
Submitted:
11 December 2023
Posted:
12 December 2023
You are already at the latest version
Abstract
Support effect is an important issue in heterogeneous catalysis while the explicit role of a catalytic support is often unclear for catalytic reactions. A systematic density functional theory computational study is reported in this paper to elucidate the effect of a model boron nitride (BN) support on the first N-H bond activation step of NH3 on Run (n =1, 2, 3) metal clusters. Geometry optimizations and energy calculations were carried out using density functional theory (DFT) calculation for intermediates and transition state from the starting materials undergoing the N-H activation process. The primary findings are summarized as follows. The involvement of the model BN support does not significantly alter the equilibrium structure of intermediates and transition state in the most favorable pathway (MFP). Moreover, the involvement of BN support decreases the free energy of activation, ΔG≠, thus improving the reaction rate constant. This improvement is more obvious at high temperatures like 673 K than low temperatures like 298 K. The BN support effect leading to the ΔG≠ decrease is most significant for the single Ru atom case among all three cases studied. Finally, the involvement of model BN may change the spin transition behavior of the reaction system during the N-H bond activation process. All these findings provide a deeper insight into the support effect on the N-H bond activation of NH3 for supported Ru catalyst in particular and for supported transition metal catalysts in general.
Keywords:
N-H bond activation of ammonia
; support effect
; boron nitride
; ruthenium
; density functional theory calculation.
1. Introduction
Bockris introduced the concept of a “hydrogen (H2) economy” envisioning an energy transition founded on the utilization of H2 as a vector for the generation of clean and environmentally sustainable energy [1]. Over recent decades, the production of H2 from various sources, its transport and storage, and finally its use have been extensively investigated [2]. H2 has assumed a prominent role in the energy sector, finding applications in stationary power generation, transportation, and as an energy vector for storing surplus electrical energy generated during off-peak periods [3]. However, one of the challenges in H2 technology today is storage and transportation. Due to the problems of physical storage methods, chemical storage methods based on using another easily transportable hydrogen-containing compound, which in turn produces H2 by chemical reaction may be more favored. Ammonia (NH3) is currently one of the most promising H2 carriers. It can form a liquid at low pressure at ambient temperature, it is easy to transport and store, and its industrial synthesis is mature. If the cost-effective production of H2 through NH3 decomposition can be achieved, it is anticipated that H2 storage and transportation via NH3 will exhibit significant technical and economic competitiveness.
In recent years, NH3 decomposition to produce H2 has received more and more attention by people in fundamental researches and industrial applications [4,5,6,7,8,9,10,11,12]. Because of the inertia of NH3, its activation and conversion to N2 and H2 has to involve catalysis. The activation of N-H bond is one of the key steps as well as the first step in the catalytic conversion of NH3. The activation mechanism of N-H bond is undoubtedly important for understanding the existing NH3 catalytic conversion processes and developing new NH3 catalytic conversion systems. Supported nickel (Ni) and supported ruthenium (Ru) catalysts are the most commonly used catalysts in fundamental research and in pilot plant [4,5,8,9,10,11,12]. Many researchers have studied the reaction kinetics or/and reaction mechanism of NH3 decomposition catalyzed by Ru- or Ni-based catalysts [12,13]. Yue et al. [14] summarized several ways of the N-H bond activation on metal catalysts. They also proposed that the difficulty of N-H bond activation of NH3 is due to the relatively large N-H bond energy and the relative activity of the lone pair of electrons on the N atom. However, these mechanistic point of views focused on reactions of organic synthesis. Piers et al. [15] reported the N-H bond activation of NH3 via reaction with low-valence molybdenum complexes of a diborate pentadentate ligand system.
In a broader scope of the NH3 decomposition mechanism on transition metal catalysts, most of researches have been carried out with a plane surface model like Pd(111) [16], Ni(111), Co(111), Fe(110) [17], Cu(111) [18], Cu(100) [19] and WC(0001) [20]. For example, Jiang et al. [16] reported that NH is the most abundant intermediate on Pd (111) surface and the dehydrogenation of NH3 is the rate-determining step in the overall reaction. However, the kinetic analysis and mechanistic studies of NH3 decomposition on Ni and Ru catalysts have not yet been led to clearly study in detail [21]. Especially, the activation of N-H bonds on the metal atom/clusters is still not well understood, at the level of elementary steps and at the molecular level [4,22,23]. For example, a well-recognized reaction pathway of NH3 decomposition can be expressed as described in Scheme 1 [24].
Scheme 1, as described by Sun in Ref. [24] and the references therein, outlines a reaction mechanism in which the symbol “∗” designates the reactive site responsible for NH3 decomposition. The initial two steps within Scheme 1 correspond to the initial activation of the first N-H bond in NH3 on a specific catalytic site. However, the comprehensive understanding of the reaction mechanism presented in Scheme 1, particularly within these initial two steps, remains elusive. This holds especially true for critical information regarding the nature of the catalytic site represented by “∗.” Several unresolved questions come to light, particularly concerning these initial two steps in Scheme 1. For example, firstly, it is not clear whether the two “*” notations in the first two steps correspond to a same metal site or two different metal sites. It should be noted that, the answer may be different when dealing with a single metal atom and with metal clusters. It is worth noting that the answer to this question may differ when considering a single metal atom as opposed to metal clusters. Secondly, there is a lack of understanding regarding how the incorporation of a catalytic support into the metal site alters the reaction behaviors. These questions are certainly important since catalytically active metal components are always resided on a frequently used support, like Al2O3, carbon nanotube, graphene and boron nitride (BN). Support effect is an important topic in heterogeneous catalysis, in particular for Ru-catalyzed hydrogen utilization processes [25], and for NH3 decomposition to obtain high-purity hydrogen [26].
To gain a clearer comprehension of the impact of catalytic support and metal cluster size on N-H bond activation behavior in metal atom/clusters, we present a DFT study of N-H bond activation of NH3 on both of the unsupported and supported Ru atom/clusters in this paper. This paper focuses on the structural, energetic, and spin multiplicity changes during the N-H bond activation process without and with the model hexagonal BN support. BN, like its C analogue, can exist in various forms like hexagonal sheets, nanotubes and nanobowls, making it owing various interesting properties and useful in the field of catalysis [27,28,29]. We found that the model support can change the structure of intermediates and transition states and decrease the reaction energy barrier. The results presented in this work offer valuable qualitative insights, contributing to a deeper understanding of the impact of catalytic support and metal cluster size on the activation of N-H bonds and other types of saturated bonds in a broader context.
2. Computational Methods and Reactant Models
The DFT calculations were performed by employing the M062X [30] exchange and correlation functionals to explore the potential energy surfaces (PESs) of the first N-H bond activation process of NH3. To better description of the long-term interaction between NH3 and Ru or BN nanosheet due to dispersion problem, the Grimme’s D3 dispersion correction [31] was applied for all DFT calculations. The activation process undergoes on the Run (n=1,2,3) clusters without and with B19N19H16 as the model BN support [32]. This model support is denoted as “-BN” appeared in a certain species notation hereafter in this paper. M062X is known to be able to provide a good description of the PES for the bond activation process on transition metal clusters [33,34] as well as for the BN involved reaction system [35]. In the present status, although DFT is hard to give quantitative explanation of the experimental data, the relative reaction barrier is much more credible [36]. The basis set information will be specified after the description of the model of reactants. All PESs were explored by optimizing the geometries in the energy minimums for the reactants, the intermediates and the products, and the first-order saddle points for transition states using the Gaussian 09 program suite (B.09 (for initial optimization) and C.01 (for final optimization and frequency analysis) versions [37,38]). Frequency analyses were performed to confirm the energy minimums and the first-order saddle points, as well as to obtain the zero-point corrected energies of the optimized geometries. Intrinsic reaction coordinate (IRC) computations [39] were performed to confirm the transition states connected the appropriate reactants and products.
Since this paper emphasizes at understanding of the support effect of model BN on the N-H bond activation, we investigated and compared the structural and energetic data for the interaction of supported and unsupported Ru metal clusters with one NH3 molecule. In order to directly understanding the role of model BN support, all of the 6 reactions interested in this paper are categorized into two types, and expressed as follows.
The first type corresponds to the unsupported cases, i.e., the reaction of NH3 with unsupported Run cluster (where n = 1, 2, or 3) to form the NH2-Run-H species, which includes the following three reactions.
NH3 + Ru1 → NH2-Ru1-H …… Rxn.
NH3 + Ru2 → NH2-Ru2-H…… Rxn.
NH3 + Ru3 → NH2-Ru3-H…… Rxn.
The second type corresponds to the supported cases, i.e., the reaction between NH3 and model BN-supported Run cluster (denoted as Run-BN, where n = 1, 2, 3) to afford NH2-Run-H-BN species, which includes the following three reactions in detail:
NH3 + Ru1-BN → NH2-Ru1-H-BN …… Rxn.
NH3 + Ru2-BN → NH2-Ru2-H-BN …… Rxn.
NH3 + Ru3-BN → NH2-Ru3-H-BN …… Rxn.
For the above 6 reactions, the key species on different PESs with a certain spin multiplicity (S) were optimized in geometries and energetically calculated. For easy description hereafter in this paper, the notations for different key species on different PESs are defined as in the following regulations.
Firstly, these key species include the starting materials (SM), the first intermediate formed from the SM, IM1, the transition state followed by IM1, TS, and the second intermediate followed by TS, IM2. The SM is actually the system of separated NH3 and one of the six Ru clusters in left side of Rxns. (1)~(6), and IM2 is actually the first N-H bond activation product of one of Rxns. (1)~(6). Secondly, since all species in all of the six reactions have even number of electrons, the PESs with different multiplicities of S = 1, 3, 5, 7… (i.e. singlet, triplet, quintet, heptet, and so on) were explored. The information about S is put in the upper-left superscript in front of a species notation to indicate its spin multiplicity. For example, 7Ru3 is a heptet Ru3 cluster, and 3TS is a triplet transition state. Therefore the possibly most complicated notation for a certain species in this paper can be expressed asS(SM, IM1, TS, or IM2)-Run-(unsup or BN), where the suffix “-unsup” stands for the unsupported case, and “-BN” stands for the model BN-supported cases. For example, 7IM2-Ru3-BN means the IM2 from the reaction of NH3 with a model BN-supported Ru3 cluster with a heptet state, and 3TS-Ru2-unsup means the TS in the reaction of NH3 with an unsupported Ru2 cluster with a triplet state under the above name regulation.
Figure 1 shows the M06X-GD3 optimized geometries of the B19N19H16 sheet and 5Ru1-BN (c) in different views. For B19N19H16, the 6-311G** basis set was used for atoms in the red circles as indicated in panels a and c in Figure 1. The 6-31G basis set was used for the residual atoms of B19N19H16. Two levels of basis set were used for describing the model BN support in order to compromise between the computational accuracy and the time expense. The SDD basis set was used for the Ru atoms [40]. The 6-311G** basis set was also used for N and H atoms in NH3. The cluster model was used for the calculations in this work. For all of the structure optimization and energy calculations, all of the atoms were allowed to relax. Hereafter in this paper all the geometries in the supported cases, only the side view of the BN support will be shown in Section 3 unless specially specified.
3. Results and Discussion
3.1. Adsorption Energy of Run Atom/Clusters on the BN Support
Before studying the support effect of BN on the N-H bond activation process, the stability of Run clusters on the BN support should be firstly examined. In general, a transition metal center can have more than one accessible spin states which can be close in energy to each other. In particular, as is well known, an isolated Ru atom have a ground state in quintet (spin multiplicity, S =5, denoted as 5Ru) state since its ground state electron configuration is [Kr]4d75s1. However, when a Ru atom interacts with other entities like a second Ru atom to form a Ru2 cluster, or an NH3 molecule for further reaction, it is possible to change its ground state spin multiplicity. The change in spin multiplicity is always not possible to increase, since the incorporation of another entity into a Ru atom lowers its symmetry. Similarly, when a Run cluster interacts with the BN support, the ground spin multiplicity may also change in principle.
