
Submitted:
07 December 2023
Posted:
08 December 2023
You are already at the latest version
Abstract
Replication stress (RS) is a characteristic state of cancer cells as they tend to exchange precision of replication for fast proliferation and increased genomic instability. To overcome the consequences of improper replication control malignant cells frequently inactivate parts of their DNA damage response (DDR) pathways (the ATM-CHK2-p53 pathway), while relying on other pathways which help to maintain replication fork stability (ATR-CHK1). This creates a dependency on the remaining DDR pathways, vulnerability to further destabilization of replication and synthetic lethality of DDR inhibitors with common oncogenic alterations such as mutations of TP53, RB1, ATM, amplifications of MYC, CCNE1 and others. The response to RS is normally limited by coordination of cell cycle, transcription and replication. Inhibition of WEE1 and PKMYT1 kinases, which prevent unscheduled mitosis entry, leads to fragility of under-replicated sites. Recent evi-dence also shows that inhibition of Cyclin-dependent kinases (CDKs), such as CDK4/6, CDK2, CDK8/19 and CDK12/13 can contribute to RS through disruption of DNA repair and replication control. Here, we review the main causes of RS in cancers as well as main therapeutic targets – ATR, CHK1, PARP and their inhibitors.
Keywords:
replication stress
; DNA damage response
; ATR
; CHK1
; PARP
; WEE1
; PKMYT1
; Cyclin dependent kinases
1. Introduction
Dysregulation of proliferation is the most recognized aspect of cancer. Tumors increase their proliferation rate through increased pro-proliferative signaling and evasion of growth suppressors. As part of these changes tumors lose tumor suppressors and activate oncogenes controlling the entry into the cell cycle, progression to replication and mitosis, and checkpoints, which monitor preparedness for these processes. In short, cancers can risk a higher rate of errors in replication and mitosis for higher rate of proliferation, which also contributes to genetic instability, required for further oncogenic progression. This state of tolerability towards increased rate of errors together with a higher pressure on replication machinery, creates a state of so-called RS (RS). RS is one of the main causes of genome instability and it has serious implications for cell survival, aging and disease development [1]. DNA RS is a cell state that may be caused by different exogenous and endogenous events occurring during DNA synthesis and resulting from defects in the replicative machinery.
The three major sources of RS in cancers originate from the most important hallmarks of cancer - the demand for higher proliferation rate, genetic instability and dysregulation of transcription [2,3]. Increased oncogenic activity of pro-proliferative proteins such as сyclin E, K-Ras, Myc, and inactivation of tumor suppressors like p53 and Rb (retinoblastoma protein), leads to decrease of replisome components activity, aberrant origin firing, and insufficient levels of dNTPs required for replication. Genetic instability caused by defects in DNA damage response (DDR) pathways such as malfunctions of p53, ATM and others leads to replication fork collapse and transcription dysregulation caused by activation of Myc and CDK2/Cyclin E, inactivation of ATR-CHK1 leads to transcription-replication conflicts [4,5].
However, by repressing certain DDR genes, tumors became strongly dependent on remaining DDR branches. Inactivation of the ATM-CHK2-p53 pathway leads to dependency on the remaining ATR-CHK1 DDR, as in the absence of both response pathways DNA repair mechanisms can not be activated, leading to cell death. The concept, when mutations beneficial to the tumor create a vulnerability to inactivation of targets which compensate for such alterations is known as synthetic lethality [6]. For example, RS can be further enhanced by chemotherapy drugs that lead to DNA damage by interfering with replication such as topoisomerase I and II inhibitors, alkylating agents and nucleoside metabolic inhibitors, and tumors that already have high levels of RS are more sensitive to these drugs [7]. Several key studies demonstrated that genetic inactivation of ATR leads to synthetic lethality in ATM or TP53 mutated tumors, paving the way to use of small-molecule ATR inhibitors (ATRi) [8,9]. Similarly other mutations that increase RS through activation of oncogenes or inactivation of tumor suppressors maintaining genome stability leave cancers vulnerable to a whole new class of drugs such as ATRi, PARP (PARPi) and CHK1 (CHK1i) inhibitors. Another strategy for targeting RS-high tumors is inhibiting activity of proteins which prevent entry into the S-phase and/or mitosis, such as WEE1 and PKMYT1 inhibitors (WEE1i, PKMYT1i), forcing cells into the next phase of the cell cycle, for which they are not prepared. Finally, recent evidence shows that both the cell cycle and transcriptional Cyclin-dependent kinases (CDKs) are implicated in coordinating replication, transcription and DNA repair. Inhibitors of CDK4/6, CDK2, CDK8/19 and CDK12/13 increase RS in a number of models and contribute to their activity shown in clinical trials.
This review focuses on such vulnerabilities caused by RS which could be used as targets for cancer therapy as well as markers for such therapies.
2. Main body
2.1. Replication stress mechanisms in cancer
2.1.1. Overview of replication control
Replication is a tightly controlled process, which ensures faithful and complete doubling of the DNA. Preparation for replication takes place in the G1 phase, after mitosis or exit from dormancy, and consists of several steps. The origins of replication are recognized by the origin recognition complex, consisting of ORC1-6 proteins, which then recruits CDC6 and Cdt1, which in turn load the replication helicase complex (MCM2-7) onto the chromatin, forming the pre-replication complex (pre-RC). This process is known as “licensing” and prepares points where replication would start [10]. The loading of additional pre-RCs onto the chromatin is prevented by increase of CDKs activity at the initiation of replication [10]. Binding of MCM2-7 induces dissociation of CDC6 and degradation of Cdt1 [11]. The pre-RC complex is then activated by phosphorylation by CDK1 and the Cdc7 kinase, and binding of Cdc45 and GINS in the S phase, forming the pre-initiation complex [12,13]. These events depend on activation of E2F1, after inactivation of Rb by CDK2/сyclin E [14]. Replication complexes are loaded on chromatin in excessive numbers to ensure complete replication. They remain dormant in normal conditions, but start firing in case of the replication fork stalling or collapsing; or the replisome being displaced by the RNA polymerase [15]. Recent evidence suggests that additional RC complexes can be assembled in late S, or even G2 phase of the cell cycle, to ensure full replication [11]. Coordination of the cell cycle progression, DNA repair and replication completion depends on the replication stress sensor – kinase ATR which slows down replication, prevents origin firing, and activates DNA damage machinery to restart replication forks in case of DNA damage during DNA synthesis. Most importantly ATR activates CHK1 and WEE1 kinases, which in turn inhibit CDKs’ activity, delaying cell cycle progression, especially entry into mitosis [16]. In case the ATR1-CHK1 pathway fails, replication forks would collapse, leading to double-strand breaks (DSBs) and activation of the ATM-CHK2-p53 pathway (Figure 1).
All of the above processes are often altered during oncogenic progression. The impairment of normal replication process, resulting in increased stalling and collapse of replication forks is defined as RS. In the following sections we would discuss how stimulation of oncogene activity and suppression of oncogenes affects the normal process of replication, leading to RS in cancer.
2.1.2. Oncogenic transformation leads to replication stress
A significant portion of oncogenes deregulated in cancers consists of components of signaling pathways required for entry into the cell cycle from quiescence. The link between dysregulation of proliferation and replication-dependent DNA damage and genetic instability was recognized in the late 1990s - early 2000s [17,18], when experiments with the introduction of active oncogenes into non-transformed cells.
A key early study by Di Micco et al [19] demonstrated that introduction of activated H-Ras into normal human fibroblasts led to burst of proliferation, followed by increased DDR and subsequent oncogene-induced senescence. DNA damage was associated both with under-replication in the S phase and re-replication due to aberrant origin firing. Knockout of DDR components such as CHK2 prevented cells from exiting the cell cycle and allowed maintenance of proliferation with accumulation of DNA damage. In parallel, induction of pro-survival signaling like ERK in Ras-mutated cancers prevents cell death and p53 activation by DDR [20]. Other mutations which upregulate Ras activity or act downstream of it such as B-Raf [21,22], EGFR [23,24] and others lead to similar effects [25].
Downstream of the signaling pathways is the cell cycle machinery (Figure 1). The proliferation signals converge on increase of CCND1 transcription. Cyclin D1 (product of CCND1) is instrumental in leading the cells into replication through phosphorylation of Rb and transcription and stabilization of E Cyclins. Despite that, overexpression of сyclin D1 does not induce DDR markers and increase of RS [26].
E Cyclins (E1 (coded by CCNE1) and E2 (CCNE2), together further referred as Cyclin E) are critical to entry into the S phase, and its deregulation is one of the main causes of RS. E Cyclins are aberrantly expressed in 21% of cancer (data from TCGA Pancancer Studies, accessed through Cbioportal.org [27]) with highest levels in breast, ovarian and endometrial cancers. Сyclin E/CDK2 complex polyphosphorylates Rb, completely inactivating it and releasing the E2F1 transcription factor. Besides that, сyclin E is involved in licensing the origins of replication and centrosome duplication [28,29]. Cyclin E phosphorylates a number of substrates like CDC6 in the origin recognition complex and other components of the pre-replicative complex - Recql and Treslin, and increases expression of CDC6, CDT1, required for loading of the pre-RC complex, and MCM genes of the pre-RC complex itself (reviewed in [30]). Despite its obvious pro-oncogenic role, сyclin E, while increasing the entry into the S phase, lowers the speed of replication, because of RS. Excessive amounts of Cyclin E diminishes MCM2-7 loading on chromatin, and induces aberrant firing of origins, causing transcription-replication conflicts [31]. Cyclin E overexpression stimulates pre-initiation complex formation, but inhibits the pre-replication complex and interferes with proper origin firing [30]. This effect can be counteracted by further mutations in DDR response. A particularly pro-oncogenic form of Cyclin E – processed low-molecular-weight Cyclin E (LMW-E) increases both entry into the S phase and its speed and has a particularly negative prognosis [32]. LMW-E increases RS tolerance through both increasing pre-replication complex assembly and DNA repair through Rad51 [33]. Notably, LMW-E-expressing tumors are particularly resistant to targeted therapy, as its complex with CDK2 is resistant to CDK inhibitor proteins such as p21 and p27 [34], while CDK2 activity decreases p27 levels through phosphorylation [35]. CCNE1 amplification is one of the main markers both for synthetic lethality in cancer models [36,37,38] and for use of drugs affecting RS in clinic [39]. Besides direct mutations in CCNE1, it can be stabilized through inactivating mutations of FBXW7, its product is a component of the Skp1-Cullin1-F-box (SCF) E3 ligase, which degrades Cyclin E. Mutations of FBXW7 additionally increase RS through stabilization of Myc [40].
2.1.3. Transcription-replication conflicts are increased in cancers
A particular mechanism of elevated RS in cancer is increase of transcription-replication conflicts. The transcription factor с-Myc also acts as a universal amplifier of transcription, increasing transcription rate for thousands of genes [41]. Activated Ras [23] and other oncogenes also increase transcriptional activity. Collisions of the replication machinery with transcription complexes [42] or R-loops formed by the RNA-DNA hybrids [43] lead to fork stalling and collapse, which activate DDR and increasing RS (Figure 2). Under normal conditions transcription and replication are spatially separated, especially in transcriptionally active regions such as nucleoli [44]. To ensure complete replication of transcribed regions, cells must activate the S-phase checkpoint (ATR-CHK1-dependent inhibition of CDK1) to prevent onset of mitosis before end of replication. Deregulation of these mechanisms increases genomic instability [11]. Transcription machinery also displaces the MCM complexes from DNA, so often transcribed genes can lose licensed origins, leading to under-replicated DNA [45]. These under replicated sites of active transcription are therefore commonly broken in mitosis and are known as common fragile sites (CFSs). Recent data suggests that new MCM complexes can be assembled in the S phase through stabilization of Cdt1 [11], but this assembly relies on activation of DNA damage response.
2.1.4. Rb/E2F pathway, transition to S phase and replication stress
Phosphorylation of Rb and subsequent cell cycle promotion to S phase by release of E2F transcription factors primarily depends on Cyclin D-CDK4/6 activity and after that on p53 and Cyclin E/A-CDK2 [14,46,47]. In vivo experiments have shown that if tumor cells originally have RB1 (gene of Rb protein) depletion, this Rb status will be obligatory for the further growth and survival. Thus, recovery of intact Rb significantly decreased tumor proliferation after continuous growth and reduced levels of CCNE2, PCNA and MCM3 genes’ expression, which is vital for cell cycle progression [48]. Mutations of RB1 are a major contributor to RS and genetic instability in general. RB1 is frequently mutated in a number of cancers (7% in total, according to TCGA database), including sarcoma, bladder, endometrial, ovarian, breast, prostate and non-small cell lung (NSCLC) cancers, and the Rb protein is inactivated by high CDK4/6 or CDK2 activity [27,49,50]. Rb mainly contributes to genetic instability through release of E2F1, which leads to accumulation of γH2AX foci and increased phosphorylation of RPA (replication protein A) (Figure 1). Rb-E2F1 complex is required for condensin II (a key player of mitotic chromosome assembly) recruitment, preventing under-replication [51]. Additionally Rb is recruited to stalled replication forks, displacing PCNA and allowing their repair [52]. E2F1 recruits DNA damage response proteins through Rb and its interactions with epigenetic modifiers such as BRG1, p300/CBP and others, maintaining genome stability and preventing RS [53]. Rb has a number of functions apparently independent of the E2F family through an extensive network of protein interactions [54]. One of the studies was devoted to the aberrant activation Rb/E2F by oncogenic proteins E6/E7 encoded by human papillomavirus HPV-16 or Cyclin E that lead to depletion of the nucleotide pool. It caused RS and DNA damage accumulation (DSBs, γH2AX foci), however Myc activated nucleotide biosynthesis that, in turn, restocked nucleotides level, rescuing the phenotype [55]. RB1 mutations seem to be indicative of sensitivity to inducers of RS, including PARP trapping inhibitors [56]. Combined mutations of RB1 and TP53 lead to resistance to any chemotherapeutics, but increased sensitivity to inductors of RS, including ATRi and PARPi [57].
2.1.5. Replication in G2 phase and transition to mitosis
WEE1 kinase is one of the most well-known key regulators of mitotic entry. Phosphorylation and subsequent inactivation of CDK1 is vital for G2/M checkpoint [58] and spindle assembly checkpoint [59]. Its antagonist, protein phosphatase CDC25, performs activation of CDK1 [60], while CDC25 activity depends on inhibiting phosphorylation by CHK1 (Figures 1,2). Besides, WEE1 has an additive role as a CDK2 inhibitor during replication [61] that is crucial for correct origin firing and replication fork progress. WEE1 deficiency in cancer cells leads to hyperactivation of CDK1 [62] and CDK2 [61], thereby leading to multiple initiation of replication origins that causes depletion of replication factors, subsequent fork slowing, accumulation of single-stranded DNA and genomic instability followed by accumulation of RPA. Furthermore, WEE1 acts as a protector of the replication fork from DNA2 nuclease [63].
2.1.6. DNA damage response and replication stress
Presence of continuous DNA damage in cells with activated oncogenes, led to investigations of precise mechanisms by which dysregulation of cell cycle control and S phase entry leads to genomic instability. DNA damage in general activates either ATR (ataxia telangiectasia and Rad3 related protein) or ATM (ataxia telangiectasia mutated) kinases, depending on the type of damage. They in turn activate CHK1 (checkpoint kinase 1) and CHK2 (checkpoint kinase 2), which block progression through the cell cycle and phosphorylate components of the DNA repair machinery, allowing the cell to reverse the damage, while not propagating it to daughter cells [64] (Figures 1,2).
The MRE11–RAD50–NBS1 (MRN) complex serves as a DNA damage sensor that identifies DNA DSB sites and binds them, initiating DNA repair. After autophosphorylation, the MRN complex induces ATM activation that is involved in regulation of the cell cycle via the checkpoint kinase CHK2. ATR is a master regulator of response to DNA single-stranded breaks (SSB), potentiates subsequent RS response and cell cycle block via CHK1. Activation of the ATR/ATM signaling pathways is a key process involved in DDR in the intra-S phase checkpoint leading to cell cycle arrest, DNA repair or apoptosis induction [65].
ATM plays a central role in DSB repair and is involved in the response to RS only indirectly. The key role in response to RS is played by the ATR kinase. Firstly, a single strand DNA sensor RPA is activated and binds to the stalled replication forks. Then, together with ATR-interacting protein (ATRIP) RPA recruits ATR kinase that in turn activates DNA topoisomerase II-binding protein 1 (TOPBP1) and phosphorylates CHK1 inducing the ATR-CHK1 pathway. It leads to S/G2 cell cycle arrest through CDK2 reduction and CDK1 phosphorylation by WEE1 and subsequently to DNA repair activation. ATR is a general responder to RS and its inhibitors will be reviewed below. Pharmacological inhibitors of ATR are widely studied as agents of synthetic lethality in the context of ATM mutations or deletion, DNA damage inducing drugs or radiotherapy where replicative stress is high. CHK1 is a serine/threonine checkpoint kinase downstream of ATR induced in response to DNA damage and RS and therefore regulates cell cycle mitotic progression. CHK1 is the effector of the intra-S and G2/M phase checkpoints [66]. CHK1 forms a complex together with checkpoint regulatory protein Claspin and is phosphorylated at Ser-345 and Ser-317 by ATR [66,67] or independently of ATR through autophosphorylation. [68,69]. CHK1 facilitates the degradation or sequestration of CDC25A phosphatases that remove the inhibitory phosphorylation of CDKs by WEE1 and allow cell cycle progression. Thus, CHK1 delays cell cycle progression until resolution of RS. Also, CHK1 plays a role in mitotic progression and mitotic exit phosphorylating CDC25 phosphatases, preventing CDK1 activation, and stopping the cell cycle in the G2 phase [66].
There are a number of studies reporting the importance of CHK1 in cancer treatment. It was demonstrated that CHK1 inhibitors exacerbate RS induced by insulin-like growth factor inhibition, inducing cancer cell death through replication catastrophe [70].
PARP family is known as a main regulator of SSB repair [71]. The mechanism consists of binding to DNA and synthesis of poly(ADP-ribose) chains to engage DNA-repairing proteins such as XRCC1 and Pol β. Thus, the PARP family is involved in base excision repair (BER), which is a major pathway in SSB repair. Also there was some evidence that PARP can promote an alternative pathway of non-homologous end joining (NHEJ) called microhomology-mediated end joining (MMEJ) [72,73]. Besides, PARP is also responsible for the avoidance of DSB by preventing transforming of SSB to DSB during DNA replication that can be a significant trigger of genomic instability and RS [74]. Hence, inhibition of PARP in cells with a decreased homologous recombination (HR) level, commonly indicating BRCA1/2 defects, may lead to the inability of SSB and DSB repair and further to programmed cell death [75]. It was demonstrated that the ATR-CHK1 pathway is vital for PARP-mediated response to DNA damage, according to their roles as restrainers of cell cycle progression by inhibition of CDK1 and CDK2 [76].
