Submitted:
15 December 2023
Posted:
15 December 2023
You are already at the latest version
Abstract
Background: Idiopathic pulmonary fibrosis (IPF) is a fatal lung disease of unknown origin, with a median survival time of ~3 years after diagnosis without anti-fibrotic therapy. It is characterized by progressive fibrosis indicated by increased collagen deposition and high numbers of fibroblasts in the lung. It was demonstrated that CCL18 induces collagen and αSMA synthesis in fibroblasts. We aimed to identify the CCL18 receptor responsible for its pro-fibrotic activities. Methods: We used a random phage display library to screen for potential CCL18-binding peptides, demonstrated its expression in human lungs and fibroblast lines by PCR and immunostaining and verified its function in cell lines. Results: We identified CCR6 (CD196) as a CCL18 receptor and found its expression in fibrotic lung tissue and on lung fibroblast lines derived from fibrotic lungs but it is almost absent in control lines and tissue. CCL18 induces receptor-internalization in a CCR6-overexpressing cell line. CCR6 blockade in primary human lung fibroblasts reduces CCL18-induced FGF2 release as well as collagen-1 and αSMA expression. Knock-down of CCR6 in a mouse fibroblast cell line abolishes the induction of collagen and -smooth muscle actin expression. Conclusion: Our data indicate that CCL18 triggers pro-fibrotic processes via CCR6, highlighting its role in fibrogenesis.
Keywords:
idiopathic pulmonary fibrosis
; fibrogenesis
; chemokine receptor
; ligand-induced internalization
; co-immunoprecipitation
; collagen
; alpha-smooth muscle actin
; FGF2
1. Introduction
Fibrogenesis is a key pathomechanism in various pulmonary diseases, particularly idiopathic pulmonary fibrosis (IPF) [1,2,3] and non-specific interstitial pneumonia (NSIP); but occurs also in end-stage disease lungs of patients with hypersensitivity pneumonitis (HP) or sarcoidosis (SAR). Fibrogenesis leads to a remodelling of the delicate lung micro-architecture into compact and rigid connective tissue. Due to the severe loss of lung function and the lack of an effective therapeutic option in IPF [4], the mean survival time has been ranging between 2.5 and 3.5 years after diagnosis [1,5,6] but some improvement is achieved with newly approved therapeutics like nintedanib and pirfenidone targeting signalling in FGF, PDGF and TGFβ pathways [7,8,9]. The prognosis of non-IPF pulmonary fibrosis is in most cases more favourable compared with IPF although no approved therapeutics exists [10,11].
Current hypotheses on the onset of fibrosis postulate epithelial damage with ongoing repair processes either due to recurrent inflammation or repeated injuries. Thus, fibrosis is regarded a result from an abnormal wound-healing process in which aberrant cross-talk between fibroblasts and epithelial cells promotes chronic fibroblast proliferation [12,13,14,15]. This causes the release of several growth factors and cytokines resulting in an increased migration and proliferation of fibroblasts. Thus, fibroblasts are the key players in pulmonary fibrosis and their enhanced extracellular matrix deposition is a hallmark in pulmonary fibrosis [12,16].
Therapeutic options are rare as anti-inflammatory therapeutic approaches like steroids or cyclophosphamide have only little impact on fibrotic processes of the lung. While these drugs are of value in distinct forms of pulmonary fibrosis characterized by an inflammatory cell composition, e.g. non-specific-interstitial pneumonia (NSIP) [6,17], they are hardly effective in IPF the most common form of pulmonary fibrosis. Currently, two new drugs are approved for the therapy of mild to moderate fibrosis in IPF which, however, slow down disease progression but do not induce a “restitutio ad integrum” [7,8].
CC-chemokine 18 (CCL18) is a chemokine with chemotactic and immuno-regulatory functions mainly released by alternatively activated macrophages [18,19,20,21,22]. Interestingly, CCL18 is unique in the chemokine system as it is present only in humans and there is currently no comparable chemokine known in rodents [23,24]. We found elevated CCL18 serum levels and increased CCL18 release by alveolar macrophages from patients suffering from pulmonary fibrosis [25]. Moreover, we could demonstrate that both, CCL18 production by bronchoalveolar lavage (BAL) cells and serum CCL18 concentrations reflect pulmonary fibrotic activity [26] and is a prognostic marker in IPF [27].