Therefore, in order to calculate the adsorption energy of the Ru clusters on BN support one needs to compare the energy of the Run clusters for both of the unsupported and supported cases with different spin multiplicities. All the species notations shown in the first column in Table 1 correspond to their ground states after a similar energy calculation process as the case of Ru1-BN. For instance, 5Ru1-BN is considered as the ground state since its energy is lower than 1Ru1-BN, 3Ru1-BN and 7Ru1-BN. 9Ru3 is considered as the ground state since its energy is lower than 1Ru3, 3Ru3, 5Ru3, 7Ru3, and 11Ru3. In addition, the model BN sheet has a singlet ground state since a triplet BN sheet has a much higher energy.
Figure 2 shows the optimized geometries of 7Ru2-BN and 9Ru3-BN (see Figure 1c as well for 5Ru1-BN). Interestingly the two Ru atoms tend to stand perpendicular rather than lie parallel to the BN plane in 7Ru2-BN. The triangular plane formed from the three Ru atoms also tend to stand perpendicular to the BN plane. Table 1 shown the adsorption energy (Ead < 0) of these Ru clusters on the BN sheet are moderately high, making the adsorption process feasible (Gad < 0).
3.2. Support Effect on the First N-H Bond Activation Process of NH3 on One Ru Atom
3.2.1. Structure
As mentioned above, since Ru1 has a quintet ground state, in this work the reaction behavior of the first N-H bond activation of NH3 on a 5Ru atom was investigated in the beginning. Figure 3a presents the optimized geometries of the species participating in the NH3 and 5Ru reaction (Rxn. (1) as defined in Section 2). The reaction commences with the isolated NH3 molecule and a 5Ru atom, serving as the starting materials (designated as 5SM-Ru1-unsup). As the reaction progresses, the NH3 molecule approaches the 5Ru atom, resulting in the formation of the first energy minimum structure, denoted as 5IM1-Ru1-unsup, with an N…Ru distance of 2.460 Å. Concurrently, the N-H bond containing the detaching H atom (referred to as Ha hereafter) experiences a slight increase in length. Subsequently, 5IM1-Ru1-unsup evolves into another intermediate, 5IM2-Ru1-unsup, via a transition state structure denoted as 5TS-Ru1-unsup. During this transition, Ha detaches from the N atom, moving closer to the Ru atom, while the N atom further approaches Ru. These structural alterations are evident in Figure 3a, where, for instance, the N…Ha distance increases from 1.016 Å in 5IM1-Ru1-unsup to 1.599 Å in 5TS-Ru1-unsup and then to 3.589 Å in 5IM2-Ru1-unsup. In the 5IM2-Ru1-unsup intermediate, the Ru-Ha bond is fully formed, as indicated by its length of 1.680 Å.
Later, the PESs in the singlet, triplet and heptet states (S = 1, 3, and 7, respectively) were also investigated in this work similar to the quintet PES case. Since the energies of the SM, IM1, TS and IM2 species on the singlet and heptet PESs are significantly higher than the corresponding species on the triplet and quintet PESs, the results related to the singlet and heptet PESs will not be reported in this paper. The geometrical characters of the key species on the triplet PES (Figure 3b) are rather similar to the ones on the quintet PES. The primary differences are in that the N…Ru and the N…Ha distances are often shorter in the triplet species than that in the quintet species for Rxn.(1). For example, the N…Ru distance is 1.985 Å in 3TS-Ru1-unsup compared to 2.094 Å in 5TS-Ru1-unsup, and the N…Ha distance is 1.444 Å in 3TS-Ru1-unsup compared to 1.599 Å in 5TS-Ru1-unsup.
As described in Section 3.1, when an isolated Ru atom resides on a model BN surface to form Ru1-BN (see Figure 1c), quintet is still the ground spin state compared to the singlet, triplet and heptet states. Similar to the unsupported case of Rxn. (1) described above, the results related to the singlet and heptet PESs will also not be reported for the supported case of Rxn. (4). Figure 3c,d show that, except the incorporation of the BN support, the geometries of the Ru…NH3 part are somewhat similar to the case of unsupported reaction system, with the main different in that the N…Ha distance in the TS for the supported case is slightly longer than that for the unsupported case. For example, the distance between the detaching atom of Ha and N atom in NH3 in 3TS-Ru1-unsup is 1.444 Å (Figure 2b), which is 1.472 Å in 3TS-Ru1-BN (Figure 2d) when the BN support is added to the reaction system. In addition, the distance between Ha and Ru atoms becomes shorter after adding the model BN support.
3.2.2. Energy Profiles
Figure 4a and Figure 4b show the relative energy profile for Rxns. (1) and (4) undergoing on the quintet and triplet PESs, respectively. For Rxn. (1), 3SM-Ru1-unsup is higher in energy than 5SM-Ru1-unsup, which is not surprising since a Ru atom has a quintet ground state. However, it is noteworthy that the intermediate IM1 has a triplet ground state rather than quintet. In another word, the ground spin state of Ru is changed during the course of a NH3 molecule approaching a Ru atom. More importantly, both the transition state TS and intermediate IM2 display lower energy on the triplet PES compared to the quintet PES. The activation energy of Rxn. (1), defined as the energy difference between TS-Ru1-unsup and IM1-Ru1-unsup, is also lower on the triplet PES (25.6 kcal/mol) than on the quintet PES (38.8 kcal/mol).
Examining the energy profiles presented in Figure 4a reveals that IM1 proceeds much easier to TS on the triplet PES than on the quintet PES for Rxn. (1) (5IM1-Ru1-unusp →5TS-Ru1-unusp vs. 3IM1-Ru1-unusp →3TS-Ru1-unusp). However, considering that 5SM-Ru1-unsup is much more stable than 3SM-Ru1-unsup in energy, in this time it is still insufficient to verify the following hypothesis, named as Hypothesis A, i.e., Rxn. (1) undergoes on the triplet PES. On the basis of energetic data, Hypothesis A is valid if Hypothesis B is valid, that is, 5IM1-Ru1-unsup can undergo spin transition to 3IM1-Ru1-unsup with a low spin transition energy (lower than 5IM1-Ru1-unsup going to 5TS-Ru1-unsup). Exact calculation of such spin transition energy, belonging to a “spin-forbidden” process problem [41,42], can be achieved by the “minimum energy at crossing point (MECP)” method. Although in this work we did not calculate the MECP value for the 5IM1-Ru1-unsup species, through two single-point energy calculations (the results shown in Figure 5, see more explanation in its figure caption as well) and logical reasoning Hypothesis B can be verified. Since the energy difference of b→b’ is the MECP, which is certainly smaller than a→a’ or c→c’ (Eb-b’ < Ea-a’, Eb-b’ < Ec-c’). From Figure 5, the energy different of points a→a’ and c→c’ are 19.6 and 5.5 kcal/mol, respectively, and thus the energy difference of b→b’, or MECP, is lower than 5.5 kcal/mol. This comparison consequently verifies that, 5IM1-Ru1-unsup (actually the same point as c) going to 3IM1-Ru1-unsup (requiring less than 5.5 kcal/mol) is much easier than it going to 5TS-Ru1-unsup (requiring 31.5 kcal/mol, see Figure 3a), that is, Hypothesis B is verified. Therefore, Hypothesis A is also verified.
The verification of Hypothesis A mentioned above shows we can use the concept of the most favorable pathway (MFP) to describe the reaction behavior of Rxn. (1). In the MFP energy profile all key species are considered only with its ground state spin multiplicity. Figure 4c shows the energy profile of the MFP for Rxn. (1) deduced from Figure 4a, and correspondingly, the optimized geometries of the key species involved in the MFP for Rxn. (1) can be seen in Figure 3e.
In this work we have performed similar verification process for all other reactions of Rxns. (2~5). All of the six reactions in this work have an MFP. Hereafter in this paper, the structures and energy profiles are only reported for the MFPs, instead of presenting the results about all of the spin states.
Similar to the unsupported case of Rxn.(1), in Figure 4d shown is the energy profile of the MFP for the BN supported case of Rxn.(4), which is deduced from the two profiles in Figure 4b, and correspondingly, the optimized geometries of the key species involved in the MFP for Rxn. (4) shown in Figure 3f. A comparison between the results in Figure 4c,d reveal that the intermediates of IM1 and IM2 are obviously stabilized by ~ 14 kcal/mol when the BN support is involved. TS is lowered by ~ 15 kcal/mol, leading to the reaction energy barrier is lowered from 25.6 kcal/mol for the Ru1-unsup case to 24.0 kcal/mol for the Ru1-BN case. The reaction barrier of Rxn. (4), i.e., the energy difference between 3IM1-Ru1-BN and 3TS-Ru1-unsup is 24.0 kcal/mol, which is rather consistent with 1.066 eV (24.6 kcal/mol) for CNT supported Ru1, as reported by Zhou et al. [43]. The results in this work and in Ref. 43 show that Ru can be more effective than Pd for N-H bond activation, since the N-H bond activation barrier is 39.4 kcal/mol on Pd(111) [16].
3.2.3. Effect of the BN Model Size
To examine the rationality of the B19N19H16 sheet as a representative model BN sheet, we examined how the reaction barrier of Rxn. (4), i.e., energy difference of 3IM1-Ru1-BN →3TS-Ru1-BN, changed with expanding and reducing the B19N19H16 sheet model. On one hand, we enlarged it to create a B26N26H18 sheet model, and on the other, we reduced it to a B15N15H14 sheet model [21]. These three distinct sheet models were employed as BN support for structural optimization and energy calculations of the intermediates and transition states involved in the N-H bond activation process. The structural characteristics of IM1 and TS derived from these three sheet models exhibited remarkable similarity, as depicted in Figure 3f. Furthermore, the energy results displayed a high degree of consistency. The reaction barrier obtained for the three BN sheet models used as supports are 24.3, 24.0, and 24.3 kcal/mol for B26N26H18, B19N19H16 and B15N15H14, respectively.
These calculations unequivocally demonstrate that the BN models used in this work possess size consistency, thus validating the rationale behind utilizing the B19N19H16 sheet model for BN support calculations. Considering the computational complexity of this work, we ultimately opted for the B19N19H16 sheet model as the BN support for the following calculations.
3.3. Support Effect on the First N-H Bond Activation Process of NH3 on a Ru2 Cluster
Similar to the results about Ru1 shown in Section 3.2, for all key species involved in the NH3 activation process on an unsupported (Rxn. (2)) and supported (Rnx. (5)) Ru2 cluster, the geometry optimization and energy calculation were performed with their spin multiplicities of 1, 3, 5, 7 and 9. A species having a quintet (S = 5) or heptet (S = 7) state is much more stable than it having a singlet, triplet or nonet state. An unsupported Ru2 cluster has a heptet ground state, i.e., 7Ru2. Figure 7a shows the optimized geometries of the key species involved in the reaction between NH3 and an unsupported 7Ru2 cluster (Rxn. (2)) through the MFP. By comparing between the structures shown in Figure 4e and Figure 7a, it is interesting to identify that, on a Ru2 cluster, when the N atom approaches one Ru atom, the detaching H atom, Ha, approaches the two Ru atoms at the same time during the N-H bond activation process. Finally, the NH2 fragment is attached to one Ru atom, and Ha is attached to two Ru atoms to form a trigonal H…Ru…Ru structure in 7IM2-Ru2-unsup.
