Submitted:
05 December 2023
Posted:
06 December 2023
You are already at the latest version
Abstract
Fibroblast growth factor receptors (FGFRs) are a family of receptor tyrosine kinases that involve in regulation of cell proliferation, survival, and development. FGFR alterations including amplifications, fusions, rearrangements, and mutations can result downstream activation of tyrosine kinases leading to tumor development. Targeting those FGFR alterations has shown to be effective in cholangiocarcinoma, urothelial carcinoma, and myeloid/lymphoid neoplasms and there are currently four FGFR inhibitors approved by the Food and Drug Administration (FDA). There have been developments of multiple agents targeting FGFR pathway including selective FGFR inhibitors, ligand traps, monoclonal antibodies, and antibody drug conjugates. However, most of these agents have variable and low responses with some intolerable toxicities and acquired resistances. This review will summarize previous clinical experiences and current developments of agents targeting FGFR pathway and discuss future directions of FGFR targeting agents.
Keywords:
FGF
; FGFR
; targeted therapy
; molecular profiling
; precision medicine
1. Introduction
Fibroblast growth factor (FGF) receptors (FGFRs) are a family of five receptor tyrosine kinases (RTKs) named FGFR 1-5. FGFRs have extracellular domain that binds to FGF ligand, transmembrane and intracellular tyrosine kinase domains except for FGFR5 that lacks intracellular tyrosine kinase (TK) domain [1,2]. Activation of FGFRs by FGFs play an essential role in cellular proliferation, survival, embryonic development, metabolism, homeostasis, tissue repair, endocrine functions and fetal organogenesis. Dysregulation in FGFR signaling pathways caused by FGFR gene amplifications, mutations and fusions lead to oncogenesis, tumor progression and angiogenesis in tumor microenvironment as well as resistance to anticancer treatment [1]. According to report of the next generation sequencing (NGS) study of 4,853 solid tumors, approximately 7.1% of cancers are caused by FGFR aberrations, including 66% by gene amplification, 26% by mutations and 8% by rearrangement [3]. As we have more understanding of the role of FGFR and its inhibitors in oncogenesis, there are gradual developments of FGFR targeted therapies in various types of cancers. FGFR inhibitors can be divided into small molecule oral tyrosine kinase inhibitors (TKIs), ligand traps, monoclonal antibodies and antibody drug conjugates [4]. Currently there are four FDA approved FGFR inhibitors in cholangiocarcinoma (CCA), urothelial tumors and myeloid/lymphoid neoplasms (MLNs). Several ongoing investigations are underway to evaluate its efficacy in other cancers and combination treatments.
2. FGF/FGFR signaling pathway
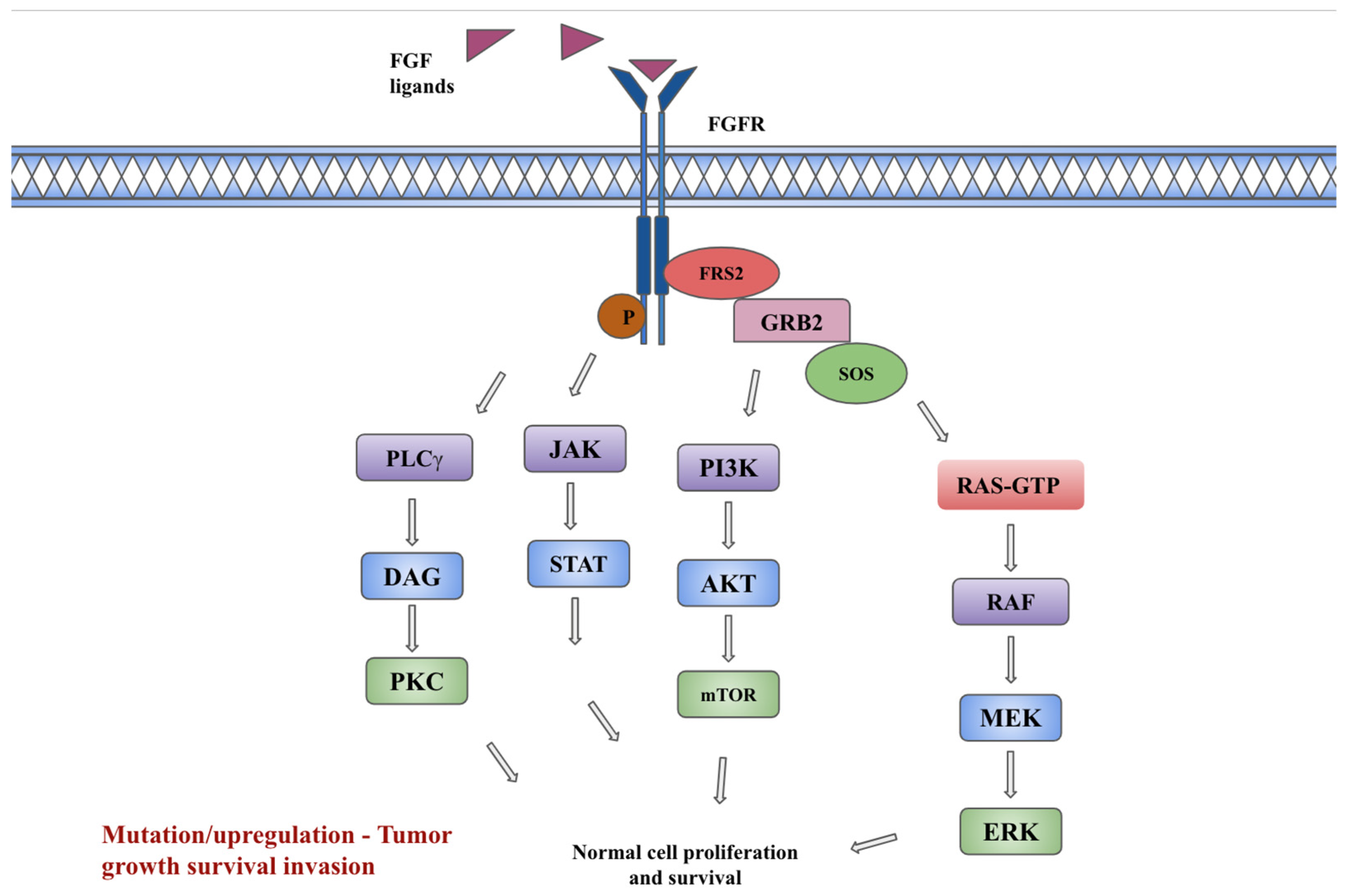
FGFs and their signaling pathway participates in a wide variety of cellular processes including cell proliferation, differentiation, tissue modeling, and angiogenesis. FGFR gene amplification, overexpression, point mutations or chromosomal translocations can lead to the development and progression of cancer [5]. In murine and mammalian genomes, 22 FGF ligands have been identified. Eighteen mammalian ligands have been identified and are divided into six subfamilies based on their phylogeny and sequence homology [5,6]. The subfamilies include 5 paracrine subfamilies, FGF1 (FGF1 and FGF2), FGF4 (FGF4, FGF5, and FGF6), FGF7 (FGF3, FGF7, FGF10, and FGF22), FGF8 (FGF8, FGF17, and FGF18), and FGF9 subfamily (FGF9, FGF16, and FGF20) and one endocrine subfamily FGF19 (FGF19, FGF21, and FGF23) [6]. FGFs show a high binding affinity for heparin and FGFRs. The binding of FGFs to the inactive monomeric FGFRs will trigger conformational changes of the receptors, resulting in dimerization and activation of the cytosolic tyrosine kinases by phosphorylating the tyrosine residues within the cytosolic tail of FGFRs. Canonical FGFs are tightly bound to heparin/heparin sulfate (HS) proteoglycans (HSPGs), which function to limit diffusion through the extracellular matrix (ECM) and serve as cofactors that regulate affinity for FGFR signaling [5].
Activation of FGFRs by FGFs cause downstream activation of RAS/RAF/MEK/ERK kinases in the MAPK (mitogen-activating protein kinase) pathway, PI3K-AKT-mTOR kinases in the PI3K (phosphoinositide 3 kinase) pathway, the JAK-STAT (Janus kinase/signal transducer and activator of transcription) pathway and protein kinase C (PKC) via phospholipase C-gamma (PLCγ) [1,7]. Regulation mechanisms to attenuate the aforementioned signaling pathways are available. The SPRY (Sprouty) family of proteins, which are MAPK phosphatases, inhibit RTK signaling by directly binding to RAF and blocking subsequent MAPK signaling. Also of note, FGF signaling activates these SPRY proteins, which may serve as a form of auto inhibition. Another negative regulation pathway is provided by a non-tyrosine kinase FGFR (FGFRL1) which can bind FGF ligands and possibly function as a decoy receptor or modulator of receptor turnover or signaling [5,8]. Intracellular signaling cascades, downstream of the signaling tyrosine kinase FGFRs, are also tightly regulated by specialized adaptor proteins such as FGFR substrate 2α (FRS2α). Each pathway regulates specific cellular behaviors such as cellular proliferation, embryonic development, endocrine homeostasis, and tissue repair. Inappropriate expression of FGF and improper activation of FGFRs are associated with various pathologic conditions, unregulated cell growth, and tumorigenesis [9].
3. FGFR Signaling diversity in cancer
Dysregulation of the FGFR pathway is associated with various human cancers and is considered an oncogenic signaling pathway. FGF oncogenic signaling mainly occurs through FGFR1-4 amplification, translocation and/or increased expression. FGFR alterations in cancers can be broadly classified into three main types of alterations including FGFR gene amplification, gene mutation and gene fusion. FGFR amplification is reported to be present in 66% of cancers with FGFR alterations, with FGFR1 amplification being the most common (42%) [3]. FGFR inhibition has been shown to reduce proliferation and induce cell death in a variety of in vitro and in vivo tumor models harboring FGFR aberrations, thus FGFR has been the target for cancer drug development [5].
3.1. Gynecologic cancers (Cervical, ovarian, and endometrial)
FGF1 amplification in ovarian cancer has been associated with promoting angiogenesis, reduced disease-free progression and overall survival (OS) [10,11]. It is also associated with chemotherapy resistance by reducing the transcriptional activity of p53 and increasing the expression of p21, leading to subsequent antiapoptotic activity in response to ovarian cancer therapies including etoposide and cisplatin [11]. Individual FGFRs including FGFR2 IIIb are overexpressed in ovarian cancer [12]. Taniguchi F et al. found that up-regulated FGFR2 expression has been shown to be potentially involved in the transformation of ovarian endometrioma to clear cell carcinoma of the ovary [13]. High levels of FGFR4 proteins have been reported in serous ovarian carcinomas and shown to be associated with poor patient survival [14].
Choi et al. studied the immunohistochemical expression of FGFR1, FGFR2, FGFR3, and FGFR4 in 336 cervical cancer patients, and confirmed that FGFR2, FGFR3, and FGFR4 expressions were important prognostic indicators in cervical cancer [15]. FGFR2 expression has been reported to be associated with cell growth and progression of cervical dysplasia.
FGFR alterations reported in endometrial cancers most frequently involve FGFR2 and thus it is thought to be a potential therapeutic molecular target [16]. In endometrial cancer cell lines harboring activating FGFR2 mutations, inhibition of FGFR kinase activity inhibited cell cycle progression, cell survival, and colony formation [16].
3.2. Gastrointestinal cancers
3.2.1. Cholangiocarcinoma
FGFR2 fusions in CCA were first identified in 2013 by Wu and colleagues when two fusions were found in patients with intrahepatic CCA (iCCA) [17]. Subsequent studies have shown that FGFR2 fusions occur nearly exclusively in iCCA compared to other biliary track cancers. The frequency of FGFR2 fusions in iCCA is estimated to be approximately 10–15% across multiple tumor genotyping studies [18,19]. FGFR2 fusions in fluke-associated and non-fluke associated CCA were 0.8% and 11.6%, respectively (p = 0.0006), suggesting that FGFR2 fusions might play a crucial role in the evolution of non-liver fluke-associated CCA, but less so in liver fluke-associated CCA [20]. FGFR2 fusions in iCCA have been associated with a better prognosis and younger age at diagnosis [19].
3.2.2. Gastric and gastroesophageal junction cancers
According to the comprehensive review by Helsten et al., FGFR1 mutations, FGFR2 amplifications and FGFR3 rearrangements are the most common FGFR alterations in gastric cancer and they may sometimes be discovered as co-occurring mutations [21]. In a study conducted in China, 5557 chinese patients with solid organ malignancies were evaluated for the presence of FGFR1-4 alterations via next generation sequencing (NGS) which included 254 cases of gastric cancer [22]. In this analysis, FGFR1-4 aberrations occurred in 12.2% of the gastric cancer samples with amplifications being the most prevalent alteration, followed by rearrangements and mutations [22]. Most frequent alterations were detected in FGFR2 gene, followed by FGFR1 gene, and to a lesser extent in FGFR3 and FGFR4 genes [22]. Another study identified 20% (5/25) of gastric cancer to carry the potentially targetable FGFR3-TACC3 (transforming acidic coiled-coil-containing protein 3) fusion [23]. In FGFR3–TACC3 fusion, the FGFR tyrosine kinase domain is fused to the TACC coiled-coil domain, resulting in constitutive activation of the fused receptor [24]. Amongst gastric cancer, FGFR2 gene amplification is the most common aberration (2–9%) which leads to FGFR2 protein overexpression and constitutive signaling of the FGFR pathway [23]. Among early-stage gastric cancer, FGFR2 amplifications are associated with higher grade tumor stage, more frequent lymph node dissemination and inferior overall survival [25,26]. In the metastatic setting, FGFR2 amplifications are also associated with inferior progression free survival (PFS) and OS in patients receiving platinum and fluoropyrimidine chemotherapy [26,27].
3.3. Urothelial cancers
In urothelial carcinoma, FGFR3 alterations have been documented in nearly 60% of low-grade noninvasive papillary urothelial carcinoma of the bladder, 35.6% of upper tract high-grade urothelial cancer, and 26.7% of overall urothelial carcinoma [28,29,30]. Base substitutions are the most common FGFR3 alterations (84%) seen in patients with urothelial cancer [31]. The most common FGFR3 alterations in advanced bladder cancer are activating missense mutations and in-frame FGFR3-TACC3 fusions [32]. The gain-of-function missense mutations in the extracellular and transmembrane domains of FGFR3 lead to ligand-independent dimerization between mutant receptors, whereas mutations in the intracellular kinase domain promote the activation of FGFR3 tyrosine kinase activity [28,33]. The missense mutations of FGFR3 are associated with higher FGFR3 mRNA and protein expression in bladder cancer [34].
