
Submitted:
23 November 2023
Posted:
24 November 2023
You are already at the latest version
Abstract
Introduction: Oral candidiasis is an opportunistic fungal infection, often caused by Candida albicans. One of the virulence factors of C. albicans is sterol 14a-demethylase or CYP51. Compounds of anthocyanins (ternatin, cyanin) and flavonoids (rutin, isoquercetin, kaempferol, myricetin, limocitrin) from the extract of the Clitoria ternatea (butterfly pea) plant are believed to have potential to inhibit the CYP51 virulence factor of C. albicans to suppress oral candidiasis. The purpose of this research was to investigate the interaction between the ligands of anthocyanins and flavonoids from C. ternatea (butterfly pea) with CYP51. Methods: The in silico study applied molecular docking to examine the molecular interactions (binding affinity) between the anthocyanin and flavonoid ligands of interest from C. ternatea and the CYP51 receptor of C. albicans. Results: Predicted interactions exist between the ligands of anthocyanins (ternatin, cyanin) and flavonoids (rutin, isoquercetin, kaempferol, myricetin, limocitrin) from C. ternatea and the CYP51 receptor of C. albicans. Conclusion: C. ternatea has the potential to provide anti-fungal agents that can inhibit or slow down the occurrence of oral candidiasis, as indicated by in silico studies. However, it is important to conduct additional research regarding the properties of the plant to determine the unknown aspects of its effects to the human body.
Keywords:
Molecular docking
; Clitoria ternatea
; CYP51 virulence factor
; Candida albicans
; oral candidiasis
; anti-fungal
INTRODUCTION
Poor oral health can significantly impact a person’s quality of life.1 Nevertheless, the importance of oral health is often overlooked. The WHO Global Oral Health Status Report (2022) estimates that oral diseases affect nearly 3.5 billion people worldwide, with 3 out of 4 individuals residing in middle-income countries.1,2 In Indonesia, the prevalence of oral diseases is relatively high. One of the oral conditions frequently experienced by the Indonesian population is candidiasis.3 According to data from UNAIDS (2017), approximately 7.700 Indonesians (2.89%) experience this problem each year.3 Oral candidiasis is an opportunistic fungal infection, often caused by Candida albicans.4 This fungus is a component of the normal oral flora in immunocompetent individuals.4,5 It mostly lives in our oral cavity as a commensal population, not a pathological one, and is found in 20-75% of the general population without causing symptoms.5 However, this fungus can become a pathogen under conditions that facilitate opportunistic infections.4 Some predisposing factors for pathological colonization of C. albicans include immunocompromised conditions, poor oral hygiene, long-term steroid use, diabetes, denture use, smoking, and xerostomia.5 Patients with candidiasis typically complain of symptoms such as tongue pain, halitosis, taste disturbances, and a burning sensation.5,6 These symptoms can adversely affect the quality of life of the patients.6
Communication between cells in C. albicans with the external environment occurs through the cell membrane.7 Sterols found in the fungal cell membrane are crucial because they provide stability to the cell and resistance to stress.7 One of the virulence factors of C. albicans is sterol 14α-demethylase or CYP51.8 CYP51 is required by C. albicans for the biosynthesis of ergosterol, which is the primary target of antifungal azole drugs.9,10 Azole antifungal drugs block ergosterol biosynthesis by inhibiting CYP51, which is located in the endoplasmic reticulum of C. albicans.8,9 However, prolonged and prophylactic use of azole drugs in clinical settings can lead to the emergence of azole-resistant fungal strains.11 This is particularly common in immunocompromised patients, HIV and AIDS patients, and cancer patients.11,12 Therefore, there is a need for new and effective means to combat C. albicans strains that are resistant to existing drugs.11
Indonesia is a country rich in biodiversity,13 with 30.000-40.000 plant species14, and around 2.500-7.500 of these are considered to have medicinal use or potential.15 One of such plants is the butterfly pea or Clitoria ternatea of the Fabaceae family, commonly known in Indonesia as “bunga telang”, named after Ternate, a Moluccan island of Indonesia.16 The plant contains a variety of active compounds, such as anthocyanin pigments responsible for the blue color of the flowers, and also flavonoids, tannins, saponins, phenols, triterpenoids, and alkaloids.17 Several anthocyanins, such as ternatin and cyanin, as well as flavonoids like rutin, isoquercetin, kaempferol, myricetin, and limocitrin, are believed to have antifungal, anti-inflammatory, anticancer, and antioxidant effects.17,18 The antifungal effect is expected to inhibit the pathological growth of C. albicans in the oral cavity by forming bonds between a soluble protein and the fungal cell wall, thereby damaging the fungal cell membrane.8,19 Additionally, it is expected to inhibit the action of the CYP51 virulence factor in Candida albicans.8 However, there is still very little research on the interaction and activity of these bioactive compounds from C. ternatea as potential anti-fungal agents.