From the Figure 7b it can be seen that the involvement of support significantly changes the stable structure of IM1 and IM2. The incorporation of the support makes 5IM1-Ru2-BN stabilized in a structure that is closer to the transition state, 5TS-Ru2-BN. The BN support also changes the position of Ha in 7IM2- Ru2-unsup, forming a structure with the NH2 fragment and Ha being at the same side, and Ha is closer to Ru-a than to Ru-b, as shown in Figure 7b.
Figure 7c shows the energy profile for activation process with the MFP on the Ru2-unsup cluster (Rxn.(2)). The SM of this reaction has a heptet ground state, and as the reaction proceeds, the energy of the reaction system is decreased by 20.1 kcal/mol to reach the first energy minimum, 5IM1-Ru2-unsup. With the low-energy spin transition, the barrier required for 5IM1-Ru2-unsup to proceed to the most favorable TS, 3TS-Ru2-unsup, is 20.6 kcal/mol. Finally, with another spin transition, the energy decreases by 30.2 kcal/mol to reach the second energy minimum of 5IM2-Ru2-unsup. Figure 7d shows the energy profile for activation process with the MFP on the Ru2-BN cluster (Rxn.(5)). Comparison between the last two panels in Figure 7, it can be seen that the involvement of the BN support leads to a slight increase of 1.0 kcal/mol in the reaction energy barrier. The reaction energy of the elementary step decreases by 3.4 kcal/mol. The reaction barrier of Rxn.(5), i.e., the energy difference between 5IM1-Ru2-BN and 5TS-Ru2-BN is 21.5 kcal/mol, which is also consistent with 0.830 eV (19.1 kcal/mol) for CNT supported Ru2, as reported by Zhou et al 32.
3.4. Support Effect on the First N-H Bond Activation Process of NH3 on a Ru3 Cluster
Similar to the results about Ru1 and Ru2 shown in Section 3.2 and Section 3.3, respectively, for all key species involved in the NH3 activation process on an unsupported (Rxn. (3)) and supported (Rnx. (6)) Ru3 cluster, the geometry optimization and energy calculation were performed with their spin multiplicities of 1, 3, 5, 7 ,9 and 11. A species having a nonet (S = 9) state is significantly more stable than one with another spin state. An unsupported Ru3 cluster has a nonet ground state, i.e., 9Ru3. Figure 9a illustrates the optimized geometries of the key species involved in the reaction between NH3 and an unsupported 9Ru3 cluster through the MFP (Rxn. (3)). Comparing the structures shown in Figure 4e, Figure 7a and Figure 8a, it can be seen that on a Ru3 cluster, similar to the cases of Ru1 and Ru2, when the N atom approaches one Ru atom, the detaching H atom, Ha approaches one of the Ru atoms at the same time during the N-H bond activation process. Finally, the NH2 fragment is attached to one Ru atom, and Ha is attached to two Ru atoms to form a trigonal H…Ru…Ru structure in 9IM2-Ru3-BN, similar as the Ru2 case.
Compared with Figure 8a, Figure 8b shows that the involvement of the BN support changes the structure of TS by changing the position of Ha atom, from being roughly at a same plane with three Ru atoms to being out of the plane. The involvement of BN also slightly changes the structure of IM2.
Figure 8c shows the energy profile for the N-H bond activation process through the MFP on the Ru3-unsup cluster (Rxn.(3)). The SM of this reaction has a nonet ground state, 9SM-Ru3-unsup, and as the reaction proceeds, the energy of the reaction system is decreased by 32.5 kcal/mol to reach the first energy minimum, 9IM1-Ru3-unsup. As the reaction continues, the energy barrier required for 9IM1-Ru3-unsup to proceed to 9TS-Ru3-unsup is 27.9 kcal/mol. Finally, the energy decreases by 35.7 kcal/mol to reach the second energy minimum, 9IM2-Ru3-unsup. Figure 8d shows the energy profile for the activation process with MFP on the Ru3-BN cluster (Rxn.(6)). From the last two panels in Figure 8, the involvement of the BN support leads to a slight decrease of 0.9 kcal/mol in the reaction energy barrier. The reaction energy of the elementary step decreases by -5.5 kcal/mol.
3.5. Further Discussion on the Support Effect for the First N-H Bond Activation of NH3 on Run (n=1,2,3) Clusters
In order to better understanding the role of the BN support with different Ru cluster sizes, the relative energy and relative free energy at 298.15 K and 673.15 K of all the key species involved in the MFP of the six reactions studied in this work are collected in Figure 9, with their corresponding SMs being chosen as the energetic reference. Based on Figure 9 the support effect on the thermodynamic aspect, kinetic aspect and the size effect aspect can be more clearly seen.
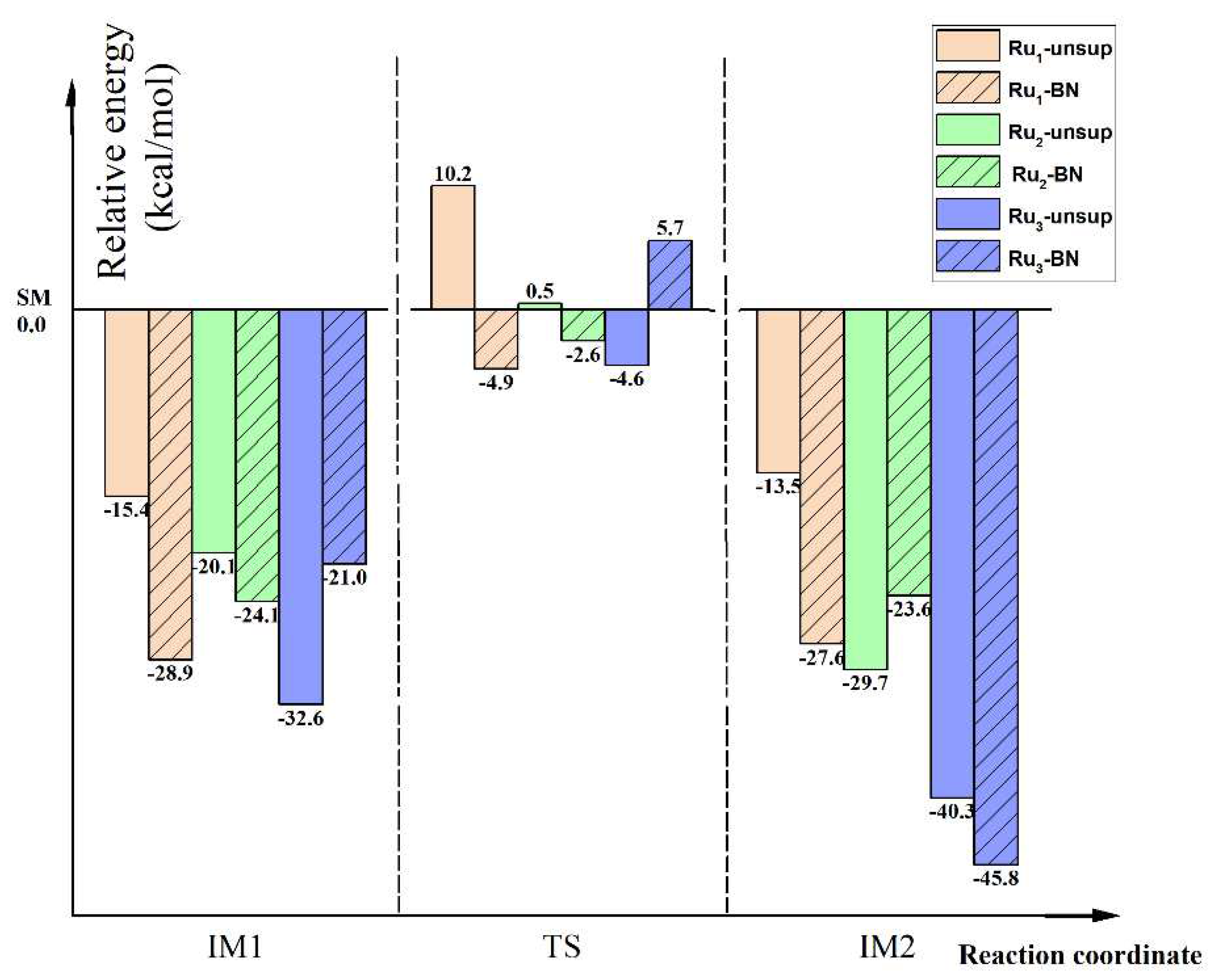
Figure 9.
Relative energy of all key species involved in the MFP of the first N-H bond activation of NH3 on unsupported and BN supported Run (n =1, 2, 3) clusters, as expressed by Rxns.(1~6).
Figure 9.
Relative energy of all key species involved in the MFP of the first N-H bond activation of NH3 on unsupported and BN supported Run (n =1, 2, 3) clusters, as expressed by Rxns.(1~6).
3.5.1. Thermodynamic Aspect of Support Effect
The incorporation of BN favors the stability of IM1 and TS for the Ru1 and Ru2 clusters, and disfavors the stability of IM1 and TS for the Ru3 cluster. Compared to the Ru1 case, the stability of these two states is less favored by BN support for the Ru2 case. From the reaction energy point of view, formation of IM2 from SM is favored by the incorporation of BN for all Run clusters.
3.5.2. Kinetic Aspect of Support Effect
Based on the transition state theory, the relationship between the rate constant (k) and the molar Gibbs free energy of activation (ΔG≠) is expressed as [33]:
where, kB is Boltzmann constant, h Planck constant, T reaction temperature and R the universal gas constant. Since this paper focuses on the support effect and the relative free energy for two similar reactions can be more reliable than the absolute free energy profile for one reaction with the status-of-the-art DFT calculation, the relative rate constant of the supported case over that of the unsupported case was calculated in this work. From Equation (1) the following equation can be easily derived:
k = (kBT/h)·exp(-ΔG≠/RT)
kBN-sup / kunsup = exp[(ΔGunsup≠-ΔGBN-sup≠) / RT]
In Equation (2) the left side term is the relative rate constant, and the subscripts “BN-sup” and “unsup” stand for the reactions involving and not involving the model support, respectively. ΔG≠ can be calculated from the relative free energy of TS over IM1 for all the reactions of Rxn. (1~6). We calculated the relative rate constants at two temperatures of 298.15 K and 673.15 K, since the former is widely concerned in general physical chemistry [44], and the latter is a typical temperature for Ru-catalyzed NH3 decomposition to H2 in practice [4,12,15,24]. By collecting the free energy data at 298.15 and 673.15 K (Table 2) for all of the TSs and IM1s in this work, and based on Equation (2), it can be easily calculated that the relative rate constants, kBN-sup/kunsup, are 20.1, 2.8 and 3.2 for Ru1, Ru2, and Ru3 clusters, respectively, at 298.15 K. These kBN-sup/kunsup values are 775, 9.4 and 272 for Ru1, Ru2, and Ru3 cases, respectively, at 673.15 K. The calculated data indicate that the involvement of the model BN support leads to a great influence on the reaction rate constant for N-H bond activation, especially for the single atom Ru catalyst, and at high reaction temperatures.
Our previous works studied the silica support effect on the C-H bond activation of ethane on a nickel oxide cluster [45]. In that work, the energy of activation of C-H bond activation of ethane on a nickel oxide cluster is increased (instead of decrease in this paper) by the involvement of the silica support. Cao et al. studied the support effect on the Pd-catalyzed semi-hydrogenation of acetylene from the structural and kinetic perspectives [46]. They found that, compared with Al2O3, the CNT support reduced the Pd0 3d binding energy and suppressed the formation of PdHx species to enhance the reaction kinetics in terms of ethylene selectivity and formation rate. These different effects on the energy of activation (consequently the reaction rate constant) may be caused by the difference of the support nature, and the computational study like this work can help researchers in rational selection of good catalytic supports.
