
Submitted:
12 December 2023
Posted:
13 December 2023
You are already at the latest version
Abstract
Colletotrichum lini is a flax fungal pathogen, comprising differently virulent strains. In this work, we sequenced the genomes of three C. lini strains of different virulence: high virulent #390-1, medium virulent #757, and low virulent #771. We obtained more than 100× genome coverage with Oxford Nanopore Technologies reads and more than 50× genome coverage with Illumina data. To construct high-quality genomes, several assembly strategies were tested. The final assemblies were obtained using the Canu – Racon ×2 – Medaka – Polca scheme and were 54.0-55.3 Mb in length, contained 26-32 contigs, had an N50 > 5 Mb and the BUSCO completeness > 96%. Comparative analysis of the constructed genomes showed high similarity between mitochondrial and nuclear genomes. However, a rearrangement event and the loss of a 0.9 Mb contig were revealed. The candidate effectors were predicted in sets of proteins received after genome annotation. The obtained genome sequences of C. lini strains extend the knowledge on the genetic diversity of Colletotrichum species and are the basis for establishing molecular mechanisms of pathogenicity of the flax pathogen and effective control of flax anthracnose.
Keywords:
Colletotrichum lini
; anthracnose
; flax pathogen
; virulence
; Nanopore sequencing
; de novo genome assembly
1. Introduction
The most important aim of the modern agriculture is to meet the food and raw material demand of the growing human population. However, yield volumes of cultivated plants depend on numerous factors [1,2]. Fungal diseases of plants are often the primary cause of the crop losses [3]. Thus, the susceptibility to various diseases can become a threat to the harvest and economic profits. The Colletotrichum genus is pathogenic to different plant species and often demonstrates severe virulence [4,5,6]. Colletotrichum lini is the causative agent of flax anthracnose [7]. The pathogen can reside in untreated flax seeds and starts the infection process in seedlings and mature plants [7]. The mature plant infected with C. lini shows stem canker and leaf spotting. For the plant seedlings, the infection can become fatal [8]. Thus, the production of two main products – flax oil and fiber – can be affected by harvest failure. In light of this fact, multifaceted studies on the fungus are highly important for anthracnose management.
Pathogenic Colletotrichum species have three main lifestyles [9] – biotrophic (hemibiotrophic), necrotrophic, and quiescent lifestyles. The Colletotrichum genus generally lacks true biotrophic species [9]. Its representatives are usually considered hemibiotrophic, as they establish the necrotrophic stage after the biotrophic one [10]. Biotrophic fungi suppress the defense mechanisms of the host and mask the hyphae to obtain nutrients from the host [11]. This stage can be indispensable for further establishment of infection and the death of host cells [10]. Necrotrophic stage implies the secretion of fungal toxins and enzymes to absorb nutrients from the dead host cells [11]. Quiescent stage is the period of dormancy of a fungus until a signal from the surrounding media is detected [9]. Then, the fungus can complete its disease cycle [9]. Along with pathogenic species, endophytic Colletotrichum strains also occur [12,13]. Endophytes live in plant tissues and receive the nutrition from a plant without causing disease symptoms. Both Colletotrichum endophytes and pathogens produce metabolites with useful bioactivities [14,15].
Colletotrichum representatives are attributed to species complexes according to intra-specific and interspecific differences in phenotype and genotype [11]. To discriminate between fungal species, genetic barcodes were applied, e.g., GAPDH, HIS3, APN2, MAT1-2-1, GAP2-IGS, ACT, CHS-1, nrITS, TUB2 [16]. However, there is no single universal barcode for all Colletotrichum species, as they demonstrate different efficiency in various species complexes [17,18]. Thus, an ITS-based approach coupled with molecular characterization was applied for Colletotrichum isolates from strawberry tissues. However, the authors observed no association with geographic origin, presence of symptoms, plant species or parts. In addition, the ITS marker failed to provide enough resolution for differentiation between C. gloeosporioides isolates [18]. Therefore, multi-locus analysis can be used for reliable results [16]. Liu et al. constructed a genome tree for 94 Colletotrichum species. The analysis of 1893 single-copy orthologs allowed to allocate the taken species in a range of species complexes [19].
Nevertheless, most comprehensive information can be extracted from full genomic sequences of the Colletotrichum species [20]. Comparative genomic analysis assists in studying the origins of pathogenicity and virulence [21]. Thus, Colletotrichum species possess a suite of potential pathogenicity genes, including effectors and CAZymes [22]. The comparison between the gene repertoire of fungal species can shed light on the difference in pathogenicity degrees of fungal isolates. Meanwhile, horizontal gene transfer events can play an important role in the evolution of pathogenicity. For instance, in C. musae, the analysis of mini-chromosome sequences revealed a set of genes which can undergo horizontal gene transfer [23].
In this study, we obtained the annotated genomes of three C. lini strains of different virulence. The comparative analysis between the obtained assemblies revealed a difference in effector gene content, a chromosome rearrangement, and the absence of a possible pathogenicity chromosome in the genome of the moderately virulent strain. The obtained data is a valuable source of information on the pathogenicity determinants of the flax anthracnose pathogen. Further in-depth research on C. lini genomes will suggest possible solutions to breeding anthracnose-resistant flax varieties.
2. Materials and Methods
2.1. Fungal Material
Fungal strains were provided as pure cultures by the Institute for Flax (Torzhok, Russia). Mycelium was provided in test tubes with potato dextrose agar. The following C. lini strains were used: highly virulent strain #390-1, moderately virulent strain #757, and lowly virulent strain #771. The virulence of the three strains was assessed by infecting two flax varieties: the resistant variety Leona and the susceptible variety Punjab. Plants were sprayed with a spore suspension (150-300 spores per cm3), and the degree of pathogenicity was estimated on the 8-9th day after infection. The type of virulence was attributed according to the ratio of affected plants. If up to 30% of plants showed the symptoms of anthracnose, the virulence was considered low; 31-50% – medium; 51% and more – high.
2.2. DNA Extraction and Purification
DNA extraction was performed according to the previously developed protocol [24,25]. The DNA was used for library preparation for both the Oxford Nanopore Technologies (ONT) and Illumina platforms. The quality and quantity of the extracted DNA were evaluated with spectrophotometry (NanoDrop 2000C, Thermo Fisher Scientific, Waltham, MA, USA) and fluorometry (Qubit 4.0, Thermo Fisher Scientific, Waltham, MA, USA).
2.3. DNA Library Preparation and Sequencing on the Oxford Nanopore Technologies and Illumina platforms
To prepare DNA libraries for sequencing on the ONT platform, the SQK-LSK109 Ligation Sequencing Kit (ONT, Oxford, UK) was used. Sequencing was performed on a PromethION instrument with the R9.4.1 flow cell (ONT, Oxford, UK).
Illumina libraries were sequenced on a NovaSeq 6000 (Illumina, San Diego, CA, USA) instrument (150+150 bp).
2.4. Genome Assembly
The obtained ONT reads were basecalled using Guppy 6.0.1 and the dna_r9.4.1_450bps_sup.cfg config file with quality filtration threshold min_qscore = 10. Porechop 0.2.4 was used for removing adapters (https://github.com/rrwick/Porechop). The obtained short Illumina reads were processed using Cutadapt 2.8 (adapters removal: -a AGATCGGAAGAG -A AGATCGGAAGAG) [26] and Trimmomatic 0.39 (filtration by length: MINLEN:50) [27]. For the highly virulent strain #390-1, draft assemblies were performed using two types of assemblers. For assembling a genome solely from ONT reads, the following assemblers were used: Canu 2.2 (-nanopore-raw; -minInputCoverage=5; -stopOnLowCoverage=5; -genomeSize=50m) [28], Flye 2.8.1 (--genome-size 50000000) [29], and Goldrush 1.0.3 (G=5e7) [30]. For a hybrid assembly from both ONT and Illumina reads, the following tools were used: Haslr 0.8a1 (-g 50m, -x nanopore), Masurca 4.1.0 (GRID_BATCH_SIZE=500000000), Spades 3.15.5, and Unicycler 0.5.0 (--mode bold) [31,32,33,34,35,36,37]. For the moderately virulent strain #757 and the lowly virulent strain #771, draft assemblies were produced by Canu 2.2 (-nanopore-raw; -minInputCoverage=5; -stopOnLowCoverage=5; -genomeSize=50m). To analyze the quality of the obtained assemblies, completeness and contiguity statistics were calculated using BUSCO 5.3.2 (glomerellales_odb10) 26 and QUAST 5.0.2, respectively [38,39]. The following reference genome was used for calculating QUAST reference-based statistics: C. higginsianum IMI 349063 (NCBI Genome, GCA_001672515.1).
