
Submitted:
30 November 2023
Posted:
30 November 2023
You are already at the latest version
Abstract
DNA extraction for downstream molecular diagnostic applications can be an expensive, time-consuming process. We devised a method to quickly extract genomic DNA from environmental samples based on sodium hydroxide lysis of cells with or without capture by magnetic beads, for subsequent PCR or quantitative PCR. The final DNA extraction method using NaOH is extremely low-cost and can be completed in 10 minutes at room temperature. NaOH extraction was effective for Gram-negative and Gram-positive bacteria in samples from air, soil, sewage, food, laboratory surfaces, and chicken cloacal swabs. The NaOH extraction method was comparable to commercial kits for extraction of DNA from pig fecal samples for 16S amplicon sequencing analyses. We demonstrate that an impinger and portable pump can efficiently capture bacteria in poultry facilities for rapid DNA extraction for quantification of total bacteria and for detection of specific species using qPCR. The air sampling and NaOH extraction procedures are well-suited for routine, high throughput screening, and for metagenomic analyses for specific pathogens, even in resource-limited situations.
Keywords:
microbiome
; bacteria
; DNA extraction
; poultry
; swine
; air sampling
Introduction
Reliable and accurate molecular diagnostics are crucial for the detection of pathogenic bacteria in clinical, food, and environmental samples [1,2,3]. In the study of bacterial genomes, one of the most important steps is the isolation of high-quality genomic DNA. There are various methods and commercial kits available for the rapid extraction of bacterial DNA from bacterial cells for polymerase chain reaction (PCR) [4,5,6,7]. To identify genomic sequences, the cells need to be lysed by heat treatment, physical, detergent, enzymes, pulverization, pH, chaotropic chemicals, or a combination of these, which is then followed by either chaotropic-based fractionation, phenol extraction, or size exclusion column purification [8,9,10,11,12]. Purification of the extracted DNA can involve precipitation or column-based nucleic acid capture. Traditionally, DNA extraction from cultured bacteria often includes expensive, and complicated enzyme combinations or costly hazardous chemicals such as phenol, or chloroform [13,14,15,16]. These methods vary in effectiveness depending on the application and the matrix from which the cells are obtained [17]. Although current techniques are well proven to produce high-quality nucleic acids (DNA or RNA), they are time-consuming, labor-intensive, and expensive to implement. In addition, the quantity of genomic DNA that they produce could be insufficient. These limitations become much more significant in settings with limited resources. As a result, the implementation of molecular testing is considerably hampered in many regions of the globe, especially in developing countries [12]. Therefore, the development of rapid, easy-to-use, and more efficient DNA extraction techniques that do not rely on complex laboratory equipment facilitates the advancement of molecular clinical detection technologies.
Recent years have seen the development of novel approaches for extracting DNA from plants [18], mouse tails [19], microbes from sediments [20,21], and fungal and oomycetes samples [22] using NaOH alone or in combination with other chemicals. Therefore, it has been proven that NaOH is capable of effectively lysing a variety of cell types from diverse biological materials within 10 minutes. The extraction would be most effective using NaOH, which would also inactivate nucleases during the extraction process. In most of these treatments, NaOH was administered along with reagents including sodium dodecyl sulfate or sodium acetate [18,23], or at elevated temperatures to disrupt cell walls, especially for Gram-positive bacteria [21,24]. Hence, these treatments complicate the protocol with additional PCR inhibitors or other time-consuming steps. Consequently, a fast DNA extraction procedure that consistently produces high DNA yields and can be applied to both Gram-positive and Gram-negative bacteria without the use of additional chemicals or enzymatic treatments would be beneficial in terms of saving time and money, while allowing for increased DNA yields.
The goal of this research was to develop an inexpensive technique for rapid lysis of bacteria that can be carried out with minimal laboratory equipment and used for subsequent DNA-based diagnostics. Our impetus was monitoring for specific bacteria in the environment during our lameness trials. In these trials we have shown that bacterial chondronecrosis with osteomyelitis (BCO) lameness can spread through the air from one pen to others [25,26]. We had previously used open agar plates for culture-based sampling of air during lameness trials [25]. However, culture-based sampling is less efficient than qPCR analysis with genus- or species-specific primers. We found that we could use a standard glass impinger to sample air for viable bacterial plate counts in our lameness trial facilities. However, extracting DNAs from these same samples was more problematic, especially for some species of concern in our experiments. We have found that simple alkaline extraction can be effective for liberation of DNA from many Gram positive and negative bacteria. The alkaline extract can either be diluted sufficiently for direct qPCR or the DNA can be directly captured from the alkaline solution using paramagnetic beads. The extraction method was effective for a variety of different environmental samples, including air, soil, sewage water, food, environmental surfaces, and chicken cloacal swabs, as well as broth and plate cultures. The optimized DNA preparation technique could be a substitute for the current expensive protocols, and commercial DNA extraction kits for the detection of various bacterial targets.
Materials and Methods
Reagents and Materials
Purified water was >18 MΩ from a Barnstead™ GenPure™ Pro Water Purification System (Thermo-Fisher Scientific, Waltham, MA, USA). Sterile pure water (SPW) was autoclaved purified water. Stocks of 1 M NaOH were prepared in SPW from solid (S318-3, Thermo-Fisher Scientific). Sterile swabs were autoclaved cotton tip swabs (Puritan LLC, Guilford, ME). Rectal samples were collected with Opti-Swab® Liquid Amies Collection & Transport System (Puritan LLC). Paramagnetic beads were Mag-Bind RXN Pure Plus (Omega Bio-Tek, Norcross, GA, USA), and PureSil-Silica Magnetic Beads (BioChain, Newark, CA, USA). The Mag-Bind RXN Pure Plus are provided in binding buffer. For the PureSil-Silica beads the bead binding buffer (BBB) was 80% isopropanol, 800 mM guanidine isothiocyanate, 8 mM TrisCl pH8, 3.52 mM Na2EDTA (BioChain). The commercial kit for DNA extraction from rectal fecal swabs was PowerLyzer PowerSoil DNA Isolation Kit (Qiagen, Hilden, Germany).
Bacterial Cultures and Growth
Bacterial strains are listed in Table 1. Bacterial stocks were stored at -80 oC in 40% glycerol. Media included: CHROMagar Orientation (CO) and CHROMagar Staphylococcus (CS) (DRG International, Inc., Springfield, NJ); tryptic soy broth (TSB) and nutrient broth (NB) (Difco Laboratories, Franklin Lakes, NJ). Difco bacteriological agar was added at 1.5% for solidification, when necessary. Overnight cultures of were diluted 1:100 in broth. Absorbance at 650 nm was used to compute the cell density based on a pre-calibrated formula:
CFU/ml = (A650 x 109) + 106
Alkaline Extraction from Environmental or Bacterial Culture Sample Preparation
For air samples: the air was collected by bubbling through 20 ml of 0.9% saline in a 30-ml autoclave-sterilized, glass impinger with sintered glass inlet tube (Chemglass Life Sciences LLC, Vineland, NJ) using a portable pipet pump for 20 minutes. Liquid samples were then centrifuged at 4400 x g for 10 minutes at 4 oC in a swinging bucket rotor. Pellets were re-suspended by vortex in 90 µl of SPW then mixed with 10 µl 1 M NaOH. See below for extraction processing.
For soil samples: the top dried layer of the ground was removed to a depth of 2–5 centimeters using a sterile spatula. The underlying moist, silt soil was collected in a sterile 50-ml conical tube. From the soil sample 50 mg was transferred into a 1.5 ml tube containing 450 µl of SPW, vortexed for 30 sec, followed by addition of 50 µl of 1 M NaOH, mixed and incubated 10 min at RT. The sample was centrifuged at 8000 x g for 5 minutes, and supernate collected for extraction processing (see below).
For cloacal or fecal swab samples: cloaca of day old chicks were swabbed using autoclaved cotton swabs. Pig rectal swabs were swirled in 450 µl of SPW in a 1.5 ml tube and mixed with 50 µl of 1 M NaOH. See below for extraction processing.
For food samples: a) the surface of a cheese block was swabbed using sterile cotton swabs then swirled in 450 µl of SPW in a 1.5 ml tube, and b) 10 mg of bread was added to 450 µl of SPW in 1.5 ml tubes. In both cases the tubes were vortexed for 30 seconds and mixed with 50 µl of 1 M NaOH. See below for extraction processing.
For environmental surfaces: sterile cotton swabs were rubbed on surfaces then swirled in 450 µl of SPW in a 1.5 ml tube which was then vortexed for 30 sec, and mixed with 50 µl of 1 M NaOH. See below for extraction processing.
For environmental liquid sampling: 1 ml sample in a 1.5 ml tube was centrifuged at 8000 x g for 10 minutes at 4 °C and the pellet resuspended in 450 µl of SPW and mixed with 50 µl of 1 M NaOH. See below for extraction processing.
Bacterial culture sampling: 10 µl from either a broth overnight cultures, or a single colony suspended in 25 µl of SPW from an overnight agar plate, was mixed with 90 µl of 100 mM NaOH. See below for extraction processing.
Extraction Processing
The NaOH extraction was allowed to proceed for 10 min at RT, and then either stored at -20 oC, directly diluted with 4 volumes of Te (10 mM TrisCl pH7.5, 0.1 mM EDTA), or processed for paramagnetic bead capture.
For Mag-Bind RXN Pure Plus beads 100 µl of NaOH extract was mixed with 100 µl of the bead suspension. For PureSil beads 100 µl of NaOH extract was mixed with 5 µl of resuspended beads and 95 µl of BBB. The extract-bead solution was mixed well and incubated at RT for 10 minutes. The beads were captured on magnetic stand, the solution was collected with a micropipettor and discarded. The beads were rinsed twice by addition of 200 µl RT 70% ethanol, the beads were resuspended with a brief vortex, then magnetic capture and solution discarded, being certain to remove all solution after the second rinse. The open tubes were allowed to air dry for 5-10 minutes. The beads were suspended in 50 µl of Te, incubated for 5 minutes at RT, then magnetic capture, and the eluate transferred to a clean tube. Eluate was either directly subjected to qPCR or stored frozen at -20 oC.
For pig rectal swabs extracted with a commercial kit, a total of 100 µl swab solution was used for the extraction with PowerLyzer PowerSoil DNA Isolation Kit (Qiagen, Hilden, Germany) according to the protocol. The extracted DNA was quantified with NanoDrop (Thermo Fisher Scientific, Wilmington, DE, USA).
