
Submitted:
08 December 2023
Posted:
13 December 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) is a frequent, debilitating and still enigmatic disease. There is a broad overlap in the symptomatology of ME/CFS and the Post-COVID Syndrome (PCS). A fraction of the PCS patients develops the full clinical picture of ME/CFS. New observations in microvessels and blood from patients suffering from PCS have appeared and include microclots and malformed pathological blood cells. Capillary blood flow is impaired not only by pathological blood components but also by prothrombotic changes in the vascular wall, endothelial dysfunction, and expression of adhesion molecules in the capillaries. These disturbances can finally cause a low capillary flow and even capillary stasis. A low cardiac stroke volume due to hypovolemia and the inability of the capacitance vessels to adequately constrict to deliver the necessary cardiac preload generate an unfavorable low precapillary perfusion pressure. Furthermore, a predominance of vasoconstrictor over vasodilator influences exists, in which sympathetic hyperactivity and endothelial dysfunction play a strong role, causing constriction of resistance vessels and of precapillary sphincters which leads to a fall in capillary pressure behind the sphincters. The interaction of these two precapillary cardiovascular mechanisms causing a low capillary perfusion pressure is hemodynamically highly unfavorable in the presence of a primary capillary stasis already caused by the pathological blood components and their interaction with the capillary wall, to severely impair organ perfusion. The detrimental coincidence of the microcirculatory with the precapillary cardiovascular disturbances may constitute the key disturbance of the Post-COVID-19 syndrome and finally lead to ME/CFS in pre-disposed patients because the interaction causes a particular kind of perfusion disturbance - capillary ischemia-reperfusion - which has a high potential of causing mitochondrial dysfunction by inducing sodium- and calcium-overload in skeletal muscles. The latter in turns worsens the vascular situation by the generation of reactive oxygen species to close a vicious cycle from which the patient can hardly escape.
Keywords:
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome
; Post-COVID Syndrome
; Long-COVID
; Precapillary cardiovascular disturbances
; Capillary disturbances
; ß2AdR dysfunction
; Mitochondrial dysfunction
; Post-Exertional Malaise
; Exercise intolerance
1. Introduction
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) is a frequent, debilitating disease and is associated with a number of syndromes or conditions, including postural orthostatic tachycardia syndrome (POTS) [1,2], orthostatic intolerance (OI) [3], and small fiber neuropathy (SFN) [1]. ME/CFS shows a broad overlap of symptomatology with the Post-COVID Syndrome (PCS). A fraction of the PCS patients develops the full picture of ME/CFS to strongly raise the prevalence of ME/CFS (designated as PCS-ME/CFS) [4].
Investigations on the causes of Long-COVID and PCS have identified pathological blood components - microclots and malformed blood cells - in patients after COVID-19 infection. Lymphocytes had a significantly decreased stiffness, monocytes and neutrophils were increased in cell size, erythrocytes were less deformable and a reduced natural killer cell function has been found in addition [5,6,7,8,9,10]. These pathological blood cells and microclots can impede capillary blood flow, even induce capillary stasis and, thus, significantly worsen organ blood supply. Capillary blood flow is not only impaired by pathological blood components but also by changes in the vascular wall, such as endothelial dysfunction (ED) and expression of adhesion molecules in the capillaries, which leads to an enhanced adhesivity of blood components to the vessel wall to aggravate the flow disturbance.
Various cardiovascular disturbances and abnormalities have been known for quite a long time in ME/CFS before COVID-19 including hypovolemia, low stroke volume and high vasoconstrictor tone [1,10,11,12,13]. Similar cardiovascular disturbances have been found in PCS or PCS-ME/CFS [1,3,10,11,12,14,15,16]. To delineate them from the newly discovered findings of pathological blood components that must disturb capillary flow, we use the term precapillary cardiovascular disturbances. Separately or jointly these different precapillary cardiovascular disturbances have the same effect. They lower the capillary perfusion pressure. In a direct comparison Long-COVID and ME/CFS patients were found to exhibit similarly impaired endothelial function, indicating a potential vascular involvement in the pathogenesis of these post-viral illnesses [17].
In the presence of a reduced capillary flow velocity caused by the pathological blood components, the inflammatory capillary wall changes and the interaction between the two, a physiological or a high capillary pressure is of outmost importance to prevent capillary stasis. The single hemodynamic disturbances and their detrimental interactions, that severely affect microcirculation and organ blood flow, will be highlighted in detail in this paper. We raise the hypothesis that the coincidence or interaction of both disturbances - primary microcirculatory capillary disturbances and precapillary cardiovascular disturbances - constitutes the key disturbance of the Post-COVID-19 syndrome and finally leads to ME/CFS in predisposed patients.
Microvascular capillary disturbances in ME/CFS of other causes than PCS have not been published apart from a report of diminished red blood cell deformability in ME/CFS [7] but investigations are ongoing. Therefore, we limit our conclusions here to ME/CFS developed out of PCS (PCS-ME/CFS).
2. Precapillary cardiovascular disturbances and Capillary disturbances in ME/CFS and PCS - The causes and effect of hypovolemia and low stroke volume
The term “precapillary cardiovascular disturbances” includes different cardiovascular disturbances, hypovolemia, capacitance vessel failure and peripheral vasoconstrictor predominance (Figure 1). Hypovolemia and capacitance vessel failure either independently or jointly reduce cardiac filling pressure. As a result of low ventricular filling, stroke volume and cardiac output significantly decrease leading to a decrease in tissue perfusion and in capillary perfusion pressure. The causes for hypovolemia are thought to be renal hyperexcretion with low renin (renin paradox) and microvascular leakage. These disturbances have been dealt with in detail in a previous paper [16]. We hypothesize that the poor energetic situation in skeletal muscle as the result of poor perfusion and mitochondrial dysfunction leads to the excessive, compensatory production of vasoactive mediators that cause renal hyperexcretion and microvascular leakage to explain hypovolemia [16] as described in section 6.
Concerning dysfunction of the capacitance vessels their contractile dysfunction could be due to three different mechanisms from a theoretical point of view:
1) Autonomic dysfunction or damage to sympathetic nerves could be a potential cause of ineffective contraction.
2) Primary dysfunction due to structural or functional disturbances of the capacitance vessels. Connective tissue disorders, also affecting the vasculature, such as the Ehlers-Danlos Syndrome (EDS) and Marfan Syndrome are related to ME/CFS [18,19,20]. In the latter large vessels are distended. In EDS peripheral veins seem visibly distended [21,22]. Orthostatic intolerance appears early on in patients with PCS [23,24,25]. In a previous publication we hypothesized that SARS-CoV2-infection affects capacitance vessels, mainly veins, directly by the virus itself or via cytokines and/or indirectly via disturbed microcirculation of the vasa vasorum of these vessels (microcirculatory disturbance) to cause orthostatic intolerance early on after the acute infection [3].
3) Failure of appropriate regulation at the vascular level. The latter could be disturbed by an impairment of vasoconstrictor mechanisms (A) or by an excess of vasodilator mechanisms (B).
3A) Autoantibodies against different vasoregulators have been found including autoantibodies against alpha1-adrenergic receptors, which is the main mechanism of venous contraction [26,27].