The importance of CCL18 in fibrotic diseases is obvious by its collagen inducing properties [28]. High levels of CCL18 are directly linked to the increased matrix deposition in pulmonary fibrosis [25]. Vice versa, we could demonstrate that collagen recognition via CD204 increases CCL18 release by alveolar macrophages [29] inducing a vicious cycle in IPF [25]. Although the pro-fibrotic properties of CCL18 are already known, the transmission of the CCL18 signals are not clear as for long time CCL18 was one of the few chemokines with unknown receptor(s).
CCL18 signalling is inhibited by pertussis toxin pointing to a conventional G-protein coupled chemokine receptor [28]. In addition, the induction of matrix production by CCL18 in human fibroblasts is triggered via the ERK-pathway [28] and PKCα [30], two signal transduction molecules which are known to be triggered by other chemokines like CCL2 or other CC chemokines via G-protein coupled chemokine receptors [31,32]. We therefore aimed to identify the receptor for CCL18 responsible for its pro-fibrotic properties. The identification of such a receptor would provide a new therapeutic target in disorders with increased levels of CCL18 like fibrosis [25] and cancer [33]. Our data suggest that the CC-chemokine Receptor 6 (CCR6) expressed on fibroblasts mediates the pro-fibrotic activity of CCL18.
2. Materials and Methods
2.1. Phage display peptide library screenings
A cyclic random phage display peptide library with seven random residues (CX7C; C = cysteine; X = random amino acid) was cloned and generated as described previously for phage [34] and for plasmid libraries in the context of the adeno-associated viral genome [35,36]. The library was screened on recombinant CCL18 as described previously for epitope mapping of monoclonal antibodies [37,38] after two-fold negative selection on irrelevant control proteins BSA and casein. The screening was done for three selection rounds. Randomly selected clones from the third panning round were sequenced (GATC, Konstanz, Germany).
2.2. Patients
Fibroblast lines were established either from surgical material, from explanted lungs (n=13), pneumectomies or lobectomies from patients suffering from squamous carcinoma (n=19) or from remaining material obtained from left-over of diagnostic biopsies of fibrotic lungs (video-assisted thoracoscopy (VATS, n=4), transbronchial biopsies (TBB, n=4) from patients suffering from idiopathic pulmonary fibrosis (IPF, n=13), non-specific interstitial pneumonitis (NSIP, n=3), sarcoidosis, (n=3) or hypersensitivity pneumonitis (HP, n=2). All of of NSIP are “non-classified NSIP” as no underlying diseases could be detected.
The study was approved by the Ethics Committee of the University Medical Centre Freiburg (vote number 276/02, renewed in 2007).
2.3. Immunohistochemistry
Tumour free lung tissues were obtained from surgery (lobectomy or pneumonectomy) due to non-small cell lung cancer; only specimens at least 7cm away from the tumour were selected as tumour free and only those confirmed by microscopy entered the study. Tissues were fixed and embedded in paraffin using the HOPE-technique [39]. Immunohistochemical detection of CC-chemokine receptor 6 (CCR6) was performed as previously described [39,40]. Primary antibody (anti hCCR-6 mouse monoclonal IgG, clone 53103, 500 µg/mL, R&D Systems, Minneapolis, Minnesota) was applied after de-paraffinization in a dilution of 1/100 for 1h at ambient temperature. Detection of signals was achieved by application of ZytoChem Plus HRP polymer system (Zytomed Systems, Berlin, Germany) according to the instructions of the manufacturer. Aminoethylcarbazole (permanent AEC, Zytomed Systems, Berlin) was used as a chromogen.
2.3. Isolation of primary fibroblasts
Lung tissue was cut in small pieces (app. 0.5 cm edge length) and placed in 6-well plates containing 1mL Quantum 333 (PAA, Pasching, Austria) with 1% penicillin/streptomycin. Quantum 333 is a fibroblast selection medium to allow the growth of the cells without the addition of foetal calf serum. Outgrowing fibroblasts were harvested by trypsinisation when they reached approximately 80% confluence, cultured in 75cm2 cell culture flasks (NUNC Thermo Fisher, Roskilde, Denmark) in Quantum 333 or DMEM with 10% FCS and subsequently split (1:3 to 1:5). The established lines were enrolled in our studies from passages 3 to ensure that no other cells (e.g. macrophages) are in our culture to 8 to prevent senescence of the cells. Phase contrast microscopy photographs of three different cell lines are depicted in Figure S1.
