
Submitted:
05 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
How crucial is big data in contemporary molecular design? In this publication we elucidate fundamental concepts and terminology in this field, critically addressing overlooked issues. We thoroughly examine the size, accessibility, quality, and structural aspects of big data alongside the primary methodologies employed for their analysis. Within chemical compounds, properties and descriptors represent two distinct data types, forming the basis for categorizing molecular big data. The primary objective of chemistry is property production, which means we are searching for novel drugs or materials rather than chemical compounds, and big data is the central issue of this philosophy. The increasing availability of data in computer-aided technology propels advancements in artificial intelligence (AI), machine learning (ML), and deep learning (DL). Accordingly, a broad chemical audience must comprehend these methods to understand data-centered chemistry. Thus, we aim to systemize big data issues through a simple illustrative framework with fundamental descriptor categories: coding, computer-generated descriptors, and property correlates. Although we employ computer-generated descriptors as big data for predictions, the measured data are irreplaceable for achieving high-quality and reliable outcomes and controlling molecular effects. The scarcity of property data remains a significant hurdle limiting comprehensive studies on the structure-property relationships within big data. Accordingly, guided by pragmatics not-so-big data is an option for drug design. We presented also a brief review of the recent big data literature.
Keywords:
big data
; not-so-big data
; molecular design
; drug design
; machine learning
; artificial intelligence
; descriptor
; computer-generated descriptors
; property
; regression
1. Introduction
The efficacy of drug design has long been a subject of debate, and doubts persist about the effectiveness of small molecule design. The Eroom Law, indicating that returns on investments in pharmaceutical drug design tend to decrease rather than increase, is widely recognized [1]. Although the emerging evidence that this trend may gradually reverse, we are still searching for novel, more efficient design methods targeting small molecule drugs. On the other hand, novel data science methods have revolutionized computer-aided research, technology and everyday life of the current society. Recent advancements have exhibited remarkable success in particular in face recognition (Deep-Face), text generation, exemplified by chat GPT, or in chess-playing machines like Deep Blue. These advancements hinge upon extensive datasets, prompting a crucial inquiry: What underlies the relatively modest efficiency in harnessing big data for drug design? To unravel this, we must first grasp the essence of big data and gauge its extent within drug design and data-centered chemistry [2,3]. As an integral facet of medicine and healthcare, molecular design operates within one of the most cutting-edge sectors of the current economy [4]. This field provides us with expansive datasets encompassing (i) patient diagnostics, (ii) treatment evaluation, (iii) wellness management, (iv) fraud and abuses, and (v) public health management. Can these multifaceted directions serve as guiding beacons for our endeavors at the core of molecular design [5].
In this publication, we undertake a comprehensive analysis of the challenges associated with harnessing large datasets to design small organic molecules for use as novel drugs or materials. We delve into the issues surrounding data availability and quality, as well as the specific types of algorithms employed in data processing. Moreover, we reviewed the basic glossary of drug design, showing that it needs to be brushed up and upgraded to modern data science standards to be understandable within the chemical and computer scientist audience, especially regarding data to big data expansion.
2. The core essence of molecular design: Matching descriptors to properties
Chemistry is a soft science where definitions are formed in specific way usually calling our intuition or previous knowledge. Let us analyze a problem of the differentiation of properties and descriptors in molecular design. IUPAC defines the property and its measurement [6] as:
Property, “A set of data elements (system, component, kind-of-property) common to a set of particular properties, e.g., substance concentration of glucose in blood plasma. Information about identification, time and result is not considered.’
Measurement, “A description of a property of a system by means of a set of specified rules, that maps the property onto a scale of specified values, by direct or 'mathematical' comparison with specified reference(s). The demand for rules makes 'measurement' a scientific concept in contrast to the mere colloquial sense of 'description'. However, in the present definition, 'measurement' has a wider meaning than given in elementary physics. Even a very incomplete description of, for instance, a patient (at a stated time) has to be given by a set of measurements, that are easier to manage and grasp.”
In turn IUPAC does not define the descriptor, a category widely used in molecular design. According to Todeschini descriptor is:
Molecular descriptor is a final result of a logic and mathematical procedure which transforms chemical information encoded with in a symbolic representation of a molecule into a useful number [7]. At the same time, we read that: Molecular descriptors are divided into two main classes: experimental measurements, such as logP, molar refractivity, dipole moment, polarizability, and, in general, physicochemical properties, and theoretical molecular descriptors” [8]
Let us try to classify the log P to the descriptors or properties. A quick analysis proves log P could be both a descriptor and a property. Log P can be both measured in an experiment and calculated. As a measured property Log P is the partition coefficient of a solute between octanol and water, at near infinite dilution. Therefore, Log P characterize the behavior of chemical substance. In turn, a large regression model allows to calculate Log P as a function of the molecular structure. In such a model Log P is a chemical descriptor, often constructed by the molecule defragmentation, e.g., the Rekker hydrophobic fragmental constants [9]
In the contemporary data-centric molecular design landscape, predominantly governed by computers and computer representations of chemical compounds [10], we can discern three fundamental types of descriptors:
- coding representations for molecular-symbols
- property correlates
- computer-generated molecular descriptors
Figure 1 briefly illustrates information size expansion while evolution from the property correlate descriptor to the computer-generated descriptors.
The log P value (calculated log P) serves as an illustration of a property correlate, where the measured property continues to hold dominance over descriptor behavior and structure. In this scenario, a single-value-descriptor is derived through regression to a corresponding measured property value. The logP can be interpreted as the predicted value replacing the partition coefficient. Conversely, multidimensional QSAR introduces a paradigm shift by offering multidimensional data simulated by the computer. Computer-generated descriptors can be both physically interpreted variables, e.g., Molecular Interaction Field, that probes the electrostatic or steric interaction between the probe situated near the molecules [15] or can take a form which cannot be easily interpreted by chemist. The latent descriptor representing the input molecular space coded by SMILES in the generative method [13] is an example of the last descriptor type. Usually, computer-generated-descriptors are high dimensional variables. The descriptor expansion is a term that can explain such data generation method [5] For example, in the CoMFA method, a single molecule represented by atomic Cartesian coordinates can be expanded to a representation even up to a hundred or thousand times its original size.
Finally, descriptors codes molecular structures. In recent methods we often use a mapping in which molecules are represented by the SMILES codes. SMILES are the best example of the coding descriptors role. We can interpret SMILES as the operators transforming molecular symbols into text. Formally, the result of the SMILES transformation is a series of the ASCII codes, i.e., the numbers, which also nicely holds the Todeschini definition, where descriptors are just useful numbers.
However, the important issue is that the classical descriptor definition focuses on the origin of the variable ignoring its modeling function in molecular design. We need to emphasize this role to understand and explain the property vs. descriptor interplay.
3. Correlation and regression fundamental data analyses methods: Predictors and criterions the basic data categories
Data analysis through correlation and regression represents a fundamental aspect of research and science. Exploring correlations allows us to uncover meaningful relationships within data, while regression allows for data compression. For instance, when we model a large data population with two variables using a simple linear equation (y = ax + b), we can replace this extensive dataset with just two parameters, a and b. Human intuition, complemented by basic mathematical operations and correlation, has enabled the creation of numerous models, such as the gravitational law. In molecular design, this approach is represented by the Hammett or Hansch QSAR equations, relating the property or activity to a certain molecule-related parameter(s).