The obtained draft assemblies of the three strains were polished with ONT reads with Racon 1.4.20 (two iterations) [40] and Medaka 1.5.0 (https://github.com/nanoporetech/medaka, accessed on 14 September 2023). Polca (Masurca 4.1.0) was used for polishing with Illumina reads [41,42]. If required, read alignment was produced with Minimap2 [43] prior to polishing.
2.5. Genome Analysis
For genome annotation, Funannotate v1.8.9 was used according to the basic assembly preparation protocol (https://funannotate.readthedocs.io/en/latest/, accessed on 14 September 2023) including cleaning up repetitive contigs, sorting the assembly by length, repeat masking, and gene prediction (https://github.com/nextgenusfs/funannotate, accessed on 14 September 2023). The received protein sequences were analyzed using KEGG (Kyoto Encyclopedia of Genes and Genomes) BlastKOALA (KEGG Orthology And Links Annotation) [44] and InterProScan [45]. The annotated genome assemblies were aligned to each other using LAST (https://gitlab.com/mcfrith/last, accessed on 14 September 2023). Tidk 0.2.31 was used for the identification of telomeric repeats and their visualization (https://github.com/tolkit/telomeric-identifier, accessed on 14 September 2023). RepeatMasker 4.1.5 was used for the identification of repeat content (https://www.repeatmasker.org/, accessed on 14 September 2023) [46]. SignalP-6.0 predicted the presence of signal peptides in protein sequences received after genome annotation (https://services.healthtech.dtu.dk/services/SignalP-6.0/, accessed on 14 September 2023) [47]. Protein sequences containing signal peptides were analyzed with EffectorP-3.0 to predict effector proteins [48]. Mega was used for sequence alignments (https://www.megasoftware.net/, accessed on 14 September 2023) [49].
3. Results
3.1. Genome Assembly and Polishing
Three strains of C. lini with different pathogenicity characteristics and close morphological traits (conidia characteristics, sporulation rate, growth rate, and mycelium color) were chosen for the study. For the highly virulent strain #390-1, we obtained 6.9 Gb of raw ONT reads with an N50 of 11.9 kb and 10 million Illumina reads (150+150 bp). For the moderately virulent strain #757, we obtained 8.7 Gb of raw ONT reads with an N50 of 6.1 kb and 23 million Illumina reads (150+150 bp). For the lowly virulent strain #771, we obtained 7.2 Gb of raw ONT data with an N50 of 5.0 kb and 25.2 million Illumina reads (150+150 bp).
To test assembling and polishing algorithms, we used sequencing data of the highly virulent strain #390-1. We obtained three draft assemblies using only long ONT reads and four draft assemblies using both long ONT and short Illumina reads (Figure 1, Supplementary Table S1). The quality of the draft genome assemblies was analyzed in terms of completeness and contiguity using BUSCO and QUAST. For each assembly, QUAST statistics were evaluated without and with a reference genome of C. higginsianum IMI 349063 (NCBI Genome, GCA_001672515.1). The contiguity of the assemblies was judged by the number of contigs, N50, and L50. The assembly completeness was evaluated by the length and percentage of complete universal single-copy orthologs inherent to an analyzed species group.
The majority of tools produced assemblies with a completeness of >90%, except Goldrush. The tool constructed an assembly with a BUSCO completeness of 41.3%. The highest assembly completeness was achieved with hybrid assemblers (Masurca and Unicycler – BUSCO completeness of 95.9% in both cases). The average length of assemblies with a completeness of more than 90% was 53.8 Mb (49.9 – 56.7 Mb). The most contiguous assemblies were performed by Canu and Masurca. These assemblies had 31 and 35 contigs, respectively. The L50 values were also close – 5 and 6, respectively. However, the N50 and N75 differed significantly between the two assemblies. The N50 values were 5.16 Mb and 2.73 Mb, and the N75 values were 4.17 Mb and 1.49 Mb, respectively. Thus, Canu assembled the genome of the best contiguity. Since the completeness of the draft assemblies can be improved by polishing, the assembly by Canu was regarded as optimal.
The optimal genome was polished according to the scheme that showed the best results in our previous studies [24,25]: polishing with long ONT reads – two rounds of Racon, one round of Medaka; polishing with high-precision Illumina reads – one round of Polca (Figure 2, Supplementary Table S2). Each round of polishing improved BUSCO completeness. The parameter reached 96.7% after Polca. Such features as the number of contigs, N50, N75, L50, and other contiguity characteristics did not change during polishing (Figure 2). The reference-based QUAST statistics, such as genomic features and covered genome fraction, which also describe the assembly completeness, increased after polishing. Indels per 100 kbp and mismatches per 100 kbp improved from 182.4 to 169.0 and from 4425.1 to 4373.7, respectively (Supplementary Table S2). It is not the absolute values of these parameters that play a key role during polishing but the general trend in improvement.
The algorithm applied for strain #390-1 was used to assemble and polish the genomes of the moderately virulent strain #757 and the lowly virulent strain #771 (assembling with Canu and polishing with Racon ×2 – Medaka – Polca). The final assemblies were 54.0-55.3 Mb in length, consisted of 26-32 contigs, and had an N50 of 5.2-5.8 Mb and an L50 of 5 (Figure 3, Supplementary Table S2). The BUSCO completeness of the final assemblies was 96.6-96.8%.
3.2. Genome Annotation and Search for Effector Proteins
To predict genetic features in the obtained assemblies, we used Funannotate, which was tailored to annotate fungal genomes (Supplementary File S1). Before annotation, the final assemblies were preprocessed by filtering out the repetitive contigs, sorting the input contigs by their size (from the longest to the shortest), relabeling the contigs, and masking repeats. Gene prediction was performed using the basic Funannotate commands. The tool annotated the majority of genes using the Pfam (~60% of genes) and BUSCO (~10% of genes) databases. After annotation, we obtained: 12891 gene models (12521 mRNAs and 370 tRNAs) for the highly virulent strain #390-1, 12520 gene models (12146 mRNAs and 374 tRNAs) for the moderately virulent strain #757, 12736 gene models (12374 mRNAs and 362 tRNAs) for the lowly virulent strain #771. All obtained protein sequences were analyzed using KEGG (Kyoto Encyclopedia of Genes and Genomes) BlastKOALA (KEGG Orthology And Links Annotation) and InterProScan. In the genome of each strain, KEGG annotated ~33% of proteins, and InterPro annotated ~80 % of proteins (Supplementary Tables S3 and S4). The largest protein categories in the KEGG annotation were associated with genetic information processing, carbohydrate metabolism, and signaling and cellular processes (Figure 4).
To identify the possible effector proteins, we determined the presence of signal peptides using SignalP and then predicted effectors from the positive hits with EffectorP (Supplementary Table S5). In the highly virulent strain #390-1, 1308 proteins contained a signal peptide, 489 (37.4%) of them were effectors: 187 (14.3%) cytoplasmic effectors and 302 (23.1%) apoplastic effectors. In the moderately virulent strain #757, 1303 proteins contained a signal peptide, 472 (36.2%) of them were effectors: 184 (14.1%) cytoplasmic effectors and 288 (22.1%) apoplastic effectors. In the lowly pathogenic strain #771, 1288 proteins contained a signal peptide, 476 (37.0%) of them were effectors: 191 (14.8%) cytoplasmic effectors and 285 (22.1%) apoplastic effectors. The predicted effectors were also analyzed with KEGG BlastKOALA and InterProScan. We searched for unique accessions in the annotations of the effector proteins. The protein was regarded as unique for a strain if it did not occur in the database annotation of the other two strains. In the KEGG BlastKOALA annotations, unique accessions were found only in the highly virulent strain #390-1 (C_lini_390-1_FUN_004740-T1, C_lini_390-1_FUN_004122-T1, C_lini_390-1_FUN_001342-T1). However, in the InterProScan annotations of effectors, 8 accessions were unique for the same strain (3 of them were annotated with KEGG) (C_lini_390-1_FUN_004637-T1, C_lini_390-1_FUN_004750-T1, C_lini_390-1_FUN_004740-T1, C_lini_390-1_FUN_004688-T1, C_lini_390-1_FUN_001551-T1, C_lini_390-1_FUN_004122-T1, C_lini_390-1_FUN_001342-T1, C_lini_390-1_FUN_009633-T1), 7 accessions – in the moderately virulent strain #757 (C_lini_757_FUN_004039-T1, C_lini_757_FUN_005381-T1, C_lini_757_FUN_010924-T1, C_lini_757_FUN_000684-T1, C_lini_757_FUN_003376-T1, C_lini_757_FUN_000430-T1, C_lini_757_FUN_000370-T1), and 2 accessions – in the lowly virulent strain #771 (C_lini_771_FUN_009418-T1, C_lini_771_FUN_009066-T1) (Supplementary Table S6). Unique accessions were related to the metabolism (catabolism) of carbohydrates and nitrogen and cell signaling pathways. This fact points to the possible role of these enzymes in the processes of plant colonization and nutrition, i.e., infection, and suggests the direction of further studies. However, other proteins related to virulence can also exist. To establish the factors of strain differentiation by virulence, further research is needed.
