
Submitted:
30 November 2023
Posted:
01 December 2023
You are already at the latest version
Abstract
This last decade, chimeric antigen receptor (CAR) T-cell therapy has become a real treatment option for patients with B-cell malignancies, while multiple efforts are being made to extend this therapy to other malignancies and broader patient populations. However, several limitations remain, including those associated with the time-consuming and highly personalized manufacturing of autologous CAR-Ts. Technologies to establish “off-the-shelf” allogeneic CAR-Ts with low alloreactivity are currently being developed, with a strong focus on gene editing technologies. Although these technologies have many advantages, they have also strong limitations including double-strand breaks in the DNA with associated multiple safety risks as well as the lack of modulation. As an alternative, non-gene editing technologies provide an interesting approach to support the development of allogeneic CAR-Ts in the future, with possibilities of fine-tuning gene expression and easy development. Here we will review the different ways allogeneic CAR-Ts can be manufactured and discuss which technologies are currently used. The biggest hurdles for successful therapy of allogeneic CAR-Ts will be summarized and finally an overview of the current clinical evidence for allogeneic CAR-Ts in comparison to its autologous counterpart will be given.
Keywords:
allogeneic
; chimeric antigen receptor
; off-the-shelf
; gene editing
1. Introduction
These last decades, immunotherapy has become an important treatment option for patients with cancer indications. Among the most promising options, T-cells engineered to express chimeric antigen receptors (CAR) aim to strengthen the power of T-cells to recognize and eliminate tumor cells in a human leukocyte antigen (HLA)-independent manner. Since 2017, six CAR-T products have been approved by the US Food and Drug Administration (FDA) in the United States and other countries, and two CAR-T products are approved in China by the National Medical Products Administration [1]. All products are aimed for patients with advanced or resistant large B-cell lymphoma, acute lymphoblastic leukemia or multiple myeloma, where outstanding results were obtained with overall response rates reaching up to 100% objective response rates in some cases [2,3]. Nevertheless, challenges remain for these cell therapies, including low durability of responses, severe adverse events, low effectiveness in the context of solid tumors and limitations due to manufacturing of a highly personalized product [2,3,4,5].
CAR-T therapies in advanced stage of development, including those marketed, are of autologous origin, whereby peripheral blood cells are taken from the individual receiving treatment to be engineered into CAR-Ts before being reinfused to the patient. The variability among patients in the initial material, due to the patient’s prior treatment and disease history, may result in disparities in efficiency or yield of the end product and lead to 2-10% manufacturing failure rates [6], resulting in treatment deprivation for a patient who has already undergone the apheresis process. Another obstacle arises from the logistics, planning and increased expenditures associated with tailored medicines which necessitate creating and releasing a single product for each patient. The manufacturing, testing and release process itself is time-consuming, and the logistical challenge in shipping cells back and forth between the treatment site and cell production facilities—which usually follows a centralized manufacturing model—poses a significant concern for individuals with rapidly progressive or aggressive cancers. The development of allogeneic and/or ‘off-the-shelf’ CAR Ts from healthy donors allows to overcome many of these limitations by contributing to scalability and direct access to CAR-T therapies, providing a readily available therapeutic solution for multiple patients (Figure 1).
Whilst allogeneic therapies are attractive new treatment opportunities, their main downside is the risk of potential life-threatening toxicity called “graft-versus-host disease” (GvHD) that is triggered by recognition of the patient’s healthy tissues by the T-cell receptor (TCR) present on the surface of allogeneic CAR-Ts. To minimize this risk, selection of T-cell sources presenting low TCR signaling capacity can be considered (see Section 3). Most often, the manufacturing process of allogeneic CAR-T therapies include an engineering step that aims to eliminate or blunt the signaling or the expression of the TCR using specific technology (see Section 4). As a result, the engineered allogeneic CAR-Ts fail to recognize the patient’s healthy tissue as foreign, thereby preventing GvHD.
Another challenge to overcome is the opposite scenario, where the patient’s immune system swiftly rejects any transferred allogeneic cell, called Host-versus-Graft (HvG) reaction, thereby limiting the persistence of allogeneic CAR-Ts. For this too, further engineering of the CAR-Ts is needed.
Here, we review potential sources of allogeneic cells for CAR-Ts and focus on advantages or inconveniences of using existing technologies to establish “off-the-shelf” allogeneic CAR-Ts with low alloreactivity, including the most studied and developed gene editing technologies, but also other non-gene editing technology alternatives.
2. Source of allogeneic cells
The potential of allogeneic CAR-T lies largely in the ability to mass-produce CAR-Ts that are as efficient and potent as their autologous counterpart. One of crucial factors in the manufacturing of allogeneic CAR-Ts lies in the source material used for the final product.
Currently the most frequently used allogeneic cell source for CAR-T manufacturing involves using peripheral blood mononuclear cells (PBMCs) from a random healthy donor. More rarely other cell sources are used like umbilical cord blood (UCB) or a renewable cell source such as induced pluripotent stem cells (iPSCs).
2.1. PBMCs
The most frequent source for the manufacturing of allogeneic CAR-Ts is PBMCs collected from healthy donors. This allows for the creation of multiple vials from a single apheresis product, that can be easily used in a very rapid and standardized manufacturing protocol [7,8]. This also allows for the generation of a bank of cells that express different human leukocyte antigen (HLA) subtypes to potentially match the donor HLA to that of the patient [9]. The selection of donors on the basis of their immune characteristics is likely to be a key factor in decreasing the heterogeneity in the final manufactured product and lower the risk of GvHD.
2.2. UCB
The use of UCB was shown to be associated with reduced incidence and severity of GvHD making it a potentially more tolerable source material than PBMCs for allogeneic T-cells, allowing for less stringent HLA-matching [10]. Furthermore, UCB is an enriched source of hematopoietic stem cells (HSCs), which are able to self-renew and can be used to differentiate into T-cells, although there is a limit to their total number [11,12]. Interestingly, T-cells isolated from UCB have a unique antigen-naive status which is probably linked to the decreased alloreactivity observed in UCB grafts [13,14]. Furthermore, UCB T-cells are characterized by impaired nuclear factor of activated T-cells (NFAT) signaling and reduced activity which most likely further contributes to the reduced GvHD [15].
However, an obvious drawback of UCB is its limited availability compared to other cell sources.
2.3. Induced pluripotent stem cells (iPSCs)
T-cells derived from iPSCs can also be used as a source of CAR-Ts [16]. In theory, iPSCs have an unlimited capacity for self-renewal, thus allowing for them to be banked and used indefinitely [17]. A bank of iPSC lines with different homozygous HLA combinations could be generated to minimize the risk of allorejection of CAR-T derived from iPSCs [18]. An advantage of using iPSCs is that CAR-T cells can be generated from a single iPSC clone with the capacity for clonal expansion and therefore the genetic modifications they undergo would be homogeneous in the final cell population [19]. However, the quality controls should be strict because undifferentiated proliferating iPSCs may compromise product safety, since they could induce important adverse effects such as teratomas [20].
iPSCs can be developed from different cell types, such as fibroblasts or lymphocytes, that are reprogrammed into a less differentiated cell by inducing the expression of specific factors. For example, Iriguchi, et al. generated iPSCs from an antigen-specific cytotoxic T-cell clone, or from TCR-transduced iPSCs, as starting material [21]. These iPSCs can then in turn be differentiated into T-cells through the addition of several differentiation drivers and/or inhibitors (SDF1α and p58 inhibitor in the above case for example) to enhance T-cell commitment. While the potential to create a large cell-bank that covers a study cohort is appealing the arduous task of T-cell differentiation and selection up to the commitment of a single positive T-cell is much more complex then the use of T-cells isolated from either PBMCs or UCB. However, while PBMCs and UCB both offer a heterogenous T-cell population of cells iPSCs are clonal and thus give rise to a homogenous T-cell population both with the advantages/disadvantages of each.
The generation of allogeneic CAR-T irrelevant of the starting material faces two major hurdles. The first is the induction of GvHD and the second is the HvG response. Each T-cell expresses a T-cell receptor (TCR), where the majority of T-cells express a TCR composed of a alpha and a beta protein chain, that can recognize HLA-peptide complexes on target cells through the direct pathway of allorecognition thus leading to GvHD [22,23] independent of the CAR.
3. How to prevent alloreactivity in CAR-Ts by selecting the right cell population?
The use of allogeneic donor T-cells (CAR or not), that still express a functioning TCR may play a role in anti-tumor effects. This has been clearly demonstrated in leukemia in a process termed graft-versus-leukemia (GvL): after allogeneic stem cell transplantation (SCT), protection from relapse is partly due to donor T-cells that recognize leukemia-specific minor antigens [24]. This may be similar with CAR-T, although the recognition of allo-antigens will likely induce GvHD, as studies assessing both acute and chronic GvHD have clearly established a central role for αβTCR in GvHD pathogenesis [25,26,27,28]. The application of SCT for example was not appreciated until T-cell-depleted grafts were assessed to eliminate GvHD [29,30]. These successfully decreased GvHD to extremely low frequencies, although the risk of opportunistic infections and relapse increased substantially [30,31]. While the role of αβTCR in GvHD development is not in doubt, the possible risk and/or benefit in the case of CAR-T therapy is not completely clear and the development of GvHD may be relatively low [32].
To avoid GvHD two main approaches exist depending on i) T-cells that have low or non-reactive TCRs (discussed in this section) or ii) engineering methods to avoid allorecognition (section 4). The αβTCR repertoire is selected in the thymus and is educated based on the ability to be tolerant to self-HLA complexes. This tolerance means that the TCR recognizes the self-HLA and responds to non-self peptide. However in the case of allorecognition the TCR recognizes both structurally similar HLA-peptide complexes and dissimilar HLA-peptide complexes, therefore allowing for the high frequency of alloreactive T-cells (1 in 103) [33]. It is these alloreactive αβTCR expressed on T-cells that drive GvHD.
The HLA locus is the most polymorphic region in the human genome, thus leading to many HLA variants in each individual. There are six HLA-class-I molecules and six HLA-class-II molecules, making the matching between donor/patient a complex issue and although decades of data from transplantation centers have shown that the most important HLAs to match are the class I HLAs A, B and class II HLA-DR [34,35], this still requires a vast bank of cells in order to produce the CAR-Ts, which renders the allogeneic manufacturability rather complicated.
3.1. Infusion of allogeneic CAR-Ts post or prior to an allogeneic transplantation
Patients treated with allogeneic SCT, can be subsequently treated with CAR-Ts generated from the same donor if they relapse. This was done in a study by Brudno, et al. where 20 patients with B-cell malignancies received CD19 CAR-Ts generated from the same donor as SCT with no chemotherapy administered before T-cell infusion. Six patients achieved complete remission and two patients achieved a partial response. No GvHD was reported [36]. These results confirmed previous observations made by other groups [37,38]. In a more recent study, 8 r/r B-ALL patients received either HLA-matched (n = 4) or HLA-haploidentical (n = 4) CD19 CAR-Ts immediately preceding an intended SCT [39]. The haploidentical CAR-Ts induced transient or no reduction in peripheral blood leukemia cells with no significant CAR-T expansion which suggests rejection. In contrast patients treated with the HLA-matched CAR-Ts exhibited higher complete response rates, although more severe toxic side effects, with no GvHD observed in either group. However, only 3 out of 8 patients reached complete response and only 2 of the 8 patients proceeded to transplant, indicating that while HLA-matched and HLA-haploidentical allogeneic CD19 CAR-Ts are feasible in r/r B-ALL before SCT, other factors besides GvHD need to be considered in clinical applications of allogeneic CAR T cell infusions.
3.2. Memory T-cells
T-cells with a specific memory phenotype are considered to have a TCR specificity directed to previously detected antigens, which are expected to be different from those of the patient receiving the CAR-T therapy. Interestingly, studies have shown that memory T-cells do not induce GvHD [40]. It is unclear why this is the case, but one possibility is the diversity of the TCR which is limited in memory T-cells, thus reducing GvHD. One manner by which to further specify the T-cell memory and TCR-specificity is through selection or the development of virus-specific T-cells (VST) as has been done in Epstein-Barr virus (EBV)-associated malignancies. Adoptive transfer of HLA partially matched EBV-specific T-cells from healthy donors has had positive results in post-transplant lymphoproliferative disease for example, with response rates of 60–70% and low incidences of toxicity or GVHD [41]. Infusion of EBV-specific T-cells has also been used in patients with Hodgkin lymphoma with good tolerance and remission rates [42,43]. The use of the viral antigens can enhance the proliferative capacity of the allogeneic CAR-Ts making them persist longer and possibly enhance their efficacy. This has been shown with cytomegalovirus (CMV)-specific CD19-CAR-Ts that had enhanced in vivo anti-tumor activity by the administration of anti-CMV vaccination [44].
However, all these methodologies require partial matching and thus require the creation of multiple cellular banks. Next to the above-mentioned options, sub-populations of T-cells can be used for the generation of allogeneic CAR-Ts.
3.3. T-cell sub-populations
T-cell sub-populations comprise a relative low percent of the circulating total T-cells (making anywhere between 0.01%–10% of the T-cells). These sub-populations include: double negative T (DNT)-cells; invariant Natural Killer T-cells (iNKT); cytokine-induced killer (CiK) cells; Mucosal-associated invariant T (MAIT)-cells and lastly γδT-cells.
3.3.1. Double negative T (DNT)-cells
DNTs are a rare subset of immune cells that express CD3 but not CD4, CD8, and CD1d-αGalCer [45,46,47]. DNTs comprise about 1 to 5% of human PBMCs and can be isolated and expanded ex-vivo under clinically compliant conditions from the peripheral blood of healthy donors [48,49]. Expanded DNTs can express either γδTCR or αβTCR, where the frequency of TCR expressing DNTs can range between 60 and 90% depending on the donor origin.
In a recent study conducted by Vasic, et al. the feasibility, safety, and efficacy of DNTs for the development of allogeneic CD19-CAR-T was assessed. The resulting allogeneic CD19-CAR DNTs had the properties of an off-the-shelf cellular therapy and were effective against CD19-expressing hematological and solid malignancies [50]. Pre-clinical studies have thus confirmed the feasibility of DNTs, but whether DNTs will actually yield good results clinically remains to be seen.
A phase I/IIa clinical trial using third-party donor-derived genetically non-modified DNTs to treat patients with relapsed/refractory acute myeloid leukemia (AML) showed that the therapy was safe and had a positive efficacy profile [51]. One major concern is regarding the cellular efficacy. Interestingly, Kang et al. have shown that, one manner by which the cellular efficacy and persistence of DNTs CARs can be enhanced, is through inhibition of the PI3K pathway during the manufacturing. Something that we and others have seen in αβT-cells as well [52,53].