Such interactions can be evaluated by using the approach of molecular docking.20 Molecular docking is a crucial method in structure-based drug design (SBDD).21 This method allows us to predict the activity of a molecule at the binding site of a target protein, both structurally and energetically, by analyzing the 3D interactions between receptors and ligands and assessing the binding affinity of these interactions.22 Additional research is needed to understand the relationship between the active compounds in C. ternatea and the CYP51 virulence factor in C. albicans causing candidiasis, as relevant data is currently scarce. This research aims to assess the potential of C. ternatea as a candidate for anti-fungal treatments.
MATERIALS AND METHODS
Research sample
Virulence factor sterol 14α-demethylase (CYP51) of the fungus C. albicans was downloaded from the Protein Data Bank website (http://www.rscb.org).
Test substances
The ligands of anthocyanin (ternatin, cyanin), and flavonoid (rutin, isoquercetin, kaempferol, myricetin, limocitrin) compounds extracted from the C. ternatea plant were downloaded from the PubChem website in 3D.sdf format with the following CID codes:
| Compounds | Pubchem CID |
| Ternatin | 5459184 |
| Cyanin | 441688 |
| Rutin | 5280805 |
| Isoquercetin | 5280804 |
| Kaempferol | 5318767 |
| Myricetin | 5281672 |
| Limocitrin | 5489485 |
Research instruments
The hardware utilized was a common personal computer, running on the Windows 11 operating system. The software tools and applications that were employed throughout the molecular docking process (downloaded for free) included the following:
| Autodock 4.2.6 and Autodock Tools v.1.5.6(The Scripps Research Institute available for download at http://www.autodock.scripps.edu/) | Used for conducting molecular docking simulations. |
| ChemDraw v.16.0 (available for download at https://www.medicogiants.com/chemdraw-16-0-free-download/) | Utilized for creating 3D structures and minimizing energy of the relevant ligand compounds. |
| BIOVIA Discovery Studio Visualizer v.21 (available for download at https://discover3ds.com/discovery-studio-visualizer-download-thank-you) | Employed for receptor and ligand purification as well as visualizing interactions between the receptor and related ligands (in 3D and 2D). |
Research procedure
Receptor and ligand preparation
The BIOVIA Discovery Studio Visualizer v.21 software was used to separate the native ligand from the CYP51 receptor, resulting in ligand.pdb and receptor.pdb files that are devoid of water and other molecules. The SMILES notation for the compounds from the C. ternatea plant was obtained from the PubChem website and then transformed into a 3D structure using the Chemdraw v.16 software.
Docking method validation
Pose validation was carried out to determine the interaction position between the CYP51 receptor and the native ligand. In the validation process, the Root Mean Square Deviation (RMSD) results are considered satisfactory for RMSD of 2 Å or less. Additionally, the grid box and grid position that align with the validated results, representing the best pose, was utilized as the grid box and grid position in the execution of molecular docking for all eight ligands of the C. ternatea compounds.
Molecular docking and data analysis
Docking on the test ligands is conducted to obtain a histogram displaying two out of the four molecular docking parameters: Gibbs energy (∆G) and inhibition concentration (IC). The selection of these two parameters was based on the principle that the smaller (the more negative) the value of ∆G of the bond, the more stable it is. Moreover, a smaller value of the inhibition concentration also signifies a more stable bond.23–25 These two parameters are well correlated, and indicate the affinity of the bond formed between the ligand and protein.23,25
RESULTS
Docking method validation results
The validation results of the pose between the native ligand and the CYP51 receptor (PDBID: 5TZ1) demonstrate the accuracy of the interactions between the ligand and the receptor. The selected pose is the 18th pose with an RMSD of 1.881 Å (Table 1). Furthermore, existence of various interactions have been confirmed between the native ligand and the CYP51 receptor, including hydrogen bonding, Van der Waals interactions, Pi interactions, hydrophobic interactions, and other interactions. Thus, the study involving the CYP51 virulence factor of C. albicans and the ligands from the C. ternatea compounds can proceed, as the method was successfully validated.