3.3. Comparative Genomic Analysis
In each assembly, we identified a mitochondrial contig by blasting the sequence of the mitochondrion from the previously obtained genome of C. lini #811 (highly virulent) [25]. The extracted mitochondrial genomes were compared using the visualization tool geneCo and aligned to each other in Mega. The mitochondrial genomes had similar sizes: 38,956-39,090 bp. Only several mismatches were observed in the multiple alignment. Besides, the annotations of the received mitochondrial genomes had no differences.
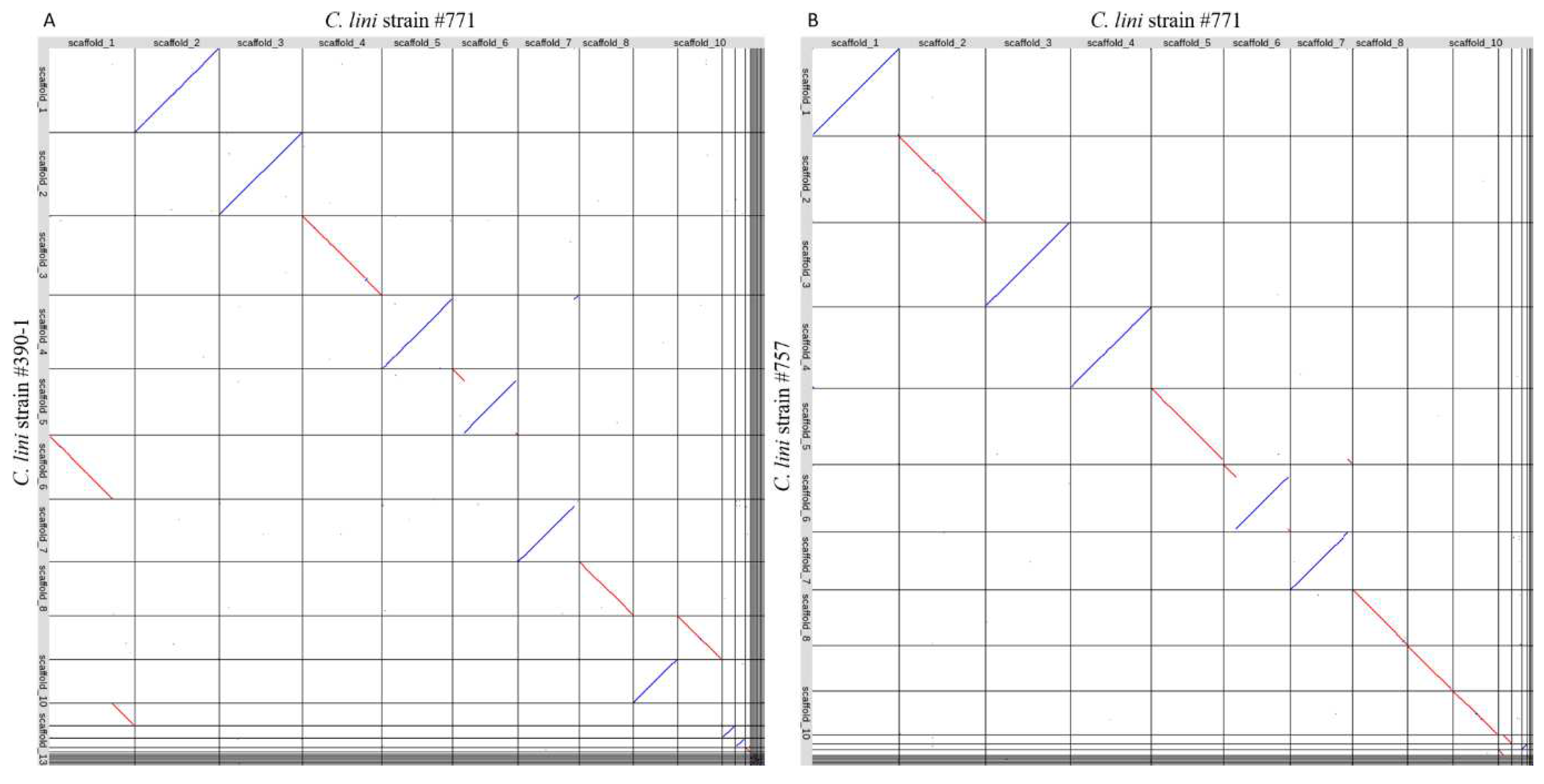
The final genome assemblies of C. lini strains were aligned to each other using LAST (Figure 5). The obtained plots showed that the majority of scaffolds of the moderately virulent strain #757 aligned to the scaffolds of the lowly virulent strain #771. However, the scaffolds of the highly virulent strain #390-1 were shifted by one. Scaffold 1 of the strain #771 consisted of scaffolds 6 and 11 of strain #390-1. In scaffold 6 in the lowly virulent strain #771, we detected a rearrangement. The beginning of scaffold 6 and a little part at the end of this scaffold aligned to scaffold 5 in strain #390-1 and scaffold 6 in strain #757 in forward orientation. Meanwhile, the middle part of scaffold 6 aligned in reverse orientation to the same scaffolds in the two other strains. Scaffold 12 in the lowly virulent strain #771 corresponded to scaffold 13 in the highly virulent strain #390-1. Yet a similar scaffold was missing from the genome of the moderately virulent strain #757. To confirm the absence of the missing scaffold, its sequence was blasted to the genome of strain #757 genome assembly. However, no significant hits were found. The missing scaffold was 0.7 Mb in size. We blasted amino acid sequences of predicted proteins from this scaffold against the NCBI database. The found protein hits were helicases.
Next, we searched for telomeric repeats (‘TTAGGG’) in the obtained genome assemblies using Tidk [50]. The output Tidk files with diagrams of the occurrence frequency of the target sequence are presented in Supplementary Figures S1, S2, and S3. High peaks at both ends of a scaffold indicated the presence of telomeric repeats. Such a scaffold could possibly be a complete chromosome. Therefore, the assembled genomes can have from 4 to 6 complete chromosomes. We also observed several scaffolds with telomeric repeats at only one end. Assuming the karyotype of n=13 [51], nearly a third or a half of the assembled contigs can be complete chromosomes. The scaffold missing from the assembly of the moderately pathogenic strain #757 (scaffold 12 in the assembly of the lowly virulent strain #771 and scaffold 13 in the assembly of the highly virulent strain #390-1) had telomeric repeats at both ends. Thus, this genomic locus is likely a complete 0.7 Mb-long chromosome and might be a minichromosome associated with pathogenicity [52].
4. Discussion
Colletotrichum species are widely distributed plant pathogens which cause significant economic losses. The representatives of the genus are actively studied, including at the level of complete genomes. At the time of writing the manuscript, 270 assemblies of Colletotrichum species were deposited in the NCBI Genome database (the size of the genomes is about 50-60 Mb). The advances in long-read sequencing technologies allowed obtaining high-quality genome assemblies of Colletotrichum species [53,54]. Thus, high-quality genomes became the basis for further molecular genetic studies. Genomics and transcriptomics of Colletotrichum species provided valuable information on the genes regulating their life cycle and the ability to produce proteins and secondary metabolites damaging plant cells [55,56,57,58,59,60,61]. To identify molecular genetic factors that determine pathogenicity, special attention is paid to research on the interaction of Colletotrichum species and their hosts [62,63,64]. Using high-quality genome assemblies of Colletotrichum species, a range of pathogenicity-associated genome regions were identified, including rapidly evolving regions in telomeres, repeat-rich pathogenicity minichromosomes, clusters of effector genes, and a number of genes co-expressing upon infection of a host [50,65,66,67].
