
Submitted:
22 November 2023
Posted:
23 November 2023
You are already at the latest version
Abstract
Colon cancer (CRC) is a prevalent malignancy that exhibits distinct differences in incidence, prognosis, and treatment responses between males and females. These disparities have long been attributed to hormonal differences, particularly the influence of estrogen signaling. This review aims to provide a comprehensive analysis of recent advances in our understanding of the molecular mechanisms underlying sex differences in colon cancer and the protective role of membrane and nuclear estrogen signaling in CRC development, progression, and therapeutic interventions. We discuss the epidemiological and molecular evidence supporting sex differences in colon cancer, followed by an exploration of the impact of estrogen in CRC through various genomic and non-genomic signaling pathways involving membrane and nuclear estrogen receptors. Furthermore, we examine the interplay between estrogen receptors and other signaling pathways, in particular the Wnt/-catenin proliferative pathway and hypoxia in shaping biological sex differences and estrogen protective actions in colon cancer. Lastly, we highlight the potential therapeutic implications of targeting estrogen signaling in the management of colon cancer and propose future research directions to address the current gaps in our understanding of this complex phenomenon.
Keywords:
colon cancer
; estrogen
; estrogen receptors
; sex differences
1. Introduction
1.1. Sex Differences/Sexual Dimorphism/Gender Differences in Cancer
Sex differences and sexual dimorphism refer to biological and morphological differences, respectively, between males and females in various traits or characteristics. While sexual dimorphism refers to the form or appearances such as height and morphology (evolutionary adaptations), sex differences are primarily associated with physiological features, and their impact on cancer is an exciting and rapidly developing area of ongoing research. These terms have been used interchangeably, and not without some misunderstanding, in the scientific literature with reference to male/female differences in cancer [1,2,3]. Gender differences, on the other hand, commonly refer to behavioural and lifestyle traits but are often confused in the with biological sex differences [4,5,6]. Several types of cancer have demonstrated differences in incidence, presentation, and outcomes between males and females [7,8]. Certain cancers are more prevalent in one sex compared to the other due to anatomical differences, for example, prostate cancer in males, and ovarian or uterine cancer in females, whereas others may be associated with hormonal and genetic factors such as breast cancer [9]. In a survey of the most recent data from the Global Cancer Observatory, it can be noted that the top 20 most common non-reproductive tissue cancers world-wide show sex differences both in incidence and mortality, with females showing lower age-standardised incidence and mortality for all cancers (excluding those of anatomical differences) and for all cancers in every global region surveyed (Figure 1). While hormone and genetic factors contribute to these differences, other non-biological risk factors can increase the likelihood of incidence and mortality including smoking and excessive alcohol consumption, particularly for lung, stomach, oesophagus and liver cancer [10,11]. Here we discuss the evidence for genomic and non-genomic biological actions of estrogen underpinning biological sex differences in colon cancer. We focus on more recent reports and try to integrate current knowledge into a holistic understanding of the role of estrogen, its receptors and molecular targets, in order to explain this very complex phenomenon for which many mechanistic questions still remain unanswered.
1.2. Sex Differences in Colon Cancer
Colon cancer, also known as colorectal cancer (CRC), is one of the most common types of cancer worldwide. The exact prevalence may vary depending on factors such as age, gender, geographical location, and lifestyle choices. According to the World Health Organization, colon cancer is the second most commonly diagnosed cancer globally and the third leading cause of cancer-related deaths (Figure 1). Sex differences showing a female advantage is a feature of CRC [12]. Although the main risk factor for CRC is age: about 90% of patients are older than 50 years, sex differences in CRC are evident in age-matched male and female patients [13]. The age-standardized mortality rate for men is 50% higher (10.8 per 100,000 person-year) than for women (7.2 per 100,000 person-year), [14].
The CRC incidence rates can differ significantly between countries but all countries show a male predominance in both incidence and mortality of CRC (Figure 2), [15]. Moreover, the trends for the next 20 years predict a continuing sex difference with more males than females affected by CRC both in its incidence and mortality (Globocan 2020, https://gco.iarc.fr, accessed on 29 July 2023), [16]. Certain risk factors can increase the likelihood of developing colon cancer, including a family history of the disease, a personal history of inflammatory bowel disease, a sedentary lifestyle, a diet high in processed meats and fat, and low in fruits, vegetables, and fibre, smoking, and excessive alcohol consumption. It is important to note that advancements in early detection and improved treatment options have contributed to higher survival rates and better outcomes for individuals diagnosed with colon cancer. Regular screenings such as colonoscopies can help detect pre-cancerous polyps or early-stage colon cancer, improving the chances of successful treatment. Biological sex differences, however, are too often a neglected factor in both clinical trials and treatment of colon cancer, although its added value to personalised medicine in incontestable [17]. Sex differences exist at multiple levels in colon cancer and women have a lower risk of developing CRC than men. Females at a younger age are less likely to die from CRC than age-matched male patients and certain types of CRC occur predominantly in women. The biological differences in CRC mortality are noticeable in the survival advantage of women during pre-menopause [18-44y] compared to men of the same age or to older women post-menopause [18]. These pre-menopausal advantages and the observations that hormone replacement therapy (HRT) may also be protective in CRC [19,20,21] indicate a role for the female sex steroid hormone estrogen in delaying the onset and reducing mortality in females with colon cancer [22,23]. Sexual disparity has also been shown in epidemiological studies to be an important factor in the site of onset and metastases in CRC [24]. Women show a higher frequency of right-sided tumours (proximal colon) than men who present more commonly with left-sided (distal colon) tumours [25]. Right-sided proximal tumours tend to be more aggressive and resistant to chemotherapy which may confound the advantages conferred by estrogen protection [25].
1.3. Estrogen and Sex Differences in Colon Cancer
Estrogen is the main sex hormone which controls physiological functions of the female reproductive system, as well as the development of secondary sexual characteristics during female puberty. The predominant circulating estrogen in humans is 17β-estradiol (E2) which is the most physiologically relevant estrogen during the female reproductive years. In human females during the reproductive years, the plasma levels of E2 fluctuate over the estrous cycle reaching peak concentrations in the follicular phase; 150 to 300 pg/ml (0.5nM – 1nM) 24 hours before ovulation, [26], while the highest likely E2 level in the plasma during pregnancy is 8ng/ml (30nM), [27]. Estrogen plasma levels fall off dramatically post-menopause [>45y] decreasing to below 30pg/ml (110pM) which is close to levels found in age-matched males (10-50pg/ml). Estrogen at sub-namolar concentrations has been shown to exert multiple sexually distinct physiological actions with a female advantage in tissues and organs outside the reproductive system including brain, skeleton, muscle, cardiovascular, immune system, intestine, kidney, liver and pancreas to influence, respectively, memory, bone strength, cardiac and skeletal muscle contractility, immunity, intestinal secretion, renal Na+ handling, blood pressure and metabolism [28]. These pleiotropic effects of estrogen are thought to underpin sex differences and reinforce female physiology, reproduction, longevity and healthy aging [29]. The beneficial biological effects of estrogen are lost and often only become apparent after the menopause [30], and may be restored with hormone replacement therapy [31] but only if started early, for example as in treating cardiovascular disease [32]. Pathophysiological effects of estrogen are observed in some cancers, notably breast, uterine and cervical cancers [33], whereas in non-reproductive organ cancers, estrogen can have protective effects against morbidity and mortality [34]. Estrogen signaling via specific estrogen receptors has emerged as a crucial factor in sex differences in the development and progression of colon cancer [12,22,23].
2. Estrogen Receptors in Colon Cancer
Estrogen receptors determine the biological sex differences, specificity and transduction of the multiple signaling responses to estrogen [35]. Probably the most important factor in sex differences in colon cancer development is ligand activation of estrogen receptors (ER), primarily ERα and ERβ. These receptors are expressed in both males and females but can have differential expression levels and activity patterns, depending on circulating levels of E2, as well as prognostic value in CRC [36]. The role of estrogen in CRC progression is dependent on the relative abundance of the ER subtypes and their hormone responsiveness. The colonic crypt epithelium expresses both ERα and ERβ [37]. Positional differential expression of ERs has been reported along the colon length and within the crypt axis. ERα is expressed more highly at the base of the crypt of the proximal colon while ERβ expression is predominant in the mid-section of the crypt and in the apical surface cells [38]. This spatial partitioning of ER isoform expression indicates antagonistic roles for ERα and ERβ in transducing differential effects of estrogen on the physiological function of the epithelial cells located at different sites along the crypt. For example, progenitor cell proliferation at the base of the crypt gives way to enterocyte differentiation in the midsection and shedding of senescent or apoptotic cells at the lumen surface. There is strong evidence that ERβ is more highly expressed in colon cancer tissues from females compared to males, while ERα expression levels can vary among the sexes [36,39]. ERβ has been proposed as a tumour suppressor in CRC and ERβ expression is selectively lost during tumour progression through methylation-dependent gene silencing [40]. Thus the ratio between ERα and ERβ expression and balance in their cell signaling may contribute to sex differences in CRC [41].
2.1. Nuclear Estrogen Receptors in Colon Cancer
The two main subtypes of ERs are ERα (encoded by estrogen receptor 1, ESR1) and ERβ (encoded by estrogen receptor 2, ESR2) which are differentially expressed in normal colon tissue as well as in colon cancer cells. These two estrogen receptor sub-types are often referred to as canonical or nuclear ERs, eliciting latent genomic responses to estrogen and working in an antagonistic fashion on cell biology [42]. The natural ligand for all ERs is the biologically active form of estrogen, 17β-estradiol (E2). Nuclear ERs play a complex role in colon cancer, influencing various aspects of tumour development and progression [34]. Colon cancer cells expressing ERs can be hormone-responsive, meaning they can respond to estrogen stimulation. Estrogen can directly bind to ERs in these cells and trigger a cascade of signaling events leading to changes in gene expression, protein synthesis, and cell behaviour (Figure 3). It is worth repeating that the specific roles and effects of ERs in colon cancer can vary depending on factors such as the subtype of ER (ERα vs ERβ), their relative expression levels, and the interplay with other signaling pathways. For example, while ERα has been associated with promoting tumour growth and progression, ERβ has been suggested to have a potentially protective or inhibitory effect on colon cancer development [43,44]. Importantly, nuclear ERs are proven prognostic and therapeutic targets in colon cancer [45,46].
2.2. Membrane Estrogen Receptors in in Colon Cancer
Membrane estrogen-sensitive receptors have been implicated in CRC [12,23,34] which unlike the classical nuclear ERs, involve estrogen-liganded membrane-initiated cell signaling pathways (Figure 3), [47]. Membrane-initiated estrogen responses in colonic epithelium are typified by rapid non-genomic actions on protein kinase cell signaling pathways, intracellular Ca2+ activity and ion channel function [48] which distinguish these events from the more latent genomic responses to estrogen in a wide range of normal and cancerous cell types. The presence and role of membrane estrogen receptors in colon cancer, and cancer in general, are a hot topic of ongoing research and have been the subject of dedicated international meetings continuously since 1992 (RRSH, FASEB SRC Meetings), [49,50,51]. The two most well-characterised membrane receptors for estrogen are membrane ERα (mERα]) [52] and G protein-coupled estrogen receptor 1 (GPER1), also known as GPR30 [53], which are discussed in detail below.
3. Genomic and non-Genomic Estrogen Signaling Pathways in Colon Cancer
Steroid hormone signaling occurs via non-genomic membrane-initiated pathways and genomic nuclear transcriptional pathways working in a coordinated fashion (Figure 3), [54,55]. Estrogen can trigger cellular signalling in both healthy and cancerous colonic epithelial cells via genomic and non-genomic mechanisms [4,14,36] in a sexually differentiated manner which also shows dependency on the stage of the estrous cycle [56]. Both genomic and non-genomic estrogen signaling pathways in the colon are cell type-specific and may differ among healthy and cancerous colonic tissues. Genomic estrogen signaling pathways are characterised by long latency [hours/days] and involve nuclear ERs that bind estrogen in the cytosol which then dimerise and translocate to the nucleus to bind to specific DNA sites to trigger synthesis of proteins which regulate cell growth, differentiation and proliferation [57]. Non-genomic estrogen signalling, on the other hand, is characterised by rapid onset of cell signaling responses [secs/mins] involving estrogen binding to a membrane receptor which through phosphorylation reactions can transactivate other membrane receptors and trigger a myriad of cellular signaling pathways impacting upon ion channels, protein kinases, and transcription factors to extend its rapid actions into genomic events [58]. The genomic and non-genomic receptor signaling pathways of estrogen in colonic crypt cells are summarised in Figure 3, showing both protective and exacerbating arms of these pathways in colon cancer.
3.1. Genomic Mechanisms of Estrogen Signaling in Colon Cancer
The genomic actions of estrogen in colon cancer are not as well-studied compared to its effects on other hormone-responsive cancers like breast or ovarian cancer. While estrogen receptors have been found in the colon and rectal tissue, the specific role and mechanisms of estrogen and ERs in colon cancer development and progression between the sexes are still not well-understood. Several studies, however, over the past 20 years have suggested potential genomic actions of estrogen in colon cancer which could underpin sex differences: Estrogen may promote or inhibit cell proliferation and reduce or stimulate cell death (apoptosis) depending on the ER subtype expression in colon cancer cells (Figure 3), [59]. E2-ERα interactions can activate signaling pathways, such as the PI3K/Akt pathway, leading to increased CRC cell growth and survival [60]. Estrogen has been implicated in promoting angiogenesis, the formation of new blood vessels, which is essential for tumour growth and metastasis [61] and can upregulate the expression of vascular endothelial growth factor (VEGF), a key factor involved in angiogenesis [62]. Estrogen may also influence epigenetic modifications in colon cancer cells [63] to effect post-translational DNA methylation patterns, histone modifications, and chromatin remodeling, potentially impacting gene expression and cellular behaviour [64]. Nuclear estrogen receptors may also play a role in inducing or influencing the process of epithelial-mesenchymal transition (EMT), [65] which is involved in tumour metastases in colon cancer [66]. Estrogen signaling through ERα can regulate the expression of genes associated with EMT, leading to increased cell motility and invasiveness in prostate and breast cancer cells [67,68] and in CRC [12]. Estrogen signaling has also been found to impact the function and activation of immune cells, including macrophages and T cells, and regulate the production of cytokines and other immune-related molecules [69] which can affect the tumour microenvironment and immune surveillance against cancer cells [70]. In this regard, estrogen via ERβ has been shown to modulate the immunogenicity of the tumour microenvironment and immune responses in colon cancer [71].
