09 (d, J = 8.3 Hz, 2H), 7.97 (d, J = 8.2 Hz, 2H), 7.55 (d, J = 8.4 Hz, 2H), 7.52 (s, 2H). 13C NMR (101 MHz, DMSO- d6) δ 166.29, 154.99, 146.05, 136.19, 133.62, 133.29, 129.58, 128.60, 127.37 (2C), 117.31. HRMS (ESI), m/z: [M+H]+ calcd. for C15H11ClN2O2S2 351.0023; found 351.0020.
4.4. Preparation and characterization of the thiazole 8
General procedure for the preparation of thiazole 8 (GP3). Following the previously reported procedure (Yamane et al., 2004), 4-(aminosulfonyl)benzenecarbothioamide 4 (0.9 mmol, 1 equiv.) was dissolved in i-PrOH (2 mL) and 3-chlorobutan-2-one 7 (1.4 mmol, 1.5 equiv.) was added. The reaction mixture was stirred at room temperature for 1 h and then stirred overnight at 82.5 °C. The solvent was evaporated under reduced pressure and the reaction mixture was diluted with cold water (5 mL). The resulting precipitate was collected by filtration, washed with water (15 mL) and air-dried at 50 °C.
4-(4,5-Dimethylthiazol-2-yl)benzenesulfonamide (8). Beige solid, yield 72%. m.p. 211-213 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.02 (d, J = 8.4 Hz, 2H), 7.89 (d, J = 8.4 Hz, 2H), 7.45 (s, 2H), 2.41 (s, 3H), 2.34 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 160.97, 150.43, 145.19, 136.61, 129.11, 127.24, 126.57, 15.24, 11.77. HRMS (ESI), m/z: [M+H]+ calcd. for C11H12N2O2S2 269.0413; found 269.0415.
4.7. Procedure for measuring MAO inhibition potency
The catalytic activities of MAO-A and MAO-B were measured by following the literature procedure (Mostert et al., 2015; Weissbach et al., 1960). Commercially available recombinant human MAO-A and MAO-B served as enzyme sources while kynuramine was used as a mixed MAO-A/B substrate. The MAO enzymes oxidize kynuramine to yield 4-hydroxyquinoline as the ultimate product. Since 4-hydroxyquinoline is fluorescent in basic medium, the reactions were terminated the end-point with the addition of sodium hydroxide and fluorescence spectrophotometry was used to quantitate 4-hydroxyquinoline.
4.8. Procedure for measuring cytotoxicity
The human cell culture lines, IMR-32 (neuroblastoma) and HS-5 (bone marrow stroma), were grown in a mixture (1:1) of RPMI-1640 and Ham's F12 media without glutamine. To the media was added FBS (10%), L-glutamine (2 mM), penicillin (50 IU/ml) and streptomycin (50 μg/mL). The test compounds were dissolved in DMSO and added to the culture media to yield a final concentration of 0.1% DMSO. As positive control (0% cell viability) the cells were exposed to sodium azide (0.1%). As negative control, cell viability was measured in the absence of the test compounds. The cytotoxicity of the test compounds was determined by the MTT [3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide] protocol (Gradiz et al., 2016; Mosmann, 1983). Cells were seeded at a concentration of 1×104 cells /200 μL in a 96-well plate and cultured at 37 °C in a humid atmosphere with 5% CO2. After 24 h of incubation, the test compounds were added to the wells at final concentrations that ranged from 0.03–100 µM and the cells were cultured under the same conditions for a further 72 h. Each test concentration was performed in triplicate. All substances were dissolved in DMSO and the final concentration of DMSO in the wells did not exceed 0.1%. For the negative control wells, DMSO was added to also yield a final concentration of 0.1%. After incubation, 20 μL of MTT (5 mg/mL) was added to each well and the plates were incubated for 2 h. The growth media was removed from the wells and 100 μL of DMSO was added to each well to dissolve the resulting formazan crystals. The absorbance was measured at a wavelength of 590 nm with a CLARIOstar (BMG Labtech) spectrophotometer. The blank consisted of culture medium.
4.9. Procedure for performing molecular docking
Molecular docking was carried out according to the procedure reported in literature (Mostert et al., 2015; Shetnev et al., 2019). The Discovery Studio 3.1 suite of life sciences software was used for all simulations and the X-ray crystal structures of MAO-A (PDB code: 2Z5X) and MAO-B (PDB code: 3PO7) bound to harmine and zonisamide, respectively, were selected as the protein models (Son et al., 2008; Binda et al., 2011).
Supplementary Materials
Supplementary data associated with this article (experimental procedures, analytical data and copies of 1H and 13C NMR spectra) can be found in the online version at Preprints.org.
Author Contributions
Conceptualization, A.Shetnev and J.P. Petzer; Methodology, A. Petzer; Software, E.V. Petersen.; Validation, E.V. Petersen., D. Lifanov; Formal Analysis, E. Shabalina; Investigation, J. Efimova; O.Rogova; E. Shabalina; Resources, E.V. Petersen; Data Curation, O.Rogova; Writing – Original Draft Preparation, J.P. Petzer; Writing – Review & Editing, A. Shetnev; Visualization, D. A. Lifanov; Supervision, M.K. Korsakov; Project Administration, E.V. Petersen; Funding Acquisition, M.K. Korsakov.
Funding
This research was supported by the Russian Science Foundation (project 22-13-20085). This work was financially supported by the National Research Foundation of South Africa [Grant specific unique reference numbers (UID) 137997 and 132168]. The Grantholders acknowledge that opinions, findings and conclusions or recommendations expressed in any publication generated by the NRF supported research are that of the authors, and that the NRF accepts no liability whatsoever in this regard.
Conflicts of Interest
The authors declare no financial and intellectual conflict of interest.