The causative agent of flax anthracnose, C. lini (syn. C. linicola) has been unfairly deprived of attention in molecular genetic studies. The species was only studied using DNA markers [25,68,69,70]. In this study, we sequenced the genomes of three C. lini strains of different virulence and conducted a comparative analysis of the obtained fungal genomes to reveal pathogenicity-associated factors. To exclude the contribution of multiple factors in further genomic analysis, we studied the strains with close morphological characteristics. The strains represented three degrees of virulence – low, medium, and high.
The combination of long ONT data and short precision Illumina reads allows obtaining high-quality genomes of the fungal pathogens [71,72,73]. In this study, we obtained from ~140× to 170× genome coverage with raw ONT reads (assuming the genome size is ~50 Mb), having an N50 from ~5 to 12 kb. Coverage with Illumina data ranged from ~60× to 150×. To construct the most contiguous and complete assemblies, two approaches were tested on the highly virulent strain #390-1. The first one was based on constructing a draft assembly from long reads and polishing it with both long and short precision reads. The second approach implied the use of hybrid assembly software, taking both ONT and Illumina data as input. We used recently developed tools and software that demonstrated optimal results in our previous studies [24,25]. The most contiguous assembly was obtained using Canu – 31 contigs, N50 = 5.2 Mb, L50 = 5. However, its completeness (92.1%) was lower than for the assemblies obtained with hybrid tools. Thus, Masurca and Unicycler assembled genomes with a BUSCO completeness of 95.9%. Since polishing can increase the parameter, the draft assembly by Canu was considered optimal. According to the scheme that showed the best results in our previous studies, the chosen draft assembly was polished using Racon ×2 – Medaka (ONT reads) and Polca (Illumina reads) [24,25]. Thus, the BUSCO completeness of the assembly rose from 92.1 to 96.7%. The final value was higher than that achieved by Masurca and Unicycler. Thus, Canu – Racon ×2 – Medaka – Polca allowed us to assemble a contiguous and complete genome. The scheme was employed to assemble the genomes of strains #757 and #771. The final genomes consisted of 26-32 contigs, had N50 values in the megabase range (5.2-5.8 Mb), and were more than 96% complete.
Thus, the obtained genomes had high contiguity. After the search for telomeric repeats and their visualization (Supplementary Figures S1, S2, and S3), we observed peaks at one or both ends of the obtained contigs. This indicated that the assembled contigs were possibly big parts or complete chromosomes. At the time of writing the manuscript, two chromosome-level assemblies were available in the NCBI database (Colletotrichum higginsianum IMI 349063 GCA_001672515.1, Colletotrichum graminicola GCA_029226625.1). The Contig N50 values of these two assemblies are 5.2 and 5 Mb, respectively. The L50 values for both assemblies are 5. In this study, we constructed contig-level assemblies for C. lini. However, the analysis of telomeric repeats suggested the presence of complete chromosomes. Thus, high coverage with long ONT reads probably allowed assembling the sequences of complete chromosomes. Besides, the contiguity of the obtained assemblies is comparable to that of the chromosome-level assemblies prior to anchoring to chromosomes.
The assemblies were annotated using Funannotate. The resulting annotations had close numbers of predicted gene models. The highly virulent strain had the highest number of gene models (#390-1) – 12891, and the moderately virulent strain (#757) had the lowest number of gene models – 12520. This correlated with the BUSCO completeness of the strains assemblies. Strain #757 had lower completeness than strain #390-1. Although the completeness of an assembly impacts the accuracy of gene prediction, the highest number of gene models in the genome of the highly virulent strain can still correlate with its high pathogenicity. In C. graminicola, ~15000 genes were predicted [74]. Thus, the number of predicted gene models for C. lini was in the order of the values from the literature data. To conduct a primary analysis of virulence genes, we searched for the encoded effector proteins in the obtained genome assemblies. Effector proteins are the small cysteine-rich proteins influencing plant cellular processes to facilitate the infection process [75]. The lowly virulent strain #771 contained the lowest number of proteins with signal sequences, i.e., potentially secreting, and the lowest number of uniquely annotated effectors (according to InterProScan. The highly virulent strain had 37.4% effectors from the potentially secreting proteins. Meanwhile, the moderately virulent strain #757 had the lowest number of effectors – 36.2%. The percentages correlated with the total numbers of the predicted gene models. Higher percentages of effector proteins are likely related to higher pathogenicity. However, the obtained results can still be prone to fluctuations in predicted values. Besides, the detection of an effector protein can also trigger plant immunity mechanisms [76]. Therefore, further research is needed to collect more information on the effector proteins of the studied fungi and elucidate true virulence mechanisms. In the genome of the lowly virulent strain, the smallest number of uniquely annotated effectors can correlate with its low virulence. In a similar study, the strain of C. scovillei was characterized by defective growth and virulence, along with a reduced number of effectors [77].
To reveal the possible effect of genome rearrangements, we performed whole-genome alignment of the three C. lini genomes with each other. In the assembly of the lowly virulent strain #771, scaffold 6 contained one big inversion. Such genome rearrangements might be crucial for the function of certain genomic regions. Small scaffold 12 (0.7 Mb) in the lowly virulent strain #771 aligned to scaffold 13 in the highly virulent strain #390-1. However, this sequence was completely missing in the genome of the moderately virulent strain #757. Since this scaffold had an increased occurrence of repeats at its ends, we assumed that it could be a small pathogenicity-associated chromosome [52]. Besides, BLAST analysis of the annotated proteins from the scaffold showed that it contained helicases, peptidases, and hydrolases. Therefore, the minichromosome can be implicated in replication events, the growth of the fungus, and necrotrophy.
In this work, using ONT and Illumina data, we obtained the first three high-quality C. lini genomes. We performed primary comparative analysis of the obtained assemblies. The difference in the number of effector proteins and the presence of a possible pathogenicity minichromosome suggested possible determinants of the high virulence. The assembled whole high-quality genome sequences created the foundation for further in-depth search for molecular determinants of pathogenicity both at the chromosome and gene levels. Such data is indispensable for the advancement of disease management techniques and conceiving new strategies for breeding resistant varieties. Moreover, the obtained high-quality genomes of C. lini expand the knowledge of the genetic diversity of the genus Colletotrichum.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: QUAST statistics of Colletotrichum lini strain #390-1 draft genome assemblies; Table S2: QUAST statistics of polished genome assemblies of Colletotrichum lini highly virulent strain #390-1, moderately virulent strain #757, and lowly virulent strain #771 polished genomes assemblies; Table S3: The full list of InterPro-annotated proteins with IP, definition, and GO terms for Colletotrichum lini highly virulent strain #390-1, moderately virulent strain #757, and lowly virulent strain #771; Table S4: The full list of KEGG-annotated proteins of Colletotrichum lini highly virulent strain #390-1, moderately virulent strain #757, and lowly virulent strain #771; Table S5: The full list of effector proteins predicted by EffectorP for Colletotrichum lini highly virulent strain #390-1, moderately virulent strain #757, and lowly virulent strain #771; Table S6: The list of InterPro annotations for unique effector proteins predicted by EffectorP for Colletotrichum lini highly virulent strain #390-1, moderately virulent strain #757, and lowly virulent strain #771; Figure S1: Telomeric repeat (TTAGGG) content along the contig sequences of Colletotrichum lini strain #390-1; Figure S2: Telomeric repeat (TTAGGG) content along the contig sequences of Colletotrichum lini strain #757; Figure S3: Telomeric repeat (TTAGGG) content along the contig sequences of Colletotrichum lini strain #771; File S1: Genome annotations for three Colletotrichum lini strains of high (#390-1), medium (#757), and low (#771) virulence in gbk and gff3 formats.
Author Contributions
Conceptualization, N.V.M. and A.A.D.; Investigation, E.M.D., E.A.S., T.D.M., T.A.R., L.P.K., R.O.N., A.A.T., D.A.Z., E.V.B., E.N.P., N.V.M., and A.A.D.; Writing, E.M.D., E.A.S., N.V.M., and A.A.D. All authors have read and agreed to the published version of the manuscript.