Quantification of Extracted DNA by qPCR-HRM
The diluted NaOH extracts or paramagnetic bead purified DNAs was quantified for total bacterial DNA using qPCR for 16S rDNA using primers 8F and 936R [27]. Reactions were run in triplicates of 20 µl in 1x Taq Buffer (50 mM Tris pH8.3, 1.25 mM MgCl2, 300 ng/µl BSA), 0.2 mM dNTPs, 0.25 µM primers, 1 X EvaGreen® Dye (Biotium, Fremont, CA, USA), 2 µl of extracted DNA and 4 U Taq Polymerase, in 96 well plates in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Cycle parameters were: 90 °C for 45 seconds, 5 cycles of 90 °C for 15 sec, 71.5 °C for 15 sec, and 72 °C for 60 seconds; followed by 35 cycles with the same parameters with a plate read after each cycle. The products were then subjected to high resolution melt (HRM) analysis (to verify that any amplification signal was not artifactual) consisting of 72° C for 180 sec, 90 °C for 60 sec, 70 °C for 120 sec, followed by a 70 °C to 90 °C melt with steps of 0.1 °C for 5 sec and plate read. The amplification and melt curves were analyzed using CFX Manager v 3.1 (Bio-Rad Laboratories).
Statistical Analyses
Student T-Test was used to compare means of Ct values from qPCR, where significant difference was assumed at P<0.05.
Direct Dilution Method
The NaOH lysate was diluted with four volumes of Te, and then 2 µl of the DNA was directly used per 20 µl PCR. The extracts were stored at -20 oC.
Paramagnetic Beads DNA Purification Procedure
NaOH lysate (100 µl) was mixed with either an equal volume of Mag-Bind RXN Pure Plus, or with 95 µl of bead binding buffer and 5 µl of PureSil-Silica beads, at RT in a 0.5 ml microfuge tube. The suspension was mixed by vortexing for 30 seconds, incubated at RT for 5 minutes, and then captured on a magnetic stand. The supernatant was pipetted off and discarded. The captured beads were rinsed twice for 1 minute with 200 µl of 70% ethanol in the stand, followed by supernate removal with a pipet. The 0.5 ml tube was then opened while on the magnetic stand for 5 minutes to air dry. The tube was removed from the magnetic stand. Elution solutions were either Te (10 mM TrisCl 0.1 mM EDTA pH7.5), TE (10 mM TrisCl 1 mM EDTA pH7.5), 10 mM Tris pH 8.0, or SPW, as indicated. The elution volume, as indicated, was added to the 0.5 ml tube, vortexed for 30 seconds, incubated at RT for 5 minutes, and then beads were captured on the magnetic stand. The eluate was then transferred to a new 1.5 ml tube then either stored at -20 oC, or directly assayed by PCR/qPCR.
Results
Air-Sampling during Bacterial Infection Lameness Trial
We had previously found that waving open agar plates for several minutes could capture 50-300 CFUs during our experiments [25,28]. The primary airborne bacteria during these experiments is typically Staphylococcus saprophyticus [25] while similar sampling on a commercial broiler farm was primarily Staphylococcus cohnii [28]. In order to improve the sampling and provide better quantitative data, we evaluated using a sterile impinger containing 20 ml of 0.9% saline, and a battery-operated pipet pump as a portable air sampler for collecting viable bacteria during our BCO lameness trials. With the portable pump and impinger, we found that total CFU increased with the length of time sampled on a given day, and generally increased with day of the experiment implying that the bacterial load in the air was increasing as the birds matured and the bacterial infection spread (Table 1). As we were interested in using qPCR to determine the prevalence of specific bacterial genera/species, we tried several standard methods for rapid liberation of PCR-ready DNA. These included boiling [29,30], sonication [31], and glass bead-beating [32]. The qPCR results were either negative or variable even though we had viable bacteria present in the sample (Table 1). Therefore, we evaluated other methods for liberating PCR-ready DNA from bacterial broth and plate cultures.

Table 1.
Air samples, bacterial counts, extraction method and qPCR results during a 56 day lameness trial in Spring 2021. Samples were collected using the impinger on the indicated day for a given sampling duration (Minutes). For each sample the resuspended pellet was divided for serial dilution for plate counts and for DNA extraction. Total CFU is the computed total viable bacteria in the original sample. Extraction indicates the method used (see text) for that sample where NaOH+MB is NaOH extraction with Magnetic Bead capture and elution. qPCR positive indicates whether that extract produced a specific product for on qPCR-HRM, where NS signifies no specific amplification based on HRM. Avg Ct 16S presents the average Ct (±SEM) from triplicate samples using 8Fx936R 16S primers where ND (Not Determined) indicates species-specific qPCR primers were used.
Table 1.
Air samples, bacterial counts, extraction method and qPCR results during a 56 day lameness trial in Spring 2021. Samples were collected using the impinger on the indicated day for a given sampling duration (Minutes). For each sample the resuspended pellet was divided for serial dilution for plate counts and for DNA extraction. Total CFU is the computed total viable bacteria in the original sample. Extraction indicates the method used (see text) for that sample where NaOH+MB is NaOH extraction with Magnetic Bead capture and elution. qPCR positive indicates whether that extract produced a specific product for on qPCR-HRM, where NS signifies no specific amplification based on HRM. Avg Ct 16S presents the average Ct (±SEM) from triplicate samples using 8Fx936R 16S primers where ND (Not Determined) indicates species-specific qPCR primers were used.
| Day | Minutes | Total CFU | Extraction | qPCR positive |
Avg Ct 16S |
|---|---|---|---|---|---|
| 17 | 3 | 4100 | Boil | Yes | ND |
| 17 | 10 | 8825 | Boil | Yes | ND |
| 20 | 15 | 12205 | Boil | NS | |
| 20 | 30 | 21050 | Boil | NS | |
| 21 | 20 | 4500 | Bead beating | NS | |
| 27 | 20 | 35200 | Sonication | NS | |
| 29 | 20 | 4000 | Sonication | NS | |
| 35 | 20 | 17200 | Sonication | NS | |
| 42 | 20 | 6000 | NaOH+sodium acetate | NS | |
| 42 | 20 | 6600 | NaOH+MB | Yes | 23.9±0.03 |
| 44 | 20 | 20000 | Boil | NS | |
| 44 | 20 | 29300 | NaOH+MB | Yes | 22.0±0.1 |
| 46 | 20 | 46400 | Boil | NS | |
| 46 | 20 | 20000 | NaOH+C2mimOAc | Yes | 20.8±0.1 |
| 48 | 20 | 16000 | C2mimOAc | NS | |
| 48 | 20 | 20000 | NaOH+MB | Yes | 23.1±0.1 |
| 50 | 20 | 8000 | NaOH+MB | Yes | 24.4±0.2 |
| 50 | 20 | 12000 | NaOH+MB | Yes | 20.5±2.4 |
| 52 | 20 | 9000 | NaOH+MB | Yes | 23.2±0.6 |
| 52 | 20 | 5000 | NaOH+MB | Yes | 23.3±0.6 |
| 54 | 20 | 12200 | NaOH+MB | Yes | 23.2±0.5 |
| 54 | 20 | 12500 | NaOH+MB | Yes | 17.4±2.0 |
| 56 | 20 | 20000 | NaOH+MB | Yes | 19.7±2.4 |
| 56 | 20 | 52900 | NaOH+MB | Yes | 21.8±0.3 |
Optimization of DNA Extraction
Broth cultures and bacterial colonies for several different genera and species from our collection were tested for DNA liberation using boiling, sonication, glass bead-beating, enzymatic extraction [33]; lipid extraction with 1-ethyl-3-methylimidazolium acetate (C2minOAc) [13]; and f) NaOH lysis with sodium acetate neutralization [34]. Bacteria species tested included: Enterococcus faecalis (3 isolates), Enterococcus cecorum, Escherichia coli (2 isolates), Escherchia fergusonii, Staphylococcus agnetis, Staphylococcus aureus (2 isolates), Staphylococcus gallinarum, Staphylococcus nepalensis, Staphylococcus simulans, Staphylococcus saprophyticus, Staphylococcus lentus, Staphylococcus equorum, Staphylococcus cohnii, Streptococcus agalactiae, and Salmonella enterica. Of the 6 methods tested, the boiling approach was the most reliable as methods b-f were largely ineffective for many of the Gram-positive organisms. Particularly problematic for the boil method were the Enterococcus isolates where from multiple boil extractions only a few gave positive PCR for 16S rDNA amplification.
NaOH lysis is a common method for bacterial lysis, especially for plasmid isolation [14], and procedures have been published for extraction of plant, human, bacterial and yeast DNA for PCR using NaOH and heat [20,24,35,36] so we investigated how to best process bacterial NaOH extracts for subsequent PCR. We first examined what the threshold concentration of NaOH for inhibition of a standard qPCR reaction for 16S rDNA using purified S. agnetis DNA as the target. The results with our PCR buffer formulation (Methods) showed no inhibition at NaOH concentrations of 0 to 5.6 mM NaOH. The average Ct values increased by about 1 cycle with 7.5 mM NaOH and by approximately 4 with 10 mM NaOH. We then tested 25, 50, 75, and 100 mM NaOH in duplicate, for 10-minute room temperature (RT) extraction of 4 x 103 CFU of broth grown S. agnetis 908 (see methods). After incubation each sample was diluted with 4 volumes of SPW and qPCR-HRM was performed in triplicate on 2 µl of the diluted sample (final NaOH concentrations in the PCR were therefore 0.5, 1, 1.5 and 2 mM). The qPCR results in Figure 1 demonstrate that all 4 concentrations of NaOH liberated DNA but that 75 and 100 mM yielded lower Ct values, consistent with more complete lysis. We chose 100 mM as an appropriate effective concentration for subsequent extractions where 5-fold dilution would reduce the concentration to 20mM NaOH with subsequent addition of 2 µl to a 20 µl PCR would result in a final concentration of 2 mM NaOH in the PCR or less than half the minimum inhibitory concentration. We compared RT extraction to extraction at 37 oC and extraction for 5, 10, 20 or 30 minutes, and found no difference for time or temperature. We added water washed S. agnetis 908 from broth overnight cultures, in 10-fold dilutions estimated at 8x10-2 to 8x105 CFU in 2 µl directly to 20 µl qPCR. Specific amplification of the 16S amplicon resulted from the reactions from 8x102 to 8x105 but the Ct values were higher for ≥ 8x104 CFU, consistent with PCR inhibition. When we instead used a similar dilution series for extraction with 100 mM NaOH followed by 5-fold dilution there was specific amplification with input of as little as 10 CFUs and no inhibition even with input of estimated 107 CFU. Thus, NaOH extraction produced a specific qPCR-HRM product from fewer input cells and minimized PCR inhibition from addition of more than one hundred times more cells.
