
Submitted:
24 November 2023
Posted:
28 November 2023
You are already at the latest version
Abstract
This comprehensive review delves into the multifaceted aspects of ERK signaling and the intricate mechanisms underlying distinct cellular fates. ERK1 and ERK2 (ERK) govern proliferation, transformation, epithelial-mesenchymal transition, differentiation, senescence, or cell death, con-tingent upon activation strength, duration, and context. The biochemical mechanisms underlying these outcomes are inadequately understood, shaped by signaling feedback and the spatial localization of ERK activation. Generally, ERK activation aligns with the Goldilocks principle in cell fate determination. Excessive or insufficient ERK activity inhibits growth, whereas moderated activation supports proliferation and survival. Unraveling the intricacies of how the degree of ERK activation dictates cell fate requires deciphering mechanisms encompassing protein stability, transcription factors downstream of ERK, and the chromatin landscape.
Keywords:
cell proliferation
; cell fate
; ERK
; senescence
; cell signaling
; EMT
; apoptosis
; pluripotency
1. Introduction
The extracellular signal-regulated kinases ERK1 and ERK2 (ERK) are the effectors of a signaling module activated by many membrane receptors to regulate cell fate (Figure 1). These kinases can control proliferation, transformation, epithelial-mesenchymal transition (EMT), differentiation, senescence, or cell death depending on the strength, duration, and context of their activation [1]. The biochemical mechanisms explaining how each different cell fate can be controlled by the same signaling module are poorly understood. ERK interacts with multiple proteins [2] and many of them are modulated by ERK-dependent phosphorylation [3]. The biological outcome is contingent upon the specific ERK targets undergoing phosphorylation, which are determined by the dynamics and spatial localization of ERK activation. ERK dynamics is controlled by both positive and negative feedback loops (Figure 1) [4] while its location depends on its activation site (membrane vs Golgi) [5,6] and the relocalization of activated ERK once they dissociate from their upstream activator complex [7]. Here, we review the distinctive aspects of ERK signaling linked to different fates as well as the mechanisms proposed to decode these signals.
2. Proliferation and survival
Cell proliferation was the first outcome linked to activation of the ERK pathway [8] and multiple ERK targets have been identified to explain this effect [3]. Initial studies on the response to ERK activation were conducted in tumor cell lines where proliferation is the default response due to their transformed state. Critical insights to understand how ERK controls proliferation came from studies of the rat adrenal pheochromocytoma cell lines (PC-12). In these cells, the epidermal growth factor (EGF) induces a transient activation of the pathway and cell proliferation while the nerve growth factor (NGF) induces sustained activation and differentiation [9].
Given the association between proliferation and transient pathway activation, it logically follows that negative feedback mechanisms operate to prevent sustained activation. As expected, EGF induced these negative feedback mechanisms in PC12 cells. They include the phosphorylation of BRAF and MEK by ERK and the downregulation of SOS, a protein that stimulates the Ras GTPase in response to the activation of membrane receptors (Figure 1) [10,11]. In contrast, NGF induced positive feedback mechanisms that sustained the activation of the pathway [11]. The positive feedback was mediated through ERK-dependent RAF activation via protein kinase C (PKC) and inhibition of RAF kinase inhibitory protein (RKIP). Blocking these positive feedback mechanisms converted the NGF response from differentiation to proliferation [11]. Recent research has confirmed the antiproliferative effect of the PKCα-RAS-ERK positive feedback within the realm of intestinal epithelial cells. [12]. The need for negative feedback in the ERK pathway to promote proliferation has been observed as well in fibroblasts lacking expression of the dual specificity phosphatase DUSP4 [13] and in lung adenocarcinoma or leukemia cells treated with DUSP6 inhibitors [14,15]. In colorectal cancers, despite the presence of KRAS and BRAF mutations in over 90% of cancer cells, phospho-ERK staining was detectable in less than 10 % of the cells [16]. This aligns with the necessity to prevent sustained elevation of ERK signaling for the maintenance of cell proliferation.
In addition to its role in cell proliferation, ERK is required for cell survival in multiple cell types [17]. Total ablation of ERK in mice completely blocked KRas tumorigenesis. However, animals did not survive the loss of both ERK1 and ERK2 [18]. In this study, the block in tumor formation could be due to a proliferation defect or cell death in tumor cells. One important mechanism of cell survival triggered by ERK involves the phosphorylation of BIM on Ser69 ultimately resulting in its ubiquitination and proteasomal degradation of this pro-apoptotic protein [19]. ERK also phosphorylates FoxO3 triggering its degradation, which results in inhibition of the expression of Bim [20].
While in the PC12 system, proliferation is linked to a momentary activation of ERK, it's important to note that time is a relative concept. A very brief pulse of ERK activation does not induce proliferation, but more prolonged stimulation does. Blenis and colleagues proposed a mechanism to explain why only long pulses of ERK activation stimulate cell proliferation. In their model, the initial ERK activation phosphorylates and stimulates transcription factors leading to the expression of unstable immediate early genes (IEG). Some IEG products are then stabilized by ERK-catalyzed phosphorylation only if the ERK pathway remains activated [21]. In the Blenis model, signal duration is decoded in two steps, first transcriptional induction and then phosphorylation and stabilization of newly transcribed proteins (Figure 2). A variation of this decoding mechanism can occur in the same molecule via multisite phosphorylation. This was described for the phosphorylation of ELK-1 by ERK where the more rapidly phosphorylated sites promote transcription by mediating interactions with the Mediator complex, but the late phosphorylation sites are inhibitory [22] (Figure 3).
Several transcription factors are activated by ERK-mediated phosphorylation (c-MYC, c-FOS, NR4A1, NR4A2, UBF, and MITF), and resistance to ERK pathway inhibitors can be mediated by reactivation of some of these transcription factors by ERK-independent pathways [23,24,25]. In addition, using multiple cell lines, it has been shown that proliferation required a relatively low amplitude but lasting ERK activation [26]. This is important for the clinical use of inhibitors of this pathway because they need to be administered at doses that can inhibit more than 85% of the output [26]. In addition, pathway inhibitors release negative feedback signals conditioning cells for a rebound and drug resistance/addiction [27,28]. Of note, tumor cells that become addicted to pathway inhibitors, proliferate with a very low level of ERK activation (only 2-3% of ERK was phosphorylated according to mass spectrometry) [28] indicating once more that anti-ERK therapies must inhibit the pathway with high efficiency to achieve an antiproliferative effect.
2.1. ERK pulses in cell proliferation and survival
The pulsatile nature of several signaling pathways allows the regulation of different outcomes using the same signaling module. This occurs through the transmission of information by modulating the amplitude, frequency, or duration of the pulses. (Figure 4). The proliferation fate in response to ERK requires frequency-modulated pulses of ERK activity as measured with a live cell ERK-reporter in cell culture [26] or a FRET (Förster resonance energy transfer)-ERK reporter in vivo [29]. Computer modeling suggested that negative feedback is central to this behavior in ERK signaling [26]. However, the precise mechanism responsible for deciphering pulses of ERK activity was unidentified. Work in yeast suggests that gene activation can result from nonoverlapping pulses of an activator transcription factor (TF) and a repressor [30]. Oscillation in the DNA binding capacity of TFs may also help to dissociate them from the large number of binding sites present in the genome that effectively act as decoys [31]. This suggests that ERK pulses are decoded by affecting the dynamics of transcription factors regulating cell proliferation (Figure 4).
Like proliferation, survival is causally linked to pulsatile ERK activation. High-frequency pulses characterize proliferation and survival in mammary acini grown in organoids while low-frequency ERK pulses are associated with cell death and lumen formation. Inducing high-frequency ERK pulses promoted the survival of lumen cells. Interestingly, survival depended on ERK pulse frequency and not on the accumulation of ERK signals because spacing ERK pulses over longer times that achieve a similar overall ERK activity did not promote survival [32].
3. Differentiation
ERK operates downstream of growth factors that play a role in development such as FGF (fibroblast growth factor), NGF (nerve growth factor), and many others [33,34]. Mouse models bearing conditional null alleles of ERK1 and ERK2 (Mapk3 and Mapk1) display craniofacial and cardiovascular defects. For the most part, the affected tissues involved derivatives of the neural crest, which constitutes a population of cells specified shortly after gastrulation. These neural crest cells give rise to multiple tissues including bones, connective tissue, melanocytes, and various nerves [35,36,37]. In PC-12 cells, which originate from the neural crest, NGF treatment induces differentiation, and as discussed above this requires sustained ERK activation and nuclear localization of ERK [2]. The ERK nuclear interactome in NGF-treated cells provided insights into how ERK regulates differentiation. Two critical targets are ERF, a suppressor of ETS transcription factor activity, and TRPS1, a suppressor of GATA transcription factors. Nuclear ERK phosphorylates ERF and TRPS1 inhibiting their activity and allowing both ETS- and GATA-mediated gene expression [2]. The data suggest that the regulation of cell differentiation by ERK depends on the integration of both duration and subcellular localization of ERK signaling. This integration provides more opportunities for regulation. Additional TFs regulating cell differentiation downstream of ERK in the PC-12 cell system were identified by studying the transcriptome after stimulation with either EGF or NFG. Short-term EGF stimulation-activated genes are regulated by E2F1, EBF1, SOX9, and SP1, while late-acting NGF-activated genes are regulated by BACH2, AP1, ETV4, and ELF2 [38]. The factors regulating the exchange of transcription factors between early and late time points are unknown. Late-acting TFs include IEG stimulated during the initial wave of gene expression, as well as TFs induced by autocrine stimulation via secreted factors such as the urokinase-type plasminogen activator (uPA) and matrix metalloproteinases (MMPs) [38]. It is tempting to speculate that fates controlled by sustained ERK activity depend on TF that do not contain inhibitory phosphorylation sites but only low-affinity, positively acting phosphorylation sites. In contrast, the proliferation fate will depend on factors such as ELK-1 (Figure 3) containing both early-acting positive phosphorylation sites and late-acting negative phosphorylation sites (Figure 3).
The duration of ERK activity was also important to determine neurogenic endoderm (transient, 30 min) or gut ectoderm (sustained, 1h-+) in flies. The role of signaling duration was demonstrated in this study by using optogenetics [39]. Interestingly, the mechanism specifying the gut ectoderm depended on cumulative ERK signaling implying a memory of ERK activity across multiple cellular divisions [39] (Figure 5).