3.2. Non-Genomic Mechanisms of Estrogen Signaling in Colon Cancer
It has been known for over two decades that estrogen can exert sexually segregated rapid non-genomic actions independent of gene transcription on cell signaling in colon [72]. Non-genomic actions of estrogen in CRC involve signaling pathways that do not require changes in gene expression or protein synthesis [12,23]. Although the non-genomic actions of estrogen in colon cancer are less well-studied compared to its genomic actions, here we outline some of the potential mechanisms (Figure 3). Activation of membrane receptors: Estrogen binding to ERα or GPER can trigger intracellular signaling cascades, including activation of protein kinase pathways, calcium mobilization, and stimulation of cyclic adenosine monophosphate (cAMP) production [73]. Rapid kinase activation: Estrogen can rapidly (secs-mins) activate various kinases, such as mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK [74]. These kinases play crucial roles in cell proliferation, survival, and migration, which are important in colon cancer progression [75]. Ion channels: Estrogen modulates the activity and expression of ion channels in colon cancer cells, such as calcium and potassium channels, leading to changes in intracellular ion concentrations and membrane potential which can affect cell proliferation, apoptosis, and migration [76,77,78]. Activation of second messenger systems: Estrogen can stimulate the production of second messengers, including cyclic adenosine monophosphate (cAMP) cyclic guanosine monophosphate (cGMP), and inositol trisphosphate (IP3), [79]. These signaling intermediates can regulate various cellular processes, including gene expression, protein phosphorylation, and intracellular calcium release [80]. It is important to note that both genomic and non-genomic actions of estrogen can contribute to the overall effects of estrogen on colon cancer cells. The balance between these actions and the interplay with other signaling pathways determine the ultimate impact of estrogen on colon cancer development and progression.
3.3. Cooperativity between Genomic and Non-Genomic Estrogen Signaling in Colon Cancer
Multiple studies in a wide variety of tissues (brain, vascular, epithelial) have shown that genomic and non-genomic cellular responses to estrogen are not mutually exclusive and can cooperate to produce synergistic effects in physiological functions and in cancer biology (Figure 3), [81]. In this context, it is important to note that a strict dichotomy does not exist between membrane non-genomic and nuclear genomic actions of estrogen, and evidence exists for cross-talk and integration of these rapid and latent pathways to amplify biological responses to estrogen [82]. Non-genomic actions of estrogen have been shown to be both permissive and potentiating for genomic responses [83]. This type of cross-talk can be rapidly initiated by estrogen actions through membrane ERs to transactivate growth factor receptor tyrosine kinases (EGF and IGF-I receptors), [84] which can produce rapid activation of ERK MAPK and phosphorylation of cytosolic ER to allow its translocation into the nucleus [85] and also cause phosphorylation and recruitment of coactivators (AP-1, Sp-1) to the nuclear transcriptosome to amplify the genomic response [86]. Estrogen signaling through membrane estrogen receptors can also involve activation (phosphorylation) of protein kinase targets to induce gene transcription and latent nuclear transcriptional activity via ERK, MAPK, c-fos, PI3K/AKT, CREB, and JNK [87,88]. Conversely, genomic actions may amplify non-genomic responses to estrogen in a sex-specific manner, for example, by stimulating the transcription of protein kinase intermediates of the membrane-initiated signaling pathways in colonocytes [89]. Thus nuclear ERs are necessary for the expression of proteins which transduce estrogen effects at the membrane [90]. An important caveat, therefore, in the debate on genomic versus non-genomic estrogen responses is the accumulating evidence that these are not stand-alone independent events but show integration and cooperativity and, if considered separately, do not encompass the full range of estrogen actions in physiology nor in cancer. Another important principle is that labelling non-genomic steroid hormone responses as being solely ‘rapid’ no longer holds true as latent genomic events may be initiated by earlier membrane-cell signalling [91,92]. Thus non-genomic and genomic actions of estrogen can be integrated in targeting cell proliferation pathways in colon cancer biology [93].
4. Estrogen Signaling via ERα and ERβ in Colon Cancer
When estrogen binds to ERα in the cytosol, the hormone receptor complex dimerizes and translocates to the nucleus where it interacts with specific DNA sequences, estrogen response elements (ERE), or non-ERE transcription factors, such as c-Jun and c-Fos of the activating protein-1 complex (AP-1), and transcription factor specificity protein 1 (SP1) and NFκB, which extend E2-ERα cell proliferation and pro-inflammatory actions in colon cancer cells [94]. ERα activation can also initiate signaling pathways that promote cell cycle progression, such as the PI3K/Akt and MAPK/ERK pathways [51]. These pathways, in turn, can stimulate cell growth and survival. From these studies we can reasonably conclude that estrogen signal transduction via ERα is pro-tumorigenic in CRC (Figure 3).
In contrast, estrogen signaling through ERβ produces anti-tumorigenic cell signaling responses in a broad range of cancers including CRC by repressing ERα transcription and activating anti-proliferative cell signaling pathways (Figure 3), [95]. ERβ upon binding to its ligand estrogen, dimerizes and translocates to the nucleus where the E2-ERβ complex transcriptionally upregulates target genes which, unlike E2-ERα, promote pro-apoptotic and anti-proliferative responses in CRC. The E2-ERβ complex binds to DNA elements such as ERE or AP1 which activate the FOXO3a gene. Activated FOXO3a in turn transcriptionally upregulates PUMA, p21, and p27 which have been shown to induce apoptosis of CRC cells [96]. ERβ also inhibits the expression of cell proliferating genes such as c-Myc and p45Skp2 [97]. Moreover, some of the EMT and metastasis genes, such as β-catenin, Slug and Twist, are inhibited by ERβ [98]. Nuclear ERβ, can also exert pro-apoptotic responses in colon cancer cells through increased Caspase-3 activity [99] and inhibit cell proliferation by locking the cell cycle in G1-S phase [100]. There is evidence that ERβ may also inhibit colon cancer cell growth through autophagy mediated by suppression of the mammalian target of rapamycin (mTOR) through Cyclin-D1 degradation [101,102]. In addition to these anti-tumorigenic actions, E2-ERβ may have immunosuppressive effects in CRC [103] as well as preserving an epithelial phenotype through stimulating the expression of tight junction proteins occludin-1 and JAMA [104] and inhibiting de-differentiation and epithelial mesenchymal transition via upregulation of E-cadherin and α-catenin while inhibiting β-catenin [105]. E2- ERβ may also induce microRNA protection by repression of the oncogenic prospero homebox 1 (PROX1) through the upregulation of miR-205 which inhibits colon cancer cell migration and invasiveness [106]. A protective role of ERβ in reducing colon crypt proliferation and inflammatory responses resulting from a high fat diet has recently been demonstrated both in male and female mice [107].
There is evidence for cross-talk between the ER sub-types, the most important for CRC being the inhibition of ERα transcription by ERβ with estrogen binding [108] and the negative regulation of the expression of ERα by ERβ [109] as colonocytes differentiate to a mature tight-junction epithelium [110]. Increased ERβ expression could also lead to ERα-ERβ hetero-dimerization, thereby skewing the expression pattern of target genes from proliferative, anti-apoptotic towards an anti-proliferative, pro-apoptotic and anti-tumorigenic profile [111]. Disruption of ERβ expression, but not of ERα, increased intestinal neoplasia and promoted tumorigenisis in APC-/- mice [112]. A combined high ERβ expression together with negative ERα expression was found to be correlated with a better prognosis for CRC patients [113]. ERβ expression predominates over ERα in the normal healthy colon and in the initial stages of adenocarcinoma with progressive loss of ERβ and increased ERα expression observed in colon biopsies in later stages of tumour development [114]. Moreover, the loss of ERβ is associated with enhanced CRC proliferation potential [115], leading to the hypothesis that high ERβ expression may not only be protective against developing CRC but also a prognostic marker and molecular target in the treatment of colon cancer [116,117]. Indeed, a predominant expression of ERβ may show sexual differentiation for its protective role in the early stages of CRC [118].
4.1. Non-Ligand Activation of Nuclear ERs in Colon Cancer
In addition to ligand-binding activation, nuclear ERs can be phosphorylated and trans-activated by EGFR activated tyrosine kinases without requiring binding to estrogen. For example, EGFR can activate the Ras/Raf/MAPK pathway, which phosphorylates ERs, resulting in dimerization and ligand-independent activation of target gene expression [119]. Moreover, the membrane HER2 receptor (Erb-B2 receptor tyrosine kinase 2) when bound to EGFR can also activate tyrosine kinases signaling pathways RAS/RAF/ERK, PIK3K/AKT/mTOR, JAK/STAT3 producing a hyperactivation of mitogenic signals leading to uncontrolled cell proliferation and tumorigenesis in CRC [120].
4.2. Membrane Estrogen Receptors mERα and mERβ in Colon Cancer
While ERα is traditionally recognized as a nuclear receptor that regulates gene expression, detailed evidence accumulated over the past 20 years has demonstrated that a small percentage (approx. 1%) of ERα can also be localized to the cell membrane in certain contexts to produce physiological and clinically relevant biological responses [121]. The membrane ERα [mERα] was first discovered in 1999 [122] and characterised in breast cancer cells by Levin and colleagues [123,124]. The molecular identity was shown to be a palmitoylated variant of the full length (66kDa) nuclear ERα receptor which allowed its tethering at the cell membrane in a caveolin-1 signalsome [125,126]. The understanding of the role of the mERα in physiology and cancer biology was hampered by the lack of specific inhibitors and agonists which could distinguish E2-mERα ligand binding from nuclear ERα signal transduction [127]. The non-genomic and rapid actions (secs/mins) of estrogen on protein kinases, intracellular calcium and ion channel activity in colonic crypts [128,129,130] distinguish it from canonical genomic E2-ERα signal transduction which shows typical long latency of hours/days to generate transcriptional responses [131]. It is important to note that the rapid membrane-initiated responses to estrogen can be elicited at physiological sub-nanomolar concentrations of estrogen such that dose-response is of little value in distinguishing non-genomic from nuclear E2-ERα actions [132]. One way to overcome this difficulty is the use of estrogen analogues which penetrate poorly, if at all, the cell membrane, or the generation of nuclear excluded ERα mutants [133]. In this regard, certain specific membrane-impeded analogues of estrogen such as E2-BSA [134] and estrogen dendrimer conjugates (EDC) [135] which do not enter the cytosol to bind nuclear ERα have been shown to replicate the rapid non-genomic actions of free unbound estrogen (17β-estradiol). The most significant advance, however, in understanding the physiological and pathological roles of mERα have resulted from the generation by the Levin group of selective mouse models with membrane-only mERα (MOER) or nuclear-only ERα (NOER) expression [136]. Mutations of the palmitoylation site of ERα have also provided a useful tool to dissect membrane-initiated and nuclear actions of estrogen [137]. These studies have shown the absolute requirement for mERα expression in transducing rapid actions of estrogen on protein kinases and tyrosine kinase signaling pathways in cancer cell proliferation but not in the development of reproductive organs and tissues [136,137,138]. The presence of ERα on the cell membrane, in addition to its primarily nuclear localization, expands the range of estrogen actions through combined non-genomic and genomic regulation of cell differentiation and proliferation through an expanded signalsome [139,140].
Regarding the role of membrane ERα in colon cancer, research in this specific area is still evolving, and the understanding of its implications is not yet fully established. However, many of the signaling pathways regulated by membrane estrogen actions in colon (EGFR, ERK-MAPK, PI3K-Akt and Wnt) are involved in cell proliferation, survival, and migration in colon cancer cells (Figure 3), [141]. For example, activation of membrane ERα in colonic epithelial cells isolated from females has been associated with the rapid activation of mitogenic signaling cascades, including the MAPK pathway, PI3K/Akt pathway, and Src kinase signaling [142,143,144], which can promote cell growth and survival. Additionally, membrane ERα has been implicated in modulating epithelial-mesenchymal transition in breast [145] and colon cancer cells [146], a process involved in CRC tumour invasion and metastasis [147].
The current data indicate that mERα is the primary endogenous ER mediator of rapid E2 responses although a membrane ERβ (mERβ) is also found co-expressed with mERα in cancer cells to regulate cell proliferation [148]. The role of mERβ is less well-studied in cancer biology although ERβ may be present at the cell membrane in a palmitoylated form in colon cancer cells to inhibit cell proliferation [149]. Studies have demonstrated that mERβ can produce rapid non-genomic actions of estrogen on ERK and JNK kinase activity when expressed in Chinese hamster ovarian cells [150] while other studies have shown E2-mERβ to rapidly activate p38 MAPK in human colon cancer cells [151]. The membrane ER subtypes appear to mimic their respective nuclear ER responses to estrogen i.e., pro-proliferative, pro-tumorigenic effects via nuclear ERα and mERα while conversely producing anti-proliferative, anti-tumorigenic effects via both nuclear ERβ and mERβ. In this way, estrogen non-genomic interactions with membrane ERα and membrane ERβ can modulate cell proliferation, apoptotic pathways and cell death in CRC [152].
4.3. Estrogen Signaling via Truncated ERs in Colon Cancer
Several splice variants of full-length ERs have been reported in various healthy tissues and cancers [153,154]. These truncated ERα and ERβ receptor proteins arise from mutations in ESR1 and ESR2 genes, but cannot form homodimers or recruit cofactors like full-length ERs [153]. Truncated ERs may form inactive heterodimers with full-length ERs [155] and collaborate with other estrogen receptors such as GPER [156] to modulate proliferative and inflammatory responses in cancer. The best studied truncated ER has been ERα36 (36kDa) in breast and gastric cancers [157,158], which transduces estrogenic non-genomic signaling to promote cell proliferation and metastatic potential [159] Apart from one study reporting decreased ERα36 mRNA expression with advanced Duke’s Stage CRC [160], the relevance of truncated ERs to estrogen signaling and sex differences in CRC is unknown and merits further investigation.