3.5.3. Cluster Size Effect
Combined the findings described in Section 3.5.1 & Section 3.5.2, one can have a different view angle of cluster size effect. For the unsupported cases, with using the SM as the energetic reference, the stability of either IM1 or IM2 increases with the order of Ru1 < Ru2 < Ru3. The involvement of BN make the stability order for the IM1 case changes to the order of Ru1 > Ru2 > Ru3, with the stability of IM2 still maintain the order of Ru1 < Ru2 < Ru3.
In the kinetic aspect, the free energy of activation follows an increasing order of Ru2 < Ru1 < Ru3, for both of the unsupported and supported cases, and thus the theoretical rate constant follows the decreasing order of Ru2 > Ru1 > Ru3. However, the degree of influence on the reaction rate constant induced by the BN support follows the order of Ru1 > Ru3 > Ru2.
3.5.4. Support Effect on the Electron Transfer from IM1 to TS
Table 3 shows the NBO charge changes of different moieties for the process of IM1 going to TS. During the IM1→TS process, Run clusters undergo electron loss, while NH2 fragments containing hydrogen atoms (Ha) experience electron gain. The involvement of the BN support lead to a decrease of the electron loss of Ru for the Ru1 case, and in contrast, an increase of the electron loss of Ru for the Ru3 case.
During the IM1→TS process, the Ha atom gains electron for all six cases. The involvement of the BN support leads to a decrease of the electron gain for the Ha atom. At the same time, as can be seen in Table 2, the involvement of the BN support leads to a decrease of N-H bond activation free energy. Put the results of NBO analysis and the reaction energy barrier together, one can further find that the trend of electron transfer of Ha is consistent with the change of reaction energy barrier. The decrease of the reaction barrier introduced by the BN support can be associable with its electron transfer behavior. Our previous works studied the silica support effect on the C-H bond activation of ethane on a nickel oxide cluster [45]. In that work, the energy barrier of C-H bond activation of ethane on a nickel oxide cluster is increased (instead of decrease in this paper) by the involvement of the silica support. Cao et al. studied the support effect on the Pd-catalyzed semi-hydrogenation of acetylene from the structural and kinetic perspectives [46]. They found that, compared with Al2O3, the CNT support reduced the Pd0 3d binding energy and suppressed the formation of PdHx species to enhance the reaction kinetics in terms of ethylene selectivity and formation rate. These different effects on the reaction energy barrier (consequently reaction rate) may be caused by the difference of support nature, and the computational study like this work can help researchers in rational selection of good catalytic supports.
3.5.5. Support Effect on the Spin Conversion Behavior for the MFPs
Table 4 shows the spin multiplicity of intermediates and transition states in the MFP on the Run-unsup and Run-BN clusters. The main changes for the spin states introduced by the incorporation of the BN support are as follows. Firstly, Table 4 shows that the ground states of all SM conform to a regular pattern, with the most favorable spin multiplicity being 2n+3 where “n” is the number of Ru atoms. The involving BN support does not change the ground states of the metal clusters. Second, the changes of the spin multiplicity of the intermediate and transition state in the MFP by the BN support is shown by the fact that the most favorable spin multiplicity is not changed for the n = 1 & 3 cases, and the most favorable spin multiplicity is changed with for the n = 2 case. No spin transition occurs at the Ru3 cases. Finally, the involving BN support changes the spin multiplicity of IM1 and IM2 at n = 2, from 7IM1 to 5IM1 and from 7IM2 to 7IM2.
The current literature shows that the reaction of NH3 decomposition to generate H2 at low temperatures is unsatisfactory. Based on the data obtained, it is reasonable to speculate that the reaction requires high temperature to induce the transition of the spin states in the intermediates and transition states. When the energies of the two spin states are close, various external perturbations like temperature, pressure and magnetic field can induce the spin state transition or crossover [47]. The spin state of the transition metal affects the magnetization strength and thermal conductivity of the material to change the thermoelectric properties of the material [48]. Inspection of the spin state data in Table 4 data shows that state crossover occurs in most cases. So it is reasonable to speculate that a suitable support may help improve the N-H bond activation rate during NH3 decomposition to generate H2 at low temperatures. In a long term, with the aid of computational tools, the findings in this paper will provide a promising direction for designing good catalyst for H2 generation from NH3 decomposition at low temperature.
3.5.6. Preliminary Orbital Analysis
In principle the ground spin multiplicity of a certain species can be explained by the relative energy of the frontier orbitals of this species, namely the highest occupied molecular orbital (HOMO), the singly occupied molecular orbital(s) (SOMO) and the lowest unoccupied molecular orbital (LUMO). In order to better understand the spin transition behavior of some key species involved in the N-H bond activation processes in this work, in the preliminary stage we focused on understanding the ground spin multiplicity of 5SM-Ru1-unsup, 3IM-Ru1-unsup, 5SM-Ru1-BN and 3IM-Ru1-BN. Figure 10 shows the relative energy/energy split of the HOMO, SOMO and LUMO of these four species as well as their orbital contours. Since NH3 is a singlet species, the orbital image of NH3 is not shown for the SMs. As is well known that a Ru atom has a quintet state, that is, having four singly occupied electrons. Figure 10a shows SOMO-1~3 orbitals of 5SM-Ru1-unsup (actually Ru atom) having Ru 4d characters are nearly degenerated, and SOMO-4 having Ru 5s character. The energy split between this Ru 5s orbital and Ru 4d in SOMO-3 is 0.11 atomic unit (a.u.). Figure 10b shows approaching of NH3 to Ru to form3IM-Ru1-unsup making the energy spit of this Ru 5s orbital over the Ru 4d orbital significantly increased (energy split of 0.21 a.u), and thus the electron in SOMO-4 in 5SM-Ru1-unsup tend to occupy SOMO-1 to form an electron pair. Therefore, SOMO-1 in 5SM-Ru1-unsup changes to a new HOMO’ in 3IM-Ru1-unsup, making it having a triplet ground state.
Figure 10c shows that when a BN support approaches the Ru atom to form SM-Ru1-BN, the energy split of the Ru 5s orbital with the Ru 4d orbital in SOMO-3 is only 0.10 a.u., making SOMO-4 still being occupied by an electron in SM-Ru1-BN, thus having a quintet state. Similar reason for why IM1-Ru1-BN having a triplet ground state can be found compared to the case of IM1-Ru1-unsup according to the energy values shown in Figure 10d.
4. Conclusions
To gain deeper insight into the influence of a BN support on the N-H bond activation reaction, the optimized geometries and energetics calculated with the DFT method for the first N-H bond activation of NH3 on unsupported Run clusters and on Run-BN clusters were compared. This DFT study provides the following primary conclusions.
(1) From a geometric standpoint, the incorporation of the BN support does not lead to obvious alterations of the structure of the intermediates and transition states involved in the most favorable pathway (MFP). This is mainly reflected by slight changes of the distance between the Ha and N atoms in NH3 in the TSs, IM1s and IM2s when the unsupported and BN-supported cases are compared.
(2) Considering thermodynamics, the formation of IM2 is favored by the presence of the BN support for all Run clusters. In contrast, the formation of IM1 is favored for the Ru1 and Ru2 cases, and disfavored for the Ru3 case by the presence of BN.
(3) In terms kinetics, the incorporation of the BN support leads to a decrease in the free energy of activation of the first N-H bond activation process of NH3, and thus can improve the reaction rate constant. The rate constant improvement induced by the BN support is more significant at high temperatures.
(4) Spin transition occurs in the MFP in Rxns. (1), (2), (4) and (5) for the Ru1 and Ru2 cases, and no spin transition occurs in the MFP in Rxns. (3) and (6) for the Ru3 cases. The incorporation of the BN support changes the spin transition behavior for the Ru2 cluster during the first N-H bond activation of NH3.
The spin transition behavior connecting to the single and gemini-Ru atom catalysts underscores the importance of considering spin transition behavior when choosing catalytic supports, particularly in the field of single atom catalysis.
Our study contributes to a deeper understanding of the N-H bond activation process in catalytic NH3 decomposition. These insights offer valuable guidance for selecting more favorable catalytic supports in order to synthesize better catalysts. We will continue to carry out further works to provide better theoretical guidance for the design of efficient catalysts for H2 production via NH3 decomposition.
Author Contributions
L. Zhao, help idea development, computational job running, data collection and analysis, manuscript writing; H. Zhuang, discussion, paper revision, picture drawing; Y. Zhang, computational job running, data collection and analysis; L. Ma, discussion and suggestion, Y. Xi, discussion and suggestion; X. Lin, idea development, job running, paper organization and revision, funding provision.
Acknowledgments
Support from the National Natural Science Foundation of China (21576291, 22003076) and from the Fundamental Research Funds for the Central Universities (23CX03007A 22CX06012A) is gratefully acknowledged.
Conflicts of Interest
The authors declare no competing financial interest.