4. Pluripotency
ERK activity also regulates pluripotency. Active ERK1/2 inhibits the `naïve’ pluripotent state of mouse embryonic stem (ES) cells and favors their differentiation [40]. Similarly, sustained ERK activity inhibits stem cells in colon crypt structures, while inhibiting ERK leads to the expansion of stem cells [41]. The combination of MEK and GSK3 inhibitors, which compose the medium formulation known as 2i, is used to maintain naïve stem cells in culture [42]. Several mechanisms inhibiting ERK in naïve pluripotent cells have been identified. Myc promotes the pluripotent cell fate by induction of the dual specificity phosphatases DUSP2 and 7 capable of inhibiting ERK activation [43]. The transcription factors PRDM14 and PRDM15 reinforce the naïve state by inhibiting FGF-ERK signaling and activating WNT signaling via R-spondin, mimicking the 2i condition [44]. ERK can be also inhibited via RSK-dependent negative feedback [45] and March5-mediated degradation of Prkar1a. The latter acts as a negative regulator of PKA, which in turn inhibits ERK by phosphorylation of RAF1 at Ser259 [46].
On the other hand, ERK inhibits the expression of pluripotency factors such as Nanog [47] via its ability to recruit PRC2 complexes and pause RNA polymerase II by phosphorylation at the CTD (C-terminal domain) at pluripotency genes [48]. ERK also regulates Nanog protein stability. In early embryos, cells experience a drop in ERK activity after mitosis. This drop leads to high levels of Nanog and the epiblast fate. The mechanism of ERK inactivation after mitosis is not fully understood but it requires the E3 ubiquitin ligase APC (anaphase-promoting complex) [33]. In sharp contrast to its inhibitory role for mouse naïve ES cells, ERK1/2 activation through FGF promotes the `primed’ pluripotent epiblast stem cell state (EpiSC) [49]. Similarly, ERK1/2 activation through FGF promotes the primed pluripotent state of human ES cells [50]. In this setting, ERK promotes the expression of genes essential for the survival and proliferation of human ES cells by binding to their promoters in association with the transcription factor ELK1 [51]. These works highlight the importance of ERK dynamics and chromatin interactions for developmental cell fate specification.
The ability of ERK to inhibit pluripotency may be relevant to cancer biology in some contexts. For example, in triple-negative breast cancer, chemotherapy resistance is associated with enrichment for cancer stem cells via reduction of ERK activity. In this case, the ERK pathway is attenuated at the level of MEK, which requires copper for its activity. Chemotherapy agents such as Carboplatin, Gemcitabine, and Paclitaxel induce copper chelation by elevating glutathione levels. Inhibition of MEK-ERK signaling reactivated FoxO3 nuclear translocation and transcriptional activation of the pluripotency factor Nanog [52]. Similarly, chemotherapy triggers DUSP9 through HIF activation, diminishing ERK signaling. This enables Nanog activation, thereby contributing to the enrichment of cancer stem cells [53].
5. Senescence
Cellular senescence in response to ERK pathway activation depends on high-intensity ERK signals [1,54,55]. In support of the link between intense ERK signals and tumor suppression, it has been shown that p53 contributes to ERK activation [56,57]. A model based on phosphorylation-dependent protein degradation was proposed based on evidence of multiple proteins degraded by the proteasome during oncogene-induced senescence [54]. The process was named SAPD (Senescence-Associated Protein Degradation). SAPD targets include several transcription factors, ribosome biogenesis factors, and mitochondrial proteins [58]. The SAPD model was further supported by the demonstration that inactivation of its targets (MYC, RSL1D1, STAT3) is sufficient to induce senescence [54,59,60]. Intriguingly, ERK-dependent protein stabilization is linked to cell cycle progression and survival (Figure 2) while ERK-dependent protein degradation is linked to both inhibition of pluripotency and senescence (Figure 6).
The activation of transcription factors is a key event to explain the cell proliferation, but also senescence, in response to ERK activation [21]. In one study, distinct genes were triggered following BRAFV600E activation in retinal epithelial cells, eliciting either proliferation or senescence based on the intensity of its activation: low levels prompted proliferation, whereas high levels induced senescence. Interestingly, gene expression changes relative to the control were calculated after considering both the time and the levels of ERK activation [61]. The data indicate that the relationship between ERK activation and cell proliferation is non-monotonic, fitting the Goldilocks principle where an intermediate level of ERK activity promotes proliferation, but higher levels block proliferation and trigger senescence (Figure 7). Intriguingly, during pancreatic tumorigenesis a similar relationship between ERK activity and proliferation was documented. Proliferating malignant tumors have moderate levels of ERK activation while benign lesions, containing senescent cells, have aberrantly high levels of activated ERK. In addition, increasing ERK activation in malignant pancreatic cancer cell lines triggered senescence and nucleolar alterations, notably the emergence of newly identified senescence-associated nucleolar foci [62]. Collectively, the recent data demonstrate that senescence triggered by prolonged or intense ERK activation entails distinct alterations in gene expression and nucleolar stress, in addition to protein degradation (Figure 8).
The decision between senescence and transformation in oncogene-expressing cells is greatly influenced by factors that reinforce or attenuate ERK signaling. This model explains why in some contexts, factors that reduce ERK signaling exhibit oncogenic properties, while inhibiting them can induce senescence and/or impede tumor progression. These oncogenic ERK-pathway attenuators include dual specificity phosphatases [14,15,63,64,65,66], the tyrosine kinase FER [67], the S/T kinase Wnk2 [68], the miRNA binder Ago2 [69,70], the heat shock protein mortalin (HSPA9/GRP75/PBP74) [71] and the lysosomal cation transporter TRPML1 [72]. An intriguing attenuation mechanism in the ERK pathway was uncovered by measuring the abundances of non-phosphorylated, monophosphorylated (pT- or pY-), and double-phosphorylated (pTpY-) ERK1 and ERK2 by mass spectrometry. It's noteworthy that solely double-phosphorylated ERK is functionally active. Tumor samples exhibited elevated levels of monophosphorylated pT-ERK1/2 in contrast to non-tumor samples, indicating a potential alteration in MEK-dependent ERK phosphorylation to prevent pathway hyperactivation [73]. Of note, some tumors retain high levels of ERK activation and still avoid senescence. This was discovered in colorectal cancer cells having KRAS G13D mutations that became resistant to MEKi. In this case, resistant cells develop an ERK-dependent epithelial-mesenchymal transition (EMT) that may inactivate senescence downstream of ERK [28].
6. EMT
ERK is an important activator of epithelial-mesenchymal transition (EMT) [74,75]. Several mechanisms have been proposed to explain how ERK activation triggers EMT. First, the kinase RPS6KA1 (Ribosomal Protein S6 Kinase A1), also known as RSK1, activates the late response transcription factor FRA1 which induces the expression of the EMT transcriptional repressors ZEB1 and ZEB2 [76,77]. Its paralog, RPS6KA3 (RSK2) also mediates EMT in response to activation of the tyrosine kinase receptor RON by macrophage-stimulating protein (MSP) in the context of cells having Ras mutations and activation of the ERK pathway [78]. Second, ERK can directly activate ZEB1, leading to the formation of a repressor complex between ZEB1 and the corepressor CtBP. The latter effectively suppresses expression of E-cadherin, a hallmark event during the process of EMT [75,79]. Before ERK stimulation, ZEB1 is inactive in a complex with the MAPK Regulated Corepressor Interacting Protein -1 (MCRIP1). However, upon phosphorylation by ERK, MCRIP dissociates from ZEB1 allowing CtBP binding [79]. These results suggest that ZEB1 can turn the high ERK response from senescence to EMT. In agreement with this idea, the knockout of Zeb1 promotes cellular senescence by alleviating the repression of the CDK inhibitors p15INK4b and p21 [80]. In a lung cancer model, initiation of Ras-dependent tumorigenesis required Zeb1 [81] likely due to the ability of the latter to block the protective senescence response triggered by Ras. Consistent with this hypothesis, in melanoma, high levels of ERK drive the expression of the senescence antagonist TWIST [82] leading to EMT [83]. These studies suggest that EMT regulators are critical components in cell fate determination downstream of ERK.
The interplay between cellular senescence and EMT in response to ERK activation has been described in several experimental models of cancer. Early, during pancreatic carcinogenesis, ERK cooperates with the TGFβ pathways to induce p21 and cell cycle arrest in benign cells. However, upon progression to pancreatic adenocarcinoma, ERK antagonizes TGFβ-induced cell cycle arrest and promotes EMT [84]. Both senescence [85,86,87] and EMT [88] are detected early during pancreatic carcinogenesis suggesting that EMT inducers may help to elude senescence. In colorectal cancer, ERK activity is heterogeneous within tumors, and cells with the highest activity localize to the tumor edge. These high-ERK cells were present both in KRAS mutated and wild-type cells, and displayed decreased proliferation markers and signs of EMT. Moreover, increasing ERK activity in colorectal cancer cells induced the EMT fate irrespective of their KRAS status. The tumor microenvironment likely regulates ERK levels in these tumors from a low basal level that promotes proliferation to a higher level that promotes EMT and cancer stem cell potential [89]. The senescence fate is likely evaded in these cases via the actions of the EMT mediators.
The context dependent EMT regulation by the ERK pathway was revealed by studying the response to MEK inhibitors (MEKi) in colorectal cancer cells. Tumors that contain the BRAFV600E mutation develop resistance and addiction to MEKi via amplification of BRAFV600E. Drug withdrawal triggers senescence or apoptosis due to hyperactive ERK signaling in BRAFV600E-expressing cells. On the other hand, tumors with the mutation KRASG13D developed resistance due to ERK-dependent EMT [28]. A similar cell fate shift from ERK-dependent apoptosis to EMT was described in lung cancer cells that acquired resistance to the antifolate pemetrexed [90]. The chromatin landscape may provide the context in which ERK activation regulates EMT. As an example, targeting KRAS mutations to different skin epithelial cells in mice only generated mesenchymal lesions from hair follicle stem cells, which already possessed accessible chromatin at EMT transcription factors binding sites [91].