3.3.2. iNKT-cells
invariant NKT-cells (iNKT) are a subset of T-cells that share morphological and functional characteristics of both NK and T-cells. They have a restricted TCR, that has a constant α-chain paired with a low-diverse β-chain. iNKTs comprise between 0.01 and 1% of the peripheral blood T-cell population and have shown not to cause GvHD in xenograft models [54,55,56]. They are restricted by CD1d, a glycolipid presenting HLA-I like molecule expressed on B-cells, antigen-presenting-cells and some epithelial cells [57,58]. The fact that iNKT-cells recognize B-cell lymphomas through CD1d makes them of particular interest for B-cell malignancies [59].
3.3.3. CIK-cells
CIK-cells are a heterogenous population of polyclonal effector T-cells that have functional NK-cell properties. They comprise between 0.01 and 1% of the peripheral blood T-cell population and can be expanded from PBMCs, bone marrow and UCB through a manufacturing process that involves the addition of cytokines like IFN-γ and IL-2 and TCR-activating antibodies [60,61]. CIK-cells have the advantage of exerting non-HLA-restricted cytotoxicity and very low alloreactivity across HLA-barriers in comparison with conventional donor lymphocyte infusion [62,63,64]. This was further confirmed by preclinical and phase I/II studies where the infusion of bulk CIK-cells population was well-tolerated [65,66,67]. In addition to the alloreactivity, the dual activity (of both NK cell receptors and TCRs) gives CiKs an added ability to mediate cytotoxicity and prevent infection, which is a major concern after CAR-T therapy. In a recent clinal trial where relapsed B-Acute lymphoblastic leukemia (B-ALL) patients were treated with CD19 CAR CIK-cells, no GvHD was observed and the cells could be detected up to 10 months after infusion [68]. The overall response rate was 61.5% (13 patients) which is in-line with its autologous counterpart.
3.3.4. Mucosal associated invariant T (MAIT) cells
MAIT-cells are primarily localized to mucosa-rich regions, comprising a fraction of T-cells distributed throughout the pulmonary (5%), hepatic (20%–40%), and intestinal (1%–2%) lamina propria, as well as peripheral circulation (1%–10%; [69,70,71]). MAIT cells have a heavily restricted TCR repertoire, that consists of TCR alpha variable (TRAV)1 combined with three kinds of TCRA junctional (TRAJ; TRAJ33, TRAJ12, TRAJ20) and a limited repertoire of β chains in humans [72]. The MAIT TCR can recognize modified derivatives from the vitamin B2 synthesis pathway presented by MHC class I-related molecule MR1 on APCs. MR1 is a conserved molecule thus making MAIT cells devoid of inducing strong GvHD in vivo [73]. This has further been shown in clinical studies where MAIT cells were positively correlated with improved survival and less allogeneic adverse events [74].
The use of MAIT cells for CAR-T has been assessed in multiple pre-clinical studies, and while their efficacy against tumor antigens was significant (as assessed with a mesothelin and a CD19 targeting CAR) a significant concern was raised based on both cellular persistence and manufacturing due to the limited cell number [75,76]. These imply that the use of MAIT cells clinically may be limited.
3.3.5. γδ.T-cells
One other subset of T-cells that is currently being used extensively in both preclinical and clinical studies are γδT-cells (reviewed elsewhere [77,78,79]) which represent 1–10% of circulating T-cells (although they are also prevalent in some epithelial tissues; [80]). The γδT-cells have a unique TCR composed of variable gamma and delta chains and recognize antigens independent of the HLA leading to low or no risk of GvHD [81,82]. It is this that has made them a popular starting material for the creation of allogeneic CAR-T and at least a dozen trials are currently underway to assess this as a viable option [77,79,83].
Several studies have shown the safety and some efficacy of γδT-cells transfusion into cancer patients, thereby relying on the HLA-independent function of γδT-cells (mediated by NKG2D for example, among others; [84,85]). These studies imply that the use of γδT-cells may prove beneficial as a CAR-T therapy. This observation has led to multiple CAR-T and TCR-based strategies employed by companies to improve the efficacy of γδT-cells for cancer immunotherapy. However, the tumor-toxicity has been limited and consistent problems with both persistence and homing in vivo has limited the translation of γδCAR-Ts.
4. ‘Off-the-shelf’ allogeneic CAR-Ts
4.1. Methods to engineer ‘off-the-shelf” allogeneic CAR-Ts
The strategies to reduce GvHD by using partial-matched allogeneic material, and/or T-cells that have low or no TCR, naturally offer good alternatives and many CAR-Ts have shown the alloreactivity to be limited or manageable. However, in most instances allogeneic cells are persistent for a very short amount of time, meaning that the lack of GvHD may be due in part to the lack of persistence. This lack of persistence is driven by multiple-factors including: i) a resurgence of the host immune response (in most instances the patients undergo lymphodepletion prior to CAR-T therapy) that in turn rejects the allogeneic cells; ii) the immune-suppressive tumor microenvironment that may inhibit T-cell proliferation as well as other factors [86]. This requires additional engineering to circumvent the host immune-response and/or the tumor microenvironment. The different methods can be divided into gene editing technologies and non-gene editing technologies.
4.1.1. Gene editing technology
The two biggest hurdles in the use of allogeneic T-cells are GvHD and HvG. The former can be avoided by eliminating the TCR, usually through the knockout (KO) of the constant domain of one of its chains (α and/or β), or by replacing some TCR subunits which impedes its antigen recognition function [87]. However, although this takes care of the alloreactivity, the cells would still be susceptible to HvG. The most common antigens driving HvG are the mismatched donor-HLA-I molecules on the donor cells. These are recognized by the patient αβT-cells that are CD8+ through the direct pathway of allorecognition. By knocking-out the common subunit β2-microglobulin (encoded by the B2M gene), the HLA-I molecule will not be expressed on the cell surface, thus making the cell susceptible to NK-cell lysis [88]. To avoid recognition by NK-cells different strategies have been developed, most commonly utilizing overexpression of a non-classical HLA-I such as HLA-E or G fusion protein to avoid lysis [89,90].
Other strategies to avoid HvG include: i) CD47 overexpression [91] and ii) CD52 KO [92]. CD47 is found on both healthy and malignant cells and regulates macrophage-mediated phagocytosis by sending a “don’t eat me” signal to the signal regulatory protein alpha receptor. Upon depletion of HLA-I on CAR-Ts, recognition by both macrophages and NK-cells is triggered. In a recent study by Hu, et al. the overexpression of CD47 in allogeneic CD19-CAR-T negated the recognition of NK and macrophages to the absence of HLA on the cell surface, thus avoiding rejection [93]. This approach is currently under investigation in a phase I clinical trial (NCT05878184).
CD52 is protein expressed on the cell surface of many immune cells such as mature lymphocytes, NK-cells, monocytes/macrophages and others [92,94]. The humanized anti-CD52 monoclonal antibody (mAb), alemtuzumab, has been widely used in clinics for the treatment of transplant patients, and B-cell chronic lymphocytic leukaemia [95,96,97]. Alemtuzumab, targets CD52+ T-cells and is capable of both complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity [96]. Therefore, CD52 KO in allogeneic CAR-Ts can be combined with Alemtuzumab to enhance the CAR persistence. Although, this will necessitate multiple infusions and close monitoring of the immune-system of each patient. This approach has been assessed in multiple clinical trials involving allogeneic CAR-Ts most notably by Allogene who have used this in combination with CD70 [98] and CD19 CAR-Ts [99].
Next to recipient CD8+ T-cells that recognize the HLA-I molecules, CD4+ T-cells recognize HLA-II molecules, that are expressed on multiple cell-types including activated T-cells [100]. Therefore, once donor CAR-Ts recognize their antigen they will upregulate the HLA-II expression and become targets for recognition by recipient CD4+ alloreactive T-cells [101] It is therefore likely that for a persistent CAR-T the removal of HLA-II becomes necessary. One strategy that can achieve this is through the removal of the CIITA gene, a HLA class II transactivator that controls HLA-II expression [102].
However, it is likely that for the success of allogeneic CAR-Ts other modifications become necessary to tackle the tumor microenvironment for example. Different strategies exist to introduce double-stranded DNA break (DSB) that allow for the editing of proteins. These breaks are subsequently repaired in error-prone pathways that can result in insertions/deletions that can disrupt open reading frames. An overview is shown in Table 1.
Zinc finger nucleases (ZFNs)—A ZFN is an artificial endonuclease that has a zinc finger protein (ZFP) fused to the cleavage domain of the FokI restriction enzyme [103]. A ZFN is targeted to cleave a chosen genomic sequence. The FokI cleavage domain needs to be dimerized to cut DNA and because the dimer-interface is weak a construct of two sets of fingers directed to neighboring sequences is needed. The cleavage-induced event by ZFN leads to a cellular repair process that mediates the efficient modification of the targeted locus. If the event is resolved via non-homologous end joining (NHEJ), it can result in small deletions or insertions, effectively leading to gene KO. If the break is resolved via a homology-directed repair (HDR), small changes or entire transgenes can be transferred, into the chromosome. Because each zinc-finger unit recognizes three nucleotides, three to six zinc finger units are needed to generate a specific DNA-binding domain.
The use of ZFNs has multiple challenges such as the specificity of ZFN binding where some fingers bind equally well to triplets other than their supposed preference. Thus off-targets can occur and it is therefore necessary to extensively test ZFNs employed in clinical trials [104,105] Furthermore, the efficient delivery of ZFNs and donor DNA will naturally be different among applications, and biological variations in the availability of particular DNA repair pathways may affect the outcome.
Current clinical trials involving ZFNs include the knockout of the CCR5 gene, which is the coreceptor for HIV-1 (e.g. NCT02388594, NCT00842634, NCT01044654, NCT01252641 or NCT02225665) [106]. ZFNs are also currently being used for the targeting of the glucocorticoid receptor in IL13Rα2-targeting CAR-T in an allogeneic setting. Where infusion of the CAR has led to dexamethasone-resistant effector activity in six patients with unresectable recurrent glioblastoma [107].
TALEN—TALEN are similar to ZFNs in that they are heterodimeric nucleases that contain a fusion between the FokI restriction enzyme and a transcription activator-like effector (TALE) DNA-binding domain. The amino-acid repeat variable di-residues (RVD) are two hypervariable amino acids that make part of the sequence that mediates the binding of TALE to DNA [108]. This greatly simplifies the TALEN design. The TALEN monomeric architecture are developed by fusing TALE domains to a sequence-specific catalytic domain derived from the homing endonuclease (HE) I-TevI, resulting in a Tev-TALE monomeric nuclease [109].
Currently multiple CAR-Ts have been developed using TALEN for the purpose of creating allogeneic CAR-Ts. TALEN has been used to knockout both TRAC and CD52 in UCART19 (a CD19 targeting CAR-T) as assessed by Allogene Therapeutics. Similarly Cellectis has assessed multiple CAR-Ts such as CD123 [110], CD22 [111] and CS1 [112] targeted CAR-Ts. In all candidates, TRAC was disrupted but multiple strategies assessed to enhance cellular persistence. Among those CD52, and B2M have been discussed previously. However an additional target is CS1 (SLAMF7) which in this instance is specifically removed to inhibit fratricide by the CAR-Ts.
MegaTALs—Are a short TALE domain that is fused to the homing endonuclease (HE). The artificial chimeric nucleases derived from HEs can be engineered to target specific sequences within the genome [113,114,115]. This fusion increases the specificity and activity of the megaTALs [116]. Currently, to our knowledge no clinical trials are utilizing MegaTALs for allogeneic CAR-Ts.
Clustered regularly interspaced short palindromic repeats (CRISPR)—The CRISPR system is derived from microbial adaptive immune system. It combines a nuclease and a short RNA. The specificity of the CRISPR system is not through the protein-DNA interaction (like the above) but rather RNA-DNA base pairing. A 20 nucleotide RNA that is complementary to the target DNA(termed single guide RNA; sgRNA) is responsible for the specificity. However, due to the system off-targets are tolerated [117,118]. The most common nuclease is Cas9 [119]. CRISPR/Cas9 is the most widely used because it has demonstrated a remarkably low rate of off-target mutagenesis in T cells [120,121]. In addition, a specific high-fidelity Cas9 mutant, called eSpCas9, did not cause any detectable off-target effect, making it an even safer technology [122,123].
CRISPR/Cas9 has been used to KO multiple targets to inhibit both GvHD and HvG. Focusing on TRAC, B2M, CD52 (as previously mentioned) but multiple preclinical studies have also shown the KO of many other genes to play a role in cellular persistence and efficacy, thus giving rise to the need of multiplexing (as reviewed by [124]). Since multiplex gene editing with Cas9 nuclease can increase the risk of chromosomal instability due to DSBs, a lot of work has gone into multiplexing with an effort to reduce this risk. Through the use of base editor technology, modifications can be made to optimize and improve the limitations of CRISPR/Cas9 [125].
Although multiplexing these in unison becomes increasingly difficult, the relative improvements seen, when such targets are removed, does imply that allogeneic CAR-Ts may need more engineering to become long-persisting CAR-Ts.
CRISPR/other Cas—The most widely used CRISPR-Cas system is the CRISPR/Cas9, however there are multiple systems that are generally divided into two classes (class 1 and 2), and subsequently subdivided into six types (types I through VI). Class 1 (types I, III and IV) systems use multiple Cas proteins while class 2 systems (types II, V and VI) use a single Cas protein [126]. The class 1 CRISPR/Cas comprise 90% of all identified CRISPR/Cas loci. The class 2 comprises the remaining 10% and is almost exclusively in bacteria [127]. Cas9 (type II) still presents challenges, mostly due to the possible off-targets and difficulty in delivering the ribonucleoprotein particle [126]. The second most utilized Cas is Cas12a (type V). It has substantial differences in comparison with Cas9 in multiple aspects. One of which is a higher gene repression in the template strand of the target DNA than SpdCas9 [128]. It may also be easier to multiplex in comparison with Cas9 [129]. However, both Cas 9 and 12a suffer from the dependence on host cell DNA repair machinery. Meaning the induction of DSB and induction of repair. Although both technologies have been used successfully to insert specific DNA into the genomic loci, their efficiency differs between cell types [130,131,132]. Furthermore, DNA repair through HDR is also related to active cell division meaning that cells that do not divide (like neurons) render the tools ineffective.
Recently, CRISPR-Cas12a was successfully used in combination with CRISPR-Cas9 to generate simultaneous genetic manipulations for the generation of allogeneic CAR-Ts. Combining both Cas12a and Cas9 led to triple-edited CAR-Ts that resulted in TCR and HLA-I/II negative CAR-Ts resistant to allogeneic stimuli [133]. However, due to the nature of DSBs explained above, and the high safety concern when multiplexing CRISPR/Cas a secondary methodology was necessary to achieve a safe CAR-T, and minimize DSBs. This technology is base-pair-editing.