Table 1.
Validation results of the native ligand poses with the CYP51 target protein.

Docking results
Ternatin interaction with CYP51
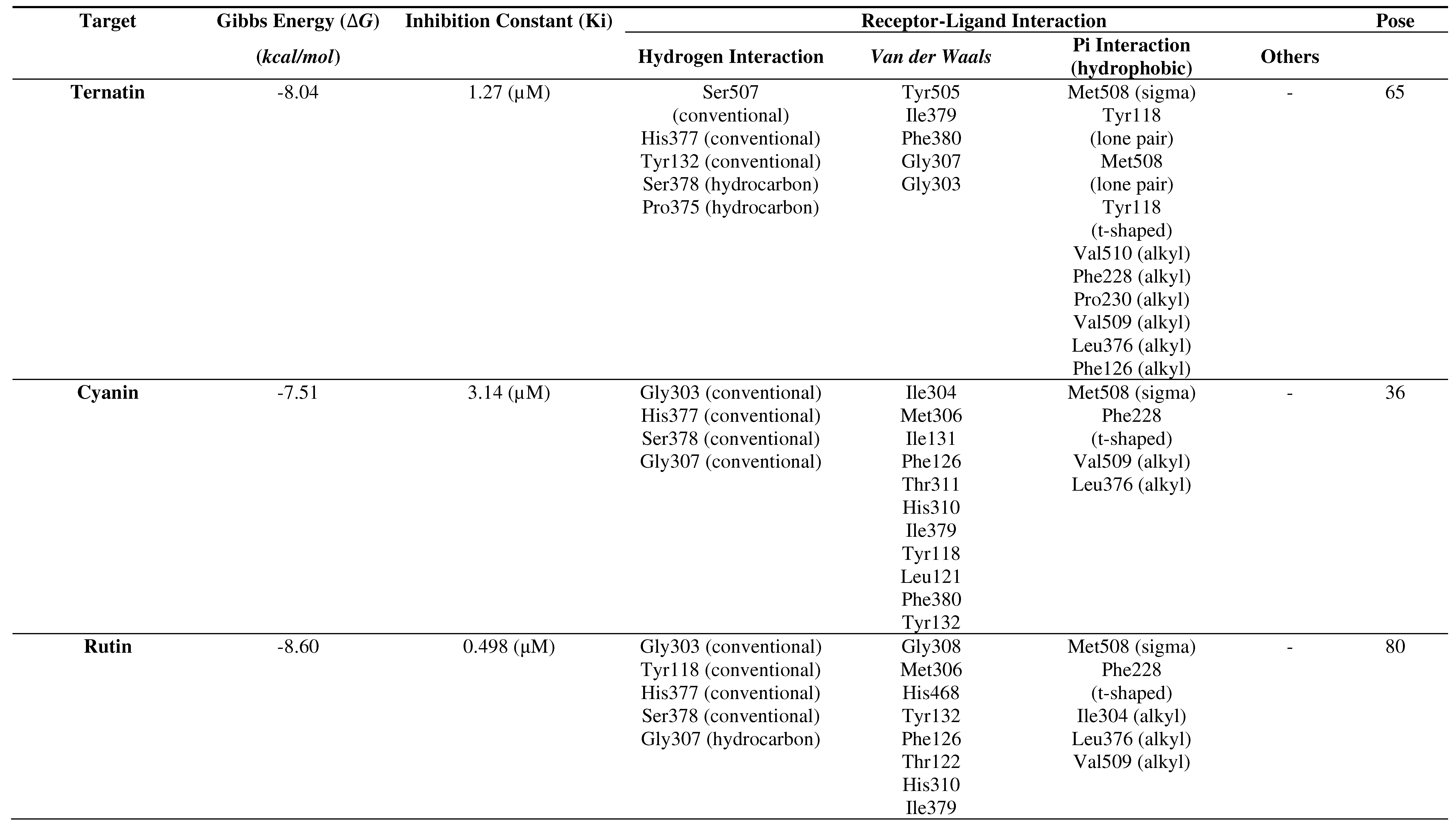
From the results of 100 molecular docking attempts to predict the ternatin pose at the binding site of CYP51 using the Autodock Tools 1.5.6 program, a histogram indicated the pose in the 65th run as the best pose with a Gibbs energy (∆G) of -8.04 kcal/mol. The pose was repeated 61 times with an average Gibbs energy (∆G) of -7.50 kcal/mol. The resulting inhibition constant (Ki) was 1.27 µM, and ternatin was bound in the coordinates: x = 66.579, y = 35.727, z = 40.937.