References
- Alborghetti, M.; Bianchini, E.; De Carolis, L.; Galli, S.; Pontieri, F.E.; Rinaldi, D. Type-B monoamine oxidase inhibitors in neurological diseases: Clinical applications based on preclinical findings. Neural Regen Res 2024, 19, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.W.; Lan, N.C.; Johnson, D.L.; Abell, C.W.; Bembenek, M.E.; Kwan, S.W.; Seeburg, P.H.; Shih, J.C. Cdna cloning of human liver monoamine oxidase a and b: Molecular basis of differences in enzymatic properties. Proc Natl Acad Sci U S A 1988, 85, 4934–4938. [Google Scholar] [CrossRef] [PubMed]
- Bala, A. A need for clinical trial: Re-purposing the monoamine oxidase inhibitors (MAO-I) for rheumatoid arthritis (RA). Inflammopharmacology 2023. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Aldeco, M.; Mattevi, A.; Edmondson, D.E. Interactions of monoamine oxidases with the antiepileptic drug zonisamide: Specificity of inhibition and structure of the human monoamine oxidase b complex. J Med Chem 2011, 54, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Newton-Vinson, P.; Hubalek, F.; Edmondson, D.E.; Mattevi, A. Structure of human monoamine oxidase b, a drug target for the treatment of neurological disorders. Nat Struct Biol 2002, 9, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase b complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J Med Chem 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Andersen, J.K. Dopaminergic neurons. Int J Biochem Cell Biol 2005, 37, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.E. Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: Biological implications. Curr Pharm Des 2014, 20, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J.; Wiberg, A.; Oreland, L.; Marcusson, J.; Winblad, B. The effect of age on the activity and molecular properties of human brain monoamine oxidase. J Neural Transm 1980, 49, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.S.; Volkow, N.D.; Wang, G.J.; Logan, J.; Pappas, N.; Shea, C.; MacGregor, R. Age-related increases in brain monoamine oxidase b in living healthy human subjects. Neurobiol Aging 1997, 18, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Gradiz, R.; Silva, H.C.; Carvalho, L.; Botelho, M.F.; Mota-Pinto, A. MIA PaCa-2 and PANC-1 – pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci. Rep. 2016, 6, 21648. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, P.; Carradori, S.; D'Agostino, I.; Campestre, C.; Petzer, J.P. An updated patent review on monoamine oxidase (mao) inhibitors. Expert Opin Ther Pat 2022, 32, 849–883. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Li, H.; Ma, Y.; Zhao, Z.; An, Q.; Zhao, J.; Shi, C. Monoamine oxidase a (maoa): A promising target for prostate cancer therapy. Cancer Lett 2023, 563, 216188. [Google Scholar] [CrossRef] [PubMed]
- Hubalek, F.; Binda, C.; Khalil, A.; Li, M.; Mattevi, A.; Castagnoli, N.; Edmondson, D.E. Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase b by specific reversible inhibitors. J Biol Chem 2005, 280, 15761–15766. [Google Scholar] [CrossRef] [PubMed]
- Kaboudin, B.; Elhamifar, D. Phosphorus pentasulfide: A mild and versatile reagent for the preparation of thioamides from nitriles. Synthesis-Stuttgart 2006, 224–226. [Google Scholar] [CrossRef]
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol 2014, 73, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Gupta, V.P.; Kumar, V. A perspective on monoamine oxidase enzyme as drug target: Challenges and opportunities. Curr Drug Targets 2017, 18, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Lum, C.T.; Stahl, S.M. Opportunities for reversible inhibitors of monoamine oxidase-a (rimas) in the treatment of depression. CNS Spectr 2012, 17, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.E.; Rigacci, S.; Borchi, E.; Bargelli, V.; Miceli, C.; Giordano, C.; Raimondi, L.; Nediani, C. Monoamine oxidase is overactivated in left and right ventricles from ischemic hearts: An intriguing therapeutic target. Oxid Med Cell Longev 2016, 2016, 4375418. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.H.; Ginovart, N.; Boovariwala, A.; Sagrati, S.; Hussey, D.; Garcia, A.; Young, T.; Praschak-Rieder, N.; Wilson, A.A.; Houle, S. Elevated monoamine oxidase a levels in the brain: An explanation for the monoamine imbalance of major depression. Arch Gen Psychiatry 2006, 63, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Mostert, S.; Petzer, A.; Petzer, J.P. Indanones as high-potency reversible inhibitors of monoamine oxidase. ChemMedChem 2015, 10, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Mulinari, S. Monoamine theories of depression: Historical impact on biomedical research. J Hist Neurosci 2012, 21, 366–392. