Submitted:
05 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
A fundamentally new synthetic approach to the synthesis of 2-aminopurine has been developed. It consists in the combination of creation of condensed polyazotic heterocyclic tetrazolopyrimidine structure, it’s transformation into triaminopyrimidine and subsequent cyclization into 2-aminopurine. The structure of the obtained compounds was established based on spectral characteristics and intermediate 5 directly by X-ray diffraction analysis.
Keywords:
tetrazololopyrimidines
; purines
; reduction
; nitro group transformations
; condensation
; reconstructive methodology
1. Introduction
The 2-Aminopurine, as structural analogue of guanine, represent a significant group of chemical materials - products of small molecules construction based on modifications of biologically relevant natural compounds [1]. These molecules include purines and their derivatives, such as nucleosides and nucleotides. [2]. The methods of structural modifications of purines and their derivatives are different, including the construction of purines, their glycosidic and/or nucleotide components. Initial studies have been limited to one of three structural modification options: heterocycle, glycoside, and phosphonate. All three techniques exhibited significant efficacy in terms of introducing innovative qualitative approaches for the development of antiviral and antitumor agents to combat related diseases and infections.
The 2-Aminopurine and its derivatives represent an important structural element for the creation of antagonists of nucleic acid monomers and their precursors. In addition, it is an intermediate in the biochemical pathways of purine and nucleic acid metabolism. It is also used as a marker to determine the activity of some enzymes involved in these processes [3].
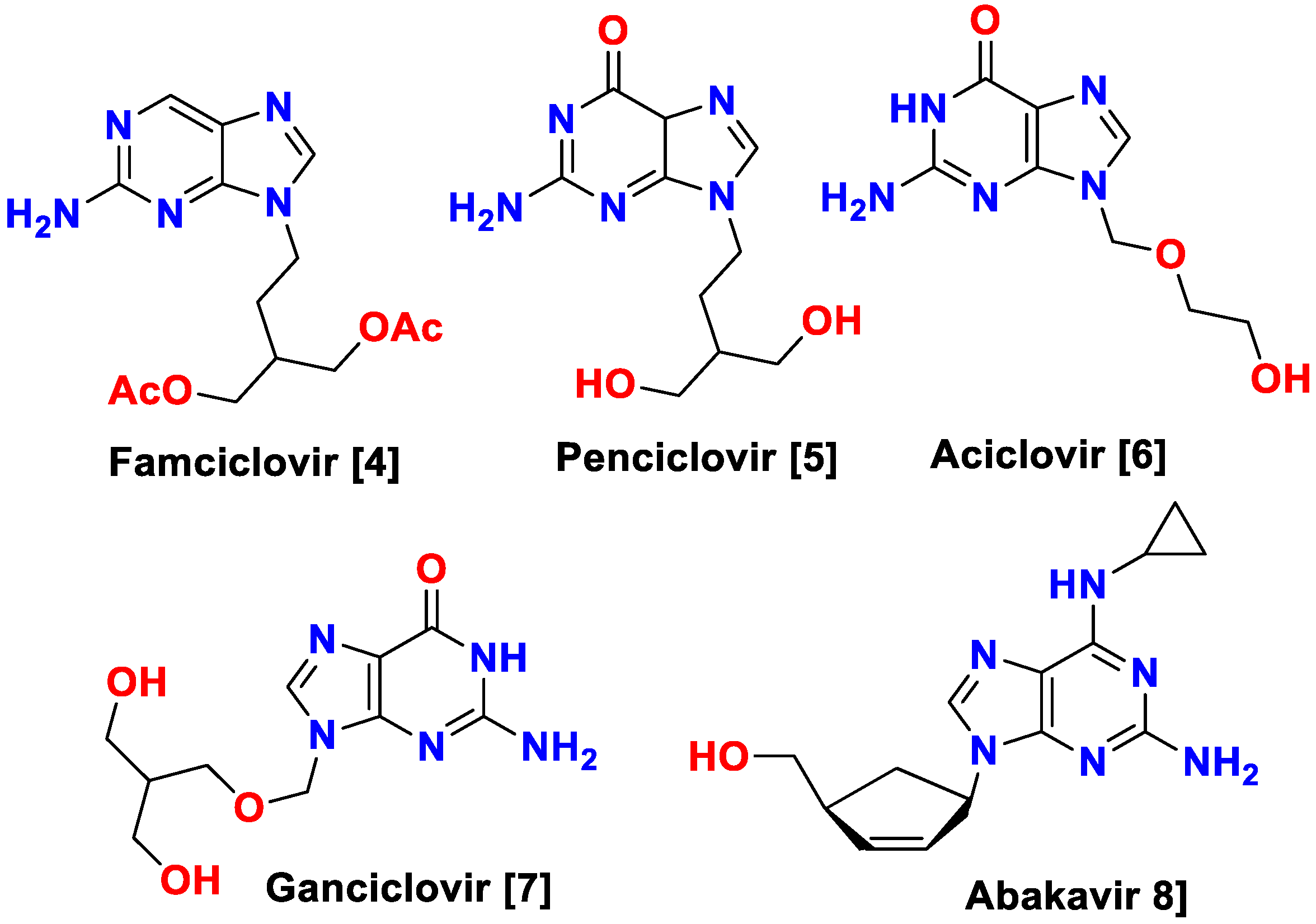
The most significant representatives of this group of antiviral drugs are 2-aminopurines and non-natural nucleosides based on them (Figure 1).
Famciclovir is an antiviral medicine used to treat genital herpes and shingles. It works by preventing the viruses that cause these diseases from multiplying [4]. Penciclovir is an antiviral medication used to treat infections caused by the herpes simplex virus. It works by preventing the virus from multiplying and can be used to treat both genital and oral herpes infections [5]. Acyclovir is an antiviral medicine used to treat infections caused by the herpes simplex virus, such as herpes on the lips and genitals. It works by blocking the reproduction of the virus [6]. Ganciclovir is a medicine used to treat viruses such as cytomegalovirus and herpes simplex virus. It works by stopping viruses from multiplying in the body [7]. Abacavir is a medicine used to treat HIV infection. It works by blocking the activity of an enzyme that is needed for the virus to multiply [8].