2.1.7. P53 and RS
P53 as one of the most crucial regulators of different biochemical pathways is predictably engaged in RS occurrence and response. P53 affects response to RS through its activities related to both DNA repair and influence on cell cycle (Figures 1,2). The first function consists of p21 induction with subsequent G1/S and G2/M arrests through binding to CDK1 and CDK2, therefore p53 is responsible for Rb phosphorylation and further cell cycle promotion to S phase [77]. Besides, p53 is also known as G2 arrest activator due to its function as an inducer of GADD45 and 14-3-3σ, proteins that impair Cyclin B/CDC2 complex. [78,79]. In cells overexpressing CCNE1 and experiencing high RS, p53 was important for preventing catastrophic mitosis through increased levels of p21, WEE1 activity and activation of APC/CCdh1. This leads to endoreduplication (mitosis bypass, leading to polyploidy) in p53-proficient cells and senescence, which is reversed in CCNE1-expressing cells [80].
P53 has multiple roles in DNA repair and response to RS. P53 participates in replication of DNA under normal conditions and in RS. Participation in DNA repair processes includes recruitment of 53BP1 protein, which is involved in a NHEJ repair and suppression of RAD51 gene, which is pivotal for HR, thereby regulating main mechanisms of DSBs repair [81,82]. On the other hand, p53 is involved in SSB repair by modulating APE1 expression, according to its participation in DNA BER [83]. P53 helps to resolve conflicts between transcription and replication, and its loss is synthetically lethal with inhibition of topoisomerase II [84]. P53 binds to both active and stalled replication forks, helping to restart stalled forks and organize DNA repair enzymes at these sites, activating MRE1 and suppressing error-prone Rad52 and POLθ-mediated repair [85,86]. P53 depletion slows replication fork progression [87], which is dependent on its transcriptional activity. This result explains increased tolerance of p53-proficient cells towards chemotherapeutics that increase RS, including gemcitabine and WEE1i [88,89]. In addition to mutations, p53 inactivation or silencing, through MDM2 hyperexpression or oncogenic activation of Ras, also increases sensitivity to RS-inducing drugs, such as ATR-CHK1 inhibitors [90]. These results point to p53 inactivation as a vulnerability for RS-inducing drugs and as a marker for their clinical use.
2.2. Small molecule inducers of replication stress
2.2.1. ATR-CHK1 inhibitors
The most important function of the ATR–CHK1 signaling pathway is to stabilize replication forks, limit the number of active origins and repair of DSB and collapsed replication forks during S phase. By repressing the origins’ firing the ATR-CHK1 pathway realizes control of the cell cycle and maintains the genomic integrity in response to DNA damage and RS (Figure 3). On the other hand, RS that is common among different types of cancers serves as a potent activator of ATR-CHK1 signaling [91]. Thus, the ATR-CHK1 pathway is considered as an attractive target for anticancer therapy.
ATRi including berzosertib (also known as M6620, VX-970) and gartisertib (M4344, VX-803) produced by Merck Serono, ceralasertib (AZD6738) by AstraZeneca, elimusertib (BAY1895344) by Bayer, camonsertib (RP-3500) by Repare Therapeutics, and ART0380 by Artios Pharma, have demonstrated high anti-cancer activity, especially in cancers with high RS level and increased ATR-CHK1 dependency, oncogenic Ras activation, CCNE1 or MYC amplification [92]. Also small molecule ATRi have been employed to improve the efficacy of DNA damage-based chemotherapy for rapid elimination of proliferating tumor cells. It was demonstrated that ATRi could improve response of cancer cells to conventional chemotherapeutic agents as studied in a rapidly growing number of clinical trials (Table 1).
Berzosertib is a potent and ATP-competitive selective small-molecule ATRi [93]. Berzosertib is currently in 1/2 phase clinical trials in combination with other anticancer treatments, specifically chemotherapeutic drugs. It showed single-agent activity, as well as synergistic activity in combination with cisplatin, especially in advanced cancers with ATM aberrations confirming synthetically lethal interaction between ATM deficiency and ATR inhibition during the phase 1 clinical studies[94,95,96]. Berzosertib is effective in the treatment of brain metastases from NSCLC enhancing the effect of radiation [97], in combination with topoisomerase I inhibitor topotecan in small cell lung cancer [98], and with cisplatin in neuroendocrine tumors [99]. Prominent results were demonstrated on chemotherapy-resistant small cell neuroendocrine cancer and high-grade serous ovarian cancer (HGSOC), which exhibit high levels of RS. Combinations of berzosertib with topoisomerase I inhibitors and gemcitabine were synergistically cytotoxic and showed durable tumor regressions and increase in progression-free survival [98,100,101]. Berzosertib demonstrated synthetic lethality in vitro and in vivo in tumor cells with ARID1 deletion or mutations, one of genes commonly altered in cancers [93,102,103].
AZ20 is a potent and selective ATRi that belongs to sulfonylmorpholinopyrimidines. AZ20 inhibits ATR and ATR-mediated phosphorylation of Chk1 in HT29 colorectal adenocarcinoma tumor cells. AZ20 demonstrated high antiproliferative activity against different neoplasms in vitro and in vivo [104,105,106]. The structure of AZ20 was optimized to create AZD6738 (ceralasertib) – another ATRi with improved preclinical physicochemical and pharmacokinetic characteristics [107,108]. Ceralasertib was active as single agent in NSCLC cell lines [109] and potentiated the cytotoxicity of cisplatin and gemcitabine in NSCLC cells, and ATM-deficient lung cancer xenografts [110]. Similar synergistic effects were demonstrated for pancreatic ductal adenocarcinoma (PDAC) [111]. Ceralasertib inhibited gemcitabine-induced CHK1 activation and prevented cell-cycle arrest, leading to the strong induction of RS markers. For instance, ATR inhibition by ceralasertib promotes sensitization to cisplatin in head and neck squamous cell carcinoma (HNSCC) regardless of presence of HPV, one of the important causes of oropharyngeal infection [112]. There is a complex interaction between HPV and DDR proteins. HPV creates aberrant DNA structures during its rapid replication thereby activating cellular RS and recruiting DDR proteins. Subsequently, activation of the ATR signaling pathway leads to the DNA repairing, facilitating viral replication and ensuring a successful viral life cycle [113]. Thus, in in vivo studies, combined treatment with cisplatin and ceralasertib exhibited greater anti-tumor effects, relative to either mono agent, against both HPV+ and HPV− xenograft models, including patient-derived xenograft (PDX) models [112]. Anti-tumor activity of ceralasertib as mono agent and with cisplatin was demonstrated on HER2-positive breast cancer in vitro [114].
Sensitivity to ceralasertib is elevated in models with increased RS such as tumors with defects in the ATM pathway or CCNE1 amplification [115]. Complete loss of ATM function in PDAC models is also critical for efficacy of ATRi/gemcitabine combinational treatment [116]. Ceralasertib also showed increased activity in other models with high genomic instability, such as a BRCA2-mutant triple-negative breast cancer (TNBC) PDX model. Moreover, ceralasertib had combinatorial efficacy with chemotherapy medications carboplatin and irinotecan and the PARP inhibitor olaparib [115]. Ceralasertib is currently being evaluated in a 1/2 phase clinical trials and one study is in a 3 phase and is aimed to check the efficacy and safety the inhibitor in combination with durvalumab in patients with locally advanced and metastatic NSCLC after progression on prior anti-programmed death ligand 1 (anti-PD-L1) therapy and platinum-based chemotherapy (NCT05450692, Table 1). It has been demonstrated that the DNA damage checkpoint plays a critical role in regulating PD-L1 expression [117]. It was demonstrated that PD-L1 expression in cancer cells is upregulated in response to DSBs and requires ATR/CHK1 kinases [118]. Anti-PD-L1 can be combined with ATR targeted drugs to improve therapeutic response to immune checkpoint blockade therapy.
Elimusertib (BAY1895344) is a ATR kinase inhibitor in 1/2 phase clinical trials that showed synergistic antitumor activity in combination with DNA damage-inducing, repair-compromising chemotherapy or radiotherapy in preclinical cancer models. Moreover, it improved antitumor efficacy of nonsteroidal androgen receptor antagonist darolutamide in hormone-dependent prostate cancer [119]. High-risk neuroblastomas are in a group of tumors with oncogene-induced RS because of MYCN amplification and frequent ALK mutations. It was shown that together with ALKi elimusertib potently inhibited cell growth, and led to complete tumor regression in mice models [120]. Elimusertib demonstrated high efficacy against aggressive uterine leiomyosarcoma harboring ATRX mutations in in vivo models [121] and ovarian and uterine carcinosarcoma cell lines and xenografts [122]. Together with PARP inhibitor elimusertib potentiated antitumor activity of HER2-targeted antibody-drug conjugates in HER2-positive cancer in vitro and in xenograft models [123]. Treatment by elimusertib together with anti-PD-L1 resulted in high antitumor activity in a syngeneic mice model with androgen-indifferent, aggressive prostate cancer [124]. The inhibitor is currently in 1/2 phase clinical trials (Table 1).
Gartisertib (M4344, VX-803) is a relatively novel ATP-competitive ATRi that is currently in clinical development. Gartisertib suppressed cancer cell proliferation at concentrations similar to elimusertib and was more potent than berzosertib and ceralasertib [125]. It, as other ATRi, is highly synergistic with a broad range of DNA-targeting anticancer agents including topoisomerase inhibitors, gemcitabine, cisplatin, and PARPi [125,126]. The anticancer activity of gartisertib was demonstrated in multiple cancer cell lines, patient-derived tumor organoids, and mouse xenograft models. In combination with ATM inhibitor M4076, gartisertib demonstrated high anti-tumor efficacy in PDX models of TNBC [127]. The inhibitor is in 1/2 phase clinical trials (Table 1).
Camonsertib (RP-3500) is a 1/2 phase clinical-stage [128] inhibitor that demonstrated high effectiveness in preclinical models as a monotherapy and in combination with PARPi olaparib or niraparib [129]. Another novel ATRi – ART0380 – is in a phase 1/2 clinical trial as a monotherapy or in combination with gemcitabine in patients with advanced or metastatic solid tumors (Table 1).
ATR inhibition is effective in combination with other drugs that induce RS. For example, ATR inhibition by ceralasertib as a single agent and in combination with either CHK1 or WEE1 inhibitors was effective in several preclinical models of Mantle cell lymphoma and diffuse large B-cell lymphoma (DLBCL) regardless of their TP53, MYC, and ATM mutational status in vitro and in vivo studies [130]. Also high antitumor activity of ATRi (ceralasertib) and WEE1i (adavosertib) was shown in non-germinal center DLBCL cell lines, characterized by high MYC expression and CDKN2A/B deletion [131]. ATR inhibition led to accumulation of 53BP1 nuclear bodies in daughter G1 cells and G1 arrest. WEE1 inhibition caused more pronounced DNA damage, inducing arrest in the S phase, and rapid induction of apoptosis. In vivo xenograft DLBCL models showed potential for effective ATRi combinations. Moreover, ATRi and CHK1i are shown to resensitize PARPi-resistant, BRCA1-deficient cancer cells to PARPi [132,133,134,135,136] that also makes ATR-CHK1 pathway an attractive target in drug resistance context.
Particular challenge for anticancer therapy is to kill cells in a quiescent or slowly growing state. So it was demonstrated that treatment with the ATRi enhanced cell apoptotic signaling induced by cisplatin in quiescent cancer cells in vitro [137].

Table 1.
Current clinical trials of ATRi.
| Compound | Study Phases | Key Indications | References |
| Berzosertib (M6620, VX-970) | Phase 2 (7 trials) | Different types of cancer, including DDR deficient and TP53 mutant tumors | NCT02595892 [100] NCT04266912 NCT03517969 NCT02567409 [138] NCT03896503 NCT04216316 NCT03641313 NCT03718091 (completed) NCT02487095 [139,140] |
| Phase 1 (9 trials) | Different types of cancer, including DDR solid tumors | NCT02723864 NCT02589522 NCT02595931 NCT05246111 NCT04266912 NCT02567422 NCT02627443 NCT04216316 NCT04052555 NCT02157792 [94,95,96,101] |
|
| Ceralasertib (AZD 6738) | NSCLC | NCT05450692 | |
| Phase 2 (28 trials) | Different types of solid tumors, including NSCLC, breast and ovarian cancers | NCT02264678 NCT04417062 NCT05061134 NCT05941897 NCT04564027 NCT05582538 NCT03801369 NCT04699838 NCT04090567 NCT03579316 NCT03878095 (suspended) NCT04239014 (withdrawn) NCT03334617 NCT03330847 NCT02937818 NCT03833440 NCT02813135 NCT04298021 NCT04298008 NCT03462342 NCT03428607 (completed) NCT03780608 NCT04065269 NCT04361825 NCT02576444 (terminated) NCT03740893 NCT03182634 NCT02664935 |
|
| Phase 1 (13 trials) | Different types of solid tumors and leukemias | NCT05469919 NCT02264678 NCT05514132 NCT03328273 NCT03022409 (completed) NCT04704661 NCT03669601 NCT02630199 NCT03770429 NCT02223923 NCT01955668 (completed) NCT03527147 (completed) |
|
| Elimusertib (BAY1895344) | Phase 2 (1 trial) | Relapsed or refractory solid tumors | NCT05071209 |
| Phase 1 (10 trials) | Different types of carcinomas and lymphomas | NCT05010096 (withdrawn) NCT03188965 (completed) NCT04095273 (completed) NCT05071209 NCT04616534 NCT04267939 NCT04491942 NCT04535401 NCT04576091 NCT04514497 |
|
| Gartisertib (M4344, VX-803) |
Phase 2 (1 trial) |
Advanced breast cancer with DDR mutations | NCT04655183 (withdrawn) |
| Phase 1 (1 trial) | Solid tumors | NCT02278250 (completed) | |
| Camonsertib (RP-3500) | Phase 1/2 (2 trials) | Advanced solid tumors | NCT04972110 NCT04497116 |
| ART0380 | Phase 2 (1 trial) | Advanced tumors | NCT05798611 |
| Phase 1/2 (1 trial) | Advanced tumors | NCT04657068 |
CHK1 inhibitors (CHK1i) have actively been investigated in different tumor models and in combinations with a variety of drugs. The most promising compounds are prexasertib by Lilly Oncology, SCH 900776 by Merck and Co., and SRA737 by Sierra Oncology Inc. all of which are in early clinical trials (Table 2).
Prexasertib (LY2606368, ACR-368) is a highly selective dual CHK1/CHK2 inhibitor that prevents CHK1 autophosphorylation, stabilizing CDC25A and increasing RS, leading to replication catastrophe and apoptosis [141] (Figure 3). It is effective in monotherapy and in combination with other replication-stress inducing agents such as PARPi, antimetabolites and platinum-based chemotherapy [142]. The FDA has granted fast track designations to prexasertib in platinum-resistant ovarian cancer and endometrial cancer as monotherapy or in combination with low-dose gemcitabine (NCT05548296). As MYCN amplification has been shown to increase RS it is considered as a possible additional biomarker for use of CHK1 inhibitors like prexasertib in neuroblastoma [143]. Prexasertib was tested either as a single agent or in combination with PARPi olaparib in serous carcinoma PDX models and in a panel of ovarian cancer cell lines[134,144,145]. Several phase 1 and 2 clinical trials are ongoing (Table 2).
SRA 737 (PNT 737, CCT245737) is a novel orally bioavailable selective CHK1i that has shown preclinical activity in MYC-amplified models of neuroblastoma [146] and lymphoma [147]. CHK1 inhibition by SRA 737 showed synthetical lethality with loss of B-family DNA polymerase function in lung and colorectal cancer cells [148].
MK-8776 (SCH 900776) a highly selective dual CHK1/2i [149]. This inhibitor was studied as a monotherapy and in combination with gemcitabine in patients with advanced solid tumors in phase 1 of clinical trials [150]. MK-8776 is capable of restoring the sensitivity for chemotherapy drugs in cancer cells that overexpress P-glycoprotein, the ABC transporters which regulate the uptake and efflux of chemotherapeutics [151].
Table 2.
Current clinical trials of CHK1i.
| Compound | Study Phases | Key Indications | References |
| Prexasertib (LY2606368, ACR-368) | Phase 2 (7 trials) | Different types of tumors, including small cell lung cancer, ovarian cancer, etc. | NCT02735980 (completed) [152] NCT03414047 (completed) NCT02203513 (terminated) NCT02873975 (completed) NCT04095221 NCT04032080 (completed) NCT05548296 |
| Phase 1 (14 trials) | Different types of solid tumors and leukemias | NCT02778126 (completed) NCT02514603 (completed) NCT03495323 (completed) NCT02860780 (completed) NCT01115790 (completed) NCT03057145 (completed) NCT04095221NCT02808650 (completed) NCT04023669 NCT05548296 NCT03735446 (terminated) NCT02649764 (completed) NCT02124148 (completed) NCT02555644 (completed) |
|
| SRA 737 | Phase 1/2 (2 trials) | Advanced solid tumors or non-Hodgkin's lymphoma | NCT02797964 [152,153] NCT02797977 |
| MK-8776 (SCH 900776) | Phase 2 (1 trial) | Leukemias | NCT00907517 (terminated) [154] |
| Phase 1 (2 trials) | Solid tumors, leukemias and lymphomas | NCT00779584 (completed) |
The ATRi and especially CHK1i drugs have been extensively developed only in recent years, and available clinical data is relatively limited, compared to approved PARPi. Toxicity remains a major hurdle for ATRi and CHKi [155]. More clinical data and basic research would hopefully allow to determine precise markers and indications for these drugs, allowing for the maximum therapeutic window.
2.2.2. PARP inhibitors
Presently many PARPi such as olaparib, rucaparib, niraparib and talazoparib are approved by the US FDA primarily for BRCA1/2-mutated tumors [172,173]. Recent data about clinical trials of these drugs are represented in Table 3. It is pivotal to verify BRCA1/2 status of patients before therapy due to the role of HR in the potency of PARPi usage. However, even BRCA1/2-mutated tumors can manifest resistance to PARPi. There are many ways to perform it such as replicative fork stabilization, HSP90-mediated BRCA1 stabilization and subsequent HR repair [174], or recently researched recruitment of protein complex shieldin: REV7, RINN1, RINN2, and RINN3 (Figure 3). This complex promotes NHEJ-dependent DNA repair by ATM-53BP1-RIF1-REV7 pathway that leads to the development of resistance to PARPi by the HR-independent pathway [175,176].
Some trials indicated that PARPi treatment may be improved by addition of ATRi [177,178]. That being the case, many up-to-date studies suggest that PARPi can be sufficiently effective against a wider variety of tumors than was thought before. Numerous clinical trials demonstrate beneficial results of PARPi treatment of pancreatic cancer [179], urothelial carcinoma [180], NCT03397394) and mesenchymal sarcomas [181].
