
Submitted:
06 December 2023
Posted:
07 December 2023
You are already at the latest version
Abstract
Delivery systems of the biologically active substance such as proanthocyanidins (PCAN) produced through the electrospinning method were designed using a blend of poly(L-lactide-co-glycolide) and poly[(R,S)-3-hydroxybutyrate]. The research involved the structural and thermal characteristics of the developed electrospun three-dimensional fiber matrix, as well as hydrolytic degradation tests performed on them. Additionally, the release profile of PCAN from the electrospun nonwoven was determined using UV–VIS spectroscopy. Approximately 30% of PCAN was released from the tested electrospun nonwoven during the initial 15-20 days of incubation. The chemical structure of water-soluble oligomers that were formed after the hydrolytic degradation of the developed delivery system was identified through electrospray ionization mass spectrometry. Oligomers of lactic acid and OLAGA oligocopolyester, as well as oligo-3-hydroxybutyrate terminated with hydroxyl and carboxyl end groups, were recognized as degradation products released into the water during the incubation time. It was also demonstrated that variations in the degradation rate of individual mat components influenced the degradation pattern, and the number of formed oligomers. The obtained results suggest that the incorporation of proanthocyanidins into the system slowed down the hydrolytic degradation process of the poly(L-lactide-co-glycolide)/poly[(R,S)-3-hydroxybutyrate] three-dimensional fiber matrix. In addition, in vitro cytotoxicity and antimicrobial studies advocate the use of PCAN for biomedical applications with promising antimicrobial activity.
Keywords:
Poly(L-lactide-co-glycolide)/poly[(R
; S)-3-hydroxybutyrate]
; electrospun nonwoven delivery sys-tem
; biodegradable polyesters
; antimicrobial
; proanthocyanidins
; periodontal
1. Introduction
Drug delivery systems (DDS) are vehicles for the transportation of therapeutic agents including antibodies, peptides, vaccines, drugs, and enzymes to safely achieve the desired therapeutic effect and improve the administration and efficacy of these pharmaceutical compounds [1,2]. Conventional drug delivery methods are the most popular route for drug administration because they have advantages, such as ease of use and a very high degree of dosage flexibility. However, systemic administration pathways also have disadvantages, such as serious side effects, adverse biodistribution, low selectivity of therapeutic effect, burst release of the drug, and damage to healthy cells [3,4]. Moreover, conventional drug delivery methods do not fully utilize the therapeutic potential of medications. Even though the use of drug delivery methods in medicine or cosmetology has been used for years, it still has great perspectives and is enjoying increasing interest. The effectiveness of the medicine increases with the development of fabrication of drug delivery carriers, which can administer accurate doses of drugs, minimize side effects, and improve healthcare treatment for patients.
Periodontitis is an extremely common chronic inflammatory disease characterized by periodontal tissue destruction, usually caused by the accumulation of pathogenic microorganisms [2,5]. According to the World Health Organization report, it is one of the world’s most widespread chronic ailments occurring after the age of 35. Recently, there has been growing interest in controlled drug delivery systems in oral infectious disease as a potential method to address all these challenges [6]. It is worth mentioning that local sustained administration of drugs into the periodontal pockets in the mouth is problematic due to the continuous secretion of saliva, which makes it hard for the administered drug to remain in the periodontal pocket, resulting in being swallowed by the patient [7]. Drug delivery devices improve the antimicrobial efficacy in the periodontal pocket and demonstrate other clinical benefits.
An important step in the preparation of such delivery systems is the selection of appropriate polymer matrix, which should be non-toxic to mammalian cells, biodegradable and flexible enough to suit the wound topography well [2,8].
The aliphatic polyesters of alfa- and beta-hydroxy acids (polylactide and its copolymers with glycolide, caprolactone, and polyhydroxybutyrate) are of great importance in this field [9]. The drugs dispersed in such matrices are released both by diffusion and by erosion of the carrier. The poly(lactic-co-glycolic acid) copolymers containing less than 30% of glycolide units are the most successfully used synthetic biodegradable polymers in the medical field. Biocompatibility, biodegradability, flexibility, and minimal side effects are the main advantages of using this polymer for such an application [10]. Recently poly(L-lactide-co-glycolide), PLGA biopolyesters in the form of implants [11], disks [12] or dental films [13] can be used in the treatment of periodontium for better local administration of antibiotics and for reducing their side effects.
At the moment, there is growing interest in incorporating bioactive substances into polymer matrices, for example in the form of electrospun mats. This simple and powerful technique of electrospinning involves preparation of polymeric fibers with different diameters, from a few microns to tens of nanometers. Nanofibers can be fabricated from natural or synthetic polymers and blends with various additives. Due to control of infection and small-size pores, electrospun mats are very beneficial for preventing the penetration of bacteria [14,15,16,17].
The aim of this study was to develop a polymer biomaterial based on poly(L-lactide-co-glycolide), PLGA and synthetic poly[(R,S)-3-hydroxybutyrate], (R,S)-PHB blends, and to conduct preliminary research towards its application in the construction of drug delivery devices intended for potential use in periodontitis treatment with the use of proanthocyanidins, PCAN extracted from Pelargonium sidoides as an active substance. Pelargonium sidoides (PSRE) is a medical plant derived from coastal regions of southern Africa and is highly valued by the native population for its curative properties. The antibacterial activities of this plant are mostly assigned to contain proanthocyanidins, which also revealed antioxidant, anti-inflammatory, anti-aging, and anticancer properties [18,19].
The strategic aspect of this research is related to the methods of incorporation of bioactive substances to polymeric carriers.
Our previous research demonstrated the possibility of obtaining polymer delivery systems for potential wound treatment application in the form of electrospun mats with flexible properties ensuring compatibility with the wound topology from the P(D,L)LA/(R,S)-PHB polymeric blend containing PCAN [20].
In this study, we focused on designing drug delivery devices to improve the antimicrobial efficacy in the periodontal pocket based on biodegradable polyesters. As a polymer carrier, a blend of bioresorbable polyesters poly(L-lactide-co-glycolide) with synthetic poly[(R,S)-3-hydroxybutyrate], (PLGA/(R,S)-PHB) in the form of the electrospun mats was used. Our intention was to develop a system that would enable controlled release of the biologically active substance PCAN throughout the defined period during therapy without the need to remove the system. The polymeric matrices due to systematic degradation after therapy should be completely bio-resorbed by the body.
These studies included the preparation of polymer blends of poly(L-lactide-co-glycolide) copolymer, PLGA with synthetic poly[(R,S)-3-hydroxybutyrate], (R,S)-PHB with a chemical composition of PLGA/(R,S)-PHB of 80/20 wt%, and examining the possibility of using the developed biomaterial for the production of delivery systems in the form of electrospun mats with the desired properties enabling loading the biologically active substance—PCAN. In the next stage, the structural and thermal characterization of the obtained PLGA/(R,S)-PHB electrospun nonwoven were investigated, and the comparative hydrolytic degradation tests of the developed systems, both with and without loaded PCAN, were carried out. The results of the hydrolytic degradation studies allowed us to determine the PCAN release profile from the elaborated system and determine the rate of the electrospun nonwovens hydrolysis dependent on PCAN presence. Additionally, the structural characterization of the degradation products of the developed polymeric carrier was performed. The cytocompatibility and antimicrobial activity of the developed system were also tested.
2. Results and Discussion
The delivery system of PCAN was designed from biodegradable polymeric blend in the form of three-dimensional fiber matrix. The electrospun nonwovens studied were prepared from the polymeric blend of poly(L-lactide-co-glycolide) and poly[(R,S)-3-hydroxybutyrate] (PLGA/(R,S)-PHB; 80 wt%/20 wt%) using electrospinning method. The electrospinning processes typically produce randomly oriented, electrospun nonwoven. Our previous studies on the polymer solution concentration showed that increase of polymer concentration causes a slower sedimentation of bioactive compound particles in syringe during electrospinning process. Very good fibers were produced using a 10% w/v polymeric blend solution in HFIP [20]. Thus this concentration was used in the production of electrospun mats from PLGA/(R,S)-PHB blend with and without proanthocyanidins. The other solution properties (density, viscosity, surface tension etc.) and operating parameters (flow rate, electric field strength and electric current flow), presents in section Electrospinning experiment were also significant parameters to obtain a desired proper size of fiber diameter.
Figure 1 and Figure 2, below, show the 1H-NMR spectrum and DSC traces of the resulting PLGA/(R,S)-PHB electrospun nonwovens.
The 1H-NMR spectrum revealed the presence of signals corresponding to the protons of lactidyl (LA), glycidyl (GA), and 3-hydroxybutyrate (3-HB) repeating units, characteristic for the blend components according to structure (Figure 1). The chemical composition of the electrospun nonwoven sample before degradation was calculated based on the integrals value of the signals 4, and 1, 3 corresponding to methyl group protons of PHB and methyl group protons of PLA as well as methylene group proton of PGA in PLGA copolymer, respectively.
The second DSC heating traces for PLGA/(R,S)-PHB three-dimensional fiber matrix without PCAN shows only one glass transition temperature, Tg with equal 38.9 °C which is located between the Tg values of blend components (see Figure 2). Thus, thermal analysis confirms that the electrospinning process does not disturb the miscibility of the blend components. In the case of the electrospun nonwoven loaded with PCAN in DSC trace, two Tg values were observed which indicates that the addition of PCAN to the PLGA/(R,S)-PHB polymer system disrupted its compatibility.
2.1. Degradation of PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN electrospun nonwoven
The polymeric electrospun nonwoven were subjected to hydrolytic degradation. The progress of mats hydrolysis was assessed by macro- and microscopic observations as well as molar mass, structure, and thermal properties changes of samples studied. The visual inspection of initial PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN nonwoven fabric as well as their changes during incubation was investigated with aid of POM microscopy and are shown in Figure 3.
The digital images, as well as POM micrographs, show that the surface of PLGA/(R,S)-PHB mats before incubation was smooth and bioactive substance was uniformly distributed in the polymer matrix (Figure 3). After degradation, pitting and cracks were observed on the surface of both types of mats, which are layered in nature. It is especially visible in the case of a mat loaded with PCAN. The morphology of the PLGA/(R,S)-PHB three-dimensional fibers matrix were also investigated with aid of SEM microscopy and results are shown in Figure 4.
As shown in SEM micrographs, the bioactive substance formed different size agglomerates, which are irregularly intercalated between the fibers (Figure 4). It is probably caused by the very poor solubility of the bioactive substance in organic solvents and the impossibility of keeping a homogenous solution used in the electrospinning process. The fibers in PLGA/(R,S)-PHB electrospun nonwoven studied possess average diameters of 2.2 µm. The PLGA/(R,S)-PHB fibers look smooth and linear which is typical for electrospun nonwoven prepared by electrospinning. The addition of PCAN caused disorganization and entanglement of fibers, which also showed different diameters varying over the entire length of a single fiber. Disordered fibers in the electrospun nonwoven have an impact on the forming of porous structures with different densities [24,25]. Appearance of the sample’s surface morphology in the higher magnification results from the gradual softening of the material during the analysis.
The changes in number-average molar mass of the samples during the degradation process were monitored by gel permeation chromatography, GPC. All investigated samples show the systematic shifting of GPC traces to a lower molar mass value with the progress of the degradation process (Figure 5).
The results of this measurement revealed that Mn values for both mats without and with PCAN decreased and reached a value at the level of 1000 g mol−1 after 71 days of incubation. The overlapping of the GPC curves may indicate differences in the degradation profile of the polymeric mat components. It is worth noting that the molar mass loss of the PLGA/(R,S)-PHB sample with PCAN is lower than for the PLGA/(R,S)-PHB mats not loaded with biological active substance. This indicates that the addition of PCAN has an influence on the behavior of three-dimensional fiber matrix of PLGA/(R,S)-PHB during degradation.