3.4. Non-small cell lung cancer
In a study by Zhou et al., a total of 10,966 patients with non-small cell lung cancer (NSCLC) received NGS of tumor specimen or cell free tumor DNA [35]. FGFR aberrations, including mutations, fusions, and gene amplifications, were detected in 1.9% (210/10,966) of the population, with more prevalence in squamous cell carcinoma of lung compared to lung adenocarcinoma [35]. More than half of the patients who carried FGFR activating and transforming mutations had concurrent dominant mutations in PI3K pathway genes, including PIK3CA and PIK3R2. This highlights an intriguing molecular feature and potential development of combination therapies targeting both FGFR and PI3K pathways in FGFR-positive NSCLC patients exhibiting activated PI3K pathways. FGFR amplification was detected in 24 patients with FGFR1 amplification being the most common alteration while FGFR fusions were detected in 0.11% of the patients [35]. It is worth noting that half of the patients with FGFR fusions also carried epidermal growth factor receptor (EGFR) aberrations [35]. A study by Ou et al. suggested that FGFR fusions may act as a mechanism of acquired resistance to EGFR inhibitors in patients who were previously treated with EGFR TKIs [36]. This sugguests that concurrent FGFR and EGFR inhibition could overcome the acquired resistance of EGFR inhibitors. A low frequency of FGF19 amplifications were also noted. As FGF19 encodes the ligand for FGFR4, FGF19 amplifications corresponds with constitutive activation of FGFR4 dependent signaling activation which can act as an oncogenic driver and a potential therapeutic target.
3.5. Breast cancers
The amplification of FGFR1 represents the most frequent genomic alteration in breast cancer, with FGFR2-4 gene amplifications being less commonly seen [37]. The genomic analysis of The Cancer Genome Atlas (TCGA) and the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) databases has confirmed that the amplification of FGFR1 is the highest among the FGFR family members, as it occurs in nearly 14% of breast cancer patients [38]. Patients with either FGFR1 overexpression or copy number gain exhibit reduced survival rates compared to the remaining cohort of patients [38]. FGFR1 amplification is also shown to be responsible for the resistant mechanisms to endocrine therapy in breast cancer via both aberrant ligand-dependent and ligand-independent signalings [39]. FGFR1 amplification and overexpression also contribute to the resistance of breast cancer cells to the CDK4/6 inhibitors used in combination with endocrine therapy in either in vitro or in vivo patient-derived xenograft models [40]. A recent study has also demonstrated that FGFR genetic aberrations could forecast the occurrence of brain metastases in breast cancer patients [41]. On the other hand, FGFR 2-4 amplifications represent approximately 1–2% of all breast cancer cases [16]. FGFR gene fusions such as FGFR3-TACC3 may trigger cancer development allowing the activation of FGFR3 tyrosine kinase [42]. The expression of FGFR3 is higher in a subset of tamoxifen-resistant estrogen receptor (ER) positive breast cancers with respect to tamoxifen-sensitive ER+ breast tumors. FGFR3 stimulation has been found to trigger resistance to tamoxifen via the activation of the PLCγ signaling cascade [43].
3.6. Glioblastoma
Yamaguchi et al. demonstrated that expression of FGFR1 increases with WHO grade in astrocytoma [44]. FGFR1α is the predominant isoform in normal brain and low-grade gliomas, while high-grade gliomas show a shift towards the expression of FGFR1β [44,45]. Loss of the FGFR1α exon increases the receptor-ligand affinity and the sensitivity of tumor cells to FGFs present in their environment, thus contributing to glioblastoma (GBM) development [46]. FGFR1 expression in malignant glioma has also been associated with increased migration of cancer cells [47]. FGFR1 signaling also promotes radioresistance in glioma cell lines through PLCγ1 and hypoxia-inducible factor 1-alpha (HIF1α) [48]. High expression of ephrin type-A receptor 4 (EPHA4) gene in glioma cells was found to potentiate FGF2–FGFR1 signaling and promote cell growth and migration through the AKT/MAPK and RAC1/CDC42 pathways, respectively [48]. In summary, FGFR1 is a key regulator of tumor growth, invasion, and therapy resistance in malignant glioma. While FGFR1 is mainly expressed on neurons, FGFR2 is the primary FGFR on astrocytes [49]. In contrast to FGFR1, FGFR2 expression decreases with glioma grade. Reduced expression of FGFR2, as well as its IIIb and IIIc isoforms, are associated with a higher tumor grade and poorer survival in glioma patients [50]. Loss of FGFR2 is associated with a loss of chromosome 10q, which in and of itself carries an unfavorable prognosis [51]. In a small subset of GBM patients, fusion of the FGFR3 and TACC3 gene also generates an oncogenic FGFR3 form [24].
3.7. Gastrointestinal stromal tumors and other soft tissue sarcomas
3.7.1. Gastrointestinal stromal tumors
In the subset of gastrointestinal stromal tumors (GIST) without any alterations of the KIT/PDGFRA/SDH/RAS-pathway, targeted sequencing of so called quadruple wild-type GIST has shown the presence of activating mutations or gene fusions involving FGFR1. One case harbored the FGFR1–Hook homolog 3 (HOOK3) fusion and two patients carried FGFR1–TACC1 fusion transcripts [52,53]. These fusions have not been directly confirmed to be oncogenic in GIST, however, they have been reported in other types of cancers. FGFR pathway has also been shown to be related to the imatinib resistance. FGF2 is overexpressed in imatinib-resistant GIST cells [54]. The interaction of FGF2 with FGFR1 and FGFR3, respectively, restores MAPK signaling during treatment with imatinib and proto-oncogene c-KIT phosphorylation in imatinib-resistant models [55]. A gain of function mutation in FGFR2 has also been suggested as an additional potential mechanism associated with imatinib resistance [56].
3.7.2. Rhabdomyosarcoma
Taylor et al. described aberrations in FGFR4 in primary human rhabdomyosarcoma (RMS) [57]. In RMS cell line models, two of these mutations were shown to promote FGFR4 autophosphorylation, STAT3 phosphorylation, activation of cell cycle and DNA replication pathways. FGFR4 mutations increased proliferation, invasion, and metastatic potential. High expression of FGFR4 mRNA was also associated with worse survival in a clinical cohort of 146 patients [57]. In alveolar RMS cells, FGFR4 stimulation induces degradation of the pro-apoptotic molecule Bcl-2-like protein 11 (BIM) and upregulation of its antagonist B-cell lymphoma-extra-large (Bcl-XL) proteins [58]. The FGFR pathway has been implicated in the development and progression of several other soft tissue sarcoma (STS) subtypes. Comprehensive analysis by Chudasama et al. ultimately revealed FGFR1 copy number gain and overexpression in leiomyosarcomas (LMS), undifferentiated pleomorphic sarcomas (UPS) and de-differentiated liposarcoma (DDLPS) as well as other sarcoma subtypes [59]. In cellular models of STS from various histologies, the MAPK signaling axis was shown to be the most critical effector pathway mediating FGFR1 signaling [59]. FGFR1 was also shown to be overexpressed in Ewing’s sarcoma, a high-grade mesenchymal malignancy of bone or soft tissue [60].
3.8. Head and Neck Cancers
FGFR aberrations are one of the most frequent RTK genomic alterations in head and neck squamous cell carcinoma (HNSCC), making FGF/FGFR axis a promising target for the development of new treatment options for HNSCC patients [61]. FGF/FGFR genomic alterations can be divided into ligand-dependent aberrations, which involves FGF genomic alterations, and ligand-independent aberrations that consist of FGFR aberrations. FGF/FGFR gene deregulation, including gene amplification, mutation, and chromosomal rearrangement, have been detected in approximately 30–50% of HNSCC patients based on the genomic analysis of TCGA [62,63]. Among them, FGFR1 gene amplification, FGF3/4/19 gene amplifications, and FGFR3 mutations are the most frequent FGF/FGFR genomic alterations [61]. FGF2 is the most well-studied FGF ligand in HNSCC. FGF2 has been reported to be highly expressed in up to 60% of HNSCC [64]. Marshall et al. noted that FGF2 are frequently co-expressed with FGFRs in the majority of HNSCC cell lines they tested, which can form an autocrine loop to drive oncogenesis [65]. FGFR1 gene amplification is predominantly detected in HPV-negative HNSCC patients and is more prevalent in laryngeal (LPSCC) and hypopharyngeal squamous cell carcinoma (HPSCC) [66]. Genomic alterations in FGFR2 are less prevalent than FGFR1 and FGFR3 in HNSCC. Unlike FGFR1 gene amplification, FGFR2 mutations are mainly enriched in HPV-positive HNSCC patients [67]. FGFR3 mutations have been implicated in about 5.8–24% of HNSCC patients. FGFR3-TACC3 fusion is also reported in about 2.5–3.7% of HNSCC patients [68]. The prognostic value of FGFR3 overexpression is also controversial and there has been accumulating evidence showing that FGFR3 expression is not related to survival. FGFR4 is much less studied than other FGFRs in HNSCC even though FGFR4 is highly expressed in 16–39% of HNSCC patients [69]. FGFR4 overexpression has been reported to be associated with poorer overall survival of HNSCC patients in several studies [70].
4. Generations of FGFR inhibitors
Given similar structure to adenosine triphosphate (ATP), oral TKIs compete for the ATP binding cleft of the kinase domain on the FGFR receptor. Reduction of tyrosine kinase phosphorylation by competitive reversible inhibition leads to the blockade of multiple downstream pathways, thus causing inhibition of cell proliferation [4]. There are high similarities in ATP binding site of intracellular kinase domains among RTK family. Thus, first generation FGFR inhibitors were non-selective inhibitors against multiple kinases (PDGFRs, VEGFRs, KIT and RET), which include ponatinib, lucitanib, dovitinib, lenvatinib [4,7]. Multi-kinase FGFR inhibitors can lead to various adverse effects due to low specificity and multi-target effects, thus leading to the development of more specific and selective TKIs for FGFR pathway [4].
Second generation FGFR inhibitors are more selective including FGFR 1-3 inhibitors (pemigatinib, infigratinib, AZD4547, Debio1347), FGFR 1-4 inhibitors (erdafitinib, rogaratinib) and selective FGFR 4 inhibitor (fisogatinib) [7]. Despite their potential anti-tumor activity, they are ineffective at overcoming commonly acquired FGFR gatekeeper mutations (FGFR1 V561M, FGFR2 V564F, FGFR3 V555M, FGFR4 V550M/L) and other mutations including FGFR1 N546K mutation and FGFR2 N550H mutations [71,72,73]. Third generation FGFR inhibitors such as futibatinib (TAS-120) can covalently bind to a highly conserved cysteine residue (Cys488 in FGFR1c) in FGFR kinase, and cannot be readily replaced by ATP, thus prolonging the duration of activity, and overcoming secondary FGFR2 resistance mutations in patients with infigratinib or Debio1347 resistance [7].
FDA approved FGFR inhibitors
The benefit of targeting FGFR has been demonstrated in urothelial cancers, CCA and MLNs. Currently there are four FDA approved FGFR inhibitors but infigratinib has been withdrawn from the market in the United States [74].
Pemigatinib is an oral selective reversible ATP competitive FGFR 1-3 TKI. FDA approved pemigatinib on April 17, 2020 as a second line for the treatment of adults with previously treated, unresectable locally advanced or metastatic CCA with FGFR2 fusion or rearrangements [75]. The approval was based on a single arm phase II FIGHT-202 trial, which evaluated patients with locally advanced unresectable or metastatic CCA with FGFR2 gene fusions or rearrangements, who progressed on at least one prior therapy [76]. Patients were divided into three cohorts including cohort 1 with FGFR2 fusions or rearrangements, cohort 2 with other FGF/FGFR alterations, and cohort 3 with no FGF/FGFR alterations. Objective response rate (ORR) was 35.5% (95% CI 26.5–45.4) in cohort 1 patients with FGFR2 fusions or rearrangements with duration of response (DOR) of 9.1 months while patients in cohort 2 and 3 had 0% ORR [76]. Most common adverse effects were hyperphosphatemia, arthralgia, stomatitis, hyponatremia, abdominal pain, fatigue, pyrexia, cholangitis, and pleural effusions [76]. Updated results from FIGHT-202 presented at ASCO 2021 showed independent, centrally confirmed durable responses and sustained tolerability; with ORR of 37.0% with median OS of 17.5 months (95% CI 14.4-22.9) with higher OS in responders (30.1 months vs 13.7 months) [77]. A phase III FIGHT 302 study is currently being evaluated to use pemigatinib as a first line treatment in comparison with gemcitabine and cisplatin in unresectable or metastatic CCA patients with FGFR2 alterations (fusions/rearrangements) [78,79].
Pemigatinib was also investigated in another landmark phase II FIGHT-203 trial in patients with relapsed/refractory (R/R) MLNs with FGFR1 rearrangements regardless of prior lines of treatment [80]. MLN with FGFR1 rearragements could present as chronic or blast phase with involvement of bone marrow and/or extramedullary disease (EMD). The most common FGFR1 fusion partner genes were 13q12/ZMYM2 (45.5%) and 22q11/BCR (24.2%). Complete responses (CR) were seen in 64.7% of evaluated patients (77.4% per Central Review Committee review). Complete cytogenic response was seen in 72.7% of patients with 83.3% CR in chronic phase patients without EMD and 38.5% in blast phase with or without EMD [80]. This led to FDA approval of pemigatinib for adults with R/R MLNs with FGFR1 rearrangements on August 26, 2022 [81]. It is a treatment option for patients with MLN with FGFR1 rearrangements ineligible for hematopoietic stem-cell transplantation (HSCT) or it may facilitate bridging to HSCT in eligible patients.
Infigratinib is another selective oral reversible ATP competitive FGFR 1-3 TKI. FDA granted accelerated approval to infigratinib on May 28, 2021, as a second line for adults with previously treated unresectable locally advanced or metastatic CCA with FGFR2 fusions or rearrangements [82]. Approval was based on the phase II CBGJ398X2204 trial of patients with advanced or metastatic CCA with FGFR genetic alterations who have received at least one prior line of treatment [82,83]. ORR was 23.1% (95% CI 15.6-32.2) including 1 CR and 24 partial responses (PR) with DOR of 5.0 months and median PFS of 7.3 months [84]. Infigratinib was also compared to gemcitabine plus cisplatin as a front-line setting in patients with advanced ormetastatic CCA with FGFR2 gene fusion/translocation in phase III PROOF trial (NCT03773302). However, the study was terminated as infigratinib was withdrawn from the market on March 31, 2023, and due to difficulties in recruiting and enrolling study participants for required confirmatory trial. Termination was not because of safety or efficacy concerns [85].
