
Submitted:
03 December 2023
Posted:
05 December 2023
You are already at the latest version
Abstract
Antimicrobial resistance is an increasing problem for the global public health. One of the strategies to combat this issue is the synthesis of novel antimicrobials through rational drug design based on extensive structure-activity relationship studies. The thiazole nucleus is a prominent feature in the structure of many authorized antimicrobials, being clubbed with different heterocycles. The purpose of this review is to study the structure-activity relationship in antimicrobial thiazoles clubbed with various heterocycles reported in the literature between 2017 and 2022, in order to offer an overview of the last years in terms of antimicrobial research and provide a helpful instrument for future research in the field.
Keywords:
thiazole
; structure-activity relationship
; antimicrobial
; heterocycles
; hybrid compounds
1. Introduction
Antimicrobial resistance (AMR) represents a major threat to the global public health and an increasing challenge to overcome. According to the latest statistics [1], AMR has caused globally 1.27 million deaths alone, with the indiscriminate use of antimicrobials, not only for human but also for livestock, representing the main cause of the problem. Pathogens with the most increased risk for developing resistance are Pseudomonas aeruginosa, Acinetobacter baumanii, and Enterobacteriales to carbapenems and to new molecular entities of newer generation cephalosporins [1].
Strategies in medicinal chemistry for fighting AMR include the total synthesis of well-established antibiotics classes, based on extensive structure-activity relationships of the preexistent antibiotic classes (for example eravacycline), optimization of the physicochemical property space (finafloxacin), knowledge of the target in order to design new scaffolds (avibactam and vaborbactam), synthesis of hybrid compounds with synergistic activity (cadazolid), re-exploration of targets of older antibiotics (griselimycins targeting DnaN, the sliding clamp of DNA polymerase), and targeting the virulence factors (elastase B, lectins A and B of P. aeruginosa, quorum-sensing, or transcriptional regulator PqsR) [2].
Thiazole is a five-membered heterocycle, part of the azoles group containing one nitrogen and one sulfur heteroatoms. It is found in a series of authorized antimicrobial drugs, such as aztreonam, various cephalosporins (including the newest cefiderocol), abafungin, isavuconazole, ravuconazole, ethaboxam, and myxothiazol, thus underlining the importance of this nucleus for the antimicrobial activity [3,4,5]. The effect of the thiazole nucleus on the biological activity of a molecule can be influenced by clubbing it directly or through a linker with other moieties with pharmacological potential, therefore creating hybrid compounds [6].
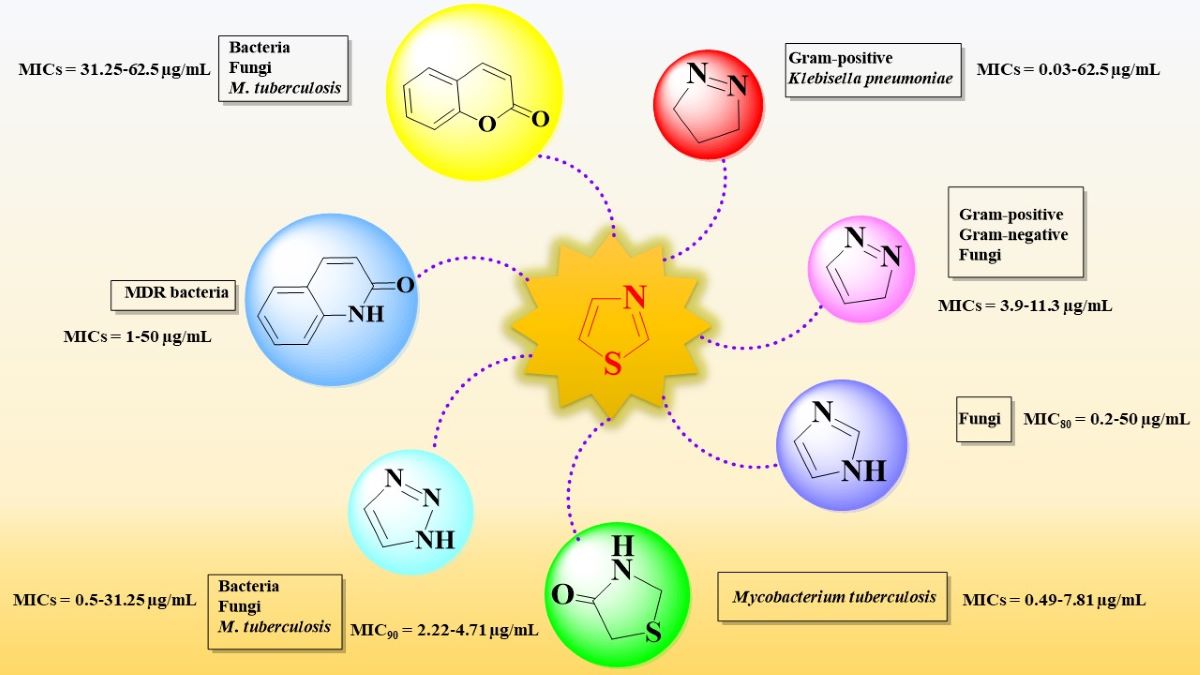
Knowledge of qualitative and quantitative structure-activity relationship studies (SAR) has great importance in a rational drug design. Therefore, this review aims to study the structure-activity relationships of novel antimicrobial thiazole clubbed with various heterocycles in hybrid compounds, reported in the last six years, as a helpful tool for further research and development of antimicrobials bearing the thiazole heterocycle. The publications were selected by interrogating Scopus, SpringerLink, PubMed, and EMBASE databases, using “Thiazole AND Hybrid AND Antimicrobial” as search syntax, filtering the results by “Classical Article” filter as article type, and setting the publication date range between 2017 and 2022. A total of 366 results from all four databases were obtained. However, it is important to mention that many of the results were indexed in two or more databases. A bibliometric analysis performed on the selected publications revealed that thiazole in antimicrobial hybrid compounds is a topic of increasing interest in the last three years (Figure 1). Therefore, 45 publications that met the eligibility criteria for this review (synthesis of hybrid compounds bearing the thiazole scaffold clubbed with other heterocycles) were discussed.
2. Structure-Activity Relationships in Antimicrobial Thiazole-based Hybrid Compounds
The selected papers were organized based on the nature of heterocycles that were clubbed with thiazole. Additionally, they were ordered by their size, thus starting with the smallest heterocycles and finishing with polyheterocyclic systems (Figure 2). This offered an overview on how the antimicrobial activity may also be affected by this aspect. It is worth mentioning that no research on antimicrobial thiazoles clubbed with three-membered heterocycles was available in the literature in the specified timeframe. Thus, the classification started with the four-membered group of heterocycles.
2.1. Thiazole Clubbed with Four-Membered Heterocyles
Four-membered heterocycles are a class of highly reactive and low stability chemical entities, classified in unsaturated heterocycles azetes, oxetenes, thietes, and their saturated derivatives azetidines, oxetanes, and thietanes. The best known and important derivative of this class is the 2-azetidinone. The chemical instability of four-membered heterocycles makes it difficult to use them extensively in medicinal chemistry, hence the reduced number of hybrid compounds available in the literature. Herein, we present the SAR studies in some antimicrobial thiazole clubbed with 2-azetidinone hybrid compounds.
2.1.1. Thiazolyl-Azetidin-2-one Hybrid Compounds
2-Azetidinone is the centerpiece of β-lactam antibiotics and is also found in some β-lactamase inhibitors, like clavulanic acid, sulbactam, and tazobactam, thus making it an essential scaffold in the development of new potential antibacterial molecules [7].
Desai et al. reported the design and synthesis of a series of 4-(quinolin-3-yl)-1-(thiazol-2-yl)-amino-azetidin-2-ones (Figure 3) with two points of variation: the sixth position of the quinoline ring (R1) and the phenyl from the fourth position of the thiazole ring (R2) [8]. Depending on each case, the quinoline ring can be unsubstituted (1-3) or substituted with a methyl group (4-7) in the sixth position. Similarly, the thiazole ring can be either unsubstituted (4) or substituted with electron-donating (1, 5, and 7) or electron-withdrawing groups (2, 3, and 6) in the fourth position [8].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative bacteria and for their antifungal activity against Candida sp. and Aspergillus sp. strains. The results were quantified as minimal inhibitory concentrations (MICs) [8]. Overall, the compounds were more effective against the Gram-negative strains compared to the Gram-positive ones. Compounds 1-4 and 6 showed the strongest antibacterial activity against Escherichia coli MTCC 443 (MICs = 100 µg/mL), while compound 5 was the most efficient against P. aeruginosa MTCC 1688 (MIC = 100 µg/mL), but in both cases the activity was inferior to chloramphenicol (MIC = 50 µg/mL) [8].
In terms of antifungal activity, compounds 5 and 7 showed inferior activity (MIC = 200 µg/mL) against Candida albicans MTCC 227, Aspergillus niger MTCC 282, and A. clavatus MTCC 1323, compared to nystatin (MIC = 100 µg/mL) [8].
SAR studies in this series showed that R2 substitution was associated with an increased antibacterial activity (Figure 3). Electron-donating groups for both R1 and R2 (5 and 7) were associated with antifungal activity [8].
The compounds are potential inhibitors of β-ketoacyl-acyl-carrier protein synthase III, which is an important enzyme in the fatty acid biosynthesis and growth of bacterium, based on the molecular docking studies. No potential mechanism for the antifungal activity was reported [8].
2.2. Thiazole Clubbed with Five-Membered Heterocycles
Five-membered heterocycles represent an extensive group of valuable structures for designing pharmacologically active compounds, including antimicrobials. Compared to four-membered heterocycles, they have a significantly greater chemical stability and versatility, which is transposed in their ubiquitarian usage in pharmaceutical research and development.
Based on the literature search of the last six years, we will discuss further the structure-activity relationships in antimicrobial hybrid compounds containing thiazole clubbed with the following five-membered heterocycles and their derivatives: pyrazoline, pyrazolinone, pyrazole, imidazole, thiazolidinone, thiazolidindione, 1,3,4-thiadiazole, 1,2,3-triazole, 1,3,4-oxadiazole, and thioxo-1,3,4-oxadiazole.
2.2.1. Thiazolyl-2-Pyrazoline Hybrid Compounds
2-Pyrazoline is an important scaffold in anti-infective drugs, possessing antibacterial, antifungal, antiviral, antiparasitic, and antituberculosis potential [9]. Herein, we present the structure-activity relationship for some series of thiazole linked with 2-pyrazoline derivatives with promising antimicrobial potential, to establish how clubbing these two heterocycles influences the biological activity of the entire molecule.
Based on the found structures, it can be concluded that there is one general scaffold consisting of both heterocycles linked directly, but in different positions. Therefore, there have been identified three types of linking patterns: the fifth position of the thiazole ring to the fifth position of 2-pyrazoline ring [10], the second position of the thiazole ring to the first position of 2-pyrazoline ring [11,12,13,14,15,16], and the fifth position of the thiazole ring to the third position of 2-pyrazoline ring [17] (Figure 4).
Cuartas et al. designed four series (8a-14a, 8b-14b, 8c-14c, 8d-14d) of 2-(N-mustard)-5-(2-pyrazolin-5-yl)-thiazoles (Figure 5), with two points of variation: the substituted phenyl from the third position of the 2-pyrazoline (R) and the first position of the 2-pyrazoline ring (R1) [10]. The compounds are bearing a nitrogen mustard moiety, known for its DNA alkylating properties. Depending on each case, R1 can be a carbonylic group (a and b) or a phenyl ring (c and d), while R can be hydrogen (14), a halogen atom (8-10), or etheric groups (11-13).
The compounds were tested for their antifungal activity against Candida sp. and Cryptococcus sp. strains and for their antibacterial activity against Gram-positive and Gram-negative strains. The results were quantified as IC50, representing the minimum concentration that inhibits 50% of growth, and MIC [10]. Compounds 8c-14c (R1 = -C6H5) and 12d-14d (R1 = -3,5-di-Cl-C6H3) showed inferior activity (IC50 = 15.6-125 µg/mL) against Cryptococcus neoformans ATCC 32264 compared to amphotericin B (IC50 = 0.50 µg/mL) [10].
In terms of antibacterial activity, the compounds showed significant effect against vancomycin-intermediate S. aureus (VISA) (MICs = 3.25-500 µg/mL), methicillin-susceptible S. aureus (MSSA ATCC 25923) (MICs = 62.5-500 µg/mL), methicillin-resistant S. aureus (MRSA ATCC 43300) (MICs = 125-500 µg/mL), and Neisseria gonorrhoeae ATCC 31426 (MICs = 125-500 µg/mL), compared to penicillin, ceftriaxone, and ciprofloxacin [10].
SAR studies revealed that the nature of the substituents on 2-pyrazoline (a-d) and phenyl (8-14) were the most important for the antibacterial and antifungal activities (Figure 5) [10]. The phenyl substituent from the first position of the 2-pyrazoline ring (c) conferred anticryptococcal activity regardless of the other substituents (8c-14c), while 3,5-dichlorophenyl substituent (d) was conditioned by electron-donating groups, particularly methoxy (12d) or trimethoxy (13d) or no substituent at all (14d) on the phenyl ring from the third position of the 2-pyrazoline ring. It is worth mentioning that 3,5-dichlorophenyl substituent is found in the structure of some important antifungal azoles like miconazole, ketoconazole, or itraconazole. However, the situation is opposite for the antibacterial activity. The 3,5-dichlorophenyl substituent abolished the antibacterial effect, while for phenyl-substituted derivatives it was very low. The best activity was observed when small substituents in R1 were present, like acetyl (a) and formyl (b). Another important aspect is that the presence of chloro (8a and 8b), bromo (9a and 9b), and fluoro (9c) substituents or no substituents (14b) increased the activity against VISA (Figure 5) [10]. No potential target was reported by the authors.
Rashdan and Abdelmonsef designed 2-(4-(1-thiazol-2-yl)-2-pyrazolin-3-yl)-1,2,3-triazol-1-yl)-1,3,4-thiadiazole, with a single point of variation: fifth position of the 2-pyrazoline ring (R) (Figure 6) [11].
These compounds were assayed for their antibacterial activity against Gram-positive and Gram-negative bacteria and for their antifungal activity against C. albicans [11]. The thiophene substituted compound 15 showed promising results against E. coli, P. aeruginosa, and S. aureus (MICs = 5-10 µg/mL), compared to ciprofloxacin (MICs = 1.25-7 µg/mL) [11].
Concerning the antifungal activity, the same compound showed identic activity to nystatin against C. albicans (MIC = 5 µg/mL) [11].
Based on the minimal structural differences between the obtained derivatives, it seems that switching from a bulkier (3-metoxy-4-hydroxy-phenyl in 16) to a smaller substituent (2-thienyl in 15) enhanced the activity (Figure 6) [11].
Additionally, compound 15 was a potent inhibitor of SARS-CoV-2 transmembrane serine protease 2 (TMPRSS2), which plays an important role in the disease propagation, based on molecular docking studies [11].
Budak et al. reported the synthesis of a series of 2-(4-(1-(thiazol-2-yl)-2-pyrazolin-3-yl)-phenyl)-methanoisoindol-1,3-dione derivatives. The obtained compounds varied by the substituent in the fifth position of the 2-pyrazoline ring (R), which can be an aryl (17-23) or hetaryl ring (24-25) (Figure 7) [12].
The compounds were tested for the antimicrobial activity against Gram-positive and Gram-negative bacteria and C. albicans ATCC 1213 [12]. The results were quantified by inhibition zone (IZ), through disk diffusion method. All compounds (17-25) showed inferior activity against S. aureus ATCC 29213 (IZs = 10-19 mm), compared to cefoperazone-sulbactam (IZs = 19-26 mm). Additionally, compound 25 was active against Proteus vulgaris KUEN 1329 (IZ = 10 mm), but inferior to the standard drug [12].
SAR studies in this series showed that aryl substitution induced better activity compared to hetaryl substitution (Figure 7). Thus, in the aryl substituted derivatives, para substitution (17-20) favorized an overall better activity compared to meta substitution (21-23). The position of R1 substituent was found to be more important than their nature, as substitution in para with both electron-withdrawing and electron-donating groups yielded similar activities [12]. In the case of compounds 24 and 25, bearing 2-thienyl and 2-furanyl substituents, a broader activity spectrum was observed for 24, which could be attributed to the higher aromaticity of 2-thienyl compared to 2-furanyl substituent, which makes it suitable for the bioisosteric substitution of phenyl rings [12]. No potential target was reported by the authors.
Mansour et al. designed three series of 2-(3-aryl-5-hetaryl-2-pyrazolin-1-yl)-thiazole derivatives (Figure 8), with three points of variation: one is the linking position to the naphthyl ring (a = 1-naphthyl or b = 2-naphthyl) from the third position of 2-pyrazoline ring and the other two are in the fourth (R1) and fifth positions (R2) of the thiazole ring [13]. Depending on each case, R1 can be a para-halogen substituted phenyl ring (26 and 27) or a methyl group (28-33), while R2 can be either hydrogen (26 and 27) or various acyl substituents (28-30) and arenediazo groups (31-33) (Figure 8) [13].
The compounds were tested for the antimicrobial activity against Gram-positive and Gram-negative bacteria and various fungal strains [13]. Most of the compounds displayed antibacterial activity against S. aureus (IZs = 0.5-2.6 mm) and antifungal activity against A. flavus (IZs = 0.5-2.3 mm), but inferior compared to amoxicillin (IZs = 2.2-3.5 mm) and griseofulvin (IZs = 2.1-3.3 mm). Only compounds 27b and 33a were active against all the tested microbial strains (S. aureus, Bacillus subtilis, K. pneumoniae, P. aeruginosa, A. fumigatus, A. flavus, Syncephalastrum racemosum, Penicillium expansum, and C. albicans) [13].
SAR studies suggest the activity depends on the nature of R1 and R2 and the linkage 2-pyrazoline ring to the naphthyl group [13]. Thus, in the 4-(p-halophenyl)-thiazolyl series (26a,b and 27a,b) (Figure 8), bromo substituent (27a and b) inactivated the compounds, while the chloro substituent (26a and b) was responsible for the antibacterial activity, apart from compound 26b, which displayed activity against all tested bacterial and fungal strains. Therefore, chloro and 2-naphthyl substituents (26b) were the best combination for highly active antimicrobials [13].
In the 5-acylthiazolyl series (28a,b, 29a,b, and 30a,b) (Figure 8), the compounds substituted with acetate (29a and 29b) or anilido (30a and 30b) groups were inactive or had a very low activity only against A. flavus (IZ = 0.5 mm) and P. expansum (IZ = 0.8 mm), while acetyl substitution (28a and 28b) yielded a moderate antimicrobial activity (IZs = 0.5-1.2 mm) [13].
Finally, in the 5-arendiazothiazolyl series (31a,b, 32a,b, and 33a,b), the activity depended more on the linkage to the naphthyl group (a or b) [13]. Thus, the best combination was between p-chlorobenzenediazo and 1-naphthyl (33a), which was the most active. Combinations between p-toluenediazo and 1-naphthyl (32a) or benzenediazo and 2-naphthyl (31b) yielded inactive molecules (Figure 8) [13]. No potential target was reported by the authors.
Using a similar scaffold, Masoud et al. [14] reported the synthesis of two more series of 2-(3-aryl-5-hetaryl-2-pyrazolin-1-yl)-thiazole derivatives, with two points of variation: (R) linked to the fifth position of 2-pyrazoline ring (c = 3,4-dimethoxyphenyl and d = 1,3-benzodioxole) and (R1) from the fifth position of thiazole ring [14]. Depending on each case, R1 can be various acyl substituents (34-36) or arenediazo groups (37-39) (Figure 8).
The antimicrobial effect was assayed using the same strains and positive controls mentioned by Mansour et al [14]. The most notable results for the antibacterial activity were obtained for compounds 36d, 38d, and 39c against S. aureus (IZs = 0.9-1.2 mm), compounds 34c, 35c, 37c, 38c, and 39c against K. pneumoniae (IZs = 1.2-2.0 mm) and P. aeruginosa (IZs = 1.0-1.8 mm) [14].
In the case of antifungal activity, the most important results were obtained for compounds 37c, 38c, and 39d against C. albicans (IZs = 1.3-2.1 mm), compound 38c against A. fumigatus (IZ = 1.2 mm) and compound 38d against A. flavus (IZ = 2.3 mm) [14].
SAR studies in these compounds suggest that acyl group substitution in position 5 of the thiazole ring (34c,d, 35c,d, and 36c,d) results in only antibacterial active compounds (Figure 8) [14]. Favorable substituents for the antibacterial activity were acetate (34c and d), anilido (35c and d), and acetyl (36c and d), which was the opposite compared to the compounds by Mansour et al [14].
Arendiazo substitution of the thiazole ring (37c,d, 38c,d, and 39c,d) expanded the spectrum against both bacterial and fungal strains. In this case, the main difference in the potence was dictated by whether if the substituent from position 5 of the 2-pyrazoline ring was 3,4-dimethoxyphenyl (c) or 1,3-benzodioxole (d), with the first being more active [14]. No potential target was reported by the authors.
By analyzing the SAR studies in the molecules synthesized by both authors (Figure 8), it seemed that the naphthyl group brought drawbacks to the compounds designed by Mansour et al., as the antimicrobial potential was reduced compared to the compounds designed by Masoud et al, who replaced it with a 3,4-dimethoxyphenyl moiety. Thus, a higher polarity should be considered when designing novel antimicrobial compounds [13,14].
Bhandare et al. reported the synthesis of some 2-(2-pyrazolin-1-yl)-thiazoles in which the fourth position of the thiazole ring was linked via a methylene bridge to various thiol- and tioether-containing azoles (1,3,4-oxadiazole and 1,2,4-triazole) (Figure 9) [15].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative strains and for their antifungal activity against Candida sp. and Aspergillus sp. strains [15]. Overall, all compounds displayed a significant antimicrobial potential. Compounds 42, 46, 48, and 49 showed similar activity (MICs = 0.5-8 µg/mL) against S. aureus ATCC 11632, S. faecalis ATCC 14506, K. pneumoniae ATCC 10031, E. coli ATCC 10536, P. aeruginosa ATCC 10145, compared to ciprofloxacin (MICs between ≤1 and >5 µg/mL) [15].
Regarding the antifungal activity, the same compounds showed similar activity against C. tropicalis ATCC 1369 and A. niger ATCC 6275, compared to fluconazole (MICs ≤ 1 µg/mL) [15].
SAR studies pointed out that the heterocyclic tetrad induced promising overall antibacterial and antifungal activities (Figure 9) [15]. The additional aryl substituent (42-46 and 48-51) in the thioether series was essential for the activity. Switching to a hetaryl substituent, 3-pyridinyl (47), decreased the activity. The substituents on aryl were also an important factor in determining the antibacterial and antifungal strengths. Thus, a small halogen substituent, like 4-fluoro (46 and 49), induced excellent activity, while larger substituents, such as 2-trifluoromethyl (44), 4-trifluoromethoxy (45), 4- or 5-chloro (43 and 50), and 4-bromo (51) significantly decreased the activity. Noteworthy, unsubstituted rings (42 and 48) also displayed important activity (Figure 9) [15].
These compounds could potentially target DNA gyrase, important for the replication of genetic material in bacteria, and Cytochrome P450 14 α-sterol demethylase, important for the synthesis of ergosterol from lanosterol, for the antifungal activity, based on the molecular docking studies [15].
Abdel-Wahab et al. designed a series of 2-(5-(3-(1,2,3-triazol-4-yl)-pyrazol-4-yl)-2-pyrazolin-1-yl)-thiazole (Figure 10) [16]. These compounds are para substituted on the phenyl ring from the third position of the 2-pyrazoline ring (R1) and have an additional substituent in the fourth position of the thiazole ring (R2).
The compounds were tested for the antimicrobial activity against Gram-positive and Gram-negative bacteria and C. albicans NRRL Y-477 [16]. Compounds 52 and 53 showed inferior activity (MIC = 50 µg/mL) against S. aureus ATCC 29213 and K. pneumoniae ATCC 13883, compared to ciprofloxacin (MIC = 25 µg/mL) [16].
Only compound 54 was active against C. albicans (MIC = 200 µg/mL), but the activity was inferior compared to clotrimazole (MIC = 25 µg/mL) [16].
SAR studies in these compounds suggest that identic substitution (52 and 53) of both 2-pyrazoline and 1,3-thiazole moieties could be responsible for the antibacterial activity (Figure 10). Compound 54, containing an additional 1-(p-tolyl)-5-methyl-1,2,3-triazol-4-yl moiety, may induce a closer resemblance to fluconazole than the rest of the compounds, by containing two triazole moieties in its structure, hence the better antifungal activity against C. albicans [16]. No potential target was reported by the authors.
Bondock and Fouda designed some 2-(N-allyl)-5-(2-pyrazolin-3-yl)-thiazole derivatives with various substituents in the first position of the 2-pyrazoline ring (55-60) (Figure 11) [17].
The compounds were tested for the antimicrobial activity against Gram-positive and Gram-negative bacteria and fungal strains [17]. All selected compounds showed similar activities (MICs = 0.03-7.81 µg/mL) against S. pneumoniae RCMB 010010 and S. epidermidis RCMB 010024, compared to ampicillin (MICs = 0.6-0.24 µg/mL). Compounds 55, 57, and 60 showed similar activity (MICs = 0.03-7.81 µg/mL) against E. coli RCMB 010052, P. vulgaris RCMB 010085, and K. pneumoniae RCMB 010093, compared to gentamycin (MICs = 0.03-1.95 µg/mL) [17].
In terms of antifungal activity, which was similar to amphotericin B (MICs = 0.12-7.81 µg/mL), compounds 56-58 and 60 showed results against A. fumigatus RCMB 02568 (MICs = 0.12-7.81 µg/mL), while only compounds 57 and 60 were active against S. racemosum RCMB 05922 (MICs = 0.24-7.81 µg/mL) [17].
According to SAR studies, substitution of 2-pyrazoline ring with a thiocarbamido group (57) favorized the overall antibacterial and antifungal activities (Figure 11), while phenylthiocarbamido group (58) only induced good activity against Gram-positive bacteria and A. fumigatus [17]. Substitution with a phenyl ring (56) induced a moderate antifungal activity (Figure 11). Further expansion of the molecule with a phenylthiazolyl moiety (59 and 60) yielded overall medium antibacterial and antifungal activities. The 4-fluorophenyl derivative (60) was more potent against S. epidermidis RCMB 010024, P. vulgaris RCMB 010085, A. fumigatus RCMB 02568, and S. racemosum RCMB 05922, compared to the compound with unsubstituted phenyl (59) [17]. No potential target was reported by the authors.
To conclude the results observed in the analyzed papers (Figure 12), clubbing thiazole with 2-pyrazoline to obtain novel antimicrobials should be considered when aiming for compounds active against Gram-positive bacterial strains, especially against S. aureus. Moderate results were observed against Gram-negative bacterial strains, except against K. pneumoniae, where the results were promising [13,14,15,16,17].
The antifungal activity was much lower compared to the antibacterial activity. Thus, this scaffold may not be suitable for designing potent antifungals.
2.2.2. Thiazolyl-Pyrazolin-3-one Hybrid Compounds
3-Pyrazolinone is an important scaffold for designing antimicrobial molecules [18], as presented further.
Abu-Melha obtained some N-(4-pyrazolin-3-one)-2-thiazolyl-hydrazonomethyl-phenoxyacetamides, with various substituents in the fourth and fifth positions of the thiazole ring (Figure 13), through a multi-step synthesis between N-(4-antipyrinyl)-2-chloroacetamide, p-hydroxybenzaldehyde, thiosemicarbazide, and various α-halogenocarbonyl compounds [19].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative strains and for their antifungal activity [19]. Compounds 61-63 showed superior antibacterial activity (MICs = 28-168 µg/mL) on S. aureus ATCC 25923, Salmonella typhimurium ATCC 14028, and E. coli ATCC 25922, compared to chloramphenicol (MICs = 143-152 µg/mL) and cephalothin (MICs = 135-229 µg/mL) [19].
In terms of antifungal activity, compounds 62 and 63 showed superior activity (MICs = 168-172 µg/mL) on C. albicans ATCC 10231, compared to cycloheximide (MIC = 254 µg/mL) [19].
Additionally, the compounds can target the penicillin binding proteins 4 (PBP4) from S. aureus and E. coli for the antibacterial activity, based on the molecular docking studies [19].
2.2.3. Thiazolyl-Pyrazole Hybrid Compounds
Pyrazole bears significant antimicrobial, anthelmintic, and anticancer properties, which makes this heterocycle an important motif when designing novel antimicrobial compounds [20]. Herein, we present the structure-activity relationship in thiazole clubbed with pyrazole compounds with promising antimicrobial potential, in order to establish how clubbing these two heterocycles influences the biological activity.
Based on the found structures, it can be concluded that there were two types of scaffolds used: one in which the thiazole and pyrazole rings are clubbed through a linker, which is a methylylidenehydrazinyl, and the other where both rings are linked directly. For the second type of scaffold, there were two possible linking positions to the pyrazole ring: one in the first position and the other in the third position of the pyrazole ring (Figure 14).
Gondru et al. [21] and Patil et al. [22] synthesized some series of 2-(pyrazol-4-yl)-methylylidenehydrazinyl-thiazoles (Figure 15). These compounds were substituted in the fourth position of thiazole ring with various aryl and hetaryl substituents (R1), in the second position of 2-pyrazoline ring with a benzothiazole or phenyl ring (R2), and in the fifth position with a coumarin (64-69) or a substituted phenyl ring (70-81) (R3) (Figure 15).
The compounds were assayed for their antibacterial activity against both Gram-positive and Gram-negative strains and for their antifungal activity against Candida sp. and Aspergillus sp. strains [21,22]. Compounds 64, 65, 67, and 69 showed inferior activities (MICs = 1.9-7.8 µg/mL) on S. aureus MTCC 96 and MTCC 2940, Micrococcus luteus MTCC 2470, K. planticola MTCC 530, E. coli MTCC 739, and P. aeruginosa MTCC 2453, compared to ciprofloxacin (MIC = 0.9 µg/mL). Compounds 70-81 showed superior activities (MICs = 3.9-18.5 µg/mL) on S. aureus, E. coli, and P. aeruginosa, compared to chloramphenicol (MICs = 24.6-32.8 µg/mL) [21,22].
Concerning the antifungal activity, compounds 64, 66, 67, and 69 showed similar activities (MICs = 7.8 µg/mL) on C. albicans MTCC 3017 compared to miconazole (MIC = 7.8 µg/mL). Compounds 70-81 showed superior activities on A. niger and C. albicans (MICs = 3.9-11.3 µg/mL) [21,22].
Additionally, the antibiofilm activity was evaluated on S. aureus and K. planticola biofilms [21]. Compound 69 presented a promising biofilm inhibition against S. aureus MTCC 96 (IC50 = 1.8 µM), while compound 67 inhibited the biofilm formation of S. aureus MTCC 2940 (IC50 = 12 µM) and K. planticola MTCC 530 (IC50 = 14 µM).
According to the structure-activity relationship study, inserting a strong electron-withdrawing group, particularly nitro, in the para position of the phenyl ring in R1 (70, 73, 76, and 79) resulted in an increased overall antibacterial and antifungal activity, while meta substitution was not as favorable (71, 74, 77, and 80) (Figure 15). The presence of benzothiazole moiety (64-69), which is known for its toxicity, could halt any future progress towards leader molecules [21,22].
Compounds 64-69 could target dehydrosqualene synthase of S. aureus, important for staphyloxanthin biosynthesis, a virulence factor. Based on the molecular docking studies, the coumarin moiety is important for binding to the Lys20 residue, through hydrogen bonds [21].
Abdel-Aziem et al. [23] and Kumar et al. [24] designed some 5-(coumarin-3-yl)-2-(pyrazol-1-yl)-thiazoles. These compounds were halo-substituted in the sixth position of coumarin ring (R1) and variously substituted in the third and fourth positions of the pyrazole ring (R2 and R3) (Figure 16).
The compounds were evaluated for their antibacterial activity, using the agar well diffusion method or MIC assay, against both Gram-positive and Gram-negative strains and for their antifungal activity against Candida sp. strains [23,24]. Compounds 82-85 showed superior activity on Enterococcus faecalis ATCC 29212, compared to chloramphenicol, while compounds 84 and 85 showed superior active P. aeruginosa ATCC 27853, compared to cephalotin [23]. Both compounds 86 and 87 showed superior activity (MICs = 15.67-31.25 µM) against S. aureus MTCC 3160, S. pyogenes MTCC 442, and E. faecalis MTCC 439, compared to kanamycin (MICs = 31.25-62.50 µM) [24].
Regarding the antifungal activity, compounds 86 and 87 showed inferior activity (MIC = 61.25 µM) against C. albicans NCPF 400034, C. keyfer NCPF 410004, C. krusei NCPF 44002, and C. parapsilosis NCPF 450002, compared to amphotericin B (MICs = 0.78-12.50 µM) [24].
SAR studies showed that small substituents, like methyl (82) and hydroxy (83), were important for the activity against E. faecalis, while the larger substituents, like trifluoromethyl (84), against P. aeruginosa (Figure 16). Moreover, only the compounds with both R1 and R3 substituents being electron-withdrawing groups (86 and 87) displayed an overall improved antibacterial and antifungal activity. No potential target was reported by the authors [23,24].
Mahmoodi and Ghodsi designed some 2-pyrazolium-thiazol-4-yl salts substituted in the fourth position of the thiazole ring with a coumarin, in the third position of pyrazolium ring with various substituted coumarins (R2-R4), and in the fifth position with various aryl and hetaryl substituents (R1) (Figure 17) [25].
The compounds were tested, using the zone inhibition method, for their antibacterial activity against both Gram-positive and Gram-negative strains and for their antifungal activity against Aspergillus sp. strains [25]. Compounds 88-94 and 96 showed inferior activities (IZs = 12-17 mm) against S. aureus, E. coli and M. luteus compared to gentamycin (IZs = 18-21 mm) [25].
Only compound 93 was active (IZs = 16-17 mm) against on A. niger and A. flavus, but inferior to fluconazole (IZ = 25 mm), in terms of antifungal activity [25].
Nevertheless, the heterogeneity of the results and the lack of activity in MIC terms make it difficult to draw conclusions about potential structure-activity relationships. No potential target was reported by the authors [25].
Nalawade et al. designed a series of 2-phenyl-5-(4-hetaryl-pyrazol-3-yl)-thiazoles (Figure 18) [26].
The compounds were tested, using the well diffusion method, for the antibacterial activity against Gram-positive and Gram-negative strains and for the antifungal activity against three types of strains [26]. All tested compounds (99-115) showed inferior activity (IZs = 9.6-14.4 mm) against E. coli and S. epidermidis, compared to streptomycin (IZs = 18.52-25.0 mm). In terms of antifungal activity, all compounds were active (IZs = 13.0-22.3 mm) against C. albicans NCIM 3100, A. niger ATCC 504, and Rhodotorula glutinis NCIM 3168, but inferior to fluconazole (IZs = 18.35-25.30 mm) and ravuconazole (IZs = 20.15-28.64 mm).
The antifungal activity was further evaluated through MIC screening [26]. Eleven compounds (99-119) emerged as promising anti A. niger agents (MICs = 31.25 µg/mL), with similar activity compared to ravuconazole (MICs = 7.81-31.25 µg/mL). Twelve compounds (100, 102-106, 110-115) were moderately active against R. glutinis (MICs = 62.5 µg/mL) and only one (101) against C. albicans (MIC = 62.5 µg/mL), but with inferior activity compared to fluconazole (MIC = 7.81 µg/mL) and ravuconazole.