Funding
The work was financially supported by the Russian Science Foundation, grant number 22-16-00169.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The generated dataset for this study can be found in the NCBI database under the BioProject accession numbers PRJNA929545.
Acknowledgments
We thank the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, EIMB RAS for providing the computing power and techniques for the data analysis. This work was performed using the equipment of EIMB RAS “Genome” center (http://www.eimb.ru/ru1/ckp/ccu_genome_ce.php, accessed on 14 September 2023).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Kumar, R.; Kumar, A.; Dhaka, R.K.; Chahar, M.; Malyan, S.K.; Singh, A.P.; Rana, A. Climate change and agriculture: impact assessment and sustainable alleviation approach using rhizomicrobiome. In Bioinoculants: Biological Option for Mitigating global Climate Change; Springer: 2023; pp. 87-114.
- Sembiring, H.; A. Subekti, N.; Erythrina; Nugraha, D.; Priatmojo, B.; Stuart, A.M. Yield gap management under seawater intrusion areas of Indonesia to improve rice productivity and resilience to climate change. Agriculture 2020, 10, 1. [Google Scholar] [CrossRef]
- Różewicz, M.; Wyzińska, M.; Grabiński, J. The most important fungal diseases of cereals—problems and possible solutions. Agronomy 2021, 11, 714. [Google Scholar] [CrossRef]
- Zakaria, L. Diversity of Colletotrichum species associated with anthracnose disease in tropical fruit crops—a review. Agriculture 2021, 11, 297. [Google Scholar] [CrossRef]
- Talhinhas, P.; Loureiro, A.; Oliveira, H. Olive anthracnose: a yield- and oil quality-degrading disease caused by several species of Colletotrichum that differ in virulence, host preference and geographical distribution. Molecular Plant Pathology 2018, 19, 1797–1807. [Google Scholar] [CrossRef]
- Singh, S.; Prasad, D.; Singh, V.P. Evaluation of fungicides and genotypes against anthracnose disease of mungbean caused by Colletotrichum lindemuthianum. International Journal of Bio-resource and Stress Management 2022, 13, 448–453. [Google Scholar] [CrossRef]
- Gruzdeviene, E.; Dabkevicius, Z. The control of flax anthracnose [Colletotrichum lini [West.] Toch.] by fungicidal seed treatment. Journal of plant protection research 2003, 43. [Google Scholar]
- Nyvall, R.F. Diseases of flax. In Field Crop Diseases Handbook; Nyvall, R.F., Ed.; Springer US: Boston, MA, 1989; pp. 251–264. [Google Scholar]
- De Silva, D.D.; Crous, P.W.; Ades, P.K.; Hyde, K.D.; Taylor, P.W.J. Life styles of Colletotrichum species and implications for plant biosecurity. Fungal Biology Reviews 2017, 31, 155–168. [Google Scholar] [CrossRef]
- Münch, S.; Lingner, U.; Floss, D.S.; Ludwig, N.; Sauer, N.; Deising, H.B. The hemibiotrophic lifestyle of Colletotrichum species. Journal of Plant Physiology 2008, 165, 41–51. [Google Scholar] [CrossRef] [PubMed]
- da Silva, L.L.; Moreno, H.L.A.; Correia, H.L.N.; Santana, M.F.; de Queiroz, M.V. Colletotrichum: species complexes, lifestyle, and peculiarities of some sources of genetic variability. Applied Microbiology and Biotechnology 2020, 104, 1891–1904. [Google Scholar] [CrossRef]
- Manamgoda, D.S.; Udayanga, D.; Cai, L.; Chukeatirote, E.; Hyde, K.D. Endophytic Colletotrichum from tropical grasses with a new species C. endophytica. Fungal Diversity 2013, 61, 107–115. [Google Scholar] [CrossRef]
- Zheng, H.; Yu, Z.; Jiang, X.; Fang, L.; Qiao, M. Endophytic Colletotrichum species from aquatic plants in southwest China. Journal of fungi 2022, 8. [Google Scholar] [CrossRef]
- Talukdar, R.; Padhi, S.; Rai, A.K.; Masi, M.; Evidente, A.; Jha, D.K.; Cimmino, A.; Tayung, K. Isolation and characterization of an endophytic fungus Colletotrichum coccodes producing tyrosol from Houttuynia cordata Thunb. using ITS2 RNA secondary structure and molecular docking study. Frontiers in bioengineering and biotechnology 2021, 9, 650247. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Shim, S.H. The fungus Colletotrichum as a source for bioactive secondary metabolites. Archives of pharmacal research 2019, 42, 735–753. [Google Scholar] [CrossRef] [PubMed]
- Vieira, W.; Bezerra, P.A.; Silva, A.C.D.; Veloso, J.S.; Camara, M.P.S.; Doyle, V.P. Optimal markers for the identification of Colletotrichum species. Molecular phylogenetics and evolution 2020, 143, 106694. [Google Scholar] [CrossRef]
- Bhunjun, C.S.; Phukhamsakda, C.; Jayawardena, R.S.; Jeewon, R.; Promputtha, I.; Hyde, K.D. Investigating species boundaries in Colletotrichum. Fungal Diversity 2021, 107, 107–127. [Google Scholar] [CrossRef]
- Van Hemelrijck, W.; Debode, J.; Heungens, K.; Maes, M.; Creemers, P. Phenotypic and genetic characterization of Colletotrichum isolates from Belgian strawberry fields. Plant Pathology 2010, 59, 853–861. [Google Scholar] [CrossRef]
- Liu, F.; Ma, Z.Y.; Hou, L.W.; Diao, Y.Z.; Wu, W.P.; Damm, U.; Song, S.; Cai, L. Updating species diversity of Colletotrichum, with a phylogenomic overview. Studies in mycology 2022, 101, 1–56. [Google Scholar] [CrossRef]
- Crouch, J.; O’Connell, R.; Gan, P.; Buiate, E.; Torres, M.F.; Beirn, L.; Shirasu, K.; Vaillancourt, L. The genomics of Colletotrichum. In Genomics of Plant-Associated Fungi: Monocot Pathogens, Dean, R.A., Lichens-Park, A., Kole, C., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2014; pp. 69–102. [Google Scholar]
- Gardiner, D.M.; McDonald, M.C.; Covarelli, L.; Solomon, P.S.; Rusu, A.G.; Marshall, M.; Kazan, K.; Chakraborty, S.; McDonald, B.A.; Manners, J.M. Comparative pathogenomics reveals horizontally acquired novel virulence genes in fungi infecting cereal hosts. 2012.