5. Estrogen Signaling via G Protein-Coupled Estrogen Receptor in Colon Cancer
G protein-coupled estrogen receptor [GPER], also known as GPR30, first reported in 2005 [161], is a seven-transmembrane receptor that mediates membrane-initiated estrogen signaling in a wide range of tissues [162]. Initially identified as an alternative estrogen receptor in breast cancer [163], GPER signaling has emerged as a significant player in colon cancer [164], contributing to various aspects of tumour progression and estrogenic cell signaling responses [165]. While many studies over the past twenty years support an estrogen-ligand receptor role for GPER [166,167], the involvement of GPER in transducing non-genomic actions of estrogen in vivo has been challenged [168,169]. Several recent studies, however, have demonstrated the functional expression of GPER, in colon cancer cells which may vary among different colon cancer subtypes and individual tumours [170]. The role of GPER in CRC has garnered much interest as its expression predominates in colon cancer after the loss of nuclear ER, in particular after the loss of ERβ which has been reported to negatively regulate GPER expression [171]. While the estrogen protective effects in CRC have been mainly attributed to ERβ, its expression is lost during CRC progression, and this raises the possibility for a role in sex differences of GPER which remains expressed after ERβ loss in CRC [172]. Chronic mucosal inflammation has been proposed as a precursor of CRC and it is interesting to note that GPER expression shows sex dependence in inflammatory bowel disease (IBD), [173] and may transduce estrogen protective effects on mucosal barrier function in IBD [174]. The potential therapeutic implications of targeting GPER signaling in cancer have been recently reviewed. This includes the use of GPER agonists or antagonists, alone or in combination with other therapies [175].
GPER has been reported to have both tumorigenic and anti-tumourigenic roles in cancer progression [176,177]. Some studies have shown a protective role for GPER in CRC [178] and its activation has been reported to inhibit cell proliferation in CRC cell lines [179]. Furthermore, a low expression of GPER in CRC was associated with poor patient survival [176]. In contrast, other studies indicate GPER to be pro-tumorigenic in CRC via estrogen activation by steroid sulfatase [180] and to stimulate cell proliferation in CRC cell lines not expressing nuclear ERs [34]. In support of this hypothesis, GPER expression was shown to be upregulated and stimulated by MAPK signaling in mycotoxin-induced growth of colon cancer cells [181]. In addition, GPER can promote chromosomal instability in CRC leading to neoplastic transformation and tumour development [182]. The impact of GPER signaling on angiogenesis may also be determinant in its tumour promoter actions [183,184]. GPER may have differential protective or exacerbating effects on CRC tumorigenesis depending the expression of ER and activation of cell proliferation signaling pathways.
5.1. Hypoxia and GPER Signaling in Colon Cancer
There is evidence that the divergent roles of GPER in CRC as being either protective or tumorigenic are dependent on the stage of the cancer and the level of hypoxia in the tumour microenvironment and on sex-specific factors influencing hypoxic signals [172]. Moreover, the actions of GPER in the regulation of vascular endothelial growth factor (VEGF) and hypoxia-inducible factor 1-alpha (HIF1α) in CRC show sex dependence (Figure 3), [172].
Hypoxia is a key factor in promoting tumour growth through cell signaling involving HIF1-α [185] and its target VEGF, which are associated with poor clinical outcomes in CRC [186]. In a detailed study of the functional consequences of estrogen actions within the hypoxic CRC cell micro-environment, Bustos et al. [172] found the pro- and anti-tumorigenic potential of GPER in CRC cell lines to be dependent on the level of oxygen exposure. Under normoxic conditions, estrogen and the GPER agonist G1 both suppressed CRC cell proliferation. Under hypoxic conditions, however, GPER activation produced the opposite functional effect with both estrogen and G1 enhancing CRC cell proliferation whereas the GPER antagonist G15 inhibited proliferation. Estrogen treatment enhanced the hypoxia-induced expression of HIF1α and VEGF, but repressed HIF1α and VEGF expression under normoxic conditions. The expression and repression of VEGF by estrogen were mediated by a GPER-dependent mechanism. Thus, GPER is essential in transducing the normoxic anti-proliferative effects of estrogen as well as its hypoxic proliferative actions in CRC cells. The latter response may be amplified by up-regulation of GPER expression following exposure to hypoxia and estrogen. Another pro-tumorigenic amplification factor in the GPER response is the estrogen-modulated gene, Ataxia Telangiectasia Mutated (ATM), which was shown to be repressed in hypoxia via GPER signalling [172]. Loss of ATM expression is associated with poor survival in CRC [187] and an increase in phosphorylated ATM protein levels has been observed in hypoxic colon cancer cells [188]. The modulation of ATM expression by GPER in low oxygen tension and the sensitivity of its expression to estrogen in CRC provides an additional mechanism for pro-tumorigenic actions of estrogen via GPER in colon cancer under hypoxic conditions. Thus it is important to take into account the CRC stage and tumour microenvironment when interpreting the role of E2-GPER interactions in colon cancer tumorgenesis, sex differences and patient survival/treatment [189].
The involvement of GPER in CRC patient survival displays clear sex differences. In a cohort of 566 CRC patient tumour samples, GPER expression significantly associated with poor survival in CRC Stages 3-4 females but not in the stage-matched male population [172]. Since an hypoxic tumour microenvironment is associated with late stages in CRC, we may conclude that sex differences in this case are underpinned by E2-GPER tumorigenic actions on HIF1α /VEGF activation and on ATM suppression under hypoxia. The anti-proliferative effects of E2-GPER signaling in normoxia may explain the observations of protective effects of GPER expression on CRC survival in the early stages of cancer development.
6. Estrogen Regulation of Wnt/β-Catenin Signaling in Colon Cancer
Wnt/β-catenin signaling plays a key role in various biological processes, including embryonic development, tissue homeostasis, and cell proliferation [190,191]. Dysregulation of the Wnt/β-catenin signaling pathway has been strongly associated with the development and progression of colon cancer [192]. The Wnt/β-catenin signaling pathway is commonly hyperactivated in colon cancer and plays a crucial role in CRC development and progression [193]. Estrogen has been shown to modulate this pathway in a sex-dependent manner in CRC [194] and in breast and endometrial cancers [195,196] through reciprocal interaction between ERα and β-catenin. In reproductive tissues estrogen promotes cell proliferation via ERα stimulation of β-catenin nuclear translocation [197]. In contrast, other studies indicate that ERα is inhibitory for Wnt/β-catenin mediated proliferation and neoplasia in non-reproductive tissues, for example in liver cancer [198]. Moreover, ERβ was reported in the latter study to have no role in estrogen modulation of Wnt/β-catenin signaling. These studies suggest that estrogen may have an inhibitory effect on Wnt/β-catenin signaling in non-reproductive tissue cancers in females, potentially contributing to sex differences in CRC incidence and outcomes. But how does this work in CRC and what are the factors which determine whether estrogen will exert a protective or exacerbating role via Wnt/β-catenin pathway signaling in CRC, in particular when the expression of ER is lost with advancing tumorigenesis? To answer this question we must first understand the possibility of crosstalk with other receptors and signaling pathways. The Wnt signaling pathway interacts with various signaling pathways, particularly K+ channels, implicated in colon cancer. Interactions between the Wnt/β-catenin pathway and the estrogen-regulated K+ ion channel KCNQ1 in CRC is a major molecular mechanism which displays sex differences in the regulation of CRC cell proliferation and epithelial mesenchymal transition [23].
6.1. Estrogen Regulation of Wnt-KCNQ1 Interactions in Colon Cancer
KCNQ1 is a voltage-gated K+ channel (Kv7.1) expressed in the basolateral membranes of colonic crypts where it functions to provide the electrical driving force for Cl- secretion [199]. In colon, the KCNQ1 channel is co-expressed with the β-regulatory subunit KCNE3 which greatly increases the ionic conductance of the channel and confers voltage and cAMP sensitivity to the channel [200]. KCNQ1 has been shown to have an anti-tumorigenic role in many gastrointestinal (GI) cancers including colon cancer [201]. Moreover, relapse-free CRC patient survival was positively associated with high KCNQ1 gene expression which displayed sex-dependence for a female advantage [202]. KCNQ1 channels modulate Wnt signaling in a wide number of GI cancers [203] and there is strong evidence for bi-directional interaction between KCNQ1 and β-catenin in normal healthy colon and in colon cancer cells [202]. Estrogen regulates KCNQ1:KCNE3 channel function by uncoupling KCNQ1 from KCNE3 via PKC-dependent phosphorylation of KCNE3 at residue Ser82 which destabilizes the KCNE3:KCNQ1 channel complex [204]. The uncoupling of KCNQ1 from KCNE3 promotes KCNQ1 endocytotic recycling in colonic crypts [205] which allows KCNQ1 to leave the plamsamembrane and bind cytosolic activated β-catenin. KCNQ1 then returns β-catenin to the cell membrane to trap it in a complex with E-cadherin at adherens junctions Figure 4, [202]. In this way, KCNQ1 anchors β-catenin at the plasma membrane, stabilizing adherens junctions, and promoting cell-cell adhesion. The association of KCNQ1 with β-catenin at the cell membrane is essential to preserve a well-differentiated epithelial phenotype by maintaining a stable KCNQ1: β-catenin:E-cadherin complex at adherens junctions and preventing epithelial mesenchymal transition [206]. Trapping β-catenin with KCNQ1 at the cell membrane has been shown to retard the nuclear translocation of β-catenin and prevent the transcriptional activation of proliferative genes [202]. These observations suggest that estrogen-regulated KCNQ1 channel quells the Wnt:β-catenin nuclear signaling pathway to suppress CRC cell proliferation and EMT, but only in females [207].
6.3. Estrogen Regulation of Wnt Receptor Oncogenic Signaling in Colon Cancer
The receptor tyrosine kinase pathway, particularly the EGFR pathway, can cross-regulate Wnt signaling in colon cancer [208]. Additionally, the Transforming Growth Factor-beta (TGF-β) pathway can modulate Wnt signaling through Smad proteins [209], all of which could amplify the cell proliferation responses in CRC. In contrast, a recent study has provided evidence for a role of GPER in protecting against CRC progression by selectively reducing the oncogenic effects of hyperactive Wnt/β-catenin signalling pathways in CRC [210]. Firstly, sex differences were observed in the gene expression of the Wnt receptor FZD1 (Frizzled 1) in Kaplan–Meier survival analyses across multiple CRC patient gene microarray datasets. High expression of FZD1 was associated with poor relapse-free survival rates in the male but not in the female CRC population. Secondly, activation of GPER with the G1 agonist prevented the Wnt pathway-induced upregulation of the JUN oncogene. These novel findings indicate a mechanistic role for GPER in protecting against CRC progression in females by selectively reducing the tumorigenic effects of Wnt/β-catenin oncogenic signalling pathways.
7. Estrogen Regulation of Epigenetic, Microbiome and Metabolic Factors in Colon Cancer
Other emerging factors underpinning sex differences in CRC include microRNAs and the gut microbiota. MicroRNAs (miRNAs) are small non-coding RNA molecules that regulate gene expression by targeting specific messenger RNAs for degradation or translational repression. Differential expression of miRNAs has been observed between males and females in CRC [211,212]. Estrogen can modulate the expression and activity of specific miRNAs, which in turn influence the expression of genes involved in CRC development [213]. These sex-specific miRNA profiles may contribute to the estrogen-mediated sex differences in CRC. This has been shown for miR-30c-5p expression which was associated with better survival in females and was downregulated in males [214]. Several miRNAs are currently under investigation for therapeutic applications in CRC [215].
Recent studies suggest that the gut microbiota can influence CRC development and responses to estrogen [216]. The composition and function of the gut microbiota can differ between males and females [217], potentially affecting estrogen metabolism and signaling. Thus, another contributory factor to sex disparities in CRC is the estrogen-gut microbiome axis [218]. Microbial metabolites produced by the gut microbiota may modulate estrogen receptor activity and alter estrogen levels, contributing to sexual differentiation in CRC [219]. In addition, E2 may alter the gut microbiota to reduce the risk of developing CRC [220].
Another potential factor contributing to CRC sex differences is estrogen metabolism. The metabolism of estrogen is an important process that can influence its bioavailability and activity. In females, estrogen is predominantly metabolized by cytochrome P450 enzymes, including CYP1A1 and CYP1B1, which convert estradiol to 2-hydroxyestradiol (2-OHE2), [221]. In males, estrogen is mainly metabolized by CYP1A2 and CYP3A4, leading to the formation of 4-hydroxyestradiol (4-OHE2), [222]. The different metabolic pathways can result in distinct estrogen metabolite profiles, which may contribute to sex variances in CRC susceptibility. There is some evidence that testosterone can exert neoplastic actions in CRC [223] and for the presence of upregulated genes on the Y chromosome that contribute to colorectal cancer in males by driving tumor invasion and aiding immune escape [41,224,225].
8. Conclusions and Perspectives
An increasing number of epidemiological and molecular studies have demonstrated a female sex advantage in colon cancer such that the incidence of CRC is lower in premenopausal females compared to males and postmenopausal females. The protective effect of estrogen against CRC has been proposed as a possible explanation for this observation.
Membrane estrogen receptors, particularly mERα, GPER and nuclear estrogen receptor ERα, ERβ, have been detected in colon cancer tissue. The presence of these receptors suggests that estrogen non-genomic and genomic signaling could have an impact on CRC tumorigenesis in a sex-specific manner. Estrogen signaling may protect against colon cancer development and growth through its effects on cell proliferation, apoptosis, cell cycle progression and epithelial differentiation. These beneficial effects of estrogen are absent in males with the added disadvantage of testosterone and Y chromosome tumorigenic actions in CRC (Figure 5). It is worth emphasising that in some cases estrogen signaling might be detrimental in CRC, as in an hypoxic tumor microenvironment which is a hallmark of late stage CRC,.
Estrogen receptor status in CRC tumors may serve as a prognostic marker, providing information on the likelihood of disease progression and patient outcomes. ER-positive tumours in females might be associated with a better prognosis compared to ER-negative tumours. Some colon cancers that express estrogen receptors may be candidates for targeted hormonal therapies. Selective estrogen receptor modulators or aromatase inhibitors, which are used in hormone receptor-positive breast cancer, have been explored in preclinical studies and early-phase clinical trials for colon cancer [226]. It is essential to note that not all colon cancers express estrogen receptors, and the clinical relevance of estrogen signaling targets may vary between individuals and tumor subtypes [227]. Therefore, identifying patients who may benefit from hormone-based therapies requires careful patient selection and further research [228].