3B) Histamine: the high prevalence of allergies and mast cell overactivation, which might even be augmented by the SARS-CoV2- infection, particularly in the gut, and the fact that histamine has a strong vasodilator effect on veins in humans [3,15,28,29,30] warrants further discussion. The most convincing evidence for the involvement of histamine is the alleviating effect of long-term symptoms of Post-COVID-19 infection by anti-histamine treatment [31,32,33,34,35]. From a theoretical point of view the dilating effect of histamine on human veins may be particularly detrimental if the capacitance vessels or veins are already weakened or structurally distended as in EDS or the Marfan-Syndrome or damaged by the mechanisms after COVID-19 infection [15]. For physical (Laplace`s law) and physiological reasons (lacking overlap of sarcomeres) the force needed for effective constriction is higher in overstretched circular structures. Histamine induces microvascular leakage. The resulting loss of plasma volume may contribute to a low cardiac filling pressure. The physiological mechanisms of venous constriction by alpha1-adrenergic stimulation required for appropriate orthostatic function and for raising the circulatory blood volume for exercise may then become quite ineffective against two simultaneously present disturbances that weaken venous contraction. Additionally, autoantibodies against alpha1-adrenergic receptors may even weaken the constrictor stimulus.
Taken together, key mechanisms, which generate a low precapillary perfusion pressure are hypovolemia and contractile disturbance of capacitance vessels. Separately or jointly, they lead to a low cardiac preload and subsequently low stroke volume. In the presence of a low cardiac filling pressure both tachycardia by shortening the cardiac filling time and diastolic dysfunction worsen ventricular filling which will be further discussed below.
2.1. The disturbed vasoconstrictor/vasodilator balance causes excessive vasoconstriction
In ME/CFS there is vast literature showing a vasoconstrictor predominance over vasodilator influences by a high sympathetic and low vagal tone and endothelial dysfunction (ED) by different mechanism that raise vascular resistance to which resistance vessels make the strongest contribution (Figure 1) [36]. This leads to a pressure fall behind the resistance vessels with the consequence that capillary perfusion and capillary perfusion pressure become low.
The interaction of disturbed cardiac filling with increased vascular resistance: it cannot be emphasized enough that the combined effect of low stroke volume together with a high vascular resistance has a detrimental effect by strongly lowering of tissue perfusion and of capillary perfusion pressure.
3. Microcirculatory disturbances in ME/CFS and PCS
Low capillary perfusion pressures as outlined above as the result of the precapillary vascular disturbances coincides with preexisting capillary stasis. The interaction of these disturbances has further detrimental effects on capillary blood flow (Figure 1). Microcirculatory disturbances and capillary stasis are primarily caused by the presence of pathological blood components mainly too large so that they hinder blood flow and induce prothrombotic change of the capillary wall (increased adhesivity). There is also a detrimental interaction of pathological blood components with prothrombotic, inflammatory capillary wall changes: Stagnation or slow flow of blood cells enhances the interaction and time for interaction with the prothrombotic wall changes, mainly expressed adhesive molecules, further slowing blood flow velocity [9]. The observations of pathological blood components include microclots, large cells, less deformable erythrocytes, decreased lymphocyte stiffness, increased monocyte cell size, large, deformable, activated neutrophils, activated monocytes and platelets. These activated cells may convert the normally antithrombotic surface of the vascular endothelium to a prothrombotic state to upregulate coagulation, stimulation of inflammatory reactions (thrombo-inflammation) [5,9,37]. Independent of the effect of inflammatory blood cells on the microvessels the vascular system including capillaries could be directly damaged by SARS-CoV-2 virus or affected by cytokines generating an inflammatory and prothrombotic vascular wall [9,10].
Due to the long-term inflammatory environment in Long-COVID (PCS), immune cells such as neutrophils, are excessively activated, and persistently degranulate maintaining inflammatory responses. This leads to the formation of fibrin amyloid microclots, which promote tissue hypoxia and impaired oxygen exchange and may additionally block the capillaries [9,10,38,39,40]. It has been described that chronic inflammation can trigger coagulation proteases to bind to protease-activated receptors on the activated endothelium to induce the synthesis and expression of cell adhesion molecules, which further promotes microcirculatory disorders [9]. Several endothelial cell-related biomarkers have been observed to be strongly correlated with COVID-19 and PCS, such as VWF and Factor VIII, as well as ET-1 and angiopoietin-2 [9]. A recent study identified a disturbed retinal microcirculation to be strongly associated with PCS and ME/CFS, which may serve as a potential marker for microcirculatory disorders [10,41]. Notably, the thrombo-inflammatory status persists months after the patient has been recovered and the virus has cleared [5,39,42]. Therefore, malformed blood cells, microclots and thrombo-inflammation can potentially impede capillary perfusion and promote a microcirculatory disorder and thus, impair the organ blood supply by capillary obstruction [9].
Figure 1.
Association of precapillary cardiovascular disturbances and capillary disturbances in ME/CFS and PCS. Capillary low flow or even stasis is triggered by malformed blood components, such as microclots, malformed leukocytes and an increased endothelial adhesivity promoted by increased expression of adhesion molecules in the capillaries. Capillary stasis is strongly aggravated by the precapillary cardiovascular disturbance generating a low capillary perfusion pressure for which a low stroke volume and high vasoconstrictor tone account for. The low stroke volume is caused by both hypovolemia and failure of capacitance vessels to adequately constrict to raise the circulating blood volume. A peculiar perfusion disturbance – capillary ischemia-reperfusion – occurs that has a high pathogenetic potential. ED: endothelial dysfunction.
Figure 1.
Association of precapillary cardiovascular disturbances and capillary disturbances in ME/CFS and PCS. Capillary low flow or even stasis is triggered by malformed blood components, such as microclots, malformed leukocytes and an increased endothelial adhesivity promoted by increased expression of adhesion molecules in the capillaries. Capillary stasis is strongly aggravated by the precapillary cardiovascular disturbance generating a low capillary perfusion pressure for which a low stroke volume and high vasoconstrictor tone account for. The low stroke volume is caused by both hypovolemia and failure of capacitance vessels to adequately constrict to raise the circulating blood volume. A peculiar perfusion disturbance – capillary ischemia-reperfusion – occurs that has a high pathogenetic potential. ED: endothelial dysfunction.
