Submitted:
16 August 2024
Posted:
20 August 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Historically, macrophages have been long implicated in the control of the severity of infectious diseases. This has been based on the observations of a higher risk of severe disease and death upon rechallenge with viral variants due to antibody dependent enhance-ment (ADE) of infection into macrophages. The question remains as to what can account for this potent heterologous protection in macrophages? Here it is argued that the elusive defense mechanism of M1-like pro-inflammatory macrophages may pertain to a novel virus anti-virus response. This system initiates with high replication of human endogenous retrovirus K102 (HERV-K102), a non-pathogenic, protector foamy retrovirus of humans which generates M1-like pro-inflammatory foamy macrophages, glycolysis, and epigenetic changes, all characteristic of trained immunity. This virus-anti-virus system kills virally infected cells by several mechanisms, amplifies the innate interferon response via ‘viral mimicry’, has many unique components that interfere with exoge-nous virus replication, and may be especially adept at neutralizing enveloped exogenous pandemic viruses, such as SARS-CoV-2 and HIV-1. The goal of this treatise is to introduce the multifaceted HERV-K102 protector system, to illustrate how SARS-CoV-2 may target the HERV-K102 protector system by ADE, and to explore how this innate defense system may be exploited for pandemic preparedness.
Keywords:
ADE
; HERV-K102
; HERV-K HML-2
; immunosenescence
; foamy macrophages
; virus-anti-virus
; pandemic preparedness
; mRNA COVID-19 vaccines
; alpha-fetoprotein antagonists
; virus strategies
; anti-viral
; trained (innate) immunity
Graphical Abstract:
1. Introduction
1.1. Single cell RNA (scRNA) Sequencing has Revealed the Importance of Innate Immunity, Especially Macrophages
Single cell RNA (scRNA) sequencing has substantially revolutionized the ability to gauge cell fate and developmental programs such as for immune cells in sickness and in health [1,2]. Such investigations have revealed the importance of innate immune mechanisms in recovery from or protection against many common diseases.
For example, an initial investigation involving 39 cancer types and about 18,000 tumors unexpectantly determined in 2015 that the gamma-delta2 (γδ2) T cells and/or other innate T cells were frequently correlated with protection against many although not all cancers [3]. This discrimination was largely based on the expression of KLRB1 (CD161) on these innate T cells. KLRB1 is a lectin-like receptor that binds Galactose -a-(1,3) Galactose which is a carbohydrate marker of non-human cells and where humans have IgM against this moiety [4]. On the other hand, FOXM1 considered a marker of poor prognosis tended to identify neutrophil populations within many cancers.
More recently, using scRNA sequencing, M1-like pro-inflammatory macrophages have been implicated in remission from acute myeloid leukemia [5], and as correlates of protection in patients with lung cancers [6].
In addition, via scRNA sequencing, monocytes/macrophages have been identified as essential to a reduced risk of SIV/SHIV acquisition in non-human primate (NHP) animal studies and were implicated in the partial vaccine efficacy against HIV-1 infection demonstrated in the RV144 trial [7]. At a more detailed level, the induction of hypoxia with inflammasome activation in CD14 monocytes has been associated with a decreased risk of SIVmac251 acquisition in non-human primate (NHP) models [8]. It should be appreciated that hypoxia and inflammasome activation results in foam cell formation in M1-like proinflammatory macrophages [9]. Accordingly, it appears, pro-inflammatory foamy macrophages provide protection against HIV-1 acquisition, a conclusion reached earlier on entirely different evidence [10,11].
Macrophages although long considered the central orchestrator of both innate and adaptive immunity, are primarily associated with innate immunity, the antigen non-specific arm of the immune system involving interferon responses and intrinsic pathogen detection systems termed pattern recognition receptors (PRRs). Dendritic cells that are the antigen presenters for adaptive immune responses are derived from a separate set of bone marrow progenitors, called the Common Lymphoid Progenitors (CLPs). Instead, macrophages (and monocytes) are derived from the Common Myeloid Progenitors (CMPs) which then generate the Granulocyte-Macrophage Progenitors (GMPs) and the Megakaryocyte-Erythroid Progenitors (MEPs) [12]. Accordingly, discussions of monocytes in the blood or macrophages in the tissues refer and pertain to innate immunity.
1.2. The Clue of Antibody Dependent Enhancement (ADE) of Infection into Macrophages
Historically a major line of evidence emphasizing the importance of macrophages in the control of infectious diseases and outcomes has been the concept of antibody dependent enhancement (ADE) of infection into monocyte/macrophages. Disease escalation involving antibodies that mediate the enhanced infection of macrophages through Fc receptors was first documented for dengue in 1967. Since that time ADE has been observed for various viral infections [13] such as measles, SARS-CoV, MERS-CoV, HIV-1, West Nile virus, Japanese encephalitis virus, Ross River virus, Ebola virus, respiratory syncytial virus, feline infectious peritonitis virus, porcine reproductive and respiratory syndrome virus, and now SARS-CoV-2 [14]. Most notably, it has been suggested that COVID-19 disease progression from moderate to severe commonly relates to ADE mediated infection of SARS-CoV-2 into monocytes/macrophages [14] (which will be examined and validated in more detail below). This view is, however, not supported by everyone, as some profess that ADE has not been observed clinically during COVID-19 infection [15].
Despite the controversy in opinion, incontrovertible evidence had emerged that even before the COVID-19 vaccines were authorized for emergency use that demonstrated IgG antibodies to spike protein were not protective but instead were associated with progression to more severe COVID-19 disease including death during natural infection [16,17,18,19,20,21,22,23,24,25,26,27,28,29,30]. Despite this unanimous finding, three papers claimed exceptions to the consensus finding based on assumptions about neutralizing activity [31,32,33]. However, these could be discounted based on technical grounds. All three excursions were based on neutralizing antibody data generated from pseudotyped virus produced in human 293T cells rather than live SARS-CoV-2 virus grown and tested in the non-human, green monkey cell line, Vero cells. As virus pseudotyped in human cells likely carries human innate immunity target antigens, the authors did not address and exclude the possibility that the neutralization they observed was due to innate neutralizing antibodies rather than those reactive with spike protein.
As also noted by Ricke [14], the main problem which led to more severe disease during natural infection was that the onset of IgG to spike protein preceded the clearance of SARS-CoV-2 by the innate immune system [34,35,36,37,38]. This earlier onset leading to more severe disease was also documented previously for spike IgG antibody to SARS-CoV-1 [39,40]. In contrast when SARS-CoV-2 was neutralized or cleared from the upper respiratory tract before the onset of spike specific IgG in the blood, this invariably resulted in mild COVID-19 disease [41].
The problem of ADE following vaccination particularly for RNA viruses is believed in part to reflect the rapidity at which new variants are generated in the host following the introduction of antibodies to spike protein that can and do readily select for new variants. For example, in immunosuppressed high-risk individuals it took 10 days on average for a neutralizing monoclonal antibody, bamlanivimab to spike protein to generate escape mutants and establish a rebound viremia [42]. Fortunately, in many instances but not all patients studied, convalescent plasma was able to provide recovery presumably related to innate neutralizing antibodies and other innate humoral immune mechanisms found in plasma.
Perhaps more importantly, a growing body of evidence implies for natural infection unless the case of COVID-19 was severe/critical, there were few IgG1 or IgG3 antibodies to spike RBD in the nasal secretions and none in the saliva [43,44]. However, with the second dose of COVID-19 vaccines (mRNA or virus vectored), these IgG1/3 antibodies to spike protein capable of mediating ADE were commonly detected at high levels in the upper respiratory tract (URT) [43,44]. Thus, the global mass vaccination with COVID-19 vaccines not only greatly increased the likelihood of the emergence of variants, but led to increased transmission and infection rates, all of which would and did prolong the pandemic beyond May 2021 [45].
Given the importance of the selection of SARS-CoV-2 variants and increased infection rates as salient proof of ADE, which in turn validates that the macrophage provides the most important correlate of protection against COVID-19, data supporting each of these statements will be briefly discussed.
1.3. Evidence from Canada for Selection of SARS-CoV-2 Variants Correlating with Vaccination Milestones
For both the Pfizer and Moderna mRNA COVID-19 vaccines, it was about 14 days after the second dose of vaccines, when the IgG antibodies to spike protein became significantly detectable in most individuals [46,47]. Also, it was known that the first dose which provoked trained innate immunity induced heterologous protection against all-cause mortality. Negative excess all-cause mortality was clearly demonstrated in countries especially those that postponed the second dose (Canada [48] and the UK) at the time that the first dose was administered [49]. For the USA a study of seven integrated health care organizations in the USA demonstrated that heterologous protection against non-COVID-19 mortality was induced upon the first dose and which was not appreciably altered upon receiving the second dose, at least for the first 7.5 months of the vaccination campaign [50].
In Canada, the selection of variants (initial emergence or when the variant became dominant) was observed [51] either: a) when the percentage of the eligible population who received two doses over those who had only received one dose exceeded 0.5 (Table 1) or b) when the negative excess all-cause mortality (heterologous protection against all-cause mortality comparing 2021 to the average for 2015 to 2019) reversed direction and started to trend positive (Figure 1). In either case these parameters are alternative ways of looking at the relative impact of the induction of ADE-promoting, spike specific IgG1/3 by the second dose relative to the heterologous protection by trained innate immunity of the first dose. Both sets of parameters examine and reflect ADE. As detailed elsewhere [51], and reproduced here by the author, when the second dose to first dose ratio exceeded 0.5, this was associated with the initial emergence of the alpha variant and was also associated with the dominance of the delta variant (Table 1). Conversely, when the negative excess all-cause mortality reversed direction (ie., the heterologous protection of trained innate immunity against mortality became less, Figure 1) this correlated with the alpha variant becoming dominant and with the initial emergence of the delta variant.
Accordingly, these tight correlations indicate a high likelihood that the COVID-19 vaccines through ADE mechanisms were causally related to selection of the alpha and delta variants. In further substantiation of this notion, the global selection of immune escape variants did not appreciably occur until the COVID-19 vaccines were introduced [49]. For clarification, the initial emergence of the alpha variant which occurred by December 2020 in the UK could have reflected that over 600,000 UK residents had taken part in vaccine clinical trials. Presumably the use of neutralizing monoclonal antibodies to spike protein and/or use of convalescent plasma may have also contributed to the earlier emergence of the alpha variant in the UK. Conversely, Canada did not participate in mRNA vaccine clinical trials. This enabled an assessment of both the emergence of the alpha and delta variants associated with mass immunization.
There is additional published data supporting the above conclusions that the COVID-19 vaccines were likely causally related to the selection of variants. SARS-CoV-2 variants sequenced from the two-dose vaccinated when compared with the unvaccinated had significantly higher percentages of immune escape variants as well as variants associated with higher infectivity [53]. Furthermore, in a separate report, boosted innate immunity (the first 6 days after the second dose) showed 100% vaccine effectiveness (VE) from April 2021 to August 2021 while during the same period the VE for adaptive immunity (7 days or longer after the second dose) declined from 78% to 48% as determined in a large integrated health system in the USA ([54]: see their Supplemental Table 5c). The latter findings are consistent with the notion that the neutralizing antibodies of innate immunity can provide sterilizing or lasting herd immunity against SARS-CoV-2, whereas adaptive immunity neutralizing antibodies cannot. In other words, as expected due to ADE, adaptive immunity vaccines particularly producing IgG1 and IgG3 antibodies to spike protein are not very useful and may be harmful to control a pandemic.
1.4. Vaccination Increased SARS-CoV-2 Symptomatic Infection Rates in the UK Putatively by ADE
In the UK most COVID-19 vaccines administered were the Pfizer-BioNTech mRNA COVID-19 vaccines. UK data consistent with the increased risk of symptomatic infection in the two dose COVID-19 vaccinated over the unvaccinated (unadjusted rates per 100,000) were first revealed in the 40 + age group [55]. This correlated with the onset of the dominance of the delta variant starting with the September 9, 2021, Week 36 Report (reporting for the previous 4-week interval) released by Public Health England. It should be noted that the UK authorities did not release informative data concerning these risks prior to September 9, 2021 [55].
Over time these weekly vaccine surveillance reports [55] indicated that as the vaccines were rolled out to younger and younger age groups the increased risk of infection in the fully vaccinated (per 100,000) over unvaccinated affected younger and younger age groups, showing a cause-and-effect trend. For example, by week 41, reported on October 14, 2021, the fully vaccinated 30 + were additionally at increased risk. By week 50 reported on December 16, 2021, the 18+ age groups were at increased risks. This is consistent with ADE in the URT enhancing symptomatic infections in the vaccinated over the unvaccinated.
Vaccination which increases the risks of symptomatic infection particularly in the younger age groups who are generally at very low or insignificant risk (when compared to those over 65) clearly is the opposite goal of the intended public health measure of mass vaccination. This dilemma was predictable based on the known characteristics of ADE and the multitude of reports that universally showed the IgG antibodies to spike protein correlated with COVID-19 disease progression [13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30] especially when they emerged prior to the clearance of SARS-CoV-2 [35,36,37,38,39,40,42] by innate immunity [41].
However due to extreme transmissibility of the omicron SARS-COV-2 variants witnessed in January 2022 and after where the results of home testing were not reported to government authorities, the UKHSA reports on infection risk of the vaccinated over the unvaccinated ceased after the end of March 2022. By the last report on March 24, 2022, [56] the risk of symptomatic infection in the three dose vaccinated over the unvaccinated (unadjusted rates per 100,000) had reached 4.0- fold in the 30-39 age group which indicated the increased risks of infection related to vaccination were not trivial (Figure 2). As a rule of thumb in medical science, anything over 1.20-fold could be clinically significant.
In the USA, Shrestha et al demonstrated that increasing doses of the COVID-19 shots were associated with an increasing risk of COVID-19 symptomatic infection in health care workers at the Cleveland Clinic [57], in agreement with and as an extension of the UK government findings. Although it has been reported that there is an isotype switch following the third COVID-19 vaccine dose where the IgG1/3 antibodies in the blood to the spike protein are converted to largely IgG4 [58] which does not contribute to ADE, clearly this does not occur in the URT where risk of infection/transmission continues to escalate at 3 or more doses (Figure 2). Note by March 2022 most of the UK ever vaccinated had already received the third dose by December 2021 [49].
Indeed, as captured by ‘Our World in Data’ [49] the population of Canada and the United States as a whole, exhibited an increase in the reproductive rate (transmission reproductive rate) associated with the second and more so, the third dose associated also with the selection of variants (Figure 3). These findings corroborate the notion that the problem of ADE with the adaptive immunity spike vaccines leading to the selection of variants and higher infectivity was in fact, experienced globally.
1.5. From First Principles Adaptive Immunity is Not Able to Control Emerging Pathogens or Pandemics
Adaptive immunity antibodies particularly to spike protein would serve to prolong the pandemic by introducing new waves and making the newer selected variants more transmissible and more infectious as just reviewed. In addition, there are other fundamental reasons as to why adaptive immunity mechanisms are not well suited to handling pandemics or emerging pathogens. The adaptive response to a pathogen never seen before requires at least 14 to 21 days before levels of IgG antibody peak in most individuals [41]. Many pathogens can kill the host before the adaptive system ramps up. For SARS-CoV-2 the time to death was commonly pegged at 10 days [59]. Thus, from first principles, it is clear innate immunity which is the first line of defense, would and does play a pivotal role in COVID-19 disease outcomes, but adaptive immunity cannot and in fact could jeopardize the innate immunity critical to recovery via ADE.
Accordingly, adaptive immunity vaccines such as the COVID-19 vaccines employing spike protein, were intrinsically unable to control SARS-CoV-2 infection at the individual and population level. This is because of the problem of ADE, which would increase transmissibility and lengthen the duration of the pandemic.
The ADE of SARS-CoV-2 in the URT, involved classical FCGR2A expression in macrophages which were labelled as the interferon responsive macrophages [60]. None of the 5 types of macrophages in the URT nasopharyngeal swab samples expressed ACE2 nor TMPRSS2 indicating entry of SARS-CoV-2 into macrophages likely related to ADE. Many groups have indicated that macrophages in the lungs of severe COVID-19 patients but not those with mild or moderate disease are infected with SARS-CoV-2 where again ACE2 and/or TMPRSS2 are not expressed [61,62,63,64,65,66]. This corroborates the notion of SARS-CoV-2 entry into macrophages by ADE causing disease progression. In a very comprehensive study Ren et al [61] uncovered in the lower respiratory tract (LRT), a novel type of ADE involving a switch from the ACE2: spike interaction to basigin (BSG): spike. This focused the infections on the macrophages and other immune cells and away from lung epithelial cells. In the URT, macrophages which were judged to be sebocytes (specialized lipid body negative foamy macrophages) lost expression of BSG upon their activation and as mentioned appeared to instead use the classical FCGR2A for ADE mediated entry of SARS-CoV-2 [60]. Thus, there are two types of ADE associated with SARS-CoV-2 infection, classical in the URT and a novel switch type in the LRT involving BSG.
1.6. Aims of the Review
Given the importance of ADE as an impediment to the development of safe and effective vaccines against emerging and pandemic viruses including SARS-CoV-2, it is important to understand what is so special about the M1-like, proinflammatory foamy macrophages and to elucidate how they generate a potent virus-anti-virus (heterologous) response. Human endogenous retrovirus K102 (HERV-K102) replication produces particles in M1-like macrophages and generates a foamy appearance when the particles bud into vacuoles (Figure 4) [10,11,67]. We were first to suggest HERV-K102 is a replication competent live virus both in vitro and in vivo [10,67] and provided evidence that its replication may protect against HIV-1 acquisition ie., generates sterilizing immunity [10,67]. Other virologists have also unwittingly isolated HERV-K HML-2 DNA containing particles at the same low levels and frequencies from HIV-1 infected patients [68] in direct support of our findings as discussed elsewhere [11]. The goals of the present paper are to ask what host innate immunity mechanisms are abrogated by ADE infection into macrophages and why does this cause and determine progression to more severe disease. Answers to these questions are relevant to all-cause mortality and thus human survival and so, are needed for preparation for future pandemics.
2. The HERV-K102 Protection System of Pro-inflammatory M1-like Foamy Macrophages
2.1. HERV-K102 is a Type 1 HERV-K HML-2 Group Member
HERV-K102 also named ERVK-7, is a member of the HERV-K (HML-2) group consisting of about 91 full length human endogenous retroviruses in the human genome [70]. HML-2 represents the most biologically active and most recently acquired of the 10 HERV-K groups in humans [71]. The letter K refers to the use of lysine-transfer RNA used to prime reverse transcription.
In the HML-2 group, there are two types of HML-2, those with (type 2) and without (type 1) a 292 nucleotide base pair region between the polymerase gene (pol) and envelope (env) genes [71]. The significance of this difference is yet to be determined. This sequence encodes Rec protein which results from an alternatively spliced transcript and is analogous to the Rev protein in HIV-1 and the Rex protein in HTLV used for encapsidation of proviral genomes in their particles [71]. HERV-K102 is a type 1 and lacks this region. The absence of Rec such as in HERV-K102 has led to the false notion that it is unlikely to be replication competent and thus has been largely ignored by virologists. However, the unconventional and non-pathogenic (foamy) spumaretroviruses also lack this domain yet remain fully replication competent [72]. Remarkably HERV-K102 has been shown to be replication competent in in vitro [10] and in vivo [11,67]. As might be anticipated, HERV-K102 fortunately exhibits all the hallmarks of the non-pathogenic category of retroviruses namely the spumaretroviruses [10,67]. Moreover, its replication has been associated with protection against HIV-1 acquisition in an HIV-1 exposed seronegative (HESN) cohort (Figure 5) [10] cementing the notion that HERV-K102 replication is not pathogenic but protective.
2.2. HERV-K102 as the Elusive Foamy Retrovirus of Humans
Spumaretroviruses, commonly referred to as foamy viruses, are complex retroviruses belonging to the subfamily Spumaretrovirinae in the family Retroviridae. Foamy viruses are unconventional non-pathogenic retroviruses belonging to the oldest of virus phylogenies estimated at 400 million years old [72,73]. Simian foamy retroviruses have been co-evolving with their primate hosts for over 60 million years [74] implying they provide major benefits to the host. While foamy viruses have been described in many species examined, until the documentation that replication competent HERV-K102 had all the hallmarks of foamy viruses [10], the foamy virus of humans had remained elusive. In other words, our research group was the first to identify that humans indeed have a protector foamy virus, namely HERV-K102 encoded on chromosome 1, at 1q22.
It should be clarified that in the literature there has been reference to a human foamy virus (HFV) which was isolated from a human nasopharyngeal cell line. However, this virus originated in chimpanzees and when this was discovered it was therefore renamed Prototype Foamy Virus (PFV) as by this time it had been well characterized [72,73].
As mentioned, HERV-K102 has all the salient features of foamy viruses as exemplified by comparison with PFV [see extensive list in Supplemental Materials in reference 10]. First and foremost, when the virus replicates in macrophages (Figure 4) particles accumulate in vacuoles giving the cells a foamy cell appearance which is the primary tell-tale sign of foamy viruses [72]. Another telltale sign of foamy viruses is that their genomes begin with “tgtg” which relates to how the genomes integrate into genomic DNA. Another distinguishing feature of foamy viruses is that the genomes are cDNA [75] which has been clearly demonstrated for HERV-K102 [67]. Rather than reverse transcribing upon entry into cells as is known for orthoretroviruses (the pathogenic retroviruses), instead foamy viruses reverse transcribe upon exit from cells [72,73,75] and so have a reverse life cycle to the orthoretroviruses. This could provide foamy viruses with a replication advantage over the orthoretroviruses which would be important for protecting the host. In addition, PFV is capable of multiple integrations in myeloid cells up to 20-fold [76] and multiple integrations into genomic DNA have also been demonstrated for HERV-K102 in vivo (Figure 5)[10] and in vitro (unpublished observations).
Somewhat unexpectantly, and despite their non-pathogenic nature, foamy viruses such as PFV can undergo cytopathic infections in some fibroblast cell lines but not others [72,73]. This was also demonstrated for HERV-K102 particles (Figure 6). In fact, it turns out PFV is oncolytic meaning it causes cell lysis when it replicates in tumor cells [77] while it merely integrates in normal cells. PFV infection also induces cell death in HIV-1 and HTLV infected cells [78] implying foamy retroviruses may help provide immune surveillance against cancers as well as virally infected cells. Along these lines, it has been suggested that foamy viruses seem to have a peculiar relationship with or companionship with lentiviruses particularly in primates [72,73]. Thus, as suspected, foamy retroviruses do perform an important role defending the host, which may help explain its co-evolution with the host [74]. However, it remains to be directly demonstrated if in fact HERV-K102 particles are oncolytic or induce lysis in virus infected cells.
The cell attachment receptor for PFV has been identified as heparan sulphate [80] which is the same for HERV-K HML-2 [81] and which is widely expressed on cells. This explains the broad spectrum of permissive cells for foamy viruses and is consistent with their potentially protective nature broadly against viral infections and tumors.
2.3. Sebocytes of Sebaceous Glands Lining the Mucosa Were Discovered to Produce HERV-K102 Particles
Another fascinating aspect of foamy viruses is that when they transmit to a new host, they replicate solely in the non-proliferating sebocytes of sebaceous glands and thus are deposited to the exterior mucosa causing no harm to the host [73]. An examination of hematoxylin and eosin-stained sections of sebaceous glands (Figure 7) [82] reveals sebocytes have the exact same morphology as the M1-like foamy macrophages producing the HERV-K102 particles (Figure 4). A search of the expressed genes of sebocytes as available through GEO Profiles [83] revealed both in vitro [84] and in vivo [85] sebocytes are positive for the major antigens of M1-like foamy macrophages [86,88] including: CD14, CD16, CD68, CD163, WDR74, TNFSF10; for myeloid specific enhancers SPI1 and CEBPB [88] which are also trained innate immunity enhancers [89,90]; for genes involved in foam cell formation (NR1H3, LDLR, SQLE, EGFR, HIF1A, BSG, SREBF1/2, PPARG, CD36) which are also implicated in the induction of trained innate immunity [90,91,92,93,94,95,96]; for genes involved in the expression of HERV-K102 full length proviral genomes (IRF1, NFKB1, VDR, IFNGR1/2, NR3C1 +/-MIF) [97,98,99], and genes associated with a novel day 6-7 apoptosis mechanism triggered in the cytoplasm (DNASE2, LAMP1, LCN2 and MX1) [92,100]. Not only do macrophages that are M1 polarized express high levels of HERV-K102 proviral transcripts [99] but ERVK-7 (HERV-K102) was constitutively expressed in sebocytes [84]. Thus, sebocytes are in fact programmed and phenotypically the same as M1-like foamy macrophages, and they respond the same way as macrophages do both in vitro [101] and in vivo [60], except they constitutively express and release HERV-K102 particles. There is no doubt that sebocytes are specialized M1-like foamy macrophages that line the mucosa. This discovery makes it very plausible that the HERV-K102 protector system is in fact the first line of defense against infectious agents anticipating them in the mucosa and so, is critical to infectious disease outcomes.