We then compared the boil and NaOH extraction methods for an E. cecorum isolate (1415) and three different isolates of E. faecalis (1558, 1570, and 1582) which had previously been problematic for boil extractions for PCR. We included S. agnetis 908 as a control. Equivalent volumes of cell extracts from each method were added to the qPCR-HRM. The results in Figure 2 show that the NaOH extraction produced Ct values lower than those from the boil. For E. faecalis 1570 there was no 16S amplification from the boil extract while the NaOH with dilution method was positive for 16S PCR. Based on ΔCt calculations [37] using the positive control of purified S. agnetis 908 genomic DNA using we compute the E. faecalis 1570 NaOH extract from 20 µl of overnight culture and dilution to a final volume of 500 µl was 3 pg/µl. Similar calculations for the NaOH extracts for the other cultures were: S. agnetis 908 680 pg/µl, E. cecorum 1415 71 pg/µl, E. faecalis 1558 16 pg/µl and E. faecalis 1572 5 pg/µl. The NaOH yield for E cecorum 1415 was more than 30x higher than the boil method.
Figure 2.
Average Ct values from qPCR-HRM for boil (gray) vs NaOH extraction (green) for duplicate samples from broth cultures Staphylococcus agnetis, Enterococcus cecorum or three isolates of Enterococcus faecalis. Overnight cultures were pelleted, and suspended in 10 volumes of sterile water. Duplicate 100 aliquots were treated for boil (10 min 100 oC in PCR machine) or NaOH (addition of 11 µl 1 M NaOH for 10 min. at room temperature). The samples were then diluted with 400 µl Te and assayed in triplicate by qPCR. Error bars are standard deviation. There was no amplification for E. faecalis 1570 for the boil extract.
Figure 2.
Average Ct values from qPCR-HRM for boil (gray) vs NaOH extraction (green) for duplicate samples from broth cultures Staphylococcus agnetis, Enterococcus cecorum or three isolates of Enterococcus faecalis. Overnight cultures were pelleted, and suspended in 10 volumes of sterile water. Duplicate 100 aliquots were treated for boil (10 min 100 oC in PCR machine) or NaOH (addition of 11 µl 1 M NaOH for 10 min. at room temperature). The samples were then diluted with 400 µl Te and assayed in triplicate by qPCR. Error bars are standard deviation. There was no amplification for E. faecalis 1570 for the boil extract.
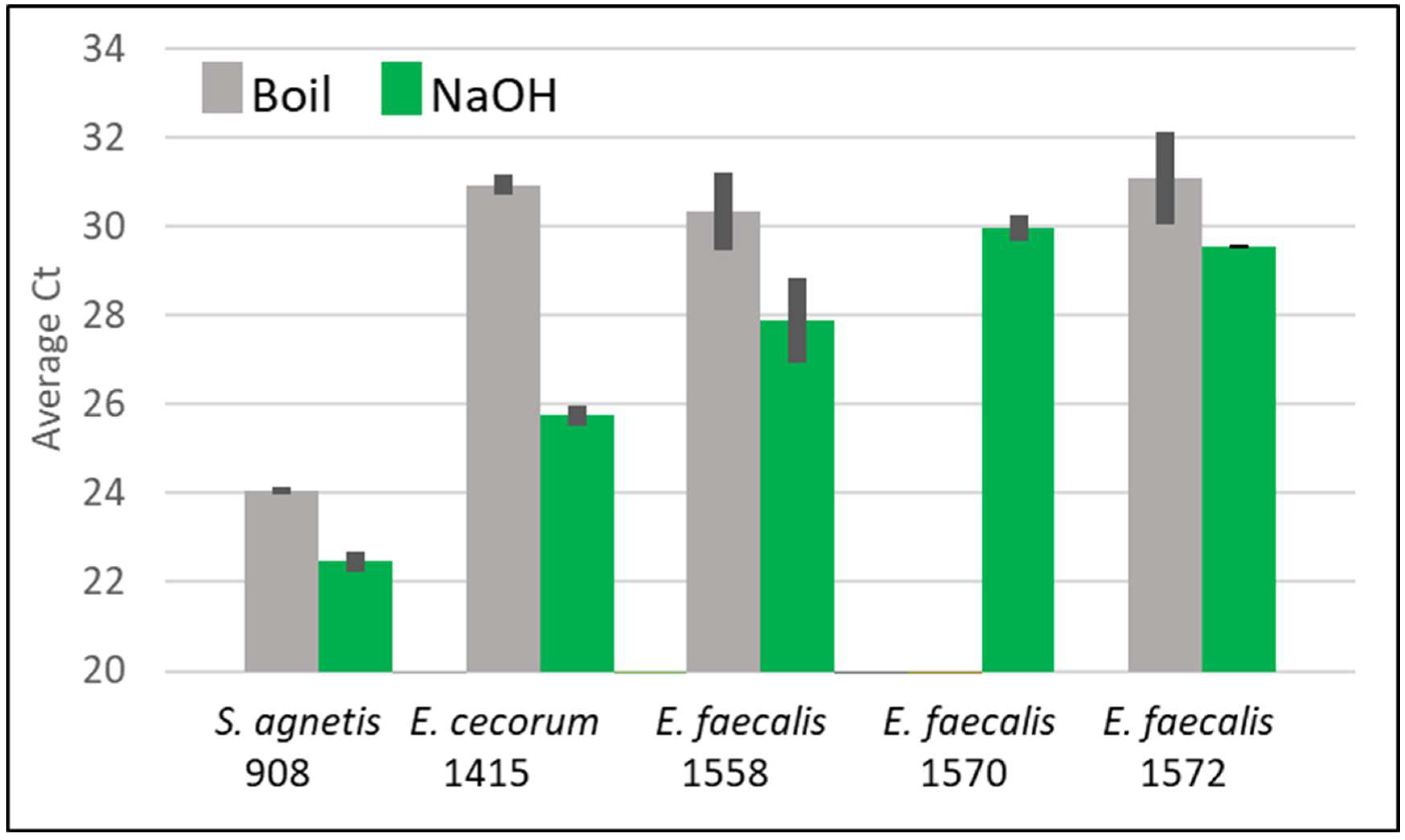
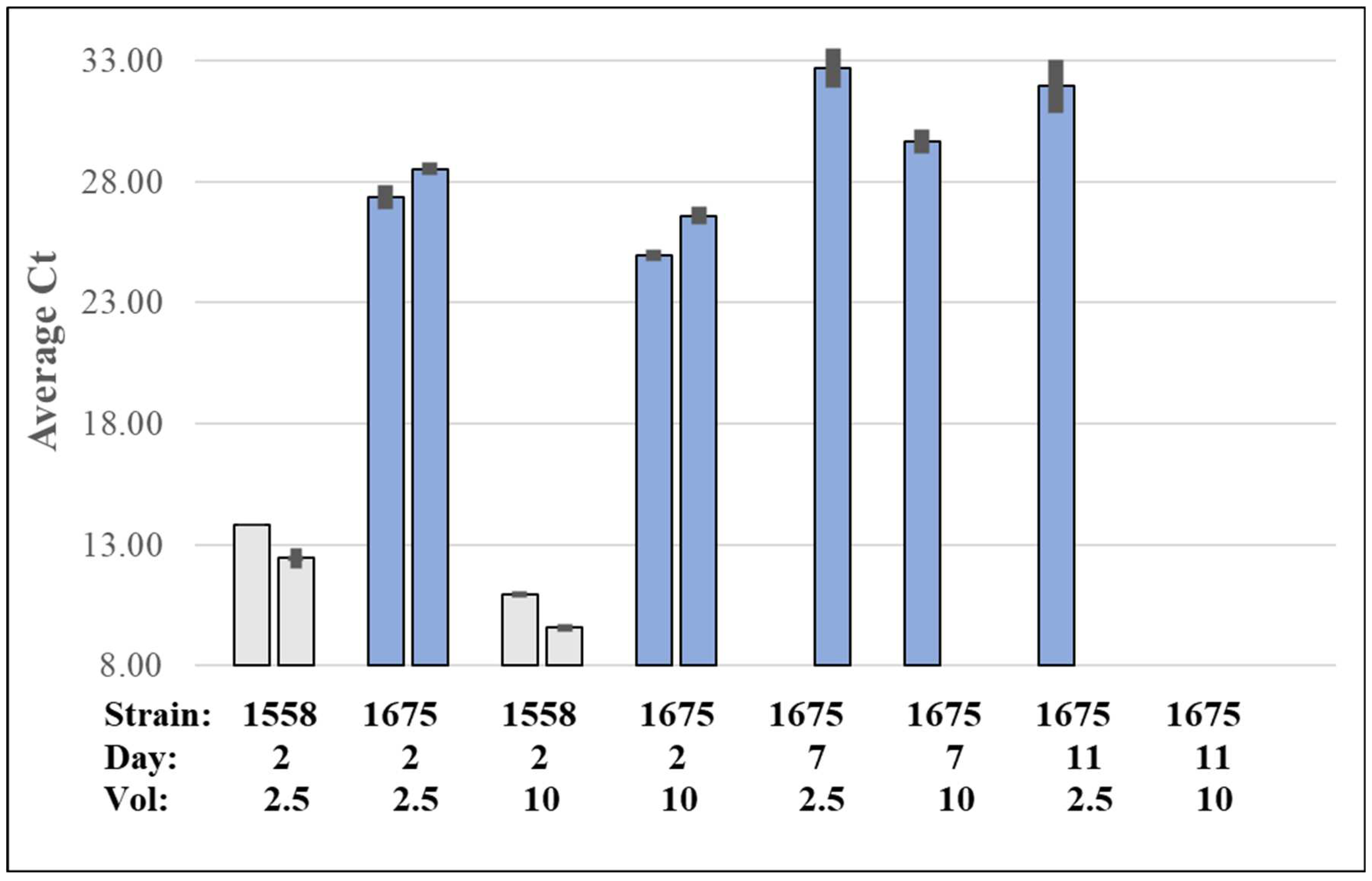
Figure 3.
Average Ct values from qPCR-HRM for NaOH extraction for duplicate samples from Enterococcus faecalis 1558 vs new and old colonies of Enterococcus cecorum 1675. Single colonies were assayed after 2 days incubation at 37 oC in 5% CO2 or from the same plates after 5 or 9 days at 4 oC. Colonies were suspended in 25 µl SPW and the indicated volume diluted to 25 µl with SPW, extracted by mixing with 25 µl 200 mM NaOH, diluted with 4 volumes of SPW then assayed by qPCR-HRM in triplicate. Error bars are standard deviation.
Figure 3.