Cyanin interaction with CYP51
From the results of 100 molecular docking attempts to predict the cyanin pose at the binding site of CYP51 using the Autodock Tools 1.5.6 program, a histogram indicated the pose in the 36th run as the best pose with a Gibbs energy (∆G) of -7.51 kcal/mol. The pose was repeated 35 times with an average Gibbs energy (∆G) of -6.25 kcal/mol. The resulting inhibition constant (Ki) was 3.14 µM, and cyanin was bound in the coordinates: x = 65.500, y = 32.810, z = 41.496.
Rutin interaction with CYP51
From the results of 100 molecular docking attempts to predict the rutin pose at the binding site of CYP51 using the Autodock Tools 1.5.6 program, a histogram indicated the pose in the 80th run as the best pose with a Gibbs energy (∆G) of -8.60 kcal/mol. The pose was repeated 8 times with an average Gibbs energy (∆G) of -7.05 kcal/mol. The resulting inhibition constant (Ki) was 497.61 nM (= 0.498 μM), and rutin was bound in the coordinates: x = 65.117, y = 32.144, z = 40.695.
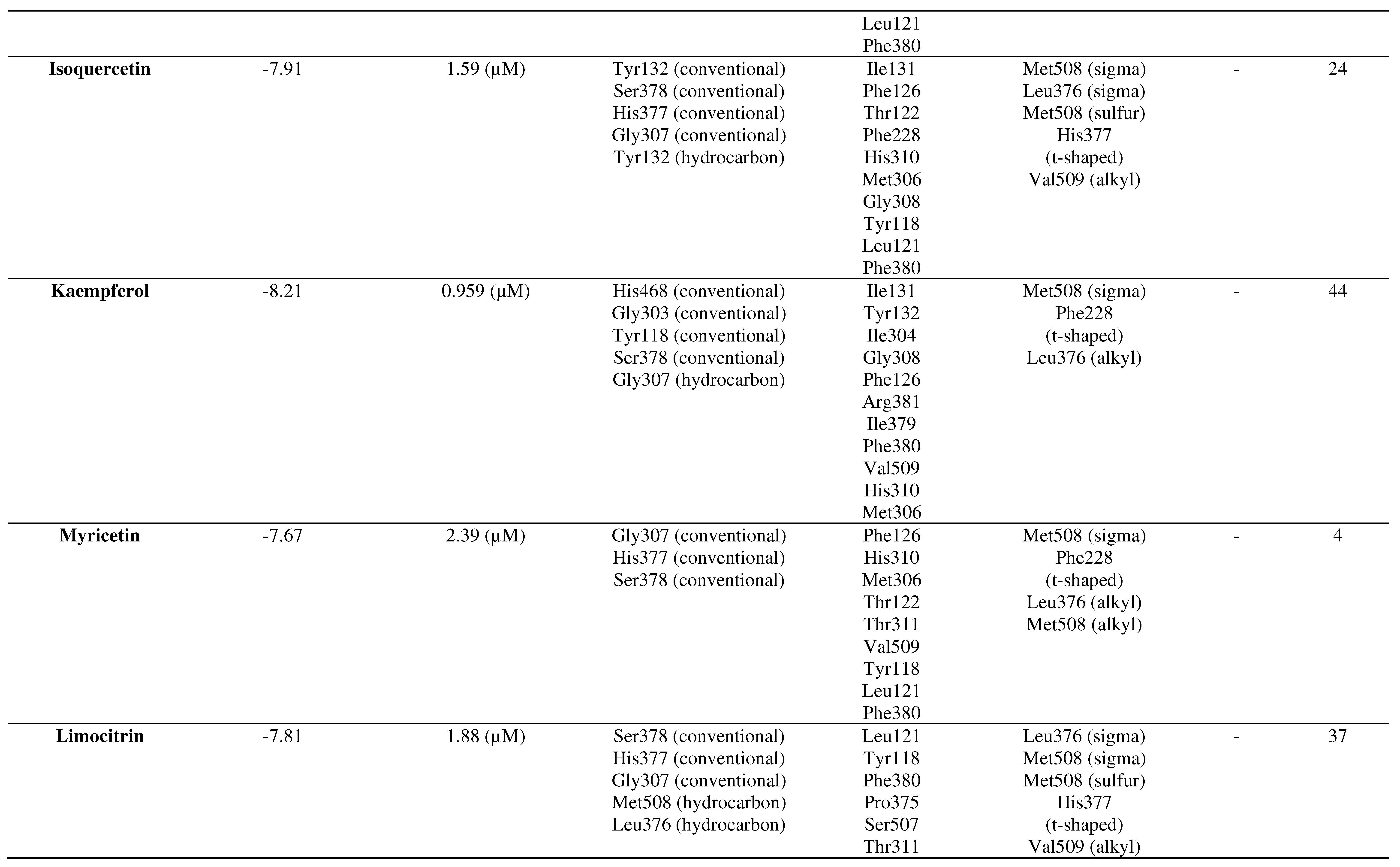
Isoquercetin interaction with CYP51
From the results of 100 molecular docking attempts to predict the isoquercetin pose at the binding site of CYP51 using the Autodock Tools 1.5.6 program, a histogram indicated the pose in the 24th run as the best pose with a Gibbs energy (∆G) of -7.91 kcal/mol. The pose was repeated 70 times with an average Gibbs energy (∆G) of -6.75 kcal/mol. The resulting inhibition constant (Ki) was 1.59 µM, and isoquercetin was bound in the coordinates: x = 65.786, y = 34.257, z = 41.150.
Kaempferol interaction with CYP51
From the results of 100 molecular docking attempts to predict the kaempferol pose at the binding site of CYP51 using the Autodock Tools 1.5.6 program, a histogram indicated the pose in the 44th run as the best pose with a Gibbs energy (∆G) of -8.21 kcal/mol. The pose was repeated 11 times with an average Gibbs energy (∆G) of -6.77 kcal/mol. The resulting inhibition constant (Ki) was 958.50 nM (= 0.959 μM), and kaempferol was bound in the coordinates: x = 65.448, y = 31.633, z = 40.578.