By understanding the correlation and regression, we can comprehend a differentiation of the descriptor and property categories. The most fundamental descriptor feature is its role in the molecular design model. QSAR is an example of a molecular design method based on regression. In regression, we manipulate independent variable values to calculate the dependent variable and model a final relationship. Illustratively, the independent variable can be called a predictor, while the dependent variable is a criterion that helps us highlight their roles [16]. The predictor manipulation allows for the predictions of unknown criterions values. In molecular design, we cannot manipulate unrestrictedly with the predictor values. If we design novel molecules, we must know a relation that maps molecular objects to predictors. The descriptor function relates molecular objects by mathematical or logic operators to their representations that computers can process as predictors. Molecules or properties can represent chemical compounds [10]. As substance properties need to be measured the molecules-related data are available for predictions. Therefore, the descriptors are linked to molecules and their characteristics.
In regression, prediction is just one facet of its utility. Another essential aspect lies in the ability of data modeling to help us grasp the interactions between variables. Now, we do not necessarily predict the criterions for new objects. Such descriptors are used, for example, in the OMICS technology, where we compress the information coded by the measured data observables. Here we usually cannot predict criterions for entirely new, unmeasured predictors, but we can explore the entire predictor-criterion space to seek out rules that elucidate the patterns within the measured experimental data. A similar rationale applies to Quantitative Property-Property Relationships (QPPR), where the properties of novel molecular entities cannot serve as predictors without prior synthesis and experimental data [17].
3. The basic glossary of big data, artificial intelligence (AI), machine learning (ML) and deep learning (DL) in molecular design
To understand the term big data, it is essential to discern the differences between traditional and big data systems. Broadly defined, data encompasses recorded information, including metadata, which refers to additional data. This broad interpretation allows for data to exist as ordered or unordered collections of values, which can be either nominal or numerical. Numerical values are further categorized into discrete numbers, intervals, or ratios. Binary Large Objects (BLOBS) represent another data type specifically designed for audio, video, and graphic files, necessitating specialized analysis methods [4].
Big data is buzzword covering large topics among the current information methods that brings enormous expectations in various technologies. With the development of computers capable of the aggregation, storing massive data we realized that we need to adapt data processing to these novel information type. As the information volume is far above human capability we need a computer (machine) for data learning, interpretation, patter identification, parsing or prediction. A term machine learning underlines this big data feature. Joshi defines big data by the memory size that is needed to store: When the size of the data is large enough such that it cannot be processed on a single machine, it is called big data. Based on the current generation computers this equates to something roughly more than 10 GB. It can go into hundreds of petabytes (1 petabyte is 1000 terabytes and 1 terabyte is 1000 gigabyte) and more [18].
Numerous definitions of big data exist, but the key distinctions between conventional and large datasets generally revolve around volume, velocity, veracity, and variety (4V). Volume pertains to the vast size of datasets, velocity to the rapid rate of information growth, and variety to the diverse forms of data. Additionally, big data is occasionally characterized by a high degree of information complexity, leading traditional methods to falter when employed for processing. A more detailed discussion of the 4V expansion to 7V the reader can find in the reference [19]. The Gonzales Garcia review is also the informatics-guided introduction to big data topics with the comprehensive recent literature review and references.
The not-so-big data is another term that appeared recently to face big data problems. Especially in molecular design we are investigating what Drumond [20] described as a middle range of a few tenths of samples which can hardly face with the big data definition. On one hand, it appears that learning entirely new concepts of forms in humans, e.g., reading characters [21]. In other words, not-so-big data is a pragmatic approach replacing big data due to better interpretability or simply when we cannot expand the data space. Chemo-, pharmaco-, pharmaco-economic substance behavior analyses were published using large data available [22,23,24,25]. In molecular design, we can suggest the term all-data available. For example, we have not more than 3000 FDA-approved NMEs. Therefore, we cannot expect to expand this data into real big data now or soon. Therefore, pragmatically, this data can replace big data in analyses. An interesting analysis of the chemical substance attractivity using not-so-big data can be found in the reference [26] and probing the drug (FDA approvals) fate on the market is another pragmatic analysis of the all-data [27,28].
Machine learning is a branch of artificial intelligence, a feature of computers enabling solving problems by autonomous learning from experience. The AI idea focus on the machine-human-like behavior. In turn latest trend in molecular design involves neural networks (NN) and deep learning (DL) as the ML branches. Especially in chemistry the terms AI and ML are often used interchangeable but in more precise definition a ML focuses on the use of data and algorithms for solving problems imitating human learning. Regression; Principal Component Analysis; Partial Least Square Analysis, are among the ML methods [29]. A good early informative introduction into computational background of these methods can be found in the references [30,31,32]
What distinguishes standard regression commonly used by statistics from ML regression is the size of the dataset used. Generally, we relate ML to large data analyses. How big should the limit data to encompass ML learning? The idea of ML was formulated already in 1950 where we are far for from the efficiency of the contemporary computers. Let us consider a question if we categorize the Hansch QSAR model [34] as the ML. The answer is negative. By mapping a single predictor against a single criterion for a relatively small data size, we were able to deduce the Hansch model. We can easily understand the meaning of this model with these two variables. In this place we should stress that it was not either easy to initially deduce this law or even to prove the Hansch drug transport model as a general law in contemporary medicinal chemistry which reveal many more specific membrane transport effects. [35] Instead, we should recognize ML as a method wherein the criterion cannot be easily explained by a function of predictors that can be readily found and interpreted by a human without the need for computer support. In ML we usually need a large population of predictors with their weights to model criterions.
Deep learning (DL), in turn, is a methodology that employs intricate multilayer neural networks for data processing. While neural networks are not the exclusive method for data analysis, they have evolved into valuable technologies, exemplified by achievements such as face recognition (Deep Face), chess-playing prowess (Deep Blue), and notably in chemical applications like retrosynthesis [36,37]. It has been suggested that serendipity plays a role in these successes [38]."
4. Big data attributes and structure
In the context of drug design, the ownership attribute of data becomes crucial, posing challenges to data availability. While recognizing the significance of sharing data (sharable data and the data-sharing problem), the pharmaceutical Research and Development (R&D) sector often faces the reality of confidential data practices. The diminishing efficiency in pharma R&D has spurred collaborative drug design projects, such as Collaborative Drug Discovery (CDD) at collaborativedrug.com, where involved collaborators share data. While data sharing among traditional pharmaceutical companies remains controversial, there is a growing conviction that such collaboration could significantly enhance efficiency in the field.
Let us now embark on identifying and defining big data sources, along with the methods employed for manipulating big data in bio- and chemoinformatics. Bio- and chemoinformatics are integral tools for in silico data processing in chemistry, pharmacy, and medicine [3Pol]. Chemoinformatics, initially defined as the amalgamation of all information resources essential for optimizing the properties of a ligand to transform it into a drug, shares similar objectives with bioinformatics but with a more biologically oriented focus. Consequently, both disciplines contribute to designing drug discovery and development tools that navigate challenges in managing and processing big data in drug design.