Erdafitinib is an oral reversible inhibitor of FGFR 1-4 which was FDA approved on April 12, 2019 as a second line for locally advanced or metastatic urothelial cancers with FGFR 2 or 3 alterations who has progressed on prior platinum containing chemotherapy including within 12 months of neoadjuvant or adjuvant chemotherapy [86]. The appproval was based on the phase II BLC2001 trial in which erdafitinib was evaluated in pretreated patients with locally advanced and unresectable or metastatic urothelial cancers with FGFR alterations [87]. Participants had FGFR3 gene mutations (R248C, S249C, G370C, Y373C) or FGFR gene fusions (FGFR3-TACC3, FGFR3-BAIAP2L1, FGFR2-BICC1, FGFR2-CASP7) [87]. ORR was 40% (95% CI, 31-50) with CR in 3% and PR in 37%. Median PFS was 5.5 months (95% CI 4.2-6.0) and median OS was 13.8 months (95% CI, 9.8 - not reached) [87]. The response rate among patients with FGFR mutations were 49% whereas response rate among those with FGFR fusions were 16% [87]. Response rates are not affected by particular FGFR mutation. There seems to be less response to immunotherapy in patients with FGFR mutations or fusions but 59% had a response to erdafitinib after failure of immunotherapy [87]. It should be noted that ORR appears higher in patients with previous exposure to immunotherapy compared to those with no exposure (59% vs 40%) [87]. Updated results in 2022 showed durable efficacy with 40% ORR at median follow-up of 24 months and manageable safety profile [88].
Erdafitinib is currently being evaluated in a confirmatory phase III THOR trial evaluating metastatic or unresectable urothelial carcinoma patients with FGFR 2/3 alterations (mutations/fusions) who progressed after 1-2 prior lines of treatment to get full FDA approval and application is being submitted as of August 2023 [89]. THOR trial divides patients into two cohorts. Cohort one compares erdafitinib vs standard of care (docetaxel or vinflunine) after at least one prior line of treatment including an anti-PD-L1 (programmed death-ligand 1) agent; while cohort two compares erdafitinib vs pembrolizumab after one prior line not containing PD-L1 agent [90,91]. 70% of patients had visceral metastases and 90% were PD-L1 low (Combined positive score - CPS <10). Loriot et al., recently reported analysis of Cohort 1 after a median follow-up of 15.9 months [90,91]. Primary end point OS was met with 12.1 months in erdafitinib group vs 7.8 months in chemotherapy group (Hazard ratio for death (HR) 0.64; 95% CI 0.47-0.88; p=0.005) with improved PFS of 5.6 vs 2.7 months (HR 0.58; 95% CI: 0.44 to 0.78; p<0.001) and improved ORR of 46% vs 12% (relative benefit, 3.94; 95% CI 2.37-6.57) in chemotherapy group [90,91]. More treatment related adverse effects leading to dose reductions were observed in erdafitinib group (66% vs 21%) but more discontinuations observed in chemotherapy group (8.1% vs 13%) and grade 3 or 4 adverse effects of central serous retinopathy were seen in 2.2% of patients in erdafitnib group [90,91]. However, no significant OS was observed cohort 2 between erdafitinib vs pembrolizumab with median OS of 10.9 months in erdafitinib vs 11.1 months in pembrolizumab group (HR 1.18, 95% CI 0.47–0.88; p = 0.18) even though erdafitinib had numerically longer PFS of 4.4 months compared to 2.7 months in pembrolizumab group (HR 0.88, 95% CI 0.70-1.10) and higher ORR of 40.0% compared to 21.6% with pembrolizumab group (95% CI 1.32-2.39; p < 0.001) [92].
Futibatinib (TAS-120) is a next generation, highly selective irreversible TKI that covalently binds FGFR 1-4 and it has been shown to overcome the acquired resistant mutations observed at progression with other FGFR inhibitors including pemigatinib and infigratinib [93]. Futibatinib had accelerated FDA approval on September 30, 2022 for previously treated adults patients with unresectable, locally advanced or metastatic intrahepatic CCA with FGFR2 fusions or rearrangements. It is the third FGFR inhibitor to get approval for patients with CCA who harbor FGFR2 fusions or rearrangements [94]. The approval is based on data from the phase II single arm FOENIX-CCA2 trial of patients with unresectable or metastatic FGFR2 fusion or rearrangement positive iCCA. ORR was 42% (95% CI, 32-52) with DOR of 9.7 months (95% CI 7.6 to 17.0) and disease control rate (DCR) of 83% [93]. Median OS was 21.7 months (95% CI, 14.5-not reached) and median PFS was 9.0 months (95% CI, 6.9-13.1) [93]. Currently, futibatinib is being compared to cisplatin and gemcitabine as a first line treatment in phase III trial of advanced CCA harboring FGFR2 gene rearrangements in the FOENIX-CCA3 trial [95].

Table 1.
Current FDA approved FGFR inhibitors with supporting trials for FDA approval.
| Drug | Trial | Phase | Study population, number (n) | ORR (%) | mDOR months | mOS months | mPFS months | FDA approval | Adverse effects |
|---|---|---|---|---|---|---|---|---|---|
|
Pemigatinib FGFR 1-3 inhibitor |
Abou-Alfa et al. FIGHT-202 trial [76,77] |
II |
Locally advanced, unresectable or metastatic CCA with FGFR2 gene fusion or rearrangements, progressed on at least one prior line of therapy. n = 146 |
35.5 | 9.1m | 17.5 |
- | April 17, 2020 as second line | Hyperphosphatemia, alopecia, dysgeusia, diarrhea, fatigue, stomatitis, dry mouth, arthralgia, hyponatremia, abdominal pain, fatigue, pyrexia, cholangitis, and pleural effusion |
| Gotlib et al. FIGHT-203 trial [80] |
II | MLNs with FGFR1 rearrangement regardless of prior lines of treatment. n = 34 | 64.7 (CR) |
Not reached | - | - | August 26, 2022 as second line | Hyperphosphatemia, alopecia, diarrhea, stomatitis, anemia, and pain in extremity | |
|
Infigratinib FGFR 1-3 inhibitor |
Javle et al. CBGJ398X2204 trial [84] |
II | Locally advanced, or metastatic CCA with FGFR2 fusions or rearrangements, progressed on at least one prior line. n = 108 | 23.1 | 5 | - | 7.3 | May 28, 2021 as second line | Hyperphosphatemia, eye disorders, hyponatremia, stomatitis, and fatigue |
|
Erdafitinib FGFR 1-4 inhibitors |
Siefker-Radtke et al. BLC2001 trial [88] |
II | Locally advanced, unresectable, or metastatic urothelial cancers with FGFR alterations, progressed on at least prior line or within 12 months after neoadjuvant or adjuvant chemotherapy. n = 99 | 40 | 5.6 | 13.8 | 5.5 | April 12, 2019 as second line | Hyperphosphatemia, stomatitis, diarrhea, and dry mouth, hyponatremia, and asthenia |
|
Futibatinib FGFR 1-4 inhibitor |
Goyal et al. FOENIX-CCA2 trial [93] |
II | Locally advanced, unresectable, or metastatic iCCA with FGFR2 fusions or rearrangements who progressed on at least one prior line. n=109 | 42 | 9.7 | 21.7 | 9.0 | September 30, 2022 as second line | Hyperphosphatemia, alopecia, dry mouth, diarrhea, dry skin, fatigue, increased aspartate aminotransferase level, and stomatitis |
5. Other FGFR inhibitors
5.1. Selective FGFR inhibitors
Several other FGFR specific inhibitors have been developed and investigated in numerous trials with variable efficacies and toxicities but none of them has FDA approval yet.
Rogaratinib (BAY1163877) is an oral FGFR 1-4 inhibitor which has shown to have favorable toxicity profile in phase I trials of solid tumors overexpressing FGFR1-3 alterations including HNSCC, NSCLC and urothelial carcinoma [96]. It was then evaluated in phase II/III randomized FORT-1 trial compared to chemotherapy (docetaxel, paclitaxel or vinflunine) in patients with locally advanced or metastatic FGFR1-3 mRNA-overexpressing urothelial carcinoma previously treated with platinum based therapy [97]. Primary end point OS was 8.3 months in rogaratinib vs 9.8 months in chemotherapy group (HR 1.11, 95% CI 0.71-1.72, p=0.67) with ORR of 20.7% in rogaratinib vs 19.3% in chemotherapy group [97]. It is currently being evaluated in phase I/II trials in urothelial carcinoma and sarcomas/GISTs (NCT03473756, NCT04595747).
Derazantinib (ARQ-087) is an oral selective FGFR 1-3 inhibitor, which has been evaluated in several phase I/II trials, including advanced/inoperable CCA with FGFR2 gene fusion with ORR of 20.7% and DCR of 82.9% [98]. In a phase II FIDES-01 trial, derazantinib was evaluated in previously treated iCCA patients with FGFR2 mutations or amplifications [99]. Interim analysis showed ORR of 8.7% with stable disease of 65.2% and DCR of 73.9% with higher responses seen in FGFR2 fusion with ORR of 21.4% and DCR of 75.7% [99]. Derazantinib was also investigated in another phase Ib/II FIDES-02 trial as a single agent in previously treated patients with metastatic urothelial cancer harboring FGFR1-3 alterations, but derazantinib monotherapy did not meet its primary end point with ORR rate of 8.2% (95% CI 2.2-19.6) based on 4 PR (all with FGFR3 S249C mutation/FGFR3-TACC3 fusion), and median DOR of 6.9 months [100]. FEDES-03 trial of derazantinib vs paclitaxel and ramucirumab or atezolizumab in human epidermal growth factor receptor (HER2) negative gastric adenocarcinoma with FGFR2 alterations was terminated due to administrative issues [101]. It is currently being evaluated in combination with atezolizumab in metastatic iCCA and other advanced tumors with FGFR2 alterations (NCT05174650).
Lirafugratinib (RLY4008) is an oral highly selective irreversible FGFR2 inhibitor with activity across FGFR2 alterations and resistance mutations [102]. It has been shown to induce responses without clinically significant off isoform toxicities (hyperphosphatemia, diarrhea), making it a potential FGFR targeted therapy [102]. It is currently being evaluated in phase I/II trial of patients with unresectable or metastatic CCA and solid tumors harboring FGFR2 gene fusion, mutation, or amplification in the REFOCUS trial (NCT04526106). It has so far shown high and durable responses in patients with FGFR2 fusion or rearrangement positive CCA patients in 3 case studies of patients from REFOCUS trial [102].
Zoligratinib (Debio1347) is an oral highly selective FGFR 1-3 inhibitor which has shown anti-tumor efficacy (ORR of 12% with 20% stable disease) and tolerability in phase I dose-escalation trial in patients with advanced solid tumors harboring FGFR alterations [103].
Fisogatinib (BLU554) is a highly potent and selective oral FGFR4 inhibitor which covalently binds a cysteine residue found in FGFR4 [104]. Fisogatinib was evaluated in phase Ib/II trial in patients with locally advanced or metastatic hepatocellular carcinoma (HCC) [104]. It showed ORR of 17% in patients with FGF19 positive patients with median DOR of 5.3 months and median PFS of 3.3 months at maximal tolerable dose of 600 mg daily [104].
Aflofanib (RPT835) is a novel selective allosteric FGFR2 inhibitor, which has been evaluated in breast, ovarian and gastric cancers [105,106]. In a phase Ib trial of previously treated patients (at least one prior line) with metastatic gastric adenocarcinoma, it showed acceptable tolerability and some clinical efficacy with ORR rate of 9.5% and DCR of 71.4% [105].
Roblitinib (FGF401) is a selective reversible covalent inhibitor of FGFR4. Roblitinib in combination with spartalizumab showed ORR Of 16% in phase I/II trial of patients with HCC or FGFR4/KLB expressing tumors [107]. Most frequent toxicities were diarrhea, increased aspartate aminotransferase and alanine aminotransferase levels [107].
Fexagratinib (AZD4547) is an oral selective FGFR1-3 inhibitor, which has been evaluated in several phase I/II studies. In the phase II trial of previously treated patients with FGFR pathway activated stage IV squamous lung cancer (Lung-MAP substudy), fexagratinib has shown to have acceptable tolerability but has poor efficacy and minimal DOR with only one PR each in patient with FGFR3 S249C mutation and in patient with FGFR1 amplification [108]. In another phase II basket trial (NCI-MATCH) of tumors including breast, urothelial and cervical cancers harboring FGFR 1-3 aberrations, fexagratinib did not meet its primary end point and demonstrated ORR of only 8% (90% CI 3–18%) with responses observed only in patients with FGFR 1-3 point mutations or fusions. Stable disease was seen in 37.5% of patients (90% CI, 25.8% to 50.4%) [109]. Fexagratinib in combination with anastrozole or letrozole showed ORR of 10% in a single arm phase II study of ER positive metastatic breast cancer patients who progressed on prior hormone therapy, meeting its primary end point. However, 20% of patients had reversible and 2% had irreversible asymptomatic retinal pigment epithelial detachments [110].
5.2. Multitarget tyrosine kinase inhibitors including FGFR
Some of the multi-TKIs also target FGFR at variable degrees. Please see Table 2 for examples of multi-TKI which have been investigated and utilized in various types of cancers.
5.3. FGFR ligand trap
FGFR ligand traps facilitate the binding and trapping of FGF ligand with the decoy receptors that expresses the extracellular kinase domain only, thus preventing FGF ligand binding to FGFR receptor and downstream activation of FGF pathway [4]. FP1039 (GSK3052230) is a soluble FGFR1 decoy receptor, formed by the fusion of the FGFR1 extra-cellular domain and the human immunoglobulin G, IgG1 Fc fragment. It has shown to be effective against FGFR2 mutated endometrial and lung cancer cells as well as mesothelioma cell lines with FGFR1 amplification in pre-clinical studies [116,117,118]. The phase 1b trial evaluated FP1039 in combination with pemetrexed and cisplatin in 36 patients with treatment-naive, unresectable malignant pleural mesothelioma at doses of 10, 15 and 20 mg/kg. It demonstrated overall ORR of 39% (95% CI 23.1-56.5), and DCR of 86% while ORR was highest at 44% (95% CI 24.4-65.1) in patients treated with 15 mg/kg of FP1039 [119].
5.4. FGFR monoclonal antibody
FGFR monoclonal antibodies are developed to target the extracellular domain of FGFR and interfere with ligand binding and receptor dimerization.