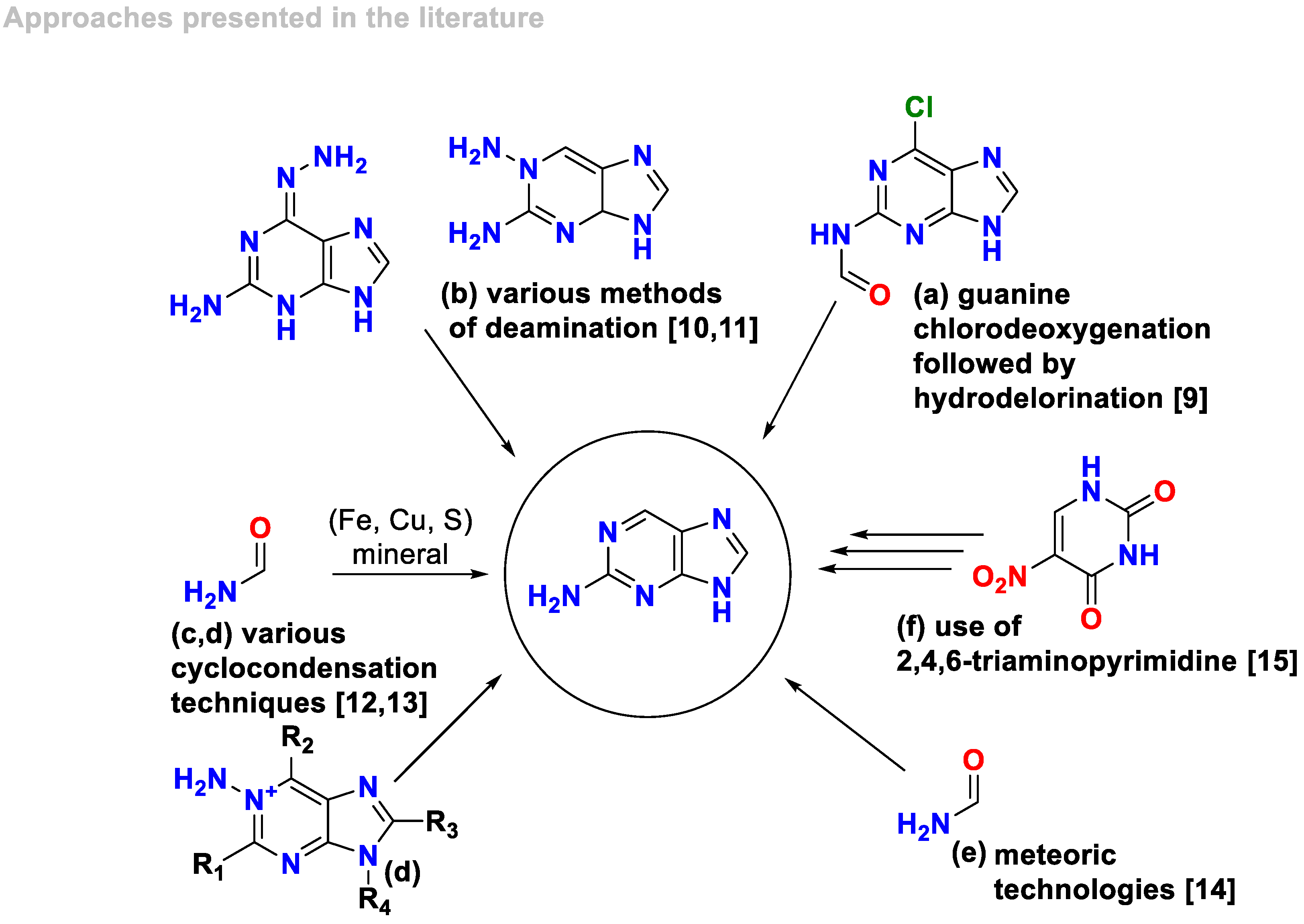
Various methods have been developed for the preparation of 2-aminopurine (Scheme 1). These methods include hydrogenation of 6-chloraminopurine: one recognized method involves the hydrogenation of 6-chloraminopurine [9]; deamination methods: several different deamination methods have been employed for the synthesis of 2-aminopurine [10,11]. These methods involve the removal of an amino group from a precursor compound to yield 2-aminopurine; cyclocondensation methods: various cyclocondensation methods have also been utilized for the synthesis of 2-aminopurine [12,13]. These methods involve the formation of a cyclic compound through the condensation of suitable precursors; meteoric technologies: meteoric technologies have been explored as a potential method for the synthesis of 2-aminopurine [14]. These technologies involve the use of meteoric materials or processes to facilitate the synthesis; use of 2,4,6-triaminopyrimidine: the use of 2,4,6-triaminopyrimidine has been employed as a starting material for the synthesis of 2-aminopurine [15].
It is important to note that these methods represent some of the recognized approaches for the preparation of 2-aminopurine, and there may be additional methods or variations available in the scientific literature.