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Okumura, M.; Ozawa, M.; Mimori, M.; Maku, T.; Shiraishi, T.; Kitagawa, T.; Takatsu, H.; Sato, T.; Komatsu, T. , et al. Effects of monotherapy with a monoamine oxidase b inhibitor on motor symptoms in parkinson's disease are dependent on frontal function. Neurol Sci 2023, 44, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Novaroli, L.; Reist, M.; Favre, E.; Carotti, A.; Catto, M.; Carrupt, P.A. Human recombinant monoamine oxidase b as reliable and efficient enzyme source for inhibitor screening. Bioorg Med Chem 2005, 13, 6212–6217. [Google Scholar] [CrossRef] [PubMed]
- Petzer, A.; Shetnev, A.; Efimova, J.; Korsakov, M.; Filimonov, S.; Petzer, P.J. Pyridazinone-substituted benzenesulfonamides demonstrate inhibition of monoamine oxidase. Letters in Drug Design & Discovery 2023, 20, 1–8. [Google Scholar]
- Pizzinat, N.; Copin, N.; Vindis, C.; Parini, A.; Cambon, C. Reactive oxygen species production by monoamine oxidases in intact cells. Naunyn Schmiedebergs Arch Pharmacol 1999, 359, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S. The two-century journey of parkinson disease research. Nat Rev Neurosci 2017, 18, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Scobie, M. Mth1 Inhibitors for Treatment of Inflammatory and Autoimmune Conditions. U.S. Patent US16/120,170, 31 August 2018. [Google Scholar]
- Shetnev, A.; Shlenev, R.; Efimova, J.; Ivanovskii, S.; Tarasov, A.; Petzer, A.; Petzer, J.P. 1,3,4-oxadiazol-2-ylbenzenesulfonamides as privileged structures for the inhibition of monoamine oxidase b. Bioorg Med Chem Lett 2019, 29, 126677. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.C. Monoamine oxidase isoenzymes: Genes, functions and targets for behavior and cancer therapy. J Neural Transm (Vienna) 2018, 125, 1553–1566. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu Rev Neurosci 1999, 22, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Son, S.Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase a at 2.2-a resolution: The control of opening the entry for substrates/inhibitors. Proc Natl Acad Sci U S A 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [PubMed]
- Sonsalla, P.K.; Wong, L.Y.; Winnik, B.; Buckley, B. The antiepileptic drug zonisamide inhibits mao-b and attenuates mptp toxicity in mice: Clinical relevance. Exp Neurol 2010, 221, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Suchting, R.; Tirumalajaru, V.; Gareeb, R.; Bockmann, T.; de Dios, C.; Aickareth, J.; Pinjari, O.; Soares, J.C.; Cowen, P.J.; Selvaraj, S. Revisiting monoamine oxidase inhibitors for the treatment of depressive disorders: A systematic review and network meta-analysis. J Affect Disord 2021, 282, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Umbarkar, P.; Singh, S.; Arkat, S.; Bodhankar, S.L.; Lohidasan, S.; Sitasawad, S.L. Monoamine oxidase-a is an important source of oxidative stress and promotes cardiac dysfunction, apoptosis, and fibrosis in diabetic cardiomyopathy. Free Radic Biol Med 2015, 87, 263–273. [Google Scholar] [CrossRef]
- Weissbach, H.; Smith, T.E.; Daly, J.W.; Witkop, B.; Udenfriend, S. A rapid spectrophotometric assay of mono-amine oxidase based on the rate of disappearance of kynuramine. J Biol Chem 1960, 235, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Yamane, T.; Mitsudera, H.; Shundoh, T. Highly regioselective direct halogenation: A simple and efficient method for preparing 4-halomethyl-5-methyl-2-aryl-1,3-thiazoles. Tetrahedron Letters 2004, 45, 69–73. [Google Scholar] [CrossRef]
- Youdim, M.B.; Bakhle, Y.S. Monoamine oxidase: Isoforms and inhibitors in parkinson's disease and depressive illness. Br J Pharmacol 2006, 147 Suppl 1, S287–296. [Google Scholar] [CrossRef]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Zisook, S. A clinical overview of monoamine oxidase inhibitors. Psychosomatics 1985, 26, 240–246, 251. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
The synthesis of primary benzenesulfonamide building blocks 2a,b and 4.
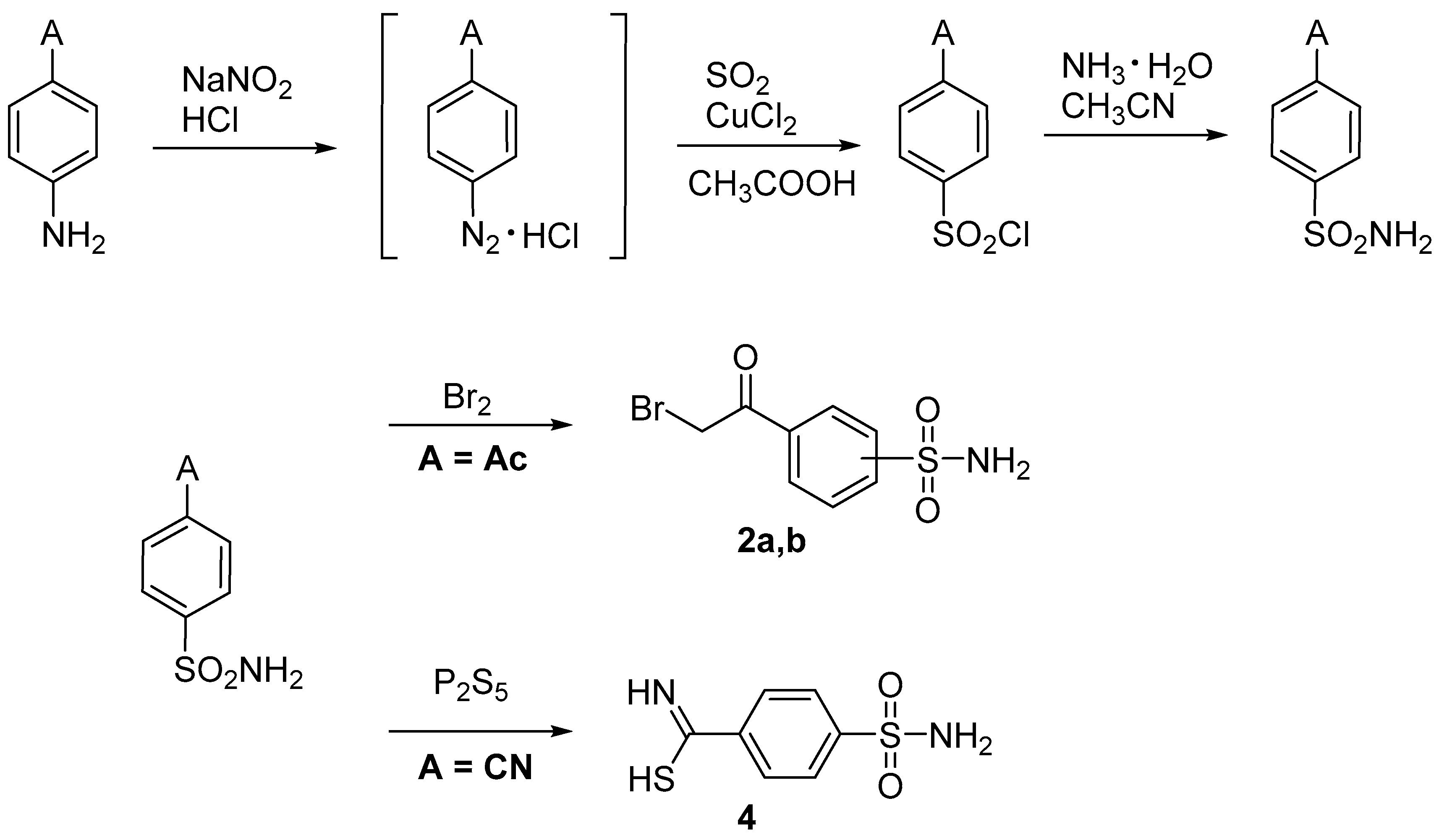
Figure 3.