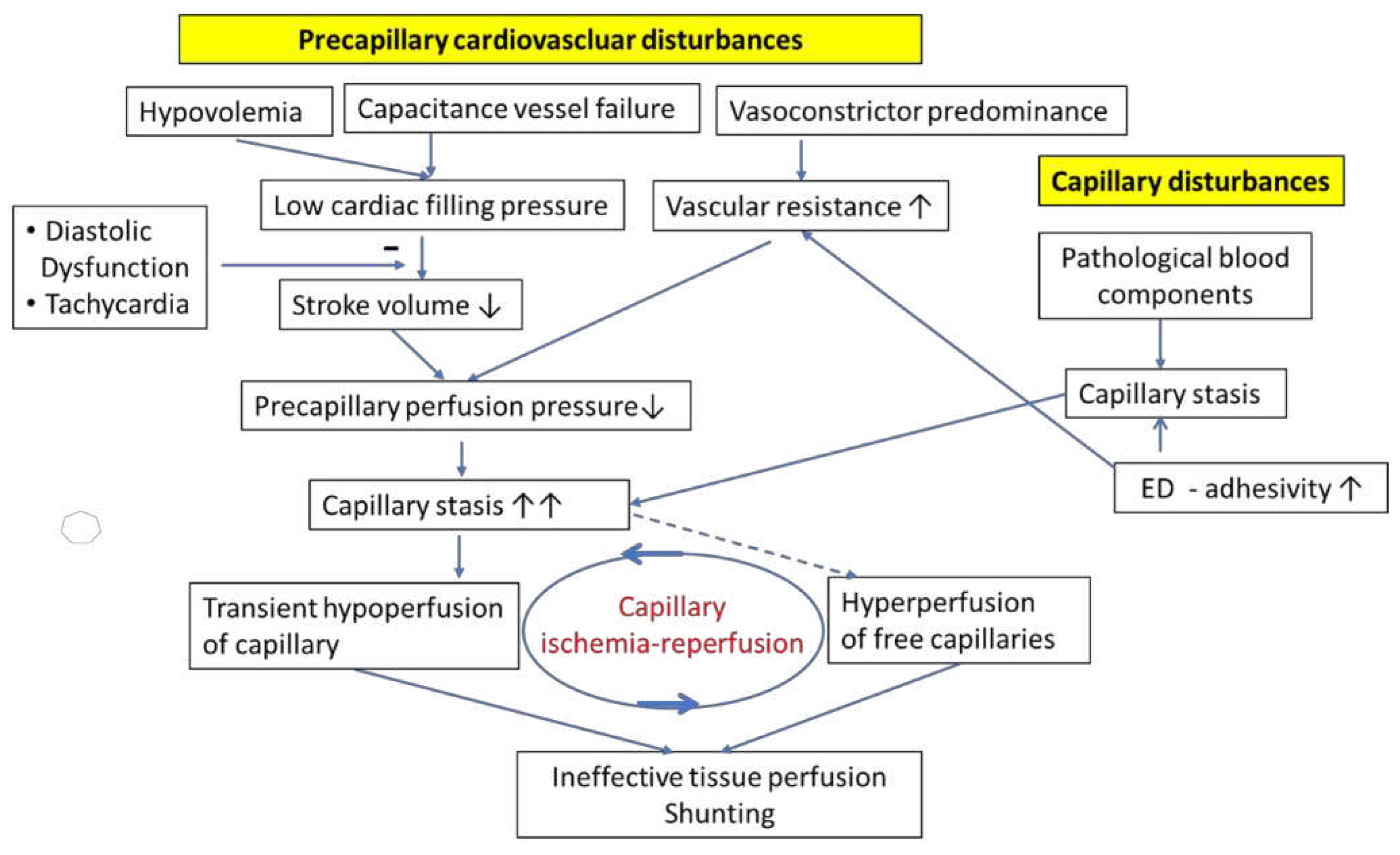
It is unlikely that microclots, whose size are larger than the capillary diameter, play a major role in capillary perfusion disorder since these microclots would get stuck in an organ and thus be removed from circulating blood with every circulatory passage. If very large microclots were common, one would expect the development of ischemic organ damage by vascular occlusions over time or the development of pulmonary hypertension if they got stuck in the pulmonary vascular system [5,39]. Although severe ischemic organ damage has not been observed in PCS-induced ME/CFS so far, permanent obstruction of capillaries, e.g., by microclots larger than capillary diameter may still occur and could account for the observation of vessel rarefaction (disappearance of capillaries) in skeletal muscle [43]. It is likely that transient capillary blood stasis is present in a fraction of the capillaries so that the free capillaries are hyperperfused, which would lead to a blood flow distribution disorder and capillary shunting (Figure 1). Peripheral left-to-right shunting was indeed observed in patients with ME/CFS [44]. After removal of the obstruction the transiently ischemic capillary would be reperfused generating the phenomenon of capillary ischemia-reperfusion. Ischemia-reperfusion is a pathomechanism with a high potential of causing a particular damage that will be extensively explained as described in section 5. Given the variety in the size of microclots and of disturbed blood cells all degrees of obstruction ranging from weak slowing of capillary flow velocity to transient ischemia seem possible and could occur simultaneously in different capillaries. Microvascular rarefaction or temporary obstruction markedly alters blood flow distribution significantly stronger than microvascular constriction [43,45]. These various disturbances might also have a significant impact on neurovascular coupling, a mechanism, which is based on the vasodilator release by active nerve cells to increase their energy supply by dilating local capillaries and precapillary sphincters, which protect capillaries from high blood pressure while maintaining the blood flow [46,47,48]. Cerebral blood flow measured over a large cerebral artery was found decreased in ME/CFS and PCS [49]. The extent of flow reduction measured over a large brain artery does not reflect the true extent of the disturbance as it does not consider the microcirculatory disturbance, the maldistribution of blood flow in the capillary system (capillaries with no flow versus hyperperfused capillaries). The severity of the resulting cerebral blood flow disturbance may explain cognitive impairment, mental fatigue and at least partially brain fog. Endothelial dysfunction found in ME/CFS and PCS certainly worsens the function of capillaries as well of precapillary vessels e.g., by raising vasoconstrictor tone of resistance vessels [17],[50,51].
4. The Interaction of the precapillary cardiovascular disturbances with the capillary disturbances – a highly unfavorable synergistic interaction in ME/CFS and PCS
To quickly overcome a capillary stasis the precapillary perfusion pressure must be high, but it is lower than normal in PCS and ME/CFS. Because it is very likely that only a fraction of the capillaries is transienly occluded and ischemic, e.g., by a microclot-induced blood stasis, other capillaries are hyperperfused and a blood flow distribution disorder may develop (capillary shunting) (Figure 1). In this situation, the low stroke volume cannot maintain a continuous capillary flow, particularly during diastole. Physiologically, capillary blood flow is continuous, also in diastole, since the energy delivered in the systole is stored in the vascular system (referred to as “Windkessel-effect”). Interestingly, a recently performed clinical study used carotid-femoral pulse wave velocity as a measurement to predict COVID-19-related cardiovascular complications and found a marked increase in arterial stiffness and an increased carotid-femoral pulse wave velocity [52]. An increase in arterial stiffness is due to a structural loss of vascular elasticity or due to a high tone of conductance vessels to diminish the Windkessel-function. The latter is not only caused by elastic storage of kinetic energy provided by the systole but also by a dynamic phenomenon – by the reflection of pulse waves that are appropriately timed to the diastole. An increase in arterial stiffness therefore is not only indicative of a loss of elasticity but also changes the time pattern of pulse wave reflection so that reflection occurs earlier leading to a loss of capillary thrust at least later in diastole [53,54]. Therefore, the determination of the carotid-femoral pulse wave velocity potentially could serve as a biomarker for COVID-19-related cardiovascular complications. Another clinical study on 256 patients suffering from ME/CFS showed a decreased cognitive efficiency, a reduced narrow pulse pressure less than 25% of systolic pressure and an increased heart rate during the orthostatic challenge, thus confirming the negative effect of the low stroke volume that causes the narrow pulse pressure impairing diastolic capillary perfusion and suggesting a negative role of tachycardia [55]. Diastolic cardiac dysfunction is expected to strongly worsen the disturbance by impairing ventricular filling and stroke volume in the presence of a low filling pressure. In a recent study on patients suffering from COVID-19 infection impaired left ventricular diastolic function and an impaired right ventricular function were echocardiographically detected [56]. Even physiological ventricular hypertrophy as it occurs in sportsmen might worsen ventricular filling under condition of a low filling pressure. Tachycardia as in POTS can easily aggravate the disturbance due to shortened filling time and reduced cardiac preload that occurs in a situation of low filling pressure. Frequent but short pressure peaks in tachycardia will not sufficiently accelerate and move forward a stagnating capillary blood column and will favor capillary stasis during diastole. A pharmacological confirmation of the negative role of tachycardia is the beneficial effect of the bradycardic drug Ivabradine in PCS patients [57,58,59,60]. None of the single hemodynamic disturbances outlined needs to be strong enough to explain the severity of the disease but all these hemodynamic disturbances acting in concert can cause transient capillary ischemia, maldistribution of capillary blood flow and induce a peculiar type of flow disturbance, namely capillary ischemia-reperfusion. This also explains the lack of a characteristic diagnostic hemodynamic parameter. Capillary ischemia-reperfusion has a particular potential to trigger the development of mitochondrial dysfunction in skeletal muscle as will be outlined in the next section.