The differentiation between descriptors and properties allows us to organize big data into distinct structures. As physical, chemical or biological experiments are expensive and chemical compounds are represented by the calculable data within the chemical space. Properties measured in physical or chemical experiments represent the initial category among them. Multidimensional QSAR (m-QSAR) is an example of regression based model where a series of chemical compounds is labeled with a single measured property. Then 3D representations of these compounds simulated in silico to provide a series of descriptor data. Accordingly, a single compound is represented by a large dimension data. Such a scenario can be categorized as descriptor expansion. However, we generate big data also by property expansion. The scarcity of measured properties results in the PE structure being predominantly large due to the numerous objects annotated with a single property type, rather than a diversity of property types. In contrast, the array of descriptors designed to characterize molecules contributes to the largeness of DE data due to the numerous descriptor variables. Furthermore, the limited availability of properties often necessitates the substitution of measured property values with predicted property values. This gives rise to a distinct data type referred to as PPA (predicted property annotation) data. The reader can compare for more details the Reference [5Pol].
Virtual screening of drug-like molecules exemplifies one of the current trends in molecular design. In one of the most expansive experiments of this nature, Graham Richards designed the screensaver project, utilizing distributed computation to simulate the docking of 3.5 million compounds to the binding sites of 14 protein targets in pursuit of optimizing molecular structures. This protocol can be characterized as descriptor expansion, where a single measured property (compound’s activity) is correlated with complex multidimensional descriptor [39]. This project has given us novel methods for dealing with enormous volumes of data, “generating an enormous number of hits (…) none of these has yet produced a candidate drug that has entered clinical trials [40].
On the other hand, the Cancer Genome Atlas represents a large-scale dataset dominated by measured properties. Collecting 150 terabytes of data on 25 diseases from 7,104 patients and 6,720 samples spanning 2006 to 2012, it showcases a rich source of information [40]. Currently, data types such as chemogenomics, genomics, and lipidomics are at the forefront, generating multidimensional measured properties for molecular design.
5. Big data availability and quality
The databases available for big data acquisition in molecular design was reviewed and critically evaluated in a number of publications [3,42,43]. Below we specified the most prominent examples.
- Chemical Abstracts Service; www.cas.org/support/documentation/cas-databases
- Reaxys; www.reaxys.com
- CheMBL; EMBL's European Bioinformatics Institute; www.ebi.ac.uk
- eMolecules; https://search.emolecules.com
- PubChem; https://pubchem.ncbi.nlm.nih.gov/
- GDP Databases; https://gdb.unibe.ch/downloads/
The illustrative examples, further discussion and other big data resources can be found in the References [44,45,46].
PubChem is one of the largest molecular database collecting 116,123,817 unique chemical structures, 310,123,085 chemical substances information, 1,627,316 bioassay data. The steady development of PubChem is discussed in the series of publications in the journal Nucleic acids research. In Figure 2 we illustrated the data use increase from 2016 to 2023 2005-2013] and the increase in the record number of the data, respectively [47,48].
Bender et al. [43] extensively examined the challenges associated with AI applications in molecular design and QSAR, particularly focusing on data availability and quality issues. We need a deeper understanding of biological systems and the generation of substantially meaningful and practical data on a larger scale. To illustrate, Bender compared data availability across different fields, citing examples such as