Synthesis of 1,3-thiazol-4-ylbenzenesulfonamide derivatives 3.
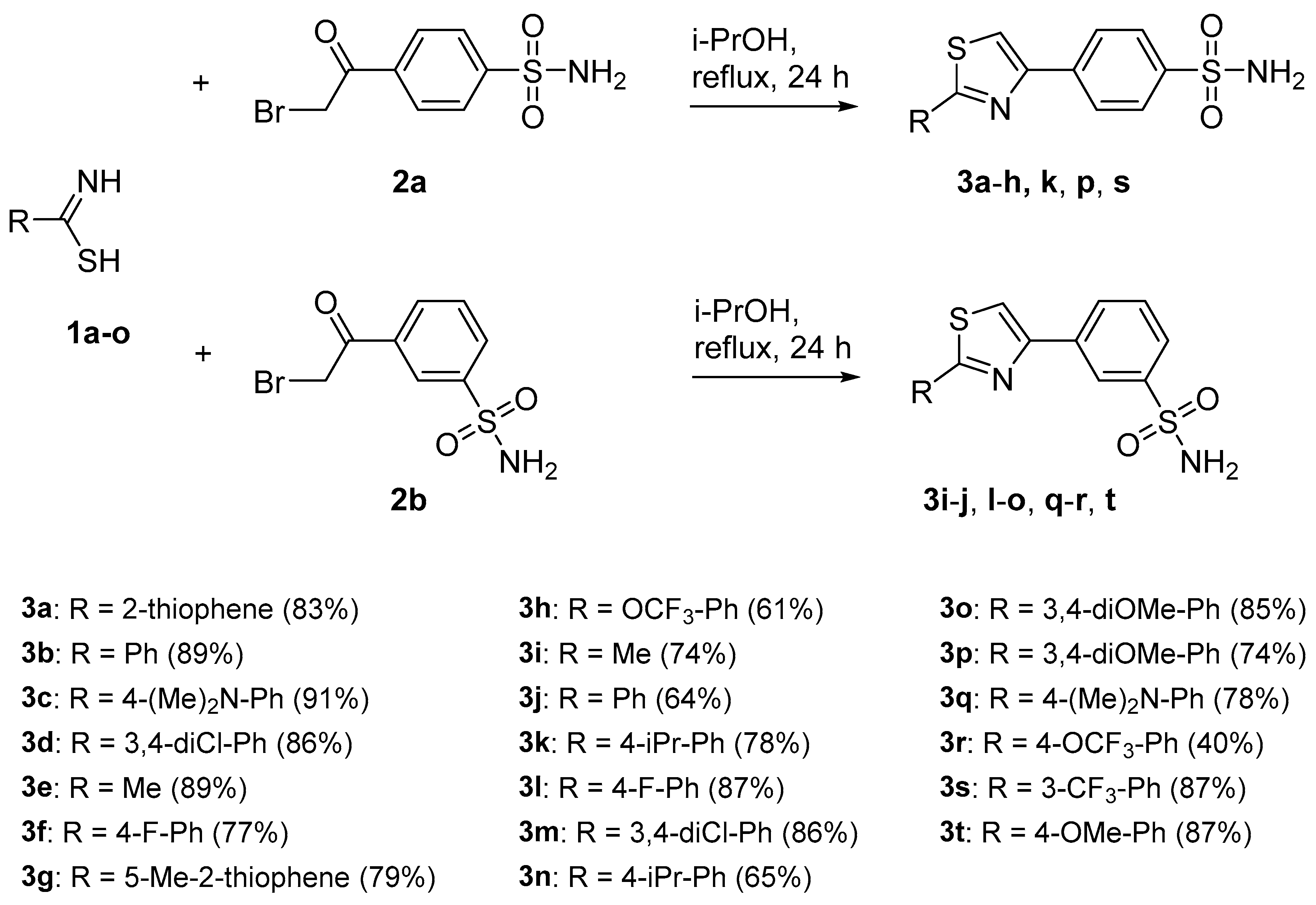
Figure 4.
Synthesis of 1,3-thiazol-2-ylbenzenesulfonamide derivatives 6 and 8.