Myricetin interaction with CYP51
From the results of 100 molecular docking attempts to predict the myricetin pose at the binding site of CYP51 using the Autodock Tools 1.5.6 program, a histogram indicated the pose in the 4th run is the best pose with a Gibbs energy (∆G) of -7.67 kcal/mol. The pose was repeated 67 times with an average Gibbs energy (∆G) of -7.01 kcal/mol. The resulting inhibition constant (Ki) was 2.39 µM, and myricetin was bound in the coordinates: x = 65.602, y = 36.097, z = 40.428.
Limocitrin interaction with CYP51
From the results of 100 molecular docking attempts to predict the limocitrin pose at the binding site of CYP51 using the Autodock Tools 1.5.6 program, a histogram indicated the pose in the 37th run as the best pose with a Gibbs energy (∆G) of -7.81 kcal/mol. The pose was repeated 46 times with an average Gibbs energy (∆G) of -6.85 kcal/mol. The resulting inhibition constant (Ki) was 1.88 µM, and limocitrin was bound in the coordinates: x = 65.549, y = 36.046, z = 40.376.
Molecular docking results of the C. ternatea compounds with the CYP51 receptor
Based on the molecular docking results, summarized in Table 2, it can be concluded that all ligands of the C. ternatea compounds interact with the CYP51 receptor (PDBID: 5TZ1) in terms of Gibbs free energy (∆G), inhibition concentration (IC), hydrogen bonding, Van der Waals interactions, Pi interactions (hydrophobic), and other interactions (Table 5.2). All of these seven ligands form stable bonds. In order from the most stable to the least stable binding, the sequence is as follows: rutin → kaempferol → ternatin → isoquercetin → limocitrin → myricetin → cyanin.
Table 2.
Molecular docking results of the C. ternatea ligands with the CYP51 receptor.