2.4. Cell lines
Rat lung epithelial cells were obtained from ATCC (Teddington, Middlesex, UK) and cultured in HAM-F12 (Lonza, Verviers, Belgium) containing 10% foetal bovine serum (FCS gold, PAA, Pasching, Austria), 10 µg/mL bovine pituitary extract (Gibco, Invitrogen, Paisley, UK), 5 µg/mL insulin (Sigma, Taufkirchen, Germany), 2.5 ng/mL insulin-like growth factor (ImmunoTools, Friesoythe, Germany), 1.25 µg/mL transferrin (Gibco, Invitrogen, Paisley, UK), and 2.5 ng/mL EGF (ImmunoTools, Friesoythe, Germany).
U937 cells are a pro-monocytic lymphoma cell line originally derived from a male patient. We found that in our laboratory cell line a large percentage of cells express CCR6. We therefore used anti-CCR6 antibodies (R&D Systems, Minneapolis, Minnesota) and anti-mouse IgG microbeads (Miltenyi. Bergisch-Gladbach, Germany) and MACS cell enrichment columns (Miltenyi) to enrich CCR6 positive U937 cells in order to establish a “U937*CCR6+” cell line (Figure S2). The cells were cultured in DMEM (Gibco, Invitrogen, Paisley, UK), containing 10% foetal bovine serum (FCS gold, PAA, Pasching, Austria) and 1% penicillin/streptomycin (Gibco, Invitrogen, Paisley, UK).
The mouse embryonal fibroblast line NIH/3T3 was obtained from ATCC and cultured in DMEM (Gibco, Invitrogen, Paisley, UK), containing 10% foetal bovine serum (FCS gold, PAA, Pasching, Austria) and 1% penicillin/streptomycin (Gibco, Invitrogen, Paisley, UK). All cells are incubated at 37°C at 5% CO2.
2.5. Flow cytometry
Fibroblasts populations were gated by forward/sideward scatter and surface expression of CCR6 was estimated using a FITC-labelled anti-human CCR6 antibody (R&D Systems, Minneapolis, MN) and a suitable IgG control. As CCR6 is a trypsin-sensitive molecule, trypsinized cells were allowed to recover in medium in polystyrene tubes (to avoid adhesion) for at least 3h at 37°C before staining.
2.6. FGF2-Determination
Fibroblasts were seeded at a density of 300.000 cells per well in a 6-well cell culture plate in Quantum 333 medium. Cells were allowed to attach overnight and the medium was replaced with 1 mL of DMEM plus 10% FCS. Cells were either left non-stimulated or were stimulated with 10 ng/mL of CCL18. To block CCR6/CCL18 interaction a blocking antibody against CCR6 (R&D systems, Wiesbaden, FRG) or an irrelevant antibody (mouse anti human IgG, R&D Systems) was added to a final concentration of 20 µg/mL in parallel cultures. After 24h of culture the supernatant was harvested and stored at -80°C until FGF2 determination. FGF2 was measured using an ELISA development kit (R&D) and performed as suggested by the supplier.
2.7. Real-time PCR
After the indicated culture period total RNA was extracted using TRIzol Reagent according to the manufacturer's protocol (Invitrogen, Karlsruhe, Germany). Total RNA was reverse-transcribed with StrataScript RT (Stratagene, CA) using oligo (dT)12-18 primers to produce cDNA according to the manufacturer's protocol. Specific primers for human CCR6, collagen 1, alpha-smooth muscle actin and GAPDH were designed using AmplifX 1.7.0 (https://inp.univ-amu.fr/en/amplifx-manage-test-and-design-your-primers-for-pcr) [41] using LocusLink and GenBank databases (National Center for Biotechnology Information; http://www.ncbi.nlm.nih.gov/LocusLink/index.html). Accession code numbers for the nucleotide sequences used to generate the respective primers and the primer sequences are depicted in Table 1.
All primers were intron-spanning and synthesized by biomers (https://www.biomers.net/). Real time PCR was performed with the iQ SYBR Green SuperMix, iCycler thermocycler, and iCycler iQ 3.0 software (Bio-Rad Laboratories GmbH, Germany) according to the manufacturer's protocol. After initial denaturation (10 minutes, 95°C) PCR was performed using an annealing time of 30 seconds at 57°C, amplification for 30 seconds, and 15 seconds at 94°C for denaturation for 45 cycles. To control for specificity of the amplification products, a melting curve analysis was performed. No amplification of nonspecific products was observed in any of the reactions. A threshold cycle value (Ct) was calculated and used to compute the relative level of specific mRNA by the following formula:
"relative expression = 2(Ct GAPDH-Ct(target))x10,000" for each cDNA sample.