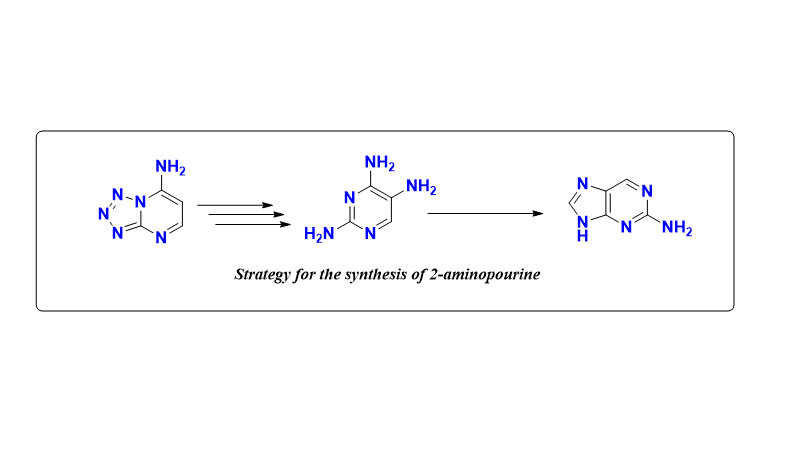
In the present work we propose a new method for the preparation of 2-aminopurine based on 2,4,6-triaminopyrimidine involving aminotetrazole and nitropyrimidine as key reagents.
2. Results
2.1. Synthesis
As a novel approach for the preparation of purine systems, we propose a synthetic strategy based on the synthesis of tetrazolopyrimidines.
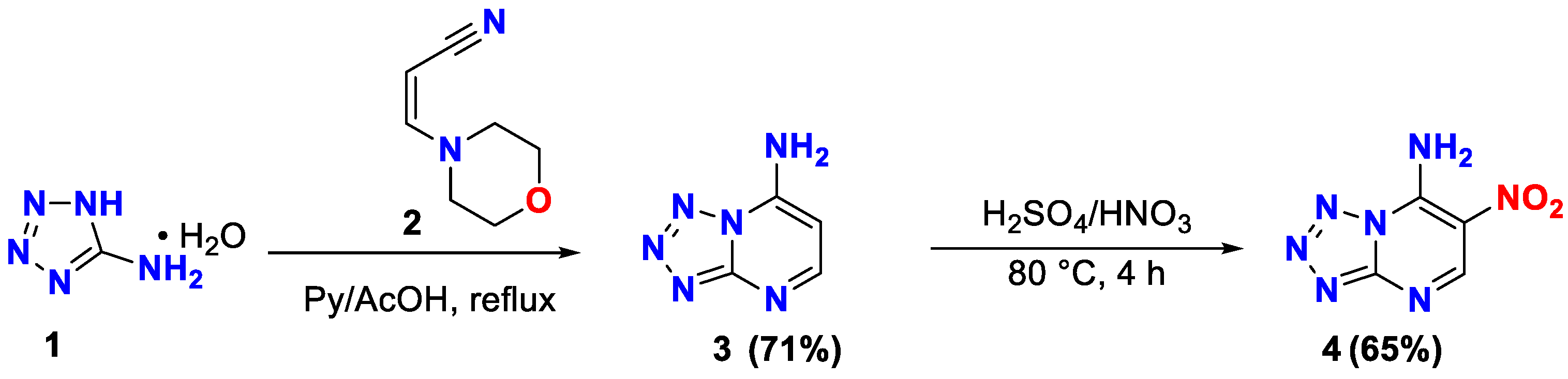
The interaction of 1H-tetrazol-5-amine 1 with morpholinoacrylonitrile 2 in a 1:1 mixture of pyridine and acetic acid leads tetrazolo[1,5-a]pyrimidine-7-amine 3 (71%), which was further converted to 6-nitrotetrazolo[1,5-a]pyrimidine-7-amine 4 (65%) by nitration (Scheme 2).
The structure of compound 4 was established based on 1H, 13C NMR spectroscopy, IR spectroscopy (Figures S1 and S5, see Supplementary Materials) and elemental analysis data. The spectrum 1H NMR of compound 4 shows a single-proton signal in the region of δ 9.01 ppm. corresponding to the resonance of the 5-H atom of the CH group, two single-proton singlet signals corresponding to the protons of the amino group in the regions δ 8.89 ppm and 8.27 ppm.
In the IR spectrum of compound 4, the valence vibration of the azide fragment can be observed, which corresponds to an intense band in the region of 2137 cm−1. This shows that 6-nitrotetrazolo[1,5-a]pyrimidine-7-amine 4 is in azide form in solid state. The absorption band in the region of 3436 cm-1 corresponds of the amino group. In the region of 1633 cm−1, 1329 cm-1 are the absorption band of nitro group.
It’s known that the nitro group can act both as a leaving group and as a cryptoform of the amino group [16]. Thus, in the case for 6-nitroazolo[1,5-a]pyrimidines containing an amino group at the 5 or 7 position, it appears possible to complete the heterocyclic system to the corresponding purine. Therefore, the next stage of the study was the development of methods for the obtaining of pyrimidine-2,4,5-triamine 6. There is a known method the hydrogenolysis of the azo bond of compounds in the presence of Pd/C in the atmosphere of hydrogen at the pressure of 3-5 bar, which is also productive, but high pressures are applied [17].
One of the synthetic strategies for obtaining of the 2-aminopurine is the synthesis of a tetrazolo[1,5-a]pyrimidine-6,7-diamine. Initially, attempt of selective reduction of the nitro group was carried out. The following reducing agents were used for this purpose: attempts to use zinc in acetic acid and in ethanol with ammonia did not lead to the formation of the desired intermediate. The typical reducing agent iron and hydrochloric acid as solvent failed to isolate 5-nitropyrimidine-2,4-diamine 5. Sodium dithionite, which played the role of reducing agent in the next step - to obtain triaminopyrimidine, was ineffective towards the selective reduction of tetrazole. In addition, H2/Pd, which is often used for the reduction of nitro groups, failed to yield an intermediate intermediate. In some cases nothing happened, while in others the tetrazole was reduced but the nitro group remained unreduced.
The construction of 5-nitropyrimidine-2,4-diamine 5 based on aminotetrazole is novel in the synthesis of nitrogenous heterocycles [18]. Thus, we have developed the following approach for the synthesis of pyrimidine-2,4,5-triamine 6 - reduction of nitrotetrazolo[1,5-a]pyrimidin-7-amine 4.