2.4. The Origins of HERV-K102 Also Provides a Major Clue to Its Purpose
Accumulating phylogenetic evidence is consistent with a potential role of HERV-K HML-2 in limiting invasion of the human genome by orthoretroviruses [102]. Ancestral HML-2 elements emerged about 10.3 million years ago (Mya) [102]. There has been a striking decline of insertions of ERVs in the last 10 million years (My) in the genomes of all sequenced hominids (great apes and gibbons), but not in old world monkeys (baboons and macaques), particularly regarding HERV-H [102]. HERV-H makes up 88% of all the ERV integrations into the human genome in the last 30 My and became extinct over the past 10 My. HERV-H is a gamma retrovirus, which integrated around 45 to 60 Mya and has about 962 copies in the human genome [103]. HERV-K, with 10 groups in the clade, only one of which is HML-2, on the other hand, entered the genome of ancestral catarrhines about 32 to 44 Mya, after the split from New World monkeys and before the split of hominids from the Old-World monkeys [104]. The sister lineages of HERV-K in most other catarrhines appear to have become extinct. Most remarkably, the HERV-K HML-2 group in humans is the only HERV-K that has continued to replicate since the origin of the catarrhines [102]. HERV-K102 is a member of the bioactive HERV-K HML-2 group and as mentioned appears to be the only known replication competent member both in vitro and in vivo [10,67]. Accordingly, since phylogenetic evidence supports an association of HERV-K HML-2 activity with protection against integration of orthoretroviruses, this substantiates the notion that modern day HERV-K102 particles, along with expression of proteins/transcripts from other HML-2 elements, might help prevent HIV-1 acquisition and provide sterilizing immunity (Figure 5).
2.4.1. On the Curious Origins of HERV-K102 in Humans
Somewhat ironically, humans apparently acquired the HERV-K102 defense mechanism from the same source of the modern HIV-1 pandemic strain; namely, chimpanzees [105], possibly between 500,000 and up to 2 Mya [70,106]. The Homo-Pan split has been estimated at 6.6 Mya [104] or earlier at 7-8 Mya [106].
As mentioned, the HERV-K HML-2 elements originated in primates about 10.3 Mya and the CERV-K102 sequence (DQ112149), which is 97% identical to HERV-K102, was estimated to have integrated into chimpanzees at a non-orthologous position about 10 (+/- 3.3) Mya [105].
Lentiviruses like HIV-1 may have been active in primates since the divergence of chimpanzees and humans [107,108]. Moreover, it has been suggested the ancestor to HIV-1 may have arisen in chimpanzees about 4 Mya [109]. Since it has been reported that subsets of chimpanzees with chronic HIV-1 infection showed progression analogous to humans, including greater expression of CD38 in CD8+ HLA-DR+ T cells [110], this raises the notion that an HERV-K102 ancestor, as a potential antidote for HIV-1 infection may have been selected through evolution in chimpanzees before it was acquired by humans. The genus Homo arose about 2 Mya [111]. Accordingly, it is possible over about a 2-million-year window or longer, the HERV-K102 ancestor may have adapted to an HIV-1 like ancestor lentivirus(es) in chimpanzees prior to its acquisition by humans. Indeed, there is genetic evidence from a 5 amino acid deletion fixed in a human orthologue of tetherin, that humans may have been afflicted by a lentivirus presence long before HIV-1, perhaps about 800,000 years ago, which may have caused changes in innate immune genes in humans [112]. Thus, the phylogenetic evidence raises the notion that HERV-K102 as a replication competent HERV-K HML-2 foamy retrovirus, may have evolved specific mechanisms to limit HIV/lentivirus replication and genome invasion in chimpanzees before being acquired in humans. Indeed, a high level of segmental duplication related to telomere like repeats, particularly at 1q21 to 1q22 in the human genome (ie., HERV-K102 is located at 1q22) have been shown to be a causative factor in primate genome evolution [113]. This finding may further substantiate the role of HERV-K102 as being critical to hominin and Homo sapiens’ survival and thus evolution.
Neanderthals and Denisovans appear to have lost HERV-K102 at the orthologous chromosome position at 1q22 (Figure 8) and both went extinct. As mentioned, the Homo species which includes Neanderthals and Denisovans, arose about 2 million years ago [111]. The divergence of Neanderthal from anatomically modern humans (AHM) had been estimated at 550 to 760 thousand years before present (TYBP) but the mitochondrial DNA assessment suggests divergence only occurred around 400 TYBP [114]. Also, from sequencing of the Y chromosome, Denisovans split about 700 TYBP from AMH, and Neanderthal from AMH around 370 TYBP [115] implying first the divergence of Denisovans and then Neanderthals. That the Neanderthal genome contains a few more nucleotides surrounding the deleted HERV-K102 at 1q22 than the Denisovans (Figure 8) may or may not be consistent with Denisovans separating earlier from AMH while Neanderthal split later.
It should be noted that the earliest modern human remains in Europe so far discovered was about 45,000 years ago [116]. Thus, at the time of HERV-K102 acquisition the common ancestor would have resided in Africa, and this makes it plausible that HERV-K102 may have been acquired from chimpanzees known to live in Africa but not Europe. The finding by Compton et al. [112] referenced above implicates an exposure to chimpanzee lentiviruses around the same time, consistent with co-mingling of humans and chimpanzees at that time that HERV-K102 crossed over and into humans. Whether lentivirus played a role in the loss of HERV-K102 in the extinct hominins and/or in their extinction remains to be determined.
Nevertheless, RNA virus epidemics likely played some role in the demise of the extinct hominins as adaptively introgressed Neanderthal genes in humans frequently feature innate immunity genes against RNA viruses [117] or immune pathways against pathogens [118]. As well, innate immune reactions with pathogens affected human evolution between 0.6 and 2 Mya [111]. Consistent with a notion of a superior innate defense system of humans versus other interacting hominins was the finding of successful admixture between humans and Neanderthals but only when the female was human. This was deduced by showing mitochondrial DNA carried from the egg (ie., female) in humans contained no introgressed Neanderthal genes [119]. However, it should be appreciated that there may have been other reasons for this one-way admixture aside from innate immunity. Nevertheless, it is tempting to speculate that at least in part, the HERV-K102 protection system may have contributed to the survival of Homo sapiens over Neanderthals given the known selection pressure from RNA viral epidemics at the time of admixture.
In summary, the phylogenetic history suggests HERV-K102 may have first co-evolved in chimpanzees to protect against lentiviruses prior to being acquired by the Homo species. The remarkable observation that HERV-K102 replication was associated with sterilizing immunity to HIV-1 in the HIV-1 exposed seronegative sex trade workers (HESN) which manifested as protection against HIV-1 acquisition (Figure 5) [10] becomes much more plausible and thus, credible.
2.5. Evidence for a Role of HERV-K HML-2 Activation in Innate Immunity
The HERV-K HML-2 group was discovered by hybridizing the cloned region of the reverse transcriptase of the Syrian hamster intracisternal A particles against a human cDNA genomic library [120]. Not long after, Lower et al, described the detection of antibodies to HERV-K HML-2 envelope (Env) in 45 % of patients with testicular cancers, 26 % of patients with lymphomas, 70 % of HIV-1 patients, and 38 % of pregnant women but in only 3% of normal healthy blood donors [121]. Since the antibodies that were found in 60 % of patients with germ cell tumors disappeared upon resection [122], these early observations were first to imply HERV-K HML-2 activity likely contributed to innate immune surveillance against tumors and pathogens. As a potential follow-up to antibodies to HERV-K HML-2 Env associated with pregnancy, HERV-K particles were subsequently identified as isolated from human placenta [123]. By 2015, Grow et al, discovered HML-2 was reactivated in human preimplantation embryos and pluripotent cells (including particles) to protect against exogenous viral infections [124]. For example, they also demonstrated that the Rec protein of type 2 HML-2 group members was found to induce IFITM1 which guides exogenous viruses into the lysosome for their destruction [124].
As a side note, these findings of the activation of HERV-K HML-2 elements during early days of conception further substantiate the possibility that the HERV-K102 particles likely played a role generating successful progeny when females of the mating admixture between Neanderthals and AMH were of human origin [119].
Presumptive evidence for HERV-K102-like particle production like Figure 4, had been published prior to our discovery. Morgan and Bodsky in 2004 described these immature HERV-K particles which budded into vacuoles in megakaryocytes in patients with essential thrombocytopenia [125]. An unknown virus appeared to be budding from the cell surface of the megakaryocytes suggesting a viral infection may have led to the induction of HERV-K immature particles putatively in the Common Myeloid Progenitor (CMP) that macrophages and megakaryocytes share [12]. Presumably, the release of the particles from the megakaryocytes would have also been through cell apoptosis, and this lysis and loss of megakaryocytes presumably would have resulted in thrombocytopenia.
The electron microscopy images of the megakaryocytes [125] also revealed the cytoplasmic ‘leopard spots’ that congregated around the vacuoles as was shown for HERV-K102 replication in the foamy macrophages (Figure 4, blue arrow in the right bottom image). These aggregates represent the telltale signature of the pre-assembly of Env with the Gag protein outside of vacuoles which is characteristic of non-pathogenic spumaviruses [69]. The pathogenic orthoretroviruses do not require the presence of Env for particle production explaining their proclivity for pseudotyping and their lack of the formation of ‘leopard spots’. On the other hand, foamy retroviruses cannot be pseudotyped [69, 72.73].
Our group was first to identify and demonstrate that HERV-K102 particles were commonly and, in many instances, highly induced by viruses in vivo. The levels frequently reached 1012 particles per ml of plasma although not in HIV-1 infected patients [10,11,67]. It was only more recently that others have confirmed that HERV-K102 full length transcripts are strongly induced upon M1-like polarization of macrophages but not when M2-like polarizing protocols were used [99]. In HIV-1 patients there seemed to be direct antagonism with HERV-K102 replication where on average there may have been only 8,200-8,300 DNA containing particles per ml of plasma and about 70 % of HIV-1 patients scoring positive [11,67,68].
Further data implying a role of HERV-K activation in innate immunity host defenses was the finding of Morozov et al [126] that the transmembrane region of HERV-K HML-2 Env suppressed adaptive immunity reactivity such as the Concanavalin-A T cell proliferative response. Along similar lines we found that PHA and IL-2 added to the IMDM media inhibited foam cell formation in the cultured cord blood mononuclear cells of Figure 4 (unpublished data). Thus, innate immunity downregulates adaptive mechanisms and the converse is also true as would be expected.
Moreover, the HERV-K HML-2 envelope transmembrane region peptides strongly induced proteins related to M1-pro-inflammatory macrophages as assessed in peripheral blood mononuclear cells (PBMCs) [126]. These cytokines and chemokines included IL-1α/β, IL-6, IL-8, CCL2-5, PLAUR, G-CSF, TNFRSF1B and MMP1. Interestingly, the latter is a zinc dependent protease involved in the breakdown of the extracellular matrix, but which interestingly, also binds and inactivates the Tat protein of HIV-1. In addition, by microarray analysis of expressed genes, Morozov et al, demonstrated mRNA of various genes associated with M1-polarization were upregulated while those for M2-polarization [87] were downregulated [126]. An exception was that CXCL10, a chemokine of M1-polarized macrophages was found to be downregulated in the M1 macrophages. Interestingly in the list of down-regulated genes, HS3ST2 a heparan sulfate (glucosamine) 3-O-sulfotransferase 2 was also downregulated potentially implying a reduced entry of HERV-K102 particles in cells that might express HERV-K Env and/or HERV-K102 particles.
2.5.1. The ‘Virus-Antivirus Properties’ Associated with HERV-K HML-2 Activity
The full gamut of how the expression of HERV-K HML-2 RNA and proteins along with HERV-K102 particles may contribute to innate immunity and protect the host in an antigen non-specific manner has not yet been realized. We have only seen the tip of the iceberg of the novel ‘virus-antivirus responses’ driven by expression of HERV-K HML-2 proteins/transcripts.
The ones that have been identified in addition to those already mentioned include the following: i) the protease of HML-2 may cut exogenous viruses in the wrong places, reducing their infectivity [127], ii) HERV-K18 but not HERV-K102 Env may pseudotype the lentivirus HIV-1 reducing its ability to target and reduce certain cell populations such as CD4 T cells [128], iii) HERV-K HML-2 Gag interferes with HIV-1 Gag again reducing infectivity [129,130], and of primary clinical significance, while many pathogenic viruses including SARS-CoV-2 may block or delay the initial interferon response of the host [131,132], iv) HERV-K102 particles could upon infection of cells and release of their genomes and particle contents into the cytoplasm trigger innate PRRs including the RIG-1/MDA5/MAVS system, and/or the cGAS-STING response [133,134,135] to regain the protective interferon and other anti-viral responses through alternative pathways. This feature of how HERV-K HML-2 endogenous elements may trigger innate PRRs to amplify interferon and other anti-viral responses has been called “viral mimicry”[136].
Additionally, there is some evidence to suggest that HERV-K LTR elements may serve as enhancers for nearby immune response genes [136]. Moreover, it is not likely a coincidence that HERV-K LTR enhancers that bind the transcription factors STAT1 and IRF1 response elements which are induced by gamma interferon (IFN-γ) are located in the vicinity of interferon (type I) stimulated genes (ISGs) [99]. Thus, HERV-K elements may serve to convert the IFN-γ response to the type I innate immunity interferon response [99,136] which may be useful when an adaptive immunity response such as virus-specific antibodies might jeopardize the survival of the host by ADE.
The fact that HERV-K102 is replication competent and can quickly replicate itself reaching 2.55 x 1011 particles per ml of plasma from zero in about 84 hours [11], indicates there can be a massive early induction and release of HERV-K102 to ensure the interferon and other innate antiviral responses prevail. In this regard, there is already evidence that HERV-K102 genomic sequences amplify the cGAS-STING response in COVID-19 patients resulting in mild disease [137].
By examining scRNA sequencing data involving the activation of human macrophages with the TEcount and Telescope software packages Russ et al. [99], were able to determine that with M1 polarization in response to LPS (TLR4) and IFN-γ proinflammatory signaling, HERV-K102 activation comprised the majority of HML-2 transcripts in direct substantiation of our work [10,11,67]. Moreover, the transcription factors Stat 1 and IRF1 critical for HERV-K102 induction by IFN-γ bound to a region called “LTR12F” which resides just upstream of the 5’ LTR of HERV-K102. Genes subsequently induced by HERV-K102 expression in M1 macrophages included via cGAS: IRF1, IRF8, SOCS3, and ICAM1; via ISREs: MX1, ISG15, IFIT1-3, USP18, OAS1-3, OASL, and ISG20; and via cGAS and ISRE: STAT1/2, IRF9, IFITM1, BST2, TAP1, SOCS1, IFI35, HLA-G, ZC3HAV1, AIM2, and TRIM69 [99]. Many of these genes are interferon stimulated genes and confirms the finding of HERV-K102 in amplifying the critical type I interferon response in vivo [136]. Furthermore, in a humanized mouse model of mild COVID-19 disease it was reported that macrophages somehow were able to amplify the interferon response critical to recovery [138].
Russ et al [99] also reported that VDR response elements and the glucocorticoid receptor response elements along with response elements for NFKB1 and IRF-1 are contained within the 5’ LTR of HERV-K102. Others have previously reported the response elements for these and other inflammatory transcription factors in the 5’ LTR of HERV-K102 [97,98].
2.6. The Concept of Innate T and B Cell Responses Against HERV-K102 Envelope
The team of F. Wang-Johanning and G.L. Johanning started their legacy of investigating the expression of HERV-K102 transcripts and proteins in common cancers with reports starting in 2001 [139] in breast cancers and lasting up until 2017. In their seminal report which used northern blots, full length sequences referred to as proviral transcripts and spliced env transcripts were detected in the cancer samples and cell lines. Sequencing of these transcripts revealed type 1 HERV-K102 was strongly induced. Two years later using RT-PCR they published that both type 1 and type 2 HERV-K HML-2 env transcripts were detected in breast cancers and were induced in breast cancer cell lines when treated with estrogen and progesterone [140]. That the 5’ LTR contains response elements for these hormones was known at the time of initial sequencing of HERV-K HML-2 [120].
In 2008 this team published a comprehensive examination of the host immune response to the expression of HERV-K102 Env. About 88% of breast cancer tumors had detectable HERV-K102 Env by immunohistology which was not detected in normal breast tissue [141]. As well about 79% of the breast cancer patients and 5 % of the normal healthy controls displayed IgG antibody to HERV-K102 Env. Furthermore, they investigated T cell responses to HERV-K102 Env surface unit antigens and found T cells proliferated and produced interferon gamma. During in vitro stimulation of PBMCs from breast cancer patients, an M1 like macrophage and a T helper 1 cytokine response were generated. T cell cytotoxic lymphocytes (CTLs) that lysed targets expressing HERV-K protein were found in breast cancer patients but not normal healthy controls [141]. This was the first time that the immunogenicity of HERV-K102 antigens had been investigated for T cell responses.
A most remarkable finding was subsequently reported by Wang-Johanning et al in 2012 [142]. In this report they showed in vitro, a single-chain variable fragment (sc-Fv) monoclonal antibody (MAb that lacked the FC domain) which reacted with HERV-K102 Env surface unit was able to directly trigger apoptosis in the breast cancer cells without the need for complement or accessory cells such as those involved in antibody mediated cytotoxicity. They confirmed that in breast cancer cells, HERV-K102 Env was directly wired to the host cell apoptosis machinery involving CIDEA, TP53, and caspase 3 and caspase 7 pathways [142].
On the heels of the Wang-Johanning et al success, interest in addressing T cell and B cell responses turned to HIV-1 patients. A T cell clone from an HIV-1 elite suppressor (an HIV-1 infected person able to control HIV-1 replication in vivo and maintain near zero levels of HIV-1 in serum without pharmaceutical intervention) recognized a peptide in HML-2 Env that was 100% identical in sequence to HERV-K102. This T cell clone was able to clear human cells infected with various monkey and human orthoretroviruses [143] showing innate T cells provide heterologous (antigen non-specific) protection against cells infected with various viruses. However, this group was not able to identify a linear peptide sequence of HERV-K102 Env surface unit where antibodies from sera from HIV-1 patients were strongly or more commonly reactive over healthy controls [144]. They did discover antibodies to the transmembrane region of HERV-K102 Env albeit of unknown significance.
In our hands we were able to determine that HIV-1 patients had significantly higher levels of antibodies to HERV-K102 surface unit peptides ML-4 and ML-5 than those with other viral infections and a higher percentage of positive reactivity (80% and 70% of the HIV-1 patients were positive for their reactivity, respectively) [67]. This was similar to that reported in breast cancers [141]. While only 1/51 healthy normal controls (2%) were judged to be positive by peptide ELISA, these were very weak and only marginally positive reactions.
We had developed a very sensitive real time quantitative PCR HERV-K102 pol ddCt method performed on DNA isolated from plasma that contained an internal probe in pol to validate a true pol amplified fragment signal [67]. Moreover, unlike traditional PCRs because we measured the relative increase in plasma of a HERV-K102 pol signal over genomic equivalents detected with an 18S RNA probe this enabled a built-in validation that the isolation of DNA and PCR methods were working. With this method which was compared with a mean ddCt ratio of 0.88 +/-0.37 for 30 normal healthy controls, it was determined that 28/37 (75.7%) of HIV-1 patients were positive for HERV-K102 particles. However, 96 % of the HIV-1 patients had a positive PCR and/or a positive serological test meaning HERV-K102 activation by these criteria was almost universal [67].
2.6.1. The Concept of SELECT Epitopes Reactive to Innate Antibodies to HERV-K102 Env
The ML-4 and ML-5 peptides had been preselected for their immunogenicity, selectivity for HERV-K102 sequences and their likelihood of being cryptic on the HERV-K102 particles [67]. We were concerned that antibodies to the cell surface HERV-K102 protein (P61567 Env) expressed on the surface of tumor cells or virally infected cells which represents the product of a splice variant [139,140], should not react with HERV-K102 particles. Otherwise, these protector antibodies would be self-defeating.
The particle associated type 1 Env encoded in the proviral transcript (P63135 Pol-Env) contains KRASTE which is absent in the type 2 Env. The ML4 sequence (KRASTEMVTPVTWMDN)[67] included the additional KRASTE leader sequence common to type 1 but not type 2 envelopes. It was possible that the addition of KRASTE could alter the conformation of the ML4 Env peptide on particles for example rendering the MVTPVTWMDN cryptic. The ML5 peptide (LETRDCKPFYTIDLNSS) [67] has a cysteine amino acid in the middle of the peptide sequence meaning depending on the conformation, the antibody binding site might be obscured by a di-sulphide bridge. It also contains an N-glycosylation site (i.e., the NxS/T motif) which could also help render the ML5 sequence cryptic such as on particles. According to Alpha-Fold software [145,146] the conformation of P61567 is quite distinct from P63135 (Figure 9).
As a side note, the fact that HERV-K102 uniquely has both the spliced Env protein sequence and a Pol-Env protein sequence associated with particles, provides further evidence substantiating that only HERV-K102 forms particles and could be replication competent.
We were not able to detect reactions of our affinity purified rabbit ML4 or ML5 antiserum with the foamy macrophages in Figure 4 by flow cytometry even when the cells were permeabilized (unpublished data). However, these antisera labelled proteins by western blotting and stained intracellular particles by immunohistology on paraffin embedded samples of the foamy macrophages [10]. Thus, positive reactions involving HERV-K102 particles were only obtained when the particle proteins had been denatured. This suggested that the ML4 and ML5 epitopes were likely cryptic on the HERV-K102 particles as had been anticipated.
As shown in Figure 10, viruses that bud from the cell surface of human cells, the so-called enveloped viruses like HIV-1 and SARS-CoV-2 (that cloak themselves in the plasma membranes of cells), pick up HERV-K102 Env in the process meaning their virions carry HERV-K102 Env. Thus, antibodies to HERV-K102 Env are most probably neutralizing antibodies that can clear and inactivate exogenous virions as well as lyse virally infected cells. One of the earliest investigations of the SARS-CoV-2 pandemic had established in mild disease that innate immunity neutralized and cleared SARS-CoV-2 from the URT prior to the onset of the spike-specific antibodies [41]. Therefore, the innate immunity SARS-CoV-2 neutralizing antibodies likely involve at least in part, antibodies to HERV-K102 Env.
Accordingly, it is tempting to speculate that monoclonal (sc-Fv) antibodies reactive with epitopes found on the P61567 HERV-K102 cell surface Env but not the particle associated P63135 pol-env, such as the ML4 and ML5 peptide sequences, could be used as novel treatments for viral infections especially against emerging and pandemic enveloped viruses. Although many of the monoclonal antibodies against spike protein may have selected for new variants causing progression of disease and/or lost activity with the onset of immune escape variants [42], there is little risk of this here as it is not the virus which encodes HERV-K102 Env and thus, there would be no viral selection. Moreover, these antibodies may also be useful for cancer patients especially those more recently diagnosed with “turbo cancer”. Turbo cancers is a colloquial term used to describe cancers arising in individuals who have been injected with the mRNA gene therapy vaccines and where progression is rapid and/or onset is in a much younger population [148].
The concept that the SARS-CoV-2 virions produced in human cells may carry HERV-K102 Env is quite intriguing. Arru et al [149] described peptides corresponding to HERV-K102 Env surface unit peptide sequences that were able to induce TNF-α in T cells (the 19-37 peptide) that is, without the requirement for MHC. Also, the 19-37 and the 109-126 peptides of HERV-K102 induced IL-6, IFN-γ, CCL2 and CCL3 in B cells. This suggests innate T cells may be activated by HERV-K102 Env protein along with presumably innate B cells. Innate T cells include NKT cells, the mucosal associated invariant T (MAIT) cells and the γδ T cells [150].
Recall that Morozov et al [126] showed that the transmembrane region of HERV-K HML-2 Env strongly induced M1-activation cytokines and chemokines but not those for M2 when tested on PBMCs. When taken together these results imply that wherever the ‘enveloped’ exogenous virus goes, because it carries HERV-K102 Env, it will activate the HERV-K102 protector system (M1-like proinflammatory foamy macrophages and the innate T and B cells) providing yet another viral anti-virus mechanism. This virus-anti-virus mechanism reveals nature to be quite ingenious with redundancy for innate immune activation relating to viral invasion.
While HERV-K HML-2 Env when expressed on tumor cells or virally infected cells appears to be an autoantigen that marks the cell for destruction, interestingly in neurons, the oppositive may be the case.
Bhat et al suggested that HERV-K HML-2 Env increased neuronal cellular viability and prevented neurotoxicity mediated by HIV-1 Vpr [151]. In contrast in amyotrophic lateral sclerosis (ALS) Garcia-Montojo et al reported that HML-2 Env protein expression in neurons was neurotoxic, but that antibodies to Env protected against neurodegeneration. Furthermore, these antibody levels correlated with survival times of patients with ALS [152]. Interestingly, TDP-43 (gene card TARDBP) accumulation, which is associated with neuropathology in ALS, also induced HERV-K102 Env expression [153]. Clearly more work is needed to resolve these discrepancies as HERV-K102 Env and/or the IgG are protective in breast and other cancers [154].
2.7. Epigenetic Control of HERV-K102 Expression and Trained Immunity
Systemic lupus erythematosus (SLE) is an autoimmune disease involving autoreactive T and B cells, immune complex disease, a systematic activation of type I interferon, and neutrophil activation with the formation of neutrophil extracellular traps (NETs). The strong induction of HERV-K102 Env RNA in whole blood (with minor contributions by HERV-K18, HERV-K106, and HERV-K115) was found in female SLE patients but not male [155]. HML-2 RNA expression correlated with a lack of epigenetic silencing and where the antibody (largely IgG2) to HERV-K102 correlated with higher interferon I ISG expression [155]. HERV-K HML-2 regulation is through epigenetic control whereby DNA methylation and repressive histone methylation suppress ERV expression. The repressive marks are maintained by the kruppel-associated box domain-zinc finger protein (KRAB-ZFP)-TRIM28 complex and human silencing complex (HUSH). Accordingly, HERV-K102 expression correlated inversely with TRIM28 expression [155].