DISCUSSION
In this study, the selected receptor is CYP51, one of the virulence factors of C. albicans, a fungus which often causes oral candidiasis. The structure of CYP51 receptor used for molecular docking validation is a complex that contains both CYP51 and the native ligand, VT1. This structure served for validation purposes before conducting molecular docking with the selected compounds of C. ternatea. The protein structure with PDBID 5TZ1, accessed via the Protein Data Bank (PDB, https://www.rscb.org/), was chosen because it fulfils the required resolution of 2 Å (or less) for validation.20
The pose validation between the CYP51 receptor and the native ligand resulted in an RMSD of 1.881 Å (< 2 Å). Therefore, it was concluded that the selected structure of CYP51 receptor can be used in the molecular docking process to observe interactions with ligands from the C. ternatea with similar grid box and grid position as for validation purposes.
The simulation results, summarized in Table 2, predicted most stable binding between the CYP51 receptor and the ligands of the included flavonoid compounds, with rutin and kaempferol as leaders of the list. The only exception was the anthocyanin compound ternatin that appeared as third on the bond stability list, while the bond to the other anthocyanin (cyanin) was the least stable of all. Increasing bond stability would likely imply more effective ability by the compound to interfere with the normal action of the CYP51 receptor and increasing antifungal potential. Low (Gibbs) free energy of binding (ΔG) is an obvious indicator of such stability, and so is also the inhibition constant Ki.23-25 Logarithm of Ki is linearly correlated with ΔG (Figure 8).
Figure 8.
Predicted free energy of binding ΔG as a function of the inhibition constant Ki for molecular
docking between C. ternatea anthocyanin/flavonoid ligands and CYP51 receptor.
Figure 8.
Predicted free energy of binding ΔG as a function of the inhibition constant Ki for molecular
docking between C. ternatea anthocyanin/flavonoid ligands and CYP51 receptor.
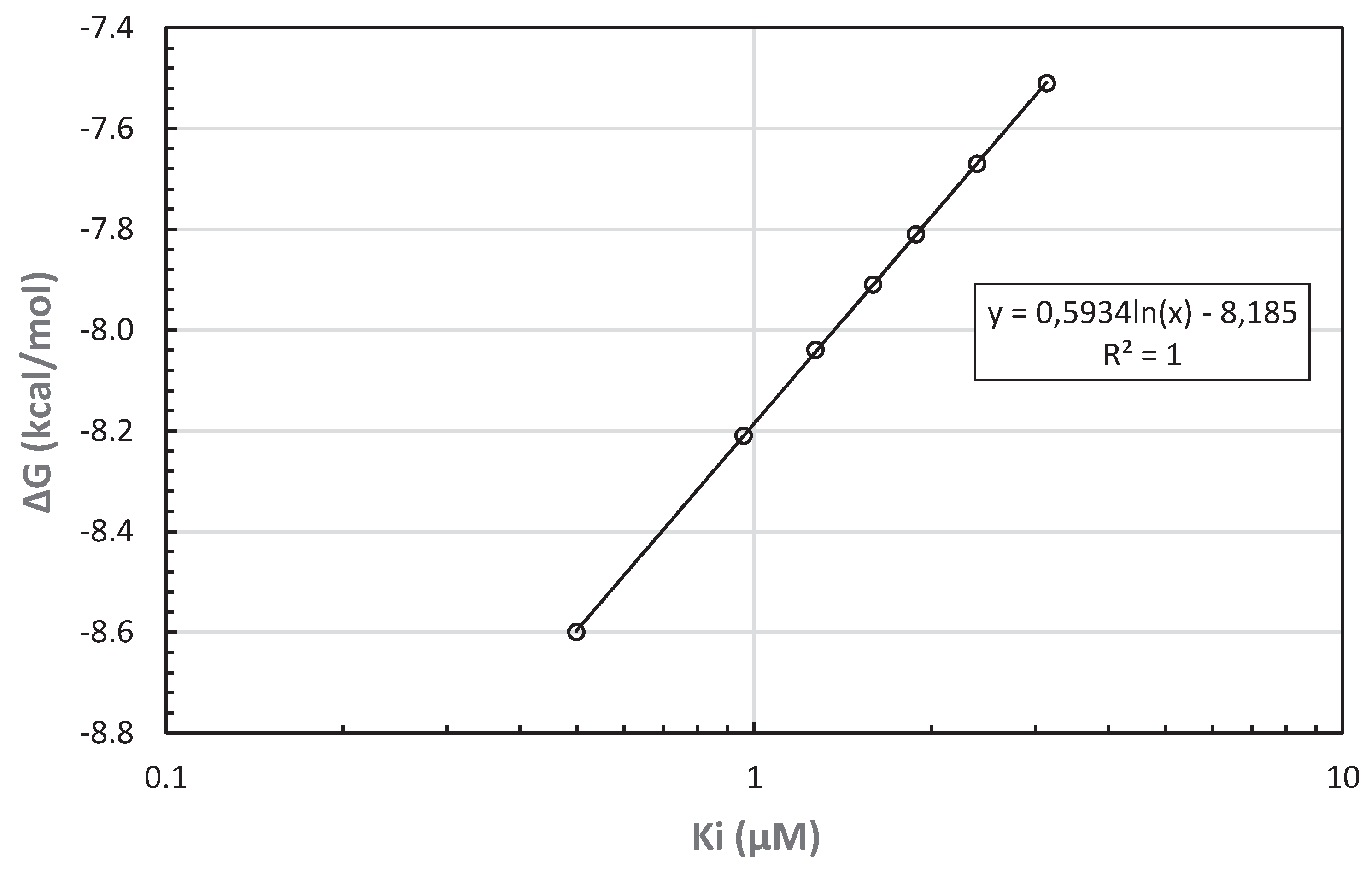