Author Contributions
Conceptualization, BJH and HMH; writing -original draft preparation, BJH; writing-reviewing and editing, BJH and HMH. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant from the RCSI Academic Fund to BJH.
Institutional Review Board Statement
Not applicable.
Informed Consent: Not applicable.
Data Availability Statement
Not applicable
Acknowledgements
The authors thank numerous collaborators and fellow researchers working in this field whose work is cited here.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rubin, J.B.; Lagas, J.S.; Broestl, L.; Sponagel, J.; Rockwell, N.; Rhee, G.; Rosen, S.F.; Chen, S.; Klein, R.S.; Imoukhuede, P.; Luo, J. Sex differences in cancer mechanisms. Biol. Sex. Differ. 2020, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Clocchiatti, A.; Cora, E.; Zhang, Y.; et al. Sexual dimorphism in cancer. Nat. Rev. Cancer 2016, 16, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Dorak, M.T. Sexual dimorphism in molecular biology of cancer, Principles of Gender-Specific Medicine [Fourth Edition], Editor[s]: Marianne J. Legato, Academic Press, 2023; Chapter 29, Pages 463-476. [CrossRef]
- Lassek, W.D.; Gaulin, S.J.C. Substantial but Misunderstood Human Sexual Dimorphism Results Mainly From Sexual Selection on Males and Natural Selection on Females. Front. Psychol. 2022, 13, 859931. [Google Scholar] [CrossRef]
- Regitz-Zagrosek, V. Sex and gender differences in health. Science & Society Series on Sex and Science. EMBO Rep. 2012, 13, 596–603. [Google Scholar] [CrossRef]
- Maney, D.L. Perils and pitfalls of reporting sex differences. Phil. Trans. R. Soc. 2016; B3712015011920150119. [Google Scholar] [CrossRef]
- Zheng, D.; Trynda, J.; Williams, C.; Vold, J.A.; Nguyen, J.H.; Harnois, D.M.; Bagaria, S.P.; McLaughlin, S.A.; Li, Z. Sexual dimorphism in the incidence of human cancers. BMC Cancer. 2019, 19, 684. [Google Scholar] [CrossRef]
- Kim, H.I.; Lim, H.; Moon, A. Sex Differences in Cancer: Epidemiology, Genetics and Therapy. Biomol Ther [Seoul]. 2018, 26, 335–342. [Google Scholar] [CrossRef]
- Rubin, J.B. The spectrum of sex differences in cancer. Trends Cancer. 2022, 8, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Jacob, L.; Freyn, M.; Kalder, M.; Dinas, K.; Kostev, K. Impact of tobacco smoking on the risk of developing 25 different cancers in the UK: a retrospective study of 422,010 patients followed for up to 30 years. Oncotarget. 2018, 9, 17420–17429. [Google Scholar] [CrossRef] [PubMed]
- Rumgay, H.; Shield, K.D.; Charvat, H.; Ferrari, P.; Sornpaisarn, B.; Obot, I. Global burden of cancer in 2020 attributable to alcohol consumption: a population-based study. Lancet Oncol 2021. [CrossRef]
- Abancens, M.; Bustos, V.; Harvey, H.; McBryan, J.; Harvey, B.J. Sexual Dimorphism in Colon Cancer. Front. Oncol. 2020, 10, 607909. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, G.; He, J.; Ren, S.; Wu, F.; Zhang, J.; Wang, F. Gender Differences in Colorectal Cancer Survival: A Meta-Analysis. Int. J. Cancer 2017, 141, 1942–1949. [Google Scholar] [CrossRef] [PubMed]
- 14. Globocan. Estimated cancer incidence, mortality and prevalence worldwide 2018. Available at: https://gco.iarc.fr/today/data/factsheets/cancers/10_8_9-Colorectum-fact-sheet.pdf; at: https; Available at: https://gco.iarc.fr/today/data/factsheets/cancers/10_8_9-Colorectum-fact-sheet.pdf.
- Global Cancer Observatory. Available online: https://gco.iarc.fr/. (accessed on 14 July 2023).
- Global Cancer Observatory. Available online: https://gco.iarc.fr/tomorrow/en/dataviz/trends?types=0_1&sexes=1_2&mode=population&group_populations=0&multiple_populations=1&multiple_cancers=1&cancers=8&populations=994&apc=cat_ca20v1.5_ca23v-1.
- Hogan, A.M.; Collins, D.; Baird, A.W.; Winter, D.C. Estrogen and gastrointestinal malignancy. Mol. Cell Endocrinol. 2009, 307, 19–24. [Google Scholar] [CrossRef]
- Koo, J.H.; Jalaludin, B.; Wong, S.K.; Kneebone, A.; Connor, S.J.; Leong, R.W. Improved survival in young women with colorectal cancer. Am. J. Gastroenterol. 2008, 103, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Rennert, G.; Rennert, H.S.; Pinchev, M.; Lavie, O.; Gruber, S.B. Use of hormone replacement therapy and the risk of colorectal cancer. J. Clin. Oncol. 2009, 27, 4542–4547. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.D.; Humphrey, L.L.; Nygren, P.; Teutsch, S.M.; Allan, J.D. Postmenopausal hormone replacement therapy: scientific review. JAMA. 2002, 288, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; Wactawski-Wende, J.; Ritenbaugh, C.; Hubbell, F.A.; Ascensao, J.; Rodabough, R.J.; Rosenberg, C.A.; Taylor, V.M.; Harris, R.; Chen, C.; Adams-Campbell, L.L.; White, E. Estrogen plus progestin and colorectal cancer in postmenopausal women. N. Engl. J. Med. 2004, 350, 991–1004. [Google Scholar] [CrossRef]
- Kennelly, R.; Kavanagh, D.O.; Hogan, A.M.; Winter, D.C. Oestrogen and the colon: potential mechanisms for cancer prevention. Lancet Oncol. 2008, 9, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, F.; Ryan, E.J.; Doherty, G.; Winter, D.C.; Sheahan, K. Estrogen receptors and their implications in colorectal carcinogenesis. Front. Oncol. 2015, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Perotti, V.; Fabiano, S.; Contiero, P.; Michiara, M.; Musolino, A.; Boschetti, L.; Cascone, G.; Castelli, M.; Tagliabue, G.; Cancer Registries Working Group. Influence of Sex and Age on Site of Onset, Morphology, and Site of Metastasis in Colorectal Cancer: A Population-Based Study on Data from Four Italian Cancer Registries. Cancers 2023, 15, 803. [Google Scholar] [CrossRef]
- Lee, M.S.; Menter, D.G.; Kopetz, S. Right Versus Left Colon Cancer Biology: Integrating the Consensus Molecular Subtypes. J. Natl. Compr. Canc Netw. 2017, 15, 411–419. [Google Scholar] [CrossRef]
- Dupon, C.; Hosseinian, A.; Kim, M.H. Simultaneous determination of plasma estrogens, androgens, and progesterone during the human menstrual cycle. Steroids. 1973, 22, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Shutt, D.A.; Smith, I.D.; Shearman, R.P. Oestrone, oestradiol-17beta and oestriol levels in human foetal plasma during gestation and at term. J. Endocrinol. 1974, 60, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Banerjee, A.; Singh, D.; Thakur, G.; Kasarpalkar, N.; Gavali, S.; Gadkar, S.; Madan, T.; Mahale, S.D.; Balasinor, N.H.; Sachdeva, G. Estradiol: A Steroid with Multiple Facets. Horm. Metab. Res. 2018, 50, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Brooks, R.C.; Garratt, M.G. Life history evolution, reproduction, and the origins of sex-dependent aging and longevity. Ann. N. Y Acad. Sci. 2017, 1389, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, K.J.; Hewitt, S.C.; Arao, Y.; Korach, K.S. Estrogen Hormone Biology. Curr. Top. Dev. Biol. [CrossRef]
- Barzi, A.; Lenz, A.M.; Labonte, M.J.; Lenz, H.J. Molecular pathways: Estrogen pathway in colorectal cancer. Clin. Cancer Res. 2013, 19, 5842–5848. [Google Scholar] [CrossRef] [PubMed]
- Hashemzadeh, M.; Haseefa, F.; Peyton, L.; Park, S.; Movahed, M.R. The effects of estrogen and hormone replacement therapy on platelet activity: a review. Am. J. Blood Res. 2022, 12, 33–42. [Google Scholar] [PubMed]
- Henderson, B.E.; Feigelson, H.S. Hormonal carcinogenesis. Carcinogenesis 2000, 21, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Das, P.K.; Saha, J.; Pillai, S.; Lam, A.K.; Gopalan, V.; Islam, F. Implications of estrogen and its receptors in colorectal carcinoma. Cancer Med. 2023, 12, 4367–4379. [Google Scholar] [CrossRef]
- Moggs, J.G.; Orphanides, G. Estrogen receptors: orchestrators of pleiotropic cellular responses. EMBO Rep. 2001, 2, 775–781. [Google Scholar] [CrossRef]
- Topi, G.; Ghatak, S.; Satapathy, S.R.; Ehrnström, R.; Lydrup, M.L.; Sjölander, A. Combined Estrogen Alpha and Beta Receptor Expression Has a Prognostic Significance for Colorectal Cancer Patients. Front Med [Lausanne]. 2022, 9, 739620. [Google Scholar] [CrossRef]
- Thomas, M.L.; Xu, X.; Norfleet, A.M.; Watson, C.S. The presence of functional estrogen receptors in intestinal epithelial cells. Endocrinology. 1993, 132, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.L.; Javid, S.H.; Carothers, A.M.; Redston, M.; Bertagnolli, M.M. Estrogen receptors α and β are inhibitory modifiers of Apc-dependent tumorigenesis in the proximal colon of Min/+ mice. Cancer Res. 2007, 67, 2366–2372. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; DiLeo, A.; Niv, Y.; Gustafsson, J.Å. Estrogen receptor beta as target for colorectal cancer prevention. Cancer Lett. 2016, 372, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Foley, E.F.; Jazaeri, A.A.; Shupnik, M.A.; Jazaeri, O.; Rice, L.W. Selective loss of estrogen receptor β in malignant human colon. Cancer Res. 2000, 60, 245–248. [Google Scholar]
- Baraibar, J. Ros, N. Saoudi, F. Salvà, A. García, M.R. Castells, J. Tabernero, E. Élez, Sex and gender perspectives in colorectal cancer, ESMO Open 2023, 8, 101204. [Google Scholar] [CrossRef]
- Matthews, J.; Gustafsson, J.A. Estrogen signaling: a subtle balance between ER alpha and ER beta. Mol. Interv. 2003, 3, 281–292. [Google Scholar] [CrossRef]
- Ditonno, I.; Losurdo, G.; Rendina, M.; Pricci, M.; Girardi, B.; Ierardi, E.; Di Leo, A. Estrogen Receptors in Colorectal Cancer: Facts, Novelties and Perspectives. Curr. Oncol. 2021, 28, 4256–4263. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; DiLeo, A.; Niv, Y.; Gustafsson, J.Å. Estrogen receptor beta as target for colorectal cancer prevention. Cancer Lett. 2016, 372, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, S.; Qiu, S.; Fiorentino, F.; et al. The prognostic and therapeutic role of hormones in colorectal cancer: a review. Mol. Biol. Rep. 2019, 46, 1477–1486. [Google Scholar] [CrossRef]
- Maingi, J.W.; Tang, S.; Liu, S.; Ngenya, W.; Bao, E. Targeting estrogen receptors in colorectal cancer. Mol. Biol. Rep. 2020, 47, 4087–4091. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Levin, E.R. Nature of functional estrogen receptors at the plasma membrane. Mol. Endocrinol. 2006, 20, 1996–2009. [Google Scholar] [CrossRef] [PubMed]
- O'Mahony, F.; Thomas, W.; Harvey, B.J. Novel female sex-dependent actions of oestrogen in the intestine. J. Physiol. 2009, 587 Pt. 21, 5039–5044. [Google Scholar] [CrossRef]
- Jacquot, Y.; de Cremoux, P.; Harvey, B.J.; Wehling, M. The Rapid Responses to Steroid Hormones meetings: An important event for steroid science. Ann Endocrinol [Paris]. 2023, 84, 235–237. [Google Scholar] [CrossRef]
- Barton, M.; Frigo, D.E.; Madak-Erdogan, Z.; Mauvais-Jarvis, F.; Prossnitz, E.R. Steroid Hormones and Receptors in Health and Disease: A Research Conference Co-Organized by FASEB and the International Committee on Rapid Responses to Steroid Hormones [RRSH], May 25-27, 2021. FASEB J. 2021, 35, e21858. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.J. Guest editorial: 11th international meeting on rapid responses to steroid hormones RRSH2018. Steroids. 2020, 154, 108552. [Google Scholar] [CrossRef]
- Levin, E.R. Plasma membrane estrogen receptors. Trends Endocrinol. Metab. 2009, 20, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Jacenik, D.; Beswick, E.J.; Krajewska, W.M.; Prossnitz, E.R. G Protein-Coupled Estrogen Receptor in Colon Function, Immune Regulation and Carcinogenesis. World J. Gastroenterol. 2019, 25, 4092–4104. [Google Scholar] [CrossRef]
- Wilkenfeld, S.R.; Lin, C.; Frigo, D.E. Communication between genomic and non-genomic signaling events coordinate steroid hormone actions. Steroids. 2018, 133, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R.; Hammes, S.R. Nuclear receptors outside the nucleus: extranuclear signalling by steroid receptors. Nat. Rev. Mol. Cell Biol. 2016, 17, 783–797. [Google Scholar] [CrossRef]
- O'Mahony, F.; Harvey, B.J. Sex and estrous cycle-dependent rapid protein kinase signaling actions of estrogen in distal colonic cells. Steroids. 2008, 73, 889–894. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Extranuclear estrogen receptor's roles in physiology: lessons from mouse models. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E133–E140. [Google Scholar] [CrossRef] [PubMed]