Structure-activity relationship in these compounds implies that the best activity against A. niger was in the presence of methyl (101-103) or fluoro (104-108 and 111) as substituents on the phenyl ring directly linked to thiazole, while bulkier substituents, such as chloro (108, 112, and 113) and bromo (109, 110, and 113-115), were associated with a lower activity (Figure 18). This was opposite in the case of anti R. glutinis activity.
The antifungal activity of these compounds can be attributed to their capacity to target lanosterol 14α-demethylase, based on the molecular docking studies. No potential target was reported by the authors for the antibacterial activity [26].
To conclude the results observed in the analyzed papers (Figure 19), clubbing thiazole with pyrazole to obtain novel antimicrobials seems to expand the activity spectrum, compared to 2-pyrazoline. Pyrazole-containing compounds displayed antibacterial activity against Gram-positive and Gram-negative strains, while the antifungal activity got increasingly better. However, it should be noted that additional structural elements, such as a hydrazine linker or a supplementary heterocycle, like coumarin or 1,2,3-triazole, could significantly influence the antimicrobial activity. For example, coumarin-containing compounds were only active against Gram-positive bacterial and fungal strains, while 1,2,3-triazole-containing compounds were potent antifungal agents.
Based on the provided results, the general structure-activity relationship studies of antimicrobial 3-(2-(pyrazol-1-yl)-thiazol-4-yl)-coumarins and 2-pyrazolium-thiazol-4-yl salts could be formulated as follows: a halogen atom, bromo or chloro, in the sixth position of the coumaryl moiety enhances, but is not essential (as observed in the pyrazolium series, Figure 17), the antimicrobial potential, while the nature of the substituents from pyrazole or pyrazolium moieties influences the spectrum. Thus, compounds containing electron-withdrawing groups had a larger span of activity compared to those containing electron-donating groups, covering both Gram-positive and Gram-negative bacterial strains, as well as fungal strains.
Nevertheless, clubbing pyrazole with 1,3-thiazole is a promising research hypothesis when designing novel antifungals.
2.2.4. Thiazolyl-Imidazole Hybrid Compounds
Imidazole is frequently present in various bioactive compounds. Besides the potent activity against fungal strains, imidazole is found in compounds with various effects, such as antibacterial, antituberculosis, antiviral, antiparasitic or anticancer [27].
Nikalje et al. reported the synthesis of some 2-(2,4,5-triphenyl-imidazol-1-yl)-thiazoles, variably substituted in the second position of imidazole ring (Figure 20) [28]. The general structure of these series was rationally designed by using the thiazole heterocycle from abafungin, isavuconazole, and ravuconazole, the imidazole heterocycle and the two phenyl rings from clotrimazole, flutrimazole, and bifonazole.
The compounds were tested for their antifungal activity against multiple strains. The activity was quantified using MIC80, which is the minimal inhibitory concentration for 80% inhibition of growth. Among the synthesized compounds, those containing either electron-donating groups (116-119) or nitro (120) on the aryl showed superior activity, in most cases, against C. albicans NCIM 3471 and C. glabrata NCYC 388 (MIC80 = 0.2-0.35 µg/mL), Fusarium oxysporum NCIM 1332 (MIC80 = 20-35 µg/mL), A. flavus NCIM 539 (MIC80 = 35-50 µg/mL), A. niger NCIM 1196 (MIC80 = 40-45 µg/mL), and C. neoformans NCIM 576 (MIC80 = 5-10 µg/mL), compared to miconazole and fluconazole (MIC80 between 0.5 µg/mL and >64 µg/mL). The antifungal activity of these compounds can be attributed to their capacity to target lanosterol 14α-demethylase, based on the molecular docking studies [28].
2.2.5. Thiazolyl-Thiazolidin-4-one Hybrid Compounds
Thiazolidin-4-one is another versatile five-membered heterocycle, used in designing novel antibacterial compounds, one important direction being the development of antituberculosis compounds [29].
Othman et al. designed a series of novel 2-(thiazol-2-yl)-N-thiazolidin-4-ones, variably substituted in the second position of thiazolidin-4-one ring (Figure 21) [30].
The compounds were tested for their antibacterial activity against sensitive (ATCC 25177 H37Ra), MDR (multidrug resistant, ATCC 35822), and XDR (extended drug resistant, RCMB 2674) strains of Mycobacterium tuberculosis and various bronchitis causing bacteria [30]. Five compounds (121-125) showed equal or inferior activity against the sensitive strain, compared to isoniazid (MIC = 0.12 µg/mL). Three compounds (121, 123, and 125) showed activity against the MDR strain (MICs = 1.95-7.81 µg/mL), and one (123) against the XDR strain (MIC = 7.81 µg/mL). Concerning the antibacterial activity, all five compounds showed similar activity (MICs = 0.48-7.81 µg/mL) against Mycoplasma pneumoniae ATCC 15531, S. pneumoniae ATCC 1659, and K. pneumoniae ATCC 43816, while four compounds (123 and 125-127) showed similar activity against Bordetella pertussis ATCC 9340 (MICs = 1.95-7.81 µg/mL), compared to azithromycin (MICs = 0.49-7.81 µg/mL).
SAR studies showed that the activity on M. tuberculosis sensitive strain was favorably influenced by the presence in para position of halo-substituents (123 and 125) or methoxy substituents (121, 122, and 124). Monosubstitution (121, 123, and 125) was associated with MDR antituberculosis activity. Supplementary, the presence of a voluminous halogen in the fourth position (123) was associated with XDR antituberculosis activity, most likely due to increased molecular lipophilicity (Figure 21) [30].
Concerning the activity against bronchitis causing bacteria, SAR studies showed that the best antibacterial activity was associated with the grafting of methoxy groups in meta position (122 and 124) (Figure 21) [30].
Molecular docking studies showed that the compounds can target the enoyl-acyl carrier protein reductase InhA of M. tuberculosis, important for the type II fatty acids biosynthesis. The inhibition is enhanced by the substituent from the fourth position of the thiazole, the carbonyl of the ester group binding to the target through an accepting hydrogen bond. The inhibitory activity on InhA was further confirmed by in vitro studies [30].
Abo-Ashur et al. reported the synthesis of a series of 2-(thiazol-2-yl)-imino-thiazolidin-4-ones (Figure 22) [31].
The compounds were tested for their antituberculosis activity, with six compounds (128-133) presenting similar activity (MICs = 0.78-3.12 µg/mL) to isoniazid (MIC = 0.78 µg/mL), against M. tuberculosis RCMB 010126 [31].
The antibacterial activity was also tested against Gram-positive and Gram-negative strains [31]. Three compounds (130, 131, and 134) showed excellent activity against S. aureus RCMB 010028 and P. aeruginosa RCMB 010043 (MICs = 0.49-0.98 µg/mL) and six compounds (130-134) against E. coli RCMB 010052 (MICs = 0.49-0.98 µg/mL), compared to ciprofloxacin (MICs = 1.95-3.90 µg/mL) [31].
With respect to the antifungal activity, which was tested against Candida and Aspergillus strains, two compounds showed superior activity (130 and 134) against A. fumigatus (MICs = 0.49 µg/mL), and four compounds (127, 130, 131, and 134) against C. albicans (MICs = 0.49 µg/mL), compared to amphotericin B (MICs = 0.98-1.95 µg/mL) [31].
SAR studies in these series suggest that a halogen (126 and 129) or methoxy (127, 131, and 132) substituent grafted on the phenyl ring (R2) is essential for the antituberculosis, antibacterial, and antifungal activities (Figure 22). Advantageous for these activities were also the bioisosteric substitution of the phenyl ring with a 3-pyridinyl (134) and the grafting of an additional morpholinyl ring (128 and 130) [31]. No potential target was reported by the authors.
2.2.6. Thiazolyl-Thiazolidindione Hybrid Compounds
Widely known for the antidiabetic activity in glitazones, the thiazolidinedione heterocycle was also used in designing new antibacterial, antifungal, antiretroviral, antituberculosis, and anticancer compounds [32].
Alegaon et al. designed a series of antimicrobial 2-(thiazolidin-2,4-dion-3-yl)-N-(thiazol-2-yl)acetamides (Figure 23). The active methylene from the fifth position of the thiazolidin-2,4,-dione ring was derivatized (R) with various aromatic and heteroaromatic aldehydes through Knoevenagel condensations [33].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative strains and for their antifungal activity against various fungal strains [33]. Five compounds (135-139) showed inferior activity (MICs = 4-32 µg/mL) against S. aureus ATCC 25923, E. faecalis ATCC 35550, E. coli ATCC 35218, and P. aeruginosa ATCC 25619, compared to ciprofloxacin (MICs = 2 µg/mL) [33].
Concerning the antifungal activity, the same compounds showed inferior activity against C. albicans ATCC 2091, A. flavus NCIM 524, A. niger ATCC 6275, and C. neoformans (clinical isolate), compared to ketoconazole (MICs = 1-2 µg/mL) [33].
SAR studies underlined that the substitution of the arylidene moiety from the fifth position of thiazolidin-2,4-dione ring with electron-donating groups, especially methoxy (136-138), enhanced the overall antibacterial and antifungal activities (Figure 23). Bioisosteric substitution of the phenyl ring with 2-furanyl (139) also favorized the antibacterial and antifungal activities. The substitution with electron-withdrawing groups, such as nitro or cyano, almost abolished both activities [33].
These compounds could potentially target the ATP binding domain of bacterial DNA gyrase B and the fungal lanosterol 14α-demethylase, according to the molecular docking studies [33].
2.2.7. Thiazolyl-1,3,4-Thiadiazole Hybrid Compounds
The versatility of 1,3,4-thiadiazole heterocycle comes from its ability to act as a hydrogen binding domain, as a two-electron donor system and as a bioisosteric replacement of thiazole. Thus, it is a valuable moiety for designing novel antimicrobial, anticancer, and antiprotozoal compounds [34].
Using the artificial intelligence, Stokes et al. repurposed an antidiabetic compound, SU-3327, which acts as a c-Jun N-terminal protein kinase (JNK) inhibitor, into a promising antibacterial molecule called halicin. Structurally, halicin (140) contains a 5-nitrothiazole and a 2-amino-1,3,4-thiadiazole, linked in the 2,5’ positions by a thioether group (Figure 24) [35,36].
Halicin displayed broad antibacterial activity against E. coli (MIC = 2 µg/mL), M. tuberculosis, carbapenem-resistant Enterobacteriaceae and A. baumanii, but no activity against P. aeruginosa [35]. Further studies for the assessment of the antibacterial potential were reported by other authors as well [36,37].
Booq et al. tested halicin with significant results against S. aureus ATCC BAA-977 (MIC = 16 µg/mL), E. coli ATCC 25922 (MIC = 32 µg/mL), A. baumanii ATCC BAA-747 (MIC = 128 µg/mL), and A. baumanii MDR isolate (MIC = 256 µg/mL) bacterial strains [36]. Hussain et al. obtained promising results when assayed halicin against strains of E. faecalis and E. faecium (MICs = 4-8 µg/mL) [37]. Moreover, halicin showed proof of antibiofilm potential, alone and also in synergism with other molecules, as reported by some authors [38,39]. Currently, there are no ongoing clinical trials for testing halicin on humans.
The unique mechanism of action, which implies the disruption of membrane electrochemical gradient, pH modification and upregulation of iron acquisition genes, makes it difficult to acquire resistance. It is also possible that halicin may act as a siderophore prior to pH alteration [35].
2.2.8. Thiazolyl-1,2,3-Triazole Hybrid Compounds
1,2,3-Triazole is the most stable among heterocycles with three adjacent nitrogen atoms and it can be found in variable bioactive compounds, including antibacterial, antifungal, antimalarial, and anticancer agents [40]. Herein, we present the structure-activity relationship in thiazole clubbed with 1,2,3-triazole compounds with promising antimicrobial potential, to establish how clubbing these two heterocycles could potentially influence the biological activity.
Based on the found structures, it can be concluded that there were two types of scaffolds used: one in which the thiazole and 1,2,3-triazole rings are linked directly, but with different linking positions (a-b) [41,42], and the other one in which the rings are clubbed through various linkers and variable linking positions (c-e) [43,44,45] (Figure 25).
Shinde et al. reported the design of novel antituberculosis 5-(1,2,3-triazol-4-yl)-thiazoles, substituted in the second position of the thiazole ring with various aryl substituents (Figure 26) [41].
The antituberculosis activity was tested against M. tuberculosis H37Ra (ATCC 25177), using rifampicin (IC50 = 0.002 µg/mL and MIC90 = 0.75 µg/mL) as reference. The activity of the compounds was quantified using both IC50 and MIC90. While most of the compounds were very active in terms of IC50 values (0.58-8.23 µg/mL) [41], only two compounds (141 and 142) were active in terms of MIC90 values (2.22 µg/mL and 4.71 µg/mL), the activity being inferior to rifampicin [41].
SAR studies show that fluoro substitution of the benzyl group linked to 1,2,3-triazole is responsible for the antituberculosis activity (Figure 26) [41]. However, this boost in the activity only took place when the other phenyl ring was either unsubstituted (141) or 3-methyl substituted (142). Double halogen substitution was associated with a decrease in the activity [41].
These compounds can potentially target enoyl acyl carrier protein reductase, which is an important enzyme in the fatty acid biosynthesis and growth of mycobacteria, based on the molecular docking studies [41].
Mahale et al. synthesized a series of 2-(1,2,3-triazol-4-yl)-thiazole derivatives, substituted on the first position of 1,2,3-triazole ring (R1) and the fifth position of thiazole ring (R2) with aryl substituents (Figure 27) [42].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative strains and for their antifungal activity against Candida sp. and Aspergillus sp. strains [42]. Compounds 144, 146, 148, and 150-154 were equally active (MIC = 0.5 µg/mL) against E. coli ATCC 25922 and S. aureus ATCC 25923, compared to streptomycin (MIC = 0.5 µg/mL).
Concerning the antifungal activity, compounds 143-145, 147, 149, and 150 were equipotent (MIC = 0.5 µg/mL) against C. albicans MTCC 2977 and A. niger MCIM 545, compared to griseofulvin (MIC = 0.5 µg/mL) [42].
SAR studies suggest that substitution with predominantly electron-withdrawing groups, like nitro and halogen atoms, provide an antibacterial effect to the molecules, while the antifungal effect can be achieved using electron-donating, such as methyl and methoxy, and electron-withdrawing groups in both positions (Figure 27). No potential target was reported by the authors [42].
Jagadale et al. designed a series of 1-(thiazol-5-yl)-2-(1,2,3-triazol-1-yl)-ethanol derivatives, substituted in the second position of the thiazole ring (R1) and the fourth position of the 1,2,3-triazole ring (R2) with various aryl substituents (Figure 28) [43].
The compounds were tested for the antibacterial activity against Gram-positive and Gram-negative strains and for the antifungal activity against various strains [43]. Compounds 156-158 showed inferior activity (MIC = 62.5 µg/mL) against S. epidermidis NCIM 2178, compared to streptomycin (MIC = 7.81 µg/mL) [43].
Regarding the antifungal activity, compounds 155 and 159-162 were inferior against A. niger (ATCC 504) compared to fluconazole (MIC = 7.81 µg/mL), but similar to ravuconazole (MIC = 31.25 µg/mL). Additionally, compound 158 displayed activity against R. glutinis (MIC = 62.5 µg/mL) too, but inferior to both reference compounds [43].
SAR studies suggest that substitution with 4-chloro (160-162) and 4-fluoro (159) in R1, as well as unsubstituted phenyl (155) are responsible for the activity against A. niger (Figure 28). Fluoro substitution of only one (156-157, and 160) or both (158) of the phenyl rings was associated with antibacterial activity against S. epidermidis. Double 4-fluorophenyl substitution induced activity against S. epidermidis, A. niger, and R. glutinis, as seen in compound 158. No potential target was reported by the authors [43].
Poonia et al. designed some 2-(1,2,3-triazol-1-yl)-N-(thiazol-2-yl)-acetamides and 2-(1,2,3-triazol-1-yl)-N-(benzothiazol-2-yl)-acetamides, para-substituted on the phenyl-ureidomethyl moiety, linked to the fourth position of 1,2,3-triazole ring (Figure 29) [44].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative strains and for their antifungal activity against Candida sp. and Rhizopus sp. strains [44]. All compounds (163-174) showed superior activity (MICs = 0.0074-0.0333 µmol/mL) against C. albicans MTCC 183 and Rhizopus oryzae MTCC, compared to fluconazole (MIC = 0.0408 µmol/mL) [44].
Concerning the antibacterial activity, four compounds (165, 170, 173, and 174) showed noteworthy activity. Compounds 165 and 170 showed inferior activity (MICs = 0.0287-0.0299 µmol/mL) against E. coli MTCC 1654 and S. aureus MTCC 3160, compared to ciprofloxacin (MIC = 0.0094 µmol/mL). Compounds 170 and 173 showed inferior activity (MICs = 0.0257-0.0299 µmol/mL) against P. fluorescens MTCC 664, compared to the reference drug, while compound 174 showed superior activity (MIC = 0.0071 µmol/mL). Lastly, compound 171 was the most active against Bacillus endophyticus (MIC = 0.0257 µmol/mL), but still inferior to the reference drug [44].
Based on the SAR studies, the insertion of thiazole or benzothiazole rings increased the antifungal activity, compared to the phenylureidopropargyl precursors and fluconazole (Figure 29) [44]. For the antibacterial activity, bromo (165, 169, and 173) and methoxy (166, 170, and 171) substitutions, as well as the annulation of the thiazole ring (171-174) were the most important [44].
These compounds act as antifungals by targeting sterol 14-α demethylase, according to the molecular docking studies [44].
Gondru et al. reported the synthesis of two series of 2-(1,2,3-triazol-4-yl-methoxybenzylidenehydrazinyl)-thiazoles, substituted on the fourth position of thiazole ring with aryl and hetaryl substituents (Figure 30) [45]. They used para-hydroxybenzaldehyde as starting material, of which the phenolic hydroxyl group was etherified with a substituted 1,2,3-triazole moiety, while the carbonylic group was derivatized to the corresponding hydrazonothiazoles [45].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative strains and for their antifungal activity against various Candida sp. and Issatchenkia sp. strains [45]. Compounds 175-181 showed inferior activity (MICs = 2.8-15.7 µM) against S. aureus MTCC-96, S. aureus MLS16 (MTCC 2940), M. luteus MTCC 2470, K. planticola MTCC 530, E. coli MTCC 739, and P. aeruginosa MTCC 2453, compared to ciprofloxacin (MIC = 2.7 µM) [45].
Concerning the antifungal activity, compounds 176-180 showed superior activity (MICs = 5.9-14.2 µM) against C. albicans MTCC 183, MTCC 854, and MTCC 3018, C. aaseri MTCC 1962, C. glabrata MTCC 3019, and Issatchenkia hanoiensis MTCC 4755, compared to miconazole (MIC = 18.7 µM) [45].
According to the SAR studies, 4-methoxyphenyl (175), benzo[f]coumarinyl (176), and 8-methoxycoumarinyl (177), 6-bromo-8-methoxycoumarinyl (178), and 8-bromocoumarinyl (179) substitutions were associated with antibacterial activity against S. aureus (Figure 30) [45]. The presence of the coumarin heterocycle was important for the antifungal activity, especially against C. albicans MTCC 183 strain. By replacing the methoxy group from the coumarin ring with an ethoxy group or introducing electron-withdrawing groups, like chloro and nitro, in both 6- and 8- positions of the coumarin, cancelled the antibacterial effect, but not the antifungal one. No potential target was reported by the authors [45].
In conclusion, 1,2,3-triazole heterocycle is a versatile moiety for designing novel antimicrobial compounds with broad activity spectrum. As observed in the presented studies, clubbing with thiazole resulted in potent compounds against a large variety of pathogen strains, including mycobacteria.
A prominent feature of the presented compounds was the presence of substituted phenyl rings, linked directly to the heterocycles or through a linker. Thus, the difference between compounds’ activity were mostly attributed to these substituents. By far, the most used substituents in these compounds were halogens, methyl and methoxy groups [41,42,43,44,45]. A summary of the structure-activity relationships in antimicrobial 1,3-thiazole clubbed with 1,2,3-triazole hybrid compounds is presented in Figure 31.
2.2.9. Thiazolyl-1,3,4-Oxadiazole Hybrid Compounds
Similarly to the previously mentioned heterocycles containing three heteroatoms, the 1,3,4-oxadiazole ring can be found in a plenitude of compounds with various biological activities, including anticancer, antibacterial, antifungal, and antiviral effects. This heterocycle can act as bioisostere for the carbonyl group and can be used in the structure of a molecule as a flat aromatic linker to ensure an adequate orientation [46].
As reported in the literature, the 5-thioxo-1,3,4-oxadiazole heterocycle can be found in a series of anticancer and antimicrobial compounds [47,48,49], thus making this heterocycle an important contender for designing novel antimicrobials.
Some series of 2-thiazolyl-5-mercapto-1,3,4-oxadiazoles, either linked through a methylene linker between the fourth position of thiazole ring and the second position of 1,3,4-oxadiazole ring (Figure 33), or directly linked between the fifth position of thiazole ring and the second position of 1,3,4-oxadiazole ring (Figure 34), were synthesized [50,51].
The compounds exist in two tautomeric forms (Figure 32). The existing data [52] show that the thione tautomer (II) is more stable in the solid state, while in solution the thiol tautomer (I) is predominant. The existence of thiol-thione tautomerism was valorized by obtaining the corresponding thioether derivatives (Figure 33) and Mannich bases (Figure 34) [50,51].
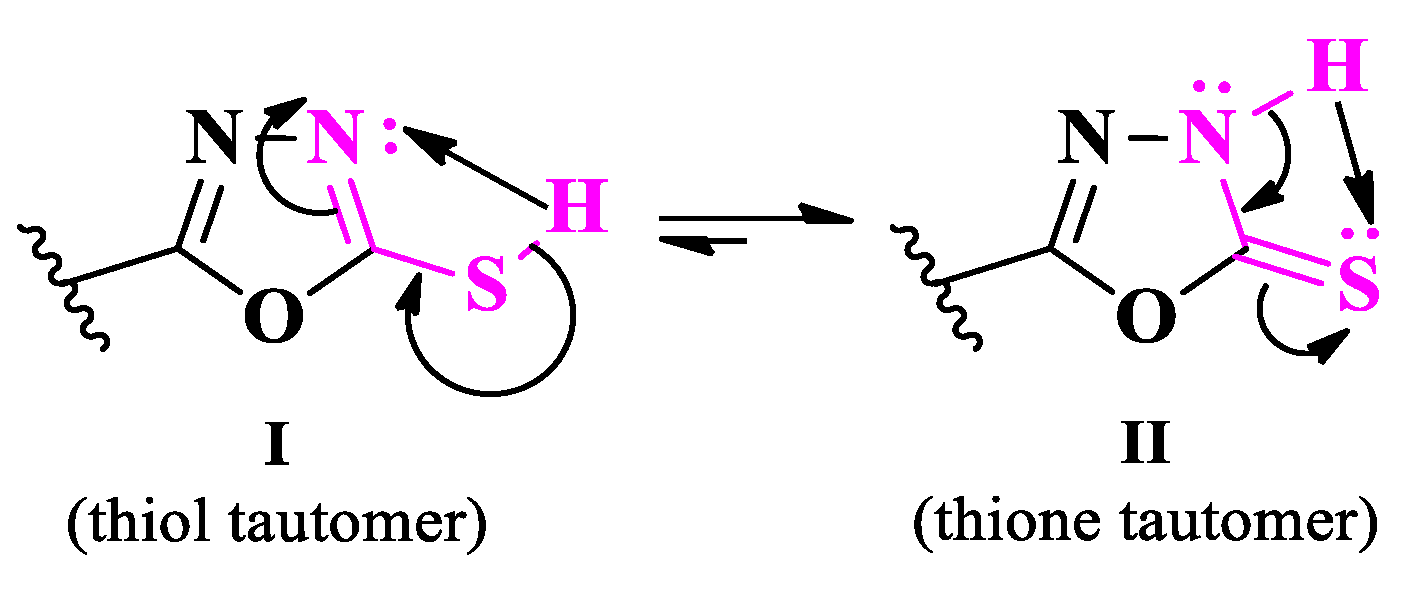
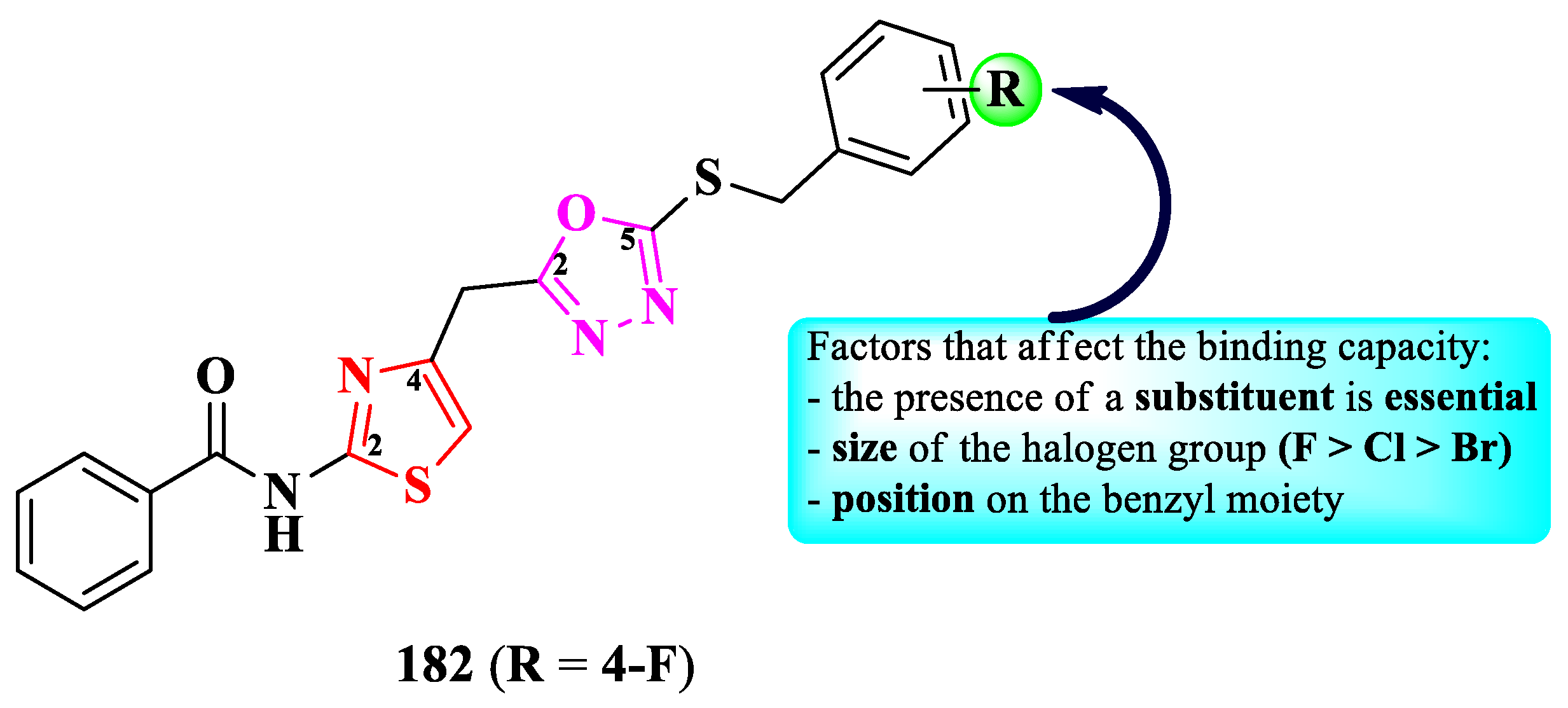
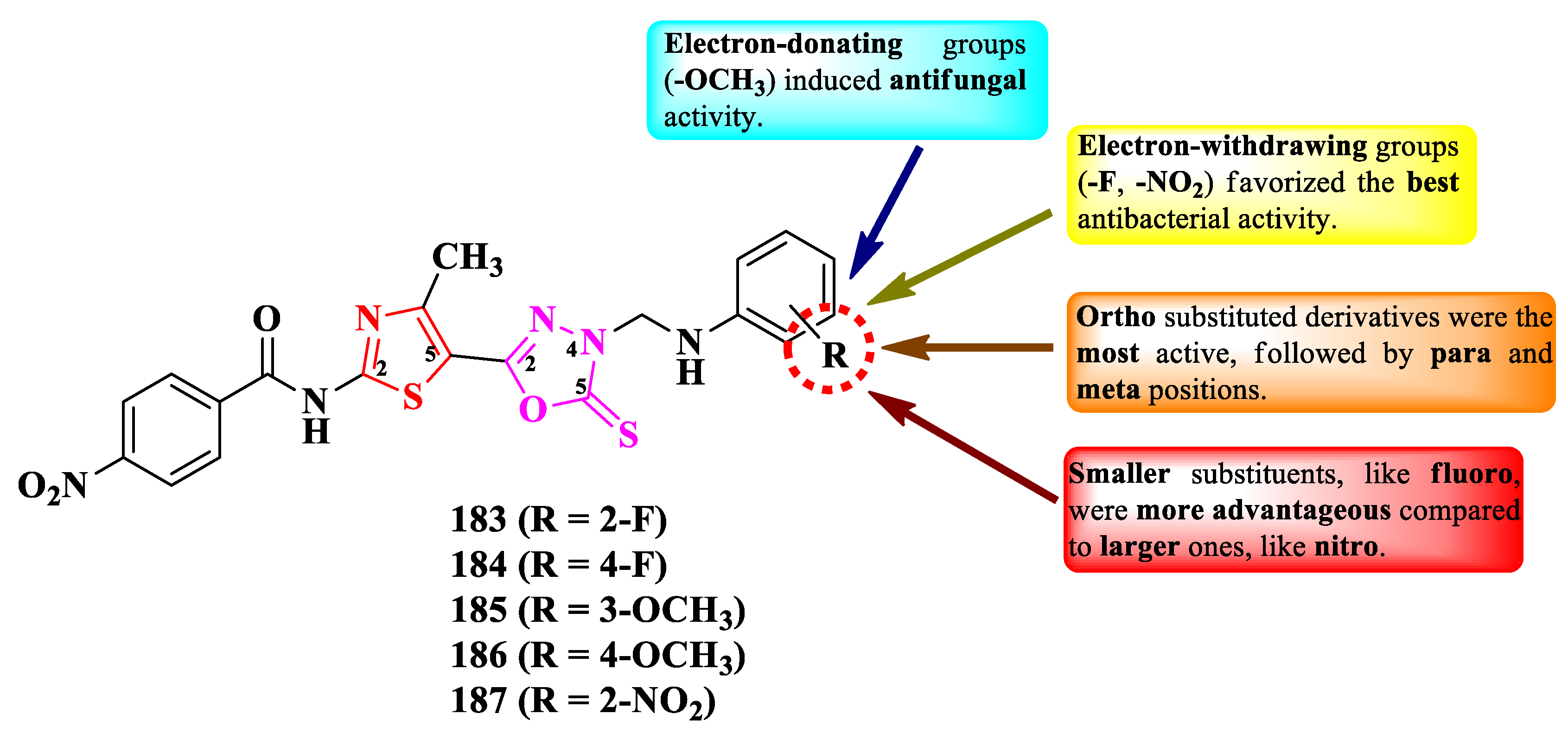
Figure 32.
The thiol-thione tautomerism [52].
Figure 32.
The thiol-thione tautomerism [52].
Figure 33.
SAR studies in 1,3-thiazole clubbed in 2-(thiazol-4-yl)-methylene-5-thio-1,3,4-oxadiazoles urease inhibitors, reported by Athar Abasi et al. [50].
Figure 33.
SAR studies in 1,3-thiazole clubbed in 2-(thiazol-4-yl)-methylene-5-thio-1,3,4-oxadiazoles urease inhibitors, reported by Athar Abasi et al. [50].
Figure 34.
SAR studies in antimicrobial 2-(thiazol-5-yl)-5-mercapto-1,3,4-oxadiazoles, reported by Desai et al. [51].
Figure 34.
SAR studies in antimicrobial 2-(thiazol-5-yl)-5-mercapto-1,3,4-oxadiazoles, reported by Desai et al. [51].
Athar Abbasi et al. described the synthesis of 2-(thiazol-4-yl)-5-thio-1,3,4-oxadiazoles, capable of urease inhibition, thus offering an alternative potential treatment to Helicobacter pylori infections (Figure 33) [50].
The inhibitory activity was tested using thiourea (IC50 = 21.11 ± 0.12 µM) as reference. Based on the results (IC50 = 2.17 ± 0.41 µM) and molecular docking studies, compound 182 (Figure 33) presented the best binding affinity (-8.40 kcal/mol), among all synthesized and tested compounds, and was able to bind to the active site of the enzyme [50].
By integrating all the obtained information, it could be concluded that the fluorine atom forms two halogen-metal bonds with the nickel active center of urease. The potential inhibition of urease was influenced by the type and position of the halogen atom, on the benzyl moiety linked to the sulfur atom. The fluorine atom in para position (182) was the most advantageous for the inhibition. For chlorine, meta position was favorable, while for bromine was either ortho or para position. One more important aspect observed was that an unsubstituted benzyl moiety yielded the weakest inhibitory capacity, therefore the presence of a halogen substituent was essential for urease inhibition [50].
Desai et al. designed a series of 2-(thiazol-5-yl)-5-mercapto-1,3,4-oxadiazole Mannich bases. These compounds are variably substituted on the anilino moiety linked to the fourth position of the 1,3,4-oxadiazole ring, through a methylene bridge (Figure 34) [51].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative bacterial strains and for their antifungal activity against Candida sp. and Aspergillus sp. strains [51]. Compounds 183, 184, and 187 showed superior activity against E. coli MTCC 443, P. aeruginosa MTCC 1688, and S. pyogenes MTCC 442, compared to ampicillin (MICs = 100-250 µg/mL) [51].
Concerning the antifungal activity, compounds 185 and 186 showed superior activity (MICs = 25-50 µg/mL) against C. albicans (MTCC 227) and A. niger (MTCC 282), compared to griseofulvin (MICs = 100-500 µg/mL) [51].
SAR studies implied that the most active antibacterial compounds had electron-withdrawing groups, such as nitro (187) and fluoro (183-184), while the most active antifungal compounds had electron-donating groups, such as methoxy (185-186) (Figure 34). No potential target was reported by the authors [51].
2.3. Thiazole Clubbed with Five-Membered Benzofused Heterocycles
Benzofusion of heterocycles leads to an increase in lipophilicity and electron-withdrawing character, which could be advantageous in drug design. Additionally, benzofused heterocycles constitute bioisostere alternatives to naphthalene, which can facilitate better binding to a potential target [53].
Based on the literature search of the last six years, we will discuss further the structure-activity relationship in antimicrobial hybrid compounds containing thiazole clubbed with the following five-membered heterocycles and their derivatives: indole, carbazole, indolin-2-one, and tetrahydroindenofuran.
2.3.1. Thiazolyl-Indole and Thiazolyl-Carbazole Hybrid Compounds
The antimicrobial properties of carbazole derivatives were known since the isolation of carbazole alkaloids from Murraya koenigii leaves [54]. Similarly, indole naturally occurs in various alkaloids with antimicrobial properties [55,56,57]. Since then, these heterocycles have been increasingly used in the design of synthetic novel antimicrobials.
Zhao et al. reported the design of a novel series of antimicrobial thiazole clubbed with either indole or carbazole hybrid compounds, containing a ferrocene scaffold (Figure 35) [58].