Image Net with 14 million entries,
Tesla cars generating 1.021 bytes of data,
CHeMBL (Release 26) with 16 million bioactivity labels,
Marketed Drugs from DrugBank v5.1.5 comprising 13,548 entries (2626 approved small molecules, 1372 approved biologics, 131 nutraceuticals, and >6363 experimental drugs), and
Gene expression data from the Open Targets Platform showing 8,462,444 associations spanning 13,818 diseases and 27,700 targets.
Bender also highlighted challenges in data acquisition for drug discovery, such as hypothesis-free protocols via emerging techniques that may not be suitable for a given purpose. For a more detailed exploration of these challenges, readers should refer to the comprehensive discussion in the references [42,43]. These challenges contribute to the frequent poor labeling of activity data with properties, making it challenging to anticipate collecting more data under various experimental conditions. The prospect of finding the right computer program to interpret cellular activities hinges on a better understanding of biology, guiding data generation for specific purposes.
Additionally, Aldrich et al. [49] emphasized potential artifacts in experiments screening for potential hits. Many false hits, particularly those classified as Pan Assay INterference compounds (PAINS) or colloidal aggregators, are often recorded rather than genuine actives against the desired targets. The authors provided a detailed list of considerations for controlling assays to eliminate PAINS and mitigate resulting artifact data.
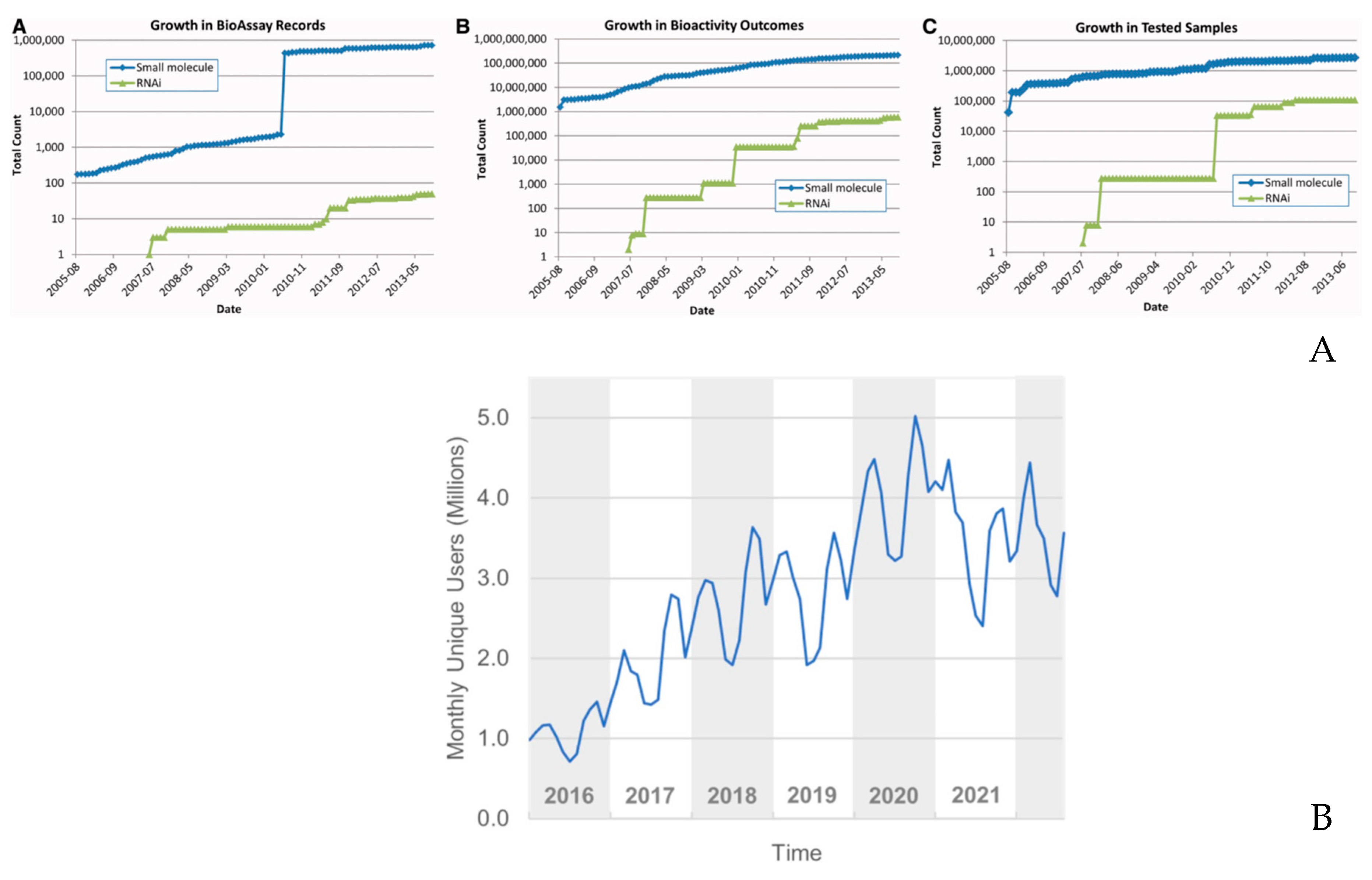
6. Recent big data studies in molecular design
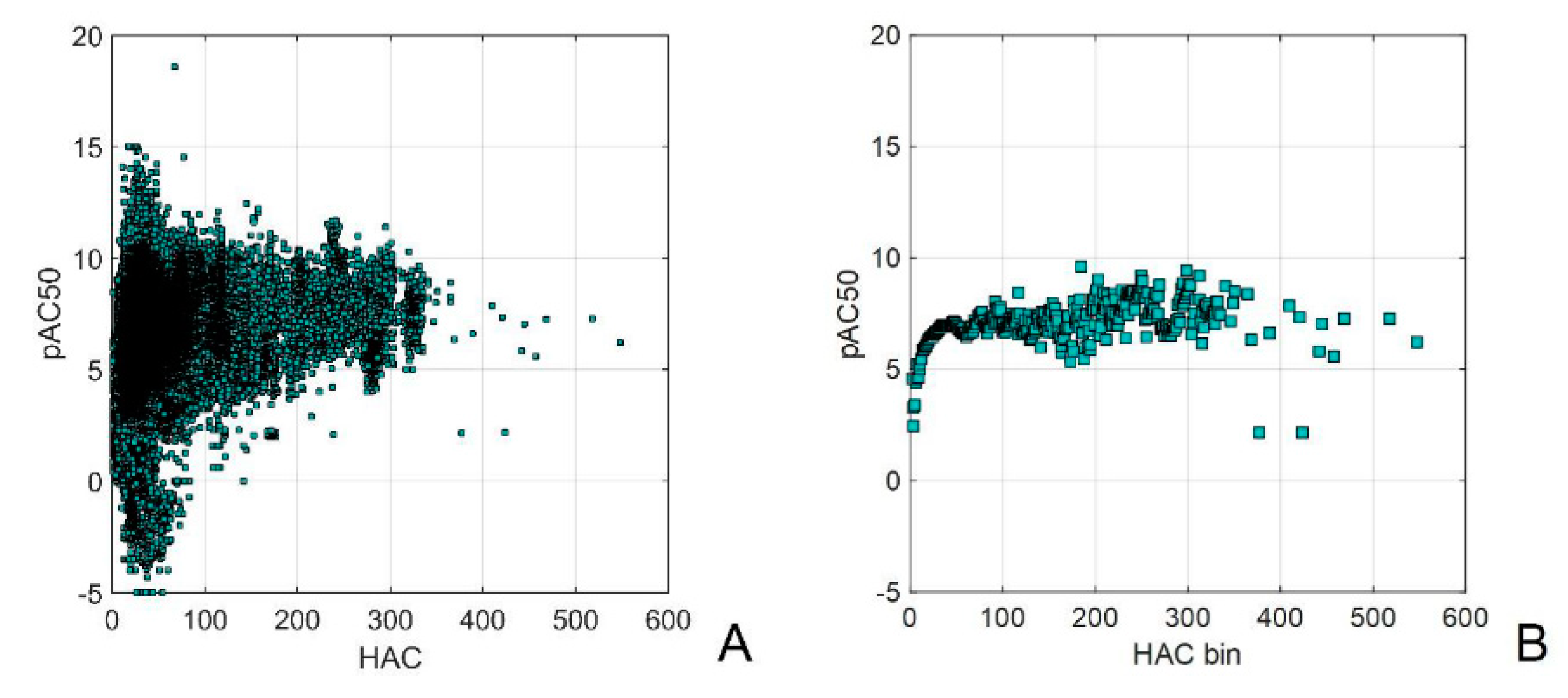
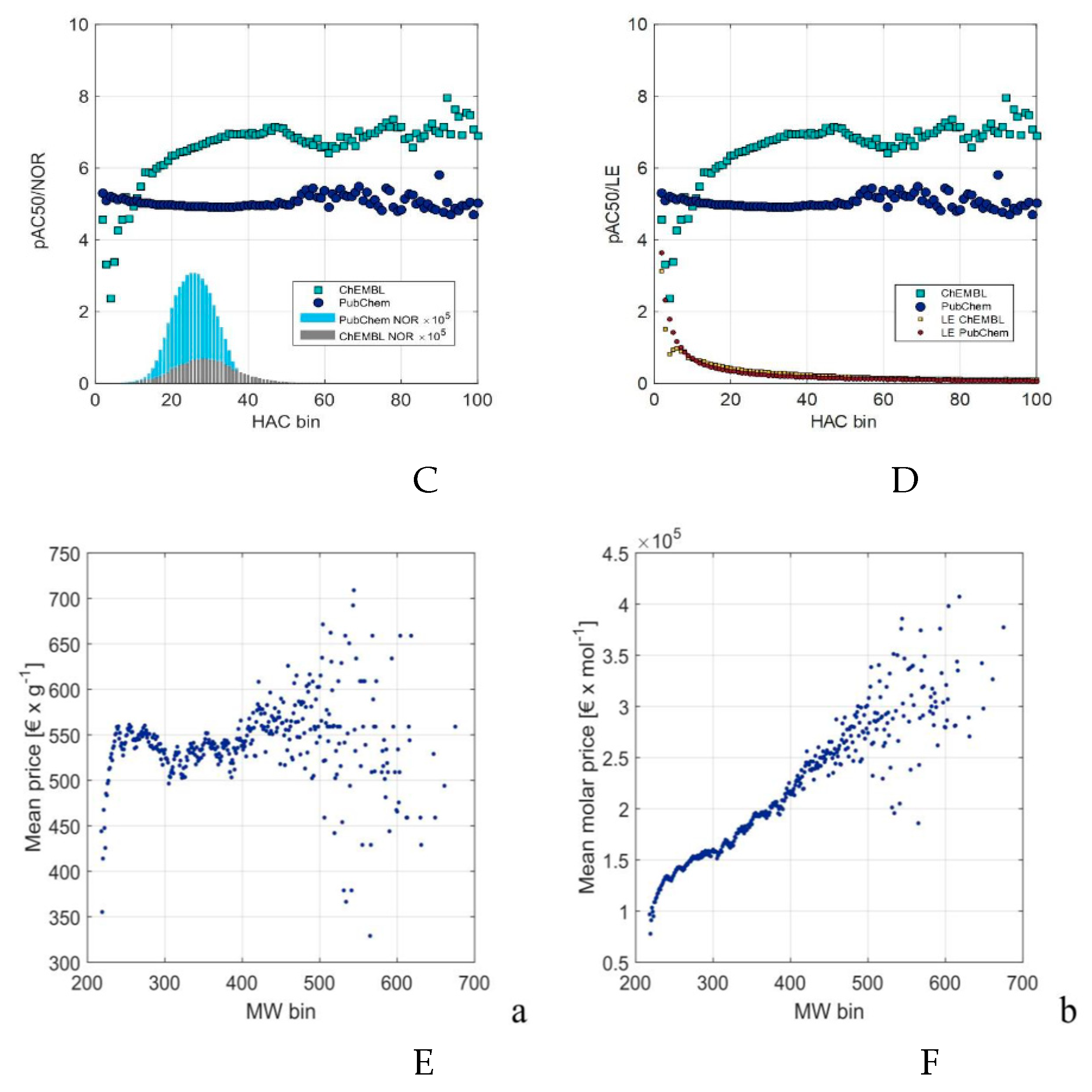
In the most general version computers should itself recognize the errors, finding the best predictors and criterions among all possible ones, where in a more general meaning predictors are input representations to be mapped to criterions. Figure 3 presents the application of the binning method for visualizing extensive molecular data gathered from PubChem, ChEMBL, or a large chemical catalog. It becomes evident that the chaotic, unbinned data lacks any discernible useful information. However, after applying binning, the ChEMBL data reveals a discernible pattern: an increase in mean biological activity with higher heavy atom counts (HAC). It's noteworthy that when the number of molecular objects is insufficient for higher HAC, this relationship starts to exhibit distortion (Figure 3C,D). The larger but not data curated PubChem database indicates no correlation between the activity and HAC (Figure 3C,D).
The reviews discussing big data problems and analyses in molecular design are available. Jing enumerate the examples of big and not-so-big data analyses that involves from 125 samples to the whole ChEMBL database that use the DL method with different descriptors [50].
Figure 3.
ChEMBL activity data plotted vs. heavy atom counts (HAC) (A); binned ChEMBL activity data vs. HAC values (B); binned ChEMBL and PubChem activity data plotted vs. HAC compared with the number of the database records (NOR) (C); ChEMBL and PubChem activity and ligand efficiency data plotted vs. HAC (D) [24] and the chemo-economic analysis of the large several million substance catalogue with weight (E) and molar (F) prices vs. binned molecular weight (MW) [22]. Graphics modified from [51].
Figure 3.
ChEMBL activity data plotted vs. heavy atom counts (HAC) (A); binned ChEMBL activity data vs. HAC values (B); binned ChEMBL and PubChem activity data plotted vs. HAC compared with the number of the database records (NOR) (C); ChEMBL and PubChem activity and ligand efficiency data plotted vs. HAC (D) [24] and the chemo-economic analysis of the large several million substance catalogue with weight (E) and molar (F) prices vs. binned molecular weight (MW) [22]. Graphics modified from [51].