- Ditonno, I.; Losurdo, G.; Rendina, M.; Pricci, M.; Girardi, B.; Ierardi, E.; Di Leo, A. Estrogen Receptors in Colorectal Cancer: Facts, Novelties and Perspectives. Curr. Oncol. 2021, 28, 4256–4263. [Google Scholar] [CrossRef] [PubMed]
- Koveitypour, Z.; Panahi, F.; Vakilian, M.; et al. Signaling pathways involved in colorectal cancer progression. Cell Biosci. 2019, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Losordo, D.W.; Isner, J.M. Estrogen and angiogenesis: A review. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Gargett, C.E.; Zaitseva, M.; Bucak, K.; Chu, S.; Fuller, P.J.; Rogers, P.A. 17-Beta-estradiol up-regulates vascular endothelial growth factor receptor-2 expression in human myometrial microvascular endothelial cells: role of estrogen receptor-alpha and -beta. J. Clin. Endocrinol. Metab. 2002, 87, 4341–4349. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Yu, M.; Markowitz, S.D. Epigenetic Alterations in the Gastrointestinal Tract: Current and Emerging Use for Biomarkers of Cancer. Gastroenterology. 2021, 160, 690–709. [Google Scholar] [CrossRef] [PubMed]
- Cignarella, A.; Boscaro, C.; Albiero, M.; Bolego, C.; Barton, M. Post-transcriptional and epigenetic regulation of estrogen signaling. J. Pharmacol. Exp. Ther. 2023. [Google Scholar] [CrossRef]
- Yoriki, K.; Mori, T.; Kokabu, T.; Matsushima, H.; Umemura, S.; Tarumi, Y.; Kitawaki, J. Estrogen-related receptor alpha induces epithelial-mesenchymal transition through cancer-stromal interactions in endometrial cancer. Sci. Rep. 2019, 9, 6697. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Datta, P.K. Regulation of EMT in Colorectal Cancer: A Culprit in Metastasis. Cancers 2017, 9, 171. [Google Scholar] [CrossRef]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Bilancio, A.; Perillo, B.; Sinisi, A.A.; Migliaccio, A.; Castoria, G. Estrogen Receptors in Epithelial-Mesenchymal Transition of Prostate Cancer. Cancers [Basel]. 2019, 11, 1418. [Google Scholar] [CrossRef]
- Bouris, P.; Skandalis, S.S.; Piperigkou, Z.; Afratis, N.; Karamanou, K.; Aletras, A.J.; Moustakas, A.; Theocharis, A.D.; Karamanos, N.K. Estrogen receptor alpha mediates epithelial to mesenchymal transition, expression of specific matrix effectors and functional properties of breast cancer cells. Matrix Biol. 2015, 43, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Orzołek, I.; Sobieraj, J.; Domagała-Kulawik, J. Estrogens, Cancer and Immunity. Cancers [Basel]. 2022, 14, 2265. [Google Scholar] [CrossRef]
- Zafari, N.; Khosravi, F.; Rezaee, Z.; Esfandyari, S.; Bahiraei, M.; Bahramy, A.; Ferns, G.A.; Avan, A. The role of the tumor microenvironment in colorectal cancer and the potential therapeutic approaches. J. Clin. Lab. Anal. 2022, 36, e24585. [Google Scholar] [CrossRef] [PubMed]
- Hases, L.; Archer, A.; Williams, C. ERβ and Inflammation. Adv. Exp. Med. Biol. 2022, 1390, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Doolan, C.M.; Condliffe, S.B.; Harvey, B.J. Rapid non-genomic activation of cytosolic cyclic AMP-dependent protein kinase activity and [Ca[2+]][i] by 17beta-oestradiol in female rat distal colon. Br. J. Pharmacol. 2000, 129, 1375–1386. [Google Scholar] [CrossRef]
- Arterburn, J.B.; Prossnitz, E.R. G Protein-Coupled Estrogen Receptor GPER: Molecular Pharmacology and Therapeutic Applications. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 295–320. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, T.; Genazzani, A.R. Non-genomic actions of sex steroid hormones. Eur. J. Endocrinol. 2003, 148, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Lundgreen, A.; Wolff, R.K. MAP kinase genes and colon and rectal cancer. Carcinogenesis. 2012, 33, 2398–2408. [Google Scholar] [CrossRef] [PubMed]
- Irnaten, M.; Blanchard-Gutton, N.; Harvey, B.J. Rapid effects of 17beta-estradiol on epithelial TRPV6 Ca2+ channel in human T84 colonic cells. Cell Calcium. 2008, 44, 441–452. [Google Scholar] [CrossRef]
- O'Mahony, F.; Alzamora, R.; Betts, V.; LaPaix, F.; Carter, D.; Irnaten, M.; Harvey, B.J. Female gender-specific inhibition of KCNQ1 channels and chloride secretion by 17beta-estradiol in rat distal colonic crypts. J. Biol. Chem. 2007, 282, 24563–24573. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Pallottini, V.; Trentalance, A. Estrogens cause rapid activation of IP3-PKC-alpha signal transduction pathway in HEPG2 cells. Biochem. Biophys. Res. Commun. 1998, 245, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Doolan, C.M.; Harvey, B.J. Modulation of cytosolic protein kinase C and calcium ion activity by steroid hormones in rat distal colon. J. Biol. Chem. 1996, 271, 8763–8767. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Marino, M. Synergism between genomic and non genomic estrogen action mechanisms. IUBMB Life. 2003, 55, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Pedram, A.; Razandi, M.; Aitkenhead, M.; Hughes, C.C.; Levin, E.R. Integration of the non-genomic and genomic actions of estrogen. Membrane-initiated signaling by steroid to transcription and cell biology. J. Biol. Chem. 2002, 277, 50768–50775. [Google Scholar] [CrossRef] [PubMed]
- Björnström, L.; Sjöberg, M. Mechanisms of Estrogen Receptor Signaling: Convergence of Genomic and Nongenomic Actions on Target Genes. Mol. Endocrinol. Baltim. Md. 2005, 19, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.B.; Franke, T.F.; Stoica, G.E.; Chambon, P.; Katzenellenbogen, B.S.; Stoica, B.A.; McLemore, M.S.; Olivo, S.E.; Stoica, A. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology. 2000, 141, 4503–4511. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; Metzger, D.; Chambon, P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995, 270, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Integration of the Extranuclear and Nuclear Actions of Estrogen. Mol. Endocrinol. 2005, 19, 1951–1959. [Google Scholar] [CrossRef]
- Madak-Erdogan, Z.; Lupien, M.; Stossi, F.; Brown, M. Genomic Collaboration of Estrogen Receptor α and Extracellular Signal-Regulated Kinase 2 in Regulating Gene and Proliferation Programs. Molecular and Cellular Biology 2011, 31, 226–236. [Google Scholar] [CrossRef]
- Hennessy, B.A.; Harvey, B.J.; Healy, V. 17beta-Estradiol rapidly stimulates c-fos expression via the MAPK pathway in T84 cells. Mol. Cell Endocrinol. 2005, 229, 39–47. [Google Scholar] [CrossRef] [PubMed]
- O'Mahony, F.; Alzamora, R.; Chung, H.L.; Thomas, W.; Harvey, B.J. Genomic priming of the antisecretory response to estrogen in rat distal colon throughout the estrous cycle. Mol. Endocrinol. 2009, 23, 1885–1899. [Google Scholar] [CrossRef] [PubMed]
- Wade, C.B.; Robinson, S.; Shapiro, R.A.; Dorsa, D.M. Estrogen receptor [ER]alpha and ERbeta exhibit unique pharmacologic properties when coupled to activation of the mitogen-activated protein kinase pathway. Endocrinology. 2001, 142, 2336–2342. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Membrane estrogen receptors signal to determine transcription factor function. Steroids. 2018, 132, 1–4. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Aitkenhead, M.; Hughes, C.C.; Levin, E.R. Integration of the non-genomic and genomic actions of estrogen. Membrane-initiated signaling by steroid to transcription and cell biology. J. Biol. Chem. 2002, 277, 50768–50775. [Google Scholar] [CrossRef]
- Barzi, A.; Lenz, A.M.; Labonte, M.J.; Lenz, H.J. Molecular pathways: Estrogen pathway in colorectal cancer. Clin. Cancer Res. 2013, 19, 5842–5848. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen signaling multiple pathways to impact gene transcription. Curr. Genomics. 2006, 7, 497–508. [Google Scholar] [CrossRef]
- Mal, R.; Magner, A.; David, J.; Datta, J.; Vallabhaneni, M.; Kassem, M.; Manouchehri, J.; Willingham, N.; Stover, D.; Vandeusen, J.; Sardesai, S.; Williams, N.; Wesolowski, R.; Lustberg, M.; Ganju, R.K.; Ramaswamy, B.; Cherian, M.A. Estrogen Receptor Beta [ERβ]: A Ligand Activated Tumor Suppressor. Front. Oncol. 2020, 10, 587386. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell. 2001, 7, 673–682. [Google Scholar] [CrossRef]
- Dey, P.; Barros, R.P.; Warner, M.; Ström, A.; Gustafsson, J.Å. Insight into the mechanisms of action of estrogen receptor β in the breast, prostate, colon, and CNS. J. Mol. Endocrinol. 2013, 51, T61–T74. [Google Scholar] [CrossRef]
- Topi, G.; Satapathy, S.R.; Dash, P.; Fred Mehrabi, S.; Ehrnström, R.; Olsson, R.; Lydrup, M.L.; Sjölander, A. Tumour-suppressive effect of oestrogen receptor β in colorectal cancer patients, colon cancer cells, and a zebrafish model. J. Pathol. 2020, 251, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Waters, C.E.; Lewis, A.E.; Langman, M.J.; Eggo, M.C. Oestrogen-induced apoptosis in colonocytes expressing oestrogen receptor beta. J. Endocrinol. 2002, 174, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Topi, G.; Satapathy, S.R.; Dash, P.; Fred Mehrabi, S.; Ehrnström, R.; Olsson, R.; Lydrup, M.L.; Sjölander, A. Tumour-suppressive effect of oestrogen receptor β in colorectal cancer patients, colon cancer cells, and a zebrafish model. J. Pathol. 2020, 251, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Martineti, V.; Picariello, L.; Tognarini, I.; Carbonell Sala, S.; Gozzini, A.; Azzari, C.; et al. ERbeta is a potent inhibitor of cell proliferation in the HCT8 human colon cancer cell line through regulation of cell cycle components. Endocr. Relat. Cancer 2005, 12, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Huang, C.; Wu, H.; Huang, J. Estrogen Receptor Beta [ERβ] Mediated-CyclinD1 Degradation via Autophagy Plays an Anti-Proliferation Role in Colon Cells. Int. J. Biol. Sci. 2019, 15, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Hases, L.; Indukuri, R.; Birgersson, M.; Nguyen-Vu, T.; Lozano, R.; Saxena, A.; et al. Intestinal estrogen receptor beta suppresses colon inflammation and tumorigenesis in both sexes. Cancer Lett. 2020, 492, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Leveque, M.; Buisson-Brenac, C.; Bueno, L.; Fioramonti, J.; Houdeau, E. Oestradiol decreases colonic permeability through oestrogen receptor beta-mediated up-regulation of occludin and junctional adhesion molecule-A in epithelial cells. J. Physiol. 2009, 587 Pt. 13, 3317–3328. [Google Scholar] [CrossRef]
- Silva, R.S.; Lombardi, A.P.G.; de Souza, D.S.; Vicente, C.M.; Porto, C.S. Activation of estrogen receptor beta [ERβ] regulates the expression of N-cadherin, E-cadherin and β-catenin in androgen-independent prostate cancer cells. Int. J. Biochem. Cell Biol. 2018, 96, 40–50. [Google Scholar] [CrossRef]
- Nguyen-Vu, T.; Wang, J.; Mesmar, F.; Mukhopadhyay, S.; Saxena, A.; McCollum, C.W.; Gustafsson, J.Å.; Bondesson, M.; Williams, C. Estrogen receptor beta reduces colon cancer metastasis through a novel miR-205 - PROX1 mechanism. Oncotarget. 2016, 7, 42159–42171. [Google Scholar] [CrossRef]
- Hases, L.; Archer, A.; Indukuri, R.; Birgersson, M.; Savva, C.; Korach-André, M.; Williams, C. High-fat diet and estrogen impacts the colon and its transcriptome in a sex-dependent manner. Sci. Rep. 2020, 10, 16160. [Google Scholar] [CrossRef]
- Matthews, J.; Wihlén, B.; Tujague, M.; Wan, J.; Ström, A.; Gustafsson, J.A. Estrogen receptor [ER] beta modulates ERalpha-mediated transcriptional activation by altering the recruitment of c-Fos and c-Jun to estrogen-responsive promoters. Mol. Endocrinol. 2006, 20, 534–543. [Google Scholar] [CrossRef]
- Gougelet, A.; Mueller, S.O.; Korach, K.S.; Renoir, J.M. Oestrogen receptors pathways to oestrogen responsive elements: the transactivation function-1 acts as the keystone of oestrogen receptor [ER]beta-mediated transcriptional repression of ERalpha. J. Steroid Biochem. Mol. Biol. 2007, 104, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Paech, K.; Webb, P.; Kuiper, G.G.; et al. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997, 277, 1508–1510. [Google Scholar] [CrossRef]