Previously, it was demonstrated that PARP inhibition therapy led to an increase of ATR and CHK1 phosphorylation, suggesting that activation of the ATR-CHK1 replication fork protection pathway is one of the main ways to save genome stability in response to PARP shortage. Hence, inhibition of ATR (ceralasertib) or CHK1 (MK-8776) in combination with olaparib led to a considerable synergistic effect that was proved by experiments on HGSOC both in vitro and in vivo [132]. Interestingly, Parmar et al. [134] demonstrated that combination therapy with olaparib and a CHK1i prexasertib was highly effective in models of ovarian and osteosarcoma cancer cells and patient-derived xenografts, resistant to PARPi monotherapy. The mechanism of synergy was associated with RAD51 depletion and replicative fork destabilization; the best effect was achieved in combination treatment of RAD51-mutated cells. Experiments on xenograft models (HGSOC) with BRCA1 mutations and without them showed that PARPi monotherapy did not cause effect, but models were sensitive to CHK1i prexasertib monotherapy. Combination of PARPi and CHK1i showed increased efficiency compared to CHK1i monotherapy.
Another growing field for PARP inhibitors usage is combination treatment with WEE1 inhibitors such as a novel small molecule adavosertib (AZD1775). It might be connected with the G2/M checkpoint, which is vital for correct DNA repair before mitosis and further cell cycle progress. Inhibition of WEE1 compromises G2 arrest, leading to abnormal exit to mitosis, and accumulation of DNA damage, leading to RS and subsequent cell death. Thus , it was recently demonstrated [182] that WEE1i adavosertib monotherapy of TNBC was effective, inhibiting RAD51-mediated HR DNA repair, and increasing quantity of γH2AX foci.cExperiments on breast cancer cell lines (MDA-MB-231, BT-549) showed synergistic effects with combination of PARPi olaparib and adavosertib. During combinational treatment, the number of DNA-damaged cells was greater than in adavosertib monotherapy; such synergy was caused by HR deficiency that noticeably enhanced PARPi impact. Besides, the same study demonstrated highly efficient synergism of tumor growth inhibition by PARPi and WEE1i combined treatment in human breast cancer xenograft models (MDA-MB-231) without significant toxicity. Similar results were also reported by [183], emphasizing G2-M arrest induced after PARP inhibition by talazoparib. PARP inhibition led to increased expression or phosphorylation of major proteins involved in S and G2 DNA damage checkpoints: Cyclin B1, Rb, WEE1, CDK1, FOXM1, CHK1, CHK2 and ATM. Interestingly, sequential therapy did not reduce the efficiency of PARPi and WEE1i combination for ovarian cancer cells in vitro and in vivo, compared to concurrent inhibition, while reducing toxicity for non-transformed cells.
Table 3.
Clinical trials of approved PARPi.
| Compound | Study Phases | Key Indications | References |
| Olaparib | Phase 4 (4 trials) | Ovarian cancer (3 trials), prostate cancer (1 trial), metastatic breast cancer (1 trial) | [156,157,158,159] |
| Phase 3 (37 trials) | Ovarian cancer (more than 30 trials), breast cancer (13 trials), prostate cancer (4 trials) | ||
| Phase 2 (more than 200 trials) |
Ovarian cancer (more than 30 trials), breast cancer (more than 30 trials), prostate cancer (more than 20 trials), lung cancer (more than 20 trials) | ||
| Phase 1 (more than 100 trials) | Ovarian cancer (more than 30 trials), breast cancer (more than 20 trials), prostate cancer (12 trials), lung cancer (10 trials) | ||
| Niraparib | Phase 4 (3 trials) | Ovarian cancer (3 trials) | [160,161,162,163] |
| Phase 3 (23 trials) | Ovarian cancer (12 trials), fallopian tube cancer (5 trials), prostate cancer (3 trials), breast cancer (2 trials), | ||
| Phase 2 (more than 100 trials) | Ovarian cancer (more than 30 trials), breast cancer (15 trials), fallopian tube cancer (9 trials) | ||
| Phase 1 (62 trials) | Ovarian cancer (19 trials), breast cancer (13 trials), prostate cancer (7 trials) | ||
| Rucaparib | Phase 3 (8 trials) | Ovarian cancer (4 trials), fallopian tube cancer (4 trials), prostate cancer 2 trials) | [164,165,166,167] |
| Phase 2 (40 trials) | Ovarian cancer (8 trials), prostate cancer (7 trials), breast cancer (4 trials), | ||
| Phase 1 (24 trials) | Ovarian cancer (7 trials), breast cancer (4 trials), prostate cancer (4 trials) | ||
| Talazoparib | Phase 3 (5 trials) | Ovarian cancer (2 trials), breast cancer (1 trial), prostate cancer (1 trial) | [168,169,170,171] |
| Phase 2 (64 trials) | Breast cancer (17 trials), prostate cancer (8 trials), ovarian cancer (4 trials) | ||
| Phase 1 (51 trials) | Breast cancer (14 trials), prostate cancer (5 trials), ovarian cancer (4 trials) |
2.2.3. WEE1 and PKMYT1 inhibitors
WEE1 and PKMYT1 (MYT1) are two protein kinases that regulate activity of CDK complexes through inhibitory phosphorylations. WEE1 inhibits activity of CDK2 at the both G1/S and G2/M transitions, while PKMYT1 is active only in the G2/M checkpoint. Both are rarely mutated in cancers and in tumors with high levels of RS they act as oncogenes, protecting cells from excessive DNA damage. Both are overexpressed in many hematological and solid tumors [209].
Currently, the most commonly investigated WEE1i is a small molecule AZD1775 (adavosertib, Table 4). Presently it is in trials as an anticancer drug for different types of solid tumors even in pediatric patients [199]. The most recent studies involved in phase I or phase 2 of clinical trials are examining WEE1i for the treatment of pancreatic, gastric, head and neck, breast, ovarian, and other tumors [194]. Many therapies with WEE1i frequently include combinations with other drugs that induce RS, such as carboplatin [184], gemcitabine [186], and PARPi like olaparib [210].
WEE1 inhibition by adavosertib in tumor cells causes acceleration of cell cycle promotion by activation of CDK2 that, in turn, leads to RS, DNA aberrations and further cell death. Other research by Lindemann and colleagues [211] suggested that DNA aberrations and RS, caused by WEE1 inhibition, could be useful for cancer treatment under conditions of DNA repair disorder. Сombined incubation with Rad51 inhibitor B02 and adavosertib manifested notable synergism, with increased markers of DNA damage (γH2AX) and RS (pRPA32), and levels of cell death, than in monotherapy. As was demonstrated in previous works [212], CHK1 phosphorylation level was decreasing, while CDK1 activity was rising that, as a result, caused accumulation of RS, indicating DSB repair shortage. Interestingly, in HPV-positive lines, an increase of p53 level was observed that can be connected with activity of E6 or E7 oncogenes [213], although p21 level was increased irrespective of HPV status in response to combination of B02 and adavosertib. In vivo experiments in mouse models of oral tongue cancer have shown that HPV-negative tumors were not sensitive to B02 and adavosertib mono- or combined therapy. In HPV-positive mice, drugs’ combination significantly inhibited growth of the tumor and substantially increased animal survival rate [211]. These findings are very important for the therapeutic aims due to the role of Rad51 in patients’ survival rate [214].
P53 status is found to play a significant role in WEE1i performance. Thus, TP53-depleted HNSCC cells demonstrate remarkable accumulation of SSB and DSB DNA damage markers and PARP1 cleavage in response to adavosertib, compared to TP53 WT (wild type) cells. Interestingly, the number of 53BP1 foci, which is an important marker of DNA damage, was lower in TP53 knockdown cells, however most of the 53BP1-positive cells did not express γH2AX. It can be considered as a mismatch between 53BP1 foci localization and DSBs [215]. Furthermore, previous study investigated that WEE1i sensitized TP53-mutated mouse xenografts to cisplatin exposure, indicating potency of adavosertib as an effective supplemental drug for TP53-mutated tumors therapy [216]. Data from the previously mentioned article [131] revealed that adavosertib and ATRi ceralasertib treatments slowed progression of the replication fork and increased origin firing. WEE1i led to activation of the ATR–CHK1 pathway and decrease of CHK1 and ATM protein expression after 24 hours. However, the combination of adavosertib and ceralasertib was not effective in vivo, resulting in tumor regressions comparable to the adavosertib single-agent.
PKMYT1 is overexpressed in a number of tumors with markers of RS and PKMYT1 inhibitors (PKMYT1i) are effective in preclinical in vitro and in vivo models. PKMYT1 inhibition was effective in MYCN-amplified neuroblastomas, but not neuroblastomas without amplification [217]. Blocking PKMYT1 activity was effective in eradication of CCNE1-amplified ovarian cancer cells, but not cell lines without amplification through preventing completion of DNA synthesis and increasing the rates of premature mitotic entry [38]. Notably PKMYT1 is overexpressed in CCNE-amplified ovarian carcinoma [218], and currently PKMYT1i RP-6306 is in clinical trials in this setting (Table 4).
Additionally, PKMYT1 and WEE1 inhibition synthetically eradicates cancers with high levels of RS such as glioma [219] and HGSOC, relatively sparing normal tissues or cancers with lower levels of RS [220]. As WEE1i are hindered by toxicity, the authors consider such combinations more selective for therapy. As PKMYT1, but not WEE1, is more important for G2/M transition during checkpoint recovery [221] its inhibitors could be important for a number of combinational therapies aimed at RS.
Table 4.
Clinical trials of WEE1 and PKMYT1 inhibitor.
| Compound | Study Phases | Key Indications | References |
| Adavosertib (AZD1775) | Phase 2 (32 trials, including 7 terminated and 1 withdrawn) | Solid tumors, harboring CCNE1 amplification, ovarian (2 studies), neuroblastoma, medulloblastoma, and rhabdomyosarcoma | [184,185,186,187,188,189,190,191,192,193] |
| Phase 1 (34 trials, including 6 terminated and 1 withdrawn) | HNSCC, uterine cancers, TNBC, pancreatic cancer, acute myeloid leukemia, glioblastoma | [194,195,196,197,198,199,200,201,202,203,204,205,206,207,208] |
2.3.4. CDK inhibitors and RS
Сyclin-dependent kinases in complexes with their Cyclins regulate several critical processes in the cell. The better-known group, consisting of CDK1-6 primarily controls the transition through stages of the cell cycle, while other CDKs, such as CDK7, CDK8/19, CDK9 and CDK12/13 are mainly involved in transcription [222]. Interestingly both groups of CDKs are implicated in RS and DNA damage response, as proper alignment of transcription and cell cycle transition is critically important to the proper replication process (Figure 3). As inhibitors of CDKs are approved by the regulator or are investigated in clinical trials, more studies focus on their impact on tumors with increased RS (Table 5).
CDK1 is the Cyclin-dependent kinase essential for cell cycle progression in S, G2 and M phases of the cell cycle and can also replace other Cyclin-dependent kinases in many models [13,223]. CDK1 inhibition compromises the BRCA1-dependent ATR and ATM DDR in the S phase, increasing sensitivity to DNA damaging agents [224]. Compromised CDK1 activity also leads to increased sensitivity to PARPi [225]. On the other hand CDK1 inhibition is an important mechanism, limiting transcription-replication conflicts [11].
As discussed above, CDK2/Cyclin E is responsible for G1/S entry by phosphorylating Rb and components of the replication machinery such as CDC6, treslin and RECQL4, as well as CDT1 and MCM complex components [30]. Amplification of the CCNE1, is common in many cancers, especially in breast and ovarian carcinomas. Increased activity of CDK2/Cyclin E, in these tumors is known to increase RS and genetic instability via several mechanisms such as shortening of the G1 phase and aberrant origin licensing [30]. Although CCNE-amplified tumors are more prone to RS they remain hard to treat using standard DNA-damaging chemotherapy regimens, especially in ovarian cancer. Several studies revealed, counterintuitively, that CDK2 participates in DNA repair [5,226], and CCNE-amplified ovarian carcinomas rely on CDK2 for DNA repair through homologous recombination, its inhibition compromises replication fork repair [5]. The same study also demonstrated that Cyclin E1 is present at the stalled forks, probably participating in repair.
CDK4/6 inhibitors (palbociclib, ribociclib, abemaciclib) are known as effective drugs for breast cancer treatment irrespective of the p53 status [227,228]. Long-term G1 arrest induced by palbociclib caused RS that was a reason for significant cell proliferation decrease due to transition to senescent state of TP53 WT cells or, in the absence of p53, by causing cells to undergo mitotic catastrophe, resulting in DNA damage [229]. This difference in cells' fate depended on the level of p21 – one of the main proteins involved in transition to a senescence condition. Thus, in WT cells, p53-induced p21 level rise, while in TP53 KO (knockout) cells induction was absent. Effect of palbociclib for TP53 WT and especially TP53 KO was ATR-dependent: ATR inhibition after CDK4/6i 7 days treatment demonstrated an increase in the number of fragmented nuclei that is a consequence of chromosome segregation errors. These data suggested that prolonged CDK4/6 inhibition led to a shortened period of replication and increased the number of cells, which enter mitosis.
Transcriptional CDKs are also implied in RS regulation. CDK8 and CDK19 are two homologous kinases which regulate initiation of transcription as part of the Mediator complex. They are required for transcription activated by a number of transcription factors, such as STATs, SMADs, beta-catenin, p53 and others [230]. Similarly to CDK2, CDK8/19 have a dual role in response to RS. One article demonstrates that CDK8/19 deletion decreases RS due to replication-transcription conflicts [231], and CDK8/19 activity is required for sensitivity to ATRi and CHK1i. On the other hand, in uterine fibroids inhibition of CDK8/19 increased the number of stalled replication forks and markers of RS, including ATR phosphorylation. This phenotype was also dependent on R-loop formation [232]. Additionally, at least, in certain cancers such as prostate carcinoma inhibition of CDK8/19 led to increased ATR-dependent RS and DNA damage by inducing aberrant G1/S transition [233]. Another recent study has demonstrated a similar increased G1/S transition in CML [234]. CDK8/19 is also required for normal origin firing during replication by interacting with the MTBP (Mdm2 p53 binding protein) complex with Treslin, and its inhibition leads to an increase in the number of fragile metaphase chromosome sites [235]. It is possible that similarly to CDK2, CDK8/19 participates in ATR-alternative repair pathways, during RS, as DNA-repair proteins such as BRCA2 and MDC1 were identified as CDK8/19 substrates [236], and inhibition of CDK8/19 increased activity of DNA-damaging agents [237].
While CDK8/19 are involved in initiation of transcription, CDK12 and CDK13 regulate transcription termination and splicing. CDK12/13 are involved in transcriptional regulation of a number of DNA damage response genes, such as BRCA1, ATR and FANC1 [238]. CDK12 expression is required for expression of core replication genes, and its inhibition delays G1/S transition and increases in the number of chromosomal aberrations. This replication-dependent DNA damage is caused by reduced processivity of RNA polymerase II in long poly-(A)-signal-rich genes [239]. CDK12 was implicated in sensitivity to PARPi [240] as well as survival of ovarian cancer cell lines irrespective of synergy with PARPi [241].
Table 5.
Clinical trials of CDK2 inhibitors in tumors with RS markers.
| Compound | Study Phases | Key Indications | References |
| BLU-222 | Phase 1/2 (1 trial) | Solid tumors, including CCNE1-amplified, ovarian carcinoma, breast cancer, endometrial and gastric cancer | NCT05252416 |
| INX-315 | Phase 1/2 (1 trial) | Solid tumors, including breast cancer who progressed on a prior CDK4/6i regimen, and CCNE1-amplified solid tumors | NCT05735080 |
| PF-07104091 | Phase 1/2 (2 trials) | Small cell lung cancer, ovarian cancer, breast cancer | NCT04553133 NCT05262400 |
| ARTS-021 | Phase 1/2 (1 trial) | CCNE1-amplified solid tumors | NCT05867251 |
| INCB123667 | Phase 1 (1 trial) | Solid tumors | NCT05238922 |
3. Conclusions and Future Directions
Tumors with increased RS are one of the most hard to treat. Basal type breast cancer and platinum-resistant ovarian carcinomas have dismal prognosis, compared to same localizations, which don’t have elevated RS. In recent years drugs that target cancers with increased RS and inhibited DNA damage response started to enter clinical trials and even progressed to the clinic (PARPi for BRCA1/2-mutated cancers). Targeted inhibitors were developed against almost every link of DDR to RS, and key proteins which safeguard cancers against replication catastrophe. These approaches have yielded a hope that survival in some of the most aggressive cancers can be radically improved. Nevertheless, the road to introduction of these medications into the clinic remains difficult, with high toxicity and small therapeutic windows leading to termination of a number of programs, and even putting some RS targets into doubt [155,242]. Basic research has demonstrated that sensitivity of cancers to inducers of RS is highly dependent on markers, with most clinical development programs recently focusing on a more narrow group of populations with such markers as CCNE and MYC amplifications, RB1 and TP53 mutations and others. Two future directions may improve the efficiency of targeting high RS cells. First, a number of inhibitors targeting more specific targets, such as Rad51 [243], can limit toxicities, seen for more broad inhibitors, such as ATRi. Second, as more clinical data becomes available, improved markers and even gene signatures [244,245] can allow precise targeting of cancers most susceptible to particular inhibitors [246] and predicting responses [247]. More sophisticated dosing schedules, with sequential treatment with DNA damaging agents and RS inducers are also a promising development [248].