References
- Bockris, J. O. M. The hydrogen economy: Its history. Int J Hydrogen Energ 2013, 38, 2579–2588. [Google Scholar] [CrossRef]
- Staffell, I.; Scamman, D.; Abad, A. V.; Balcombe, P.; Dodds, P. E.; Ekins, P.; Shah, N.; Ward, K. R. The role of hydrogen and fuel cells in the global energy system. Energ Environ Sci 2019, 12, 463–491. [Google Scholar] [CrossRef]
- Pandev, M.; Lucchese, P.; Mansilla, C.; Le Duigou, A.; Abrashev, B.; Vladikova, D. Hydrogen Economy: the future for a sustainable and green society. Bulg Chem Commun 2017, 49, 84–92. [Google Scholar]
- Lucentini, I.; Garcia, X.; Vendrell, X.; Llorca, J. Review of the Decomposition of Ammonia to Generate Hydrogen. Industrial & Engineering Chemistry Research 2021, 60, 18560–18611. [Google Scholar] [CrossRef]
- Yi, Y.; Wang, L.; Guo, Y.; Sun, S.; Guo, H. Plasma-Assisted ammonia decomposition over Fe–Ni alloy catalysts for CO x -Free hydrogen. AIChE Journal 2018. [Google Scholar] [CrossRef]
- Hu, Z.-P.; Weng, C.-C.; Chen, C.; Yuan, Z.-Y. Two-dimensional mica nanosheets supported Fe nanoparticles for NH3 decomposition to hydrogen. Molecular Catalysis 2018, 448, 162–170. [Google Scholar] [CrossRef]
- Hu, Z.-P.; Chen, L.; Chen, C.; Yuan, Z.-Y. Fe/ZSM-5 catalysts for ammonia decomposition to COx-free hydrogen: Effect of SiO2/Al2O3 ratio. Molecular Catalysis 2018, 455, 14–22. [Google Scholar] [CrossRef]
- Pinzón, M.; Ruiz-López, E.; Romero, A.; de la Osa, A. R.; Sánchez, P.; de Lucas-Consuegra, A. Electrochemical activation of Ru catalyst with alkaline ion conductors for the catalytic decomposition of ammonia. Molecular Catalysis 2021, 511. [Google Scholar] [CrossRef]
- Li, G.; Yu, X.; Lei, Z.; Yin, F.; Zhang, H.; He, X. Preparation of Lanthanum Hexaaluminate Supported Nickel Catalysts for Hydrogen Production by Ammonia Decomposition. Catalysis Letters 2022. [Google Scholar] [CrossRef]
- Maleki, H.; Bertola, V. Co–Ce–Al–O mesoporous catalysts for hydrogen generation via ammonia decomposition. Int J Hydrogen Energ 2022. [Google Scholar] [CrossRef]
- Qiu, Y.; Fu, E.; Gong, F.; Xiao, R. Catalyst support effect on ammonia decomposition over Ni/MgAl2O4 towards hydrogen production. Int J Hydrogen Energ 2022, 47, 5044–5052. [Google Scholar] [CrossRef]
- Liu, P.; Sun, L.; Zhang, Z.; Wang, X.; Zhang, Y.; Yang, X. Hydrogen production from ammonia decomposition catalyzed by Ru nano-particles in alkaline molecular sieves under photothermal conditions. Molecular Catalysis 2023, 543. [Google Scholar] [CrossRef]
- Chellappa, A. S.; Fischer, C. M.; Thomson, W. J. Ammonia decomposition kinetics over Ni-Pt/Al2O3 for PEM fuel cell applications. Appl Catal a-Gen 2002, 227, 231–240. [Google Scholar] [CrossRef]
- Hengwei. , Y. C. Z. K. W. J. Z. z. B. N-H bond activation of ammonia for catalytic organic reactions (chinese). Chemical World 2019, 60, 553–560. [Google Scholar] [CrossRef]
- Almquist, C. C.; Removski, N.; Rajeshkumar, T.; Gelfand, B. S.; Maron, L.; Piers, W. E. Spontaneous Ammonia Activation Through Coordination-Induced Bond Weakening in Molybdenum Complexes of a Dianionic Pentadentate Ligand Platform. Angew Chem Int Ed Engl 2022, 61, e202203576. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Pan, Q.; Li, M.; Yan, T.; Fang, T. Density functional theory study on direct catalytic decomposition of ammonia on Pd (111) surface. Applied Surface Science 2014, 292, 494–499. [Google Scholar] [CrossRef]
- Duan, X.; Ji, J.; Qian, G.; Fan, C.; Zhu, Y.; Zhou, X.; Chen, D.; Yuan, W. ; Ammonia decomposition on Fe(110), Co(111) and Ni(111) surfaces: A density functional theory study. Journal of Molecular Catalysis A: Chemical 2012, 357, 81–86. [Google Scholar] [CrossRef]
- Jiang, Z.; Qin, P.; Fang, T. ; Mechanism of ammonia decomposition on clean and oxygen-covered cu (1 1 1) surface: a dft study. Chemical Physics 2014, 445, 59–67. [Google Scholar] [CrossRef]
- Jiang, Z.; Fang, T. ; Probing the effect of Pd coverage towards NH3 decomposition on Cu(100) surface. Chemical Physics Letters 2019, 729, 30-31. [Google Scholar] [CrossRef]
- Rao, X.; Lou, Y.; Zhou, Y.; Zhang, J.; Zhong, S. ; First-principles insights into ammonia decomposition on WC (0001) surface terminated by W and C. Applied Surface Science 2021, 566, 150635. [Google Scholar] [CrossRef]
- Takahashi, A.; Fujitani, T. , Kinetic Analysis of Decomposition of Ammonia over Nickel and Ruthenium Catalysts. Journal of Chemical Engineering of Japan, 2016, 49, 22–28. [Google Scholar] [CrossRef]
- Nakamura, I.; Fujitani, T. Role of metal oxide supports in NH3 decomposition over Ni catalysts. Appl Catal a-Gen 2016, 524, 45–49. [Google Scholar] [CrossRef]
- Garcia-Garcia, F. R.; Guerrero-Ruiz, A.; Rodriguez-Ramos, I.; Goguet, A.; Shekhtman, S. O.; Hardacre, C. TAP studies of ammonia decomposition over Ru and Ir catalysts. Phys Chem Chem Phys 2011, 13, 12892–12899. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Jiang, Q.; Zhao, D.; Cao, T.; Sha, H.; Zhang, C.; Song, H.; Da, Z. Ammonia as hydrogen carrier: Advances in ammonia decomposition catalysts for promising hydrogen production. Renewable and Sustainable Energy Reviews 2022, 169. [Google Scholar] [CrossRef]
- Zielinski, M.; Janiszewska, E.; Drewniak, A.; Pietrowski, M. ; Methanation of CO2 over Ruthenium Supported on Alkali-Modified Silicalite-1 Catalysts. Molecules 2023, 28, 6376. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Chen, C.; Bian, C.; Ren, J.; Liu, J.; Huang, W. ; Enhanced Ammonia Decomposition by Tuning the Support Properties of Ni/GdxCe1-xO2-δ at 600 ◦C. Molecules 2023, 28, 2750. [Google Scholar] [CrossRef] [PubMed]
- Anota, E. C.; Cocoletzi, G. H.; Tapia, A. M. G. Armchair Boron Nitride nanotubes—heterocyclic molecules interactions: A computational description. Open Chem. 2015, 13, 734–742. [Google Scholar] [CrossRef]
- Bautista, M. C. F.; Cortés-Arriagada, D.; Shakerzadeh, E.; Anota, E. C. Acetylsalicylic acid interaction with Boron nitride nanostructures – A density functional analysis. Journal of Molecular Liquids 2022, 355, 118980. [Google Scholar] [CrossRef]
- Anota, E. C. 2D boron nitride incorporating homonuclear boron bonds: stabilized in neutral, anionic and cationic charge. SN Applied Sciences 2022, 4, 295. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts 2007, 120, 215-241. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. The Journal of Chemical Physics 2010, 132. [Google Scholar] [CrossRef] [PubMed]
- Larijani, H. T.; Jahanshahi, M.; Ganji, M. D.; Kiani, M. H. Computational studies on the interactions of glycine amino acid with graphene, h-BN and h-SiC monolayers. Physical Chemistry Chemical Physics 2017, 19, 1896–1908. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Saeed, M.; Mehboob, M. Y.; Khan, S. U.; Usman Khan, M.; Adnan, M.; Ahmed, M.; Iqbal, J.; Ayub, K. Density functional theory study of palladium cluster adsorption on a graphene support. RSC Advances 2020, 10, 20595–20607. [Google Scholar] [CrossRef] [PubMed]
- Granatier, J.; Lazar, P.; Prucek, R.; Šafářová, K.; Zbořil, R.; Otyepka, M.; Hobza, P. Interaction of Graphene and Arenes with Noble Metals. The Journal of Physical Chemistry C 2012, 116, 14151–14162. [Google Scholar] [CrossRef]
- Shakourian-Fard, M.; Kamath, G.; Jamshidi, Z. Trends in Physisorption of Ionic Liquids on Boron-Nitride Sheets. The Journal of Physical Chemistry C 2014, 118, 26003–26016. [Google Scholar] [CrossRef]
- Kozuch, S.; Shaik, S. How to conceptualize catalytic cycles? The energetic span model. Acc Chem Res 2017, 44, 101–110. [Google Scholar] [CrossRef] [PubMed]
- [Gaussian 09B01] M. J. Frisch, G. W. T., H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010.
- M. J. Frisch, G. W. T., H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J.Fox. Gaussian 09, Revision D.01. Gaussian, Inc, 2013.
- Gonzalez, C.; Schlegel, H. B. An improved algorithm for reaction path following. J Chem Phys 90:2154. Journal of Chemical Physics 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- K. L. Schuchardt, B. T. D., T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li, T.L. Windus,. Basis set exchange: a community database for computational sciences. J. Chem. Inf. Model 2007, 47, 1045–1052. [CrossRef]
- Harvey, J. N.; Aschi, M. Spin-forbidden dehydrogenation of methoxy cation: a statistical view. Physical Chemistry Chemical Physics 1999, 1, 5555–5563. [Google Scholar] [CrossRef]
- Poli, R.; Harvey, J. N. Spin forbidden chemical reactions of transition metal compounds. New ideas and new computational challenges. Chemical Society Reviews 2002, 32, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Lin, S.; Guo, H. First-Principles Insights into Ammonia Decomposition Catalyzed by Ru Clusters Anchored on Carbon Nanotubes: Size Dependence and Interfacial Effects. The Journal of Physical Chemistry C 2018, 122, 9091–9100. [Google Scholar] [CrossRef]
- P. Atkins, J. d. P. Atkins’ Physical Chemistry (Seventh Edition). Oxford University Press 2022. DOI: in chapter 27, molecular reaction dynamics, 944-976.
- Lin, X.; Xi, Y.; Phillips, D. L.; Guo, W. The effect of a silica support: a density functional theory study of the C-H bond activation of ethane on a nickel oxide cluster. Journal of Physical Organic Chemistry 2016, 29, 134–144. [Google Scholar] [CrossRef]
- Cao, Y.; Ge, X.; Li, Y.; Si, R.; Sui, Z.; Zhou, J.; Duan, X.; Zhou, X. Structural and Kinetics Understanding of Support Effects in Pd-Catalyzed Semi-Hydrogenation of Acetylene. Engineering 2021, 7, 103–110. [Google Scholar] [CrossRef]
- Gütlich, P.; Garcia, Y.; Goodwin, H. A. Spin crossover phenomena in Fe(II) complexes. Chemical Society Reviews 2000, 29, 419–427. [Google Scholar] [CrossRef]
- Terasaki, I.; Shibasaki, S.; Yoshida, S.; Kobayashi, W. Spin State Control of the Perovskite Rh/Co Oxides. Materials 2010, 3, 786–799. [Google Scholar] [CrossRef]
Scheme 1.
A reaction mechanism described by Sun in Ref. 24 and the references therein.
Figure 1.
Vertical (a) and side (b) views of the model BN of a B19N19H16 sheet, and the5Ru1-BN model (c). The Ru atom is located close to the indicated N atom with a distance of 3.562 Å. For the B and N atom in the cycle high level basis set of 6-311G** was used for calculation as described in the text. Color for each atoms: N(blue), H(white), Ru(green), B(pink), see the inset over panel (b).
Figure 1.
Vertical (a) and side (b) views of the model BN of a B19N19H16 sheet, and the5Ru1-BN model (c). The Ru atom is located close to the indicated N atom with a distance of 3.562 Å. For the B and N atom in the cycle high level basis set of 6-311G** was used for calculation as described in the text. Color for each atoms: N(blue), H(white), Ru(green), B(pink), see the inset over panel (b).
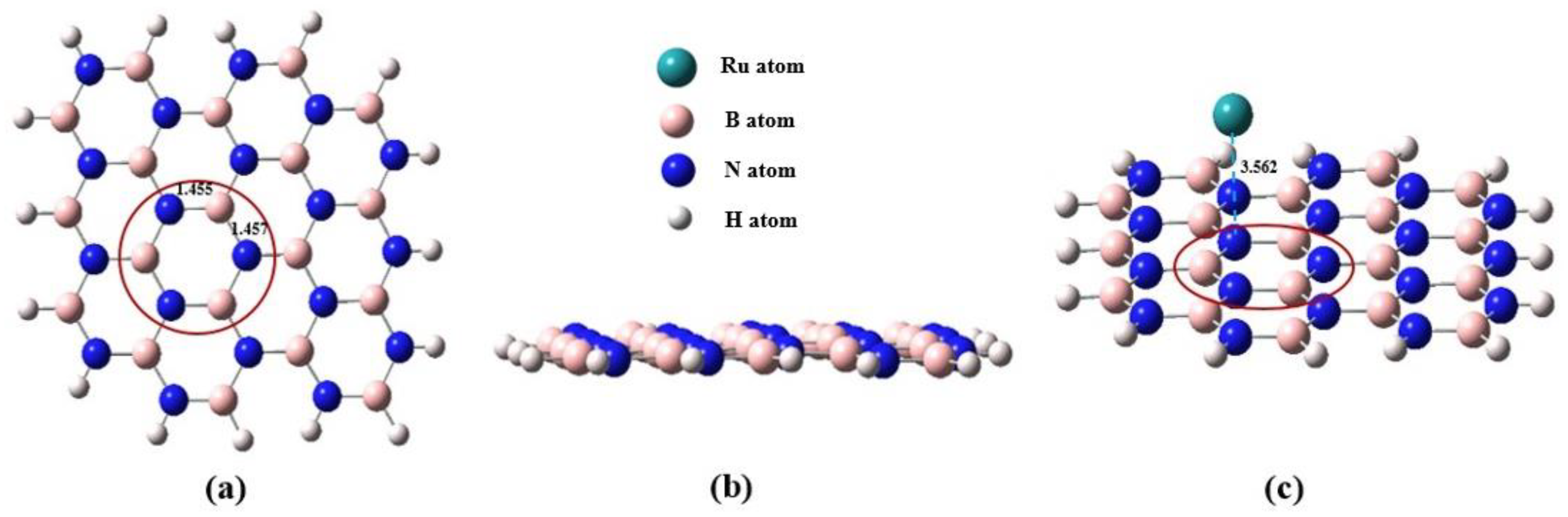
Figure 2.