- Cho, N.L.; Javid, S.H.; Carothers, A.M.; Redston, M.; Bertagnolli, M.M. Estrogen receptors alpha and beta are inhibitory modifiers of Apc-dependent tumorigenesis in the proximal colon of min/+ mice. Cancer Res. 2007, 67, 2366–2372. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, A.G.; Oikarinen, S.I.; Bynoté, K.K.; Marttinen, M.; Rafter, J.J.; Gustafsson, J.A.; Roy, S.K.; Pitot, H.C.; Korach, K.S.; Lubahn, D.B.; Mutanen, M.; Gould, K.A. Disruption of estrogen receptor signaling enhances intestinal neoplasia in Apc[Min/+] mice. Carcinogenesis. 2009, 30, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Topi, G.; Ghatak, S.; Satapathy, S.R.; Ehrnström, R.; Lydrup, M.L.; Sjölander, A. Combined Estrogen Alpha and Beta Receptor Expression Has a Prognostic Significance for Colorectal Cancer Patients. Front Med [Lausanne]. 2022, 9, 739620. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.L.; Javid, S.H.; Carothers, A.M.; Redston, M.; Bertagnolli, M.M. Estrogen receptors alpha and beta are inhibitory modifiers of Apc-dependent tumorigenesis in the proximal colon of min/+ mice. Cancer Res. 2007, 67, 2366–2372. [Google Scholar] [CrossRef]
- Xie, L.Q.; Yu, J.P.; Luo, H.S. Expression of estrogen receptor beta in human colorectal cancer. World J. Gastroenterol. 2004, 10, 214–217. [Google Scholar] [CrossRef]
- Williams, C.; DiLeo, A.; Niv, Y.; Gustafsson, J.Å. Estrogen receptor beta as target for colorectal cancer prevention. Cancer Lett. 2016, 372, 48–56. [Google Scholar] [CrossRef]
- Stevanato Filho, P.R.; Aguiar Júnior, S.; Begnami, M.D.; Ferreira, F.O.; Nakagawa, W.T.; Spencer, R.M.S.B.; et al. Estrogen receptor β as a prognostic marker of tumor progression in colorectal cancer with familial adenomatous polyposis and sporadic polyps. Pathol. Oncol. Res. 2018, 24, 533–540. [Google Scholar] [CrossRef]
- Herichova, I.; Reis, R.; Hasakova, K.; Vician, M.; Zeman, M. Sex-dependent regulation of estrogen receptor beta in human colorectal cancer tissue and its relationship with clock genes and VEGF-A expression. Physiol. Res. 2019, 68 (Suppl. 3), :S297–S305. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R. Levin, Bidirectional Signaling between the Estrogen Receptor and the Epidermal Growth Factor Receptor. Molecular Endocrinology 2003, 17, 309–317. [Google Scholar] [CrossRef]
- Ahcene Djaballah, S.; Daniel, F.; Milani, A.; Ricagno, G.; Lonardi, S. HER2 in Colorectal Cancer: The Long and Winding Road From Negative Predictive Factor to Positive Actionable Target. Am. Soc. Clin. Oncol. Educ. Book. 2022, 42, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Arnal, J.F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; Katzenellenbogen, J. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef] [PubMed]
- Norfleet, A.M.; Thomas, M.L.; Gametchu, B.; Watson, C.S. Estrogen receptor-alpha detected on the plasma membrane of aldehyde-fixed GH3/B6/F10 rat pituitary tumor cells by enzyme-linked immunocytochemistry. Endocrinology. 1999, 140, 3805–3814. [Google Scholar] [CrossRef] [PubMed]
- Pedram, A.; Razandi, M.; Levin, E.R. Nature of functional estrogen receptors at the plasma membrane. Mol. Endocrinol. 2006, 20, 1996–2009. [Google Scholar] [CrossRef]
- Levin, E.R. Plasma membrane estrogen receptors. Trends Endocrinol. Metab. 2009, 20, 477–482. [Google Scholar] [CrossRef]
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-dependent estrogen receptor alpha membrane localization: regulation by 17beta-estradiol. Mol Biol Cell. 2005, 16, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Ascenzi, P. Steroid hormone rapid signaling: the pivotal role of S-palmitoylation. IUBMB Life. 2006, 58, 716–719. [Google Scholar] [CrossRef]
- Evinger, A.J., 3rd; Levin, E.R. Requirements for estrogen receptor alpha membrane localization and function. Steroids. 2005, 70, 361–363. [Google Scholar] [CrossRef]
- Condliffe, S.B.; Doolan, C.M.; Harvey, B.J. 17beta-oestradiol acutely regulates Cl- secretion in rat distal colonic epithelium. J. Physiol. 2001, 530 Pt. 1, 47–54. [Google Scholar] [CrossRef]
- McNamara, B.; Winter, D.C.; Cuffe, J.; Taylor, C.; O'Sullivan, G.C.; Harvey, B.J. Rapid activation of basolateral potassium transport in human colon by oestradiol. Br. J. Pharmacol. 2000, 131, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Doolan, C.M.; Harvey, B.J. A Galphas protein-coupled membrane receptor, distinct from the classical oestrogen receptor, transduces rapid effects of oestradiol on [Ca2+]i in female rat distal colon. Mol. Cell Endocrinol. 2003, 199, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Arnal, J.F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; Katzenellenbogen, J. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Horkeby, K.; Henning, P.; Wu, J.; Lawenius, L.; Engdahl, C.; Gupta, P.; Movérare-Skrtic, S.; Nilsson, K.H.; Levin, E.; Ohlsson, C.; Lagerquist, M.K. Membrane estrogen receptor α signaling modulates the sensitivity to estradiol treatment in a dose- and tissue- dependent manner. Sci. Rep. 2023, 13, 9046. [Google Scholar] [CrossRef] [PubMed]
- Stefkovich, M.L.; Arao, Y.; Hamilton, K.J.; Korach, K.S. Experimental models for evaluating non-genomic estrogen signaling. Steroids. 2018, 133, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Pappas, T.C.; Gametchu, B.; Watson, C.S. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. FASEB J. 1995, 9, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Harrington, W.R.; Kim, S.H.; Funk, C.C.; Madak-Erdogan, Z.; Schiff, R.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol. Endocrinol. 2006, 20, 491–502. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Kim, J.K.; O'Mahony, F.; Lee, E.Y.; Luderer, U.; Levin, E.R. Developmental phenotype of a membrane only estrogen receptor alpha [MOER] mouse. J. Biol. Chem. 2009, 284, 3488–3495. [Google Scholar] [CrossRef]
- Adlanmerini, M.; Solinhac, R.; Abot, A.; Fabre, A.; Raymond-Letron, I.; Guihot, A.L.; Boudou, F.; Sautier, L.; Vessières, E.; Kim, S.H.; Lière, P.; Fontaine, C.; Krust, A.; Chambon, P.; Katzenellenbogen, J.A.; Gourdy, P.; Shaul, P.W.; Henrion, D.; Arnal, J.F.; Lenfant, F. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc. Natl. Acad. Sci. U S A. 2014, 111, E283–E290. [Google Scholar] [CrossRef]
- Abot, A.; Fontaine, C.; Buscato, M.; Solinhac, R.; Flouriot, G.; Fabre, A.; Drougard, A.; Rajan, S.; Laine, M.; Milon, A.; Muller, I.; Henrion, D.; Adlanmerini, M.; Valéra, M.C.; Gompel, A.; Gerard, C.; Péqueux, C.; Mestdagt, M.; Raymond-Letron, I.; Knauf, C.; Ferriere, F.; Valet, P.; Gourdy, P.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; Lenfant, F.; Greene, G.L.; Foidart, J.M.; Arnal, J.F. The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor α modulation, uncoupling nuclear and membrane activation. EMBO Mol. Med. 2014, 6, 1328–1346. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Rapid signaling by steroid receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1425–R1430. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Fiocchetti, M.; Busonero, C.; Fernandez, V.S.; Montalesi, E.; Cipolletti, M.; Pallottini, V.; Marino, M. The extra-nuclear interactome of the estrogen receptors: implications for physiological functions. Mol. Cell Endocrinol. 2021, 538, 111452. [Google Scholar] [CrossRef] [PubMed]
- Koveitypour, Z.; Panahi, F.; Vakilian, M.; et al. Signaling pathways involved in colorectal cancer progression. Cell Biosci. 2019, 9, 97. [Google Scholar] [CrossRef]
- Doolan, C.M.; Harvey, B.J. Modulation of cytosolic protein kinase C and calcium ion activity by steroid hormones in rat distal colon. J. Biol. Chem. 1996, 271, 8763–8767. [Google Scholar] [CrossRef] [PubMed]
- Winter, D.C.; Taylor, C.; CO'Sullivan, G.; Harvey, B.J. Mitogenic effects of oestrogen mediated by a non-genomic receptor in human colon. Br. J. Surg. 2000, 87, 1684–1689. [Google Scholar] [CrossRef]
- Doolan, C.M.; Condliffe, S.B.; Harvey, B.J. Rapid non-genomic activation of cytosolic cyclic AMP-dependent protein kinase activity and [Ca[2+]][i] by 17beta-oestradiol in female rat distal colon. Br. J. Pharmacol. 2000, 129, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Salazar, J.E.; Posadas-Rodríguez, P.; Lazzarini-Lechuga, R.C.; et al. Membrane-Initiated Estradiol Signaling of Epithelial-Mesenchymal Transition-Associated Mechanisms Through Regulation of Tight Junctions in Human Breast Cancer Cells. HORM CANC 2014, 5, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Sabouni, E.; Nejad, M.M.; Mojtabavi, S.; Khoshduz, S.; Mojtabavi, M.; Nadafzadeh, N.; Nikpanjeh, N.; Mirzaei, S.; Hashemi, M.; Aref, A.R.; Khorrami, R.; Nabavi, N.; Ertas, Y.N.; Salimimoghadam, S.; Zandieh, M.A.; Rahmanian, P.; Taheriazam, A.; Hushmandi, K. Unraveling the function of epithelial-mesenchymal transition [EMT] in colorectal cancer: Metastasis, therapy response, and revisiting molecular pathways. Biomed. Pharmacother. 2023, 160, 114395. [Google Scholar] [CrossRef]
- Sabouni, E.; Nejad, M.M.; Mojtabavi, S.; Khoshduz, S.; Mojtabavi, M.; Nadafzadeh, N.; Nikpanjeh, N.; Mirzaei, S.; Hashemi, M.; Aref, A.R.; Khorrami, R.; Nabavi, N.; Ertas, Y.N.; Salimimoghadam, S.; Zandieh, M.A.; Rahmanian, P.; Taheriazam, A.; Hushmandi, K. Unraveling the function of epithelial-mesenchymal transition [EMT] in colorectal cancer: Metastasis, therapy response, and revisiting molecular pathways. Biomed. Pharmacother. 2023, 160, 114395. [Google Scholar] [CrossRef]
- Marino, M.; Ascenzi, P. Membrane association of estrogen receptor alpha and beta influences 17beta-estradiol-mediated cancer cell proliferation. Steroids. 2008, 73, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Galluzzo, P.; Caiazza, F.; Moreno, S.; Marino, M. Role of ERbeta palmitoylation in the inhibition of human colon cancer cell proliferation. Endocr. Relat. Cancer. 2007, 14, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Razandi, M.; Pedram, A.; Greene, G.L.; Levin, E.R. Cell membrane and nuclear estrogen receptors [ERs] originate from a single transcript: studies of ERα and ERβ expressed in Chinese hamster ovary cells. Mol. Endocrinol. 1999, 13, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Galluzzo, P.; Leone, S.; Acconcia, F.; Ascenzi, P. Nitric oxide impairs the 17β-estradiol-induced apoptosis in human colon adenocarcinoma cells. Endocr. Relat. Cancer. 2006, 13, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Totta, P.; Ogawa, S.; Cardillo, I.; Inoue, S.; Leone, S.; Trentalance, A.; Muramatsu, M.; Marino, M. Survival versus apoptotic 17beta-estradiol effect: Role of ER alpha and ER beta activated non-genomic signaling. J. Cell. Physiol. 2005, 203, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Cui, H. Estrogen Receptor Alpha Splice Variants, Post-Translational Modifications, and Their Physiological Functions. Cells. 2023, 12, 895. [Google Scholar] [CrossRef] [PubMed]
- Langdon, S.P. Estrogen Receptor Signaling in Cancer. Cancers [Basel]. 2020, 12, 2744. [Google Scholar] [CrossRef] [PubMed]
- Resnick, E.M.; Schreihofer, D.A.; Periasamy, A.; Shupnik, M.A. Truncated estrogen receptor product-1 suppresses estrogen receptor transactivation by dimerization with estrogen receptors alpha and beta. J. Biol. Chem. 2000, 275, 7158–7166. [Google Scholar] [CrossRef]
- Notas, G.; Panagiotopoulos, A.; Vamvoukaki, R.; Kalyvianaki, K.; Kiagiadaki, F.; Deli, A.; Kampa, M.; Castanas, E. ERα36-GPER1 Collaboration Inhibits TLR4/NFκB-Induced Pro-Inflammatory Activity in Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 7603. [Google Scholar] [CrossRef]
- Pagano, M.T.; Ortona, E.; Dupuis, M.L. A Role for Estrogen Receptor alpha36 in Cancer Progression. Front Endocrinol [Lausanne]. 2020, 11, 506. [Google Scholar] [CrossRef]
- Deng, H.; Huang, X.; Fan, J.; Wang, L.; Xia, Q.; Yang, X.; et al. . A variant of estrogen receptor-alpha, ER-alpha36 is expressed in human gastric cancer and is highly correlated with lymph node metastasis. Oncol Rep. 24:171–6. 10. 3892. [Google Scholar]
- Thomas, C.; Gustafsson, J.Å. Estrogen receptor mutations and functional consequences for breast cancer. Trends Endocrinol. Metab. 2015, 26, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Teng, R.; Wang, Q.; Zhang, X.; Wang, H.; Wang, Z.; Cao, J.; Teng, L. Transcriptional analysis of estrogen receptor alpha variant mRNAs in colorectal cancers and their matched normal colorectal tissues. J. Steroid Biochem. Mol. Biol. 2008, 112, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; et al. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Edward, J. Filardo, Peter Thomas, Minireview: G Protein-Coupled Estrogen Receptor-1, GPER-1: Its Mechanism of Action and Role in Female Reproductive Cancer, Renal and Vascular Physiology, Endocrinology 2012, 153, 2953–2962. [CrossRef]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology.
