
Submitted:
03 December 2023
Posted:
05 December 2023
You are already at the latest version
Abstract
For the first time, this study identified seven potential ligands with high binding energies through the docking approach against the Crystal Structure of human Melanoma-associated antigen B1 (MAGEB1). The ligands—Imatinib, Bafatinib, Diosmin, Ginkgetin, Bilobetin, Amentoflavone, and Hypericin—showing strong binding energies around -10 kcal/mol, indicate a potentially robust interaction with the target protein. Moreover, the additional step of predicting toxicity properties using the pKCSM server provides valuable insights into the safety profiles of these ligands. From these results, the parameters for Diosmin: —Max. tolerated dose (human),- Oral Rat Acute Toxicity (LD50), and - Oral Rat Chronic Toxicity (LOAEL)—suggest a favorable safety profile, making it a potential candidate for further consideration against Melanoma. Moving forward, it's advisable to conduct further experimental validations, including in vitro and in vivo studies, to confirm the efficacy and safety of Diosmin and other potential ligands. Consideration of pharmacokinetics, metabolism, and specific mechanisms of action will contribute to a comprehensive understanding of these compounds' potential as therapeutic agents.
Keywords:
Melanoma
; pKCSM
; Docking approach
; Diosmin
; MAGEB1
1. Introduction
Melanoma is a type of skin cancer that arises from cells called melanocytes. These cells are responsible for the production of melanin, the pigment responsible for coloring the skin, hair and eyes. Melanoma can occur when melanocytes undergo genetic mutations that lead to uncontrolled growth and proliferation of cells [1,2].It is important to note that melanoma is the most dangerous type of skin cancer because of its ability to quickly spread to other organs via the lymphatic system or bloodstream. Therefore, early diagnosis and timely treatment are crucial to improve the chances of recovery [3].Risk factors for developing melanoma include excessive UV exposure, genetic predisposition, having many atypical moles, and immunosuppression. Prevention, which includes using sunscreen, limiting sun exposure and regularly self-examining your skin, is key to reducing your risk of developing melanoma. If you have a suspected skin lesion, it is important to see a doctor for evaluation and possible early intervention[1,2,3,4]. The present study focusing on the Crystal Structure of human Melanoma-associated antigen B1 (MAGEB1) [5] using molecular docking and virtual screening. Generally, these are computational approaches commonly employed in silico to predict the interactions between molecules and target proteins. Indeed, Molecular docking involves predicting the preferred orientation of one molecule to a second when bound to each other to form a stable complex [6,7]. In the context of drug discovery and development, it helps researchers understand how a potential drug or ligand might interact with a specific target protein, in this case, the MAGEB1 protein associated with melanoma.
Virtual screening is another computational technique that involves the rapid in silico assessment of a large number of compounds to identify those with potential biological activity against a specific target[8]. In the context of this study, virtual screening was used to screen hundreds of drugs and natural substances to identify those that have the highest likelihood of binding to MAGEB1.
By combining these computational techniques, we can potentially identify molecules that show promise in interacting with the MAGEB1 protein. However, it's crucial to recognize that these computational predictions need to be validated through experimental studies, such as in vitro and in vivo assays, to confirm the actual biological activity and safety of the identified compounds.
2. Material and Methods
Crystal Structure of human Melanoma-associated antigen B1 (MAGEB1) was taken by Protein Data Bank ( PDB Code 6R7T)and was accurately prepared before to perform rigid Blind Docking analysis by Autodock Vina [9] with Pyrx program [10].Drugs and natural compounds were taken by PUBCHEM Database in 3D format and they were prepared before to perform Docking analysis. The binding Center of Grid box to perform docking is: X (-18.2422), Y( 20.9925 ), Z ( -11.0192) with Size X ( 54.2067010403) , Size Y ( 52.8891197491) and Size Z( 43.4450090027).
3. Results and Discussion
The main objective is to identify potential ligands that show a high binding affinity with the MAGEB1 target through a virtual screening process[9,10].
The initial virtual screening involved 100 compounds, and seven potential ligands were subsequently selected based on their most negative energy affinity to the MAGEB1 target. These selected ligands include Imatinib, Amentoflavone, Bilobetin, Ginkgetin, Bafetinib, Diosmin, and Hypericin, with energy affinity ranging between -9.9 kcal/mol and -9.7 kcal/mol. Next, attention was focused on these seven compounds for further repeatability testing via docking. General Speaking, this additional step is important to confirm and consolidate the results obtained from the initial virtual screening. Repeatability testing is critical to ensuring results are consistent and reliable. The main results of this work that all seven substances, (Imatinib, Amentoflavone, Bilobetin, Ginkgetin, Bafetinib, Diosmin and Hypericin), have continued to show high binding energy with the MAGEB1 protein even after repeatability tests using docking is certainly interesting. This suggests that these molecules might have a strong affinity and stable interaction with the target protein with energy affinity ranging between -9.9 kcal/mol and -9.7 kcal/mol. Table 1 shows the docking results of selected potential ligands with MAGEB1 target by Autodock Vina [9] with Pyrx program [10].
However, it is important to consider several aspects before drawing definitive conclusions:
Experimental validation: Although docking is a useful tool for predicting molecular interactions, the results need to be validated through in vitro experimental experiments. Tests such as binding assays, functional assays, or X-ray crystallography experiments can confirm the effectiveness of binding.