List of abbreviations
| Anti-PD-L1 | anti-programmed death ligand 1 |
| ATM | ataxia telangiectasia mutated |
| ATMi | ATM inhibitor(s) |
| ATR | ataxia telangiectasia and Rad3 related protein |
| ATRi | ATR inhibitor(s) |
| BER | base excision repair |
| CDK(s) | Cyclin dependent kinase(s) |
| CHK | checkpoint kinase |
| CHK1i | CHK1 inhibitor(s) |
| DLBCL | diffuse large B-cell lymphoma |
| DDR | DNA damage response |
| DSB(s) | double-strand DNA break(s) |
| HGSOC | high-grade serous ovarian cancer |
| HNSCC | head and neck squamous cell carcinoma |
| HPV | human papilloma virus |
| HR | homologous recombination |
| KO | knockout |
| LMW-E | low-molecular-weight Cyclin E |
| NHEJ | non-homologous end-joining repair |
| NSCLC | non-small cell lung cancer |
| PARP | poly (ADP-ribose) polymerase |
| PARPi | PARP inhibitor(s) |
| PDAC | pancreatic ductal adenocarcinoma |
| PDX | patient-derived xenograft model(s) |
| PKMYT1i | PKMYT1 inhibitor(s) |
| RPA | replication protein A |
| RS | replication stress |
| SSB | single-strand DNA breaks |
| TNBC | triple-negative breast cancer |
| WEE1i | WEE1 inhibitor(s) |
| WT | wild type |
Author Contributions
Conceptualization, A.V.B. and V.V.T.; writing—original draft preparation, A.I.K., Y.E.A. and V.V.T., writing—review and editing, A.V.B., A.I.K. and V.V.T.; visualization, V.V.T.; funding acquisition, V.V.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation, grant number #22-24-00990.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication Stress and Cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Saxena, S.; Zou, L. Hallmarks of DNA Replication Stress. Mol. Cell 2022, 82, 2298–2314. [Google Scholar] [CrossRef]
- Brison, O.; El-Hilali, S.; Azar, D.; Koundrioukoff, S.; Schmidt, M.; Nähse, V.; Jaszczyszyn, Y.; Lachages, A.-M.; Dutrillaux, B.; Thermes, C.; et al. Transcription-Mediated Organization of the Replication Initiation Program across Large Genes Sets Common Fragile Sites Genome-Wide. Nat. Commun. 2019, 10, 5693. [Google Scholar] [CrossRef]
- Brown, V.E.; Moore, S.L.; Chen, M.; House, N.; Ramsden, P.; Wu, H.-J.; Ribich, S.; Grassian, A.R.; Choi, Y.J. CDK2 Regulates Collapsed Replication Fork Repair in CCNE1-Amplified Ovarian Cancer Cells via Homologous Recombination. NAR Cancer 2023, 5, zcad039. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic Lethality and Cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Zhang, J.; Chan, D.W.; Lin, S.-Y. Exploiting DNA Replication Stress as a Therapeutic Strategy for Breast Cancer. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective Killing of ATM- or p53-Deficient Cancer Cells through Inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Menezes, D.L.; Holt, J.; Tang, Y.; Feng, J.; Barsanti, P.; Pan, Y.; Ghoddusi, M.; Zhang, W.; Thomas, G.; Holash, J.; et al. A Synthetic Lethal Screen Reveals Enhanced Sensitivity to ATR Inhibitor Treatment in Mantle Cell Lymphoma with ATM Loss-of-Function. Mol. Cancer Res. 2015, 13, 120–129. [Google Scholar] [CrossRef]
- Arias, E.E.; Walter, J.C. Strength in Numbers: Preventing Rereplication via Multiple Mechanisms in Eukaryotic Cells. Genes Dev. 2007, 21, 497–518. [Google Scholar] [CrossRef]
- Brison, O.; Gnan, S.; Azar, D.; Koundrioukoff, S.; Melendez-Garcia, R.; Kim, S.-J.; Schmidt, M.; El-Hilali, S.; Jaszczyszyn, Y.; Lachages, A.-M.; et al. Mistimed Origin Licensing and Activation Stabilize Common Fragile Sites under Tight DNA-Replication Checkpoint Activation. Nat. Struct. Mol. Biol. 2023, 30, 539–550. [Google Scholar] [CrossRef]
- Heller, R.C.; Kang, S.; Lam, W.M.; Chen, S.; Chan, C.S.; Bell, S.P. Eukaryotic Origin-Dependent DNA Replication in Vitro Reveals Sequential Action of DDK and S-CDK Kinases. Cell 2011, 146, 80–91. [Google Scholar] [CrossRef]
- Suski, J.M.; Ratnayeke, N.; Braun, M.; Zhang, T.; Strmiska, V.; Michowski, W.; Can, G.; Simoneau, A.; Snioch, K.; Cup, M.; et al. CDC7-Independent G1/S Transition Revealed by Targeted Protein Degradation. Nature 2022, 605, 357–365. [Google Scholar] [CrossRef]
- Kim, S.; Leong, A.; Kim, M.; Yang, H.W. CDK4/6 Initiates Rb Inactivation and CDK2 Activity Coordinates Cell-Cycle Commitment and G1/S Transition. Sci. Rep. 2022, 12, 16810. [Google Scholar] [CrossRef]
- Blow, J.J.; Ge, X.Q.; Jackson, D.A. How Dormant Origins Promote Complete Genome Replication. Trends Biochem. Sci. 2011, 36, 405–414. [Google Scholar] [CrossRef]
- Lemmens, B.; Hegarat, N.; Akopyan, K.; Sala-Gaston, J.; Bartek, J.; Hochegger, H.; Lindqvist, A. DNA Replication Determines Timing of Mitosis by Restricting CDK1 and PLK1 Activation. Mol. Cell 2018, 71, 117–128. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Fikaris, A.J.; Lewis, A.E.; Abulaiti, A.; Tsygankova, O.M.; Meinkoth, J.L. Ras Triggers Ataxia-Telangiectasia-Mutated and Rad-3-Related Activation and Apoptosis through Sustained Mitogenic Signaling*. J. Biol. Chem. 2006, 281, 34759–34767. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Giovanni Nuciforo, P.; Bensimon, A.; et al. Oncogene-Induced Senescence Is a DNA Damage Response Triggered by DNA Hyper-Replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Murcia, L.; Clemente-Ruiz, M.; Pierre-Elies, P.; Royou, A.; Milán, M. Selective Killing of RAS-Malignant Tissues by Exploiting Oncogene-Induced DNA Damage. Cell Rep. 2019, 28, 119–131. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of Nucleotide Metabolism Underlies the Establishment and Maintenance of Oncogene-Induced Senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef]
- Klotz-Noack, K.; Klinger, B.; Rivera, M.; Bublitz, N.; Uhlitz, F.; Riemer, P.; Lüthen, M.; Sell, T.; Kasack, K.; Gastl, B.; et al. SFPQ Depletion Is Synthetically Lethal with BRAFV600E in Colorectal Cancer Cells. Cell Rep. 2020, 32, 108184. [Google Scholar] [CrossRef]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased Global Transcription Activity as a Mechanism of Replication Stress in Cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef]
- Struve, N.; Hoffer, K.; Weik, A.-S.; Riepen, B.; Krug, L.; Cetin, M.H.; Burmester, J.; Ott, L.; Liebing, J.; Gatzemeier, F.; et al. Increased Replication Stress and R-Loop Accumulation in EGFRvIII-Expressing Glioblastoma Present New Therapeutic Opportunities. Neurooncol Adv 2022, 4, vdab180. [Google Scholar] [CrossRef]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef]
- Tort, F.; Bartkova, J.; Sehested, M.; Orntoft, T.; Lukas, J.; Bartek, J. Retinoblastoma Pathway Defects Show Differential Ability to Activate the Constitutive DNA Damage Response in Human Tumorigenesis. Cancer Res. 2006, 66, 10258–10263. [Google Scholar] [CrossRef]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023. [Google Scholar] [CrossRef]
- Spruck, C.H.; Won, K.A.; Reed, S.I. Deregulated Cyclin E Induces Chromosome Instability. Nature 1999, 401, 297–300. [Google Scholar] [CrossRef]
- Chen, Z.; Indjeian, V.B.; McManus, M.; Wang, L.; Dynlacht, B.D. CP110, a Cell Cycle-Dependent CDK Substrate, Regulates Centrosome Duplication in Human Cells. Dev. Cell 2002, 3, 339–350. [Google Scholar] [CrossRef]
- Fagundes, R.; Teixeira, L.K. Cyclin E/CDK2: DNA Replication, Replication Stress and Genomic Instability. Front Cell Dev Biol 2021, 9, 774845. [Google Scholar] [CrossRef]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; García, P.; Helleday, T.; Petermann, E. Increased Replication Initiation and Conflicts with Transcription Underlie Cyclin E-Induced Replication Stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef]
- Nanos-Webb, A.; Jabbour, N.A.; Multani, A.S.; Wingate, H.; Oumata, N.; Galons, H.; Joseph, B.; Meijer, L.; Hunt, K.K.; Keyomarsi, K. Targeting Low Molecular Weight Cyclin E (LMW-E) in Breast Cancer. Breast Cancer Res. Treat. 2012, 132, 575–588. [Google Scholar] [CrossRef]
- Li, M.; Tsavachidis, S.; Wang, F.; Bui, T.; Nguyen, T.D.T.; Luo, L.; Multani, A.S.; Bondy, M.L.; Hunt, K.K.; Keyomarsi, K. Low-Molecular-Weight Cyclin E Deregulates DNA Replication and Damage Repair to Promote Genomic Instability in Breast Cancer. Oncogene 2022, 41, 5331–5346. [Google Scholar] [CrossRef]
- Akli, S.; Zheng, P.-J.; Multani, A.S.; Wingate, H.F.; Pathak, S.; Zhang, N.; Tucker, S.L.; Chang, S.; Keyomarsi, K. Tumor-Specific Low Molecular Weight Forms of Cyclin E Induce Genomic Instability and Resistance to p21, p27, and Antiestrogens in Breast Cancer. Cancer Res. 2004, 64, 3198–3208. [Google Scholar] [CrossRef]
- Sheaff, R.J.; Groudine, M.; Gordon, M.; Roberts, J.M.; Clurman, B.E. Cyclin E-CDK2 Is a Regulator of p27Kip1. Genes Dev. 1997, 11, 1464–1478. [Google Scholar] [CrossRef]
- Chen, X.; Low, K.-H.; Alexander, A.; Jiang, Y.; Karakas, C.; Hess, K.R.; Carey, J.P.W.; Bui, T.N.; Vijayaraghavan, S.; Evans, K.W.; et al. Cyclin E Overexpression Sensitizes Triple-Negative Breast Cancer to Wee1 Kinase Inhibition. Clin. Cancer Res. 2018, 24, 6594–6610. [Google Scholar] [CrossRef]
- Chen, X.; Yang, D.; Carey, J.P.W.; Karakas, C.; Albarracin, C.; Sahin, A.A.; Arun, B.K.; Guray Durak, M.; Li, M.; Kohansal, M.; et al. Targeting Replicative Stress and DNA Repair by Combining PARP and Wee1 Kinase Inhibitors Is Synergistic in Triple Negative Breast Cancers with Cyclin E or BRCA1 Alteration. Cancers 2021, 13. [Google Scholar] [CrossRef]
- Gallo, D.; Young, J.T.F.; Fourtounis, J.; Martino, G.; Álvarez-Quilón, A.; Bernier, C.; Duffy, N.M.; Papp, R.; Roulston, A.; Stocco, R.; et al. CCNE1 Amplification Is Synthetic Lethal with PKMYT1 Kinase Inhibition. Nature 2022, 604, 749–756. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; da Costa, A.A.B.A.; Gulhan, D.; Lee, E.K.; Cheng, S.-C.; Hendrickson, A.E.W.; Kochupurakkal, B.; Kolin, D.L.; Kohn, E.C.; Liu, J.F.; et al. A Replication Stress Biomarker Is Associated with Response to Gemcitabine versus Combined Gemcitabine and ATR Inhibitor Therapy in Ovarian Cancer. Nat. Commun. 2021, 12, 5574. [Google Scholar] [CrossRef]
- Sailo, B.L.; Banik, K.; Girisa, S.; Bordoloi, D.; Fan, L.; Halim, C.E.; Wang, H.; Kumar, A.P.; Zheng, D.; Mao, X.; et al. FBXW7 in Cancer: What Has Been Unraveled Thus Far? Cancers 2019, 11. [Google Scholar] [CrossRef]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. C-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef]
- Mirkin Ekaterina, V.; Mirkin Sergei, M. Replication Fork Stalling at Natural Impediments. Microbiol. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef]
- Smirnov, E.; Borkovec, J.; Kováčik, L.; Svidenská, S.; Schröfel, A.; Skalníková, M.; Švindrych, Z.; Křížek, P.; Ovesný, M.; Hagen, G.M.; et al. Separation of Replication and Transcription Domains in Nucleoli. J. Struct. Biol. 2014, 188, 259–266. [Google Scholar] [CrossRef]
- Sugimoto, N.; Maehara, K.; Yoshida, K.; Ohkawa, Y.; Fujita, M. Genome-Wide Analysis of the Spatiotemporal Regulation of Firing and Dormant Replication Origins in Human Cells. Nucleic Acids Res. 2018, 46, 6683–6696. [Google Scholar] [CrossRef]
- Rubin, S.M.; Gall, A.-L.; Zheng, N.; Pavletich, N.P. Structure of the Rb C-Terminal Domain Bound to E2F1-DP1: A Mechanism for Phosphorylation-Induced E2F Release. Cell 2005, 123, 1093–1106. [Google Scholar] [CrossRef]
- Burke, J.R.; Hura, G.L.; Rubin, S.M. Structures of Inactive Retinoblastoma Protein Reveal Multiple Mechanisms for Cell Cycle Control. Genes Dev. 2012, 26, 1156–1166. [Google Scholar] [CrossRef]
- Doan, A.; Arand, J.; Gong, D.; Drainas, A.P.; Shue, Y.T.; Lee, M.C.; Zhang, S.; Walter, D.M.; Chaikovsky, A.C.; Feldser, D.M.; et al. RB Depletion Is Required for the Continuous Growth of Tumors Initiated by Loss of RB. PLoS Genet. 2021, 17, e1009941. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Ishak, C.A.; Coschi, C.H.; Roes, M.V.; Dick, F.A. Disruption of CDK-Resistant Chromatin Association by pRB Causes DNA Damage, Mitotic Errors, and Reduces Condensin II Recruitment. Cell Cycle 2017, 16, 1430–1439. [Google Scholar] [CrossRef]
- Braden, W.A.; Lenihan, J.M.; Lan, Z.; Luce, K.S.; Zagorski, W.; Bosco, E.; Reed, M.F.; Cook, J.G.; Knudsen, E.S. Distinct Action of the Retinoblastoma Pathway on the DNA Replication Machinery Defines Specific Roles for Cyclin-Dependent Kinase Complexes in Prereplication Complex Assembly and S-Phase Progression. Mol. Cell. Biol. 2006, 26, 7667–7681. [Google Scholar] [CrossRef]
- Manickavinayaham, S.; Velez-Cruz, R.; Biswas, A.K.; Chen, J.; Guo, R.; Johnson, D.G. The E2F1 Transcription Factor and RB Tumor Suppressor Moonlight as DNA Repair Factors. Cell Cycle 2020, 19, 2260–2269. [Google Scholar] [CrossRef]
- Sanidas, I.; Morris, R.; Fella, K.A.; Rumde, P.H.; Boukhali, M.; Tai, E.C.; Ting, D.T.; Lawrence, M.S.; Haas, W.; Dyson, N.J. A Code of Mono-Phosphorylation Modulates the Function of RB. Mol. Cell 2019, 73, 985–1000. [Google Scholar] [CrossRef]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide Deficiency Promotes Genomic Instability in Early Stages of Cancer Development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef]
- Zamalloa, L.G.; Pruitt, M.M.; Hermance, N.M.; Gali, H.; Flynn, R.L.; Manning, A.L. RB Loss Sensitizes Cells to Replication-Associated DNA Damage after PARP Inhibition by Trapping. Life Sci Alliance 2023, 6. [Google Scholar] [CrossRef]
- Nyquist, M.D.; Corella, A.; Coleman, I.; De Sarkar, N.; Kaipainen, A.; Ha, G.; Gulati, R.; Ang, L.; Chatterjee, P.; Lucas, J.; et al. Combined TP53 and RB1 Loss Promotes Prostate Cancer Resistance to a Spectrum of Therapeutics and Confers Vulnerability to Replication Stress. Cell Rep. 2020, 31, 107669. [Google Scholar] [CrossRef]
- Donker, L.; Houtekamer, R.; Vliem, M.; Sipieter, F.; Canever, H.; Gómez-González, M.; Bosch-Padrós, M.; Pannekoek, W.-J.; Trepat, X.; Borghi, N.; et al. A Mechanical G2 Checkpoint Controls Epithelial Cell Division through E-Cadherin-Mediated Regulation of Wee1-Cdk1. Cell Rep. 2022, 41, 111475. [Google Scholar] [CrossRef]
- Serpico, A.F.; Febbraro, F.; Pisauro, C.; Grieco, D. Compartmentalized Control of Cdk1 Drives Mitotic Spindle Assembly. Cell Rep. 2022, 38, 110305. [Google Scholar] [CrossRef]
- Timofeev, O.; Cizmecioglu, O.; Settele, F.; Kempf, T.; Hoffmann, I. Cdc25 Phosphatases Are Required for Timely Assembly of CDK1-Cyclin B at the G2/M Transition*. J. Biol. Chem. 2010, 285, 16978–16990. [Google Scholar] [CrossRef]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.Y.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-Dependent Kinase Suppression by WEE1 Kinase Protects the Genome through Control of Replication Initiation and Nucleotide Consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef]
- Moiseeva, T.N.; Qian, C.; Sugitani, N.; Osmanbeyoglu, H.U.; Bakkenist, C.J. WEE1 Kinase Inhibitor AZD1775 Induces CDK1 Kinase-Dependent Origin Firing in Unperturbed G1- and S-Phase Cells. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 23891–23893. [Google Scholar] [CrossRef]
- Elbæk, C.R.; Petrosius, V.; Benada, J.; Erichsen, L.; Damgaard, R.B.; Sørensen, C.S. WEE1 Kinase Protects the Stability of Stalled DNA Replication Forks by Limiting CDK2 Activity. Cell Rep. 2022, 38, 110261. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA Damage Checkpoint Kinases in Cancer. Expert Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the Biology and Clinical Potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef]
- Zhang, Y.; Hunter, T. Roles of Chk1 in Cell Biology and Cancer Therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef]
- Okita, N.; Minato, S.; Ohmi, E.; Tanuma, S.-I.; Higami, Y. DNA Damage-Induced CHK1 Autophosphorylation at Ser296 Is Regulated by an Intramolecular Mechanism. FEBS Lett. 2012, 586, 3974–3979. [Google Scholar] [CrossRef]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef]
- Wu, X.; Seraia, E.; Hatch, S.B.; Wan, X.; Ebner, D.V.; Aroldi, F.; Jiang, Y.; Ryan, A.J.; Bogenrieder, T.; Weyer-Czernilofsky, U.; et al. CHK1 Inhibition Exacerbates Replication Stress Induced by IGF Blockade. Oncogene 2022, 41, 476–488. [Google Scholar] [CrossRef]