The 1H-NMR spectra of PLGA/(R,S)-PHB electrospun nonwoven without and with biological active substance, PCAN before and after degradation time are shown in Figure 6.
The resulting 1H-NMR spectra show the signals of protons characteristic for the LA, GA, and 3-HB repeating units according to the structure presented in Figure 1. In addition, in the spectra obtained after 71 days of degradation, proton signals were detected, characteristic of low-molecular-mass OHB and OLA oligomers terminated with hydroxyl and carboxyl end groups which are formed during hydrolyses of the polymeric mats components the (R,S)-PHB and PLGA, respectively [26]. The presence of proton signals of OHB and OLA oligomers with a simultaneous change in the area of proton signals of GA units (due to the formation of OLAGA copolyester oligomers) indicates the progress of the hydrolysis process. Random cleavage of the polymer chains leads to a decrease in the molar mass of the components of the blend. This was confirmed by the GPC results (see Figure 5). It should also be noted that no signals from PCAN were observed in the obtained 1H-NMR spectra. This is because PCAN does not dissolve in CDCl3.
Moreover, on the basis of 1H-NMR spectra recorded after a certain period of degradation, changes in the chemical composition of electrospun nonwovens PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN were also estimated and the results are presented in Figure 7.
The (R,S)-PHB component content in three-dimensional fiber matrix samples remaining after a specific incubation period was calculated based on the integrals value of signals corresponding to protons of methyl groups of both 3-HB repeating units and OHB oligomers formed during degradation [26].
Figure 7 shows that the content of the PLGA component in the tested mats without and with PCAN decreases while the content of the PHB component increases. However, it was observed that changes in the chemical composition of the PLGA/(R,S)-PHB electrospun nonwoven started from the beginning of the incubation process, while in the case of the PCAN-loaded mat, a decrease in the PLGA content was observed after 51 days of degradation. The lower degradation rate of the PLGA component in this case is probably related to the presence of PCAN, which interferes with the penetration of water into the electrospun nonwoven.
Figure 8 presents the second DSC heating traces for PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN electrospun nonwoven samples remaining after 71days of hydrolytic degradation.
A decrease in Tg from 38.9 °C to 17.9 °C was observed for the sample without PCAN, after 71 days of their incubation. In contrast, for the sample with PCAN, a decrease of Tg value from 47 °C to 28.9 °C was observed (see Figure 2 and Figure 8). The observed differences in the thermal properties of the tested mats probably result from the different rates of hydrolytic degradation of the loaded and unloaded PCAN mats, leading to the formation in the degradation process of a different amount of oligomers, which remain in the tested mats. The PLGA/(R,S)-PHB electrospun nonwoven degraded faster, and oligomers formed acts as plasticizers of the system. In the case of PLGA/(R,S)-PHB/PCAN electrospun nonwoven loaded with PCAN the biologically active substance located between fibers of the polymer matrix probably hinders the absorption of water, which slows down the hydrolysis process, and the observed amount of low-molar mass oligomeric degradation products formed was lower. The presence of PCAN between the fibers inside the electrospun nonwoven was confirmed with the SEM analysis (see Figure 4). The progress of the hydrolytic degradation of PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN three-dimensional fiber matrix was also monitored by the mass loss measurements.
Figure 9 shows a mass loss for the PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN electrospun nonwoven observed during degradation carried out in the water.
The observed mass loss of the tested samples during their incubation in water is related to the release of low-molar mass degradation products of the mats polymer components into the water environment, as well as, in the case of the PLGA/(R,S)-PHB/PCAN electrospun nonwoven, by the release of the bioactive substance contained therein. At the first stage of incubation up to 51 days a higher mass loss was observed in the case of the PLGA/(R,S)-PHB/PCAN electrospun mat samples than for PLGA/(R,S)-PHB ones. A significant increase in the mass loss of PLGA/(R,S)-PHB/PCAN sample after 51 days of incubation can be connected to the fact that the three-dimensional fiber matrix start to disintegrate after this incubation time, and the release of PCAN is facilitated. The observed violent increase in mass loss after 51 days of incubation may additionally arise from the increased rate of degradation of the matrix themselves, which is facilitated by less amount of PCAN contained therein. The above observations were confirmed by the results obtained from the 1H-NMR analysis. At first stage, the differences in mass loss of the electrospun nonwoven samples result mainly from the release of a biological active substance, PCAN. In the second stage, starting on day 51, due to the increased migration of low-molar mass degradation products from the mats into the water, for both electrospun nonwoven samples greater mass loss was observed. Wherein in the case of PLGA/(R,S)-PHB electrospun nonwoven mass loss was systematically higher than for the PLGA/(R,S)-PHB/PCAN samples; after 71 days of degradation, mass loss of 51.1% and 38.5% was observed for the PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN samples, respectively. Thus, PCAN contained in the electrospun nonwoven slows down degradation, which may be due to the presence of physical interactions polymer-PCAN such as ion-ion attraction/repulsion, hydrogen bonding, and van der Waals forces which can significantly alter the degradation time and hinder the release of oligomeric degradation products of the three-dimensional fiber matrix into the degradation environment.
2.2. Release study of PCAN from PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN electrospun nonwoven
Figure 10 shows a release profile of PCAN from PLGA/(R,S)-PHB/PCAN electrospun nonwoven during incubation in the water.
Comparing the mass loss of the tested mat and the amount of released PCAN, from the beginning of incubation, the PCAN release process is accompanied by the degradation of the polymer mat and the release of polymeric degradation products into the degradation medium. At the beginning of the incubation a rapid release of PCAN from the mat up to the level of 20 wt%, then from the 15th day a gradual, more moderate release of PCAN reached about 50% after 51 days of incubation. The observed further increase in the amount of PCAN released after 51 days is because the three-dimensional fiber matrix starts to disintegrate after this incubation time. The increased release of low-molar mass oligomeric degradation products from the samples into the degradation environment (Figure 9) is accompanied by a facilitated release of the PCAN occluded inside the fiber matrix.
Depending on the type of preparation polymer and bioactive compound, sample procedure, and degradation conditions, the kinetics of release may be influenced by one or more physical and chemical phenomena [27]. In this case, the biological active substance is released by diffusion from the polymer matrix into the degradation medium. With the progressive release of PCAN and microenvironmental changes in pH inside the matrix caused by OHB, OLA, and OGA oligomers formed, the degradation process of the PLGA/(R,S)-PHB/PCAN fibers matrix should be less disturbed over time, however, the degradation of this samples was still slower than the electrospun nonwoven without PCAN.
2.3. ESI-MS study of the degradation products released from PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN electrosun nonwoven
The chemical structure of the low-molar mass oligomeric degradation products which were released from the PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN three-dimensional fiber matrix into the degradation medium was determined with the aid of ESI-mass spectrometry. The ESI-mass spectra recorded for the water solutions collected after 71 days of incubation of PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN samples are shown in Figure 11a and Figure 11b, respectively.
The oligomers of the lactic acid OLA and OLAGA copolyester oligomers as well the oligomers of poly(3-hydroxybutyrate), OHB terminated with hydroxyl and carboxyl end groups were identified as low-molar mass degradation products released into the water during the incubation of the polymeric mats studied. The ESI-mass spectrum recorded for the water solution collected after 71 days of incubation of PLGA/(R,S)-PHB sample shown in Figure 11a, is less complicated. One maximum can be distinguished in it with the visible major series of ions at m/z 557, 643, 729, 815, 901, 987 (with mass increment between the signals equal to 86 Da). Those signals represent sodium adducts of 3-HB oligomers terminated with hydroxyl and carboxyl end groups. The structure of oligomers visible on ESI-mass spectra was confirmed with aid of ESI-MSn experiments. The conducted tests confirmed that the PLGA matrix component degrades faster than the PHB component. During the incubation, PLGA copolyester systematically degrades to oligomers with lower and lower molar mass and, consequently, lactic and glycolic acids. Therefore, 3-HB oligomers dominated the solutions after an increasingly longer period of degradation. The two major maxima of singly charged positive ions were observed on the ESI-mass spectrum recorded for the solution collected after incubation of the PLGA/(R,S)-PHB/PCAN sample and presented in Figure 11b. The main series of ions at m/z 257, 328, 401, 473, 545, and 617 (with a mass spacing of 72 Da) located in the area of the first maximum correspond to the sodium adducts of lactic acid oligomers with hydroxyl and carboxyl end groups. Moreover, in the range of the first maximum, the ions representing sodium adducts of copolyester oligomers with hydroxyl and carboxyl end groups can be also noticed. At the second maximum located in the mass range of m/z 600-1000, the visible major series of ions at m/z 557, 643, 729, 815, 901, 987 (with mass increment between the signals equal 86 Da) corresponds to sodium adducts of 3-HB oligomers terminated with hydroxyl and carboxyl end groups.
It is worth noting that the mass spectrometry analysis confirmed that the fiber matrix containing the biological active substance PCAN decomposes more slowly than the samples without the addition of this substance. The slower degradation of the three-dimensional fiber matrix is probably due to the interactions between the PCAN found on the fibers of the electrospun nonwoven, the polymer components, and the resulting degradation products. Difficult access of water to the interior of the electospun nonwoven caused by the presence of PCAN slows down the hydrolysis process and hinders the release of oligomeric degradation products into the degradation environment.
2.4. Cytocompatibility test (MTT assay)
Cytocompatibility stands as a fundamental attribute of a material when it comes to its utilisation in the biomedical sector. In this current research, the in vitro MTT assay was employed to assess the cytocompatibility of PLGA/(R,S)-PHB electrospun nonwoven, both without and with the 20 wt% addition of biological active substance, PCAN. The MTT assay results indicated varying levels of cytocompatibility of PLGA/(R,S)-PHB with the selected cell lines: U251MG, MSTO, PANC 1. MSTO demonstrated the highest cell viability (90.91 ± 6.54%) when exposed to conditioned DMEM media followed by PANC 1 and U251MG (Table 1). These results confirmed that the U251MG cell line is sensitive to PLGA/(R,S)-PHB.
When cell lines were exposed (in vitro) to DMEM conditioned with PLGA/(R,S)-PHB/PCAN the viability was significantly reduced (p <0.05) for all three cell lines. MSTO cell line demonstrated highest cell viability (%) out of all the three tested cell lines (Table 1). These findings are in accordance with our previous study where the cytotoxicity of P(D,L)LA/(R,S)-PHB with 20 wt% of PCAN was reported [20]. When PCAN containing electrospun nonwoven are produced with PLGA/(R,S)-PHB, the viability of MSTO cell lines was recorded to be better than P(D,L)LA/(R,S)-PHB with 20 wt% of PCAN. These findings confirm that PCAN at this concentration (20 wt%) demonstrates the cytotoxic effect in the in vitro test settings but different cell lines respond differently with MSTO being highest viable vs. PANC 1 being least (Table 1).
PCAN possess an established antioxidative characteristic that affects numerous signaling pathways including, nuclear factor erythroid 2-related factor 2 (Nrf2), mitogen-activated protein kinase (MAPK), nuclear factor-kB (NF-κB), and phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) [28]. The decrease in cell viability, as shown in Table 1 and Figure 12, might be linked to the free radical scavenging capacity of PCAN, which can impact signaling pathways [29]. Nonetheless, it is important to note that PCAN has demonstrated safety profile advocating its safe application in clinical medicine [28]. Furthermore, PCAN has a strong antimicrobial property advocating the application of these electrospun nonwoven in periodontal pockets [20]. However, further work is required to evaluate the behavior (in vivo) of these electrospun nonwoven before their clinical application as a drug delivery system for periodontal application to control infections.