Average Ct values from qPCR-HRM for NaOH extraction for duplicate samples from Enterococcus faecalis 1558 vs new and old colonies of Enterococcus cecorum 1675. Single colonies were assayed after 2 days incubation at 37 oC in 5% CO2 or from the same plates after 5 or 9 days at 4 oC. Colonies were suspended in 25 µl SPW and the indicated volume diluted to 25 µl with SPW, extracted by mixing with 25 µl 200 mM NaOH, diluted with 4 volumes of SPW then assayed by qPCR-HRM in triplicate. Error bars are standard deviation.
We speculated that the NaOH extraction method would be much more useful if it could be combined with a simple enrichment step. Others had shown that genomic DNA could be captured on silica spin columns from ethanol-NaOH lysis solutions [24]. We found that we could capture DNA from our NaOH lysates using the Mag-Bind RXN Pure Plus, a commercial preparation of paramagnetic beads in unspecified binding buffer. We compared paramagnetic bead purification versus direct dilution, for extraction from duplicate samples of broth cultures of 3 Gram-positive and 3 Gram-negative bacteria (Figure 4). For these comparisons the elution from the paramagnetic beads used the same volume as the final volume from direct dilution. The qPCR Ct values for the extractions from the Gram-negative species were nearly equivalent with both methods, while for the Gram-positive the paramagnetic bead capture gave consistently lower Ct values indicative of reduced PCR inhibition.
The NaOH extraction and magnetic bead capture was employed on selected air samples from days 42, 44, and 48-56 days and all gave positive qPCR-HRM results for 16S rDNA (Table 1), demonstrating that this method is highly effective for extraction from these air samples. The NaOH extraction and magnetic bead capture was then used for a second BCO challenge experiment where for days 21-37 we compared computed total CFU to the Ct values for days 21-37 of air sampling (Table 2). The total CFU increases through day 37 and the qPCR Ct values decrease from day 21-37 as expected with increasing bacterial load in the air. Therefore, the system we describe using a portable pump and sterile impinger is a low-cost, effective system for collecting not only viable bacteria, but can also be efficiently extracted for qPCR analysis of total bacteria or can be subjected to species-specific qPCR to detect particular bacteria of interest.
Optimized Extraction Applied to Diverse Environmental Samples
As NaOH extraction was a reliable method for obtaining bacterial DNA from the air samples, we tested whether this method was also applicable to other environmental samples (Table 3; Figure 5). The bacterial samples include soil; chicken cloacal swabs; food samples; laboratory surfaces; pond water and sewage. Soil specimens, collected on the University of Arkansas campus, were suspended in 100 mM NaOH with or without 5% saline. The results in Table 3 show that the inclusion of 5% saline during the extraction resulted in increased Ct values (P=0.02)and purification with paramagnetic beads gives superior amplification compared to the direct dilution (P=0.019). Chicken cloacal swabs, collected at the University of Arkansas Poultry Research farm, were extracted followed by paramagnetic bead capture or direct dilution. Eight total swabs were individually processed with four purified by paramagnetic bead capture and four directly diluted for triplicate qPCR-HRM for each swab extracted. Ct values from the paramagnetic purified extracts were lower but not significant (P=0.13). NaOH extraction was applied to swabs from cheese slices, and bread. While none of the swabs from bread gave specific amplification of bacterial 16S, the extracts from swabs from cheese purified with paramagnetic bead gave specific amplification whereas no amplification resulted from the direct dilution. This may results from PCR inhibitors since alternative methods of DNA extraction from foods with high fat and protein content typically result in the release of significant amounts of PCR inhibitors [38] which may be removed during the paramagnetic bead capture. NaOH extractions from swab samples from four different laboratory surfaces were extracted with 100 mM NaOH followed by either paramagnetic bead capture for two, or direct dilution for the other two, prior to qPCR-HRM. All four samples gave specific 16S amplification with paramagnetic bead capture somewhat lower. Sewage and pond water samples collected included: a) incoming sewage from the Paul R. Noland Wastewater Treatment facility and b) pond water samples from Clarence Craft Park, both in Fayetteville, AR. Pellets from these samples were resuspended in SPW and brought to 100 mM with NaOH. Separate aliquots of each sample were subjected to either direct dilution or paramagnetic bead capture prior to qPCR-HRM. The Ct values (Figure 5) for the sewage plant effluent were lower, than for the pond water samples, and although paramagnetic bead capture gave lower Ct values than direct dilution that was not always the case.
Optimized Extraction use for Fecal Microbiome
Fecal microbiome is an increasingly important focus in domestic animal health and human medicine [39,40,41,42,43]. We compared our NaOH extraction method to a commercial kit for extraction of DNA suitable for PCR amplification of the V4 region of 16S rDNA for microbiome analyses from swine fecal swab samples. The analysis was for 6 samples including two different growth ages using duplicate swabs for the two extraction methods. Paired swabs were extracted either using a commercial kit, or the NaOH extraction with paramagnetic bead capture. Quantification of the PCR products from the NaOH extraction and NanoDrop results from kit extraction are provided in Table 4. The NaOH extraction with magnetic bead capture was comparable, if not superior, to the commercial kit based on the quantification of PCR products (Table 4). The PCR product sequences from the two DNA extraction methods were analyzed using Qiime2-2021.11, denoised by Deblur, and annotated with Silva (version 138) database [44]. Figure 6 shows the alpha and beta diversity for the swab microbiome. Kruskal-Wallis test results show no significant differences between alpha diversities for the two extraction methods (see P values in Figure 6). Analysis of similarity (ANOSIM) conducted in qiime2 also shows microbiome communities of these two methods were very similar. The swine gut microbiome structure based on Bray-Curtis distance (Fig 6C) and membership as measured by Jaccard distance (Fig 6D) were separated by animal growth stage for lactation versus nursery (ANOSIM, R=0.962, P=0.003 for Bray-Curtis distance; R=0.998, P=0.001 for Jaccard distance), but not by DNA extraction methods (ANOSIM, R=-0.081, P==0.767 for Bray-Curtis distance; R=-0.120, P=0.832 for Jaccard distance). Figure 7 shows the comparisons of the microbiome composition at the phylum and genus level for the pairs of swabs from the two ages. Although there are differences between individual pigs for the minor phyla, especially at 10 days of age, the paired samples show similar distributions and frequencies whether extracted with the kit or with the NaOH with paramagnetic bead capture. There are more differences between the extraction methods for the genus level analyses but the data clearly support the NaOH extraction with magnetic bead capture as an inexpensive alternative for high-throughput microbiome analyses.
As the bulk of our paramagnetic bead capture used the Mag-Bind RXN Pure Plus which consists of paramagnetic beads supplied in a binding buffer we repeated our NaOH extractions from bacterial culture samples using PureSil-Silica paramagnetic beads and a defined bead binding buffer (Methods). The average Ct values for the PureSil-Silica were 25.1±0.3, while the Mag-Bind RXN Pure Plus Ct values were 30.1±0.3. Thus, either source can bind DNA but there may be some differences in either capacity or efficiency of capture under alkaline conditions, with the PureSil beads and defined bead binding buffer apparently recovering more DNA from equivalent aliquots. Additionally, with the PureSil beads and a separate buffer we could examine different ratios of binding buffer to NaOH. First, we determined that a 1:1 mixture of 100 mM NaOH and the defined bead binding buffer is pH 12.9. Therfore, the DNA capture is clearly under high pH. We then examined DNA recovery using ratios of binding buffer to 100 mM NaOH of 0:1, 0.25:1, 0.5:1, 0.75:1 and 1:1, followed by ethanol rinses, elution in Te, and qPCR quantification. While some DNA was captured even at 0:1 (Ct =33.5±0.1 with specific HRM), increasing binding buffer increased yield: 0.25:1 Ct=23.5±0.5, 0.5:1 Ct=21.2±0.2, 0.75:1 Ct=20.9±2.0 and 1:1 Ct=20.8±1.0. Size selection of double-stranded fragments has been reported through the ratio of binding buffer under neutral pH [45,46]. Given that our quantification is based on qPCR for chromosomal markers it appears that 0.5:1 binding buffer to 100 mM NaOH is sufficient to capture the larger DNAs in this extraction procedure. For smaller fragments it may be better to use 1:1 or higher ratios.
Discussion
The use of 100 mM NaOH to lyse bacteria and extract bacterial DNA from several different environmental samples presents an inexpensive method. Many of these extracts can be directly subjected to qPCR by 5 fold dilution where the final DNA sample is less than or equal to 1/10th of the qPCR volume. For some samples the sensitivity is improved through capture of the liberated DNA using silica-coated paramagnetic beads. Shi et al. [47] determined that DNA has two major binding mechanisms for silica at pH>3: through the phosphate backbone and hydrophobic binding of the DNA bases. Single-stranded nucleic acids bind to silica with greater affinity than double-stranded nucleic acids because the hydrophobic bases are exposed and because single-stranded DNA is more flexible than double-stranded DNA, maximizing contacts. Typically, nucleic acids are driven onto silica using chaotropic salts that dehydrate the nucleic acid and silica, but some amino acid buffers have been also demonstrated to comparably facilitate nucleic acid binding to silica [48]. We have used a recommended bead binding buffer (BBB- see methods) where a 50:50 mixture of 100 mM NaOH and BBB is pH 12.9. Surprisingly, some DNA will bind to the beads in 100 mM NaOH with no addition of BBB. However, more DNA is recovered from bacterial lysates by addition of at least 0.5:1 BBB to 100 mM NaOH. Presumably alternative binding buffers could also be compatible for DNA capture from high pH solutions.
We compared the alkaline lysis method with different rapid methods for DNA extraction from bacterial cells collected from air samples. These included rapid boiling; sonication; bead-beating; enzyme digestion; and C2minOAc. The 100 mM NaOH protocol was almost always successful for broth bacterial cultures, and somewhat less successful for toothpick samples of bacterial colonies. We had previously used a 10 minute 100 oC treatment for toothpick samples of bacterial colonies with routine success but that suffered from over or under-sampling of the colony [26,49]. The 100 mM NaOH extraction seems to be much less susceptible to “over-load”. We also successfully used the 100 mM NaOH extraction protocol to extract DNA from samples from air, garden soil, chicken cloaca, food, environmental surfaces, sewage, pond water, and pig rectal swabs. This technique provides a high degree of reliability, short incubation time, and produces readily amplifiable PCR substrates. NaOH stocks are stable at ambient temperature and the extraction requires no organic solvents, inhibitory compounds, expensive enzymes, or special equipment (shakers, mixers, heaters, etc.). Of note, assessment of animal microbiome is related to animal nutrition and health, and is a research topic of many scientists [39,40,44,50,51,52,53,54]. Efficient DNA extraction from specimens such as fecal swabs is critical to reveal microbial composition and structures. Interestingly, the paired swab microbiomes illustrated by the two DNA extraction methods, a commercial kit, or the NaOH extraction, were very similar in terms of community composition, overall alpha and beta diversities. Thus, the inexpensive NaOH-magnetic bead extraction technique could be used to replace the more expensive and time-consuming commercial kit(s), for microbiome studies.