Side view of the model BN supported 7Ru2 (a, 7Ru2-BN) and 9Ru3 (b, 9Ru3-BN) clusters. Key distances are indicated in Å.
Figure 2.
Side view of the model BN supported 7Ru2 (a, 7Ru2-BN) and 9Ru3 (b, 9Ru3-BN) clusters. Key distances are indicated in Å.
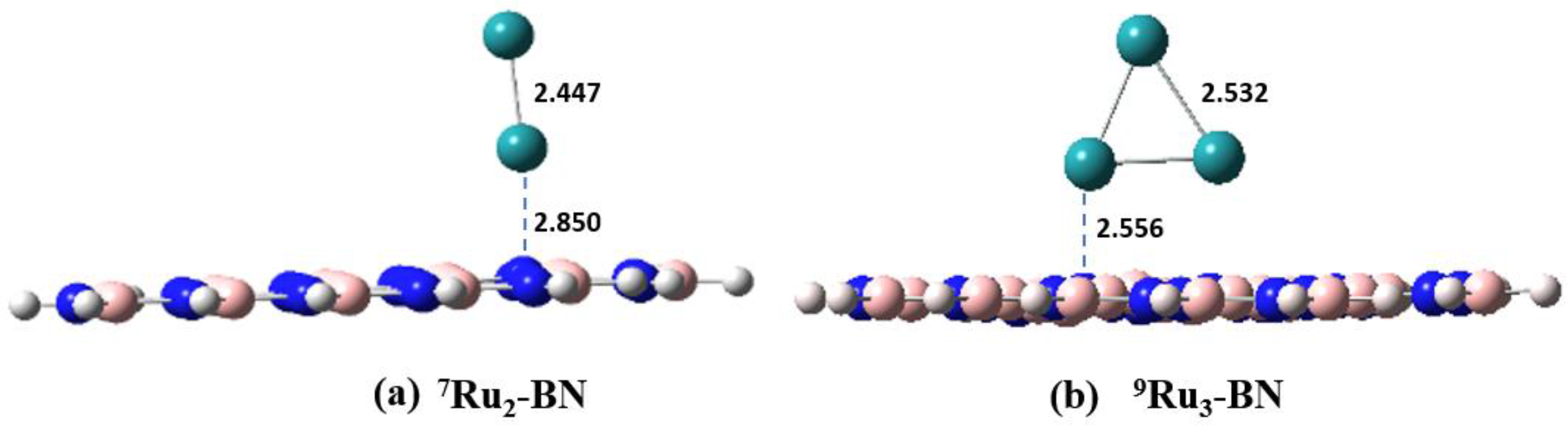
Figure 3.
Shown are the optimized geometries of the key species involved in the first N-H bond activation of NH3 on unsupported Ru1 (Rxn.(1) with suffix of -Ru1-unsup) and the model BN-supported Ru1 (Rxn.(4) with suffix of -Ru1-BN). Panels (a) and (b) illustrate the unsupported cases on the quintet and triplet PES, respectively, while (c) and (d) demonstrate the supported cases on the quintet and triplet PES. Panel (e) show the cases of Rxn. (1) with the most favorable pathway (MFP), and (f) for Rxn. (4). All distances are indicated in Å.
Figure 3.
Shown are the optimized geometries of the key species involved in the first N-H bond activation of NH3 on unsupported Ru1 (Rxn.(1) with suffix of -Ru1-unsup) and the model BN-supported Ru1 (Rxn.(4) with suffix of -Ru1-BN). Panels (a) and (b) illustrate the unsupported cases on the quintet and triplet PES, respectively, while (c) and (d) demonstrate the supported cases on the quintet and triplet PES. Panel (e) show the cases of Rxn. (1) with the most favorable pathway (MFP), and (f) for Rxn. (4). All distances are indicated in Å.
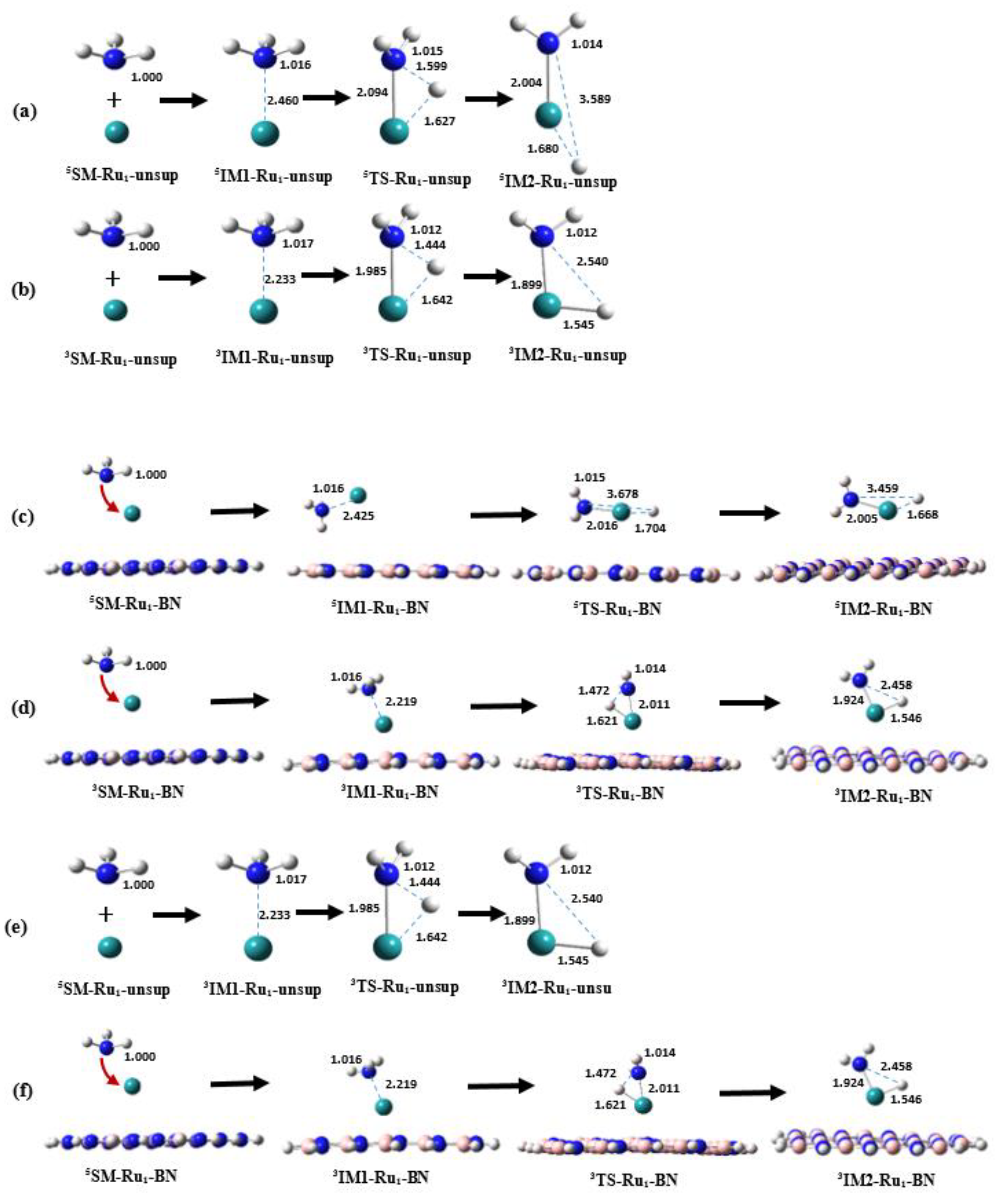
Figure 4.
Shown is the relative energy profiles for the first N-H bond activation of NH3 on one Ru atom for the unsupported (Rxn. (1) with suffix of -Ru1-unsup) and supported (Rxn.(4) with suffix of -Ru1-BN). The relative energy profiles on the triplet (green) and quintet (blue) PESs are included in panel (a) for Rxn.(1) and in panel (b) for Rxn. (4). on Ru1-unsup (a) and Ru1-BN (b). The relative energy profiles of the most favorable pathway (MPF) are shown in panel (c) for Rxn.(1) and in panel (d) for Rxn.(4).
Figure 4.
Shown is the relative energy profiles for the first N-H bond activation of NH3 on one Ru atom for the unsupported (Rxn. (1) with suffix of -Ru1-unsup) and supported (Rxn.(4) with suffix of -Ru1-BN). The relative energy profiles on the triplet (green) and quintet (blue) PESs are included in panel (a) for Rxn.(1) and in panel (b) for Rxn. (4). on Ru1-unsup (a) and Ru1-BN (b). The relative energy profiles of the most favorable pathway (MPF) are shown in panel (c) for Rxn.(1) and in panel (d) for Rxn.(4).
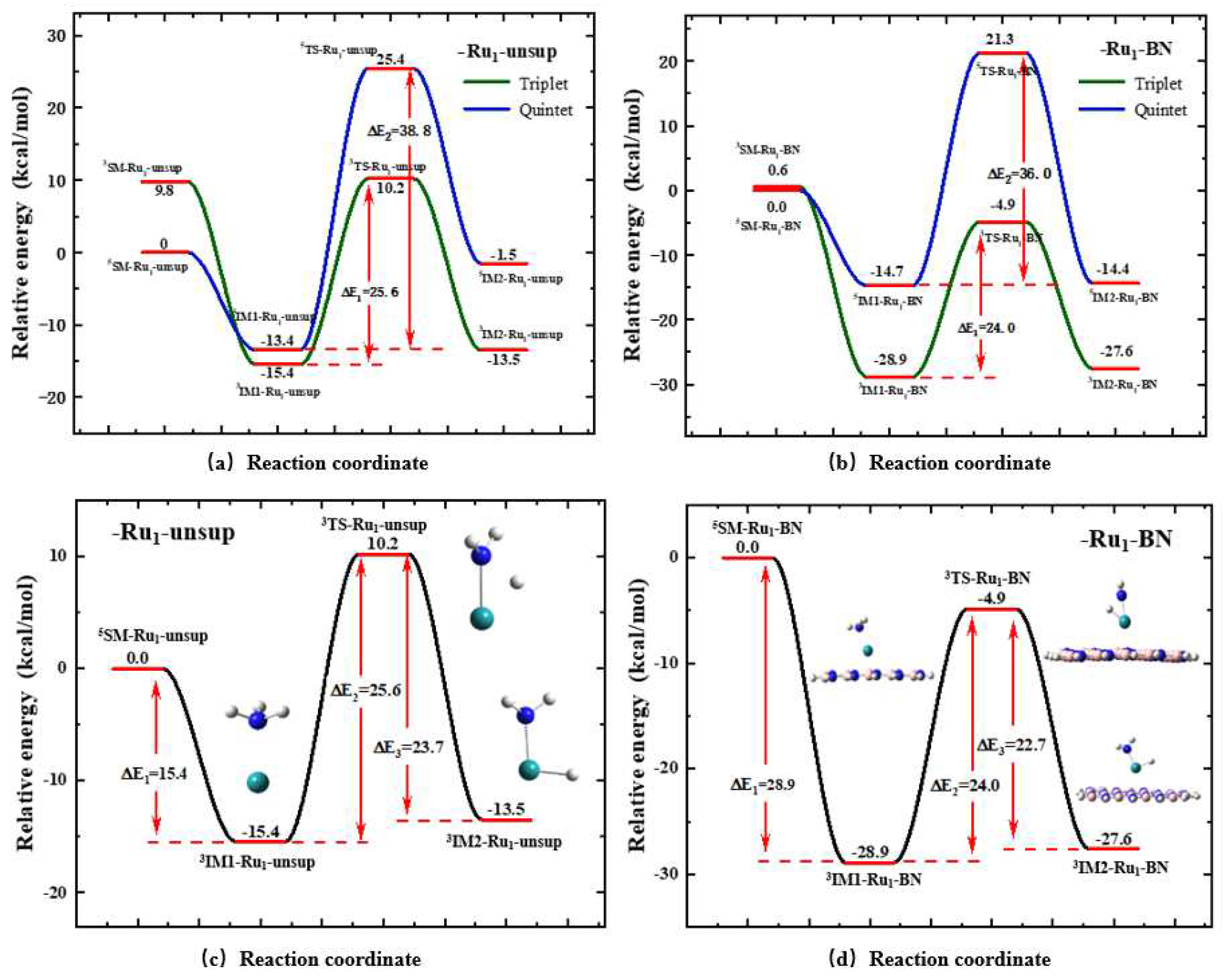
Figure 5.