- Woodhouse, B.C.; Dianova, I.I.; Parsons, J.L.; Dianov, G.L. Poly(ADP-Ribose) Polymerase-1 Modulates DNA Repair Capacity and Prevents Formation of DNA Double Strand Breaks. DNA Repair 2008, 7, 932–940. [Google Scholar] [CrossRef]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku Compete for Repair of DNA Double Strand Breaks by Distinct NHEJ Pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Howard, S.M.; Yanez, D.A.; Stark, J.M. DNA Damage Response Factors from Diverse Pathways, Including DNA Crosslink Repair, Mediate Alternative End Joining. PLoS Genet. 2015, 11, e1004943. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Caron, M.-C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.-P.; Langelier, M.-F.; et al. Poly(ADP-Ribose) Polymerase-1 Antagonizes DNA Resection at Double-Strand Breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef]
- Gralewska, P.; Gajek, A.; Rybaczek, D.; Marczak, A.; Rogalska, A. The Influence of PARP, ATR, CHK1 Inhibitors on Premature Mitotic Entry and Genomic Instability in High-Grade Serous BRCAMUT and BRCAWT Ovarian Cancer Cells. Cells 2022, 11. [Google Scholar] [CrossRef]
- Engeland, K. Cell Cycle Regulation: p53-p21-RB Signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Peng, C.Y.; Graves, P.R.; Thoma, R.S.; Wu, Z.; Shaw, A.S.; Piwnica-Worms, H. Mitotic and G2 Checkpoint Control: Regulation of 14-3-3 Protein Binding by Phosphorylation of Cdc25C on Serine-216. Science 1997, 277, 1501–1505. [Google Scholar] [CrossRef]
- Wang, X.W.; Zhan, Q.; Coursen, J.D.; Khan, M.A.; Kontny, H.U.; Yu, L.; Hollander, M.C.; O’Connor, P.M.; Fornace, A.J., Jr; Harris, C.C. GADD45 Induction of a G2/M Cell Cycle Checkpoint. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 3706–3711. [Google Scholar] [CrossRef]
- Zeng, J.; Hills, S.A.; Ozono, E.; Diffley, J.F.X. Cyclin E-Induced Replicative Stress Drives p53-Dependent Whole-Genome Duplication. Cell 2023, 186, 528–542. [Google Scholar] [CrossRef]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Ashworth, A.; Silva, A. p53 Modulates Homologous Recombination by Transcriptional Regulation of the RAD51 Gene. EMBO Rep. 2006, 7, 219–224. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Ho, T.L.F.; Hariharan, A.; Goh, H.C.; Wong, Y.L.; Verkaik, N.S.; Lee, M.Y.; Tam, W.L.; van Gent, D.C.; Venkitaraman, A.R.; et al. Rapid Recruitment of p53 to DNA Damage Sites Directs DNA Repair Choice and Integrity. Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2113233119. [Google Scholar] [CrossRef]
- Poletto, M.; Legrand, A.J.; Fletcher, S.C.; Dianov, G.L. p53 Coordinates Base Excision Repair to Prevent Genomic Instability. Nucleic Acids Res. 2016, 44, 3165–3175. [Google Scholar] [CrossRef]
- Yeo, C.Q.X.; Alexander, I.; Lin, Z.; Lim, S.; Aning, O.A.; Kumar, R.; Sangthongpitag, K.; Pendharkar, V.; Ho, V.H.B.; Cheok, C.F. p53 Maintains Genomic Stability by Preventing Interference between Transcription and Replication. Cell Rep. 2016, 15, 132–146. [Google Scholar] [CrossRef]
- Hampp, S.; Kiessling, T.; Buechle, K.; Mansilla, S.F.; Thomale, J.; Rall, M.; Ahn, J.; Pospiech, H.; Gottifredi, V.; Wiesmüller, L. DNA Damage Tolerance Pathway Involving DNA Polymerase ι and the Tumor Suppressor p53 Regulates DNA Replication Fork Progression. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4311–E4319. [Google Scholar] [CrossRef]
- Roy, S.; Tomaszowski, K.-H.; Luzwick, J.W.; Park, S.; Li, J.; Murphy, M.; Schlacher, K. p53 Orchestrates DNA Replication Restart Homeostasis by Suppressing Mutagenic RAD52 and POLθ Pathways. Elife 2018, 7. [Google Scholar] [CrossRef]
- Klusmann, I.; Rodewald, S.; Müller, L.; Friedrich, M.; Wienken, M.; Li, Y.; Schulz-Heddergott, R.; Dobbelstein, M. p53 Activity Results in DNA Replication Fork Processivity. Cell Rep. 2016, 17, 1845–1857. [Google Scholar] [CrossRef]
- Kranz, D.; Dobbelstein, M. Nongenotoxic p53 Activation Protects Cells against S-Phase-Specific Chemotherapy. Cancer Res. 2006, 66, 10274–10280. [Google Scholar] [CrossRef]
- Li, Y.; Saini, P.; Sriraman, A.; Dobbelstein, M. Mdm2 Inhibition Confers Protection of p53-Proficient Cells from the Cytotoxic Effects of Wee1 Inhibitors. Oncotarget 2015, 6, 32339–32352. [Google Scholar] [CrossRef]
- Segeren, H.A.; van Liere, E.A.; Riemers, F.M.; de Bruin, A.; Westendorp, B. Oncogenic RAS Sensitizes Cells to Drug-Induced Replication Stress via Transcriptional Silencing of P53. Oncogene 2022, 41, 2719–2733. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and Consequences of Replication Stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Gilad, O.; Nabet, B.Y.; Ragland, R.L.; Schoppy, D.W.; Smith, K.D.; Durham, A.C.; Brown, E.J. Combining ATR Suppression with Oncogenic Ras Synergistically Increases Genomic Instability, Causing Synthetic Lethality or Tumorigenesis in a Dosage-Dependent Manner. Cancer Res. 2010, 70, 9693–9702. [Google Scholar] [CrossRef]
- Gorecki, L.; Andrs, M.; Rezacova, M.; Korabecny, J. Discovery of ATR Kinase Inhibitor Berzosertib (VX-970, M6620): Clinical Candidate for Cancer Therapy. Pharmacol. Ther. 2020, 210, 107518. [Google Scholar] [CrossRef]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; de Miguel Luken, M.J.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef]
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 Study of the ATR Inhibitor Berzosertib (formerly M6620, VX-970) Combined with Gemcitabine ± Cisplatin in Patients with Advanced Solid Tumours. Br. J. Cancer 2021, 125, 510–519. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Wesolowski, R.; Devoe, C.; Lord, S.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R.; Yap, T.A. Phase 1 Study of the ATR Inhibitor Berzosertib in Combination with Cisplatin in Patients with Advanced Solid Tumours. Br. J. Cancer 2021, 125, 520–527. [Google Scholar] [CrossRef]
- Baschnagel, A.M.; Elnaggar, J.H.; VanBeek, H.J.; Kromke, A.C.; Skiba, J.H.; Kaushik, S.; Abel, L.; Clark, P.A.; Longhurst, C.A.; Nickel, K.P.; et al. ATR Inhibitor M6620 (VX-970) Enhances the Effect of Radiation in Non-Small Cell Lung Cancer Brain Metastasis Patient-Derived Xenografts. Mol. Cancer Ther. 2021, 20, 2129–2139. [Google Scholar] [CrossRef]
- Thomas, A.; Takahashi, N.; Rajapakse, V.N.; Zhang, X.; Sun, Y.; Ceribelli, M.; Wilson, K.M.; Zhang, Y.; Beck, E.; Sciuto, L.; et al. Therapeutic Targeting of ATR Yields Durable Regressions in Small Cell Lung Cancers with High Replication Stress. Cancer Cell 2021, 39, 566–579. [Google Scholar] [CrossRef]
- Saito, Y.D.; Li, Z.; Lustberg, M.; Grenade, C.; Wesolowski, R. Remarkable Response to a Novel ATR Inhibitor in a Patient with Poorly Differentiated Neuroendocrine Carcinoma. Cancer Treat Res Commun 2018, 16, 9–12. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cheng, S.-C.; Wahner Hendrickson, A.E.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus Gemcitabine versus Gemcitabine Alone in Platinum-Resistant High-Grade Serous Ovarian Cancer: A Multicentre, Open-Label, Randomised, Phase 2 Trial. Lancet Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef]
- Plummer, R.; Dean, E.; Arkenau, H.-T.; Redfern, C.; Spira, A.I.; Melear, J.M.; Chung, K.Y.; Ferrer-Playan, J.; Goddemeier, T.; Locatelli, G.; et al. A Phase 1b Study Evaluating the Safety and Preliminary Efficacy of Berzosertib in Combination with Gemcitabine in Patients with Advanced Non-Small Cell Lung Cancer. Lung Cancer 2022, 163, 19–26. [Google Scholar] [CrossRef]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR Inhibitors as a Synthetic Lethal Therapy for Tumours Deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 2018, 23, 882–897. [Google Scholar] [CrossRef]
- Foote, K.M.; Blades, K.; Cronin, A.; Fillery, S.; Guichard, S.S.; Hassall, L.; Hickson, I.; Jacq, X.; Jewsbury, P.J.; McGuire, T.M.; et al. Discovery of 4-{4-[(3R)-3-Methylmorpholin-4-Yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-Yl}-1H-Indole (AZ20): A Potent and Selective Inhibitor of ATR Protein Kinase with Monotherapy in Vivo Antitumor Activity. J. Med. Chem. 2013, 56, 2125–2138. [Google Scholar] [CrossRef]
- Liu, S.; Ge, Y.; Wang, T.; Edwards, H.; Ren, Q.; Jiang, Y.; Quan, C.; Wang, G. Inhibition of ATR Potentiates the Cytotoxic Effect of Gemcitabine on Pancreatic Cancer Cells through Enhancement of DNA Damage and Abrogation of Ribonucleotide Reductase Induction by Gemcitabine. Oncol. Rep. 2017, 37, 3377–3386. [Google Scholar] [CrossRef]
- Ma, J.; Li, X.; Su, Y.; Zhao, J.; Luedtke, D.A.; Epshteyn, V.; Edwards, H.; Wang, G.; Wang, Z.; Chu, R.; et al. Mechanisms Responsible for the Synergistic Antileukemic Interactions between ATR Inhibition and Cytarabine in Acute Myeloid Leukemia Cells. Sci. Rep. 2017, 7, 41950. [Google Scholar] [CrossRef]
- Jones, C.D.; Blades, K.; Foote, K.M.; Guichard, S.M.; Jewsbury, P.J.; McGuire, T.; Nissink, J.W.; Odedra, R.; Tam, K.; Thommes, P.; et al. Abstract 2348: Discovery of AZD6738, a Potent and Selective Inhibitor with the Potential to Test the Clinical Efficacy of ATR Kinase Inhibition in Cancer Patients. Cancer Res. 2013, 73, 2348–2348. [Google Scholar] [CrossRef]
- Foote, K.M.; Nissink, J.W.M.; McGuire, T.; Turner, P.; Guichard, S.; Yates, J.W.T.; Lau, A.; Blades, K.; Heathcote, D.; Odedra, R.; et al. Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. J. Med. Chem. 2018, 61, 9889–9907. [Google Scholar] [CrossRef]
- Vendetti, F.P.; Lau, A.; Schamus, S.; Conrads, T.P.; O’Connor, M.J.; Bakkenist, C.J. The Orally Active and Bioavailable ATR Kinase Inhibitor AZD6738 Potentiates the Anti-Tumor Effects of Cisplatin to Resolve ATM-Deficient Non-Small Cell Lung Cancer in Vivo. Oncotarget 2015, 6, 44289–44305. [Google Scholar] [CrossRef]
- Dunne, V.; Ghita, M.; Small, D.M.; Coffey, C.B.M.; Weldon, S.; Taggart, C.C.; Osman, S.O.; McGarry, C.K.; Prise, K.M.; Hanna, G.G.; et al. Inhibition of Ataxia Telangiectasia Related-3 (ATR) Improves Therapeutic Index in Preclinical Models of Non-Small Cell Lung Cancer (NSCLC) Radiotherapy. Radiother. Oncol. 2017, 124, 475–481. [Google Scholar] [CrossRef]
- Wallez, Y.; Dunlop, C.R.; Johnson, T.I.; Koh, S.-B.; Fornari, C.; Yates, J.W.T.; Bernaldo de Quirós Fernández, S.; Lau, A.; Richards, F.M.; Jodrell, D.I. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol. Cancer Ther. 2018, 17, 1670–1682. [Google Scholar] [CrossRef]
- Leonard, B.C.; Lee, E.D.; Bhola, N.E.; Li, H.; Sogaard, K.K.; Bakkenist, C.J.; Grandis, J.R.; Johnson, D.E. ATR Inhibition Sensitizes HPV- and HPV+ Head and Neck Squamous Cell Carcinoma to Cisplatin. Oral Oncol. 2019, 95, 35–42. [Google Scholar] [CrossRef]
- Bristol, M.L.; Das, D.; Morgan, I.M. Why Human Papillomaviruses Activate the DNA Damage Response (DDR) and How Cellular and Viral Replication Persists in the Presence of DDR Signaling. Viruses 2017, 9. [Google Scholar] [CrossRef]
- Kim, H.-J.; Min, A.; Im, S.-A.; Jang, H.; Lee, K.H.; Lau, A.; Lee, M.; Kim, S.; Yang, Y.; Kim, J.; et al. Anti-Tumor Activity of the ATR Inhibitor AZD6738 in HER2 Positive Breast Cancer Cells. Int. J. Cancer 2017, 140, 109–119. [Google Scholar] [CrossRef]
- Wilson, Z.; Odedra, R.; Wallez, Y.; Wijnhoven, P.W.G.; Hughes, A.M.; Gerrard, J.; Jones, G.N.; Bargh-Dawson, H.; Brown, E.; Young, L.A.; et al. ATR Inhibitor AZD6738 (Ceralasertib) Exerts Antitumor Activity as a Monotherapy and in Combination with Chemotherapy and the PARP Inhibitor Olaparib. Cancer Res. 2022, 82, 1140–1152. [Google Scholar] [CrossRef]
- Dunlop, C.R.; Wallez, Y.; Johnson, T.I.; Bernaldo de Quirós Fernández, S.; Durant, S.T.; Cadogan, E.B.; Lau, A.; Richards, F.M.; Jodrell, D.I. Complete Loss of ATM Function Augments Replication Catastrophe Induced by ATR Inhibition and Gemcitabine in Pancreatic Cancer Models. Br. J. Cancer 2020, 123, 1424–1436. [Google Scholar] [CrossRef]
- Mouw, K.W.; Konstantinopoulos, P.A. From Checkpoint to Checkpoint: DNA Damage ATR/Chk1 Checkpoint Signalling Elicits PD-L1 Immune Checkpoint Activation. Br. J. Cancer 2018, 118, 933–935. [Google Scholar]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA Double-Strand Break Repair Pathway Regulates PD-L1 Expression in Cancer Cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Lücking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bömer, U.; Moosmayer, D.; Eberspächer, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef]
- Szydzik, J.; Lind, D.E.; Arefin, B.; Kurhe, Y.; Umapathy, G.; Siaw, J.T.; Claeys, A.; Gabre, J.L.; Van den Eynden, J.; Hallberg, B.; et al. ATR Inhibition Enables Complete Tumour Regression in ALK-Driven NB Mouse Models. Nat. Commun. 2021, 12, 6813. [Google Scholar] [CrossRef]
- Harold, J.; Bellone, S.; Manavella, D.D.; Mutlu, L.; McNamara, B.; Hartwich, T.M.P.; Zipponi, M.; Yang-Hartwich, Y.; Demirkiran, C.; Verzosa, M.S.; et al. Elimusertib (BAY1895344), a Novel ATR Inhibitor, Demonstrates in Vivo Activity in ATRX Mutated Models of Uterine Leiomyosarcoma. Gynecol. Oncol. 2023, 168, 157–165. [Google Scholar] [CrossRef]
- Manavella, D.D.; McNamara, B.; Harold, J.; Bellone, S.; Hartwich, T.M.P.; Yang-Hartwich, Y.; Mutlu, L.; Zipponi, M.; Demirkiran, C.; Verzosa, M.S.; et al. Ovarian and Uterine Carcinosarcomas Are Sensitive in Vitro and in Vivo to Elimusertib, a Novel Ataxia-Telangiectasia and Rad3-Related (ATR) Kinase Inhibitor. Gynecol. Oncol. 2023, 169, 98–105. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Fu, H.; Yao, Q.; Wang, L.; Lou, L. Combined Inhibition of PARP and ATR Synergistically Potentiates the Antitumor Activity of HER2-Targeting Antibody-Drug Conjugate in HER2-Positive Cancers. Am. J. Cancer Res. 2023, 13, 161–175. [Google Scholar]
- Tang, Z.; Pilié, P.G.; Geng, C.; Manyam, G.C.; Yang, G.; Park, S.; Wang, D.; Peng, S.; Wu, C.; Peng, G.; et al. ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer. Clin. Cancer Res. 2021, 27, 4898–4909. [Google Scholar] [CrossRef]
- Jo, U.; Senatorov, I.S.; Zimmermann, A.; Saha, L.K.; Murai, Y.; Kim, S.H.; Rajapakse, V.N.; Elloumi, F.; Takahashi, N.; Schultz, C.W.; et al. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells With Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol. Cancer Ther. 2021, 20, 1431–1441. [Google Scholar] [CrossRef]
- Seidel, P.; Rubarth, A.; Zodel, K.; Peighambari, A.; Neumann, F.; Federkiel, Y.; Huang, H.; Hoefflin, R.; Adlesic, M.; Witt, C.; et al. ATR Represents a Therapeutic Vulnerability in Clear Cell Renal Cell Carcinoma. JCI Insight 2022, 7. [Google Scholar] [CrossRef]
- Turchick, A.; Zimmermann, A.; Chiu, L.-Y.; Dahmen, H.; Elenbaas, B.; Zenke, F.T.; Blaukat, A.; Vassilev, L.T. Selective Inhibition of ATM-Dependent Double-Strand Break Repair and Checkpoint Control Synergistically Enhances the Efficacy of ATR Inhibitors. Mol. Cancer Ther. 2023, 22, 859–872. [Google Scholar] [CrossRef]
- Yap, T.A.; Fontana, E.; Lee, E.K.; Spigel, D.R.; Højgaard, M.; Lheureux, S.; Mettu, N.B.; Carneiro, B.A.; Carter, L.; Plummer, R.; et al. Camonsertib in DNA Damage Response-Deficient Advanced Solid Tumors: Phase 1 Trial Results. Nat. Med. 2023, 29, 1400–1411. [Google Scholar] [CrossRef]
- Roulston, A.; Zimmermann, M.; Papp, R.; Skeldon, A.; Pellerin, C.; Dumas-Bérube, É.; Dumais, V.; Dorich, S.; Fader, L.D.; Fournier, S.; et al. RP-3500: A Novel, Potent, and Selective ATR Inhibitor That Is Effective in Preclinical Models as a Monotherapy and in Combination with PARP Inhibitors. Mol. Cancer Ther. 2022, 21, 245–256. [Google Scholar]
- Restelli, V.; Lupi, M.; Chilà, R.; Vagni, M.; Tarantelli, C.; Spriano, F.; Gaudio, E.; Bertoni, F.; Damia, G.; Carrassa, L. DNA Damage Response Inhibitor Combinations Exert Synergistic Antitumor Activity in Aggressive B-Cell Lymphomas. Mol. Cancer Ther. 2019, 18, 1255–1264. [Google Scholar] [CrossRef]
- Young, L.A.; O’Connor, L.O.; de Renty, C.; Veldman-Jones, M.H.; Dorval, T.; Wilson, Z.; Jones, D.R.; Lawson, D.; Odedra, R.; Maya-Mendoza, A.; et al. Differential Activity of ATR and WEE1 Inhibitors in a Highly Sensitive Subpopulation of DLBCL Linked to Replication Stress. Cancer Res. 2019, 79, 3762–3775. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR Inhibition Disrupts Rewired Homologous Recombination and Fork Protection Pathways in PARP Inhibitor-Resistant BRCA-Deficient Cancer Cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.-B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Färkkilä, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Gralewska, P.; Gajek, A.; Marczak, A.; Mikuła, M.; Ostrowski, J.; Śliwińska, A.; Rogalska, A. PARP Inhibition Increases the Reliance on ATR/CHK1 Checkpoint Signaling Leading to Synthetic Lethality-An Alternative Treatment Strategy for Epithelial Ovarian Cancer Cells Independent from HR Effectiveness. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR Inhibition Overcomes PARP Inhibitor and Platinum Resistance in Ovarian Cancer Models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- Hutcherson, R.J.; Kemp, M.G. ATR Kinase Inhibition Sensitizes Quiescent Human Cells to the Lethal Effects of Cisplatin but Increases Mutagenesis. Mutat. Res. 2019, 816-818, 111678. [Google Scholar] [CrossRef]