2.8. Stable transfection of RLE-6TN
As the human CCR6 vector (pORF9-hCCR6, InvivoGen, San Diego, California) does not have a Sal I or Sma I cleavage site, the vector was first cut with EcoRV and a Sal I linker was integrated into the cleavage site. In order to create a GFP fusion protein, a part of the coding sequence of CCR6 was re-synthesized together with a Sma I cleavage site, the stop codon at the C-terminal end was removed and ligated back into the original vector. Only then the cleavage was carried out with Sal I and Sma I.
3-5x105 293T-cells were transfected with pCDNL B**, pLTR VSVG and pNL CEV CCR6*GFP using FuGeneHD (Roche Diagnostics, Penzberg). After 24h the medium was replaced and after 48h virus supernatant was used to infect RLE-6TN.
Stable transfected RLE-CCR6*GFP cells were separated from wild-type cells by fluorescence activated cell sorting and limiting dilution. The purity of the cells was 99% and was stable during the culture periods.
2.9. Knock-down of CCR6 in NIH/3T3 cells by CRISPR/Cas9
Primer sequences for the generation of CRISPR/Cas9 were selected from the gRNA design tool by ThermoFischer (ThermoFisher, Waltham, Massachusetts, USA) (thermofisher.com/crisprdesign) and synthesized by biomers (https://www.biomers.net/). The following primer sequences were used: ATAATCATCCGTTCCAAAGT and GTAGACGTCAGTCATGGATC. The gRNA was then synthesized and transcribed using the GeneArt Precision gRNA Synthesis Kit (Invitrogen/ThermoFisher, Waltham, Massachusetts, USA). GeneArt Platinum Cas9 Nuclease protein (Invitrogen/ThermoFisher) and the gRNAs were then used to transfect the cells using the Lipofectamine™ CRISPRMAX™ Transfection Reagent (Invitrogen/ThermoFisher) according to the protocol provided by the manufacturer. After transfection the cells were cultured for two days, harvested and cloned by limiting dilution. Clones were picked and expanded and analysed for CCR6 expression. CCR6-negative clones were expanded and used for the experiments.
2.10. Receptor internalization
Receptor internalization was estimated using flow cytometry and fluorescence microscopy. CCR6 is a trypsin-sensitive receptor thus, it is lost during the detachment of adherent cells using trypsin. To avoid artefacts by damaging CCR6 by trypsinization, we used the non-adherent U967*CCR6+ cell line. The cells were incubated with CCL18 or CCL20 in varying concentrations ranging from 0.03 to 1000 nM in 30µl of culture medium containing 100,00 cells for 20 minutes and immediately fixed by addition of 200µl of cold paraformaldehyde (4%, Sigma, Taufkirchen, Germany) for 5 minutes on ice. The cells were then washed by the addition of 1 mL PBS and centrifugation. This step was performed twice. Subsequently the cells were stained with a FITC-labelled anti-human CCR6 antibody (R&D Systems, Minneapolis, MN) and analysed using a FACS Calibur (Becton Dickinson, Heidelberg, Germany) and FlowJo for data analysis (Becton Dickinson, Heidelberg, Germany).
For fluorescence microscopy RLE-CCR6*GFP were seeded on chamber slides and cultured over night at 37°C and 5% CO2 to allow to adhere. The next day the medium was discarded and replaced by medium containing 10 ng/mL of CCL18. As control we added CXCL10 in the same concentration. After the indicated time points, the cells were fixed and embedded in Vecta shield containing DAPI (Vector Laboratories, Newark, CA, United States) and analysed using a fluorescence microscope (Olympus, Hamburg, Germany).