Vofatamab (B-701, MFGR1877S) is a fully human IgG1 monoclonal antibody against FGFR3 [120]. Evaluation of vofatamab in phase I trials of R/R multiple myeloma patients with t(4;14) translocation causing FGFR3 overexpression and advanced solid tumors showed tolerable safety profile but without impressive response [121,122]. Stable disease was best response achieved in 6/14 myeloma patients and 9/26 patients in advanced solid tumors including 5 patients with urothelial carcinoma, 2 patients with adenoid cystic carcinoma and 2 patients with carcinoid tumors [121,122]. In the phase Ib/II FIERCE-21 trial, vofatamab was tolerated either as monotherapy or in combination with docetaxel in previously treated metastatic urothelial cancer patients (at least one prior line with chemotherapy including taxanes) with FGFR 3 alterations (mutations/fusions). However, it showed minimal single-agent activity with ORR of 11% in heavily pretreated patients [120]. The most common adverse events were decreased appetite, diarrhea, asthenia, hypotension and increased creatinine [120]. The phase Ib/II FIERCE-22 trial of vofatamab in combination with pembrolizumab in previously treated metastatic urothelial cancers with FGFR alterations showed ORR of 30% (more than reported response of 20% to immune check point inhibitors (ICI)). It may be due to the fact that FGFR inhibition could enhance antigen expression and antigen T-cell clonality, making ICI more effective [123]. Another novel agent [225Ac]-FPI-1966 is a targeted alpha therapeutic that comprised of vofatamab, a bifunctional chelate, and actinium-225, an alpha particle emitting radionuclide and it is currently being investigated in phase 1 trial of advanced solid tumors (NCT05363605) with FGFR3 alterations.
Bemarituzumab (FPA144) is a first-in-class recombinant FGFR2b targeting humanized IgG1 kappa monoclonal antibody [124]. It binds to the third immunoglobulin region of the FGFR2b receptor, blocks the activation of FGFR2b and downstream FRS2 (fibroblast growth factor receptor substrate 2) phosphorylation as well as enhances the antibody-dependent cellular toxicity against tumor cells that express FGFR2b [124]. In the phase II FIGHT trial of bemarituzumab in combination with mFOLFOX6 (modified 5-fluorouracil, leucovorin, and oxaliplatin) as a first line in FGFR2b positive advanced gastric/gastroesophageal (GE) junction adenocarcinomas, patients with FGFR2b overexpression irrespective of circulating DNA gene amplification showed improved OS. Bemarituzumab plus chemotherapy arm had better ORR (53% vs 40%) and better OS (19.2 months vs 13.5 months) compared to chemotherapy alone arm [125]. OS was even higher with 25.4 months in a subset of patients with ≥10% FGFR2b overexpression by immunohistochemistry (2/3+) compared to 11 months in chemotherapy alone group [125]. Analysis of PFS was reported in 2022, with median PFS of 9.5 months in bemarituzumab and chemotherapy group vs 7.4 months in chemotherapy alone group, which met its primary end point but was not statistically significant [124]. However, it showed promising clinical efficacy [124]. The most common grade 3 or worse adverse effects were neutropenia, cornea disorder, and stomatitis. However hyperphosphatemia or retinal detachments were not seen as much as in other FGFR-TKIs [124]. Currently, bemarituzumab in combination with mFOLFOX is being compared to mFOLFOX alone in untreated, unresectable, locally advanced or metastatic FGFR2b overexpressed gastric or GE junction adenocarcinoma in the phase III FORTITUDE-101 trial (NCT05052801). In the phase III FORTITUTE-102 trial (NCT05111626), bemarituzumab with mFOLFOX and nivolumab is being compared to mFOLFOX and nivolumab in previously untreated advanced gastric and GE junction cancer with FGFR2b overexpression [126].
5.5. Antibody drug conjugates
LY3076226 is an antibody drug conjugate (ADC) comprised of human IgG1 monoclonal antibody against FGFR3 linked to the cytotoxic payload, maytansine derivative ravtansine (DM4) [117]. LY3076226 was evaluated in first in human phase I trial of patients with advanced or metastatic cancer. It was well tolerated with no dose limiting toxicities and mostly grade 1 or 2 adverse effects but lacks efficacy (ORR 0%) [117].
Aprutumab ixadotin (BAY1187982) is another ADC which consists of a fully human anti-FGFR2 monoclonal antibody attached to an auristatin-like cytotoxic payload [127]. Even though it showed efficacy with inhibition or regression of gastric and breast cancer xenograft models leading to phase I trial, it was terminated early due to poor tolerability with dose limiting toxicities including proteinuria, nephropathy, thrombocytopenia and epithelial microcytosis [127]. Therefore, there is a need for improved clinical models to predict the effects of investigational ADCs and their metabolites in humans during preclinical development.
Another ADC comprised of tetravalent anti-FGFR 1 antibody (T-Fc) linked to cytotoxic drug, monomethyl auristatin E (MMAE, a tubulin inhibitor) linked via lysosomally cleavable dipeptide, valine-citrulline (vc), has been recently under development [128]. T-Fc mediates the clustering of FGFR1, leading to the uptake of FGFR1-T-Fc complexes by the induction of clathrin-independent endocytic routes and they have shown to have effective drug delivery and internalization by FGFR1 producing cells leading to the cells’ death [128]. There is potential for development of ADC with highly effective internalization into FGFR producing cells and effective killing of cancer cells with tolerable toxicity.
6. Resistance mechanisms to FGF/FGFR pathway inhibitors
The underlying mechanisms that potentiate FGF/FGFR signaling pathway related resistance can be associated with various factors. These resistance mechanisms include overexpression of ligands and receptors, downregulation of negative regulators, epithelial-mesenchymal transformation, nuclear translocation and activation of downstream signaling [129]. Mutations at the gatekeeper residue of FGFR is one of the first mechanism of resistance to FGFR inhibitors. Gene amplification translates into FGFR overexpression, causing receptor accumulation and continuous activation of the down-stream signaling pathways, including ligands-dependent and independent pathways [130]. Additionally, increased FGF expression from the tumor cell or microenvironment can overstimulate FGFR and saturate their down-stream signaling pathways. Furthermore, FGFR inhibition can propagate negative feedback mechanisms that lead to downstream inhibitor resistance. FGFR inhibition induced the activation of Erb-B2 receptor tyrosine kinase 3 (ERBB2) and to a lesser extent EGFR. As a consequence, PI3K-AKT signaling is activated, thereby possibly blunting the effects of FGFR inhibitors [131].
Another resistance mechanism is thought to be that FGF/FGFR signaling may contribute to epithelial-mesenchymal transition (EMT), which is the morphological changes defined by cells becoming more spindle shaped, leading to more potential resistance. Grygielewicz and colleagues observed these morphological changes in the gastric cancer cell line SNU-16 (FGFR2 amplification), following chronic exposure to medications like infigratinib [132]. Another mechanism for resistance is nuclear translocation of FGF or FGFR. FGFR-TKI resistance events can be directly propagated by FGFR gene fusion. Kim and colleagues identified a novel FGFR2-ACSL5 fusion from a metastatic gastric cancer patient with FGFR2 amplification through RNA sequencing [131,133]. Intriguingly, at the beginning of FGFR inhibitor treatment, the patient showed strong sensitivity to the study drug, and no FGFR2-ACSL5 fusion was found in vivo. However, as exposure persisted, drug resistance was detected along with the FGFR2-ACSL5 fusion gene. Gene fusions can also indirectly lead to FGFR inhibitor resistance. With JHDM1D-BRAF fusion, constructive dimerization of the fusion protein is enhanced, and is accompanied by the activation of downstream MAPK pathway. This led to the disappearance of FGFR2 phosphorylation, and a decrease in FGFR2 expression [71]. Further mechanisms of resistance include downregulation of negative feedback proteins, such as SPRY, which leads to continuous activation of FGF/FGFR signaling.
7. Future Therapeutic Combinations with FGF/FGFR Inhibitors
Given that a single agent FGFR inhibition treatment could cause intrinsic and acquired resistance, the combination of FGFR inhibitors with chemotherapy, immunotherapy and targeted therapy combination could be considered for both synergistic effect and reduction of drug resistance development [73]. FGFR inhibitors can enhance tumor sensitivity to chemotherapy drugs including irinotecan, paclitaxel, 5-fluorouracil, and etoposide in human oncogenic cells with aberrant FGFR activation in vitro studies [134]. Thus, combination of chemotherapy with FGFR inhibitors is a consideration.
Dovitinib was investigated in combination with fulvestrant in postmenopausal women with hormone receptor (HR) positive, HER2 negative, FGFR (FGFR1, FGFR2, or FGF3) amplified breast cancer who progressed on endocrine therapy in a phase II trial [135]. This trial showed that its combination with fulvestrant in patients with FGFR amplification had significantly better PFS of 10.9 months in dovitinib arm vs 5.5 months in placebo [135]. Prolonged estrogen deprivation in breast cancer cells can upregulate FGFR1 together with FGF3, FGF4 and FGF19 due to co-amplification of the FGFR gene and those located in the 11q13 region [136]. FGFR1 amplification and treatment-induced FGFR1 overexpression occur preferentially in patients with letrozole-resistant HR-positive breast cancer [136].
On the other hand, combination of FGFR inhibitors with mTOR inhibitors can be considered in some patients who progressed on FGFR inhibitors due to mutations in FGFR kinase domain. There seems to be upregulation of the PI3K/AKT/mTOR signaling pathway in cells harboring the FGFR2 p.E565A mutation and thus combination therapy with FGFR and mTOR inhibitors may be considered to overcome resistance to FGFR inhibition [72]. Synergic activity of both FGFR and mTOR inhibitors have been demonstrated in cells harboring HNSCC, lung cancer, and HCC [137,138]. Analysis of circulating tumor DNA (ctDNA) from patients enrolled in MONALEESA-2 trial of ribociclib demonstrated that patients with FGFR1 amplification exhibited a shorter PFS compared to patients with wild type FGFR1, thus suggesting FGFR1 as a mechanism of drug resistance to CDK 4/6 inhibitors and hormone therapy [139]. FGFR multi-TKI lucitanib has shown promising activity in overcoming that resistance, thus there is potential for the combination use of CDK4/6 inhibitors with FGFR inhibitors in breast cancers with FGFR pathway alterations [139]. In addition, combination of erdafitinib, palbociclib and fulvestrant has resulted in complete responses in FGFR1-amplified, HR positive patient-derived-xenografts [139,140]. Further investigations in this combination led to an ongoing phase Ib clinical trial evaluating a combination treatment of fulvestrant, palbociclib, and erdafitinib in endocrine-resistant HR positive, HER2 negative metastatic breast cancer patients with FGFR amplifications (NCT03238196). Futibatinib is also currently being evaluated as either alone or in combination with fulvestrant in metastatic breast cancer patients with FGFR alterations in NCT04024436.
Combination of erdafitinib and anti-PD1 (programmed cell death protein 1) therapy in an indigenous FGFR2K660N/p53mutant lung cancer mouse model demonstrated that combination treatment led to significant tumor regression and improved survival when compared to either treatment alone [141]. The enhanced antitumor activity was supposed to be due to decreased expression of PD1, increased T cell infiltration, T-cell clone proliferation and alteration of the tumor microenvironment by immunological changes mediated by erdafitinib [141]. Patients with FGFR alterations seem to be less responsive to immunotherapy but 59% of those patients with prior immunotherapy failure had responded to erdafitinib in the BLC2001 trial of erdafitinib [87]. Non-T-cell inflamed subtype of urothelial carcinoma with FGFR3 mutations was found to have low to absent CD8+ T-cells in the tumor microenvironment resulting in resistance to ICI monotherapy [142]. Investigation of derazantinib in combination with atezolizumab in patients with FGFR altered urothelial cancers in phase I/II FIDES-02 trial (NCT04045613) was recently completed and results are pending [143].
8. Mechanism and management of the most relevant toxicity
The toxicity profile of non-selective FGFR inhibitors is similar to that of VEGFR TKIs which include fatigue, anorexia, pyrexia, diarrhea, arthralgia, liver toxicity, hypertension, proteinuria, thrombotic microangiopathy, and hypothyroidism. Selective FGFR TKIs can cause hyperphosphatemia, nail disorder with onycholysis, alopecia, mucosal dryness, mucositis, dry eye, conjunctivitis, keratitis, asymptomatic retinal pigment epithelial detachment, osteoarticular pains, myalgias and muscle cramps [144].
Hyperphosphatemia is a very common adverse effect of FGFR inhibitors because FGFR1 signaling pathway is a fundamental mechanism to limit the phosphate reabsorption in the proximal renal tubule by inhibiting the phosphate co-transporters [145]. In addition, FGF23 blocks the conversion from 25-hydroxy vitamin D to 1,25-dihydroxy vitamin D in normal physiology. Therefore, FGFR inhibitors increase the 1-25-dihydroxy vitamin D and increase the phosphate absorption from the intestine [145]. Serum phosphate level needs to be monitored closely. Phosphate binding therapy and diet modification are routinely used to lower the phosphate level. Grade 1 toxicity is defined as a sharp rise of 25% above baseline level (3.5-5.5 mg/dl). Grade 2 is defined as 5.5-6.9 mg/dl while grade 3 is 7-9.9 mg/dl and grade 4 is >10 mg/dl. FGFR inhibitor treatment needs to be held when patients have adverse effects of grade 3 or 4. Acetazolamide can be used in severe cases [145].
Diarrhea is another common adverse effect of FGFR inhibitors because FGFR pathway regulates bile acid production by a negative feedback mechanism. Bile acids are shown to stimulate the FGF19/FGFR4/ERK1/2 signaling pathway which in turns cause negative feedback mechanism. FGFR inhibitors affect this process, resulting in increased production of bile acid, increased gastrointestinal motility and secretion [145]. Supportive measures with intravenous or oral fluid replacement, probiotics, anti-diarrheal medications, and electrolytes corrections are recommended [145].
Fatigue is also commonly reported but its mechanism is not well understood. Skin, nail, and mucosal changes are also associated with FGFR inhibitors. Topical steroid cream, moisturizers, oral hygiene, and non-alcohol containing mouthwash are recommended [76,146]. Ocular toxicity such as retinal detachment, central serous retinopathy, dry eyes and cataracts, are significant adverse effects of FGFR inhibitors to be monitored [76,87]. Baseline pretreatment comprehensive eye exam is recommended, and symptomatic patients need to be monitored closely. FGFR inhibitors should be discontinued if ocular toxicities are of grade 3 or higher. Ocular toxicities are reversible upon discontinuation of treatment and thus close monitoring and timely discontinuation of FGFR inhibitor is important [147]. FGFR inhibitors can be restarted at a lower dose if grade 1-2 ocular toxicities resolve [145].
9. Landscape of ongoing investigations, challenging issues, and future directions
Uehara et al., reported that patients who harbor FGFR alterations have worse OS than those without and 94% of those who harbor FGFR alterations also have other genomic co-alterations, including TP53 axis (70%), cell cycle (58%), PI3K pathways (55%) and receptor kinases and MAPK pathways (65%) [2].
FGFR inhibitors have shown efficacy in tumors with FGFR alterations in several clinical trials including patients with hematological, iCCA, lung cancers, and urothelial carcinomas. Challenges faced so far are difficulties in patient selection, molecular detection of FGFR alterations, acquired resistance of FGFR inhibitors and management of toxicities [7]. For instance, for gastric cancer, tumor heterogeneity is a challenge, affecting the accuracy of FGFR2 amplification or overexpression and clinical applications for therapeutic targeting. As tissue heterogeneity poses challenges for molecular diagnostic testing, ctDNA is currently under investigation as a potential modality with comparable rates of detection to tissue based methods as seen in the GOZILA study [148].