Base-pair editing—Base editing involves the use of the CRISPR-Cas9 (or other Cas) together with avoidance of DNA DSBs during genetic modification. Fusing, a single-strand DNA (ssDNA) deaminase enzyme to a catalytically inactive Cas9 variant leads to there being only a ssDNA cut (nick). The Cas9-mediated nicking of the genomic DNA means that a short stretch of ssDNA is exposed to the attached deaminase that can convert the selected bases within their target window [134]. Many improvements have been conducted since the report on cytosine base editors (CBE), that have yielded novel base editors that reduce unwanted byproducts, can improve the targeting scope and allow the editing of different bases [135]. Currently four possible transition mutations can be installed C→T, A→G, T→C and G→A.
The added safety and possibility to multiplex gene KO through CRISPRs makes this approach very interesting for CAR-Ts. A proof of concept for the approach was shown by Diorio C, et al. using an allogeneic CD7 CAR-T for T-cell acute lymphoblastic leukemia (T-ALL). Here, base-editing was used in combination with CRISPR-Cas9 to target four genes namely: CD52 (to enable lymphodepletion with alemtuzumab); TRAC (removal of the TCRα chain, GvHD); CD7 (to inhibit fratricide) and PDCD1 (PD1-receptor—an immune-checkpoint inhibitor) successfully [136], currently under clinical evaluation (NCT05885464). Importantly, the CD7 CAR-Ts functioned well and showed no detectable translocations or karyotypic abnormalities. Similar base-pair edited CD7 CAR-Ts were assessed in a phase-I clinical trial. Preliminary results reported one patient to be in leukemic remission, one that received SCT while in remission and the third developed an opportunistic fatal fungal infection. Other adverse events included cytokine release syndrome and multilineage cytopenia [137].
4.1.2. Non-gene editing
The biggest concern with gene editing is the complexity involved in removing multiple genes (multiplexing), while keeping the safety concerns to a minimum. We developed two non-gene edited approaches based on i) a TCR inhibitory molecule (TIM) that upon incorporation with the T-cell DNA, competes with TCR elements rendering the TCR unresponsive [8]. This approach was used together with an NKG2D-based CAR and assessed in metastatic colorectal cancer [138]; ii) the use of a miRNA scaffold targeting CD3ζ, which has led to a complete abolishment of the TCR from the cells [139]. This approach was assessed in a phase-I clinical trial using a BCMA targeting CAR-T in a relapse/refractory multiple myeloma patient cohort.
Another approach includes intracellular retention of TCR/HLA-I to prevent GvHD/HvG. There are multiple methods to retain components in the endoplasmatic reticulum (ER) which includes using a peptide (such as KDEL) that associated with the ER-retention domain. Then by combining said peptide with an scFv targeting the TCR for example all TCRs will be retained in the ER [140].
While the argument for the removal of the TCR is clear, it is unclear which factors govern cellular persistence in an allogeneic setting. While the usual suspects (HLA-I/II) naturally play a role, other proteins are possibly involved in HvG. Furthermore, other cellular processes such as metabolic regulation, may affect cellular persistence in an allogeneic setting. Current results suggest that additional modifications are needed to achieve success in an allogeneic setting. In this regard the ability to multiplex multiple targets simultaneously becomes a key factor. While, this is complex in the gene editing approaches, this is relatively simple in a non-gene edited approach. Multiple groups have either combined miRNA, siRNA like sequences in an effort to inhibit multiple target-sequences together either through a natural scaffold or a synthetic one [141,142,143,144]. We have recently developed a microRNA (miRNA)-based multiplex shRNA platform, obtained by combining highly efficient miRNA scaffolds into a chimeric cluster [145]. We were able to deliver up to four shRNA-like sequences (in a plug-and-play manner) in a single vector containing the CAR and four different shRNA-like sequences targeting: CD3ζ (GvHD), B2M (HLA-I/HvG), and additional combinations of either CIITA (HLA-II/HvG), CD95 (Fas receptor/inhibit apoptosis), LAG-3(Immune-checkpoint inhibitor) and/or CD28(co-stimulation, reduction/persistence). Interestingly, we discovered that the modulation of genes rather than gene KO is essential for certain targets (such as B2M, where a clear balance exists between removal of the HLA-I and recognition by NK-cells and the minimal expression needed to avoid NK-cell lysis and/or T-cell mediated activation), making the method a good and easy-to-use tool for certain targets.
4.2. Clinical experience with ‘off-the-shelf’ allogeneic CAR-Ts
4.2.1. Successes to date
Several off-the-shelf allogeneic CAR-T products are currently under clinical evaluation in Phase I or Phase I/II studies by several groups (Table 2).
Most experience to date is obtained with an allogeneic universal anti-CD19 CAR-T product (UCART19/ALLO-501) which is genome-edited with TALEN technology to simultaneously disrupt TRAC and CD52 genes. While TRAC is targeted to prevent GvHD risk, CD52 gene knockout aims to protect allogeneic CAR-Ts from rejection by alemtuzumab/ALLO-647, an anti-CD52 antibody used as an additional lymphodepleting agent [117]. UCART19/ALLO-501 was evaluated in two completed Phase I studies in pediatric (PALL study, NCT02808442) and adult (CALM study, NCT02746952) populations with relapsed or refractory (r/r) B-cell acute lymphoblastic leukemia (B-ALL) [147,171], initiated following successful therapy in 2 infants with r/r B-ALL who had relapsed after a first allo-SCT [172]. UCART19/ALLO-501 is still under evaluation in adult patients with r/r large B-cell lymphoma (LBCL) or follicular lymphoma (FL) in the ALPHA study (NCT03939026). Globally, UCART19/ALLO-501 induced antileukemic activity with an overall response rate (ORR) of 48% in these heavily pretreated populations and exhibited a manageable safety profile with moderate cytokine release syndrome (CRS) events and minimal—but still some (8% of patients)—grade 1 acute cutaneous GvHD. Two deaths in the CALM trial were considered to be associated with UCART19, and both were reported as dose-limiting toxicity [171]. To note, between 3% and 6% of UCART19/ALLO-501 cells had translocation-associated karyotype abnormalities, without suggestion of adverse effects [171]. An evolution of UCART19/ALLO-501 was also developed and referred to as ALLO-501A, in which the safety switch rituximab recognition was removed, and is currently evaluated in the ALPHA2 (NCT04416984) and EXPAND (NCT05714345) studies. Data with optimal lymphodepletion regimen confirmed a good anti-tumor efficacy with an ORR of 67% across both ALPHA and ALPHA2 studies and no GvHD reported [173].
The same TALEN technology is used in the universal anti-CD123 (UCART123) and anti-CD22 (UCART22) CAR-T product candidates and evaluated in two Phase I studies involving adult patients with relapsed or refractory B-ALL (NCT04150497, BALLI-01 study) [152] or relapsed/refractory acute myeloid leukemia (NCT03190278, AMELI-01 study) [110], respectively and in the anti-BCMA CAR-T product ALLO-715 currently under evaluation in the Phase 1 UNIVERSAL study involving refractory/relapsed adult multiple myeloma patients [150]. At the optimal lymphodepleting regimen, 70.8% of patients had an objective response. The median duration of response was 8.3 months and no cases of GvHD were reported.
The ARCUS genome editing technology is used in the anti-CD19 allogeneic CAR-T product (PBCAR0191, Azercabtagene zapreleucel or Azer-Cel) currently under evaluation at in a Phase I/II study involving relapsed/refractory non-Hodgkin lymphoma (NHL) and B-ALL patients (NCT03666000) and showed promising results. Azer-Cel Achieved 83% ORR, 61% complete response (CR) rate with 55% durable response among evaluable patients who had relapsed following autologous CAR-T therapy (n=18) and 58% ORR overall (n=61) and no GvHD was reported [155]. The PBCAR19B product, an anti-CD19 targeting allogeneic CAR-T designed to evade immune rejection by host T-cell and NK cells, was evaluated in a Phase I study (NCT04649112) and achieved 71% ORR and 43% CR rate [155].
CRISPR/Cas9 is also used in the CD19-targeting CTX110 product evaluated in the phase-1 CARBON trial (NCT04035434) in patients with r/r NHL. 67% ORR was observed at the highest dose-level [159]. No cases of GvHD were reported despite a high HLA mismatch between donors and patients [159]. The only case of Grade 3 or higher immune effector cell-associated neurotoxicity syndrome (ICANS) was in a patient with concurrent HHV-6 (Human Herpes Virus). Administration of a second CTX110 infusion was well tolerated and demonstrated evidence of further clinical benefit. CTX-130, an anti-CD70 allogeneic CAR-T evaluated in the COBALT-LYM study (NCT05722418) in patients with T-cell lymphoma and in the COBALT-RCC study (NCT04502446) in patients with advanced clear cell renal cell carcinoma (RCC). At the highest dose-level, ORR was 71% in patients with T-cell lymphoma [162] and there was no report of GvHD in none of the 17 evaluated patients. Results in the RCC population showed an ORR of 8% (n=13) with one patient who experienced a durable complete response maintained at 18+ months and no GvHD reported [161]. The anti-CD19 CAR-T product CB-010, engineered via CRISPR/Cas9 to knockout both TRAC and PD-1, to reduce T cell exhaustion, is evaluated in the ANTLER Phase 1 study (NCT04637763) in patients with B-NHL and showed 94% ORR across all dose-levels, which rivals autologous products [158] and no GvHD was observed (n=16).
CAR T-cell products using non-gene editing technologies were also evaluated in clinic. CYAD-101 is an allogeneic CAR-T candidate engineered to co-express a CAR based on NKG2D, a receptor recognizing 8 different stress ligands, and an inhibitory peptide interfering with the signaling by the endogenous TCR complex. CYAD-101 was evaluated in the alloSHRINK phase-I study in patients with unresectable metastatic colorectal cancer (NCT03692429). 25 patients received three infusions of CYAD-101 after standard preconditioning chemotherapy (FOLFOX or FOLFIRI). No dose-limiting toxicity or GvHD were reported, nor patient discontinuation due to treatment-related adverse events or treatment-related adverse events greater than Grade 3. Results also showed two patients achieved a partial response (13% ORR), including one patient with a KRAS-mutation, and nine patients (60%) reached a stable disease [138,168]. In CYAD-211, a miRNA-based shRNA approach was used to silence the mRNA coding for the CD3ζ component of the TCR, co-expressed with an anti-BCMA CAR in the CYAD-211 product, which was evaluated in the phase-I IMMUNICY-1 trial (NCT04613557) for the treatment of patients with r/r multiple myeloma. Clinical activity from 12 patients in the dose-escalation segment of the IMMUNICY-1 trial was encouraging with three patients achieving partial response (PR), while eight patients had stable disease (SD). Overall, CYAD-211 was well tolerated with no dose limiting toxicity (DLT), GvHD nor neurotoxicity at the 3 dose-levels [169]. A CD19-targeting allogeneic CAR-T using intracellular retention of membrane proteins to prevent TCR expression at the surface, ThisCART19A, was evaluated in a Phase I study (NCT04384393) in patients with NHL [140]. Data over the first 8 patients demonstrated no evidence of GvHD reaction and encouraging activity (75% ORR).
Finally, the iPSC-derived CAR T-cell product candidate FT819 targeting CD19 was evaluated in a Phase I study (NCT04629729) in patients with B-cell malignancies demonstrated a tolerable safety profile with no reported DLT or GvHD in the 12 evaluated patients [170].
4.2.2. Challenges to overcome
In the clinical setting, some allogeneic candidates have reached objective response rates similar to those observed with their autologous counterparts, and, apart from two patients (1 infant and 1 adult) presenting with Grade I acute skin GvHD that was easily controlled [146], preliminary data from any of those studies showed no evidence of acute GvHD. Therefore, despite earlier concerns, the modifications made to prevent GvHD in allogeneic ‘off-the-shelf’ CAR-Ts seem sufficient to reduce drastically this risk.
In contrast, the engraftment of the allogeneic CAR-Ts has been stymied to some extent by host rejection, mediated by the recognition of non-self HLA molecules on the donor T-cell membrane and is clearly the main concern of allogeneic CAR-Ts as this limit their activity and duration of responses. For example, in the CALM study, although expansion of the CAR-Ts, similar to those observed with autologous CAR-Ts, was observed from day 8 to day 14 after infusion, a rapid decline was observed in most patients by day 28 [174], and limited duration of response. Cellular kinetics was also limited beyond day 28 with ALLO-715 [150], UCART123) [152], UCART122 [110], CYAD-101 [138] and CYAD-211 [139]. As a first solution, a deeper lymphodepletion through a more intense preconditioning regimen is generally used as an approach to improve the allogeneic CAR-T persistence. In addition, strategies to increase the dose of cells—either by using higher dose-levels at first infusion, or by using multiple infusions, have been proposed but do not fully counteract the allorejection. The CALM study evaluated different lymphodepleting regimen (fludarabine 30 mg/m²x3 [days-7 to day-5] and cyclophosphamide 500 mg/m²x3 [day-4 to day-2] with or without alemtuzumab 1 mg/kg, 40 or 60 mg flat doses [day-7 to day-3]) and different cell doses (from 6x106 to 2.4x108 CAR-Ts per infusion). Nevertheless, although the dose of alemtuzumab, and altogether the intensity of the lymphodepleting regimen, had a positive impact on cell persistence post infusion, it also increased the risk of infectious complications. In the UNIVERSAL study, one dose-limiting toxicity of grade 5 fungal pneumonia related to lymphodepletion was reported [150]. A second (consolidation) dose of ALLO-501/ALLO-501A was therefore proposed in the ALPHA and ALPHA2 studies around day 30 after first infusion to further maintain peripheral blood levels of CAR-Ts beyond day 28, with the aim to improve duration of responses [148,149]. Clinical hold was also reported for UCART123 in the ABC study, after a fatality event [153], which led the Data Safety Monitoring Board to recommend lowering the dose of UCART123 cells and capping cyclophosphamide to a total dose of 4g over 3 days. Similarly, one grade 5 event of multifocal pneumonia after ALLO-715 infusion was considered to be related to progressive myeloma and the conditioning regimen [150].