- Lelwala, R.V.; Korhonen, P.K.; Young, N.D.; Scott, J.B.; Ades, P.K.; Gasser, R.B.; Taylor, P.W.J. Comparative genome analysis indicates high evolutionary potential of pathogenicity genes in Colletotrichum tanaceti. PloS one 2019, 14, e0212248. [Google Scholar] [CrossRef]
- Wang, H.; Huang, R.; Ren, J.; Tang, L.; Huang, S.; Chen, X.; Fan, J.; Li, B.; Wang, Q.; Hsiang, T.; et al. The evolution of mini-chromosomes in the fungal genus Colletotrichum. mBio 2023, 14, e0062923. [Google Scholar] [CrossRef]
- Krasnov, G.S.; Pushkova, E.N.; Novakovskiy, R.O.; Kudryavtseva, L.P.; Rozhmina, T.A.; Dvorianinova, E.M.; Povkhova, L.V.; Kudryavtseva, A.V.; Dmitriev, A.A.; Melnikova, N.V. High-quality genome assembly of Fusarium oxysporum f. sp. lini. Frontiers in genetics 2020, 11, 959. [Google Scholar] [CrossRef] [PubMed]
- Sigova, E.A.; Pushkova, E.N.; Rozhmina, T.A.; Kudryavtseva, L.P.; Zhuchenko, A.A.; Novakovskiy, R.O.; Zhernova, D.A.; Povkhova, L.V.; Turba, A.A.; Borkhert, E.V.; et al. Assembling quality genomes of flax fungal pathogens from Oxford Nanopore Technologies data. Journal of fungi 2023, 9. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 2011, 17, 3. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome research 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nature Biotechnology 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Wong, J.; Coombe, L.; Nikolić, V.; Zhang, E.; Nip, K.M.; Sidhu, P.; Warren, R.L.; Birol, I. Linear time complexity de novo long read genome assembly with GoldRush. Nature Communications 2023, 14, 2906. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome research 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nature biotechnology 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Wong, J.; Coombe, L.; Nikolic, V.; Zhang, E.; Nip, K.M.; Sidhu, P.; Warren, R.L.; Birol, I. Linear time complexity de novo long read genome assembly with GoldRush. Nat Commun 2023, 14, 2906. [Google Scholar] [CrossRef]
- Haghshenas, E.; Asghari, H.; Stoye, J.; Chauve, C.; Hach, F. HASLR: Fast Hybrid Assembly of Long Reads. iScience 2020, 23, 101389. [Google Scholar] [CrossRef]
- Zimin, A.V.; Puiu, D.; Luo, M.C.; Zhu, T.; Koren, S.; Marcais, G.; Yorke, J.A.; Dvorak, J.; Salzberg, S.L. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res 2017, 27, 787–792. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes de novo assembler. Current protocols in bioinformatics 2020, 70, e102. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS computational biology 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome research 2017, 27, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sovic, I.; Nagarajan, N.; Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 2017, 27, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Salzberg, S.L. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PLoS computational biology 2020, 16, e1007981. [Google Scholar] [CrossRef]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. Journal of molecular biology 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 2004, 5, 4.10. 11–4.10. 14. [Google Scholar] [CrossRef] [PubMed]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature biotechnology 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Molecular plant-microbe interactions : MPMI 2022, 35, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Molecular biology and evolution 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Gan, P.; Hiroyama, R.; Tsushima, A.; Masuda, S.; Shibata, A.; Ueno, A.; Kumakura, N.; Narusaka, M.; Hoat, T.X.; Narusaka, Y.; et al. Telomeres and a repeat-rich chromosome encode effector gene clusters in plant pathogenic Colletotrichum fungi. Environmental Microbiology 2021, 23, 6004–6018. [Google Scholar] [CrossRef]
- Taga, M.; Tanaka, K.; Kato, S.; Kubo, Y. Cytological analyses of the karyotypes and chromosomes of three Colletotrichum species, C. orbiculare, C. graminicola and C. higginsianum. Fungal Genetics and Biology 2015, 82, 238–250. [Google Scholar] [CrossRef]
- Wang, H.; Huang, R.; Ren, J.; Tang, L.; Huang, S.; Chen, X.; Fan, J.; Li, B.; Wang, Q.; Hsiang, T.; et al. The evolution of mini-chromosomes in the fungal genus Colletotrichum. mBio 2023, 14, e00629–00623. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.; Wang, Y.; Hao, Y.; Yao, Y.; Wang, Y.; Zhang, K.; Tan, X.; Li, Z.; Wang, W. Genome sequence resource for Colletotrichum scovillei, the cause of anthracnose disease of chili. Molecular plant-microbe interactions : MPMI 2021, 34, 122–126. [Google Scholar] [CrossRef]
- Liang, X.; Cao, M.; Li, S.; Kong, Y.; Rollins, J.A.; Zhang, R.; Sun, G. Highly contiguous genome resource of Colletotrichum fructicola generated using long-read sequencing. Molecular plant-microbe interactions : MPMI 2020, 33, 790–793. [Google Scholar] [CrossRef]
- O'Connell, R.J.; Thon, M.R.; Hacquard, S.; Amyotte, S.G.; Kleemann, J.; Torres, M.F.; Damm, U.; Buiate, E.A.; Epstein, L.; Alkan, N.; et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nature genetics 2012, 44, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Gan, P.; Ikeda, K.; Irieda, H.; Narusaka, M.; O'Connell, R.J.; Narusaka, Y.; Takano, Y.; Kubo, Y.; Shirasu, K. Comparative genomic and transcriptomic analyses reveal the hemibiotrophic stage shift of Colletotrichum fungi. The New phytologist 2013, 197, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Gan, P.; Narusaka, M.; Kumakura, N.; Tsushima, A.; Takano, Y.; Narusaka, Y.; Shirasu, K. Genus-wide comparative genome analyses of Colletotrichum species reveal specific gene family losses and gains during adaptation to specific infection lifestyles. Genome biology and evolution 2016, 8, 1467–1481. [Google Scholar] [CrossRef]
- Gan, P.; Narusaka, M.; Tsushima, A.; Narusaka, Y.; Takano, Y.; Shirasu, K. Draft genome assembly of Colletotrichum chlorophyti, a pathogen of herbaceous plants. Genome announcements 2017, 5. [Google Scholar] [CrossRef]
- Gan, P.; Tsushima, A.; Narusaka, M.; Narusaka, Y.; Takano, Y.; Kubo, Y.; Shirasu, K. Genome sequence resources for four phytopathogenic fungi from the Colletotrichum orbiculare species complex. Molecular plant-microbe interactions : MPMI 2019, 32, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wang, B.; Dong, Q.; Li, L.; Rollins, J.A.; Zhang, R.; Sun, G. Pathogenic adaptations of Colletotrichum fungi revealed by genome wide gene family evolutionary analyses. PloS one 2018, 13, e0196303. [Google Scholar] [CrossRef]
- Jayawardena, R.; Bhunjun, C.S.; Gentekaki, E.; Hyde, K.; Promputtha, I. Colletotrichum: lifestyles, biology, morpho-species, species complexes and accepted species. mycosphere 2021, 12, 519–669. [Google Scholar] [CrossRef]
- Yan, Y.; Yuan, Q.; Tang, J.; Huang, J.; Hsiang, T.; Wei, Y.; Zheng, L. Colletotrichum higginsianum as a model for understanding host(-)pathogen interactions: a review. International journal of molecular sciences 2018, 19. [Google Scholar] [CrossRef]
- Abreha, K.B.; Ortiz, R.; Carlsson, A.S.; Geleta, M. Understanding the sorghum-Colletotrichum sublineola interactions for enhanced host resistance. Frontiers in plant science 2021, 12, 641969. [Google Scholar] [CrossRef]
- Boufleur, T.R.; Ciampi-Guillardi, M.; Tikami, I.; Rogerio, F.; Thon, M.R.; Sukno, S.A.; Massola Junior, N.S.; Baroncelli, R. Soybean anthracnose caused by Colletotrichum species: Current status and future prospects. Mol Plant Pathol 2021, 22, 393–409. [Google Scholar] [CrossRef]
- Dallery, J.F.; Lapalu, N.; Zampounis, A.; Pigne, S.; Luyten, I.; Amselem, J.; Wittenberg, A.H.J.; Zhou, S.; de Queiroz, M.V.; Robin, G.P.; et al. Gapless genome assembly of Colletotrichum higginsianum reveals chromosome structure and association of transposable elements with secondary metabolite gene clusters. BMC genomics 2017, 18, 667. [Google Scholar] [CrossRef] [PubMed]
- Plaumann, P.L.; Schmidpeter, J.; Dahl, M.; Taher, L.; Koch, C. A dispensable chromosome is required for virulence in the hemibiotrophic plant pathogen Colletotrichum higginsianum. Frontiers in microbiology 2018, 9, 1005. [Google Scholar] [CrossRef] [PubMed]
- Bhadauria, V.; MacLachlan, R.; Pozniak, C.; Cohen-Skalie, A.; Li, L.; Halliday, J.; Banniza, S. Genetic map-guided genome assembly reveals a virulence-governing minichromosome in the lentil anthracnose pathogen Colletotrichum lentis. The New phytologist 2019, 221, 431–445. [Google Scholar] [CrossRef]
- Novakovskiy, R.O.; Dvorianinova, E.M.; Rozhmina, T.A.; Kudryavtseva, L.P.; Gryzunov, A.A.; Pushkova, E.N.; Povkhova, L.V.; Snezhkina, A.V.; Krasnov, G.S.; Kudryavtseva, A.V.; et al. Data on genetic polymorphism of flax (Linum usitatissimum L.) pathogenic fungi of Fusarium, Colletotrichum, Aureobasidium, Septoria, and Melampsora genera. Data in brief 2020, 31, 105710. [Google Scholar] [CrossRef]
- Latunde-Dada, A.O.; Lucas, J.A. Localized hemibiotrophy in Colletotrichum: cytological and molecular taxonomic similarities among C. destructivum, C. linicola and C. truncatum. Plant Pathology 2007, 56, 437–447. [Google Scholar] [CrossRef]
- Crouch, J.A.; O'Connell, R.; Gan, P.; Buiate, E.; Torres, M.; Beirn, L.; Shirasu, K.; Vaillancourt, L. The Genomics of Colletotrichum; 2014.