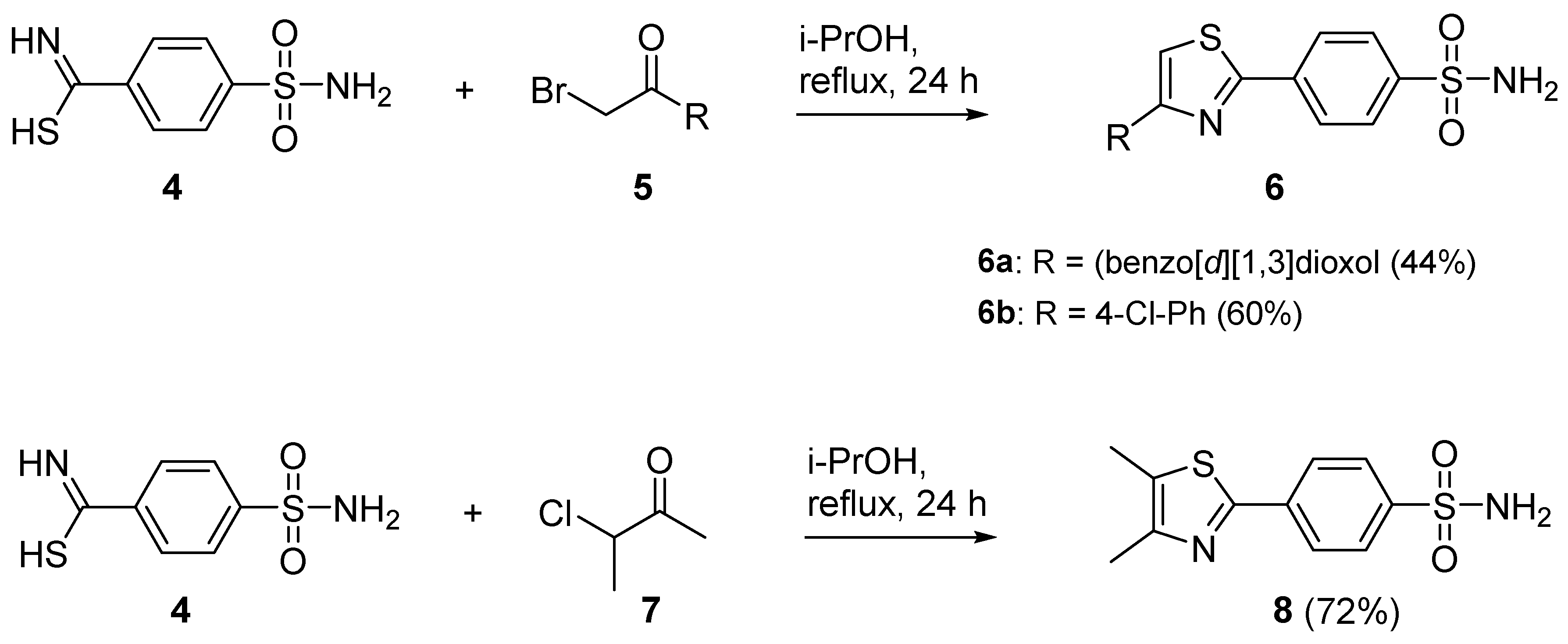
Figure 5.
Sigmoidal graphs constructed to measure IC50 values for the inhibition of MAO-B by 3j (filled circles), 3l (open circles) and 3m (triangles). The graphs are representative of triplicate experiments.
Figure 5.
Sigmoidal graphs constructed to measure IC50 values for the inhibition of MAO-B by 3j (filled circles), 3l (open circles) and 3m (triangles). The graphs are representative of triplicate experiments.
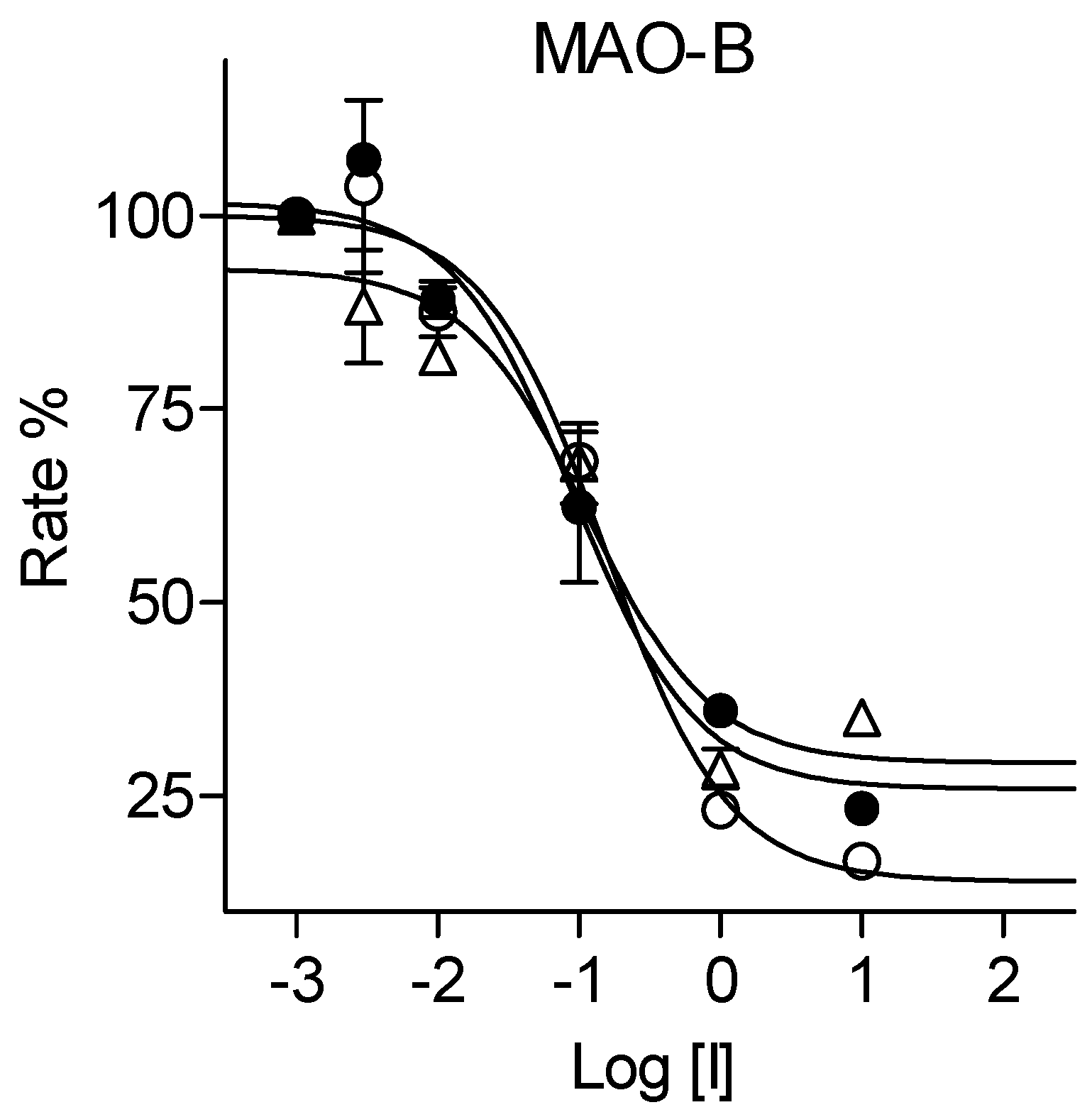
Figure 6.