The full range of ΔG values for all seven ligands and CYP51 receptor is 12.7% of the corresponding ΔG for the most stable bond (with rutin). The range of Ki, however, is nearly an order of magnitude and therefore comparable to the range of ΔG only on logarithmic scale. The linear correlation between ln(Ki) and ΔG is here perfect (R2 = 1), as expected for chemical reactions.
The limitations of the study are related to the unconfirmed predicted interaction results between the C. ternatea compounds and CYP51 virulence factor, as well as the experimentally unconfirmed toxicity of C. ternatea to the human body.
CONCLUSION
The results and discussions of the conducted research indicate that interactions between the ligands of the C. ternatea compounds and the CYP51 receptor are feasible. The presence of interactions between the ligands and the receptor can be observed through the binding affinity marked by Gibbs free energy (∆G), inhibition concentration (IC), hydrogen bonding, and Van der Waals interactions occurring between the ligand of the C. ternatea compounds and the CYP51 receptor. Among the seven ligands of the C. ternatea plant, the ligand with the highest binding affinity to the CYP51 receptor is rutin with ∆G = -8.60 kcal/mol and IC = 498 nM. To address the limitations of this study, further research is needed through in vitro and in vivo testing to validate the molecular docking results. Toxicity testing on the C. ternatea plant is also necessary to assess the potentially adverse effects that the plant may have to the human body.
Author Contributions
All authors have the same contributions.
Data Availability
The datasets generated and/or analyzed during the current study are available in: Protein Data Bank Website (http://www.rscb.org) with PDBID: 5TZ1; PubChem Website (https://pubchem.ncbi.nlm.nih.gov/) with PubChem CID: Ternatin: 5459184; Cyanin: 441688; Rutin: 5280805; Isoquercetin: 5280804; Kaempferol: 5318767; Myricetin: 5281672; Limocitrin: 5489485.
References
- Global Status Report WHO, 2022. Available from: http://apps.who.int/bookorders.
- Oral Health Indonesia 2022 country profile [cited 2023 Jun 18]. Available from: https://www.who. 2022.
- Wahyuningsih, R.; Adawiyah, R.; Sjam, R.; Prihartono, J.; Ayu Tri Wulandari, E.; Rozaliyani, A. , et al. Serious fungal disease incidence and prevalence in Indonesia. Mycoses. 2021 Oct 25;64(10):1203–12.