Shown is a simple schematic illustration for the landscape of the energy surface near the energy minimum of triplet and quintet IM1-Ru1-unsup in Rxn. (1). Point a, the energy minimum on the triplet PES, represents the energy of optimized 3IM1-Ru1-unsup. Point a′ represents the energy of IM1-Ru1-unsup in quintet state while having the same geometry as point a. The energy difference of a and a’ is the Frank-Condon excitation energy (EFC) at point a. Point b represents the geometry and the energy of 3IM1-Ru1-unsup where the spin transition occurs with the largest probability. The energy difference of b and b’ represents the minimum energy at cross point (MECP). Point c is the energy of optimized 5IM1-Ru1-unsup. Point c′ represents the energy of IM1-Ru1-unsup in triplet state while having the same geometry as point c. The energy difference of c and c’ is the EFC at point c, with the geometry of optimized 5IM1-Ru1-unsup.
Figure 5.
Shown is a simple schematic illustration for the landscape of the energy surface near the energy minimum of triplet and quintet IM1-Ru1-unsup in Rxn. (1). Point a, the energy minimum on the triplet PES, represents the energy of optimized 3IM1-Ru1-unsup. Point a′ represents the energy of IM1-Ru1-unsup in quintet state while having the same geometry as point a. The energy difference of a and a’ is the Frank-Condon excitation energy (EFC) at point a. Point b represents the geometry and the energy of 3IM1-Ru1-unsup where the spin transition occurs with the largest probability. The energy difference of b and b’ represents the minimum energy at cross point (MECP). Point c is the energy of optimized 5IM1-Ru1-unsup. Point c′ represents the energy of IM1-Ru1-unsup in triplet state while having the same geometry as point c. The energy difference of c and c’ is the EFC at point c, with the geometry of optimized 5IM1-Ru1-unsup.

Figure 6.
Shown are the optimized geometries of IM1 and TS involved in the first N-H bond activation of NH3 on the varied model BN-supported Ru1 (Rxn.(4) with different sizes of BN model sheet). (a) 3IM1-Ru1-BN with a B26N26H18 model in vertical view, (b) 3IM1-Ru1-BN with a B26N26H18 model in side view, (c) 3TS-Ru1-BN with a B26N26H18 model in side view, (d) 3IM1-Ru1-BN with a B15N15H14 model in vertical view, (e) 3IM1-Ru1-BN with a B15N15H14 model in side view, and (f) 3TS-Ru1-BN with a B15N15H14 model in side view.
Figure 6.
Shown are the optimized geometries of IM1 and TS involved in the first N-H bond activation of NH3 on the varied model BN-supported Ru1 (Rxn.(4) with different sizes of BN model sheet). (a) 3IM1-Ru1-BN with a B26N26H18 model in vertical view, (b) 3IM1-Ru1-BN with a B26N26H18 model in side view, (c) 3TS-Ru1-BN with a B26N26H18 model in side view, (d) 3IM1-Ru1-BN with a B15N15H14 model in vertical view, (e) 3IM1-Ru1-BN with a B15N15H14 model in side view, and (f) 3TS-Ru1-BN with a B15N15H14 model in side view.
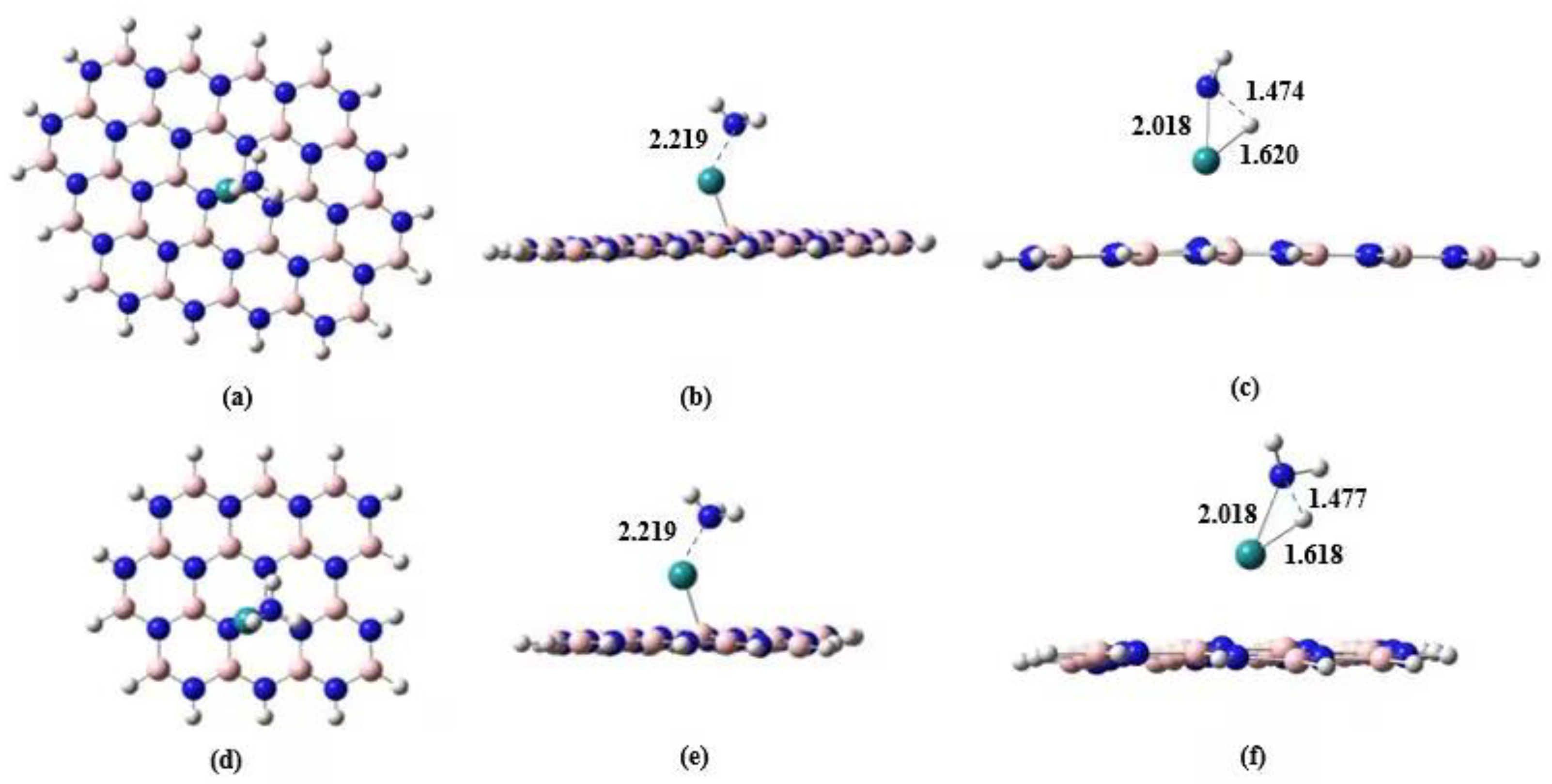
Figure 7.
Shown are the optimized geometries of the key species involved in the first N-H bond activation of NH3 on unsupported Ru2 (Rxn.(2) with suffix of –Ru2-unsup) and the model BN-supported Ru2 (Rxn.(5) with suffix of –Ru2-BN) cluster and their relative energy profiles. Panel (a) is for Rxn.(2) with the most favorable pathway (MFP), and (b) for Rxn.(5) with the most favorable pathway (MFP). The key distances are indicated in Å. The two Ru atoms are named as Ru-a (indicated with *) and Ru-b in this part for more convenient elaboration. Panels (c) and (d) are the relative energy profiles for the first N-H bond activation of NH3 on a Ru2-unsup (Rxn.(2)) and on a Ru2-BN cluster (Rxn.(5)) with the most favorable pathway (MFP).
Figure 7.
Shown are the optimized geometries of the key species involved in the first N-H bond activation of NH3 on unsupported Ru2 (Rxn.(2) with suffix of –Ru2-unsup) and the model BN-supported Ru2 (Rxn.(5) with suffix of –Ru2-BN) cluster and their relative energy profiles. Panel (a) is for Rxn.(2) with the most favorable pathway (MFP), and (b) for Rxn.(5) with the most favorable pathway (MFP). The key distances are indicated in Å. The two Ru atoms are named as Ru-a (indicated with *) and Ru-b in this part for more convenient elaboration. Panels (c) and (d) are the relative energy profiles for the first N-H bond activation of NH3 on a Ru2-unsup (Rxn.(2)) and on a Ru2-BN cluster (Rxn.(5)) with the most favorable pathway (MFP).
Figure 8.
Shown are the optimized geometries of the key species involved in the first N-H bond activation of NH3 on unsupported Ru3 (Rxn.(3) with suffix of –Ru3-unsup) and the model BN-supported Ru3 (Rxn.(6) with suffix of –Ru3-BN) cluster and their relative energy profiles. Panel (a) is for Rxn.(3) with the most favorable pathway (MFP), and (b) for Rxn.(6) with the MFP. Ru atom indicated with “*” is closed to the N atom in NH3. Panels (c) and (d) are the relative energy profiles for the first N-H bond activation of NH3 on a Ru3-unsup (Rxn.(3)) and on a Ru3-BN cluster (Rxn.(6)) with the most favorable pathway (MFP).
Figure 8.
Shown are the optimized geometries of the key species involved in the first N-H bond activation of NH3 on unsupported Ru3 (Rxn.(3) with suffix of –Ru3-unsup) and the model BN-supported Ru3 (Rxn.(6) with suffix of –Ru3-BN) cluster and their relative energy profiles. Panel (a) is for Rxn.(3) with the most favorable pathway (MFP), and (b) for Rxn.(6) with the MFP. Ru atom indicated with “*” is closed to the N atom in NH3. Panels (c) and (d) are the relative energy profiles for the first N-H bond activation of NH3 on a Ru3-unsup (Rxn.(3)) and on a Ru3-BN cluster (Rxn.(6)) with the most favorable pathway (MFP).