One disadvantage of NaOH DNA extraction might be in converting the dsDNA to ssDNA, which cannot be quantified using the high sensitivity quantification methods, such as fluorimetry. However, the liberated DNA can be easily quantified using qPCR. The NaOH method of DNA extraction has significant importance when extracting DNA from any bacterial species that cannot be cultivated, as might be present in sampling air, soil, sewage or feces. The extracted DNA can be utilized for conventional or quantitative PCR with relative ease, and it may also be useful for other molecular diagnostics such as LAMP or nanopore sequencing. The immediate extract appears to be stable and so are directly applicable to sampling in resource-constrained environments.
In summary, the liberation of genomic DNA from bacteria using 100 mM NaOH followed by purification using paramagnetic beads or a 1:5 dilution in Te offers many benefits over conventional enzymatic techniques and commercial kits. The proposed technique is distinguished by the fact that it requires as few as one incubation step of ten minutes, and no centrifugation. Even with magnetic bead capture the method can be completed in less than one hour. We have routinely scaled up the NaOH extraction and magnetic bead capture to process 100 µl of overnight stationary Staphylococcal cultures to produce 1 ml at 10-30 ng/µl, based on ΔΔCt for qPCR vs control DNA [37]. Thus, avoiding larger culture volumes, multiple centrifugations, long incubation times, expensive enzymes, and organic solvents [15]. This simple extraction method combined with an inexpensive air-sampling system, as we have described, should be readily employed in agricultural systems to detect and monitor viral, bacterial, and fungal pathogens for rapid response to critical diseases.
Author Contributions
AS developed the extraction techniques and wrote the initial draft of the manuscript, BZ produced and analyzed the swine microbiome data. AA, JZ and DR secured funding and oversaw the research. DR was responsible for the final draft of the manuscript with contributions from AS, BZ, AA, and JZ. All authors approved the final manuscript.
Acknowledgements
This research was funded by: the Arkansas Biosciences Institute, the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000; and the Ginny Lewis Fund of the Fulbright College of Arts and Sciences at the University of Arkansas.
Conflicts of Interest
The authors declare that they have no competing interests. The funders played no role in the design of the experiments, the interpretation of the data, the writing of this manuscript, or the decision to publish this manuscript.
References
- Kralik, P.; Ricchi, M. A Basic Guide to Real Time PCR in Microbial Diagnostics: Definitions, Parameters, and Everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef]
- Deshmukh, R.A.; Joshi, K.; Bhand, S.; Roy, U. Recent developments in detection and enumeration of waterborne bacteria: a retrospective minireview. Microbiologyopen 2016, 5, 901–922. [Google Scholar] [CrossRef]
- Deurenberg, R.H., E. Bathoorn, M.A. Chlebowicz, N. Couto, M. Ferdous, S. García-Cobos, A.M. Kooistra-Smid, E.C. Raangs, S. Rosema, and A.C. Veloo, Application of next generation sequencing in clinical microbiology and infection prevention. J Biotechnol 2017. 243 16-24.
- Sambrook, J., E.F. Fritsch, and T. Maniatis, Molecular cloning: a laboratory manual. 1989, Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press.
- Maloy, S.R., Experimental techniques in bacterial genetics. 1990: Jones & Bartlett Learning.
- Nelson, J.E.; Krawetz, S.A. Purification of cloned and genomic DNA by guanidine thiocyanate/isobutyl alcohol fractionation. Anal. Biochem. 1992, 207, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Ausubel, F., R. Brent, R. Kingston, D. Moore, J. Seidman, J. Smith, and K. Struhl, Phenol: chloroform extraction. Current Protocols in Molecular Biology. New York, John Wiley & Sons, Inc 1994. 2 3.
- Kolm, C.; Martzy, R.; Brunner, K.; Mach, R.L.; Krska, R.; Heinze, G.; Sommer, R.; Reischer, G.H.; Farnleitner, A.H. A Complementary Isothermal Amplification Method to the U.S. EPA Quantitative Polymerase Chain Reaction Approach for the Detection of Enterococci in Environmental Waters. Environ. Sci. Technol. 2017, 51, 7028–7035. [Google Scholar] [CrossRef]
- Chapela, M.-J.; Garrido-Maestu, A.; Cabado, A.G. Detection of foodborne pathogens by qPCR: A practical approach for food industry applications. Cogent Food Agric. 2015, 1, 1013771. [Google Scholar] [CrossRef]
- Law, J.W.-F.; Ab Mutalib, N.-S.; Chan, K.-G.; Lee, L.-H. Rapid methods for the detection of foodborne bacterial pathogens: principles, applications, advantages and limitations. Front. Microbiol. 2015, 5, 770. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C., S. Nogueira, M. Gadanho, and S. Chaves, Chapter 7 - DNA extraction: finding the most suitable method, in Molecular Microbial Diagnostic Methods, N. Cook, M. D'Agostino, and K.C. Thompson, Editors. 2016, Academic Press: San Diego. p. 135-154.
- Martzy, R.; Bica-Schröder, K.; Pálvölgyi, M.; Kolm, C.; Jakwerth, S.; Kirschner, A.K.T.; Sommer, R.; Krska, R.; Mach, R.L.; Farnleitner, A.H.; et al. Simple lysis of bacterial cells for DNA-based diagnostics using hydrophilic ionic liquids. Sci. Rep. 2019, 9, 13994. [Google Scholar] [CrossRef]
- Martzy, R.; Bica-Schröder, K.; Pálvölgyi, M.; Kolm, C.; Jakwerth, S.; Kirschner, A.K.T.; Sommer, R.; Krska, R.; Mach, R.L.; Farnleitner, A.H.; et al. Simple lysis of bacterial cells for DNA-based diagnostics using hydrophilic ionic liquids. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ish-Horowicz, D.; Burke, J. Rapid and efficient cosmid cloning. Nucleic Acids Res. 1981, 9, 2989–2898. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.W.; Iandolo, J.J. Rapid isolation of DNA from Staphylococcus aureus. Appl. Environ. Microbiol. 1983, 46, 283–285. [Google Scholar] [CrossRef]
- Pitcher, D.G.; Saunders, N.A.; Owen, R.J. Rapid extraction of bacterial genomic DNA with guanidium thiocyanate. Lett. Appl. Microbiol. 1989, 8, 151–156. [Google Scholar] [CrossRef]
- Reischer, G.H.; Haider, J.M.; Sommer, R.; Stadler, H.; Keiblinger, K.M.; Hornek, R.; Zerobin, W.; Mach, R.L.; Farnleitner, A.H. Quantitative microbial faecal source tracking with sampling guided by hydrological catchment dynamics. Environ. Microbiol. 2008, 10, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qi, M.; Cutler, A.J. A simple method of preparing plant samples for PCR. Nucleic Acids Res. 1993, 21, 4153–4154. [Google Scholar] [CrossRef] [PubMed]
- Truett, G.; Heeger, P.; Mynatt, R.; Truett, A.; Walker, J.; Warman, M.; Meeker, N.D.; Hutchinson, S.A.; Ho, L.; Trede, N.S.; et al. Preparation of PCR-Quality Mouse Genomic DNA with Hot Sodium Hydroxide and Tris (HotSHOT). BioTechniques 2000, 29, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Kouduka, M.; Suko, T.; Morono, Y.; Inagaki, F.; Ito, K.; Suzuki, Y. A new DNA extraction method by controlled alkaline treatments from consolidated subsurface sediments. FEMS Microbiol. Lett. 2012, 326, 47–54. [Google Scholar] [CrossRef]
- Morono, Y., T. Terada, T. Hoshino, and F. Inagaki, Hot-alkaline DNA extraction method for deep-subseafloor archaeal communities. Appl Environ Microbiol 2014. 80 1985-1994.
- Osmundson, T.W., C.A. Eyre, K.M. Hayden, J. Dhillon, and M.M. Garbelotto, Back to basics: An evaluation of N a OH and alternative rapid DNA extraction protocols for DNA barcoding, genotyping, and disease diagnostics from fungal and oomycete samples. Mol Ecol Resour 2013. 13 66-74.
- Park, H.J.; Oh, S.; Vinod, N.; Ji, S.; Noh, H.B.; Koo, J.M.; Lee, S.H.; Kim, S.C.; Lee, K.-S.; Choi, C.W. Characterization of Chemically-Induced Bacterial Ghosts (BGs) Using Sodium Hydroxide-Induced Vibrio parahaemolyticus Ghosts (VPGs). Int. J. Mol. Sci. 2016, 17, 1904. [Google Scholar] [CrossRef]
- Vingataramin, L.; Frost, E.H. A single protocol for extraction of gDNA from bacteria and yeast. BioTechniques 2015, 58, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Alrubaye, A., N.S. Ekesi, A. Hasan, D.A. Koltes, R. Wideman Jr, and D. Rhoads, Chondronecrosis with osteomyelitis in broilers: Further defining a bacterial challenge model using standard litter flooring and protection with probiotics. Poult. Sci. 2020. 99 6474-6480.
- Alrubaye, A.A.; Ekesi, N.S.; Hasan, A.; Elkins, E.; Ojha, S.; Zaki, S.; Dridi, S.; Wideman, R.F.; Rebollo, M.A.; Rhoads, D.D. Chondronecrosis with osteomyelitis in broilers: further defining lameness-inducing models with wire or litter flooring to evaluate protection with organic trace minerals. Poult. Sci. 2020, 99, 5422–5429. [Google Scholar] [CrossRef]
- Baker, G.C.; Smith, J.J.; Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods 2003, 55, 541–555. [Google Scholar] [CrossRef]
- Ekesi, N.S.; Dolka, B.; Alrubaye, A.A.; Rhoads, D.D. Analysis of genomes of bacterial isolates from lameness outbreaks in broilers. Poult. Sci. 2021, 100, 101148. [Google Scholar] [CrossRef]
- Holmes, D.S.; Quigley, M. A rapid boiling method for the preparation of bacterial plasmids. Anal. Biochem. 1981, 114, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Trkov, M. and G. Avgustin, An improved 16S rRNA based PCR method for the specific detection of Salmonella enterica. Int J Food Microbiol 2003. 80 67-75.
- Zhang, L.; Foxman, B.; Gilsdorf, J.R.; Marrs, C.F. Bacterial genomic DNA isolation using sonication for microarray analysis. BioTechniques 2005, 39, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Teng, F., S.S. Darveekaran Nair, P. Zhu, S. Li, S. Huang, X. Li, J. Xu, and F. Yang, Impact of DNA extraction method and targeted 16S-rRNA hypervariable region on oral microbiota profiling. Sci Rep 2018. 8 16321.
