Submitted:
29 November 2023
Posted:
30 November 2023
You are already at the latest version
Abstract
Lyme disease is the most common tick-borne illness in the United States, Europe, and Asia. Borrelia burgdorferi, a spirochete bacterium transmitted by the tick vector Ixodes scapularis, causes Lyme disease in the U.S. If untreated, Lyme arthritis, carditis, and meningitis will occur. Given the absence of a human Lyme disease vaccine, we developed a vaccine using the rabies virus vaccine vector, BNSP333, and an outer surface borrelial protein BBI39. To incorporate BBI39 into the RABV virion, we generated a chimeric BBI39 antigen, BBI39RVG, by fusing it with the final amino acids of the RABV glycoprotein. Here we demonstrated that BBI39RVG antigen was incorporated into the RABV virion, and mice vaccinated with our RABV-BBI39RVG vaccine induced high amounts of BBI39-specific antibodies which are maintained long-term. The BBI39 antibodies neutralized Borrelia in vaccinated mice when challenged with Borrelia burgdorferi by either syringe injection or infected ticks, and reduced Lyme disease pathology.
Keywords:
Lyme disease vaccine
; borrelia burgdorferi
; rabies virus
; viral vaccine vectors
Background
Lyme disease (LD), also called Lyme borreliosis, is the most common vector-borne illness in the United States (U.S), Europe, and Asia, with approximately 476,000 estimated new cases diagnosed annually in the U.S. alone [1]. Cases have increased by 59% in the last year, per the Centers for Disease Control and Prevention (CDC) [2]. The etiologic agent of LD is the spirochete pathogen Borrelia burgdorferi (Bb) and the tick vector Ixodes scapularis, or the deer tick. Bb begins infection at the tick bite site in the host’s skin. A bull’s-eye rash, known as erythema migrans, can appear on the skin due to the spread of Bb from the initial bite site. Symptoms of early-stage disease mimic flu-like symptoms such as fever, headache, fatigue, muscle and joint aches, and swollen lymph nodes [3]. However, 20-30% of patients do not display the rash, which can lead to misdiagnosis [4]. Frequent misdiagnoses further exacerbate disease into late-stage LD where Bb travels to distant organs, including the joints, heart, and central nervous system [4,5]. This can cause complications including Lyme arthritis, carditis, and meningitis [5]. Symptoms include arthritis with severe joint pain and swelling, heart palpitations, and inflammation of the brain and spinal cord [4]. Currently, the only treatment for LD is antibiotics; however, antibiotics are only effective during the early-stage LD [6]. Also, after treatment and Bb clearance, patients can still develop Post-treatment Lyme Disease Syndrome (PTLDS), a chronic inflammatory disease in patients previously diagnosed with LD [7]. These complications indicate a need for a human LD vaccine.
Despite efforts to create a vaccine [8,9], no effective FDA-approved human LD vaccine exists. The rise in global temperatures has caused the tick population to double in the past two decades [10]. The CDC predicts that a continued rise in the tick population will increase LD cases [10]. Additionally, the economic burden of LD is significant; the current annual cost of treatment is estimated to be $1.3 million per year for the U.S. healthcare system [3]. A rise in cases will continue to increase costs, whereas a preventative vaccine would provide a long-term solution for preventing LD [10].
Previously, the FDA approved an LD vaccine, LYMErix, a recombinant OspA protein vaccine adjuvanted with alum [11]. OspA has been shown to protect against Bb, and other strains of Borrelia. LYMErix decreased LDA rates by 76% within the first year on the market [11]. However, there was a reported linkage of OspA-specific serum IgG to an epitope on the human leukocyte function-associated antigen-1 (hLFA1) [12], which was predicted to cause arthritis in vaccinated patients [13]. Despite studies concluding that autoimmunity induced by the LYMErix vaccine was insignificant, vaccine sales declined, which resulted in the removal of the vaccine from the market [10]. Nonetheless, OspA remains a target antigen in LD vaccines, such as in Vanguard crLyme [14], a dog vaccine, and VLA15, a human vaccine developed by Pfizer currently in phase III clinical trials (NCT05477524) [8].
To avoid the possible autoimmune effects, we developed a non-OspA LD vaccine with the protective outer surface protein, BBI39 [15]. BBI39 is an outer surface protein produced on Bb while residing in the tick and early host infection in the skin [15]. This compares to OspA, which is solely produced while Bb resides in the tick and is downregulated when entering the host [16]. A previous study found that mice vaccinated with the recombinant protein, BBI39, induced BBI39-specific antibodies, depleted borrelial load, and reduced LD pathogenesis in the heart and joints [15]. Therefore, we used BBI39 as our target antigen for our rabies virus (RABV) vectored LD vaccine.
To create an effective LD vaccine, a highly immunogenic vaccine vector is crucial. Rabies virus (RABV) vaccine vectors produce a long-lasting humoral immune response [17]. These vaccine vectors are safe because they are highly attenuated and can be made into inactivated vaccines [18,19,20,21]. This study utilized the recombinant RABV vector BNSP333, derived from the SAD-B19, an attenuated wildlife rabies vaccine strain [22]. BNSP333 has been further attenuated by an arginine-to-glutamine mutation at amino acid position 333 of the RABV glycoprotein (G). This mutation further reduces the neurovirulence of the RABV vector and increases its safety profile [23,24]. BNSP333 has a simple genome with only five proteins, making it easy to manipulate and allowing for the addition of stably expressed foreign antigens [25]. The RABV vaccine is highly immunogenic; it induces long-term immunity and a type-1 biased immune response, making it an ideal vector for protection against Bb [26,27]. RABV has been used as a vaccine vector against various infectious diseases including SARS-CoV-2 (CoraVax) [21], Lassa virus (LassaRab) [28], Ebola virus (FiloRab1) [29], and Crimean Congo Hemorrhagic Fever Virus [30]. Some of these vaccines have been tested in nonhuman primates (NHPs) and human studies, further describing the safety and efficacy of this vaccine vector [31].
In this study, we utilized the attenuated RABV vaccine vector with the borrelial outer surface protein BBI39. We demonstrate that the RABV vector successfully incorporated the chimeric BBI39RVG antigen into the RABV virion. Mice immunized with our candidate vaccine, BNSP333-BBI39RVG, induced high and long-term anti-BBI39 antibody titers with a Th-1 biased immune response, compared to the recombinant protein vaccine. In addition, we studied the adjuvant effects of PHAD-SE. Finally, we show that vaccinated mice inhibit Bb infection during both syringe inoculum and via Bb-infected tick challenge, which is induced by borreliacidal antibodies. We show that the RABV vectored BBI39RVG vaccine is an ideal vaccine candidate against LD.
Materials and Methods:
Borrelia burgdorferi, Cell Lines, Mice, and Ticks
Borrelia burgdorferi strain B31 grown in Barbour-Stoenner-Kelly-H (BSK-H) medium complete (supplemented with 6% rabbit serum) (Sigma-Aldrich B8291) was used in this study. Bacteria were grown at 33°C without CO2 in 5 mL bacterial culture tubes. Ixodes scapularis ticks were maintained in a colony within the Pal lab at University of Maryland. Ticks were subjected to microinjection of Bb to perform challenge experiments with infected ticks. BSR and Vero (CCL81) cells were obtained from ATCC and cultured in 1X DMEM (Corning Cat# 10-013-CV) supplemented with 5% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Cells were maintained at 37°C with 5% CO2. Six- to eight-week-old C3H/HeN mice were purchased from Charles River Laboratories. All animal experiments were performed under the guidelines of the Institutional Animal Care and Use Committee and Institutional Bio-safety Committee of Thomas Jefferson University.
cDNA Molecular Cloning of Vaccine Vectors
We inserted BBI39 and BBI39RVG, both synthesized by Genscript in pUC57 vectors, in BsiWI and NheI restriction sites of BNSP333 RABV vaccine vector [20,32] by T4 Ligation (New England Biolabs catalog #: M0202). This included BNSP333-BBI39 and BNSP333-BBI39RVG. BBI39RVG included the final 51 amino acids of the ectodomain (ED51), transmembrane domain (TM), and cytoplasmic domain (CD) of RABV-G, all in the gene synthesis. JM109 E. coli cells were used during molecular cloning under ampicillin resistance. Once plasmids were synthesized, they were sent out for sequencing to Azenta. We utlized forward (5′-GGAGGTCGACTAAAGAGATCTCACATAC-3′) and reverse (5′-TTCTTCAGCCATCTCAAGATCGGCCAGAC) primers for sequencing of BBI39 between RABV-N and RABV-P. WE also used, forward (5′-GTTATGGTGCCATTAAACCGCTG-3′) and reverse (5′-TCTCCAGGATCGATCGAGCATCTT-3′) primers to sequence RABV-G to determine if the 333 mutation was still viable in the glycoprotein before virus recovery.
Recovery of Recombinant Rabies Viruses
Recombinant RABV vectors were recovered on BSR cells in the above-listed conditions. X-tremeGENE 9 transfection reagent (Roche Diagnostics) in Opti-MEM was utilized to transfect BSR cells in 6-well plates with full-length BNSP333 cDNA along with T7 RNA polymerase, RABV nucleoprotein, phosphoprotein, glycoprotein, and polymerse cDNA plasmids. Supernatants were harvested on day 4 post-transfection and overlayed on seeded BSR cells in 12-well plates. After 48 hours, cells were subjected to fix by acetone and staining with a GFP stain against RABV-N (FujiRebio, Cat# 800-092).
Viral Growth, Titration, Purification, and Inactivation
Once viruses were recovered, they were grown on Vero CCL81 cells in viral production serum-free medium (VP-SFM) (Thermo Fisher Scientific) supplemented with 5% Glutamax, 1% penicillin-streptomycin, and 1% HEPES buffer. Cells were infected at a MOI of 0.01 in either T175 flask or 2-stack chambers (Corning). Supernatant collections occurred 5 days post-infection and every 3 days afterward, for a total of 17 days. For titration, RABVs were overlayed on VeroCCL81 cells with a starting dilution of 1:10 and diluted 10-fold in a 96-well plate in triplicate. After 48 hours, cells were fixed with acetone and stained with RABV-N GFP stain to determine the foci forming units (FFU)/mL by fluorescence microscopy.
Viruses were filtered through 0.45 µm PES membrane filters (Nalgene) and concentrated down to 50 mL for purification. Viruses were purified over 20% sucrose cushion and ultracentrifuged at 25,000 rpm in a SW32 rotor for 1.5 hours. Viral particles were resuspended in TEN buffer + 2% sucrose and inactivated with 50 µL per mg of particles with β-propiolactone (BPL, Millipore Sigma, Cat# P5648) in cold culture grade water. The level of inactivation was verified by inoculating Vero CCL81 cells over 3 passages with 10 µg of BPL-inactivated virions.
Immunofluorescence
Vero CCL81 cells were seeded on coverslips in 1x DMEM supplemented with 5% FBS and 1% penicillin-streptomycin at 5x105 cells per well. On the same day, cells were infected with BNSP333-BBI39RVG, BNSP333-BBI39, and BNSP333 at an MOI of 0.05. Cells were kept at 34°C for 72 hours and then stained. Cells were fixed in 2% paraformaldehyde (PFA) in 1X PBS for 30 min for surface-stained cells and 15 min for intracellular-stained cells. Then, 2% PFA and 0.01% TritonX were added to intracellular stained cells for another 15 min. After fixing, cells were blocked in PBS with 5% FBS for 1 hour. Following washing with PBS three times, cells were stained with 1:200 primary antibody for 1 hour at room temperature (RT) in PBS with 1% FBS. Primary antibodies included polyclonal mouse anti-BBI39 IgG and human anti-RABV-G 4C12 (provided by Scott Dessain, Lankenau Institute for Medical Research, Wynnewood, PA). Cells were washed with PBS and incubated with secondary antibody in PBS with 1% FBS for 45 min at RT. Secondary antibodies utilized for fluorescent staining were anti-mouse Cy3 and anti-human Cy2. Stained cells were then washed 5 times with PBS and mounted on slides with mounting media containing DAPI (ProLong™ Glass Antifade Mountant, Invitrogen, cat#: P36980). Slides were stored for 24-48 hours in the dark at RT to dry. Slides were visualized with a Nikon A1R confocal microscope. Images were analyzed by Fuji. Red (Cy3) BBI39 staining was changed to magenta by FIJI ImageJ.