In their comprehensive review, Tripathi et al. highlighted that pharmaceutical giants such as Sanofi, Merck, Takeda, and Bayer have initiated and are maintaining collaborations with AI companies. In this publication we can also find the broad variety software tools supporting AI based drug discovery. The comparison of the scope of this software with the ML computational tool clearly indicates the broad AI scope. This involves, for example, GoPubMed (PubMed search engine with the text mining tool), DeepNeuralNetQSAR (the Python-based system assisting in the detection of molecular activity), etc. [45].
The appliaction of DL for the prediction of the protein folding is especially worth of mentioning. AlphaFold is the recent software offering high accuracy of the predictions. The prediction of protein-protein interactions is a related problem. For the more detailed discussion, available software and modeling accuracy the reader can compare Reference [45].
6. Conclusion
The principal goal of chemistry lies in property production, indicating a pursuit of novel drugs or materials over chemical compound generation, with big data at the forefront of this paradigm. Since chemistry is about property production, chemists need to understand novel data-oriented methods better. Here, we provide an elementary introduction to such methods. We assess the dimensions, accessibility, quality, and structural aspects of big data, exploring primary methodologies for its analysis. In chemical compounds, properties, and descriptors emerge as distinct data types, forming the foundation for categorizing molecular big data. The classification of descriptors as predictors while properties as criterions, allows for a better understanding of the individual variable role played in molecular design.
The escalating availability of data in computer-aided technology propels advancements in artificial intelligence (AI), machine learning (ML), and deep learning (DL).
Consequently, a broad chemical audience must grasp these methodologies to comprehend data-centric chemistry. We aimed to systematize big data challenges through an illustrative framework encompassing fundamental descriptor categories: coding, computer-generated descriptors and property correlates. While computer-generated descriptors serve nowadays as substantial big data in predictions, measured data remains indispensable for ensuring high-quality and reliable outcomes and controlling molecular effects. The scarcity of property data poses a significant obstacle, constraining comprehensive studies on structure-property relationships within big data. Guided by pragmatics, the consideration of not-so-big data becomes a viable option for drug design. Additionally, we concisely specified recent review literature on big data, highlighting key insights in this evolving field.
Funding
The research activities co-financed by the funds granted under the Research Excellence Initiative of the University of Silesia in Katowice.
Conflicts of Interest
The author declares no conflict of interest.
References
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Williams, W.L.; Zeng, L.; Gensch, T.; Sigman, M.S.; Doyle, A.G.; Anslyn, E.V. The Evolution of Data-Driven Modeling in Organic Chemistry. ACS Central Sci. 2021, 7, 1622–1637. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J. Chemoinformatics: From Chemical Art to Chemistry in Silico. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 601–618. ISBN 978-0-12-811432-2. [Google Scholar]
- Maheshwari, A. Data Analytics Made Accessible; Amazon Digital Services: Seattle, USA, 2014. [Google Scholar]
- Polanski, J. Big data in structure-property studies—From definitions to models. In Advances in QSAR Modeling: Applications in Pharmaceutical, Chemical, Food, Agricultural and Environmental Sciences; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Olesen, H. Properties and units in the clinical laboratory sciences-I. Syntax and semantic rules (IUPAC-IFCC Recommendations 1995). Pure Appl. Chem. 1995, 67, 1563–1574. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Handbook of molecular descriptors; Wiley-VCH: Weinheim, 2000. [Google Scholar] [CrossRef]
- Consonni, V.; Todeschini, R. Molecular descriptors. In Recent advances in QSAR studies; Puzyn, T., et al., Eds.; Springer: Dordrecht, 2010; pp. 29–102. [Google Scholar]
- Rekker, R.F.; Mannhold, R. Calculation of Drug Lipophilicity. The Hydrophobic Fragmental Constant Approach, VCH, Weinheim, 1992.