Despite the efficacy of FGFR inhibitors, acquired resistance occurs due to the devolvement of secondary mutations in FGFR kinase domain, making it resistant to infigratinib or pemigatinib. Third generation irreversible FGFR inhibitors such as futibatinib can overcome those mutations but cysteine mutation could still occur [7]. Acquired mutations in the kinase domain could be avoided by the development of FGFR kinase allosteric or specific fusion partner inhibitors, such as TACC3-targeting inhibitors (BO-264) which can inhibit the growth of cells harboring FGFR3-TACC3 fusions [149]. In addition, FGFR and fusion partners can be degraded by FGFR-targeting proteolysis targeting chimera (PROTACs), thus avoiding inhibitor-induced acquired mutation [7].
On the other hand, we could consider the combination of FGFR inhibitors with mTOR inhibitors in some patients who progressed on FGFR inhibitors due to mutations in FGFR kinase domain. As discussed earlier, there is upregulation of the PI3K/AKT/mTOR signaling pathway in cells harboring the FGFR2 p.E565A mutation. Thus combination therapy of FGFR and mTOR inhibitors may overcome resistance to FGFR inhibition [72]. Given acquired mutations and resistance, there is a need for a more comprehensive understanding of mutations that develop in response to FGFR inhibition and sensitivity of current inhibitors to develop novel inhibitors.
In addition, future investigations into combination treatments with immunotherapy and optimal sequencing of immunotherapy and FGFR inhibitors in FGFR-altered cancers are also needed given that ORR of erdafitinib was higher in patients with urothelial cancers previously exposed to ICI. FGFR monoclonal antibody bemarituzumab is currently under a few phase III clinical trials to be used as first line treatment in gastric or GE junction cancers. However, further advances in ligand traps and ADCs are not well developed yet.
The landscape of FGFR inhibitors has been evolved over the last decade with the approval of FGFR inhibitors in recent years and more to come with several ongoing clinical trials as of this writing (refer to Table 3 for summary of selected current ongoing clinical trials).
10. Conclusions
FGFR inhibitors have been shown to have efficacy across various types of cancers with FGFR alterations and currently several approved and novel FGFR inhibitors are being investigated in various clinical trials, either alone or in combinations with other therapies. Despite their limitations due to limited responses, acquired resistances and intolerable toxicities, they have potential in treating various cancers with FGFR alterations and overcoming resistance to other anti-cancer treatments. Thus, further investigations are needed to develop FGFR inhibitors that have better and durable responses with more tolerable toxicities and ability to overcome acquired resistance. Further developments of FGFR inhibitors will pave the way for personalized medicine in which individualized treatments are given based on molecular profiling of tumors.
Figure 1.
FGF/FGFR and downstream signaling pathway.
Author Contributions
First author contributed to this manuscript’s conceptualization, writing, making tables and figures, editing, and reviewing. All authors contributed to writing and reviewing. All authors have read and agreed to the published version of the manuscript.
Funding
This review did not receive any funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are thankful for this special issue.
Conflicts of Interest
B.P participates as consulting or advisory role at Janssen and receives research funding from TSCAN Therapeutics (Inst), Immune-Onc Therapeutics (Inst). Other authors declare no conflict of interests.
References
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nature Reviews Cancer 2017, 17, 318-332. [CrossRef]
- Uehara, Y.; Ikeda, S.; Kim, K.H.; Lim, H.J.; Adashek, J.J.; Persha, H.E.; Okamura, R.; Lee, S.; Sicklick, J.K.; Kato, S.; et al. Targeting the FGF/FGFR axis and its co-alteration allies. ESMO Open 2022, 7, 100647. [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clinical Cancer Research 2016, 22, 259-267. [CrossRef]
- Gordon, A.; Johnston, E.; Lau, D.K.; Starling, N. Targeting FGFR2 Positive Gastroesophageal Cancer: Current and Clinical Developments. OncoTargets and Therapy 2022, Volume 15, 1183-1196. [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol 2015, 4, 215-266. [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov 2009, 8, 235-253. [CrossRef]
- Chen, L.; Zhang, Y.; Yin, L.; Cai, B.; Huang, P.; Li, X.; Liang, G. Fibroblast growth factor receptor fusions in cancer: opportunities and challenges. Journal of Experimental & Clinical Cancer Research 2021, 40. [CrossRef]
- Teven, C.M.; Farina, E.M.; Rivas, J.; Reid, R.R. Fibroblast growth factor (FGF) signaling in development and skeletal diseases. Genes Dis 2014, 1, 199-213. [CrossRef]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduction and Targeted Therapy 2020, 5, 181. [CrossRef]
- Birrer, M.J.; Johnson, M.E.; Hao, K.; Wong, K.K.; Park, D.C.; Bell, A.; Welch, W.R.; Berkowitz, R.S.; Mok, S.C. Whole genome oligonucleotide-based array comparative genomic hybridization analysis identified fibroblast growth factor 1 as a prognostic marker for advanced-stage serous ovarian adenocarcinomas. J Clin Oncol 2007, 25, 2281-2287. [CrossRef]
- Smith, G.; Ng, M.T.; Shepherd, L.; Herrington, C.S.; Gourley, C.; Ferguson, M.J.; Wolf, C.R. Individuality in FGF1 expression significantly influences platinum resistance and progression-free survival in ovarian cancer. Br J Cancer 2012, 107, 1327-1336. [CrossRef]
- Steele, I.A.; Edmondson, R.J.; Bulmer, J.N.; Bolger, B.S.; Leung, H.Y.; Davies, B.R. Induction of FGF receptor 2-IIIb expression and response to its ligands in epithelial ovarian cancer. Oncogene 2001, 20, 5878-5887. [CrossRef]
- Taniguchi, F.; Itamochi, H.; Harada, T.; Terakawa, N. Fibroblast growth factor receptor 2 expression may be involved in transformation of ovarian endometrioma to clear cell carcinoma of the ovary. Int J Gynecol Cancer 2013, 23, 791-796. [CrossRef]
- Zaid, T.M.; Yeung, T.L.; Thompson, M.S.; Leung, C.S.; Harding, T.; Co, N.N.; Schmandt, R.S.; Kwan, S.Y.; Rodriguez-Aguay, C.; Lopez-Berestein, G.; et al. Identification of FGFR4 as a potential therapeutic target for advanced-stage, high-grade serous ovarian cancer. Clin Cancer Res 2013, 19, 809-820. [CrossRef]
- Choi, C.H.; Chung, J.Y.; Kim, J.H.; Kim, B.G.; Hewitt, S.M. Expression of fibroblast growth factor receptor family members is associated with prognosis in early stage cervical cancer patients. J Transl Med 2016, 14, 124. [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin Cancer Res 2016, 22, 259-267. [CrossRef]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov 2013, 3, 636-647. [CrossRef]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427-1434. [CrossRef]
- Jain, A.; Borad, M.J.; Kelley, R.K.; Wang, Y.; Abdel-Wahab, R.; Meric-Bernstam, F.; Baggerly, K.A.; Kaseb, A.O.; Al-Shamsi, H.O.; Ahn, D.H.; et al. Cholangiocarcinoma With FGFR Genetic Aberrations: A Unique Clinical Phenotype. JCO Precis Oncol 2018, 2, 1-12. [CrossRef]
- Kongpetch, S.; Jusakul, A.; Lim, J.Q.; Ng, C.C.Y.; Chan, J.Y.; Rajasegaran, V.; Lim, T.H.; Lim, K.H.; Choo, S.P.; Dima, S.; et al. Lack of Targetable FGFR2 Fusions in Endemic Fluke-Associated Cholangiocarcinoma. JCO Glob Oncol 2020, 6, 628-638. [CrossRef]
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: biologic and clinical implications. Cancer Metastasis Rev 2015, 34, 479-496. [CrossRef]
- Gu, W.; Yang, J.; Wang, Y.; Xu, J.; Wang, X.; Du, F.; Hu, X.; Guo, H.; Song, C.; Tao, R.; et al. Comprehensive identification of FGFR1-4 alterations in 5 557 Chinese patients with solid tumors by next-generation sequencing. Am J Cancer Res 2021, 11, 3893-3906.
- Costa, R.; Carneiro, B.A.; Taxter, T.; Tavora, F.A.; Kalyan, A.; Pai, S.A.; Chae, Y.K.; Giles, F.J. FGFR3-TACC3 fusion in solid tumors: mini review. Oncotarget 2016, 7, 55924-55938. [CrossRef]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231-1235. [CrossRef]
- Su, X.; Zhan, P.; Gavine, P.R.; Morgan, S.; Womack, C.; Ni, X.; Shen, D.; Bang, Y.J.; Im, S.A.; Ho Kim, W.; et al. FGFR2 amplification has prognostic significance in gastric cancer: results from a large international multicentre study. Br J Cancer 2014, 110, 967-975. [CrossRef]
- Matsumoto, K.; Arao, T.; Hamaguchi, T.; Shimada, Y.; Kato, K.; Oda, I.; Taniguchi, H.; Koizumi, F.; Yanagihara, K.; Sasaki, H.; et al. FGFR2 gene amplification and clinicopathological features in gastric cancer. Br J Cancer 2012, 106, 727-732. [CrossRef]
- Jung, E.J.; Jung, E.J.; Min, S.Y.; Kim, M.A.; Kim, W.H. Fibroblast growth factor receptor 2 gene amplification status and its clinicopathologic significance in gastric carcinoma. Hum Pathol 2012, 43, 1559-1566. [CrossRef]
- Tomlinson, D.C.; Hurst, C.D.; Knowles, M.A. Knockdown by shRNA identifies S249C mutant FGFR3 as a potential therapeutic target in bladder cancer. Oncogene 2007, 26, 5889-5899. [CrossRef]
- AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017, 7, 818-831. [CrossRef]
- Sfakianos, J.P.; Cha, E.K.; Iyer, G.; Scott, S.N.; Zabor, E.C.; Shah, R.H.; Ren, Q.; Bagrodia, A.; Kim, P.H.; Hakimi, A.A.; et al. Genomic Characterization of Upper Tract Urothelial Carcinoma. Eur Urol 2015, 68, 970-977. [CrossRef]
- Ross, J.S.; Wang, K.; Khaira, D.; Ali, S.M.; Fisher, H.A.; Mian, B.; Nazeer, T.; Elvin, J.A.; Palma, N.; Yelensky, R.; et al. Comprehensive genomic profiling of 295 cases of clinically advanced urothelial carcinoma of the urinary bladder reveals a high frequency of clinically relevant genomic alterations. Cancer 2016, 122, 702-711. [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540-556.e525. [CrossRef]
- di Martino, E.; Tomlinson, D.C.; Williams, S.V.; Knowles, M.A. A place for precision medicine in bladder cancer: targeting the FGFRs. Future Oncol 2016, 12, 2243-2263. [CrossRef]
- Tomlinson, D.C.; Baldo, O.; Harnden, P.; Knowles, M.A. FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J Pathol 2007, 213, 91-98. [CrossRef]
- Zhou, Z.; Liu, Z.; Ou, Q.; Wu, X.; Wang, X.; Shao, Y.; Liu, H.; Yang, Y. Targeting FGFR in non-small cell lung cancer: implications from the landscape of clinically actionable aberrations of FGFR kinases. Cancer Biol Med 2021, 18, 490-501. [CrossRef]
- Ou, S.I.; Horn, L.; Cruz, M.; Vafai, D.; Lovly, C.M.; Spradlin, A.; Williamson, M.J.; Dagogo-Jack, I.; Johnson, A.; Miller, V.A.; et al. Emergence of FGFR3-TACC3 fusions as a potential by-pass resistance mechanism to EGFR tyrosine kinase inhibitors in EGFR mutated NSCLC patients. Lung Cancer 2017, 111, 61-64. [CrossRef]
- Reis-Filho, J.S.; Simpson, P.T.; Turner, N.C.; Lambros, M.B.; Jones, C.; Mackay, A.; Grigoriadis, A.; Sarrio, D.; Savage, K.; Dexter, T.; et al. FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin Cancer Res 2006, 12, 6652-6662. [CrossRef]
- Santolla, M.F.; Vivacqua, A.; Lappano, R.; Rigiracciolo, D.C.; Cirillo, F.; Galli, G.R.; Talia, M.; Brunetti, G.; Miglietta, A.M.; Belfiore, A.; et al. GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells toward Breast Tumor Progression. Cells 2019, 8. [CrossRef]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 2010, 70, 2085-2094. [CrossRef]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun 2019, 10, 1373. [CrossRef]
- Xie, N.; Tian, C.; Wu, H.; Yang, X.; Liu, L.; Li, J.; Xiao, H.; Gao, J.; Lu, J.; Hu, X.; et al. FGFR aberrations increase the risk of brain metastases and predict poor prognosis in metastatic breast cancer patients. Ther Adv Med Oncol 2020, 12, 1758835920915305. [CrossRef]
- Chew, N.J.; Nguyen, E.V.; Su, S.P.; Novy, K.; Chan, H.C.; Nguyen, L.K.; Luu, J.; Simpson, K.J.; Lee, R.S.; Daly, R.J. FGFR3 signaling and function in triple negative breast cancer. Cell Commun Signal 2020, 18, 13. [CrossRef]
- Tomlinson, D.C.; Knowles, M.A.; Speirs, V. Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int J Cancer 2012, 130, 2857-2866. [CrossRef]
- Yamaguchi, F.; Saya, H.; Bruner, J.M.; Morrison, R.S. Differential expression of two fibroblast growth factor-receptor genes is associated with malignant progression in human astrocytomas. Proc Natl Acad Sci U S A 1994, 91, 484-488. [CrossRef]
- Morrison, R.S.; Yamaguchi, F.; Saya, H.; Bruner, J.M.; Yahanda, A.M.; Donehower, L.A.; Berger, M. Basic fibroblast growth factor and fibroblast growth factor receptor I are implicated in the growth of human astrocytomas. J Neurooncol 1994, 18, 207-216. [CrossRef]
- Wang, F.; Kan, M.; Yan, G.; Xu, J.; McKeehan, W.L. Alternately spliced NH2-terminal immunoglobulin-like Loop I in the ectodomain of the fibroblast growth factor (FGF) receptor 1 lowers affinity for both heparin and FGF-1. J Biol Chem 1995, 270, 10231-10235. [CrossRef]
- Fukai, J.; Yokote, H.; Yamanaka, R.; Arao, T.; Nishio, K.; Itakura, T. EphA4 promotes cell proliferation and migration through a novel EphA4-FGFR1 signaling pathway in the human glioma U251 cell line. Mol Cancer Ther 2008, 7, 2768-2778. [CrossRef]
- Gouazé-Andersson, V.; Delmas, C.; Taurand, M.; Martinez-Gala, J.; Evrard, S.; Mazoyer, S.; Toulas, C.; Cohen-Jonathan-Moyal, E. FGFR1 Induces Glioblastoma Radioresistance through the PLCγ/Hif1α Pathway. Cancer Res 2016, 76, 3036-3044. [CrossRef]
- Miyake, A.; Hattori, Y.; Ohta, M.; Itoh, N. Rat oligodendrocytes and astrocytes preferentially express fibroblast growth factor receptor-2 and -3 mRNAs. J Neurosci Res 1996, 45, 534-541. [CrossRef]
- Ohashi, R.; Matsuda, Y.; Ishiwata, T.; Naito, Z. Downregulation of fibroblast growth factor receptor 2 and its isoforms correlates with a high proliferation rate and poor prognosis in high-grade glioma. Oncol Rep 2014, 32, 1163-1169. [CrossRef]
- Daido, S.; Takao, S.; Tamiya, T.; Ono, Y.; Terada, K.; Ito, S.; Ouchida, M.; Date, I.; Ohmoto, T.; Shimizu, K. Loss of heterozygosity on chromosome 10q associated with malignancy and prognosis in astrocytic tumors, and discovery of novel loss regions. Oncol Rep 2004, 12, 789-795.