Cell Lysate Preparation for Western Blot
For infected cell lysates, 1x106 BSR cells were infected at an MOI of 5 for 72 hours at 34°C in a 6-well plate. Cells were washed twice with cold PBS and 1 mL of RIPA lysis buffer + 1X protease inhibitor (ThermoFisher Halt™ Protease Inhibitor Cocktail 100X, cat#: 78430) was added to lyse infected cells. After 5 minutes of lysis, cells were centrifuged at 14,000 rpm for 1 minute. The supernatant was then subjected to a BCA assay to determine the concentration of the proteins from the cell lysates. Finally, the concentration of cell lysates was adjusted to 10 µg/µL in urea sample buffer containing 2-mercaptoethanol.
Western Blot
Infected cell lysates, recombinant proteins, and purified viral particles were subjected to Western Blot analysis. Lysates and particles were denatured in urea sample buffer and reduced with 2-mercaptoethanol. Samples were boiled for 10 minutes at 95°C. 30 µg of cell lysates, 20 ng of BBI39 recombinant protein, and 1 µg of sucrose purified virions were separated on the gel. Gels were run at 150 V for about 1.5 hours in 1X Laemmli buffer. Proteins were transferred to nitrocellulose membranes for 1 hour at 90 V in 1X Towbin transfer buffer. Electrophoresis of gels and transfer of proteins were done using BioRad western blot equipment. After transfer, membranes were blocked with 5% milk in 1X PBST for 1 hour at RT. Primary antibodies used for probing included polyclonal mouse anti-BBI39 IgG and human anti-RABV-G 4C12 (provided by Scott Dessain, Lankenau Institute for Medical Research, Wynnewood, PA). Blots were probed overnight in 5% BSA in PBS at 4°C. The following day, blots were probed with secondary antibody, which included horseradish peroxidase (HRP)-conjugated anti-mouse (Jackson ImmunoResearch, 115–035–146) at 1:5000 or human IgG (SouthernBiotech, 2040–05) at 1:20,000 diluted in 1X PBST for 1 hour at RT. Proteins were detected with SuperSignal West Dura Chemiluminescent substrate (Thermofisher cat#: A38554) and imaged on a FlourChem R system (ProteinSimple).
Production of Recombinant Proteins for Western Blot, Immunizations, and ELISA
BBI39 was purified using a plasmid from the Utpal Pal laboratory in University Park, Maryland. BBI39 plasmid transformed into JM109 E. coli cells and grew into a 1 L bacterial culture in LB broth with ampicillin. Bacteria were induced with 0.3 mM of ITPG overnight at RT. The following day, bacteria were centrifuged at 4000 rpm for 30 minutes at 4°C. The pellet was resuspended in 20 mL of PBS with 1% TritonX. Then, for further lysis, 0.1 mg/mL of Lysozyme was added to the resuspension. After a 30-minute incubation on ice, bacterial cells were further lysed by sonication for 7 minutes. Lysed bacterial cells were centrifuged at 4000 rpm for 30 minutes at 4°C. BBI39 was purified using Glutathione Agarose (Pierce™, Thermofisher cat#: 16100). The GST tag was removed by 80 units of precision protease (GenScript cat#: Z02799) overnight at 4°C in PCB buffer (50 mM Tris pH 7.0, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, mixed in water). Purification of the protein was further characterized by Western Blot, listed above.
RABV glycoprotein (G) was produced by stripping the glycoprotein from rVSV-ΔG-RABV-G-GFP virions. BEAS-2B cells were infected with rVSV-ΔG-RABV-G-GFP in OptiPRO SFM at an MOI of 0.01. Once all the cells were lysed, supernatants were concentrated and ultracentrifuged through a 20% sucrose cushion at 25,000 rpm for 1.5 hours at 4°C. Viral pellets were then resuspended in β-Octyl-glucopyranoside (OGP) and stripped by ultracentrifugation at 45,000 rpm for 1.5 hours in an SW55Ti rotor at 4°C. Supernatants were collected, frozen in small aliquots, and characterized by Western Blot and ELISA.
Immunizations
Groups of five 6–8-week-old female and male C3H/HeN mice purchased from Charles River Laboratories were immunized intramuscularly (I.M.) with 10 µg of inactivated RABV virions or 1 × 104 ffu/mL of live attenuated virus. Five females were used for Figure 3b,c. Five females and Five males were used for Figure 2d,e, separately vaccinated from Figure 3b,c. Inactivated vaccines were formulated either unadjuvanted or with 5 µg of synthetic monophosphoryl Lipid-A (MPLA), 3D(6 A)-PHAD, in a squalene oil-in-water emulsion (PHAD-SE) adjuvant. Each immunization contained 100 µL, with 50 µL injected into each hind leg of each mouse. All mice were primed on day 0 and boosted on day 28. Serum for further testing was collected by retroorbital bleeds while mice were under isoflurane anesthesia on days 0, 14, 28, and 56 for short-term experiments with the addition of days 112, 168, and 224 for long-term experiments.
anti-BBI39 and anti-RABV-G ELISA
Total and isotype subclass IgG antibody responses were determined by indirect ELISA. We purified recombinant RABV-G and BBI39, described above, and utilized these proteins to coat Immulon 4 HBX 96-well flat-bottom microtiter plates. Plates were coated with antigens overnight at 4°C with in 15 mM Na2CO3, 35 mM NaHCO3 coating buffer. BBI39 antigen was utilized at 500 ng/well and RABV-G at 50 ng/well. Post-incubation, plates were washed three times with PBS containing 0.05% Tween20 (PBST) and blocked in 5% milk for 2 hours at RT. Plates were washed and primary antibody dilution buffer, containing 0.5% BSA and 0.05% NaN3 in PBST, was added at 100 µL per plate. Mouse sera, at 1:50 starting dilution, was further diluted 3-fold down the plates. Plates were incubated overnight at 4°C with primary antibody. The following day, plates were washed and 100 µL of secondary antibody diluted in PBST was added. Secondary antibodies used in this study were horseradish peroxidase-conjugated goat anti-mouse IgG-Fc (Jackson ImmunoResearch, Cat# 115-005-008); IgG2a (Jackson ImmunoResearch Cat# 115-035-206); or IgG1 (Jackson ImmunoResearch Cat# 115-035-205). All secondary antibodies were diluted to a concentration of 80 ng/mL for BBI39 ELISAs and 25 ng/mL for RABV-G ELISAs. After incubation for 2 hours at RT, plates were washed and then developed with 200 µL/well of o-Phenylenediamine Dihydrochloride substrate (ThermoFisher) for 15 minutes. The reaction was stopped with 3M H2SO4. The optical density (OD) was determined at 490 nm (experimental) and 630 nm (background) on a BioTek ELx800 plate reader on Gen5 software to determine the delta values between the experimental and background readings. ELISA data were analyzed in GraphPad Prism 9 software to determine the EC50 values of antibodies in the mouse sera.
Borreliacidal Assay
Borrelia burgdorferi, strain B31, was seeded at 1x105 spirochetes/mL in 96-well round-bottom plates with a 1:10 dilution of heat-inactivated mouse sera or 100 ug/mL of LA-2 antibody (Absolute Antibody, Ab01070-3.0) with 1:10 guinea pig complement sera (Sigma S1639) diluted in BSK-H complete media (Sigma-Aldrich B8291) for a total of 100 uL. Mouse sera were heat-inactivated at 56°C for 30 minutes and tested in duplicate. Borrelia was incubated at 33°C with mouse sera for 48 hours in a 96-well round-bottom plate. 1 µL of each well was added to 1 mL of BSK media in Eppendorf tubes. After 7 days, each replicate was counted under a Nikon dark-field microscope with a Petroff-Hausser counting chamber (Hausser Scientific, Cat#: 3900). Borrelial counts were analyzed in GraphPad Prism 9 software to determine if mouse sera inhibited bacterial growth by borreliacidal activity.
RABV Neutralization by Rapid Fluorescent-Focus Inhibition Test (RFFIT)
RFFITs were performed to identify RABV-neutralizing antibodies as described previously [21]. Mouse sera were heat inactivated at 56°C for 30 minutes. BSR cells were seeded at 25,000 cells/well and cultured in DMEM with 5% FBS and 1% penicillin-streptomycin, in 96-well flat-bottom plates. Individual mouse sera, collected at day 56, were diluted 3-fold with a starting dilution of 1:50. The WHO standard of rabies IgG was used at a starting dilution of 2 international units (IU)/mL. After dilution of sera, CVS11, a challenge virus strain of rabies virus, was added to each well at a titer to achieve 90% infection of BSR cells. The virus and antibody mixture was incubated in a 96-well round-bottom plate for 1 hour at 34°C. After incubation, 105 µL of sera/virus mixture was added to BSR cells and incubated at 34°C for 24 hours. Cells were fixed with 80% acetone and stained with FAD stain against RABV nucleoprotein. Stained cells were assessed for the percentage of viral infection by fluorescent microscopy. The Reed-Muench method was utilized to calculate 50% endpoint titers. Titers were converted to IU/mL by comparison to the WHO standard.
Borrelia burgdorferi Challenge by Needle Injection
Immunized and unimmunized mice were subjected to challenge with Borrelia burgdorferi, strain B31, by needle route. Mice (Figure 2d,e) were injected with 1x105 spirochetes/100 µL intradermally with an insulin needle. Bacteria were grown in BSK-H complete medium (Sigma-Aldrich B8291) at 33°C before being counted by Nikon dark-field microscopy with a Petroff-Hausser counting chamber (Hausser Scientific, Cat#: 3900). Bacteria were diluted in BSK media in Eppendorf tubes and kept at RT before injection.
21 days post-infection, skin (ear), tibiotarsi joints, heart, and bladders were harvested from infected mice aseptically and were subjected to RNA extraction and qPCR (described below) or for further analysis under dark-field microscopy. Organs were placed in 1 mL BSK-H medium and incubated at 33°C. Every 2 weeks, for 8 weeks, organs were analyzed under dark-field microscopy for qualitative analysis of Borrelia in each organ’s supernatant.
Blood was also collected from challenged mice and subjected to Western Blot analysis to determine whether each mouse was successfully challenged with Borrelia. Borrelia burgdorferi was grown in 50 mL to 1x108 spirochetes/mL. Bacteria were centrifuged at 4000 x g for 20 minutes at 4°C. Borrelial pellets were washed five times with 1 mL of PBS. After each wash, the lysates were centrifuged in Eppendorf tubes at 14,000 x g for 1 minute. Borrelial lysates were subjected to BCA assay to determine the final concentration. Lysates were diluted to a final concentration of 1.5 µg/10 µL in 1X urea sample buffer. Aliquots were frozen at -80°C or used for Western Blot analysis. Lysates were denatured for 10 minutes at 95°C and gels were run and transferred as described above. For primary antibody, individual mouse sera were diluted to 1:1000 in 5% BSA and added to strips of nitrocellulose membrane with Borrelial lysates transferred on each strip. Primary antibody was incubated overnight and further processed as listed above. Individual strips were imaged at the same time to test whether challenged or unchallenged mouse sera responded to borrelial lysates on blots.
Borrelia burgdorferi Challenge by Ixodes scapularis
Groups of 6 mice were immunized as described earlier. On day 56 post-primary immunization, mice were challenged with infected Ixodes scapularis nymphs (5 ticks/mouse). After 3 weeks of infection, mice were euthanized and Bb burden within mouse organs was assessed by qRT-PCR (described below). Skin, heart, and joints were cultured in BSK-H media described above.
RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
Organs from challenged mice were harvested and placed in 1 mL TRIzol Reagent in 2mL RNase/DNase free Omni tubes (Omni International) which contained beads for homogenization. Bladders were collected in Omni tubes with 1.4 mm ceramic beads (Cat#: 19-627D). Hearts and skin (ear) were collected in Omni tubes with 2.8 ceramic beads (Cat#: 19-628D). Joints were collected in Omni tubes with 2.8 metal beads (Cat#: 19-620D). Tubes were homogenized by Omni Bead Ruptor for 90 seconds. Homogenates were frozen at -80°C for RNA extraction. RNA was extracted from whole organs using TRIzol Reagent phase separation protocol. RNA extraction was done using the PureLink RNA Mini Kit (Ambion). The quantity and quality of RNA extracted was measured using NanoDrop (Thermofisher).
Borrelia was quantified by RT-qPCR. FlaB and mouse B-actin primer probes were designed for use with TaqMan Fast Virus 1 Step Master Mix reagent (ThermoFisher) using 5 µL of extracted RNA from each mouse organ. Primer probes were ordered from ThermoFisher. FlaB was amplified by forward primer (5’-TTGCTGATCAAGCTCAATATAACCA-3’) and reverse primer (5’-GCATCGCTTTCAGGGTCTCAA-3’) with a probe quenched with FAM fluorescent dye (5’-AGAACAGCTGAAGAGCTTGGAATGCAGCCTGCAAAAATTAACACA-3’). Mouse B-actin was amplified by forward primer (5’-AGAGGGAAATCGTGCGTGAC-3’) and reverse primer (5’-ACGGCCAGGTCATCACTATTG-3’) with a probe quenched with VIC fluorescent dye (5’-CAAAGAGAAGCTGTGCTATGTTGCTCTAGACTTCGAGCAGGAGAT-3’). The reaction was set up for a fast-cycling mode with the following cycling protocol: 1 cycle for 5 minutes at 50 °C, 1 cycle for 20 seconds at 95 °C, and 40 cycles of 95 °C for 3 seconds and 60 °C for 30 seconds. The reactions were run on Step One Plus qPCR machine.