Specificity: It is important to evaluate the specificity of these molecules towards the MAGEB1 protein. The specificity of the binding is critical to avoid unwanted side effects on other proteins in the body.
Pharmacokinetics and toxicity: The pharmacokinetics (absorption, distribution, metabolism and elimination) and toxicity of these molecules must be evaluated to ensure that they can be safely developed as drugs.
Mechanism of action: Understanding the mechanism of action of these molecules can provide crucial information on their efficacy and potential therapeutic impact.
The following step of this work focused on investigation of selected best ligands to narrow down the list for further investigation. The research was evaluated toxicity through theoretical studies with the pKCSM server[11], a valuable step in prioritizing compounds that may have a better safety profile. The parameters provided by pKCSM, such as AMES toxicity, Max. tolerated dose, hERG inhibition, oral toxicity in rats, hepatotoxicity, skin sensitization, and toxicity in T.Pyriformis, cover key aspects of safety evaluation[11].
The toxicity parameters you mentioned, such as AMES toxicity, Max. tolerated dose (MTD), inhibition of hERG I and hERG II, acute oral toxicity in rats, chronic oral toxicity in rats, hepatotoxicity, skin sensitization and toxicity in T.Pyriformis , cover several dimensions of safety and can be instrumental in selecting compounds for subsequent development. Let's look briefly at some of these parameters[11]:
- -
- AMES toxicity: Evaluates the mutagenic potential of molecules. A negative result is preferable, as it suggests a lower probability of causing genetic mutations.
- -