- Shi, E.; Chmielecki, J.; Tang, C.M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 actionable alterations in "Wild-Type" gastrointestinal stromal tumors. J Transl Med 2016, 14, 339. [CrossRef]
- Pantaleo, M.A.; Urbini, M.; Indio, V.; Ravegnini, G.; Nannini, M.; De Luca, M.; Tarantino, G.; Angelini, S.; Gronchi, A.; Vincenzi, B.; et al. Genome-Wide Analysis Identifies MEN1 and MAX Mutations and a Neuroendocrine-Like Molecular Heterogeneity in Quadruple WT GIST. Mol Cancer Res 2017, 15, 553-562. [CrossRef]
- Javidi-Sharifi, N.; Traer, E.; Martinez, J.; Gupta, A.; Taguchi, T.; Dunlap, J.; Heinrich, M.C.; Corless, C.L.; Rubin, B.P.; Druker, B.J.; et al. Crosstalk between KIT and FGFR3 Promotes Gastrointestinal Stromal Tumor Cell Growth and Drug Resistance. Cancer Res 2015, 75, 880-891. [CrossRef]
- Li, F.; Huynh, H.; Li, X.; Ruddy, D.A.; Wang, Y.; Ong, R.; Chow, P.; Qiu, S.; Tam, A.; Rakiec, D.P.; et al. FGFR-Mediated Reactivation of MAPK Signaling Attenuates Antitumor Effects of Imatinib in Gastrointestinal Stromal Tumors. Cancer Discov 2015, 5, 438-451. [CrossRef]
- Boichuk, S.; Galembikova, A.; Dunaev, P.; Valeeva, E.; Shagimardanova, E.; Gusev, O.; Khaiboullina, S. A Novel Receptor Tyrosine Kinase Switch Promotes Gastrointestinal Stromal Tumor Drug Resistance. Molecules 2017, 22. [CrossRef]
- Taylor, J.G.t.; Cheuk, A.T.; Tsang, P.S.; Chung, J.Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest 2009, 119, 3395-3407. [CrossRef]
- Wachtel, M.; Rakic, J.; Okoniewski, M.; Bode, P.; Niggli, F.; Schäfer, B.W. FGFR4 signaling couples to Bim and not Bmf to discriminate subsets of alveolar rhabdomyosarcoma cells. Int J Cancer 2014, 135, 1543-1552. [CrossRef]
- Chudasama, P.; Renner, M.; Straub, M.; Mughal, S.S.; Hutter, B.; Kosaloglu, Z.; Schweßinger, R.; Scheffler, M.; Alldinger, I.; Schimmack, S.; et al. Targeting Fibroblast Growth Factor Receptor 1 for Treatment of Soft-Tissue Sarcoma. Clin Cancer Res 2017, 23, 962-973. [CrossRef]
- Agelopoulos, K.; Richter, G.H.; Schmidt, E.; Dirksen, U.; von Heyking, K.; Moser, B.; Klein, H.U.; Kontny, U.; Dugas, M.; Poos, K.; et al. Deep Sequencing in Conjunction with Expression and Functional Analyses Reveals Activation of FGFR1 in Ewing Sarcoma. Clin Cancer Res 2015, 21, 4935-4946. [CrossRef]
- Bao, Y.; Gabrielpillai, J.; Dietrich, J.; Zarbl, R.; Strieth, S.; Schröck, F.; Dietrich, D. Fibroblast growth factor (FGF), FGF receptor (FGFR), and cyclin D1 (CCND1) DNA methylation in head and neck squamous cell carcinomas is associated with transcriptional activity, gene amplification, human papillomavirus (HPV) status, and sensitivity to tyrosine kinase inhibitors. Clin Epigenetics 2021, 13, 228. [CrossRef]
- Tillman, B.N.; Yanik, M.; Birkeland, A.C.; Liu, C.J.; Hovelson, D.H.; Cani, A.K.; Palanisamy, N.; Carskadon, S.; Carey, T.E.; Bradford, C.R.; et al. Fibroblast growth factor family aberrations as a putative driver of head and neck squamous cell carcinoma in an epidemiologically low-risk patient as defined by targeted sequencing. Head Neck 2016, 38 Suppl 1, E1646-1652. [CrossRef]
- Brands, R.C.; Knierim, L.M.; De Donno, F.; Steinacker, V.; Hartmann, S.; Seher, A.; Kübler, A.C.; Müller-Richter, U.D.A. Targeting VEGFR and FGFR in head and neck squamous cell carcinoma in vitro. Oncol Rep 2017, 38, 1877-1885. [CrossRef]
- Nayak, S.; Goel, M.M.; Makker, A.; Bhatia, V.; Chandra, S.; Kumar, S.; Agarwal, S.P. Fibroblast Growth Factor (FGF-2) and Its Receptors FGFR-2 and FGFR-3 May Be Putative Biomarkers of Malignant Transformation of Potentially Malignant Oral Lesions into Oral Squamous Cell Carcinoma. PLoS One 2015, 10, e0138801. [CrossRef]
- Marshall, M.E.; Hinz, T.K.; Kono, S.A.; Singleton, K.R.; Bichon, B.; Ware, K.E.; Marek, L.; Frederick, B.A.; Raben, D.; Heasley, L.E. Fibroblast growth factor receptors are components of autocrine signaling networks in head and neck squamous cell carcinoma cells. Clin Cancer Res 2011, 17, 5016-5025. [CrossRef]
- Kim, E.K.; Cho, Y.A.; Koh, Y.W.; Shin, H.A.; Cho, B.C.; Yoon, S.O. Prognostic implications of Fibroblast growth factor receptor 1 (FGFR1) gene amplification and protein overexpression in hypopharyngeal and laryngeal squamous cell carcinoma. BMC Cancer 2020, 20, 348. [CrossRef]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res 2015, 21, 632-641. [CrossRef]
- Yuan, L.; Liu, Z.H.; Lin, Z.R.; Xu, L.H.; Zhong, Q.; Zeng, M.S. Recurrent FGFR3-TACC3 fusion gene in nasopharyngeal carcinoma. Cancer Biol Ther 2014, 15, 1613-1621. [CrossRef]
- Koole, K.; Clausen, M.J.; van Es, R.J.; van Kempen, P.M.; Melchers, L.J.; Koole, R.; Langendijk, J.A.; van Diest, P.J.; Roodenburg, J.L.; Schuuring, E.; et al. FGFR Family Members Protein Expression as Prognostic Markers in Oral Cavity and Oropharyngeal Squamous Cell Carcinoma. Mol Diagn Ther 2016, 20, 363-374. [CrossRef]
- Shi, S.; Li, X.; You, B.; Shan, Y.; Cao, X.; You, Y. High Expression of FGFR4 Enhances Tumor Growth and Metastasis in Nasopharyngeal Carcinoma. J Cancer 2015, 6, 1245-1254. [CrossRef]
- Yue, S.; Li, Y.; Chen, X.; Wang, J.; Li, M.; Chen, Y.; Wu, D. FGFR-TKI resistance in cancer: current status and perspectives. Journal of Hematology & Oncology 2021, 14, 23. [CrossRef]
- Krook, M.A.; Lenyo, A.; Wilberding, M.; Barker, H.; Dantuono, M.; Bailey, K.M.; Chen, H.-Z.; Reeser, J.W.; Wing, M.R.; Miya, J.; et al. Efficacy of FGFR Inhibitors and Combination Therapies for Acquired Resistance in FGFR2-Fusion Cholangiocarcinoma. Molecular Cancer Therapeutics 2020, 19, 847-857. [CrossRef]
- Mahapatra, S.; Jonniya, N.A.; Koirala, S.; Ursal, K.D.; Kar, P. The FGF/FGFR signalling mediated anti-cancer drug resistance and therapeutic intervention. J Biomol Struct Dyn 2023, 1-25. [CrossRef]
- Helsinn to Withdraw NDA for Cholangiocarcinoma. Available online: https://www.fdanews.com/articles/209797-helsinn-to-withdraw-nda-for-cholangiocarcinoma (accessed on November 6, 2023).
- FDA, P. FDA grants accelerated approval to pemigatinib for cholangiocarcinoma with an FGFR2 rearrangement or fusion. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pemigatinib-cholangiocarcinoma-fgfr2-rearrangement-or-fusion (accessed on 08/27/2023).
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. The lancet oncology 2020, 21, 671-684. [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.M.; Melisi, D.; Al-Rajabi, R.M.d.T.; Paulson, A.S.; Borad, M.J.; Gallinson, D.H.; Murphy, A.G.; et al. Pemigatinib for previously treated locally advanced/metastatic cholangiocarcinoma (CCA): Update of FIGHT-202. Journal of Clinical Oncology 2021, 39, 4086-4086. [CrossRef]
- Bekaii-Saab, T.S.; Valle, J.W.; Cutsem, E.V.; Rimassa, L.; Furuse, J.; Ioka, T.; Melisi, D.; Macarulla, T.; Bridgewater, J.A.; Wasan, H.S.; et al. FIGHT-302: Phase III study of first-line (1L) pemigatinib (PEM) versus gemcitabine (GEM) plus cisplatin (CIS) for cholangiocarcinoma (CCA) with FGFR2 fusions or rearrangements. Journal of Clinical Oncology 2020, 38, TPS592-TPS592. [CrossRef]
- Bekaii-Saab, T.S.; Valle, J.W.; Cutsem, E.V.; Rimassa, L.; Furuse, J.; Ioka, T.; Melisi, D.; Macarulla, T.; Bridgewater, J.; Wasan, H.; et al. FIGHT-302: first-line pemigatinib vs gemcitabine plus cisplatin for advanced cholangiocarcinoma with FGFR2 rearrangements. Future Oncology 2020, 16, 2385-2399. [CrossRef]
- Gotlib, J.; Kiladjian, J.-J.; Vannucchi, A.; Rambaldi, A.; Reiter, A.; Shomali, W.; George, T.I.; Patel, J.L.; Colucci, P.; Walker, C.; et al. A Phase 2 Study of Pemigatinib (FIGHT-203; INCB054828) in Patients with Myeloid/Lymphoid Neoplasms (MLNs) with Fibroblast Growth Factor Receptor 1 (FGFR1) Rearrangement (MLN FGFR1). Blood 2021, 138, 385. [CrossRef]
- FDA, P. FDA approves pemigatinib for relapsed or refractory myeloid/lymphoid neoplasms with FGFR1 rearrangement. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pemigatinib-relapsed-or-refractory-myeloidlymphoid-neoplasms-fgfr1-rearrangement#:~:text=On%20August%2026%2C%202022%2C%20the,receptor%201%20(FGFR1)%20rearrangement. (accessed on 08/27/2023).
- FDA, I. FDA grants accelerated approval to infigratinib for metastatic cholangiocarcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-infigratinib-metastatic-cholangiocarcinoma (accessed on.
- Javle, M.M.; Kelley, R.K.; Springfeld, C.; Abou-Alfa, G.K.; Macarulla, T.; Tanasanvimon, S.; Goyal, L.; Borbath, I.; Bitzer, M.; Yong, W.-P.; et al. A phase II study of infigratinib in previously treated advanced/metastatic cholangiocarcinoma with FGFR gene fusions/alterations. Journal of Clinical Oncology 2021, 39, TPS356-TPS356. [CrossRef]
- Javle, M.M.; Roychowdhury, S.; Kelley, R.K.; Sadeghi, S.; Macarulla, T.; Waldschmidt, D.T.; Goyal, L.; Borbath, I.; El-Khoueiry, A.B.; Yong, W.-P.; et al. Final results from a phase II study of infigratinib (BGJ398), an FGFR-selective tyrosine kinase inhibitor, in patients with previously treated advanced cholangiocarcinoma harboring an FGFR2 gene fusion or rearrangement. Journal of Clinical Oncology 2021, 39, 265-265. [CrossRef]
- discontinuation, I. Helsinn Therapeutics – Discontinuation of Truseltiq® (infigratinib). Available online: https://professionals.optumrx.com/content/dam/optum3/professional-optumrx/news/rxnews/drug-withdrawls/drugwithdrawal_truseltiq_2022-1117.pdf (accessed on 08/27/2023).
- FDA, E. FDA grants accelerated approval to erdafitinib for metastatic urothelial carcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-erdafitinib-metastatic-urothelial-carcinoma (accessed on.
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. New England Journal of Medicine 2019, 381, 338-348. [CrossRef]
- Siefker-Radtke, A.O.; Necchi, A.; Park, S.H.; García-Donas, J.; Huddart, R.A.; Burgess, E.F.; Fleming, M.T.; Rezazadeh Kalebasty, A.; Mellado, B.; Varlamov, S.; et al. Efficacy and safety of erdafitinib in patients with locally advanced or metastatic urothelial carcinoma: long-term follow-up of a phase 2 study. Lancet Oncol 2022, 23, 248-258. [CrossRef]
- Janssen Submits Supplemental New Drug Application to the U.S. Food and Drug Administration Seeking Full Approval of BALVERSA® (erdafitinib) for the Treatment of Patients with Locally Advanced or Metastatic Urothelial Carcinoma and Selected Fibroblast Growth Factor Receptor Gene Alterations. Available online: https://www.jnj.com/janssen-submits-supplemental-new-drug-application-to-the-u-s-food-and-drug-administration-seeking-full-approval-of-balversa-erdafitinib-for-the-treatment-of-patients-with-locally-advanced-or-metastatic-urothelial-carcinoma-and-selected-fibroblast-growth-factor-receptor-gene-alterations (accessed on 11/6/2023).