Lineweaver-Burk plots for the inhibition of human MAO-B by 3j. From a replot of the slopes versus inhibitor concentration, a Ki value of 0.21 µM was estimated.
Figure 6.
Lineweaver-Burk plots for the inhibition of human MAO-B by 3j. From a replot of the slopes versus inhibitor concentration, a Ki value of 0.21 µM was estimated.
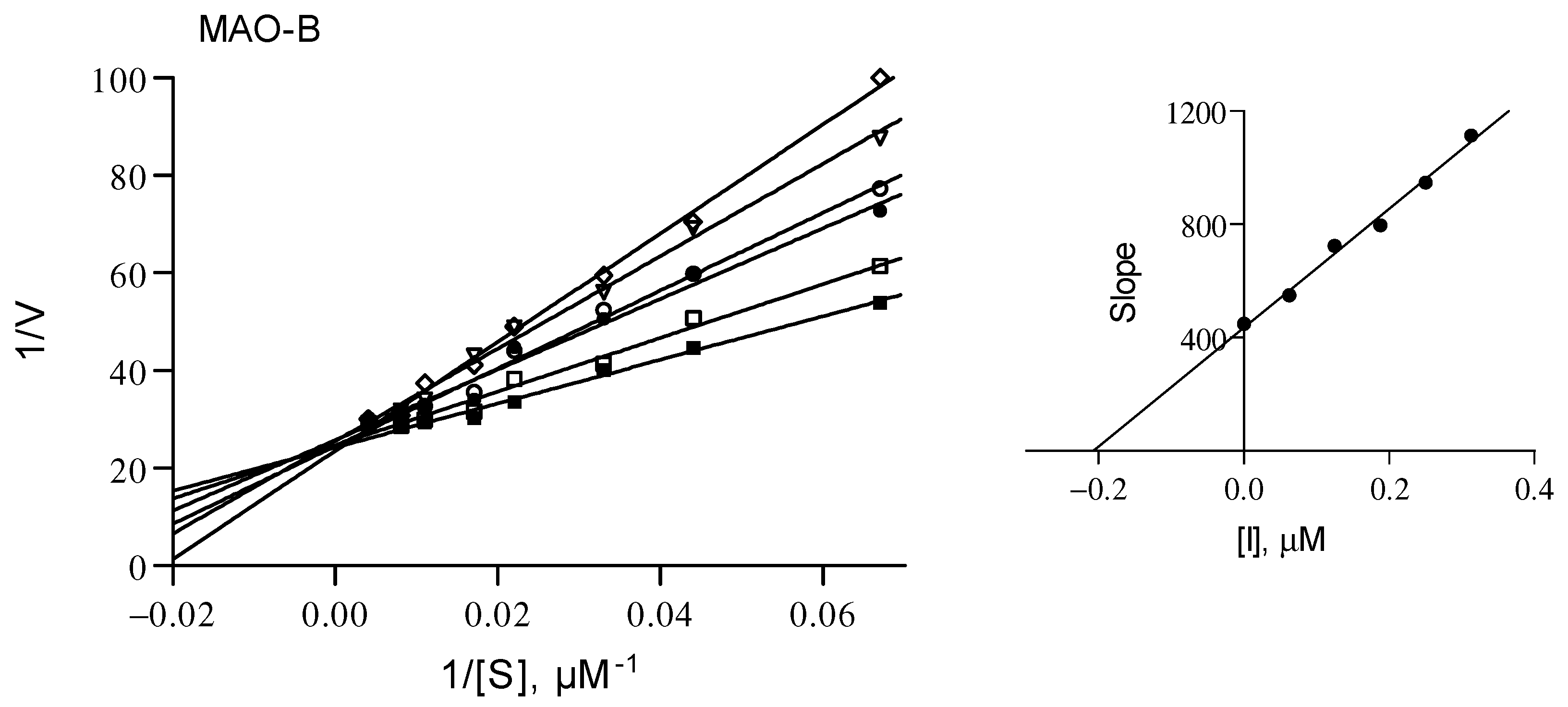
Figure 7.
Cell viability after treatment of IMR-32 and HS-5 cell lines with compounds 3j and 3m (0.5–100 µM).
Figure 7.
Cell viability after treatment of IMR-32 and HS-5 cell lines with compounds 3j and 3m (0.5–100 µM).