- Schuster, J.E.; Fisher, B.T. Candidiasis. Pediatric Transplant and Oncology Infectious Diseases [Internet]. 2023 [cited 2023 Sep 17]; 195-205.e3. Available from: https://www.ncbi.nlm.nih. 29 May 5606. [Google Scholar]
- Taylor, M.; Brizuela, M.; Raja, A. Oral Candidiasis. StatPearls [Internet]. 2023 Jul 4 [cited 2023 Sep 17]; Available from: https://www.ncbi.nlm.nih. 5452. [Google Scholar]
- Hato, H.; Sakata K ichiro Sato, J.; Hasebe, A.; Yamazaki, Y.; Kitagawa, Y. Factor associated with oral candidiasis caused by co-infection of Candida albicans and Candida glabrata: A retrospective study. J Dent Sci. 2022, 17, 1458–1461. [Google Scholar] [CrossRef] [PubMed]
- Talapko, J.; Juzbašić, M.; Matijević, T.; Pustijanac, E.; Bekić, S.; Kotris, I. , et al. Candida albicans—The Virulence Factors and Clinical Manifestations of Infection. Journal of Fungi. 2021, 7, 79. [Google Scholar] [PubMed]
- Hargrove, T.Y.; Friggeri, L.; Wawrzak, Z.; Qi, A.; Hoekstra, W.J.; Schotzinger, R.J. , et al. Structural analyses of Candida albicans sterol 14α-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. Journal of Biological Chemistry. 2017, 292, 6728–43. [Google Scholar] [PubMed]
- Warrilow, A.G.S.; Martel, C.M.; Parker, J.E.; Melo, N.; Lamb, D.C.; Nes, W.D.; et al. Azole Binding Properties of Candida albicans Sterol 14-α Demethylase (CaCYP51). Antimicrob Agents Chemother. 2010, 54, 4235–45. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.L. The Multifunctional Fungal Ergosterol. mBio. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.A.; Diekema, D.J. Epidemiology of Invasive Candidiasis: a Persistent Public Health Problem. Clin Microbiol Rev. 2007, 20, 133–63. [Google Scholar] [CrossRef] [PubMed]
- SAME AS 11.
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.B.; Kent, J. Biodiversity hotspots for conservation priorities. Nature. 2000, 403, 853–8. [Google Scholar] [CrossRef] [PubMed]
- Hamid, A.; Sitepu, D. An understanding of native herbal medicine in Indonesia. Ind Crops Res J. 1990, 3. [Google Scholar]
- Elfahmi Woerdenbag, H.J.; Kayser, O. Jamu: Indonesian traditional herbal medicine towards rational phytopharmacological use. J Herb Med. 2014, 4, 51–73. [Google Scholar] [CrossRef]
- Oguis, G.K.; Gilding, E.K.; Jackson, M.A.; Craik, D.J. Butterfly Pea (Clitoria ternatea), a Cyclotide-Bearing Plant With Applications in Agriculture and Medicine. Front Plant Sci. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Multisona, R.R.; Shirodkar, S.; Arnold, M.; Gramza-Michalowska, A. Clitoria ternatea Flower and Its Bioactive Compounds: Potential Use as Microencapsulated Ingredient for Functional Foods. Applied Sciences 2023, 13, 2134. [Google Scholar] [CrossRef]
- Jeyaraj, E.J.; Lim, Y.Y.; Choo, W.S. Extraction methods of butterfly pea (Clitoria ternatea) flower and biological activities of its phytochemicals. J Food Sci Technol. 2021, 58, 2054–67. [Google Scholar] [CrossRef] [PubMed]
- Cowan, M.M. Plant Products as Antimicrobial Agents. Clin Microbiol Rev. 1999, 12, 564–82. [Google Scholar] [CrossRef]
- Stanzione, F.; Giangreco, I.; Cole, J.C. Use of molecular docking computational tools in drug discovery. In 2021. p. 273–343.
- Kuntz, I.; Blaney, J.; Oatley, S.; Langridge, R.; Ferrin, T. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982, 161, 269–88. [Google Scholar] [CrossRef]
- McConkey, B.; Sobolev, V.; Edelman, M. The performance of current methods in ligand-protein docking. Curr Sci. 2002, 83, 845–55. [Google Scholar]
- Wan, S.; Bhati, A.P.; Zasada, S.J.; Coveney, P.V. Rapid, accurate, precise and reproducible ligand–protein binding free energy prediction. Interface Focus. 2020, 10, 20200007. [Google Scholar] [CrossRef] [PubMed]
- Du X, Li Y, Xia YL, Ai SM, Liang J, Sang P, et al. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int J Mol Sci. 2016, 17, 144.
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004, 3, 935–49. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