Alternative methods to prevent allorejection are currently developed and some of them were already evaluated in clinical trials. The PBCAR19B product, was engineered to knock-down β2M (beta-2 microglobulin) and express an HLA-E transgene to prevent allorejection [155]. Preliminary clinical results provide proof-of-concept that these modifications appeared to be effective in delaying recovery of host T- and NK-cells. Similarly, CTX-130, an anti-CD70 allogeneic CAR-T is modified to disrupt β2M and CD70 genes to reduce allorejection and fratricide, has reported a durable complete response in a patient with RCC [161], which may suggest the approach is indeed improving the activity of allogeneic CAR-Ts even in solid tumors. CB-011, an anti-BCMA allogeneic CAR-T engineered with CRISPR/CAS12a to KO not only TRAC but also β2M and co-express a β2M-HLA-E fusion peptide is currently evaluated in the CaMMouflage Phase 1 study, and has demonstrated promising preclinical data leading to significant improvement of anti-tumor activity durability [158].
Finally, safety risks related to the use of gene editing technologies are still a big concern. Chromosomal abnormality was reported in a single patient who received a consolidation dose of ALLO-501A, which caused a clinical hold of several months of all studies with similar technology [149]. Investigations concluded that the chromosomal abnormality was unrelated to TALEN gene editing or manufacturing process but raised the question of safety concern of gene edited cellular therapies.
5. Conclusions
Undoubtedly, more extensive research is required to prove the superior clinical efficacy of allogeneic CAR-Ts compared to their approved autologous counterparts, particularly in treating solid cancer which has limited therapeutic options.
However, current evidence suggests that allogeneic CAR Ts can efficiently overcome major hindrances that restrict access to CAR-T therapy to a wider patient population. This feasible approach stems from the emergence of suitable techniques that interrupt the endogenous TCR and mitigate GvHD, which is the primary risk for toxicity in allogeneic T-cell treatment. Genetic ablation of TCRα through targeted gene editing techniques has become popular in this field. However, there are potential drawbacks related to double-strand DNA breaks and the manufacturing complexities which may impact cell fitness and/or yield. A noteworthy substitute exists in the form of non-gene editing technologies, which warrant further exploration because they offer potentially safer and more adaptable choices for manufacturing next-generation CAR-Ts. Nevertheless, although those approaches seem promising to prevent the risk of GvHD, rejection of the cells following post-infusion, via HvG reaction; is the greatest challenge as of today.
Figure 2.
Safe and effective engineered off-the-shelf allogeneic CAR-Ts require technologies to target (and downregulate or disrupt) genes involved in alloreactivity and allorejection.
Figure 2.
Safe and effective engineered off-the-shelf allogeneic CAR-Ts require technologies to target (and downregulate or disrupt) genes involved in alloreactivity and allorejection.
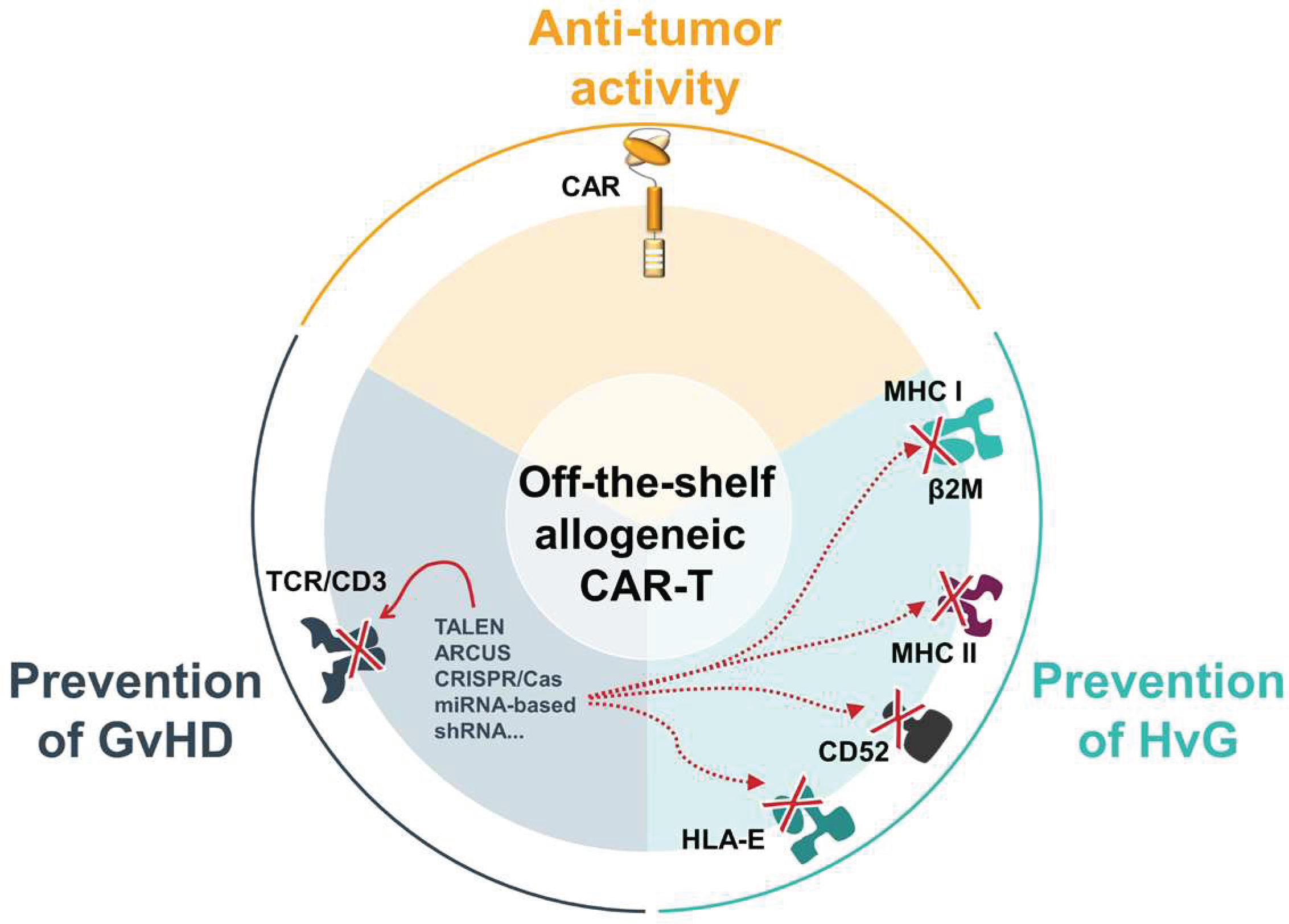
Hence, the ideal “off-the-shelf’ allogenic CAR-T not only needs to prevent GvHD but also HvG via diverse modifications like down-regulation or disruption of genes involved in allorejection, like β2M, CIITA, or CD52. It is therefore imperative to further invest in developing technologies allowing safe administration of allogeneic CAR-Ts while improving their persistence and their efficacy and maintaining a favorable safety profile.
References
- Mitra, A.; Barua, A.; Huang, L.; Ganguly, S.; Feng, Q.; He, B. From Bench to Bedside: The History and Progress of CAR T Cell Therapy. Front. Immunol. 2023, 14, 1188049. [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. Long-Term Outcomes Following CAR T Cell Therapy: What We Know so Far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [CrossRef]
- Rotte, A.; Frigault, M.J.; Ansari, A.; Gliner, B.; Heery, C.; Shah, B. Dose–Response Correlation for CAR-T Cells: A Systematic Review of Clinical Studies. J. Immunother. Cancer 2022, 10, e005678. [CrossRef]
- Ruella, M.; Korell, F.; Porazzi, P.; Maus, M.V. Mechanisms of Resistance to Chimeric Antigen Receptor-T Cells in Haematological Malignancies. Nat. Rev. Drug Discov. 2023, 1–20. [CrossRef]
- Daei Sorkhabi, A.; Mohamed Khosroshahi, L.; Sarkesh, A.; Mardi, A.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Baradaran, B. The Current Landscape of CAR T-Cell Therapy for Solid Tumors: Mechanisms, Research Progress, Challenges, and Counterstrategies. Front. Immunol. 2023, 14, 1113882. [CrossRef]
- Martinez-Cibrian, N.; Español-Rego, M.; Pascal, M.; Delgado, J.; Ortiz-Maldonado, V. Practical Aspects of Chimeric Antigen Receptor T-Cell Administration: From Commercial to Point-of-Care Manufacturing. Front. Immunol. 2022, 13.
- Abraham-Miranda, J.; Menges, M.; Atkins, R.; Mattie, M.; Kanska, J.; Turner, J.; Hidalgo-Vargas, M.J.; Locke, F.L. CAR-T Manufactured from Frozen PBMC Yield Efficient Function with Prolonged in Vitro Production. Front. Immunol. 2022, 13, 1007042. [CrossRef]
- Michaux, A.; Mauën, S.; Breman, E.; Dheur, M.-S.; Twyffels, L.; Saerens, L.; Jacques-Hespel, C.; Gauthy, E.; Agaugué, S.; Gilham, D.E.; et al. Clinical Grade Manufacture of CYAD-101, a NKG2D-Based, First in Class, Non-Gene-Edited Allogeneic CAR T-Cell Therapy. J. Immunother. Hagerstown Md 1997 2022, 45, 150–161. [CrossRef]
- Tang, T.C.Y.; Xu, N.; Nordon, R.; Haber, M.; Micklethwaite, K.; Dolnikov, A. Donor T Cells for CAR T Cell Therapy. Biomark. Res. 2022, 10, 14. [CrossRef]
- Eapen, M.; Rocha, V.; Sanz, G.; Scaradavou, A.; Zhang, M.-J.; Arcese, W.; Sirvent, A.; Champlin, R.E.; Chao, N.; Gee, A.P.; et al. Effect of Graft Source on Unrelated Donor Haemopoietic Stem-Cell Transplantation in Adults with Acute Leukaemia: A Retrospective Analysis. Lancet Oncol. 2010, 11, 653–660. [CrossRef]
- Boyd, N.; Cartledge, K.; Cao, H.; Evtimov, V.; Pupovac, A.; Trounson, A.; Boyd, R. “Off-the-Shelf” Immunotherapy: Manufacture of CD8+ T Cells Derived from Hematopoietic Stem Cells. Cells 2021, 10, 2631. [CrossRef]
- Van Caeneghem, Y.; De Munter, S.; Tieppo, P.; Goetgeluk, G.; Weening, K.; Verstichel, G.; Bonte, S.; Taghon, T.; Leclercq, G.; Kerre, T.; et al. Antigen Receptor-Redirected T Cells Derived from Hematopoietic Precursor Cells Lack Expression of the Endogenous TCR/CD3 Receptor and Exhibit Specific Antitumor Capacities. OncoImmunology 2017, 6, e1283460. [CrossRef]
- Kwoczek, J.; Riese, S.B.; Tischer, S.; Bak, S.; Lahrberg, J.; Oelke, M.; Maul, H.; Blasczyk, R.; Sauer, M.; Eiz-Vesper, B. Cord Blood-Derived T Cells Allow the Generation of a More Naïve Tumor-Reactive Cytotoxic T-Cell Phenotype. Transfusion (Paris) 2018, 58, 88–99. [CrossRef]
- Liu, D.-D.; Hong, W.-C.; Qiu, K.-Y.; Li, X.-Y.; Liu, Y.; Zhu, L.-W.; Lai, W.-X.; Chen, H.-; Yang, H.-Q.; Xu, L.-H.; et al. Umbilical Cord Blood: A Promising Source for Allogeneic CAR-T Cells. Front. Oncol. 2022, 12, 944248. [CrossRef]
- Kadereit, S.; Mohammad, S.F.; Miller, R.E.; Woods, K.D.; Listrom, C.D.; McKinnon, K.; Alali, A.; Bos, L.S.; Iacobucci, M.L.; Sramkoski, M.R.; et al. Reduced NFAT1 Protein Expression in Human Umbilical Cord Blood T Lymphocytes. Blood 1999, 94, 3101–3107.
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of Tumor-Targeted Human T Lymphocytes from Induced Pluripotent Stem Cells for Cancer Therapy. Nat. Biotechnol. 2013, 31, 928–933. [CrossRef]
- Themeli, M.; Rivière, I.; Sadelain, M. New Cell Sources for T Cell Engineering and Adoptive Immunotherapy. Cell Stem Cell 2015, 16, 357–366. [CrossRef]
- Ueda, T.; Kaneko, S. In Vitro Differentiation of T Cell: From CAR-Modified T-IPSC. Methods Mol. Biol. Clifton NJ 2019, 2048, 85–91. [CrossRef]
- Wang, B.; Iriguchi, S.; Waseda, M.; Ueda, N.; Ueda, T.; Xu, H.; Minagawa, A.; Ishikawa, A.; Yano, H.; Ishi, T.; et al. Generation of Hypoimmunogenic T Cells from Genetically Engineered Allogeneic Human Induced Pluripotent Stem Cells. Nat. Biomed. Eng. 2021, 5, 429–440. [CrossRef]
- Moradi, S.; Mahdizadeh, H.; Šarić, T.; Kim, J.; Harati, J.; Shahsavarani, H.; Greber, B.; Moore, J.B. Research and Therapy with Induced Pluripotent Stem Cells (IPSCs): Social, Legal, and Ethical Considerations. Stem Cell Res. Ther. 2019, 10, 341. [CrossRef]
- Iriguchi, S.; Yasui, Y.; Kawai, Y.; Arima, S.; Kunitomo, M.; Sato, T.; Ueda, T.; Minagawa, A.; Mishima, Y.; Yanagawa, N.; et al. A Clinically Applicable and Scalable Method to Regenerate T-Cells from IPSCs for off-the-Shelf T-Cell Immunotherapy. Nat. Commun. 2021, 12, 430. [CrossRef]
- Wood, K.J.; Goto, R. Mechanisms of Rejection: Current Perspectives. Transplantation 2012, 93, 1–10. [CrossRef]
- Alhaj Hussen, K.; Michonneau, D.; Biajoux, V.; Keita, S.; Dubouchet, L.; Nelson, E.; Setterblad, N.; Le Buanec, H.; Bouaziz, J.-D.; Guimiot, F.; et al. CD4+CD8+ T-Lymphocytes in Xenogeneic and Human Graft-versus-Host Disease. Front. Immunol. 2020, 11, 579776. [CrossRef]
- Ringdén, O.; Karlsson, H.; Olsson, R.; Omazic, B.; Uhlin, M. The Allogeneic Graft-versus-Cancer Effect. Br. J. Haematol. 2009, 147, 614–633. [CrossRef]
- Champlin, R.E.; Passweg, J.R.; Zhang, M.J.; Rowlings, P.A.; Pelz, C.J.; Atkinson, K.A.; Barrett, A.J.; Cahn, J.Y.; Drobyski, W.R.; Gale, R.P.; et al. T-Cell Depletion of Bone Marrow Transplants for Leukemia from Donors Other than HLA-Identical Siblings: Advantage of T-Cell Antibodies with Narrow Specificities. Blood 2000, 95, 3996–4003.