2.11. Co-immunoprecipitation
For co-immunoprecipitation (Co-IP) 107 RLE-6TN CCR6*GFP cells were cultured overnight in a petri dish to let them adhere and subsequently loaded with 500ng/mL recombinant human CCL18 (Immunotools, Friesoyte, Germany) for 10 and 30 minutes. After incubation loaded cells were gently lysed using the lysing buffer provided by the manufacturer and CCR6*GFP was isolated using the isolation kit for GFP-tagged proteins according the manufacturers protocol (Miltenyi, Bergisch Gladbach, Germany). The isolated CCR6*GFP and CCR6*GFP/CCL18 complexes were analysed by routine western blot. In brief: isolated proteins were boiled at 95°C for 5 minutes in equal volumes of loading buffer (0,5M Tris-HCl pH 6.8, 2% SDS, 0,05% bromophenol blue, 20% 2-mercaptoethanol, 10% Glycerol) and subjected to 12% sodium dodecyl-sulphate–PAGE, separated by electrophoresis and transferred to a polyvinylidene difluoride membrane (PVDF). After blocking for 2h in Tris-buffered saline (TBS) containing 5% non-fat dry milk, the membranes were incubated with primary antibody against CCR6 (CKR6/C20, Santa Cruz, California) and anti-CCL18 (R&D Systems, Minneapolis, MN) both diluted 1:700 with TBS at 4°C overnight. Visualization was performed using appropriate secondary antibodies labelled with IRDye 800CW or IRDye 700CW (Li-COR Bioscience, Bad Homburg, Germany) diluted 1:10000-1:20000 for 2 h and scanned using Odyssey system (Li-COR Bioscience) according to the manufactures instructions.
2.12. Statistical analysis
Data are shown as median, box plots or bar charts. For comparison of independent samples Mann-Whitney test were performed using StatView for Windows (SAS Institute, Cary, NC, USA). Values of p≤0.05 were considered statistically significant.
3. Results
3.1. Identification of a CCL18-binding peptide using phage display
To identify peptides binding to CCL18 by phage display we used a random CX7C phage peptide library (C indicates cysteine; X indicates any amino acid) and selected the binding phages on immobilized CCL18. Competent E. coli were added to the bound phages for uptake and expansion. Phages were specifically enriched on CCL18 after three rounds of selection. Fifteen clones of the phages derived from the selection on CCL18 were sequenced and two of them were found to display variants of a CCR6 motif (CTCGCGACTGGTGTGGTGTTT (AL121935) and TGGTGAGCTGGAGTCATCAGA (NM_031409)).
3.2. CCR6 expression in human fibrotic lung tissue
To identify CCR6 expression in human lungs we performed immune-histochemical studies on tissue sections taken from lungs of patients with IPF and lung tissue from lobectomies afar from the tumour. Analysing lung tissue from IPF patients revealed intense CCR6 expression in bronchial epithelia (Figure 1A), hyperplastic alveolar epithelial cells type II (Figure 1B), fibroblasts (Figure 1C), and macrophages (Figure 1D). In contrast, in tumour-free lung from a bronchial carcinoma patient (Figure 1E) no CCR6 expression could be detected.
Fibroblasts are regarded as important drivers in fibrotic lung diseases [42] and it is described that CCL18 fosters collagen and αSMA expression in fibroblasts [28,43]. Thus, although we found CCR6 expression on several cell types within our fibrotic lung sections, we concentrate here on the role of the CCR6/CCL18 interaction in fibroblasts.
3.3. CCR6 expression in human primary fibroblast lines
Next, we determined the mRNA levels of CCR6 in cultured human primary fibroblast lines isolated from lungs of patients with varying pulmonary diseases. Highest expression was found in fibroblast lines derived from lungs of patients suffering from IPF and non-specific interstitial pneumonia (NSIP) (Figure 2). Three out of four IPF fibroblast lines and two of three NSIP lines expressed increased levels of CCR6 mRNA. The median was 3-fold increased, in extremes we found a 10-20-fold increase. Fibroblast lines derived from lungs of patients suffering from squamous carcinoma expressed only background levels of CCR6 mRNA (Figure 2).
Flow cytometric analysis of the fibroblast lines (Figure 3B) revealed detectable CCR6 expression on the surface of fibroblast lines derived from patients suffering from NSIP (92±6% positive cells) and IPF (53±33% positive cells, Figure 3B). In only two out of 13 IPF lines no CCR6 surface expression (<5% positive cells) could be detected. Of note, fibroblast lines established from lungs of patients suffering from end-stage sarcoidosis or hypersensitivity pneumonitis with fibrotic lesions different from IPF did not show relevant CCR6 expression corresponding with the low frequency of fibrosis in these disorders. CCR6 expression in control fibroblasts lines was detected on a very low percentage of cells (<8%; Figure 2B). Remarkably, increased CCR6 expression of fibroblasts from fibrotic lungs remained increased throughout all cell culture passages (tested up to passage 12, Figure S3).
Interestingly, splitting the results according to the sampling method revealed that fibroblast lines grown from TBB express the lowest levels of CCR6, lines established from explant lungs disclosed the highest CCR6 expression. Lines established from VATS samples disclosed an intermediate expression level, however, none of these differences reached statistical significance most likely due to the low number of samples (Figure S4).