The antibacterial activity was assayed against S. aureus, E. coli, and P. aeruginosa, using ciprofloxacin as reference (MIC = 15.625 µg/mL). Only three compounds (188-190) showed good activity against E. coli (MIC = 31.25 µg/mL), while the activities against the other strains were poor (MICs = 125-250 µg/mL) [58].
SAR studies in these series suggest that the introduction of a second heterocycle on the 4-ferrocenylthiazole scaffold, indole or carbazole, significantly decreased the antibacterial activity (Figure 35), compared to the 4-ferrocenyl-2-N-anilinothiazole series. No potential target was reported by the authors [58].
2.3.2. Thiazolyl-Indolin-2-one Hybrid Compounds
Indolin-2-one derivatives are ubiquitous compounds found in the human body. They possess various biological activities, including antioxidant, antimicrobial, and antiproliferative activities [59].
Starting from 5-(piperazin-1-yl)-sulfonylindolin-2,3-dione, Alzahrani et al. obtained some clubbed thiazole Schiff bases and thiazolin-4-one compounds as dihydrofolate reductase (DHFR) inhibitors and anti-quorum sensing (QS) agents (Figure 36) [60].
They derivatized the carbonyl group from the third position of indolin-2,3-dione by either condensation with 2-N-anilino-thiazolin-4-one or with different aminothiazoles and hydrazonothiazoles (Figure 36, 191-194) [60].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative bacterial strains and for their antifungal activity against C. albicans ATCC 10213 [60]. Compounds 191, 193, and 194 showed superior activity (MICs = 1.9-15.6 µg/ml) against S. aureus ATCC 25923 and ATCC 29213, E. faecalis ATCC 29212, E. coli ATCC 35218, P. aeruginosa ATCC 9027 and PAR.36, and S. typhimurium ATCC 14028, compared to levofloxacin (MICs = 8.1-130 µg/mL). Furthermore, compounds 191, 192, and 194 inhibited the E. faecalis QS system at low concentrations and presented IC50 values of DHFR inhibition in the submicromolar range (0.04-0.28 µM), which were similar to methotrexate (IC50 = 0.061 µM) [60].
Regarding the antifungal activity, as it was expected, the compounds had inferior activity against C. albicans (192, MIC = 62.5 µg/ml), compared to nystatin (MIC = 3.9 µg/mL) [60].
SAR studies in these series can be constructed on four levels. The first level is the nature of the bond between the indolin-2-one ring and the thiazolin-4-one- or thiazole-containing fragments (Figure 36). Compound 191, containing an ethenyl bond (-C=C-), showed antibacterial activity against S. aureus and P. aeruginosa (MIC = 12.5 µg/mL), while compounds 192-194, containing an imine bond (-C=N-), showed both antibacterial and antifungal activities against a broader spectrum, including E. faecalis, E. coli, S. typhimurium, and C. albicans [60].
The second level is based on the nature of the azole heterocycle. As observed on the previous level, the compound containing a thiazolin-4-one ring had a narrower activity spectrum compared to the compounds containing the thiazole ring (Figure 36) [60].
The third level is based on the distance between the indolin-2-one ring and the thiazole ring. As the distance between the two rings grows, so does the antibacterial activity. Compound 192, in which the rings are directly linked through an imine bond, had the weakest activity among all three imino compounds (MIC = 62.5 µg/mL). Compound 193 had registered better activity when the imino bond was swapped with a hydrazono one (MIC = 7.8 µg/mL), while introducing a benzensulfonamide moiety in compound 194 resulted in the best antibacterial activity (MICs = 1.9-7.8 µg/mL). An additional sulfonamide group will increase the antibacterial activity, which is the last level of the SAR studies in these series (Figure 36) [60].
2.3.3. Thiazolyl-Tetrahydroindenofuran Hybrid Compounds
Indenofurans are rare occurring in nature compounds, found in Anisodus tanguticus species. These compounds are mostly known for their antioxidant properties, but there have been reports about their antibacterial potential [61,62].
Adole et al. designed a series of 2-(2-tetrahydroindenofuranylidene)-hydrazinylthiazoles, substituted in the fourth position of the thiazole ring with various aryl groups (Figure 37) [63].
The compounds were tested for the antibacterial activity against Gram-negative strains and for the antifungal activity against various strains [63]. Compounds 195-197 showed inferior activity (MICs = 15.62-31.25 µg/mL) against E. coli and Shigella boydii, compared to ampicillin (MICs = 1.95-3.9 µg/mL).
Concerning the antifungal activity, compounds 195-199 showed similar activity (MICs = 1.95-15.62 µg/mL) against R. oryzae, Mucor mucedo, A. niger, and C. albicans, compared to fluconazole (MICs = 0.97-1.95 µg/mL) [63].
SAR studies in these compounds underline that the para substitution with bulky substituents was associated with overall good antibacterial and antifungal activities (Figure 37). The meta substitution may be useful in enhancing the antifungal effect, compound 198 being twice or thrice more active than the para substituted compounds. It is also worth mentioning that fluoro substitution (199) is beneficial for the antifungal activity, but not for the antibacterial [63]. Switching from phenyl to naphthyl in the fourth position of thiazole ring decreased the overall antibacterial and antifungal activities and it should be avoided. No potential target was reported by the authors [63].
2.4. Thiazole Clubbed with Six-Membered Heterocycles
The six-membered heterocyclic compounds are numerous and versatile in terms of structure and bioactivity. Based on the literature research in the last years, only thiazole clubbed with pyridine antimicrobials were reported [64,65,66,67]. Thus, we present the SAR studies only for them.
2.4.1. Thiazolyl-Pyridine Hybrid Compounds
Pyridine is a privileged heterocycle found in a large variety of antimicrobial compounds, either as a sole heterocycle or together with other heterocycles. There have been reports that noted an additional heterocycle to pyridine may have the potential to enhance its biological activity [68].
Muluk et al. and Patil et al. designed some 2-(4-pyridyl)-thiazoles as potential DNA gyrase and lumazine synthase inhibitors [64,65,66]. These compounds are substituted in the fourth position of the thiazole ring with aryl or hetaryl substituents, linked to the thiazole ring via an acylhydrazonomethylene or α,β-unsaturated carbonyl linker (R2) (Figure 38). There is a supplementary substituent in the second position of the pyridine ring, which is ethyl or propyl, depending on the starting thiocarbamide fragment (-CN=S) for the Hantzsch synthesis, ethionamide or prothionamide [64,65,66].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative strains and for their antifungal activity against Candida sp. and Aspergillus sp. strains [64,65,66]. Compounds 200-204, 208, and 209 showed inferior activity (MICs = 18-170 µg/mL) against S. typhi ATCC 9207, E. coli ATCC 8739, E. aerogenes ATCC 13048, S. aureus ATCC 6538, and P. aeruginosa, compared to tetracycline (MICs = 3.0-20 µg/mL) and streptomycin (MIC = 30 µg/mL) [64,65,66].
Concerning the antifungal activity, the same compounds showed inferior activity (MICs = 60-280 µg/mL) against C. albicans ATCC 10231, compared to fluconazole (MIC = 30 µg/mL) [64,65,66].
SAR showed that by bioisosteric substitution of the phenyl ring with a furan heterocycle (204 and 208) (Figure 38), the antibacterial and antifungal activities increased. Substitution with electron-withdrawing groups was favorable for the overall antibacterial and antifungal activities. The presence of a 2-nitro group (200) was favorable for the activity on E. aerogenes and the 3,5-dichloro substitution (203) for the activity on C. albicans, this being in agreement with the presence of this substituent in some important antifungal azoles like miconazole, ketoconazole, or itraconazole. The elimination of the methyl group from the fifth position of the thiazole ring (R3) did not affect the overall antibacterial and antifungal activities [64,65,66].
In order to establish the importance of the position by which the pyridine ring is linked to the thiazole one, Eryılmaz et al. synthesized two series of 2-(2/4-pyridyl)-thiazoles, substituted in the fourth position of the thiazole ring with aryl or hetaryl substituents (Figure 38) [67].
The compounds were tested for the antibacterial activity against Gram-positive and Gram-negative strains and for the antifungal activity against C. albicans ATCC 10231 [67]. Both 2-pyridyl (213-217a) and 4-pyridyl (213-217b) series showed inferior activity (MICs = 0.01-5.7 mM) against E. coli W3110, P. aeruginosa ATCC 27853, and S. aureus ATCC 6538P, compared to cefepime (MICs = 0.001-0.06 mM) and amikacin (MICs = 0.01-0.02 mM). The 4-pyridyl series (b) showed better activity (MICs = 0.01-4.7 mM) compared to 2-pyridyl series (a) (MICs = 1.2-5.7 mM) [67].
Concerning the antifungal activity, both series showed superior activity against C. albicans compared to fluconazole, the 4-pyridyl series showed better activity (MICs = 0.15-1.2 mM) than 2-pyridyl series (MICs = 0.15-2.8 mM) [67].
Based on these results, 4-pyridyl had a better influence on the antibacterial and antifungal activities compared to 2-pyridyl. This can be due to the better capacity of the compounds to interact with a potential target through hydrogen bonds or ionic bonds, which may be difficult in 2-pyridyl series, due to the steric hinderances [67].
Some of the compounds can express their antibacterial activity by targeting DNA gyrase, important for the replication of genetic material in bacteria, while some of the compounds express the antifungal activity by targeting lumazine synthase, a key enzyme in the biosynthesis of riboflavin in fungi [64,65].
In conclusion, clubbing thiazole with pyridine to design potent antimicrobials in the presented cases depends mostly on the nature of the used substituents, which were mainly electron-withdrawing substituents (nitro and halogens). Based on the obtained results, the spectrum is narrower and the activity is lower compared to hybrid compounds containing five-membered heterocycles, such as pyrazole or triazole. Thus, clubbing an additional heterocycle may be helpful, however there was no similar study performed in the selected timeframe for this paper to support our hypothesis.
2.5. Thiazole Clubbed with Six-Membered Benzofused Heterocycles
Based on the literature search of the last six years, we will discuss further the structure-activity relationship in antimicrobial hybrid compounds containing thiazole clubbed with the following six-membered heterocycles and their derivatives: coumarin, flavone, quinoline, quinoline-2(1H)-one, and quinazolin-4(3H)-one.
2.5.1. Thiazolyl-Coumarin Hybrid Compounds
The coumarin heterocycle is found in some authorized antimicrobials (novobiocin and chlorobiocin) [69], thus making this moiety veritable for designing novel antimicrobials. Herein, we present the SAR studies in antimicrobial thiazole clubbed with coumarin compounds.
Yusufzai et al. reported the design and synthesis of some bis-coumarin derivatives (series I). In this series, the 4-(3-coumaryl)-thiazoles are linked to a second coumarin heterocycle by a methylylidenhydrazinyl bridge in the second position of the thiazole ring (Figure 39) [70].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative strains and for their antituberculosis activity against the H37Rv strain (ATCC 25618) [70]. Compounds 218-220, 222, 224, 226, and 227 showed similar activities (MICs = 31.25-62.5 µg/mL) against E. coli, E. aerogenes, S. typhi, S. pneumoniae, and S. aureus, compared to streptomycin (MICs = 31.25-62.5 µg/mL), kanamycin (MICs = 62.5-125 µg/mL), and vancomycin (MICs = 31.25-250 µg/mL) [70].
Regarding the antituberculosis activity, compounds 218-220, 222-225, 227, and 228 showed inferior activity (MIC = 50 µg/mL) compared to isoniazid (MIC = 0.0781 µg/mL) [70].
SAR studies in these compounds suggest that the substitution of the second coumarin unit (R2) with a nitro group (221 and 226) abolished the antituberculosis activity and severely decreased the antibacterial activity (Figure 39) [70]. Bromo (227) and methoxy (218-220 and 223-225) substitutions on the same coumarin were advantageous for both activities, while hydroxy substitution (228) was by far the best for the activity against all tested strains (MICs = 31.25-62.5 µg/mL). Coumarin heterocycles may also have a significant contribution to the activity, as all compounds displayed different degrees of potency, but none of them was completely inactive. No potential target was reported by the authors [70].
Another attempt to obtain urease inhibitors against H. pylori was reported by Salar et al., who designed a series of 4-(3-coumaryl)-thiazoles, in which they replaced the second coumarin heterocycle with an aromatic structure (series II) (Figure 39) [71].
The inhibitory capacity of the compounds was compared to acetohydroxamic acid (IC50 = 27 ± 0.5 µM) [71]. The most active compound (229) had a better inhibitory activity than acetohydroxamic acid (IC50 = 16.29 ± 1.1 µM). Other results worth mentioning were registered by compounds 230 (IC50 = 76.41 ± 0.1 µM), 231 (IC50 = 77.67 ± 1.5 µM), and 232 (IC50 = 71.21 ± 1.6 µM) [71].
Structure-activity relationships in this series reflect the importance of fluoro, chloro, hydroxy, and methoxy substitutions for the urease inhibition, as well as their relative position on the aryl ring (Figure 39). Thus, 2-fluoro (231), 2-hydroxy (232), 2-hydroxy-5-fluoro (229), and 2-chloro-3-methoxy (231) substitutions were the most advantageous for the inhibition [71].
Molecular docking studies further emphasized the capacity of these compounds to inhibit urease by binding to its active site [71].
Hu et al. designed two series of antibacterial 2-(8-(1-oxoprop-2-en-2-yl)-oxycoumarinyl)-thiazoles, substituted on the α,β-unsaturated carbonyl linker with aryl or hetaryl substituents (Figure 40) [72].
The compounds were tested for the antibacterial activity against Gram-positive and Gram-negative strains. Compounds 237-242 showed superior activity (MICs = 0.004-0.0016 mM) against MRSA (S. aureus N315), E. coli ATCC 25922, and E. faecalis, compared to norfloxacin (MICs = 0.025-0.050 mM) [72].
SAR studies in these series of compounds underline the selectivity confined by the two major substituents, phenyl and indole, towards MRSA, E. coli, and E. faecalis (Figure 40) [72]. The substituents on these rings influence the antibacterial activity. In the phenyl series, 4-nitro (234) and 2,4-dichloro (233) groups influenced the activity towards MRSA, while 4-methyl (236) against E. coli. The 4-methoxy (235) substitution was the most advantageous against both strains [72].
In the indole series, small alkyl chains (241 and 242) selectively influenced the activity against E. faecalis. As reported by the authors, the bis-N-(2-hydroxyethyl) groups are important for the development of some antiparasitic and antifungal drugs (Figure 40) [72].
These compounds can express the antibacterial activity by targeting DNA gyrase, based on the molecular docking studies [72].
In conclusion, the clubbing of thiazole with coumarin resulted in very potent antibacterial and antifungal agents against a broad spectrum of pathogen strains, including even MRSA and mycobacteria.
The broad spectrum was also demonstrated in other hybrid compounds bearing a coumarin heterocycle, previously mentioned in this paper [21,23,24,25,45].
The coumarin heterocycle should be taken into consideration when designing novel antibacterials against problematic strains, like MRSA.
2.5.2. Thiazolyl-Flavone Hybrid Compounds
Flavones are part of the large family of flavonoids, a widely distributed class of natural polyphenolic compounds. These impressive compounds exhibit a large variety of biological activities, including antimicrobial, antioxidant, and anticancer activities, thus constituting an attractive scaffold for drug design [73].
Zhao et al. reported the design and synthesis of a series of antistaphylococcal 2-(7-aminoethoxyflavonyl)-thiazoles, substituted on the fifth position of the thiazole ring with various aryl substituents (Figure 41) [74].
The compounds were tested for the antistaphylococcal activity against 40 clinical isolates (DPHS001 to DPHS040) of S. aureus, one MRSA strain (ATCC 43300), and one MSSA strain (ATCC 29213). Out of the 40 clinical isolates, 35 were mecA-positive (DPHS001 to DPSH035), (mecA = the gene complex responsible for altered penicillin-binding protein (PBP2a) synthesis and methicillin resistance), and 5 were mecA-negative (DPHS036 to DPHS040) [74].
Only three compounds (238-240) were active against the tested clinical isolates. The antibacterial activity was higher against MSSA and mecA-negative strains (MICs = 31.2-125 µg/mL) than against MRSA and mecA-positive strains (MICs = 31.2-500 µg/mL. The most active was compound 238, with good activity against all tested strains (MICs = 31.2-125 µg/mL) [74].
SAR studies in these compounds showed that for the nitro derivatives, 4-nitro substitution (240) increased the antistaphyloccocal activity, while 2- and 3-nitro substitutions inactivated the compounds (Figure 41) [74]. For the chloro derivatives, 2-chloro substitution (238) induced activity against all tested strains, while 3-chloro (239) only against MSSA and mecA-negative strains. 4-Chloro and other substituents, such as fluoro, hydroxy, and methyl inactivated the compounds [74].
These compounds express their antistaphylococcal activity by targeting DNA gyrase. Their inhibitory capacity was evaluated in comparison to novobiocin (IC50 = 0.8-3.23 µg/mL) [74]. Six compounds presented promising inhibitory capacity: IC50 = 23.45-38 µg/mL against DPHS001 DNA gyrase and IC50 = 12.21-31.34 µg/mL against DPHS0036 DNA gyrase. Molecular docking studies showed that 4-nitro derivative 240 was able to block the ATP binding site of DNA gyrase [74].
2.5.3. Thiazolyl-Quinoline and Thiazolyl-Quinolone Hybrid Compounds
Quinoline and quinolinone are heterocycles found in some antimalarial drugs, such as quinine, and antibacterial compounds, as part of the fluoroquinolones class. Thus, they should be taken into consideration when designing novel antimicrobial compounds [75]. Herein, we report the structure-activity relationship of antimicrobial thiazoles clubbed with quinolines and quinoline-2(1H)-ones.
Ammar et al. reported the synthesis of some 2-(quinolin-3-yl-methylenehydrazinyl)-thiazoles, substituted in the fourth and fifth positions of the thiazole ring (Figure 42) [76].
The compounds were tested for their antibacterial activity against sensitive and multidrug resistant bacterial strains and against some fungal strains [76]. Compounds 241-243 showed superior activity (MICs = 0.97-27.77 µg/mL) against S. aureus, E. faecalis, E. coli, P. aeruginosa, and S. typhi sensitive strains, compared to tetracycline (MICs = 15.62-62.5 µg/mL) [76]. Regarding the multidrug resistant strains, these compounds showed similar activity against S. aureus (ATCC 33591), E. coli (BAA-196), and P. aeruginosa (BAA-2111), compared to vancomycin (MICs = 0.78-1.95 µg/mL) and ciprofloxacin (MICs = 1.38-3.9 µg/mL) [76].
Concerning the antifungal activity, compounds 241-243 showed similar activity (MICs = 7.81-62.5 µg/mL) against C. albicans and F. oxysporum, compared to amphotericin B (MICs = 15.62-31.25 µg/mL) [76].
SAR studies in these compounds underline the importance of a substituent in para position of the phenyl ring from the fourth position of thiazole ring, as the unsubstituted thiazole compound had low antibacterial and antifungal activities (Figure 42) [76]. The nature of the substituent was as important too, since the hydroxy derivative (242) had better activity compared to the chloro derivative (241). The importance of electron-donating groups can be seen in compound 243, where the phenyl ring was replaced with methyl [76].
These compounds express their antibacterial activity by targeting DHFR. Compound 242 showed better inhibitory capacity (IC50 = 10.02 ± 1.05 µM) than trimethoprim (IC50 = 18.25 ± 0.65 µM) [76].
Litim et al. designed some 2-(quinolin-3-yl)- and 2-(quinolinon-3-yl)-thiazoles, in which the heterocycles are linked by an α-aminophosphonate group (Figure 43). The obtained compounds are substituted on the hexatomic heterocycles with methyl or methoxy groups and on the fifth position of the thiazole ring with a phenyl or coumarinyl ring. Additionally, the quinoline derivatives are substituted on the third position with hydroxy or chloro substituents [77].
The compounds were tested for their antibacterial activity against sensitive and drug resistant bacterial strains and for their antifungal activity against C. albicans [77]. Compounds 244-250 showed similar or lower activity (MICs = 0.25-128 µg/mL) against S. aureus ciprofloxacin resistant (cipro R), ATCC 25923, and ATCC 6538, E. faecalis vancomycin resistant (vanco R) and ATCC 29212, E. coli ATCC 25922, ESBL (extended spectrum beta-lactamase), cipro R, colistin resistant (mcr-1), and ATCC 8739, P. aeruginosa VIM-2 and ATCC 9027, A. baumanii NDM-1 and OXA-23, E. cloacae fosfomycin resistant (fos R), K. pneumoniae (carbapenem-resistant and carbapenem-sensitive), Serratia marcescens, S. typhi 14028, and Citrobacter sp., compared to imipenem, ciprofloxacin, and amikacin (MIC = 2 µg/mL) [77].
Concerning the antifungal activity, compounds 244-250 showed similar or lower activity (MICs = 0.25-32 µg/mL) against C. albicans compared to fluconazole (MIC = 2 µg/mL) [77].
According to the SAR studies, quinolone compounds showed better activity (MICs = 0.25-256 µg/mL) than quinoline derivatives (MICs = 0.25-512 µg/mL) (Figure 43) [77]. The inhibitory effect was also related to the type of substituent on the α-aminophosphonate moiety, with 5-(3-coumarinyl)-thiazole being more active than 5-phenylthiazole substituent [77].
The presence of both 2-hydroxyquinoline and 5-(coumarin-3-yl)-thiazole moieties in the same molecule (compounds 248-249) favorized the antibacterial activity against E. faecalis vanco R and A. baumanni NDM 1. The combination between quinolone and 5-phenylthiazole moieties (247) showed potential activity against E. faecalis ATCC 29212, E. cloacae fos R, and K. pneumoniae carbapenemase-producing bacteria, while the combination between quinolone and 5-(coumarin-3-yl)-thiazole moieties (244-246) favorized the antibacterial activity against E. faecalis vanco R and E. coli ESBL. No potential target was reported by the authors [77].
2.5.4. Thiazolyl-Quinazolones Hybrid Compounds
Quinazolones are found in compounds with a wide range of activities, including antibacterial, antifungal, anti-HIV, antimalarial, and antituberculosis activities [78]. Herein, we report the structure-activity relationship in antimicrobial thiazoles clubbed with quinazolones.
Desai et al. designed and synthesized antibacterial 2-(quinazolon-3-yl)-thiazole Mannich bases, directly linked to a substituted 5-thioxo-1,3,4-oxadiazole heterocycle (series I), as DNA gyrase inhibitors as it was predicted by the molecular docking studies (Figure 44) [79].
In a similar manner, Wang et al. reported the identification of thiazole quinazolones, in which the two heterocycles are linked by an α,β-unsaturated carbonyl group (series II), as lactate dehydrogenase (LDH) inhibitors (Figure 44) [80].
The inactivation of LDH by compound 254 was supported by molecular docking studies. Important for the interaction with the target were the 7-chloro substituent, which formed hydrophobic bonds with Ile241 residue, the thiazole ring, which formed π-alkyl bonds with Ile25 and Val30 residues, and the two carbonyl groups and sulfur atom, which interacted through hydrogen bonds with Asn137 residue [80].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative strains. Compounds 251-253 showed inferior activity (MICs = 50-150 µg/mL) against S. aureus MTCC-96, E. coli MTCC-443, and P. aeruginosa MTCC-1668, compared to gentamycin (MICs = 0.05-1 µg/mL) [79]. Compound 254 showed superior activity (MIC = 1 µg/mL) against E. coli and P. aeruginosa compared to norfloxacin (MIC = 4 µg/mL) [80].
Compound 254 produced inappreciable hemolysis, compared to the other tested compounds [80]. It showed synergistic effect in combination with norfloxacin and displayed different mechanisms of action including the blocking of biofilm formation, favorizing the ROS generation in the bacterial cell, distortion of the bacterial metabolism by lactate dehydrogenase (LDH) inactivation, and intercalating into the bacterial DNA, which all could be responsible for a low bacterial resistance [80].
2.6. Thiazole Clubbed with Condensed Heterocycles
Condensed heterocycles are moieties that result from fusing two or more heterocycles into compact structures with enhanced properties. Based on the literature research, examples of condensed heterocycles clubbed with thiazole are quinuclidine, thiazolopyrimidines, pyrido-thiazolopyrimidines, and pyrazolo-thiazolopyrimidines [81,82]. Herein, we report the SAR studies of antimicrobial thiazole clubbed with condensed heterocycles.
2.6.1. Thiazolyl-Quinuclidine Hybrid Compounds
Quinuclidine is found in the structures of natural compounds extracted from Cinchona species as alkaloids, as well as in semisynthetic authorized antibacterials, namely quinupristin [83].
Łączkowski et al. designed a series of 2-(quinuclidin-3-ylidene)-hydrazonothiazoles, with a large potential of biological activities, including antibacterial and antifungal ones. These compounds are substituted in the fourth position of the thiazole ring with various aryl substituents (Figure 45) [81].
The compounds were tested for their antibacterial activity against Gram-positive and Gram-negative strains and for their antifungal activity against Candida sp. strains [81]. Compounds 255-260 showed similar or inferior activity (MICs = 0.48-500 µg/mL) against S. aureus ATCC 6538, ATCC 25923, and ATCC 43300, S. epidermidis ATCC 12228, M. luteus ATCC 10240, E. coli ATCC 25922, P. mirabilis ATCC 12453, K. pneumoniae ATCC 13883, S. thyphimurium ATCC 14028, and B. bronchiseptica ATCC 4617, compared to ciprofloxacin (MICs = 0.004-0.98 µg/mL) [81].
Regarding the antifungal activity, compounds 255-260 showed inferior activity (MICs = 1.95-125 µg/mL) against C. albicans ATCC 2091 and ATCC 10231, C. parapsilosis ATCC 22019, C. glabrata ATCC 90030, and C. krusei ATCC 14243, compared to nystatin (MICs = 0.24-0.48 µg/mL) [81].
According to SAR studies, substitutions with halogens (255, 256, 258, and 259), large electron-withdrawing groups (260), and methyl (257) were the most advantageous for the overall antibacterial and antifungal activities (Figure 45) [81]. The bromo (263) substitution induced the best antifungal activity, the iodo substitution (264) induced the best activity against Gram-positive bacterial strains, and trifluoromethyl group (265) favorized the overall antibacterial and antifungal activities. The substitution with larger groups, such as amides, sulfonamides, or large heterocycles like coumarin decreased the activity, while additional substitutions on the thiazole ring cancelled the effect [81].
These compounds express their antibacterial activity by targeting DNA gyrase and the antifungal activity by targeting secretory aspartyl proteinase, important for the fungal pathogenesis, based on the molecular docking studies [81].
2.6.2. Thiazole Clubbed with Polyheterocyclic Systems
An important strategy for the design of bioactive compounds is represented by the obtention of some polyheterocyclic systems, as they combine the biological activities of each of their components into compact structures.
Abdel-Latif et al. designed and synthesized thiazole-based polyheterocyclic systems, containing pyrimidine, pyridine, triazole, benzimidazole, and pyrazole heterocycles, as new antibacterial compounds (Figure 46) [82].
Starting from 3-cyano-thiazolo[3,2-a]pyrimidinone (261), they obtained some polyheterocyclic systems by annelation with pyrido[2’,3’:3,4]pyrazolo[1,5-a]pyrimidin-4-amine (a), benzo[4,5]imidazo[1,2-a]pyrimidin-4-amine (b), pyrazolo[1,5-a]pyrimidin-7-amine (c), [1,2,4]triazolo[3,4-a]pyrimidin-5-amine (d), 4,5-dihydro-1H-pyrazol-3-amine (e), and pyridines-2,6-diamine substituted in the ninth position (f, 262-263). Other series contained isolated heterocycles as thiazol-2-yl-pyridinone derivatives (g) (Figure 46) [82].
The activity of the compounds was assayed against S. aureus and E. coli, using the agar diffusion method and determination of the inhibition zone, compared to ampicillin (IZs = 24-27 mm, 100%) [82]. Compounds 261-263 showed similar activity with the reference against S. aureus (IZs = 20-22 mm, 83.3-91.7%) and E. coli (IZs = 22-23 mm, 81.48-85.18%) [82].
Despite the heterogeneity of the synthesized molecules, some SAR studies can be underlined based on their antibacterial potential (Figure 46) [82]. A first difference can be noticed based on how the thiazole is linked to the rest of the system. Condensed thiazolo[3,2-a]pyrimidinones (a-f) displayed stronger activity than isolated thiazol-2-yl-pyridinones (g) [82].
Concerning the polycondensed heterocyclic systems, differences occur in the number of annulated rings. The activity decreased by supplementary annulation. The best results were obtained for the bicyclic compound 261 (IZ = 22 mm and 91,7% against S. aureus, while IZ = 23 mm and 85,18% against E. coli), with tetra- (c, d) and pentacyclic (a, b) compounds having very low or no activity (Figure 46) [82]. In the tricyclic compounds (e, f), pyrazolo-thiazolo[3,2-a]pyrimidinone (e) derivatives, containing azole rings, had lower activity compared to the pyrido-thiazolo[3,2-a]pyrimidinone (f) derivatives, containing azine rings (Figure 46) [82].
The activity was also influenced by the nature of the substituents present in the pyrido-thiazolo[3,2-a]pyrimidinone derivatives. Compound 262, containing a 9-cyano substituent, displayed better activity (IZ = 21 mm and 87,5% against S. aureus, while IZ = 23 mm and 85,18% against E. coli) than compound 263, containing a 9-ethoxycarbonyl substituent (IZ = 20 mm and 83,3% against S. aureus, while IZ = 22 mm and 81,48% against E. coli) (Figure 46). No potential target was reported by the authors [82].
3. Conclusions
Thiazole is an important heterocycle for drug design, not only for antibacterials and antifungals, but also for many other pharmacological classes. Approaching a rational design in novel drugs development allows for a faster progress and effectiveness. Therefore, a rational approach for drug discovery is the optimization based on structure-activity relationships.
Based on the observations drawn in this review, it is easy to notice that each compound behaves differently based on its structure. However, they can exhibit the same pharmacological effect but with different potency. Thus, this work aimed to collect and visualize the SAR studies in some of the reported antimicrobial scaffolds of hybrid thiazolic compounds, as a mean to observe the structural heterogeneity that can induce the antimicrobial activity.
Thiazole clubbing with various heterocycles had different results depending on the other heterocycle(s). Pyrazoline-clubbed antimicrobials expressed activity against Gram-positive bacteria and K. pneumoniae, while using pyrazole the activity was extended against fungal strains, similar with imidazole. Hybrid thiazoles clubbed with thiazolidinone were promising antituberculosis compounds, while clubbing with triazole covered all three types of activity, antibacterial, antifungal, and antituberculosis. A lower potency was observed in hybrid compounds with pyridine, but this was not transposable to the benzofused heterocycles quinoline and quinolone. Similarly to triazole, coumarin is another veritable scaffold for designing novel antimicrobials. Also, high importance should be allocated to the substituents used in each scaffold.
The observations made in this work could provide a valuable scientific material in the design of novel antimicrobials in future research.
Author Contributions
Conceptualization, D.U., B.T. and O.O.; writing—original draft preparation, D.U. and B.T.; writing—review and editing, D.U., B.T., C.N., I.I., G.M., I.O. and O.O.; visualization, D.U., B.T. and O.O.; supervision, O.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Allel, K.; Day, L.; Hamilton, A.; Lin, L.; Furuya-Kanamori, L.; Moore, C.E.; Van Boeckel, T.; Laxminarayan, R.; Yakob, L. Global antimicrobial-resistance drivers: an ecological country-level study at the human–animal interface. Lancet Planet. Heal. 2023, 7, e291–e303. [Google Scholar] [CrossRef] [PubMed]
- Walesch, S.; Birkelbach, J.; Jézéquel, G.; Haeckl, F.P.J.; Hegemann, J.D.; Hesterkamp, T.; Hirsch, A.K.H.; Hammann, P.; Müller, R. Fighting antibiotic resistance—strategies and (pre)clinical developments to find new antibacterials. EMBO Rep. 2023, 24, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Borcea, A.-M.; Ionuț, I.; Crișan, O.; Oniga, O. An Overview of the Synthesis and Antimicrobial, Antiprotozoal, and Antitumor Activity of Thiazole and Bisthiazole Derivatives. Molecules 2021, 26, 624. [Google Scholar] [CrossRef] [PubMed]
- Mishra, I.; Mishra, R.; Mujwar, S.; Chandra, P.; Sachan, N. A retrospect on antimicrobial potential of thiazole scaffold. J. Heterocycl. Chem. 2020, 57, 2304–2329. [Google Scholar] [CrossRef]
- Petrou, A.; Fesatidou, M.; Geronikaki, A. Thiazole ring—a biologically active scaffold. Molecules 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, H.C. Coumarin-1,2,3-triazole Hybrid Molecules: An Emerging Scaffold for Combating Drug Resistance. Curr. Top. Med. Chem. 2021, 21, 737–752. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, B.; Almendros, P.; Aragoncillo, C. Highly reactive 4-membered ring nitrogen-containing heterocycles: Synthesis and properties. Curr. Opin. Drug Discov. Dev. 2010, 13, 685–697. [Google Scholar]
- Desai, N.C.; Harsora, J.P.; Monapara, J.D.; Khedkar, V.M. Synthesis, Antimicrobial Capability and Molecular Docking of Heterocyclic Scaffolds Clubbed by 2-Azetidinone, Thiazole and Quinoline Derivatives. Polycycl. Aromat. Compd. 2021, 0, 1–15. [Google Scholar] [CrossRef]
- Kumar, S.; Bawa, S.; Drabu, S.; Kumar, R.; Gupta, H. Biological Activities of Pyrazoline Derivatives -A Recent Development. Recent Pat. Antiinfect. Drug Discov. 2009, 4, 154–163. [Google Scholar] [CrossRef]
- Cuartas, V.; Robledo, S.M.; Vélez, I.D.; Crespo, M. del P.; Sortino, M.; Zacchino, S.; Nogueras, M.; Cobo, J.; Upegui, Y.; Pineda, T.; et al. New thiazolyl-pyrazoline derivatives bearing nitrogen mustard as potential antimicrobial and antiprotozoal agents. Arch. Pharm. 2020; 353. [Google Scholar] [CrossRef]
- Rashdan, H.R.M.; Abdelmonsef, A.H. Towards Covid-19 TMPRSS2 enzyme inhibitors and antimicrobial agents: Synthesis, antimicrobial potency, molecular docking, and drug-likeness prediction of thiadiazole-triazole hybrids. J. Mol. Struct. 2022, 1268, 133659. [Google Scholar] [CrossRef]
- Budak, Y.; Kocyigit, U.M.; Gürdere, M.B.; Özcan, K.; Taslimi, P.; Gülçin, İ.; Ceylan, M. Synthesis and investigation of antibacterial activities and carbonic anhydrase and acetyl cholinesterase inhibition profiles of novel 4,5-dihydropyrazol and pyrazolyl-thiazole derivatives containing methanoisoindol-1,3-dion unit. Synth. Commun. 2017, 47, 2313–2323. [Google Scholar] [CrossRef]
- Mansour, E.; Aboelnaga, A.; Nassar, E.M.; Elewa, S.I. A new series of thiazolyl pyrazoline derivatives linked to benzo[1,3]dioxole moiety: Synthesis and evaluation of antimicrobial and anti-proliferative activities. Synth. Commun. 2020, 50, 368–379. [Google Scholar] [CrossRef]
- Masoud, D.M.; Azzam, R.A.; Hamdy, F.; Mekawey, A.A.I.; Abdel-Aziz, H.A. Synthesis of Some Novel Pyrazoline-Thiazole Hybrids and Their Antimicrobial Activities. J. Heterocycl. Chem. 2019, 56, 3030–3041. [Google Scholar] [CrossRef]
- Bhandare, R.R.; Munikrishnappa, C.S.; Suresh Kumar, G.V.; Konidala, S.K.; Sigalapalli, D.K.; Vaishnav, Y.; Chinnam, S.; Yasin, H.; Al-karmalawy, A.A.; Shaik, A.B. Multistep synthesis and screening of heterocyclic tetrads containing furan, pyrazoline, thiazole and triazole (or oxadiazole) as antimicrobial and anticancer agents. J. Saudi Chem. Soc. 2022, 26, 101447. [Google Scholar] [CrossRef]
- Abdel-Wahab, B.F.; Khidre, R.E.; Mohamed, H.A.; El-Hiti, G.A. A Simple Process for the Synthesis of Novel Pyrazolyltriazole and Dihydropyrazolylthiazole Derivatives as Antimicrobial Agents. Arab. J. Sci. Eng. 2017, 42, 2441–2448. [Google Scholar] [CrossRef]
- Bondock, S.; Fouda, A.M. Synthesis and evaluation of some new 5-(hetaryl)thiazoles as potential antimicrobial agents. Synth. Commun. 2018, 48, 561–573. [Google Scholar] [CrossRef]
- Vijesh, A.M.; Isloor, A.M.; Isloor, S.; Shivananda, K.N.; Shyma, P.C.; Arulmoli, T. Synthesis of some new pyrazolone derivatives as potent antimicrobial agents. Der Pharma Chem. 2011, 3, 454–463. [Google Scholar]
- Abu-Melha, S. Molecular modeling and docking studies of new antimicrobial antipyrine-thiazole hybrids. Arab. J. Chem. 2022, 15, 103898. [Google Scholar] [CrossRef]
- Jamwal, A.; Javed, A.; Bhardwaj, V. A review on Pyrazole derivatives of pharmacological potential. J. Pharm. BioSci 2013, 3, 114–123. [Google Scholar]
- Gondru, R.; Sirisha, K.; Raj, S.; Gunda, S.K.; Kumar, C.G.; Pasupuleti, M.; Bavantula, R. Design, Synthesis, In Vitro Evaluation and Docking Studies of Pyrazole-Thiazole Hybrids as Antimicrobial and Antibiofilm Agents. ChemistrySelect 2018, 3, 8270–8276. [Google Scholar] [CrossRef]
- Patil, S.V.; Suryavanshi, M.B.; Nagargoje, D.R.; Kokate, S.V. Synthesis and Antimicrobial Evaluation of Some New Pyrazole Derivatives Containing Thiazole Scaffolds. In Proceedings of the ECSOC-25; MDPI: Basel Switzerland, noiembrie 15 2021; Vol. 17, p. 46.