- Abdelhakim, H.; Abdel-Azim, H.; Saad, A. Role of Aβ T Cell Depletion in Prevention of Graft versus Host Disease. Biomedicines 2017, 5, 35. [CrossRef]
- Biernacki, M.A.; Sheth, V.S.; Bleakley, M. T Cell Optimization for Graft-versus-Leukemia Responses. JCI Insight 2020, 5, e134939, 134939. [CrossRef]
- Rådestad, E.; Wikell, H.; Engström, M.; Watz, E.; Sundberg, B.; Thunberg, S.; Uzunel, M.; Mattsson, J.; Uhlin, M. Alpha/Beta T-Cell Depleted Grafts as an Immunological Booster to Treat Graft Failure after Hematopoietic Stem Cell Transplantation with HLA-Matched Related and Unrelated Donors. J. Immunol. Res. 2014, 2014, 578741. [CrossRef]
- Korngold, R.; Sprent, J. Lethal Graft-versus-Host Disease after Bone Marrow Transplantation across Minor Histocompatibility Barriers in Mice. Prevention by Removing Mature T Cells from Marrow. J. Exp. Med. 1978, 148, 1687–1698.
- Maraninchi, D.; Gluckman, E.; Blaise, D.; Guyotat, D.; Rio, B.; Pico, J.L.; Leblond, V.; Michallet, M.; Dreyfus, F.; Ifrah, N. Impact of T-Cell Depletion on Outcome of Allogeneic Bone-Marrow Transplantation for Standard-Risk Leukaemias. Lancet Lond. Engl. 1987, 2, 175–178. [CrossRef]
- Ho, V.T.; Soiffer, R.J. The History and Future of T-Cell Depletion as Graft-versus-Host Disease Prophylaxis for Allogeneic Hematopoietic Stem Cell Transplantation. Blood 2001, 98, 3192–3204. [CrossRef]
- Anwer, F.; Shaukat, A.-A.; Zahid, U.; Husnain, M.; McBride, A.; Persky, D.; Lim, M.; Hasan, N.; Riaz, I.B. Donor Origin CAR T Cells: Graft versus Malignancy Effect without GVHD, a Systematic Review. Immunotherapy 2017, 9, 123–130. [CrossRef]
- Felix, N.J.; Allen, P.M. Specificity of T-Cell Alloreactivity. Nat. Rev. Immunol. 2007, 7, 942–953. [CrossRef]
- Mangum, D.S.; Caywood, E. A Clinician’s Guide to HLA Matching in Allogeneic Hematopoietic Stem Cell Transplant. Hum. Immunol. 2022, 83, 687–694. [CrossRef]
- Yeung, M.Y.; Coates, P.T.; Li, P.K.-T. Kidney Organ Allocation System: How to Be Fair. Semin. Nephrol. 2022, 42, 151274. [CrossRef]
- Brudno, J.N.; Somerville, R.P.T.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 1112–1121. [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-Derived CD19-Targeted T Cells Cause Regression of Malignancy Persisting after Allogeneic Hematopoietic Stem Cell Transplantation. Blood 2013, 122, 4129–4139.
- Cruz, C.R.Y.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of Donor-Derived CD19-Redirected Virus-Specific T Cells for B-Cell Malignancies Relapsed after Allogeneic Stem Cell Transplant: A Phase 1 Study. Blood 2013, 122, 2965–2973.
- Jin, X.; Cao, Y.; Wang, L.; Sun, R.; Cheng, L.; He, X.; Xiao, X.; Jiang, Y.; Li, Q.; Zhang, H.; et al. HLA-Matched and HLA-Haploidentical Allogeneic CD19-Directed Chimeric Antigen Receptor T-Cell Infusions Are Feasible in Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia before Hematopoietic Stem Cell Transplantation. Leukemia 2020, 34, 909–913. [CrossRef]
- Melenhorst, J.J.; Leen, A.M.; Bollard, C.M.; Quigley, M.F.; Price, D.A.; Rooney, C.M.; Brenner, M.K.; Barrett, A.J.; Heslop, H.E. Allogeneic Virus-Specific T Cells with HLA Alloreactivity Do Not Produce GVHD in Human Subjects. Blood 2010, 116, 4700–4702. [CrossRef]
- O’Reilly, R.J.; Prockop, S.; Hasan, A.N.; Koehne, G.; Doubrovina, E. Virus-Specific T-Cell Banks for “off the Shelf” Adoptive Therapy of Refractory Infections. Bone Marrow Transplant. 2016, 51, 1163–1172. [CrossRef]
- Savoldo, B.; Rooney, C.M.; Di Stasi, A.; Abken, H.; Hombach, A.; Foster, A.E.; Zhang, L.; Heslop, H.E.; Brenner, M.K.; Dotti, G. Epstein Barr Virus Specific Cytotoxic T Lymphocytes Expressing the Anti-CD30 Artificial Chimeric T-Cell Receptor for Immunotherapy of Hodgkin Disease. Blood 2007, 110, 2620–2630. [CrossRef]
- Curran, K.J.; Sauter, C.S.; Kernan, N.A.; Prockop, S.E.; Boulad, F.; Perales, M.; Giralt, S.A.; Riviere, I.; Wang, X.; Boelens, J.-J.; et al. Durable Remission Following “Off-the-Shelf” Chimeric Antigen Receptor (CAR) T-Cells in Patients with Relapse/Refractory (R/R) B-Cell Malignancies. Biol. Blood Marrow Transplant. 2020, 26, S89. [CrossRef]
- Wang, X.; Diamond, D.J.; Forman, S.J.; Nakamura, R. Development of CMV-CD19 Bi-Specific CAR T Cells with Post-Infusion in Vivo Boost Using an Anti-CMV Vaccine. Int. J. Hematol. 2021, 114, 544–553. [CrossRef]
- Young, K.J.; Kay, L.S.; Phillips, M.J.; Zhang, L. Antitumor Activity Mediated by Double-Negative T Cells. Cancer Res. 2003, 63, 8014–8021.
- Lee, J.-B.; Vasic, D.; Kang, H.; Fang, K.K.-L.; Zhang, L. State-of-Art of Cellular Therapy for Acute Leukemia. Int. J. Mol. Sci. 2021, 22, 4590. [CrossRef]
- Chen, B.; Lee, J.B.; Kang, H.; Minden, M.D.; Zhang, L. Targeting Chemotherapy-Resistant Leukemia by Combining DNT Cellular Therapy with Conventional Chemotherapy. J. Exp. Clin. Cancer Res. CR 2018, 37, 88. [CrossRef]
- Lee, J.B.; Kang, H.; Fang, L.; D’Souza, C.; Adeyi, O.; Zhang, L. Developing Allogeneic Double-Negative T Cells as a Novel Off-the-Shelf Adoptive Cellular Therapy for Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2241–2253. [CrossRef]
- Lee, J.; Minden, M.D.; Chen, W.C.; Streck, E.; Chen, B.; Kang, H.; Arruda, A.; Ly, D.; Der, S.D.; Kang, S.; et al. Allogeneic Human Double Negative T Cells as a Novel Immunotherapy for Acute Myeloid Leukemia and Its Underlying Mechanisms. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 370–382. [CrossRef]
- Vasic, D.; Lee, J.B.; Leung, Y.; Khatri, I.; Na, Y.; Abate-Daga, D.; Zhang, L. Allogeneic Double-Negative CAR-T Cells Inhibit Tumor Growth without off-Tumor Toxicities. Sci. Immunol. 2022, 7, eabl3642. [CrossRef]
- Baolin, T.; Lee, J.; Cheng, S.; Yao, W.; Wang, D.; Tu, M.; Xiang, Z.; Geng, L.; Wang, M.; Qiang, P.; et al. Safety and Efficacy of Ex Vivo Expanded Healthy Donor-Derived Double Negative T Cells for the Treatment of AML Relapsed after Allogeneic Stem Cell Transplantation: A First in-Human Phase I/IIa Clinical Trial. Blood 2020, 136, 1–2. [CrossRef]
- Kang, H.; Lee, J.B.; Khatri, I.; Na, Y.; D’Souza, C.; Arruda, A.; Minden, M.D.; Zhang, L. Enhancing Therapeutic Efficacy of Double Negative T Cells against Acute Myeloid Leukemia Using Idelalisib. Cancers 2021, 13, 5039. [CrossRef]
- Breman, E.; Demoulin, B.; Agaugué, S.; Mauën, S.; Michaux, A.; Springuel, L.; Houssa, J.; Huberty, F.; Jacques-Hespel, C.; Marchand, C.; et al. Overcoming Target Driven Fratricide for T Cell Therapy. Front. Immunol. 2018, 9, 2940. [CrossRef]
- Leveson-Gower, D.B.; Olson, J.A.; Sega, E.I.; Luong, R.H.; Baker, J.; Zeiser, R.; Negrin, R.S. Low Doses of Natural Killer T Cells Provide Protection from Acute Graft-versus-Host Disease via an IL-4-Dependent Mechanism. Blood 2011, 117, 3220–3229. [CrossRef]
- Schneidawind, D.; Baker, J.; Pierini, A.; Buechele, C.; Luong, R.H.; Meyer, E.H.; Negrin, R.S. Third-Party CD4+ Invariant Natural Killer T Cells Protect from Murine GVHD Lethality. Blood 2015, 125, 3491–3500. [CrossRef]
- Yang, J.; Gao, L.; Liu, Y.; Ren, Y.; Xie, R.; Fan, H.; Qian, K. Adoptive Therapy by Transfusing Expanded Donor Murine Natural Killer T Cells Can Suppress Acute Graft-versus-Host Disease in Allogeneic Bone Marrow Transplantation. Transfusion (Paris) 2010, 50, 407–417. [CrossRef]
- Bendelac, A.; Savage, P.B.; Teyton, L. The Biology of NKT Cells. Annu. Rev. Immunol. 2007, 25, 297–336. [CrossRef]
- Salio, M.; Silk, J.D.; Jones, E.Y.; Cerundolo, V. Biology of CD1- and MR1-Restricted T Cells. Annu. Rev. Immunol. 2014, 32, 323–366. [CrossRef]
- Rotolo, A.; Caputo, V.S.; Holubova, M.; Baxan, N.; Dubois, O.; Chaudhry, M.S.; Xiao, X.; Goudevenou, K.; Pitcher, D.S.; Petevi, K.; et al. Enhanced Anti-Lymphoma Activity of CAR19-INKT Cells Underpinned by Dual CD19 and CD1d Targeting. Cancer Cell 2018, 34, 596-610.e11. [CrossRef]
- Cappuzzello, E.; Vigolo, E.; D’Accardio, G.; Astori, G.; Rosato, A.; Sommaggio, R. How Can Cytokine-Induced Killer Cells Overcome CAR-T Cell Limits. Front. Immunol. 2023, 14, 1229540. [CrossRef]
- Lv, Z.; Luo, F.; Chu, Y. Strategies for Overcoming Bottlenecks in Allogeneic CAR-T Cell Therapy. Front. Immunol. 2023, 14.