The epigenetic control of HERV-K102 expression is a segue into a discussion on trained (innate) immunity.
The term trained immunity (TI) was first coined by Professor Mihai Netea and colleagues in 2011 to describe the enhancement of a secondary innate immunity response after a primary infection or vaccination [156]. Unlike adaptive immunity, TI lacks specificity for any pathogen-specific antigens and thus, invariably involves heterologous protection or cross-protection against unrelated pathogens. TI was initially invoked to explain how vaccination with the live Bacillus Calmette–Guérin (BCG) vaccine (attenuated Mycobacterium bovis) in West Africa decreased childhood mortality from several different pathogens including tuberculosis. The protection furnished by the BCG vaccine was mediated by macrophages. Since in humans, macrophages cannot replicate (unlike mice), this has led to the concept of central TI in the hematopoietic stem and progenitor cells (HSPCs) [157,158,159]. Accordingly, TI involves peripheral (monocytes-macrophages) and central (myeloid HSPCs) compartments. Most notably the memory aspect of TI involves a metabolic switch to glycolysis and epigenetic changes. These involve changes in the histone methylation and acetylation in chromatin providing access to macrophage lineage and inflammatory genes [159].
TI refers to a short-term enhancement (usually 3 to 12 months) of the release of cytokines (TNF-α, IL-1β, IL-6) from M1-like macrophages upon rechallenge but which is also associated with enhanced heterologous anti-microbial and anti-tumor activity in vivo. How TI in proinflammatory macrophages relates to enhanced pathogen and tumor control in vivo remains to be fully elucidated, however. As well, recent evidence indicates TI in macrophages occurs with or without the induction of inflammatory cytokines [160,161] presenting somewhat of a conundrum for the functional definition of TI.
Commonly used inducers of trained immunity include microbial products such as beta glucan and muramyl dipeptide, but also oxLDL, uric acid, the BCG vaccine, other live vaccines, and/or exposures to viruses [159]. The differences in epigenetic marks by the different TI inducers are reviewed elsewhere [162]. The myeloid specific enhancers SPI1 and CEBPB [88] are in fact trained immunity specific enhancers [89,90].
The metabolic and epigenetic changes associated with TI are mediated via the Akt/mTOR/HIF-1a pathway [89,163] but where insulin resistance promotes an anti-inflammatory M2-like phenotype [164,165]. Glycolysis drives the PI3K/Akt/mTOR/HIF-1α pathway [169]. In M1 macrophages activation leads to the accumulation of succinate in the Krebs cycle which leads to the stabilization of the transcription factor (TF) HIF-1α which induces the transcription of glycolysis genes [163]. In contrast M2 macrophages primarily use oxidative phosphorylation.
TI associates with the induction of glycolysis which is similar to the Warburg effect described for tumors. At the risk of being an oversimplification, a reason why glycolysis is needed for tumorigenesis (the Warburg effect) is so the mitochondria can produce the substrate acetyl-CoA/citrate (from glycolysis) needed for cholesterol production through the mevalonate pathway [166]. Replenishing of the cell surface membrane and other membranes in the cell via higher cholesterol production would be needed in order to support tumor proliferation. Indeed, mevalonate initiates DNA synthesis and cell proliferation [166]. In human monocyte derived macrophages, which incidentally do not proliferate, excess cholesterol would be needed and utilized instead for foam cell formation pertaining to the replication of the protector foamy virus HERV-K102. Hence, glycolysis is linked to TI to support the generation of foam (HERV-K102 particles and vacuoles) during macrophage training.
TI involves foam cell formation in M1 macrophages [93]. Most notably the mevalonate (cholesterol) pathway is needed for TI in the monocyte/macrophage lineage as statins which inhibit HMG-CoA reductase (HMGCR) block TI induction [94]. Trained macrophages uptake lipids such as oxLDL through OLR1 to form foam cells and produce high levels of TNF-α, IL-6, IL-8, and IL-18 upon secondary challenge [167] associated with glycolysis [96,168]. Interestingly, SARS-COV-2 infection disrupts the mevalonate pathway [169] showing that it directly targets foam cell formation and thus, trained immunity and thus, HERV-K102 particle production.
Hypoxia inducible factor 1 alpha (HIF-1α) plays a key role in initiating and promoting the formation of foam cells in macrophages [170]. NF-κβ1 which induces the inflammatory response in macrophages is required for the transcription of HIF-1α [171]. A critical role of HIF-1α in foam cell formation in macrophages was demonstrated by the inhibition of foam cell formation by small interfering RNAs against HIF-1α [172]. Thus, it is very clear TI critically involves the induction of foam in M1-like proinflammatory macrophages related to NF-κβ1 and HIF-1α expression.
The PI3K/Akt/mTOR pathway plays a role in autophagy, apoptosis, metabolism and cell growth but is commonly hyperactivated in tumors where it contributes to malignant potential [173]. It is also hyperactivated upon SARS-CoV-2 infection such as in the hepatocellular cell line Huh7 associated with ERBB2 hyperactivity and alpha-fetoprotein (AFP) mRNA and protein expression [174 and Prof. Ujjwal Neogi, personal communication]. In macrophages, this pathway is used by the epidermal growth factor receptor (EGFR) to generate foam cell formation [175,176]. For example, gene deletion of EGFR in macrophages in murine models limits the production of IL-6 and TNF-α, reduces lipid uptake by reducing the expression of the scavenger receptor CD36 and inhibits the development of atherosclerosis which involves foamy macrophages [177]. Similarly for human macrophages, the EGFR is activated by TLR4 and disruption of TLR4 or EGFR reduced inflammation and foam cell formation [175,176,177]. Triggering TLR4 activates HIF-1α, IRF1, VDR, S100A9 and NR3C1 (the glucocorticoid receptor) while downregulating PPARG and IFNGR1 [178]. EGFR antagonists were also shown to block oxLDL induction of inflammation and foam cell formation with down-regulation of IL-6 and TNF-α [177].
It should be noted that while the PI3K/Akt/mTOR pathway induces foam cell formation associated with basigin (BSG/CD147) expression in macrophages such as induced by oxLDL, interfering with NFκβ1 does not block foam cell formation [91]. This means the inflammatory component such as by NFκβ1 is not an absolute requirement for TI as has been recently noted by others [160,161]. Perhaps foam cell formation involving HERV-K102 particle production in foamy macrophages would better define and capture the essence of TI.
Recent evidence shows that upon LPS and IFN-γ triggering of the M1 phenotype, AFP transfection into MO macrophages co-induces CD163 and IL-10 which are considered markers of M2 [179]. However, CD163, IL-10 and the MI/M2 phenotype characterize the foamy macrophages illustrated in Figure 4 which exhibit high levels of vacuoles and particles [86]. This may further substantiate the role of AFP in the training of macrophages associated with foam cell formation.
As mentioned, drivers of myeloid differentiation are the TFs SPI1 and CEBPB [88] which also drive TI [89,90]. SpI1 is considered a pioneering transcription factor (also called lineage determining transcription factors) that opens up the chromatin in this case for macrophage differentiation which enables inflammation TFs to bind to response elements in the appropriate genes [180]. Macrophage inflammation involves networks of signal transducer and activator of transcription (STAT) factors, interferon response factors (IRFs) and NF-κβ TFs [180]. Monocytes exposed to LPS and interferon gamma (IFN-γ) undergo classical M1-like (pro-inflammatory) macrophage activation with upregulation of SPI1, IRF1, IRF5, IRF8, STAT1, STAT2 and NF-κβ1. The alternative activation with IL4 and IL13 generates M2-like (anti-inflammatory) cells featuring SPI1, IRF4, STAT6, KDM6B, PPARγ, PPARδ, and CEBPB [180]. However, PPARγ plays an important role in the differentiation of monocyte to foamy macrophages [181] and CEBPB is a TI enhancer. Accordingly, the macrophages conferring TI express M1 and M2-like markers including CEBPB and PPARG which are associated with foam cell formation. As just mentioned, AFP expression may also contribute to M2 marker expression in the inflammatory M1-like macrophages.
It is noteworthy that IRF1 and NF-κβ1 bind to two interferon stimulated response elements (ISREs) in the promoter of HERV-K102 which is found in the 5’ long terminal repeat (LTR) [97,98]. TNF-α is a potent activator of canonical NF-κβ1 transcription factor activity while IFN-γ and TNF-α synergize to activate IRF1. Indeed, newer evidence confirms that M1 polarization of macrophages in humans explicitly in response to IFN-γ signalling is promoted via HERV-K102 expression [99]. More recently the key role of IFN-γ in TI induction in vivo in response to BCG vaccination was affirmed by scRNA sequencing [167].
In summary, TI critically involves EGFR/TLR4 induced foam cell formation in M1-like proinflammatory macrophages. LPS which triggers TLR4 along with IFN-γ induces M1-like proinflammatory foamy macrophages which is strongly associated with HERV-K102 proviral induction [99]. The evidence is incontrovertible that it is HERV-K102 replication in macrophages which generates trained (innate) immunity.
Figure 11 provides an illustration of the main cellular players involved in the HERV-K102 protector system and their targets. Thus, it is probable that TI heterologous protection extends to the innate T and B cells that recognize HERV-K102 Env. Indeed, Ren et al indicated that T and B cells were also infected by SARS-CoV-2 associated with COVID-19 severity and that BSG, TFRC (transferrin receptor) and interferon stimulated genes (ISGs) correlated with SARS-CoV-2 viral RNA in the different cell types [61]. BSG is an alternative or secondary receptor for SARS-CoV-2 entry into cells [182] and antibodies to BSG (CD147) block SARS-CoV-2 infection in vitro [182,183] and in vivo in BSG humanized mice [183]. The notion that SARS-CoV-2 specifically interacts with BSG was corroborated when the amino acids involved in mutual binding were mapped [184]. Curiously, in the PBMCs, CEBPB and SPI1 (trained immunity enhancers thought to be specific to M1-like foamy macrophages ) were also expressed in virus positive T cells and B cells [61]. This might imply that the innate T and B cells which express BSG may share some of the trained immunity programming of foamy macrophages.
In summary, HERV-K HML-2 activation and HERV-K102 particle production in foamy macrophages constitute a ‘virus-anti-virus response’ analogous to fighting fire with fire. Not only does the ‘virus mimicry’ aspect help to amplify the interferon response through PRRs but innate T cells and antibodies that recognize HERV-K102 Env expressed on the surface of virus-infected cells are generated which kill virus infected cells. Indeed, in a twisted turn of events it seems HERV-K102 Env expressed on the SARS-CoV-2 virions may alternatively stimulate M1-macrophages as well as innate T cell and B cell activation. Most significantly, the innate antibodies to HERV-K102 Env are postulated to neutralize and clear SARS-CoV-2 in mild cases as reported by Wolfel et al [41]. This feature helps to explain how boosting of innate immunity in the first 6 days following the second dose (before the adaptive IgG antibodies to spike protein are made) can be associated with sterilizing immunity (100 % VE) [54] which is probably related to a recall or memory release of the antibody to HERV-K102 Env.
2.8. When Things Go Wrong with the HERV-K102 Protector System: Immunosenescence
It is equally important to discuss what happens when the HERV-K102 system fails and under what circumstances.
Clinical research concerning the study of putative HERV-K102/foamy macrophage immune parameters based on plasma amino acid profiling algorithms performed at Immune System Management led to the publication of the new immunosenescence paradigm (of macrophages) in 2015 [185]. The new paradigm was then separately validated for cardiovascular disease [186].
In brief, the new immunosenescence paradigm (of macrophages) was simply defined as the failed lytic release of HERV-K102 particles from the foamy macrophages (Figure 12) which was causally related to alpha-fetoprotein (AFP) activity. The 67 kD AFP receptor which mediates the effects of AFP in macrophages was identified and characterized by 1991 [187]. In addition, active AFP or the AFP agonist monoclonal antibodies to the 67 kD AFPr blocked apoptosis in human macrophages [187,188]. Since dehydroepiandrosterone (DHEA) binds and inactivates AFP [185] and it was established that cortisol induces AFP expression in humans [189], meant that as DHEA levels declined with age (faster in males) the cortisol/DHEA ratio would increase placing the host at higher risk of immunosenescence of macrophages and thus, disease. Most notably, high cortisol and low DHEA are commonly associated with chronic disease initiation and progression. For example, after adjustment for age, the cortisol/DHEAS ratio correlated with all-cause, cancer and non-cancer mortality as shown in a prospective study of 4255 Vietnam army veterans over 15 years and where higher cortisol/DHEA ratios were associated with increased risk of death [190].
As shown in Figure 12 cortisol is proposed to also induce HERV-K102 expression to generate the foamy macrophages as its LTR contains glucocorticoid response elements [97,98,99]. Oddly, it appears high dose dexamethasone may not induce HERV-K HML-2 such as tested in a breast cancer cell line [192]. However, whether methylprednisolone would induce these protector transcripts should be investigated since dexamethasone failed to reverse the SARS-CoV-2 interference of the IFN type 1 and IFN-γ innate responses while methylprednisolone did [193,194]. Moreover, methylprednisolone was associated with a much lower number needed to treat (nnt=5) indicating higher proficiency. Alternatively, macrophage migration inhibitory factor (MIF) which is known to counteract the anti-inflammatory effects of glucocorticosteroids, and which was inducible at low levels of dexamethasone but suppressed at higher levels [195], potentially might additionally influence HERV-K102 proviral expression in macrophages secondary to corticosteroids. However, the differences between dexamethasone and methylprednisolone activation of HERV-K102 particle production requires further evaluation.
Critically, when AFP binds its receptor, it triggers a negative signal which abrogates any incoming signal whether it is for apoptosis, differentiation, proliferation, activation, adherence etc. [187]. AFP is well established to be intrinsically immunosuppressive, but this was corroborated with AFP agonistic monoclonal antibodies to the 67 Kd AFP receptor [187]. By the time the 67 kD AFP receptor becomes expressed, the macrophages are already expressing and releasing the pro-inflammatory cytokines, TNF-α, IL-1β and IL-6. Thus, AFP prevents the ability to downregulate the expression of pro-inflammatory cytokines. Immunosenescence involves immunosuppression, a simultaneous pro-inflammatory state and apoptosis resistance, all mediated by AFP binding to its receptor [187,188]. Accordingly, the use of anti-inflammatories which are immunosuppressive, would only contribute to the problem of immunosenescence of macrophages and would not address the cause of disease: namely, AFP activity. Only AFP antagonists such as zinc, DHEA (or better, 7-keto-DHEA which cannot be converted to sex hormones), genistein, and more recently ivermectin [191], are able to prevent and reverse immunosenescence. It is well appreciated that therapies can be curative if they target the cause of disease such as immunosenescence by AFP antagonists rather than just treating symptoms of disease such as inflammation with anti-inflammatories (which are immunosuppressive).
3. Effects of ADE on the Launch of the Critical HERV-K102 Protector System in M1-like Foamy Macrophages
Figure 13 attempts to illustrate the harm of ADE mediated entry of SARS-CoV-2 into the M1-like foamy macrophages that produce the HERV-K102 particles [10,67,99]. First, the entry of SARS-CoV-2 into the monocytes/macrophages causes them to convert to M2-like polarized macrophages with lipid bodies [196] and would no longer express HERV-K102 transcripts [99]. Second, the S2 component of the spike protein interferes with p53 and BRCA which blocks apoptosis [197]. In this state of apoptosis resistance, SARS-CoV-2 is able to replicate and release virions by budding through the plasma membrane [196]. Apoptosis resistance means any HERV-K102 particles made are not being released by lysis, and so SARS-CoV-2 entry into M1-like macrophages directly and indirectly causes immunosenescence. Also p53 down modulates AFP [198] so the loss of p53 function would mean more AFP would be expressed again contributing to immunosenescence.
However, in the presence of adequate vitamin D3, the conversion of the M1-like lipid body negative foamy macrophages to the M2-like lipid body positive foamy macrophages [196] would not occur [165]. Second, as demonstrated in a Mycobacterium tuberculosis model involving macrophages, the addition of vitamin D3 upregulated the genes known to mediate M1-like polarization and HERV-K102 induction, including CD14, IL-8, IRG1 and CD163 expression, as well as foam cell formation [200]. While the mRNA for VDR was unaltered, the addition of Vitamin D3 nevertheless strongly induced VDR protein [200]. Thus, due to the presence of VDR response elements in the 5’ LRT, HERV-K102 expression and M1-like polarization [99] are likely maintained by vitamin D3.
It should be noted that Ren et al [61] established that of the 63 identified different cell types in bronchioalveolar lavage fluid (BALF) specimens, only the CD14+CD16+ monocytes and their progeny, the WDR74 positive macrophages (putatively producing HERV-K102 particles based on differentially expressed transcription factors) had highly activated VDR. In BALF, the WDR74 macrophages which were not induced in healthy uninfected controls were critical to recovery from mild-moderate COVID-19 but were lost with progression to severe disease consistent with ADE. The progenitors of the WDR74 macrophages the CD14+CD16+ (intermediate type) monocytes that are present in the BALF of healthy controls were similarly induced in progression to and recovery from moderate disease. Although they were depleted with progression to severe disease, they were highly activated in those who recovered. Autopsies of patients who died from COVID-19 revealed the inflammatory monocytes in lung samples expressed macrophage-associated markers such as CD68 and CD163 [66] suggestive of emergency myelopoiesis. Thus, while the WDR74 macrophages per se did not promote recovery from severe COVID-19 their temporary expedited replacements (inflammatory monocytes) did. Interestingly, in the BALF, the γδ2 T cells, which were present in healthy uninfected controls and expanded with progression and recovery from moderate disease, remained depleted during progression to severe disease and during recovery [61].
In view of the above it is not surprising that optimal levels of vitamin D3 of at least 50 ng/ml of serum could prevent the onset of COVID-19 severity, as recently concluded from a systematic review and meta-analysis [201]. In addition to the prevention of COVID-19 mortality with >50 ng/ml vitamin D3 in plasma, SARS-CoV-2 infection could be prevented, and hypertension averted [202]. The latter implies vitamin D3 prevents or reverses immunosenescence raising the possibility that vitamin D3 might indirectly antagonize AFP activity. Moreover, the risk of death from myocardial infarction, cancer, type 2 diabetes, Alzheimer’s disease, and more generally, all-cause mortality could be significantly decreased with vitamin D levels above 50 ng/ml [202]. These findings corroborate the notion that the HERV-K102 protective innate system that launches in M1-like foamy macrophages [99] which is strongly favored by optimal vitamin D3 levels plays a major role in human survival at the individual and population levels, including perhaps, phylogenetically (Figure 8).
4. Incontrovertible Evidence for ADE As an Impediment to the Safety and Effectiveness of the Adaptive Immunity COVID-19 Vaccines
In Table 2 the UK monthly mortality rate data released by the Office for National Statistics (ONS) on July 6, 2022 [203], has been recompiled into summaries of ratios of vaccinated mortality rates per 100,000 person-years over the same for the unvaccinated by month for all ages (14 plus), and categorized by all-cause, COVID-19 specific and non-COVID-19 mortality ratios covering January 1, 2021, to May 31, 2022. For this, the individual rates per 100,000 person years for each subcategory of vaccination and duration were separately added up to provide a total as the total for the ever vaccinated provided by the ONS appeared to be erroneous. Apparently, deaths before 2 weeks following an immunization dose may have been reclassified as unvaccinated as articulated by Fenton et al [204] and which ONS confirmed on January 20, 2023 [205].
Upon examination of Table 2, first one notes that the only month in which the all-cause mortality showed vaccination benefit over risk was in February 2021 (ratio of ever vaccinated rates over unvaccinated of 0.61) but not in any other month from January 1, 2021, to May 31, 2022. Overall, the all-cause mortality test for risk versus benefit for the entire period was 6.37 with a p value of 0.0001 meaning that there were over six-fold higher all-cause deaths per 100,000 person years in the vaccinated over the unvaccinated which was highly significant. This also matched the results of the randomized clinical trials where none of the COVID-19 vaccines met the requirement for a statistically significant benefit over risk as demonstrated on all-cause mortality [206]. So overall the risks of COVID-19 vaccination did outweigh the benefits for the entire UK population measured over January 1, 2021 to May 31, 2022 with the exception of February 2021.
In February 2021 but not at any other time in the mass vaccination campaign in the UK, the benefit of vaccination surprisingly outweighed the risks. However, upon closer examination the reason for the benefit over risk in February 2021 was because of those vaccinated by the end of February 2021, 95.6 % had only received the first dose of vaccine but not yet the second (only 531,525 had received the second dose or about 4.4 % of the 11,976,296 vaccinated mostly elders) [207]. Recall that the first dose did not generate spike-specific IgG antibodies [46,47] but was known to generate heterologous protection against non-COVID-19 mortality [50]. In other words, it was likely the trained (innate) immunity that offered protection against COVID-19, non-COVID-19 and all-cause mortality in February 2021 in England.
As a separate issue, the reason why boosters were needed every 3-6 months was likely because trained immunity is temporary usually lasting 3 to 12 months [159]. As well lingering spike protein from the mRNA vaccine may have abrogated trained immunity potentially also via ADE or possibly by binding to TLRs. For example, a recombinant vaccine spike fragment (which lacks a trypsin cleavage site) was found in 50 % of post-vaccination plasma samples at 69 days and with a maximum of 187 days [208]. As well, spike protein (non RBD) may trigger the TLR4/MyD88 activation pathway in macrophages leading to abnormal inflammation [209] potentially related to immunosenescence. In any case internalized spike protein through S2 interference with p53 [197], would have created immunosenescence (Figure 12) abrogating trained innate immunity. Therefore, it is possible that critical protection against all-cause mortality by trained innate immunity may have been lost by persistent spike protein expression related to the use of mRNA gene therapy vaccines. Clearly this possibility needs to be investigated further.
Indeed, since in January 2021 the all-cause mortality test was 1.39, it could be reasonably argued that the COVID-19 vaccines should have been removed from the global market by the first week in February 2021 for which Dr. Peter McCullough concurred based on the data in Table 2 [210]. By a year later in February 2022, the all-cause mortality risk versus benefit ratio for the COVID-19 vaccines grew to a ratio of 11.04. This indicated with time the risk of mortality associated with the COVID-19 vaccines was becoming far worse rather than better. Thus, it is very clear from this comprehensive population data of England that the vaccines were not very good at saving lives from COVID-19, except for February 2021. The fact that the non-COVID-19 mortality rates in the vaccinated were exceptionally higher than the unvaccinated (Table 2) provides irrefutable proof that the Pfizer-BioNTech COVID-19 mRNA vaccine was toxic and deadly [211]. There were high proportions of sudden death following vaccination linked to myocarditis as determined through autopsies for the mRNA COVID-19 vaccines [212,213], and abnormal clotting [211]. Other longer term issues of concern stemming from the COVID-19 mRNA vaccines are the associated ‘turbo cancers’[148].
It is doubtful that the spike-specific IgG antibodies could have helped anyone survive because a priori, they caused progression of disease [16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,34,35,36,37,38]. The UK population mortality results in Table 2 strongly corroborate and endorse this point that ADE was and remains a serious problem for disease associated with COVID-19 vaccines. This mortality data further corroborates the evidence cited earlier about immune escape variant selection and a higher risk of infection pertaining to ADE. Despite the notion that the third mRNA vaccine dose was associated with conversion of the spike-specific IgG1/3 which promoted ADE, to the spike-specific IgG4 which does not mediate ADE [58], it is possible that: i) this conversion did not occur in the URT and, ii) the lipid nanoparticles may have inadvertently targeted myeloid cells due to phagocytosis regardless.
5. Future Directions
The notion of developing innate immunity vaccines particularly for pandemic preparedness is not new given the general lack of success for adaptive immunity vaccines related to the problem of ADE. Based on BCG vaccines which enhance trained immunity, others are trying to further optimize trained immunity vaccines such as IMM-101 which involves heat killed Mycobacterium obuense. This vaccine induces M1 polarization of macrophages [214] and thus would induce HERV-K102 particle production [99]; and may promote the Vγ9Vδ2 T cells which help recover from SARS-CoV-2 [214] as well as cancers [3]. Indeed, in addition to identifying M1-like foamy macrophages in recovery from COVID-19 [61], several papers have directly confirmed the role of HERV-K102 proviral expression or Env antibodies in recovery from COVID-19 [137,147,215,216]. In addition, HERV-K HML-2 elements played a role in recovery from COVID-19 in children [217] and others have provided indirect evidence corroborating HERV-K102 activation in recovery [218,219].
In terms of pandemic planning, the safest, most practical, and most effective means to prevent severe disease from pandemic viruses (and intracellular pathogens like Mycobacterium tuberculosis) is to top up the vitamin D3 levels by supplements and when possible, by suntanning. In addition, the benefits of 15-minute daily exposures to near infrared rays for 7 days on COVID-19 recovery included a faster: i) reduction in the systolic and diastolic blood pressure, ii) reduction in neutrophils and leukocytosis, and iii) increased lymphocytes [220]. Note that the earlier normalization of hypertension by near infrared light exposures indicates a faster reversal of the immunosenescence caused by SARS-CoV-2 which would greatly benefit the host and diminish all-cause mortality. In addition, the prevention and reversal of immunosenescence using AFP antagonists (zinc, genistein/isoflavones, 7-keto-DHEA, and ivermectin) especially in those with co-morbidities may also help prevent moderate as well as more severe disease from developing.