- Abdel-Aziem, A.; Baaiu, B.S.; Elbazzar, A.W.; Elabbar, F. A facile synthesis of some novel thiazoles, arylazothiazoles, and pyrazole linked to thiazolyl coumarin as antibacterial agents. Synth. Commun. 2020, 50, 2522–2530. [Google Scholar] [CrossRef]
- Kumar, S.; Saini, V.; Maurya, I.K.; Sindhu, J.; Kumari, M.; Kataria, R.; Kumar, V. Design, synthesis, DFT, docking studies and ADME prediction of some new coumarinyl linked pyrazolylthiazoles: Potential standalone or adjuvant antimicrobial agents. PLoS ONE 2018, 13, e0196016. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodi, N.O.; Ghodsi, S. Thiazolyl-pyrazole-biscoumarin synthesis and evaluation of their antibacterial and antioxidant activities. Res. Chem. Intermed. 2017, 43, 661–678. [Google Scholar] [CrossRef]
- Nalawade, J.; Shinde, A.; Chavan, A.; Patil, S.; Suryavanshi, M.; Modak, M.; Choudhari, P.; Bobade, V.D.; Mhaske, P.C. Synthesis of new thiazolyl-pyrazolyl-1,2,3-triazole derivatives as potential antimicrobial agents. Eur. J. Med. Chem. 2019, 179, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Kant, V. A Review on Biological Activity of Imidazole and Thiazole Moieties and their Derivatives. Sci. Int. 2013, 1, 253–260. [Google Scholar] [CrossRef]
- Nikalje, A.P.G.; Tiwari, S.V.; Sarkate, A.P.; Karnik, K.S. Imidazole-thiazole coupled derivatives as novel lanosterol 14-α demethylase inhibitors: ionic liquid mediated synthesis, biological evaluation and molecular docking study. Med. Chem. Res. 2018, 27, 592–606. [Google Scholar] [CrossRef]
- Jain, A.K.; Vaidya, A.; Ravichandran, V.; Kashaw, S.K.; Agrawal, R.K. Recent developments and biological activities of thiazolidinone derivatives: A review. Bioorganic Med. Chem. 2012, 20, 3378–3395. [Google Scholar] [CrossRef]
- Othman, D.I.A.; Hamdi, A.; Abdel-Aziz, M.M.; Elfeky, S.M. Novel 2-arylthiazolidin-4-one-thiazole hybrids with potent activity against Mycobacterium tuberculosis. Bioorg. Chem. 2022, 124, 105809. [Google Scholar] [CrossRef] [PubMed]
- Abo-Ashour, M.F.; Eldehna, W.M.; George, R.F.; Abdel-Aziz, M.M.; Elaasser, M.M.; Abou-Seri, S.M.; Abdel Gawad, N.M. Synthesis and Biological Evaluation of 2-Aminothiazole-Thiazolidinone Conjugates as Potential Antitubercular Agents. Future Med. Chem. 2018, 10, 1405–1419. [Google Scholar] [CrossRef]
- Sucheta; Tahlan, S.; Verma, P.K. Biological potential of thiazolidinedione derivatives of synthetic origin. Chem. Cent. J. 2017, 11, 1–29. [Google Scholar] [CrossRef]
- Alegaon, S.G.; U, V.; Alagawadi, K.R.; Kumar, D.; Kavalapure, R.S.; Ranade, S.D.; Priya A, S.; Jalalpure, S.S. Synthesis, Molecular Docking and ADME Studies of Thiazole-Thiazolidinedione Hybrids as Antimicrobial Agents. J. Biomol. Struct. Dyn. 2022, 40, 6211–6227. [Google Scholar] [CrossRef] [PubMed]
- Bhuva, H.; Sahu, D.; Shah, B.; Modi, C.; Patel, M.B. Biological Profile of Thiadiazole. Pharmacologyonline 2011, 1, 528–543. [Google Scholar]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackerman, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702.e13. [Google Scholar] [CrossRef]
- Booq, R.Y.; Tawfik, E.A.; Alfassam, H.A.; Alfahad, A.J.; Alyamani, E.J. Assessment of the Antibacterial Efficacy of Halicin against Pathogenic Bacteria. Antibiotics 2021, 10, 1480. [Google Scholar] [CrossRef]
- Hussain, Z.; Pengfei, S.; Yimin, L.; Shasha, L.; Zehao, L.; Yifan, Y.; Linhui, L.; Linying, Z.; Yong, W. Study on antibacterial effect of halicin (SU3327) against Enterococcus faecalis and Enterococcus faecium. Pathog. Dis. 2022, 80, ftac037. [Google Scholar] [CrossRef]
- Higashihira, S.; Simpson, S.J.; Collier, C.D.; Natoli, R.M.; Kittaka, M.; Greenfield, E.M. Halicin Is Effective Against Staphylococcus aureus Biofilms In Vitro. Clin. Orthop. Relat. Res. 2022, 480, 1476–1487. [Google Scholar] [CrossRef] [PubMed]
- van Gent, M.E.; van der Reijden, T.J.K.; Lennard, P.R.; de Visser, A.W.; Schonkeren-Ravensbergen, B.; Dolezal, N.; Cordfunke, R.A.; Drijfhout, J.W.; Nibbering, P.H. Synergism between the Synthetic Antibacterial and Antibiofilm Peptide (SAAP)-148 and Halicin. Antibiotics 2022, 11, 1–15. [Google Scholar] [CrossRef]
- Asif, M. Pharmacological activities of Triazole analogues as antibacterial, antifungal, antiviral agents. Pharm. Sci. Asia 2017, 44, 59–74. [Google Scholar] [CrossRef]
- Shinde, V.; Mahulikar, P.; Mhaske, P.C.; Chakraborty, S.; Choudhari, A.; Phalle, S.; Choudhari, P.; Sarkar, D. Synthesis and antimycobacterial evaluation of new 5-(1-benzyl-1H-1,2,3-triazol-4-yl)-4-methyl-2-arylthiazole derivatives. Med. Chem. Res. 2019, 28, 805–819. [Google Scholar] [CrossRef]
- Mahale, K.A.; Gosavi, K.S.; Gaikwad, N.D.; Bholay, A.D.; Patil, S.V. Thiazole Substituted [1,2,3] Triazole: Synthesis and Antimicrobial Evaluation. Indian J. Chem. 2022, 61, 640–649. [Google Scholar] [CrossRef]
- Jagadale, S.; Chavan, A.; Shinde, A.; Sisode, V.; Bobade, V.D.; Mhaske, P.C. Synthesis and antimicrobial evaluation of new thiazolyl-1,2,3-triazolyl-alcohol derivatives. Med. Chem. Res. 2020, 29, 989–999. [Google Scholar] [CrossRef]
- Poonia, N.; Lal, K.; Kumar, A.; Kumar, A.; Sahu, S.; Baidya, A.T.K.; Kumar, R. Urea-Thiazole/Benzothiazole Hybrids with a Triazole Linker: Synthesis, Antimicrobial Potential, Pharmacokinetic Profile and in Silico Mechanistic Studies. Mol. Divers. 2022, 26, 2375–2391. [Google Scholar] [CrossRef] [PubMed]
- Gondru, R.; Kanugala, S.; Raj, S.; Ganesh Kumar, C.; Pasupuleti, M.; Banothu, J.; Bavantula, R. 1,2,3-triazole-thiazole hybrids: Synthesis, in vitro antimicrobial activity and antibiofilm studies. Bioorganic Med. Chem. Lett. 2021, 33, 127746. [Google Scholar] [CrossRef] [PubMed]
- Glomb, T.; Świątek, P. Antimicrobial Activity of 1,3,4-Oxadiazole Derivatives. Int. J. Mol. Sci. 2021, 22, 6979. [Google Scholar] [CrossRef] [PubMed]
- Glomb, T.; Szymankiewicz, K.; Świątek, P. Anti-cancer activity of derivatives of 1,3,4-oxadiazole. Molecules 2018, 23, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, M.M.; Comin, M.; Duarte, T.S.; Foglio, M.A.; De Carvalho, J.E.; Do Carmo Vieira, M.; Formagio, A.S.N. Synthesis, antiproliferative activity and molecular properties predictions of galloyl derivatives. Molecules 2015, 20, 5360–5373. [Google Scholar] [CrossRef] [PubMed]
- Savariz, F.C.; Formagio, A.S.N.; Barbosa, V.A.; Foglio, M.A.; de Carvalho, J.E.; Duarte, M.C.T.; Dias Filho, B.P.; Sarragiotto, M.H. Synthesis, antitumor and antimicrobial activity of novel 1-substituted phenyl-3-[3-alkylamino(methyl)-2-thioxo-1,3,4-oxadiazol-5-yl] β-carboline derivatives. J. Braz. Chem. Soc. 2010, 21, 288–298. [Google Scholar] [CrossRef]
- Athar Abbasi, M.; Raza, H.; Aziz-ur-Rehman; Zahra Siddiqui, S.; Adnan Ali Shah, S.; Hassan, M.; Seo, S.Y. Synthesis of novel N-(1,3-thiazol-2-yl)benzamide clubbed oxadiazole scaffolds: Urease inhibition, Lipinski rule and molecular docking analyses. Bioorg. Chem. 2019, 83, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.C.; Bhatt, N.B.; Joshi, S.B. Synthetic modifications in ethyl 2-amino-4-methylthiazole-5-carboxylate: 3D QSAR analysis and antimicrobial study. Synth. Commun. 2019, 49, 1055–1066. [Google Scholar] [CrossRef]
- Tiperciuc, B.G. Design and development of new azoles heterocycles with biological potential [habilitation thesis]. [Cluj-Napoca: Romania]: “Iuliu Hațieganu” University of Medicine and Pharmacy; 2021. Chapter 1.6.1., Thiazol-5-yl-azoles-5-thiones; pp. 45–7.
- Meanwell, N.A. Chapter Five - A Synopsis of the Properties and Applications of Heteroaromatic Rings in Medicinal Chemistry. In: Advances in Heterocyclic Chemistry; Scriven, E.F. V, Ramsden, C.A.B.T.-A. in H.C., Ed.; Academic Press, 2017; Vol. 123, pp. 245–361 ISBN 0065-2725.
- Patil, S.A.; Patil, S.A.; Ble-González, E.A.; Isbel, S.R.; Hampton, S.M.; Bugarin, A. Carbazole Derivatives as Potential Antimicrobial Agents. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Hanafy, M.S.; Matter, M.A.; Asker, M.S.; Rady, M.R. Production of indole alkaloids in hairy root cultures of Catharanthus roseus L. and their antimicrobial activity. South African J. Bot. 2016, 105, 9–18. [Google Scholar] [CrossRef]
- Yu, H.F.; Qin, X.J.; Ding, C.F.; Wei, X.; Yang, J.; Luo, J.R.; Liu, L.; Khan, A.; Zhang, L.C.; Xia, C.F.; et al. Nepenthe-Like Indole Alkaloids with Antimicrobial Activity from Ervatamia chinensis. Org. Lett. 2018, 20, 4116–4120. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Ding, C.F.; Deng, S.Y.; Gao, W.; Tan, B.Y.; Wu, H.; Guo, Y.; Song, J.F.; Zhang, L.C.; Zhang, R.P.; et al. Monoterpene indole N-oxide alkaloids from Tabernaemontana corymbosa and their antimicrobial activity. Fitoterapia 2022, 158, 105178. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Liu, Y.; Li, Y.; Chen, Y. A green synthesis and antibacterial activity of ferrocene-based thiazole derivatives in choline chloride/glycerol eutectic solvent. RSC Adv. 2022, 12, 22054–22059. [Google Scholar] [CrossRef] [PubMed]
- Ashok, D.; Gundu, S.; Aamate, V.K.; Devulapally, M.G. Microwave-assisted synthesis, antioxidant and antimicrobial evaluation of 2-indolinone-based bis-1,2,3-triazole derivatives. Mol. Divers. 2018, 22, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.Y.; Ammar, Y.A.; Abu-Elghait, M.; Salem, M.A.; Assiri, M.A.; Ali, T.E.; Ragab, A. Development of novel indolin-2-one derivative incorporating thiazole moiety as DHFR and quorum sensing inhibitors: Synthesis, antimicrobial, and antibiofilm activities with molecular modelling study. Bioorg. Chem. 2022, 119, 105571. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.W.; Zhao, H.Y.; Zhu, H.; Peng, C.; Zhou, Q.M.; Xiong, L. Novel Indane Derivatives with Antioxidant Activity from the Roots of Anisodus tanguticus. Molecules 2023, 28, 1–10. [Google Scholar] [CrossRef]
- Obafemi, C.A.; Adelani, P.O.; Fadare, O.A.; Akinpelu, D.A.; Famuyiwa, S.O. Synthesis, crystal structure and in vitro antibacterial activity of 2,3a,8b-trihydroxy-3-(thiophen-2-ylcarbonyl)-2-(trifluoromethyl)-2,3,3a, 8b-tetrahydro-4H-indeno[1,2-b]furan-4-one. J. Mol. Struct. 2013, 1049, 429–435. [Google Scholar] [CrossRef]
- Adole, V.A.; More, R.A.; Jagdale, B.S.; Pawar, T.B.; Chobe, S.S. Efficient Synthesis, Antibacterial, Antifungal, Antioxidant and Cytotoxicity Study of 2-(2-Hydrazineyl)thiazole Derivatives. ChemistrySelect 2020, 5, 2778–2786. [Google Scholar] [CrossRef]
- Muluk, M.B.; Phatak, P.S.; Pawar, S.B.; Dhumal, S.T.; Rehman, N.N.M.A.; Dixit, P.P.; Choudhari, P.B.; Haval, K.P. Synthesis, antimicrobial, and antioxidant activities of new pyridyl- and thiazolyl-bearing carbohydrazides. J. Chinese Chem. Soc. 2019, 66, 1507–1517. [Google Scholar] [CrossRef]
- Muluk, M.B.; Ubale, A.S.; Dhumal, S.T.; Rehman, N.N.M.A.; Dixit, P.P.; Kharat, K.K.; Choudhari, P.B.; Haval, K.P. Synthesis, anticancer and antimicrobial evaluation of new pyridyl and thiazolyl clubbed hydrazone scaffolds. Synth. Commun. 2020, 50, 243–255. [Google Scholar] [CrossRef]
- Patil, P.S.; Kasare, S.L.; Badar, A.D.; Kulkarni, R.S.; Dixit, P.P.; Kulkarni, J.A.; Choudhari, P.B.; Haval, K.P. Synthesis, Antimicrobial Evaluation, and Molecular Docking Study of New Thiazole-5-phenylpropenone Derivatives. Russ. J. Gen. Chem. 2020, 90, 1523–1528. [Google Scholar] [CrossRef]
- Eryılmaz, S.; Türk Çelikoğlu, E.; İdil, Ö.; İnkaya, E.; Kozak, Z.; Mısır, E.; Gül, M. Derivatives of Pyridine and Thiazole Hybrid: Synthesis, DFT, Biological Evaluation via Antimicrobial and DNA Cleavage Activity. Bioorg. Chem. 2020, 95. [Google Scholar] [CrossRef] [PubMed]
- Marinescu, M.; Popa, C.V. Pyridine Compounds with Antimicrobial and Antiviral Activities. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Al-Majedy, Y.K.; Kadhum, A.A.H.; Al-Amiery, A.A.; Mohamad, A.B. Coumarins: The antimicrobial agents. Syst. Rev. Pharm. 2016, 8, 62–70. [Google Scholar] [CrossRef]
- Yusufzai, S.K.; Osman, H.; Khan, M.S.; Mohamad, S.; Sulaiman, O.; Parumasivam, T.; Gansau, J.A.; Johansah, N. Noviany Design, characterization, in vitro antibacterial, antitubercular evaluation and structure–activity relationships of new hydrazinyl thiazolyl coumarin derivatives. Med. Chem. Res. 2017, 26, 1139–1148. [Google Scholar] [CrossRef]
- Salar, U.; Qureshi, B.; Khan, K.M.; Lodhi, M.A.; Ul-Haq, Z.; Khan, F.A.; Naz, F.; Taha, M.; Perveen, S.; Hussain, S. Aryl hydrazones linked thiazolyl coumarin hybrids as potential urease inhibitors. J. Iran. Chem. Soc. 2022, 19, 1221–1238. [Google Scholar] [CrossRef]
- Hu, Y.; Hu, C.; Pan, G.; Yu, C.; Ansari, M.F.; Yadav Bheemanaboina, R.R.; Cheng, Y.; Zhou, C.; Zhang, J. Novel chalcone-conjugated, multi-flexible end-group coumarin thiazole hybrids as potential antibacterial repressors against methicillin-resistant Staphylococcus aureus. Eur. J. Med. Chem. 2021, 222, 113628. [Google Scholar] [CrossRef]
- Leonte, D.; Ungureanu, D.; Zaharia, V. Flavones and Related Compounds: Synthesis and Biological Activity. Molecules 2023, 28, 6528. [Google Scholar] [CrossRef]
- Zhao, G.; Lan, D.; Qi, G. Design and development of some thiazole-based flavanoids as novel antibacterial against pathogens causing surgical site infection for possible benefit in bone trauma via inhibition of DNA gyrase. Chem. Biol. Drug Des. 2017, 90, 778–790. [Google Scholar] [CrossRef]
- Kumar, S.; Bawa, S.; Gupta, H. Biological Activities of Quinoline Derivatives. Mini-Reviews Med. Chem. 2010, 9, 1648–1654. [Google Scholar] [CrossRef] [PubMed]
- Ammar, Y.A.; El-Hafez, S.M.A.A.; Hessein, S.A.; Ali, A.M.; Askar, A.A.; Ragab, A. One-Pot Strategy for Thiazole Tethered 7-Ethoxy Quinoline Hybrids: Synthesis and Potential Antimicrobial Agents as Dihydrofolate Reductase (DHFR) Inhibitors with Molecular Docking Study. J. Mol. Struct. 2021, 1242. [Google Scholar] [CrossRef]
- Litim, B.; Djahoudi, A.; Meliani, S.; Boukhari, A. Synthesis and potential antimicrobial activity of novel α-aminophosphonates derivatives bearing substituted quinoline or quinolone and thiazole moieties. Med. Chem. Res. 2022, 31, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Alsibaee, A.M.; Al-Yousef, H.M.; Al-Salem, H.S. Quinazolinones, the Winning Horse in Drug Discovery. Molecules 2023, 28. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.; Shihory, N.; Khasiya, A.; Pandit, U.; Khedkar, V. Quinazoline clubbed thiazole and 1,3,4-oxadiazole heterocycles: synthesis, characterization, antibacterial evaluation, and molecular docking studies. Phosphorus, Sulfur Silicon Relat. Elem. 2021, 196, 569–577. [Google Scholar] [CrossRef]
- Wang, J.; Ansari, M.F.; Zhou, C.H. Identification of Unique Quinazolone Thiazoles as Novel Structural Scaffolds for Potential Gram-Negative Bacterial Conquerors. J. Med. Chem. 2021, 64, 7630–7645. [Google Scholar] [CrossRef] [PubMed]
- Łączkowski, K.Z.; Landowska, K.; Biernasiuk, A.; Sałat, K.; Furgała, A.; Plech, T.; Malm, A. Synthesis, biological evaluation and molecular docking studies of novel quinuclidinone derivatives as potential antimicrobial and anticonvulsant agents. Med. Chem. Res. 2017, 26, 2088–2104. [Google Scholar] [CrossRef]
- Abdel-Latif, E.; Almatari, A.S.; Abd-ElGhani, G.E. Synthesis and Antibacterial Evaluation of Some New Thiazole-Based Polyheterocyclic Ring Systems. J. Heterocycl. Chem. 2019, 56, 1978–1985. [Google Scholar] [CrossRef]
- Radman Kastelic, A.; Odžak, R.; Pezdirc, I.; Sović, K.; Hrenar, T.; Čipak Gašparović, A.; Skočibušić, M.; Primožič, I. New and Potent Quinuclidine-Based Antimicrobial Agents. Molecules 2019, 24, 1–17. [Google Scholar] [CrossRef]
Figure 1.
(a) Bibliometric graphic of the indexed results obtained from Scopus, SpringerLink, PubMed, and EMBASE databases; (b) Bibliometric graphic of the selected publications for this review, based on the publication year.
Figure 1.
(a) Bibliometric graphic of the indexed results obtained from Scopus, SpringerLink, PubMed, and EMBASE databases; (b) Bibliometric graphic of the selected publications for this review, based on the publication year.
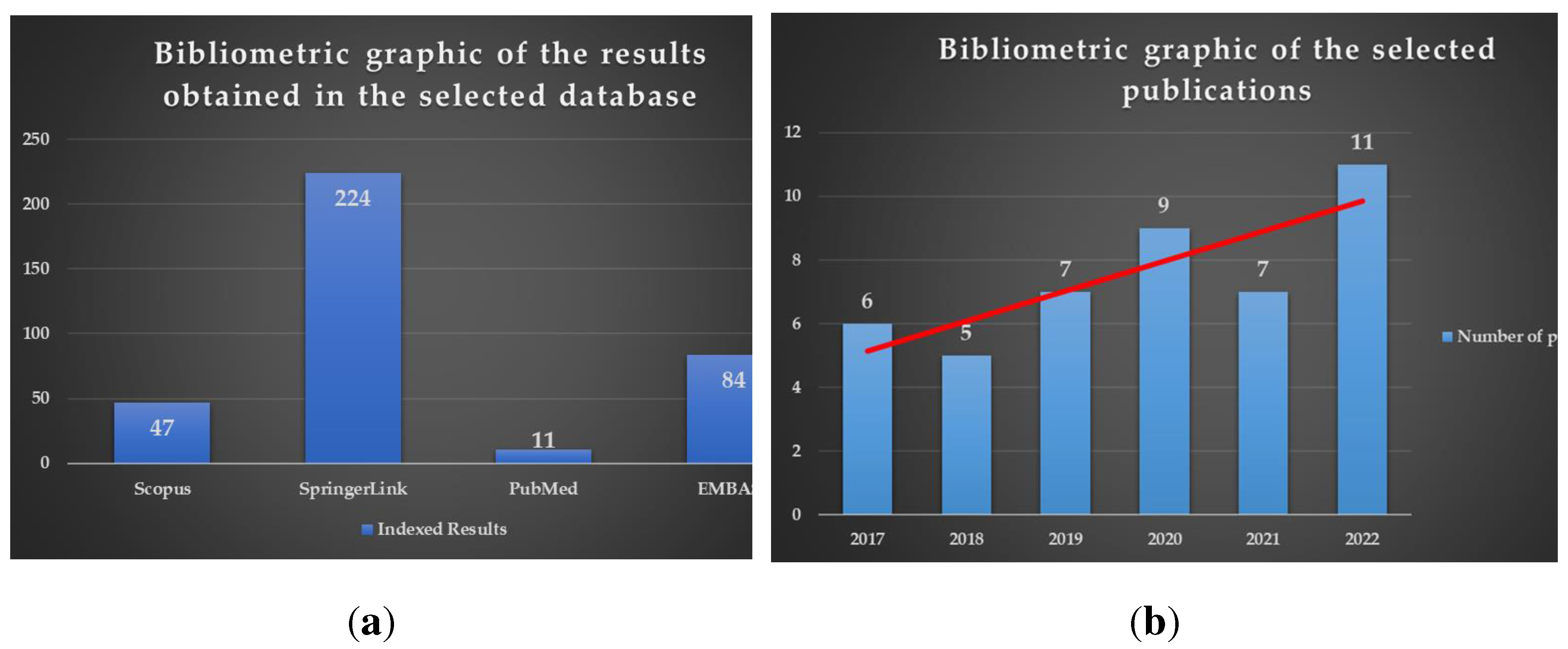
Figure 2.
Summary of the antimicrobial thiazole-based hybrid compounds from the selected papers.
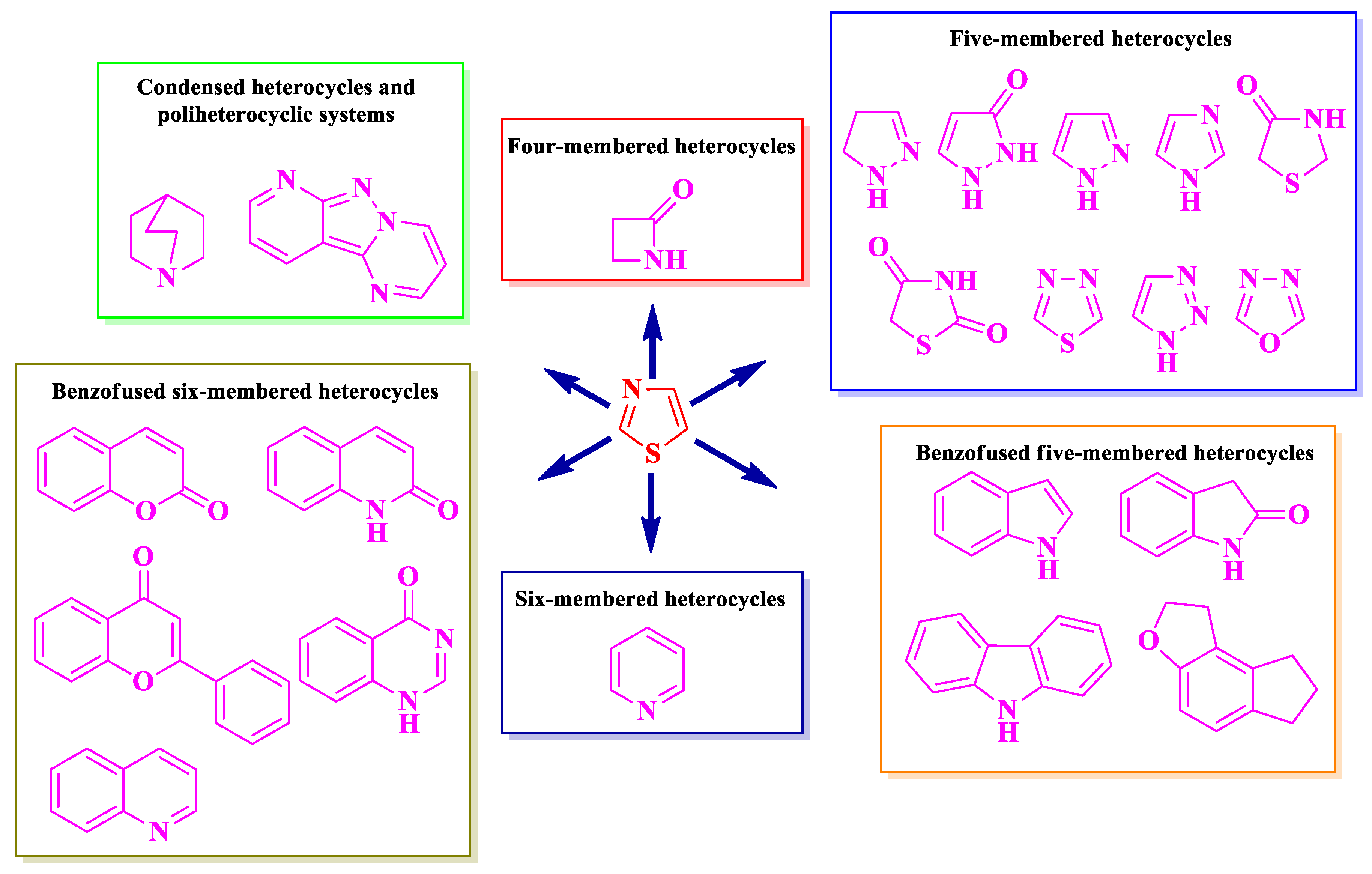
Figure 3.
SAR studies in antimicrobial 4-(quinolin-3-yl)-1-(thiazol-2-yl)-amino-azetidin-2-one derivatives, reported by Desai et al. [8].
Figure 3.
SAR studies in antimicrobial 4-(quinolin-3-yl)-1-(thiazol-2-yl)-amino-azetidin-2-one derivatives, reported by Desai et al. [8].
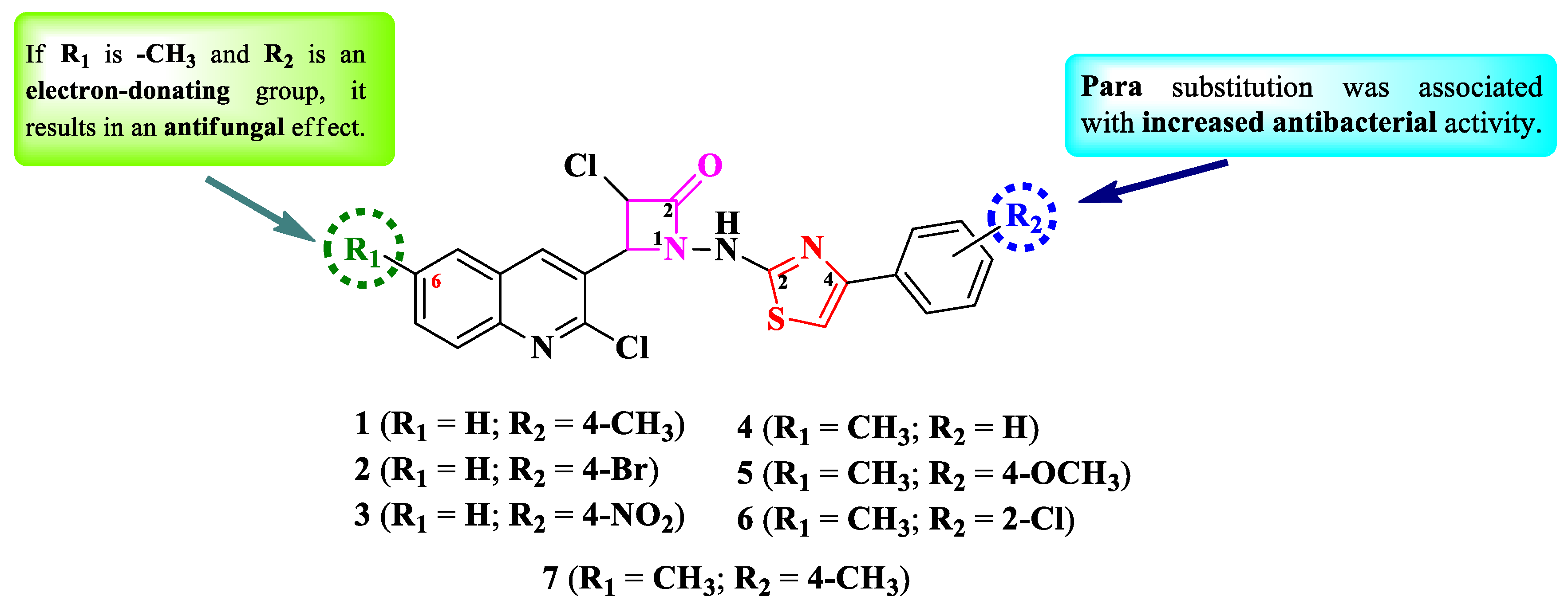
Figure 4.
The general structures for the series discussed, containing thiazole and 2-pyrazoline rings [10,11,12,13,14,15,16,17].
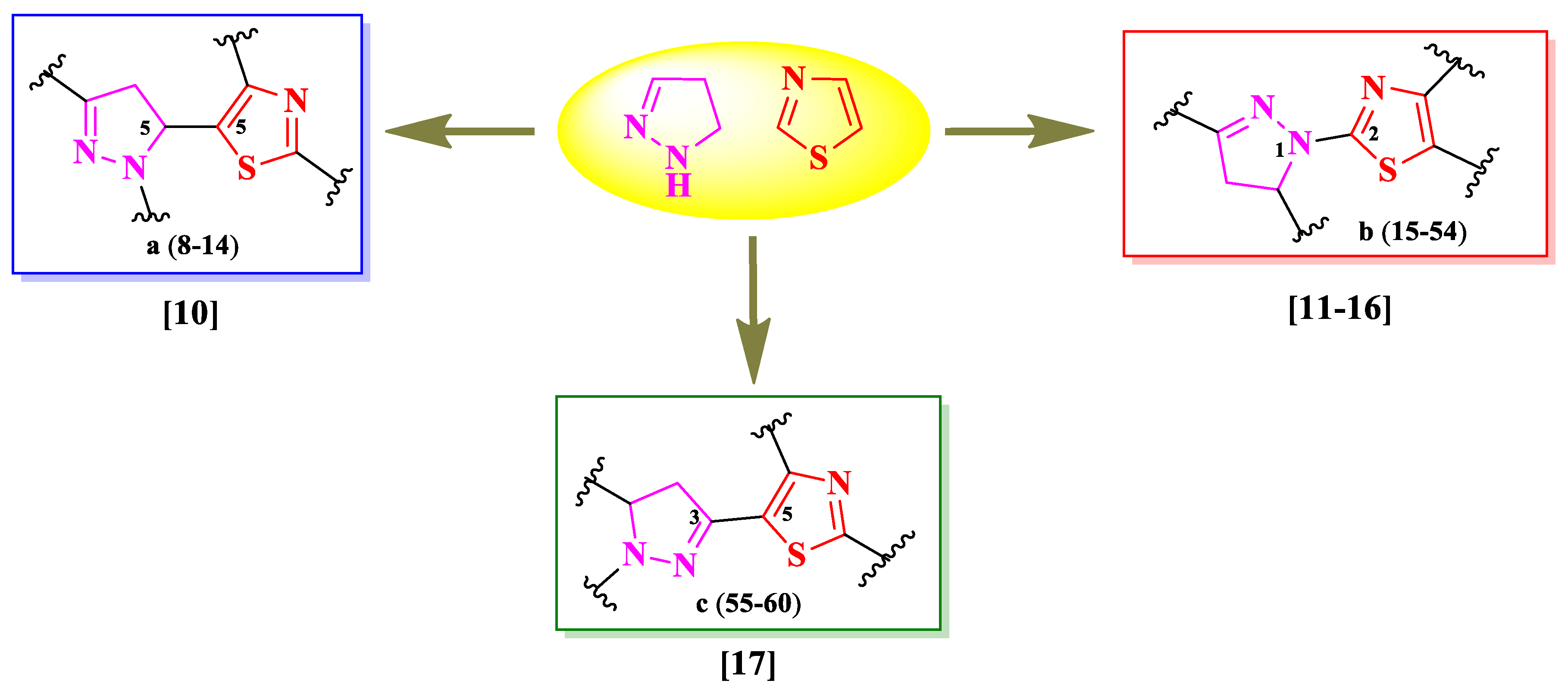
Figure 5.