5. Mitochondrial dysfunction
Energy depletion in skeletal muscle in ME/CFS is the consequence of the combined effect of perfusion disturbances and mitochondrial dysfunction according to our hypothesis (Figure 2). This can explain low oxygen consumption on CPET which can be considered a biomarker in patients with ME/CFS [61]. Finally, both disturbances trigger each other as will be outlined in this section. First, we explain how the perfusion disturbances could trigger mitochondrial dysfunction in skeletal muscle and why the particular type of perfusion disturbance, namely capillary ischemia-reperfusion, plays an important role.
We have already used the experimental ischemia-reperfusion injury paradigm to explain mitochondrial dysfunction in a previous publication [62]. The recognition that capillary ischemia-reperfusion could occur as the consequence of the recently recognized capillary flow disturbances now allows to more precisely work out the pathomechanisms involved and to recognize possible differences. In both conditions it is finally sodium-overload that indirectly causes damage: At a certain level of sodium, called the reverse mode threshold, the sodium-calcium exchanger (NCX) changes its transport direction importing calcium instead of exporting it. This leads to calcium overload and cellular and mitochondrial damage. We do not claim that the mechanisms of sodium loading are the same in all aspects in both conditions of ischemia-reperfusion although we have identified capillary ischemia-reperfusion as the type of perfusion disturbance with the highest potential to cause mitochondrial dysfunction. We assume that a number of different disturbances – not only vascular disturbances - jointly raise intramuscular sodium in PCS and ME/CFS to finally cause calcium-overload and damage. Capillary ischemia-reperfusion in ME/CFS is certainly less severe than the experimental ischemia-reperfusion situation in which a large vessel is totally occluded for a short time. In the classical experimental ischemia-reperfusion injury paradigm a sudden and massively activated sodium-proton-exchanger (NHE1) during earliest reperfusion leads to extremely fast sodium loading and overload. There is overt tissue damage in the classical ischemia paradigm and mitochondrial dysfunction while in ME/CFS there is no such tissue damage but evidence of mitochondrial disturbance. The experimental ischemia-reperfusion paradigm is enforced in general anesthesia which causes tissue damage. Awake ME/CFS patients, however, would stop muscle work at a degree of effort that would cause clear tissue damage so that mainly mitochondrial dysfunction occurs. In the experimental ischemia-reperfusion injury paradigm the short occlusion of a vessel would not already cause damage by the short ischemia itself. It is sudden reperfusion that causes damage by sodium-induced calcium overload [63,64]. During total vascular occlusion intravascular protons accumulate to almost totally inhibit NHE1 so that it cannot export protons any more against the high extracellular proton concentration that has accumulated in the occluded vessel during ischemia. With reperfusion vascular proton concentration suddenly becomes normal. The NHE1, which is driven only by ion gradients, then becomes unleashed and extremely active for a short time only. It is then strongly driven by the high intracellular proton concentration generated by the anaerobic metabolism during ischemia and accumulated in the cell due to the inhibition of the NHE1 by the high proton concentration in the totally occluded vessel. In early reperfusion the transiently very high NHE1 activity causes sodium loading, rather in seconds than minutes. The sodium-potassium-ATPase (Na+/K+-ATPase), which exports intracellular sodium for the import of potassium [65], cannot cope with such a sudden high sodium load – its activity is impaired by low ATP due to ischemia - so that sodium rises to cross the reverse mode threshold of the NCX to cause calcium overload for a brief moment. This is sufficient to cause damage.
A rise in intracellular sodium has indeed been demonstrated in skeletal muscles of patients with ME/CFS in an MRI study with 23-Na+ [66]. Depending on the muscle type sodium was already elevated at rest before exercise or was found elevated only after exercise. Undoubtedly, continuous malperfusion already contributes to sodium loading via anaerobic metabolism to which mitochondrial dysfunction contributes as soon as it has developed. Anaerobic metabolism raises cellular proton production that are extruded by NHE1. This causes continuous sodium influx and raises intracellular sodium. The reperfusion-induced sodium-loading strongly adds to the metabolically and hypoperfusion-induced intracellular rise in sodium to cause or predispose to calcium overload. Presumably, enhanced sodium loading is not enough to cause a rise in sodium to an extent that it causes calcium overload by reversing the transport mode of the NCX. Another disturbance is needed.
Figure 2.
Proposed mechanisms of induction of the mitochondrial dysfunction as a result of the combined effect of perfusion disturbances which are already shown in Figure 1 and an insufficient rise of the Na+/K+-ATPase activity in skeletal muscle. Mitochondrial dysfunction impairs perfusion via ROS and favors itself by lowering the Na+/K+-ATPase activity via ROS and low ATP. The poor energetic situation leads to the excessive production and spillover of vasoactive algesic mediators that can reach every organ to cause symptoms and induce hypovolemia. ED: endothelial dysfunction.
Figure 2.
Proposed mechanisms of induction of the mitochondrial dysfunction as a result of the combined effect of perfusion disturbances which are already shown in Figure 1 and an insufficient rise of the Na+/K+-ATPase activity in skeletal muscle. Mitochondrial dysfunction impairs perfusion via ROS and favors itself by lowering the Na+/K+-ATPase activity via ROS and low ATP. The poor energetic situation leads to the excessive production and spillover of vasoactive algesic mediators that can reach every organ to cause symptoms and induce hypovolemia. ED: endothelial dysfunction.
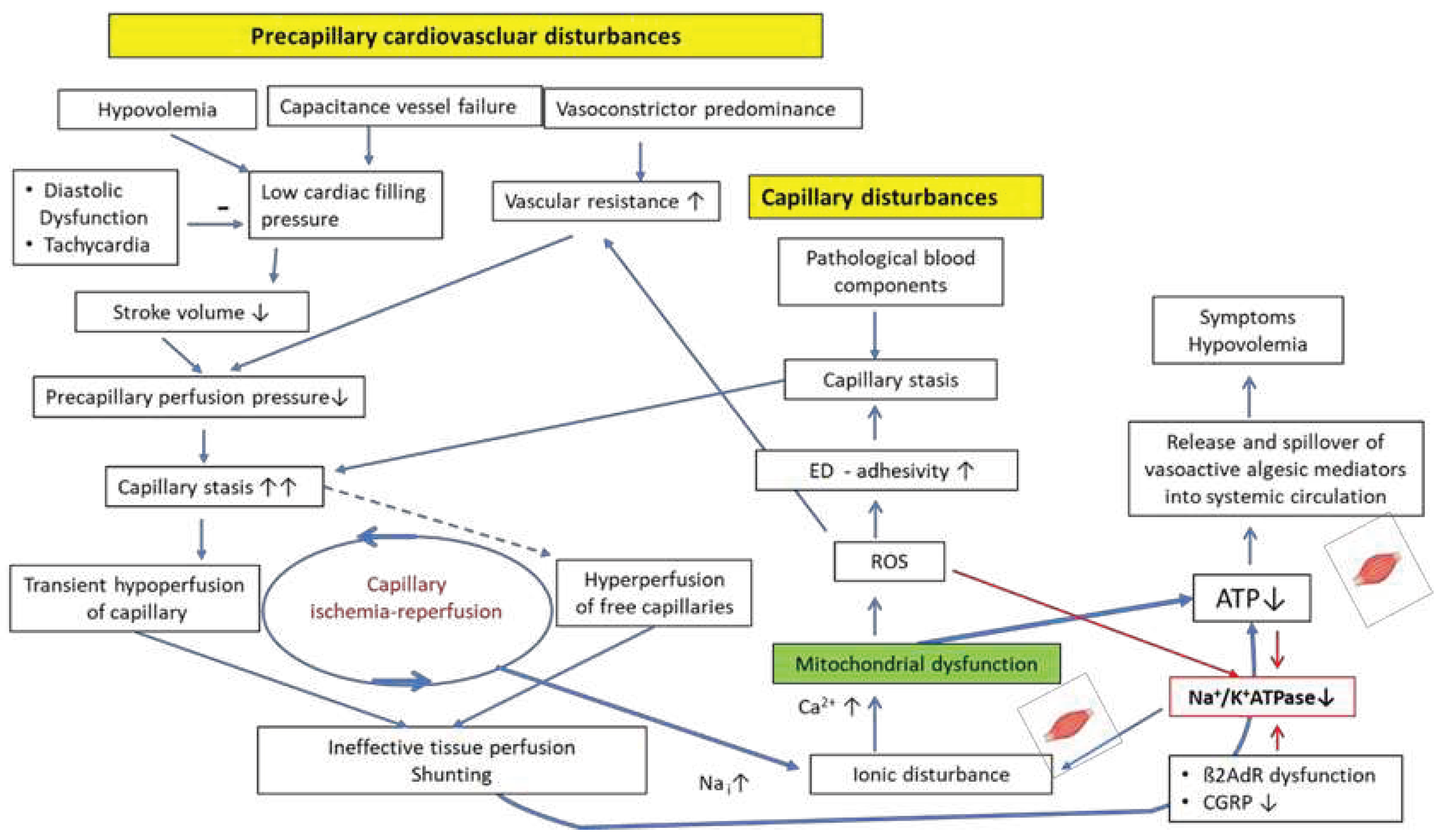
In ME/CFS intramuscular sodium loading is strongly aggravated by a disturbed function of Na+/K+-ATPase diminishing cellular sodium export. The latter physiologically exports three sodium ions for the import of two potassium ions [65].