Public health authorities should consider providing free annual vitamin D3 testing (and retesting when indicated). They need to provide encouragement to go outdoors, safely suntan and to closely monitor serum vitamin D3 levels. People can also reduce their risks of pandemic severity by employing various lifestyle improvements of maintaining an ideal weight, no or low sugar exposures, avoiding trans fats, being outdoors, exercising, reducing stress, and maintaining a normal blood pressure. In addition to Vitamin D3 and a multi-vitamin supplement, AFP antagonists such as zinc, genistein, 7 keto-DHEA (only legal in the USA) could be taken daily as a prophylactic. For those who may be at higher risk of poor outcomes, ivermectin which may also be an AFP antagonist [191] might be used for early treatment along with other measures of the early treatment protocols [193,221]. Note as mentioned, methylprednisolone is preferred over dexamethasone as dexamethasone failed to reverse the SARS-CoV-2 interference of the IFN type 1 and IFN-γ innate responses while methylprednisolone did [193,194]. Interestingly, it had been previously reported that high dose dexamethasone does not induce HERV-K102 transcripts in vitro [192] implying methylprednisolone might activate HERV-K102 proviral expression and replication. Clearly this also needs further investigation.
Finally, it is proposed that monoclonal sc-Fv antibodies that recognize the cell surface HERV-K102 Env (P61567 Env protein) but not the Env on HERV-K102 particles (P63135 Pol-Env protein), may provide novel therapeutics not only against pandemic viruses but also cancers. Unlike the monoclonal antibodies to the spike protein, here there is little concern for loss of monoclonal antibody effectiveness related to the emergence of viral variants (Figures 10,11).
6. Summary and Conclusions
Many novel findings of relevance to understanding the adverse effects of ADE on survival of humans to pandemic viruses are uncovered in this treatise.
First, overwhelming evidence strongly implies that HERV-K102 particle production in M1-like proinflammatory macrophages constitutes the core attributes of trained (innate) immunity related to epigenetic changes and glycolysis in macrophages. Accordingly, ADE mediated infection of macrophages abrogates this potent protector mechanism and thus, it seems many human pathogens target HERV-K102. Second, for the first time, it is clarified that sebocytes in sebaceous glands that line the mucosa are specialized lipid body negative foamy macrophages that constitutively produce and release HERV-K102 particles by lysis. This validates that the HERV-K102 protector response is not only critical but always active at the front lines constituting critical host defense. Third, how trained (innate) immunity may generate a reduction in all-cause mortality (by reversing immunosenescence of macrophages which is causally related to chronic disease [185,186]) has been suggested. Fourth, a novel virus-anti-virus innate response has been uncovered and expanded upon which reveals how the body attempts to overcome pathogen mediated down-modulation of the protective interferon innate immunity response. This is by ‘virus mimicry’ [136] amplified by high replication of HERV-K102 particles which enter normal cells and then integrate into the cell’s genome additionally ‘arming the genome’ with extra copies. Fifth, a more comprehensive explanation of how optimal blood levels of vitamin D3 can promote survival against pandemic viruses, as well as reduce all-cause mortality, is revealed, and involves the preservation of the HERV-K102 protection system of M1-like foamy macrophages. Six, novel evidence is provided implying the HERV-K102 protection system may be germane to overall human survival given the loss of HERV-K102 at the orthologous chromosomal positions in the extinct hominins. Finally, not only do HERV-K102 and/or M1-like macrophages appear to mediate recovery from mild-moderate COVID-19 disease, but it was previously reported that HERV-K102 high replication and integration was associated with resistance to HIV-1 acquisition [10] providing evidence of sterile immunity against pandemic viruses.
All in all, the evidence shows that pandemic enveloped RNA viruses like SARS-CoV-2 and/or HIV-1 target HERV-K102 particle production in foamy macrophages. In COVID-19 disease, severe pathogenesis appears to relate to ADE of infection into macrophages but where optimal levels of vitamin D3 prevents the transition to more severe disease [222]. This is accomplished by blocking the transition of the LB-FMs producing the protective HERV-K102 particles to LB+FMs [165] the latter that generate and secrete high levels of SARS-CoV-2 virions [196]. There is also much hope for the development innate immunity vaccines to pre-induce the HERV-K102 protector system in the most vulnerable. However, for most adults, the reversal /prevention of immunosenescence in adults, especially elders, and maintaining vitamin D3 levels over 50 ng/ml would be main strategies to help prepare for the next pandemic.
There may be one additional strategy to prevent the next pandemic. In a direct comparison of viral evolution, SARS-CoV-2 was found to be adapted to humans upon its emergence whereas SARS-CoV-1 was not [223] suggesting a lab-leak versus zoonotic source (respectively) of these coronavirus pandemics. Allegedly, the Wuhan Institute of Virology could have serial passaged a SARS-CoV-2 progenitor virus through humanized mice using the new model published in October 2019 by Baric and colleagues [224]. These along with other arguments [225] strengthen the notion of a lab-leak source of SARS-CoV-2 including the notion presented here that SARS-CoV-2 targets a protection system only found in humans. This implies there was selection within the human immune system prior to release. Thus, a primary way to help deal with pandemics would be by banning gain-of-function virology research which in the USA, the House has recently approved [226].
7. Patents
The Public Health Agency of Canada (PHAC) does not allow patents to be assigned to inventors to prevent conflicts of interest. Patent applications for the discovery of the replicative activity of HERV-K102 in vivo and in vitro and that this replication may be associated with innate immunity protection against pandemic viruses, were filed in the USA (March 2005), Canada (March 2005), and worldwide (March 2006). All patent applications have since been abandoned. Anyone is therefore able to use the technology without liability of patent interference.
- Cdn Patent Application 2,501,301 March 18, 2005:Patent number CA 2673395 issued October 22, 2013, for screening methods.
- US Patent Application 60/663,263 March 21, 2005:Patent 7,964,341 issued June 21, 2011, for screening methods.
- PCT Application CA2006/000397 March 20, 2006:PCT: WO 2006/096985
Author Contributions
The author M.P.L. contributed solely to the work.
Funding
This review article and research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
I would like to acknowledge Dr. Danuta Showronski for her scientific input which led to the deferral of the second dose of the COVID-19 mRNA vaccines in Canada which undoubtedly, saved many lives.
Conflicts of Interest
The author declares no conflict of interest. As mentioned above, according to Federal Government policies, inventors of patentable discoveries must assign their interests to the Federal Government of Canada to avoid conflicts of interest.
References
- Yuan GC, Cai L, Elowitz M, et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017 May 8;18(1):84. [CrossRef]
- Gao S, Yan L, Wang R, et al. Tracing the temporal-spatial transcriptome landscapes of the human fetal digestive tract using single-cell RNA-sequencing. Nat Cell Biol. 2018 Jun;20(6):721-734. [CrossRef]
- Gentles AJ, Newman AM, Liu CL, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015 Aug;21(8):938-945. [CrossRef]
- https://en.wikipedia.org/wiki/Galactose-alpha-1,3-galactose.
- Mumme H, Thomas BE, Bhasin SS, et al. Single-cell analysis reveals altered tumor microenvironments of relapse- and remission-associated pediatric acute myeloid leukemia. Nat Commun. 2023 Oct 5;14(1):6209. [CrossRef]
- Zhang JT, Zhang J, Wang SR, et al. Spatial downregulation of CD74 signatures may drive invasive component development in part-solid lung adenocarcinoma. iScience. 2023 Aug 21;26(10):107699. [CrossRef]
- Shangguan S, Ehrenberg PK, Geretz A, et al. Monocyte-derived transcriptome signature indicates antibody-dependent cellular phagocytosis as a potential mechanism of vaccine-induced protection against HIV-1. Elife. 2021 Sep 17;10:e69577. [CrossRef]
- Vaccari M, Fourati S, Gordon SN, et al. HIV vaccine candidate activation of hypoxia and the inflammasome in CD14+ monocytes is associated with a decreased risk of SIVmac251 acquisition. Nat Med. 2018 Jun;24(6):847-856. [CrossRef]
- Thomas C, Leleu D, Masson D. Cholesterol and HIF-1α: dangerous liaisons in atherosclerosis. Front Immunol. 2022 Mar 21;13:868958. [CrossRef]
- Laderoute MP, Larocque LJ, Giulivi A, Diaz-Mitoma F. Further evidence that human endogenous retrovirus K102 is a replication competent foamy virus that may antagonize HIV-1 replication. Open AIDS J. 2015 Dec 7;9:112-22. [CrossRef]
- Laderoute MP. Clues to finding correlates of risk/protection for HIV-1 vaccines [version 2; peer review: 2 approved with reservations] F1000 Research 2018, 6:868. [CrossRef]
- Hussein HAM, Thabet AAA, Mohamed TIA, et al. Phenotypical changes of hematopoietic stem and progenitor cells in COVID-19 patients: Correlation with disease status. Cent Eur J Immunol. 2023;48(2):97-110. [CrossRef]
- Sawant J, Patil A, Kurle S. A review: understanding molecular mechanisms of antibody-dependent enhancement in viral infections. Vaccines (Basel). 2023 Jul 14;11(7):1240. [CrossRef]
- Ricke, D.O. Two different antibody-dependent enhancement (ADE) risks for SARS-CoV-2 antibodies. Front Immunol. 2021 Feb 24;12:640093. [CrossRef]
- Gartlan C, Tipton T, Salguero FJ, Sattentau Q, Gorringe A, Carroll MW. Vaccine-associated enhanced disease and pathogenic human coronaviruses. Front Immunol. 2022 Apr 4;13:882972. [CrossRef]
- Zohar T, Alter G. Dissecting antibody-mediated protection against SARS-CoV-2. Nat Rev Immunol. 2020 Jul;20(7):392-394. [CrossRef]
- Chen W, Zhang J, Qin X, et al. SARS-CoV-2 neutralizing antibody levels are correlated with severity of COVID-19 pneumonia. Biomed Pharmacother. 2020 Oct;130:110629. [CrossRef]
- Chen X, Pan Z, Yue S, et al. Disease severity dictates SARS-CoV-2-specific neutralizing antibody responses in COVID-19. Sig Transduct Target Ther. 2020 5, 180. [CrossRef]
- Hashem AM, Algaissi A, Almahboub SA, et al. Early humoral response correlates with disease severity and outcomes in COVID-19 patients. Viruses. 2020 Dec 4;12(12):1390. [CrossRef]
- Zhao J, Yuan Q, Wang H, et al. Antibody responses to SARS-CoV-2 in patients with novel coronavirus disease 2019. Clin Infect Dis. 2020 Nov 19;71(16):2027-2034. [CrossRef]
- Shrock E, Fujimura E, Kula T, et al. Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science. 2020 Nov 27;370(6520):eabd4250. [CrossRef]
- Choteau M, Scohy A, Messe S, et al. Development of SARS-CoV2 humoral response including neutralizing antibodies is not sufficient to protect patients against fatal infection. Sci Rep. 2022 Feb 8;12(1):2077. [CrossRef]
- Legros V, Denolly S, Vogrig M, et al. A longitudinal study of SARS-CoV-2-infected patients reveals a high correlation between neutralizing antibodies and COVID-19 severity. Cell Mol Immunol. 2021 Feb;18(2):318-327. [CrossRef]
- Ren L, Fan G, Wu W, et al. Antibody responses and clinical outcomes in adults hospitalized with severe coronavirus disease 2019 (COVID-19): a post hoc analysis of LOTUS China trial. Clin Infect Dis. 2021 May 18;72(10):e545-e551. [CrossRef]
- Wang K, Fan G, Wu W, et al. Longitudinal dynamics of the neutralizing antibody response to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Clin Infect Dis. 2021 Aug 2;73(3):e531-e539. [CrossRef]
- Xu X, Nie S, Wang Y, et al. Dynamics of neutralizing antibody responses to SARS-CoV-2 in patients with COVID-19: an observational study. Signal Transduct Target Ther. 2021 May 18;6(1):197. [CrossRef]
- Gao L, Zhou J, Yang S, et al. The dichotomous and incomplete adaptive immunity in COVID-19 patients with different disease severity. Sig Transduct Target Ther. 2021 6, 113. [CrossRef]
- Huang AT, Garcia-Carreras B, Hitchings MDT, et al. A systematic review of antibody mediated immunity to coronaviruses: kinetics, correlates of protection, and association with severity. Nat Commun. 2020 Sep 17;11(1):4704. [CrossRef]
- Nair S, Chen X. Biology of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the humoral immunoresponse: a systematic review of evidence to support global policy-level actions and research. Glob Health J. 2022 Mar;6(1):38-43. [CrossRef]
- Wu F, Liu M, Wang A, et al. Evaluating the association of clinical characteristics with neutralizing antibody levels in patients who have recovered from mild COVID-19 in Shanghai, China. JAMA Intern Med. 2020 Oct 1;180(10):1356-1362. [CrossRef]
- Dispinseri S, Secchi M, Pirillo MF, et al. Neutralizing antibody responses to SARS-CoV-2 in symptomatic COVID-19 is persistent and critical for survival. Nat Commun. 2021 May 11;12(1):2670. [CrossRef]
- Garcia-Beltran WF, Lam EC, Astudillo MG, et al. COVID-19-neutralizing antibodies predict disease severity and survival. Cell. 2021 Jan 21;184(2):476-488.e11. [CrossRef]
- Gilbert PB, Montefiori DC, McDermott AB, et al. Immune correlates analysis of the mRNA-1273 COVID-19 vaccine efficacy clinical trial. Science. 2022 Jan 7;375(6576):43-50. [CrossRef]
- Zohar T, Loos C, Fischinger S, et al. Compromised humoral functional evolution tracks with SARS-CoV-2 mortality. Cell. 2020 Dec 10;183(6):1508-1519.e12. [CrossRef]
- Tetro, JA. Is COVID-19 receiving ADE from other coronaviruses? Microbes Infect. 2020 Mar;22(2):72-73. [CrossRef]
- Moorlag SJCFM, Taks E, Ten Doesschate T, et al. Efficacy of Bacillus Calmette-Guérin vaccination against respiratory tract infections in the elderly during the Covid-19 pandemic. Clin Infect Dis. 2022 Mar 5:ciac182. [CrossRef]
- Bigay J, Le Grand R, Martinon F, Maisonnasse P. Vaccine-associated enhanced disease in humans and animal models: lessons and challenges for vaccine development. Front Microbiol. 2022 Aug 10;13:932408. [CrossRef]
- Munitz A, Edry-Botzer L, Itan M, et al. Rapid seroconversion and persistent functional IgG antibodies in severe COVID-19 patients correlates with an IL-12p70 and IL-33 signature. Sci Rep. 2021 Feb 10;11(1):3461. [CrossRef]
- Ho, M.S., Chen W.J., Chen H.Y., Lin S.F., Wang W.C., Di J. Neutralizing antibody response and SARS severity. Emerg Infect Dis. 2005;11:1730–1737.
- Liu L, Wei Q, Lin Q, et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight. 2019 Feb 21;4(4):e123158. [CrossRef]
- Wolfel R, Corman VM, Guggemos W, et al. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020 May;581(7809):465-469. [CrossRef]
- Jensen B, Luebke N, Feldt T, et al. Emergence of the E484K mutation in SARS-COV-2-infected immunocompromised patients treated with bamlanivimab in Germany. Lancet Reg Health Eur. 2021 Sep;8:100164. [CrossRef]
- Guerrieri M, Francavilla B, Fiorelli D, et al. Nasal and salivary mucosal humoral immune response elicited by mRNA BNT162b2 COVID-19 vaccine compared to SARS-CoV-2 natural infection. Vaccines (Basel). 2021 Dec 18;9(12):1499. [CrossRef]
- Aksyuk AA, Bansal H, Wilkins D, et al. AZD1222-induced nasal antibody responses are shaped by prior SARS-CoV-2 infection and correlate with virologic outcomes in breakthrough infection. Cell Rep Med. 2022 Dec 15:100882. [CrossRef]
- Kistler KE, Huddleston J, Bedford T. Rapid and parallel adaptive mutations in spike S1 drive clade success in SARS-CoV-2. Cell Host Microbe. 2022 Apr 13;30(4):545-555.e4. [CrossRef]
- Walsh EE, Frenck RW Jr, Falsey AR, et al. Safety and immunogenicity of two RNA-based Covid-19 vaccine candidates. N Engl J Med. 2020 Dec 17;383(25):2439-2450. [CrossRef]
- Chu L, McPhee R, Huang W, et al. A preliminary report of a randomized controlled phase 2 trial of the safety and immunogenicity of mRNA-1273 SARS-CoV-2 vaccine. Vaccine. 2021 May 12;39(20):2791-2799. [CrossRef]
- https://www.cbc.ca/news/health/vaccine-dose-delay-canada-covid-19-research-1.5965996#.
- https://ourworldindata.org.
- Xu S, Huang R, Sy LS, et al. COVID-19 vaccination and non–COVID-19 mortality risk — seven integrated health care organizations, United States, December 14, 2020–July 31, 2021. MMWR Morb Mortal Wkly Rep 2021;70:1520–1524. [CrossRef]
- Laderoute MP. Did the Second Dose of mRNA COVID-19 Vaccines Select for the Alpha and Delta Variants Which Prolonged the Pandemic? June 30, 2022. https://hervk102.substack.com/p/did-the-second-dose-of-mrna-covid.
- https://science.gc.ca/site/science/en/blogs/science-health/surviving-heat-impacts-2021-western-heat-dome-canada.
- Servellita V, Morris MK, Sotomayor-Gonzalez A, et al. Predominance of antibody-resistant SARS-CoV-2 variants in vaccine breakthrough cases from the San Francisco Bay Area, California. Nat Microbiol. 2022 Feb;7(2):277-288. [CrossRef]
- Tartof SY, Slezak JM, Fischer H, Hong V, Ackerson BK, Ranasinghe ON, et al. Effectiveness of mRNA BNT162b2 COVID-19 vaccine up to 6 months in a large integrated health system in the USA: a retrospective cohort study. Lancet. 2021 Oct 16;398(10309):1407-1416. [CrossRef]
- UK: COVID-19 Weekly Surveillance Reports by Public Health England: COVID-19 Vaccine Surveillance Reports 2021 Weeks 19-38 at https://www.gov.uk/government/publications/covid-19-vaccine-surveillance-report and by UK Health Security Agency: COVID-19 Surveillance Reports for 2021 Week 39 to 2023 Week 23 at https://www.gov.uk/government/publications/covid-19-vaccine-weekly-surveillance-reports.
- https://assets.publishing.service.gov.uk/media/623c6ef0d3bf7f6ab9fe28bf/Vaccine-surveillance-reportweek-12.pdf.
- Shrestha NK, Burke PC, Nowacki AS, Simon JF, Hagen A, Gordon SM. Effectiveness of the coronavirus disease 2019 bivalent vaccine. Open Forum Infect Dis. 2023 Apr 19;10(6):ofad209. [CrossRef]
- Irrgang P, Gerling J, Kocher K, et al. Class switch toward noninflammatory, spike-specific IgG4 antibodies after repeated SARS-CoV-2 mRNA vaccination. Sci Immunol. 2023 Jan 27;8(79):eade2798. [CrossRef]
- https://ourworldindata.org/explorers/coronavirus-data-explorer? The case fatality rate (CFR) is the ratio between confirmed deaths and confirmed cases. Our rolling-average CFR is calculated as the ratio between the 7-day average number of deaths and the 7-day average number of cases 10 days earlier.
- Ziegler CGK, Miao VN, Owings AH, et al. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell. 2021 Sep 2;184(18):4713-4733.e22. [CrossRef]
- Ren X, Wen W, Fan X, et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell. 2021 Apr 1;184(7):1895-1913.e19. [CrossRef]
- Bost P, Giladi A, Liu Y, et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell. 2020 Jun 25;181(7):1475-1488.e12. [CrossRef]
- Chua RL, Lukassen S, Trump S, et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat Biotechnol. 2020 Aug;38(8):970-979. [CrossRef]
- Wendisch D, Dietrich O, Mari T, et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell. 2021 Dec 22;184(26):6243-6261.e27. [CrossRef]
- Grant RA, Morales-Nebreda L, Markov NS, et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature. 2021 Feb;590(7847):635-641. [CrossRef]
- Delorey TM, Ziegler CGK, Heimberg G, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature. 2021 Jul;595(7865):107-113. [CrossRef]
- Laderoute MP, Giulivi A, Larocque L, et al. The replicative activity of human endogenous retrovirus K102 (HERV-K102) with HIV viremia. AIDS. 2007 Nov 30;21(18):2417-24. [CrossRef]
- Bhardwaj N, Maldarelli F, Mellors J, Coffin JM. HIV-1 infection leads to increased transcription of human endogenous retrovirus HERV-K (HML-2) proviruses in vivo but not to increased virion production. J Virol. 2014 Oct;88(19):11108-20. [CrossRef]
- Yu SF, Eastman SW, Linial ML. Foamy virus capsid assembly occurs at a pericentriolar region through a cytoplasmic targeting/retention signal in Gag. Traffic. 2006 Aug;7(8):966-77. [CrossRef]
- Subramanian RP, Wildschutte JH, Russo C, Coffin JM. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology. 2011 Nov 8;8:90. [CrossRef]
- van der Kuyl, AC. HIV infection and HERV expression: a review. Retrovirology. 2012 Jan 16;9:6. [CrossRef]
- Linial, ML. Foamy viruses are unconventional retroviruses. J Virol. 1999 Mar;73(3):1747-55. [CrossRef]
- Murray SM, Linial ML. Simian foamy virus co-infections. Viruses. 2019 Sep 27;11(10):902. [CrossRef]
- Switzer WM, Salemi M, Shanmugam V, Gao F, Cong M.E, Kuiken C, et al. Ancient co-speciation of simian foamy viruses and primates. Nature. 2005;434:376–380. [CrossRef]
- Yu SF, Sullivan MD, Linial ML. Evidence that the human foamy virus genome is DNA. J Virol. 1999 Feb;73(2):1565-72. [CrossRef] [PubMed]
- Meiering CD, Comstock KE, Linial ML. Multiple integrations of human foamy virus in persistently infected human erythroleukemia cells. J Virol. 2000 Feb;74(4):1718-26. [CrossRef]
- Heinkelein M, Hoffmann U, Lücke M, et al. Experimental therapy of allogeneic solid tumors induced in athymic mice with suicide gene-transducing replication-competent foamy virus vectors. Cancer Gene Ther. 2005 Dec;12(12):947-53. [CrossRef]
- Mikovits JA, Hoffman PM, Rethwilm A, Ruscetti FW. In vitro infection of primary and retrovirus-infected human leukocytes by human foamy virus. J Virol. 1996 May;70(5):2774-80. [CrossRef]
- Laderoute MP. What you need to know about the HERV-K102 innate immunity protector system of macrophages against RNA pandemic viruses. hervk102.substack.com, January 24, 2022. https://hervk102.substack.com/p/what-you-need-to-know-about-the-herv.
- Plochmann K, Horn A, Gschmack E, et al. Heparan sulfate is an attachment factor for foamy virus entry. J Virol. 2012 Sep;86(18):10028-35. [CrossRef]
- Robinson-McCarthy LR, McCarthy KR, Raaben M, et al. Reconstruction of the cell entry pathway of an extinct virus. PLoS Pathog. 2018 Aug 6;14(8):e1007123. [CrossRef]
- https://commons.wikimedia.org/wiki/File:Insertion_of_sebaceous_glands_into_hair_shaft_x10.jpg.
- https://www.ncbi.nlm.nih.gov/geoprofiles/.