- Max. tolerated dose (MTD): Indicates the maximum dose that can be tolerated without causing serious toxic effects. This is important for estimating the safety of the doses used.
- -
- Inhibition of hERG I and hERG II: Inhibition of these ion channels may be associated with heart problems. A low inhibition potential is desirable.
- -
- Oral toxicity in rats: LD50 indicates the lethal dose for 50% of rats. LOAEL represents the lowest exposure level at which adverse effects are observed.
Hepatotoxicity: Evaluates the harmful potential of the molecule for the liver.
- -
- Skin sensitization: Tests whether the substance can cause allergic skin reactions.
- -
- Toxicity in T.Pyriformis: Evaluates the toxicity to aquatic organisms.
Table 2 shows the comparison of these parameters offer a comprehensive overview of the safety of the molecules under investigation and can guide decisions on the selection of the most promising substances for further development phases. However, it is always essential to consider that predictive models may have limitations, and therefore it is advisable to validate experimental results whenever possible.
A detailed comparative analysis of the toxic effects of the selected ligands, including Imatinib, Amentoflavone, Hypericin, Ginkgetin, Diosmin, Bafetinib, and Bilobetin were showed in Table . The parameters considered, such as Oral Rat Chronic Toxicity (LOAEL), Oral Rat Acute Toxicity (LD50), and Max. tolerated dose (human), provide valuable insights into the potential safety profiles of these compounds. From these results by pKCSM , it seems that Diosmin has shown high values for Oral Rat Chronic Toxicity (LOAEL), Oral Rat Acute Toxicity (LD50), and Max. tolerated dose (human). These high values suggest that Diosmin may have a lower potential for toxicity