- Nishimura, R.; Baker, J.; Beilhack, A.; Zeiser, R.; Olson, J.A.; Sega, E.I.; Karimi, M.; Negrin, R.S. In Vivo Trafficking and Survival of Cytokine-Induced Killer Cells Resulting in Minimal GVHD with Retention of Antitumor Activity. Blood 2008, 112, 2563–2574. [CrossRef]
- Sangiolo, D.; Martinuzzi, E.; Todorovic, M.; Vitaggio, K.; Vallario, A.; Jordaney, N.; Carnevale-Schianca, F.; Capaldi, A.; Geuna, M.; Casorzo, L.; et al. Alloreactivity and Anti-Tumor Activity Segregate within Two Distinct Subsets of Cytokine-Induced Killer (CIK) Cells: Implications for Their Infusion across Major HLA Barriers. Int. Immunol. 2008, 20, 841–848. [CrossRef]
- Wu, X.; Schmidt-Wolf, I.G.H. An Alternative Source for Allogeneic CAR T Cells With a High Safety Profile. Front. Immunol. 2022, 13, 913123. [CrossRef]
- Rettinger, E.; Huenecke, S.; Bonig, H.; Merker, M.; Jarisch, A.; Soerensen, J.; Willasch, A.; Bug, G.; Schulz, A.; Klingebiel, T.; et al. Interleukin-15-Activated Cytokine-Induced Killer Cells May Sustain Remission in Leukemia Patients after Allogeneic Stem Cell Transplantation: Feasibility, Safety and First Insights on Efficacy. Haematologica 2016, 101, e153-156. [CrossRef]
- Wang, S.; Zhang, H.; Liu, C.; Jiao, X.; Liu, D.; Du, W.; He, Y.; Zhang, Z.; Wu, X.; Wang, J.; et al. Human Leukocyte Antigen-Haploidentical Donor-Derived Cytokine-Induced Killer Cells Are Safe and Prolong the Survival of Patients with Advanced Non-Small Cell Lung Cancer. Oncol. Lett. 2014, 8, 2727–2733. [CrossRef]
- Introna, M.; Lussana, F.; Algarotti, A.; Gotti, E.; Valgarsdottir, R.; Micò, C.; Grassi, A.; Pavoni, C.; Ferrari, M.L.; Delaini, F.; et al. Phase II Study of Sequential Infusion of DLI and Cytokine Induced Killer Cells for Patients Relapsed after AlloHSCT. Biol. Blood Marrow Transplant. 2017. [CrossRef]
- Magnani, C.F.; Gaipa, G.; Lussana, F.; Belotti, D.; Gritti, G.; Napolitano, S.; Matera, G.; Cabiati, B.; Buracchi, C.; Borleri, G.; et al. Sleeping Beauty-Engineered CAR T Cells Achieve Antileukemic Activity without Severe Toxicities. J. Clin. Invest. 2020, 130, 6021–6033. [CrossRef]
- Legoux, F.; Salou, M.; Lantz, O. MAIT Cell Development and Functions: The Microbial Connection. Immunity 2020, 53, 710–723. [CrossRef]
- Treiner, E.; Duban, L.; Bahram, S.; Radosavljevic, M.; Wanner, V.; Tilloy, F.; Affaticati, P.; Gilfillan, S.; Lantz, O. Selection of Evolutionarily Conserved Mucosal-Associated Invariant T Cells by MR1. Nature 2003, 422, 164–169. [CrossRef]
- Martin, E.; Treiner, E.; Duban, L.; Guerri, L.; Laude, H.; Toly, C.; Premel, V.; Devys, A.; Moura, I.C.; Tilloy, F.; et al. Stepwise Development of MAIT Cells in Mouse and Human. PLoS Biol. 2009, 7, e54. [CrossRef]
- Chen, Z.; Wang, H.; D’Souza, C.; Sun, S.; Kostenko, L.; Eckle, S.B.G.; Meehan, B.S.; Jackson, D.C.; Strugnell, R.A.; Cao, H.; et al. Mucosal-Associated Invariant T-Cell Activation and Accumulation after in Vivo Infection Depends on Microbial Riboflavin Synthesis and Co-Stimulatory Signals. Mucosal Immunol. 2017, 10, 58–68. [CrossRef]
- Tourret, M.; Talvard-Balland, N.; Lambert, M.; Ben Youssef, G.; Chevalier, M.F.; Bohineust, A.; Yvorra, T.; Morin, F.; Azarnoush, S.; Lantz, O.; et al. Human MAIT Cells Are Devoid of Alloreactive Potential: Prompting Their Use as Universal Cells for Adoptive Immune Therapy. J. Immunother. Cancer 2021, 9, e003123. [CrossRef]
- Li, Y.-R.; Zhou, K.; Wilson, M.; Kramer, A.; Zhu, Y.; Dawson, N.; Yang, L. Mucosal-Associated Invariant T Cells for Cancer Immunotherapy. Mol. Ther. J. Am. Soc. Gene Ther. 2023, 31, 631–646. [CrossRef]
- Bohineust, A.; Tourret, M.; Derivry, L.; Caillat-Zucman, S. Mucosal-Associated Invariant T (MAIT) Cells, a New Source of Universal Immune Cells for Chimeric Antigen Receptor (CAR)-Cell Therapy. Bull. Cancer (Paris) 2021, 108, S92–S95. [CrossRef]
- Li, Y.-R.; Brown, J.; Yu, Y.; Lee, D.; Zhou, K.; Dunn, Z.S.; Hon, R.; Wilson, M.; Kramer, A.; Zhu, Y.; et al. Targeting Immunosuppressive Tumor-Associated Macrophages Using Innate T Cells for Enhanced Antitumor Reactivity. Cancers 2022, 14, 2749. [CrossRef]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer Immunotherapy with Γδ T Cells: Many Paths Ahead of Us. Cell. Mol. Immunol. 2020, 17, 925–939. [CrossRef]
- Mensurado, S.; Blanco-Domínguez, R.; Silva-Santos, B. The Emerging Roles of Γδ T Cells in Cancer Immunotherapy. Nat. Rev. Clin. Oncol. 2023, 20, 178–191. [CrossRef]
- Jhita, N.; Raikar, S.S. Allogeneic Gamma Delta T Cells as Adoptive Cellular Therapy for Hematologic Malignancies. Explor. Immunol. 2022, 2, 334–350. [CrossRef]
- Saura-Esteller, J.; de Jong, M.; King, L.A.; Ensing, E.; Winograd, B.; de Gruijl, T.D.; Parren, P.W.H.I.; van der Vliet, H.J. Gamma Delta T-Cell Based Cancer Immunotherapy: Past-Present-Future. Front. Immunol. 2022, 13, 915837. [CrossRef]
- Rozenbaum, M.; Meir, A.; Aharony, Y.; Itzhaki, O.; Schachter, J.; Bank, I.; Jacoby, E.; Besser, M.J. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front. Immunol. 2020, 11, 1347. [CrossRef]
- Nishimoto, K.P.; Barca, T.; Azameera, A.; Makkouk, A.; Romero, J.M.; Bai, L.; Brodey, M.M.; Kennedy-Wilde, J.; Shao, H.; Papaioannou, S.; et al. Allogeneic CD20-Targeted Γδ T Cells Exhibit Innate and Adaptive Antitumor Activities in Preclinical B-Cell Lymphoma Models. Clin. Transl. Immunol. 2022, 11, e1373. [CrossRef]
- Ma, L.; Feng, Y.; Zhou, Z. A Close Look at Current Γδ T-Cell Immunotherapy. Front. Immunol. 2023, 14, 1140623. [CrossRef]
- Xu, Y.; Xiang, Z.; Alnaggar, M.; Kouakanou, L.; Li, J.; He, J.; Yang, J.; Hu, Y.; Chen, Y.; Lin, L.; et al. Allogeneic Vγ9Vδ2 T-Cell Immunotherapy Exhibits Promising Clinical Safety and Prolongs the Survival of Patients with Late-Stage Lung or Liver Cancer. Cell. Mol. Immunol. 2021, 18, 427–439. [CrossRef]
- Kakimi, K.; Matsushita, H.; Murakawa, T.; Nakajima, J. Γδ T Cell Therapy for the Treatment of Non-Small Cell Lung Cancer. Transl. Lung Cancer Res. 2014, 3, 23.
- López-Cantillo, G.; Urueña, C.; Camacho, B.A.; Ramírez-Segura, C. CAR-T Cell Performance: How to Improve Their Persistence? Front. Immunol. 2022, 13, 878209. [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. “Off-the-Shelf” Allogeneic CAR T Cells: Development and Challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [CrossRef]
- Duygu, B.; Olieslagers, T.I.; Groeneweg, M.; Voorter, C.E.M.; Wieten, L. HLA Class I Molecules as Immune Checkpoints for NK Cell Alloreactivity and Anti-Viral Immunity in Kidney Transplantation. Front. Immunol. 2021, 12, 680480. [CrossRef]
- Jasinski-Bergner, S.; Eckstein, M.; Taubert, H.; Wach, S.; Fiebig, C.; Strick, R.; Hartmann, A.; Seliger, B. The Human Leukocyte Antigen G as an Immune Escape Mechanism and Novel Therapeutic Target in Urological Tumors. Front. Immunol. 2022, 13.
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, A.G.; Hanafi, L.-A.; Clegg, D.O.; et al. HLA-E-Expressing Pluripotent Stem Cells Escape Allogeneic Responses and Lysis by NK Cells. Nat. Biotechnol. 2017, 35, 765–772. [CrossRef]
- van Duijn, A.; Van der Burg, S.H.; Scheeren, F.A. CD47/SIRPα Axis: Bridging Innate and Adaptive Immunity. J. Immunother. Cancer 2022, 10, e004589. [CrossRef]
- Zhao, Y.; Su, H.; Shen, X.; Du, J.; Zhang, X.; Zhao, Y. The Immunological Function of CD52 and Its Targeting in Organ Transplantation. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. Al 2017, 66, 571–578. [CrossRef]
- Hu, X.; Manner, K.; DeJesus, R.; White, K.; Gattis, C.; Ngo, P.; Bandoro, C.; Tham, E.; Chu, E.Y.; Young, C.; et al. Hypoimmune Anti-CD19 Chimeric Antigen Receptor T Cells Provide Lasting Tumor Control in Fully Immunocompetent Allogeneic Humanized Mice. Nat. Commun. 2023, 14, 2020. [CrossRef]
- Domagała, A.; Kurpisz, M. CD52 Antigen--a Review. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2001, 7, 325–331.
- Weaver, T.A.; Kirk, A.D. Alemtuzumab. Transplantation 2007, 84, 1545. [CrossRef]
- Zent, C.S.; Secreto, C.R.; LaPlant, B.R.; Bone, N.D.; Call, T.G.; Shanafelt, T.D.; Jelinek, D.F.; Tschumper, R.C.; Kay, N.E. Direct and Complement Dependent Cytotoxicity in CLL Cells from Patients with High-Risk Early-Intermediate Stage Chronic Lymphocytic Leukemia (CLL) Treated with Alemtuzumab and Rituximab. Leuk. Res. 2008, 32, 1849–1856. [CrossRef]
- Golay, J.; Manganini, M.; Rambaldi, A.; Introna, M. Effect of Alemtuzumab on Neoplastic B Cells. Haematologica 2004, 89, 1476–1483.
- Panowski, S.H.; Srinivasan, S.; Tan, N.; Tacheva-Grigorova, S.K.; Smith, B.; Mak, Y.S.L.; Ning, H.; Villanueva, J.; Wijewarnasuriya, D.; Lang, S.; et al. Preclinical Development and Evaluation of Allogeneic CAR T Cells Targeting CD70 for the Treatment of Renal Cell Carcinoma. Cancer Res. 2022, 82, 2610–2624. [CrossRef]
- Lauron, E.J.; Zhang, K.; Sanchez, J.; Ramanathan, V.; Nguyen, D.; Nguyen, G.; Sasu, B.; Sommer, C. 279 Preclinical Evaluation of Allogeneic CD19 CAR T Cells Expressing an Anti-Rejection CD70 CAR. J. Immunother. Cancer 2023, 11. [CrossRef]
- Holling, T.M.; van der Stoep, N.; Quinten, E.; van den Elsen, P.J. Activated Human T Cells Accomplish MHC Class II Expression through T Cell-Specific Occupation of Class II Transactivator Promoter III. J. Immunol. Baltim. Md 1950 2002, 168, 763–770. [CrossRef]
- Ali, J.M.; Bolton, E.M.; Bradley, J.A.; Pettigrew, G.J. Allorecognition Pathways in Transplant Rejection and Tolerance. Transplantation 2013, 96, 681–688. [CrossRef]
- Ramia, E.; Chiaravalli, A.M.; Bou Nasser Eddine, F.; Tedeschi, A.; Sessa, F.; Accolla, R.S.; Forlani, G. CIITA-Related Block of HLA Class II Expression, Upregulation of HLA Class I, and Heterogeneous Expression of Immune Checkpoints in Hepatocarcinomas: Implications for New Therapeutic Approaches. Oncoimmunology 2018, 8, 1548243. [CrossRef]
- Li, L.; Wu, L.P.; Chandrasegaran, S. Functional Domains in Fok I Restriction Endonuclease. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 4275–4279.
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.-L.; et al. Establishment of HIV-1 Resistance in CD4+ T Cells by Genome Editing Using Zinc-Finger Nucleases. Nat. Biotechnol. 2008, 26, 808–816. [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome Editing with Engineered Zinc Finger Nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [CrossRef]
- Chattong, S.; Chaikomon, K.; Chaiya, T.; Tangkosakul, T.; Palavutitotai, N.; Anusornvongchai, T.; Manotham, K. Efficient ZFN-Mediated Stop Codon Integration into the CCR5 Locus in Hematopoietic Stem Cells: A Possible Source for Intrabone Marrow Cell Transplantation. AIDS Res. Hum. Retroviruses 2018, 34, 575–579. [CrossRef]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-Shelf, Steroid-Resistant, IL13Rα2-Specific CAR T Cells for Treatment of Glioblastoma. Neuro-Oncol. 2022, 24, 1318–1330. [CrossRef]
- Bogdanove, A.J.; Voytas, D.F. TAL Effectors: Customizable Proteins for DNA Targeting. Science 2011, 333, 1843–1846. [CrossRef]
- Beurdeley, M.; Bietz, F.; Li, J.; Thomas, S.; Stoddard, T.; Juillerat, A.; Zhang, F.; Voytas, D.F.; Duchateau, P.; Silva, G.H. Compact Designer TALENs for Efficient Genome Engineering. Nat. Commun. 2013, 4, 1762. [CrossRef]
- Sallman, D.A.; DeAngelo, D.J.; Pemmaraju, N.; Dinner, S.; Gill, S.; Olin, R.L.; Wang, E.S.; Stark, E.; Korngold, A.; Figliola, C.; et al. AMELI-01: A Phase I Trial of UCART123v1.2, an Anti-CD123 Allogeneic CAR-T Cell Product, in Adult Patients with Relapsed or Refractory (R/R) CD123+ Acute Myeloid Leukemia (AML). Mol. Ther. 2023, 31, 51, abstract 94. [CrossRef]
- Aranda-Orgilles, B.; Chion-Sotinel, I.; Skinner, J.; Grudman, S.; Mumford, B.; Dixon, C.; Postigo Fernandez, J.; Erler, P.; Duchateau, P.; Gouble, A.; et al. Preclinical Evidence of an Allogeneic Dual CD20xCD22 CAR to Target a Broad Spectrum of Patients with B-Cell Malignancies. Cancer Immunol. Res. 2023, 11, 946–961. [CrossRef]
- Korst, C.L.B.M.; Bruins, W.S.C.; Cosovic, M.; Verkleij, C.P.M.; Twickler, I.; Le Clerre, D.; Chion-Sotinel, I.; Zweegman, S.; Galetto, R.; Mutis, T.; et al. Preclinical Activity of Allogeneic CS1-Specific CAR T-Cells (UCARTCS1) in Multiple Myeloma. Blood 2022, 140, 4215–4216. [CrossRef]
- Zaslavskiy, M.; Bertonati, C.; Duchateau, P.; Duclert, A.; Silva, G.H. Efficient Design of Meganucleases Using a Machine Learning Approach. BMC Bioinformatics 2014, 15, 191. [CrossRef]
- Werther, R.; Hallinan, J.P.; Lambert, A.R.; Havens, K.; Pogson, M.; Jarjour, J.; Galizi, R.; Windbichler, N.; Crisanti, A.; Nolan, T.; et al. Crystallographic Analyses Illustrate Significant Plasticity and Efficient Recoding of Meganuclease Target Specificity. Nucleic Acids Res. 2017, 45, 8621–8634. [CrossRef]