- Polanski, J.; Gasteiger, J. Computer Representation of Chemical Compounds. In Handbook of Computational Chemistry; Leszczynski, J., Kaczmarek-Kedziera, A., Puzyn, T., Papadopoulos, M.G., Reis, H., Shukla, M.K.K., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1997–2039. ISBN 978-3-319-27281-8. [Google Scholar]
- Barbas, C.F.; Heine, A.; Zhong, G.; Hoffmann, T.; Gramatikova, S.; Björnestedt, R.; List, B.; Anderson, J.; Stura, E.A.; Wilson, I.A.; et al. Immune Versus Natural Selection: Antibody Aldolases with Enzymic Rates But Broader Scope. Science 1997, 278, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Hopfinger, A.J.; Wang, S.; Tokarski, J.S.; Jin, B.; Albuquerque, M.; Madhav, P.J.; Duraiswami, C. Construction of 3D-QSAR Models Using the 4D-QSAR Analysis Formalism. J. Am. Chem. Soc. 1997, 119, 10509–10524. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Central Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Taigman, Y.; Yang, M.; Ranzato, M.A.; Wolf, L. Deepface: Closing the gap to human-level performance in face verification. In Proceedings of the IEEE conference on computer vision and pattern recognition; 2014; pp. 1701–1708. [Google Scholar]
- Mannhold, R.; Kubinyi, H.; Folkers, G. Molecular interaction fields: Applications in drug discovery and ADME prediction; John Wiley & Sons: Hoboken, NJ, USA, 2006; ISBN 978-3-527-60767-9. [Google Scholar]
- Palmer, P.B.; Oʼconnell, D.G. Research Corner: Regression Analysis for Prediction: Understanding the Process. Cardiopulm. Phys. Ther. J. 2009, 20, 23–26. [Google Scholar] [CrossRef]
- DeJongh, J.; Verhaar, H.J.M.; Hermens, J.L.M. A quantitative property-property relationship (QPPR) approach to estimate in vitro tissue-blood partition coefficients of organic chemicals in rats and humans. Arch. Toxicol. 1997, 72, 17–25. [Google Scholar] [CrossRef]
- Joshi, A.V. Machine learning and artificial intelligence, 2nd ed.; Springer: Cham, Switzerland, 2023. [Google Scholar]
- García, C.G.; Álvarez-Fernández, E. What Is (Not) Big Data Based on Its 7Vs Challenges: A Survey. Big Data Cogn. Comput. 2022, 6, 158. [Google Scholar] [CrossRef]
- Drumond, T.F.; Viéville, T.; Alexandre, F. Bio-inspired Analysis of Deep Learning on Not-So-Big Data Using Data-Prototypes. Front. Comput. Neurosci. 2019, 12, 100. [Google Scholar] [CrossRef]
- Leroy, A. L'apprentissage de la lecture chez les jeunes enfants: Acquisition des lettres de l'alphabet et maturité mentale. Enfance 1967, 20, 27–55. [Google Scholar] [CrossRef]
- Polanski, J.; Kucia, U.; Duszkiewicz, R.; Kurczyk, A.; Magdziarz, T.; Gasteiger, J. Molecular descriptor data explain market prices of a large commercial chemical compound library. Sci. Rep. 2016, 6, 28521. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Pedrys, A.; Duszkiewicz, R.; Gasteiger, J. Scoring Ligand Efficiency: Potency, Ligand Efficiency and Product Ligand Efficiency within Big Data Landscape. Lett. Drug Des. Discov. 2019, 16, 1258–1263. [Google Scholar] [CrossRef]
- Polanski, J.; Duszkiewicz, R.; Pedrys, A.; Gasteiger, J. Scoring ligand efficiency. Acta Pol. Pharm.-Drug Res. 2019, 76, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Duszkiewicz, R. Property representations and molecular fragmentation of chemical compounds in QSAR modeling. Chemom. Intell. Lab. Syst. 2020, 206. [Google Scholar] [CrossRef]
- Sung, B.; Park, K.-M.; Park, C.G.; Kim, Y.-H.; Lee, J.; Jin, T.-E. What drives researcher preferences for chemical compounds? Evidence from conjoint analysis. PLoS ONE 2023, 18, e0294576. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Bogocz, J.; Tkocz, A. The analysis of the market success of FDA approvals by probing top 100 bestselling drugs. J. Comput. Mol. Des. 2016, 30, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Bogocz, J.; Tkocz, A. Top 100 bestselling drugs represent an arena struggling for new FDA approvals: Drug age as an efficiency indicator. Drug Discov. Today 2015, 20, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Dou, B.; Zhu, Z.; Merkurjev, E.; Ke, L.; Chen, L.; Jiang, J.; Zhu, Y.; Liu, J.; Zhang, B.; Wei, G.-W. Machine Learning Methods for Small Data Challenges in Molecular Science. Chem. Rev. 2023, 123, 8736–8780. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A. Machine-learning approaches in drug discovery: Methods and applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A. Deep learning in drug discovery: Opportunities, challenges and future prospects. Drug Discov. Today 2019, 24, 2017–2032. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Schneider, N.; Segler, M.; Walters, W.P.; Engkvist, O.; Rodrigues, T. Evaluation guidelines for machine learning tools in the chemical sciences. Nat. Rev. Chem. 2022, 6, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Hoekman, D.; Gao, H. Comparative QSAR: Toward a Deeper Understanding of Chemicobiological Interactions. Chem. Rev. 1996, 96, 1045–1076. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.J.; Nicolson, G.L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef]
- Mikolajczyk, A.; Zhdan, U.; Antoniotti, S.; Smolinski, A.; Jagiello, K.; Skurski, P.; Harb, M.; Puzyn, T.; Polanski, J. Retrosynthesis from transforms to predictive sustainable chemistry and nanotechnology: A brief tutorial review. Green Chem. 2023, 25, 2971–2991. [Google Scholar] [CrossRef]
- Polanski, J. Unsupervised Learning in Drug Design from Self-Organization to Deep Chemistry. Int. J. Mol. Sci. 2022, 23, 2797. [Google Scholar] [CrossRef] [PubMed]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef]
- Richards, W.G. COMPUTER-AIDED DRUG DISCOVERY AND DEVELOPMENT (CADDD): In silico-chemico-biological approach. Nature Reviews Drug Discovery 2002, 1, 551–555. [Google Scholar] [CrossRef]
- CLARE SANSOM, People power. Available online: www.chemistryworld.com/features/people-power/5818.article (accessed on 1 December 2023).
- TCGA, National Cancer Institute. Available online: https://www.cancer.gov/ccg/research/genome-sequencing/tcga (accessed on 2 December 2023).