- Loriot, Y.; Matsubara, N.; Park, S.H.; Huddart, R.A.; Burgess, E.F.; Houede, N.; Banek, S.; Laguerre, B.; Guadalupi, V.; Ku, J.H.; et al. Phase 3 THOR study: Results of erdafitinib (erda) versus chemotherapy (chemo) in patients (pts) with advanced or metastatic urothelial cancer (mUC) with select fibroblast growth factor receptor alterations (<i>FGFRalt</i>). Journal of Clinical Oncology 2023, 41, LBA4619-LBA4619. [CrossRef]
- Loriot, Y.; Matsubara, N.; Park, S.H.; Huddart, R.A.; Burgess, E.F.; Houede, N.; Banek, S.; Guadalupi, V.; Ku, J.H.; Valderrama, B.P.; et al. Erdafitinib or Chemotherapy in Advanced or Metastatic Urothelial Carcinoma. New England Journal of Medicine 2023. [CrossRef]
- Romero, D. THOR provides new data on the efficacy of erdafitinib. Nature Reviews Clinical Oncology 2023. [CrossRef]
- Goyal, L.; Meric-Bernstam, F.; Hollebecque, A.; Valle, J.W.; Morizane, C.; Karasic, T.B.; Abrams, T.A.; Furuse, J.; Kelley, R.K.; Cassier, P.A.; et al. Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma. N Engl J Med 2023, 388, 228-239. [CrossRef]
- FDA, F. FDA grants accelerated approval to futibatinib for cholangiocarcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-futibatinib-cholangiocarcinoma (accessed on 08/27/2023).
- Borad, M.J.; Bridgewater, J.A.; Morizane, C.; Shroff, R.T.; Oh, D.-Y.; Moehler, M.H.; Furuse, J.; Benhadji, K.A.; He, H.; Valle, J.W. A phase III study of futibatinib (TAS-120) versus gemcitabine-cisplatin (gem-cis) chemotherapy as first-line (1L) treatment for patients (pts) with advanced (adv) cholangiocarcinoma (CCA) harboring fibroblast growth factor receptor 2 (FGFR2) gene rearrangements (FOENIX-CCA3). Journal of Clinical Oncology 2020, 38, TPS600-TPS600. [CrossRef]
- Schuler, M.; Cho, B.C.; Sayehli, C.M.; Navarro, A.; Soo, R.A.; Richly, H.; Cassier, P.A.; Tai, D.; Penel, N.; Nogova, L.; et al. Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol 2019, 20, 1454-1466. [CrossRef]
- Sternberg, C.N.; Petrylak, D.P.; Bellmunt, J.; Nishiyama, H.; Necchi, A.; Gurney, H.; Lee, J.L.; van der Heijden, M.S.; Rosenbaum, E.; Penel, N.; et al. FORT-1: Phase II/III Study of Rogaratinib Versus Chemotherapy in Patients With Locally Advanced or Metastatic Urothelial Carcinoma Selected Based on FGFR1/3 mRNA Expression. J Clin Oncol 2023, 41, 629-639. [CrossRef]
- Mazzaferro, V.; El-Rayes, B.F.; Droz dit Busset, M.; Cotsoglou, C.; Harris, W.P.; Damjanov, N.; Masi, G.; Rimassa, L.; Personeni, N.; Braiteh, F.; et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. British Journal of Cancer 2019, 120, 165-171. [CrossRef]
- Javle, M.M.; Abou-Alfa, G.K.; Macarulla, T.; Personeni, N.; Adeva, J.; Bergamo, F.; Malka, D.; Vogel, A.; Knox, J.J.; Evans, T.R.J.; et al. Efficacy of derazantinib in intrahepatic cholangiocarcinoma patients with FGFR2 mutations or amplifications: Interim results from the phase 2 study FIDES-01. Journal of Clinical Oncology 2022, 40, 427-427. [CrossRef]
- Necchi, A.; Todenhöfer, T.; Deville, J.-L.; Häckl, M.; Marszewska, M.; McKernan, P.; Saulay, M.; Engelhardt, M.; Santis, M.D. Efficacy and safety of derazantinib (DZB) in patients with metastatic urothelial carcinoma (mUC) with activating FGFR1/2/3 genetic aberrations (GA): Results from the phase 1b/2 FIDES-02 study. Journal of Clinical Oncology 2023, 41, 501-501. [CrossRef]
- Available online: clinicaltrials.gov. (accessed on 11/18/2023).
- Subbiah, V.; Sahai, V.; Maglic, D.; Bruderek, K.; Touré, B.B.; Zhao, S.; Valverde, R.; O'Hearn, P.J.; Moustakas, D.T.; Schönherr, H.; et al. RLY-4008, the First Highly Selective FGFR2 Inhibitor with Activity across FGFR2 Alterations and Resistance Mutations. Cancer Discovery 2023, 13, 2012-2031. [CrossRef]
- Voss, M.H.; Hierro, C.; Heist, R.S.; Cleary, J.M.; Meric-Bernstam, F.; Tabernero, J.; Janku, F.; Gandhi, L.; Iafrate, A.J.; Borger, D.R.; et al. A Phase I, Open-Label, Multicenter, Dose-escalation Study of the Oral Selective FGFR Inhibitor Debio 1347 in Patients with Advanced Solid Tumors Harboring FGFR Gene Alterations. Clin Cancer Res 2019, 25, 2699-2707. [CrossRef]
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.-W.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.-Y.; et al. First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discovery 2019, 9, 1696-1707. [CrossRef]
- Tjulandin, S.; Statsenko, G.; Artamonova, E.; Vladimirova, L.Y.; Besova, N.; Mochalova, A.; Rykov, I.; Moiseyenko, V.; Utyashev, I.A.; Iugai, S.; et al. A first-in-human phase 1b study of a novel allosteric extracellular FGFR2 inhibitor alofanib in patients with refractory metastatic gastric cancer. Journal of Clinical Oncology 2022, 40, 304-304. [CrossRef]
- Tyulyandina, A.; Harrison, D.; Yin, W.; Stepanova, E.; Kochenkov, D.; Solomko, E.; Peretolchina, N.; Daeyaert, F.; Joos, J.B.; Van Aken, K.; et al. Alofanib, an allosteric FGFR2 inhibitor, has potent effects on ovarian cancer growth in preclinical studies. Invest New Drugs 2017, 35, 127-133. [CrossRef]
- Chan, S.L.; Schuler, M.; Kang, Y.K.; Yen, C.J.; Edeline, J.; Choo, S.P.; Lin, C.C.; Okusaka, T.; Weiss, K.H.; Macarulla, T.; et al. A first-in-human phase 1/2 study of FGF401 and combination of FGF401 with spartalizumab in patients with hepatocellular carcinoma or biomarker-selected solid tumors. J Exp Clin Cancer Res 2022, 41, 189. [CrossRef]
- Aggarwal, C.; Redman, M.W.; Lara, P.N., Jr.; Borghaei, H.; Hoffman, P.; Bradley, J.D.; Newman, A.J., 3rd; Feldman, M.J.; Minichiello, K.; Miao, J.; et al. SWOG S1400D (NCT02965378), a Phase II Study of the Fibroblast Growth Factor Receptor Inhibitor AZD4547 in Previously Treated Patients With Fibroblast Growth Factor Pathway-Activated Stage IV Squamous Cell Lung Cancer (Lung-MAP Substudy). J Thorac Oncol 2019, 14, 1847-1852. [CrossRef]
- Chae, Y.K.; Hong, F.; Vaklavas, C.; Cheng, H.H.; Hammerman, P.; Mitchell, E.P.; Zwiebel, J.A.; Ivy, S.P.; Gray, R.J.; Li, S.; et al. Phase II Study of AZD4547 in Patients With Tumors Harboring Aberrations in the FGFR Pathway: Results From the NCI-MATCH Trial (EAY131) Subprotocol W. Journal of Clinical Oncology 2020, 38, 2407-2417. [CrossRef]
- Coombes, R.C.; Badman, P.D.; Lozano-Kuehne, J.P.; Liu, X.; Macpherson, I.R.; Zubairi, I.; Baird, R.D.; Rosenfeld, N.; Garcia-Corbacho, J.; Cresti, N.; et al. Results of the phase IIa RADICAL trial of the FGFR inhibitor AZD4547 in endocrine resistant breast cancer. Nature Communications 2022, 13, 3246. [CrossRef]
- Huynh, H.; Chow, P.K.; Tai, W.M.; Choo, S.P.; Chung, A.Y.; Ong, H.S.; Soo, K.C.; Ong, R.; Linnartz, R.; Shi, M.M. Dovitinib demonstrates antitumor and antimetastatic activities in xenograft models of hepatocellular carcinoma. J Hepatol 2012, 56, 595-601. [CrossRef]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer. New England Journal of Medicine 2015, 372, 621-630. [CrossRef]
- Hui, R.; Pearson, A.; Cortes, J.; Campbell, C.; Poirot, C.; Azim, H.A., Jr.; Fumagalli, D.; Lambertini, M.; Daly, F.; Arahmani, A.; et al. Lucitanib for the Treatment of HR(+)/HER2(-) Metastatic Breast Cancer: Results from the Multicohort Phase II FINESSE Study. Clin Cancer Res 2020, 26, 354-363. [CrossRef]
- Hayashi, H.; Makiyama, A.; Okumura, N.; Yasufuku, I.; Saigo, C.; Takeuchi, T.; Miyazaki, T.; Tanaka, Y.; Matsuhashi, N.; Murase, K.; et al. Gastric carcinosarcoma with FGFR2 amplification under long-term control with pazopanib: a case report and literature review. BMC Gastroenterology 2022, 22, 360. [CrossRef]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. Ponatinib: a novel multi-tyrosine kinase inhibitor against human malignancies. Onco Targets Ther 2019, 12, 635-645. [CrossRef]
- Harding, T.C.; Long, L.; Palencia, S.; Zhang, H.; Sadra, A.; Hestir, K.; Patil, N.; Levin, A.; Hsu, A.W.; Charych, D.; et al. Blockade of Nonhormonal Fibroblast Growth Factors by FP-1039 Inhibits Growth of Multiple Types of Cancer. Science Translational Medicine 2013, 5, 178ra139-178ra139, doi:doi:10.1126/scitranslmed.3005414.
- Kollmannsberger, C.; Britten, C.D.; Olszanski, A.J.; Walker, J.A.; Zang, W.; Willard, M.D.; Radtke, D.B.; Farrington, D.L.; Bell-Mcguinn, K.M.; Patnaik, A. A phase 1 study of LY3076226, a fibroblast growth factor receptor 3 (FGFR3) antibody–drug conjugate, in patients with advanced or metastatic cancer. Investigational New Drugs 2021, 39, 1613-1623. [CrossRef]
- Blackwell, C.; Sherk, C.; Fricko, M.; Ganji, G.; Barnette, M.; Hoang, B.; Tunstead, J.; Skedzielewski, T.; Alsaid, H.; Jucker, B.M.; et al. Inhibition of FGF/FGFR autocrine signaling in mesothelioma with the FGF ligand trap, FP-1039/GSK3052230. Oncotarget 2016, 7, 39861-39871. [CrossRef]
- Van Brummelen, E.M.J.; Levchenko, E.; Dómine, M.; Fennell, D.A.; Kindler, H.L.; Viteri, S.; Gadgeel, S.; López, P.G.; Kostorov, V.; Morgensztern, D.; et al. A phase Ib study of GSK3052230, an FGF ligand trap in combination with pemetrexed and cisplatin in patients with malignant pleural mesothelioma. Investigational New Drugs 2020, 38, 457-467. [CrossRef]
- Necchi, A.; Castellano, D.E.; Mellado, B.; Pang, S.; Urun, Y.; Park, S.H.; Vaishampayan, U.N.; Currie, G.; Abella-Dominicis, E.; Pal, S.K. Fierce-21: Phase II study of vofatmab (B-701), a selective inhibitor of FGFR3, as salvage therapy in metastatic urothelial carcinoma (mUC). Journal of Clinical Oncology 2019, 37, 409-409. [CrossRef]
- Trudel, S.; Bergsagel, P.L.; Singhal, S.; Niesvizky, R.; Comenzo, R.L.; Bensinger, W.I.; Lebovic, D.; Choi, Y.; Lu, D.; French, D.; et al. A Phase I Study of the Safety and Pharmacokinetics of Escalating Doses of MFGR1877S, a Fibroblast Growth Factor Receptor 3 (FGFR3) Antibody, in Patients with Relapsed or Refractory t(4;14)-Positive Multiple Myeloma. Blood 2012, 120, 4029. [CrossRef]
- ODonnell, P.; Goldman, J.; Gordon, M.; Shih, K.; Choi, Y.; Lu, D.; Kabbarah, O.; Ho, W.; Rooney, I.; Lam, E. 621 a phase I dose-escalation study of MFGR1877S, a human monoclonal anti-fibroblast growth factor receptor 3 (FGFR3) antibody, in patients (pts) with advanced solid tumors. European Journal of Cancer 2012, 191-192.