2.5. Antibacterial activity (Disc Diffusion Assay, DDA)
Periodontitis is a chronic infection that affects the supporting tissues of the teeth. This can lead to gingival inflammation, gingival attachment loss, alveolar bone resorption, and can lead to tooth loss [30]. One stage full-mouth disinfection procedure is one of the non-invasive therapeutic strategies employed to treat periodontitis [31]. Attributing to antioxidative, anti-inflammatory and antimicrobial activities, proanthocyanidins (PCAN) have attracted a wide research interest for their biomedical applications including in the periodontal treatment [31,32]. Our group has previously reported the antimicrobial properties of PCAN from Pelargonium sidoides [20]. In the current study, the work was extended by testing the antimicrobial properties of varied concentrations of PCAN, ranging from 2.5% to 20% w/v. The test (in vitro) results revealed that PCAN at 2.5% w/v concentration is capable of inhibiting gram-positive Staphylococcus aureus. At higher concentrations, the antimicrobial activity increased (Figure 13), with 20% exhibiting the highest antimicrobial activity. These results support the application of PCAN to control infections in periodontal disease.
3. Materials and Methods
3.1. Materials
Poly(L-lactide-co-glycolide), PLGA was obtained via ring-opening polymerization of L-lactide and glycolide (both monomers HUIZHOU Foryou Medical Devices Co., Ltd., China) in the presence of biocompatible zirconium (IV) acetylacetonateZr(Acac)4 initiator (Sigma Aldrich, Merck KGaA, Germany). The molar ratio of Zr(Acac)4 to the sum of moles of monomers was equal to 1:600. The process was performed in bulk at 130 °C for 24 h and then at 115 °C for 72 h in an argon atmosphere. Unreacted monomers were removed from the bulk by dissolution in chloroform and precipitation in methanol (both Sigma Aldrich, Merck KGaA, Germany). Finally, the purified material was dried under a vacuum (20 mb; 20 °C) to constant weight [21]. ThePLGA copolymer with Mwof104 000 gmol-1and dispersity 2.48 was obtained. The molar ratio of L-lactide to glycolide units in the copolymer was 70 to 30mol% as determined from 1H-NMR spectra. Glass transition temperature,Tg determined by DSC was 58 °C. Poly[(R,S)-3-hydroxybutyrate], (R,S)-PHB was synthesized by bulk polymerization of (R,S)-β-butyrolactone, using tetrabutylammonium acetate as the initiator. The (R,S)-PHB with Mnof 1000 g mol-1and dispersity 1.2 was used [22]. Poly(L-actide-co-glycolide) and poly[(R,S)-3-hydroxybutyrate] blend with weight ratio equal 80 wt% to 20 wt% was prepared by mixed appropriate amounts of PLGA and (R,S)-PHB in hexafluoroisopropanol(HFIP,Sigma-Aldrich Chemie GmbH, Steinheim, Germany), directly before electrospinning experiment.
The U251MG glioblastoma cell line, was obtained from ATCC (UK), along with the MSTO mesothelioma and PANC 1 pancreatic ductal adenocarcinoma cell lines. Ringer solution (1/4 strength) tablets were purchased from Lab M (UK) and prepared according to the manufacturer’s protocol. Thiazolyl blue tetrazolium bromide (MTT) and sodium bicarbonate were acquired from Sigma-Aldrich (UK). For the preparation of Sorensen’s glycine buffer, NaCl (5.8 g/L) and glycine (7.6 g/L) were used and sourced from Sigma-Aldrich, (UK). Dimethyl sulfoxide (DMSO) was purchased from Alfa Aesar (UK). Trypsin was purchased from Lonza (Belgium). Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), L-glutamine, and antibiotic Antimycotic (comprising 10,000 units/mL penicillin, 10,000 µg/mL streptomycin and 25 µg/mL amphotericin B) were obtained from Gibco (UK).
Staphylococcus aureus (NCIMB 6571) was obtained from the University of Wolverhampton culture collection, maintained at − 20 °C in a lyophilised form. Stock culture was resuscitated on sterile tryptone soy agar (TSA) and sterilised by autoclaving (Priorclave, England), prior to use.
3.2. Electrospinning experiment
The electrospinning method (TL-Pro-BM electrospinning unit, TongLi Tech, China) was used for the preparation of PLGA/(R,S)-PHB three-dimensional fiber matrix, with and without proanthocyanidins, PCAN. The concentration of PLGA/(R,S)-PHB solution for the electrospinning was 10% w/v in HFIP. To prepare the suspension for electrospinning PCAN, 20% in weight ratio to the blend, was ground in a mortar and then added as a powder to the polymer solution. A microfibrous mat was formed under the following process parameters. The difference in voltage potentials between the spinning nozzle (steel needle G22) and the fiber collector (a steel mandrel with a diameter of 27 mm, rotating at a speed of 400 RPM) was 30 kV and the distance between mentioned parts of the electrospinning unit was 23 cm. The suspension was dosed to the spinning nozzle by syringe pump (PHD Ultra 4400, Harvard Apparatus, USA) with a flow rate of 3 ml per hour. To limit the sedimentation of PCAN, vertical setting of the tubeless spinneret was applied. Also, suspension was stirred inside the container with the magnetic stirrer. Electrospinning was carried out in 20 °C, at relative humidity equal to 30%.
3.3. Hydrolytic degradation under laboratory condition
For the degradation experiments three-dimensional fiber matrix of PLGA/(R,S)-PHB, with and without proanthocyanidins were incubated at 37 °C (± 0.5 °C) in screw-capped vials with an air-tight PTFE/silicone septum, containing 10 mL of demineralized water (pH = 6.1) The solid samples of electrospun mats were withdrawn from the test environment after specific time intervals such as: 1, 3, 5, 7, 9, 11, 15, 19, 23, 27, 31, 35, 39, 43, 47, 51, 55, 65, 71, and 85 days. The incubation of the samples was run in triplicate. After a predetermined degradation time, the samples were separated from the degradation medium, washed with demineralized water and dried, first on filter paper and then under a vacuum at a temperature of 25 °C to a constant mass. The changes in the molar mass, structure, thermal properties, and mass as well as the visual and microscopic examination of surface of mats before and after degradation, were determined. The percentage mass loss of electrospun nonwoven after degradation, Δmd, was evaluated by comparing the dry mass (md) at the specific incubation time intervals with the initial mass (m0).
Additionally, degradation media were collected, and then analyzed by means of ESI-MSin negative ion mode to evaluate the progress of hydrolytic degradation and by UV–VIS to evaluate the release of the bioactive substance (PCAN) from electrospun nonwoven studied.
3.4. Biological studies
3.4.1. Cytocompatibility study (In vitro MTT assay)
In the current study, the effect of the three-dimensional fiber matrix (without and with PCAN) on cell viability was investigated in vitro using mammalian cancer cell lines U251MG (humanglioblastoma), MSTO (human mesothelioma), and PANC1 (human pancreatic ductal adenocarcinoma) following the similar protocol previously reported by our group [20]. Each cell line was cultured in Dulbecco’s Modified Eagle Medium (DMEM medium) containing 4.5 g/L glucose, supplemented with fetal bovine serum (10%), antibiotic Antimycotic (1%), L-glutamine (2 mM) and incubated at 37 °C in a humidity incubator with 5% CO2. Briefly, 25,000 cells per well were seeded in 24 well plates for 24 h at 37 °C in a 5% CO2 incubator. The media was conditioned with electrospun mats of PLGA/(R,S)-PHB or PLGA/(R,S)-PHB/20% PCAN (1 cm2 electrospun mats in 10 mL DMEM) overnight under agitated conditions at 4 °C. The cells were then exposed to this media conditioned with PLGA/(R,S)-PHB or PLGA/(R,S)-PHB/20% PCAN. The in vitro cytotoxic impact of the samples on the selected cell lines was assessed using the standard MTT cytotoxicity assay. This involved adding a 5 mg/mL MTT solution (obtained from Sigma, UK) to all the wells, incubating for 2 h, and then dissolving the formazan crystals using a mixture of DMSO and Sorensen’s glycine buffer at pH 10.5. Following a 24 h incubation period, the morphology of cells, confluence of the cell monolayer, and cell viability were observed microscopically using an inverted light microscope (Nikon, Japan) and representative optical photomicrographs of cells were captured at 10x magnification. Cell viability was calculated using the mean (n = 4) absorbance measured and the results were statistically analysed by ANOVA with a Tukey’s multi comparisons test using GraphPad Prism (version 7.02).
3.4.2. Antibacterial activity (Disc Diffusion Assay, DDA)
The antimicrobial activity of PCAN extract obtained from Pelargonium sidoides was investigated against S. aureus using the disc diffusion assay. Sterile inert cellulosic discs (8 mm) were aseptically loaded with varied concentrations (2.5%, 5%, 10% and 20% w/v) of aqueous PCAN under constant agitated conditions (180 rpm) in an orbital shaker overnight (Innova® 43, USA). This loading process was undertaken in the dark at 37 °C. PBS-loaded discs (8 mm) were used as control. PCAN-loaded discs and the control discs were placed on TSA plates spread with an overnight culture of S. aureus and incubated at 37 °C for 24 h and zone of inhibition (ZOI) were measured. Data are presented as mean values of three experiments (n = 3) and error bars represent standard deviation.
3.5. Measurements
3.5.1. Polarized Optical Microscopy (POM)
The three-dimensional fibers matrix before and after degradation were observed with a polarized optical microscope Zeiss (Opton-Axioplan) equipped with a Nikon Coolpix 4500 color digital camera.
3.5.2. Scanning Electron Microscopy (SEM)
SEM of the three-dimensional fiber matrix before and after degradation was conducted using a Quanta 250 FEG (FEI Company, Fremont, CA, USA) high-resolution environmental scanning electron microscope operated at 5 kV acceleration voltage. The samples without coating were analyzed under a low vacuum (80 Pa).
3.5.3. Gel Permeation Chromatography (GPC) analysis
The GPC experiments for the three-dimensional fiber matrix after 1, 15, 31, 51 and 71 days of incubation were conducted in chloroform solution at 35 °C and a flow rate of 1 mL/min using a VE 1122 solvent delivery system (Viscotek, Malvern, UK) with two Mixed C PL-gel styragel columns in series and a Shodex SE 61 refractive index detector (Showa Denko, Munich, Germany/Japan). A volume of 10 μl of sample solutions in CHCl3 (concentration 0.5% m/v) were injected into the system. Polystyrene standards with low dispersity were used to generate a calibration curve.
3.5.4. Nuclear magnetic resonance 1H-NMR spectroscopy
1H-NMR spectra of PLGA, (R,S)-PHB and three-dimensional fiber matrix before and remaining after 1, 15, 31, 51, 65 and 71 days of incubation were recorded using a Bruker-Advance spectrometer operating at 600 MHz (Bruker BioSpin GmbH, Rheinstetten, Germany)with Bruker TOPSPIN 2.0 software using CDCl3 as the solvent and tetramethylsilane (TMS) as the internal standard. Spectra were obtained with 64 scans, a 11 μs pulse width, and a 2.66 s acquisition time.
3.5.5. DSC analysis
DSC analysis of PLGA, (R,S)-PHB and three-dimensional fiber matrix before and remaining after 71 days of incubation were conducted by means of the TA-DSC Q2000 apparatus (TA Instruments, Newcastle, DE, USA) as described previously in [23]. The calorimetric trace (second heating run) was acquired from -50 °C to 200 °C at the heating rate of 20 °C min-1, under nitrogen atmosphere (flow rate = 50 mL min−1) for 10 mg of sample. The instrument was calibrated with high purity indium. The glass transition temperature value (Tg) was taken as the midpoint of the heat capacity step change observed at the second run.
3.5.6. Electrospray ionization mass spectrometry (ESI-MSn) analysis
ESI-MS analysis was performed using a Finnigan LCQ ion trap mass spectrometer (Thermo Fisher Scientific Inc., San Jose, CA, USA). The degradation media containing degradation products were frozen and lyophilized. Then samples were dissolved in chloroform and PCAN was removed by filtration. The solutions of degradation products were dissolved in a chloroform/methanol (1:1 v/v) system and were introduced to the ESI source by continuous infusion using the instrument syringe pump at a rate of 5µL/min. The ESI source of the LCQ was operating at 4.5 kV and the capillary heater was set to 200 °C. Nitrogen was used as the nebulizing gas. The analyses were performed in the negative-ion mode.