in these specific contexts, making it a candidate that could be well-tolerated by the human organism. It showed a Max. tolerated dose(human) with a value of 0.565 (logmg/kg/day) ( See below Table 1).
It's crucial to interpret these results in the context of the specific experimental conditions and the relevance of these toxicity parameters to human health. Additionally, the actual concentrations and dosages used in your experiments and their relationship to these toxicity values should be carefully considered.
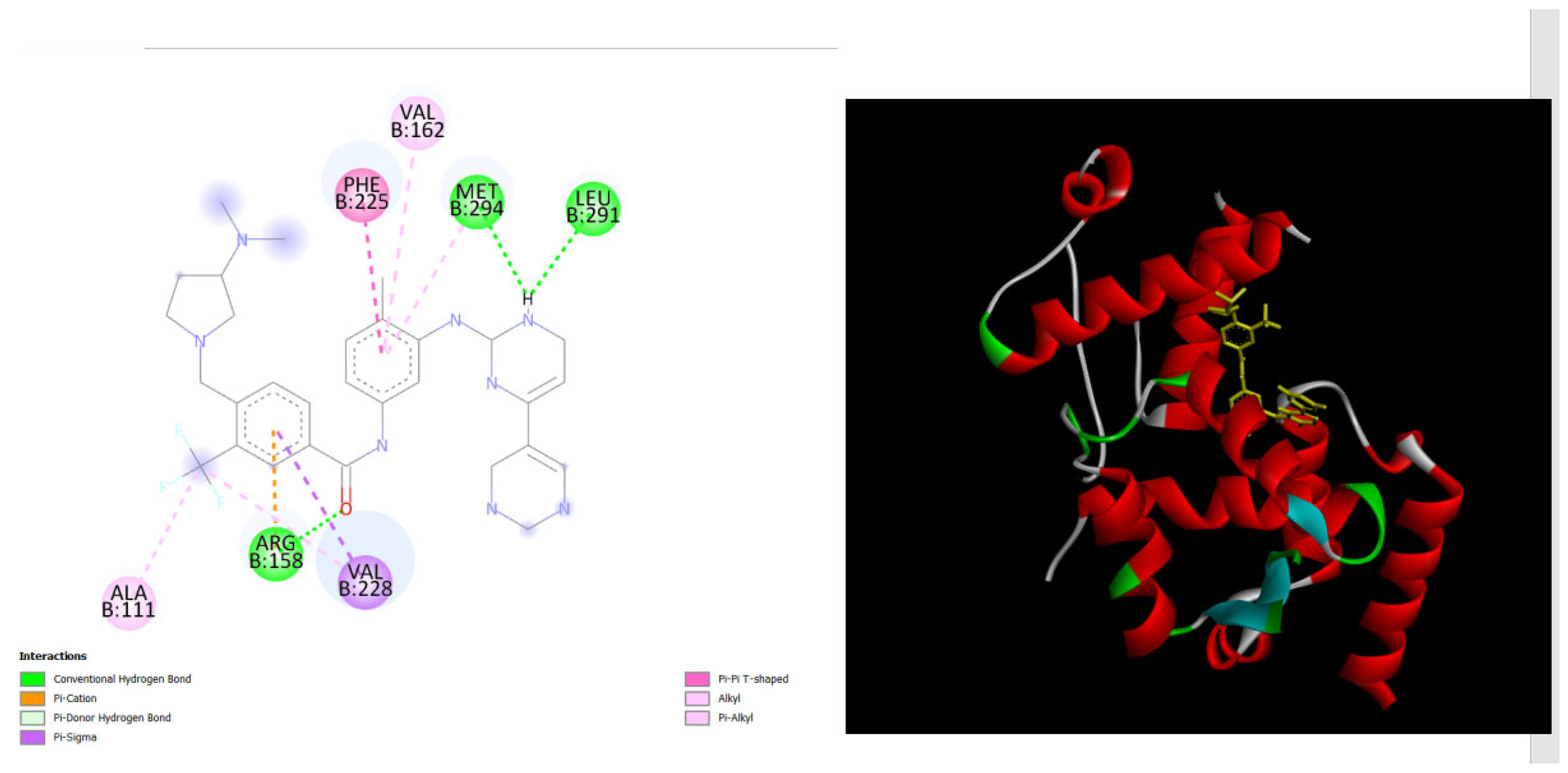
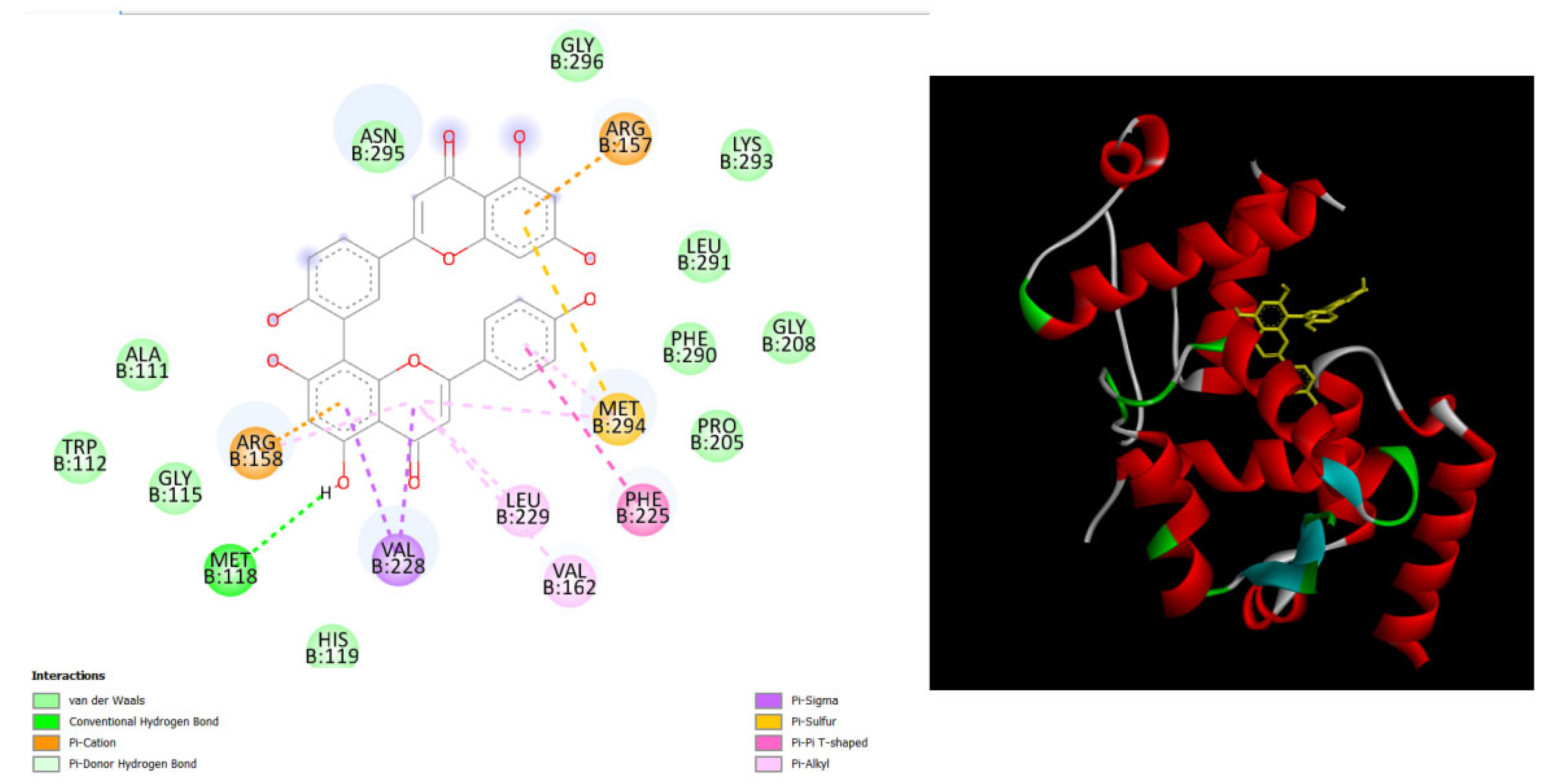
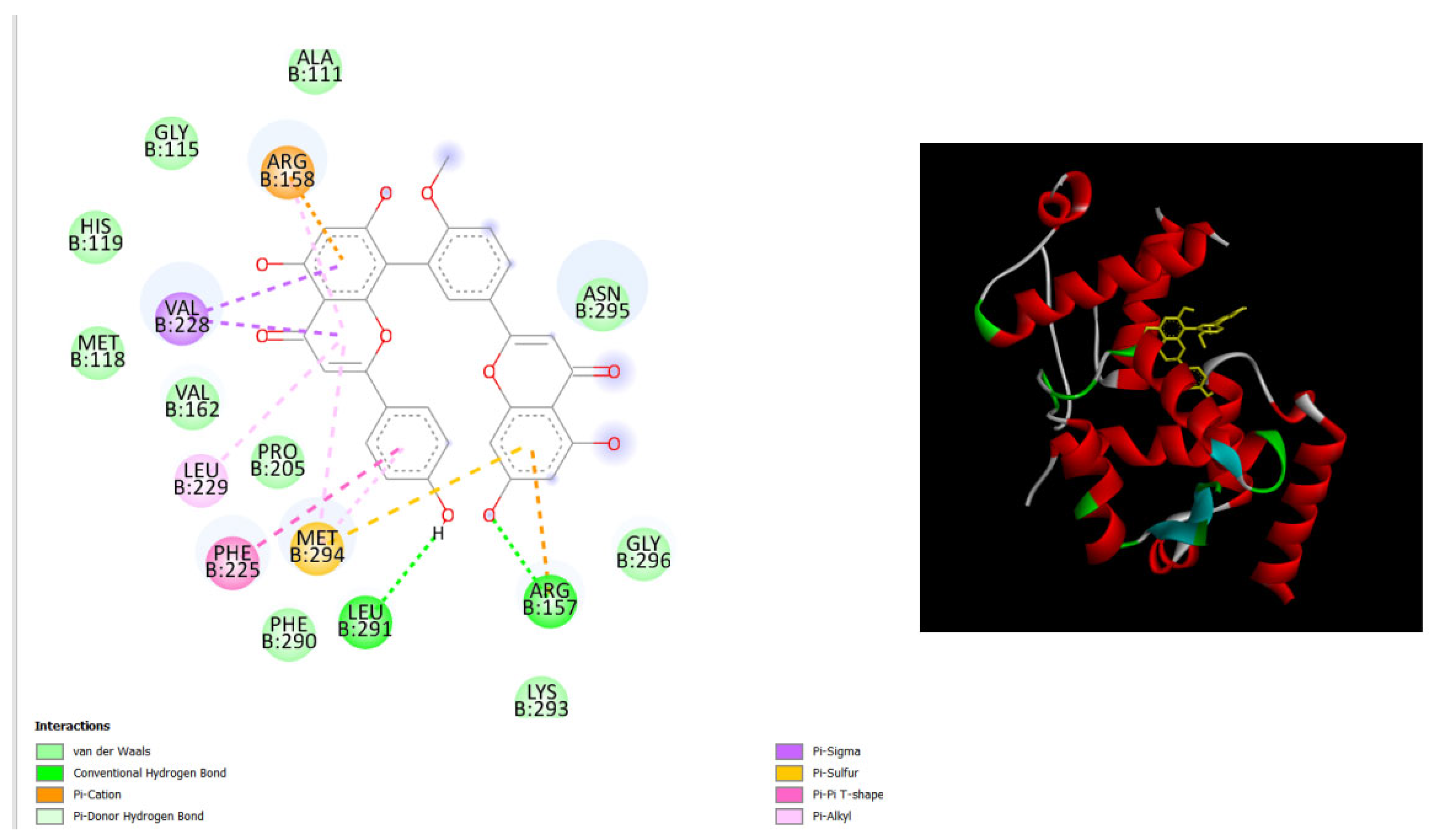
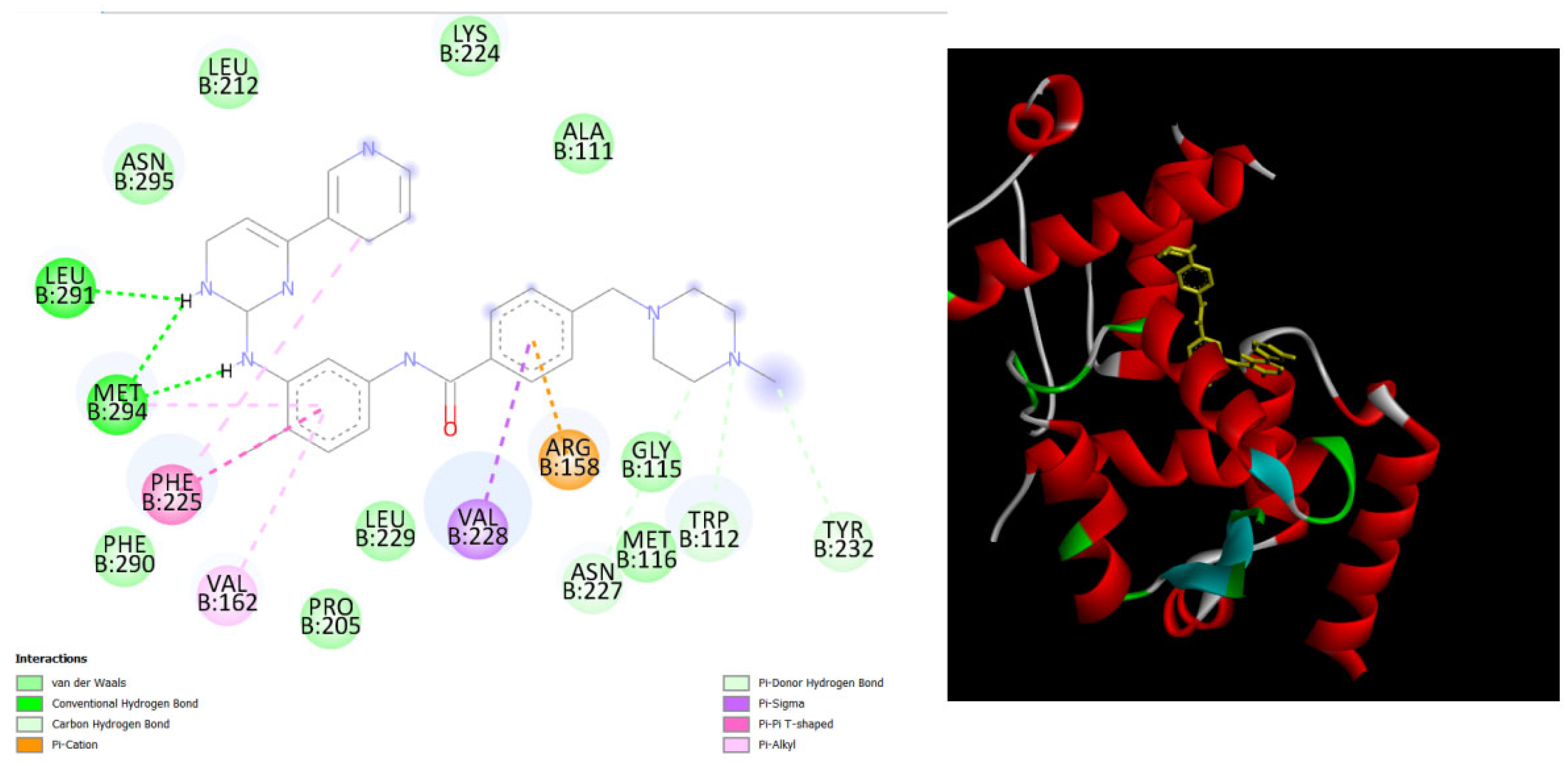
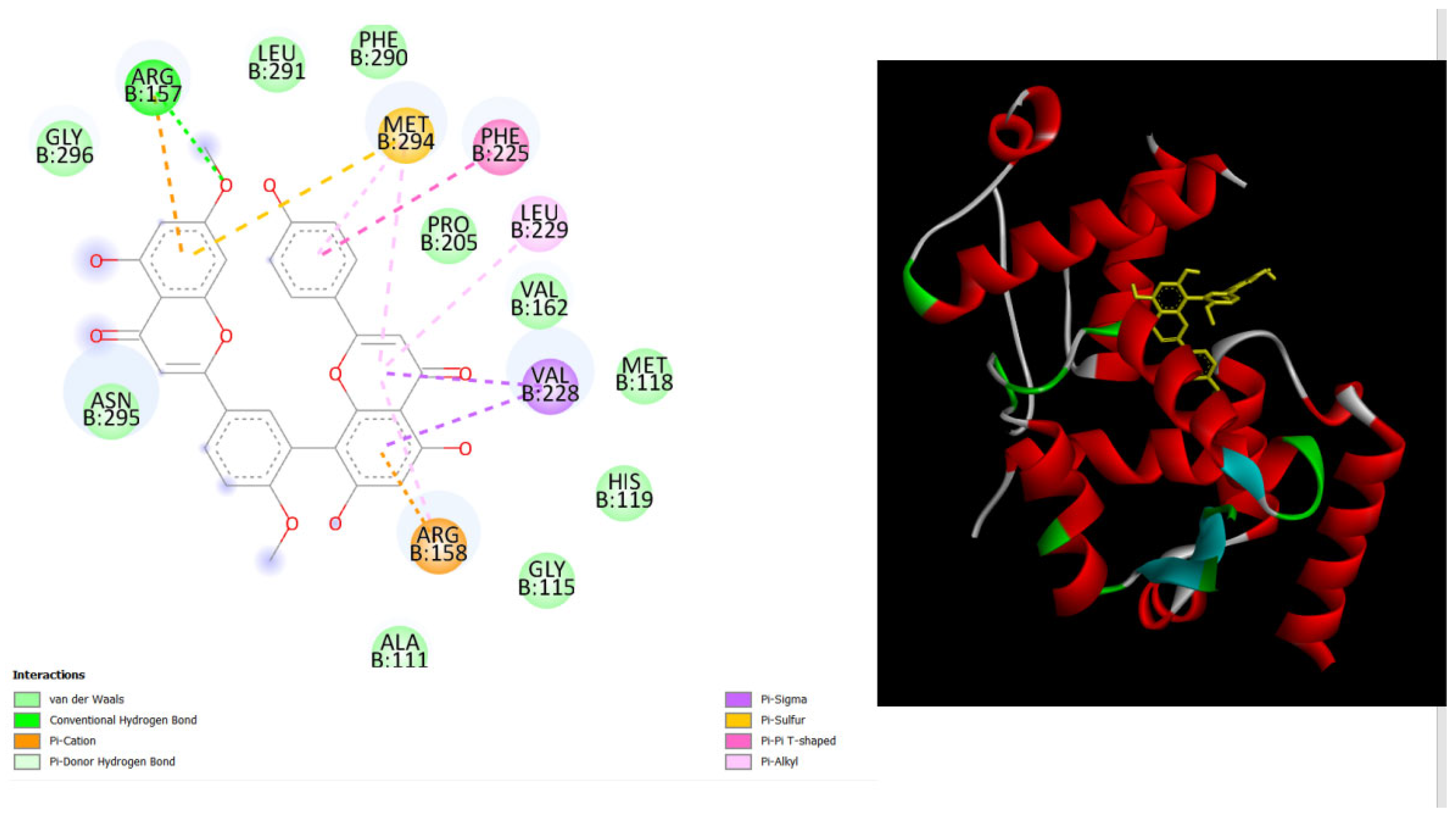
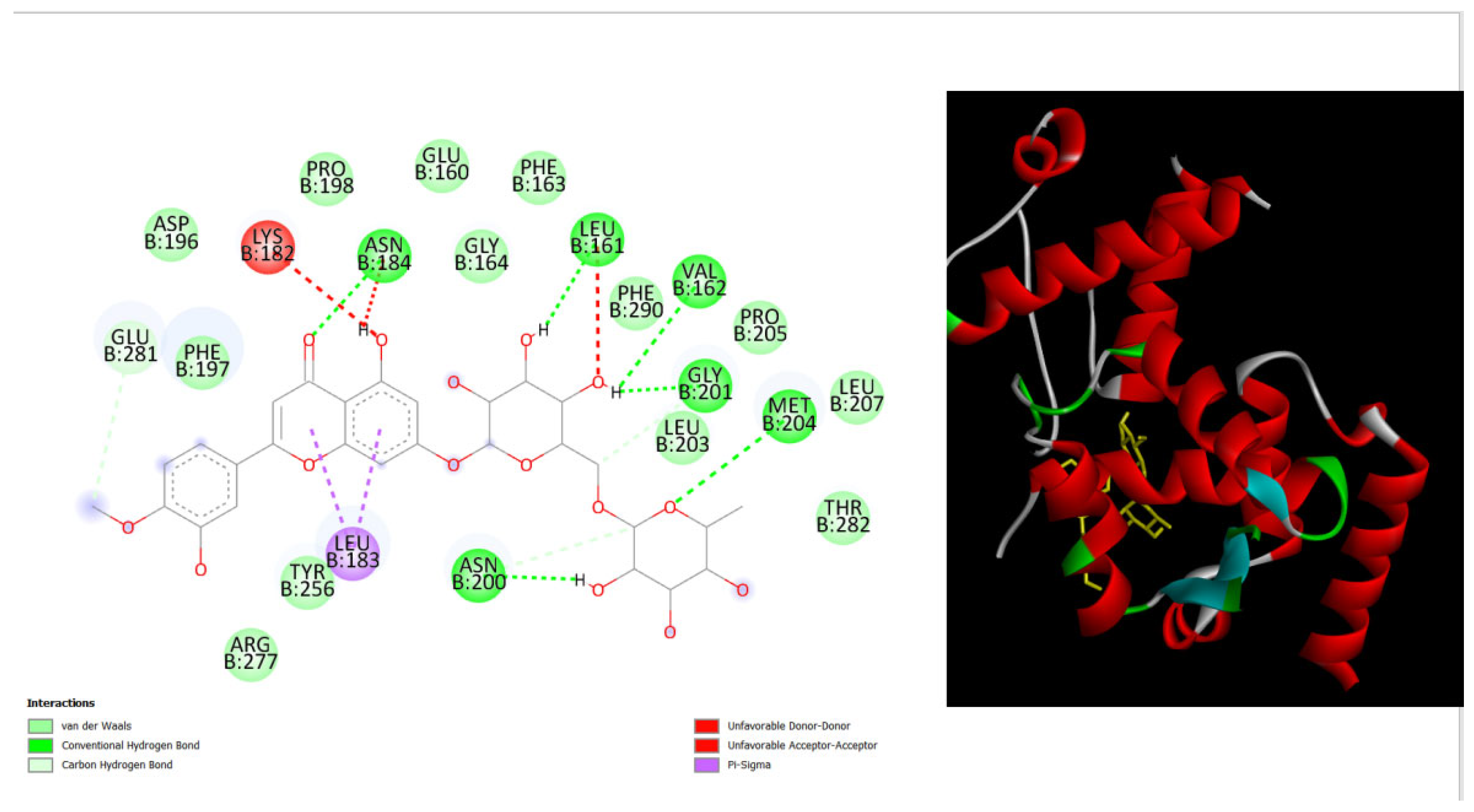
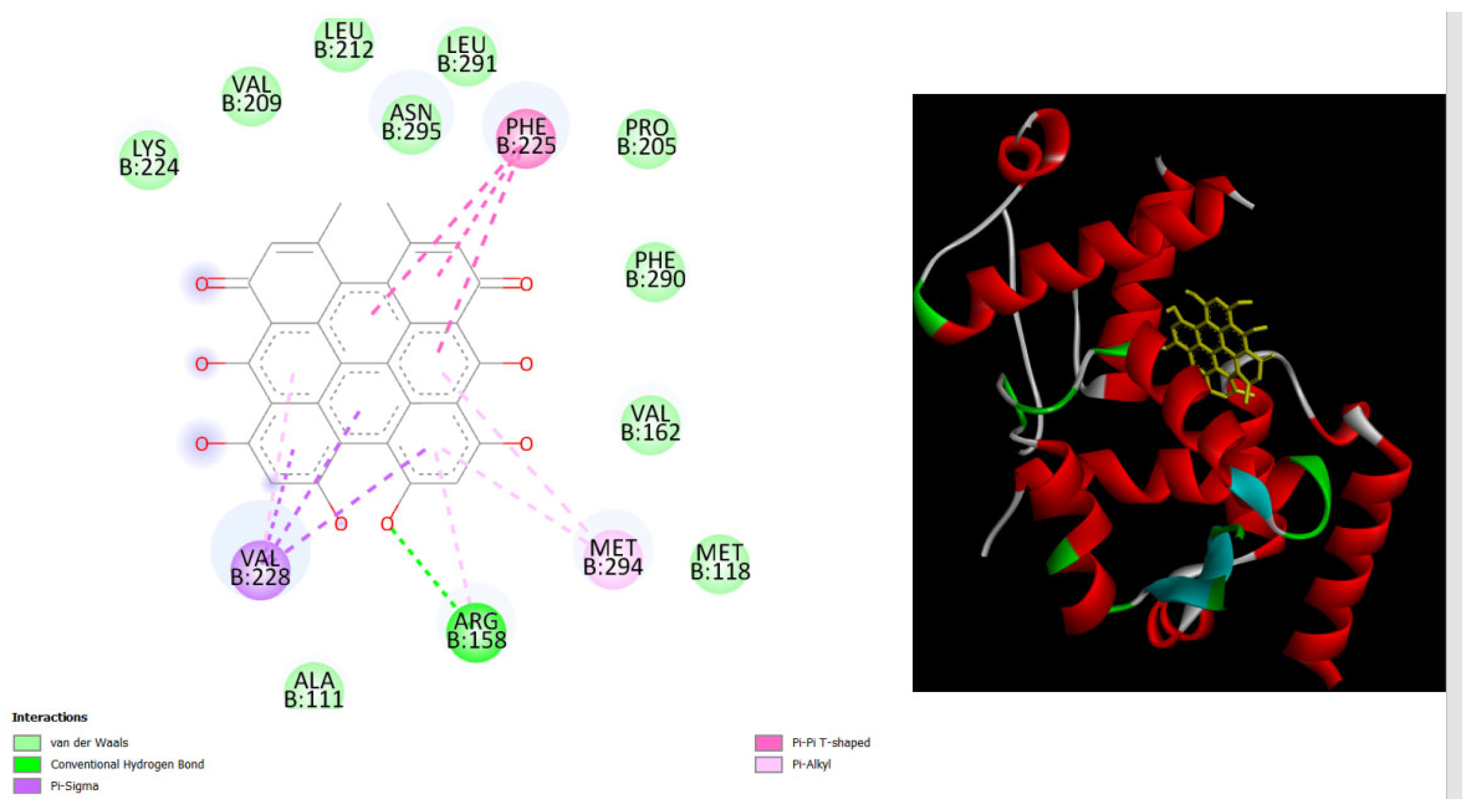

Table 1.
shows the comparison of best binding energies of selected potential ligands with Melanoma-associated antigen B1 by Autodock Vina with Pyrx program. Docking analysis was evaluated by Blind Docking approach.
Table 1.
shows the comparison of best binding energies of selected potential ligands with Melanoma-associated antigen B1 by Autodock Vina with Pyrx program. Docking analysis was evaluated by Blind Docking approach.
| Compounds | Binding Energy (1) (Kcal/mol) | Binding Energy (2) (Kcal/mol) |
|---|---|---|
| Hypericin | -9.9 | -9.9 |
| Amentoflavone | -9.8 | -9.9 |
| Imatinib | -9.9 | -9.8 |
| Bilobetin | -9.8 | -9.8 |
| Ginkgetin | -9.9 | -10 |
| Bafetinib | -9.7 | -9.7 |
| Diosmin | -9.7 | -9.7 |
Figure 1.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Bafetinib within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between the target and Bafetinib. Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Bafetinib.