7. Apoptosis
Sustained ERK signaling can also trigger apoptosis, a fate observed in cells treated with different chemotherapeutic drugs or during neuronal cell death [92]. ERK activation also promotes cell death in response to low glucose through its regulation of GCN2/eIF2a/ATF4-dependent expression of pro-apoptotic molecules [93]. The apoptosis fate in response to ERK activation also depends on reaching a threshold of high ERK activity as reported for senescence [94] (Figure 8). Interestingly, cell death in response to supraphysiological ERK activation was partially dependent on secreted factors [95]. Of interest, apoptosis induced by high ERK activity upon inhibition of DUSP6 was exploited to obtain a therapeutic response against chronic lymphocytic leukemia [96].
8. Conclusions
Cell fate in response to activated ERK pathway is context dependent. The intensity, duration, and frequency of the ERK pulses convey information decoded by transcription factors, chromatin, and perhaps other effector molecules. The utilization of heightened ERK signaling for therapeutic purposes should incorporate a strategy to prevent epithelial-mesenchymal transition (EMT). Conversely, approaches aiming to reduce ERK signaling should steer clear of promoting an enrichment of tumor stem cells. One promising strategy is the combination of MEK inhibitors with CDK4 inhibitors. This drug combo triggered senescence in KRAS mutant lung cancer cells by engaging the retinoblastoma pathway [97]. Given that MEK inhibition in other tumors leads to the development of drug-resistant cancer stem cells [52,98], these findings imply a potential role for CDK4 inhibitors in treatments directed at the ERK pathway.
Funding
This research was funded by CIHR, MOP11151 to G.F and CRS in partnership with CIHR (# 178660) to M.M. G.F. and is supported by the CIBC chair for breast cancer research.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Deschenes-Simard, X.; Kottakis, F.; Meloche, S.; Ferbeyre, G. ERKs in cancer: friends or foes? Cancer Res 2014, 74, 412-419. [CrossRef]
- von Kriegsheim, A.; Baiocchi, D.; Birtwistle, M.; Sumpton, D.; Bienvenut, W.; Morrice, N.; Yamada, K.; Lamond, A.; Kalna, G.; Orton, R.; et al. Cell fate decisions are specified by the dynamic ERK interactome. Nat Cell Biol 2009, 11, 1458-1464. [CrossRef]
- Xue, L.; Wang, P.; Cao, P.; Zhu, J.K.; Tao, W.A. Identification of extracellular signal-regulated kinase 1 (ERK1) direct substrates using stable isotope labeled kinase assay-linked phosphoproteomics. Mol Cell Proteomics 2014, 13, 3199-3210. [CrossRef]
- Ryu, H.; Chung, M.; Dobrzynski, M.; Fey, D.; Blum, Y.; Lee, S.S.; Peter, M.; Kholodenko, B.N.; Jeon, N.L.; Pertz, O. Frequency modulation of ERK activation dynamics rewires cell fate. Mol Syst Biol 2015, 11, 838. [CrossRef]
- Inder, K.; Hancock, J.F. System output of the MAPK module is spatially regulated. Commun Integr Biol 2008, 1, 178-179. [CrossRef]
- Herrero, A.; Casar, B.; Colon-Bolea, P.; Agudo-Ibanez, L.; Crespo, P. Defined spatiotemporal features of RAS-ERK signals dictate cell fate in MCF-7 mammary epithelial cells. Mol Biol Cell 2016, 27, 1958-1968. [CrossRef]
- Kholodenko, B.N.; Hancock, J.F.; Kolch, W. Signalling ballet in space and time. Nat Rev Mol Cell Biol 2010, 11, 414-426. [CrossRef]
- Boulton, T.G.; Nye, S.H.; Robbins, D.J.; Ip, N.Y.; Radziejewska, E.; Morgenbesser, S.D.; DePinho, R.A.; Panayotatos, N.; Cobb, M.H.; Yancopoulos, G.D. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 1991, 65, 663-675. [CrossRef]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179-185, doi:0092-8674(95)90401-8 [pii].
- Hernandez, M.A.; Patel, B.; Hey, F.; Giblett, S.; Davis, H.; Pritchard, C. Regulation of BRAF protein stability by a negative feedback loop involving the MEK-ERK pathway but not the FBXW7 tumour suppressor. Cellular signalling 2016, 28, 561-571. [CrossRef]
- Santos, S.D.; Verveer, P.J.; Bastiaens, P.I. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat Cell Biol 2007, 9, 324-330. [CrossRef]
- Kaur, N.; Lum, M.A.; Lewis, R.E.; Black, A.R.; Black, J.D. A novel antiproliferative PKCalpha-Ras-ERK signaling axis in intestinal epithelial cells. J Biol Chem 2022, 298, 102121. [CrossRef]
- Lawan, A.; Al-Harthi, S.; Cadalbert, L.; McCluskey, A.G.; Shweash, M.; Grassia, G.; Grant, A.; Boyd, M.; Currie, S.; Plevin, R. Deletion of the dual specific phosphatase-4 (DUSP-4) gene reveals an essential non-redundant role for MAP kinase phosphatase-2 (MKP-2) in proliferation and cell survival. J Biol Chem 2011, 286, 12933-12943. [CrossRef]
- Unni, A.M.; Harbourne, B.; Oh, M.H.; Wild, S.; Ferrarone, J.R.; Lockwood, W.W.; Varmus, H. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. eLife 2018, 7. [CrossRef]
- Shojaee, S.; Caeser, R.; Buchner, M.; Park, E.; Swaminathan, S.; Hurtz, C.; Geng, H.; Chan, L.N.; Klemm, L.; Hofmann, W.K.; et al. Erk Negative Feedback Control Enables Pre-B Cell Transformation and Represents a Therapeutic Target in Acute Lymphoblastic Leukemia. Cancer Cell 2015, 28, 114-128. [CrossRef]
- Holck, S.; Bonde, J.; Pedersen, H.; Petersen, A.A.; Chaube, A.; Nielsen, H.J.; Larsson, L.I. Localization of active, dually phosphorylated extracellular signal-regulated kinase 1 and 2 in colorectal cancer with or without activating BRAF and KRAS mutations. Hum Pathol 2016, 54, 37-46. [CrossRef]
- Finlay, D.; Healy, V.; Furlong, F.; O'Connell, F.C.; Keon, N.K.; Martin, F. MAP kinase pathway signalling is essential for extracellular matrix determined mammary epithelial cell survival. Cell death and differentiation 2000, 7, 302-313. [CrossRef]
- Blasco, R.B.; Francoz, S.; Santamaria, D.; Canamero, M.; Dubus, P.; Charron, J.; Baccarini, M.; Barbacid, M. c-Raf, but Not B-Raf, Is Essential for Development of K-Ras Oncogene-Driven Non-Small Cell Lung Carcinoma. Cancer Cell 2011, 19, 652-663. [CrossRef]
- Dehan, E.; Bassermann, F.; Guardavaccaro, D.; Vasiliver-Shamis, G.; Cohen, M.; Lowes, K.N.; Dustin, M.; Huang, D.C.; Taunton, J.; Pagano, M. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol Cell 2009, 33, 109-116. [CrossRef]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol 2008, 10, 138-148. [CrossRef]
- Murphy, L.O.; Smith, S.; Chen, R.H.; Fingar, D.C.; Blenis, J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol 2002, 4, 556-564.
- Mylona, A.; Theillet, F.X.; Foster, C.; Cheng, T.M.; Miralles, F.; Bates, P.A.; Selenko, P.; Treisman, R. Opposing effects of Elk-1 multisite phosphorylation shape its response to ERK activation. Science 2016, 354, 233-237. [CrossRef]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138-142. [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 2000, 14, 2501-2514.