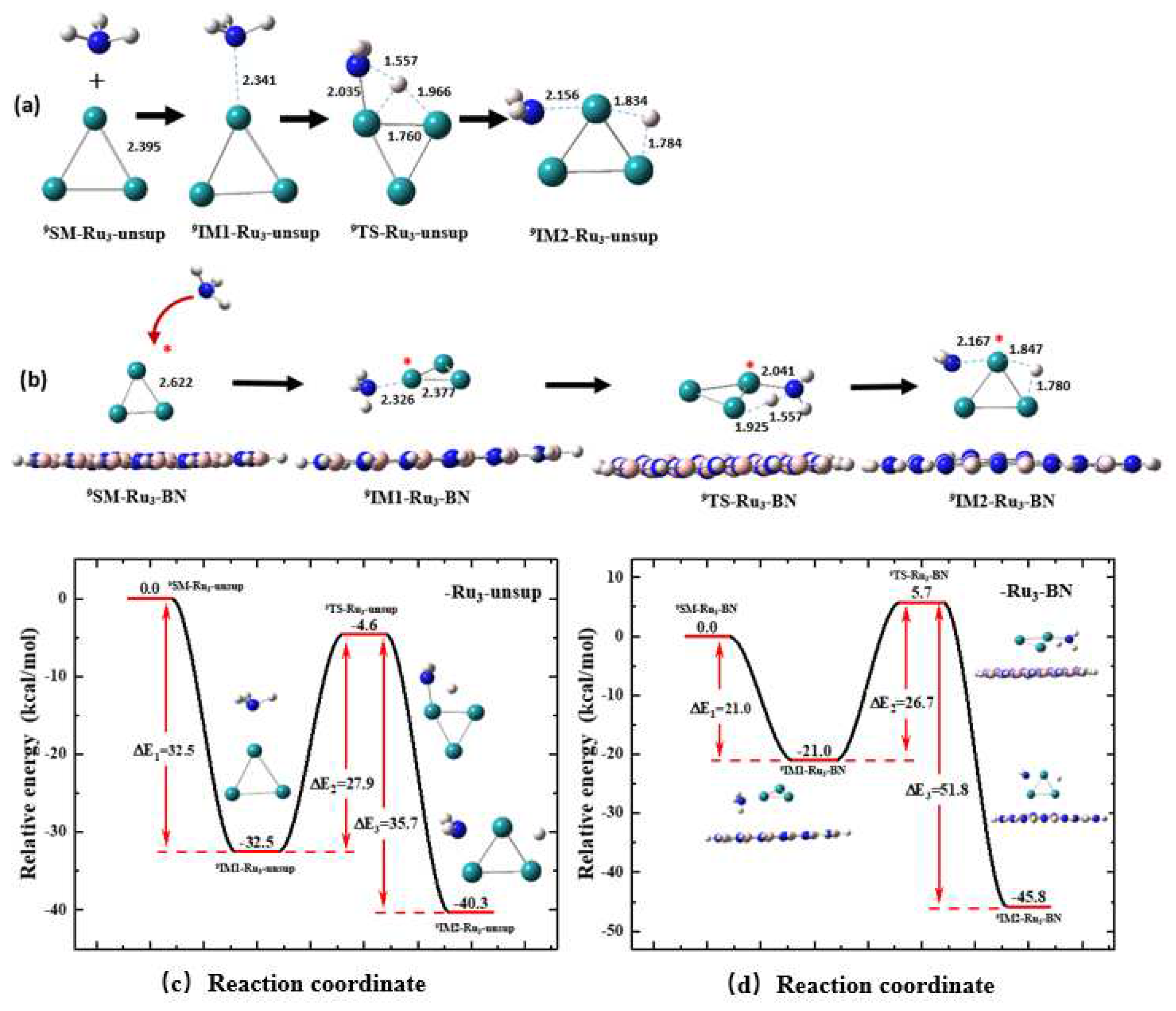
Figure 10.
The molecular orbital contours and the relative orbital energy (in atomic unit, a.u.) of some selected frontier orbitals (see the text for detail) of 5SM-Ru1-unsup (a), 3IM-Ru1-unsup (b), 5SM-Ru1-BN (c) and 3IM-Ru1-BN (d). For the relative orbital energies, the HOMO energy is selected as the energetic reference (0 atomic unit, a.u.). Since NH3 is a singlet species, the orbital image of NH3 is not shown for the SMs.
Figure 10.
The molecular orbital contours and the relative orbital energy (in atomic unit, a.u.) of some selected frontier orbitals (see the text for detail) of 5SM-Ru1-unsup (a), 3IM-Ru1-unsup (b), 5SM-Ru1-BN (c) and 3IM-Ru1-BN (d). For the relative orbital energies, the HOMO energy is selected as the energetic reference (0 atomic unit, a.u.). Since NH3 is a singlet species, the orbital image of NH3 is not shown for the SMs.
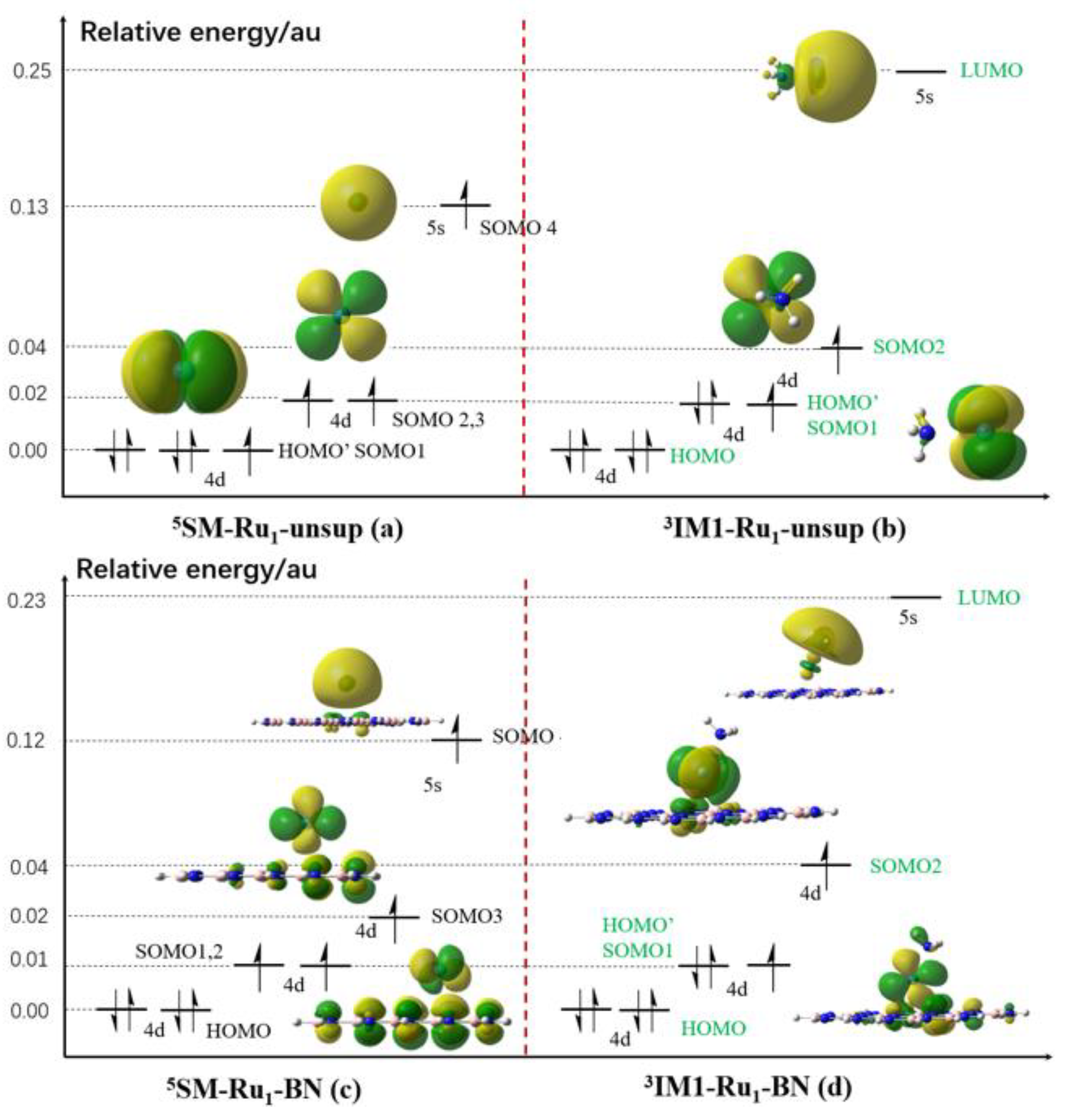

Table 1.
Adsorption energy (Ead) and free energy at 298 K (Gad) of a Run cluster on the BN support. All the spin multiplicity indicated on the upper left of a species notation corresponds to the ground spin state.
Table 1.
Adsorption energy (Ead) and free energy at 298 K (Gad) of a Run cluster on the BN support. All the spin multiplicity indicated on the upper left of a species notation corresponds to the ground spin state.
| Adsorption process | Ead (kcal/mol) | Gad (kcal/mol, 298 K) |
|---|---|---|
| 5Ru1 + 1BN → 5Ru1-BN | -11.7 | -4.4 |
| 7Ru2 + 1BN → 7Ru2-BN | -29.0 | -6.4 |
| 9Ru3 + 1BN → 9Ru3-BN | -32.2 | -10.6 |
Table 2.
Free energy of activations, ΔG≠ (in kcal/mol), at 298.15 and 673.15 K for all the six N-H bond activation reactions (Rxns. 1~6) studied in this work.
Table 2.
Free energy of activations, ΔG≠ (in kcal/mol), at 298.15 and 673.15 K for all the six N-H bond activation reactions (Rxns. 1~6) studied in this work.
| N-H activation of NH3 on | Unsupported, 298.15 K | BN-supported, 298.15 K | Unsupported, 673.15 K | BN-supported, 673.15 K |
|---|---|---|---|---|
| Ru1 atom | 26.0 | 24.1 | 25.8 | 16.9 |
| Ru2 cluster | 21.8 | 21.1 | 24.0 | 21.0 |
| Ru3 cluster | 29.8 | 28.9 | 32.5 | 25.0 |
Table 3.
NBO charge change of different moieties between the most favorable IM1 and TS involved in the first N-H bond activation process of NH3 on Run-unsup and Run-BN clusters (IM1→TS).
Table 3.
NBO charge change of different moieties between the most favorable IM1 and TS involved in the first N-H bond activation process of NH3 on Run-unsup and Run-BN clusters (IM1→TS).
| NBO charge change of different moieties | |||||
|---|---|---|---|---|---|
| Ha | N | NH2 | Run | B19N19H16 | |
| Ru1-unsup | -0.214 | -2.207 | -2.204 | 0.418 | 0.000 |
| Ru1@BN | -0.190 | 0.021 | -0.055 | 0.086 | 0.159 |
| Ru2-unsup | -0.283 | -0.037 | -0.056 | 0.339 | 0.000 |
| Ru2@BN | -0.251 | -0.098 | -0.146 | 0.372 | 0.025 |
| Ru3-unsup | -0.274 | -0.024 | -0.058 | 0.332 | 0.000 |
| Ru3@BN | -0.240 | -0.087 | -0.130 | 0.417 | -0.047 |
Table 4.
Spin multiplicity of intermediates and transition states in most favorable pathways for the first N-H bond activation of NH3 with and without the BN support.
Table 4.
Spin multiplicity of intermediates and transition states in most favorable pathways for the first N-H bond activation of NH3 with and without the BN support.
| most favorable spin state | ||||
|---|---|---|---|---|
| SM | IM1 | TS | IM2 | |
| Ru1-unsup | 5 | 3 | 3 | 3 |
| Ru1-BN | 5 | 3 | 3 | 3 |
| Ru2-unsup | 7 | 7 | 5 | 7 |
| Ru2-BN | 7 | 5 | 5 | 5 |
| Ru3-unsup | 9 | 9 | 9 | 9 |
| Ru3-BN | 9 | 9 | 9 | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