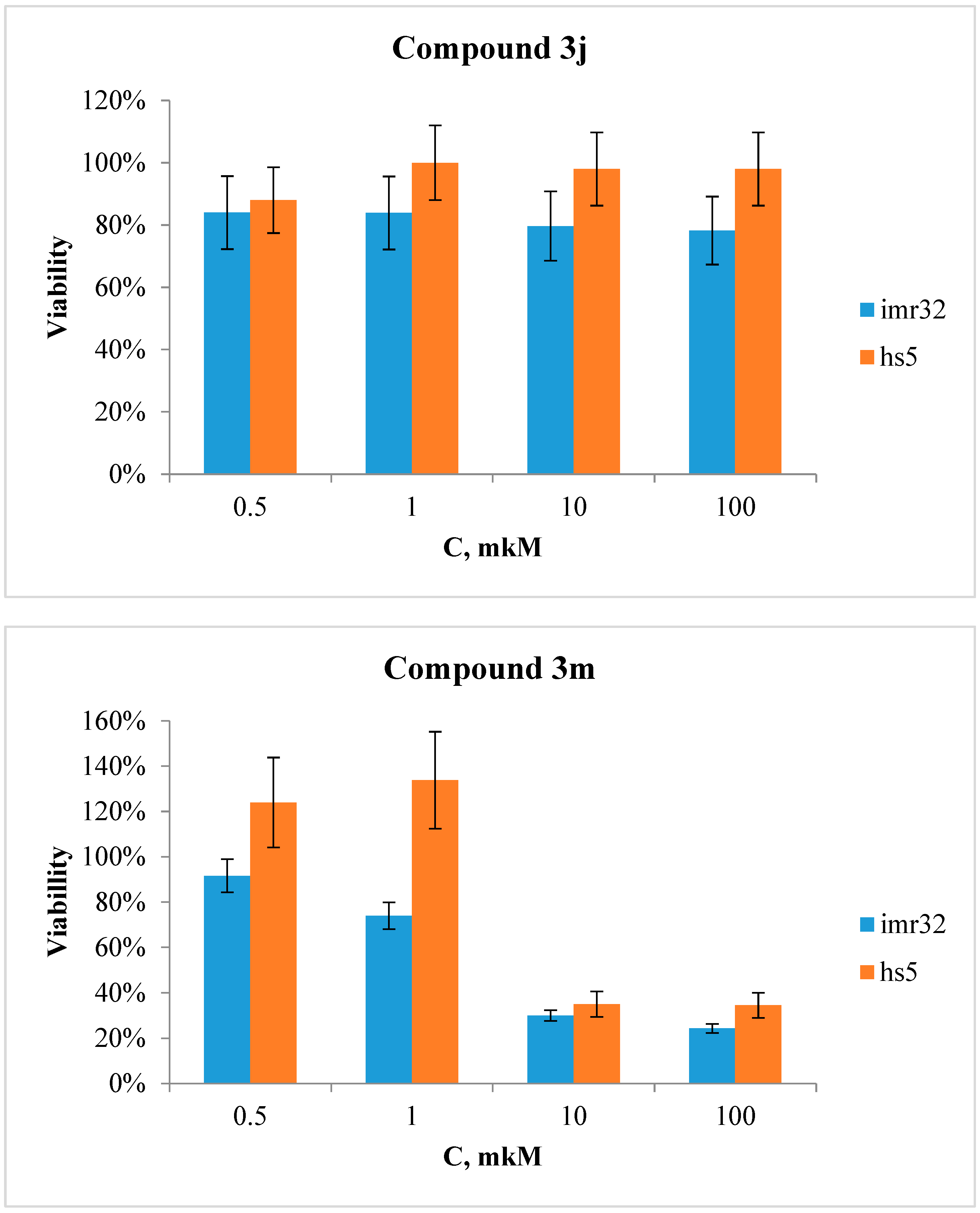
Figure 8.
The orientation of zonisamide (cyan) in the in the active site of MAO-B as determined by X-ray crystallography (PDB code: 3PO7) as well as the predicted orientation of 3m (magenta).
Figure 8.
The orientation of zonisamide (cyan) in the in the active site of MAO-B as determined by X-ray crystallography (PDB code: 3PO7) as well as the predicted orientation of 3m (magenta).
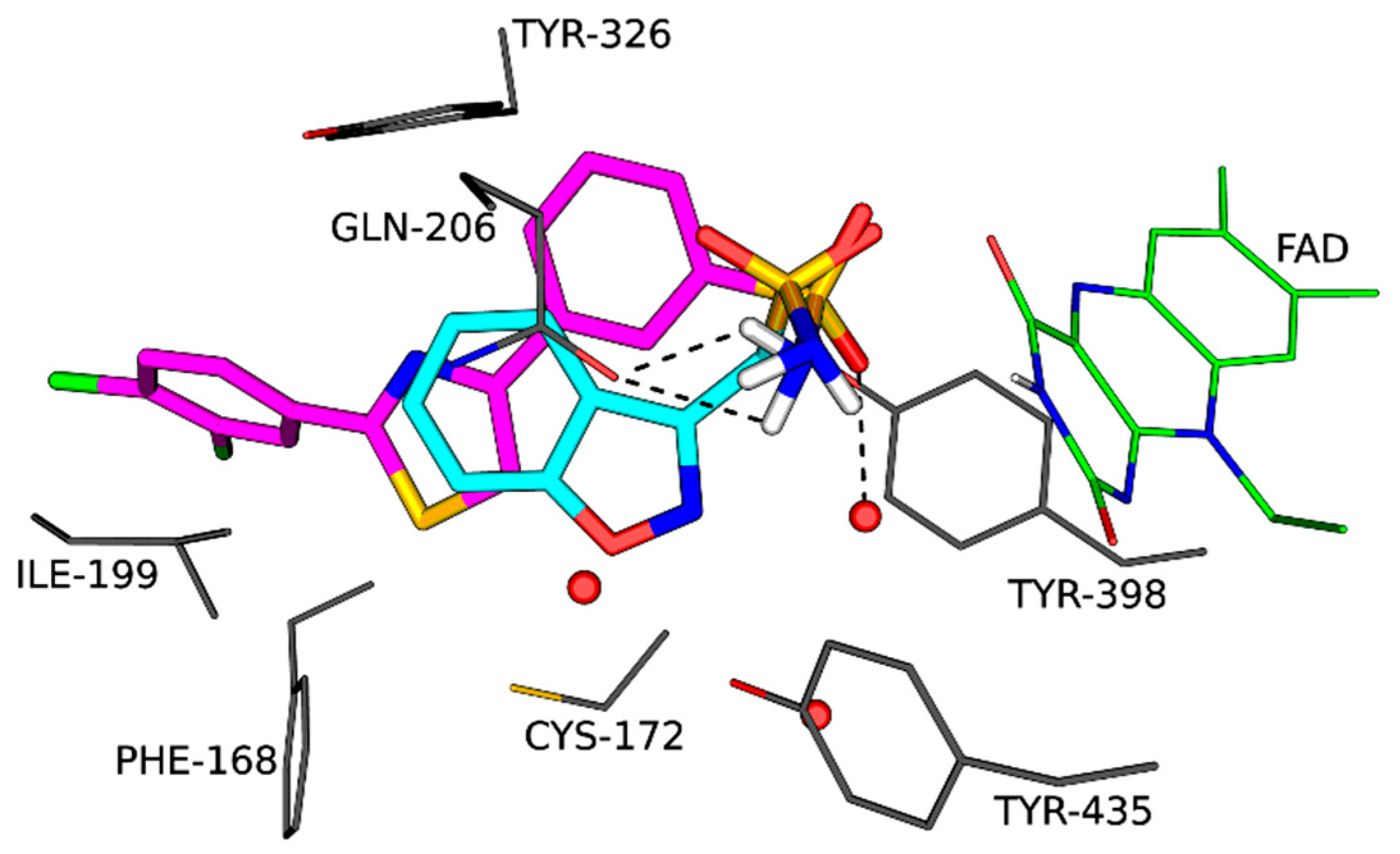

Table 1.