- Qiu, Y.A.; Xiong, J.; Yu, T. Role of G Protein-Coupled Estrogen Receptor in Digestive System Carcinomas: A Minireview. Onco Targets Ther. 2021, 14, 2611–2622. [Google Scholar] [CrossRef] [PubMed]
- Jacenik, D.; Beswick, E.J.; Krajewska, W.M.; Prossnitz, E.R. G protein-coupled estrogen receptor in colon function, immune regulation and carcinogenesis. World J. Gastroenterol. 2019, 25, 4092–4104. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Barton, M. The G protein-coupled oestrogen receptor GPER in health and disease: an update. Nat. Rev. Endocrinol. 2023, 19, 407–424. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. G protein-coupled receptor 30: estrogen receptor or collaborator? Endocrinology. 2009, 150, 1563–1565. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Liu, D. Does GPER Really Function as a G Protein-Coupled Estrogen Receptor in vivo? Front Endocrinol [Lausanne]. 2020, 11, 148. [Google Scholar] [CrossRef]
- Jacenik, D.; Krajewska, W.M. Significance of G Protein-Coupled Estrogen Receptor in the Pathophysiology of Irritable Bowel Syndrome, Inflammatory Bowel Diseases and Colorectal Cancer. Front Endocrinol [Lausanne] 2020, 11, 390. [Google Scholar] [CrossRef]
- Girgert, R.; Emons, G.; Gründker, C. Estrogen Signaling in ERα-Negative Breast Cancer: ERβ and GPER. Front Endocrinol [Lausanne]. 2019, 9, 781. [Google Scholar] [CrossRef]
- Bustos, V.; Nolan, Á.M.; Nijhuis, A.; Harvey, H.; Parker, A.; Poulsom, R.; et al. . GPER mediates differential effects of estrogen on colon cancer cell proliferation and migration under normoxic and hypoxic conditions. Oncotarget. 2017, 8, 84258–84275. [Google Scholar] [CrossRef] [PubMed]
- Jacenik, D.; Cygankiewicz, A.I.; Mokrowiecka, A.; Małecka-Panas, E.; Fichna, J.; Krajewska, W.M. Sex- and Age-Related Estrogen Signaling Alteration in Inflammatory Bowel Diseases: Modulatory Role of Estrogen Receptors. Int. J. Mol. Sci. 2019, 20, 3175. [Google Scholar] [CrossRef] [PubMed]
- van der Giessen, J.; van der Woude, C.J.; Peppelenbosch, M.P.; Fuhler, G.M. A Direct Effect of Sex Hormones on Epithelial Barrier Function in Inflammatory Bowel Disease Models. Cells 2019, 8, 261. [Google Scholar] [CrossRef] [PubMed]
- Rouhimoghadam, M.; Lu, A.S.; Salem, A.K.; Filardo, E.J. Therapeutic Perspectives on the Modulation of G-Protein Coupled Estrogen Receptor, GPER, Function. Front Endocrinol [Lausanne]. 2020, 11, 591217. [Google Scholar] [CrossRef] [PubMed]
- Skrzypczak, M.; Goryca, K.; Rubel, T.; Paziewska, A.; Mikula, M.; Jarosz, D. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability [published correction appears in PLoS One. 2010, 5. Ostrowsk, Jerzy [corrected to Ostrowski, Jerzy]]. PloS One Erratum: https://doi.org/10.1371/annotation/8c585739-a354-4fc9-a7d0-d5ae26fa06ca. 2010, 5, e13091. [Google Scholar] [CrossRef]
- Jung, J. Role of G Protein-Coupled Estrogen Receptor in Cancer Progression. Toxicological Res. 2019, 35, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Pepermans, R.A.; Sharma, G.; Prossnitz, E.R. G Protein-Coupled Estrogen Receptor in Cancer and Stromal Cells: Functions and Novel Therapeutic Perspectives. Cells 2021, 10, 672. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, Z.; Jiang, G.; Zhou, Y.; Yang, X.; Huang, H.; et al. Epigenetic down regulation of G protein-coupled estrogen receptor [GPER] functions as a tumor suppressor in colorectal cancer. Mol. Cancer 2017, 16, 87. [Google Scholar] [CrossRef]
- Gilligan, L.C.; Rahman, H.P.; Hewitt, A.M.; Sitch, A.J.; Gondal, A.; Arvaniti, A.; Taylor, A.E.; Read, M.L.; Morton, D.G.; Foster, P.A. Estrogen Activation by Steroid Sulfatase Increases Colorectal Cancer Proliferation via GPER. J. Clin. Endocrinol. Metab. 2017, 102, 4435–4447. [Google Scholar] [CrossRef]
- Lo, E.K.K.; Lee, J.C.Y.; Turner, P.C.; et al. Low dose of zearalenone elevated colon cancer cell growth through G protein-coupled estrogenic receptor. Sci. Rep. 2021, 11, 7403. [Google Scholar] [CrossRef] [PubMed]
- Bühler, M.; Fahrländer, J.; Sauter, A.; Becker, M.; Wistorf, E.; Steinfath, M.; Stolz, A. GPER1 links estrogens to centrosome amplification and chromosomal instability in human colon cells. Life Sci. Alliance. 2022, 6, e202201499. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Sims, A.H.; Maggiolini, M.; Sotgia, F.; Lisanti, M.P.; Clarke, R.B. GPER mediates the angiocrine actions induced by IGF1 through the HIF-1α/VEGF pathway in the breast tumor microenvironment. Breast Cancer Res. 2017, 19, 129. [Google Scholar] [CrossRef]
- Pepermans, R.A.; Sharma, G.; Prossnitz, E.R. G Protein-Coupled Estrogen Receptor in Cancer and Stromal Cells: Functions and Novel Therapeutic Perspectives. Cells 2021, 10, 672. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer. 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Hou, M.; Guan, Y.; Jiang, M.; Yang, Y.; Gou, H. Expression of HIF-1alpha and VEGF in colorectal cancer: association with clinical outcomes and prognostic implications. BMC Cancer. 2009, 9, 432. [Google Scholar] [CrossRef] [PubMed]
- Beggs, A.; Domingo, E.; McGregor, M.; Presz, M.; Johnstone, E.; Midgley, R.; Kerr, D.; Oukrif, D.; Novelli, M.; Abulafi, M.; Hodgson, S.V.; Fadhil, W.; Ilyas, M.; Tomlinson, I.P. Loss of expression of the double strand break repair protein ATM is associated with worse prognosis in colorectal cancer and loss of Ku70 expression is associated with CIN. Oncotarget. 2012, 3, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- El-Khatib, M.; Geara, F.; Haddadin, M.; Gali-Muhtasib, H. Cell death by the quinoxaline dioxide DCQ in human colon cancer cells is enhanced under hypoxia and is independent of p53 and p21. Radiat Oncol. 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Zafari, N.; Khosravi, F.; Rezaee, Z.; Esfandyari, S.; Bahiraei, M.; Bahramy, A.; Ferns, G.A.; Avan, A. The role of the tumor microenvironment in colorectal cancer and the potential therapeutic approaches. J. Clin. Lab. Anal. 2022, 36, e24585. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell. 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Schatoff, E.M.; Leach, B.I.; Dow, L.E. Wnt Signaling and Colorectal Cancer. Curr. Colorectal. Cancer Rep. 2017, 13, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt signaling in colorectal cancer: pathogenic role and therapeutic target. Mol. Cancer. 2022, 21, 144. [Google Scholar] [CrossRef]
- Song, H.; Sontz, R.A.; Vance, M.J.; Morris, J.M.; Sheriff, S.; Zhu, S.; Duan, S.; Zeng, J.; Koeppe, E.; Pandey, R.; Thorne, C.A.; Stoffel, E.M.; Merchant, J.L. High-fat diet plus HNF1A variant promotes polyps by activating β-catenin in early-onset colorectal cancer. JCI Insight. 2023, 8, e167163. [Google Scholar] [CrossRef]
- Kouzmenko, A.P.; Takeyama, K.; Ito, S.; Furutani, T.; Sawatsubashi, S.; Maki, A.; Suzuki, E.; Kawasaki, Y.; Akiyama, T.; Tabata, T.; Kato, S. Wnt/beta-catenin and estrogen signaling converge in vivo. J. Biol. Chem. 2004, 279, 40255–40258. [Google Scholar] [CrossRef]
- Xiong, W.; Zhang, L.; Yu, L.; Xie, W.; Man, Y.; Xiong, Y.; Liu, H.; Liu, Y. Estradiol promotes cells invasion by activating β-catenin signaling pathway in endometriosis. Reproduction. 2015, 150, 507–516. [Google Scholar] [CrossRef]
- Hou X; et al. , Canonical Wnt Signaling Is Critical to Estrogen-Mediated Uterine Growth. Mol. Endocrinol. 2004, 18, 3035–3049. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Pasini, E.; Pastrello, C.; Angeli, M.; Baciu, C.; Abovsky, M.; Coffee, A.; Adeyi, O.; Kotlyar, M.; Jurisica, I. Estrogen Receptor 1 Inhibition of Wnt/β-Catenin Signaling Contributes to Sex Differences in Hepatocarcinogenesis. Front. Oncol. 2021, 11, 777834. [Google Scholar] [CrossRef]
- Abbott, G.W. Biology of the KCNQ1 Potassium Channel New. J. Sci. 2014, 26. [Google Scholar] [CrossRef]
- Schroeder, B.C.; Waldegger, S.; Fehr, S.; Bleich, M.; Warth, R.; Greger, R.; Jentsch, T.J. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 2000, 403, 196–199. [Google Scholar] [CrossRef]
- Than, B.L.; Goos, J.A.; Sarver, A.L.; O'Sullivan, M.G.; Rod, A.; Starr, T.K.; Fijneman, R.J.; Meijer, G.A.; Zhao, L.; Zhang, Y.; Largaespada, D.A.; Scott, P.M.; Cormier, R.T. The role of KCNQ1 in mouse and human gastrointestinal cancers. Oncogene 2014, 33, 3861–3868. [Google Scholar] [CrossRef] [PubMed]
- Rapetti-Mauss, R.; Bustos, V.; Thomas, W.; McBryan, J.; Harvey, H.; Lajczak, N.; Madden, S.F.; Pellissier, B.; Borgese, F.; Soriani, O.; Harvey, B.J. Bidirectional KCNQ1:β-catenin interaction drives colorectal cancer cell differentiation. Proc. Natl. Acad. Sci. USA 2017, 114, 4159–4164. [Google Scholar] [CrossRef] [PubMed]
- Shorthouse, D.; Zhuang, L.; Rahrmann, E.P.; Kosmidou, C.; Wickham Rahrmann, K.; Hall, M.; Greenwood, B.; Devonshire, G.; Gilbertson, R.J.; Fitzgerald, R.C.; Hall, B.A. KCNQ potassium channels modulate Wnt activity in gastro-oesophageal adenocarcinomas. Life Sci. Alliance. 2023, 6, e202302124. [Google Scholar] [CrossRef] [PubMed]
- Alzamora, R.; O'Mahony, F.; Bustos, V.; Rapetti-Mauss, R.; Urbach, V.; Cid, L.P.; Sepúlveda, F.V.; Harvey, B.J. Sexual dimorphism and oestrogen regulation of KCNE3 expression modulates the functional properties of KCNQ1 K⁺ channels. J. Physiol. 2011, 589 Pt. 21, 5091–5107. [Google Scholar] [CrossRef]
- Rapetti-Mauss, R.; O'Mahony, F.; Sepulveda, F.V.; Urbach, V.; Harvey, B.J. Oestrogen promotes KCNQ1 potassium channel endocytosis and post-endocytic trafficking in colonic epithelium. J. Physiol. 2013, 591, 2813–2831. [Google Scholar] [CrossRef] [PubMed]
- Rapetti-Mauss, R.; Borgese, F.; Harvey, B.J.; Soriani, O. KCNQ1: a new regulator of the epithelio-mesenchymal transition in colorectal cancers. Med Sci [Paris]. 2018, 34, 21–24. [Google Scholar] [CrossRef]
- Rapetti-Mauss, R.; Berenguier, C.; Allegrini, B.; Soriani, O. Interplay Between Ion Channels and the Wnt/β-Catenin Signaling Pathway in Cancers. Front. Pharmacol. 2020, 11, 525020. [Google Scholar] [CrossRef]
- Hu, T.; Li, C. Convergence between Wnt-β-catenin and EGFR signaling in cancer. Mol. Cancer. 2010, 9, 236. [Google Scholar] [CrossRef]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef]
- Abancens, M.; Harvey, B.J.; McBryan, J. GPER Agonist G1 Prevents Wnt-Induced JUN Upregulation in HT29 Colorectal Cancer Cells. Int. J. Mol. Sci. 2022, 23, 12581. [Google Scholar] [CrossRef]
- Hasáková, K.; Bezakova, J.; Vician, M.; Reis, R.; Zeman, M.; Herichova, I. Gender-dependent expression of leading and passenger strand of miR-21 and miR-16 in human colorectal cancer and adjacent colonic tissues. Physiol. Res. 2017, 66 (Suppl. 4), S575–S82. [Google Scholar] [CrossRef] [PubMed]
- Hamfjord, J.; Stangeland, A.M.; Hughes, T.; Skrede, M.L.; Tveit, K.M.; Ikdahl, T.; Kure, E.H. Differential expression of miRNAs in colorectal cancer: comparison of paired tumor tissue and adjacent normal mucosa using high-throughput sequencing. PLoS One. 2012, 7, e34150. [Google Scholar] [CrossRef] [PubMed]
- Massaro, C.; Safadeh, E.; Sgueglia, G.; Stunnenberg, H.G.; Altucci, L.; Dell'Aversana, C. MicroRNA-Assisted Hormone Cell Signaling in Colorectal Cancer Resistance. Cells. 2020, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Herichova, I.; Reis, R.; Hasakova, K.; Vician, M. Downregulation of miR-30c-5p expression in colorectal cancer tissue is sex-dependent. Physiol Res. 2020, 69 (Suppl. 3), S479–S487. [Google Scholar] [CrossRef] [PubMed]
- Mehrgou, A.; Ebadollahi, S.; Seidi, K.; Ayoubi-Joshaghani, M.H.; Ahmadieh Yazdi, A.; Zare, P.; Jaymand, M.; Jahanban-Esfahlan, R. Roles of miRNAs in Colorectal Cancer: Therapeutic Implications and Clinical Opportunities. Adv. Pharm. Bull. 2021, 11, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Unno, T.; Kim, B.Y.; Park, M.S. Sex Differences in Gut Microbiota. World J. Mens. Health. 2020, 38, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Markle, J.G.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; von Bergen, M.; McCoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. 2013, 339, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen-gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef]
- Song, C.H.; Kim, N.; Nam, R.H.; Choi, S.I.; Lee, H.N.; Surh, Y.J. 17β-Estradiol supplementation changes gut microbiota diversity in intact and colorectal cancer-induced ICR male mice. Sci. Rep. 2020, 10, 12283. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef]
- 221. Reference 221.