- Stefanovsky, V.Y.; Moss, T. The splice variants of UBF differentially regulate RNA polymerase I transcription elongation in response to ERK phosphorylation. Nucleic Acids Res 2008, 36, 5093-5101. [CrossRef]
- Albeck, J.G.; Mills, G.B.; Brugge, J.S. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol Cell 2013, 49, 249-261. [CrossRef]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668-682. [CrossRef]
- Sale, M.J.; Balmanno, K.; Saxena, J.; Ozono, E.; Wojdyla, K.; McIntyre, R.E.; Gilley, R.; Woroniuk, A.; Howarth, K.D.; Hughes, G.; et al. MEK1/2 inhibitor withdrawal reverses acquired resistance driven by BRAF(V600E) amplification whereas KRAS(G13D) amplification promotes EMT-chemoresistance. Nat Commun 2019, 10, 2030. [CrossRef]
- Muta, Y.; Fujita, Y.; Sumiyama, K.; Sakurai, A.; Taketo, M.M.; Chiba, T.; Seno, H.; Aoki, K.; Matsuda, M.; Imajo, M. Composite regulation of ERK activity dynamics underlying tumour-specific traits in the intestine. Nat Commun 2018, 9, 2174. [CrossRef]
- Lin, Y.; Sohn, C.H.; Dalal, C.K.; Cai, L.; Elowitz, M.B. Combinatorial gene regulation by modulation of relative pulse timing. Nature 2015, 527, 54-58. [CrossRef]
- Wang, Z.; Potoyan, D.A.; Wolynes, P.G. Molecular stripping, targets and decoys as modulators of oscillations in the NF-kappaB/IkappaBalpha/DNA genetic network. Journal of the Royal Society, Interface / the Royal Society 2016, 13. [CrossRef]
- Ender, P.; Gagliardi, P.A.; Dobrzynski, M.; Frismantiene, A.; Dessauges, C.; Hohener, T.; Jacques, M.A.; Cohen, A.R.; Pertz, O. Spatiotemporal control of ERK pulse frequency coordinates fate decisions during mammary acinar morphogenesis. Dev Cell 2022, 57, 2153-2167 e2156. [CrossRef]
- Pokrass, M.J.; Ryan, K.A.; Xin, T.; Pielstick, B.; Timp, W.; Greco, V.; Regot, S. Cell-Cycle-Dependent ERK Signaling Dynamics Direct Fate Specification in the Mammalian Preimplantation Embryo. Dev Cell 2020, 55, 328-340 e325. [CrossRef]
- Byun, M.R.; Kim, A.R.; Hwang, J.H.; Kim, K.M.; Hwang, E.S.; Hong, J.H. FGF2 stimulates osteogenic differentiation through ERK induced TAZ expression. Bone 2014, 58, 72-80. [CrossRef]
- Fremin, C.; Saba-El-Leil, M.K.; Levesque, K.; Ang, S.L.; Meloche, S. Functional Redundancy of ERK1 and ERK2 MAP Kinases during Development. Cell reports 2015, 12, 913-921. [CrossRef]
- Newbern, J.M.; Li, X.; Shoemaker, S.E.; Zhou, J.; Zhong, J.; Wu, Y.; Bonder, D.; Hollenback, S.; Coppola, G.; Geschwind, D.H.; et al. Specific functions for ERK/MAPK signaling during PNS development. Neuron 2011, 69, 91-105. [CrossRef]
- Parada, C.; Han, D.; Grimaldi, A.; Sarrion, P.; Park, S.S.; Pelikan, R.; Sanchez-Lara, P.A.; Chai, Y. Disruption of the ERK/MAPK pathway in neural crest cells as a potential cause of Pierre Robin sequence. Development (Cambridge, England) 2015, 142, 3734-3745. [CrossRef]
- Offermann, B.; Knauer, S.; Singh, A.; Fernandez-Cachon, M.L.; Klose, M.; Kowar, S.; Busch, H.; Boerries, M. Boolean Modeling Reveals the Necessity of Transcriptional Regulation for Bistability in PC12 Cell Differentiation. Front Genet 2016, 7, 44. [CrossRef]
- Johnson, H.E.; Toettcher, J.E. Signaling Dynamics Control Cell Fate in the Early Drosophila Embryo. Dev Cell 2019, 48, 361-370 e363. [CrossRef]
- Silva, J.; Smith, A. Capturing pluripotency. Cell 2008, 132, 532-536. [CrossRef]
- Pond, K.W.; Morris, J.M.; Alkhimenok, O.; Varghese, R.P.; Cabel, C.R.; Ellis, N.A.; Chakrabarti, J.; Zavros, Y.; Merchant, J.L.; Thorne, C.A.; et al. Live-cell imaging in human colonic monolayers reveals ERK waves limit the stem cell compartment to maintain epithelial homeostasis. eLife 2022, 11. [CrossRef]
- Mulas, C.; Kalkan, T.; von Meyenn, F.; Leitch, H.G.; Nichols, J.; Smith, A. Defined conditions for propagation and manipulation of mouse embryonic stem cells. Development (Cambridge, England) 2019, 146. [CrossRef]
- Chappell, J.; Sun, Y.; Singh, A.; Dalton, S. MYC/MAX control ERK signaling and pluripotency by regulation of dual-specificity phosphatases 2 and 7. Genes Dev 2013, 27, 725-733. [CrossRef]
- Mzoughi, S.; Zhang, J.; Hequet, D.; Teo, S.X.; Fang, H.; Xing, Q.R.; Bezzi, M.; Seah, M.K.Y.; Ong, S.L.M.; Shin, E.M.; et al. PRDM15 safeguards naive pluripotency by transcriptionally regulating WNT and MAPK-ERK signaling. Nat Genet 2017, 49, 1354-1363. [CrossRef]
- Nett, I.R.; Mulas, C.; Gatto, L.; Lilley, K.S.; Smith, A. Negative feedback via RSK modulates Erk-dependent progression from naive pluripotency. EMBO Rep 2018, 19. [CrossRef]
- Gu, H.; Li, Q.; Huang, S.; Lu, W.; Cheng, F.; Gao, P.; Wang, C.; Miao, L.; Mei, Y.; Wu, M. Mitochondrial E3 ligase March5 maintains stemness of mouse ES cells via suppression of ERK signalling. Nat Commun 2015, 6, 7112. [CrossRef]
- Hamilton, W.B.; Brickman, J.M. Erk signaling suppresses embryonic stem cell self-renewal to specify endoderm. Cell reports 2014, 9, 2056-2070. [CrossRef]
- Tee, W.W.; Shen, S.S.; Oksuz, O.; Narendra, V.; Reinberg, D. Erk1/2 activity promotes chromatin features and RNAPII phosphorylation at developmental promoters in mouse ESCs. Cell 2014, 156, 678-690. [CrossRef]
- Brons, I.G.; Smithers, L.E.; Trotter, M.W.; Rugg-Gunn, P.; Sun, B.; Chuva de Sousa Lopes, S.M.; Howlett, S.K.; Clarkson, A.; Ahrlund-Richter, L.; Pedersen, R.A.; et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 2007, 448, 191-195. [CrossRef]
- Tesar, P.J.; Chenoweth, J.G.; Brook, F.A.; Davies, T.J.; Evans, E.P.; Mack, D.L.; Gardner, R.L.; McKay, R.D. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 2007, 448, 196-199. [CrossRef]
- Goke, J.; Chan, Y.S.; Yan, J.; Vingron, M.; Ng, H.H. Genome-wide kinase-chromatin interactions reveal the regulatory network of ERK signaling in human embryonic stem cells. Mol Cell 2013, 50, 844-855. [CrossRef]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc Natl Acad Sci U S A 2015, 112, E4600-4609. [CrossRef]
- Lu, H.; Tran, L.; Park, Y.; Chen, I.; Lan, J.; Xie, Y.; Semenza, G.L. Reciprocal Regulation of DUSP9 and DUSP16 Expression by HIF1 Controls ERK and p38 MAP Kinase Activity and Mediates Chemotherapy-Induced Breast Cancer Stem Cell Enrichment. Cancer Res 2018, 78, 4191-4202. [CrossRef]
- Deschenes-Simard, X.; Gaumont-Leclerc, M.F.; Bourdeau, V.; Lessard, F.; Moiseeva, O.; Forest, V.; Igelmann, S.; Mallette, F.A.; Saba-El-Leil, M.K.; Meloche, S.; et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev 2013, 27, 900-915. [CrossRef]
- Deschenes-Simard, X.; Parisotto, M.; Rowell, M.C.; Le Calve, B.; Igelmann, S.; Moineau-Vallee, K.; Saint-Germain, E.; Kalegari, P.; Bourdeau, V.; Kottakis, F.; et al. Circumventing senescence is associated with stem cell properties and metformin sensitivity. Aging Cell 2019, e12889. [CrossRef]
- Aguilar-Martinez, E.; Morrisroe, C.; Sharrocks, A.D. The ubiquitin ligase UBE3A dampens ERK pathway signalling in HPV E6 transformed HeLa cells. PloS one 2015, 10, e0119366. [CrossRef]
- Lee, S.W.; Fang, L.; Igarashi, M.; Ouchi, T.; Lu, K.P.; Aaronson, S.A. Sustained activation of Ras/Raf/mitogen-activated protein kinase cascade by the tumor suppressor p53. Proc Natl Acad Sci U S A 2000, 97, 8302-8305. [CrossRef]
- Deschenes-Simard, X.; Lessard, F.; Gaumont-Leclerc, M.F.; Bardeesy, N.; Ferbeyre, G. Cellular senescence and protein degradation: Breaking down cancer. Cell Cycle 2014, 13, 1840-1858. [CrossRef]
- Lessard, F.; Igelmann, S.; Trahan, C.; Huot, G.; Saint-Germain, E.; Mignacca, L.; Del Toro, N.; Lopes-Paciencia, S.; Le Calve, B.; Montero, M.; et al. Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat Cell Biol 2018, 20, 789-799. [CrossRef]
- Igelmann, S.; Lessard, F.; Uchenunu, O.; Bouchard, J.; Fernandez-Ruiz, A.; Rowell, M.C.; Lopes-Paciencia, S.; Papadopoli, D.; Fouillen, A.; Ponce, K.J.; et al. A hydride transfer complex reprograms NAD metabolism and bypasses senescence. Mol Cell 2021, 81, 3848-3865 e3819. [CrossRef]
- Chen, J.Y.; Hug, C.; Reyes, J.; Tian, C.; Gerosa, L.; Frohlich, F.; Ponsioen, B.; Snippert, H.J.G.; Spencer, S.L.; Jambhekar, A.; et al. Multi-range ERK responses shape the proliferative trajectory of single cells following oncogene induction. Cell reports 2023, 42, 112252. [CrossRef]
- Rowell, M.C.; Deschenes-Simard, X.; Lopes-Paciencia, S.; Le Calve, B.; Kalegari, P.; Mignacca, L.; Fernandez-Ruiz, A.; Guillon, J.; Lessard, F.; Bourdeau, V.; et al. Targeting ribosome biogenesis reinforces ERK-dependent senescence in pancreatic cancer. Cell cycle (Georgetown, Tex 2023, 1-22. [CrossRef]
- Lin, Y.K.; Wu, W.; Ponce, R.K.; Kim, J.W.; Okimoto, R.A. Negative MAPK-ERK regulation sustains CIC-DUX4 oncoprotein expression in undifferentiated sarcoma. Proc Natl Acad Sci U S A 2020, 117, 20776-20784. [CrossRef]