- MacLeod, D.T.; Antony, J.; Martin, A.J.; Moser, R.J.; Hekele, A.; Wetzel, K.J.; Brown, A.E.; Triggiano, M.A.; Hux, J.A.; Pham, C.D.; et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 949–961. [CrossRef]
- Boissel, S.; Jarjour, J.; Astrakhan, A.; Adey, A.; Gouble, A.; Duchateau, P.; Shendure, J.; Stoddard, B.L.; Certo, M.T.; Baker, D.; et al. MegaTALs: A Rare-Cleaving Nuclease Architecture for Therapeutic Genome Engineering. Nucleic Acids Res. 2014, 42, 2591–2601. [CrossRef]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-Cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-Cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [CrossRef]
- Hussain, W.; Mahmood, T.; Hussain, J.; Ali, N.; Shah, T.; Qayyum, S.; Khan, I. CRISPR/Cas System: A Game Changing Genome Editing Technology, to Treat Human Genetic Diseases. Gene 2019, 685, 70–75. [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [CrossRef]
- Georgiadis, C.; Preece, R.; Nickolay, L.; Etuk, A.; Petrova, A.; Ladon, D.; Danyi, A.; Humphryes-Kirilov, N.; Ajetunmobi, A.; Kim, D.; et al. Long Terminal Repeat CRISPR-CAR-Coupled “Universal” T Cells Mediate Potent Anti-Leukemic Effects. Mol. Ther. 2018. [CrossRef]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2255–2266. [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-Fidelity CRISPR–Cas9 Nucleases with No Detectable Genome-Wide off-Target Effects. Nature 2016, 529, 490–495. [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally Engineered Cas9 Nucleases with Improved Specificity. Science 2016, 351, 84–88. [CrossRef]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the Next-Generation of CAR T-Cells with CRISPR-Cas9 Gene Editing. Mol. Cancer 2022, 21, 78. [CrossRef]
- Webber, B.R.; Lonetree, C.; Kluesner, M.G.; Johnson, M.J.; Pomeroy, E.J.; Diers, M.D.; Lahr, W.S.; Draper, G.M.; Slipek, N.J.; Smeester, B.A.; et al. Highly Efficient Multiplex Human T Cell Engineering without Double-Strand Breaks Using Cas9 Base Editors. Nat. Commun. 2019, 10, 5222. [CrossRef]
- Nishimasu, H.; Nureki, O. Structures and Mechanisms of CRISPR RNA-Guided Effector Nucleases. Curr. Opin. Struct. Biol. 2017, 43, 68–78. [CrossRef]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and Evolution of Class 2 CRISPR–Cas Systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [CrossRef]
- Kim, S.K.; Kim, H.; Ahn, W.-C.; Park, K.-H.; Woo, E.-J.; Lee, D.-H.; Lee, S.-G. Efficient Transcriptional Gene Repression by Type V-A CRISPR-Cpf1 from Eubacterium Eligens. ACS Synth. Biol. 2017, 6, 1273–1282. [CrossRef]
- Campa, C.C.; Weisbach, N.R.; Santinha, A.J.; Incarnato, D.; Platt, R.J. Multiplexed Genome Engineering by Cas12a and CRISPR Arrays Encoded on Single Transcripts. Nat. Methods 2019, 16, 887–893. [CrossRef]
- Jasin, M.; Rothstein, R. Repair of Strand Breaks by Homologous Recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [CrossRef]
- Schmid-Burgk, J.L.; Höning, K.; Ebert, T.S.; Hornung, V. CRISPaint Allows Modular Base-Specific Gene Tagging Using a Ligase-4-Dependent Mechanism. Nat. Commun. 2016, 7, 12338. [CrossRef]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In Vivo Genome Editing via CRISPR/Cas9 Mediated Homology-Independent Targeted Integration. Nature 2016, 540, 144–149. [CrossRef]
- Glaser, V.; Flugel, C.; Kath, J.; Du, W.; Drosdek, V.; Franke, C.; Stein, M.; Pruß, A.; Schmueck-Henneresse, M.; Volk, H.-D.; et al. Combining Different CRISPR Nucleases for Simultaneous Knock-in and Base Editing Prevents Translocations in Multiplex-Edited CAR T Cells. Genome Biol. 2023, 24, 89. [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome Editing with CRISPR-Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat. Biotechnol. 2020, 38, 824–844. [CrossRef]
- Diorio, C.; Murray, R.; Naniong, M.; Barrera, L.; Camblin, A.; Chukinas, J.; Coholan, L.; Edwards, A.; Fuller, T.; Gonzales, C.; et al. Cytosine Base Editing Enables Quadruple-Edited Allogeneic CART Cells for T-ALL. Blood 2022, 140, 619–629. [CrossRef]
- Chiesa, R.; Georgiadis, C.; Syed, F.; Zhan, H.; Etuk, A.; Gkazi, S.A.; Preece, R.; Ottaviano, G.; Braybrook, T.; Chu, J.; et al. Base-Edited CAR7 T Cells for Relapsed T-Cell Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2023, 0, null. [CrossRef]
- Prenen, H.; Dekervel, J.; Hendlisz, A.; Anguille, S.; Awada, A.; Cerf, E.; Lonez, C.; Breman, E.; Dheur, M.-S.; Alcantar-Orozco, E.; et al. Updated Data from AlloSHRINK Phase I First-in-Human Study Evaluating CYAD-101, an Innovative Non-Gene Edited Allogeneic CAR-T in MCRC. J. Clin. Oncol. 2021, 39, 74–74. [CrossRef]
- Lonez, C.; Bolsee, J.; Huberty, F.; Nguyen, T.; Jacques-Hespel, C.; Demoulin, B.; Flament, A.; Breman, E. 285 Proof-of-Concept of a Non-Gene Editing Technology Using ShRNA down-Regulation to Engineer and Optimize CAR T-Cell Functionality. J. Immunother. Cancer 2023, 11. [CrossRef]
- Wang, X.; Xue, L.; Sujun Li, Qian Fan, Kaichun Liu, Rong Jin, Xiaohui Yang, Tao Wang, Ling He, Jun Li Preliminary Analyses of a Non-Gene-Editing Allogentic CAR-T in CD19+ Relapsed or Refractory Non-Hodgin’s Lymphoma. EHA Libr. 2022, 357128, S264.
- Liu, Y.P.; Haasnoot, J.; ter Brake, O.; Berkhout, B.; Konstantinova, P. Inhibition of HIV-1 by Multiple SiRNAs Expressed from a Single MicroRNA Polycistron. Nucleic Acids Res. 2008, 36, 2811–2824. [CrossRef]
- Choi, J.-G.; Bharaj, P.; Abraham, S.; Ma, H.; Yi, G.; Ye, C.; Dang, Y.; Manjunath, N.; Wu, H.; Shankar, P. Multiplexing Seven MiRNA-Based ShRNAs to Suppress HIV Replication. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 310–320. [CrossRef]
- Wang, T.; Xie, Y.; Tan, A.; Li, S.; Xie, Z. Construction and Characterization of a Synthetic MicroRNA Cluster for Multiplex RNA Interference in Mammalian Cells. ACS Synth. Biol. 2016, 5, 1193–1200. [CrossRef]
- Du, X.; Cai, Y.; Xi, W.; Zhang, R.; Jia, L.; Yang, A.; Zhao, J.; Yan, B. Multi-target Inhibition by Four Tandem ShRNAs Embedded in Homo- or Hetero-miRNA Backbones. Mol. Med. Rep. 2018, 17, 307–314. [CrossRef]
- Rossi, M.; Steklov, M.; Huberty, F.; Nguyen, T.; Marijsse, J.; Jacques-Hespel, C.; Najm, P.; Lonez, C.; Breman, E. Efficient ShRNA-Based Knockdown of Multiple Target Genes for Cell Therapy Using a Chimeric MiRNA Cluster Platform. Mol. Ther.—Nucleic Acids 2023, 34, 102038. [CrossRef]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-Edited, Donor-Derived Allogeneic Anti-CD19 Chimeric Antigen Receptor T Cells in Paediatric and Adult B-Cell Acute Lymphoblastic Leukaemia: Results of Two Phase 1 Studies. Lancet Lond. Engl. 2020, 396, 1885–1894. [CrossRef]
- Benjamin, R.; Jain, N.; Maus, M.V.; Boissel, N.; Graham, C.; Jozwik, A.; Yallop, D.; Konopleva, M.; Frigault, M.J.; Teshima, T.; et al. UCART19, a First-in-Class Allogeneic Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy for Adults with Relapsed or Refractory B-Cell Acute Lymphoblastic Leukaemia (CALM): A Phase 1, Dose-Escalation Trial. Lancet Haematol. 2022, 9, e833–e843. [CrossRef]
- Neelapu, S.S.; Nath, R.; Munoz, J.; Tees, M.; Miklos, D.B.; Frank, M.J.; Malik, S.A.; Stevens, D.; Shin, C.R.; Balakumaran, A.; et al. ALPHA Study: ALLO-501 Produced Deep and Durable Responses in Patients with Relapsed/Refractory Non-Hodgkin’s Lymphoma Comparable to Autologous CAR T. Blood 2021, 138, 3878. [CrossRef]
- Lekakis, L.J.; Locke, F.L.; Tees, M.; Neelapu, S.S.; Malik, S.A.; Hamadani, M.; Frank, M.J.; Popplewell, L.L.; Abramson, J.S.; de Vos, S.; et al. ALPHA2 Study: ALLO-501A Allogeneic CAR T in LBCL, Updated Results Continue to Show Encouraging Safety and Efficacy with Consolidation Dosing. Blood 2021, 138, 649. [CrossRef]
- Mailankody, S.; Matous, J.V.; Chhabra, S.; Liedtke, M.; Sidana, S.; Oluwole, O.O.; Malik, S.; Nath, R.; Anwer, F.; Cruz, J.C.; et al. Allogeneic BCMA-Targeting CAR T Cells in Relapsed/Refractory Multiple Myeloma: Phase 1 UNIVERSAL Trial Interim Results. Nat. Med. 2023, 29, 422–429. [CrossRef]
- Srour, S.; Kotecha, R.; Curti, B.; Chahoud, J.; Drakaki, A.; Tang, L.; Goyal, L.; Prashad, S.; Szenes, V.; Norwood, K.; et al. Abstract CT011: A Phase 1 Multicenter Study (TRAVERSE) Evaluating the Safety and Efficacy of ALLO-316 Following Conditioning Regimen in Pts with Advanced or Metastatic Clear Cell Renal Cell Carcinoma (CcRCC). Cancer Res. 2023, 83, CT011. [CrossRef]
- Boissel, N.; Chevallier, P.; Curran, K.; Schiller, G.; Liu, H.; Larson, R.; Deangelo, D.J.; Mear, J.-B.; Grupp, S.; Baruchel, A.; et al. P1408: Updated Results of the Phase I BALLI-01 Trial of UCART22, an Anti-CD22 Allogeneic CAR-t Cell Product, in Patients with Relapsed or Refractory (r/r) CD22+ B-Cell Acute Lymphoblastic Leukemia (B-ALL). HemaSphere 2023, 7, e323373f. [CrossRef]
- Cellectis Cellectis to Showcase Clinical Data from AMELI-01 and Preclinical Data from UCARTCS1 at ASH 2022 Available online: https://www.cellectis.com/en/press/cellectis-to-showcase-clinical-data-from-ameli-01-and-preclinical-data-from-ucartcs1-at-ash-2022/ (accessed on 22 November 2023).
- Patel, K.K.; Bharathan, M.; Siegel, D.; et al. UCARTCS1A, an Allogeneic CAR T-Cell Therapy Targeting CS1 in Patients with Relapsed/Refractory Multiple Myeloma (RRMM): Preliminary Translational Results from a First-in-Human Phase I Trial (MELANI-01).; Virtual, May 11 2021; Vol. 29, p. 59 abstract 118.
- Precision BioSciences Provides Update on Allogeneic CAR T Programs and Regulatory Path Forward Available online: https://investor.precisionbiosciences.com/node/9926/pdf (accessed on 22 November 2023).
- Precision Biosciences Corporate Presentation 2022 Available online: https://investor.precisionbiosciences.com/static-files/717d82d7-d304-4428-ba22-1a19c965d8d5 (accessed on 22 November 2023).
- Precision BioSciences Reports Third Quarter 2021 Financial Results and Provides Business Update Available online: https://investor.precisionbiosciences.com/node/8986/pdf (accessed on 22 November 2023).
- Caribou Biosciences Corporate Presentation Nov 2023 Available online: https://investor.cariboubio.com/static-files/3168b9f6-48dd-4e44-8103-df5ff9013b6b (accessed on 22 November 2023).
- McGuirk, J.P.; Tam, C.S.; Kröger, N.; Riedell, P.A.; Murthy, H.S.; Ho, P.J.; Maakaron, J.E.; Waller, E.K.; Awan, F.T.; Shaughnessy, P.J.; et al. CTX110 Allogeneic CRISPR-Cas9-Engineered CAR T Cells in Patients (Pts) with Relapsed or Refractory (R/R) Large B-Cell Lymphoma (LBCL): Results from the Phase 1 Dose Escalation Carbon Study. Blood 2022, 140, 10303–10306. [CrossRef]
- CRISPR Therapeutics Corporate Presentation Jun 2022 Available online: https://crisprtx.gcs-web.com/static-files/5c256eb5-4982-4e47-b463-79eff954e622 (accessed on 23 November 2023).
- Pal, S.; Tran, B.; Haanen, J.; Hurwitz, M.; Sacher, A.; Agarwal, N.; Tannir, N.; Budde, E.; Harrison, S.; Klobuch, S.; et al. 558 CTX130 Allogeneic CRISPR-Cas9–Engineered Chimeric Antigen Receptor (CAR) T Cells in Patients with Advanced Clear Cell Renal Cell Carcinoma: Results from the Phase 1 COBALT-RCC Study. J. Immunother. Cancer 2022, 10. [CrossRef]
- Iyer, S.P.; Sica, R.A.; P. Joy Ho, Boyu Hu, Jasmine Zain, Anca Prica, Wen-Kai Weng, Youn H. Kim, Michael S. Khodadoust, M. Lia Palomba, Francine M. Foss, Kimberly Tipton, Erika L. Cullingford, Qiuling He, Anjali Sharma, Steven M. Horwitz The COBALT-LYM Study of Ctx130: A Phase 1 Dose Escalation Study of CD70-Targeted Allogeneic CRISPR-Cas9–Engineered CAR T Cells in Patients with Relapsed/Refractory (r/r) T-Cell Malignancies. EHA Libr. 2022, 357126, S262.
- Hu, Y.; Zhou, Y.; Zhang, M.; Ge, W.; Li, Y.; Yang, L.; Wei, G.; Han, L.; Wang, H.; Yu, S.; et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2021, 27, 2764–2772. [CrossRef]
- Li, S.; Yuan, Z.; Lin Liu, Yu Li, Sersch Martina, Jia Liu, Zhimin Li, Xinxin Wang, Jiaping He, Wei Zhao, Lianjun Shen, Xi Zhang, Sanbin Wang Early Results of a Safety and Efficacy Study of Allogeneic TRuUCARTM GC502 in Patients with Relapsed/Refractory b-Cell Acute Lymphoblastic Leukemia (R/R B-ALL). EHA Libr. 2022, 357233, P370.
- Ghobadi, A.; Aldoss, I.; Maude, S.; S Wayne, A.; Bhojwani, D.; Bajel, A.; Dholaria, B.; Faramand, R.; Mattison, R.; Rettig, M.; et al. P356: Phase 1/2 Dose-Escalation Study of Anti-CD7 Allogenic CAR-T Cell in Relapsed or Refractory (r/r) T-Cell Acute Lymphoblastic Leukemia/Lymphoblastic Lymphoma (T-ALL/LBL). HemaSphere 2023, 7, e1789302. [CrossRef]
- Dholaria, B. Early Safety Results of P-BCMA-ALLO1, a Fully Allogeneic Chimeric Antigen Receptor T-Cell (CAR-T), in Patients with Relapsed / Refractory Multiple Myeloma (RRMM). 2023.