- Pal, S.K.; Frankel, P.H.; Mortazavi, A.; Milowsky, M.; Vaishampayan, U.; Parikh, M.; Lyou, Y.; Weng, P.; Parikh, R.; Teply, B.; et al. Effect of Cisplatin and Gemcitabine With or Without Berzosertib in Patients With Advanced Urothelial Carcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol 2021, 7, 1536–1543. [Google Scholar] [CrossRef]
- Thomas, A.; Redon, C.E.; Sciuto, L.; Padiernos, E.; Ji, J.; Lee, M.-J.; Yuno, A.; Lee, S.; Zhang, Y.; Tran, L.; et al. Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1594–1602. [Google Scholar] [CrossRef]
- Takahashi, N.; Desai, P.A.; Sciuto, L.; Nichols, S.; Steinberg, S.M.; Thomas, A. Targeting Genomic Instability in Extrapulmonary Small Cell Neuroendocrine Cancers: A Phase II Study with ATR Inhibitor Berzosertib and Topotecan. J. Clin. Orthod. 2022, 40, 8518–8518. [Google Scholar] [CrossRef]
- King, C.; Diaz, H.B.; McNeely, S.; Barnard, D.; Dempsey, J.; Blosser, W.; Beckmann, R.; Barda, D.; Marshall, M.S. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther. 2015, 14, 2004–2013. [Google Scholar] [CrossRef]
- Angius, G.; Tomao, S.; Stati, V.; Vici, P.; Bianco, V.; Tomao, F. Prexasertib, a Checkpoint Kinase Inhibitor: From Preclinical Data to Clinical Development. Cancer Chemother. Pharmacol. 2020, 85, 9–20. [Google Scholar] [CrossRef]
- Keller, K.M.; Eleveld, T.F.; Schild, L.; van den Handel, K.; van den Boogaard, M.; Amo-Addae, V.; Eising, S.; Ober, K.; Koopmans, B.; Looijenga, L.; et al. Chromosome 11q Loss and MYCN Amplification Demonstrate Synthetic Lethality with Checkpoint Kinase 1 Inhibition in Neuroblastoma. Front. Oncol. 2022, 12, 929123. [Google Scholar] [CrossRef]
- Nair, J.; Huang, T.-T.; Murai, J.; Haynes, B.; Steeg, P.S.; Pommier, Y.; Lee, J.-M. Resistance to the CHK1 Inhibitor Prexasertib Involves Functionally Distinct CHK1 Activities in BRCA Wild-Type Ovarian Cancer. Oncogene 2020, 39, 5520–5535. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Lee, J.-M.; Gao, B.; Miller, R.; Lee, J.-Y.; Colombo, N.; Vergote, I.; Credille, K.M.; Young, S.R.; McNeely, S.; et al. A Phase 2 Study of Prexasertib (LY2606368) in Platinum Resistant or Refractory Recurrent Ovarian Cancer. Gynecol. Oncol. 2022. [Google Scholar] [CrossRef]
- Osborne, J.D.; Matthews, T.P.; McHardy, T.; Proisy, N.; Cheung, K.-M.J.; Lainchbury, M.; Brown, N.; Walton, M.I.; Eve, P.D.; Boxall, K.J.; et al. Multiparameter Lead Optimization to Give an Oral Checkpoint Kinase 1 (CHK1) Inhibitor Clinical Candidate: (R)-5-((4-((Morpholin-2-Ylmethyl)amino)-5-(trifluoromethyl)pyridin-2-Yl)amino)pyrazine-2-Carbonitrile (CCT245737). J. Med. Chem. 2016, 59, 5221–5237. [Google Scholar] [CrossRef]
- Walton, M.I.; Eve, P.D.; Hayes, A.; Henley, A.T.; Valenti, M.R.; De Haven Brandon, A.K.; Box, G.; Boxall, K.J.; Tall, M.; Swales, K.; et al. The Clinical Development Candidate CCT245737 Is an Orally Active CHK1 Inhibitor with Preclinical Activity in RAS Mutant NSCLC and Eµ-MYC Driven B-Cell Lymphoma. Oncotarget 2016, 7, 2329–2342. [Google Scholar] [CrossRef]
- Rogers, R.F.; Walton, M.I.; Cherry, D.L.; Collins, I.; Clarke, P.A.; Garrett, M.D.; Workman, P. CHK1 Inhibition Is Synthetically Lethal with Loss of B-Family DNA Polymerase Function in Human Lung and Colorectal Cancer Cells. Cancer Res. 2020, 80, 1735–1747. [Google Scholar] [CrossRef]
- Zhou, Z.-R.; Yang, Z.-Z.; Wang, S.-J.; Zhang, L.; Luo, J.-R.; Feng, Y.; Yu, X.-L.; Chen, X.-X.; Guo, X.-M. The Chk1 Inhibitor MK-8776 Increases the Radiosensitivity of Human Triple-Negative Breast Cancer by Inhibiting Autophagy. Acta Pharmacol. Sin. 2017, 38, 513–523. [Google Scholar] [CrossRef]
- Daud, A.I.; Ashworth, M.T.; Strosberg, J.; Goldman, J.W.; Mendelson, D.; Springett, G.; Venook, A.P.; Loechner, S.; Rosen, L.S.; Shanahan, F.; et al. Phase I Dose-Escalation Trial of Checkpoint Kinase 1 Inhibitor MK-8776 as Monotherapy and in Combination with Gemcitabine in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2015, 33, 1060–1066. [Google Scholar] [CrossRef]
- Cui, Q.; Cai, C.-Y.; Wang, J.-Q.; Zhang, S.; Gupta, P.; Ji, N.; Yang, Y.; Dong, X.; Yang, D.-H.; Chen, Z.-S. Chk1 Inhibitor MK-8776 Restores the Sensitivity of Chemotherapeutics in P-Glycoprotein Overexpressing Cancer Cells. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Jin, T.; Xu, L.; Wang, P.; Hu, X.; Zhang, R.; Wu, Z.; Du, W.; Kan, W.; Li, K.; Wang, C.; et al. Discovery and Development of a Potent, Selective, and Orally Bioavailable CHK1 Inhibitor Candidate: 5-((4-((3-Amino-3-Methylbutyl)amino)-5-(trifluoromethyl)pyrimidin-2-Yl)amino)picolinonitrile. J. Med. Chem. 2021, 64, 15069–15090. [Google Scholar] [CrossRef]
- Kristeleit, R.; Plummer, R.; Jones, R.; Carter, L.; Blagden, S.; Sarker, D.; Arkenau, T.; Evans, T.R.J.; Danson, S.; Symeonides, S.N.; et al. A Phase 1/2 Trial of SRA737 (a Chk1 Inhibitor) Administered Orally in Patients with Advanced Cancer. Br. J. Cancer 2023, 129, 38–45. [Google Scholar] [CrossRef]
- Karp, J.E.; Thomas, B.M.; Greer, J.M.; Sorge, C.; Gore, S.D.; Pratz, K.W.; Smith, B.D.; Flatten, K.S.; Peterson, K.; Schneider, P.; et al. Phase I and Pharmacologic Trial of Cytosine Arabinoside with the Selective Checkpoint 1 Inhibitor Sch 900776 in Refractory Acute Leukemias. Clin. Cancer Res. 2012, 18, 6723–6731. [Google Scholar] [CrossRef]
- Martorana, F.; Da Silva, L.A.; Sessa, C.; Colombo, I. Everything Comes with a Price: The Toxicity Profile of DNA-Damage Response Targeting Agents. Cancers 2022, 14. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in Patients with Metastatic Castration-Resistant Prostate Cancer with DNA Repair Gene Aberrations (TOPARP-B): A Multicentre, Open-Label, Randomised, Phase 2 Trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- Penson, R.T.; Valencia, R.V.; Cibula, D.; Colombo, N.; Leath, C.A., 3rd; Bidziński, M.; Kim, J.-W.; Nam, J.H.; Madry, R.; Hernández, C.; et al. Olaparib Versus Nonplatinum Chemotherapy in Patients With Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J. Clin. Oncol. 2020, 38, 1164–1174. [Google Scholar] [CrossRef]
- Kindler, H.L.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Overall Survival Results From the POLO Trial: A Phase III Study of Active Maintenance Olaparib Versus Placebo for Germline BRCA-Mutated Metastatic Pancreatic Cancer. J. Clin. Oncol. 2022, 40, 3929–3939. [Google Scholar] [CrossRef]
- Senkus, E.; Delaloge, S.; Domchek, S.M.; Conte, P.; Im, S.-A.; Xu, B.; Armstrong, A.; Masuda, N.; Fielding, A.; Robson, M.; et al. Olaparib Efficacy in Patients with Germline BRCA-Mutated, HER2-Negative Metastatic Breast Cancer: Subgroup Analyses from the Phase III OlympiAD Trial. Int. J. Cancer 2023, 153, 803–814. [Google Scholar] [CrossRef]
- Park, J.; Lim, M.C.; Lee, J.-K.; Jeong, D.H.; Kim, S.I.; Choi, M.C.; Kim, B.-G.; Lee, J.-Y. A Single-Arm, Phase II Study of Niraparib and Bevacizumab Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer Patients Previously Treated with a PARP Inhibitor: Korean Gynecologic Oncology Group (KGOG 3056)/NIRVANA-R Trial. J. Gynecol. Oncol. 2022, 33, e12. [Google Scholar] [CrossRef]
- Smith, M.R.; Scher, H.I.; Sandhu, S.; Efstathiou, E.; Lara, P.N., Jr; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; Ståhl, O.; et al. Niraparib in Patients with Metastatic Castration-Resistant Prostate Cancer and DNA Repair Gene Defects (GALAHAD): A Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2022, 23, 362–373. [Google Scholar] [CrossRef]
- Narayan, V.; Torres, A.; Seger, P.; Aaron, M.; Louie, J.; Lee, D.; Haas, N.B.; Takvorian, S.U.; Maxwell, K.N.; Domchek, S.M. A Phase II Single-Arm Trial of Niraparib in Platinum-Sensitive Metastatic Castration-Resistant Prostate Cancer with DNA Repair Defects (PLATPARP). J. Clin. Orthod. 2023, 41, TPS291–TPS291. [Google Scholar] [CrossRef]
- Starks, D.; Rojas-Espaillat, L.; Meissner, T.; Elsey, R.; Xu, B.; Koenen, M.; Feng, S.; VanOosbree, A.; Slunecka, J.; Lee, J.; et al. A Phase 1 Evaluation of the Safety and Tolerability of Niraparib in Combination with Everolimus in Advanced Ovarian and Breast Cancers. Cancer Med. 2023, 12, 18654–18665. [Google Scholar] [CrossRef]
- Monk, B.J.; Coleman, R.L.; Fujiwara, K.; Wilson, M.K.; Oza, A.M.; Oaknin, A.; O’Malley, D.M.; Lorusso, D.; Westin, S.N.; Safra, T.; et al. ATHENA (GOG-3020/ENGOT-ov45): A Randomized, Phase III Trial to Evaluate Rucaparib as Monotherapy (ATHENA-MONO) and Rucaparib in Combination with Nivolumab (ATHENA-COMBO) as Maintenance Treatment Following Frontline Platinum-Based Chemotherapy in Ovarian Cancer. Int. J. Gynecol. Cancer 2021, 31, 1589–1594. [Google Scholar] [CrossRef]
- Patsouris, A.; Diop, K.; Tredan, O.; Nenciu, D.; Gonçalves, A.; Arnedos, M.; Sablin, M.-P.; Jézéquel, P.; Jimenez, M.; Droin, N.; et al. Rucaparib in Patients Presenting a Metastatic Breast Cancer with Homologous Recombination Deficiency, without Germline BRCA1/2 Mutation. Eur. J. Cancer 2021, 159, 283–295. [Google Scholar] [CrossRef]
- Monk, B.J.; Parkinson, C.; Lim, M.C.; O’Malley, D.M.; Oaknin, A.; Wilson, M.K.; Coleman, R.L.; Lorusso, D.; Bessette, P.; Ghamande, S.; et al. A Randomized, Phase III Trial to Evaluate Rucaparib Monotherapy as Maintenance Treatment in Patients With Newly Diagnosed Ovarian Cancer (ATHENA-MONO/GOG-3020/ENGOT-ov45). J. Clin. Oncol. 2022, 40, 3952–3964. [Google Scholar] [CrossRef]
- Fizazi, K.; Piulats, J.M.; Reaume, M.N.; Ostler, P.; McDermott, R.; Gingerich, J.R.; Pintus, E.; Sridhar, S.S.; Bambury, R.M.; Emmenegger, U.; et al. Rucaparib or Physician’s Choice in Metastatic Prostate Cancer. N. Engl. J. Med. 2023, 388, 719–732. [Google Scholar] [CrossRef]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib Monotherapy in Metastatic Castration-Resistant Prostate Cancer with DNA Repair Alterations (TALAPRO-1): An Open-Label, Phase 2 Trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Gruber, J.J.; Gross, W.; McMillan, A.; Ford, J.M.; Telli, M.L. A Phase II Clinical Trial of Talazoparib Monotherapy for PALB2 Mutation-Associated Advanced Breast Cancer. J. Clin. Orthod. 2021, 39, TPS1109–TPS1109. [Google Scholar] [CrossRef]
- Gruber, J.J.; Afghahi, A.; Timms, K.; DeWees, A.; Gross, W.; Aushev, V.N.; Wu, H.-T.; Balcioglu, M.; Sethi, H.; Scott, D.; et al. A Phase II Study of Talazoparib Monotherapy in Patients with Wild-Type BRCA1 and BRCA2 with a Mutation in Other Homologous Recombination Genes. Nat Cancer 2022, 3, 1181–1191. [Google Scholar] [CrossRef]
- Yap, T.A.; Bardia, A.; Dvorkin, M.; Galsky, M.D.; Beck, J.T.; Wise, D.R.; Karyakin, O.; Rubovszky, G.; Kislov, N.; Rohrberg, K.; et al. Avelumab Plus Talazoparib in Patients With Advanced Solid Tumors: The JAVELIN PARP Medley Nonrandomized Controlled Trial. JAMA Oncol 2023, 9, 40–50. [Google Scholar]
- Javle, M.; Shacham-Shmueli, E.; Xiao, L.; Varadhachary, G.; Halpern, N.; Fogelman, D.; Boursi, B.; Uruba, S.; Margalit, O.; Wolff, R.A.; et al. Olaparib Monotherapy for Previously Treated Pancreatic Cancer With DNA Damage Repair Genetic Alterations Other Than Germline BRCA Variants: Findings From 2 Phase 2 Nonrandomized Clinical Trials. JAMA Oncol 2021, 7, 693–699. [Google Scholar] [CrossRef]
- Hussain, M.H.A.; Kocherginsky, M.; Agarwal, N.; Zhang, J.; Adra, N.; Paller, C.J.; Picus, J.; Reichert, Z.R.; Szmulewitz, R.Z.; Tagawa, S.T.; et al. BRCAAWAY: A Randomized Phase 2 Trial of Abiraterone, Olaparib, or Abiraterone + Olaparib in Patients with Metastatic Castration-Resistant Prostate Cancer (mCRPC) with DNA Repair Defects. J. Clin. Orthod. 2022, 40, 5018–5018. [Google Scholar] [CrossRef]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.-C.; Choi, Y.-E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of Mutant BRCA1 Protein Confers PARP Inhibitor and Platinum Resistance. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 17041–17046. [Google Scholar] [CrossRef]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 Counteracts DNA Double-Strand Break Resection and Affects PARP Inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988. [Google Scholar] [CrossRef]
- McMullen, M.; Karakasis, K.; Loembe, B.; Dean, E.; Parr, G.; Oza, A.M. DUETTE: A Phase II Randomized, Multicenter Study to Investigate the Efficacy and Tolerability of a Second Maintenance Treatment in Patients with Platinum-Sensitive Relapsed Epithelial Ovarian Cancer, Who Have Previously Received poly(ADP-Ribose) Polymerase (PARP) Inhibitor Maintenance Treatment. Int. J. Gynecol. Cancer 2020, 30, 1824–1828. [Google Scholar] [CrossRef]
- Drewett, L.; Lucey, R.; Pinilla, K.A.; Grybowicz, L.; Wulff, J.; Dayimu, A.; Demiris, N.; Vallier, A.-L.; Qian, W.; Machin, A.; et al. PARTNER: A Randomized, Phase II/III Trial to Evaluate the Safety and Efficacy of the Addition of Olaparib to Platinum-Based Neoadjuvant Chemotherapy in Patients with Triple-Negative And/or Germline BRCA-Mutated Breast Cancer. J. Clin. Orthod. 2022, 40, TPS619–TPS619. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Rodriguez-Moreno, J.F.; de Velasco, G.; Bravo Fernandez, I.; Alvarez-Fernandez, C.; Fernandez, R.; Vazquez-Estevez, S.; Virizuela, J.A.; Gajate, P.; Font, A.; Lainez, N.; et al. Impact of the Combination of Durvalumab (MEDI4736) plus Olaparib (AZD2281) Administered prior to Surgery in the Molecular Profile of Resectable Urothelial Bladder Cancer: NEODURVARIB Trial. J. Clin. Orthod. 2020, 38, 542–542. [Google Scholar] [CrossRef]
- Eder, J.P.; Doroshow, D.B.; Do, K.T.; Keedy, V.L.; Sklar, J.S.; Glazer, P.; Bindra, R.; Shapiro, G.I. Clinical Efficacy of Olaparib in IDH1/IDH2-Mutant Mesenchymal Sarcomas. JCO Precis Oncol 2021, 5, 466–472. [Google Scholar] [CrossRef]
- Ha, D.-H.; Min, A.; Kim, S.; Jang, H.; Kim, S.H.; Kim, H.-J.; Ryu, H.S.; Ku, J.-L.; Lee, K.-H.; Im, S.-A. Antitumor Effect of a WEE1 Inhibitor and Potentiation of Olaparib Sensitivity by DNA Damage Response Modulation in Triple-Negative Breast Cancer. Sci. Rep. 2020, 10, 9930. [Google Scholar] [CrossRef]
- Fang, Y.; McGrail, D.J.; Sun, C.; Labrie, M.; Chen, X.; Zhang, D.; Ju, Z.; Vellano, C.P.; Lu, Y.; Li, Y.; et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity While Maintaining Efficacy. Cancer Cell 2019, 35, 851–867. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.J.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef]
- Oza, A.M.; Estevez-Diz, M.; Grischke, E.-M.; Hall, M.; Marmé, F.; Provencher, D.; Uyar, D.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-Enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-Sensitive TP53-Mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Wahner Hendrickson, A.E.; et al. Adavosertib plus Gemcitabine for Platinum-Resistant or Platinum-Refractory Recurrent Ovarian Cancer: A Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef]
- Liu, J.F.; Xiong, N.; Campos, S.M.; Wright, A.A.; Krasner, C.; Schumer, S.; Horowitz, N.; Veneris, J.; Tayob, N.; Morrissey, S.; et al. Phase II Study of the WEE1 Inhibitor Adavosertib in Recurrent Uterine Serous Carcinoma. J. Clin. Oncol. 2021, 39, 1531–1539. [Google Scholar] [CrossRef]
- Seligmann, J.F.; Fisher, D.J.; Brown, L.C.; Adams, R.A.; Graham, J.; Quirke, P.; Richman, S.D.; Butler, R.; Domingo, E.; Blake, A.; et al. Inhibition of WEE1 Is Effective in TP53- and RAS-Mutant Metastatic Colorectal Cancer: A Randomized Trial (FOCUS4-C) Comparing Adavosertib (AZD1775) With Active Monitoring. J. Clin. Oncol. 2021, 39, 3705–3715. [Google Scholar] [CrossRef]
- Takebe, N.; Naqash, A.R.; O’Sullivan Coyne, G.; Kummar, S.; Do, K.; Bruns, A.; Juwara, L.; Zlott, J.; Rubinstein, L.; Piekarz, R.; et al. Safety, Antitumor Activity, and Biomarker Analysis in a Phase I Trial of the Once-Daily Wee1 Inhibitor Adavosertib (AZD1775) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3834–3844. [Google Scholar] [CrossRef]