It turned out that sodium sulfide nonahydrate aqueous in alcoholic medium undergoes reduction of the tetrazole fragment to the amino group, which leads to the preparation of 5-nitropyrimidine-2,4-diamine 5 (32%) (Scheme 3. Route a).
A selection of conditions was required to make the yields of the compounds preparative. The conditions were selected to make the yields of the target product preparative. The results obtained clearly demonstrated that the best activating reagent was triphenylphosphine (Scheme 3. Route b) when used in an amount of 1.0 equivalent in acetic acid for 2 h, leads 5-nitropyrimidine-2,4-diamine 5 (77%) (Table 1, entry 4).
The 1H spectrum of 5-nitropyrimidine-2,4-diamine 5 shows a single-proton signal in the region of 8.84 ppm corresponding to the resonance of the 6-H-atom of the pyrimidine fragment, and two two-proton doublets corresponding to the protons of the amino group (δ = 7.98 ppm; 7.45 ppm) (Figures S3, see Supplementary Materials). In addition, the structure of 5-nitropyrimidine-2,4-diamine 5 was finally determined by X-ray diffraction analysis (Figure 2).
Nitro compounds are important building blocks for further important transformations [19].
In the next step, we attempted the reduction of the nitro group to obtain the target pyrimidine-2,4,5-triamine 6. Initially iron in acidic conditions, then tin in hydrochloric acid, after that H2/Pd. For the reduction of the nitro group, sodium dithionite proved to be a suitable reagent. As a result, pyrimidine-2,4,5-triamine 6 was obtained in 59% yield (Table 2, entry 1).
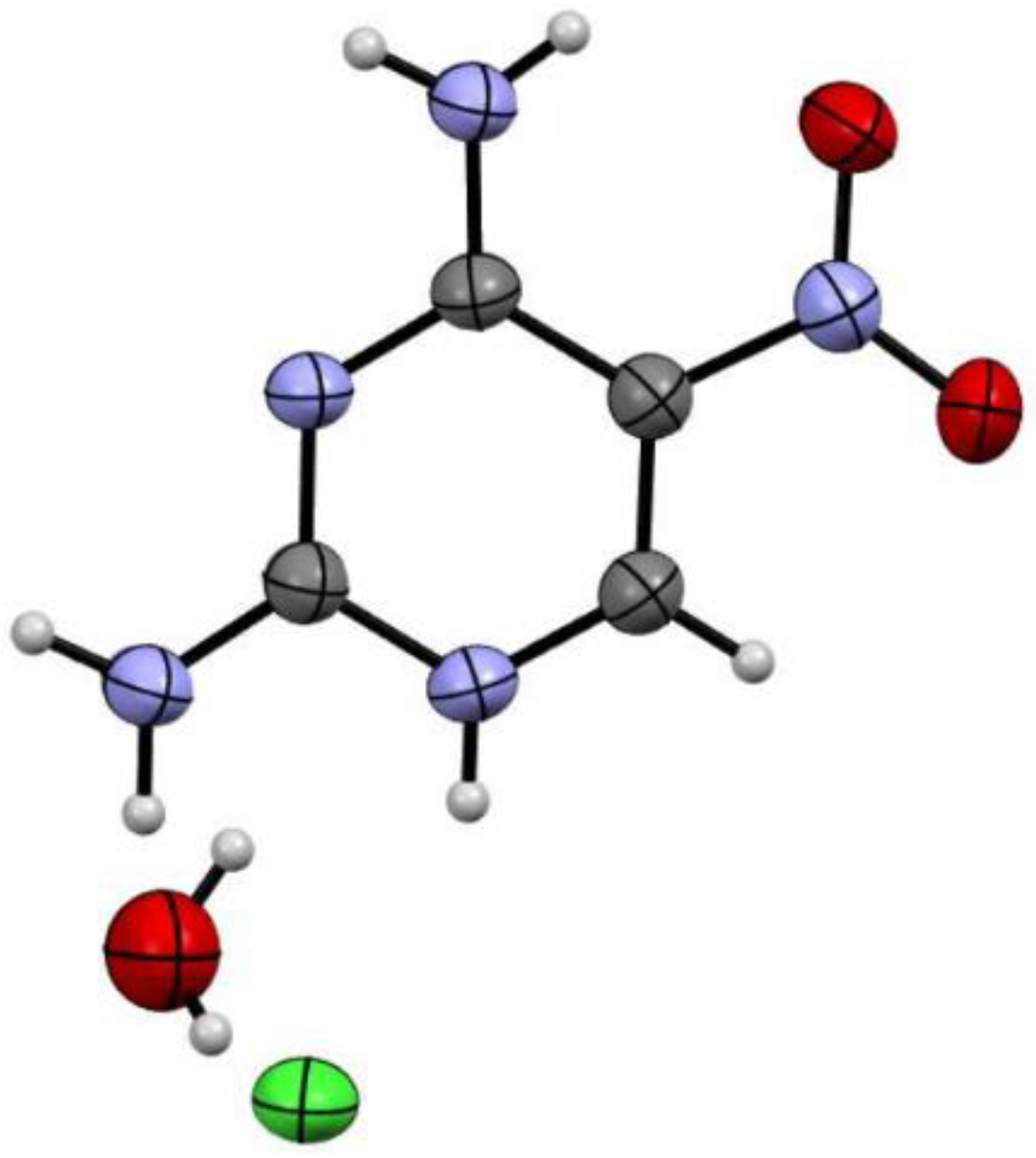
The final step of 2-aminopurine synthesis on the is the construction of the imidazole cycle. Despite the simplicity of the reaction, different derivatives of vicinal diamines, may require different conditions [9,10,11,12,13,14].
Traube synthesis is a classic method to purines through annulation of the imidazole fragment into ortho-diaminopyrimidines [20]. This method was not applicable to us. For the final cyclisation of pyrimidine-2,4,5-triamine 6 to 2-aminopurine 8 we used different formylation reagents (Table 3). The use triethylorthoformate, acetic acid as catalyst and N-formylmorpholine as formylation agent in formic acid didn’t lead to the preparation of the target product 8. The results obtained clearly demonstrated that the best reagents were triethylorthoformate and acetic anhydride (Scheme 4), 2-aminopurine 8 was obtained in yield 41%.