- Ingram, K.; Samson, S.C.; Zewdu, R.; Zitnay, R.G.; Snyder, E.L.; Mendoza, M.C. NKX2-1 controls lung cancer progression by inducing DUSP6 to dampen ERK activity. Oncogene 2022, 41, 293-300. [CrossRef]
- Ito, T.; Young, M.J.; Li, R.; Jain, S.; Wernitznig, A.; Krill-Burger, J.M.; Lemke, C.T.; Monducci, D.; Rodriguez, D.J.; Chang, L.; et al. Paralog knockout profiling identifies DUSP4 and DUSP6 as a digenic dependence in MAPK pathway-driven cancers. Nat Genet 2021, 53, 1664-1672. [CrossRef]
- Kidger, A.M.; Rushworth, L.K.; Stellzig, J.; Davidson, J.; Bryant, C.J.; Bayley, C.; Caddye, E.; Rogers, T.; Keyse, S.M.; Caunt, C.J. Dual-specificity phosphatase 5 controls the localized inhibition, propagation, and transforming potential of ERK signaling. Proc Natl Acad Sci U S A 2017, 114, E317-E326. [CrossRef]
- Sangrar, W.; Shi, C.; Mullins, G.; LeBrun, D.; Ingalls, B.; Greer, P.A. Amplified Ras-MAPK signal states correlate with accelerated EGFR internalization, cytostasis and delayed HER2 tumor onset in Fer-deficient model systems. Oncogene 2015, 34, 4109-4117. [CrossRef]
- Moniz, S.; Verissimo, F.; Matos, P.; Brazao, R.; Silva, E.; Kotelevets, L.; Chastre, E.; Gespach, C.; Jordan, P. Protein kinase WNK2 inhibits cell proliferation by negatively modulating the activation of MEK1/ERK1/2. Oncogene 2007, 26, 6071-6081. [CrossRef]
- Shankar, S.; Tien, J.C.; Siebenaler, R.F.; Chugh, S.; Dommeti, V.L.; Zelenka-Wang, S.; Wang, X.M.; Apel, I.J.; Waninger, J.; Eyunni, S.; et al. An essential role for Argonaute 2 in EGFR-KRAS signaling in pancreatic cancer development. Nat Commun 2020, 11, 2817. [CrossRef]
- Tien, J.C.; Chugh, S.; Goodrum, A.E.; Cheng, Y.; Mannan, R.; Zhang, Y.; Wang, L.; Dommeti, V.L.; Wang, X.; Xu, A.; et al. AGO2 promotes tumor progression in KRAS-driven mouse models of non-small cell lung cancer. Proc Natl Acad Sci U S A 2021, 118. [CrossRef]
- Wu, P.K.; Hong, S.K.; Park, J.I. Steady-State Levels of Phosphorylated Mitogen-Activated Protein Kinase Kinase 1/2 Determined by Mortalin/HSPA9 and Protein Phosphatase 1 Alpha in KRAS and BRAF Tumor Cells. Mol Cell Biol 2017, 37. [CrossRef]
- Kasitinon, S.Y.; Eskiocak, U.; Martin, M.; Bezwada, D.; Khivansara, V.; Tasdogan, A.; Zhao, Z.; Mathews, T.; Aurora, A.B.; Morrison, S.J. TRPML1 Promotes Protein Homeostasis in Melanoma Cells by Negatively Regulating MAPK and mTORC1 Signaling. Cell reports 2019, 28, 2293-2305 e2299. [CrossRef]
- Iwamoto, N.; D'Alessandro, L.A.; Depner, S.; Hahn, B.; Kramer, B.A.; Lucarelli, P.; Vlasov, A.; Stepath, M.; Bohm, M.E.; Deharde, D.; et al. Context-specific flow through the MEK/ERK module produces cell- and ligand-specific patterns of ERK single and double phosphorylation. Sci Signal 2016, 9, ra13. [CrossRef]
- Buonato, J.M.; Lazzara, M.J. ERK1/2 blockade prevents epithelial-mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res 2014, 74, 309-319. [CrossRef]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 2013, 24, 466-480. [CrossRef]
- Doehn, U.; Hauge, C.; Frank, S.R.; Jensen, C.J.; Duda, K.; Nielsen, J.V.; Cohen, M.S.; Johansen, J.V.; Winther, B.R.; Lund, L.R.; et al. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol Cell 2009, 35, 511-522. [CrossRef]
- Shin, S.; Dimitri, C.A.; Yoon, S.O.; Dowdle, W.; Blenis, J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell 2010, 38, 114-127. [CrossRef]
- Ma, Q.; Guin, S.; Padhye, S.S.; Zhou, Y.Q.; Zhang, R.W.; Wang, M.H. Ribosomal protein S6 kinase (RSK)-2 as a central effector molecule in RON receptor tyrosine kinase mediated epithelial to mesenchymal transition induced by macrophage-stimulating protein. Molecular cancer 2011, 10, 66. [CrossRef]
- Ichikawa, K.; Kubota, Y.; Nakamura, T.; Weng, J.S.; Tomida, T.; Saito, H.; Takekawa, M. MCRIP1, an ERK substrate, mediates ERK-induced gene silencing during epithelial-mesenchymal transition by regulating the co-repressor CtBP. Mol Cell 2015, 58, 35-46. [CrossRef]
- Liu, Y.; El-Naggar, S.; Darling, D.S.; Higashi, Y.; Dean, D.C. Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development (Cambridge, England) 2008, 135, 579-588. [CrossRef]
- Liu, Y.; Lu, X.; Huang, L.; Wang, W.; Jiang, G.; Dean, K.C.; Clem, B.; Telang, S.; Jenson, A.B.; Cuatrecasas, M.; et al. Different thresholds of ZEB1 are required for Ras-mediated tumour initiation and metastasis. Nat Commun 2014, 5, 5660. [CrossRef]
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008, 14, 79-89.
- Weiss, M.B.; Abel, E.V.; Mayberry, M.M.; Basile, K.J.; Berger, A.C.; Aplin, A.E. TWIST1 is an ERK1/2 effector that promotes invasion and regulates MMP-1 expression in human melanoma cells. Cancer Res 2012, 72, 6382-6392. [CrossRef]
- Principe, D.R.; Diaz, A.M.; Torres, C.; Mangan, R.J.; DeCant, B.; McKinney, R.; Tsao, M.S.; Lowy, A.; Munshi, H.G.; Jung, B.; et al. TGFbeta engages MEK/ERK to differentially regulate benign and malignant pancreas cell function. Oncogene 2017, 36, 4336-4348. [CrossRef]
- Deschenes-Simard, X.; Rowell, M.C.; Ferbeyre, G. Metformin turns off the metabolic switch of pancreatic cancer. Aging (Albany NY) 2019, 11, 10793-10795. [CrossRef]
- Caldwell, M.E.; DeNicola, G.M.; Martins, C.P.; Jacobetz, M.A.; Maitra, A.; Hruban, R.H.; Tuveson, D.A. Cellular features of senescence during the evolution of human and murine ductal pancreatic cancer. Oncogene 2012, 31, 1599-1608. [CrossRef]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernandez-Porras, I.; Canamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011, 19, 728-739. [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349-361. [CrossRef]
- Blaj, C.; Schmidt, E.M.; Lamprecht, S.; Hermeking, H.; Jung, A.; Kirchner, T.; Horst, D. Oncogenic Effects of High MAPK Activity in Colorectal Cancer Mark Progenitor Cells and Persist Irrespective of RAS Mutations. Cancer Res 2017, 77, 1763-1774. [CrossRef]
- Chiu, L.Y.; Hsin, I.L.; Yang, T.Y.; Sung, W.W.; Chi, J.Y.; Chang, J.T.; Ko, J.L.; Sheu, G.T. The ERK-ZEB1 pathway mediates epithelial-mesenchymal transition in pemetrexed resistant lung cancer cells with suppression by vinca alkaloids. Oncogene 2017, 36, 242-253. [CrossRef]
- Latil, M.; Nassar, D.; Beck, B.; Boumahdi, S.; Wang, L.; Brisebarre, A.; Dubois, C.; Nkusi, E.; Lenglez, S.; Checinska, A.; et al. Cell-Type-Specific Chromatin States Differentially Prime Squamous Cell Carcinoma Tumor-Initiating Cells for Epithelial to Mesenchymal Transition. Cell stem cell 2017, 20, 191-204 e195. [CrossRef]
- Sugiura, R.; Satoh, R.; Takasaki, T. ERK: A Double-Edged Sword in Cancer. ERK-Dependent Apoptosis as a Potential Therapeutic Strategy for Cancer. Cells 2021, 10. [CrossRef]
- Shin, S.; Buel, G.R.; Wolgamott, L.; Plas, D.R.; Asara, J.M.; Blenis, J.; Yoon, S.O. ERK2 Mediates Metabolic Stress Response to Regulate Cell Fate. Mol Cell 2015, 59, 382-398. [CrossRef]
- Hong, S.K.; Wu, P.K.; Park, J.I. A cellular threshold for active ERK1/2 levels determines Raf/MEK/ERK-mediated growth arrest versus death responses. Cellular signalling 2018, 42, 11-20. [CrossRef]
- Leung, G.P.; Feng, T.; Sigoillot, F.D.; Geyer, F.C.; Shirley, M.D.; Ruddy, D.A.; Rakiec, D.P.; Freeman, A.K.; Engelman, J.A.; Jaskelioff, M.; et al. Hyperactivation of MAPK Signaling Is Deleterious to RAS/RAF-mutant Melanoma. Mol Cancer Res 2019, 17, 199-211. [CrossRef]
- Ecker, V.; Brandmeier, L.; Stumpf, M.; Giansanti, P.; Moreira, A.V.; Pfeuffer, L.; Fens, M.; Lu, J.; Kuster, B.; Engleitner, T.; et al. Negative feedback regulation of MAPK signaling is an important driver of chronic lymphocytic leukemia progression. Cell reports 2023, 42, 113017. [CrossRef]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.C.; Ho, Y.J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416-1422. [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628-632. [CrossRef]
Figure 1.
The Extracellular Signal-Regulated Kinase (ERK) signaling module is controlled by negative and positive feedback signals that determine the intensity and duration of the signal. The module is activated by ligand binding to membrane receptors (R) that activate the GTPase RAS via adaptor proteins (A) such as Sos (Son of sevenless). The signaling module includes a kinase cascade formed by RAF, MEK, and ERK (Adapted from Biorender).
Figure 1.
The Extracellular Signal-Regulated Kinase (ERK) signaling module is controlled by negative and positive feedback signals that determine the intensity and duration of the signal. The module is activated by ligand binding to membrane receptors (R) that activate the GTPase RAS via adaptor proteins (A) such as Sos (Son of sevenless). The signaling module includes a kinase cascade formed by RAF, MEK, and ERK (Adapted from Biorender).