SAR studies in antimicrobial 2-(N-mustard)-5-(2-pyrazolin-5-yl)-thiazole derivatives, reported by Cuartas et al [10].
Figure 5.
SAR studies in antimicrobial 2-(N-mustard)-5-(2-pyrazolin-5-yl)-thiazole derivatives, reported by Cuartas et al [10].
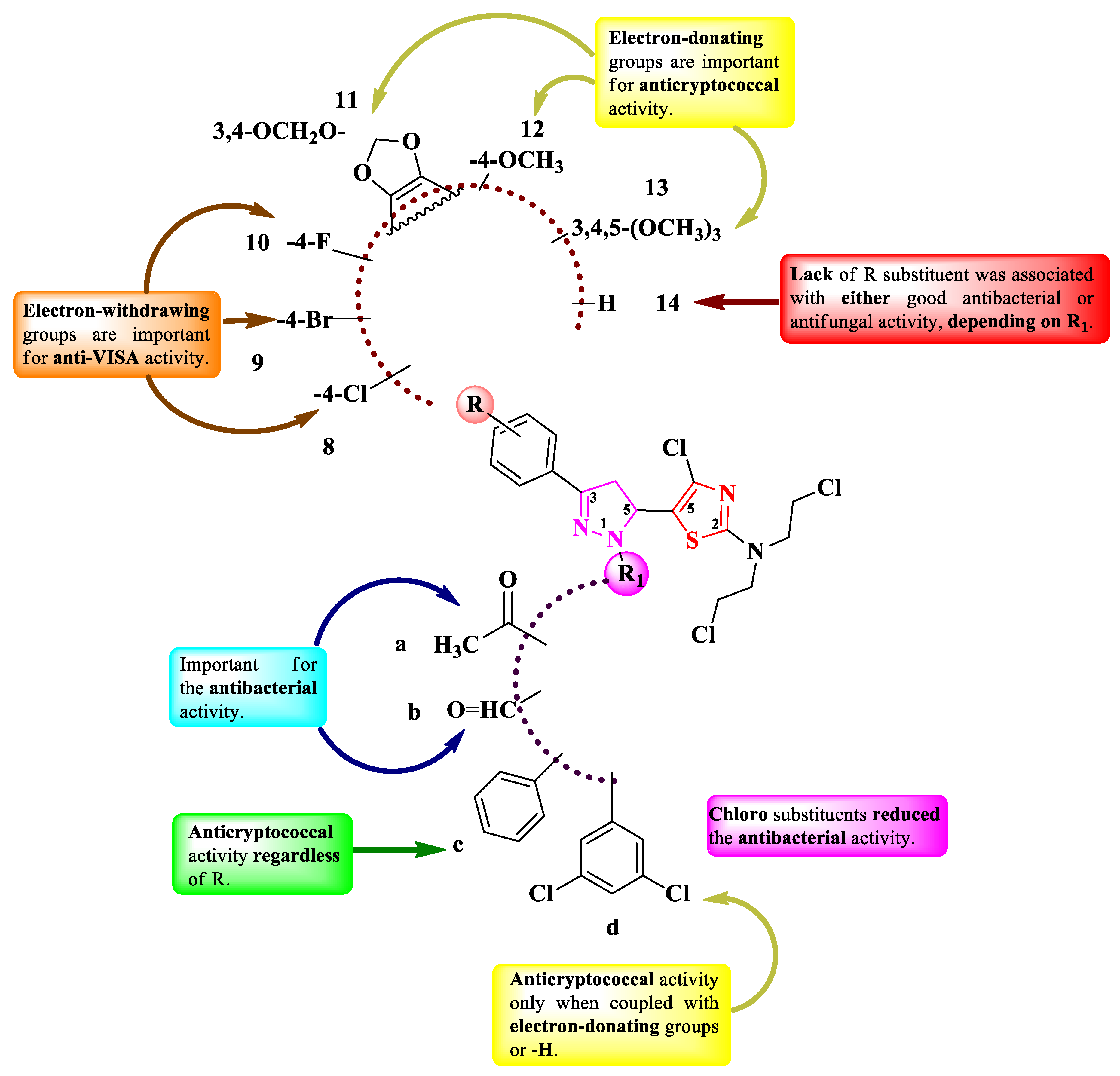
Figure 6.
SAR study of antimicrobial 2-(4-(1-thiazol-2-yl)-2-pyrazolin-3-yl)-1,2,3-triazol-1-yl)-1,3,4-thiadiazole derivatives, reported by Rashdan and Abdelmonsef [11].
Figure 6.
SAR study of antimicrobial 2-(4-(1-thiazol-2-yl)-2-pyrazolin-3-yl)-1,2,3-triazol-1-yl)-1,3,4-thiadiazole derivatives, reported by Rashdan and Abdelmonsef [11].
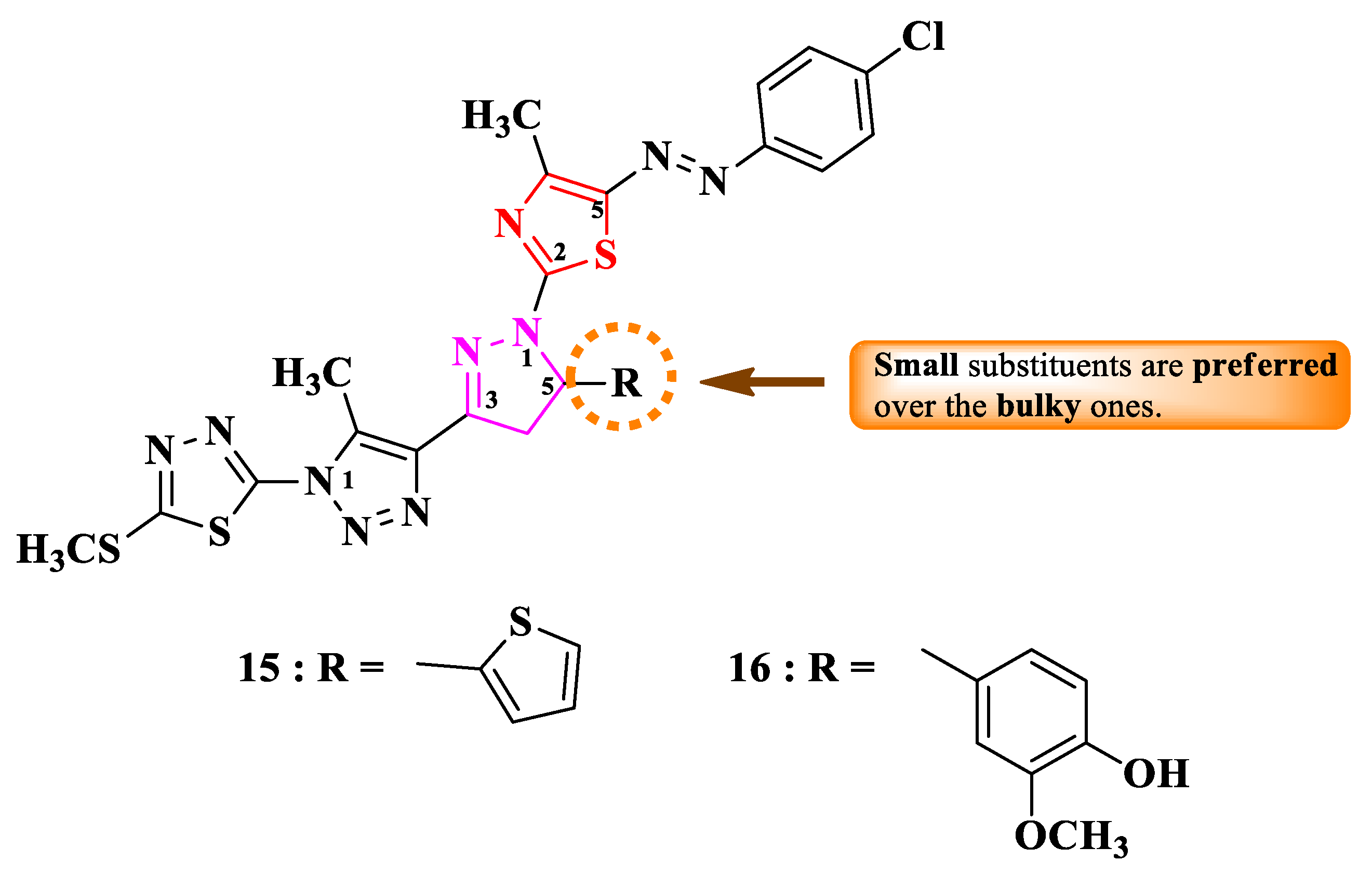
Figure 7.
SAR studies in antimicrobial 2-(4-(1-(thiazol-2-yl)-2-pyrazolin-3-yl)-phenyl)-methano-isoindol-1,3-dione derivatives, reported by Budak et al [12].
Figure 7.
SAR studies in antimicrobial 2-(4-(1-(thiazol-2-yl)-2-pyrazolin-3-yl)-phenyl)-methano-isoindol-1,3-dione derivatives, reported by Budak et al [12].
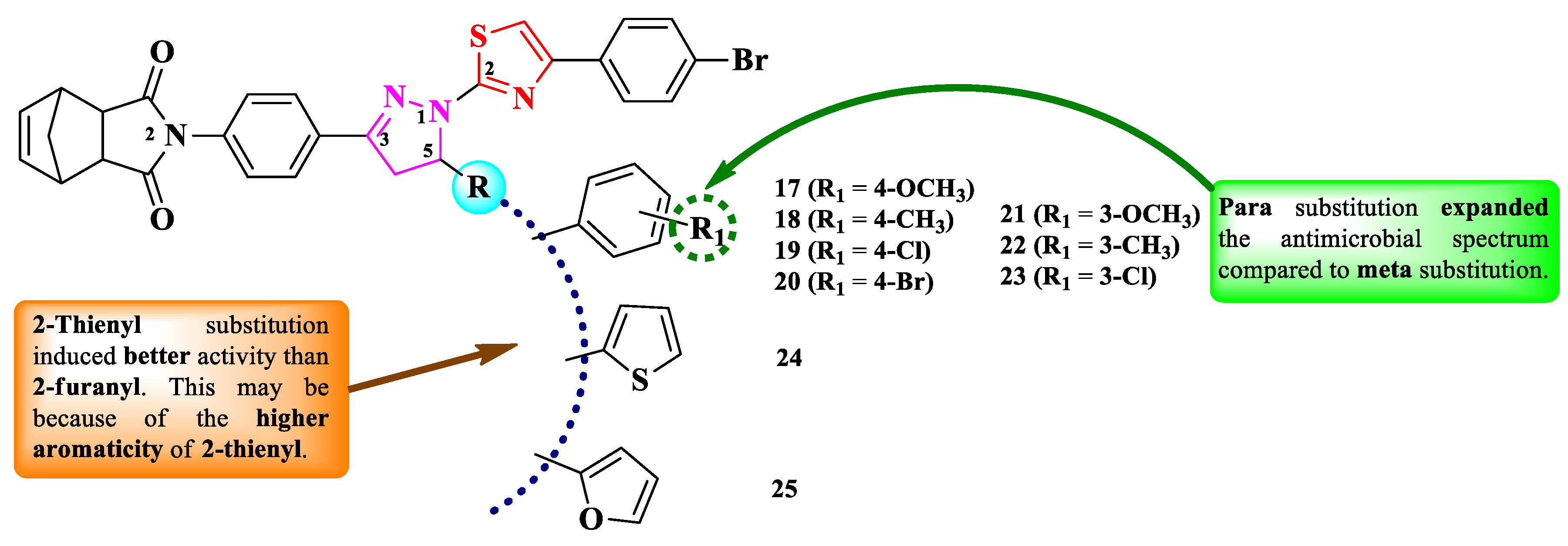
Figure 8.
SAR studies in the series of antimicrobial 2-(3-aryl-5-hetaryl-2-pyrazolin-1-yl)-thiazole derivatives, synthesized by Mansour et al. [13] and Masoud et al. [14].
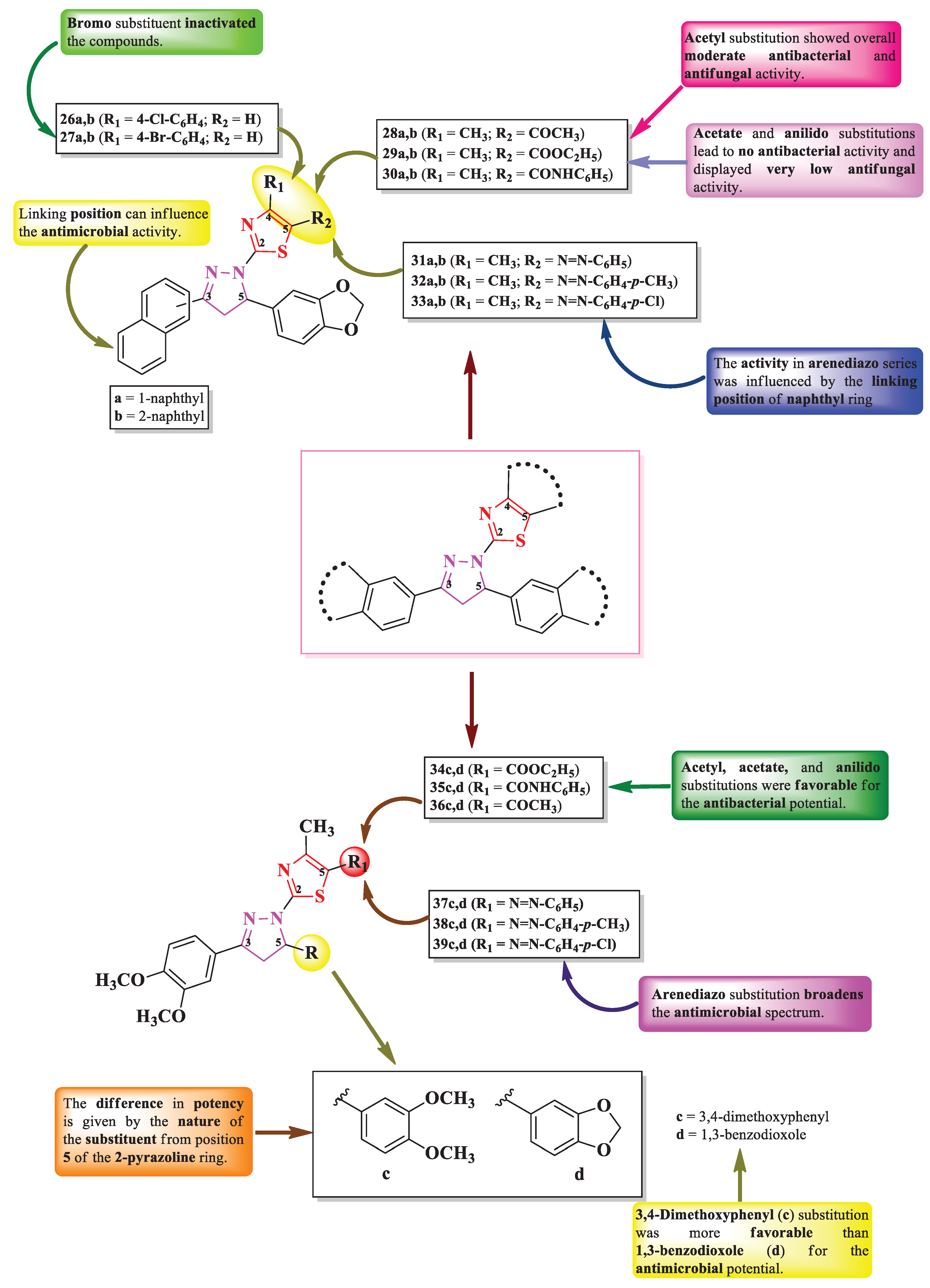
Figure 9.
SAR studies in antimicrobial 2-(5-(furan-2-yl)-2-pyrazolin-1-yl)-4-methylhetarylthio-thiazole derivatives, reported by Bhandare et al [15].
Figure 9.
SAR studies in antimicrobial 2-(5-(furan-2-yl)-2-pyrazolin-1-yl)-4-methylhetarylthio-thiazole derivatives, reported by Bhandare et al [15].
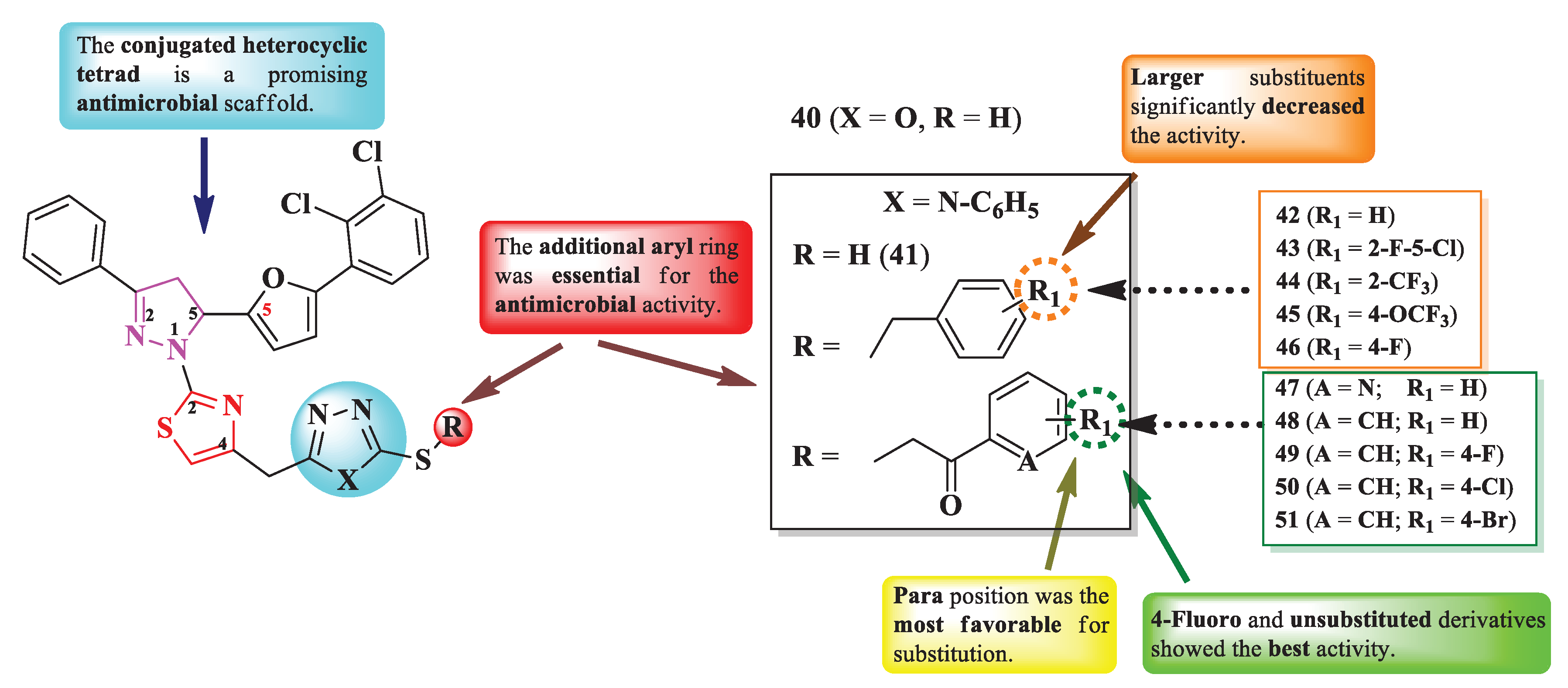
Figure 10.
Structure-activity relationships in antimicrobial 2-(5-(3-(1,2,3-triazol-4-yl)-pyrazol-4-yl)-2-pyrazolin-1-yl)-thiazole derivatives, reported by Abdel-Wahab et al [16].
Figure 10.
Structure-activity relationships in antimicrobial 2-(5-(3-(1,2,3-triazol-4-yl)-pyrazol-4-yl)-2-pyrazolin-1-yl)-thiazole derivatives, reported by Abdel-Wahab et al [16].
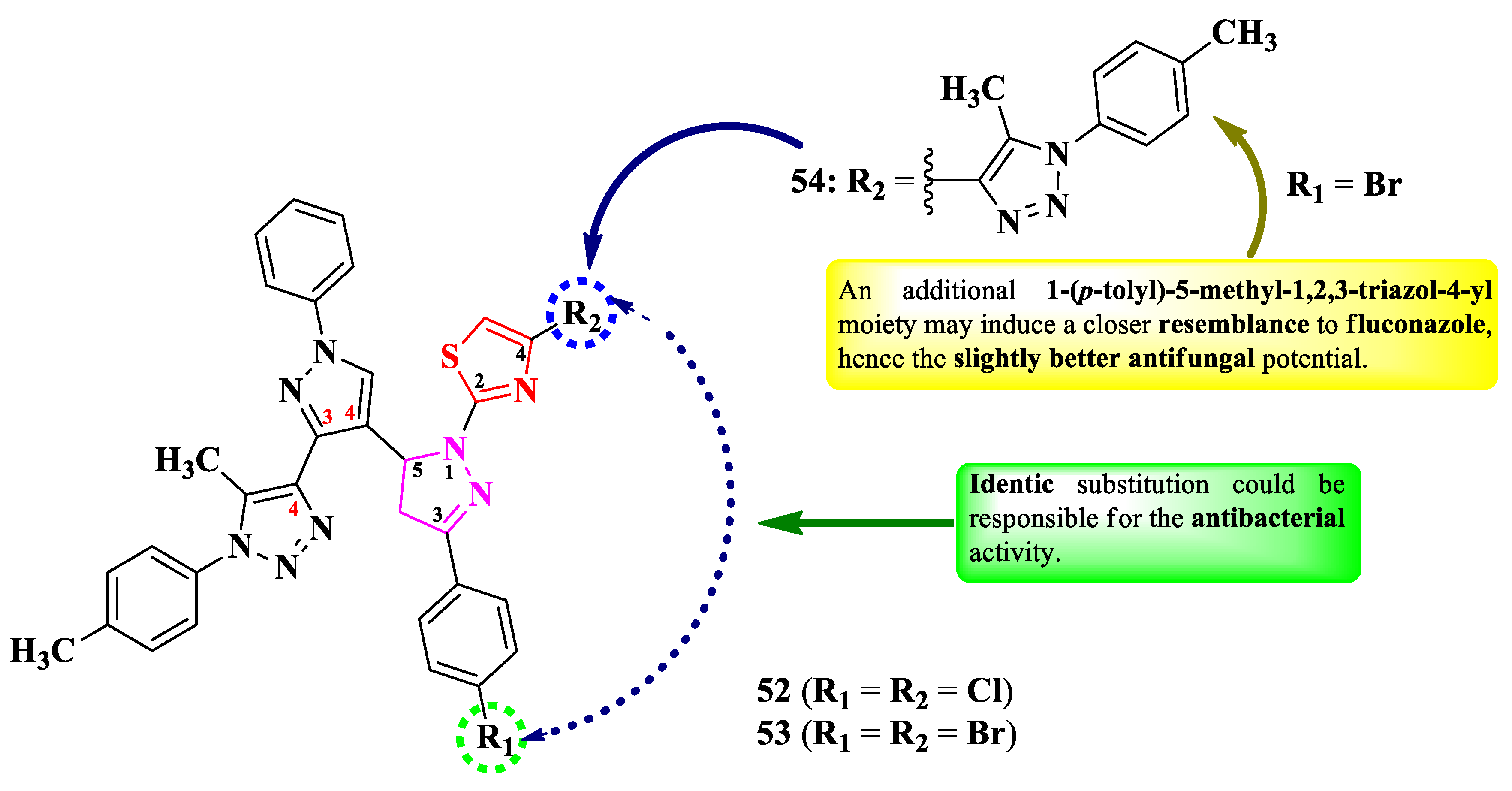
Figure 11.
SAR studies in antimicrobial 2-(N-allyl)-5-(2-pyrazolin-3-yl)-thiazole derivatives, reported by Bondock and Fouda [17].
Figure 11.
SAR studies in antimicrobial 2-(N-allyl)-5-(2-pyrazolin-3-yl)-thiazole derivatives, reported by Bondock and Fouda [17].
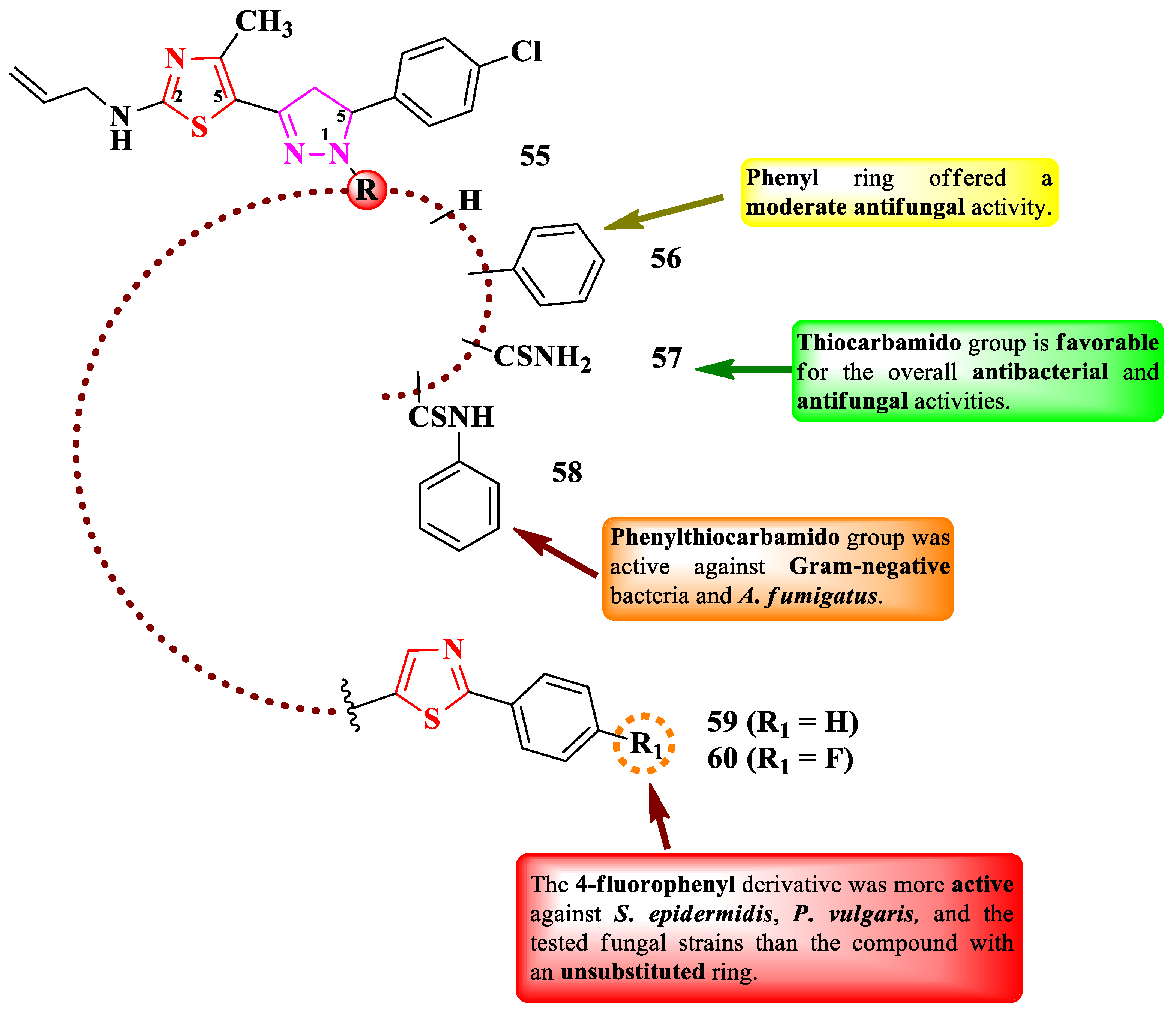
Figure 12.
Endpoints of designing novel antimicrobial thiazole clubbed with 2-pyrazoline hybrid compounds.
Figure 12.
Endpoints of designing novel antimicrobial thiazole clubbed with 2-pyrazoline hybrid compounds.
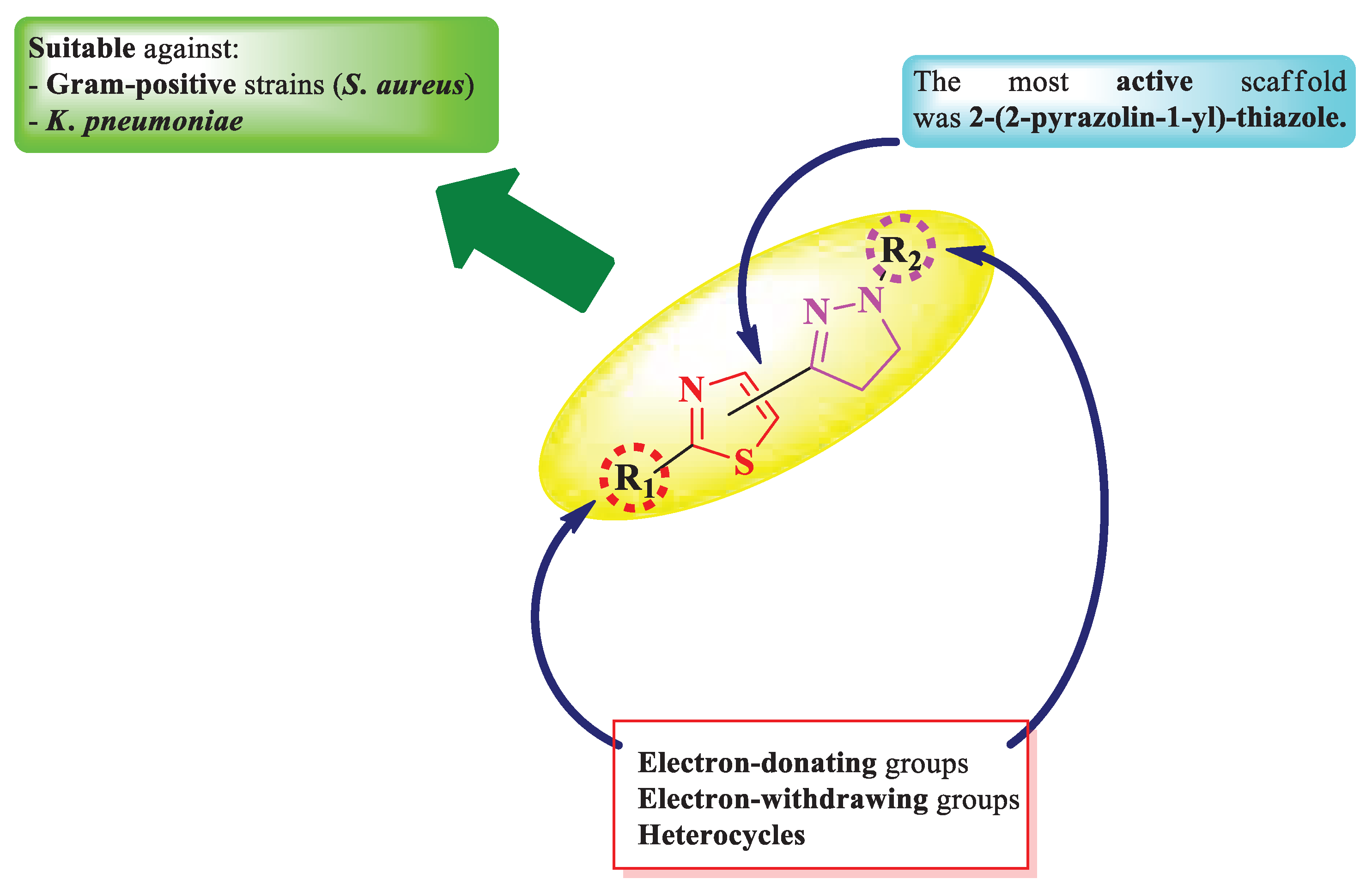
Figure 13.
Antimicrobial N-(4-pyrazolin-3-one)-2-thiazolyl-hydrazonomethyl-phenoxyacetamides, reported by Abu-Melha [19].
Figure 13.
Antimicrobial N-(4-pyrazolin-3-one)-2-thiazolyl-hydrazonomethyl-phenoxyacetamides, reported by Abu-Melha [19].
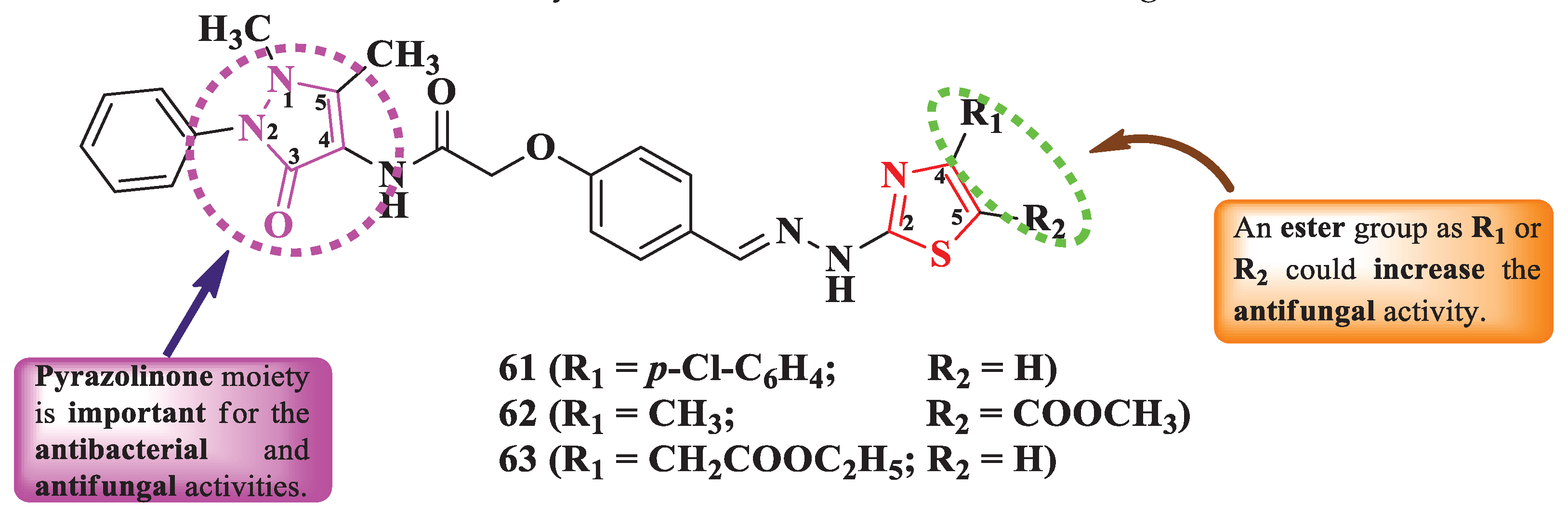
Figure 14.