2.2. Crystallography
According XRD data, two independent molecules of the compound are crystallized with a molecule H2O in the centrosymmetric space group of the triclinic system. In the result, the structurally independent unit C4H7ClN5O2.5 was used for all calculation. The geometry of the independent heterocyclic molecules has an insignificant distinguishes, however, all bond distances and angles are near to expectations. Both molecules are planar, with insignificant dihedral angles between NO2-group and plane of the heterocycle. The general geometry of the molecules was shown on the Figure 3. The crystal packing is ordered by the strong system of the H-bonds (Table S1, Supplementary Materials).
3. Materials and Methods
3.1. Chemical Experiment
Commercial reagents were obtained from Sigma-Aldrich, Acros Organics, or Alfa Aesar and used without any preprocessing. All workup and purification procedures were carried out using analytical-grade solvents. One-dimensional 1H- and 13C-NMR spectra were acquired on a Bruker DRX-400 instrument (Karlsruhe, Germany) (400 and 101 MHz, respectively) or a Bruker Avance NEO 600 instrument (151 MHz), utilizing DMSO-d6 as solvents and an external reference, respectively. Chemical shifts are expressed in δ (parts per million, ppm) values, and coupling constants are expressed in hertz (Hz). The following abbreviations are used for the multiplicity of NMR signals: s, singlet; d, doublet. IR spectra were recorded on a Bruker α spectrometer equipped with a ZnSe ATR accessory. Elemental analysis was performed on a PerkinElmer PE 2400 elemental analyzer (Waltham, MA, USA). Melting points were determined on a Stuart SMP3 (Staffordshire, UK) and are uncorrected. The monitoring of the reaction progress was performed using TLC on Sorbfil plates (Imid LTD, Russia, Krasnodar) (the eluent is EtOAc).
Procedure for the synthesis of tetrazolo[1,5-a]pyrimidine-7-amine (3). To a solution of 1H-tetrazol-5-amine 1 (0.01 mol, 1 eq.) in an equimolar mixture of pyridine (10 mL)/acetic acid (14 mL) was added morpholineacrylonitrile 2 (0.01 mol, 1 eq.). The resulting mixture was stirred at 135 °C for 4 h. The resulting precipitate is filtered off and washed with 20 mL EtOH and dried in air. Beige powder (yield 71%). m. p. 274-276 °С. FT-IR (neat) νmax (cm-1): 3108, 3287. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 6.58 (1H, d, J = 7.4 Hz, 5-H), 7.76 (2H, s, NH2), 8.92 (1H, d, J = 7.4, 5-H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm) 104.3, 132.8, 155.5, 162.2 Calcd for C4H4N6: C 35.30, H 2.96, N, 61.74; found: C 35.37, H 2.91, N 61.72.
Procedure for the synthesis of 6-nitrotetetrazolo[1,5-a]pyrimidine-7-amine (4).
To concentrated nitric acid 3.75 mL (0.09 mol) cooled to 0-5 °C concentrated sulphuric acid 28.94 mL (0.54 mol) was added). To the resulting mixture tetrazolo[1,5-a]pyrimidine-7-amine 3 was added in small portions. The reaction mixture was heated for 4 h at 80 °C, then cooled to room temperature, poured on ice and neutralized with aqueous ammonia solution (25%) (80 mL) to pH=7. The resulting precipitate was filtered off, washed thoroughly with water (40 mL). White powder (yield 65%). m. p. 233-235 °С. FT-IR (neat) νmax (cm-1): 1329, 1633, 2137, 3436. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 8.27 (1H, s, NH2), 8.89 (1H, s, NH2), 9.01 (1H, s, 5-H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm) 124.3, 157.3, 157.9, 163.8 Calcd for C4H3N7O2: C 26.53, H 1.67, N, 54.14; found: C 26.44, H 1.75, N 54.09.
Procedure for the synthesis of 5-nitropyrimidine-2,4-diamine (5a). To 6-nitrotetrazolo[1,5-a]pyrimidin-7-amine 4 (4,9 mmol) was added 10 mL of ethyl alcohol. Sodium sulfide (4,9 mmol) was added to the resulting mixture in small portions at 60 °C and stirred, then cooled at room temperature. The resulting precipitate was filtered off, washed thoroughly with water (20 mL).
Procedure for the synthesis of 5-nitropyrimidine-2,4-diamine (5b). To acetic acid (10 mL) was added of 6-nitrotetrazolo[1,5-a]pyrimidin-7-amine 4 (2,8 mmol). The reaction mixture was stirred at 130 °C, upon reaching this temperature, triphenylphosphine (2,8 mmol) was added to the mixture in small portions and endure for one hour. Then 0.1 mL of water was added, cooled after one hour, the obtained precipitate was filtered, washed with 20 mL of ethyl alcohol. Pale yellow powder (yield 77%). m. p. > 300 °С. FT-IR (neat) νmax (cm-1): 1375, 1625, 3417. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 7.46 (2H, d, J = 56 Hz, 5-H), 7.97 (2H, d, J = 32 Hz, 1-H), 8.84 (1H, s, 6-H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm) 120.1, 157.9, 158.5, 163.9 Calcd for C4H5N5O2: C 30.95, H 3.22, N, 45.20; found: C 30.98, H 3.31, N 45.07.