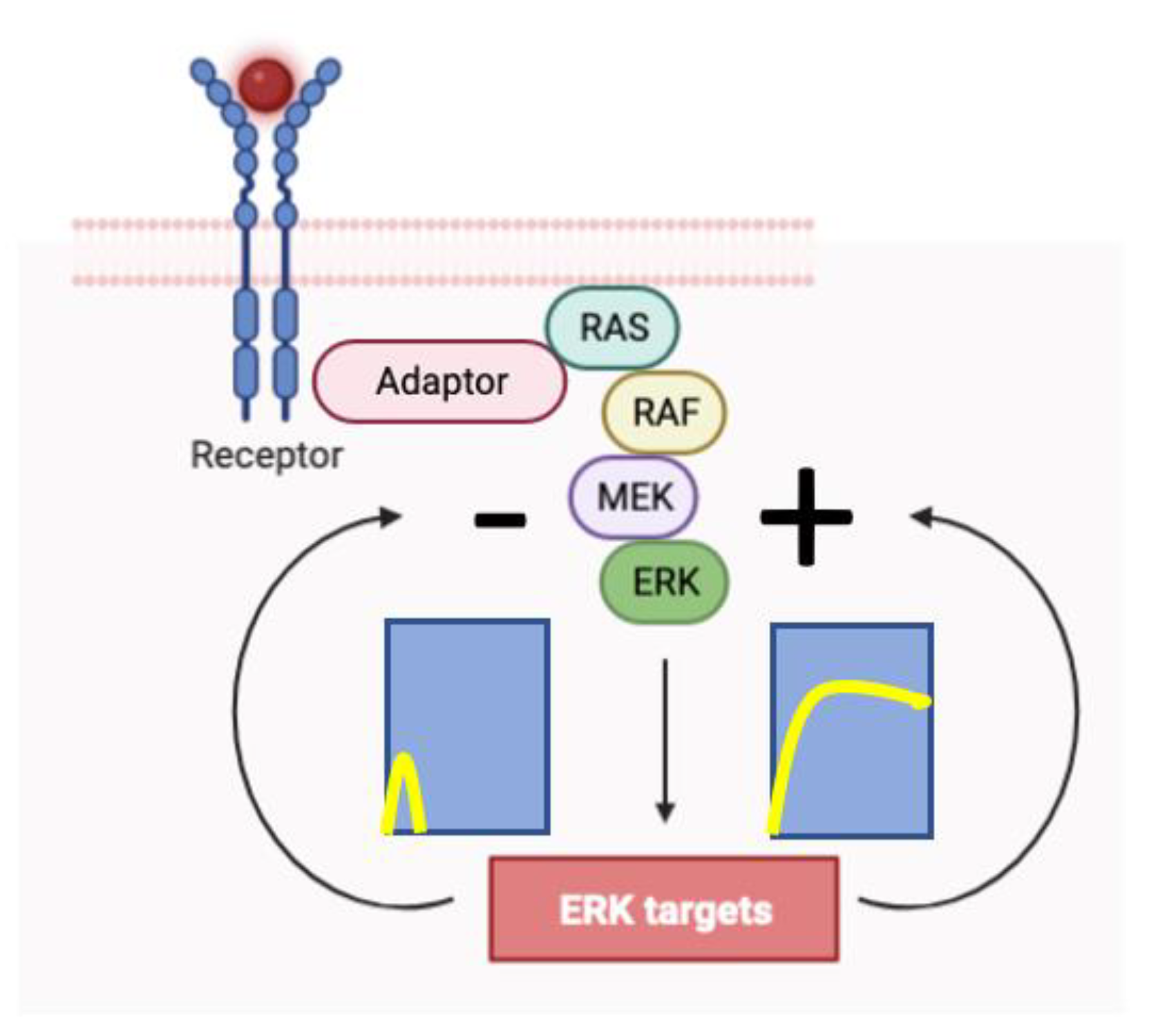
Figure 2.
Decoding ERK-signaling duration by integrating ERK-dependent IEG transcription (early effect) with ERK-dependent IEG stabilization (late effect). (a) IEG are regulated by unstable transcription factors phosphorylated by ERK at sites that drive protein degradation. (b) Sustained ERK activity stabilizes IEG by further phosphorylation events. These TFs control cell cycle genes.
Figure 2.
Decoding ERK-signaling duration by integrating ERK-dependent IEG transcription (early effect) with ERK-dependent IEG stabilization (late effect). (a) IEG are regulated by unstable transcription factors phosphorylated by ERK at sites that drive protein degradation. (b) Sustained ERK activity stabilizes IEG by further phosphorylation events. These TFs control cell cycle genes.
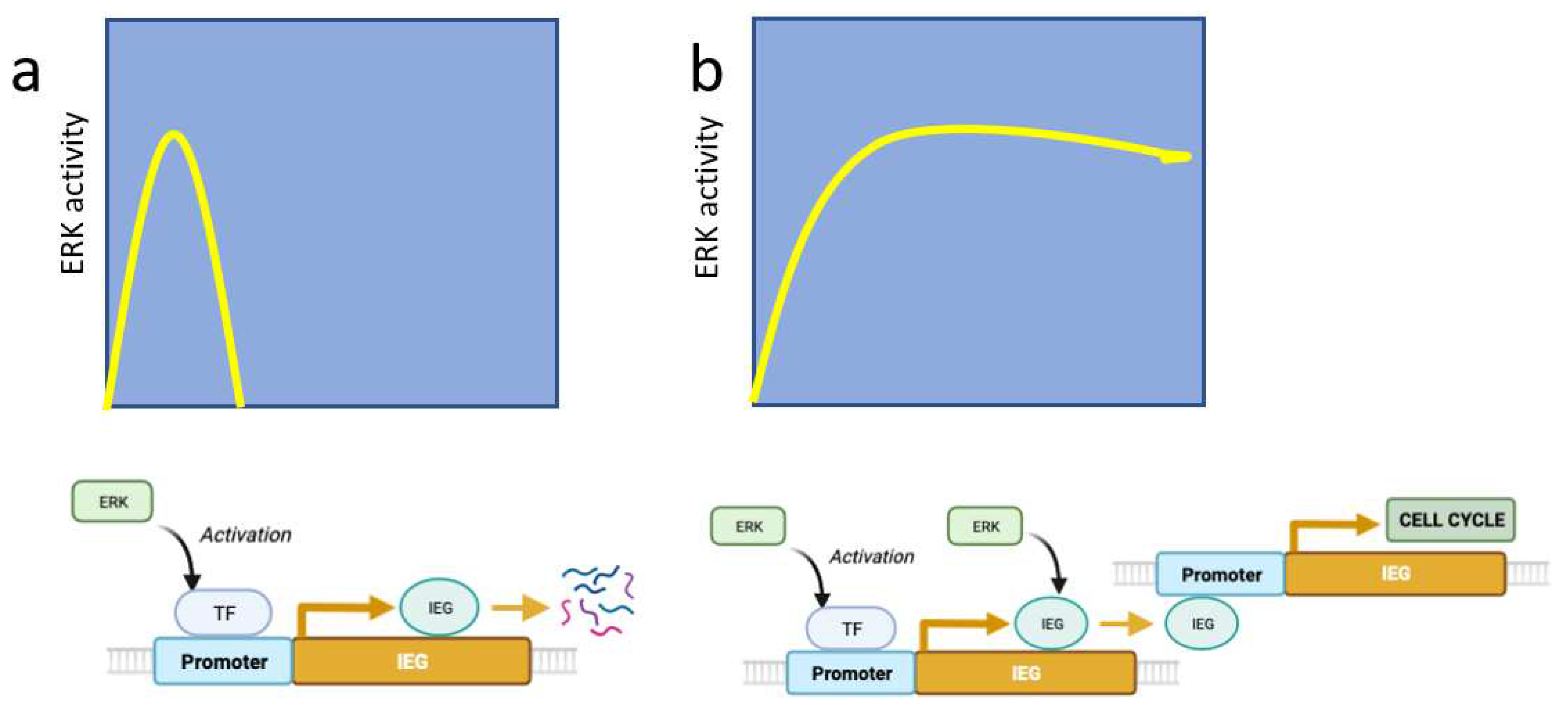
Figure 3.
Decoding ERK-signaling duration and intensity by multisite phosphorylation. In this model, originally described for Elk1 phosphorylation, prolonged ERK signals are required to phosphorylate low-affinity inhibitory sites that turn-off early early-acting transcription factors. It is plausible that lasting ERK signals may also activate other transcription factors having low-affinity activation sites.
Figure 3.
Decoding ERK-signaling duration and intensity by multisite phosphorylation. In this model, originally described for Elk1 phosphorylation, prolonged ERK signals are required to phosphorylate low-affinity inhibitory sites that turn-off early early-acting transcription factors. It is plausible that lasting ERK signals may also activate other transcription factors having low-affinity activation sites.
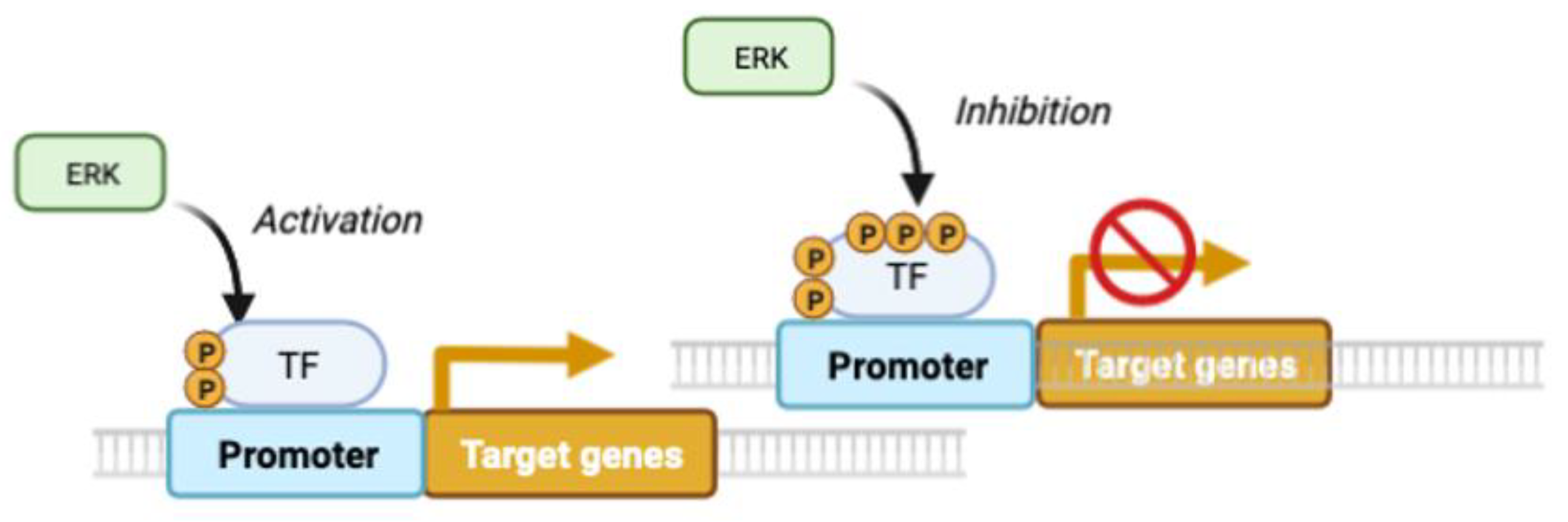
Figure 4.