3.5.7. Determination of the PCAN release profile by Ultraviolet–visible spectroscopy (UV–VIS)
The amount of biological active substance, PCAN released into the medium from polymeric mats was measured as a function of time. The quantitative assessment of the released PCAN was conducted using UV–VIS Spectrophotometry (Spectrophotometer Spark 10 M, TECAN, Mannedorf, Switzerland). The concentrations at [mg/mL] of PCAN released into the water after a specified period of time were determined based on the calibration curve according to procedure described previously in [20]. A linear calibration curve was generated using 6 diluted PCAN aqueous solutions with a concentration in the range of 0.0–0.5 mg/ml.
4. Conclusions
Based on the polymeric blend of PLGA copolymer and synthetic poly[(R,S)-3-hydroxybutyrate] a novel cytocompatible and antimicrobial system for controlled release of the bioactive substance from Pelargonium sidoides was developed. To maintain an optimized level of release of active substances, in the disease-affected periodontal region, for a sustained prolonged period, biodegradable polymers were used. The developed system of controlled delivery of PCAN, through the systematic degradation of the polymer matrix after periodontitis therapy, should be completely resorbed by the body. It can also be expected that this novel system can be, in the future, used to deliver drugs in a controlled manner in oral infectious diseases. The results support the potential application of this system for the long-term delivery of various biologically active substances to tissues at the wound site.
Author Contributions
Conceptualization, G.A.; formal analysis, M.Z., M.M. and H.J.; investigation, M.Z., M.P., A.G. and G.T.; resources, M.P. and J.W.; writing—original draft preparation, W.S., M.Z., I.R. and G.A.; writing—review and editing, G.A.; supervision, G.A. and M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by UM0-2016/22/Z/STS/00692 PELARGODONT Project financed under the M-ERA.NET.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This work was supported by UM0-2016/22/Z/STS/00692 PELARGODONT Project financed under the M-ERA.NET Programme of Horizon 2020.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Anselmo, A.C.; Mitragotri, S. An overview of clinical and commercial impact of drug delivery systems. J. Control. Release 2014, 190, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Zięba, M.; Chaber, P.; Duale, K.; Martinka Maksymiak, M.; Basczok, M.; Kowalczuk, M.; Adamus, G. Polymeric carriers for delivery systems in the treatment of chronic periodontal disease. Polymers 2020, 12, 1574. [Google Scholar] [CrossRef] [PubMed]
- Zamani, F.; Jahanmard, F.; Ghasemkhah, F.; Amjad-Iranagh, S.; Bagherzadeh, R.; Amani-Tehran, M.; Latifi, M. Chapter 7—Nanofibrous and nanoparticle materials as drug-delivery systems. In Micro and Nano Technologies, Nanostructures for Drug Delivery; Andronescu, E. Grumezescu, A.M. Eds.; Elsevier, 2017, pp. 239–270, ISBN 9780323461436.
- Swain, G.P.; Patel, S.; Gandhi, J.; Shah, P. Development of moxifloxacin hydrochloride loaded in-situ gel for the treatment of periodontitis: In-vitro drug release study and antibacterial activity. J. Oral Biol. Craniofac Res. 2019, 9, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.; Patel, R.R.; Yadav, S.K.; Kumar, N.; Chaurasia, S.; Ajmal, G.; Mishra, P.K.; Mishra,B. Development, optimization and evaluation of tinidazole functionalized electrospun poly(ε-caprolactone) nanofiber membranes for the treatment of periodontitis. RSC Adv. 2016, 6, 100214–100229. [Google Scholar] [CrossRef]
- Hamed, R.; Abu Rezeq, A.; Tarawneh, O. Development of hydrogels, oleogels, and bigels as local drug delivery systems for periodontitis. Drug Dev. Ind. Pharm. 2018, 44, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Sun, Y.; Chen, Y.; Liu, Y.; Tang, C.; Qu, X. Injectable adhesive hydrogel through a microcapsule cross-link for periodontitis treatment. ACS Appl. Bio Mater. 2019, 2, 5985–5994. [Google Scholar] [CrossRef] [PubMed]
- Rieger, K.A.; Bircha, N.P.; Schiffman, J.D. Designing electrospun nanofiber mats to promote wound healing—a review. J. Mater. Chem. B 2013, 1, 4531–4541. [Google Scholar] [CrossRef] [PubMed]
- Rydz, J.; Sikorska, W.; Kyulavska, M.; Christova, D. Polyester-based (bio)degradable polymers as environmentally friendly materials for sustainable development. Int. J. Mol. Sci. 2015, 16, 564–596. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Pr’eat, V. PLGA-based nanoparticles: an overview of biomedical applications. JCR 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Do, M.P.; Neut, C.; Delcourt, E.; Seixas Certo, T.; Siepmann, J.; Siepmann, F. In situ forming implants for periodontitis treatment with improved adhesive properties. Eur. J. Pharm. Biopharm. 2014, 88, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.-Y.; Chen, D.W.; Hu, C.-C.; Hsieh, Y.-T.; Liu, S.-J.; Chan, E.-C. In vitro and in vivo investigation of drug-eluting implants for the treatment of periodontal disease. AAPS Pharm. Sci. Tech. 2011, 12, 1110–1115. [Google Scholar] [CrossRef]
- Ahuja, A.; Ali, J.; Rahman, S. Biodegradable periodontal intrapocket device containing metronidazole and amoxycillin: formulation and characterisation. Die Pharmazie 2006, 61, 25–29. [Google Scholar] [PubMed]
- Fang, J.; Niu, H.; Lin, T.; Wang, X. Applications of electrospun nanofibers. Chin. Sci. Bull. 2008, 53, 2265–2286. [Google Scholar] [CrossRef]
- Wang, P.; Mele, E. Effect of antibacterial plant extracts on the morphology of electrospun poly(lactic acid) fibres. Materials 2018, 11, 923. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.Y.; Wen, J.; Yim, E.K.F.; Leong, K.W. Sustained release of proteins from electrospun biodegradable. Biomacromolecules 2005, 6, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- Gizaw, M. Electrospun fibers as a dressing material for drug and biological agent delivery in wound healing applications. Bioengineering 2018, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Kayser, O.; Kolodziej, H. Antibacterial activity of extracts and constituents of Pelargonium sidoides and Pelargonium reniforme. Planta Med. 1997, 6, 508–510. [Google Scholar] [CrossRef]
- Savickiene, N. , Jekabsone, A., Raudone, L., Abdelgeliel, A. S., Cochis, A., Rimondini, L.; Viškelis, P. Efficacy of proanthocyanidins from Pelargonium sidoides root extract in reducing P. gingivalis viability while preserving oral commensal S. salivarius. Materials 2018, 11, 1499. [Google Scholar] [CrossRef] [PubMed]
- Zięba, M.; Włodarczyk, J.; Gupta, A.; Pastusiak, M.; Chaber, P.; Janeczek, H.; Musioł, M.; Sikorska, W.; Kaczmarczyk, B.; Radecka, I.; Kowalczuk, M.; Savickas, A.; Savickiene, N.; Adamus, G. Bioresorbable electrospun mats of poly(D,L)-lactide/poly[(R,S)-3-hydroxybutyrate] blends for potential use in the treatment of difficult-to-heal skin wounds. Eur. Polym. J. 2021, 147, 110334. [Google Scholar] [CrossRef]
- Dobrzynski, P.; Kasperczyk, J.; Janeczek, H.; Bero, M. Synthesis of biodegradable copolymers with the use of low toxic zirconium compounds. 1. Copolymerization of glycolide with L-Lactide initiated by Zr(Acac)4. Macromolecules 2001, 34, 5090–5098. [Google Scholar] [CrossRef]
- Kurcok, P.; Śmiga,M. ; Jedliński, Z. β-Butyrolactone polymerization initiated with tetrabutylammonium carboxylates: A novel approach to biomimetic polyester synthesis. J. Polym. Sci. Part A: Pol. Chem. 2002, 40, 2184–2189. [Google Scholar] [CrossRef]
- Rydz, J.; Sikorska, W.; Musioł, M.; Janeczek, H.; Włodarczyk, J.; Misiurska-Marczak, M.; Łęczycka, J.; Kowalczuk, M. 3D-Printed polyester-based prototypes for cosmetic applications—Future directions at the forensic engineering of advanced polymeric materials. Materials 2019, 12, 994. [Google Scholar] [CrossRef] [PubMed]
- Maruccia, E.; Ferrari, S.; Bartoli, M.; Lucherini, L.; Meligrana, G.; Pirri, C.F.; Saracco, G.; Gerbaldi, C. Effect of thermal stabilization on PAN-derived electrospun carbon nanofibers for CO2 capture. Polymers 2021, 13, 4197. [Google Scholar] [CrossRef] [PubMed]
- Achille, C.; Sundaresh, S.; Chu, B.; Hadjiargyrou, M. Cdk2 silencing via a DNA/PCL electrospun scaffold suppresses proliferation and increases death of breast cancer cells. PLoS ONE 2012, 7, e52356. [Google Scholar] [CrossRef] [PubMed]
- Musioł, M.; Sikorska, W.; Adamus, G.; Janeczek, H.; Kowalczuk, M.; Rydz, J. (Bio)degradable polymers as a potential material for food packaging: studies on the (bio)degradation process of PLA/(R,S)-PHB rigid foils under industrial composting conditions. Eur. Food Res. Technol. 2016, 242, 815–823. [Google Scholar] [CrossRef]
- Zarzycki, R.; Modrzejewska, Z.; Nawrotek, K. Drug release from hydrogel matrice. Ecol. Chem. Eng. S. 2010, 17, 117–136. [Google Scholar]
- Mannino, G.; Chinigò, G.; Serio, G.; Genova, T.; Gentile, C.; Munaron, L.; Bertea, C.M. Proanthocyanidins and where to find them: A meta-analytic approach to investigate their chemistry, biosynthesis, distribution, and effect on human health. Antioxidants 2021, 10, 1229. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83, Erratum in: Microbiol. Mol. Biol. Rev. 2012, 76, 496. PMID: 21372320; PMCID: PMC3063353. [Google Scholar] [CrossRef]
- Preshaw, P.M. ; Detection and diagnosis of periodontal conditions amenable to prevention. BMC Oral Health 2015, 15 (Suppl 1), S5. [Google Scholar] [CrossRef]
- Alkimavičienė, E.; Pušinskaitė, R.; Basevičienė, N.; Banienė, R. , Savickienė, N. and Pacauskienė, I.M. Efficacy of Proanthocyanidins in Nonsurgical Periodontal Therapy. International dental journal 2023, 73, 195–204, Epub 2022 Sep 24. PMID: 36167610; PMCID: PMC10023589.. [Google Scholar] [CrossRef]
- Cos, P.; De Bruyne, T.; Hermans, N.; Apers, S.; Berghe, D.V.; Vlietinck, A.J. Proanthocyanidins in health care: current and new trends. Curr Med Chem. 2004, 11, 1345–1359. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
1H-NMR spectrum of PLGA/(R,S)-PHB electrospun nonwoven obtained.