- Siefker-Radtke, A.O.; Currie, G.; Abella, E.; Vaena, D.A.; Kalebasty, A.R.; Curigliano, G.; Tupikowski, K.; Andric, Z.G.; Lugowska, I.; Kelly, W.K. FIERCE-22: Clinical activity of vofatamab (V) a FGFR3 selective inhibitor in combination with pembrolizumab (P) in WT metastatic urothelial carcinoma, preliminary analysis. Journal of Clinical Oncology 2019, 37, 4511-4511. [CrossRef]
- Wainberg, Z.A.; Enzinger, P.C.; Kang, Y.-K.; Qin, S.; Yamaguchi, K.; Kim, I.-H.; Saeed, A.; Oh, S.C.; Li, J.; Turk, H.M.; et al. Bemarituzumab in patients with FGFR2b-selected gastric or gastro-oesophageal junction adenocarcinoma (FIGHT): a randomised, double-blind, placebo-controlled, phase 2 study. The lancet oncology 2022, 23, 1430-1440. [CrossRef]
- Catenacci, D.V.T.; Kang, Y.-K.; Saeed, A.; Yamaguchi, K.; Qin, S.; Lee, K.-W.; Kim, I.-H.; Oh, S.C.; Li, J.; Turk, H.M.; et al. FIGHT: A randomized, double-blind, placebo-controlled, phase II study of bemarituzumab (bema) combined with modified FOLFOX6 in 1L FGFR2b+ advanced gastric/gastroesophageal junction adenocarcinoma (GC). Journal of Clinical Oncology 2021, 39, 4010-4010. [CrossRef]
- Smyth, E.C.; Chao, J.; Muro, K.; Yen, P.; Yanes, R.E.; Zahlten-Kumeli, A.; Rha, S.Y. Trial in progress: Phase 3 study of bemarituzumab + mFOLFOX6 versus placebo + mFOLFOX6 in previously untreated advanced gastric or gastroesophageal junction (GEJ) cancer with FGFR2b overexpression (FORTITUDE-101). Journal of Clinical Oncology 2022, 40, TPS4164-TPS4164. [CrossRef]
- Kim, S.-B.; Meric-Bernstam, F.; Kalyan, A.; Babich, A.; Liu, R.; Tanigawa, T.; Sommer, A.; Osada, M.; Reetz, F.; Laurent, D.; et al. First-in-Human Phase I Study of Aprutumab Ixadotin, a Fibroblast Growth Factor Receptor 2 Antibody–Drug Conjugate (BAY 1187982) in Patients with Advanced Cancer. Targeted Oncology 2019, 14, 591-601. [CrossRef]
- Poźniak, M.; Porębska, N.; Krzyścik, M.A.; Sokołowska-Wędzina, A.; Jastrzębski, K.; Sochacka, M.; Szymczyk, J.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. The cytotoxic conjugate of highly internalizing tetravalent antibody for targeting FGFR1-overproducing cancer cells. Molecular Medicine 2021, 27. [CrossRef]
- Kas, S.M.; de Ruiter, J.R.; Schipper, K.; Schut, E.; Bombardelli, L.; Wientjens, E.; Drenth, A.P.; de Korte-Grimmerink, R.; Mahakena, S.; Phillips, C.; et al. Transcriptomics and Transposon Mutagenesis Identify Multiple Mechanisms of Resistance to the FGFR Inhibitor AZD4547. Cancer Res 2018, 78, 5668-5679. [CrossRef]
- Zhou, Y.; Wu, C.; Lu, G.; Hu, Z.; Chen, Q.; Du, X. FGF/FGFR signaling pathway involved resistance in various cancer types. J Cancer 2020, 11, 2000-2007. [CrossRef]
- Wang, L.; Šuštić, T.; Leite de Oliveira, R.; Lieftink, C.; Halonen, P.; van de Ven, M.; Beijersbergen, R.L.; van den Heuvel, M.M.; Bernards, R.; van der Heijden, M.S. A Functional Genetic Screen Identifies the Phosphoinositide 3-kinase Pathway as a Determinant of Resistance to Fibroblast Growth Factor Receptor Inhibitors in FGFR Mutant Urothelial Cell Carcinoma. Eur Urol 2017, 71, 858-862. [CrossRef]
- Grygielewicz, P.; Dymek, B.; Bujak, A.; Gunerka, P.; Stanczak, A.; Lamparska-Przybysz, M.; Wieczorek, M.; Dzwonek, K.; Zdzalik, D. Epithelial-mesenchymal transition confers resistance to selective FGFR inhibitors in SNU-16 gastric cancer cells. Gastric Cancer 2016, 19, 53-62. [CrossRef]
- Kim, S.Y.; Ahn, T.; Bang, H.; Ham, J.S.; Kim, J.; Kim, S.T.; Jang, J.; Shim, M.; Kang, S.Y.; Park, S.H.; et al. Acquired resistance to LY2874455 in FGFR2-amplified gastric cancer through an emergence of novel FGFR2-ACSL5 fusion. Oncotarget 2017, 8, 15014-15022. [CrossRef]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: the current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052-16074. [CrossRef]
- Musolino, A.; Campone, M.; Neven, P.; Denduluri, N.; Barrios, C.H.; Cortes, J.; Blackwell, K.; Soliman, H.; Kahan, Z.; Bonnefoi, H.; et al. Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR+, HER2− breast cancer that had progressed during or after prior endocrine therapy. Breast Cancer Research 2017, 19. [CrossRef]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nature Reviews Clinical Oncology 2019, 16, 105-122. [CrossRef]
- Singleton, K.R.; Hinz, T.K.; Kleczko, E.K.; Marek, L.A.; Kwak, J.; Harp, T.; Kim, J.; Tan, A.C.; Heasley, L.E. Kinome RNAi Screens Reveal Synergistic Targeting of MTOR and FGFR1 Pathways for Treatment of Lung Cancer and HNSCC. Cancer Research 2015, 75, 4398-4406. [CrossRef]
- Scheller, T.; Hellerbrand, C.; Moser, C.; Schmidt, K.; Kroemer, A.; Brunner, S.M.; Schlitt, H.J.; Geissler, E.K.; Lang, S.A. mTOR inhibition improves fibroblast growth factor receptor targeting in hepatocellular carcinoma. British Journal of Cancer 2015, 112, 841-850. [CrossRef]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nature Communications 2019, 10. [CrossRef]
- Sobhani, N.; Fassl, A.; Mondani, G.; Generali, D.; Otto, T. Targeting Aberrant FGFR Signaling to Overcome CDK4/6 Inhibitor Resistance in Breast Cancer. Cells 2021, 10, 293. [CrossRef]
- Palakurthi, S.; Kuraguchi, M.; Zacharek, S.J.; Zudaire, E.; Huang, W.; Bonal, D.M.; Liu, J.; Dhaneshwar, A.; DePeaux, K.; Gowaski, M.R.; et al. The Combined Effect of FGFR Inhibition and PD-1 Blockade Promotes Tumor-Intrinsic Induction of Antitumor Immunity. Cancer Immunology Research 2019, 7, 1457-1471. [CrossRef]
- Sweis, R.F.; Spranger, S.; Bao, R.; Paner, G.P.; Stadler, W.M.; Steinberg, G.; Gajewski, T.F. Molecular Drivers of the Non-T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol Res 2016, 4, 563-568. [CrossRef]
- Abdul-Karim, R.M.; Chaudhry, A.; Patrikidou, A.; Gonzalez, A.F.; Racca, F.; Loriot, Y.; Pouessel, D.; Deville, J.-L.; Lee, H.J.; Cantero, F.; et al. Derazantinib (DZB) in combination with atezolizumab (AZB) in patients with solid tumors: Results from the dose-finding phase Ib substudy of FIDES-02. Journal of Clinical Oncology 2021, 39, 437-437. [CrossRef]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.C. Targeting FGFR Signaling in Cancer. Clin Cancer Res 2015, 21, 2684-2694. [CrossRef]
- Kommalapati, A.; Tella, S.H.; Borad, M.; Javle, M.; Mahipal, A. FGFR Inhibitors in Oncology: Insight on the Management of Toxicities in Clinical Practice. Cancers (Basel) 2021, 13. [CrossRef]
- Bensinger, W.; Schubert, M.; Ang, K.-K.; Brizel, D.; Brown, E.; Eilers, J.G.; Elting, L.; Mittal, B.B.; Schattner, M.A.; Spielberger, R.; et al. NCCN Task Force Report: Prevention and Management of Mucositis in Cancer Care. Journal of the National Comprehensive Cancer Network J Natl Compr Canc Netw 2008, 6, S-1-S-21. [CrossRef]
- Prensky, C.; Marlow, E.; Gupta, M.; Sales, C.; Kiss, S.; D’Amico, D.J. Reversible Macular Lesions in the Setting of Oral Pan-Fibroblast Growth Factor Inhibitor for the Treatment of Bladder Cancer. Journal of VitreoRetinal Diseases 2018, 2, 111-114. [CrossRef]
- Jogo, T.; Nakamura, Y.; Shitara, K.; Bando, H.; Yasui, H.; Esaki, T.; Terazawa, T.; Satoh, T.; Shinozaki, E.; Nishina, T.; et al. Circulating Tumor DNA Analysis Detects FGFR2 Amplification and Concurrent Genomic Alterations Associated with FGFR Inhibitor Efficacy in Advanced Gastric Cancer. Clinical Cancer Research 2021, 27, 5619-5627. [CrossRef]
- Akbulut, O.; Lengerli, D.; Saatci, O.; Duman, E.; Seker, U.O.S.; Isik, A.; Akyol, A.; Caliskan, B.; Banoglu, E.; Sahin, O. A Highly Potent TACC3 Inhibitor as a Novel Anticancer Drug Candidate. Mol Cancer Ther 2020, 19, 1243-1254. [CrossRef]
Table 2.
Multi-target tyrosine kinase inhibitors.
| Multi-TKIs | Targets |
|---|---|
| Dovitinib (TKI258) | FGFR, VEGFR, PDGFR inhibitor [111] |
| Lenvatinib (E7080) | VEGFR 1-3, FGFR 1-4, PDGFR α, RET, KIT [112] |
| Lucatinib (E-3810) | FGFR1-2, VEGFR1-3, and PDGFRα-β [113] |
| Pazopanib (GW786034) | FGFR 1-2, VEGFR 1-3, PDGFRα-β, C-kit (stem cell factor receptor) [114] |
| Ponatinib (AP24534) | FGFR 1-4, VEGFR2, PDGFRα, c-SRC, c-Kit, FLT3, RET [115] |
Table 3.
Selected current clinical trials.
| Agent | NCT | Status | Conditions | Phase |
|---|---|---|---|---|
| Pemigatinib | NCT03914794 | R | Non-muscle invasive bladder cancer (NMIBC) with recurrent low or intermediate risk tumors (as neoadjuvant) | II |
| Pemigatinib | NCT03011372 | A, NR | Previously treated myeloid/lymphoid neoplasms with FGFR1 rearrangement (FIGHT-203) | II |
| Pemigatinib vs Gemcitabine +cisplatin |
NCT03656536 | R | Untreated unresectable or metastatic cholangiocarcinoma with FGFR2 rearrangement (FIGHT-302) | III |
| Pemigatinib | NCT05565794 | R | Intrahepatic cholangiocarcinoma with FGFR2 gene mutation, rearrangement, or translocation after curative local therapy | II |
| Pemigatinib | NCT05267106 |
R | Previously treated glioblastoma or other primary central nervous system tumors harboring activating FGFR1-3 alterations (FIGHT-209) | II |
| Pemigatinib | NCT05253807 | A, NR | Relapsed or refractory advanced non-small cell lung cancer with an FGFR alteration (FIGHT-210) | II |
| Pemigatinib | NCT04659616 | R | Acute myeloid leukemia after initial induction chemotherapy with adverse or intermediate risk cytogenetics | I |
| Futibatinib | NCT04189445 | A, NR | Previously treated advanced or metastatic solid tumors, gastric or gastroesophageal cancers, myeloid or lymphoid neoplasms with FGFR 1-4 rearrangements | II |
| Futibatinib +/- fulvestrant |
NCT04024436 | A, NR | Previously treated metastatic breast cancer with FGFR1 and FGFR2 amplification | II |
| Futibatinib vs gemcitabine + cisplatin |
NCT04093362 | A, NR | Previously untreated advanced cholangiocarcinoma harboring FGFR2 gene rearrangements (FOENIX-CCA3) | III |
| Futibatinib | NCT05727176 | R | Previously treated advanced cholangiocarcinoma with FGFR2 fusion or rearrangement (FOENIX-CCA4) | II |
| Futibatinib + pembrolizumab |
NCT04828486 | R | Advanced or metastatic hepatocellular carcinoma with FGF19 expression | II |
| Futibatinib + pembrolizumab |
NCT04601857 | R | Advanced or metastatic urothelial carcinoma who are not candidates to receive a platinum-based treatment regimen | II |
| Futibatinib + pembrolizumab |
NCT05036681 | R | Previously treated locally advanced or metastatic microsatellite stable endometrial carcinoma | II |
| Infigratinib | NCT04233567 | A, NR | Previously treated advanced or metastatic solid tumors, cholangiocarcinoma and refractory malignant solid neoplasm with FGFR gene mutations | II |
| Infigratinib | NCT04228042 | A, NR | Renal pelvis and upper tract urothelial cancer as neoadjuvant treatment | I/II |
| Erdafitinib | NCT04917809 | R | Recurrent non-Invasive Bladder Cancer with FGFR3 gene mutation after treatment with instillations of BCG or chemotherapy into the bladder | II |
| Erdafitinib | NCT04083976 |
A, NR | Advanced or metastatic solid tumors with FGFR alterations (mutations or gene fusions) after at least one prior line of systemic therapy (RAGNAR) | II |
| Erdafitinib | NCT04754425 | R | Castration-resistant prostate cancer after progressed on second generation androgen receptor targeting agents | II |
| Erdafitinib + fulvestrant + palbociclib | NCT03238196 | A, NR | Previously treated HR positive HER2 negative FGFR amplified metastatic breast cancer | I |
| Fisogatinib | NCT02508467 | A, NR | Hepatocellular carcinoma with FGF19 expression with or without prior tyrosine kinase inhibitors | I |
| Derazantinib + atezolizumab |
NCT05174650 | R | Previously treated advanced or metastatic intrahepatic cholangiocarcinoma with FGFR2 fusions/rearrangements | II |
| Lirafugratinib (RLY4008) |
NCT04526106 |
R | Previously treated unresectable/metastatic intrahepatic cholangiocarcinoma and other advanced tumors with FGFR2 alterations (REFOCUS) | I/II |
| Dovitinib + PARP inhibitor Stenoparib (2X-121) |
NCT05571969 |
R | Advanced Solid Tumors | I |
| Rogaratinib (BAY1163877) + atezolizumab |
NCT03473756 | A, NR | Metastatic or locally advanced cisplatin ineligible patients with urothelial carcinoma and FGFR 1 or 3 alterations (FORT-2) | Ib/II |
| Rogaratinib | NCT04595747 | A, NR | Previously treated advanced or metastatic sarcoma with FGFR 1-4 alterations and in patients with SDH-deficient Gastrointestinal Stromal Tumor (GIST) | II |
| Fexagratinib AZD4547 + tislelizumab |
NCT05775874 | R | Locally advanced or metastatic urothelial cancer with FGFR2/3 alterations in Chinese patient population | II |
| Bemarituzumab |
NCT05325866 | R | Refractory or relapsed advanced or metastatic solid tumors with FGFR2b overexpression after at least one prior line (FORTITUDE-301) | I |
| Bemarituzumab+ mFOLFOX6+ nivolumab vs mFOLFOX6+ nivolumab |
NCT05111626 | R | Untreated advanced or metastatic gastric and gastroesophageal junction (GEJ) cancer with FGFR2b overexpression (FORTITUDE-102) | III |
| Bemarituzumab+ + mFOLFOX vs mFOLFOX |
NCT05052801 | R | Previously treated advanced or metastatic Gastric or GEJ Cancers with FGFR2b overexpression (FORTITUDE-101) | III |
| Bemarituzumab +anti-cancer therapy |
NCT05267470 | A, NR | Advanced or metastatic squamous lung cancer with FGFR2b overexpression (FORTITUDE-201) | I |
A = Active, NR = Non-recruiting, R = Recruiting. Retrieved from clinicaltrials.gov [101].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