- Zhao, J.; Carmody, L.A.; Kalikin, L.M.; Li, J.; Petrosino, J.F.; Schloss, P.D.; Young, V.B.; LiPuma, J.J. Impact of Enhanced Staphylococcus DNA Extraction on Microbial Community Measures in Cystic Fibrosis Sputum. PLOS ONE 2012, 7, e33127. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, V.P.; Zhang, X.; Morono, Y.; Inagaki, F.; Wang, F. A Modified SDS-Based DNA Extraction Method for High Quality Environmental DNA from Seafloor Environments. Front. Microbiol. 2016, 7, 986. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qi, M.; Cutler, A.J. A simple method of preparing plant samples for PCR. Nucleic Acids Res. 1993, 21, 4153–4154. [Google Scholar] [CrossRef]
- Rudbeck, L.; Dissing, J. Rapid, Simple Alkaline Extraction of Human Genomic DNA from Whole Blood, Buccal Epithelial Cells, Semen and Forensic Stains for PCR. BioTechniques 1998, 25, 588–592. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Rossen, L.; Nørskov, P.; Holmstrøm, K.; Rasmussen, O.F. Inhibition of PCR by components of food samples, microbial diagnostic assays and DNA-extraction solutions. Int. J. Food Microbiol. 1992, 17, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.S.; Harkins, D.M.; Nelson, K.E. Advances in Microbiome Research for Animal Health. Annu. Rev. Anim. Biosci. 2021, 9, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Woodhams, D.C.; Martino, C.; Allaband, C.; Mu, A.; Javorschi-Miller-Montgomery, S.; Suchodolski, J.S.; Knight, R. Engineering the microbiome for animal health and conservation. Exp. Biol. Med. 2019, 244, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Firek, B.; Baker, R.; Burstein, D.; Soenjoyo, K.; Thomas, B.C.; Morowitz, M.; Banfield, J.F. Identical bacterial populations colonize premature infant gut, skin, and oral microbiomes and exhibit different in situ growth rates. Genome Res. 2017, 27, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.; Wilson, J.; Guthrie, A.; Cookson, K.; Vancraeynest, D.; Schaeffer, J.; Moody, R.; Clark, S. New issues and science in broiler chicken intestinal health: Emerging technology and alternative interventions. J. Appl. Poult. Res. 2015, 24, 257–266. [Google Scholar] [CrossRef]
- A Gilbert, J.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Verrow, S., M. Blair, B. Packard, and W. Godfrey. Gel-Free Size Selection Using SPRIselect For Next Generation Sequencing. [pdf] 2023 [cited 2023 7/28/2023]. Available online: https://ls.beckmancoulter.co.jp/files/appli_note/Gel_Free_Using_SPRIselect.pdf.
- Oberacker, P.; Stepper, P.; Bond, D.M.; Höhn, S.; Focken, J.; Meyer, V.; Schelle, L.; Sugrue, V.J.; Jeunen, G.-J.; Moser, T.; et al. Bio-On-Magnetic-Beads (BOMB): Open platform for high-throughput nucleic acid extraction and manipulation. PLOS Biol. 2019, 17, e3000107. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Shin, Y.K.; Hassanali, A.A.; Singer, S.J. DNA Binding to the Silica Surface. J. Phys. Chem. B 2015, 119, 11030–11040. [Google Scholar] [CrossRef] [PubMed]
- Vandeventer, P.E.; Mejia, J.; Nadim, A.; Johal, M.S.; Niemz, A. DNA Adsorption to and Elution from Silica Surfaces: Influence of Amino Acid Buffers. J. Phys. Chem. B 2013, 117, 10742–10749. [Google Scholar] [CrossRef]
- Al-Rubaye, A.A.K.; Ekesi, N.S.; Zaki, S.; Emami, N.K.; Wideman, R.F.; Rhoads, D.D. Chondronecrosis with osteomyelitis in broilers: Further defining a bacterial challenge model using the wire flooring model. Poult. Sci. 2017, 96, 332–340. [Google Scholar] [CrossRef]
- Gand, M.; Bloemen, B.; Vanneste, K.; Roosens, N.H.C.; De Keersmaecker, S.C.J. Comparison of 6 DNA extraction methods for isolation of high yield of high molecular weight DNA suitable for shotgun metagenomics Nanopore sequencing to detect bacteria. BMC Genom. 2023, 24, 438. [Google Scholar] [CrossRef]
- Wang, X.; Howe, S.; Wei, X.; Deng, F.; Tsai, T.; Chai, J.; Xiao, Y.; Yang, H.; Maxwell, C.V.; Li, Y.; et al. Comprehensive Cultivation of the Swine Gut Microbiome Reveals High Bacterial Diversity and Guides Bacterial Isolation in Pigs. mSystems 2021, 6, e00477–21. [Google Scholar] [CrossRef]
- Trudeau, S.; Thibodeau, A.; Côté, J.-C.; Gaucher, M.-L.; Fravalo, P. Contribution of the Broiler Breeders’ Fecal Microbiota to the Establishment of the Eggshell Microbiota. Front. Microbiol. 2020, 11, 666. [Google Scholar] [CrossRef] [PubMed]
- Rexroad, C.; Vallet, J.; Matukumalli, L.K.; Reecy, J.; Bickhart, D.; Blackburn, H.; Boggess, M.; Cheng, H.; Clutter, A.; Cockett, N.; et al. Genome to Phenome: Improving Animal Health, Production, and Well-Being – A New USDA Blueprint for Animal Genome Research 2018–2027. Front. Genet. 2019, 10, 327. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Sun, C.; Yuan, J.; Yang, N. Gut metagenomic analysis reveals prominent roles of Lactobacillus and cecal microbiota in chicken feed efficiency. Sci. Rep. 2017, 7, 45308. [Google Scholar] [CrossRef]
Figure 1.
Average Ct values for different NaOH concentrations (mM) for extraction from duplicate aliquots of 4x103 CFUs of a S. agnetis 908 broth culture followed by dilution with 4 volumes of Te, and then qPCR-HRM. Data plotted are average for triplicate qPCR. Error bars are standard deviation.
Figure 1.
Average Ct values for different NaOH concentrations (mM) for extraction from duplicate aliquots of 4x103 CFUs of a S. agnetis 908 broth culture followed by dilution with 4 volumes of Te, and then qPCR-HRM. Data plotted are average for triplicate qPCR. Error bars are standard deviation.
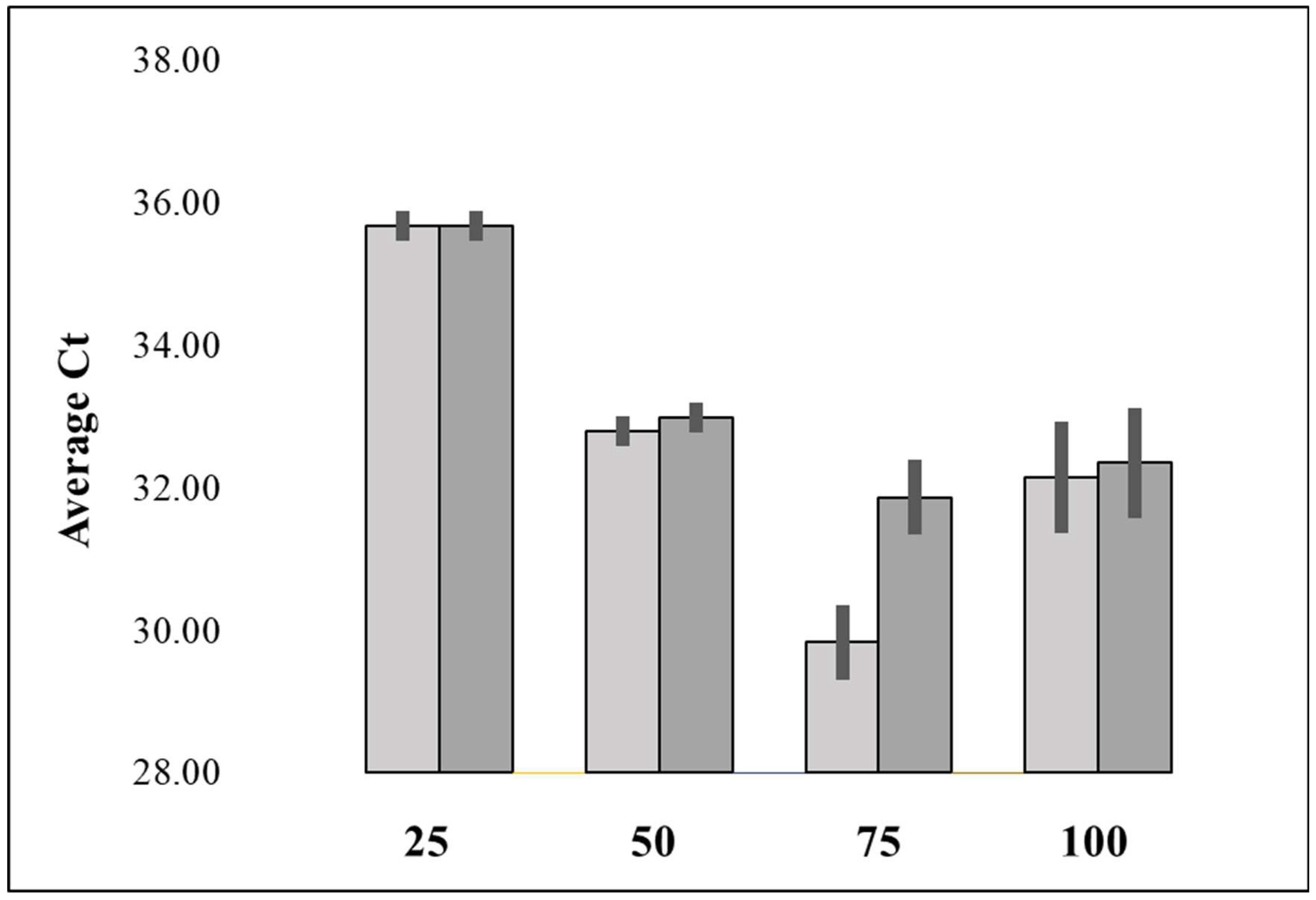
Figure 4.
Average Ct values for NaOH extracts from broth cultures of 3 Gram-positive (Staphylococcus aureus 1516, Staphylococcus agnetis 908, Enterococcus faecalis 1558) and 3 Gram-negative species (Escherichia coli 1409, Escherichia fergusonii 1412, Salmonella enterica 1414). Extracts were processed by paramagnetic bead capture (open bars) or by dilution with 4 volumes of Te (gray bars). Final samples for each method were adjusted to the same volume and assayed by qPCR-HRM in triplicate. Error bars are standard deviation.