The IC50 values for the inhibition of MAO by 1,3-thiazolylbenzenesulfonamide derivatives (3, 6, 8) as well as those of reference inhibitors.
Table 1.
The IC50 values for the inhibition of MAO by 1,3-thiazolylbenzenesulfonamide derivatives (3, 6, 8) as well as those of reference inhibitors.
| ID | Structure | IC50 (µM ± SD)a | ||
|---|---|---|---|---|
| MAO-A | MAO-B | SIb | ||
| 3a | 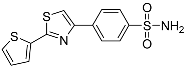 |
11.9 ± 1.49 | 13.8 ± 1.04 | 0.9 |
| 3b | 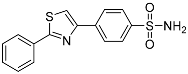 |
23.3 ± 1.88 | 6.62 ± 0.162 | 3.5 |
| 3c | 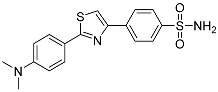 |
32.6 ± 3.80 | NIc | – |
| 3d | 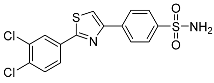 |
88.4 ± 25.3 | 0.407 ± 0.061 | 217 |
| 3e | 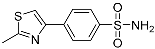 |
NIc | 79.1 ± 15.2 | – |
| 3f | 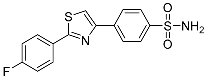 |
18.7 ± 0.226 | 0.484 ± 0.140 | 39 |
| 3g | 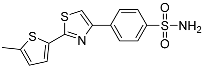 |
42.7 ± 3.66 | 9.85 ± 4.95 | 4.3 |
| 3h | 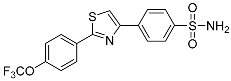 |
NIc | 30.5 ± 0.552 | – |
| 3i | 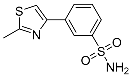 |
57.7 ± 4.32 | 79.6 ± 12.4 | 0.7 |
| 3j | 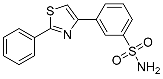 |
28.7 ± 1.46 | 0.103 ± 0.0027 | 279 |
| 3k | 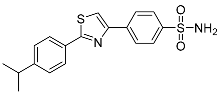 |
NIc | 12.6 ± 0.021 | – |
| 3l | 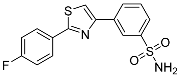 |
21.9 ± 1.10 | 0.193 ± 0.028 | 113 |
| 3m | 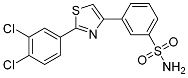 |
17.2 ± 0.212 | 0.172 ± 0.049 | 100 |
| 3n | 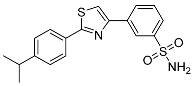 |
45.9 ± 8.64 | 3.60 ± 3.28 | 13 |
| 3o | 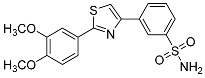 |
NIc | NIc | – |
| 3p | 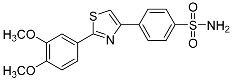 |
NIc | 77.1 ± 47.6 | – |
| 3q | 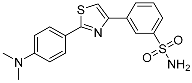 |
47.4 ± 8.03 | 0.404 ± 0.106 | 117 |
| 3r | 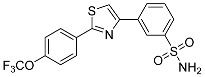 |
56.2 ± 8.25 | 29.0 ± 2.96 | 1.9 |
| 3s | 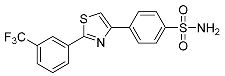 |
NIc | 40.5 ± 4.40 | – |
| 3t | 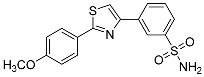 |
29.4 ± 3.06 | 2.80 ± 0.324 | 11 |
| 6a | 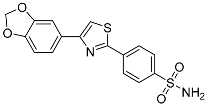 |
50.0 ± 3.50 | 58.3 ± 10.7 | 0.9 |
| 6b | 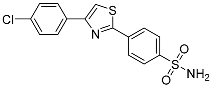 |
NIc | 0.412 ± 0.021 | – |
| 8 | 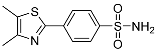 |
NIc | NIc | – |
| Toloxatoned | 1.67 ± 0.418 | – | – | |
| Safinamided | – | 0.240 ± 0.080 | – | |
a Values are given as the mean ± SD of triplicate determinations. b Selectivity index (SI) = IC50(MAO-A)/IC50(MAO-B). c No inhibition (NI) observed at maximum tested concentration of 100 µM. d Reference inhibitor.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