- Sánchez-Alcoholado, L.; Ramos-Molina, B.; Otero, A.; Laborda-Illanes, A.; Ordóñez, R.; Medina, J.A.; Gómez-Millán, J.; Queipo-Ortuño, M.I. The Role of the Gut Microbiome in Colorectal Cancer Development and Therapy Response. Cancers 2020, 12, 1406. [Google Scholar] [CrossRef] [PubMed]
- Roshan, M.H.; Tambo, A.; Pace, N.P. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016, 7, 22. [Google Scholar] [CrossRef]
- Li, J.; Lan, Z.; Liao, W.; et al. Histone demethylase KDM5D upregulation drives sex differences in colon cancer. Nature 2023, 619, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. How the Y chromosome makes some cancers more deadly for men. Nature. 2023, 618, 898. [Google Scholar] [CrossRef] [PubMed]
- Mahbub, A.A. Therapeutic Strategies and Potential Actions of Female Sex Steroid Hormones and Their Receptors in Colon Cancer Based on Preclinical Studies. Life [Basel]. 2022, 12, 605. [Google Scholar] [CrossRef] [PubMed]
- Andrei, P.; Battuello, P.; Grasso, G.; Rovera, E.; Tesio, N.; Bardelli, A. Integrated approaches for precision oncology in colorectal cancer: The more you know, the better. Semin. Cancer Biol. 2022, 84, 199–213. [Google Scholar] [CrossRef]
- Kim, H.I.; Lim, H.; Moon, A. Sex Differences in Cancer: Epidemiology, Genetics and Therapy. Biomol Ther [Seoul]. 2018, 26, 335–342. [Google Scholar] [CrossRef]
Figure 1.
Age-standardized incidence and mortality rates for all cancers per 100,000 EU, USA and Canada. Survey of most recent data from the Global Cancer Observatory Globocan 2020, https://gco.iarc.fr/overtime/en/dataviz/bars?sexes=1_2&sort_by=value2&mode=cancer ,accessed on 29 July 2023].
Figure 1.
Age-standardized incidence and mortality rates for all cancers per 100,000 EU, USA and Canada. Survey of most recent data from the Global Cancer Observatory Globocan 2020, https://gco.iarc.fr/overtime/en/dataviz/bars?sexes=1_2&sort_by=value2&mode=cancer ,accessed on 29 July 2023].

Figure 2.
Age-standardized incidence and mortality rates for colon cancer per 100,000 across WHO countries. Survey of most recent data from the Global Cancer Observatory, Colon Cancer 1989-2012, accessed on 29 July 2023]. https://gco.iarc.fr/overtime/en/dataviz/bars?sexes=1_2&sort_by=value2&cancers=5&types=1.
Figure 2.
Age-standardized incidence and mortality rates for colon cancer per 100,000 across WHO countries. Survey of most recent data from the Global Cancer Observatory, Colon Cancer 1989-2012, accessed on 29 July 2023]. https://gco.iarc.fr/overtime/en/dataviz/bars?sexes=1_2&sort_by=value2&cancers=5&types=1.
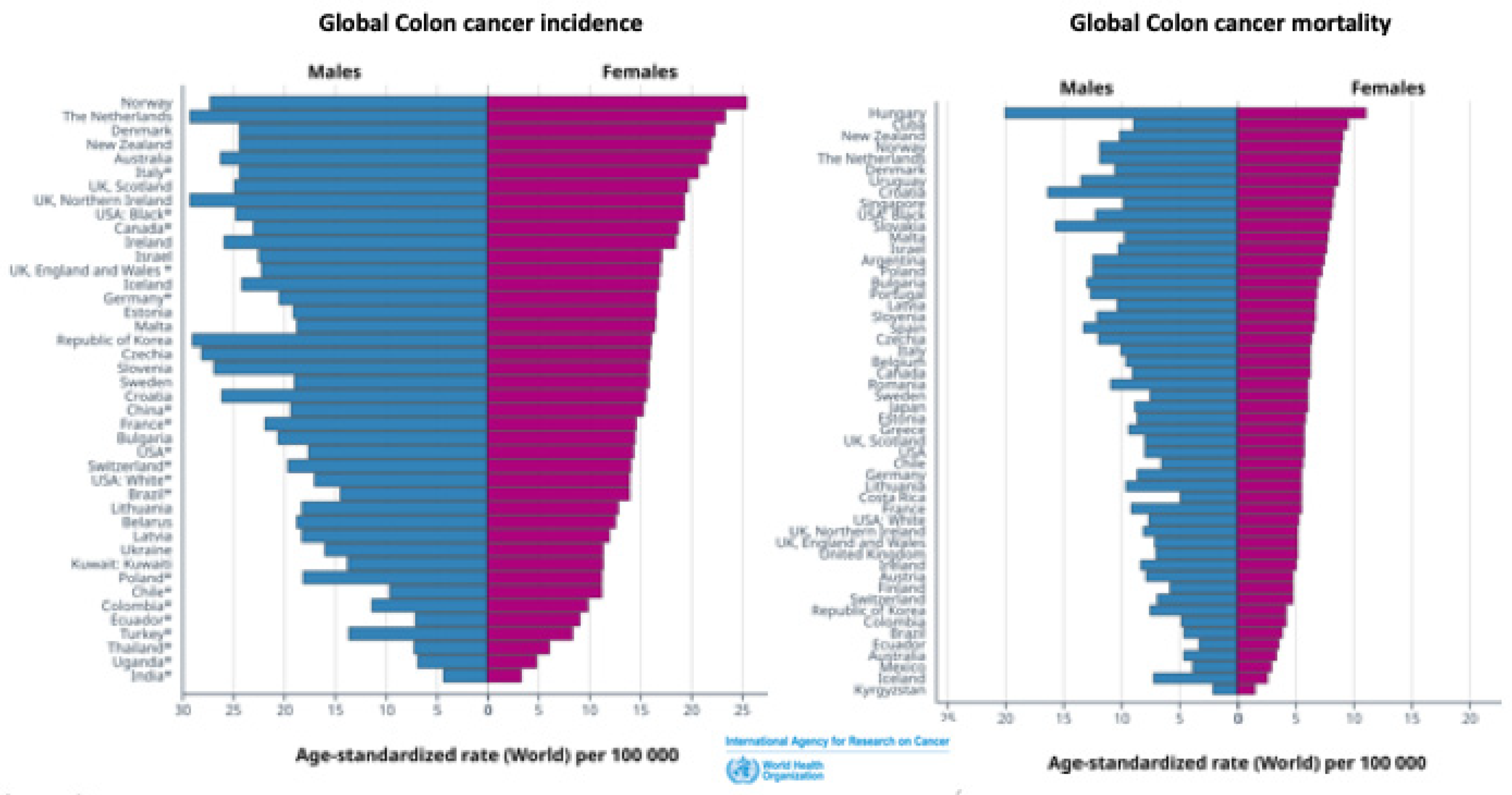
Figure 3.
Genomic and non-genomic estrogen (E2) signaling pathways in colon cancer. In the genomic pathway, estrogen interacts with estrogen receptors in the cytosol (cERα and cERβ) which translocate to the nucleus to activate (nERα) or repress (nERβ) cell proliferative genes. ERβ can also inhibit ERα nuclear transcription. Non-genomic to genomic cross-talk can occur when ERα transactivates EGFR to initiate cell proliferative signalling via MAPK, PI3K/Akt. Membrane estrogen receptors mERα and GPER signal through non-genomic pathways to activate protein kinases, calcium mobilization or intracellular pH, which in turn modulate ion channel activity or activate transcription factors such as CREB or HIF1-α and VEGF which can in turn cross-talk to genomic signaling pathways to increase the synthesis of protein kinases or ion channels. GPER activation can have dual effects on CRC cell proliferation depending on oxygen levels in the tumour microenvironment. In normoxia, E2-GPER inhibits HIF1-α, VEGF and c-Jun transcription of proliferative genes whereas in hypoxia E2-GPER activates these HIF1-α proliferative pathways. In addition Wnt/β-catenin proliferative pathways can be inhibited by the estrogen regulated ion channel KCNQ1 to trap β-catenin at adherens junctions and impede nuclear translocation of β-catenin. Red and green squares encompass signaling pathways which exacerbate CRC or protect against CRC, respectively. .
Figure 3.
Genomic and non-genomic estrogen (E2) signaling pathways in colon cancer. In the genomic pathway, estrogen interacts with estrogen receptors in the cytosol (cERα and cERβ) which translocate to the nucleus to activate (nERα) or repress (nERβ) cell proliferative genes. ERβ can also inhibit ERα nuclear transcription. Non-genomic to genomic cross-talk can occur when ERα transactivates EGFR to initiate cell proliferative signalling via MAPK, PI3K/Akt. Membrane estrogen receptors mERα and GPER signal through non-genomic pathways to activate protein kinases, calcium mobilization or intracellular pH, which in turn modulate ion channel activity or activate transcription factors such as CREB or HIF1-α and VEGF which can in turn cross-talk to genomic signaling pathways to increase the synthesis of protein kinases or ion channels. GPER activation can have dual effects on CRC cell proliferation depending on oxygen levels in the tumour microenvironment. In normoxia, E2-GPER inhibits HIF1-α, VEGF and c-Jun transcription of proliferative genes whereas in hypoxia E2-GPER activates these HIF1-α proliferative pathways. In addition Wnt/β-catenin proliferative pathways can be inhibited by the estrogen regulated ion channel KCNQ1 to trap β-catenin at adherens junctions and impede nuclear translocation of β-catenin. Red and green squares encompass signaling pathways which exacerbate CRC or protect against CRC, respectively. .
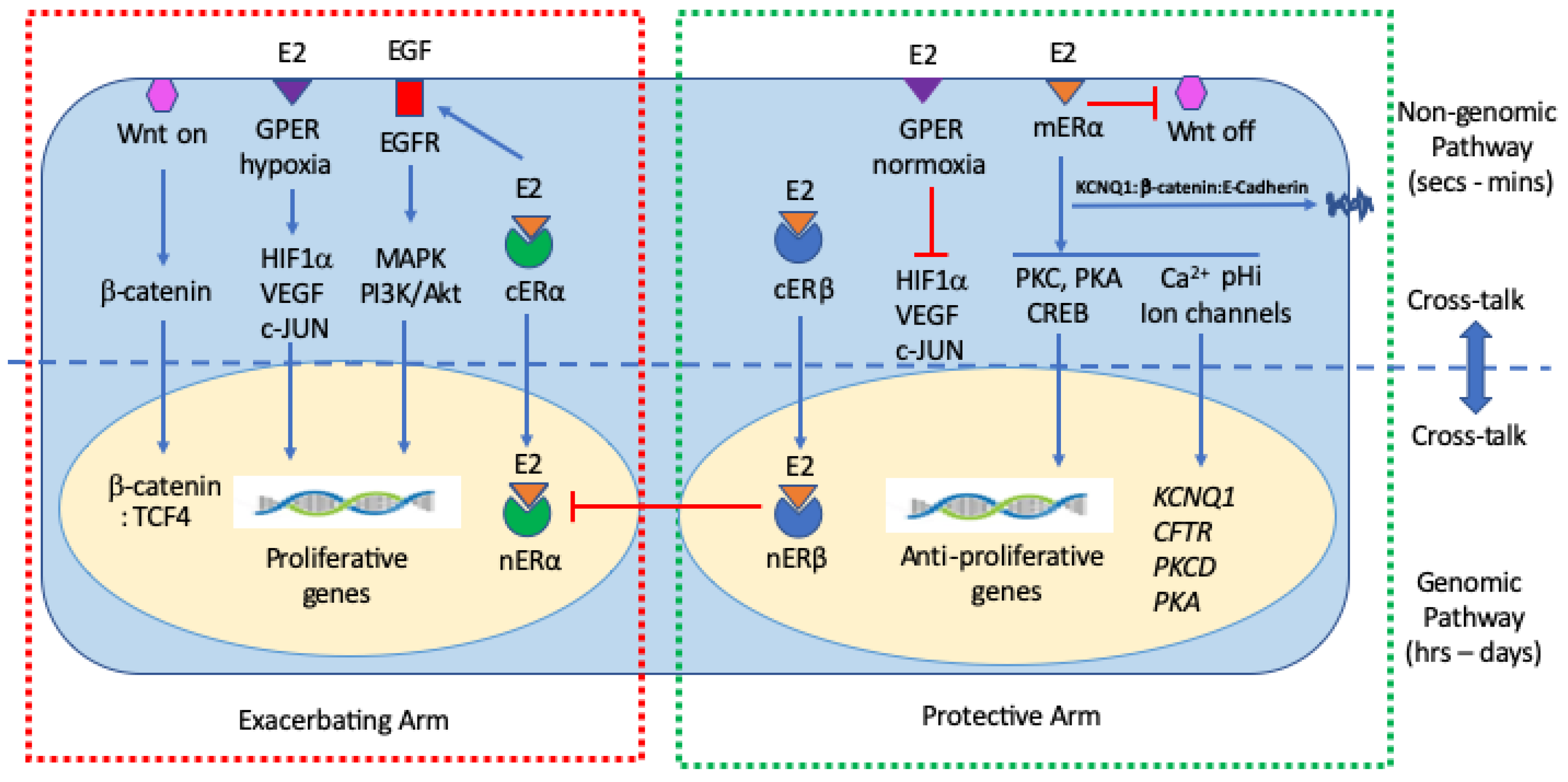
Figure 4.
KCNQ1 anchors β-catenin at the plasma membrane, stabilizing adherens junctions and promoting cell-cell adhesion to maintain a highly differentiated, tight junction epithelial phenotype in healthy colon. Estrogen uncouples KCNQ1 from KCNE3 allowing the channel protein to enter an endocytotic recycling pathway and capture activated β-catenin in the cytosol, returning it to the cell membrane in association with E-cadherin. The trapping of β-catenin at the plasma membrane prevents its nuclear translocation and suppresses the Wnt pathway activation of cell proliferation genes as well as inhibiting Epithelial Mesenchymal Transition. Adapted from References [202,204,205].
Figure 4.
KCNQ1 anchors β-catenin at the plasma membrane, stabilizing adherens junctions and promoting cell-cell adhesion to maintain a highly differentiated, tight junction epithelial phenotype in healthy colon. Estrogen uncouples KCNQ1 from KCNE3 allowing the channel protein to enter an endocytotic recycling pathway and capture activated β-catenin in the cytosol, returning it to the cell membrane in association with E-cadherin. The trapping of β-catenin at the plasma membrane prevents its nuclear translocation and suppresses the Wnt pathway activation of cell proliferation genes as well as inhibiting Epithelial Mesenchymal Transition. Adapted from References [202,204,205].

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