The general structures for the series discussed, containing thiazole and pyrazole rings [21,22,23,24,25,26].
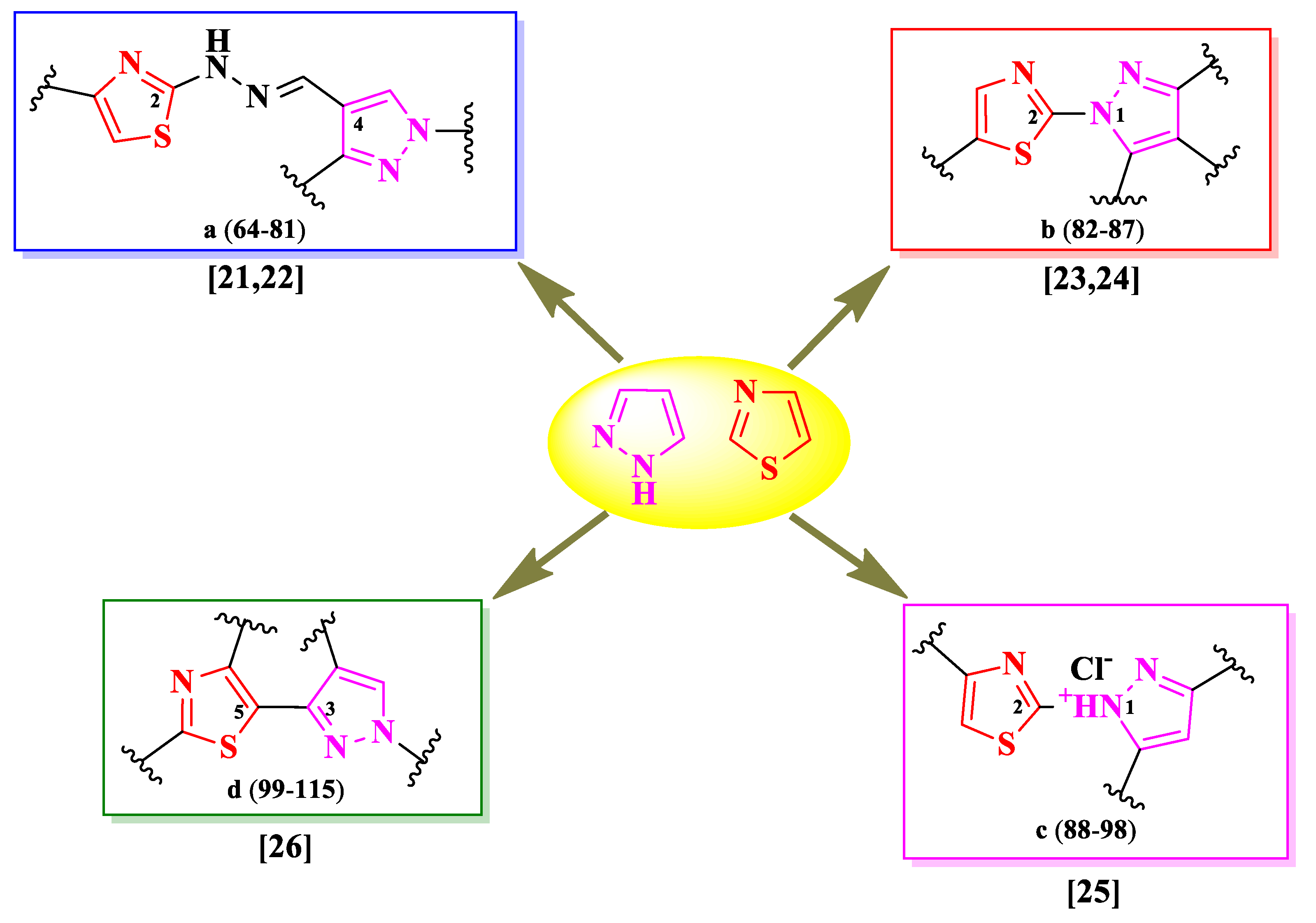
Figure 15.
SAR studies in antimicrobial 2-(pyrazol-4-yl)-methylylidenehydrazinyl-thiazole derivatves, reported by Gondru et al. [21] and Patil et al. [22].
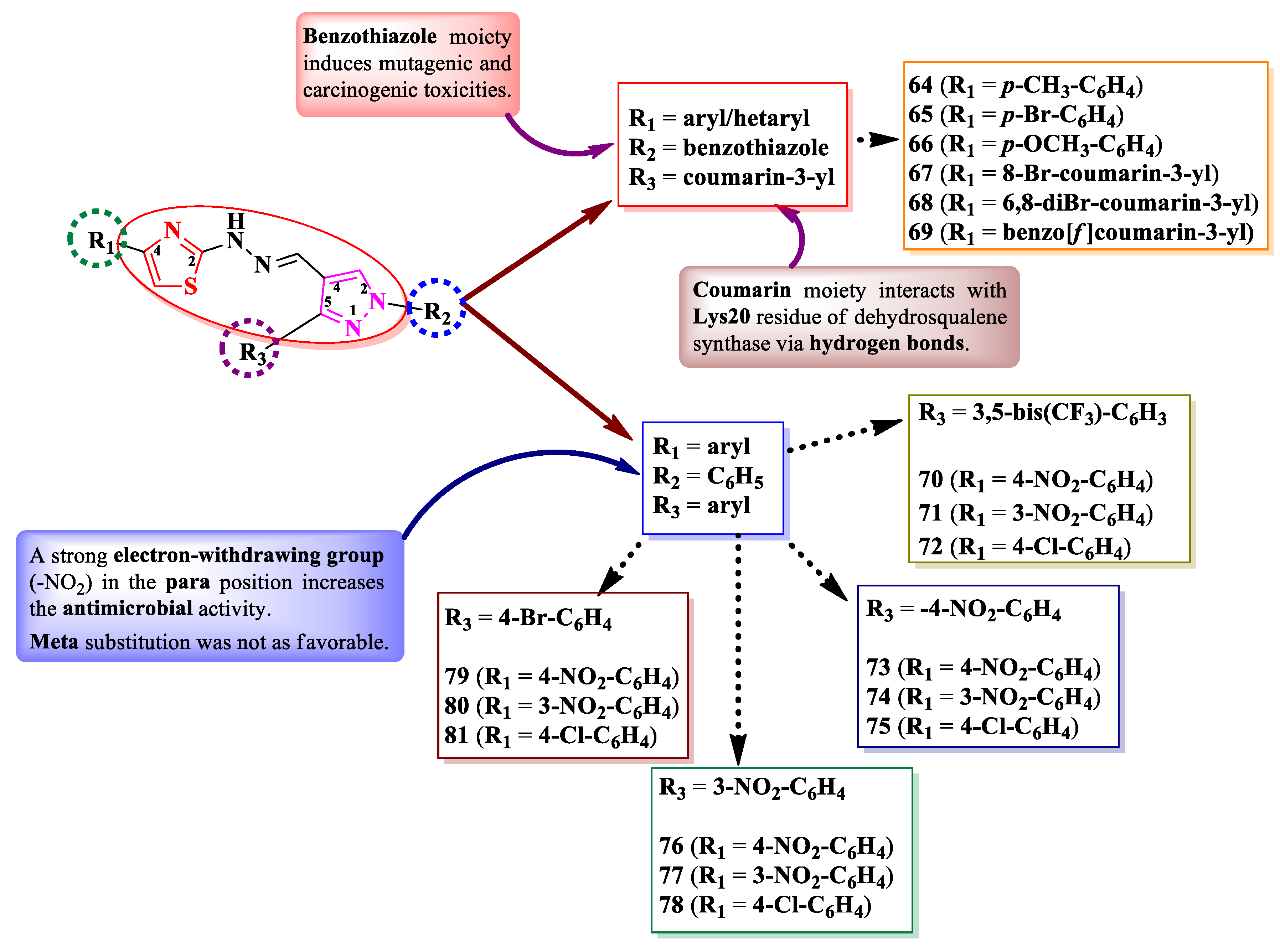
Figure 16.
SAR studies in antimicrobial 5-(coumarin-3-yl)-2-(pyrazol-1-yl)-thiazole derivatives, reported by Abdel-Aziem et al. [23] and Kumar et al. [24].
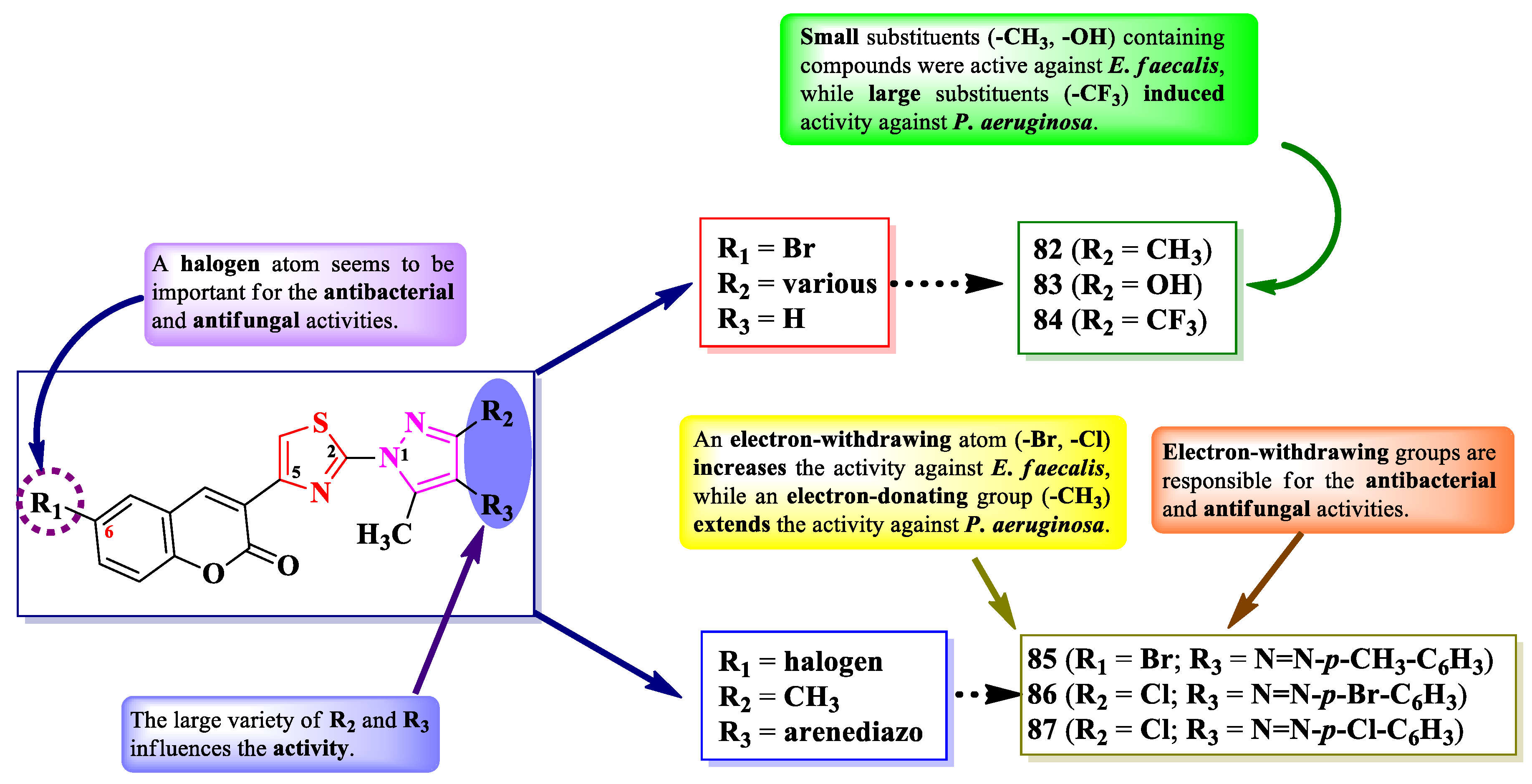
Figure 17.
Antimicrobial 2-pyrazolium-thiazol-4-yl salts, reported Mahmoodi and Ghodsi [25].
Figure 17.
Antimicrobial 2-pyrazolium-thiazol-4-yl salts, reported Mahmoodi and Ghodsi [25].
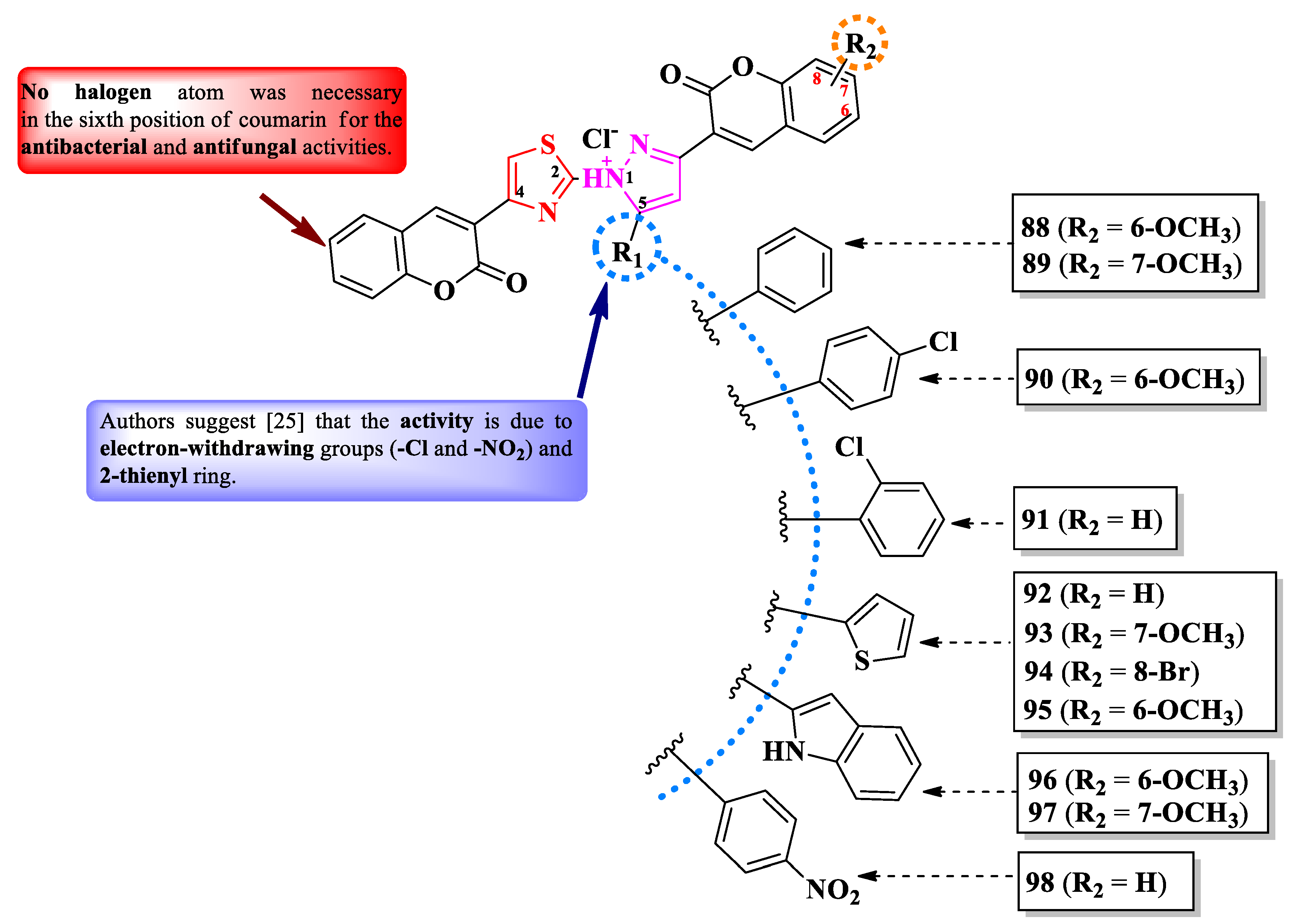
Figure 18.
SAR studies in antimicrobial 2-phenyl-5-(4-hetaryl-pyrazol-3-yl)-thiazoles, reported by Nalawade et al. [26].
Figure 18.
SAR studies in antimicrobial 2-phenyl-5-(4-hetaryl-pyrazol-3-yl)-thiazoles, reported by Nalawade et al. [26].
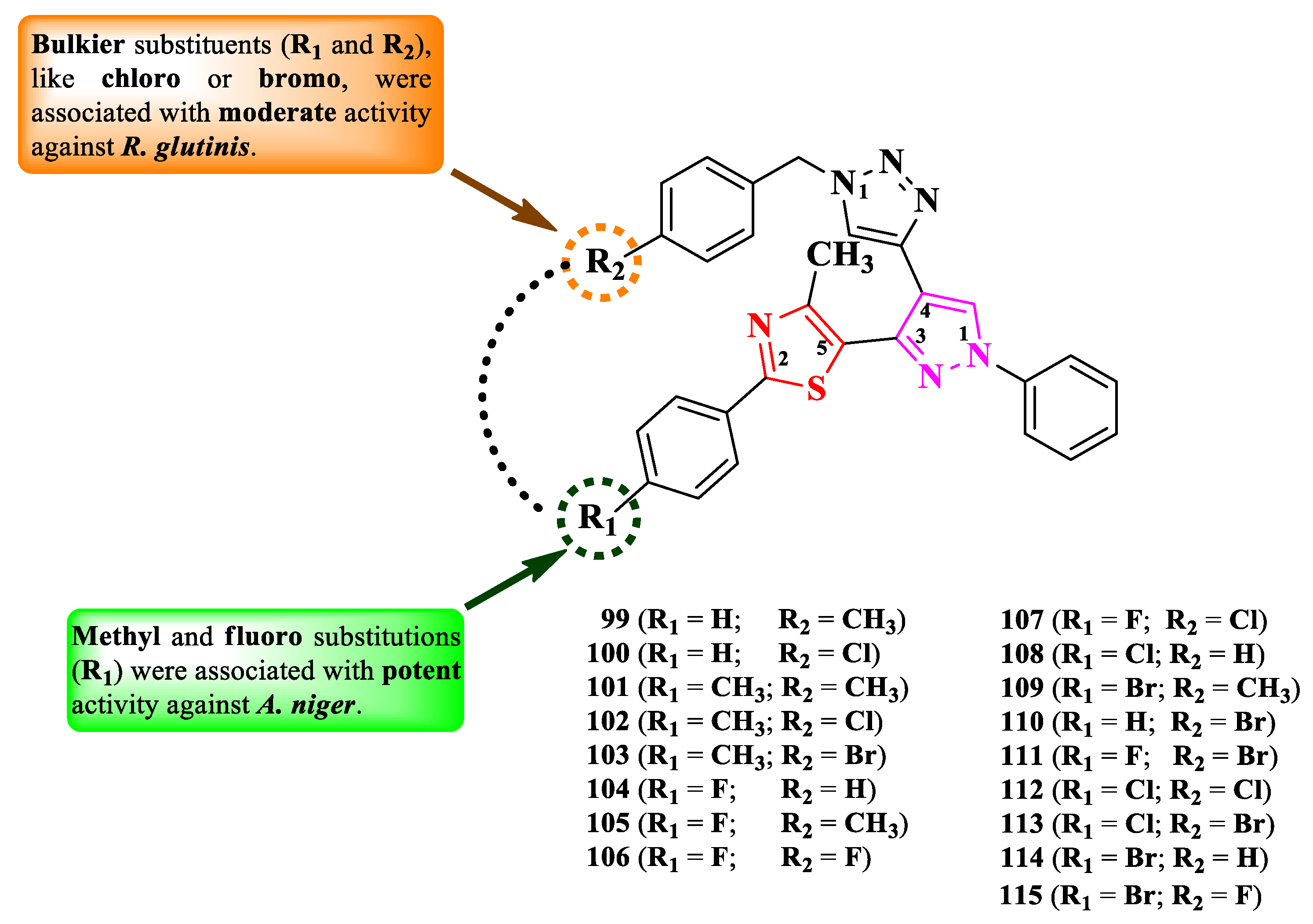
Figure 19.
Endpoints of designing novel antimicrobial thiazole clubbed with pyrazole hybrid compounds.
Figure 19.
Endpoints of designing novel antimicrobial thiazole clubbed with pyrazole hybrid compounds.
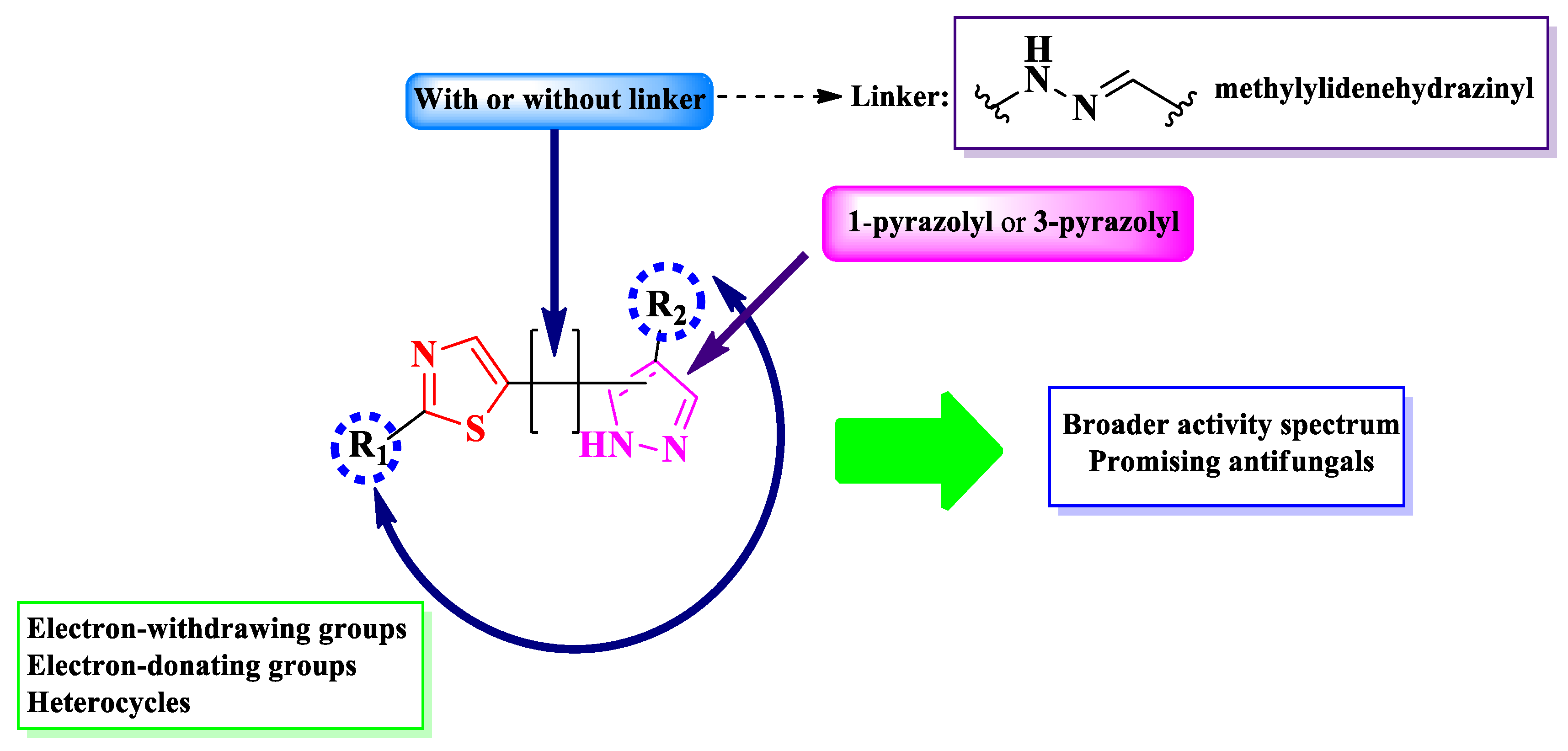
Figure 20.
Design of antifungal 2-(2,4,5-triphenyl-imidazol-1-yl)-thiazoles, reported by Nikalje et al. [28].
Figure 20.
Design of antifungal 2-(2,4,5-triphenyl-imidazol-1-yl)-thiazoles, reported by Nikalje et al. [28].
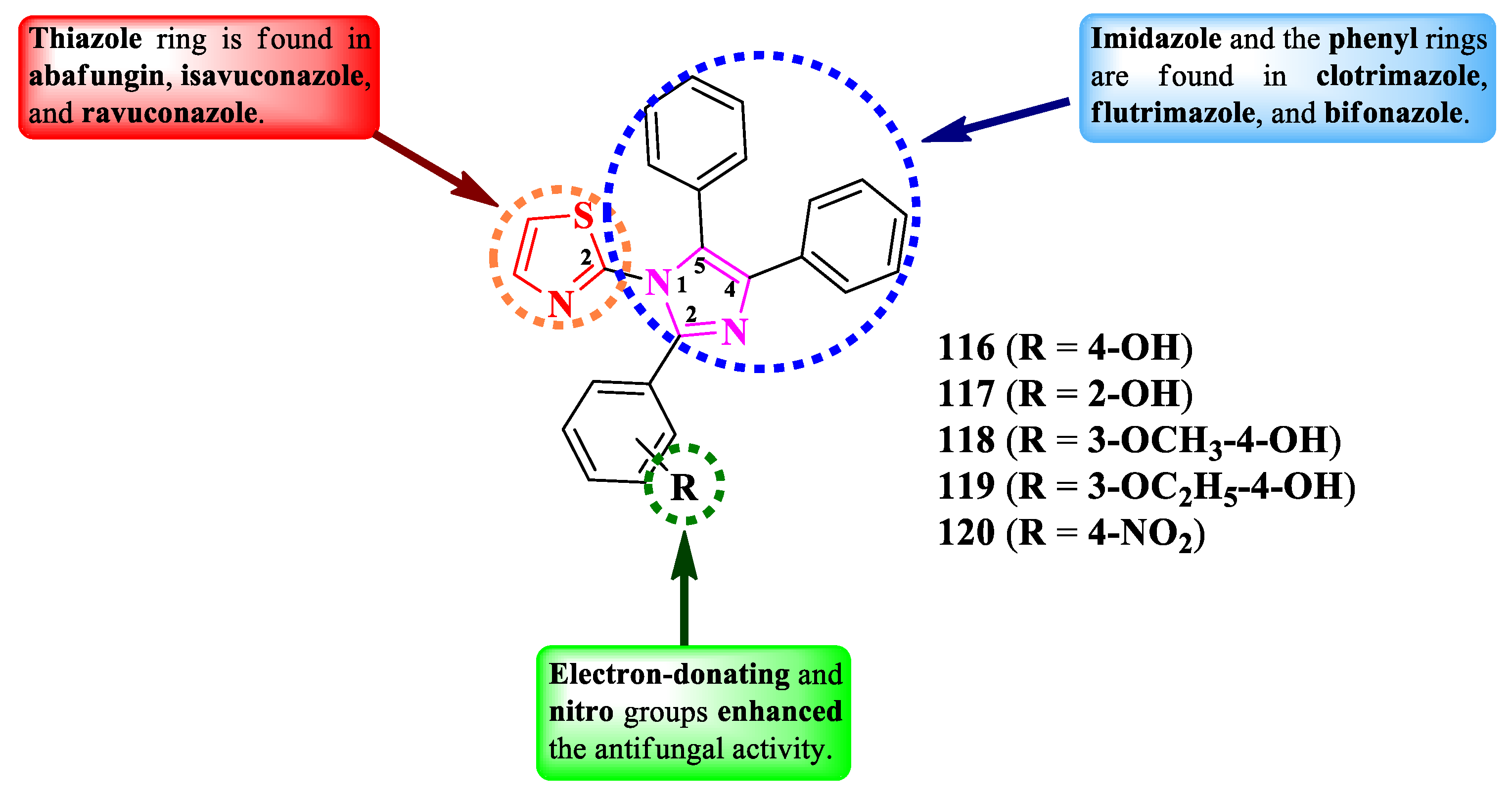
Figure 21.
SAR studies in antibacterial 2-(thiazol-2-yl)-N-thiazolidin-4-one derivatives, reported by Othman et al. [30].
Figure 21.
SAR studies in antibacterial 2-(thiazol-2-yl)-N-thiazolidin-4-one derivatives, reported by Othman et al. [30].
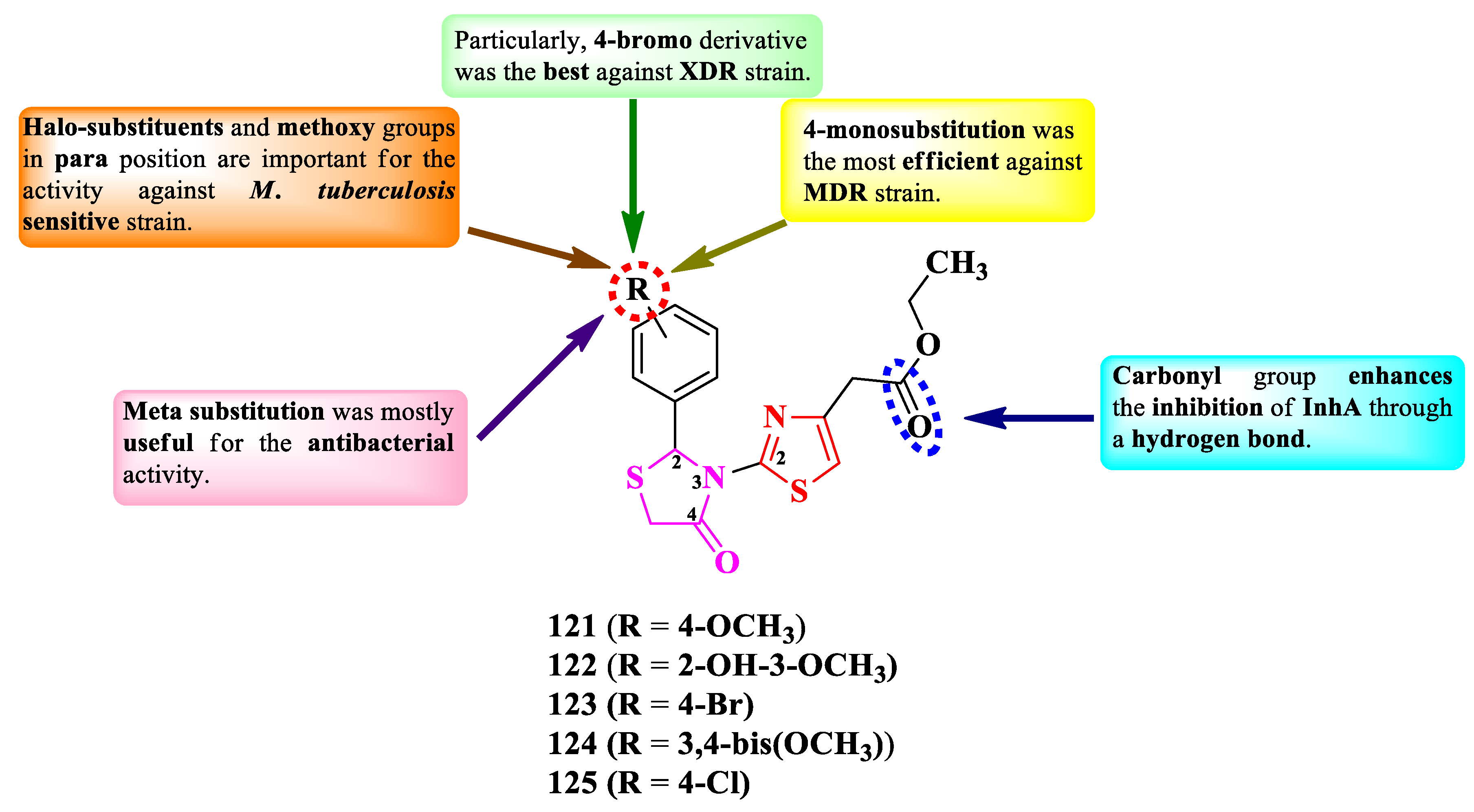
Figure 22.
SAR studies in antimicrobial 2-(thiazol-2-yl)-imino-thiazolidin-4-one derivatives, reported by Abo-Ashur et al. [31].
Figure 22.
SAR studies in antimicrobial 2-(thiazol-2-yl)-imino-thiazolidin-4-one derivatives, reported by Abo-Ashur et al. [31].
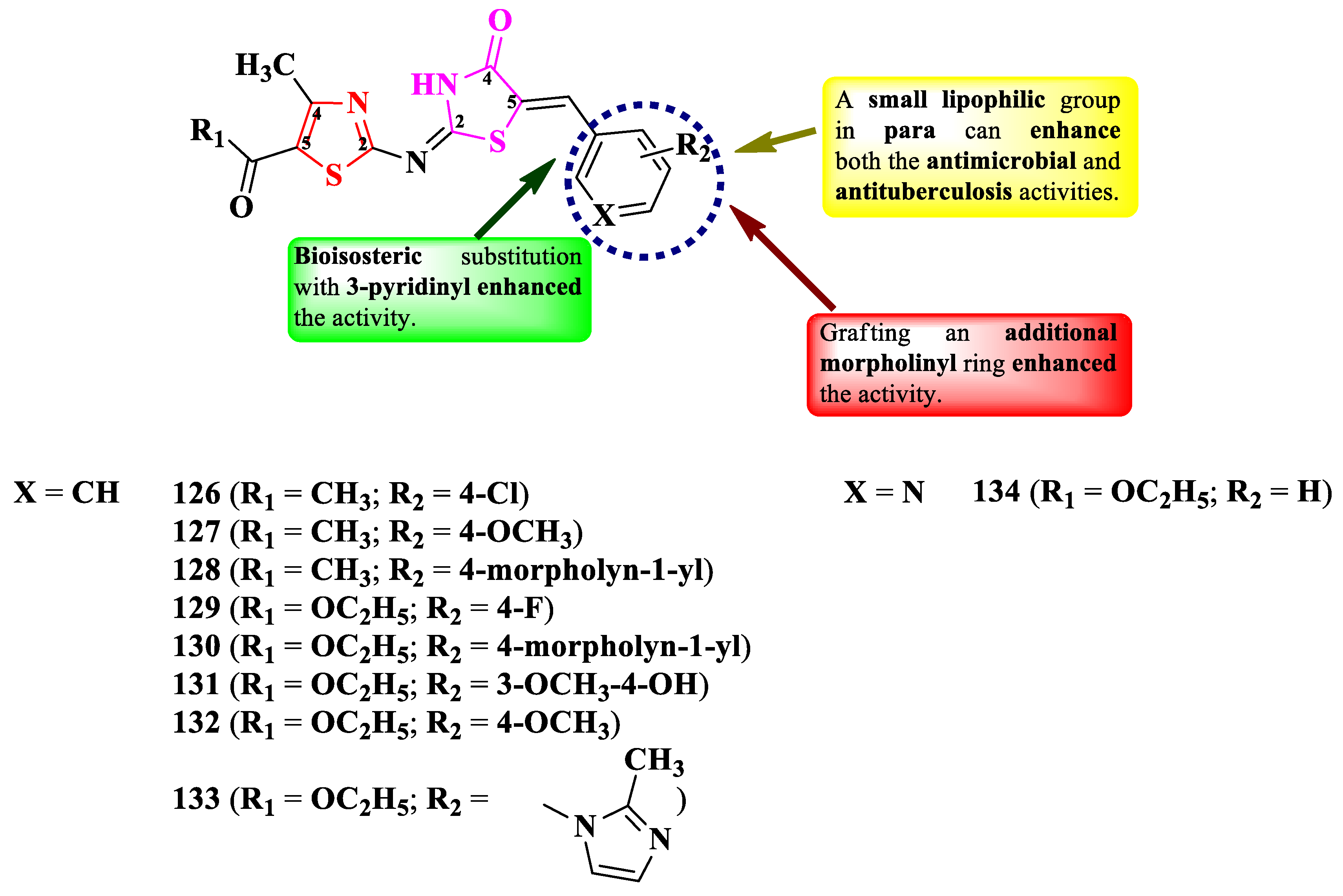
Figure 23.