Figure 4.
Average Ct values for NaOH extracts from broth cultures of 3 Gram-positive (Staphylococcus aureus 1516, Staphylococcus agnetis 908, Enterococcus faecalis 1558) and 3 Gram-negative species (Escherichia coli 1409, Escherichia fergusonii 1412, Salmonella enterica 1414). Extracts were processed by paramagnetic bead capture (open bars) or by dilution with 4 volumes of Te (gray bars). Final samples for each method were adjusted to the same volume and assayed by qPCR-HRM in triplicate. Error bars are standard deviation.
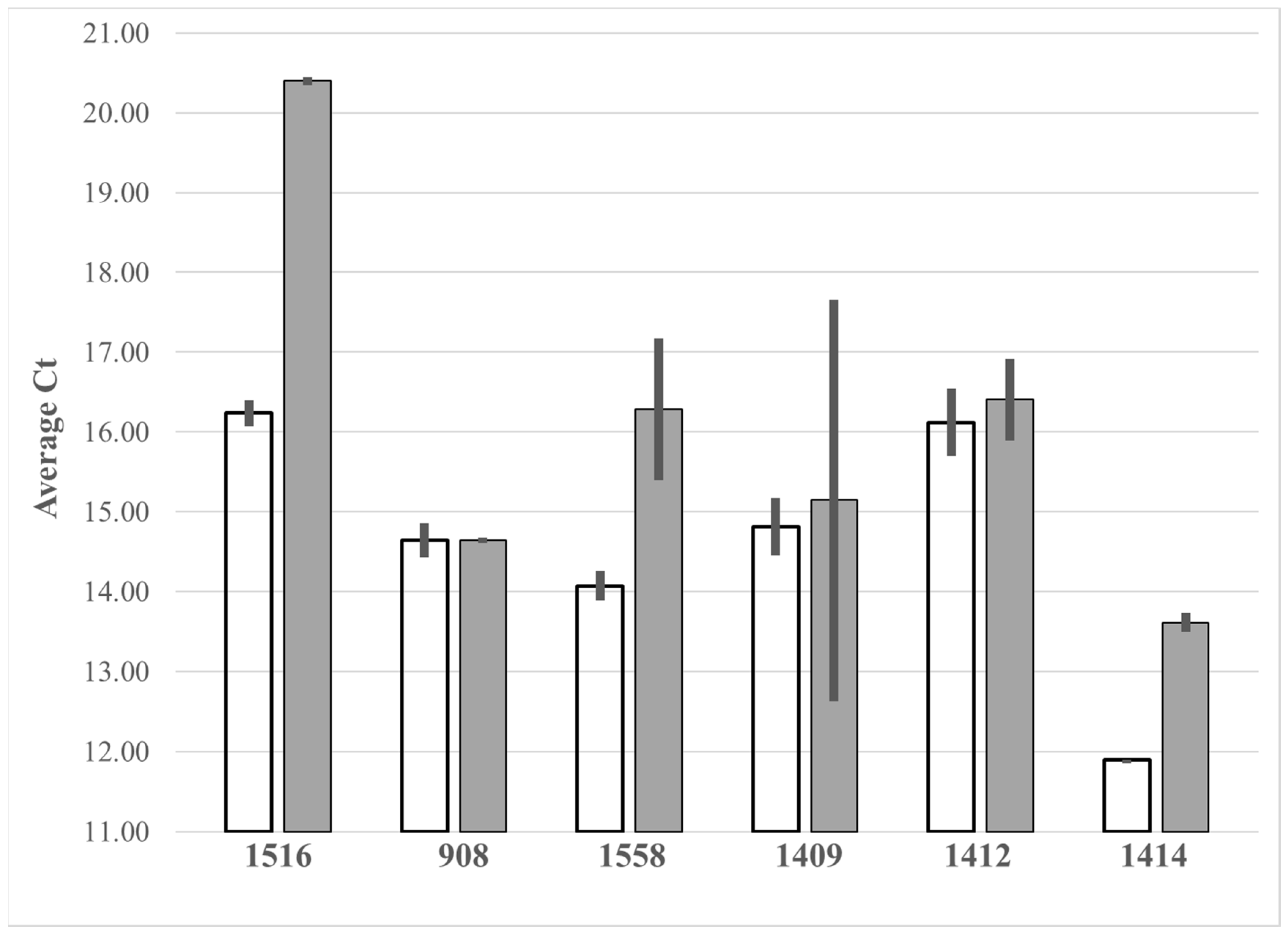
Figure 5.
Average Ct values for qPCR from NaOH extracts from three samples from sewage water (A-C) or pond water (D-F). Aliquots from each extract were subjected to paramagnetic bead purification (open bars) or dilution with 4 volumes of Te (gray bars). Final samples for each method were adjusted to the same volume and assayed by qPCR-HRM in triplicate. Error bars are standard deviation.
Figure 5.
Average Ct values for qPCR from NaOH extracts from three samples from sewage water (A-C) or pond water (D-F). Aliquots from each extract were subjected to paramagnetic bead purification (open bars) or dilution with 4 volumes of Te (gray bars). Final samples for each method were adjusted to the same volume and assayed by qPCR-HRM in triplicate. Error bars are standard deviation.
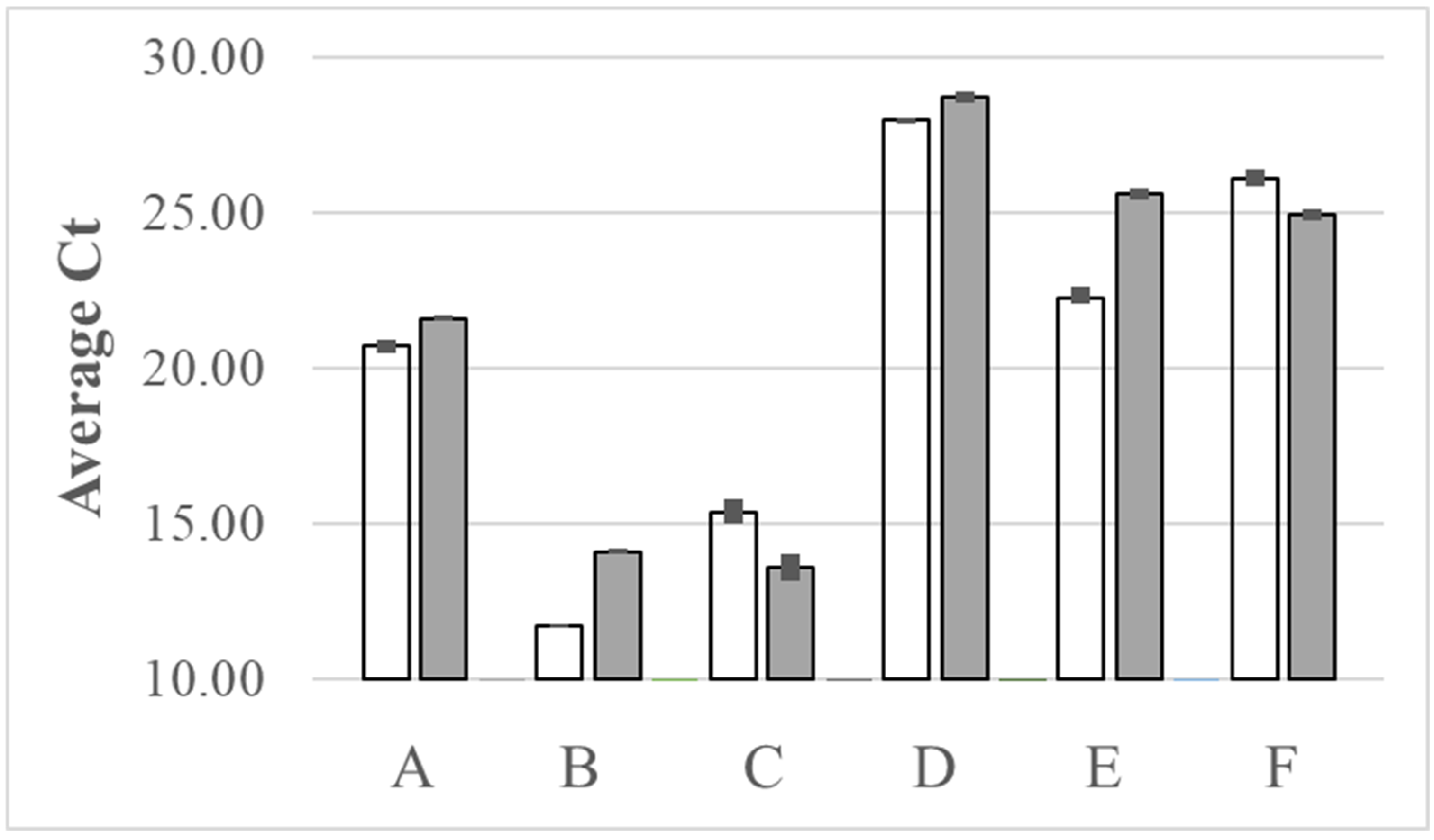
Figure 6.
Microbiome alpha diversity (A, B) and beta diversity (C, D) from six pig fecal samples extracted either with a kit (circle) or the NaOH (triangle) method. Samples numbers are same as for Table 4, and the paired samples are represented with the same color and connected with a line.
Figure 6.
Microbiome alpha diversity (A, B) and beta diversity (C, D) from six pig fecal samples extracted either with a kit (circle) or the NaOH (triangle) method. Samples numbers are same as for Table 4, and the paired samples are represented with the same color and connected with a line.