Thus, due to the disturbed function of Na+/K+-ATPase the intramuscular sodium can already rise as soon as cellular sodium influx occurs as it is the case in the beginning of exercise (via sodium-dependent transport processes such as via NHE1 and by opening of sodium channels during the process of excitation).
During muscle work Na+/K+-ATPase requires 10-20-fold stimulation as there is a strong sodium import by excitatory sodium channels and ion transport mechanisms [65,67]. The only hormonal stimuli of the Na+/K+-ATPase in the exercising muscle apart from the rising sodium concentration itself are ß2-adrenergic receptors (ß2AdR) and calcitonin-gene related peptide (CGRP) [65,67,68]. Deficiency of the only two mediators - ß2AdR and CGRP - that activate Na+/K+-ATPase during exercise, can be considered a major cause of exercise intolerance in conjunction with perfusion disturbances and mitochondrial dysfunction whereby all these three disturbances are interrelated in vicious cycles. ß2AdR dysfunction is assumed to be present in ME/CFS due to autoantibodies and/or desensitization to chronic stress in ME/CFS (high sympathetic tone) as the ß2AdR is the most sensitive adrenergic receptor to desensitization [69].
Small fiber neuropathy (SFN) occurs in part of the ME/CFS and PCS-patients [44,70,71,72]. The sensory nerves that contain and release neuropeptides such as CGRP are primarily unmyelinated sensory C-fibers and myelinated Aδ-fibers [73]. The importance of CGRP and its release from sensory nerves for the activation of the Na+/K+-ATPase and for diminishing muscle fatigue has been experimentally demonstrated under conditions of stimulation and inhibition [74,75]. After degeneration of these fibers, by which CGRP is produced, there should be a deficiency in CGRP that adds to the stimulatory deficit of the Na+/K+-ATPase during exercise already caused by ß2AdR dysfunction to aggravate and fix the disturbance. These neuropeptides released from the sensory nerves like Substance P and CGRP are also vasodilatory so that their deficit as a result of SFN may contribute to muscular malperfusion. Deficiency in CGRP certainly is no necessity because only part of the patients develops SFN. However, once SFN has developed it may contribute to fixing the disturbance. Na+/K+-ATPase activity is not only not sufficiently activated during exercise but even inhibited. At mitochondrial calcium levels higher than required for the stimulation of ATP production the mitochondrium produces reactive oxygen species (ROS) [76]. ROS generated as a consequence of mitochondrial dysfunction inhibit while reduced ATP levels may further weaken its activity [65,67]. Increased ROS production with evidence for inhibition of the Na+/K+-ATPase activity were indeed found in patients with ME/CF [77]. Cortisol, aldosterone, and triiodothyronine (T3) stimulate the expression of the Na+/K+-ATPase [67]. At least cortisol and aldosterone are rather in the low normal range in ME/CFS or PCS [78,79,80].
Finally, we explain how mitochondrial dysfunction affects perfusion to close another vicious cycle of mutual triggering. Mitochondrial dysfunction produces ROS to cause endothelial dysfunction and to promote endothelial cell inflammatory, activation of coagulation, and adhesivity [81]. The latter enhances vasoconstrictor influences and favors microcirculatory flow disturbances to further impair perfusion. Finally, via low ATP levels and the generation of ROS that inhibit Na+/K+-ATPase [82] and, via an anaerobic metabolism that produces more protons to raise intracellular sodium by the NHE1, mitochondrial dysfunction favors itself [14,83,84,85]. The energetic disturbance is not severe enough to cause organ damage but limits a rise in physical and mental performance (exercise intolerance). Even worse, at a certain level of exercise, the individual post-exertional malaise threshold (PEM) threshold, intracellular sodium in skeletal muscle rises to reach the reverse mode threshold of the NCX. This causes calcium overload to trigger and renew mitochondrial damage. Thus, during exercise the functional damage reproduces itself, keeping the patients captured in a vicious cycle from which they can hardly escape.
6. The consequences of critical energetic situation in skeletal muscle
The critical energetic situation in skeletal muscle leads to an excessive metabolically driven local generation of vasodilatory tissue mediators with algesic and inflammatory properties for a compensatory rise in skeletal muscle blood flow [14]. Mediators like bradykinin, prostaglandins, prostacyclin, and adenosine raising blood flow are physiologically meant to act locally only by their very short half-lives. Due to their excessive production following the poor metabolic situation in the large body muscle mass, however, spillover into the systemic circulation occurs. Any organ can be reached by them to produce the confusing myriad of symptoms including pain, spasms and edema as a result of their physiological actions. These mediators also cause renal hyperexcretion and induce microvascular leakage causing hypovolemia (see section 2) (Figure 2) [14]. Their particular physiological renal actions prevent a compensatory rise in renin for repletion of the vascular system, thereby explaining the paradox that renin does not rise with hypovolemia [79]. This leads to low stroke volume as explained above and causes orthostatic stress. The latter may be the greatest stressor that is involved in aggravating autonomic dysfunction and in desensitization of ß2AdR to close the vicious circle.
Conclusions
The most severe complication of the Post-COVID Syndrome (PCS) is the development of ME/CFS. Even in the absence of the full criteria defining ME/CFS, PCS and ME/CFS show a broad overlap in the symptomatology so that common mechanisms can be assumed to be operative. We hypothesize that precapillary cardiovascular disturbances that include hypovolemia and the failure of the capacitance vessels to adequately contract, act in concert to reduce ventricular filling and thereby stroke volume. This, together with the predominance of vasoconstrictor mechanisms, that excessively constrict resistance vessels, reduces tissue perfusion and diminishes the capillary perfusion pressure to severely aggravate a preexisting capillary stasis. Capillary stasis is primarily caused by malformed blood components, microclots and prothrombotic capillary wall changes as wells as by their concerted interaction. Vascular and capillary disturbances are favored by excessive reactive oxygen species (ROS) production and oxidative stress resulting from mitochondrial dysfunction. They reduce the availability of the vasodilator nitric oxide, affect the endothelial cellular barrier and enhance endothelial adhesivity. Both - precapillary and capillary disturbance - have a highly synergistic detrimental effect on tissue perfusion. Together they cause capillary ischemia, capillary ischemia-reperfusion, which favors the development of mitochondrial dysfunction in skeletal muscle, and poor tissue perfusion. These disturbances are not severe enough to cause organ damage, but they prevent an adequate rise in physical and mental performance causing exercise intolerance. Even worse, when attempting to raise the level of effort and crossing the individual post-exertional malaise (PEM) threshold, the functional disturbances that cause these limitations in exercise capacity, are triggered and renewed. Thus, the functional damage reproduces itself, keeping the patients captured in a vicious cycle from which they can hardly escape.