Procedure for the synthesis of pyrimidine-2,4,5-triamine (6). To 5-nitropyrimidine-2,4-diamine 5 (3,6 mmol) was added 10 mL of water, stirred at 80 °C, sodium dithionite (7,2 mmol) was added for 3-5 minutes, then stirred at 60 °C, after which sodium carbonate (0.018 mol) was added. The reaction mixture was evaporated at a rotary evaporator, crushed and extracted with isopropyl alcohol (30 mL). The resulting precipitate was filtered off (the procedure was repeated 3 times in total). The filtrate was evaporated at the rotary evaporator and dried in air. White powder (yield 59%). m. p. 248-250 °С. FT-IR (neat) νmax (cm-1): 1329, 1359, 1563, 3271. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 3.59 (1H, s, NH2), 4.95 (1H, s, NH2), 5.86 (1H, s, NH2), 7.22 (1H, s, 6-H). 13C-NMR (151 MHz, DMF-d7) δ (ppm) 118.7, 140.9, 156.6, 158.1 Calcd for C4H7N5: C 38.39, H 5.64, N, 55.97; found: C 38.31, H 5.60, N 56.09.
Procedure for the synthesis of 2-aminopurine (8). To pyrimidine-2,4,5-triamine 6 (0,4 mmol) was added triethylorthoformate 3.0 mL (0.018 mol) and acetic anhydride 1.0 mL (0.01 mol) boiled at 145 °C for 1.5 h, the reaction mixture was cooled and evaporated. A solution of NaOH (0.02 mol) in 5 mL of water was added, boiled for 10 min, the resulting solution was cooled and hydrochloric acid 35% 1.6 mL (0.02 mol) was added, the pH was brought to neutral with CH3COONa 4M 10 mL. The resulting solution was evaporated at the evaporator, then 15 mL acetone was added, the precipitate formed was filtered off, the filtrate was evaporated at the evaporator. The product was purified by flash chromatography with a mixture of CHCl3 and MeOH (8:2). Pale yellow powder (yield 41%). m. p. 280-282 °С. FT-IR (neat) νmax (cm-1): 1614, 1663, 3215. 1H-NMR (400 MHz, DMSO-d6) δ (ppm) 6.29 (2H, s, NH2), 8.03 (1H, s, 8-H), 8.56 (1H, s, 6-H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm) 125.4, 144.1, 156.6, 158.4 Calcd for C5H5N5: C 44.44, H 3.73, N, 51.83; found: C 44.32, H 3.82, N 51.86.
3.2. Crystallography Experiment
The XRD analyses were carried out using equipment of the Center for Joint Use “Spectroscopy and Analysis of Organic Compounds” at the Postovsky Institute of Organic Synthesis of the Russian Academy of Sciences (Ural Branch). The experiment was accomplished on the automated X-ray diffractometer «Xcalibur 3» with CCD detector on standard procedure (МoKα-irradiation, graphite monochromator, ω-scans with 1o step at Т= 295(2) К). Empirical absorption correction was applied. The solution and refinement of the structures were accomplished with using Olex program package [21]. The structures were solved by method of the intrinsic phases in ShelXT program and refined by ShelXL by full-matrix least-squared method for non-hydrogen atoms [22]. The H-atoms at C-H bonds were placed in the calculated positions, H-atoms of the O-H and N-H bonds were solved by direct method and were refined independently in isotropic approximation.
Crystal Data for C4H7ClN5O2.5 (M = 200.60 g/mol): triclinic, space group P-1, a = 5.5123(3) Å, b = 9.8766(5) Å, c = 15.7380(10) Å, α = 73.518(5)°, β = 88.104(5)°, γ = 86.787(4)°, V = 820.19(8) Å3, Z = 4, T = 295(2) K, μ(MoKα) = 0.443 mm-1, Dcalc = 1.624 g/cm3, 8459 reflections measured (7.406° ≤ 2Θ ≤ 60.94°), 4484 unique [Rint = 0.0451, Rsigma = 0.0649] which were used in all calculations. The final R1 = 0.0600, wR2 = 0.1431 (I > 2σ(I)) and R1 = 0.0955, wR2 = 0.1835 (all data). GooF = 1.041. Largest diff. peak/hole 0.41/-0.50.
The XRD data were deposited in the Cambridge Structural Database with number CCDC 2308999. This data can be requested free of charge wia www.ccdc.cam.ac.uk.
4. Conclusions
A new method for the preparation of 2-aminopurine as an important compound has been established and an important conceptual synthetic principle has been utilised. The method includes cyclisation, nitro-formation (functionalisation), and reduction with destruction of the initial heterocycle. Targeted final cyclisation. We propose this synthetic sequence as a reconstructive methodology in the synthesis of heterocycles.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figures S1–S4: 1H- and 13C-NMR spectra of compounds 3-6; Figures S5–S8 IR spectra of compounds 3-6.
Author Contributions
Synthesis, A.O.N., V.V.F. and E.B.G.; methodology, V.V.F., E.N.U. and V.L.R.; writing—original draft preparation, A.O.N., V.V.F. and S.V.A.; writing—review and editing, A.O.N., E.N.U., S.V.A. and V.L.R.; visualization, V.A.I. and D.N.L; crystallographic investigation, P.A.S.; supervision, E.N.U., V.V.F. and V.L.R.; project administration, V.L.R. All authors have read and agreed to the published version of the manuscript.
Funding
The research was financially supported by the Russian Science Foundation (Project No. 23-23-00642).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The team of authors would like to thank the Laboratory for Comprehensive Research and Expert Evaluation of Organic Materials under the direction of O.S. Eltsov.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 3-6 and 8 are available from the authors.