- Nelson AM, Zhao W, Gilliland KL, Zaenglein AL, Liu W, Thiboutot DM. Neutrophil gelatinase-associated lipocalin mediates 13-cis retinoic acid-induced apoptosis of human sebaceous gland cells. J Clin Invest. 2008 Apr;118(4):1468-78. [CrossRef]
- Nelson AM, Zhao W, Gilliland KL, Zaenglein AL, Liu W, Thiboutot DM. Isotretinoin temporally regulates distinct sets of genes in patient skin. J Invest Dermatol. 2009 Apr;129(4):1038-42. [CrossRef]
- Stec M, Weglarczyk K, Baran J, et al. Expansion and differentiation of CD14+CD16(-) and CD14+ +CD16+ human monocyte subsets from cord blood CD34+ hematopoietic progenitors. J Leukoc Biol. 2007 Sep;82(3):594-602. [CrossRef]
- Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006 Nov 15;177(10):7303-11. [CrossRef]
- Maslova A, Ramirez RN, Ma K, et al. Immunological genome project. deep learning of immune cell differentiation. Proc Natl Acad Sci U S A. 2020 Oct 13;117(41):25655-25666. [CrossRef]
- Saeed S, Quintin J, Kerstens HH, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014 Sep 26;345(6204):1251086. [CrossRef]
- Arts RJW, Joosten LAB, Netea MG. The potential role of trained immunity in autoimmune and autoinflammatory disorders. Front Immunol. 2018 Feb 20;9:298. [CrossRef]
- Lv JJ, Wang H, Cui HY, et al. Blockade of macrophage CD147 protects against foam cell formation in atherosclerosis. Front Cell Dev Biol. 2021 Jan 8;8:609090. [CrossRef]
- Zhang MF, Cai XL, Jing KP, et al. Differentiation model establishment and differentiation-related protein screening in primary cultured human sebocytes. Biomed Res Int. 2018 Apr 5;2018:7174561. [CrossRef]
- Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. 2014 Aug;34(8):1731-8. [CrossRef]
- Bekkering S, Arts RJW, Novakovic B, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. 2018 Jan 11;172(1-2):135-146.e9. [CrossRef]
- Wang J, Liu YM, Hu J, Chen C. Trained immunity in monocyte/macrophage: novel mechanism of phytochemicals in the treatment of atherosclerotic cardiovascular disease. Front Pharmacol. 2023 Feb 21;14:1109576. [CrossRef]
- Keating ST, Groh L, Thiem K, et al. Rewiring of glucose metabolism defines trained immunity induced by oxidized low-density lipoprotein. J Mol Med (Berl). 2020 Jun;98(6):819-831. [CrossRef]
- Manghera M, Ferguson-Parry J, Lin R, Douville RN. NF-κB and IRF1 induce endogenous retrovirus K expression via interferon-stimulated response elements in its 5' long terminal repeat. J Virol. 2016 Sep 29;90(20):9338-49. [CrossRef]
- Manghera M, Douville RN. Endogenous retrovirus-K promoter: a landing strip for inflammatory transcription factors? Retrovirology. 2013 Feb 9;10:16. [CrossRef]
- Russ E, Mikhalkevich N, Iordanskiy S. Expression of human endogenous retrovirus group K (HERV-K) HML-2 correlates with immune activation of macrophages and type I interferon response. Microbiol Spectr. 2023 Mar 2;11(2):e0443822. [CrossRef]
- Fischer H, Fumicz J, Rossiter H, et al. Holocrine secretion of sebum is a unique DNase2-dependent mode of programmed cell death. J Invest Dermatol. 2017 Mar;137(3):587-594. [CrossRef]
- Törőcsik D, Kovács D, Póliska S, et al.. Genome wide analysis of TLR1/2- and TLR4-activated SZ95 sebocytes reveals a complex immune-competence and identifies serum amyloid A as a marker for activated sebaceous glands. PLoS One. 2018 Jun 21;13(6):e0198323. [CrossRef]
- Magiorkinis G, Blanco-Melo D, Belshaw R. The decline of human endogenous retroviruses: extinction and survival. Retrovirology. 2015 Feb 2;12:8. [CrossRef]
- Chuong EB, Elde NC, Feschotte C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. 2016 Mar 4;351(6277):1083-7. [CrossRef]
- Kim YJ, Han K. Endogenous retrovirus-mediated genomic variations in chimpanzees. Mob Genet Elements. 2015 Feb 3;4(6):1-4. [CrossRef]
- Romano CM, Ramalho RF, Zanotto PM. Tempo and mode of ERV-K evolution in human and chimpanzee genomes. Arch Virol. 2006 Nov;151(11):2215-28. [CrossRef]
- Langergraber KE, Prüfer K, Rowney C,et al. Generation times in wild chimpanzees and gorillas suggest earlier divergence times in great ape and human evolution. Proc Natl Acad Sci U S A. 2012 Sep 25;109(39):15716-21. [CrossRef]
- Katzourakis A, Pereira V, Tristem M. Effects of recombination rate on human endogenous retrovirus fixation and persistence. J Virol. 2007 Oct;81(19):10712-7. [CrossRef]
- Sawyer SL, Emerman M, Malik HS. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004 Sep;2(9):E275. [CrossRef]
- Gifford RJ. Viral evolution in deep time: lentiviruses and mammals. Trends Genet. 2012 Feb;28(2):89-100. [CrossRef]
- O'Neil SP, Novembre FJ, Hill AB, et al. Progressive infection in a subset of HIV-1-positive chimpanzees. J Infect Dis. 2000 Oct;182(4):1051-62. [CrossRef]
- Khan N, de Manuel M, Peyregne S, et al. Multiple genomic events altering hominin SIGLEC biology and innate immunity predated the common ancestor of humans and archaic hominins. Genome Biol Evol. 2020 Jul 1;12(7):1040-1050. [CrossRef]
- Compton AA, Malik HS, Emerman M. Host gene evolution traces the evolutionary history of ancient primate lentiviruses. Philos Trans R Soc Lond B Biol Sci. 2013 Aug 12;368(1626):20120496. [CrossRef]
- Murphy WJ, Frönicke L, O'Brien SJ, Stanyon R. The origin of human chromosome 1 and its homologs in placental mammals. Genome Res. 2003 Aug;13(8):1880-8. [CrossRef]
- Posth C, Wißing C, Kitagawa K, et al. Deeply divergent archaic mitochondrial genome provides lower time boundary for African gene flow into Neanderthals. Nat Commun. 2017 Jul 4;8:16046. [CrossRef]
- Petr HC, Wißing C, Kitagawa K, et al. The evolutionary history of Neanderthal and Denisovan Y chromosomes. Science. 2020 Sep 25;369(6511):1653-1656. [CrossRef]
- Hajdinjak M, Mafessoni F, Skov L, et al. Initial upper palaeolithic humans in Europe had recent Neanderthal ancestry. Nature. 2021 Apr;592(7853):253-257. [CrossRef]
- Enard D, Petrov DA. Evidence that RNA viruses drove adaptive introgression between neanderthals and modern humans. Cell. 2018 Oct 4;175(2):360-371.e13. [CrossRef]
- Gouy A, Excoffier L. Polygenic patterns of adaptive introgression in modern humans are mainly shaped by response to pathogens. Mol Biol Evol. 2020 May 1;37(5):1420-1433. [CrossRef]
- Bücking R, Cox MP, Hudjashov G, Saag L, Sudoyo H, Stoneking M. Archaic mitochondrial DNA inserts in modern day nuclear genomes. BMC Genomics. 2019 Dec 26;20(1):1017. [CrossRef]
- Ono M. Molecular cloning and long terminal repeat sequences of human endogenous retrovirus genes related to types A and B retrovirus genes. J Virol. 1986 Jun;58(3):937-44. [CrossRef]
- Löwer R, Löwer J, Kurth R. The viruses in all of us: characteristics and biological significance of human endogenous retrovirus sequences. Proc Natl Acad Sci U S A. 1996 May 28;93(11):5177-84. [CrossRef]
- Bannert N, Kurth R. Retroelements and the human genome: new perspectives on an old relation. Proc Natl Acad Sci U S A. 2004 Oct 5;101 Suppl 2(Suppl 2):14572-9. [CrossRef]
- Simpson GR, Patience C, Löwer R, et al. Endogenous D-type (HERV-K) related sequences are packaged into retroviral particles in the placenta and possess open reading frames for reverse transcriptase. Virology. 1996 Aug 15;222(2):451-6. [CrossRef]
- Grow EJ, Flynn RA, Chavez SL, et al. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature. 2015 Jun 11;522(7555):221-5. [CrossRef]
- Morgan D, Brodsky I. Human endogenous retrovirus (HERV-K) particles in megakaryocytes cultured from essential thrombocythemia peripheral blood stem cells. Exp Hematol. 2004 Jun;32(6):520-5. [CrossRef]
- Morozov VA, Dao Thi VL, Denner J. The transmembrane protein of the human endogenous retrovirus--K (HERV-K) modulates cytokine release and gene expression. PLoS One. 2013 Aug 7;8(8):e70399. [CrossRef]
- Padow M, Lai L, Fisher RJ, et al. Analysis of human immunodeficiency virus type 1 containing HERV-K protease. AIDS Res Hum Retroviruses. 2000 Dec 10;16(18):1973-80. [CrossRef]
- Brinzevich D, Young GR, Sebra R, et al. HIV-1 interacts with human endogenous retrovirus K (HML-2) envelopes derived from human primary lymphocytes. J Virol. 2014 Jun;88(11):6213-23. [CrossRef]
- Monde K, Contreras-Galindo R, Kaplan MH, Markovitz DM, Ono A. Human endogenous retrovirus K Gag coassembles with HIV-1 Gag and reduces the release efficiency and infectivity of HIV-1. J Virol. 2012 Oct;86(20):11194-208. [CrossRef]
- Monde K, Terasawa H, Nakano Y, et al. Molecular mechanisms by which HERV-K Gag interferes with HIV-1 Gag assembly and particle infectivity. Retrovirology. 2017 Apr 26;14(1):27. [CrossRef]
- Minkoff JM, TenOever B. Innate immune evasion strategies of SARS-CoV-2. Nat Rev Microbiol. 2023 Jan 11:1–17. [CrossRef]
- Su HC, Jing H, Zhang Y, Casanova JL. Interfering with interferons: a critical mechanism for critical COVID-19 pneumonia. Annu Rev Immunol. 2023 Apr 26;41:561-585. [CrossRef]
- Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci. 2008 Nov;1143:1-20. [CrossRef]
- Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. 2020 Sep;20(9):537-551. [CrossRef]
- Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021 Sep;21(9):548-569. [CrossRef]
- Russ E, Iordanskiy S. Endogenous retroviruses as modulators of innate immunity. Pathogens. 2023 Jan 19;12(2):162. [CrossRef]
- Guo Y, Yang C, Liu Y, et al. High expression of HERV-K (HML-2) might stimulate interferon in COVID-19 patients. Viruses 2022, 14, 996. [CrossRef]
- Kenney DJ, O'Connell AK, Turcinovic J, et al. Humanized mice reveal a macrophage-enriched gene signature defining human lung tissue protection during SARS-CoV-2 infection. Cell Rep. 2022 Apr 19;39(3):110714. [CrossRef]
- Wang-Johanning F, Frost AR, Johanning GL, et al. Expression of human endogenous retrovirus K envelope transcripts in human breast cancer. Clin Cancer Res. 2001 Jun;7(6):1553-60.
- Wang-Johanning F, Frost AR, Jian B, Epp L, Lu DW, Johanning GL. Quantitation of HERV-K env gene expression and splicing in human breast cancer. Oncogene. 2003 Mar 13;22(10):1528-35. [CrossRef]
- Wang-Johanning F, Radvanyi L, Rycaj K, et al. Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res. 2008 Jul 15;68(14):5869-77. [CrossRef]
- Wang-Johanning F, Rycaj K, Plummer JB, et al. Immunotherapeutic potential of anti-human endogenous retrovirus-K envelope protein antibodies in targeting breast tumors. J Natl Cancer Inst. 2012 Feb 8;104(3):189-210. [CrossRef]
- Jones RB, Garrison KE, Mujib S, et al. HERV-K-specific T cells eliminate diverse HIV-1/2 and SIV primary isolates. J Clin Invest. 2012 Dec;122(12):4473-89. [CrossRef]
- Michaud HA, de Mulder M, SenGupta D, et al. Trans-activation, post-transcriptional maturation, and induction of antibodies to HERV-K (HML-2) envelope transmembrane protein in HIV-1 infection. Retrovirology. 2014 Jan 28;11:10. [CrossRef]
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021 Aug;596(7873):583-589. [CrossRef]
- Varadi M, Velankar S. The impact of AlphaFold Protein Structure Database on the fields of life sciences. Proteomics. 2022 Nov 16:e2200128. [CrossRef]
- Apostolou E, Rizwan M, Moustardas P, et al. Saliva antibody-fingerprint of reactivated latent viruses after mild/asymptomatic COVID-19 is unique in patients with myalgic-encephalomyelitis/chronic fatigue syndrome. Front Immunol. 2022 Oct 20;13:949787. [CrossRef]
- Risch H. Rise in Aggressive ‘Turbo Cancers” -And especially Among Younger People. September 11, 2023. https://www.theepochtimes.com/epochtv/dr-harvey-risch-rise-in-aggressive-turbo-cancers-and-especially-among-younger-people-atlnow-5489582?utm_source=prtnrhard&utm_campaign=vigilantf&src_src=prtnrhard&src_cmp=vigilantf.
- Arru G, Galleri G, Deiana GA, et al. HERV-K modulates the immune response in ALS patients. Microorganisms. 2021 Aug 23;9(8):1784. [CrossRef]
- Lo Presti E, De Gaetano A, Pioggia G, Gangemi S. Comprehensive analysis of the ILCs and unconventional T cells in virus infection: profiling and dynamics associated with COVID-19 disease for a future monitoring system and therapeutic opportunities. Cells. 2022 Feb 4;11(3):542. [CrossRef]
- Bhat RK, Rudnick W, Antony JM, et al. Human endogenous retrovirus-K(II) envelope induction protects neurons during HIV/AIDS. PLoS One. 2014 Jul 2;9(7):e97984. [CrossRef]
- Garcia-Montojo M, Simula ER, Fathi S, et al. Antibody response to HML-2 may be protective in amyotrophic lateral sclerosis. Ann Neurol. 2022 Nov;92(5):782-792. [CrossRef]
- Simula ER, Arru G, Zarbo IR, Solla P, Sechi LA. TDP-43 and HERV-K envelope-specific immunogenic epitopes are recognized in ALS patients. Viruses. 2021 Nov 18;13(11):2301. [CrossRef]
- Xue B, Sechi LA, Kelvin DJ. Human endogenous retrovirus K (HML-2) in health and disease. Front Microbiol. 2020 Jul 17;11:1690. [CrossRef]
- Tokuyama M, Gunn BM, Venkataraman A, et al. Antibodies against human endogenous retrovirus K102 envelope activate neutrophils in systemic lupus erythematosus. J Exp Med. 2021 Jul 5;218(7):e20191766. [CrossRef]
- Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe. 2011 May 19;9(5):355-61. [CrossRef]
- Mitroulis I, Ruppova K, Wang B, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. 2018 Jan 11;172(1-2):147-161.e12. [CrossRef]
- Cirovic B, de Bree LCJ, Groh L, et al. BCG vaccination in humans elicits trained immunity via the hematopoietic progenitor compartment. Cell Host Microbe. 2020 Aug 12;28(2):322-334.e5. [CrossRef]
- Al B, Suen TK, Placek K, Netea MG. Innate (learned) memory, J Allerg Clin Immunol. 2023 Jun 14. [CrossRef]
- Zhang B, Moorlag SJ, Dominguez-Andres J, et al. Single-cell RNA sequencing reveals induction of distinct trained-immunity programs in human monocytes. J Clin Invest. 2022 Apr 1;132(7):e147719. [CrossRef]
- Li W, Moorlag SJCFM, Koeken VACM, et al. A single-cell view on host immune transcriptional response to in vivo BCG-induced trained immunity. Cell Rep. 2023 May 30;42(5):112487. [CrossRef]
- Drummer C 4th, Saaoud F, Shao Y, et al. Trained immunity and reactivity of macrophages and endothelial cells. Arterioscler Thromb Vasc Biol. 2021 Mar;41(3):1032-1046. [CrossRef]
- Cheng SC, Quintin J, Cramer RA, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014 Sep 26;345(6204):1250684. [CrossRef]
- Ieronymaki E, Daskalaki MG, Lyroni K, Tsatsanis C. Insulin signaling and insulin resistance facilitate trained immunity in macrophages through metabolic and epigenetic changes. Front Immunol. 2019 Jun 12;10:1330. [CrossRef]
- Oh J, Weng S, Felton SK, et al. 1,25(OH)2 vitamin D inhibits foam cell formation and suppresses macrophage cholesterol uptake in patients with type 2 diabetes mellitus. Circulation. 2009 Aug 25;120(8):687-98. [CrossRef]
- Coleman PS, Parlo RA. Warburg's ghost-cancer's self-sustaining phenotype: the aberrant carbon flux in cholesterol-enriched tumor mitochondria via deregulated cholesterogenesis. Front Cell Dev Biol. 2021 Mar 12;9:626316. [CrossRef]
- Arts RJW, Moorlag SJCFM, Novakovic B, et al. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe. 2018;23:89–100.e5. [CrossRef]
- Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. 2019 Jul 3;10:1462. [CrossRef]
- Gomez Marti JL, Wells A, Brufsky AM. Dysregulation of the mevalonate pathway during SARS-CoV-2 infection: An in silico study. J Med Virol. 2021 Apr;93(4):2396-2405. [CrossRef]
- Gao L, Chen Q, Zhou X, Fan L. The role of hypoxia-inducible factor 1 in atherosclerosis. J Clin Pathol. 2012 Oct;65(10):872-6. [CrossRef]
- Rius J, Guma M, Schachtrup C, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008 Jun 5;453(7196):807-11. [CrossRef]
- Jiang G, Li T, Qiu Y, Rui Y, Chen W, Lou Y. RNA interference for HIF-1alpha inhibits foam cells formation in vitro. Eur J Pharmacol. 2007 May 21;562(3):183-90. [CrossRef]
- Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014 Feb;13(2):140-56. [CrossRef]
- Appelberg S, Gupta S, Svensson Akusjärvi S, et al. Dysregulation in Akt/mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. Emerg Microbes Infect. 2020 Dec;9(1):1748-1760. [CrossRef]
- Orekhov AN, Oishi Y, Nikiforov NG, et al. Modified LDL particles activate inflammatory pathways in monocyte-derived macrophages: transcriptome analysis. Curr Pharm Des. 2018;24(26):3143-3151. [CrossRef]
- Zeboudj L, Giraud A, Guyonnet L, et al. Selective EGFR (epidermal growth factor receptor) deletion in myeloid cells limits atherosclerosis-brief report. Arterioscler Thromb Vasc Biol. 2018 Jan;38(1):114-119. [CrossRef]
- Wang L, Huang Z, Huang W, et al. Inhibition of epidermal growth factor receptor attenuates atherosclerosis via decreasing inflammation and oxidative stress. Sci Rep. 2017 Apr 4;8:45917. [CrossRef]
- Tallam A, Perumal TM, Antony PM, et al. Gene regulatory network inference of immunoresponsive gene 1 (IRG1) identifies interferon regulatory factor 1 (IRF1) as its transcriptional regulator in mammalian macrophages. PLoS One. 2016 Feb 12;11(2):e0149050. [CrossRef]
- Zhang M, Liu K, Zhang Q, et al. Alpha fetoprotein promotes polarization of macrophages towards M2-like phenotype and inhibits macrophages to phagocytize hepatoma cells. Front Immunol. 2023 Feb 23;14:1081572.
- Platanitis E, Decker T. Regulatory networks involving STATs, IRFs, and NFκB in inflammation. Front Immunol. 2018 Nov 13;9:2542. [CrossRef]
- Tan L, Lu J, Liu L, Li L. Fatty acid binding protein 3 deficiency limits atherosclerosis development via macrophage foam cell formation inhibition. Exp Cell Res. 2021 Oct 1;407(1):112768. [CrossRef]
- Wang K, Chen W, Zhang Z, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther. 2020 Dec 4;5(1):283. [CrossRef]
- Geng J, Chen L, Yuan Y, et al. CD147 antibody specifically and effectively inhibits infection and cytokine storm of SARS-CoV-2 and its variants delta, alpha, beta, and gamma. Signal Transduct Target Ther. 2021 Sep 25;6(1):347. [CrossRef]
- Helal MA, Shouman S, Abdelwaly A, et al. Molecular basis of the potential interaction of SARS-CoV-2 spike protein to CD147 in COVID-19 associated-lymphopenia. J Biomol Struct Dyn. 2022 Feb;40(3):1109-1119. [CrossRef]
- Laderoute, M.P. A new paradigm about HERV-K102 particle production and blocked release to explain cortisol mediated immunosenescence and age-associated risk of chronic disease. Discov Med. 2015 Dec;20(112):379-91.
- Laderoute, M. The paradigm of immunosenescence in atherosclerosis-cardiovascular disease (ASCVD). Discov Med. 2020 Jan-Feb;29(156):41-51.
- Laderoute MP. The Characterization of a Novel, Widespread, PNA-Reactive Tumor Associated Antigen; The Alpha-fetoprotein Receptor/ Binding Protein. Ph.D. Thesis, University of Alberta, Edmonton, Alberta, Canada, January 7, 1991. https://era.library.ualberta.ca/items/6f548eb6-49a2-456c-b472-41f68976077f.
- Laderoute MP, Pilarski LM. The inhibition of apoptosis by alpha-fetoprotein (AFP) and the role of AFP receptors in anti-cellular senescence. Anticancer Res. 1994 Nov-Dec;14(6B):2429-38.
- Nakabayashi H, Koyama Y, Sakai M, Li HM, Wong NC, Nishi S. Glucocorticoid stimulates primate but inhibits rodent alpha-fetoprotein gene promoter. Biochem Biophys Res Commun. 2001 Sep 14;287(1):160-72. [CrossRef]
- Phillips AC, Carroll D, Gale CR, Lord JM, Arlt W, Batty GD. Cortisol, DHEA sulphate, their ratio, and all-cause and cause-specific mortality in the Vietnam Experience Study. Eur J Endocrin. 2010 Aug; 163(2):285-292. doi.org/10.1530/EJE-10-0299.
- Laderoute M. Ivermectin may prevent and reverse immunosenescence by antagonizing alpha-fetoprotein and downmodulating PI3K/Akt/mTOR hyperactivity. Open Heart, April 29, 2021. https://openheart.bmj.com/content/8/1/e001655.responses#ivermectin-may-prevent-and-reverse-immunosenescence-by-antagonizing-alpha-fetoprotein-and-downmodulating-pi3k-akt-mtor-hyperactivity.
- Ono M, Kawakami M, Ushikubo H. Stimulation of expression of the human endogenous retrovirus genome by female steroid hormones in human breast cancer cell line T47D. J Virol. 1987 Jun;61(6):2059-62. [CrossRef]
- Kory P, Meduri GU, Iglesias J, Varon J, Cadegiani FA, Marik PE. "MATH+" Multi-Modal Hospital Treatment Protocol for COVID-19 Infection: Clinical and Scientific Rationale. J Clin Med Res. 2022 Feb;14(2):53-79. [CrossRef]
- Draghici S, Nguyen TM, Sonna LA, et al. COVID-19: disease pathways and gene expression changes predict methylprednisolone can improve outcome in severe cases. Bioinformatics. 2021 Sep 9;37(17):2691-2698. [CrossRef]
- Leech M, Metz C, Hall P, et al. Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 1999 Aug;42(8):1601-8. [CrossRef]
- Dias SSG, Soares VC, Ferreira AC, et al. Lipid droplets fuel SARS-CoV-2 replication and production of inflammatory mediators. PLoS Pathog. 2020 Dec 16;16(12):e1009127. [CrossRef]
- Singh N, Bharara Singh A. S2 subunit of SARS-CoV-2 interacts with tumor suppressor protein p53 and BRCA: an in silico study. Transl Oncol. 2020 Oct;13(10):100814. [CrossRef]
- Lee KC, Crowe AJ, Barton MC. p53-mediated repression of alpha-fetoprotein gene expression by specific DNA binding. Mol Cell Biol. 1999 Feb;19(2):1279-88. [CrossRef]
- Desterke C, Turhan AG, Bennaceur-Griscelli A, Griscelli F. PPARγ cistrome repression during activation of lung monocyte-macrophages in severe COVID-19. iScience. 2020 Oct 23;23(10):101611. [CrossRef]
- Verway M, Bouttier M, Wang TT, et al. Vitamin D induces interleukin-1β expression: paracrine macrophage epithelial signaling controls M. tuberculosis infection. PLoS Pathog. 2013;9(6):e1003407. [CrossRef]
- Borsche L, Glauner B, von Mendel J. COVID-19 mortality risk correlates inversely with vitamin D3 status, and a mortality rate close to zero could theoretically be achieved at 50 ng/ml 25(OH)D3: results of a systematic review and meta-analysis. Nutrients. 2021 Oct 14;13(10):3596. [CrossRef]
- Grant WB, Al Anouti F, Boucher BJ, et al. A narrative review of the evidence for variations in serum 25-hydroxyvitamin D concentration thresholds for optimal health. Nutrients. 2022 Feb 2;14(3):639. [CrossRef]
- Office for National Statistics (UK). Deaths involving COVID-19 by vaccination status, England: Deaths occurring between 1 January 2021 and 31 May 2022. Age-standardised mortality rates and raw death numbers for deaths involving COVID-19 by vaccination status, broken down by age and /or sex group. https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/deaths/bulletins/deathsinvolvingcovid19byvaccinationstatusengland/deathsoccurringbetween1january2021and31may2022.
- Fenton N, Neil M, Craig C, McLachlan S. What the ONS mortality COVID-19 surveillance data can tell us about vaccine safety and efficacy, November 10, 2022 preprint. https://d7694293-ffb8-4ed0-a014-.3581d49070e4.usrfiles.com/ugd/d76942_bd64e0ebb4754477afd3d9f00bb3dc0f.pdf.
- Fenton N, Campbell J. Is ONS data on mortality by vaccine status fit for purpose? https://www.youtube.com/watch?v=W-N-17_j_44, January 22, 2023.
- Graña C, Ghosn L, Evrenoglou T, et al. Efficacy and safety of COVID-19 vaccines. Cochrane Database Syst Rev. 2022 Dec 7;12(12):CD015477. [CrossRef]
- https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/968513/Weekly_Flu_and_COVID-19_report_w10.pdf. Note: page 73/81; Table 10. Cumulative data up to week 9, 2021 for week ending March 7.
- Brogna C, Cristoni S, Marino G, et al. Detection of recombinant spike protein in the blood of individuals vaccinated against SARS-CoV-2: possible molecular mechanisms. Proteomics Clin Appl. 2023 Nov;17(6):e2300048. [CrossRef]
- Sahanic S, Hilbe R, Dünser C, et al. SARS-CoV-2 activates the TLR4/MyD88 pathway in human macrophages: a possible correlation with strong pro-inflammatory responses in severe COVID-19. Heliyon. 2023 Nov 17;9(11):e21893. [CrossRef]
- McCullough, Peter: at U.S. Sen. Ron Johnson Roundtable Discussion. COVID-19 Vaccines: What they are, how they work, and possible causes of injuries. December 7, 2022. https://twitter.com/SenRonJohnson/status/160050489272825037. https://rumble.com/v1ze4d0-covid-19-vaccines-what-they-are-how-they-work-and-possible-causes-of-injuri.html.