Author Contributions
K.J.W. and M.L. conceived the idea and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Not applicable
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank Klaus Steinmeyer, Sanofi, for continued scientific discussion and his critical review of the manuscript and valuable suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dixit, N.M.; Churchill, A.; Nsair, A.; Hsu, J.J. Post-Acute COVID-19 Syndrome and the cardiovascular system: What is known? Am Heart J Plus 2021, 5, 100025. [Google Scholar] [CrossRef] [PubMed]
- Yong, S.J.; Liu, S. Proposed subtypes of post-COVID-19 syndrome (or long-COVID) and their respective potential therapies. Rev Med Virol 2022, 32, e2315. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.J.; Lohn, M. Orthostatic Intolerance after COVID-19 Infection: Is Disturbed Microcirculation of the Vasa Vasorum of Capacitance Vessels the Primary Defect? Medicina (Kaunas) 2022, 58. [Google Scholar] [CrossRef] [PubMed]
- Legler, F.; Meyer-Arndt, L.; Modl, L.; Kedor, C.; Freitag, H.; Stein, E.; Hoppmann, U.; Rust, R.; Wittke, K.; Siebert, N.; et al. Long-term symptom severity and clinical biomarkers in post-COVID-19/chronic fatigue syndrome: results from a prospective observational cohort. EClinicalMedicine 2023, 63, 102146. [Google Scholar] [CrossRef]
- Kubankova, M.; Hohberger, B.; Hoffmanns, J.; Furst, J.; Herrmann, M.; Guck, J.; Krater, M. Physical phenotype of blood cells is altered in COVID-19. Biophys J 2021, 120, 2838–2847. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Berk, M.; Galecki, P.; Maes, M. The emerging role of autoimmunity in myalgic encephalomyelitis/chronic fatigue syndrome (ME/cfs). Mol Neurobiol 2014, 49, 741–756. [Google Scholar] [CrossRef]
- Saha, A.K.; Schmidt, B.R.; Wilhelmy, J.; Nguyen, V.; Abugherir, A.; Do, J.K.; Nemat-Gorgani, M.; Davis, R.W.; Ramasubramanian, A.K. Red blood cell deformability is diminished in patients with Chronic Fatigue Syndrome. Clin Hemorheol Microcirc 2019, 71, 113–116. [Google Scholar] [CrossRef]
- Eaton-Fitch, N.; du Preez, S.; Cabanas, H.; Staines, D.; Marshall-Gradisnik, S. A systematic review of natural killer cells profile and cytotoxic function in myalgic encephalomyelitis/chronic fatigue syndrome. Syst Rev 2019, 8, 279. [Google Scholar] [CrossRef]
- Turner, S.; Khan, M.A.; Putrino, D.; Woodcock, A.; Kell, D.B.; Pretorius, E. Long COVID: pathophysiological factors and abnormalities of coagulation. Trends Endocrinol Metab 2023, 34, 321–344. [Google Scholar] [CrossRef]
- Nunes, J.M.; Kruger, A.; Proal, A.; Kell, D.B.; Pretorius, E. The Occurrence of Hyperactivated Platelets and Fibrinaloid Microclots in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Pharmaceuticals (Basel) 2022, 15. [Google Scholar] [CrossRef]
- Natelson, B.H.; Brunjes, D.L.; Mancini, D. Chronic Fatigue Syndrome and Cardiovascular Disease: JACC State-of-the-Art Review. J Am Coll Cardiol 2021, 78, 1056–1067. [Google Scholar] [CrossRef]
- Chilazi, M.; Duffy, E.Y.; Thakkar, A.; Michos, E.D. COVID and Cardiovascular Disease: What We Know in 2021. Curr Atheroscler Rep 2021, 23, 37. [Google Scholar] [CrossRef]
- Duffy, E.; Chilazi, M.; Cainzos-Achirica, M.; Michos, E.D. Cardiovascular Disease Prevention During the COVID-19 Pandemic: Lessons Learned and Future Opportunities. Methodist Debakey Cardiovasc J 2021, 17, 68–78. [Google Scholar] [CrossRef]
- Wirth, K.; Scheibenbogen, C. A Unifying Hypothesis of the Pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): Recognitions from the finding of autoantibodies against ss2-adrenergic receptors. Autoimmun Rev 2020, 19, 102527. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.J.; Lohn, M. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) and Comorbidities: Linked by Vascular Pathomechanisms and Vasoactive Mediators? Medicina (Kaunas) 2023, 59. [Google Scholar] [CrossRef]
- Wirth, K.J.; Scheibenbogen, C.; Paul, F. An attempt to explain the neurological symptoms of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J Transl Med 2021, 19, 471. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, M.; Sanal-Hayes, N.E.M.; Hayes, L.D.; Berry, E.C.; Sculthorpe, N.F. People With Long COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) Exhibit Similarly Impaired Vascular Function. LID - S0002-9343(23)00609-5 [pii] LID -. [CrossRef]
- Kuorilehto, T.; Poyhonen, M.; Keski-Nisula, L.; Laurikka, J.; Salenius, J.P. [Vascular malformations associated with Marfan syndrome, Ehlers-Danlos syndrome and neurofibromatosis 1 and their surgical treatment]. Duodecim 2011, 127, 2280–2286. [Google Scholar]
- Meester, J.A.N.; Verstraeten, A.; Schepers, D.; Alaerts, M.; Van Laer, L.; Loeys, B.L. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann Cardiothorac Surg 2017, 6, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.; Chang, C.; Gershwin, M.E. Ehlers-Danlos Syndrome: Immunologic contrasts and connective tissue comparisons. J Transl Autoimmun 2021, 4, 100077. [Google Scholar] [CrossRef]
- Roma, M.; Marden, C.L.; De Wandele, I.; Francomano, C.A.; Rowe, P.C. Postural tachycardia syndrome and other forms of orthostatic intolerance in Ehlers-Danlos syndrome. Auton Neurosci 2018, 215, 89–96. [Google Scholar] [CrossRef]
- Rowe, P.C.; Barron, D.F.; Calkins, H.; Maumenee, I.H.; Tong, P.Y.; Geraghty, M.T. Orthostatic intolerance and chronic fatigue syndrome associated with Ehlers-Danlos syndrome. J Pediatr 1999, 135, 494–499. [Google Scholar] [CrossRef]
- Campen, C.; Rowe, P.C.; Visser, F.C. Orthostatic Symptoms and Reductions in Cerebral Blood Flow in Long-Haul COVID-19 Patients: Similarities with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Medicina (Kaunas) 2021, 58. [Google Scholar] [CrossRef] [PubMed]
- van Campen, C.; Rowe, P.C.; Visser, F.C. Deconditioning does not explain orthostatic intolerance in ME/CFS (myalgic encephalomyelitis/chronic fatigue syndrome). J Transl Med 2021, 19, 193. [Google Scholar] [CrossRef] [PubMed]
- van Campen, C.; Verheugt, F.W.A.; Rowe, P.C.; Visser, F.C. Orthostatic chronotropic incompetence in patients with myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). IBRO Neurosci Rep 2023, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Muller-Ruchholtz, E.R.; Losch, H.M.; Grund, E.; Lochner, W. Effect of alpha adrenergic receptor stimulation on integrated systemic venous bed. Pflugers Arch 1977, 370, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Coupar, I.M. The effect of isoprenaline on adrenoceptors in human saphenous vein. Br J Pharmacol 1970, 39, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Dachman, W.D.; Bedarida, G.; Blaschke, T.F.; Hoffman, B.B. Histamine-induced venodilation in human beings involves both H1 and H2 receptor subtypes. J Allergy Clin Immunol 1994, 93, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Muller-Schweinitzer, E. On the pharmacology of venous smooth muscle from dog and man. Folia Haematol Int Mag Klin Morphol Blutforsch 1979, 106, 690–704. [Google Scholar] [PubMed]