Histological Analysis
Joints from challenged mice were collected from each group 3 weeks after infection. Joints were fixed in 4% paraformaldehyde (PFA) and stained with hematoxylin-eosin (H&E) stain. Signs of arthritis were evaluated by as described elsewhere in a blinded manner [33].
Statistical Analysis
For ELISA, log-transformed 50% effective concentration (EC50) values were determined by plotting against delta OD value (OD [490nm]-OD [630nm]) on GraphPad Prism 9 software. For all statistical analyses, one-way ANOVA with post-hoc Tukey HSD test was performed on log-transformed data.
Results
BBI39 Vaccine Designs
In this study, we utilized the well-established attenuated RABV vector BNSP333 [21,27,28,29,30,31] (Figure 1a). The gene encoding the borrelial antigen, BBI39, was inserted in BNSP333 at the second position between the RABV nucleoprotein (N) and phosphoprotein (P) and named BNSP333-BBI39 (Figure 1b). This vaccine determined the immunogenicity of BBI39 after immunization with the live viral vector BNSP333-BBI39. We also developed the BNSP333-BBI39RVG vaccine, to incorporate a chimeric BBI39 protein into the RABV virion. This vaccine can be produced as a killed vaccine due to the incorporation of the antigen into the RABV virion. The chimeric BBI39 antigen contained the RABV-G Igκ signal sequence (SS) fused to the 3’ end of BBI39 nucleotide sequence, followed by the sequences for the last 51 amino acids of the ectodomain (ED51), the transmembrane domain (TM), and cytoplasmic domain (CD) of RABV-G (Figure 1c). These sections are called RVG tail. We previously showed that these additions promote translocation of the foreign protein to the endoplasmic reticulum (ER) and transport to the surface of an infected cell, allowing for BBI39 incorporation into the budding RABV virion [21,30].
Characterization of Newly Developed BBI39 Vaccines
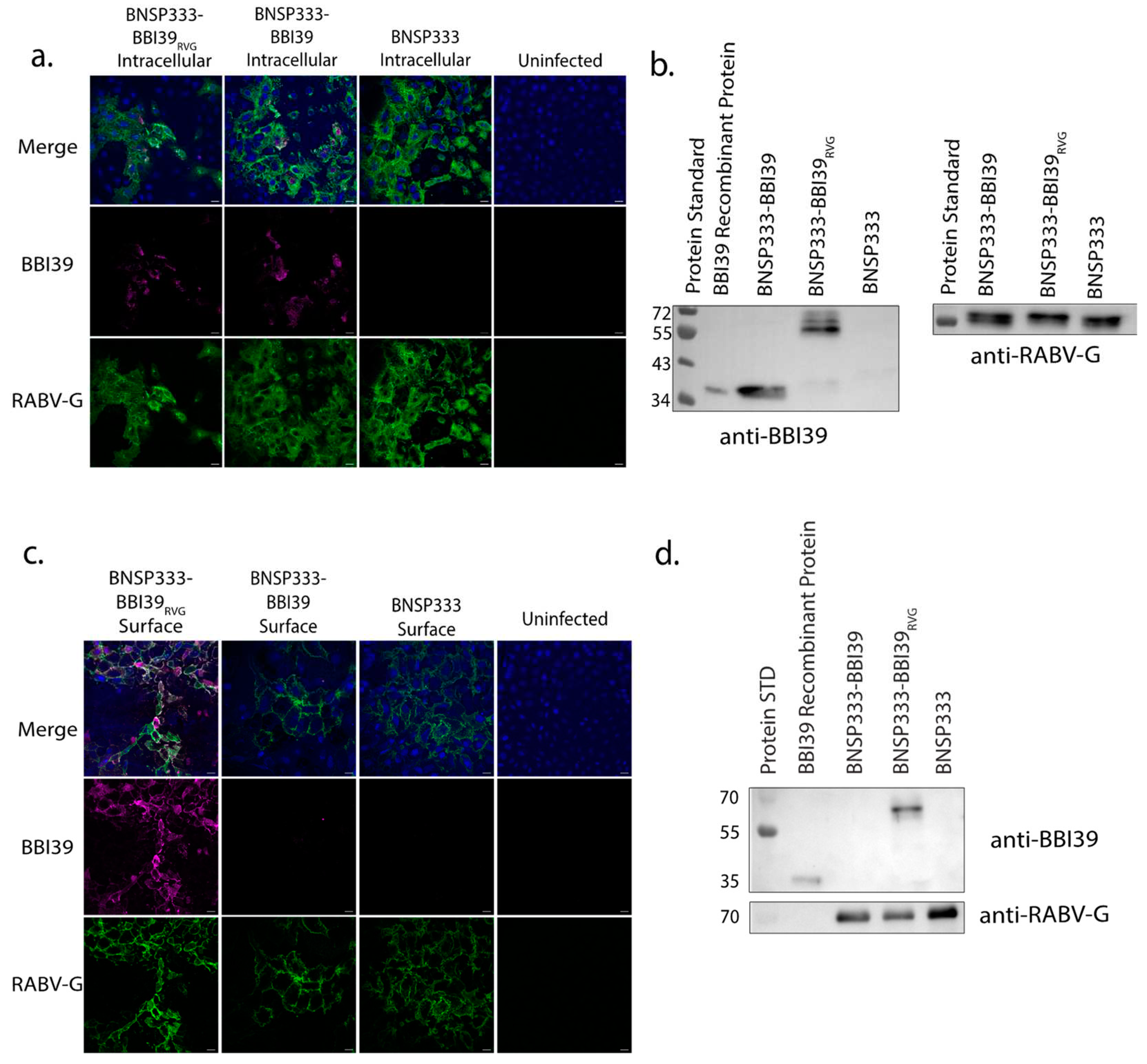
Infectious recombinant viruses were recovered by previously established methods [34,35]. To confirm BBI39 expression, we characterized these viruses by immunofluorescence staining and western blotting analysis. First, we conducted intracellular immunostaining of infected cells for BBI39 protein (magenta) and RABV-G (green) with their respective antibodies (Figure 2a). BBI39 was expressed intracellularly by both BNSP333-BBI39RVG and BNSP333-BBI39 viruses. In addition, proteins of infected cell lysates were separated by SDS-PAGE and probed for both BBI39 and RABV-G by western blot. We confirmed expression of BBI39 by both viruses, BNSP333-BBI39RVG (50 kDa) and BNSP333-BBI39 (30 kDa) (Figure 2b). This includes RABV-G at about 65 kDa (Figure 2b).
To verify the transport of BBI39 to the surface of an infected cell by the RVG tail, we performed another immunofluorescence assay with surface-stained cells. BNSP333-BBI39RVG successfully translocased BBI39 to the surface of the cell, but BNSP333-BBI39 did not (Figure 2c).
After successful translocation of BBI39 to the surface of the cell, the protein was incorporated into the budding BNSP333-BBI39RVG viral virion. Sucrose-purified virions were separated on an SDS-PAGE protein gel and probed for by western blotting. SDS-PAGE protein gel displayed all RABV proteins (L, G, N, P, and M) within the RABV virion (Supplementary Figure S1). Western immunoblotting detected incorporation of BBI39 and RABV-G in BNSP333-BBI39RVG virions; however, BNSP333-BBI39 did not incorporate BBI39 (Figure 2d). While both viruses produce BBI39 intracellularly (Figure 2a,b), only the chimeric BBI39RVG protein, expressed by BNSP333-BBI39RVG, was detected on the surface of the infected cells, and incorporated into the RABV virion (Figure 2c,d).
Figure 2.
RABV can incorporate the chimeric borrelial protein BBI39RVG. Characterization of RABV-vectored vaccine viruses by immunofluorescence (a,c) and western blot (b,d). For immunofluorescence, VEROCCL81 cells were infected at MOI 0.05 for 72 hours and fixed and permeabilized for intracellular (a) and fixed only for surface staining (c). Cells were stained with human anti-RABV-G (4C12) (green) and polyclonal mouse anti-BBI39 (magenta) and mounted with mounting media containing DAPI (blue). Images were taken at 40X magnification, and scale bars represent 10 μm. Western blot of cell lysates (b) of VERO-CCL81 cells infected at an MOI of 0.05 for 72 hours were probed for BBI39 and RABV-G. Western blots of sucrose-purified virions (d) were probed for BBI39 and RABV-G. Blots were probed with either polyclonal mouse-anti-BBI39 (produced by the Pal lab at University of Maryland) or 4C12 human anti-RABV-G monoclonal antibody (provided by Scott Dessain, Lankenau Institute for Medical Research, Wynnewood, PA) (b,d).
Figure 2.
RABV can incorporate the chimeric borrelial protein BBI39RVG. Characterization of RABV-vectored vaccine viruses by immunofluorescence (a,c) and western blot (b,d). For immunofluorescence, VEROCCL81 cells were infected at MOI 0.05 for 72 hours and fixed and permeabilized for intracellular (a) and fixed only for surface staining (c). Cells were stained with human anti-RABV-G (4C12) (green) and polyclonal mouse anti-BBI39 (magenta) and mounted with mounting media containing DAPI (blue). Images were taken at 40X magnification, and scale bars represent 10 μm. Western blot of cell lysates (b) of VERO-CCL81 cells infected at an MOI of 0.05 for 72 hours were probed for BBI39 and RABV-G. Western blots of sucrose-purified virions (d) were probed for BBI39 and RABV-G. Blots were probed with either polyclonal mouse-anti-BBI39 (produced by the Pal lab at University of Maryland) or 4C12 human anti-RABV-G monoclonal antibody (provided by Scott Dessain, Lankenau Institute for Medical Research, Wynnewood, PA) (b,d).
Immunogenicity of RABV-BBI39 Vaccines
After characterization of the recombinant viruses, we studied the immunogenicity of the vaccines in C3H/HeN mice, a well-characterized model for studying LD in mice [15]. When these mice are infected with Borrelia, they show symptoms of LD observed in humans, such as Lyme arthritis and carditis [15].
Five C3H/HeN mice per group were immunized with 104 foci forming units (ffu) of live BNSP333-BBI39 or BNSP333-BBI39RVG. Since BNSP33-BBI39RVG incorporates BBI39 into the virion, we also tested it as an inactivated vaccine, both with and without the adjuvant PHAD-SE [26]. Several studies have demonstrated that PHAD-SE induces a Th1-biased immune response [26,27]. This should induce borreliacidal antibodies that can bind complement and protect against Borrelia burgdorferi, as shown in other studies [36,37,38]. Mice were immunized on day 0 and boosted on day 28 (Figure 3a). As a positive control, we included the previously tested recombinant BBI39 protein, which depleted Bb in a previous tick and syringe challenge [15]. Since BBI39 protein immunization was previously adjuvanted with Freund’s adjuvant, an adjuvant not sanctioned for human use, we included the adjuvant PHAD-SE. Finally, as a RABV vector control, we used FiloRab1 [26], a well-studied BNSP333 vectored vaccine incorporating the Ebola virus glycoprotein. Serum was collected on days 0, 14, 28, and 56 post-initial immunization, and analyzed for the presence of IgG antibodies against BBI39 and RABV-G by ELISA (Figure 3b,c).
Figure 3.
BNSP333-BBI39RVG elicits anti-BBI39 and anti-RABV-G antibodies with type-1 biased immunity. ELISA anti-BBI39 and anti-RABV-G antibody half maximal effective concentration (EC50) titers from vaccinated mouse sera. (a) vaccination schedule of C3H/HeN mice. Mice were primed on day 0 and boosted on day 28 with RABV-based vaccines. Blood drops shown to collect sera for ELISA testing. Anti-BBI39 (b) and anti-RABV-G (c) total IgG EC50 titers from day 14-56 determined from individual mouse serum ELISA curves (n=5 females). (d) Day 56 EC50 titers with inactivated groups only, demonstrating female and male total IgG titers (n=10/vaccine group, 5 females and 5 males). Day 56 anti-BBI39 IgG isotype ELISAs. Statistics are represented by the color of the bars from each vaccinated group. Error bars represent the SEM. Statistics were calculated by one-way ANOVA with post hoc Tukey’s test of log-transformed data. p > (ns), p < 0.0332 (*), p < 0.0021 (**), p < 0.0002 (***), p < 0.0001 (****).