- Madariaga, A.; Mitchell, S.A.; Pittman, T.; Wang, L.; Bowering, V.; Kavak, N.; Quintos, J.; Chang, K.; Ramsahai, J.; Karakasis, K.; et al. Patient Self-Reporting of Tolerability Using PRO-CTCAE in a Randomized Double-Blind, Placebo-Controlled Phase II Trial Comparing Gemcitabine in Combination with Adavosertib or Placebo in Patients with Platinum Resistant or Refractory Epithelial Ovarian Carcinoma. Gynecol. Oncol. 2022, 167, 226–233. [Google Scholar] [CrossRef]
- Cole, K.A.; Ijaz, H.; Surrey, L.F.; Santi, M.; Liu, X.; Minard, C.G.; Maris, J.M.; Voss, S.; Reid, J.M.; Fox, E.; et al. Pediatric Phase 2 Trial of a WEE1 Inhibitor, Adavosertib (AZD1775), and Irinotecan for Relapsed Neuroblastoma, Medulloblastoma, and Rhabdomyosarcoma. Cancer 2023, 129, 2245–2255. [Google Scholar] [CrossRef]
- Embaby, A.; Kutzera, J.; Geenen, J.J.; Pluim, D.; Hofland, I.; Sanders, J.; Lopez-Yurda, M.; Beijnen, J.H.; Huitema, A.D.R.; Witteveen, P.O.; et al. WEE1 Inhibitor Adavosertib in Combination with Carboplatin in Advanced TP53 Mutated Ovarian Cancer: A Biomarker-Enriched Phase II Study. Gynecol. Oncol. 2023, 174, 239–246. [Google Scholar] [CrossRef]
- Fu, S.; Yao, S.; Yuan, Y.; Previs, R.A.; Elias, A.D.; Carvajal, R.D.; George, T.J.; Yuan, Y.; Yu, L.; Westin, S.N.; et al. Multicenter Phase II Trial of the WEE1 Inhibitor Adavosertib in Refractory Solid Tumors Harboring CCNE1 Amplification. J. Clin. Oncol. 2023, 41, 1725–1734. [Google Scholar] [CrossRef]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J. Clin. Oncol. 2015, 33, 3409–3415. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.J.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.A.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef]
- Li, J.; Wu, J.; Bao, X.; Honea, N.; Xie, Y.; Kim, S.; Sparreboom, A.; Sanai, N. Quantitative and Mechanistic Understanding of AZD1775 Penetration across Human Blood-Brain Barrier in Glioblastoma Patients Using an IVIVE-PBPK Modeling Approach. Clin. Cancer Res. 2017, 23, 7454–7466. [Google Scholar] [CrossRef]
- Méndez, E.; Rodriguez, C.P.; Kao, M.C.; Raju, S.; Diab, A.; Harbison, R.A.; Konnick, E.Q.; Mugundu, G.M.; Santana-Davila, R.; Martins, R.; et al. A Phase I Clinical Trial of AZD1775 in Combination with Neoadjuvant Weekly Docetaxel and Cisplatin before Definitive Therapy in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 2740–2748. [Google Scholar] [CrossRef]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2019, 37, 2643–2650. [Google Scholar] [CrossRef]
- Cole, K.A.; Pal, S.; Kudgus, R.A.; Ijaz, H.; Liu, X.; Minard, C.G.; Pawel, B.R.; Maris, J.M.; Haas-Kogan, D.A.; Voss, S.D.; et al. Phase I Clinical Trial of the Wee1 Inhibitor Adavosertib (AZD1775) with Irinotecan in Children with Relapsed Solid Tumors: A COG Phase I Consortium Report (ADVL1312). Clin. Cancer Res. 2020, 26, 1213–1219. [Google Scholar] [CrossRef]
- Kato, H.; de Souza, P.; Kim, S.-W.; Lickliter, J.D.; Naito, Y.; Park, K.; Kumar, S.; Mugundu, G.M.; Bang, Y.-J. Safety, Pharmacokinetics, and Clinical Activity of Adavosertib in Combination with Chemotherapy in Asian Patients with Advanced Solid Tumors: Phase Ib Study. Target. Oncol. 2020, 15, 75–84. [Google Scholar] [CrossRef]
- Kong, A.; Good, J.; Kirkham, A.; Savage, J.; Mant, R.; Llewellyn, L.; Parish, J.; Spruce, R.; Forster, M.; Schipani, S.; et al. Phase I Trial of WEE1 Inhibition with Chemotherapy and Radiotherapy as Adjuvant Treatment, and a Window of Opportunity Trial with Cisplatin in Patients with Head and Neck Cancer: The WISTERIA Trial Protocol. BMJ Open 2020, 10, e033009. [Google Scholar] [CrossRef]
- Någård, M.; Ah-See, M.-L.; So, K.; Vermunt, M.; Thistlethwaite, F.; Labots, M.; Roxburgh, P.; Ravaud, A.; Campone, M.; Valkenburg-van Iersel, L.; et al. Effect of Food on the Pharmacokinetics of the WEE1 Inhibitor Adavosertib (AZD1775) in Patients with Advanced Solid Tumors. Cancer Chemother. Pharmacol. 2020, 86, 97–108. [Google Scholar] [CrossRef]
- Chera, B.S.; Sheth, S.H.; Patel, S.A.; Goldin, D.; Douglas, K.E.; Green, R.L.; Shen, C.J.; Gupta, G.P.; Moore, D.T.; Grilley Olson, J.E.; et al. Phase 1 Trial of Adavosertib (AZD1775) in Combination with Concurrent Radiation and Cisplatin for Intermediate-Risk and High-Risk Head and Neck Squamous Cell Carcinoma. Cancer 2021, 127, 4447–4454. [Google Scholar] [CrossRef]
- Keenan, T.E.; Li, T.; Vallius, T.; Guerriero, J.L.; Tayob, N.; Kochupurakkal, B.; Davis, J.; Pastorello, R.; Tahara, R.K.; Anderson, L.; et al. Clinical Efficacy and Molecular Response Correlates of the WEE1 Inhibitor Adavosertib Combined with Cisplatin in Patients with Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2021, 27, 983–991. [Google Scholar] [CrossRef]
- Bauer, T.M.; Moore, K.N.; Rader, J.S.; Simpkins, F.; Mita, A.C.; Beck, J.T.; Hart, L.; Chu, Q.; Oza, A.; Tinker, A.V.; et al. A Phase Ib Study Assessing the Safety, Tolerability, and Efficacy of the First-in-Class Wee1 Inhibitor Adavosertib (AZD1775) as Monotherapy in Patients with Advanced Solid Tumors. Target. Oncol. 2023, 18, 517–530. [Google Scholar] [CrossRef]
- Falchook, G.S.; Sachdev, J.; Imedio, E.R.; Kumar, S.; Mugundu, G.M.; Jenkins, S.; Chmielecki, J.; Jones, S.; Spigel, D.R.; Johnson, M. A Phase Ib Study of Adavosertib, a Selective Wee1 Inhibitor, in Patients with Locally Advanced or Metastatic Solid Tumors. Invest. New Drugs 2023, 41, 493–502. [Google Scholar] [CrossRef]
- Gonzalez-Ochoa, E.; Milosevic, M.; Corr, B.; Abbruzzese, J.L.; Girda, E.; Miller, R.W.; Croke, J.; Mackay, H.; Lee, Y.C.; Bowering, V.; et al. A Phase I Study of the Wee1 Kinase Inhibitor Adavosertib (AZD1775) in Combination with Chemoradiation in Cervical, Upper Vaginal, and Uterine Cancers. Int. J. Gynecol. Cancer 2023, 33, 1208–1214. [Google Scholar] [CrossRef]
- Shafer, D.; Kagan, A.B.; Rudek, M.A.; Kmieciak, M.; Tombes, M.B.; Shrader, E.; Bandyopadhyay, D.; Hudson, D.; Sankala, H.; Weir, C.; et al. Phase 1 Study of Belinostat and Adavosertib in Patients with Relapsed or Refractory Myeloid Malignancies. Cancer Chemother. Pharmacol. 2023, 91, 281–290. [Google Scholar] [CrossRef]
- Ghelli Luserna di Rorà, A.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 Family Business: Regulation of Mitosis, Cancer Progression, and Therapeutic Target. J. Hematol. Oncol. 2020, 13, 126. [Google Scholar] [CrossRef]
- Li, B.T.; Hamilton, E.P.; Wang, J.S.; Falchook, G.S.; Oza, A.M.; Rodrigo Imedio, E.; Kumar, S.; Mugundu, G.M.; De Bruin, E.; Spigel, D.R.; et al. 1785P Phase Ib Expansion Study of Adavosertib plus Olaparib in Patients with Extensive-Stage or Relapsed Small Cell Lung Cancer. Ann. Oncol. 2020, 31, S1035. [Google Scholar] [CrossRef]
- Lindemann, A.; Patel, A.A.; Tang, L.; Tanaka, N.; Gleber-Netto, F.O.; Bartels, M.D.; Wang, L.; McGrail, D.J.; Lin, S.-Y.; Frank, S.J.; et al. Combined Inhibition of Rad51 and Wee1 Enhances Cell Killing in HNSCC Through Induction of Apoptosis Associated With Excessive DNA Damage and Replication Stress. Mol. Cancer Ther. 2021, 20, 1257–1269. [Google Scholar] [CrossRef]
- Tanaka, N.; Patel, A.A.; Tang, L.; Silver, N.L.; Lindemann, A.; Takahashi, H.; Jaksik, R.; Rao, X.; Kalu, N.N.; Chen, T.-C.; et al. Replication Stress Leading to Apoptosis within the S-Phase Contributes to Synergism between Vorinostat and AZD1775 in HNSCC Harboring High-Risk TP53 Mutation. Clin. Cancer Res. 2017, 23, 6541–6554. [Google Scholar] [CrossRef]
- Rampias, T.; Sasaki, C.; Weinberger, P.; Psyrri, A. E6 and e7 Gene Silencing and Transformed Phenotype of Human Papillomavirus 16-Positive Oropharyngeal Cancer Cells. J. Natl. Cancer Inst. 2009, 101, 412–423. [Google Scholar] [CrossRef]
- Qiao, G.-B.; Wu, Y.-L.; Yang, X.-N.; Zhong, W.-Z.; Xie, D.; Guan, X.-Y.; Fischer, D.; Kolberg, H.-C.; Kruger, S.; Stuerzbecher, H.-W. High-Level Expression of Rad51 Is an Independent Prognostic Marker of Survival in Non-Small-Cell Lung Cancer Patients. Br. J. Cancer 2005, 93, 137–143. [Google Scholar] [CrossRef]
- Diab, A.; Kao, M.; Kehrli, K.; Kim, H.Y.; Sidorova, J.; Mendez, E. Multiple Defects Sensitize p53-Deficient Head and Neck Cancer Cells to the WEE1 Kinase Inhibition. Mol. Cancer Res. 2019, 17, 1115–1128. [Google Scholar] [CrossRef]
- Moser, R.; Xu, C.; Kao, M.; Annis, J.; Lerma, L.A.; Schaupp, C.M.; Gurley, K.E.; Jang, I.S.; Biktasova, A.; Yarbrough, W.G.; et al. Functional Kinomics Identifies Candidate Therapeutic Targets in Head and Neck Cancer. Clin. Cancer Res. 2014, 20, 4274–4288. [Google Scholar] [CrossRef]
- Chayka, O.; D’Acunto, C.W.; Middleton, O.; Arab, M.; Sala, A. Identification and Pharmacological Inactivation of the MYCN Gene Network as a Therapeutic Strategy for Neuroblastic Tumor Cells. J. Biol. Chem. 2015, 290, 2198–2212. [Google Scholar] [CrossRef]
- Xuan, Z.-H.; Wang, H.-P.; Zhang, X.-N.; Chen, Z.-X.; Zhang, H.-Y.; Gu, M.-M. PKMYT1 Aggravates the Progression of Ovarian Cancer by Targeting SIRT3. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 5259–5266. [Google Scholar] [CrossRef]
- Toledo, C.M.; Ding, Y.; Hoellerbauer, P.; Davis, R.J.; Basom, R.; Girard, E.J.; Lee, E.; Corrin, P.; Hart, T.; Bolouri, H.; et al. Genome-Wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells. Cell Rep. 2015, 13, 2425–2439. [Google Scholar] [CrossRef]
- Benada, J.; Bulanova, D.; Azzoni, V.; Petrosius, V.; Ghazanfar, S.; Wennerberg, K.; Sørensen, C.S. Synthetic Lethal Interaction between WEE1 and PKMYT1 Is a Target for Multiple Low-Dose Treatment of High-Grade Serous Ovarian Carcinoma. NAR Cancer 2023, 5, zcad029. [Google Scholar] [CrossRef]
- Chow, J.P.H.; Poon, R.Y.C. The CDK1 Inhibitory Kinase MYT1 in DNA Damage Checkpoint Recovery. Oncogene 2013, 32, 4778–4788. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-Dependent Kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell Cycle, CDKs and Cancer: A Changing Paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Johnson, N.; Cai, D.; Kennedy, R.D.; Pathania, S.; Arora, M.; Li, Y.-C.; D’Andrea, A.D.; Parvin, J.D.; Shapiro, G.I. Cdk1 Participates in BRCA1-Dependent S Phase Checkpoint Control in Response to DNA Damage. Mol. Cell 2009, 35, 327–339. [Google Scholar] [CrossRef]
- Johnson, N.; Li, Y.-C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised CDK1 Activity Sensitizes BRCA-Proficient Cancers to PARP Inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef]
- Maude, S.L.; Enders, G.H. Cdk Inhibition in Human Cells Compromises chk1 Function and Activates a DNA Damage Response. Cancer Res. 2005, 65, 780–786. [Google Scholar] [CrossRef]
- Im, S.-A.; Lu, Y.-S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.-S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316. [Google Scholar] [CrossRef]
- Pandey, K.; Katuwal, N.B.; Park, N.; Hur, J.; Cho, Y.B.; Kim, S.K.; Lee, S.A.; Kim, I.; Lee, S.-R.; Moon, Y.W. Combination of Abemaciclib Following Eribulin Overcomes Palbociclib-Resistant Breast Cancer by Inhibiting the G2/M Cell Cycle Phase. Cancers 2022, 14. [Google Scholar] [CrossRef]
- Crozier, L.; Foy, R.; Mouery, B.L.; Whitaker, R.H.; Corno, A.; Spanos, C.; Ly, T.; Gowen Cook, J.; Saurin, A.T. CDK4/6 Inhibitors Induce Replication Stress to Cause Long-Term Cell Cycle Withdrawal. EMBO J. 2022, 41, e108599. [Google Scholar] [CrossRef]
- Chen, M.; Li, J.; Zhang, L.; Wang, L.; Cheng, C.; Ji, H.; Altilia, S.; Ding, X.; Cai, G.; Altomare, D.; et al. CDK8 and CDK19: Positive Regulators of Signal-Induced Transcription and Negative Regulators of Mediator Complex Proteins. Nucleic Acids Res. 2023, 51, 7288–7313. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Urban, V.; Muñoz-Martínez, F.; Ayestaran, I.; Thomas, J.C.; de Renty, C.; O’Connor, M.J.; Forment, J.V.; Galanty, Y.; Jackson, S.P. Loss of Cyclin C or CDK8 Provides ATR Inhibitor Resistance by Suppressing Transcription-Associated Replication Stress. Nucleic Acids Res. 2021, 49, 8665–8683. [Google Scholar] [CrossRef]
- Muralimanoharan, S.; Shamby, R.; Stansbury, N.; Schenken, R.; de la Pena Avalos, B.; Javanmardi, S.; Dray, E.; Sung, P.; Boyer, T.G. Aberrant R-Loop-Induced Replication Stress in MED12-Mutant Uterine Fibroids. Sci. Rep. 2022, 12, 6169. [Google Scholar] [CrossRef]
- Nakamura, A.; Nakata, D.; Kakoi, Y.; Kunitomo, M.; Murai, S.; Ebara, S.; Hata, A.; Hara, T. CDK8/19 Inhibition Induces Premature G1/S Transition and ATR-Dependent Cell Death in Prostate Cancer Cells. Oncotarget 2018, 9, 13474–13487. [Google Scholar] [CrossRef]
- Khamidullina, A.I.; Yastrebova, M.A.; Bruter, A.V.; Nuzhina, J.V.; Vorobyeva, N.E.; Khrustaleva, A.M.; Varlamova, E.A.; Tyakht, A.V.; Abramenko, Y.E.; Ivanova, E.S.; et al. CDK8/19 Inhibition Attenuates G1 Arrest Induced by BCR-ABL Antagonists and Accelerates Death of Chronic Myelogenous Leukemia Cells. bioRxiv, 5592. [Google Scholar]
- Köhler, K.; Sanchez-Pulido, L.; Höfer, V.; Marko, A.; Ponting, C.P.; Snijders, A.P.; Feederle, R.; Schepers, A.; Boos, D. The Cdk8/19-Cyclin C Transcription Regulator Functions in Genome Replication through Metazoan Sld7. PLoS Biol. 2019, 17, e2006767. [Google Scholar] [CrossRef]
- Poss, Z.C.; Ebmeier, C.C.; Odell, A.T.; Tangpeerachaikul, A.; Lee, T.; Pelish, H.E.; Shair, M.D.; Dowell, R.D.; Old, W.M.; Taatjes, D.J. Identification of Mediator Kinase Substrates in Human Cells Using Cortistatin A and Quantitative Phosphoproteomics. Cell Rep. 2016, 15, 436–450. [Google Scholar] [CrossRef]
- Chen, B.; Wen, P.; Hu, G.; Gao, Y.; Qi, X.; Zhu, K.; Chen, S.; Wu, L.; Xu, A.; Zhao, G. Antagonizing CDK8 Sensitizes Colorectal Cancer to Radiation Through Potentiating the Transcription of e2f1 Target Gene apaf1. Front Cell Dev Biol 2020, 8, 408. [Google Scholar] [CrossRef]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 Complex Maintains Genomic Stability via Regulation of Expression of DNA Damage Response Genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef]
- Chirackal Manavalan, A.P.; Pilarova, K.; Kluge, M.; Bartholomeeusen, K.; Rajecky, M.; Oppelt, J.; Khirsariya, P.; Paruch, K.; Krejci, L.; Friedel, C.C.; et al. CDK12 Controls G1/S Progression by Regulating RNAPII Processivity at Core DNA Replication Genes. EMBO Rep. 2019, 20, e47592. [Google Scholar] [CrossRef]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-Wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef]
- Coelho, R.; Tozzi, A.; Disler, M.; Lombardo, F.; Fedier, A.; López, M.N.; Freuler, F.; Jacob, F.; Heinzelmann-Schwarz, V. Overlapping Gene Dependencies for PARP Inhibitors and Carboplatin Response Identified by Functional CRISPR-Cas9 Screening in Ovarian Cancer. Cell Death Dis. 2022, 13, 909. [Google Scholar] [CrossRef]
- Kong, A.; Mehanna, H. WEE1 Inhibitor: Clinical Development. Curr. Oncol. Rep. 2021, 23, 107. [Google Scholar] [CrossRef]
- Shkundina, I.S.; Gall, A.A.; Dick, A.; Cocklin, S.; Mazin, A.V. New RAD51 Inhibitors to Target Homologous Recombination in Human Cells. Genes 2021, 12. [Google Scholar] [CrossRef]
- Guerrero Llobet, S.; Bhattacharya, A.; Everts, M.; Kok, K.; van der Vegt, B.; Fehrmann, R.S.N.; van Vugt, M.A.T.M. An mRNA Expression-Based Signature for Oncogene-Induced Replication-Stress. Oncogene 2022, 41, 1216–1224. [Google Scholar] [CrossRef]
- Takahashi, N.; Kim, S.; Schultz, C.W.; Rajapakse, V.N.; Zhang, Y.; Redon, C.E.; Fu, H.; Pongor, L.; Kumar, S.; Pommier, Y.; et al. Replication Stress Defines Distinct Molecular Subtypes across Cancers. Cancer Res Commun 2022, 2, 503–517. [Google Scholar] [CrossRef]
- Dreyer, S.B.; Upstill-Goddard, R.; Paulus-Hock, V.; Paris, C.; Lampraki, E.-M.; Dray, E.; Serrels, B.; Caligiuri, G.; Rebus, S.; Plenker, D.; et al. Targeting DNA Damage Response and Replication Stress in Pancreatic Cancer. Gastroenterology 2021, 160, 362–377. [Google Scholar] [CrossRef]
- Huang, R.-H.; Hong, Y.-K.; Du, H.; Ke, W.-Q.; Lin, B.-B.; Li, Y.-L. A Machine Learning Framework Develops a DNA Replication Stress Model for Predicting Clinical Outcomes and Therapeutic Vulnerability in Primary Prostate Cancer. J. Transl. Med. 2023, 21, 20. [Google Scholar] [CrossRef]
- Benada, J.; Ejlertsen, B.; Sørensen, C.S. Overcoming Treatment Toxicity through Sequential Therapy. Cancer Cell 2019, 35, 821–822. [Google Scholar]
Figure 1.