- Weinstock, L.B.; Brook, J.B.; Walters, A.S.; Goris, A.; Afrin, L.B.; Molderings, G.J. Mast cell activation symptoms are prevalent in Long-COVID. Int J Infect Dis 2021, 112, 217–226. [Google Scholar] [CrossRef]
- Glynne, P.; Tahmasebi, N.; Gant, V.; Gupta, R. Long COVID following mild SARS-CoV-2 infection: characteristic T cell alterations and response to antihistamines. J Investig Med 2022, 70, 61–67. [Google Scholar] [CrossRef]
- Mashauri, H.L. Covid-19 Histamine theory: Why antihistamines should be incorporated as the basic component in Covid-19 management? Health Sci Rep 2023, 6, e1109. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Fuhler, G.M.; Pan, Y. Could Histamine H1 Receptor Antagonists Be Used for Treating COVID-19? Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.D.; Lambert, N.; Downs, C.A.; Abrahim, H.; Hughes, T.D.; Rahmani, A.M.; Burton, C.W.; Chakraborty, R. Antihistamines for Postacute Sequelae of SARS-CoV-2 Infection. J Nurse Pract 2022, 18, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Salvucci, F.; Codella, R.; Coppola, A.; Zacchei, I.; Grassi, G.; Anti, M.L.; Nitisoara, N.; Luzi, L.; Gazzaruso, C. Antihistamines improve cardiovascular manifestations and other symptoms of long-COVID attributed to mast cell activation. Front Cardiovasc Med 2023, 10, 1202696. [Google Scholar] [CrossRef] [PubMed]
- Sivri, F.; Turkoz, I.; Sencan, M.; Icen, Y.K.; Aksoy, F.; Ceyhan, B.O. Does COVID-19 Cause Non-Dipper Hypertension? Angiology 2023, 33197231209584. [Google Scholar] [CrossRef] [PubMed]
- Blitshteyn, S.; Brinth, L.; Hendrickson, J.E.; Martinez-Lavin, M. Autonomic dysfunction and HPV immunization: an overview. Immunol Res 2018, 66, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc Diabetol 2021, 20, 172. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Venter, C.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B. Prevalence of readily detected amyloid blood clots in 'unclotted' Type 2 Diabetes Mellitus and COVID-19 plasma: a preliminary report. Cardiovasc Diabetol 2020, 19, 193. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Venter, C.; Laubscher, G.J.; Kotze, M.J.; Oladejo, S.O.; Watson, L.R.; Rajaratnam, K.; Watson, B.W.; Kell, D.B. Prevalence of symptoms, comorbidities, fibrin amyloid microclots and platelet pathology in individuals with Long COVID/Post-Acute Sequelae of COVID-19 (PASC). Cardiovasc Diabetol 2022, 21, 148. [Google Scholar] [CrossRef]
- Schlick, S.; Lucio, M.; Wallukat, G.; Bartsch, A.; Skornia, A.; Hoffmanns, J.; Szewczykowski, C.; Schroder, T.; Raith, F.; Rogge, L.; et al. Post-COVID-19 Syndrome: Retinal Microcirculation as a Potential Marker for Chronic Fatigue. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Iba, T.; Connors, J.M.; Levy, J.H. What Role Does Microthrombosis Play in Long COVID? Semin Thromb Hemost 2023. [Google Scholar] [CrossRef]
- Nusz, D.J.; White, D.C.; Dai, Q.; Pippen, A.M.; Thompson, M.A.; Walton, G.B.; Parsa, C.J.; Koch, W.J.; Annex, B.H. Vascular rarefaction in peripheral skeletal muscle after experimental heart failure. Am J Physiol Heart Circ Physiol 2003, 285, H1554–1562. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Arevalo, C.; Oliveira, R.K.F.; Faria-Urbina, M.; Felsenstein, D.; Oaklander, A.L.; Systrom, D.M. Insights From Invasive Cardiopulmonary Exercise Testing of Patients With Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. CHEST 2021, 160, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Greene, A.S.; Tonellato, P.J.; Lui, J.; Lombard, J.H.; Cowley, A.W., Jr. Microvascular rarefaction and tissue vascular resistance in hypertension. Am J Physiol 1989, 256, H126–131. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.Y.; Barnden, L.R.; Kwiatek, R.A.; Bhuta, S.; Hermens, D.F.; Lagopoulos, J. Neuroimaging characteristics of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): a systematic review. J Transl Med 2020, 18, 335. [Google Scholar] [CrossRef]
- Shan, Z.Y.; Mohamed, A.Z.; Andersen, T.; Rendall, S.; Kwiatek, R.A.; Fante, P.D.; Calhoun, V.D.; Bhuta, S.; Lagopoulos, J. Multimodal MRI of myalgic encephalomyelitis/chronic fatigue syndrome: A cross-sectional neuroimaging study toward its neuropathophysiology and diagnosis. Front Neurol 2022, 13, 954142. [Google Scholar] [CrossRef] [PubMed]
- Adingupu, D.D.; Soroush, A.; Hansen, A.; Twomey, R.; Dunn, J.F. Reduced Cerebrovascular Oxygenation in Individuals with Post-Acute COVID-19 Syndrome (PACS) ("long COVID"). Adv Exp Med Biol 2023, 1438, 211–216. [Google Scholar] [CrossRef] [PubMed]
- van Campen, C.; Rowe, P.C.; Visser, F.C. Blood Volume Status in ME/CFS Correlates With the Presence or Absence of Orthostatic Symptoms: Preliminary Results. Front Pediatr 2018, 6, 352. [Google Scholar] [CrossRef] [PubMed]
- Guven, G.; Hilty, M.P.; Ince, C. Microcirculation: Physiology, Pathophysiology, and Clinical Application. Blood Purif 2020, 49, 143–150. [Google Scholar] [CrossRef]
- Charfeddine, S.; Ibn Hadj Amor, H.; Jdidi, J.; Torjmen, S.; Kraiem, S.; Hammami, R.; Bahloul, A.; Kallel, N.; Moussa, N.; Touil, I.; et al. Long COVID 19 Syndrome: Is It Related to Microcirculation and Endothelial Dysfunction? Insights From TUN-EndCOV Study. Front Cardiovasc Med 2021, 8, 745758. [Google Scholar] [CrossRef]
- Jannasz, I.; Pruc, M.; Rahnama-Hezavah, M.; Targowski, T.; Olszewski, R.; Feduniw, S.; Petryka, K.; Szarpak, L. The Impact of COVID-19 on Carotid-Femoral Pulse Wave Velocity: A Systematic Review and Meta-Analysis. J Clin Med 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Mali, W.; Doevendans, P.A. Form and function, both matter. Neth Heart J 2015, 23, 312–313. [Google Scholar] [CrossRef] [PubMed]
- O'Rourke, M. Arterial stiffness, systolic blood pressure, and logical treatment of arterial hypertension. Hypertension 1990, 15, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Day, H.; Yellman, B.; Hammer, S.; Rond, C.; Bell, J.; Abbaszadeh, S.; Stoddard, G.; Unutmaz, D.; Bateman, L.; Vernon, S.D. Cognitive impairment in post-acute sequelae of COVID-19 and short duration myalgic encephalomyelitis patients is mediated by orthostatic hemodynamic changes. Front Neurosci 2023, 17, 1203514. [Google Scholar] [CrossRef] [PubMed]