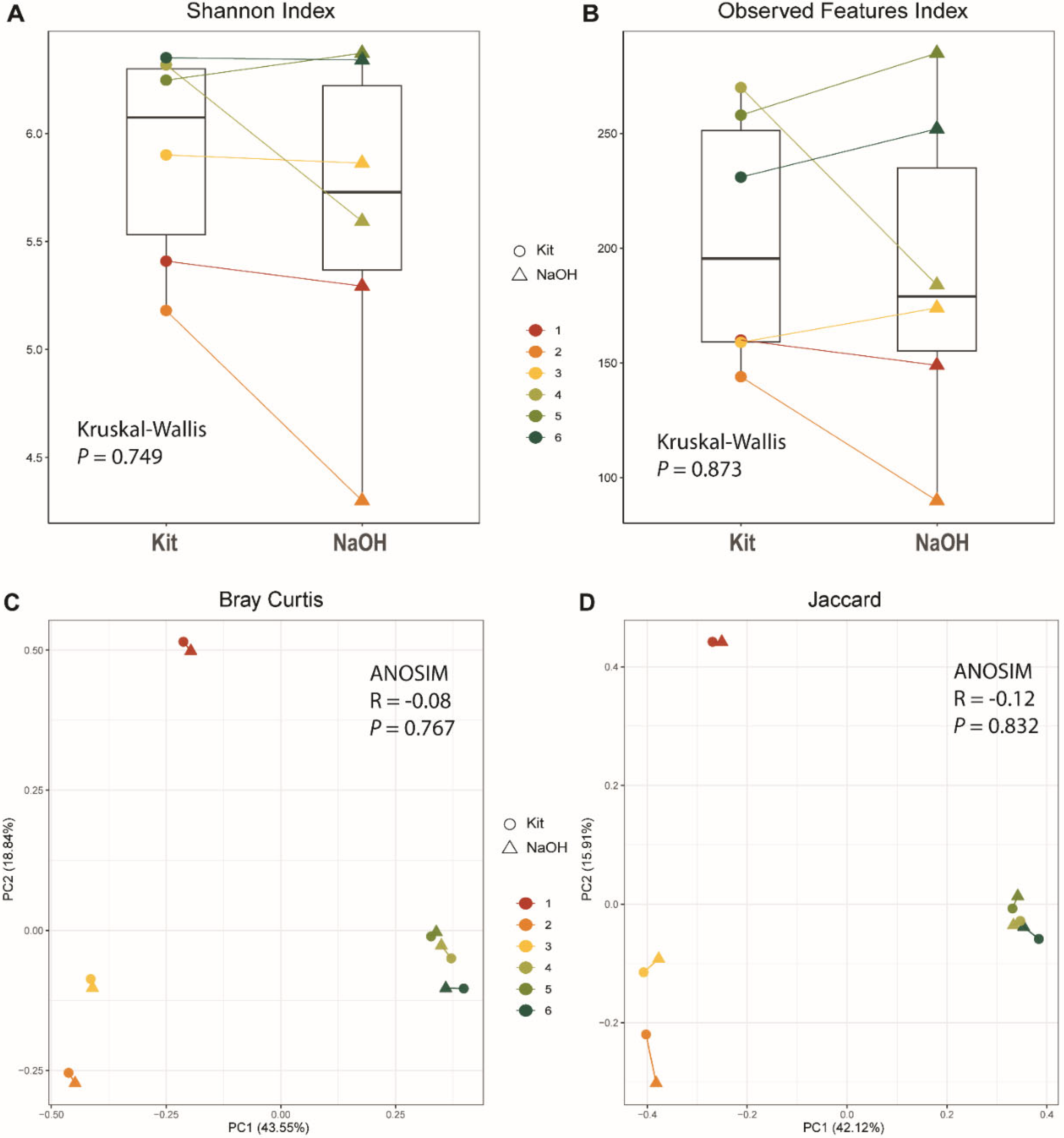
Figure 7.
Microbiome composition by Phylum (A) or Genus (B) from six pig fecal samples extracted either with a kit or the NaOH method. Sample numbers (top of paired bars) are as for Table 4, and the extraction method is indicated at the bottom.
Figure 7.
Microbiome composition by Phylum (A) or Genus (B) from six pig fecal samples extracted either with a kit or the NaOH method. Sample numbers (top of paired bars) are as for Table 4, and the extraction method is indicated at the bottom.
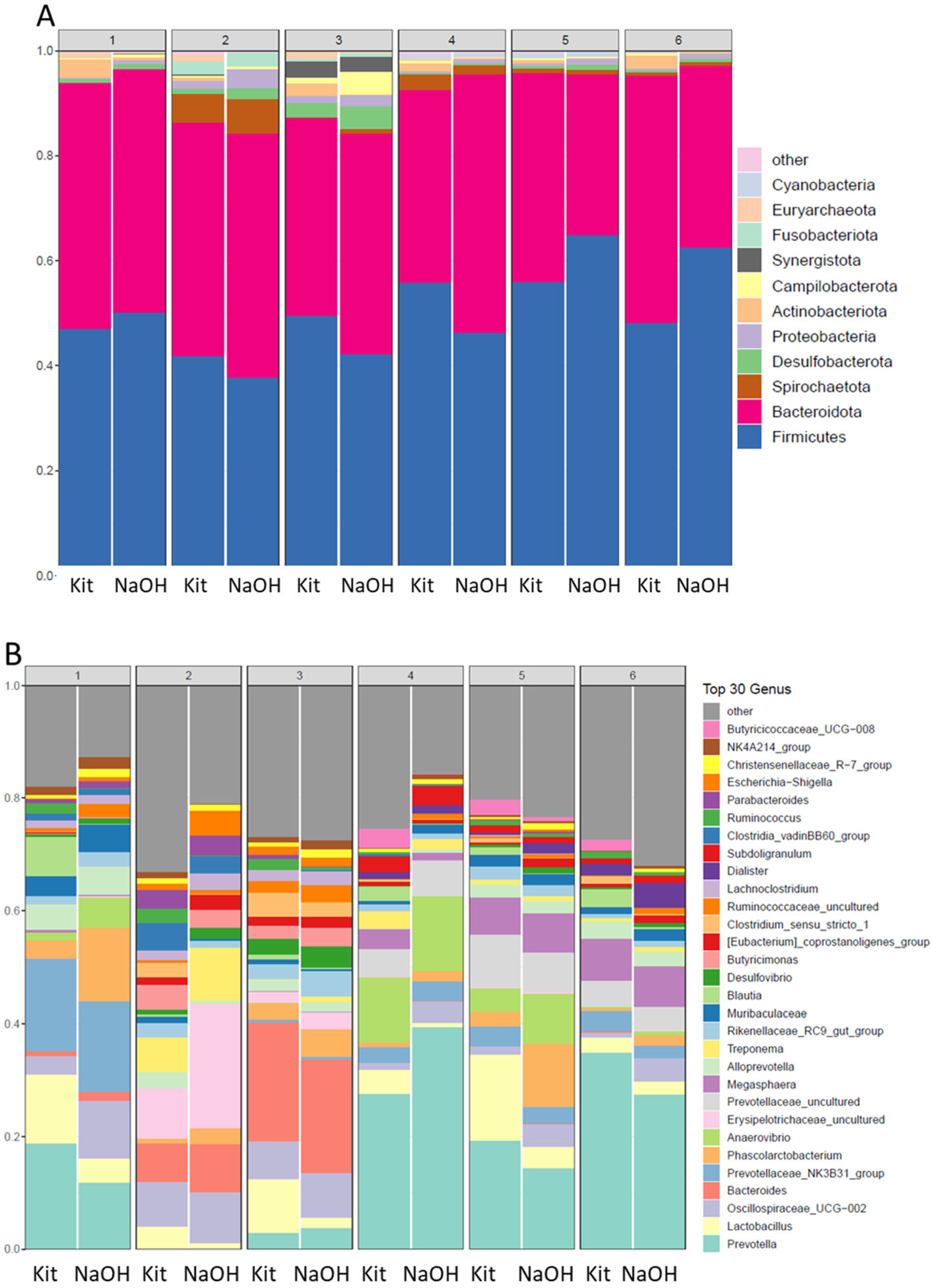
Table 2.
Air samples, bacterial counts, and qPCR Ct values during a 56 day lameness trial with all samples were 20 minutes and were extracted using 100 mM NaOH and paramagnetic bead capture. Columns are as for Table 1 except that for the qPCR the average of triplicate Ct values with SEM.
Table 2.
Air samples, bacterial counts, and qPCR Ct values during a 56 day lameness trial with all samples were 20 minutes and were extracted using 100 mM NaOH and paramagnetic bead capture. Columns are as for Table 1 except that for the qPCR the average of triplicate Ct values with SEM.
| Day | Total CFU | Average Ct±SEM |
|---|---|---|
| 21 | 340 | 22.3±0.4 |
| 21 | 1520 | 20.5±0.2 |
| 23 | 1260 | 19.5±0.2 |
| 23 | 2010 | 18.3±0.1 |
| 25 | 2080 | 16.0±0.01 |
| 25 | 3640 | 17.4±0.1 |
| 27 | 4580 | 20.1±0.01 |
| 27 | 3650 | 19.3±0.01 |
| 37 | 11200 | 16.0±0.1 |
| 37 | 12800 | 15.8±0.02 |
Table 3.
Ct values for qPCR from environmental samples extracted with 100 mM NaOH as in Methods, with MB (magnetic bead) or Dil (direct dilution) prior to qPCR-HRM in triplicate with 16S primers, where NS signifies no specific amplification based on HRM.
Table 3.
Ct values for qPCR from environmental samples extracted with 100 mM NaOH as in Methods, with MB (magnetic bead) or Dil (direct dilution) prior to qPCR-HRM in triplicate with 16S primers, where NS signifies no specific amplification based on HRM.
| Sample | MB/Dil | qPCR-HRM Ct±SEM |
|---|---|---|
| Soil with 5% saline | Dil | 30.4±0.04 |
| MB | 31.2±0.3 | |
| Soil with H2O | Dil | 27.5±0.2 |
| MB | 25.8±0.1 | |
| Four Chicken Cloacal Swabs | Dil | 21.9±1.4 |
| MB | 22.7±2.2 | |
| Cheese | Dil | NS |
| MB | 19.6±0.07 | |
| Bread | MB/Dil | NS |
| Lab surface 1 | Dil | 30.4±0.1 |
| Lab surface 2 | MB | 26.6±0.3 |
| Lab surface 3 | Dil | 32.3±0.4 |
| Lab surface 4 | MB | 29.8±0.4 |
Table 4.
DNA quantification from duplicate fecal swabs from each of six pigs of two growth stages, day 10 lactation or day 59 end of nursery. Pairs of swabs were extracted either with a commercial kit (Kit), or the NaOH extraction with magnetic bead capture (NaOH+MB). NaOH extraction was quantified by qPCR, Kit extraction was quantified with NanoDrop.
Table 4.
DNA quantification from duplicate fecal swabs from each of six pigs of two growth stages, day 10 lactation or day 59 end of nursery. Pairs of swabs were extracted either with a commercial kit (Kit), or the NaOH extraction with magnetic bead capture (NaOH+MB). NaOH extraction was quantified by qPCR, Kit extraction was quantified with NanoDrop.
| ID | Pig age (day) | PCR product (ng/µl) | |
| Kit | NaOH+MB | ||
| 1 | 10 | 241 | 27 |
| 2 | 10 | 68 | 5739 |
| 3 | 10 | 22 | 410 |
| 4 | 59 | 55 | 52 |
| 5 | 59 | 82 | 216 |
| 6 | 59 | 94 | 150 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