Decoding ERK pulses by pulses of transcription factor activation. (a) Pulses of ERK activities are transmitted to effector molecules such as transcription factors. (b) Modulating the frequency, amplitude, and duration of ERK activity pulses can control different outcomes.
Figure 4.
Decoding ERK pulses by pulses of transcription factor activation. (a) Pulses of ERK activities are transmitted to effector molecules such as transcription factors. (b) Modulating the frequency, amplitude, and duration of ERK activity pulses can control different outcomes.
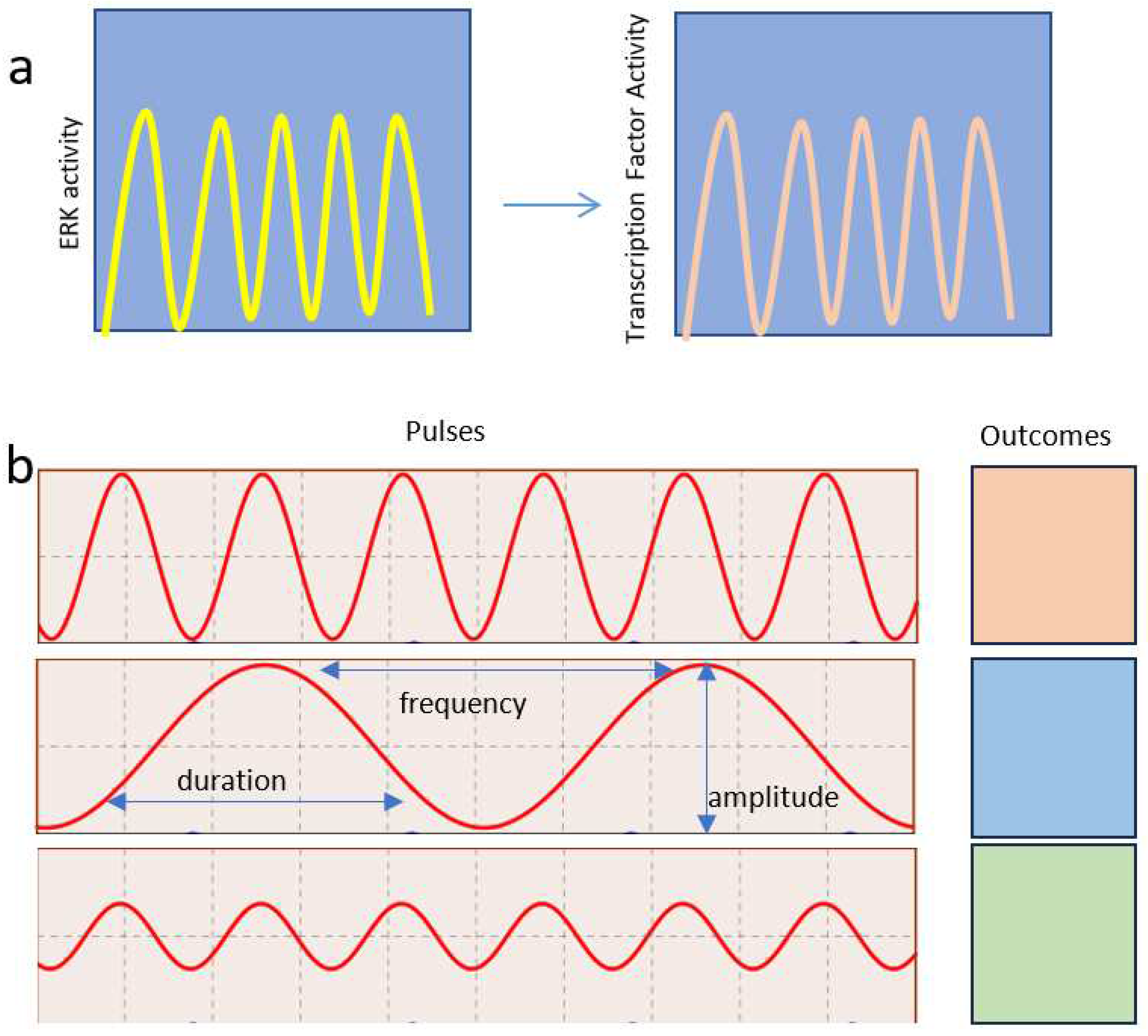
Figure 5.
Decoding ERK-signaling intensity by cumulative ERK activation. Fate 2 can be attained by a lasting ERK induction or the accumulation of the same signal after several pulses of activation (based on Johnson and Toettcher). This model implies that ERK signaling is somehow remembered in cells receiving short pulses of ERK induction.
Figure 5.
Decoding ERK-signaling intensity by cumulative ERK activation. Fate 2 can be attained by a lasting ERK induction or the accumulation of the same signal after several pulses of activation (based on Johnson and Toettcher). This model implies that ERK signaling is somehow remembered in cells receiving short pulses of ERK induction.
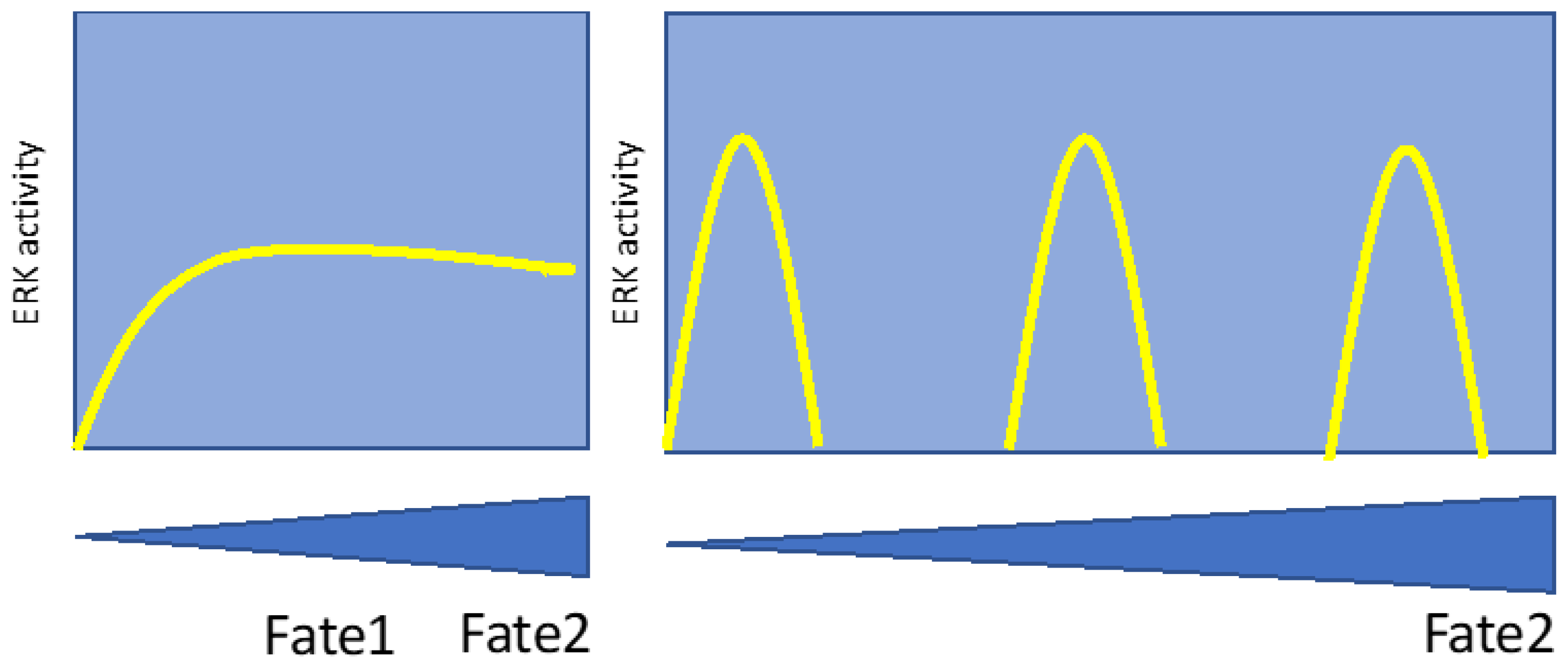
Figure 6.
Decoding ERK signal intensity by protein degradation. In this model, high-intensity ERK signals trigger the degradation of many ERK targets by coupling protein phosphorylation to ubiquitin-mediated protein degradation. Prolonged or intense ERK signals are required to increase the stoichiometry of phosphorylation so subsequent degradation effectively reduces protein levels. This was described in senescent cells as SAPD (senescence-associated protein degradation) but also in mouse ES cells that lose pluripotency upon ERK activation. It is unknown whether SAPD required sustained high ERK signals (red curve) or a burst of high ERK activity (yellow curve).
Figure 6.
Decoding ERK signal intensity by protein degradation. In this model, high-intensity ERK signals trigger the degradation of many ERK targets by coupling protein phosphorylation to ubiquitin-mediated protein degradation. Prolonged or intense ERK signals are required to increase the stoichiometry of phosphorylation so subsequent degradation effectively reduces protein levels. This was described in senescent cells as SAPD (senescence-associated protein degradation) but also in mouse ES cells that lose pluripotency upon ERK activation. It is unknown whether SAPD required sustained high ERK signals (red curve) or a burst of high ERK activity (yellow curve).