Figure 3.
BNSP333-BBI39RVG elicits anti-BBI39 and anti-RABV-G antibodies with type-1 biased immunity. ELISA anti-BBI39 and anti-RABV-G antibody half maximal effective concentration (EC50) titers from vaccinated mouse sera. (a) vaccination schedule of C3H/HeN mice. Mice were primed on day 0 and boosted on day 28 with RABV-based vaccines. Blood drops shown to collect sera for ELISA testing. Anti-BBI39 (b) and anti-RABV-G (c) total IgG EC50 titers from day 14-56 determined from individual mouse serum ELISA curves (n=5 females). (d) Day 56 EC50 titers with inactivated groups only, demonstrating female and male total IgG titers (n=10/vaccine group, 5 females and 5 males). Day 56 anti-BBI39 IgG isotype ELISAs. Statistics are represented by the color of the bars from each vaccinated group. Error bars represent the SEM. Statistics were calculated by one-way ANOVA with post hoc Tukey’s test of log-transformed data. p > (ns), p < 0.0332 (*), p < 0.0021 (**), p < 0.0002 (***), p < 0.0001 (****).
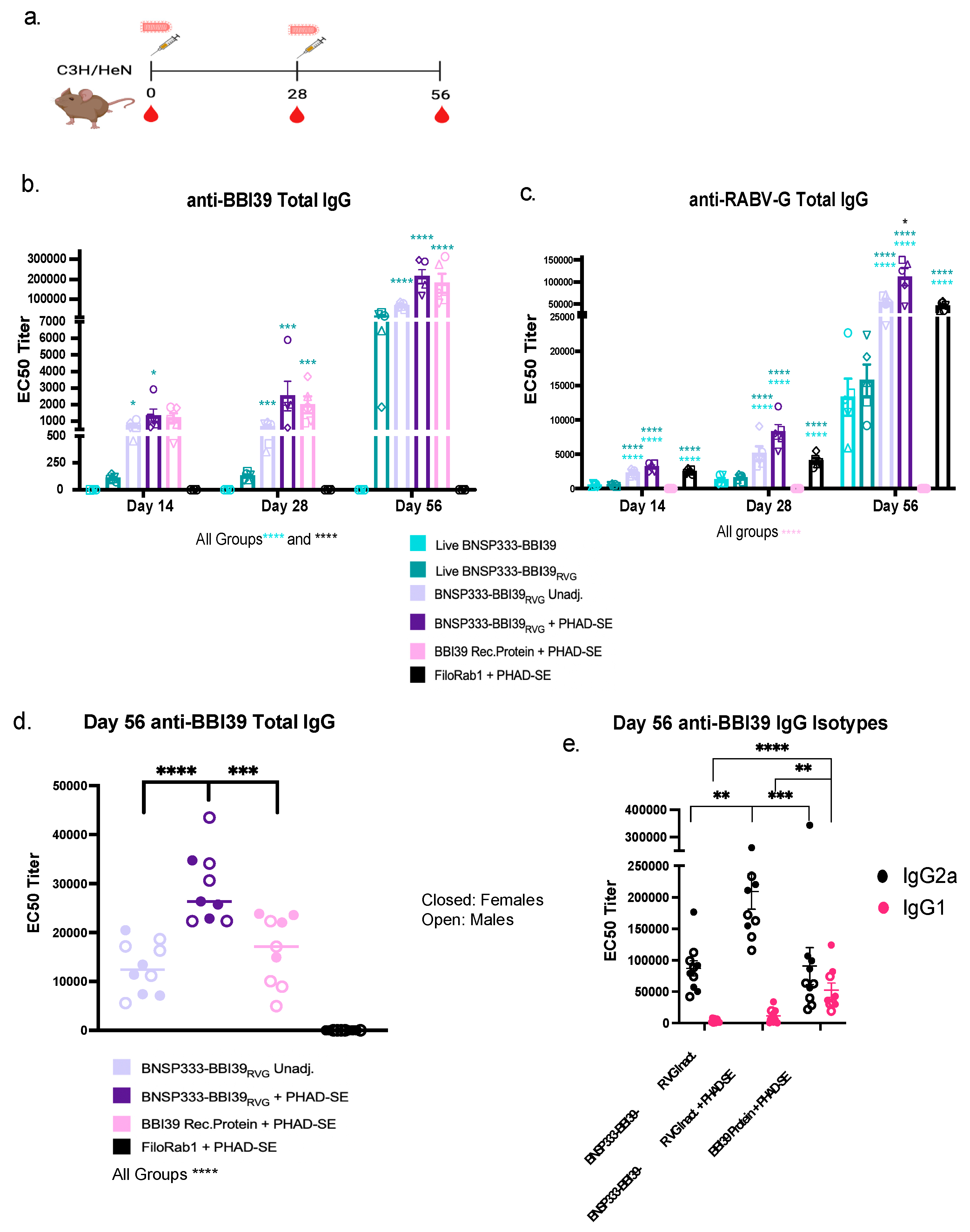
The detection of anti-BBI39 IgG responses show induction of antibody responses as early as 14 days after the first immunization in both the inactivated vaccine groups (BNSP33-BBI39RVG, with and without adjuvant) and the recombinant protein BBI39 group, with 1-fold lower titer seen in the live BNSP333-BBI39RVG group (Figure 3b). BBI39-specific antibody responses were not observed in the live BNSP333-BBI39 vaccine group (Figure 3b). These responses demonstrated a slight 1-fold increase in adjuvanted groups on day 28 compared to day 14. Serum titers increased almost 100-fold on day 56, post-boost, for all groups vaccinated with BNSP333-BBI39RVG and recombinant protein immunized groups. Overall, on day 56 BNSP333-BBI39RVG + PHAD-SE vaccinated mice had the highest BBI39 and RABV-G IgG titers compared to all other groups (Figure 3b,c). Live BNSP333-BBI39 vaccinated mice continue to not elicit anti-BBI39 antibodies after boost on day 56; however, anti-RABV-G antibodies confirmed a successful vaccination with BNSP333-BBI39 (Figure 3c). This demonstrates that cell surface expression and incorporation of BBI39 in the RABV virion is critical to induce an antibody response against the borrelial antigen. Since BNSP333-BBI39 did not elicit antibodies against BBI39, this vaccine was excluded from further studies.
Overall, inactivated BNSP333-BBI39RVG + PHAD-SE induced a higher anti-BBI39 and anti-RABV-G IgG responses than recombinant protein BBI39 + PHAD-SE immunized mice (Figure 3b–d). In addition, to analyze sex as a scientific variable, we included female and male mice in our study (n=10) (Figure 3d). We detected a 1.5-fold difference between females and males in the recombinant protein immunized groups and less than 1-fold in the RABV vectored vaccinated groups (Supplementary Figure S2a), but these differences were not statistically significant. However, the stronger humoral immune responses induced by the RABV-based vectors compared to the recombinant protein vaccine (Figure 3b–d) indicated potential benefits of the RABV vector platform compared to the recombinant protein vaccine platform.
Finally, we analyzed the type of antibody responses by BBI39 IgG isotype responses (Figure 3e). We detected IgG2a, a type-1 associated immune response antibody, and IgG1, a type-2 associated antibody by ELISA. As previously demonstrated [26,27], BNSP333-BBI39RVG + PHAD-SE induced a higher IgG2a response than the unadjuvanted and recombinant protein immunized groups (Figure 3e). The protein-vaccinated mice demonstrated a balanced response between IgG2a and IgG1, whereas RABV-vectored vaccines, both with and without adjuvant, induced a more significant skew towards a type-1 associated immune response, previously shown to protect against Borrelia [36,37]. (Figure 3e).
Neutralization of Borrelia burgdorferi and Rabies Virus by BBI39 Vaccines
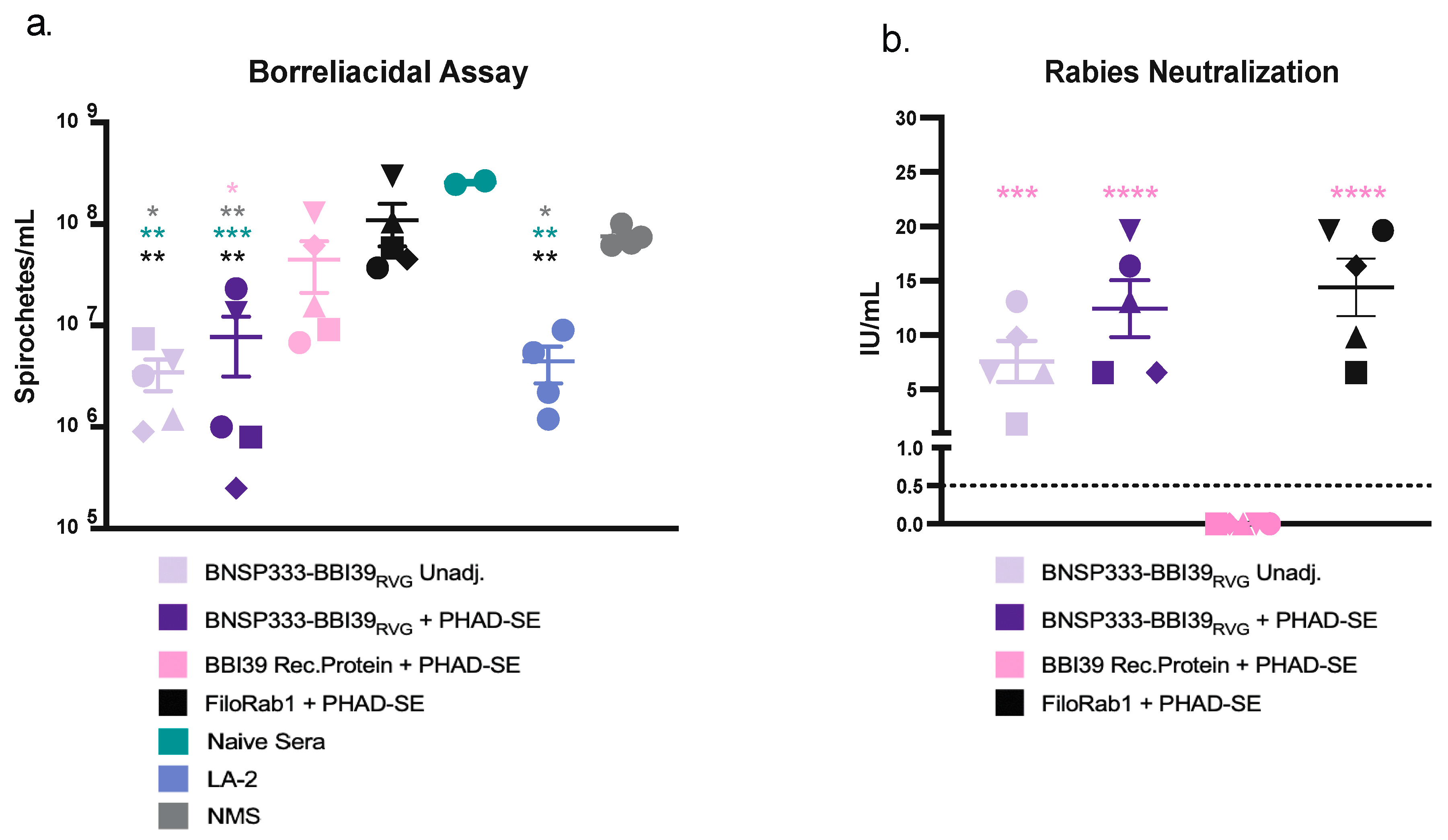
Next, we determined if these antibodies were functional against both Borrelia burgdorferi and RABV. We conducted a borreliacidal assay to determine neutralization of Bb and found that mouse sera against BBI39, with the addition of guinea pig complement, delayed the growth of Borrelia in culture by nearly 10-fold. A significantly greater growth delay was found in the RABV vectored groups, especially with the addition of PHAD-SE (Figure 4a). This inhibition similarly compares to the positive control, LA-2 antibody, which is a protective monoclonal antibody [39]. However, the sera from recombinant protein immunized mice, even with adjuvant, did not significantly deplete Bb in vitro compared to FiloRab1 vaccinated mice, naïve mice, and no mouse sera (NMS) (Figure 4a). These data indicate the potential benefits of the RABV vector platform compared to recombinant protein vaccines.