- Seneff S, Nigh G, Kyriakopoulos AM, McCullough PA. Innate immune suppression by SARS-CoV-2 mRNA vaccinations: The role of G-quadruplexes, exosomes, and microRNAs. Food Chem Toxicol. 2022 Apr 15;164:113008. [CrossRef]
- Hulscher N, Hodkinson R, Makis W, McCullough P. Autopsy proven fatal COVID-19 vaccine-induced myocarditis. Preprints 2023, 2023071198. [CrossRef]
- Schwab C, Domke LM, Hartmann L, Stenzinger A, Longerich T, Schirmacher P. Autopsy-based histopathological characterization of myocarditis after anti-SARS-CoV-2-vaccination. Clin Res Cardiol. 2023 Mar;112(3):431-440. [CrossRef]
- Kleen TO, Galdon AA, MacDonald AS, Dalgleish AG. Mitigating coronavirus induced dysfunctional immunity for at-risk populations in COVID-19: trained immunity, BCG and "new old friends". Front Immunol. 2020 Sep 4;11:2059. [CrossRef]
- Desingu PA, Nagarajan K. Unveiling HERV-K113-ENV as SARS-CoV-2 severity admissible biomarker by mining transcriptome data. J Med Virol. 2023 Jan;95(1):e28149. [CrossRef]
- Tovo PA, Garazzino S, Daprà V, et al. COVID-19 in children: expressions of type I/II/III interferons, TRIM28, SETDB1, and endogenous retroviruses in mild and severe cases. Int J Mol Sci. 2021 Jul 13;22(14):7481. [CrossRef]
- Temerozo JR, Fintelman-Rodrigues N, Dos Santos MC, et al. Human endogenous retrovirus K in the respiratory tract is associated with COVID-19 physiopathology. Microbiome. 2022 Apr 22;10(1):65. [CrossRef]
- Utrero-Rico A, González-Cuadrado C, Chivite-Lacaba M, et al. Alterations in circulating monocytes predict COVID-19 severity and include chromatin modifications still detectable six months after recovery. Biomedicines. 2021 Sep 17;9(9):1253. [CrossRef]
- Lieberman NAP, Peddu V, Xie H, et al. In vivo antiviral host transcriptional response to SARS-CoV-2 by viral load, sex, and age. PLoS Biol. 2020 Sep 8;18(9):e3000849. [CrossRef]
- Pereira PC, de Lima CJ, Fernandes AB, Zângaro RA, Villaverde AB. Cardiopulmonary and hematological effects of infrared LED photobiomodulation in the treatment of SARS-COV2. J Photochem Photobiol B. 2023 Jan;238:112619. [CrossRef]
- McCullough PA, Alexander PE, Armstrong R, et al. Multifaceted highly targeted sequential multidrug treatment of early ambulatory high-risk SARS-CoV-2 infection (COVID-19). Rev Cardiovasc Med. 2020 Dec 30;21(4):517-530. [CrossRef]
- Wimalawansa SJ. Infections and autoimmunity-the immune system and vitamin D: a systematic review. Nutrients. 2023 Sep 2;15(17):3842. [CrossRef]
- Zhan SH, Deverman BE, Chan YA. SARS-CoV-2 is well adapted for humans. What does this mean for reemergence? BioRxiv May 2 2020 (preprint). https://www.biorxiv.org/content/10.1101/2020.05.01.073262v1.
- Wahl A, De C, Abad Fernandez M, et al. Precision mouse models with expanded tropism for human pathogens. Nat Biotechnol. 2019 Oct;37(10):1163-1173. [CrossRef]
- Maxmen A, Mallaaty S. The COVID lab-leak hypothesis: what scientists do and don’t know. Nature, June 8, 2021. https://www.nature.com/articles/d41586-021-01529-3.
- Kaiser, J. House approves ban on gain-of-function pathogen research. 15 Nov 2023. https://www.science.org/content/article/house-approves-ban-gain-function-pathogen-research.
Figure 1.
Negative excess all-cause mortality in spring 2021 (February 10 to April 28) in Canada associated with postponement of second COVID-19 vaccine dose by up to 120 days [48]. Negative excess mortality relative to the average mortality per week for 2015 to 2019 is highlighted in turquoise as provided by Our World in Data [49]. Bright green highlights pertain to emergence and dominance of the alpha variant (emerged February 22, 2021 and became dominant by March 22, 2021 but never exceeded 57% of the total variants); hot pink highlights pertain to emergence (May 3, 2021) and dominance of the delta variant (July 12, 2021) and reached 99% of the total variants by Aug 30, 2021; and the gamma variant appearance is shown in grey and never reached higher than 26%. Mass vaccination started in Canada on December 22, 2020. There were no variants of concern selected prior to December 22, 2020, implicating mass vaccination as the cause. Note that the peak in excess all-cause mortality recorded on July 4 pertained to 619 heat wave related deaths from June 25 to July 1, 2021, in British Columbia [52] and thus are immaterial to the discussion. Note. All of the data provided [49] is freely available for both academic and commercial use under Creative Commons Attribution 4.0 (CC-BY 4.0) licence terms.
Figure 1.
Negative excess all-cause mortality in spring 2021 (February 10 to April 28) in Canada associated with postponement of second COVID-19 vaccine dose by up to 120 days [48]. Negative excess mortality relative to the average mortality per week for 2015 to 2019 is highlighted in turquoise as provided by Our World in Data [49]. Bright green highlights pertain to emergence and dominance of the alpha variant (emerged February 22, 2021 and became dominant by March 22, 2021 but never exceeded 57% of the total variants); hot pink highlights pertain to emergence (May 3, 2021) and dominance of the delta variant (July 12, 2021) and reached 99% of the total variants by Aug 30, 2021; and the gamma variant appearance is shown in grey and never reached higher than 26%. Mass vaccination started in Canada on December 22, 2020. There were no variants of concern selected prior to December 22, 2020, implicating mass vaccination as the cause. Note that the peak in excess all-cause mortality recorded on July 4 pertained to 619 heat wave related deaths from June 25 to July 1, 2021, in British Columbia [52] and thus are immaterial to the discussion. Note. All of the data provided [49] is freely available for both academic and commercial use under Creative Commons Attribution 4.0 (CC-BY 4.0) licence terms.
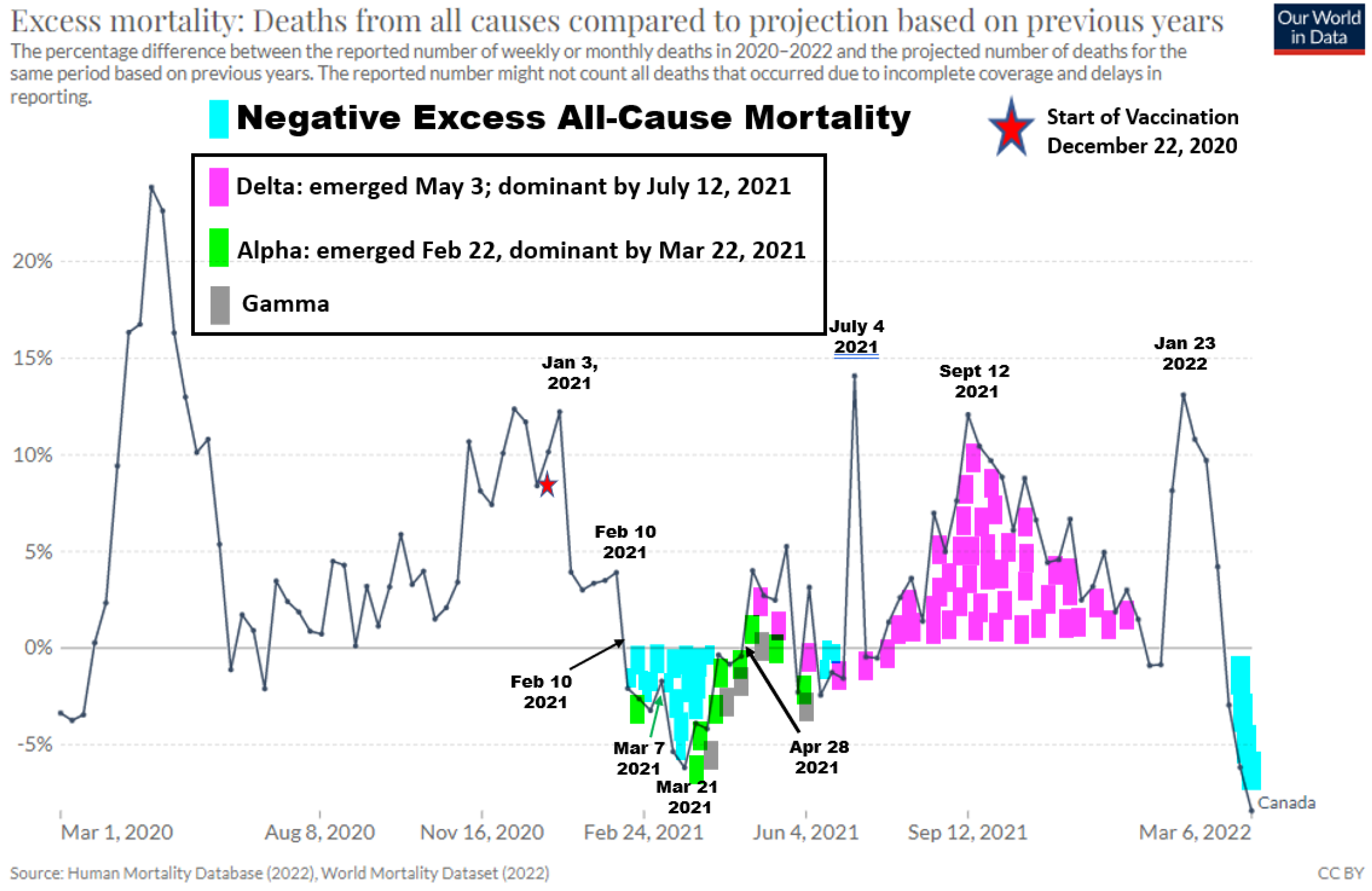
Figure 2.
The United Kingdom Health Security Agency’s COVID-19 vaccine surveillance report for week 12 reported on March 24, 2022, showed in all age groups over the age of 18 that the vaccinated were at increased risk of symptomatic infection [56]. Note. CC-By 4.0 from https://www.nationalarchives.gov.uk/doc/open-government-licence/version/3/andreferencing bulletin.
Figure 2.
The United Kingdom Health Security Agency’s COVID-19 vaccine surveillance report for week 12 reported on March 24, 2022, showed in all age groups over the age of 18 that the vaccinated were at increased risk of symptomatic infection [56]. Note. CC-By 4.0 from https://www.nationalarchives.gov.uk/doc/open-government-licence/version/3/andreferencing bulletin.

Figure 3.
The effective reproduction rate ‘R’ of SARS-CoV-2 clearly indicates increased risk of transmission with two and more so, three doses of the adaptive COVID-19 vaccine for Canada and the United States. Note. All of the data provided by Our World in Data [49] is freely available for both academic and commercial use under Creative Commons Attribution 4.0 (CC-BY 4.0) licence terms.
Figure 3.
The effective reproduction rate ‘R’ of SARS-CoV-2 clearly indicates increased risk of transmission with two and more so, three doses of the adaptive COVID-19 vaccine for Canada and the United States. Note. All of the data provided by Our World in Data [49] is freely available for both academic and commercial use under Creative Commons Attribution 4.0 (CC-BY 4.0) licence terms.
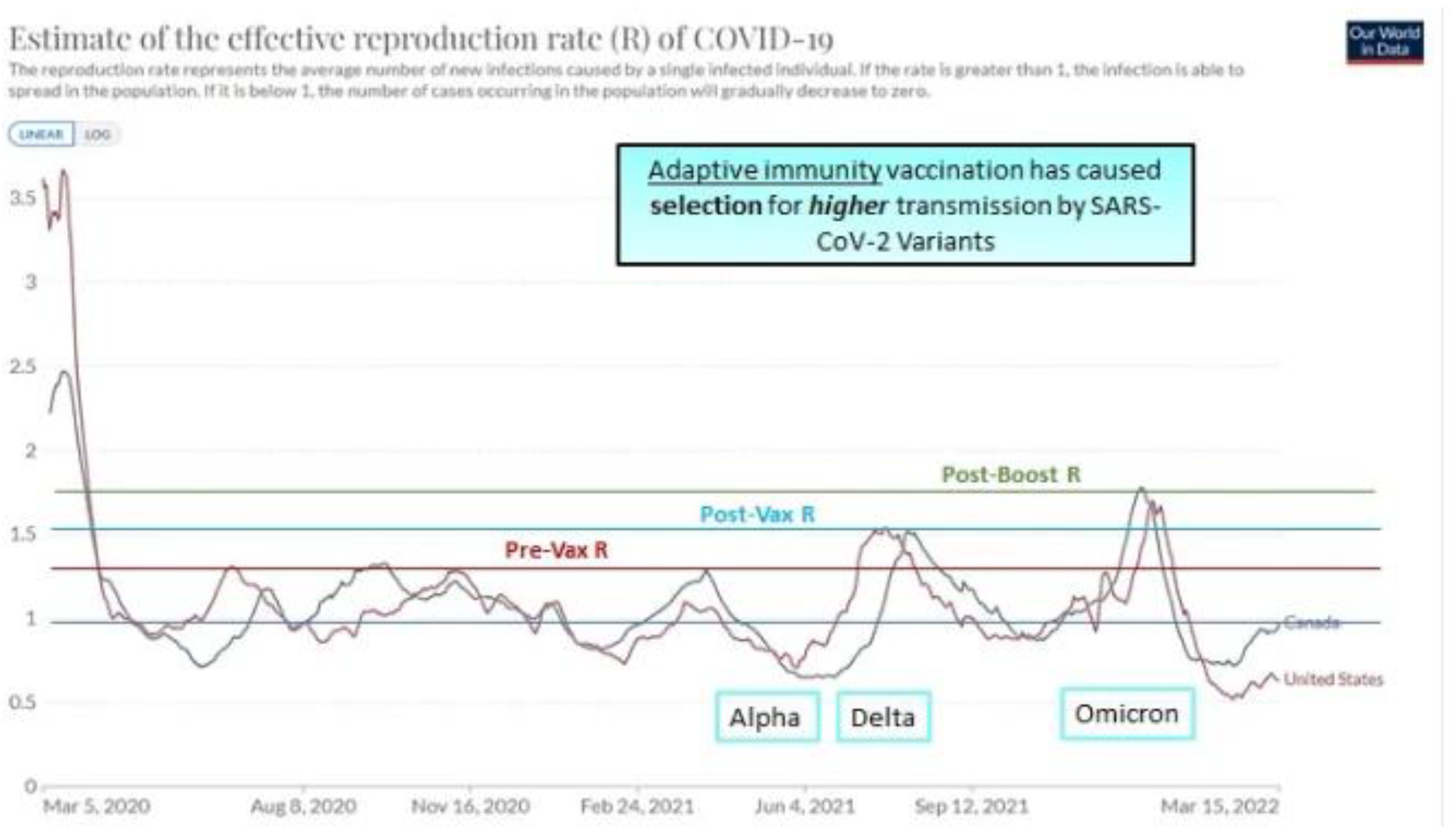
Figure 4.
HERV-K102 particle production in cord blood mononuclear cells (CB) cultured in IMDM media [10,11,67]. Left image: H&E staining of a CB sample (cytospin slide) showing the dominant presence of highly vacuolated foamy macrophages amongst normal small lymphocytes (400X). Note the presence of a rare multinucleated giant macrophage. Right image top: Electron microscopy of vacuolating CB cells day 11 at 1500X. Right image bottom: At 100,000X immature particles (the centers are not condensed) can be seen in the vacuoles which averaged about 100 nm. Envelope spikes are also noted. No cell surface budding was observed by EM. Instead, release of the particles occurred on day 6-7 by lysis. Note that when CB was cultured in traditional RPMI this aborted foam cell formation. Also, the addition of IL-2 and PHA aborted foam cell formation in IMDM cultures implying adaptive immunity may downmodulate HERV-K102 particle production. Blue arrow in right bottom image points to the preassembly of envelope with gag as aggregates in the trans-golgi network which then bud into the vacuoles [69]. This assembly which absolutely requires envelope protein is characteristic of foamy retroviruses but not orthoretroviruses. Note. Reproduced from “Clues to finding correlates of risk/protection for HIV-1 vaccines”, by Laderoute MP. 2018;6:868 [11]. CC BY Author.
Figure 4.
HERV-K102 particle production in cord blood mononuclear cells (CB) cultured in IMDM media [10,11,67]. Left image: H&E staining of a CB sample (cytospin slide) showing the dominant presence of highly vacuolated foamy macrophages amongst normal small lymphocytes (400X). Note the presence of a rare multinucleated giant macrophage. Right image top: Electron microscopy of vacuolating CB cells day 11 at 1500X. Right image bottom: At 100,000X immature particles (the centers are not condensed) can be seen in the vacuoles which averaged about 100 nm. Envelope spikes are also noted. No cell surface budding was observed by EM. Instead, release of the particles occurred on day 6-7 by lysis. Note that when CB was cultured in traditional RPMI this aborted foam cell formation. Also, the addition of IL-2 and PHA aborted foam cell formation in IMDM cultures implying adaptive immunity may downmodulate HERV-K102 particle production. Blue arrow in right bottom image points to the preassembly of envelope with gag as aggregates in the trans-golgi network which then bud into the vacuoles [69]. This assembly which absolutely requires envelope protein is characteristic of foamy retroviruses but not orthoretroviruses. Note. Reproduced from “Clues to finding correlates of risk/protection for HIV-1 vaccines”, by Laderoute MP. 2018;6:868 [11]. CC BY Author.
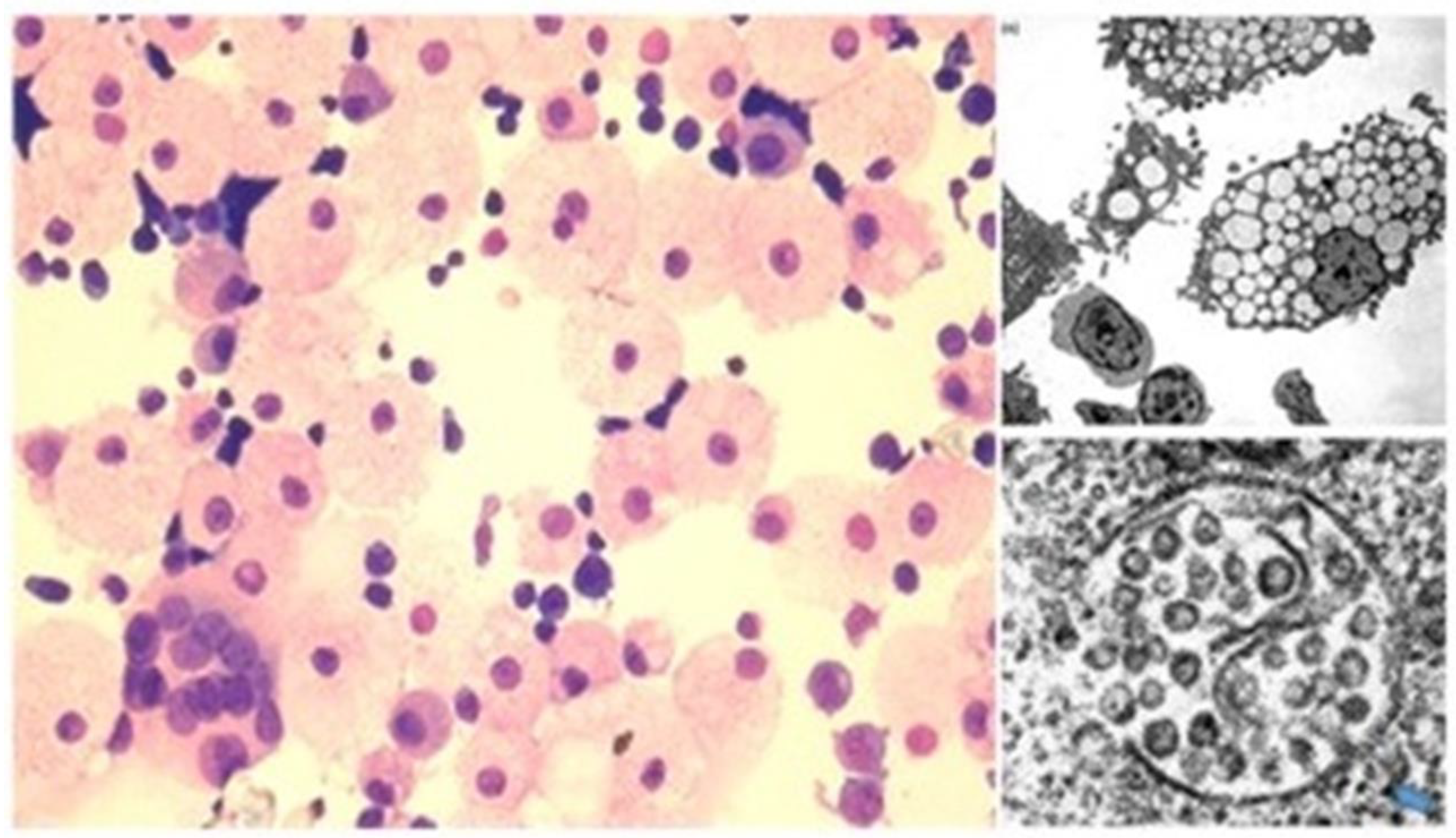
Figure 5.
Evidence that HERV-K102 replicative activity is associated with sterilizing immunity (resistance to HIV-1 acquisition) in a HESN cohort [10]. DNA was isolated from plasma. The ddCt real-time PCR method employed the 18S RNA gene to control for genomic equivalents and used uracil N-glycosylase (UNG) in the PCR buffer to digest the cDNA genomes of HERV-K102 particles which left only genomic DNA in the DNA extracted from plasma. The mean ddCt qPCR ratio (for HERV-K102 pol) of 30 normal healthy controls was 0.88 +/-0.37. In contrast, on average for the female Commercial Sex Workers (CSW) HIV-1 Exposed Seronegative (HESN) cohort it was 4.21 +/- 0.73, with a p<0.0005. This represents a substantial 5-fold increase in genomic integration over normal healthy controls. The ratio for the total HIV-1 patients or as stratified based on the use of antiretroviral therapy (T) or not (NT), was not statistically different from the normal healthy controls. Note. Reproduced from “Further evidence that human endogenous retrovirus K102 is a replication competent foamy virus that may antagonize HIV-1 replication.”, by Laderoute MP, Larocque LJ, Giulivi A, Diaz-Mitoma F. Open AIDS J. 2015;9:112-22 [10]. CC BY Author.
Figure 5.
Evidence that HERV-K102 replicative activity is associated with sterilizing immunity (resistance to HIV-1 acquisition) in a HESN cohort [10]. DNA was isolated from plasma. The ddCt real-time PCR method employed the 18S RNA gene to control for genomic equivalents and used uracil N-glycosylase (UNG) in the PCR buffer to digest the cDNA genomes of HERV-K102 particles which left only genomic DNA in the DNA extracted from plasma. The mean ddCt qPCR ratio (for HERV-K102 pol) of 30 normal healthy controls was 0.88 +/-0.37. In contrast, on average for the female Commercial Sex Workers (CSW) HIV-1 Exposed Seronegative (HESN) cohort it was 4.21 +/- 0.73, with a p<0.0005. This represents a substantial 5-fold increase in genomic integration over normal healthy controls. The ratio for the total HIV-1 patients or as stratified based on the use of antiretroviral therapy (T) or not (NT), was not statistically different from the normal healthy controls. Note. Reproduced from “Further evidence that human endogenous retrovirus K102 is a replication competent foamy virus that may antagonize HIV-1 replication.”, by Laderoute MP, Larocque LJ, Giulivi A, Diaz-Mitoma F. Open AIDS J. 2015;9:112-22 [10]. CC BY Author.