SAR studies in antimicrobial 2-(thiazolidin-2,4-dion-3-yl)-N-(thiazol-2-yl)acetamides, reported by Alegaon et al. [33].
Figure 23.
SAR studies in antimicrobial 2-(thiazolidin-2,4-dion-3-yl)-N-(thiazol-2-yl)acetamides, reported by Alegaon et al. [33].
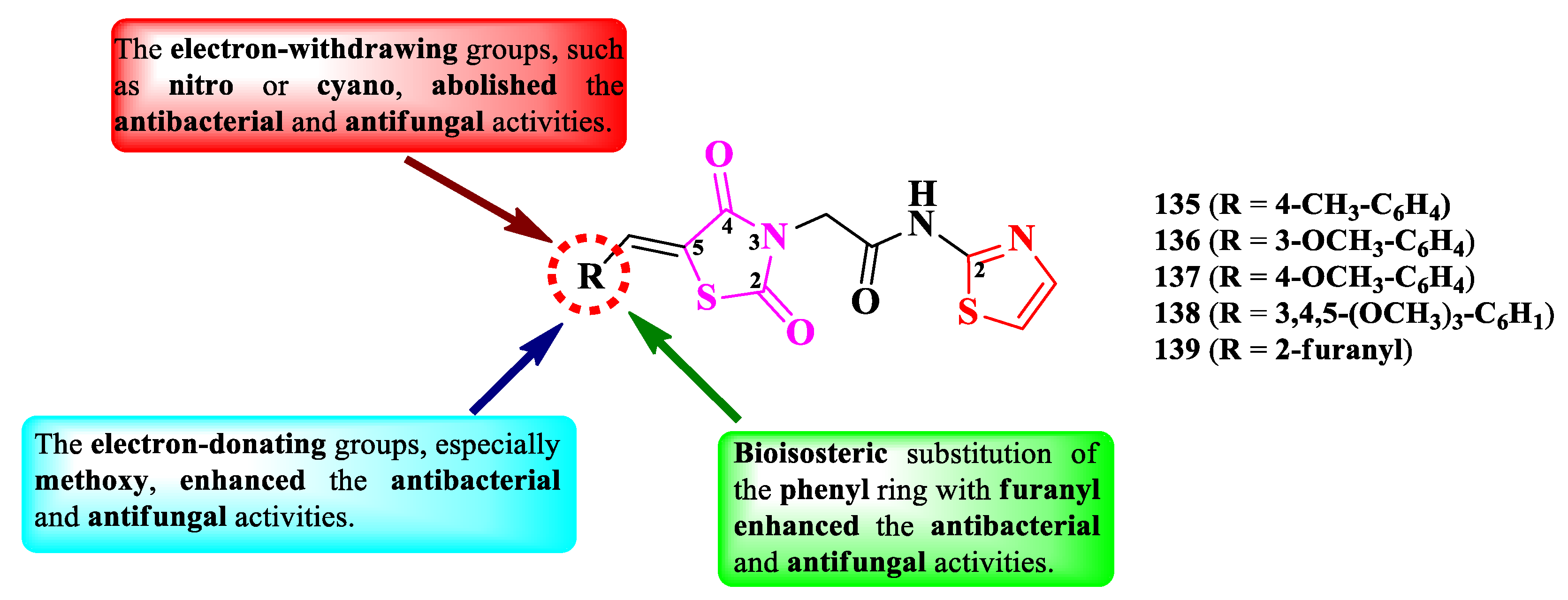
Figure 24.
Structure of halicin.
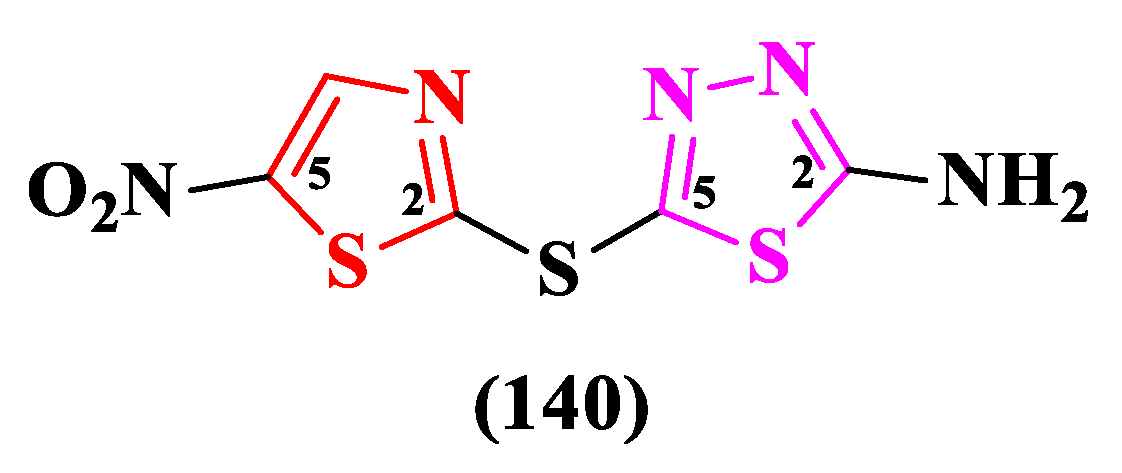
Figure 25.
The general structures for the series discussed, containing thiazole and 1,2,3-triazole rings [41,42,43,44,45].
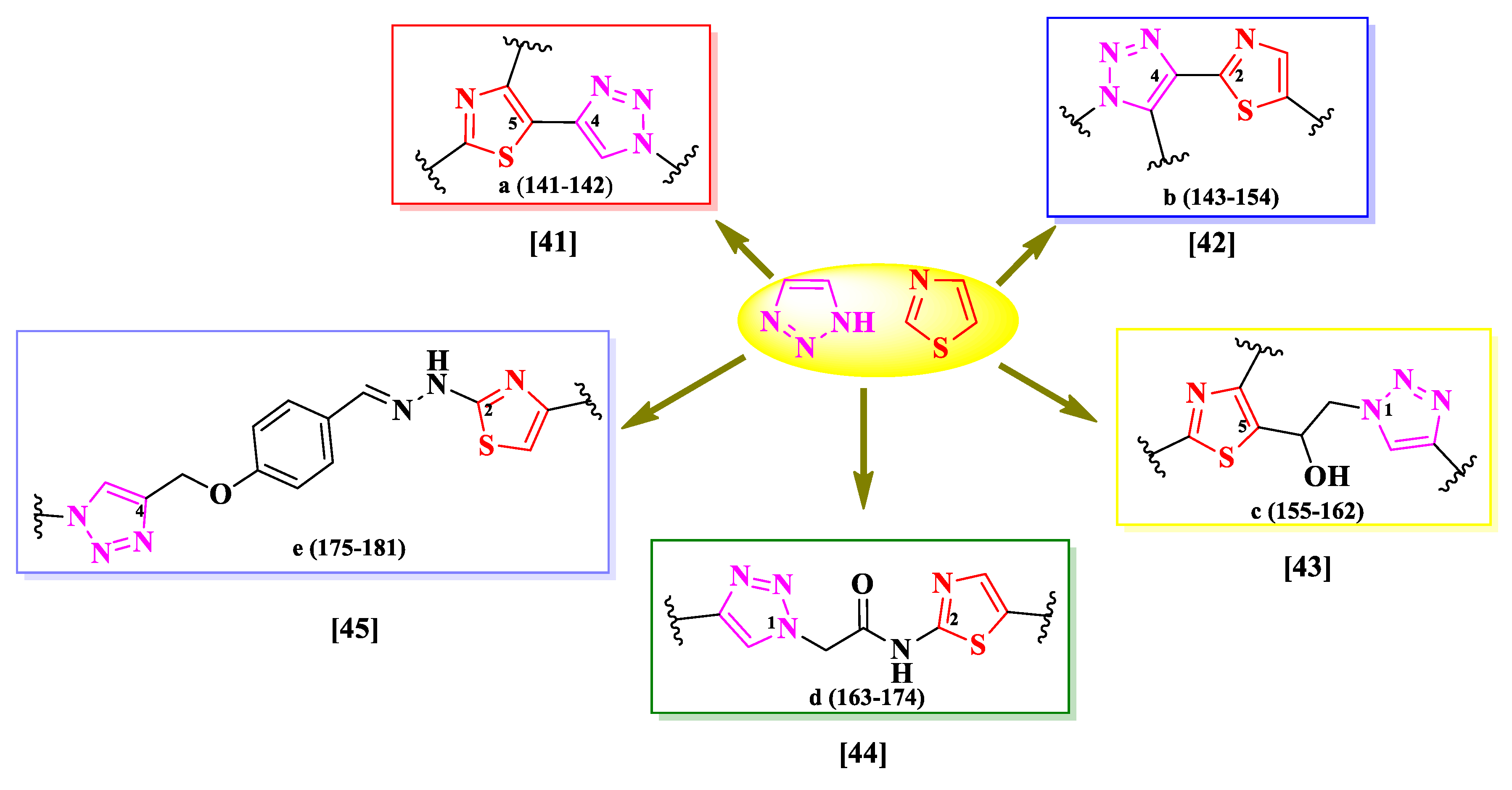
Figure 26.
SAR studies in antituberculosis 5-(1,2,3-triazol-4-yl)-thiazoles, reported by Shinde et al. [41].
Figure 26.
SAR studies in antituberculosis 5-(1,2,3-triazol-4-yl)-thiazoles, reported by Shinde et al. [41].
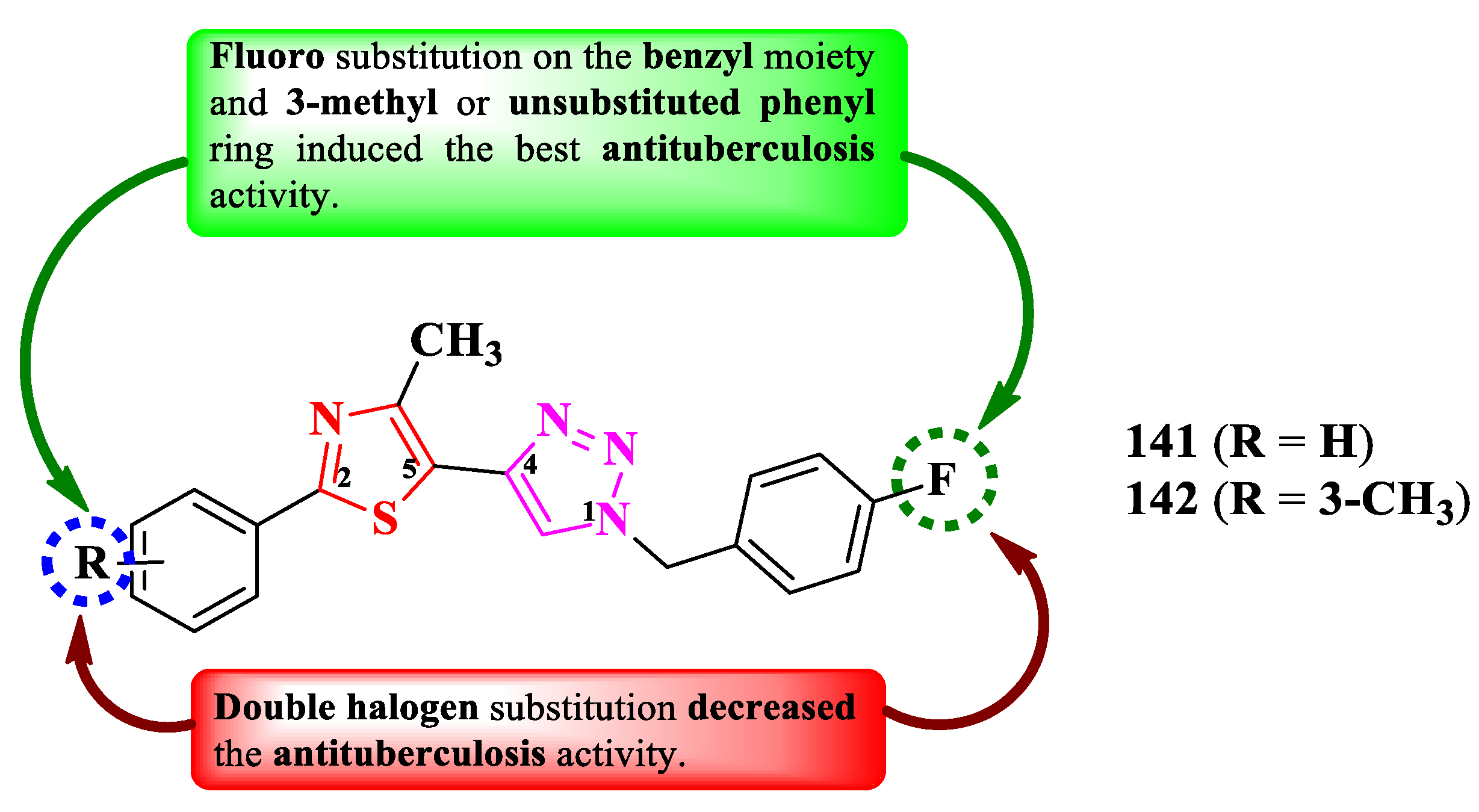
Figure 27.
SAR studies in antimicrobial 2-(1,2,3-triazol-4-yl)-thiazole derivatives, reported by Mahale et al. [42].
Figure 27.
SAR studies in antimicrobial 2-(1,2,3-triazol-4-yl)-thiazole derivatives, reported by Mahale et al. [42].
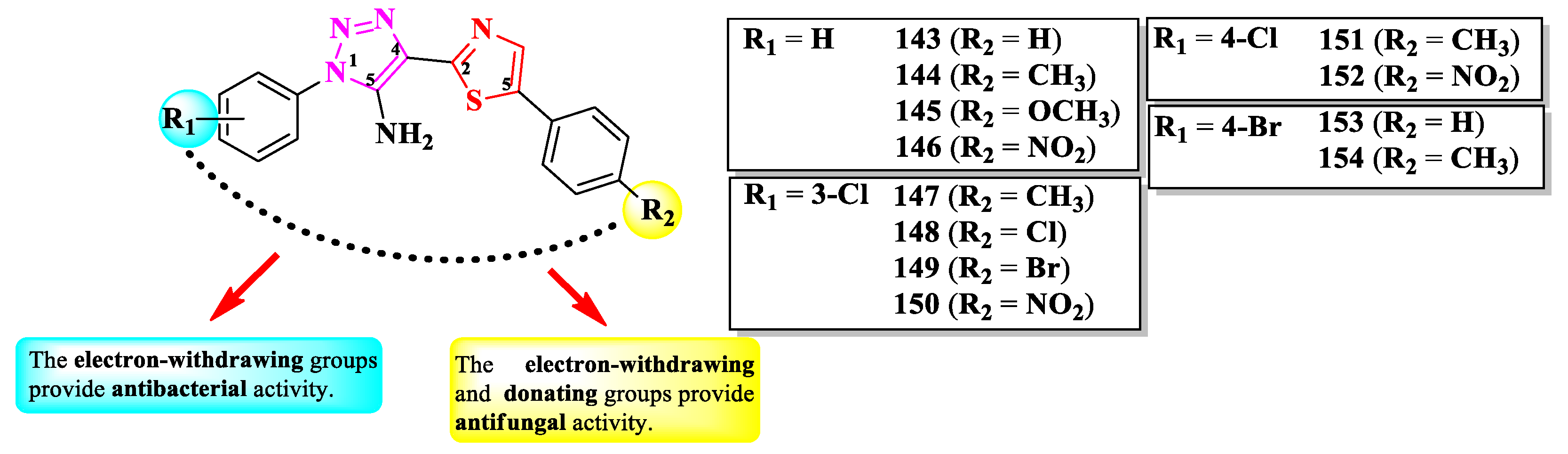
Figure 28.
SAR studies in antimicrobial 1-(thiazol-5-yl)-2-(1,2,3-triazol-1-yl)-ethanol derivatives, reported by Jagadale et al. [43].
Figure 28.
SAR studies in antimicrobial 1-(thiazol-5-yl)-2-(1,2,3-triazol-1-yl)-ethanol derivatives, reported by Jagadale et al. [43].
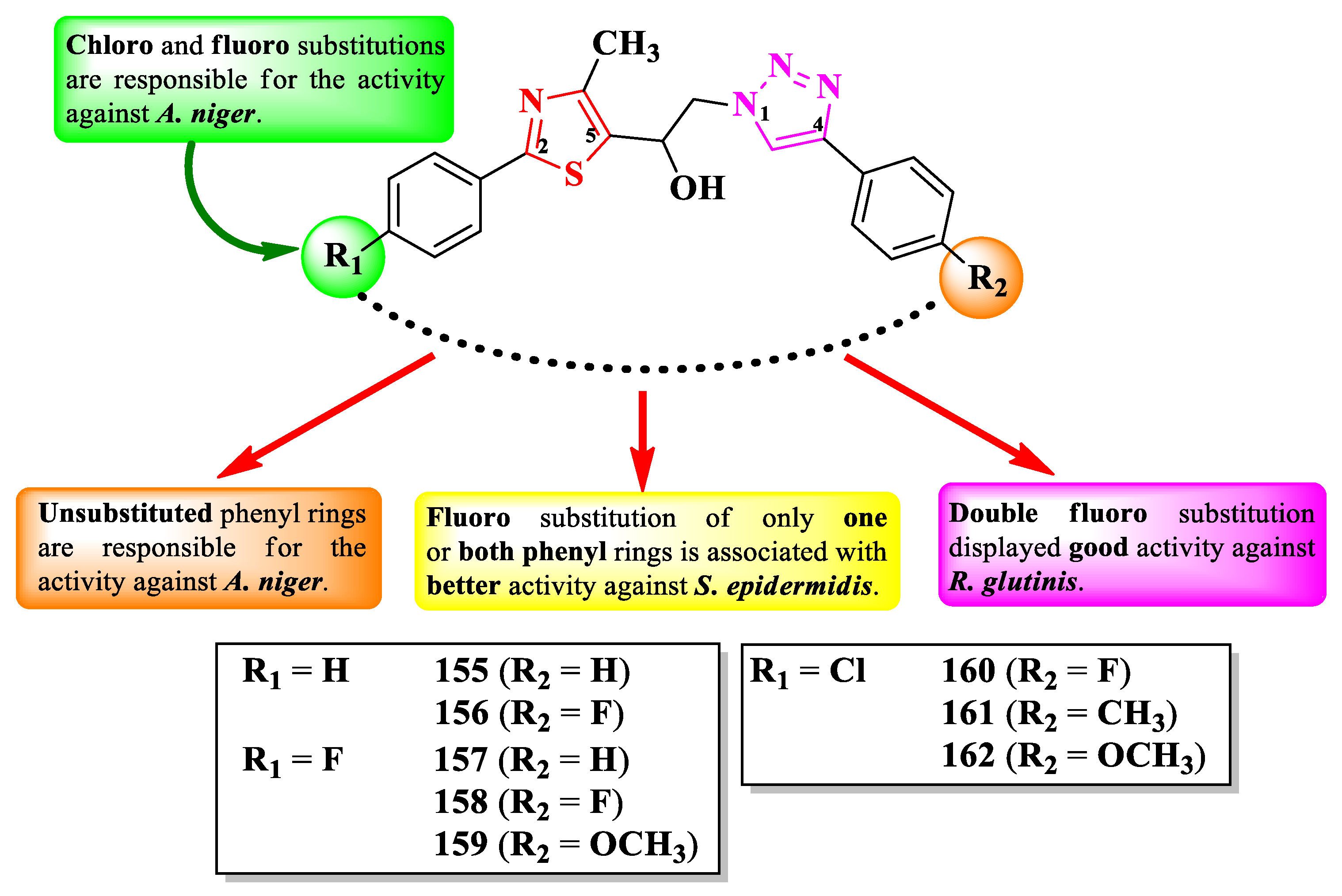
Figure 29.
SAR studies in antimicrobial 2-(1,2,3-triazol-1-yl)-N-(thiazol-2-yl)-acetamides and 2-(1,2,3-triazol-1-yl)-N-(benzothiazol-2-yl)-acetamides, reported by Poonia et al. [44].
Figure 29.
SAR studies in antimicrobial 2-(1,2,3-triazol-1-yl)-N-(thiazol-2-yl)-acetamides and 2-(1,2,3-triazol-1-yl)-N-(benzothiazol-2-yl)-acetamides, reported by Poonia et al. [44].
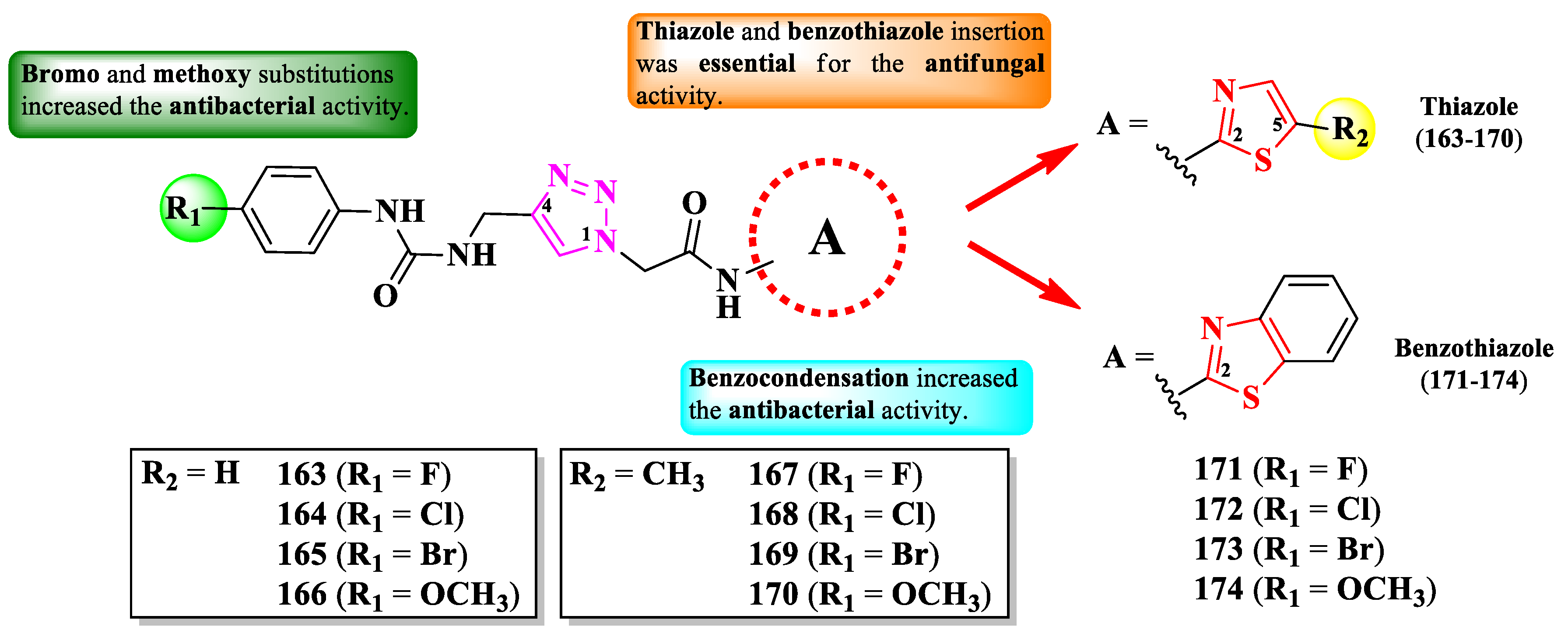
Figure 30.
SAR studies in antimicrobial 2-(1,2,3-triazol-4-yl-methoxybenzylidenehydrazinyl)-thiazoles, reported by Gondru et al. [45].
Figure 30.
SAR studies in antimicrobial 2-(1,2,3-triazol-4-yl-methoxybenzylidenehydrazinyl)-thiazoles, reported by Gondru et al. [45].
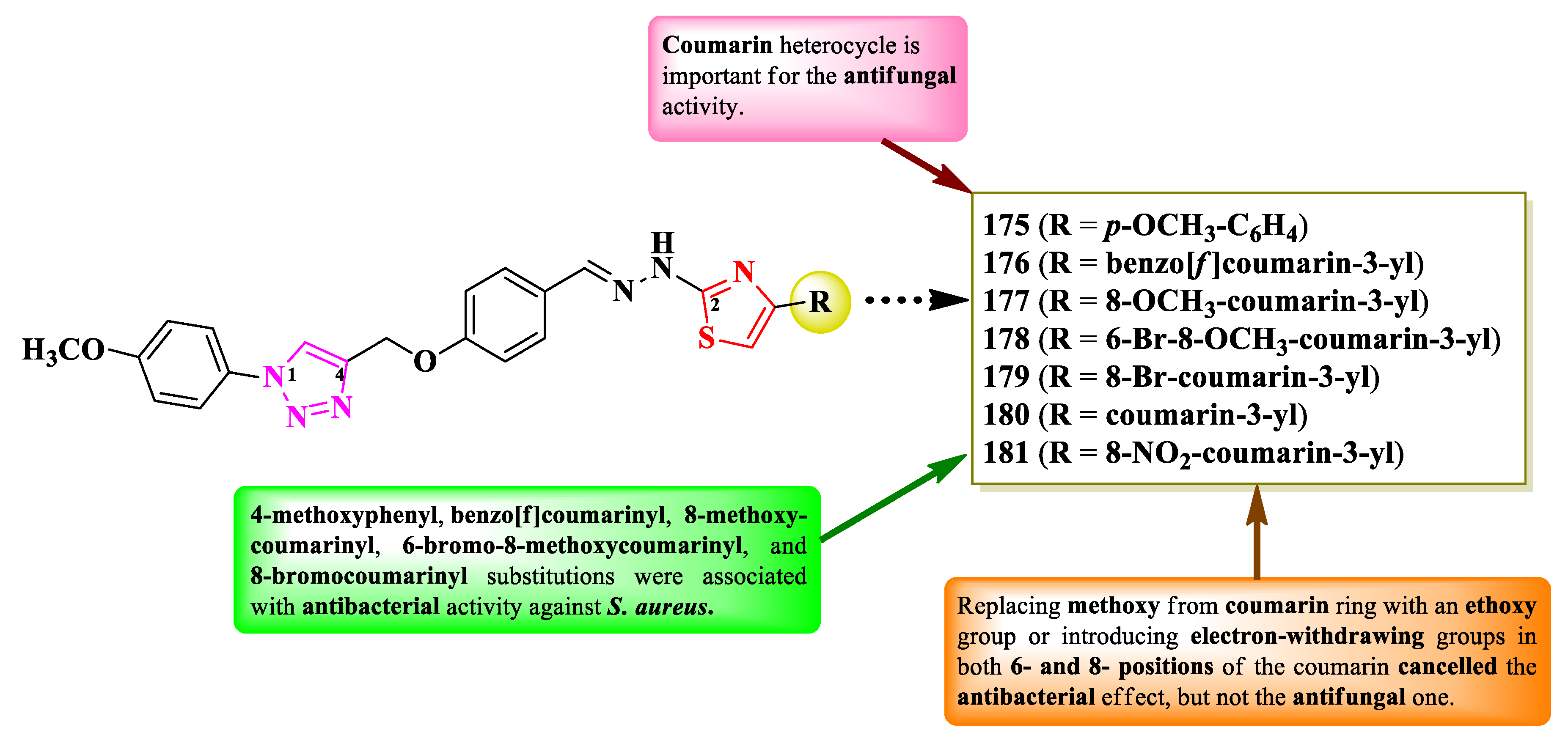
Figure 31.
Endpoints of designing novel antimicrobial 1,3-thiazole clubbed with 1,2,3-triazole hybrid compounds.
Figure 31.
Endpoints of designing novel antimicrobial 1,3-thiazole clubbed with 1,2,3-triazole hybrid compounds.
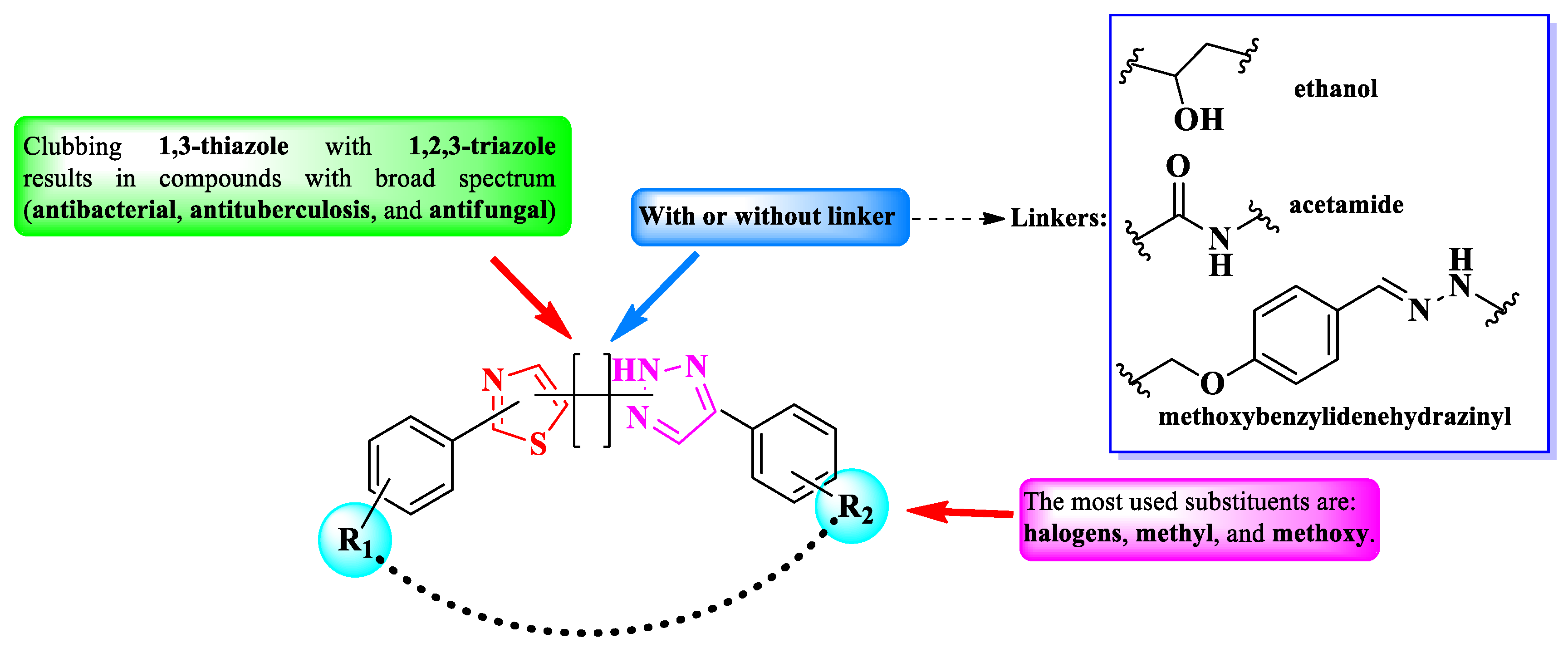
Figure 35.
SAR studies in antibacterial 4-ferrocenylthiazoles clubbed with indole or carbazole, reported by Zhao et al. [58].
Figure 35.
SAR studies in antibacterial 4-ferrocenylthiazoles clubbed with indole or carbazole, reported by Zhao et al. [58].
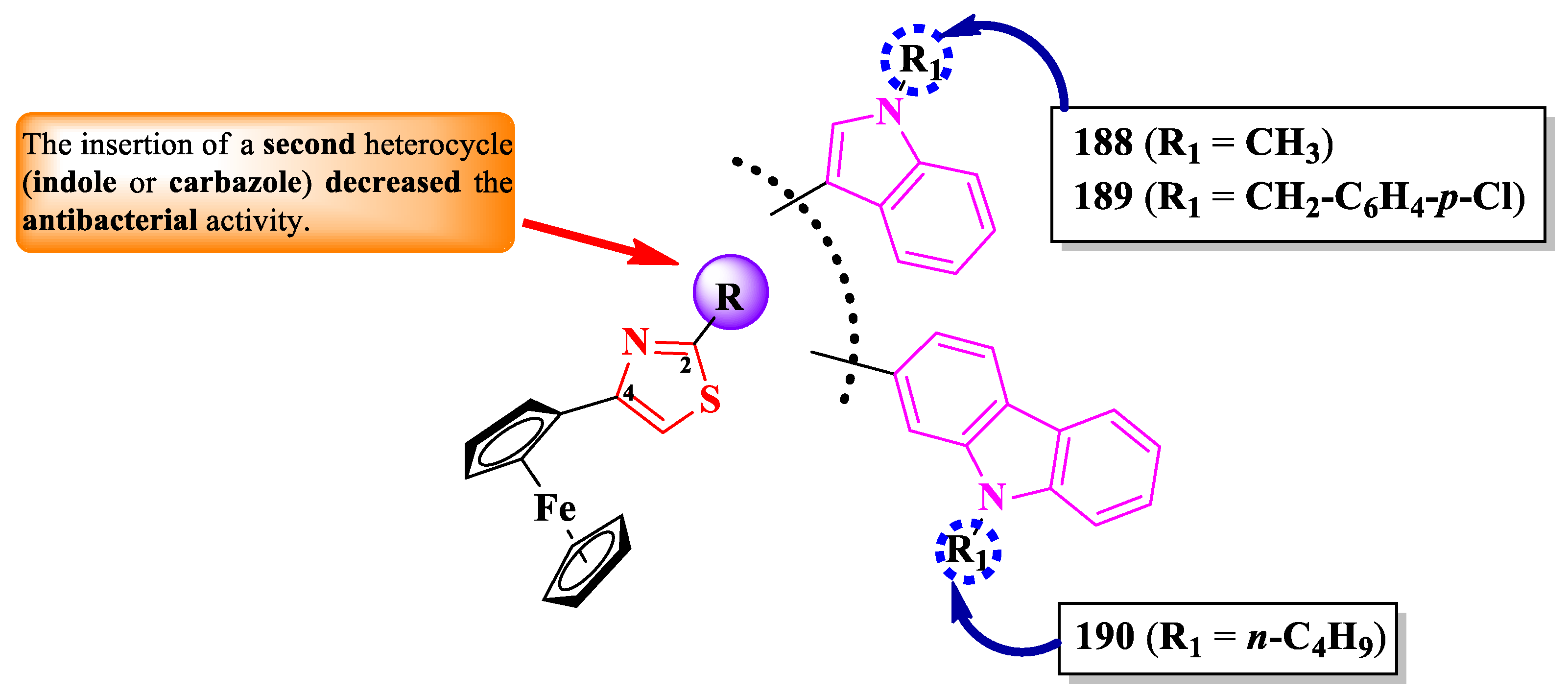
Figure 36.
SAR studies in antimicrobial thiazole clubbed with indolin-2-one compounds, reported by Alzahrani et al. [60].