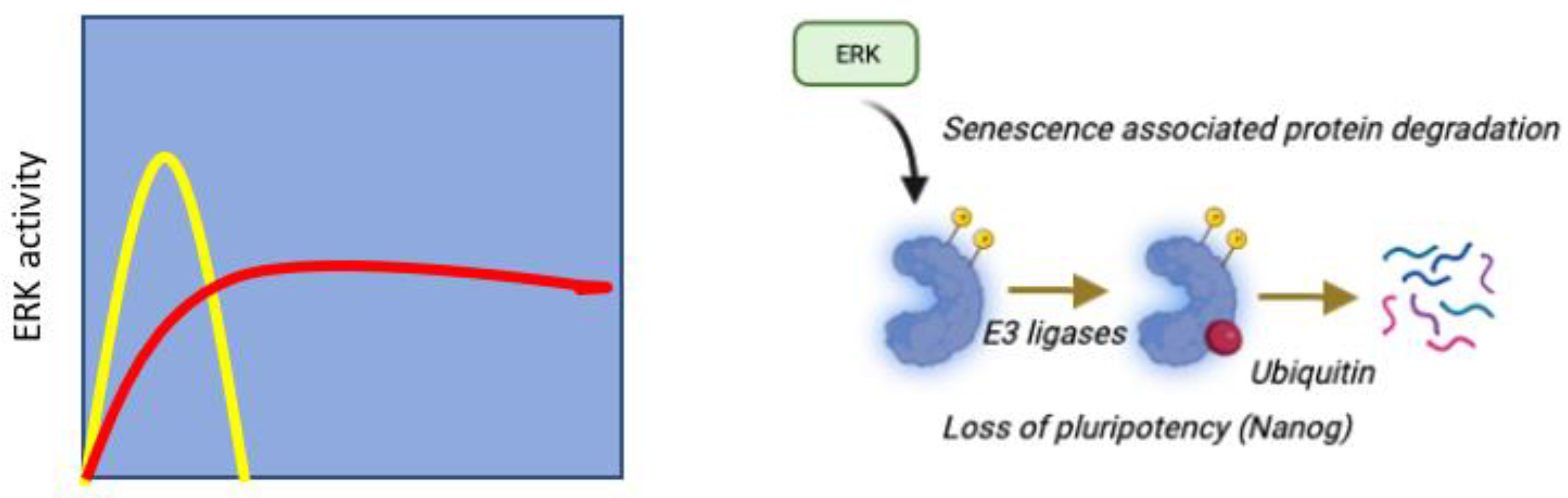
Figure 7.
Non-monotonic relationship between ERK activity and cell proliferation. The Goldilocks effect.
Figure 7.
Non-monotonic relationship between ERK activity and cell proliferation. The Goldilocks effect.
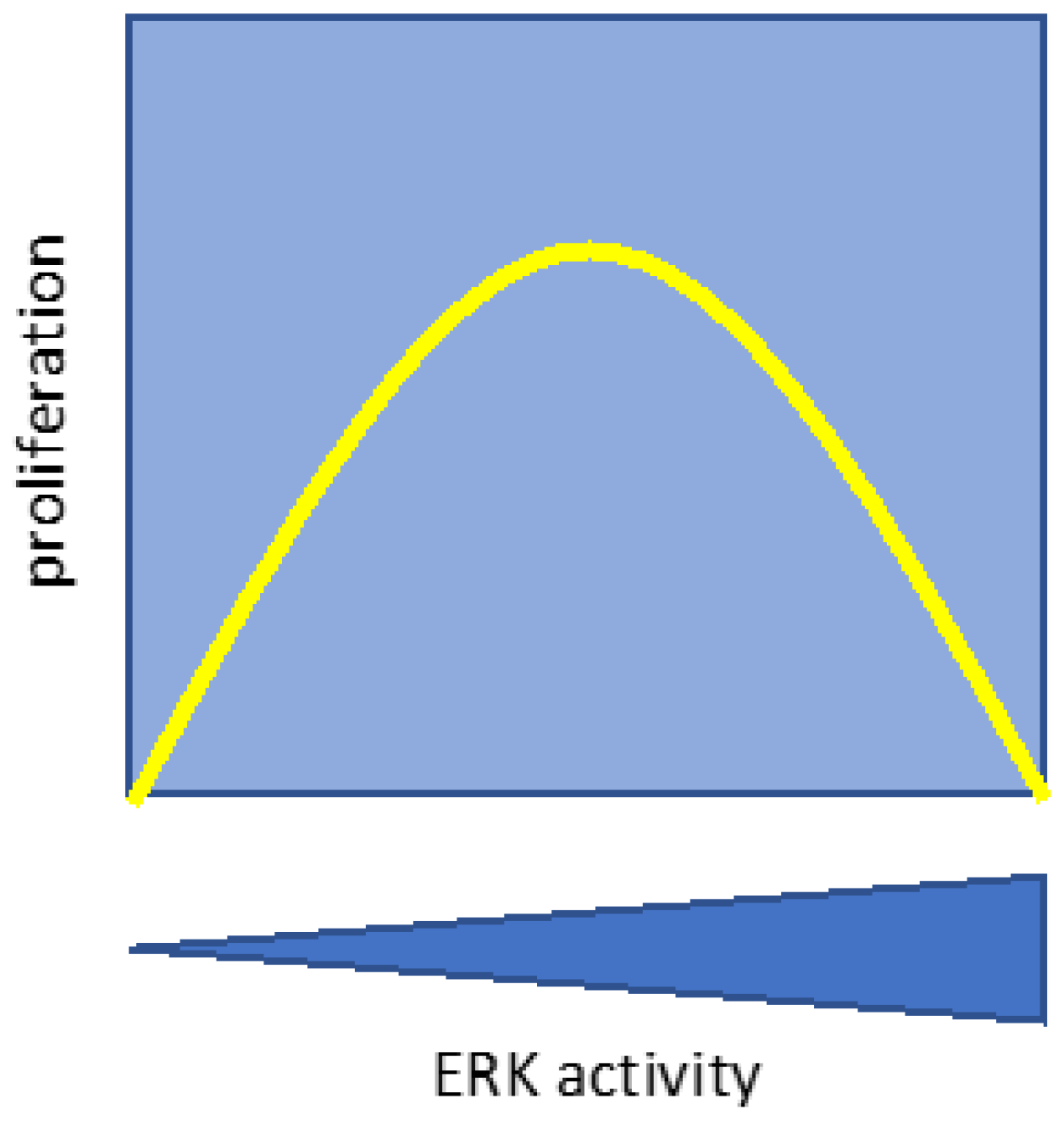
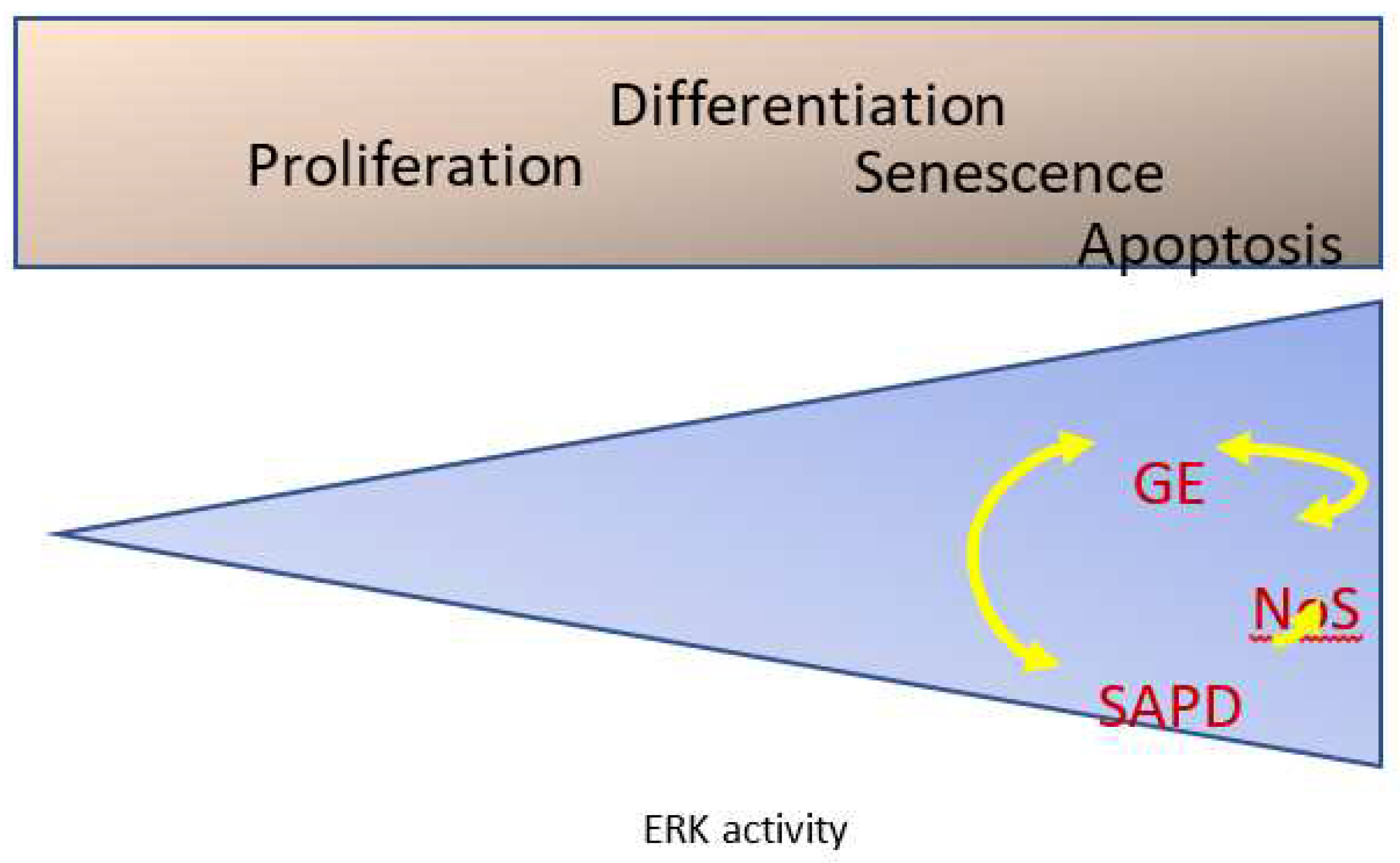
Figure 8.
Decoding intense ERK signaling to trigger senescence. Aberrantly high Erk activation triggers protein degradation (SAPD), changes in gene expression (GE), and nucleolar stress (NoS).
Figure 8.
Decoding intense ERK signaling to trigger senescence. Aberrantly high Erk activation triggers protein degradation (SAPD), changes in gene expression (GE), and nucleolar stress (NoS).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