Figure 1.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Bafetinib within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between the target and Bafetinib. Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Bafetinib.
Figure 2.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Amentoflavone within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between target and Amentoflavone. Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Amentoflavone.
Figure 2.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Amentoflavone within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between target and Amentoflavone. Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Amentoflavone.
Figure 3.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Bilobetin within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between target and Bilobetin. Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Bilobetin.
Figure 3.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Bilobetin within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between target and Bilobetin. Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Bilobetin.
Figure 4.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Imatinib within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between target and Imatinib. Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Imatinib.
Figure 4.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Imatinib within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between target and Imatinib. Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Imatinib.
Figure 5.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Ginkgetin within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between target and Ginkgetin. Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Ginkgetin.
Figure 5.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Ginkgetin within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between target and Ginkgetin. Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Ginkgetin.
Figure 6.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Diosmin within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between target and Diosmin. Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Diosmin.
Figure 6.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Diosmin within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between target and Diosmin. Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Diosmin.
Figure 7.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Hypericin within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between target and Hypericin.Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Hypericin.
Figure 7.
displays the docking outcomes of with Melanoma-associated antigen B1 in conjunction with Hypericin within the Ligand Binding Site, as analyzed by Autodock Vina through Pyrx program. On the left side, 2D diagrams illustrate the residue interactions between target and Hypericin.Meanwhile, the right side exhibits the Ligand Binding Site of the protein, highlighting the specific location of Hypericin.
Table 2.
Shows the comparison of predicted toxicity parameters with Imatinib, Amentoflavone, Hypericin, Diosmin, Ginkgetin, Bilobetin and Bafetinib, through pKCSM Server.
Table 2.
Shows the comparison of predicted toxicity parameters with Imatinib, Amentoflavone, Hypericin, Diosmin, Ginkgetin, Bilobetin and Bafetinib, through pKCSM Server.
| Compounds | AMES toxicity |
Max. tolerated dose(human) (logmg/kg/day) |
hERG I inhibitor | hERG II inhibitor | Oral Rat Acute Toxicity (LD50) (mol/kg) | Oral Rat Chronic Toxicity (LOAEL) (log mg/kg_bw/day) |
Hepatotoxicity |
|---|---|---|---|---|---|---|---|
| Hypericin | no | 0.438 | no | yes | 2.482 | 2.421 | no |
| Amentoflavone | no | 0.438 | no | yes | 2.527 | 3.572 | no |
| Imatinib | no | 0.317 | no | yes | 2.9 | 1.409 | no |
| Bilobetin | no | 0.437 | no | yes | 2.56 | 2.217 | no |
| Ginkgetin | no | 0.437 | no | yes | 2.733 | 2.475 | no |
| Bafetinib | no | 0.383 | no | yes | 2.85 | 1.321 | no |
| Diosmin | no | 0.565 | no | yes | 2.512 | 3.343 | no |
4. Conclusion
This groundbreaking study has identified seven potential ligands with strong binding energies, around -10 kcal/mol, in a docking analysis against the Crystal Structure of human Melanoma-associated antigen B1 (MAGEB1). The ligands, including Imatinib, Bafatinib, Diosmin, Ginkgetin, Bilobetin, Amentoflavone, and Hypericin, demonstrate a robust interaction with the target protein, marking a significant advancement in the field. Additionally, the study employs the pKCSM server to predict toxicity properties, providing valuable insights into the safety profiles of the ligands. Notably, Diosmin exhibits favorable toxicity parameters—Max. tolerated dose (human), Oral Rat Acute Toxicity (LD50), and Oral Rat Chronic Toxicity (LOAEL)—positioning it as a potential candidate for further exploration against Melanoma. To progress the research, the study recommends additional experimental validations, encompassing in vitro and in vivo studies, to confirm the efficacy and safety of Diosmin and other promising ligands.
References
- Schadendorf, D., Fisher, D. E., Garbe, C., Gershenwald, J. E., Grob, J. J., Halpern, A., ... & Hauschild, A. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 1–20.
- Schadendorf, D.; van Akkooi, A.C.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. The Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Miller, A.J.; Mihm, M.C., Jr. Melanoma. New England Journal of Medicine 2006, 355, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Coit, D. G., Andtbacka, R., Bichakjian, C. K., Dilawari, R. A., DiMaio, D., Guild, V., ... & Wong, M. K. Melanoma. Journal of the National Comprehensive Cancer Network 2009, 7, 250–275. [CrossRef]
- Xiao, J.; Chen, H.S. Biological functions of melanoma-associated antigens. World J. Gastroenterol. 2004, 10, 1849. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput. -Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Mehrotra, R.J. An overview of molecular docking. JSM Chem 2016, 4, 1024–1028. [Google Scholar]
- Giordano, D.; Biancaniello, C.; Argenio, M.A.; Facchiano, A. Drug design by pharmacophore and virtual screening approach. Pharmaceuticals 2022, 15, 646. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Chem. Biol. Methods Protoc. 2015; 243–250. [Google Scholar]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