- Oh, D.; Henry, J.; Baranda, J.; et al. Development of an Allogeneic CAR-T Targeting MUC1-C (MUC1, Cell Surface Associated, C-Terminal) for Epithelial Derived Tumors. Immuno-Oncol. Technol. 2022, 16, 46P. [CrossRef]
- Prenen, H.; Dekervel, J.; Anguille, S.; Hendlisz, A.; Michaux, A.; Sotiropoulou, P.A.; Gilham, D.E.; Mauen, S.; Snykers, S.; Cerf, E.; et al. CYAD-101: An Innovative Non-Gene Edited Allogeneic CAR-T for Solid Tumor Cancer Therapy. J. Clin. Oncol. 2020, 38, 3032–3032. [CrossRef]
- Al-Homsi, A.-S.; Anguille, S.; Deeren, D.; Nishihori, T.; Meuleman, N.; Abdul-Hay, M.; Morgan, G.J.; Brayer, J.; Braun, N.; Lonez, C.; et al. Immunicy-1: Targeting BCMA with Cyad-211 to Establish Proof of Concept of an ShRNA-Based Allogeneic CAR T Cell Therapy Platform. Blood 2021, 138, 2817. [CrossRef]
- Mehta, A.; Farooq, U.; Chen, A.; McGuirk, J.P.; Ly, T.; Wong, L.; Cooley, S.; Valamehr, B.; Elstrom, R.; Chu, Y.-W.; et al. Interim Phase I Clinical Data of FT819-101, a Study of the First-Ever, Off-the-Shelf, IPSC-Derived TCR-Less CD19 CAR T-Cell Therapy for Patients with Relapsed/Refractory B-Cell Malignancies. Blood 2022, 140, 4577–4578. [CrossRef]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-Edited, Donor-Derived Allogeneic Anti-CD19 Chimeric Antigen Receptor T Cells in Paediatric and Adult B-Cell Acute Lymphoblastic Leukaemia: Results of Two Phase 1 Studies. Lancet Lond. Engl. 2020, 396, 1885–1894. [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular Remission of Infant B-ALL after Infusion of Universal TALEN Gene-Edited CAR T Cells. Sci. Transl. Med. 2017, 9. [CrossRef]
- Allogene Therapeutics Presents Updated ALLO-501/501A Phase 1 Data in Large B Cell Lymphoma at the American Society of Clinical Oncology (ASCO) Annual Meeting | Allogene Therapeutics Available online: https://ir.allogene.com/news-releases/news-release-details/allogene-therapeutics-presents-updated-allo-501501a-phase-1-data/ (accessed on 24 November 2023).
- Dupouy, S.; Marchiq, I.; Derippe, T.; Almena-Carrasco, M.; Jozwik, A.; Fouliard, S.; Adimy, Y.; Geronimi, J.; Graham, C.; Jain, N.; et al. Clinical Pharmacology and Determinants of Response to UCART19, an Allogeneic Anti-CD19 CAR-T Cell Product, in Adult B-Cell Acute Lymphoblastic Leukemia. Cancer Res. Commun. 2022, 2, 1520–1531. [CrossRef]
Figure 1.
Overview of autologous versus allogeneic CAR-T manufacturing process from peripheral blood mononuclear cells.
Figure 1.
Overview of autologous versus allogeneic CAR-T manufacturing process from peripheral blood mononuclear cells.
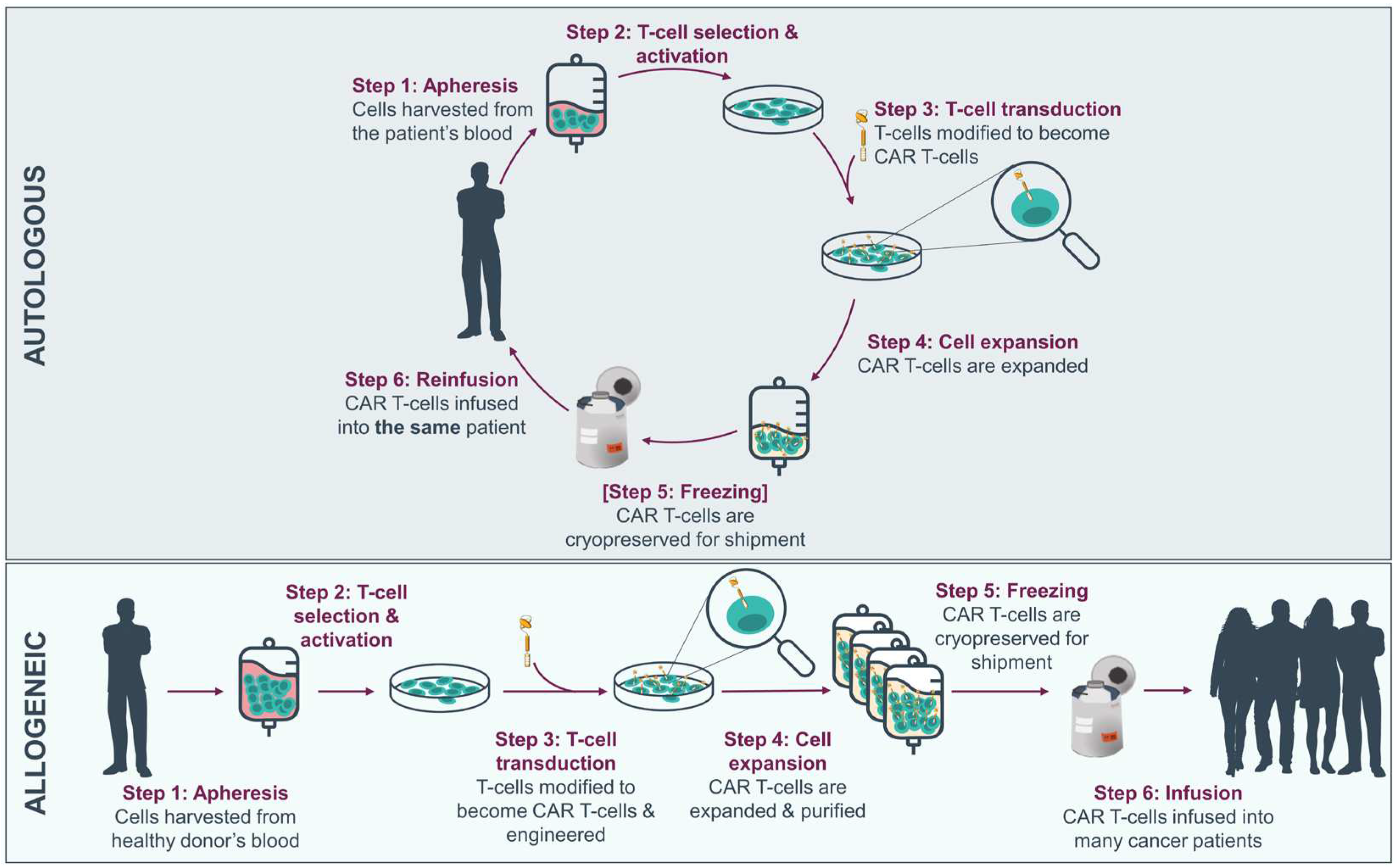

Table 1.
Gene editing technologies used to engineer allogeneic CAR-Ts.
| ▪ | ZFN | TALEN | CRISPR/Cas9 | CRISPR/Cas12a | Base-editing |
|---|---|---|---|---|---|
| ▪ Recognition site | Zinc-finger protein | RVD tandem repeat region TALE protein | Two RNA molecules (guideRNA and tracrRNA) | Single-stranded guide RNA | CRISPR/Cas dependent (Cas sequence + base-editor mRNA) |
| ▪ Modification pattern | Fok1 nuclease | Fok1 nuclease | Cas9 nuclease | Cas12a nuclease | Four possible transition mutations: C→T A→G T→C G→A |
| ▪ Target sequence size | 9-18bp | 14-20bp | 20bp- guide + PAM sequence | 20bp- guide + PAM sequence | CRISPR/Cas dependent |
| ▪ Specificity | Small number of positional mismatches | Small number of positional mismatches | Positional/multiple consecutive mismatches | Positional/multiple consecutive mismatches | CRISPR/Cas dependent |
| ▪ Targeting limitations | Difficult to target non-G-rich sites | 5′ targeted base must be a T for each TALEN monomer | Recognizes 3′ G-rich Must precede a PAM sequence of 3-5nt |
Recognizes 5′ T-rich Must precede a PAM sequence of 3-4nt |
CRISPR/Cas dependent |
| ▪ Engineering | Requires substantial protein engineering | Requires complex molecular cloning methods | Uses standard cloning procedures | Uses standard cloning procedures | Uses standard cloning procedures |
| ▪ Delivering | Easy due to small size | Difficult due to large size | Moderate to difficult due to large size of SpCas9 | Moderate to difficult due to large size of FnCas12a | Difficult due to large site and added complexity |
PAM: protospacer adjacent motif
Table 2.
Engineered allogeneic ‘off-the-shelf’ CAR-Ts with published clinical experience.
| Allogeneic engineering Technology | Target antigen | Strategy for GvHD | Strategy for HvG | Product Name | Developers | Trial Names, Phase and Number |
|---|---|---|---|---|---|---|
| αβ T-cells(from PBMCs) | ||||||
| TALEN | CD19 | Disruption of TRAC | Disruption of CD52 and use of anti-CD52 | ALLO-501 / UCART19 | Cellectis; Allogene Therapeutics | CALM Phase 1 [146,147] NCT02746952 PALL Phase 1 [146] NCT02808442 ALPHA Phase 1 [148] NCT03939026 |
| CD19 | Disruption of TRAC | Disruption of CD52 and use of anti-CD52 | ALLO-501A | Cellectis; Allogene Therapeutics | ALPHA 2 Phase 1/2 [149] NCT04416984 |
|
| BCMA | Disruption of TRAC | Disruption of CD52 and use of anti-CD52 | ALLO-715 | Allogene Therapeutics; Cellectis SA | UNIVERSAL Phase 1 [150] NCT04093596 |
|
| CD70 | Disruption of TRAC | Disruption of CD52 and use of anti-CD52 CD70 CAR designed to avoid fratricide |
ALLO-316 | Allogene Therapeutics; Cellectis SA | TRAVERSE Phase 1 [151] NCT04696731 |
|
| CD123 | Disruption of TRAC | Disruption of CD52 and use of anti-CD52 | UCART123 | Cellectis SA | AMELI-01 Phase 1 [110] NCT03190278 Phase 1 NCT04106076 ABC123 Phase 1 NCT03203369 |
|
| CD22 | Disruption of TRAC | Disruption of CD52 and use of anti-CD52 | UCART22 | Cellectis SA | BALLI-01 Phase 1 [152] NCT04150497 |
|
| SLAMF7 | Disruption of TRAC | Disruption of CS1 gene to avoid fratricide | UCARTCS1 | Cellectis SA | MELANI-01 Phase 1 [153,154] NCT04142619 | |
| ARCUS | CD19 | Disruption of TCR | - | PBCAR0191 / Azercabtagene zapreleucel | Precision BioSciences | Phase 1/2 [155] NCT03666000 |
| CD19 | Disruption of TCR | shRNA against β2M and HLA-E transgene | PBCAR19B | Precision BioSciences, Inc | Phase 1 [155] NCT04649112 |
|
| BCMA | Disruption of TCR | - | PBCAR269A | Precision BioSciences | Phase 1 [156] NCT04171843 |
|
| CD20 | Disruption of TCR | - | PBCAR20A | Precision BioSciences | Phase 1/2 [157] NCT04030195 |
|
| CRISPR/Cas9 | CD19 | Disruption of TRAC | - | CB-010 | Caribou Biosciences | ANTLER Phase 1 [158] NCT04637763 |
| CD19 | Disruption of TRAC | Disruption of β2M | CTX110 | CRISPR Therapeutics | CARBON Phase 1/2 [159] NCT04035434 | |
| BCMA | Disruption of TRAC | Disruption of β2M | CTX120 | CRISPR Therapeutics | Phase 1 [160] NCT04244656 |
|
| CD70 | Disruption of TRAC | Disruption of β2M + CD70 disruption to avoid fratricide | CTX130 | CRISPR Therapeutics | COBALT-RCC Phase 1 [161] NCT04438083 COBALT-LYM Phase 1 [162] NCT04502446 |
|
| CD19 | Disruption of TRAC | Disruption of CD52 and use of anti-CD52 | CTA101 | Nanjing Bioheng Biotech | Phase 1 [163] NCT04154709 NCT04227015 |
|
| CD19/CD7 | Disruption of TRAC | CD7 disruption to avoid fratricide | GC502 | Gracell Biotechnology Inc. | Early Phase 1 [164] NCT05105867 |
|
| CD7 | Disruption of TRAC | CD7 disruption to avoid fratricide | WU CART 007 | Wugen | Phase 1/2 [165] NCT04984356 |
|
| Cas-CLOVER™ | BCMA | Disruption of TCR beta chain 1 | Disruption of β2M | P-BCMA-ALLO1 | Poseida Therapeutics | Phase 1 [166] NCT04960579 |
| FKBP12; MUC1-C | Disruption of TCR | Disruption of β2M | P-MUC1C-ALLO1 | Poseida Therapeutics | Phase 1 [167] NCT05239143 |
|
| Base-pair editing | CD7 | Disruption of TRAC | Disruption of CD52 and CD7 to avoid fratricide | BE-CAR7 | Great Ormond Street Hospital | Phase 1 [137] ISRCTN15323014 |
| Peptide-based (TIM8) | NKG2DL | Negative competition with CD3ζ | - | CYAD-101 | Celyad Oncology | alloSHRINK Phase 1 [138,168] NCT03692429 CYAD-101-002 Phase 1 NCT04991948 |
| miRNA-based shRNA | BCMA | Knock-down of CD3ζ | - | CYAD-211 | Celyad Oncology | IMMUNICY-1 Phase 1 [169] NCT04613557 |
| Non-gene editing | CD19 | Intracellular retention of TCR/CD3 complex via KDEL-tagged anti-CD3 scFv | Decreasing surface HLA-A and HLA-B by HCMV US11 protein | ThisCART19 cells | Fundamenta Therapeutics | Phase 1 [140] NCT04384393 |
| cytolytic T-lymphocytes (from PBMCs) | ||||||
| Zinc-Finger Nuclease | IL13-zetakine | Disruption of the glucocorticoid receptor | Use of dexamethasone | GRm13Z40-2 | City of Hope | Phase 1 [107] NCT01082926 |
| αβ T-cells(from iPSCs) | ||||||
| CRISPR/Cas | CD19 | Disruption of TRAC | - | FT819 | Fate Therapeutics | Phase 1 [170] NCT04629729 |
iPSC: Induced pluripotent stem cells; PBMC: peripheral blood mononuclear cells
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