Coordination of cell cycle and replication stress response. Activity of Cyclin-dependent kinases (CDK) in each stage of the cell cycle is critical for stepwise preparation to replication. Pre-replication (MCM) complexes are assembled during late mitosis and G1. Beginning of transcription leads to degradation of Cdt1 preventing further MCM loading onto chromatin. Under conditions of single-strand DNA damage or fork-stalling the ATR-CHK1 response prevents further cell cycle transition. ATM-CHK2-p53 similarly blocks cell cycle after fork-collapse or DNA double-strand breaks. DNA damage response and WEE1/PKMYT1 ensure that mitosis does not start before replication is completed. The ATM-CHK2-p53 pathway is frequently mutated in tumors, and inhibition of ATR-CHK1 and WEE1/PKMYT1 compromises remaining response to replication stress, leading to unrepaired DNA damage and cell death.
Figure 1.
Coordination of cell cycle and replication stress response. Activity of Cyclin-dependent kinases (CDK) in each stage of the cell cycle is critical for stepwise preparation to replication. Pre-replication (MCM) complexes are assembled during late mitosis and G1. Beginning of transcription leads to degradation of Cdt1 preventing further MCM loading onto chromatin. Under conditions of single-strand DNA damage or fork-stalling the ATR-CHK1 response prevents further cell cycle transition. ATM-CHK2-p53 similarly blocks cell cycle after fork-collapse or DNA double-strand breaks. DNA damage response and WEE1/PKMYT1 ensure that mitosis does not start before replication is completed. The ATM-CHK2-p53 pathway is frequently mutated in tumors, and inhibition of ATR-CHK1 and WEE1/PKMYT1 compromises remaining response to replication stress, leading to unrepaired DNA damage and cell death.
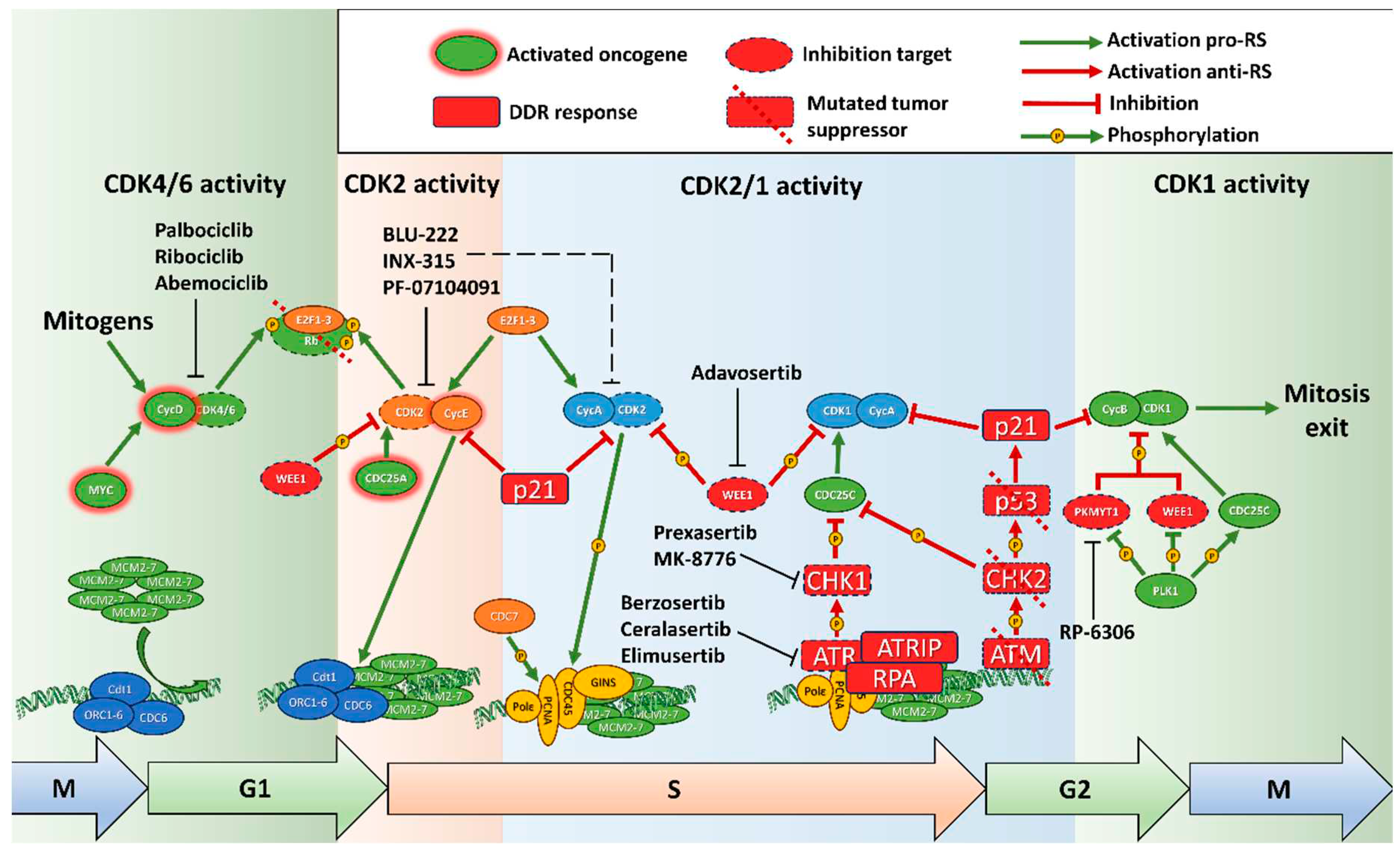
Figure 2.
Sources of replication stress in cancer. Increased activity of oncogenes and CDK2 leads to reduced MCM complex loading to chromatin during a shortened G1, preventing complete replication. Increased activity of CDKs during replication activates excessive and aberrant origin firing, depleting dNTPs and RPA proteins and causing re-replication. Deregulated transcription intensifies collisions between RNA polymerases and DNA replication machinery and displaces MCM complexes from transcriptionally active genes. DNA damage response is activated by fork stalling and fork collapse and slows active forks, activates nearby dormant origins and blocks distant origins. This ensures orderly completion of replication, while transition to mitosis is prevented by inhibition of CDK A/CyclinB1.
Figure 2.
Sources of replication stress in cancer. Increased activity of oncogenes and CDK2 leads to reduced MCM complex loading to chromatin during a shortened G1, preventing complete replication. Increased activity of CDKs during replication activates excessive and aberrant origin firing, depleting dNTPs and RPA proteins and causing re-replication. Deregulated transcription intensifies collisions between RNA polymerases and DNA replication machinery and displaces MCM complexes from transcriptionally active genes. DNA damage response is activated by fork stalling and fork collapse and slows active forks, activates nearby dormant origins and blocks distant origins. This ensures orderly completion of replication, while transition to mitosis is prevented by inhibition of CDK A/CyclinB1.
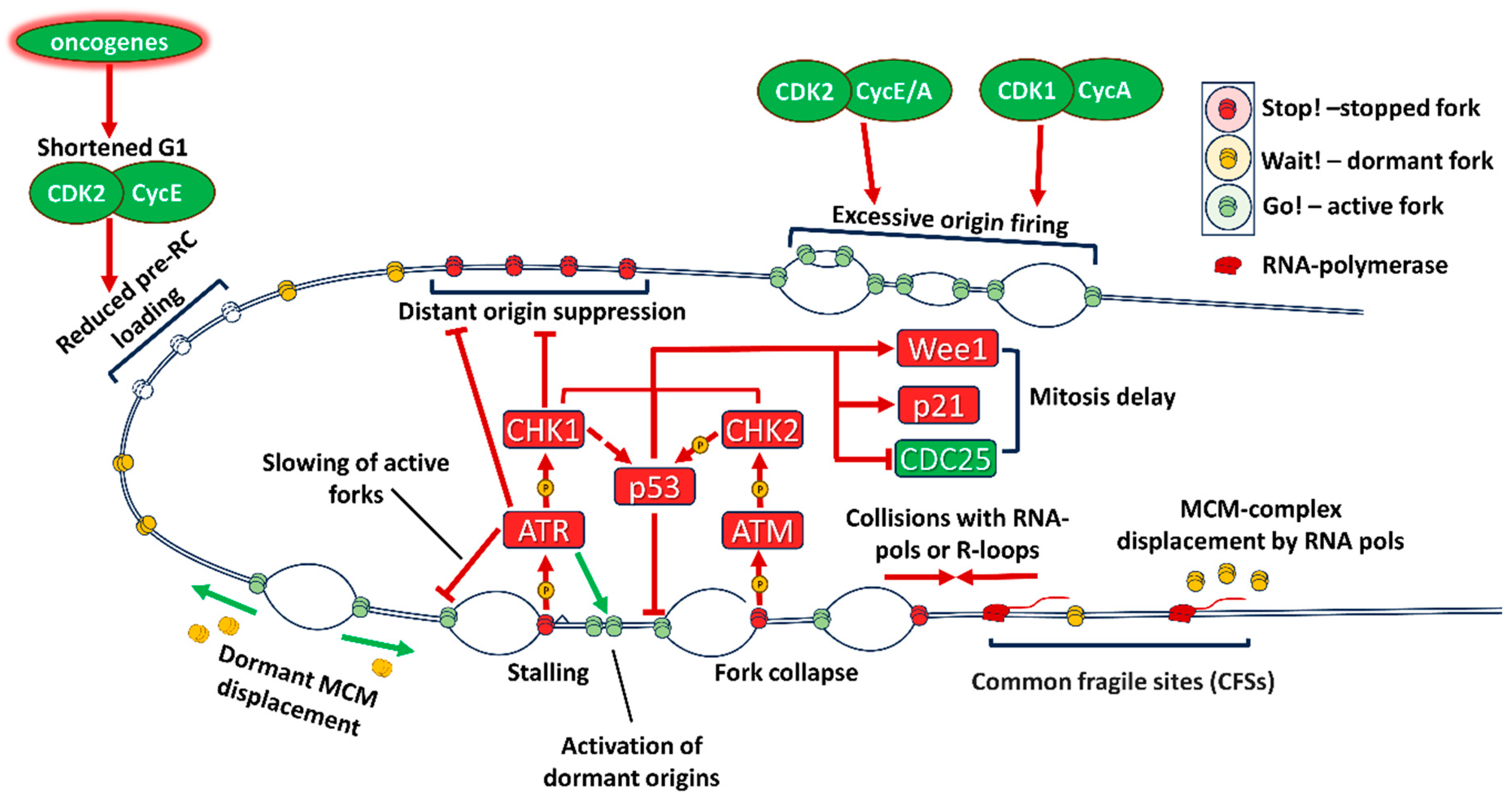
Figure 3.
Small-molecule inducers of replication stress. DNA damage during replication induces ATR and ATM responses and DNA repair (homologous recombination through BRCA1/RAD51, fork protection and stabilization through RAD51 or FANC2, non-homologous end joining through though 53BP1, PARP-dependent DNA repair and others). Mutations in ATM-CHK2-p53 reduce response to DSBs, and inhibition of ATR-CHK1, RAD51 and PARP compromises replication fork rescue, causing irreparable replication fork collapse. Inhibition of WEE1 and PKMYT1 prevents replication completion, causing DNA damage in mitosis and cell death through mitotic catastrophe.
Figure 3.
Small-molecule inducers of replication stress. DNA damage during replication induces ATR and ATM responses and DNA repair (homologous recombination through BRCA1/RAD51, fork protection and stabilization through RAD51 or FANC2, non-homologous end joining through though 53BP1, PARP-dependent DNA repair and others). Mutations in ATM-CHK2-p53 reduce response to DSBs, and inhibition of ATR-CHK1, RAD51 and PARP compromises replication fork rescue, causing irreparable replication fork collapse. Inhibition of WEE1 and PKMYT1 prevents replication completion, causing DNA damage in mitosis and cell death through mitotic catastrophe.
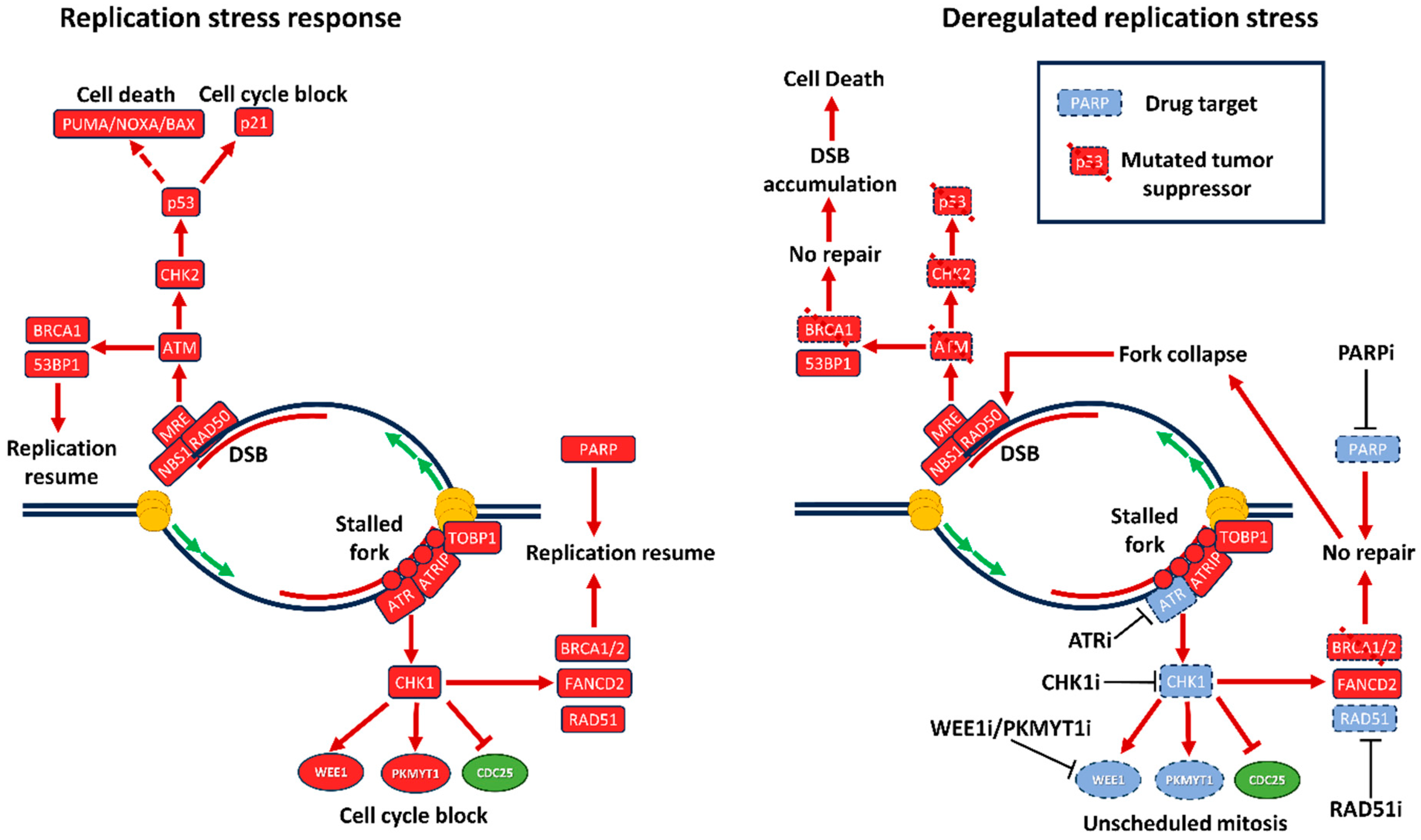
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