Figure 36.
SAR studies in antimicrobial thiazole clubbed with indolin-2-one compounds, reported by Alzahrani et al. [60].
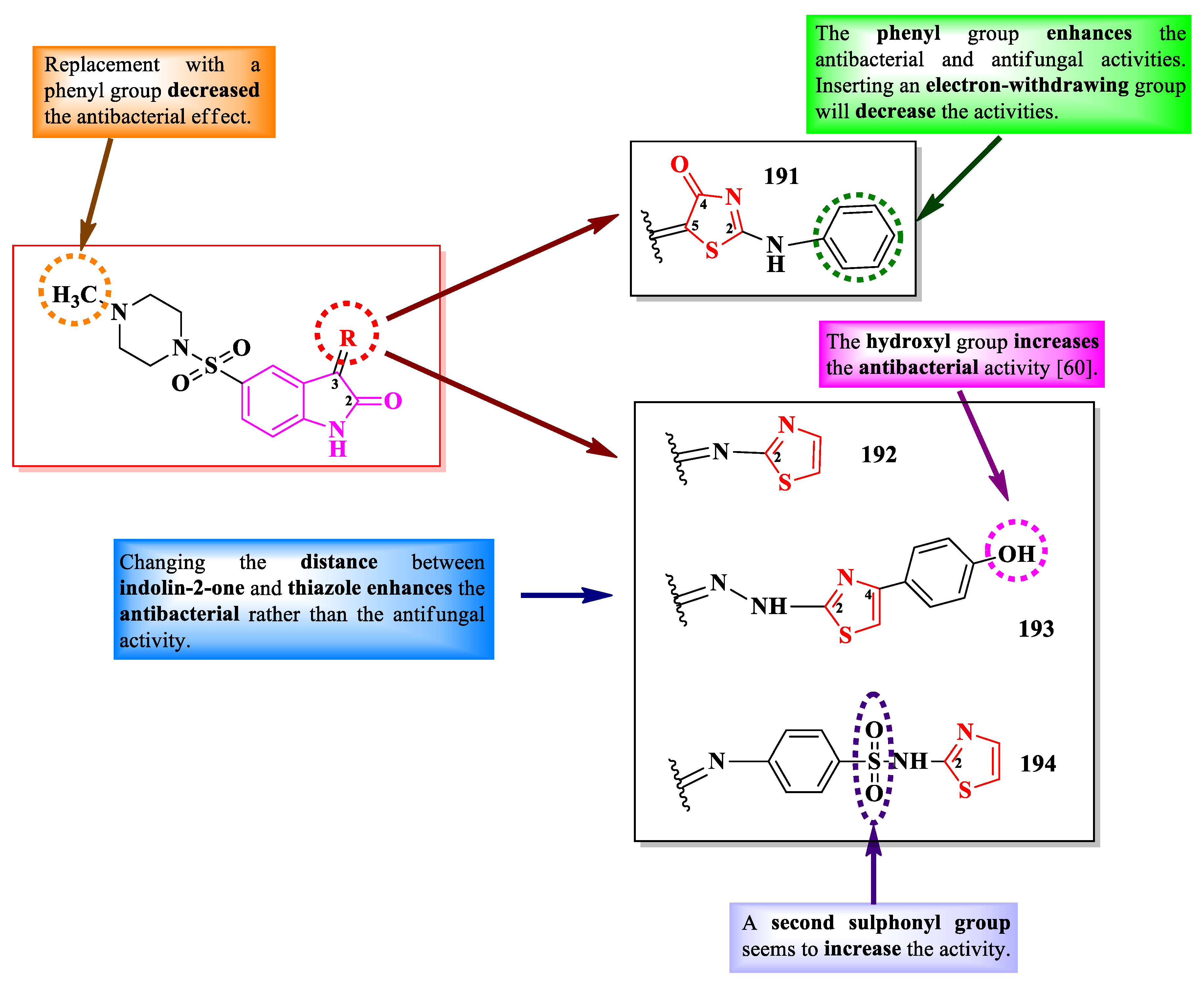
Figure 37.
Structure-activity relationships in antimicrobial 2-(2-tetrahydroindenofuranylidene)-hydrazinylthiazoles, reported by Adole et al. [63].
Figure 37.
Structure-activity relationships in antimicrobial 2-(2-tetrahydroindenofuranylidene)-hydrazinylthiazoles, reported by Adole et al. [63].
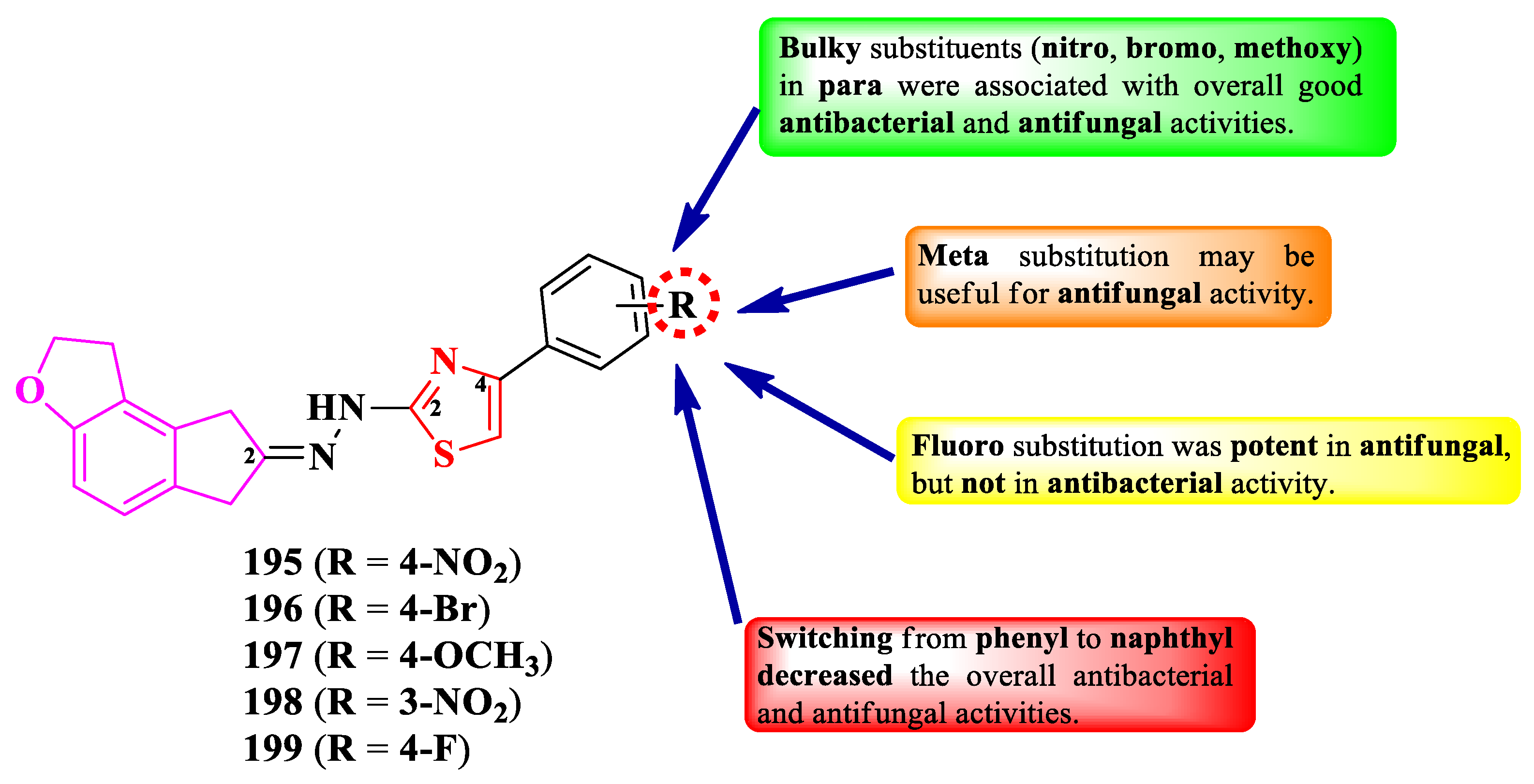
Figure 38.
Structure-activity relationships in antimicrobial 2-pyridyl-thiazoles, reported by Muluk et al. [64,65], Patil et al. [66], and Eryılmaz et al. [67].
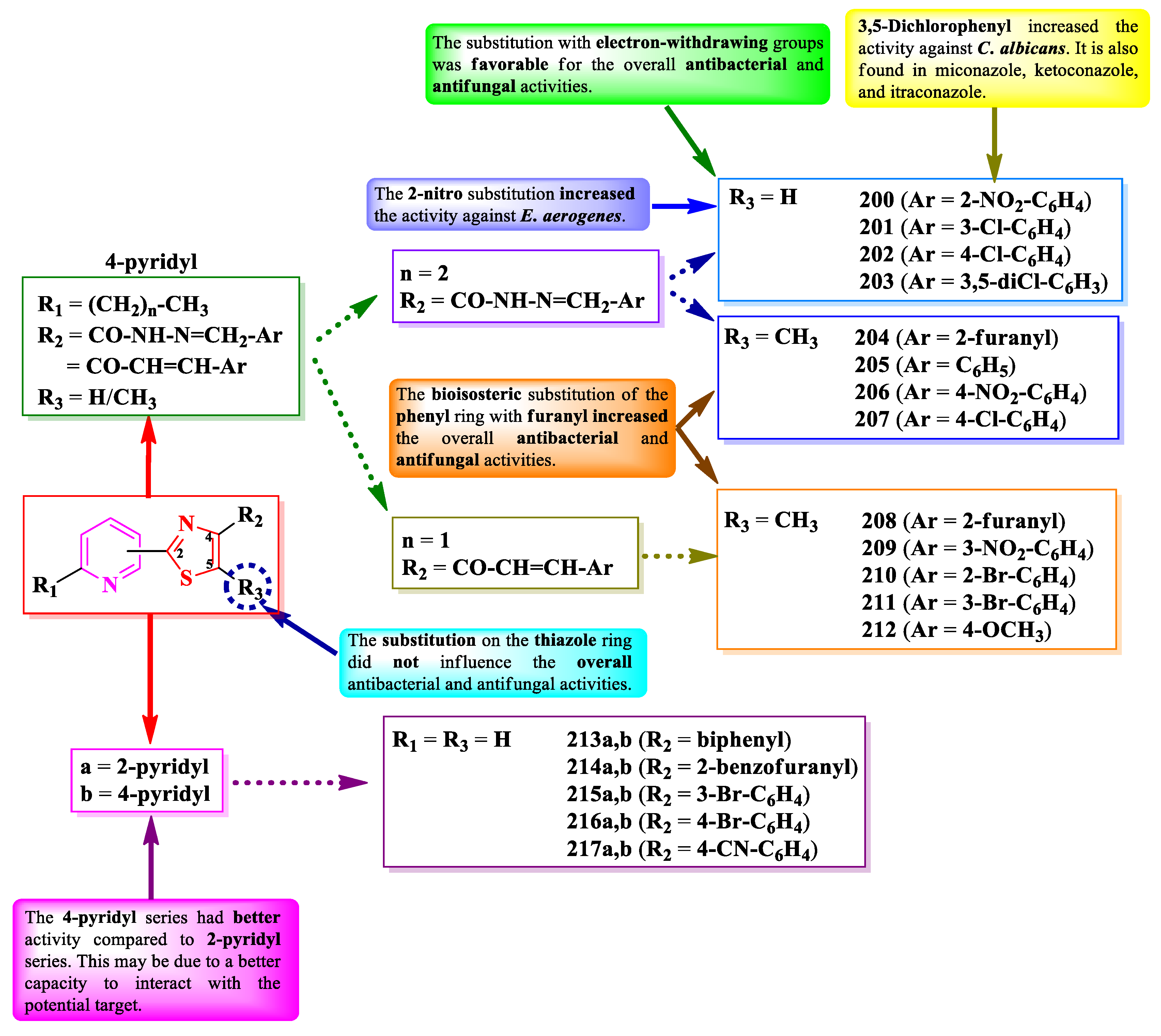
Figure 39.
SAR studies in antimicrobial 4-(3-coumaryl)-2-hydrazinylthiazoles, reported by Yusufzai et al. [70] and Salar et al. [71].
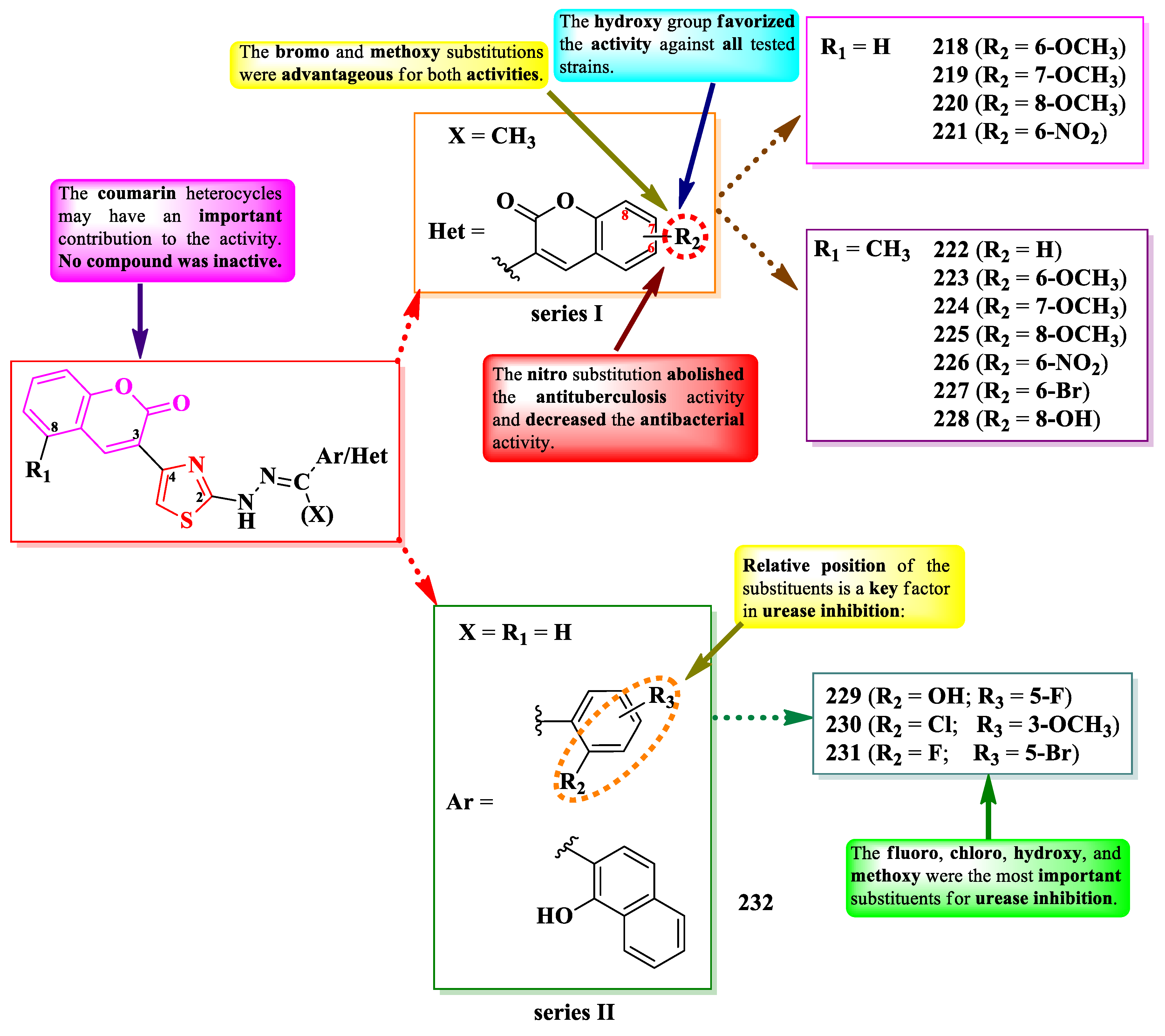
Figure 40.
SAR studies in antibacterial 2-(8-(1-oxoprop-2-en-2-yl)-oxycoumarinyl)-thiazoles, reported by Hu et al. [72].
Figure 40.
SAR studies in antibacterial 2-(8-(1-oxoprop-2-en-2-yl)-oxycoumarinyl)-thiazoles, reported by Hu et al. [72].
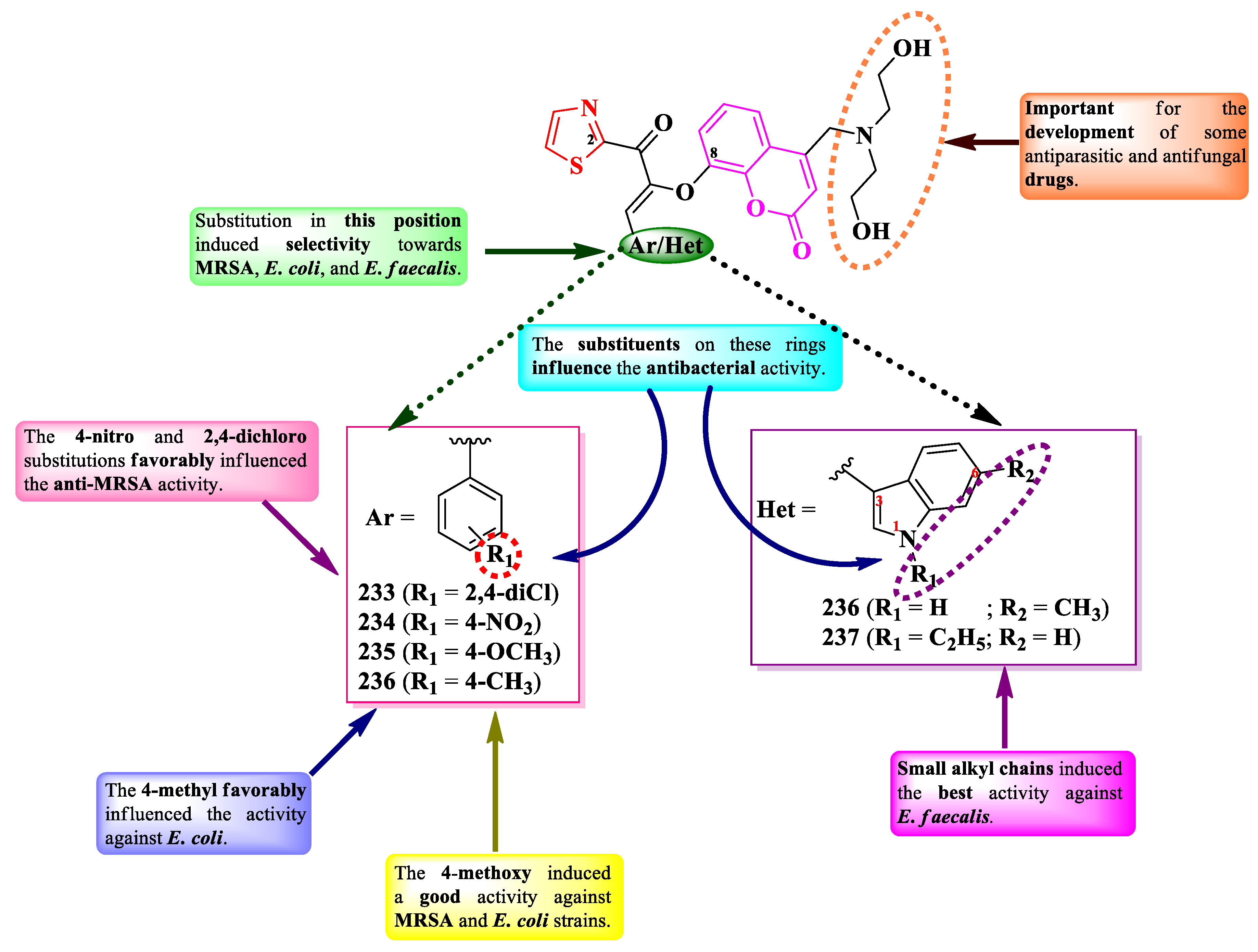
Figure 41.
SAR studies in antistaphylococcal 2-(7-aminoethoxyflavonyl)-thiazoles, reported by Zhao et al. [74].
Figure 41.
SAR studies in antistaphylococcal 2-(7-aminoethoxyflavonyl)-thiazoles, reported by Zhao et al. [74].
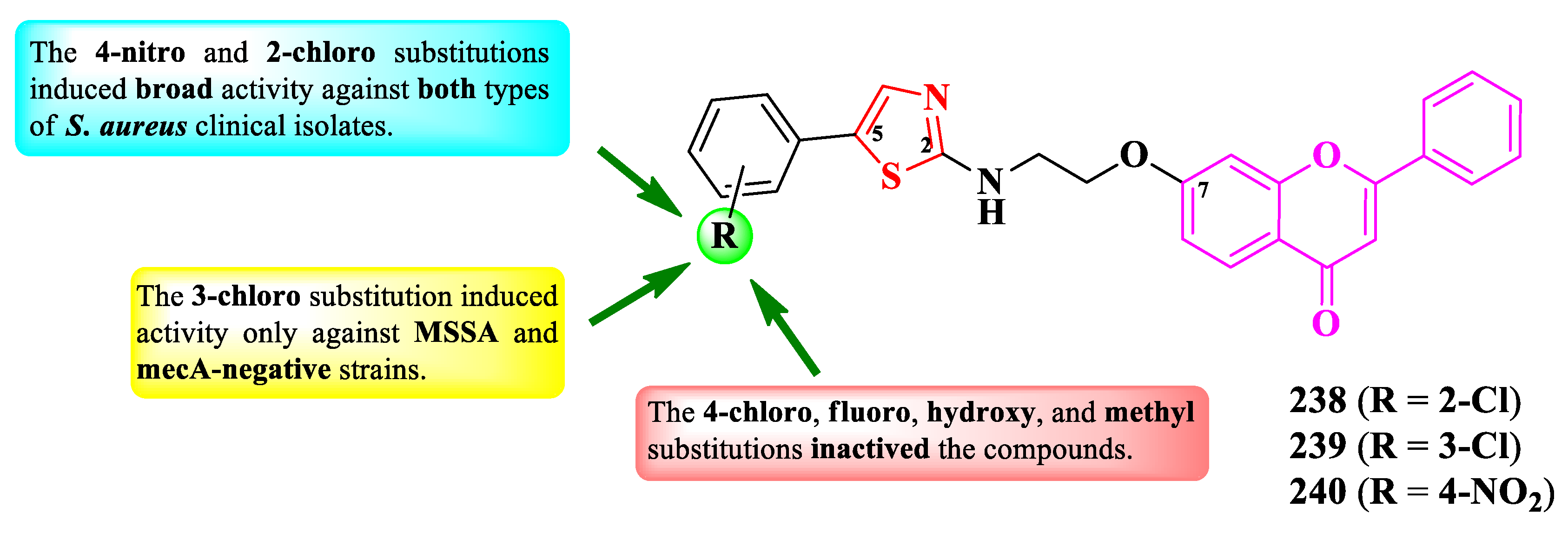
Figure 42.
SAR studies in antimicrobial 2-(quinolin-3-yl-methylenehydrazinyl)-thiazoles, reported by Ammar et al. [76].
Figure 42.
SAR studies in antimicrobial 2-(quinolin-3-yl-methylenehydrazinyl)-thiazoles, reported by Ammar et al. [76].
Figure 43.
SAR studies in antimicrobial 2-(quinolin-3-yl)- and 2-(quinolinon-3-yl)-thiazoles, reported by Litim et al. [77].
Figure 43.
SAR studies in antimicrobial 2-(quinolin-3-yl)- and 2-(quinolinon-3-yl)-thiazoles, reported by Litim et al. [77].
Figure 44.
SAR studies in antibacterial quinazolonyl-thiazoles, reported by Desai et al. [79] and Wang et al. [80].
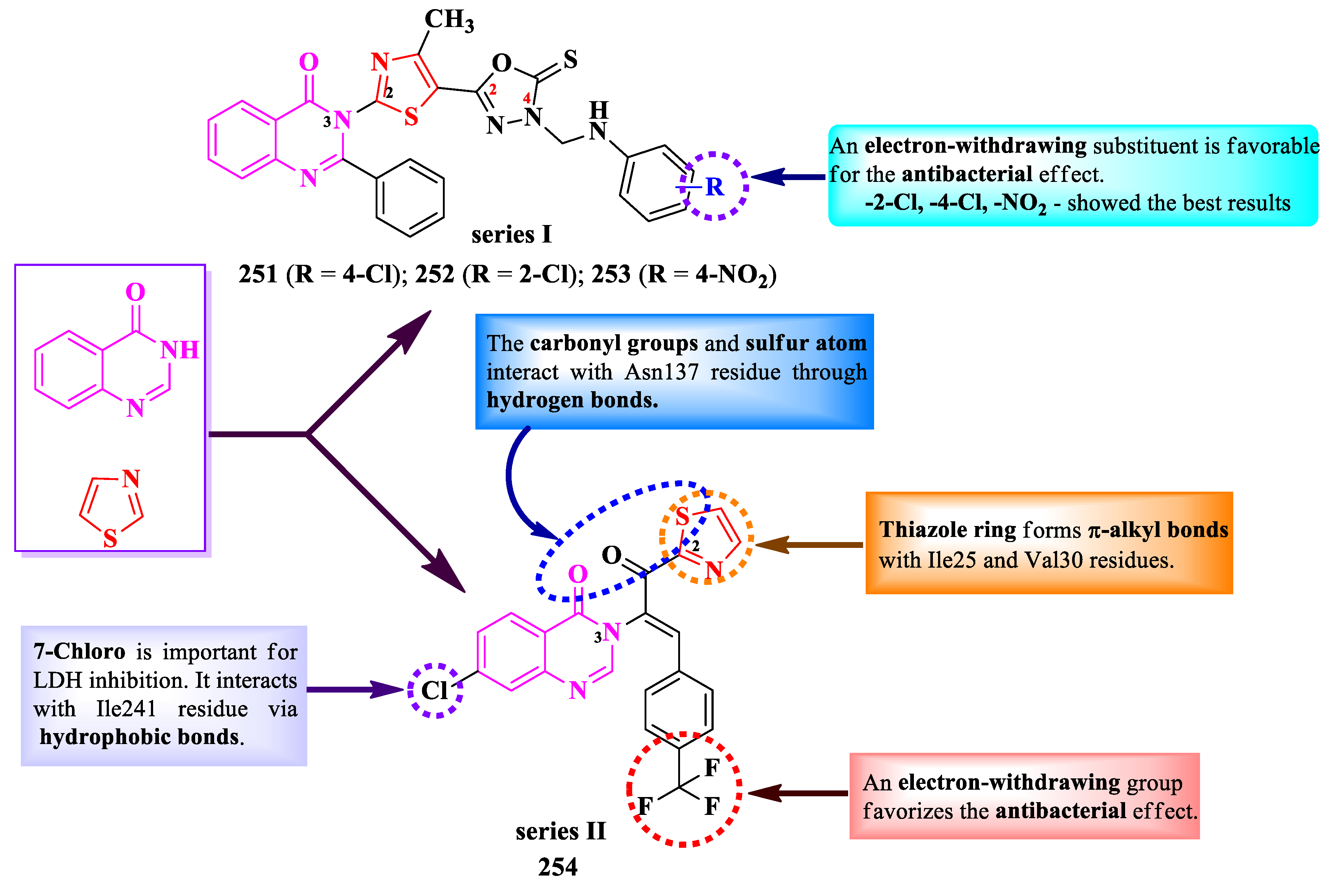
Figure 45.
SAR studies in antimicrobial 2-(quinuclidin-3-ylidene)-hydrazonothiazoles, reported by Łączkowski et al. [81].
Figure 45.
SAR studies in antimicrobial 2-(quinuclidin-3-ylidene)-hydrazonothiazoles, reported by Łączkowski et al. [81].
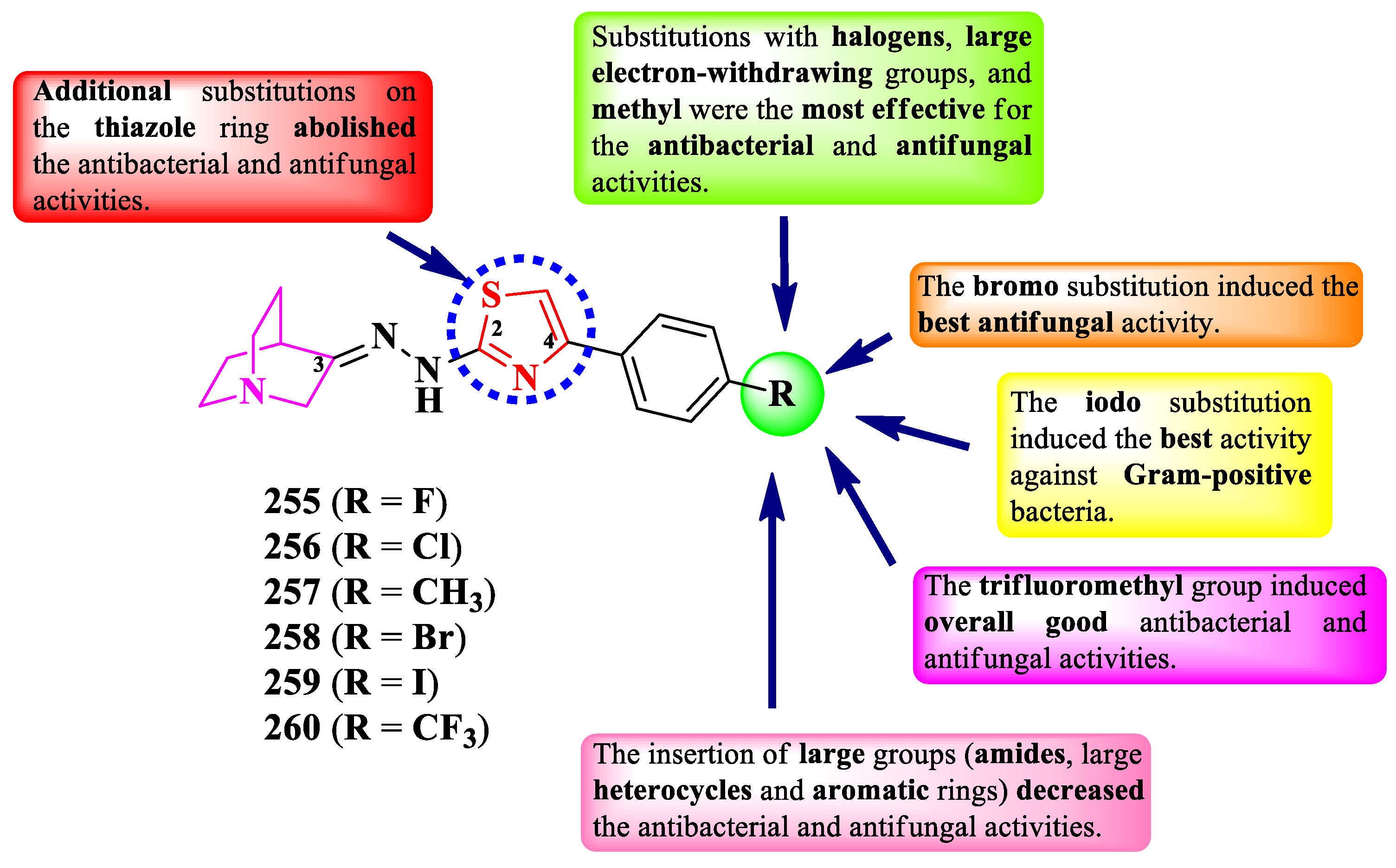
Figure 46.
SAR studies in antibacterial 1,3-thiazole clubbed with polyheterocyclic systems, reported by Abdel-Latif et al. [82].
Figure 46.
SAR studies in antibacterial 1,3-thiazole clubbed with polyheterocyclic systems, reported by Abdel-Latif et al. [82].
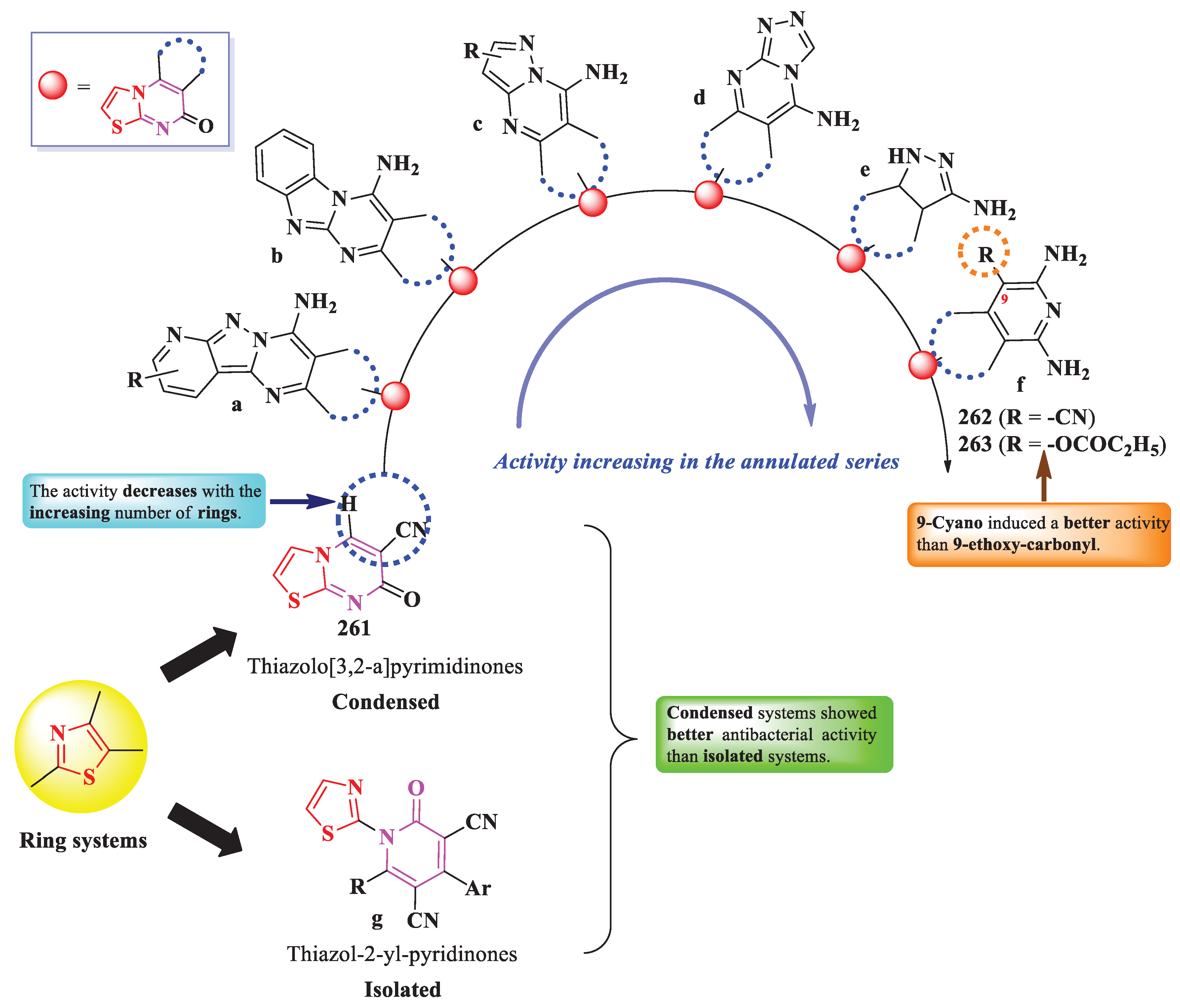
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