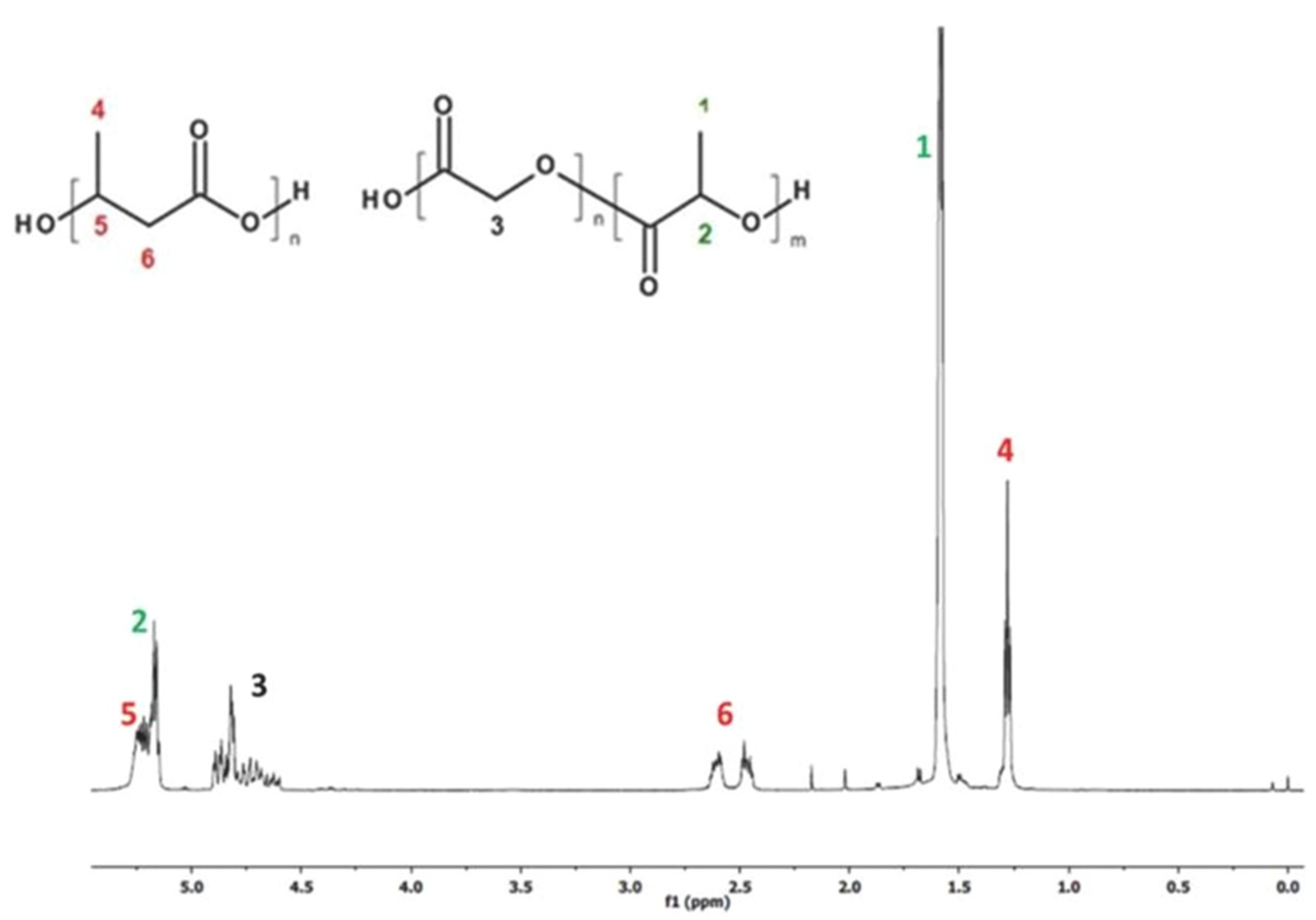
Figure 2.
The DSC traces (second heating) for PLGA/(R,S)-PHB electrospun nonwovens with and without PCAN together with traces of PLGA and (R,S)-PHB blend components.
Figure 2.
The DSC traces (second heating) for PLGA/(R,S)-PHB electrospun nonwovens with and without PCAN together with traces of PLGA and (R,S)-PHB blend components.
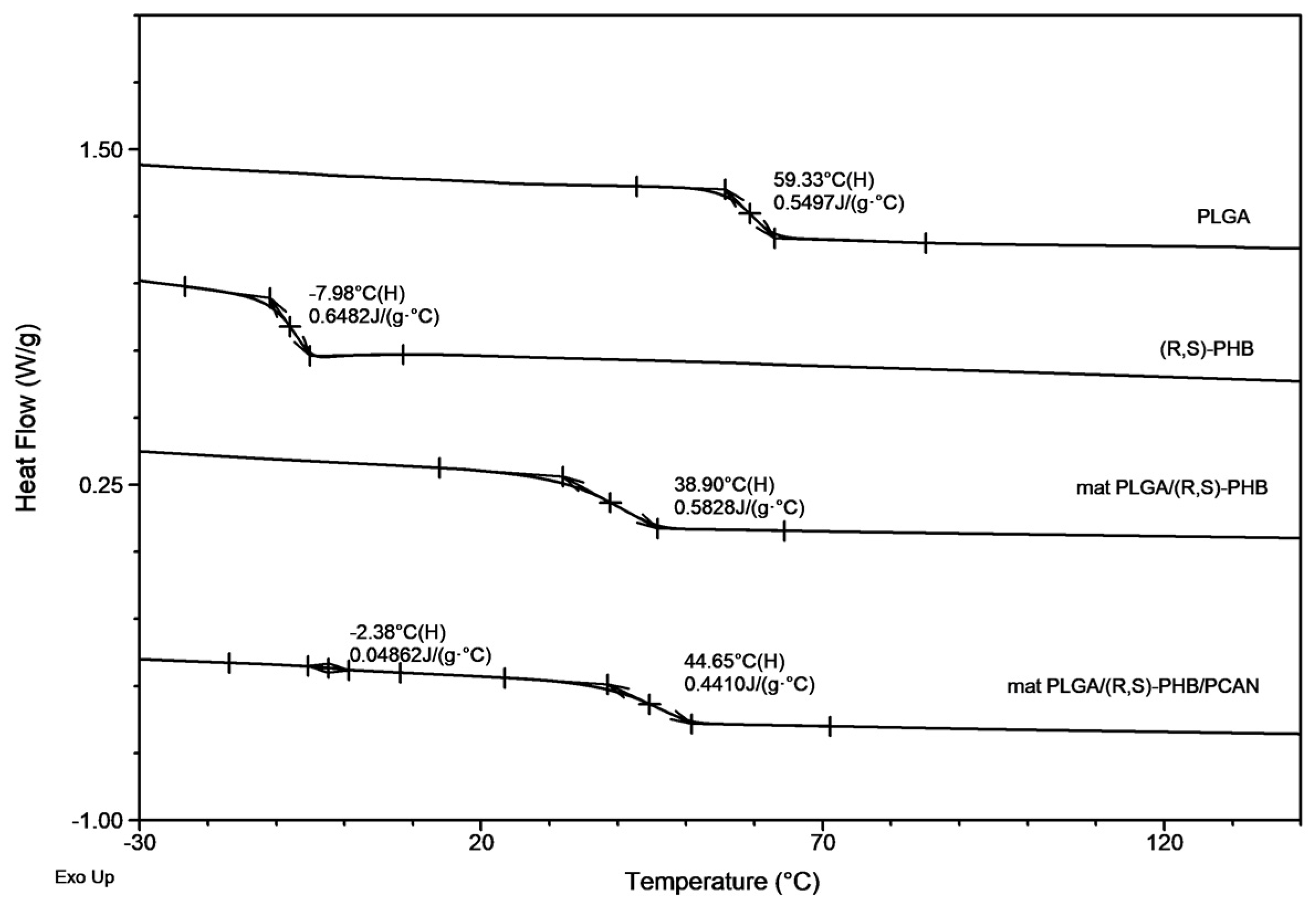
Figure 3.
Digital imagines and POM micrographs of PLGA/(R,S)-PHB electrospun nonwoven without and with PCAN before and after 71 days of incubation.
Figure 3.
Digital imagines and POM micrographs of PLGA/(R,S)-PHB electrospun nonwoven without and with PCAN before and after 71 days of incubation.

Figure 4.
SEM micrographs of PLGA/(R,S)-PHB three-dimensional fiber matrix without (A) and with (B) biological active substance before incubation.
Figure 4.
SEM micrographs of PLGA/(R,S)-PHB three-dimensional fiber matrix without (A) and with (B) biological active substance before incubation.
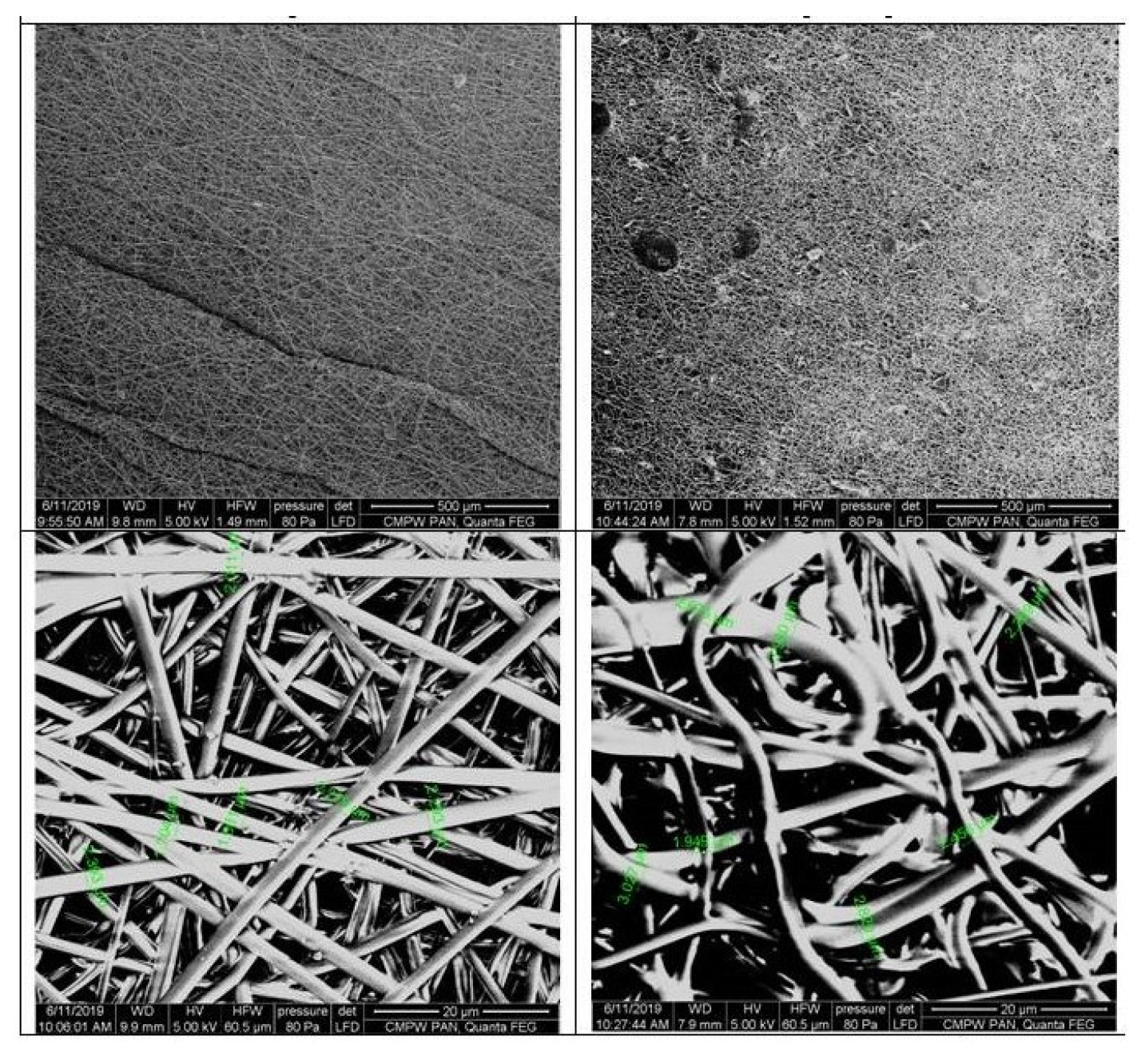
Figure 5.
The GPC traces for PLGA/(R,S)-PHB electrospun nonwoven (A) without and (B) with biological active substance—PCAN during degradation process.