- Wang, Y.; Xu, W.-T.; Lu, R.-S.; Chen, M.; Liu, J.; Sun, X.-Q.; Zhang, Y.-M. Genome sequence resource for Colletotrichum gloeosporioides, an important pathogenic fungus causing anthracnose of Dioscorea alata. Plant Disease 2023, 107, 893–895. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, G.S.; Pushkova, E.N.; Novakovskiy, R.O.; Kudryavtseva, L.P.; Rozhmina, T.A.; Dvorianinova, E.M.; Povkhova, L.V.; Kudryavtseva, A.V.; Dmitriev, A.A.; Melnikova, N.V. High-quality genome assembly of Fusarium oxysporum f. sp. lini. Frontiers in genetics 2020, 11, 959. [Google Scholar] [CrossRef]
- Sigova, E.A.; Pushkova, E.N.; Rozhmina, T.A.; Kudryavtseva, L.P.; Zhuchenko, A.A.; Novakovskiy, R.O.; Zhernova, D.A.; Povkhova, L.V.; Turba, A.A.; Borkhert, E.V.; et al. Assembling Quality Genomes of Flax Fungal Pathogens from Oxford Nanopore Technologies Data. Journal of fungi 2023, 9, 301. [Google Scholar] [CrossRef]
- Becerra, S.; Baroncelli, R.; Boufleur, T.R.; Sukno, S.A.; Thon, M.R. Chromosome-level analysis of the Colletotrichum graminicola genome reveals the unique characteristics of core and minichromosomes. Frontiers in microbiology 2023, 14, 1129319. [Google Scholar] [CrossRef]
- Ma, W.; Wang, Y.; McDowell, J. Focus on effector-triggered susceptibility. Molecular Plant-Microbe Interactions® 2018, 31, 5–5. [Google Scholar] [CrossRef]
- Cui, H.; Tsuda, K.; Parker, J.E. Effector-triggered immunity: from pathogen perception to robust defense. Annual Review of Plant Biology 2015, 66, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, D.-K.; Chuang, S.-C.; Chen, C.-Y.; Chao, Y.-T.; Lu, M.-Y.J.; Lee, M.-H.; Shih, M.-C. Comparative genomics of three Colletotrichum scovillei strains and genetic analysis revealed genes involved in fungal growth and virulence on chili pepper. Frontiers in microbiology 2022, 13, 818291. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
QUAST and BUSCO statistics of the highly virulent C. lini #390-1 draft genome assemblies. BUSCO: C – complete, D – duplicated, F – fragmented (the glomerellales_odb10 dataset). The used colors indicate estimations of the value quality: from bright green (best) to bright red (worst).
Figure 1.
QUAST and BUSCO statistics of the highly virulent C. lini #390-1 draft genome assemblies. BUSCO: C – complete, D – duplicated, F – fragmented (the glomerellales_odb10 dataset). The used colors indicate estimations of the value quality: from bright green (best) to bright red (worst).
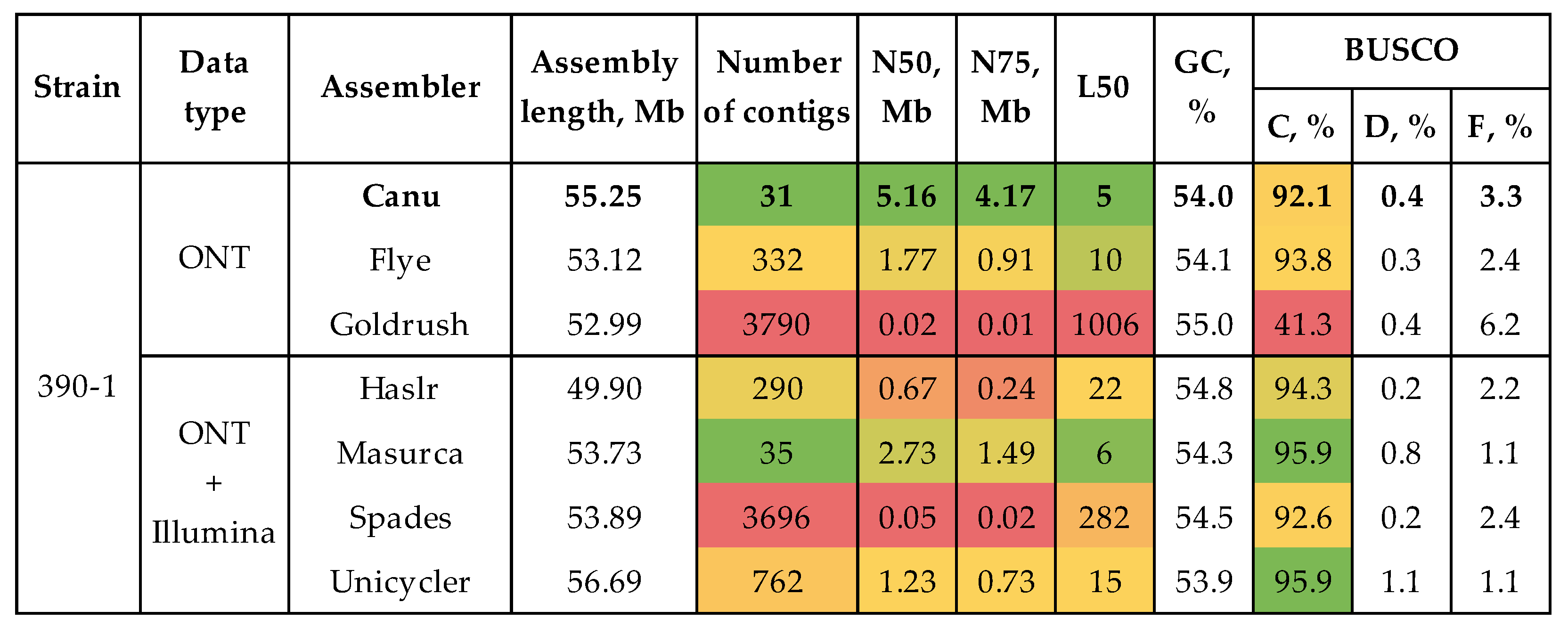
Figure 2.
QUAST and BUSCO statistics of the polished genome assemblies of the highly virulent strain C. lini #390-1. BUSCO: C – complete, D – duplicated, F – fragmented (the glomerellales_odb10 dataset). The used colors indicate estimations of the value quality: from bright green (best) to bright red (worst).
Figure 2.
QUAST and BUSCO statistics of the polished genome assemblies of the highly virulent strain C. lini #390-1. BUSCO: C – complete, D – duplicated, F – fragmented (the glomerellales_odb10 dataset). The used colors indicate estimations of the value quality: from bright green (best) to bright red (worst).
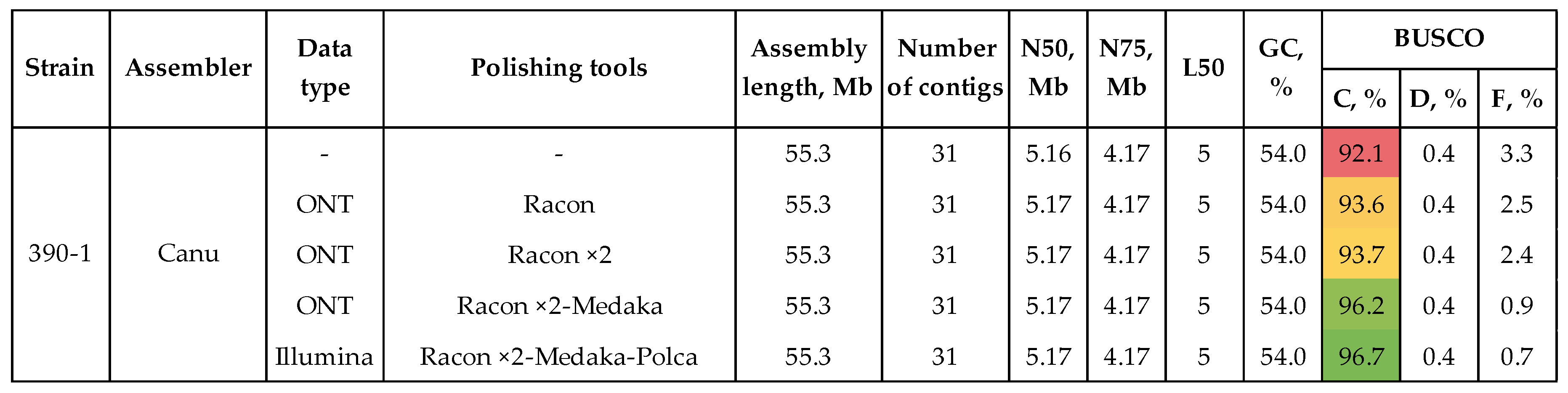
Figure 3.