References
- Nagatsugi, F.; Uemura, K.; Nakashima, S.; Maeda, M.; Sasaki, S. 2-Aminopurine Derivatives with C6-Substituted Olefin as Novel Cross-Linking Agents and the Synthesis of the Corresponding β-Phosphoramidite Precursors. Tetrahedron 1997, 53, 3035–3044. [Google Scholar] [CrossRef]
- Ts’o, P.O.P. Basic Principles in Nucleic Acid Chemistry V1; Elsevier Science: Oxford, UK, 1974; ISBN 9780323144001. [Google Scholar]
- Jean, J.M.; Hall, K.B. 2-Aminopurine Fluorescence Quenching and Lifetimes: Role of Base Stacking. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.M.; Wagstaff, A.J. Famciclovir: A Review of Its Pharmacological Properties and Therapeutic Efficacy in Herpesvirus Infections. Drugs 1995, 50, 396–415. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.R.; Safrin, S.; Kern, E.R. Penciclovir: A Review of Its Spectrum of Activity, Selectivity, and Cross-Resistance Pattern. Antivir Chem Chemother 1993, 4, 3–11. [Google Scholar] [CrossRef]
- Wagstaff, A.J.; Faulds, D.; Goa, K.L. Aciclovir: A Reappraisal of Its Antiviral Activity, Pharmacokinetic Properties and Therapeutic Efficacy. Drugs 1994, 47, 153–205. [Google Scholar] [CrossRef] [PubMed]
- Matthews, T.; Boehme, R. Antiviral Activity and Mechanism of Action of Ganciclovir. Clinical Infectious Diseases 1988, 10, S490–S494. [Google Scholar] [CrossRef] [PubMed]
- Melroy, J.; Nair, V. The antiviral activity, mechanism of action, clinical significance and resistance of abacavir in the treatment of pediatric AIDS. Curr Pharm Des. 2005, 11, 3847–3852. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Shi, C. ; Yang. S.; Wang, F. Patent CN116063306, 5 May 2023. [Google Scholar]
- Guo, H.; Rao, W.; Niu, H.; Wang, D.; Qu, G. Dehalogenations of 6-Chloropurine, 6-Chloropurine Nucleosides and 6-Bromopurine Nucleosides under Microwave Irradiation in Water. Chinese J. Chem. 2010, 18, 361–364. [Google Scholar]
- Jongejan, H.; Kos, N.J.; Van Der Plas, H.C. A 15 N Study of the Ammonia-induced Deamination of 1-aminopurinium Salts. Recl. Trav. Chim. Pays-Bas 1986, 105, 337–340. [Google Scholar] [CrossRef]
- Saladino, R.; Neri, V.; Crestini, C.; Costanzo, G.; Graciotti, M.; Di Mauro, E. Synthesis and Degradation of Nucleic Acid Components by Formamide and Iron Sulfur Minerals. J. Am. Chem. Soc. 2008, 130, 15512–15518. [Google Scholar] [CrossRef] [PubMed]
- Kos, N.J.; Jongejan, H.; Van Der Plas, H.C. Deamination, Involving Ring Opening, in Reactions of 1-Aminopurinium Mesitylenesulfonates with Methanolic Amonia. Tetrahedron 1987, 43, 4841–4848. [Google Scholar] [CrossRef]
- Saladino, R.; Botta, G.; Delfino, M.; Di Mauro, E. Meteorites as Catalysts for Prebiotic Chemistry. Chemistry A European J 2013, 19, 16916–16922. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, Z.; Bakar, M.; Din, E.; Rani, N.; Salleg, N.; Leong, G.; Ling, L.; Aiyub, Z. Synthesis and fluorescence characteristic of 2-substituted and 6-substituted purines. Malaysian J. Sci. 2008, 12, 251–256. [Google Scholar]
- Lyapustin, D.N.; Fedotov, V.V.; Ulomsky, E.N.; Rusinov, V.L.; Chupakhin, O.N. Recent Advances in the Chemistry of Two-Carbon Nitro-Containing Synthetic Equivalents. RUSS CHEM REV 2023, 92, RCR5077. [Google Scholar] [CrossRef]
- Gazizov, D.A.; Fedotov, V.V.; Chistyakov, K.A.; Gorbunov, E.B.; Rusinov, G.L.; Charushin, V.N. Access to Azolopyrimidine-6,7-Diamines as a Valuable “Building-Blocks” to Develop New Fused Heteroaromatic Systems. Tetrahedron 2021, 89, 132172. [Google Scholar] [CrossRef]
- Zubarev, V.Yu.; Ostrovskii, V.A. Methods for the Synthesis of Mono- and Polynuclear NH-Tetrazoles. (Review). Chem Heterocycl Compd 2000, 36, 759–774. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Aksenov, D.A.; Aksenov, N.A.; Skomorokhov, A.A.; Aleksandrova, E.V.; Rubin, M. Preparation of Spiro[Indole-3,5′-Isoxazoles] via Grignard Conjugate Addition/Spirocyclization Sequence. RSC Adv. 2021, 11, 1783–1793. [Google Scholar] [CrossRef] [PubMed]
- Hannser, A.; Stumer, C. Organic Syntheses Based on Name Reactions; Publisher: Pergamon, The Netherlands, 2002; p. 443. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2 : A Complete Structure Solution, Refinement and Analysis Program. J Appl Crystallogr 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr C Struct Chem 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Aminopurine-based medicines.
Scheme 1.
Literary approaches to 2-aminopurine.
Scheme 2.
Preparation of 6-nitrotetrazolo[1,5-a]-pyrimidine-7 amine 4.
Scheme 3.
Synthesis of pyrimidine-2,4,5-triamine 6.
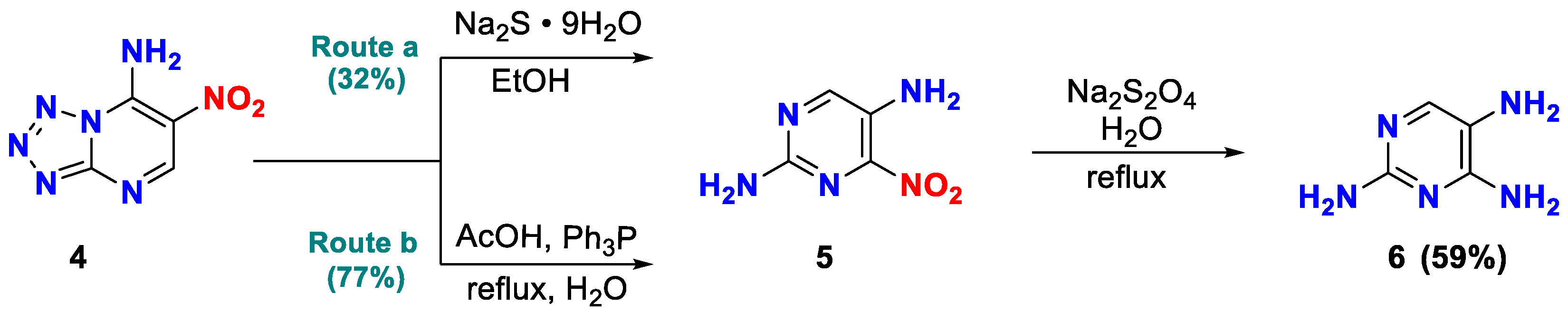
Figure 2.