- Bender, A.; Cortés-Ciriano, I. Artificial intelligence in drug discovery: What is realistic, what are illusions? Part 1: Ways to make an impact, and why we are not there yet. Drug Discov. Today 2020, 26, 511–524. [Google Scholar] [CrossRef]
- Bender, A.; Cortes-Ciriano, I. Artificial intelligence in drug discovery: What is realistic, what are illusions? Part 2: A discussion of chemical and biological data. Drug Discov. Today 2021, 26, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ciallella, H.L.; Aleksunes, L.M.; Zhu, H. Advancing computer-aided drug discovery (CADD) by big data and data-driven machine learning modeling. Drug Discov. Today 2020, 25, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.K.; Nath, A.; Singh, T.P.; Ethayathulla, A.S.; Kaur, P. Evolving scenario of big data and Artificial Intelligence (AI) in drug discovery. Mol. Divers. 2021, 25, 1439–1460. [Google Scholar] [CrossRef] [PubMed]
- Vo, A.H.; Van Vleet, T.R.; Gupta, R.R.; Liguori, M.J.; Rao, M.S. An Overview of Machine Learning and Big Data for Drug Toxicity Evaluation. Chem. Res. Toxicol. 2019, 33, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; A Shoemaker, B.; A Thiessen, P.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2022, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Suzek, T.; Zhang, J.; Wang, J.; He, S.; Cheng, T.; Shoemaker, B.A.; Gindulyte, A.; Bryant, S.H. PubChem BioAssay: 2014 update. Nucleic Acids Res. 2013, 42, D1075–D1082. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, C.; Bertozzi, C.; Georg, G.I.; Kiessling, L.; Lindsley, C.; Liotta, D.; Merz, K.M.; Schepartz, A.; Wang, S. The Ecstasy and Agony of Assay Interference Compounds. ACS Central Sci. 2017, 3, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Bian, Y.; Hu, Z.; Wang, L.; Xie, X.-Q.S. Deep Learning for Drug Design: An Artificial Intelligence Paradigm for Drug Discovery in the Big Data Era. AAPS J. 2018, 20, 1–10. [Google Scholar] [CrossRef]
- Duszkiewicz, R. Ocena liganda jako potencjalnego leku na próbie wybranych baz wielkich danych. PhD Thesis. Available online: https://rebus.us.edu.pl/bitstream/20.500.12128/18293/1/Duszkiewicz_Ocena_liganda_jako_potencjalnego_leku.pdf (accessed on 2 December 2023).
- Brown, N.; Cambruzzi, J.; Cox, P.J.; Davies, M.; Dunbar, J.; Plumbley, D.; Sellwood, M.A.; Sim, A.; Williams-Jones, B.I.; Zwierzyna, M.; et al. Big Data in Drug Discovery. Prog Med Chem. 2018, 57, 277–356. [Google Scholar] [CrossRef]
- Zhu, H. Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 573–589. [Google Scholar] [CrossRef]
- Prieto-Martínez, F.D.; López-López, E.; Juárez-Mercado, K.E.; Medina-Franco, J.L. Computational drug design methods—Current and future perspectives. In silico drug design Kunal Roy (Ed.); Academic Press: London, 2019; pp. 19–44. [Google Scholar] [CrossRef]
- Tripathi, N.; Goshisht, M.K.; Sahu, S.K.; Arora, C. Applications of artificial intelligence to drug design and discovery in the big data era: A comprehensive review. Mol. Divers. 2021, 25, 1643–1664. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ding, J.; Pan, L.; Cao, D.; Jiang, H.; Ding, X. Artificial intelligence facilitates drug design in the big data era. Chemom. Intell. Lab. Syst. 2019, 194, 103850. [Google Scholar] [CrossRef]
- Zhao, L.; Ciallella, H.L.; Aleksunes, L.M.; Zhu, H. Advancing computer-aided drug discovery (CADD) by big data and data-driven machine learning modeling. Drug Discov. Today 2020, 25, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Antolin, A.A.; Al-Lazikani, B. Transforming cancer drug discovery with Big Data and AI. Expert Opin. Drug Discov. 2019, 14, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; He, H.; Luo, H.; Zhang, T.; Jiang, J. Artificial intelligence and big data facilitated targeted drug discovery. Stroke Vasc. Neurol. 2019, 4, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 2019, 19, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, K. Combinatorics, Big Data, Neural Network & AI for Medicinal Chemistry & Drug Administration. Lett. Drug Des. Discov. 2021. [Google Scholar] [CrossRef]
- Kavidopoulou, A.; Syrigos, K.N.; Makrogkikas, S.; Dlamini, Z.; Hull, R.; Marima, R.; Lolas, G. AI and Big Data for Drug Discovery. In Trends of Artificial Intelligence and Big Data for E-Health; Springer International Publishing: Cham, 2023; pp. 121–138. [Google Scholar]
- Doherty, T.; Yao, Z.; Khleifat, A.A.; Tantiangco, H.; Tamburin, S.; Albertyn, C.; Thakur, L.; Llewellyn, D.J.; Oxtoby, N.P.; Lourida, I.; et al. Artificial intelligence for dementia drug discovery and trials optimization. Alzheimer's Dement. 2023, 19, 5922–5933. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S. The emerging roles of artificial intelligence in chemistry and drug design. In Data-Driven Technologies and Artificial Intelligence in Supply Chain; CRC Press: Boca Raton, FL, USA, 2024; pp. 158–173. [Google Scholar]
- Wu, X.; Li, W.; Tu, H. Big data and artificial intelligence in cancer research. Trends Cancer 2024, 10, 147–160. [Google Scholar] [CrossRef]
- Dou, B.; Zhu, Z.; Merkurjev, E.; Ke, L.; Chen, L.; Jiang, J.; Zhu, Y.; Liu, J.; Zhang, B.; Wei, G.-W. Machine Learning Methods for Small Data Challenges in Molecular Science. Chem. Rev. 2023, 123, 8736–8780. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Y.; Han, L. Bioinformatics: Advancing biomedical discovery and innovation in the era of big data and artificial intelligence. Innov. Med. 2023, 1. [Google Scholar] [CrossRef]
- Sinha, K.; Ghosh, N.; Sil, P.C. A Review on the Recent Applications of Deep Learning in Predictive Drug Toxicological Studies. Chem. Res. Toxicol. 2023, 36, 1174–1205. [Google Scholar] [CrossRef] [PubMed]
- Danel, T.; Łęski, J.; Podlewska, S.; Podolak, I.T. Docking-based generative approaches in the search for new drug candidates. Drug Discov. Today 2023, 28, 103439. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Song, Y.; Wang, H.; Zhang, X.; Wang, M.; He, J.; Li, S.; Zhang, L.; Li, K.; Cao, L. Advances of Artificial Intelligence in Anti-Cancer Drug Design: A Review of the Past Decade. Pharmaceuticals 2023, 16, 253. [Google Scholar] [CrossRef]
- Tsagkaris, C.; Corriero, A.C.; Rayan, R.A.; Moysidis, D.V.; Papazoglou, A.S.; Alexiou, A. Success stories in computer-aided drug design. In Computational Approaches in Drug Discovery, Development and Systems Pharmacology; Academic Press: Cambridge, MA, USA, 2023; pp. 237–253. [Google Scholar]
- Xiao, L.; Zhang, Y. AI-driven smart pharmacology. Intell. Pharm. 2023, 1, 179–182. [Google Scholar] [CrossRef]
- Seo, S.; Lee, J.W. Applications of Big Data and AI-Driven Technologies in CADD (Computer-Aided Drug Design). In Computational Drug Discovery and Design; Springer: New York, USA, 2023; pp. 295–305. [Google Scholar]
Figure 1.