We also analyzed the potential of the RABV-vectored vaccines to neutralize infectious RABV in a rapid fluorescent-focus inhibition test (RFFIT). Protection against both Borrelia and RABV would be another advantage of this vaccine. Serum from all RABV virus-vectored vaccinated mice contained a neutralizing titer of above 0.5 IU/mL, the WHO accepted protective antibody level against rabies (Figure 4b). BNSP333-BBI39RVG + PHAD-SE adjuvant induced 3-fold higher neutralizing titers compared to unadjuvanted groups. These levels compared to the control group, FiloRab1 + PHAD-SE, which was previously shown to produce high neutralization effects against RABV [26]. Therefore, the RABV-based BBI39 vaccine can induce both borreliacidal and RABV-neutralizing antibodies in vitro.
Figure 4.
BNSP333-BBI39RVG can successfully neutralize Borrelia burgdorferi and RABV in vitro. (a) Borreliacidal assay conducted with serum from vaccinated mice (5 female mice per group), naïve sera, LA-2 protective monoclonal antibody, and no mouse sera (NMS). Bb titers counted 7 days after addition of mouse sera and guinea pig complement sera to culture. (b) Rabies neutralization assay (RFFIT) containing serum from vaccinated mice. IU/mL titers were calculated by comparison to WHO standard. Statistics are represented by the color of the bars from each vaccinated group. Error bars represent the SEM. Statistics were calculated by one-way ANOVA with post hoc Tukey’s test of log-transformed data. p > (ns), p < 0.0332 (*), p < 0.0021 (**), p < 0.0002 (***), p < 0.0001 (****).
Figure 4.
BNSP333-BBI39RVG can successfully neutralize Borrelia burgdorferi and RABV in vitro. (a) Borreliacidal assay conducted with serum from vaccinated mice (5 female mice per group), naïve sera, LA-2 protective monoclonal antibody, and no mouse sera (NMS). Bb titers counted 7 days after addition of mouse sera and guinea pig complement sera to culture. (b) Rabies neutralization assay (RFFIT) containing serum from vaccinated mice. IU/mL titers were calculated by comparison to WHO standard. Statistics are represented by the color of the bars from each vaccinated group. Error bars represent the SEM. Statistics were calculated by one-way ANOVA with post hoc Tukey’s test of log-transformed data. p > (ns), p < 0.0332 (*), p < 0.0021 (**), p < 0.0002 (***), p < 0.0001 (****).
Reduction of Borrelia burgdorferi in Vaccinated Mice by Syringe Inoculation
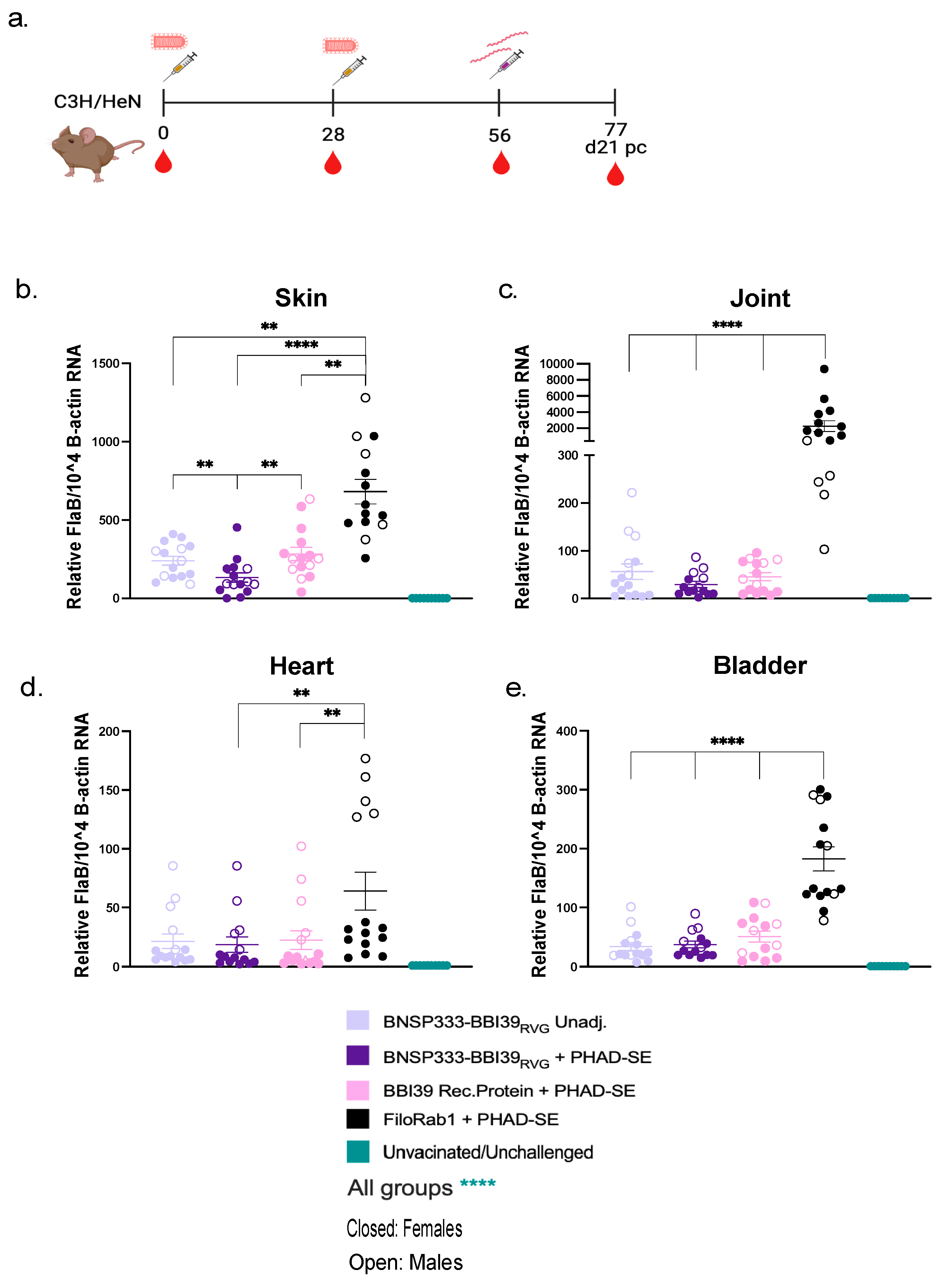
To determine the ability of this vaccine to prevent LD and borrelial infection, we conducted a syringe inoculum challenge experiment with Borrelia burgdorferi strain B31. Vaccinated and unvaccinated C3H mice, female and male (n=10), were injected intradermally with 1x105 spirochetes/mL by needle injection on day 56 (Figure 5a). Successful Bb infection was confirmed by western blot of Borrelia lysates and probing with individual mouse sera (Supplementary Figure S3a-c). At 21 days post-infection, joints, hearts, skin, and bladder were collected from infected mice to determine borrelial RNA load by qRT-PCR. For analysis, the FlaB CT, for detecting Bb load, values were normalized to mouse Beta-actin. Vaccinated mice with BNSP333-BBI39RVG, with and without PHAD-SE, and BBI39 recombinant protein showed a significant reduction of borrelial load in all organs when compared to FiloRab1 mice (Figure 5b–e). BNSP333-BBI39RVG + PHAD-SE showed a significant decrease of Bb in the skin compared to unadjuvanted BNSP333-BBI39RVG and recombinant protein vaccinated mice (Figure 5b). The skin was the only organ with significant differences between vaccinated groups. Since BBI39 is only produced while Bb is in the tick and early host infection, we expected to see significant differences in vaccinated groups. However, BNSP333-BBI39RVG + PHAD-SE trended lower in Bb loads in the joint and heart than the other vaccinated groups; however, this did not reach significance (Figure 5c,d). All vaccinated mice successfully depleted Bb in hearts, joints, skin, and bladder when compared to FiloRab1 vaccinated mice.
We also observed reduced Borrelia in infected mouse organs by culture in BSK medium. 15/15 mice contained Bb in hearts, joints, and skin from FiloRab1 vaccinated mice. However, we observed a depletion in vaccinated mice as displayed in Supplementary Figure S3d. The BNSP333-BBI39RVG, adjuvanted and unadjuvanted, vaccinated mice consistently had less Bb in mouse organs compared to the recombinant protein immunized mice. This parallels to the qRT-PCR data (Figure 5).
Figure 5.
BNSP333-BBI39RVG can successfully deplete Borrelia burgdorferi in mice. (a) Schedule Borrelia burgdorferi challenge post-vaccination. Mice (5 female and 5 male) were challenged on day 56 and infected organs were collected on day 21 post-challenge. Skin (b), joint (c), heart (d), and bladder (e) qPCR after challenge. Statistics are represented by the color of the bars from each vaccinated group. FlaB from Borrelia burgdorferi is normalized to mouse beta-actin. Error bars represent the SEM. Statistics were calculated by one-way ANOVA with post hoc Tukey’s test of log-transformed data. p > (ns), p < 0.0332 (*), p < 0.0021 (**), p < 0.0002 (***), p < 0.0001 (****).
Figure 5.
BNSP333-BBI39RVG can successfully deplete Borrelia burgdorferi in mice. (a) Schedule Borrelia burgdorferi challenge post-vaccination. Mice (5 female and 5 male) were challenged on day 56 and infected organs were collected on day 21 post-challenge. Skin (b), joint (c), heart (d), and bladder (e) qPCR after challenge. Statistics are represented by the color of the bars from each vaccinated group. FlaB from Borrelia burgdorferi is normalized to mouse beta-actin. Error bars represent the SEM. Statistics were calculated by one-way ANOVA with post hoc Tukey’s test of log-transformed data. p > (ns), p < 0.0332 (*), p < 0.0021 (**), p < 0.0002 (***), p < 0.0001 (****).
BNSP333-BBI39RVG Vaccine Protects Against B. burgdorferi Transmission and Disease in an Infected-Tick Challenge
We demonstrated that BNSP333-BBI39RVG vaccinated mice can successfully reduce Bb burden by syringe inoculum. To assess the protective efficacy, we completed a challenge via natural route of spirochete transmission via I. scapularis ticks. BNSP333-BBI39RVG + PHAD-SE vaccinated and FiloRab1 control mice (n=6/vaccine group) were subjected to the infected tick challenge using Bb infected ticks (n= 5 nymphs/mouse) on day 56 post-vaccination (Figure 6a). Skin and joints from infected mice were collected 21 days post-challenge for qRT-PCR for borrelial burden. The data in Figure 6b,c show that BNSP333-BBI39RVG immunization reduced Bb burden in mouse skin and joints compared to the control group, FiloRab1, during infected tick challenge. The RABV-based vaccine reduced Bb burden more than the recombinant protein vaccine. In addition, the mouse joints from tick challenge were analyzed for arthritis by histopathology. BNSP333-BBI39RVG vaccinated mouse joints showed lower arthritis scores than recombinant protein and FiloRab1 vaccinated mice. (Figure 6d,e). Therefore, we observed BNSP333-BBI39RVG + PHAD-SE can successfully reduce LD pathology in infected tick challenge.