X-ray diffraction analysis of compound 5-nitropyrimidine-2,4-diamine 5.
Scheme 4.
Synthesis of 2-aminopurine 8.
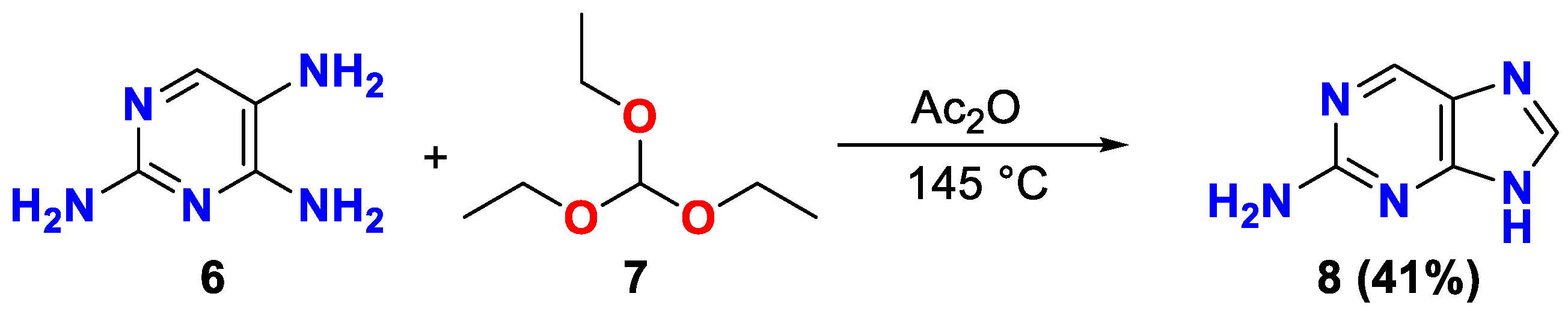
Figure 3.
The compound in the thermal ellipsoid of the 50% probability level.

Table 1.
Optimization for preparation to 5-nitropyrimidine-2,4-diamine 5 1.
| No. | Solvent 2 | Reducing Agent | X, equiv | Reaction Condition 3 |
Yield, % 5 |
|---|---|---|---|---|---|
| entry 1 | EtOH | Na2S•9H2O | 1.0 | 60 °C, 0.1 h | 32% |
| entry 2 | HCl | Fe | 8.0 | 60 °C, 2 h | - |
| entry 3 | AcOH | Zn | 10.0 | reflux, 2 h | - |
| entry 4 | AcOH | Ph3P | 1.0 | reflux, 2 h | 77% |
| entry 5 | AcOH | Cu | 1.0 | reflux, 0.2 h | - |
| entry 6 | THF | Ph3P | 1.0 | reflux, 2 h | - |
| entry 7 | H2O | Na2S2O4 | 8.0 | reflux, 0.2 h | - |
| entry 8 | NH4OH, EtOH | Zn | 4.0 | 70 °C, 1 h | - |
| entry 9 | EtOH | 6 H2/Pd | - | 4 50 °C, 4 h | - |
1 Reaction conditions: 4 (4,9 mmol); 2 amount of solvent—10 mL; 3 conventional heating with an oil bath; 4 autoclave; 5 isolated yield; and 6 10% Pd/C (5 wt %).
Table 2.
Optimization for preparation to pyrimidine-2,4,5-triamine 6 1.
| No. | Solvent 2 | Reducing Agent |
X, equiv | Reaction Condition 3 |
Yield, % 5 |
|---|---|---|---|---|---|
| entry 1 | H2O | Na2S2O4 | 6.0 | reflux, 0.2 h | 59 |
| entry 2 | TEOF, AcOH | Fe | 10.0 | 130 °C, 5 h | - |
| entry 3 | HСl | Sn | 2.0 | 110 °C, 1 h | - |
| entry 4 | DMF | 6 H2/Pd | 0.05 | 4 100 °C, 4 h | - |
1 Reaction conditions: 5 (4,9 mmol); 2 amount of solvent—10 mL; 3 conventional heating with an oil bath; 4 autoclave; 5 isolated yield; and 6 10% Pd/C (5 wt %).
Table 3.
Optimization for preparation to 2-aminopurine 8 1.
| No. | Solvent 2 | Formylation agent |
X, equiv | Reaction Condition 3 |
Yield, % 4 |
|---|---|---|---|---|---|
| entry 1 | - | HCOOH | 45.0 | reflux, 3 h | - |
| entry 2 | - | TEOF | 45.0 | reflux, 3h | - |
| entry 3 | AcOH | TEOF | 3.0 | reflux, 3 h | - |
| entry 4 | AcOH | TEOF | 5.0 | reflux, 3 h | - |
| entry 5 | N-Formylmorpholine | HCOOH | 6.5 | reflux, 3 h | - |
| entry 6 | Ac2O | TEOF | 25.0 | reflux, 2 h | 41 |
1 Reaction conditions: 6 (0,4 mmol); 2 amount of solvent—3 mL; 3 conventional heating with an oil bath; and 4 isolated yield.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