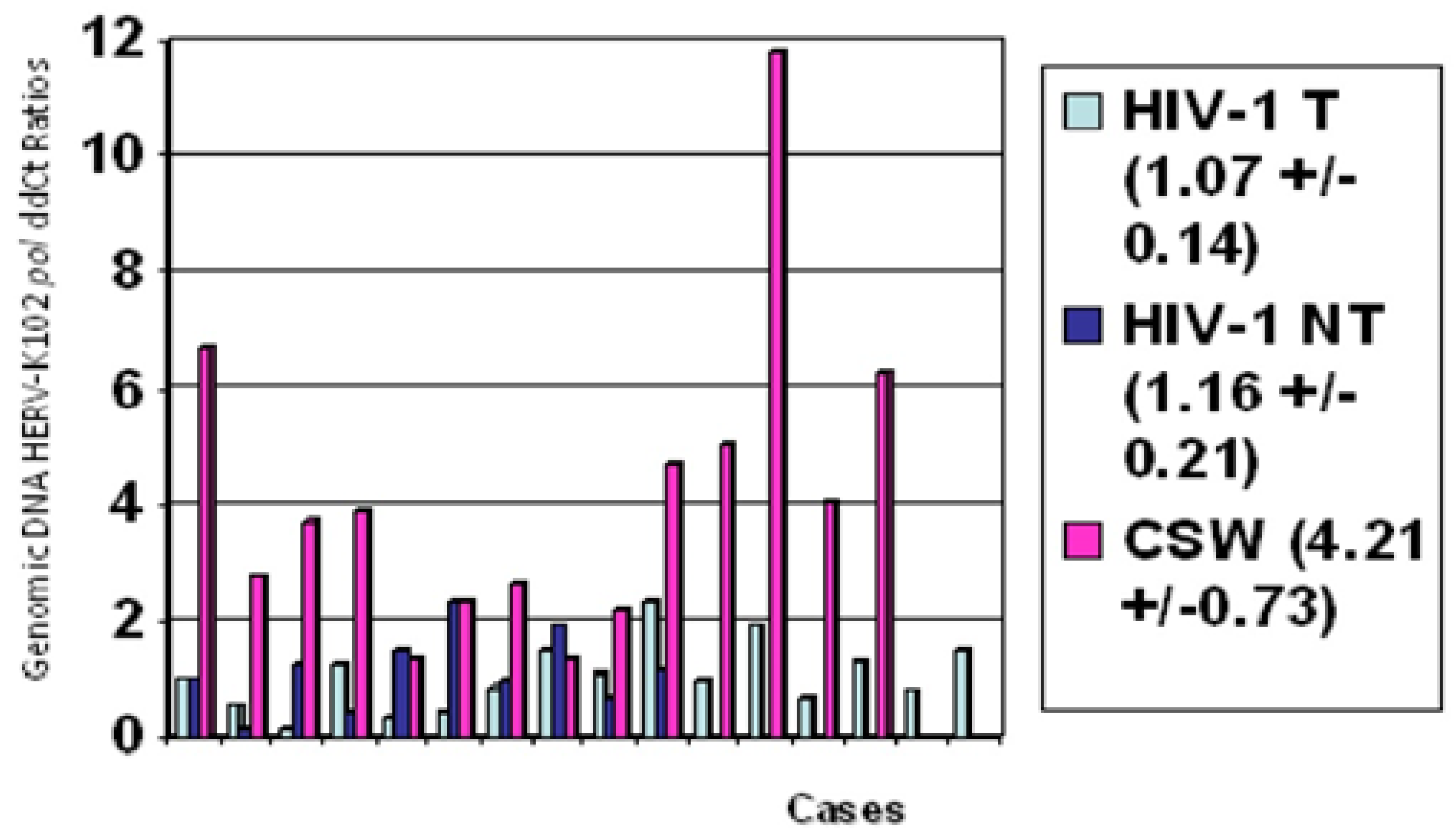
Figure 6.
HERV-K102 particles liberated from cultured cord blood mononuclear cells (CB) by freeze-thaw lysis (but not from supernatants) may rapidly induce cell lysis at 24 hours but not at 16 hours in susceptible cells. HFL-1 = human fetal lung fibroblasts; MRC-5 = human fetal lung fibroblast; Vero = green monkey kidney epithelial cells. Representative of four full sets of data conducted in duplicate. Estimated multiplicity of infection (MOI) of 2. Supernatants from the cultured CB did not induce lysis as no HERV-K102 particles bud through the cell surface of the foamy macrophages by electron microscopy and because significant cell death did not occur until day 7. Freeze-thaw treated uncultured CB (day 0) which does not exhibit HERV-K102 particles does not lyse the MRC-5 cells, but day 4 cultured CB does. This demonstrates that the particles mediate lysis and is not a non-specific factor of the culture media. Note. From: Laderoute MP. What you need to know about the HERV-K102 innate immunity protector system of macrophages against RNA pandemic viruses. hervk102.substack.com, January 24, 2022. https://hervk102.substack.com/p/what-you-need-to-know-about-the-herv [79].
Figure 6.
HERV-K102 particles liberated from cultured cord blood mononuclear cells (CB) by freeze-thaw lysis (but not from supernatants) may rapidly induce cell lysis at 24 hours but not at 16 hours in susceptible cells. HFL-1 = human fetal lung fibroblasts; MRC-5 = human fetal lung fibroblast; Vero = green monkey kidney epithelial cells. Representative of four full sets of data conducted in duplicate. Estimated multiplicity of infection (MOI) of 2. Supernatants from the cultured CB did not induce lysis as no HERV-K102 particles bud through the cell surface of the foamy macrophages by electron microscopy and because significant cell death did not occur until day 7. Freeze-thaw treated uncultured CB (day 0) which does not exhibit HERV-K102 particles does not lyse the MRC-5 cells, but day 4 cultured CB does. This demonstrates that the particles mediate lysis and is not a non-specific factor of the culture media. Note. From: Laderoute MP. What you need to know about the HERV-K102 innate immunity protector system of macrophages against RNA pandemic viruses. hervk102.substack.com, January 24, 2022. https://hervk102.substack.com/p/what-you-need-to-know-about-the-herv [79].
Figure 7.
A human sebaceous gland [82] demonstrating sebocytes have the distinctive morphology of LB-FMs. Sebocytes are programmed to cell death on day 6/7 [92] which generates sebum. This cell lysis involves a novel cytoplasmic induced apoptosis around day 6-7 involving DNASE2 and LAMP1 [100]. These cells have the identical distinct morphology of the LB-FMs of Figure 4 and differentially express the same DEGS including ERVK-7 (HERV-K102), BSG, VDR, CD14, CD16, CD68, TP53, both in vivo and in vitro [84,85]. Thus, sebocytes constitutively express and release HERV-K102 particles providing a first line of innate immunity defense at the mucosa. This directly corroborates the significance of the HERV-K102 front-line response to emerging and/or pandemic pathogens. Note. From https://commons.wikimedia.org/wiki/File:Insertion_of_sebaceous_glands_into_hair_shaft_x10.jpg [82]. This work has been released into the public domain by its author, Kilbad. This applies worldwide. CC-BY 4.0.
Figure 7.
A human sebaceous gland [82] demonstrating sebocytes have the distinctive morphology of LB-FMs. Sebocytes are programmed to cell death on day 6/7 [92] which generates sebum. This cell lysis involves a novel cytoplasmic induced apoptosis around day 6-7 involving DNASE2 and LAMP1 [100]. These cells have the identical distinct morphology of the LB-FMs of Figure 4 and differentially express the same DEGS including ERVK-7 (HERV-K102), BSG, VDR, CD14, CD16, CD68, TP53, both in vivo and in vitro [84,85]. Thus, sebocytes constitutively express and release HERV-K102 particles providing a first line of innate immunity defense at the mucosa. This directly corroborates the significance of the HERV-K102 front-line response to emerging and/or pandemic pathogens. Note. From https://commons.wikimedia.org/wiki/File:Insertion_of_sebaceous_glands_into_hair_shaft_x10.jpg [82]. This work has been released into the public domain by its author, Kilbad. This applies worldwide. CC-BY 4.0.
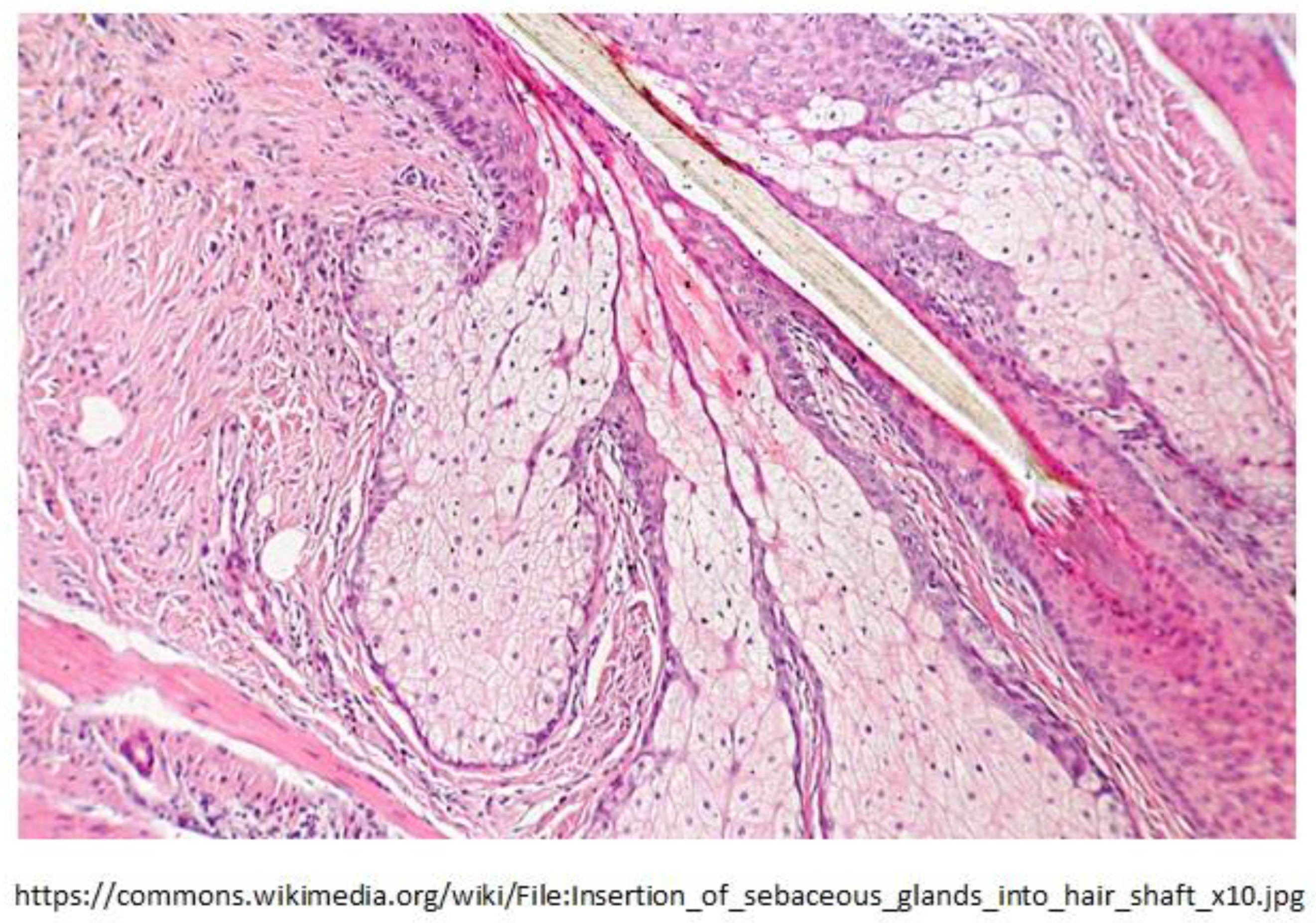
Figure 8.
Evidence for past integration and excision of HERV-K102 at orthologous positions (1q22) in the Neanderthal and Denisovan genomes. Direct repeats (gggatg) flank the orthologous HERV-K102 sequence in the human and extinct hominin genomes. The nucleotides marked in red were missing from the Denisovan orthologous position but present in the Neanderthal genome. Most of the HERV-K102 provirus was missing in both extinct hominins with only a few nucleotides corresponding to the ends of the LTRs remaining intact. “n” stands for missing nucleotides. Inquiry was made of the Altai Neanderthal or Denisovan genome at http://bioinf.eva.mpg.de/fetchseq/ on chromosome 1 strand at 155,596,423 to 155,605,644. AF164610 GenBank LTR flanking sequences used: 5’ LTR sequence = AF095801 and 3’ LTR sequence = AF095802. Note. From Laderoute MP. What you need to know about the HERV-K102 innate immunity protector system of macrophages against RNA pandemic viruses. hervk102.substack.com, January 24, 2022. https://hervk102.substack.com/p/what-you-need-to-know-about-the-herv [79].
Figure 8.
Evidence for past integration and excision of HERV-K102 at orthologous positions (1q22) in the Neanderthal and Denisovan genomes. Direct repeats (gggatg) flank the orthologous HERV-K102 sequence in the human and extinct hominin genomes. The nucleotides marked in red were missing from the Denisovan orthologous position but present in the Neanderthal genome. Most of the HERV-K102 provirus was missing in both extinct hominins with only a few nucleotides corresponding to the ends of the LTRs remaining intact. “n” stands for missing nucleotides. Inquiry was made of the Altai Neanderthal or Denisovan genome at http://bioinf.eva.mpg.de/fetchseq/ on chromosome 1 strand at 155,596,423 to 155,605,644. AF164610 GenBank LTR flanking sequences used: 5’ LTR sequence = AF095801 and 3’ LTR sequence = AF095802. Note. From Laderoute MP. What you need to know about the HERV-K102 innate immunity protector system of macrophages against RNA pandemic viruses. hervk102.substack.com, January 24, 2022. https://hervk102.substack.com/p/what-you-need-to-know-about-the-herv [79].
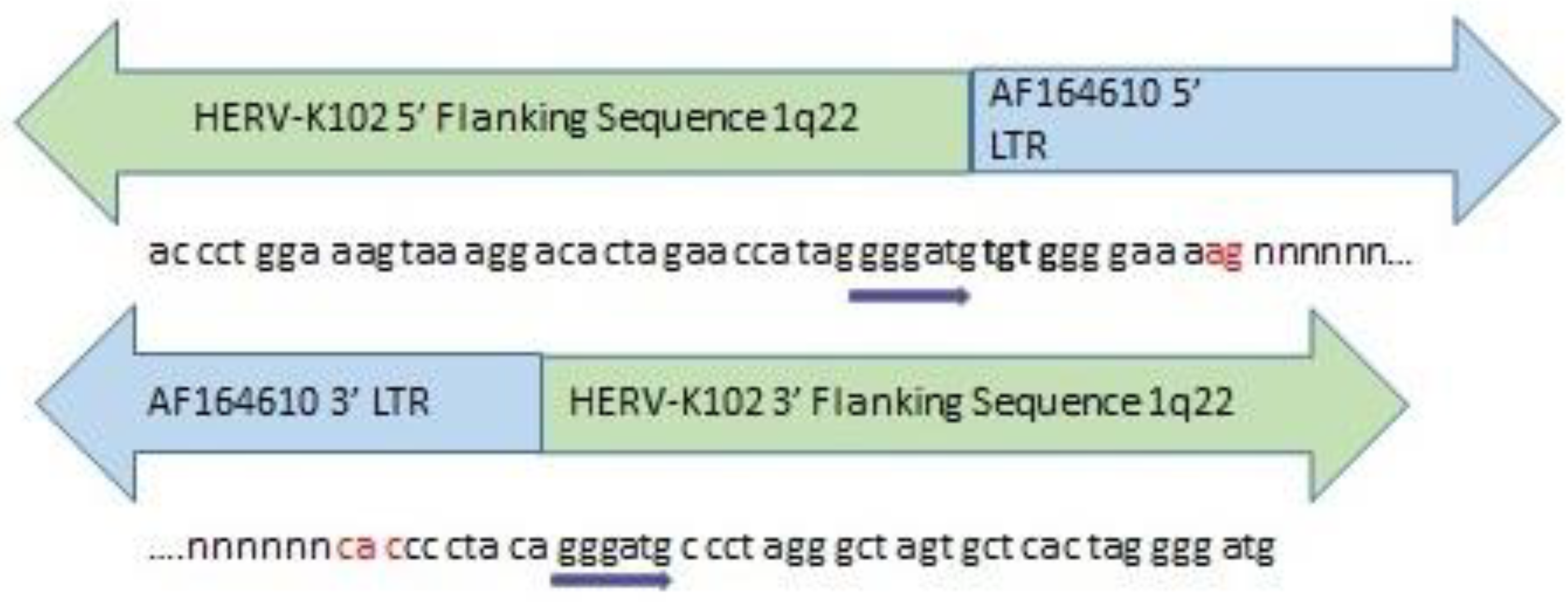
Figure 9.
3-D visualization of HERV-K102 Env by AlphaFold potentially reveals distinct conformations between the particle associated Env (P63135, left panel) and those putatively expressed at the cell surface of virally infected or tumor cells (P61567, right panel). The envelope protein (Env) of HERV-K102 becomes enzymatically cleaved into a transmembrane and a surface unit (TM and SU, respectively) domain at least when associated with particles. The conformation of the HERV-K102 Env protein after this cleavage maturation of particles has not been considered in the AlphaFold software [145,146] but could make the differences more pronounced. Nevertheless, here the view is that SU domain sits on top of the TM domain (still connected) as if one was looking down at the protein from the top of the plasma membrane. The P63135 (pol-env) Env produced from the expression of the proviral genomic sequence (of particles) begins with an additional KRASTE sequence which is not present in the P61567 sequence (spliced envelope). Yellow arrows indicate the location of the ML4 peptide epitope. Pink arrows indicate the location of the C amino acid in the ML5 peptide epitope. The orange arrows indicate the location of a C amino acid that may be closest to the C amino acid in the ML5 peptide that might be available for a di-sulphide bridge which may change the conformation of HERV-K102 Env. Note on the left side for the putative particle associated Env (P63135) the C amino acid of the ML5 peptide is very proximal to C1042 potentially consistent with a di-sulphide bridge. In contrast at the right, for the putative cell surface Env (P61567), the C amino acid of the ML5 peptide appears to be far away from the closest C amino acid at C171 consistent with the lack of a di-sulphide bridge. Thus, it is possible that the ML4 and ML5 epitopes on the cell surface Env are accessible but not on the particles, although this needs to be conclusively shown. Here it is proposed that the ML4 (the 5’ start of the Env protein) and ML5 epitopes are cryptic on the particles (left) but accessible on the cell surface of the virally infected or tumor cell (right). This conjecture is being made is because an antibody to HERV-K102 Env that can directly trigger apoptosis in virally infected or cancer cells [142] would not provide much good for the host if the same antibody cleared the protector HERV-K102 particles. From: https://alphafold.ebi.ac.uk/. Note. All of the data provided is freely available for both academic and commercial use under Creative Commons Attribution 4.0 (CC-BY 4.0) licence terms. Jumper, J et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021) [145]. Varadi, M et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research (2022) [146].
Figure 9.
3-D visualization of HERV-K102 Env by AlphaFold potentially reveals distinct conformations between the particle associated Env (P63135, left panel) and those putatively expressed at the cell surface of virally infected or tumor cells (P61567, right panel). The envelope protein (Env) of HERV-K102 becomes enzymatically cleaved into a transmembrane and a surface unit (TM and SU, respectively) domain at least when associated with particles. The conformation of the HERV-K102 Env protein after this cleavage maturation of particles has not been considered in the AlphaFold software [145,146] but could make the differences more pronounced. Nevertheless, here the view is that SU domain sits on top of the TM domain (still connected) as if one was looking down at the protein from the top of the plasma membrane. The P63135 (pol-env) Env produced from the expression of the proviral genomic sequence (of particles) begins with an additional KRASTE sequence which is not present in the P61567 sequence (spliced envelope). Yellow arrows indicate the location of the ML4 peptide epitope. Pink arrows indicate the location of the C amino acid in the ML5 peptide epitope. The orange arrows indicate the location of a C amino acid that may be closest to the C amino acid in the ML5 peptide that might be available for a di-sulphide bridge which may change the conformation of HERV-K102 Env. Note on the left side for the putative particle associated Env (P63135) the C amino acid of the ML5 peptide is very proximal to C1042 potentially consistent with a di-sulphide bridge. In contrast at the right, for the putative cell surface Env (P61567), the C amino acid of the ML5 peptide appears to be far away from the closest C amino acid at C171 consistent with the lack of a di-sulphide bridge. Thus, it is possible that the ML4 and ML5 epitopes on the cell surface Env are accessible but not on the particles, although this needs to be conclusively shown. Here it is proposed that the ML4 (the 5’ start of the Env protein) and ML5 epitopes are cryptic on the particles (left) but accessible on the cell surface of the virally infected or tumor cell (right). This conjecture is being made is because an antibody to HERV-K102 Env that can directly trigger apoptosis in virally infected or cancer cells [142] would not provide much good for the host if the same antibody cleared the protector HERV-K102 particles. From: https://alphafold.ebi.ac.uk/. Note. All of the data provided is freely available for both academic and commercial use under Creative Commons Attribution 4.0 (CC-BY 4.0) licence terms. Jumper, J et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021) [145]. Varadi, M et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research (2022) [146].
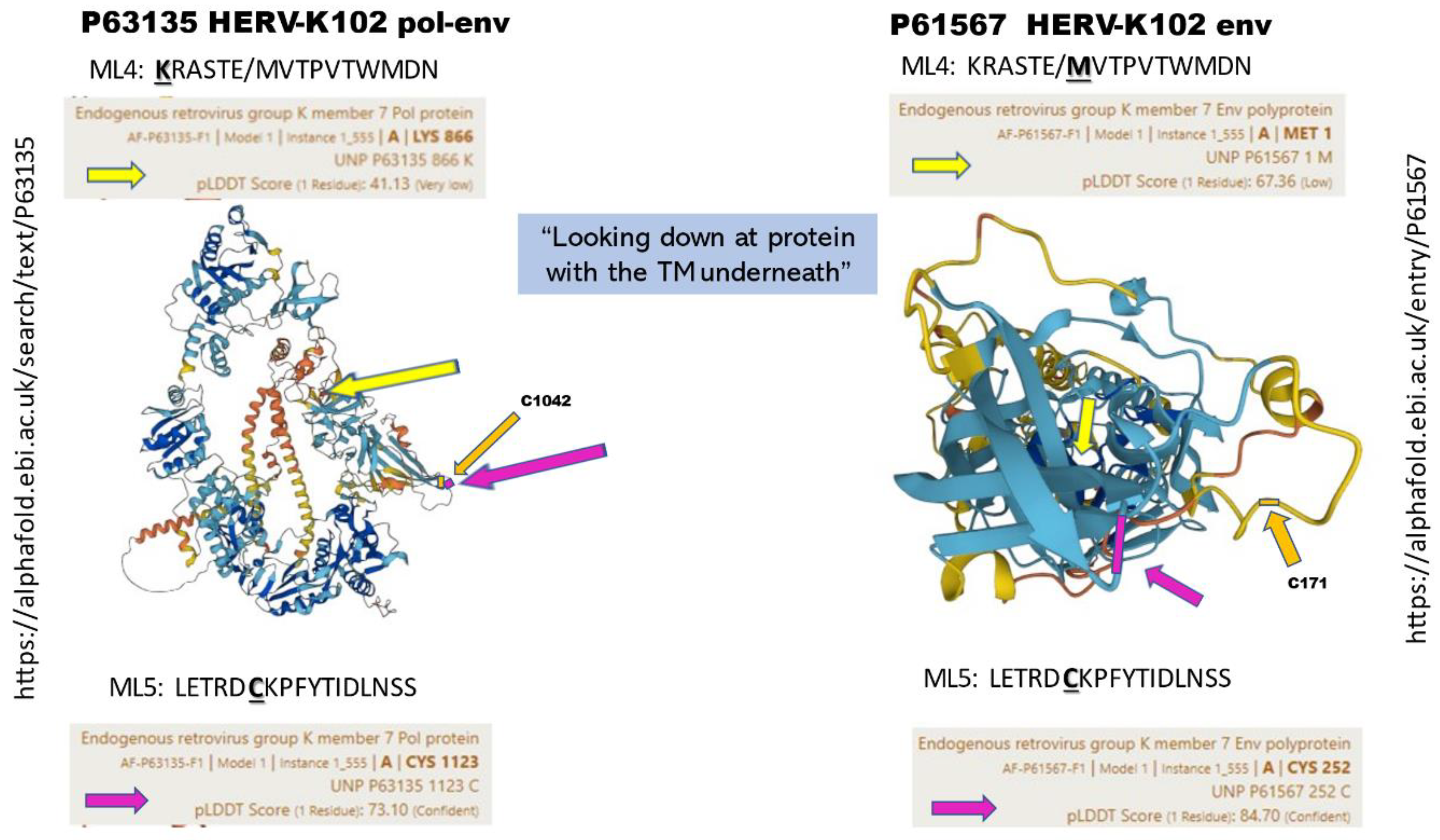
Figure 10.
Antibodies to HERV-K102 envelope may neutralize and clear SARS-CoV-2 virions that bud from the surface of human cells but not from other species. Virus infected cells or tumor cells express HERV-K102 envelope protein on their cell surface which serves to induce clearance or apoptosis by envelope specific T cells and/or antibodies [141,143]. Antibodies to HERV-K102 Env have been demonstrated in HIV-1 patients and those with other viral infections [67]. In patients with mild COVID disease, both IgA and IgG antibodies reactive with HERV-K102/HERV-K18 envelope are universally detected in saliva [147]. Viruses like SARS-CoV-2 and HIV-1 that bud through the cell surface (the so-called enveloped RNA viruses) likely contain HERV-K102 envelope (or 95- 97% identical HML-2 Env) in their virions which they acquire during the budding process. Thus, in mild COVID-19, the innate antibodies to HML-2 Env can neutralize and clear the virions in the upper respiratory tract before the onset of IgG to spike protein as was reported by Wolfel et al [41]. See text for discussion of ML4 and ML5 cryptic epitopes on the HERV-K102 particles but which were revealed by denaturation. Note. New image created by author using cell and antibody icons from Biorender.com.
Figure 10.
Antibodies to HERV-K102 envelope may neutralize and clear SARS-CoV-2 virions that bud from the surface of human cells but not from other species. Virus infected cells or tumor cells express HERV-K102 envelope protein on their cell surface which serves to induce clearance or apoptosis by envelope specific T cells and/or antibodies [141,143]. Antibodies to HERV-K102 Env have been demonstrated in HIV-1 patients and those with other viral infections [67]. In patients with mild COVID disease, both IgA and IgG antibodies reactive with HERV-K102/HERV-K18 envelope are universally detected in saliva [147]. Viruses like SARS-CoV-2 and HIV-1 that bud through the cell surface (the so-called enveloped RNA viruses) likely contain HERV-K102 envelope (or 95- 97% identical HML-2 Env) in their virions which they acquire during the budding process. Thus, in mild COVID-19, the innate antibodies to HML-2 Env can neutralize and clear the virions in the upper respiratory tract before the onset of IgG to spike protein as was reported by Wolfel et al [41]. See text for discussion of ML4 and ML5 cryptic epitopes on the HERV-K102 particles but which were revealed by denaturation. Note. New image created by author using cell and antibody icons from Biorender.com.