RABV-BBI39 Vaccines are Immunogenic and Induce Long-Term Protection
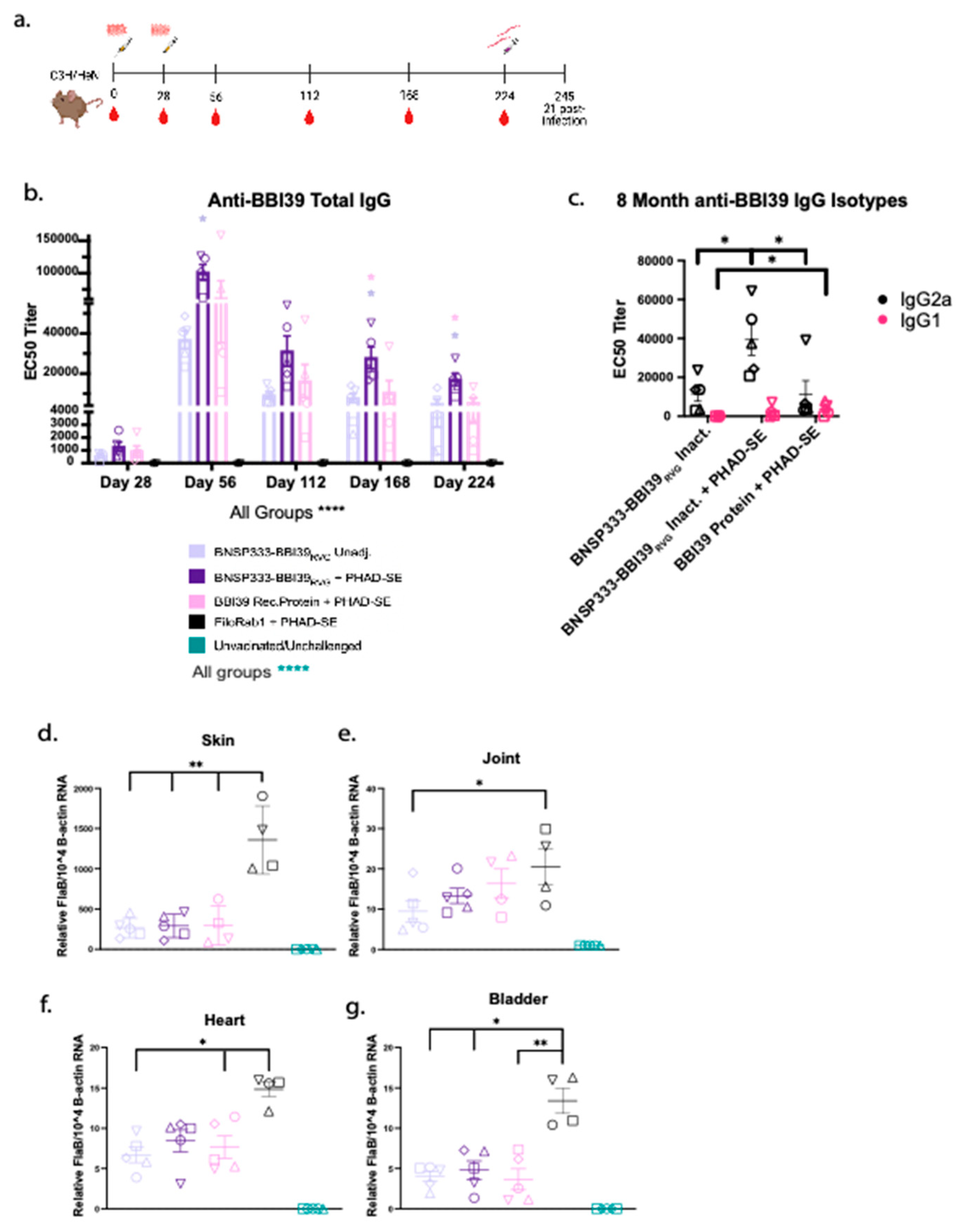
Vaccines must provide long-term efficacy; therefore, we conducted a long-term vaccination and syringe challenge experiment with BBI39-vaccinated mice. Antibody EC50 titers were determined by ELISA until 8 months post-initial vaccination (Figure 7a). We noted about 1-fold depletion of anti-BBI39 antibodies after day 56; however, they remained steady from 4-8 months post-immunization. We observed the lowest wanning antibody titers in the BNSP333-BBI39RVG + PHAD-SE group and the highest in the recombinant protein vaccinated group after 8 months (Figure 7b). RABV-vectored groups maintained a high skew towards a type-1 immune response, with high IgG2a for vaccines with and without adjuvant. This compares to the recombinant protein with a more balanced immune response, seen previously at 2 months (Figure 3e). Of note, we saw little to no IgG1 in BNSP333-BBI39RVG unadjuvanted vaccinated mice (Figure 7c).
Long-term vaccinated mice were subjected to Bb challenge by syringe inoculation. As described earlier, 21 days post-challenge, infected organs were collected and analyzed by qRT-PCR (Figure 7d–g) We observed lower borrelial loads (skin, joint, and heart) from all BBI39 vaccinated mice with consistently lower Bb load in BNSP333-BBI39RVG, adjuvanted and unadjuvanted, vaccinated mice compared to recombinant protein immunized mice (Figure 7 d–f). Adjuvanted and unadjuvanted mice displayed similar reduction of Bb; however, in unadjuvanted mice, Bb loads trended lower in all organs (Figure 7 d–f,g).
Finally, the presence of Bb from infected mouse organs was observed by dark-field microscopy in BSK culture (Supplementary Figure S4a). We observed a reduction of Borrelia in infected mouse organs in all mice vaccinated with BBI39 vaccines compared to FiloRab1 control, as displayed in Supplementary Figure S4a.
Our results show that antibody titers and type-1 biased immunity are higher in the RABV immunized mice, with and without adjuvant, when compared to the recombinant protein immunization. Further leading to lower Bb burden in BNSP333-BBI39RVG vaccinated mice compared to both recombinant protein and unvaccinated (FiloRab1) mice.
Discussion
In this study we utilized BBI39 in the RABV vaccine vector, BNSP333. We found that BBI39 can be produced incorporated into the RABV virion with the addition of RVG tail. The incorporation of BBI39 in BNSP333-BBI39RVG induces anti-BBI39 IgG antibodies in vaccinated mice, however the unincorporated vaccine, BNSP333-BBI39, did not. All RABV vectored vaccines produced anti-RABV-G antibodies. BNSP333-BBI39RVG, especially with the adjuvant PHAD-SE, can induce a type-1 associated immune response via the change in antibody isotypes. This leads to Borrelia and RABV neutralization in vitro. However, the recombinant protein vaccine, although adjuvanted with PHAD-SE, induces a more balanced type1/2 immune responses, which was seen in this study and previous studies [15,40]. BNSP333-BBI39RVG vaccinated mice successfully depleted Bb in syringe and tick challenges, more than the recombinant protein immunized mice, and eliminated LD pathogenesis.
In previous studies, OspA was the most utilized vaccine antigen [8,14,36,40,41]. While OspA is protective, it was found to contain a similar epitope to the human leukocyte function-associated antigen-1 (hLFA1), indicating the possibility of autoantibody development LYMErix vaccination [12]. Although these findings were insignificant, sales in LYMErix decreased dramatically, and the company removed the vaccine from the market in 2002. To prevent the potential for autoantibody development, we utilized BBI39, another surface protein on Borrelia protective against LD [15].
Previous platforms utilized to create an LD vaccine include recombinant protein(s) [8,14,15,36,41], viral vectors [42], DNA [43], and mRNA vaccines [40,44]. However, recombinant proteins have been highly utilized for LD vaccine platforms including LYMErix [41], VLA15 [8], and Vanguard crLyme [14]. However, recombinant proteins have low and short-lived immunogenicity requiring adjuvant and multiple inoculations with yearly revaccination [45]. Viral vaccine vectors, including RABV BNSP333, have been widely studied as efficacious, long-term options [27,28,31], but have rarely been studied as an LD vaccine. One group attempted to use Newcastle disease virus (NDV) as a vaccine vector for OspC [42], another protective borrelial antigen. However, this vaccine vector amounted to low antibody titers in mice and insignificant depletion of Bb in various organs. Another concern of utilizing a viral vector for a LD vaccine is that the Bb antigen is glycosylated differently by Borrelia than mammalian cells, how a viral vector is developed. This could possibly change the immune response to a bacterial protein induced by a viral vector [46]. Nonetheless, we developed an RABV vectored vaccine utilizing BBI39. In this study, we showed that borrelial antigen, BBI39, needs to be incorporated into the RABV virion to elicit high titer anti-BBI39 antibodies. When the borrelial antigen does not include the RVG tail, BBI39 is not incorporated into the RABV virion. Without incorporation, the protein stays inside the cell due to rabies’ non-cytolytic nature and the antigen is not presented to the immune response to elicit antibodies. With the addition of the RVG tail to BBI39RVG, the antigen is incorporated into the RABV virion, and high titer antibodies are elicited. This protects against Bb and inhibition of pathogenesis of LD. We observed glycosylation of BBI39; however, this antigen still protected against Bb in both syringe and tick challenge. In fact, we saw even greater protection from the viral vector vaccinated mice compared to recombinant protein.
In addition, the long-term efficacy of BNSP333 with BBI39 and other vaccine antigens [27] demonstrates an ideal platform against LD. The previous study on the BBI39 recombinant protein did not show long-term immunization experiments [15]; however, our study showed greater wanning immunity of the recombinant protein up to 8 months post-vaccination. In addition to wanning immunity from recombinant proteins, mRNA have shown wanning immunity to various antigens, requiring the need for multiple boosts [47]. Long-term vaccination and challenge studies were not completed in the mRNA LD vaccine study [40,48]. However, we showed with one boost of the BNSP333-BBI39RVG vaccine, long-term antibody responses were maintained 8-months in vaccinated mice. In addition, a previous study showed that BNSP333 can maintain long-term, up to 1 year, antibody titers and produce antibody-secreting cells in the spleen and bone marrow after only one boost, both with and without adjuvant [26]. Viral vectors, including RABV, are great candidates in the development a LD vaccine.
Previous studies demonstrated that antibodies are the main correlate of protection to prevent LD [49,50]. Type-1 associated antibodies, such as IgG2a are ideal for Bb neutralization [36,38]. We saw a greater IgG2a induction and Bb neutralization in vitro from BNSP333-BBI39RVG + PHAD-SE vaccinated mice than the recombinant protein immunization with adjuvant. Therfore, a viral vector (RABV) with addition of adjuvant provided greater protection against Bb in vaccinated mice compared to recombinant protein vaccinated mice. These data compares to a previous study utilizing PHAD-liposome particle bound with OspA demonstrated high antibody titers, a skew towards Th1 immunity, and borreliacidal effects which resulted in depletion of Bb in ticks [36]. Although a challenge study was not conducted, these results compare to the immunogenicity and borreliacidal activities in our study. This study also found long-term effects from their vaccine, aligning with our study. However, alum is widely used in the formulation of LD vaccines, including LYMErix [41] and VLA15 [8] but, alum is known to induce a type-2 associated immune response [26], which is not ideal for Bb protection [51]. The induction of a type-1 associated immune response from the viral vector and adjuvant PHAD-SE is an ideal formulation for developing a successful LD vaccine.
In this study, we utilized a syringe challenge and infected tick challenge. This compares to other studies that only utilized syringe inoculation [40]. The immune evasion strategies by the tick’s salivary proteins, among other strategies utilized by Bb to enter the host, are not present in a syringe challenge. Although syringe challenge is most feasible when a tick colony is not available, this should be considered when developing other LD vaccines. In our syringe challenge, we identified significant protection against BNSP333-BBI39RVG. However, in tick challenge, Bb is depleted much less. Therefore, tick challenge is ideal when analyzing the efficacy of a potential LD vaccine.
To further study BNSP333-BBI39RVG as a vaccine candidate, other mouse models of vaccination should be studied, such as C57BL/6 and BALB/C mice, which are less inflammatory mouse models compared to C3H/HeN. This could further analyze differences between adjuvanted and unadjuvanted groups. In addition, non-human primates should be utilized for protective efficacy with this vaccine. Further preclinical testing to determine the inhibition of disease pathogenesis, such as arthritis and carditis, should be completed. This includes long-term challenge experiments or keeping vaccinated and challenged mice for longer periods than 21 days post-challenge. This could show if the lower borrelial burdens in vaccinated mice will continue to prevent LD pathogenesis. Since LD is highly inflammatory, the immune profile of vaccinated and unvaccinated mice, such as cytokine profiles after vaccination and challenge, should also be studied. Greater understanding of the impact on antibody titers and passive transfer experiments will elucidate the necessity of specific antibody titers during Bb infection to prevent LD. Testing this vaccine against other strains of Borrelia such as Borrelia afzelii and Borrelia garinii, which are strains seen in LD patients in Europe and Asia, may determine whether BBI39 vaccination is conserved for these strains. Finally, since we did not see sterile immunity with the BNSP333-BBI39RVG vaccine in either challenge experiments, future research could study different borrelial antigens in combination with the BBI39 vaccine. Targeting the bacteria with different surface proteins could induce higher immunity against Bb. Studied would incorporate the foreign antigen into the RABV virion and use adjuvant PHAD-SE to create another RABV-based Bb vaccine. Other antigens studied for LD vaccines, such as OspA without the hLFA epitope, OspC, and other previously studied Bb or tick antigens could be tested in another vaccine.
In this study, we created a vaccine that targets BBI39 on the surface of Bb. Antibodies elicited against this vaccine contain neutralizing functions against both Bb and RABV, further depleting Bb in vaccinated mice. These results will help further research to develop an effective human LD vaccine.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
S.R., B.B., and K.V. performed experiments. S.R., C.K., U.P., and M.J.S. conceptualized experiments. S.R., M.J.S., D.K., and B.B. prepared the manuscript. M.J.S. and U.P. provided guidance and funding.
Data Availability
All data are available upon request from the corresponding authors.
Acknowledgments
We would like to thank Dr. Xiuli Yang for the valuable contribution in scoring and reviewing histopathological studies. Catherine Yankowski, PhD, Drishya Kurup, PhD, and Liz Declan for critical reading and editing of this manuscript. Kathryn Nassar, Gabrielle Scher, PhD, Christoph Wirblich, PhD, and Jose Villagomez for reagents and support during experiments. We thank the Thomas Jefferson University Bioimaging Core for help with confocal images. This work is supported by NIH grant R01AI54542. M.S. and U.P. were in receipt of the grant.
Competing Interests
The authors state no competing interests.