The evolution from property correlate descriptor (lipophilicity) (A) [11] to computer-generated descriptors: physically interpretable 4D QSAR grid cell occupancy descriptor (B) [12], and latent SMILES based DL generated human-uninterpretable generative AI model, presented as a schematic representation (C) [13]. For the better understanding, in the bottom line we can see how face recognition information is stored within the network weights by the DEEP FACE DL system in the computer-generated face descriptor. We can see some face features but the black-box character increases with the decreasing neural network size [14]. Graphics modified from [11,12,13,14].
Figure 1.
The evolution from property correlate descriptor (lipophilicity) (A) [11] to computer-generated descriptors: physically interpretable 4D QSAR grid cell occupancy descriptor (B) [12], and latent SMILES based DL generated human-uninterpretable generative AI model, presented as a schematic representation (C) [13]. For the better understanding, in the bottom line we can see how face recognition information is stored within the network weights by the DEEP FACE DL system in the computer-generated face descriptor. We can see some face features but the black-box character increases with the decreasing neural network size [14]. Graphics modified from [11,12,13,14].
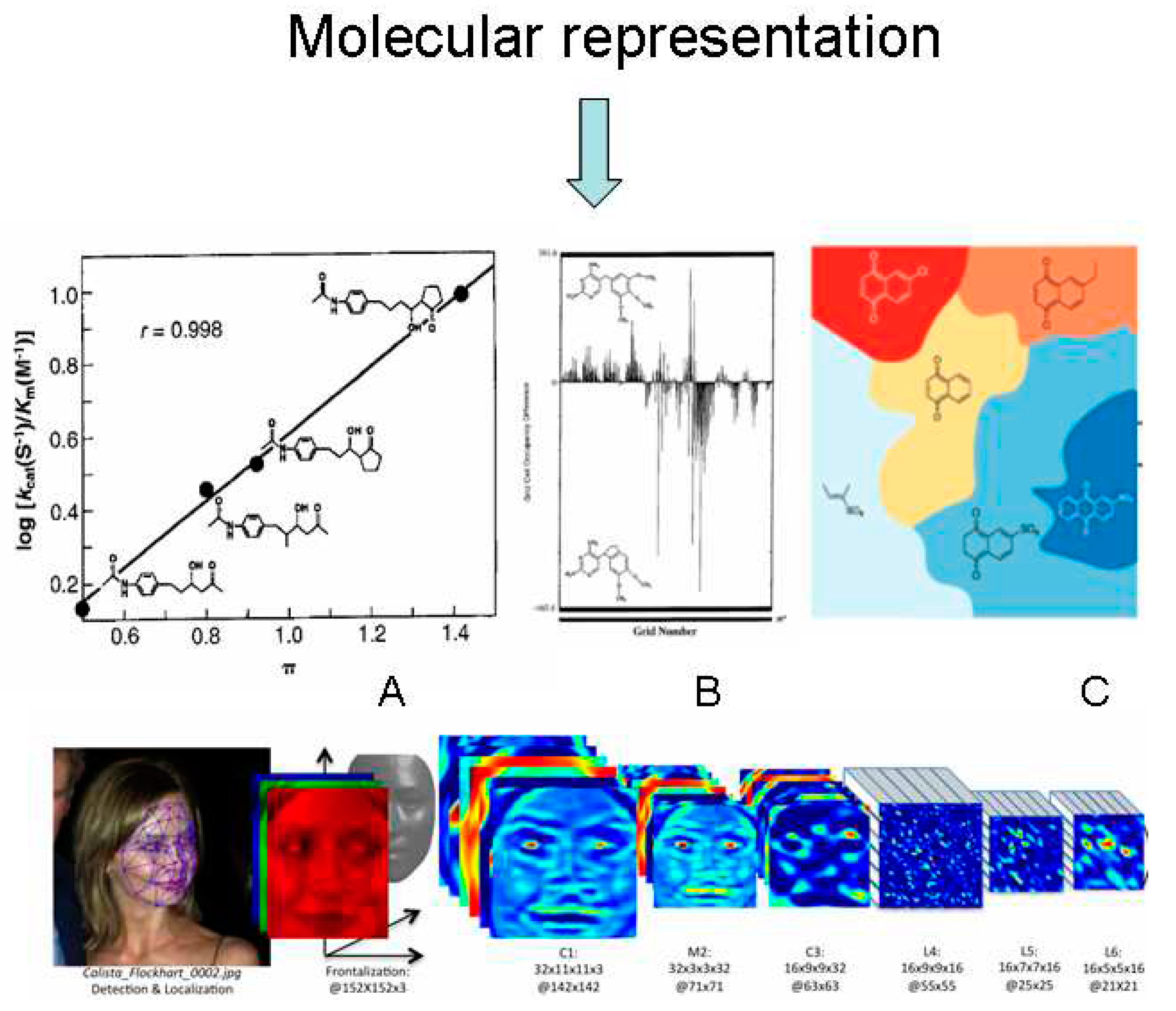

Table 1.
Some example reviews on AI applications in molecular design.
| Entry | Specific topics | Reference |
|---|---|---|
| 1 | Application in protein folding, protein-protein interaction, virtual screening, QSAR, ADME, toxicity, drug repurposing, de novo design | [55] |
| 2 | Application in protein folding, protein-protein interaction, virtual screening, QSAR, ADME, toxicity, drug repurposing, de novo design | [56] |
| 3 | Application in toxicity | [46] |
| 4 | Application in QSAR, drug discovery studies, de- novo design, drug interactions, drug repurposing | [57] |
| 5 | Integrating big-knowledge on drug targets, particularly in cancer research | [58] |
| 6 | Precision medicine | [59] |
| 7 | Generating datasets, generating new hypotheses, optimization | [60] |
| 8 | Methods for big data analyses | [61] |
| 9 | Computational drug modeling to assess compounds for their biological and toxicological effects via neural networks and similarity modeling | [62] |
| 10 | Small molecule–based drug design and development | [63] |
| 11 | Growth and distribution of AI-related chemistry | [64] |
| 12 | A machine learning based intelligent service platform, designed to integrate cancer big data and employ AI algorithms for personalized health management | [65] |
| 13 | ML in not-so-big data processing | [66] |
| 14 | AI in biomedical data processing | [67] |
| 15 | DL in toxicity | [68] |
| 16 | Docking-based generative models | [69] |
| 17 | AI-based methods in anti-cancer drug design. | [70] |
| 18 | ML, big data | [71] |
| 19 | AI driven drug discovery | [72] |
| 20 | Processing and applications of big data in molecular design | [73] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