QUAST and BUSCO statistics of the final genome assemblies of C. lini strains #390-1, #757, and #771. BUSCO: C – complete, D – duplicated, F – fragmented (the glomerellales_odb10 dataset). Green color is used to highlight the most important statistics.
Figure 3.
QUAST and BUSCO statistics of the final genome assemblies of C. lini strains #390-1, #757, and #771. BUSCO: C – complete, D – duplicated, F – fragmented (the glomerellales_odb10 dataset). Green color is used to highlight the most important statistics.
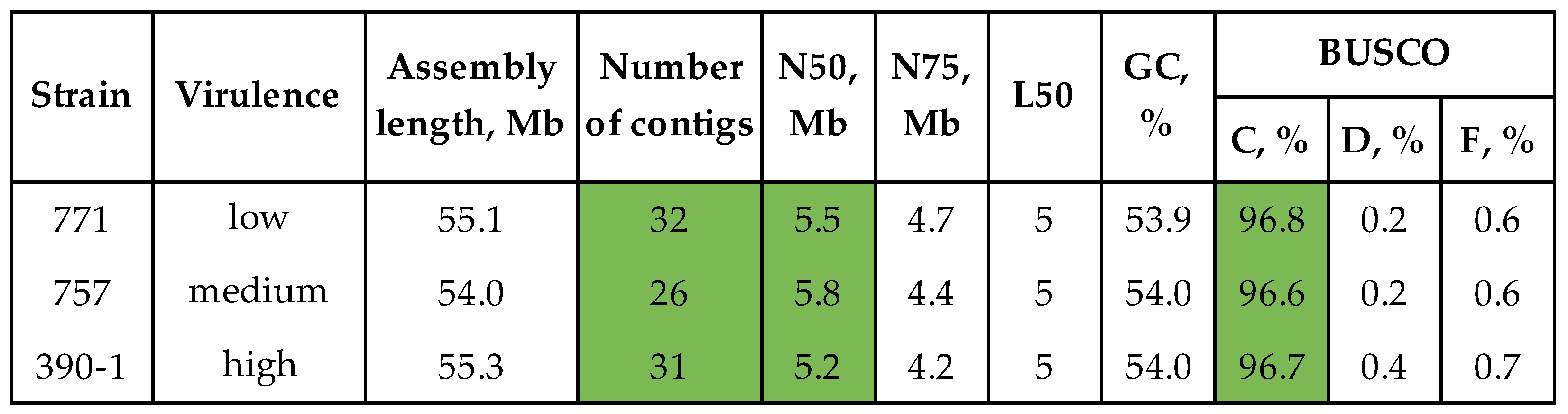
Figure 4.
KEGG BlastKOALA statistics of the annotated proteins for the C. lini highly virulent strain #390-1, moderately virulent strain #757, and lowly virulent strain #771.
Figure 4.
KEGG BlastKOALA statistics of the annotated proteins for the C. lini highly virulent strain #390-1, moderately virulent strain #757, and lowly virulent strain #771.
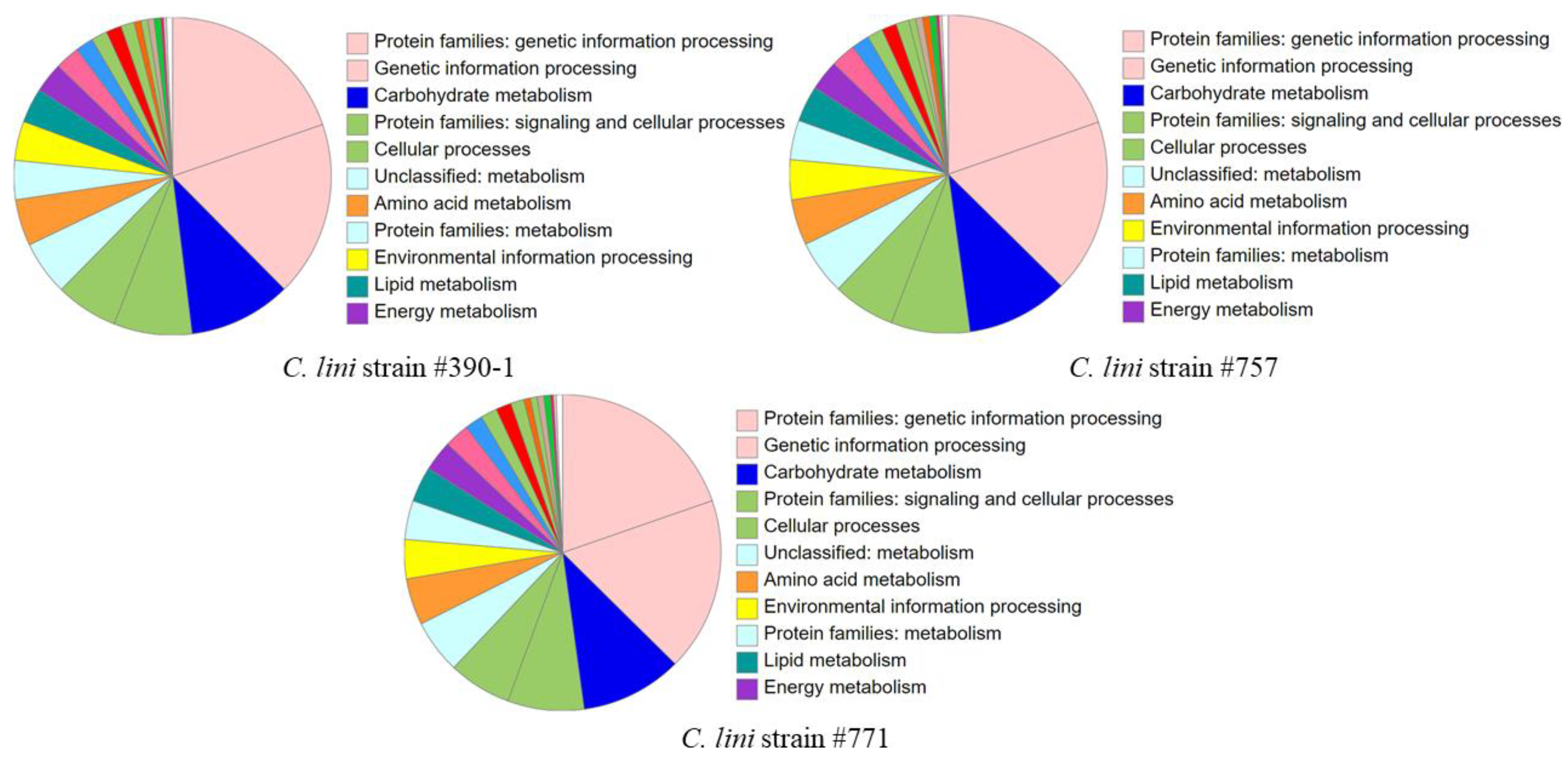
Figure 5.
The results of (A) the alignment of the genome assemblies of the lowly virulent strain #771 and the highly virulent strain #390-1; (B) the alignment of the genome assemblies of the lowly virulent strain #771 and the moderately virulent strain #757. Red lines indicate the forward orientation of the aligned sequences, and blue lines indicate the reverse orientation of the aligned sequences.
Figure 5.
The results of (A) the alignment of the genome assemblies of the lowly virulent strain #771 and the highly virulent strain #390-1; (B) the alignment of the genome assemblies of the lowly virulent strain #771 and the moderately virulent strain #757. Red lines indicate the forward orientation of the aligned sequences, and blue lines indicate the reverse orientation of the aligned sequences.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