These data have been presented in a poster and oral presentation at American Society of Virology conference in July 2022 and American Society of Microbiology (Mirobe) conference in June 2023.
References
- Kugeler, K.J.; Schwartz, A.M.; Delorey, M.J.; Mead, P.S.; Hinckley, A.F. Estimating the Frequency of Lyme Disease Diagnoses, United States, 2010-2018. Emerg Infect Dis 2021, 27, 616–619. [Google Scholar] [CrossRef] [PubMed]
- How many people get Lyme disease? Lyme Disease [Web Page] 2021.
- Adrion, E.R.; Aucott, J.; Lemke, K.W.; Weiner, J.P. Health care costs, utilization and patterns of care following Lyme disease. PLoS One 2015, 10, e0116767. [Google Scholar] [CrossRef] [PubMed]
- Control, C.f.D. Signs and Symptoms of Untreated Lyme Disease. Lyme Disease [Web Page] 2021 May 6, 2021]; Available from: https://www.cdc.gov/lyme/signs_symptoms/index.html.
- Steere, A.C.; Coburn, J.; Glickstein, L. The emergence of Lyme disease. J. Clin. Invest. 2004, 113, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Control, C.f.D. Treatment for erythema migrans. Lyme Disease [Web Page] 2020 May 6, 2021]; Available from: https://www.cdc.gov/lyme/treatment/index.html.
- Control, C.f.D. Post-treatment Lyme Disease Syndrome. Lyme Disease [Web Page] 2019 May 6, 2021]; Available from: https://www.cdc.gov/lyme/postlds/index.html.
- Comstedt, P.; Schuler, W.; Meinke, A.; Lundberg, U. The novel Lyme borreliosis vaccine VLA15 shows broad protection against Borrelia species expressing six different OspA serotypes. PLoS One 2017, 12, e0184357. [Google Scholar] [CrossRef] [PubMed]
- Kamp, H.D.; Swanson, K.A.; Wei, R.R.; Dhal, P.K.; Dharanipragada, R.; Kern, A.; Sharma, B.; Sima, R.; Hajdusek, O.; Hu, L.T. , et al. Design of a broadly reactive Lyme disease vaccine. NPJ Vaccines 2020, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- O'Bier, N.S.; Hatke, A.L.; Camire, A.C.; Marconi, R.T. Human and Veterinary Vaccines for Lyme Disease. Curr. Issues Mol. Biol. 2021, 42, 191–222. [Google Scholar] [CrossRef] [PubMed]
- Steere, A.C.; Sikand, V.K.; Meurice, F.; Parenti, D.L.; Fikrig, E.; Schoen, R.T.; Nowakowski, J.; Schmid, C.H.; Laukamp, S.; Buscarino, C. , et al. Vaccination against Lyme disease with recombinant Borrelia burgdorferi outer-surface lipoprotein A with adjuvant. Lyme Disease Vaccine Study Group. N. Engl. J. Med. 1998, 339, 209–215. [Google Scholar] [CrossRef]
- Gross, D.M.; Forsthuber, T.; Tary-Lehmann, M.; Etling, C.; Ito, K.; Nagy, Z.A.; Field, J.A.; Steere, A.C.; Huber, B.T. Identification of LFA-1 as a candidate autoantigen in treatment-resistant Lyme arthritis. Science 1998, 281, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Ball, R.; Shadomy, S.V.; Meyer, A.; Huber, B.T.; Leffell, M.S.; Zachary, A.; Belotto, M.; Hilton, E.; Bryant-Genevier, M.; Schriefer, M.E. , et al. HLA type and immune response to Borrelia burgdorferi outer surface protein a in people in whom arthritis developed after Lyme disease vaccination. Arthritis Rheum. 2009, 60, 1179–1186. [Google Scholar] [CrossRef]
- Marconi, R.T.; Garcia-Tapia, D.; Hoevers, J.; Honsberger, N.; King, V.L.; Ritter, D.; Schwahn, D.J.; Swearingin, L.; Weber, A.; Winkler, M.T.C. , et al. VANGUARD(R)crLyme: A next generation Lyme disease vaccine that prevents B. burgdorferi infection in dogs. Vaccine X 2020, 6, 100079. [Google Scholar] [CrossRef]
- Singh, P.; Verma, D.; Backstedt, B.T.; Kaur, S.; Kumar, M.; Smith, A.A.; Sharma, K.; Yang, X.; Azevedo, J.F.; Gomes-Solecki, M. , et al. Borrelia burgdorferi BBI39 Paralogs, Targets of Protective Immunity, Reduce Pathogen Persistence Either in Hosts or in the Vector. J. Infect. Dis. 2017, 215, 1000–1009. [Google Scholar] [CrossRef]
- Pal, U.; de Silva, A.M.; Montgomery, R.R.; Fish, D.; Anguita, J.; Anderson, J.F.; Lobet, Y.; Fikrig, E. Attachment of Borrelia burgdorferi within Ixodes scapularis mediated by outer surface protein A. J. Clin. Invest. 2000, 106, 561–569. [Google Scholar] [CrossRef]
- Gomes-Solecki, M.; Arnaboldi, P.M.; Backenson, P.B.; Benach, J.L.; Cooper, C.L.; Dattwyler, R.J.; Diuk-Wasser, M.; Fikrig, E.; Hovius, J.W.; Laegreid, W. , et al. Protective Immunity and New Vaccines for Lyme Disease. Clin. Infect. Dis. 2020, 70, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.E.; Koser, M.; Xiao, S.; Siler, C.; McGettigan, J.P.; Calkins, C.; Pomerantz, R.J.; Dietzschold, B.; Schnell, M.J. Rabies virus glycoprotein as a carrier for anthrax protective antigen. Virology 2006, 353, 344–356. [Google Scholar] [CrossRef]
- Mustafa, W.; Al-Saleem, F.H.; Nasser, Z.; Olson, R.M.; Mattis, J.A.; Simpson, L.L.; Schnell, M.J. Immunization of mice with the non-toxic HC50 domain of botulinum neurotoxin presented by rabies virus particles induces a strong immune response affording protection against high-dose botulinum neurotoxin challenge. Vaccine 2011, 29, 4638–4645. [Google Scholar] [CrossRef]
- Kurup, D.; Wirblich, C.; Feldmann, H.; Marzi, A.; Schnell, M.J. Rhabdovirus-based vaccine platforms against henipaviruses. J. Virol. 2015, 89, 144–154. [Google Scholar] [CrossRef]
- Kurup, D.; Wirblich, C.; Ramage, H.; Schnell, M.J. Rabies virus-based COVID-19 vaccine CORAVAX induces high levels of neutralizing antibodies against SARS-CoV-2. NPJ Vaccines 2020, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Vos, A.; Neubert, A.; Aylan, O.; Schuster, P.; Pommerening, E.; Muller, T.; Chivatsi, D.C. An update on safety studies of SAD B19 rabies virus vaccine in target and non-target species. Epidemiol. Infect. 1999, 123, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Mebatsion, T. Extensive attenuation of rabies virus by simultaneously modifying the dynein light chain binding site in the P protein and replacing Arg333 in the G protein. J. Virol. 2001, 75, 11496–11502. [Google Scholar] [CrossRef]
- Papaneri, A.B.; Wirblich, C.; Cann, J.A.; Cooper, K.; Jahrling, P.B.; Schnell, M.J.; Blaney, J.E. A replication-deficient rabies virus vaccine expressing Ebola virus glycoprotein is highly attenuated for neurovirulence. Virology 2012, 434, 18–26. [Google Scholar] [CrossRef]
- Gomme, E.A.; Wanjalla, C.N.; Wirblich, C.; Schnell, M.J. Rabies virus as a research tool and viral vaccine vector. Adv. Virus Res. 2011, 79, 139–164. [Google Scholar] [CrossRef] [PubMed]
- Yankowski, C.; Kurup, D.; Wirblich, C.; Schnell, M.J. Effects of adjuvants in a rabies-vectored Ebola virus vaccine on protection from surrogate challenge. NPJ Vaccines 2023, 8, 10. [Google Scholar] [CrossRef]
- Yankowski, C.; Wirblich, C.; Kurup, D.; Schnell, M.J. Inactivated rabies-vectored SARS-CoV-2 vaccine provides long-term immune response unaffected by vector immunity. NPJ Vaccines 2022, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Abreu-Mota, T.; Hagen, K.R.; Cooper, K.; Jahrling, P.B.; Tan, G.; Wirblich, C.; Johnson, R.F.; Schnell, M.J. Non-neutralizing antibodies elicited by recombinant Lassa-Rabies vaccine are critical for protection against Lassa fever. Nat. Commun. 2018, 9, 4223. [Google Scholar] [CrossRef] [PubMed]
- Willet, M.; Kurup, D.; Papaneri, A.; Wirblich, C.; Hooper, J.W.; Kwilas, S.A.; Keshwara, R.; Hudacek, A.; Beilfuss, S.; Rudolph, G. , et al. Preclinical Development of Inactivated Rabies Virus-Based Polyvalent Vaccine Against Rabies and Filoviruses. J. Infect. Dis. 2015, 212 (Suppl. 2), S414–S424. [Google Scholar] [CrossRef]
- Scher, G.; Bente, D.A.; Mears, M.C.; Cajimat, M.N.B.; Schnell, M.J. GP38 as a vaccine target for Crimean-Congo hemorrhagic fever virus. NPJ Vaccines 2023, 8, 73. [Google Scholar] [CrossRef]
- Johnson, R.F.; Kurup, D.; Hagen, K.R.; Fisher, C.; Keshwara, R.; Papaneri, A.; Perry, D.L.; Cooper, K.; Jahrling, P.B.; Wang, J.T. , et al. An Inactivated Rabies Virus-Based Ebola Vaccine, FILORAB1, Adjuvanted With Glucopyranosyl Lipid A in Stable Emulsion Confers Complete Protection in Nonhuman Primate Challenge Models. J. Infect. Dis. 2016, 214 (Suppl. 3), S342–S354. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, J.P.; Pomerantz, R.J.; Siler, C.A.; McKenna, P.M.; Foley, H.D.; Dietzschold, B.; Schnell, M.J. Second-generation rabies virus-based vaccine vectors expressing human immunodeficiency virus type 1 gag have greatly reduced pathogenicity but are highly immunogenic. J. Virol. 2003, 77, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Qin, J.; Promnares, K.; Kariu, T.; Anderson, J.F.; Pal, U. Novel microbial virulence factor triggers murine lyme arthritis. J. Infect. Dis. 2013, 207, 907–918. [Google Scholar] [CrossRef]
- Schnell, M.J.; Buonocore, L.; Kretzschmar, E.; Johnson, E.; Rose, J.K. Foreign glycoproteins expressed from recombinant vesicular stomatitis viruses are incorporated efficiently into virus particles. Proc. Natl. Acad. Sci. USA 1996, 93, 11359–11365. [Google Scholar] [CrossRef]
- Mebatsion, T.; Schnell, M.J.; Cox, J.H.; Finke, S.; Conzelmann, K.K. Highly stable expression of a foreign gene from rabies virus vectors. Proc. Natl. Acad. Sci. USA 1996, 93, 7310–7314. [Google Scholar] [CrossRef]