Figure 5.
The GPC traces for PLGA/(R,S)-PHB electrospun nonwoven (A) without and (B) with biological active substance—PCAN during degradation process.
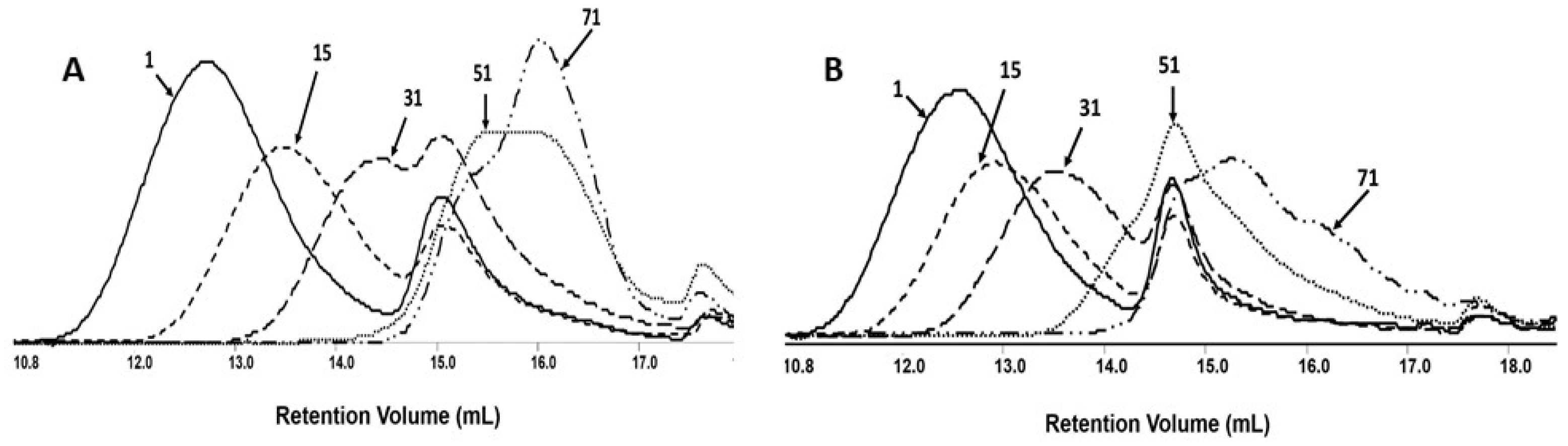
Figure 6.
The 1H-NMR spectra of the three-dimensional fiber matrix: (1) PLGA/(R,S)-PHB before degradation as well as (2) and (3) PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN after 71 days of incubation in water, respectively.
Figure 6.
The 1H-NMR spectra of the three-dimensional fiber matrix: (1) PLGA/(R,S)-PHB before degradation as well as (2) and (3) PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN after 71 days of incubation in water, respectively.
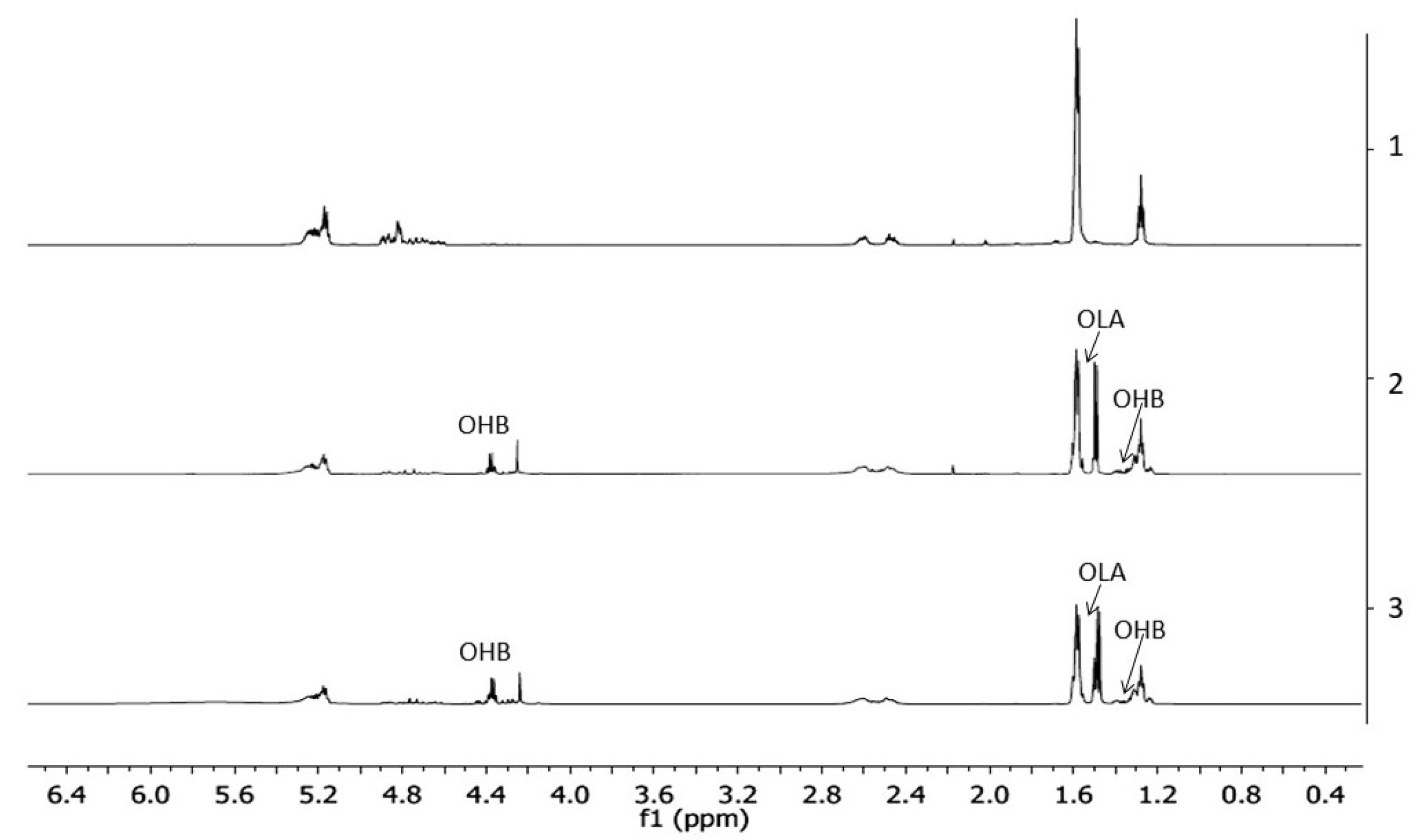
Figure 7.
The changes in the chemical composition of (A) PLGA/(R,S)-PHB and (B) PLGA/(R,S)-PHB/PCAN three-dimensional fiber matrix together with the progress of degradation process estimated based on 1H-NMR spectra (blue and green bars represent PLGA and (R,S)-PHB mat components, respectively).
Figure 7.
The changes in the chemical composition of (A) PLGA/(R,S)-PHB and (B) PLGA/(R,S)-PHB/PCAN three-dimensional fiber matrix together with the progress of degradation process estimated based on 1H-NMR spectra (blue and green bars represent PLGA and (R,S)-PHB mat components, respectively).
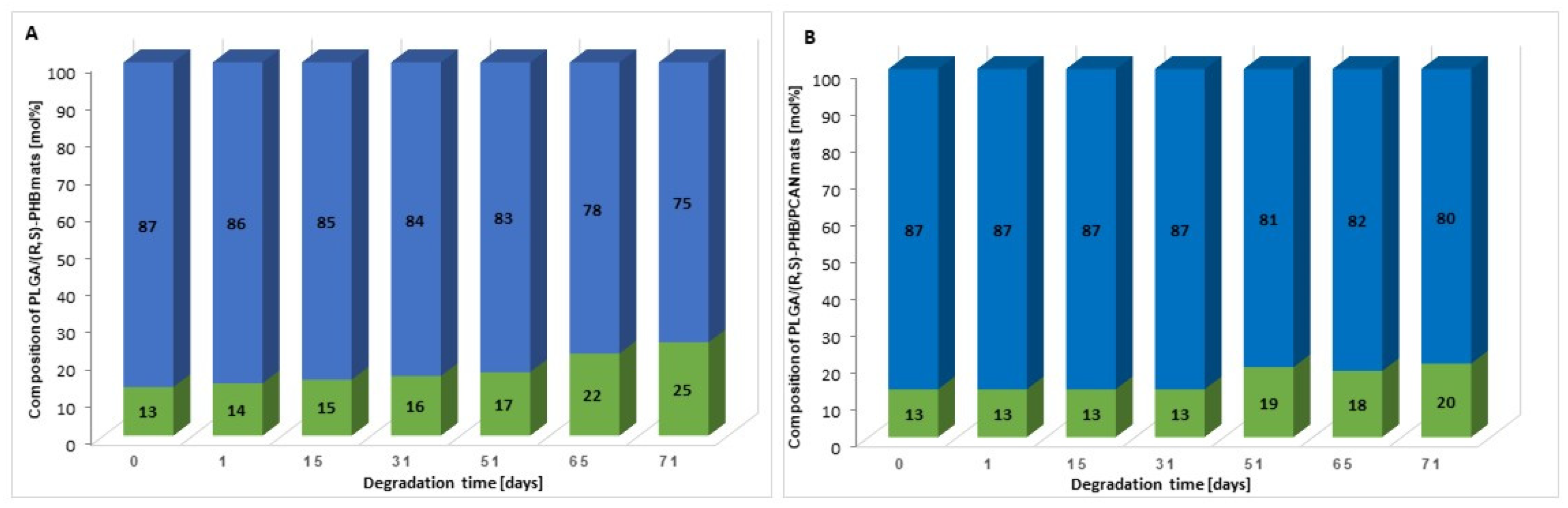
Figure 8.
The second DSC heating traces for three-dimensional fibers matrix samples remaining after 71 days of hydrolytic degradation in water.
Figure 8.
The second DSC heating traces for three-dimensional fibers matrix samples remaining after 71 days of hydrolytic degradation in water.