Figure 11.
HERV-K102 particles may utilize heparan sulphate [81] to enter SARS-CoV-2 infected cells while innate antibodies and T cells recognize HERV-K102/HML-2 envelope (Env) and may directly trigger apoptosis. HERV-K102 particles enter cells through its receptor heparan sulphate which is widely expressed on most cell types [81] and is like other non-pathogenic foamy viruses. Entry of HERV-K102 particles into normal cells results in activation of the interferon system [136] and integration (arming) [10] while in virus-infected cells or tumors entry is proposed to be associated with lysis (see Figure 6 for cytopathic effects of HERV-K102 particles). Most likely the relevant HERV-K HML-2 Env antibodies are to HERV-K102 specific epitopes like ML4 and ML5 accessible on the cell surface Env (P61567) but which are proposed to be cryptic on the particles (P63135 pol-env). Note. New image created by author using cell and antibody icons from Biorender.com.
Figure 11.
HERV-K102 particles may utilize heparan sulphate [81] to enter SARS-CoV-2 infected cells while innate antibodies and T cells recognize HERV-K102/HML-2 envelope (Env) and may directly trigger apoptosis. HERV-K102 particles enter cells through its receptor heparan sulphate which is widely expressed on most cell types [81] and is like other non-pathogenic foamy viruses. Entry of HERV-K102 particles into normal cells results in activation of the interferon system [136] and integration (arming) [10] while in virus-infected cells or tumors entry is proposed to be associated with lysis (see Figure 6 for cytopathic effects of HERV-K102 particles). Most likely the relevant HERV-K HML-2 Env antibodies are to HERV-K102 specific epitopes like ML4 and ML5 accessible on the cell surface Env (P61567) but which are proposed to be cryptic on the particles (P63135 pol-env). Note. New image created by author using cell and antibody icons from Biorender.com.
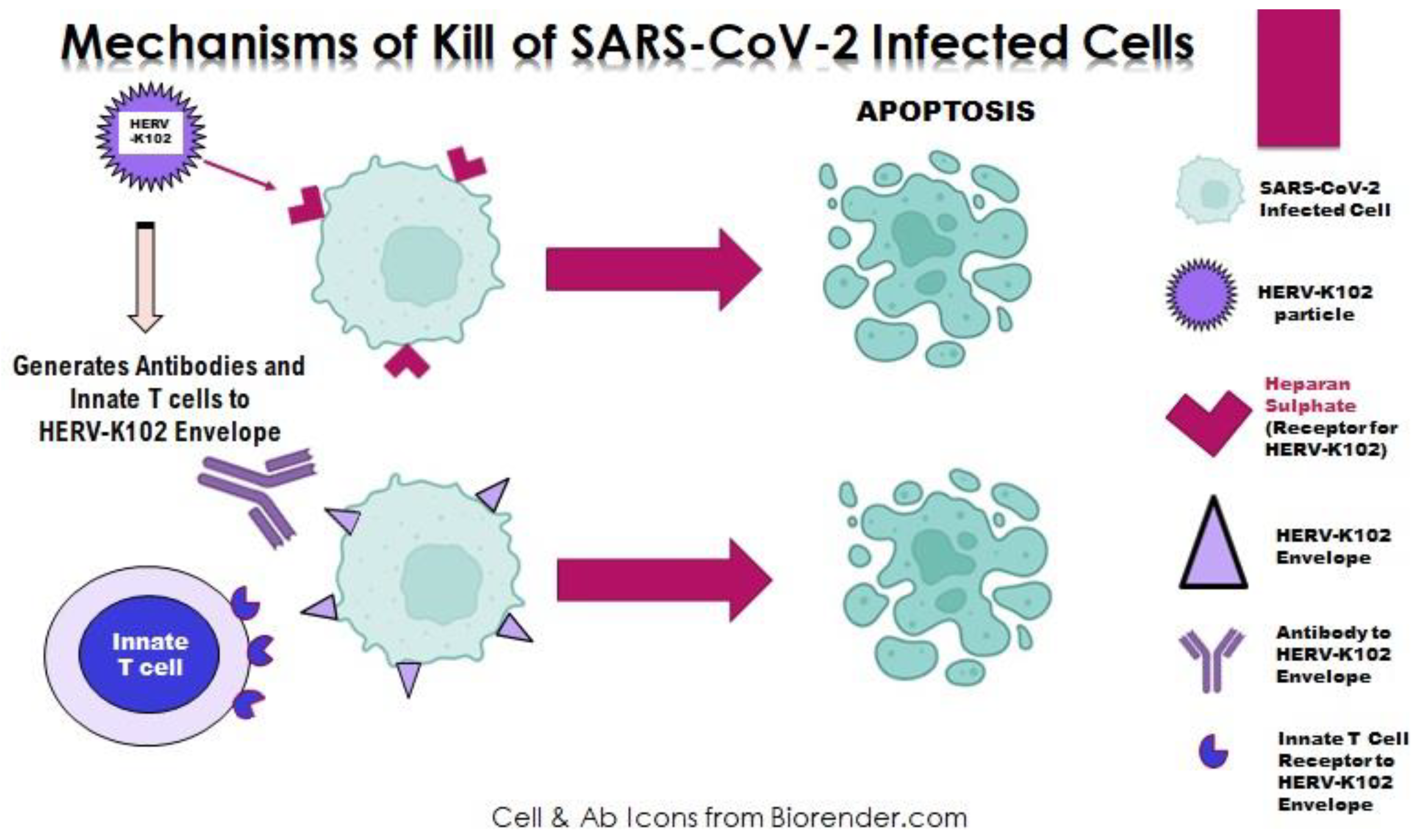
Figure 12.
The new immunosenescence paradigm, 2015 [185]. In the new immunosenescence paradigm, active alpha-fetoprotein (AFP) was proposed to mediate immunosenescence of macrophages, defined as the failed lytic release of HERV-K102 particles from foamy macrophages [185,186]. AFP blocks apoptosis in macrophages [187,188]. The activity of AFP depends in part on the DHEA/cortisol ratio which diminishes with age and/or stress. This is because cortisol induces AFP while DHEA binds and renders AFP inactive [185]. Thus, with age and/or stress there will be more active AFP in the system and a higher risk of immunosenescence. Flavonoids and zinc may reverse immunosenescence by binding and inhibiting AFP activity and thus, will appear to have antiviral properties. Recent evidence is also consistent with ivermectin reversing immunosenescence [191]. Note. Reproduced with permission from “A new paradigm about HERV-K102 particle production and blocked release to explain cortisol mediated immunosenescence and age-associated risk of chronic disease”, by Laderoute MP. Discov Med. 2015;20:379-91 [185]. © 2023 Discovery Medicine.
Figure 12.
The new immunosenescence paradigm, 2015 [185]. In the new immunosenescence paradigm, active alpha-fetoprotein (AFP) was proposed to mediate immunosenescence of macrophages, defined as the failed lytic release of HERV-K102 particles from foamy macrophages [185,186]. AFP blocks apoptosis in macrophages [187,188]. The activity of AFP depends in part on the DHEA/cortisol ratio which diminishes with age and/or stress. This is because cortisol induces AFP while DHEA binds and renders AFP inactive [185]. Thus, with age and/or stress there will be more active AFP in the system and a higher risk of immunosenescence. Flavonoids and zinc may reverse immunosenescence by binding and inhibiting AFP activity and thus, will appear to have antiviral properties. Recent evidence is also consistent with ivermectin reversing immunosenescence [191]. Note. Reproduced with permission from “A new paradigm about HERV-K102 particle production and blocked release to explain cortisol mediated immunosenescence and age-associated risk of chronic disease”, by Laderoute MP. Discov Med. 2015;20:379-91 [185]. © 2023 Discovery Medicine.
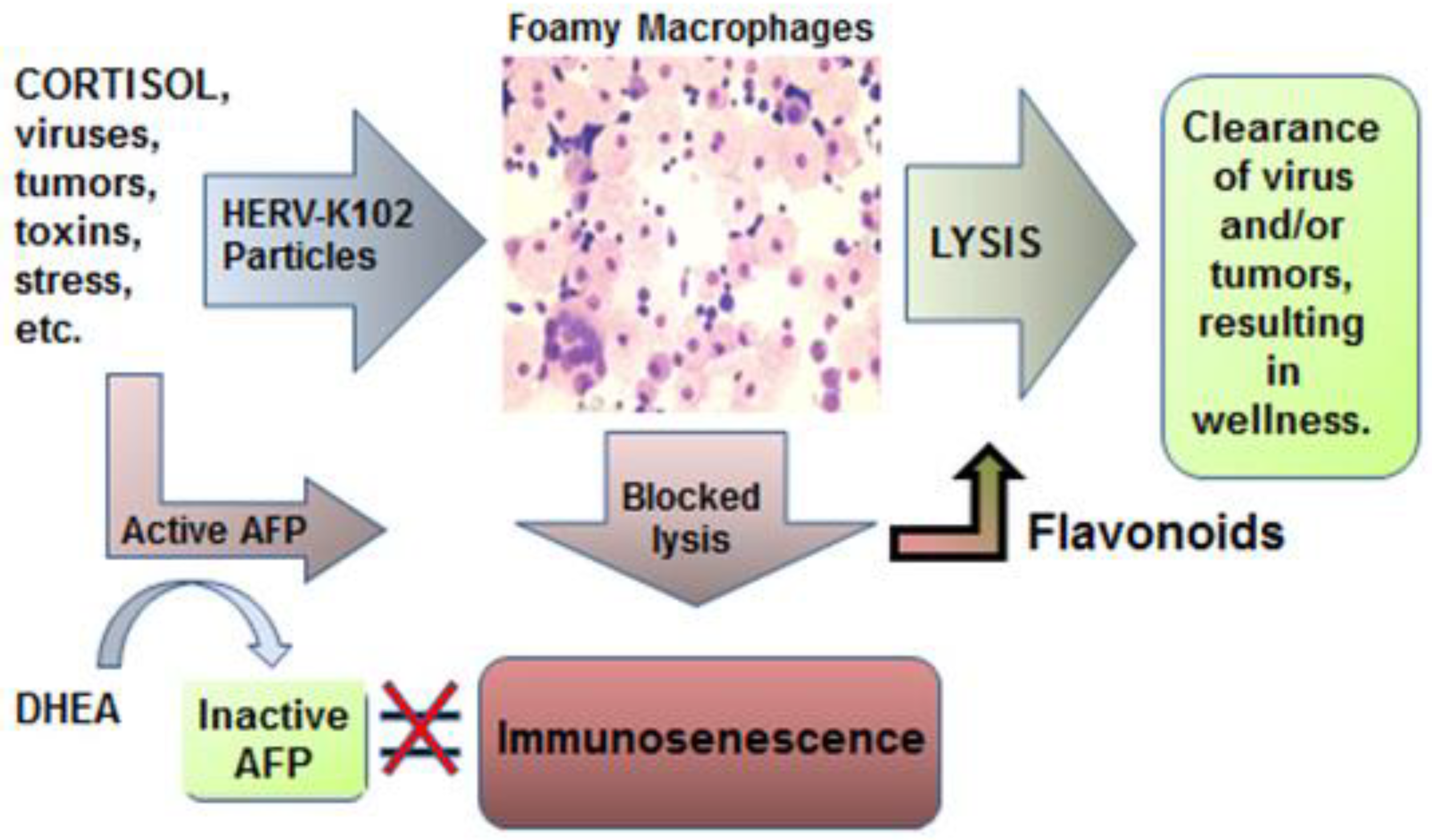
Figure 13.
SARS-CoV-2 infection of the LB-FMs by ADE causes the transition to LB+FMs [196] blocked by the activated VDR [171]. SARS-CoV-2 converts the M1-like protector foamy macrophages (LB-FMs) to M2-like LB+FMs which upregulates PPARA [61], and where the latter becomes a factory for SARS-CoV-2 replication [196]. When the levels of vitamin D3 are sufficient, the activated vitamin D receptor (VDR) can prevent the phosphorylation of MAPK8 blocking the transition from LB-FMs to LB+FMs [165]. Note it is possible that ivermectin may do the same as MAPK8 is on the ivermectin-protein string interaction network [191]. SARS-CoV-2 infection of the LB-FMs by ADE blocks programmed cell death [197] abrogating the release of the HERV-K102 particles and at the same time inducing immunosenescence of the macrophages which exacerbates chronic illness. The conversion to LB+FMs also does the same; but now SARS-CoV-2 particles are produced and released by cell surface budding and where NFκβ1 acts as the transcription factor for the release of IL1β, TNF-α, and CXCL8 [61]. Note also that SARS-CoV-2 represses (epigenetically) the PPARγ-NR3C1-RXRA cistrome via SUMO1 which interacts with SARS-CoV-2 nucleoprotein and where SUMO1 is a partner of PPARγ [199]. This also contributes to uncontrolled inflammation. Overall, this hijacking of the critical innate defense mechanism also creates an immunologically priviledged site [196] possibly secondary to the fact that foamy macrophages may not express the spliced HERV-K102 envelope on their cell surface (unpublished flow cytometry data on the LB-FMs). Note. Image is newly created by author but incorporates an image of one LB-FM excised from Figure 1A of “Further evidence that human endogenous retrovirus K102 is a replication competent foamy virus that may antagonize HIV-1 replication.”, by Laderoute MP, Larocque LJ, Giulivi A, Diaz-Mitoma F. Open AIDS J. 2015;9:112-22 [10]. CC BY Author. .
Figure 13.
SARS-CoV-2 infection of the LB-FMs by ADE causes the transition to LB+FMs [196] blocked by the activated VDR [171]. SARS-CoV-2 converts the M1-like protector foamy macrophages (LB-FMs) to M2-like LB+FMs which upregulates PPARA [61], and where the latter becomes a factory for SARS-CoV-2 replication [196]. When the levels of vitamin D3 are sufficient, the activated vitamin D receptor (VDR) can prevent the phosphorylation of MAPK8 blocking the transition from LB-FMs to LB+FMs [165]. Note it is possible that ivermectin may do the same as MAPK8 is on the ivermectin-protein string interaction network [191]. SARS-CoV-2 infection of the LB-FMs by ADE blocks programmed cell death [197] abrogating the release of the HERV-K102 particles and at the same time inducing immunosenescence of the macrophages which exacerbates chronic illness. The conversion to LB+FMs also does the same; but now SARS-CoV-2 particles are produced and released by cell surface budding and where NFκβ1 acts as the transcription factor for the release of IL1β, TNF-α, and CXCL8 [61]. Note also that SARS-CoV-2 represses (epigenetically) the PPARγ-NR3C1-RXRA cistrome via SUMO1 which interacts with SARS-CoV-2 nucleoprotein and where SUMO1 is a partner of PPARγ [199]. This also contributes to uncontrolled inflammation. Overall, this hijacking of the critical innate defense mechanism also creates an immunologically priviledged site [196] possibly secondary to the fact that foamy macrophages may not express the spliced HERV-K102 envelope on their cell surface (unpublished flow cytometry data on the LB-FMs). Note. Image is newly created by author but incorporates an image of one LB-FM excised from Figure 1A of “Further evidence that human endogenous retrovirus K102 is a replication competent foamy virus that may antagonize HIV-1 replication.”, by Laderoute MP, Larocque LJ, Giulivi A, Diaz-Mitoma F. Open AIDS J. 2015;9:112-22 [10]. CC BY Author. .

Table 1.
The two over one dose ratios for COVID-19 vaccination in Canada: December 22, 2020 to August 1, 2021 (from Our World in Data [49]).
Table 1.
The two over one dose ratios for COVID-19 vaccination in Canada: December 22, 2020 to August 1, 2021 (from Our World in Data [49]).
| DATE | % At Least One Dose | % Two Doses | % One Dose | Two / One RATIO | EVENTS | NOTES |
| 22-Dec | 0.071 | None | 0.071 | N/A | ||
| 29-Dec | 0.19 | None | 0.19 | N/A | ||
| 03-Jan | 0.3 | None | 0.30 | N/A | EACM Decreases | |
| 10-Jan | 0.84 | 0.1 | 0.74 | 0.135 | ||
| 17-Jan | 1.5 | 0.6 | 0.9 | 0.667 | EACM temporarily flattened | |
| 24-Jan | 2.03 | 0.15 | 1.88 | 0.080 | ||
| 31-Jan | 2.3 | 0.3 | 2 | 0.150 | ||
| 07-Feb | 2.4 | 0.47 | 1.93 | 0.244 | ||
| 08-Feb | 2.4 | 0.5 | 1.9 | 0.263 | ||
| 10-Feb | 2.5 | 0.6 | 1.9 | 0.316 | Enters Neg EACM | |
| 14-Feb | 2.59 | 0.81 | 1.78 | 0.455 | ||
| 21-Feb | 2.9 | 1.1 | 1.8 | 0.611 | ||
| 22-Feb | 3 | 1.2 | 1.8 | 0.667 | Alpha Emerges | |
| 25-Feb | 3.2 | 1.3 | 1.9 | 0.684 | ||
| 26-Feb | 3.4 | 1.4 | 2 | 0.700 | ||
| 27-Feb | 3.5 | 1.4 | 2.1 | 0.667 | NACI Intervention around Feb 27 | |
| 28-Feb | 3.63 | 1.41 | 2.22 | 0.635 | ||
| 07-Mar | 4.85 | 1.52 | 3.33 | 0.456 | Uptick in Neg EACM | |
| 08-Mar | 5.1 | 1.6 | 3.5 | 0.457 | ||
| 11-Mar | 5.67 | 1.59 | 4.08 | 0.390 | ||
| 14-Mar | 6.5 | 1.6 | 4.9 | 0.327 | ||
| 15-Mar | 6.8 | 1.6 | 5.2 | 0.308 | ||
| 21-Mar | 8.83 | 1.7 | 7.13 | 0.238 | Lowest Neg EACM(Nadir) | |
| 22-Mar | 9.2 | 1.7 | 7.5 | 0.227 | Alpha Dominates | |
| 28-Mar | 11.81 | 1.81 | 10 | 0.181 | Uptick in Neg EACM | |
| 04-Apr | 15.07 | 1.92 | 13.15 | 0.146 | ||
| 11-Apr | 19.04 | 2.19 | 16.85 | 0.130 | Uptick in Neg EACM | |
| 18-Apr | 24 | 2.5 | 21.5 | 0.116 | ||
| 19-Apr | 25 | 2.5 | 22.5 | 0.111 | ||
| 25-Apr | 29.18 | 2.75 | 26.43 | 0.104 | ||
| 26-Apr | 30 | 2.8 | 27.2 | 0.103 | ||
| 28-Apr | 31 | 2.9 | 28.1 | 0.103 | Exit Neg EACM | |
| 02-May | 33.58 | 3.05 | 30.53 | 0.100 | Delta Emerges | |
| 03-May | 34 | 3.1 | 30.9 | 0.100 | ||
| 09-May | 39 | 3.4 | 35.6 | 0.096 | ||
| 16-May | 45 | 3.8 | 41.2 | 0.092 | ||
| 17-May | 46 | 3.9 | 42.1 | 0.093 | Max Alpha at 59% | |
| 23-May | 51 | 4.05 | 46.95 | 0.086 | ||
| 30-May | 56.69 | 5.45 | 51.24 | 0.106 | ||
| 02-Jun | 58.8 | 6.11 | 52.69 | 0.116 | ||
| 11-Jun | 63.87 | 10.82 | 53.05 | 0.204 | ||
| 14-Jun | 64.86 | 13.11 | 51.75 | 0.253 | ||
| 20-Jun | 66.29 | 18.85 | 47.44 | 0.397 | ||
| 26-Jun | 67.38 | 26.47 | 40.91 | 0.647 | ||
| 04-Jul | 68.31 | 35.02 | 33.29 | 1.052 | 50% of the Eligible Receive 2nd Dose | |
| 12-Jul | 69.27 | 44.33 | 24.94 | 1.777 | Delta Dominant | |
| 19-Jul | 69.99 | 50.61 | 19.38 | 2.611 | 50% Fully Vaccinated | |
| 25-Jul | 71 | 55 | 16 | 3.438 | ||
| 01-Aug | 71 | 59 | 12 | 4.917 |
NACI = National Advisory Committee on Immunization (Canada). EACM=Excess All-Cause Mortality. Neg= negative. N/A=not applicable. Max Alpha at 59% means the maximal portion of the sequenced variants was recorded at 59 percent. Note. All of the data provided [49] is freely available for both academic and commercial use under Creative Commons Attribution 4.0 (CC-BY 4.0) licence terms.
Table 2.
Office for National Statistics (ONS) UK mortality rates per 100,000 person-years: re-compiled rate ratios* of ever vaccinated (ever vax) over unvaccinated (unvax) January 1, 2021 to May 31, 2022.
Table 2.
Office for National Statistics (ONS) UK mortality rates per 100,000 person-years: re-compiled rate ratios* of ever vaccinated (ever vax) over unvaccinated (unvax) January 1, 2021 to May 31, 2022.
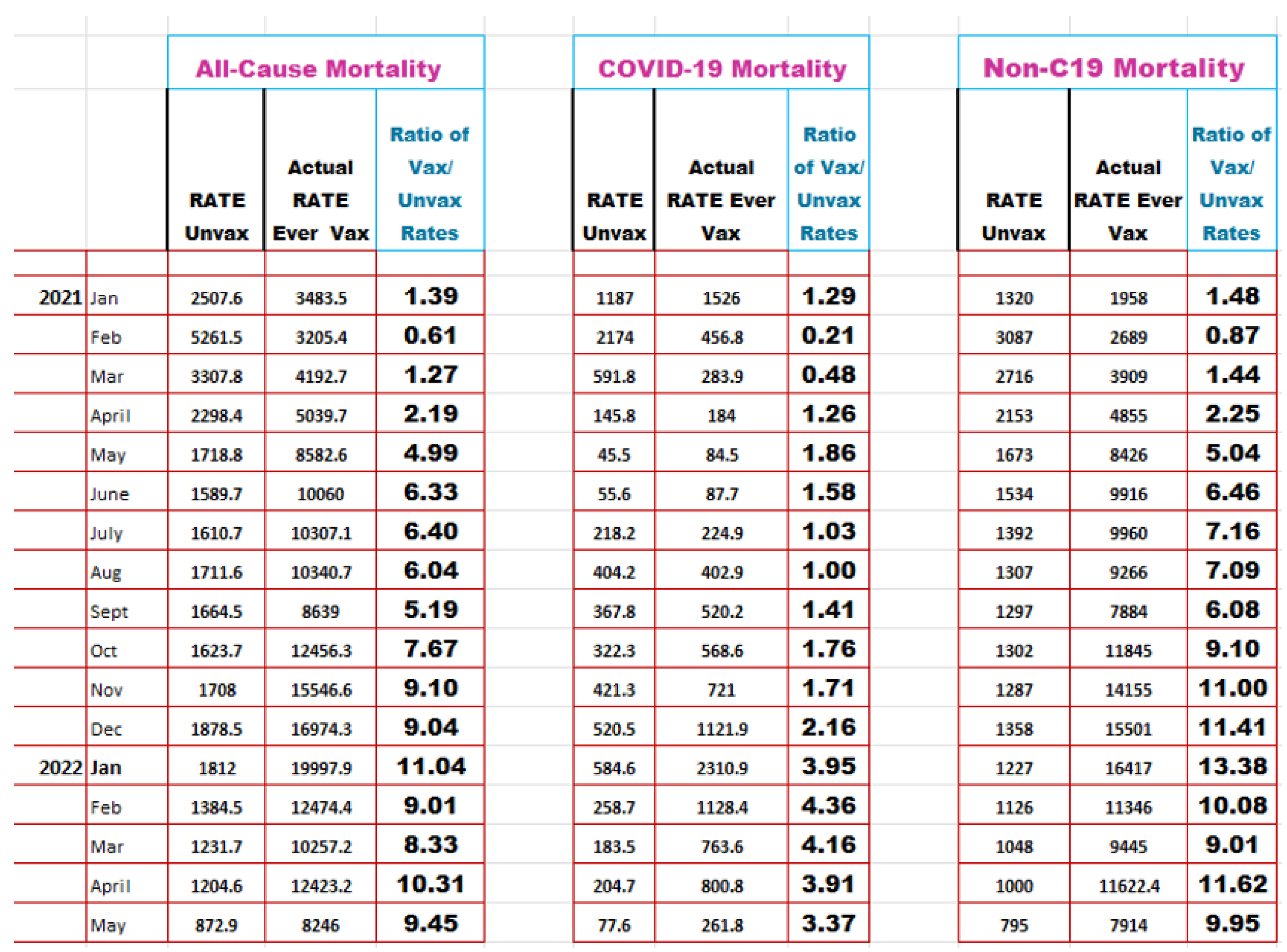 |
*Rates are per 100,000 Person-Years, and were re-compiled where all the rates for ever vaccinated were added up to provide totals (as the Ever Vaccinated total deaths reported by the ONS were erroneously undercalculated for unknown reasons). From: https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/deaths/bulletins/deathsinvolvingcovid19byvaccinationstatusengland/deathsoccurringbetween1january2021and31may2022 [203]. Note. CC-By 4.0 from https://www.nationalarchives.gov.uk/doc/open-government-licence/version/3/ and referencing bulletin “Office for National Statistics (ONS), released 6 July 2022, ONS website, statistical bulletin, Deaths involving COVID-19 by vaccination status, England: deaths occurring between 1 January 2021 and 31 May 2022”.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