- Federizon, J.; Frye, A.; Huang, W.C.; Hart, T.M.; He, X.; Beltran, C.; Marcinkiewicz, A.L.; Mainprize, I.L.; Wills, M.K.B.; Lin, Y.P. , et al. Immunogenicity of the Lyme disease antigen OspA, particleized by cobalt porphyrin-phospholipid liposomes. Vaccine 2020, 38, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Callister, S.M.; Schell, R.F.; Case, K.L.; Lovrich, S.D.; Day, S.P. Characterization of the borreliacidal antibody response to Borrelia burgdorferi in humans: a serodiagnostic test. J. Infect. Dis. 1993, 167, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Zhi, H.; Xie, J.; Skare, J.T. The Classical Complement Pathway Is Required to Control Borrelia burgdorferi Levels During Experimental Infection. Front. Immunol. 2018, 9, 959. [Google Scholar] [CrossRef] [PubMed]
- Shandilya, S.; Kurt Yilmaz, N.; Sadowski, A.; Monir, E.; Schiller, Z.A.; Thomas, W.D., Jr.; Klempner, M.S.; Schiffer, C.A.; Wang, Y. Structural and molecular analysis of a protective epitope of Lyme disease antigen OspA and antibody interactions. J. Mol. Recognit. 2017, 30. [Google Scholar] [CrossRef] [PubMed]
- Pine, M.; Arora, G.; Hart, T.M.; Bettini, E.; Gaudette, B.T.; Muramatsu, H.; Tombacz, I.; Kambayashi, T.; Tam, Y.K.; Brisson, D. , et al. Development of an mRNA-lipid nanoparticle vaccine against Lyme disease. Mol. Ther. 2023, 31, 2702–2714. [Google Scholar] [CrossRef]
- Sigal, L.H.; Zahradnik, J.M.; Lavin, P.; Patella, S.J.; Bryant, G.; Haselby, R.; Hilton, E.; Kunkel, M.; Adler-Klein, D.; Doherty, T. , et al. A vaccine consisting of recombinant Borrelia burgdorferi outer-surface protein A to prevent Lyme disease. Recombinant Outer-Surface Protein A Lyme Disease Vaccine Study Consortium. N. Engl. J. Med. 1998, 339, 216–222. [Google Scholar] [CrossRef]
- Xiao, S.; Kumar, M.; Yang, X.; Akkoyunlu, M.; Collins, P.L.; Samal, S.K.; Pal, U. A host-restricted viral vector for antigen-specific immunization against Lyme disease pathogen. Vaccine 2011, 29, 5294–5303. [Google Scholar] [CrossRef] [PubMed]
- Pfeifle, A.; Thulasi Raman, S.N.; Lansdell, C.; Zhang, W.; Tamming, L.; Cecillon, J.; Laryea, E.; Patel, D.; Wu, J.; Gravel, C. , et al. DNA lipid nanoparticle vaccine targeting outer surface protein C affords protection against homologous Borrelia burgdorferi needle challenge in mice. Front. Immunol. 2023, 14, 1020134. [Google Scholar] [CrossRef] [PubMed]
- Sajid, A.; Matias, J.; Arora, G.; Kurokawa, C.; DePonte, K.; Tang, X.; Lynn, G.; Wu, M.J.; Pal, U.; Strank, N.O. , et al. mRNA vaccination induces tick resistance and prevents transmission of the Lyme disease agent. Sci. Transl. Med. 2021, 13, eabj9827. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, I.P.; Leite, L.C. Recombinant vaccines and the development of new vaccine strategies. Braz. J. Med. Biol. Res. 2012, 45, 1102–1111. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, J.; Shen, C.; Wang, G.; Lu, Z.; Zeng, D.; Gao, Y.; Chen, H.; Xia, N.; Chen, Y. Vaccination with Deglycosylated Modified Hemagglutinin Broadly Protects against Influenza Virus Infection in Mice and Ferrets. Vaccines 2022, 10, 1304. [Google Scholar] [CrossRef]
- Naaber, P.; Tserel, L.; Kangro, K.; Sepp, E.; Jurjenson, V.; Adamson, A.; Haljasmagi, L.; Rumm, A.P.; Maruste, R.; Karner, J. , et al. Dynamics of antibody response to BNT162b2 vaccine after six months: a longitudinal prospective study. Lancet Reg. Health Eur. 2021, 10, 100208. [Google Scholar] [CrossRef] [PubMed]
- Matias, J.; Kurokawa, C.; Sajid, A.; Narasimhan, S.; Arora, G.; Diktas, H.; Lynn, G.E.; DePonte, K.; Pardi, N.; Valenzuela, J.G. , et al. Tick immunity using mRNA, DNA and protein-based Salp14 delivery strategies. Vaccine 2021, 39, 7661–7668. [Google Scholar] [CrossRef] [PubMed]
- McKisic, M.D.; Barthold, S.W. T-cell-independent responses to Borrelia burgdorferi are critical for protective immunity and resolution of lyme disease. Infect. Immun. 2000, 68, 5190–5197. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Stehle, T.; Museteanu, C.; Siebers, A.; Gern, L.; Kramer, M.; Wallich, R.; Simon, M.M. Therapeutic passive vaccination against chronic Lyme disease in mice. Proc. Natl. Acad. Sci. USA 1997, 94, 12533–12538. [Google Scholar] [CrossRef]
- Munson, E.L.; Du Chateau, B.K.; Jobe, D.A.; Lovrich, S.D.; Callister, S.M.; Schell, R.F. Production of borreliacidal antibody to outer surface protein A in vitro and modulation by interleukin-4. Infect. Immun. 2000, 68, 5496–5501. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
RABV vectored vaccine constructs containing BBI39. (a) Rabies virus (RABV) vectored vaccine constructs containing BBI39. BNSP333 is the parental RABV vector, with rabies nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G), and RNA-dependent RNA polymerase (L). Highlighted in the glycoprotein is the signal peptide (Ig-kappa sequence) (green), the final 51 amino acids of the ectodomain (brown), transmembrane domain (black) and the cytoplasmic domain (blue). (*) Indicates a point mutation at amino acid position 333 from arginine to glutamic acid. (b,c) RABV-based constructs containing BBI39. (b) Non-anchored construct with BBI39 placed between RABV-N and RABV-P, creating BNSP333-BBI39. (c) BBI39RVG with the RABV-G additions to the gene, making BNSP333-BBI39RVG.
Figure 1.
RABV vectored vaccine constructs containing BBI39. (a) Rabies virus (RABV) vectored vaccine constructs containing BBI39. BNSP333 is the parental RABV vector, with rabies nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G), and RNA-dependent RNA polymerase (L). Highlighted in the glycoprotein is the signal peptide (Ig-kappa sequence) (green), the final 51 amino acids of the ectodomain (brown), transmembrane domain (black) and the cytoplasmic domain (blue). (*) Indicates a point mutation at amino acid position 333 from arginine to glutamic acid. (b,c) RABV-based constructs containing BBI39. (b) Non-anchored construct with BBI39 placed between RABV-N and RABV-P, creating BNSP333-BBI39. (c) BBI39RVG with the RABV-G additions to the gene, making BNSP333-BBI39RVG.
Figure 6.
BNSP333-BBI39RVG can deplete Borrelia burgdorferi in mice after infected tick challenge. (a) Schedule Borrelia burgdorferi challenge post-vaccination with infected ticks. Mice (3 female and 3 male) were challenged on day 56 and infected organs were collected on day 14 post-challenge to perform skin (b) and joint (c) qPCR after challenge. FlaB from Borrelia burgdorferi is normalized to mouse beta-actin. Joints were subjected to histology analysis demonstrated by arthritis scores (d) and H&E staining (e). Scores were done in a blinded manner. Error bars represent the SEM. Statistics were calculated by one-way ANOVA with post hoc Tukey’s test of log-transformed data. p > (ns), p < 0.0332 (*), p < 0.0021 (**), p < 0.0002 (***), p < 0.0001 (****).
Figure 6.
BNSP333-BBI39RVG can deplete Borrelia burgdorferi in mice after infected tick challenge. (a) Schedule Borrelia burgdorferi challenge post-vaccination with infected ticks. Mice (3 female and 3 male) were challenged on day 56 and infected organs were collected on day 14 post-challenge to perform skin (b) and joint (c) qPCR after challenge. FlaB from Borrelia burgdorferi is normalized to mouse beta-actin. Joints were subjected to histology analysis demonstrated by arthritis scores (d) and H&E staining (e). Scores were done in a blinded manner. Error bars represent the SEM. Statistics were calculated by one-way ANOVA with post hoc Tukey’s test of log-transformed data. p > (ns), p < 0.0332 (*), p < 0.0021 (**), p < 0.0002 (***), p < 0.0001 (****).
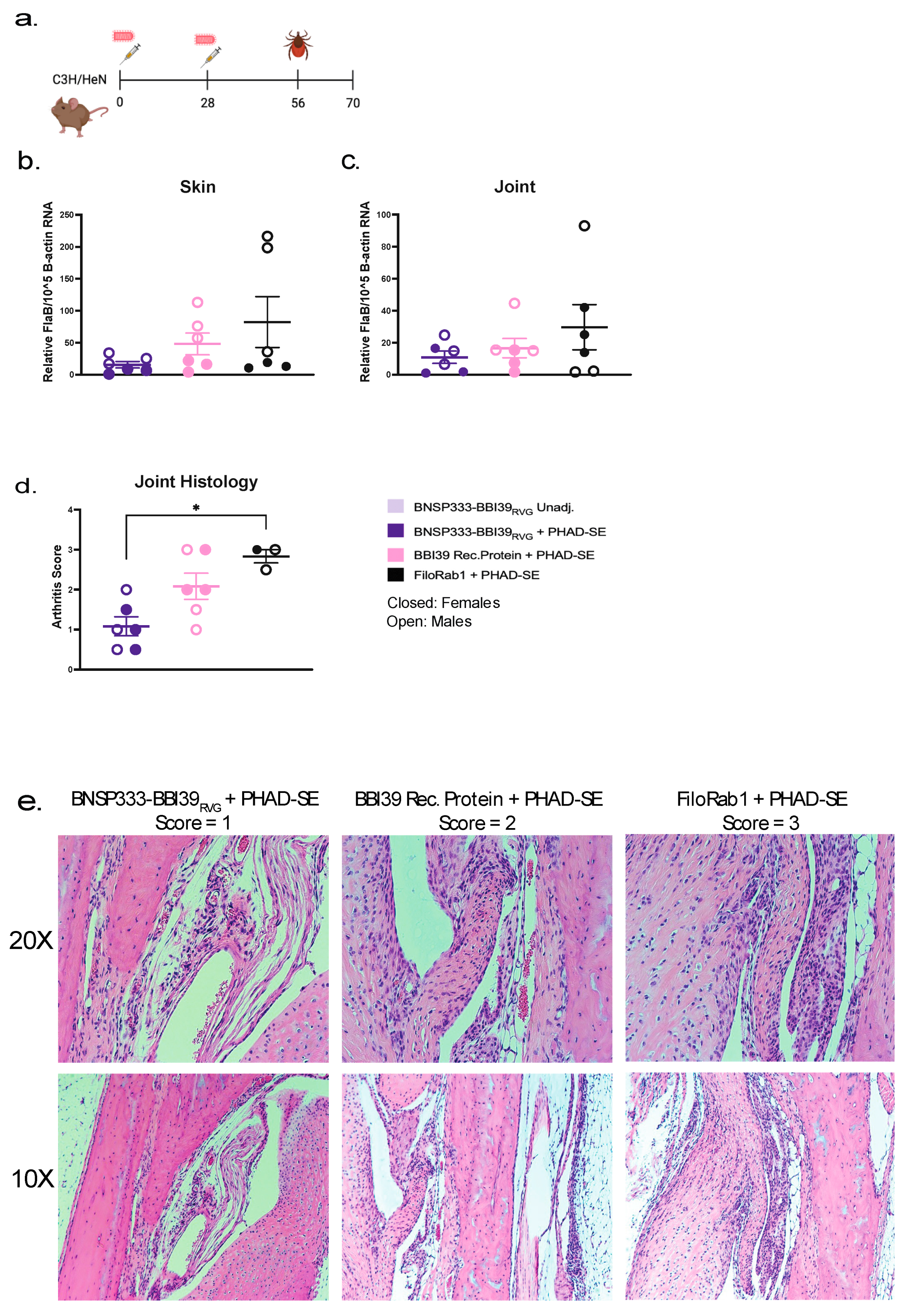
Figure 7.
BNSP333-BBI39RVG elicits anti-BBI39 antibodies and protect vaccinated mice long-term. (a) Vaccination and challenge schedule for long-term study. (b) anti-BBI39 total IgG antibody titers over time from day 28 to day 224 (8 months post-initial vaccination) (n=5 female mice/vaccine group). Statistics are represented by the color of the bars from each vaccinated group. (c) anti-BBI39 IgG isotypes 8 months post-vaccination. qPCR of infected mouse organs: Skin (d), joint (e), heart (f), and bladder (g). FlaB from Borrelia burgdorferi is normalized to mouse beta-actin. Error bars represent the SEM. Statistics were calculated by one-way ANOVA with post hoc Tukey’s test of log-transformed data. p > (ns), p < 0.0332 (*), p < 0.0021 (**), p < 0.0002 (***), p < 0.0001 (****).
Figure 7.
BNSP333-BBI39RVG elicits anti-BBI39 antibodies and protect vaccinated mice long-term. (a) Vaccination and challenge schedule for long-term study. (b) anti-BBI39 total IgG antibody titers over time from day 28 to day 224 (8 months post-initial vaccination) (n=5 female mice/vaccine group). Statistics are represented by the color of the bars from each vaccinated group. (c) anti-BBI39 IgG isotypes 8 months post-vaccination. qPCR of infected mouse organs: Skin (d), joint (e), heart (f), and bladder (g). FlaB from Borrelia burgdorferi is normalized to mouse beta-actin. Error bars represent the SEM. Statistics were calculated by one-way ANOVA with post hoc Tukey’s test of log-transformed data. p > (ns), p < 0.0332 (*), p < 0.0021 (**), p < 0.0002 (***), p < 0.0001 (****).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