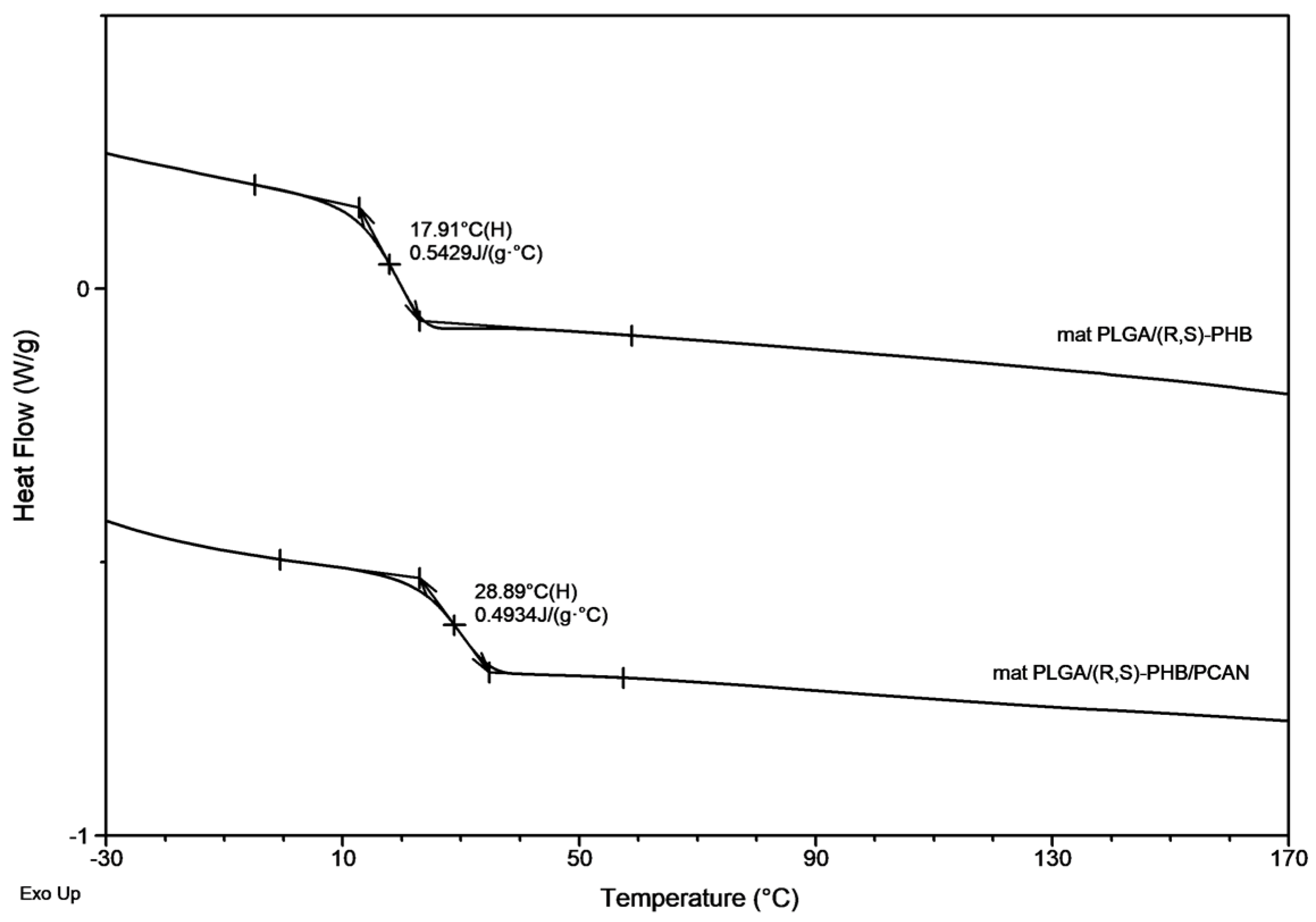
Figure 9.
Mass loss of PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN three-dimensional fiber matrix samples together with the progress of their hydrolytic degradation in water.
Figure 9.
Mass loss of PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN three-dimensional fiber matrix samples together with the progress of their hydrolytic degradation in water.
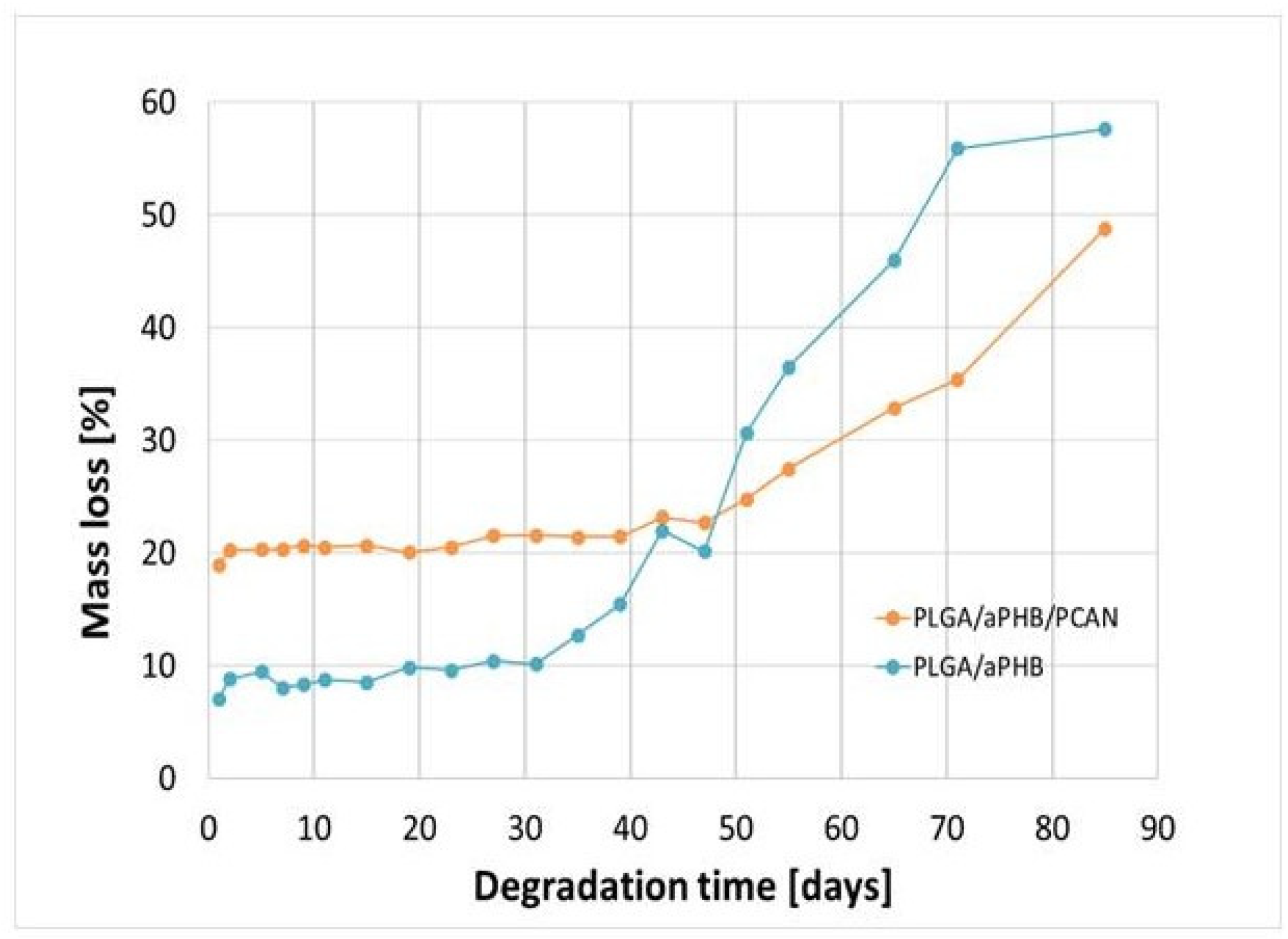
Figure 10.
Release profile of PCAN from PLGA/(R,S)-PHB/PCAN electrospun nonwoven during incubation in water.
Figure 10.
Release profile of PCAN from PLGA/(R,S)-PHB/PCAN electrospun nonwoven during incubation in water.
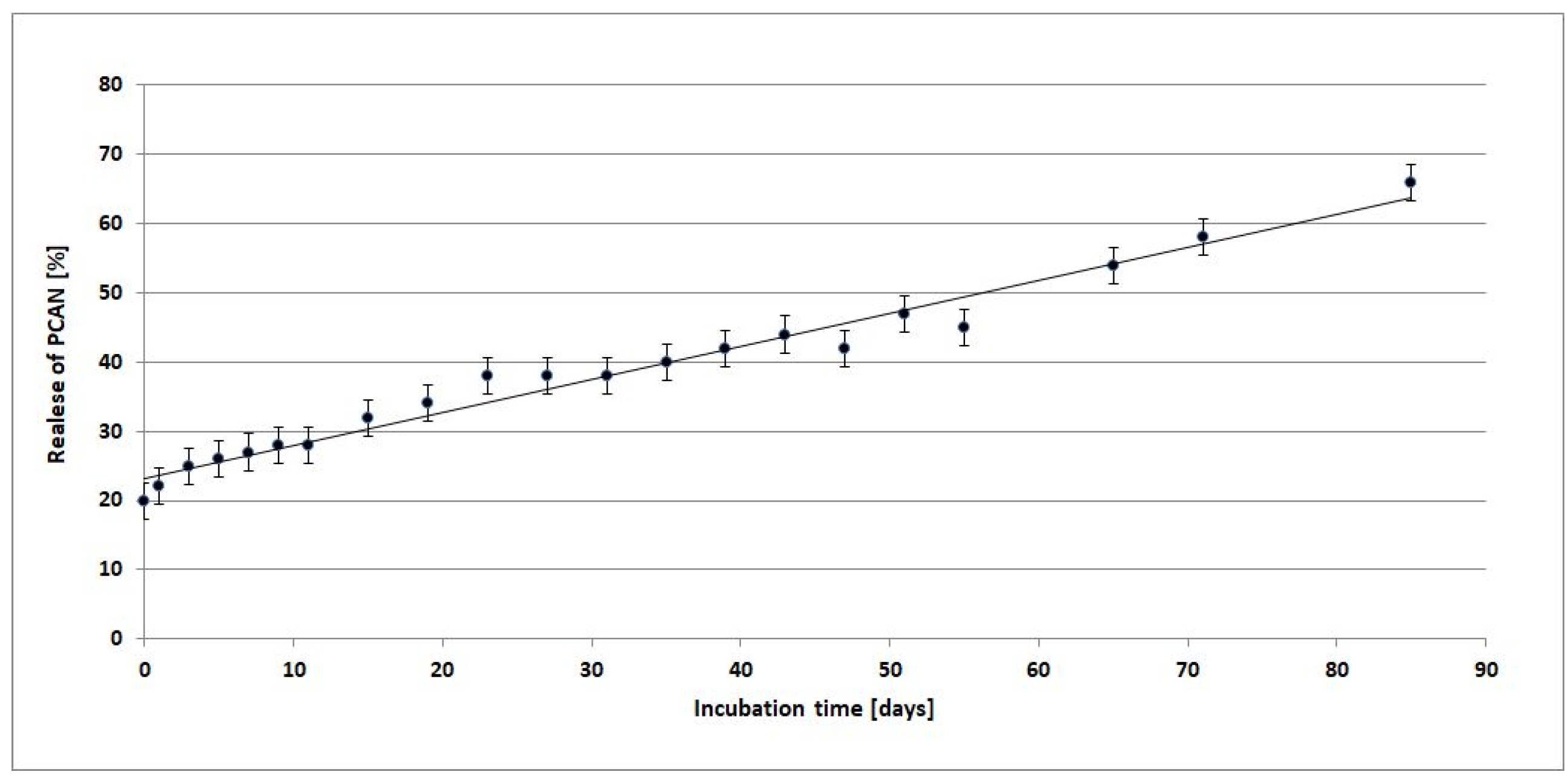
Figure 11.
ESI-mass spectra collected after 71 days incubation in water of (a) PLGA/(R,S)-PHB and (b) PLGA/(R,S)-PHB/PCAN three-dimensional fiber matrix samples.
Figure 11.
ESI-mass spectra collected after 71 days incubation in water of (a) PLGA/(R,S)-PHB and (b) PLGA/(R,S)-PHB/PCAN three-dimensional fiber matrix samples.