- Tas, S.; Tas, U. Effects of COVID-19 on the Autonomic Cardiovascular System: Heart Rate Variability and Turbulence in Recovered Patients. Tex Heart Inst J 2023, 50. [Google Scholar] [CrossRef]
- Delle Donne, G.; Roses Noguer, F.; Till, J.; Salukhe, T.; Prasad, S.K.; Daubeney, P.E.F. Ivabradine in Postural Orthostatic Tachycardia Syndrome: Preliminary Experience in Children. Am J Cardiovasc Drugs 2018, 18, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Tahir, F.; Bin Arif, T.; Majid, Z.; Ahmed, J.; Khalid, M. Ivabradine in Postural Orthostatic Tachycardia Syndrome: A Review of the Literature. Cureus 2020, 12, e7868. [Google Scholar] [CrossRef] [PubMed]
- Ruzieh, M.; Sirianni, N.; Ammari, Z.; Dasa, O.; Alhazmi, L.; Karabin, B.; Grubb, B. Ivabradine in the treatment of postural tachycardia syndrome (POTS), a single center experience. Pacing Clin Electrophysiol 2017, 40, 1242–1245. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, M.; Jacob, G. The Effect of Ivabradine on the Heart Rate and Sympathovagal Balance in Postural Tachycardia Syndrome Patients. Rambam Maimonides Med J 2015, 6. [Google Scholar] [CrossRef]
- Lacasa, M.; Launois, P.; Prados, F.; Alegre, J.; Casas-Roma, J. Unsupervised cluster analysis reveals distinct subtypes of ME/CFS patients based on peak oxygen consumption and SF-36 scores. Clinical Therapeutics. [CrossRef]
- Wirth, K.J.; Scheibenbogen, C. Pathophysiology of skeletal muscle disturbances in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). J Transl Med 2021, 19, 162. [Google Scholar] [CrossRef]
- Avkiran, M.; Gross, G.; Karmazyn, M.; Klein, H.; Murphy, E.; Ytrehus, K. Na+/H+ exchange in ischemia, reperfusion and preconditioning. Cardiovasc Res 2001, 50, 162–166. [Google Scholar] [CrossRef]
- Karmazyn, M.; Sawyer, M.; Fliegel, L. The Na(+)/H(+) exchanger: a target for cardiac therapeutic intervention. Curr Drug Targets Cardiovasc Haematol Disord 2005, 5, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T. Na+-K+ Pump Regulation and Skeletal Muscle Contractility. Physiological Reviews 2003, 83, 1269–1324. [Google Scholar] [CrossRef] [PubMed]
- Petter, E.; Scheibenbogen, C.; Linz, P.; Stehning, C.; Wirth, K.; Kuehne, T.; Kelm, M. Muscle sodium content in patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J Transl Med 2022, 20, 580. [Google Scholar] [CrossRef] [PubMed]
- Pirkmajer, S.; Chibalin, A.V. Na,K-ATPase regulation in skeletal muscle. Am J Physiol Endocrinol Metab 2016, 311, E1–E31. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T. Quantification of Na+,K+ pumps and their transport rate in skeletal muscle: Functional significance. Journal of General Physiology 2013, 142, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Cotecchia, S.; Stanasila, L.; Diviani, D. Protein-protein interactions at the adrenergic receptors. Curr Drug Targets 2012, 13, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Oaklander, A.L.; Nolano, M. Scientific Advances in and Clinical Approaches to Small-Fiber Polyneuropathy: A Review. JAMA Neurol 2019. [Google Scholar] [CrossRef]
- Abrams, R.M.C.; Simpson, D.M.; Navis, A.; Jette, N.; Zhou, L.; Shin, S.C. Small fiber neuropathy associated with SARS-CoV-2 infection. Muscle & Nerve 2022, 65, 440–443. [Google Scholar] [CrossRef]
- Oaklander, A.L.; Mills, A.J.; Kelley, M.; Toran, L.S.; Smith, B.; Dalakas, M.C.; Nath, A. Peripheral Neuropathy Evaluations of Patients With Prolonged Long COVID. Neurol Neuroimmunol Neuroinflamm 2022, 9. [Google Scholar] [CrossRef]
- Brain, S.D.; Cox, H.M. Neuropeptides and their receptors: innovative science providing novel therapeutic targets.
- Nielsen, O.B.; Hilsted L Fau - Clausen, T.; Clausen, T. Excitation-induced force recovery in potassium-inhibited rat soleus muscle.
- Jacobs, L.M.C.; Wintjens, M.; Nagy, M.; Willems, L.; Ten Cate, H.; Spronk, H.M.H.; van Kuijk, S.M.J.; Ghossein-Doha, C.; Netea, M.G.; Groh, L.A.; et al. Biomarkers of sustained systemic inflammation and microvascular dysfunction associated with post-COVID-19 condition symptoms at 24 months after SARS-CoV-2-infection. Front Immunol 2023, 14, 1182182. [Google Scholar] [CrossRef] [PubMed]
- Gunter, T.E.; Yule, D.I.; Gunter, K.K.; Eliseev, R.A.; Salter, J.D. Calcium and mitochondria. FEBS Lett 2004, 567, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Jammes, Y.; Adjriou, N.; Kipson, N.; Criado, C.; Charpin, C.; Rebaudet, S.; Stavris, C.; Guieu, R.; Fenouillet, E.; Retornaz, F. Altered muscle membrane potential and redox status differentiates two subgroups of patients with chronic fatigue syndrome. J Transl Med 2020, 18, 173. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.A.-O.; Wood, J.; Jaycox, J.R.; Dhodapkar, R.A.-O.; Lu, P.A.-O.X.; Gehlhausen, J.R.; Tabachnikova, A.; Greene, K.; Tabacof, L.; Malik, A.A.; et al. Distinguishing features of long COVID identified through immune profiling.
- Miwa, K. Down-regulation of renin–aldosterone and antidiuretic hormone systems in patients with myalgic encephalomyelitis/chronic fatigue syndrome. Journal of cardiology 2017, 69, 684–688. [Google Scholar] [CrossRef]
- Su, Y.; Yuan, D.; Chen, D.G.; Ng, R.H.; Wang, K.; Choi, J.; Li, S.; Hong, S.; Zhang, R.; Xie, J.; et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 2022, 185, 881–895.e820. [Google Scholar] [CrossRef]
- Joffre, J.; Hellman, J. Oxidative Stress and Endothelial Dysfunction in Sepsis and Acute Inflammation. Antioxid Redox Signal 2021, 35, 1291–1307. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kennedy, D.J.; Yan, Y.; Shapiro, J.I. Reactive Oxygen Species Modulation of Na/K-ATPase Regulates Fibrosis and Renal Proximal Tubular Sodium Handling. Int J Nephrol 2012, 2012, 381320. [Google Scholar] [CrossRef]
- Myhill, S.; Booth, N.E.; McLaren-Howard, J. Chronic fatigue syndrome and mitochondrial dysfunction. Int J Clin Exp Med 2009, 2, 1–16. [Google Scholar] [PubMed]
- Rutherford, G.; Manning, P.; Newton, J.L. Understanding Muscle Dysfunction in Chronic Fatigue Syndrome. J Aging Res 2016, 2016, 2497348. [Google Scholar] [CrossRef]
- Fluge, O.; Mella, O.; Bruland, O.; Risa, K.; Dyrstad, S.E.; Alme, K.; Rekeland, I.G.; Sapkota, D.; Rosland, G.V.; Fossa, A.; et al. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight 2016, 1, e89376. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