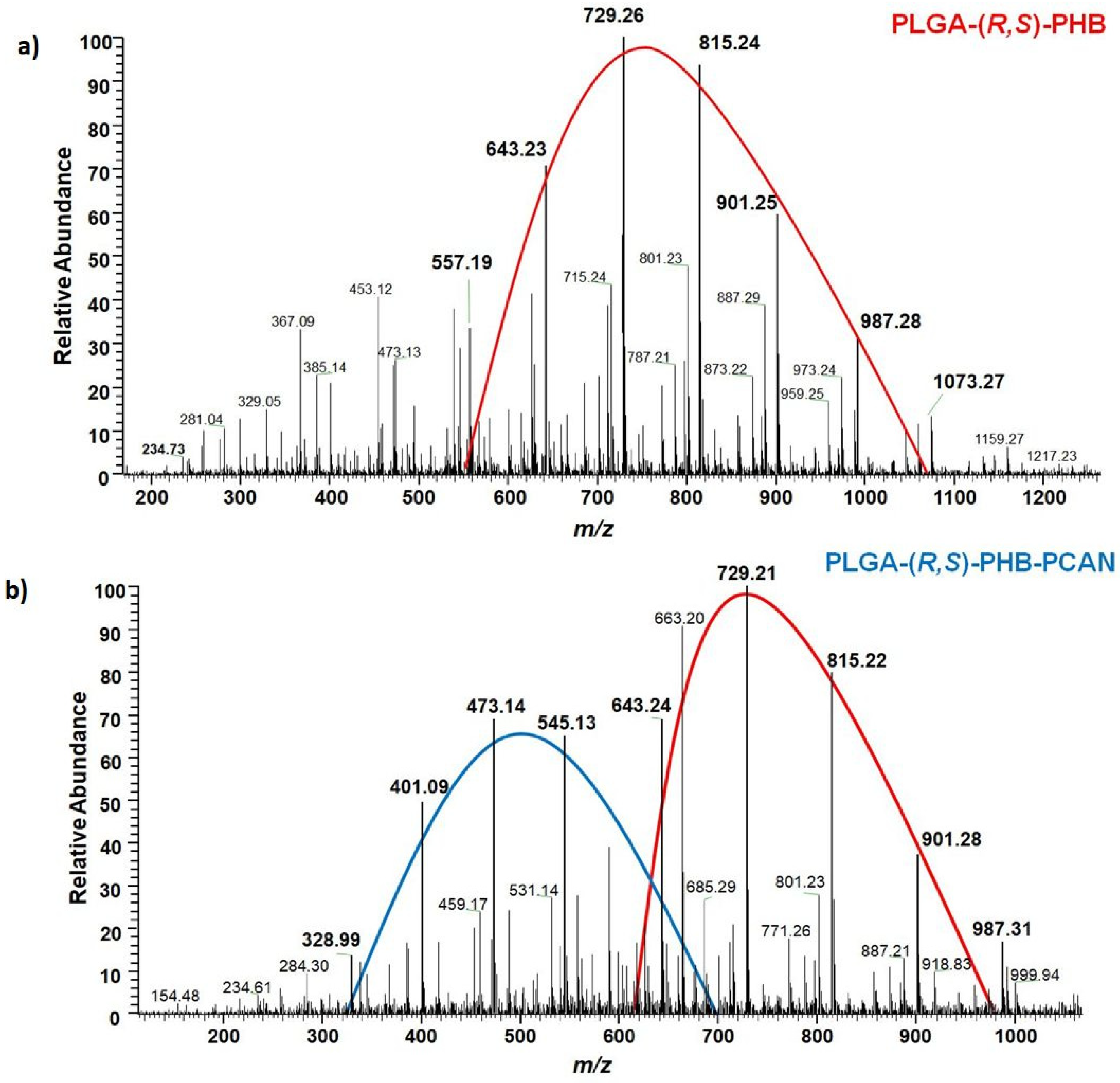
Figure 12.
Cytocompatibility test results. Representative optical photomicrographs of cells captured at 10x magnification after 24 h exposure to PLGA/(R,S)-PHB fiber matrix without and with 20 wt% of PCAN.
Figure 12.
Cytocompatibility test results. Representative optical photomicrographs of cells captured at 10x magnification after 24 h exposure to PLGA/(R,S)-PHB fiber matrix without and with 20 wt% of PCAN.
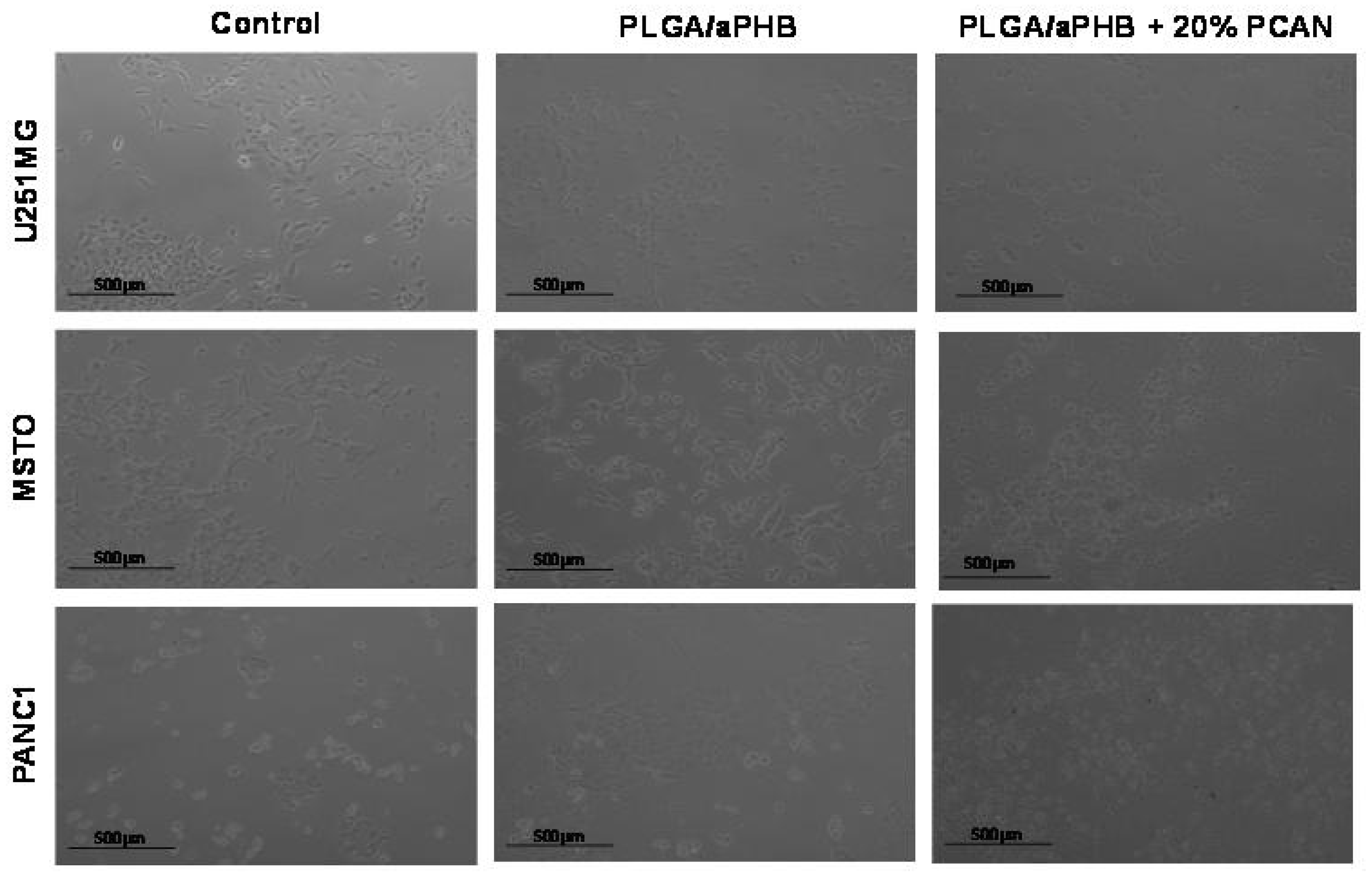
Figure 13.
Antimicrobial activity for PCAN against S. aureus at 24 h assessed by measuring ZOI during the Disc Diffusion Assay.
Figure 13.
Antimicrobial activity for PCAN against S. aureus at 24 h assessed by measuring ZOI during the Disc Diffusion Assay.
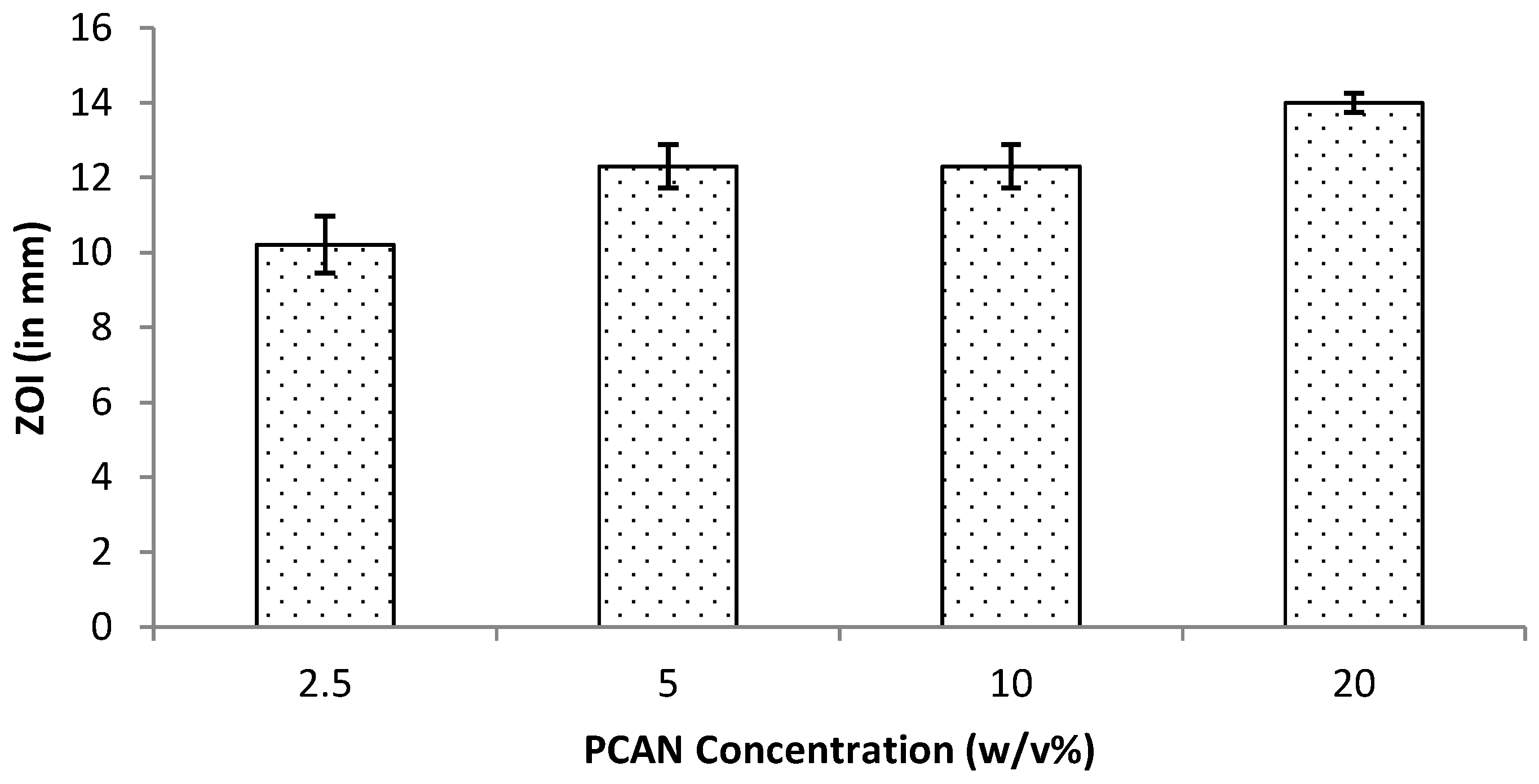

Table 1.
The MTT assay results demonstrating the cell viability of the selected cell lines following a 24 h exposure to media conditioned with PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN, loaded with 20 wt% of PCAN.
Table 1.
The MTT assay results demonstrating the cell viability of the selected cell lines following a 24 h exposure to media conditioned with PLGA/(R,S)-PHB and PLGA/(R,S)-PHB/PCAN, loaded with 20 wt% of PCAN.
| %Cell viability | PLGA/(R,S)-PHB | PLGA/(R,S)-PHB/PCAN |
|---|---|---|
| U251MG1 | 64.50 ± 1.88% | 34.19 ± 3.38% |
| MSTO 1 | 90.91 ± 6.54% | 70.43 ± 3.66% |
| PANC 1 1 | 88.62 ± 7.49% | 23.73 ± 5.77% |
1 Cell line.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