3.4. CCL18 binding to CCR6
To verify that CCR6 is indeed a receptor for CCL18 we conducted several experiments to demonstrate Receptor binding and to demonstrate the functional interaction of CCL18 and CCR6.
3.4.1. CCL18 binds to CCR6 (Co-IP)
In order to verify binding of CCL18 to CCR6 we co-precipitated CCL18 bound to CCR6 by isolation of the CCL18/CCR6*GFP complex using anti GFP beads. RLE-CCR6*GFP cells were loaded with CCL18 and the CCL18/CCR6*GFP complex was gently isolated from the lysed cells using anti-GFP beads and analysed by Western Blot. Anti-CCR6 antibodies detected CCR6 in both, CCL18 loaded and non-loaded cells (Figure 4, lane 2, 3, and 4: red bands). In contrast, CCL18 was detected when added directly or in CCL18-loaded cells after 30 minutes of incubation (Figure 4, lane 4 bottom: green band).
CCL18 induced internalization of CCR6
The RLE-CCR6*GFP cells disclose a strong CCR6 expression as well as a bright green fluorescence (Figure S5). In fluorescence microscopy, the CCR6*GFP complex appears as a clear membrane associated expression pattern of CCR6 (Figure 5A, upper panel). In addition, there is also a light intracellular distribution of CCR6*GFP visible intracellularly. This corresponds to an increase of CCR6 mean fluorescence intensity when we add a membrane-permeabilisation step in the CCR6-staining procedure of the fibroblasts (Figure S6).
Stimulation of the RLE-CCR6*GFP with CCL18 (10ng/ml) induced a time-dependent internalization of the CCR6*GFP complex (Figure 5A, mid panels). After 5 minutes, the CCR6*GFP complex was seen as green dots or as a ring intracellularly. After 20 Minutes the CCR6*GFP complex was distributed diffusely within the cells. This internalization demonstrates that CCL18 triggers CCR6. In contrast, an irrelevant chemokine (CXCL10) did not induce CCR6*GFP internalization (Figure 4A, lower panel).
Flow cytometric analysis of CCR6 expression using U937*CCR6+ cells revealed a dose dependent decrease of CCR6 expression with increasing concentrations of CCL18 (Figure 5B). As CCL20 was for long time the only known chemokine ligand for CCR6, we compared the CCR6 internalization induced by these two chemokines. Of interest, a 10-fold higher concentration of CCL18 was required to reach the same amount of receptor internalisation compared with CCL20 (Figure 5B).
3.5. Functional Analysis of CCR6
3.5.1. CCL18 induced activation of human primary lung fibroblasts
Fibroblasts derived from lungs of controls released only low levels of FGF2. CCL18 stimulated FGF2 release in these cells only marginally (Figure 6A, grey bars). In contrast, fibroblast lines derived from fibrotic lungs release increased levels of FGF2 that is further up-regulated by CCL18 (Figure 6A, dark bars).
Blockade of CCR6 by a blocking antibody exerted no effect on CCL18 stimulated FGF2 release by non-fibrotic fibroblast lines. In contrast, in fibroblast lines derived from fibrotic lungs blockade of CCR6 with a blocking antibody abrogated CCL18 induced FGF2 up-regulation almost completely. No blocking effects was seen using an irrelevant IgG isotype control antibody.
As reported [25,28], CCL18 induces the expression of collagen and aSMA. We, therefore, determined whether the induction of these molecules is also CCR6 dependent. As shown in Figure 6B (upper panel) CCL18 up-regulated collagen 1 expression in three of four fibrotic-fibroblast lines analysed. This induction was inhibited in all three CCL18 reactive fibroblast lines by the blockade of CCR6 using an inhibitory antibody. The same result was obtained for the CCL18 induced up-regulation of αSMA. Most interestingly, the same cell line which failed to increase collagen 1 expression by CCL18 stimulation also failed to up-regulate αSMA (Figure 6B, lower panel).
3.5.2. CCL18 induced activation of wild type and CCR6 KO NIH 3T3 cells
The mouse fibroblast line NIH 3T3 also expresses high levels of CCR6 (Figure 7 upper panel) whereas the KO clone discloses no difference to the control antibody indicating that there is no CCR6 expression (Figure 7 lower panel).
In wild type cells CCL18 induced a dose-dependent increase in collagen 1A (Col1A, Figure 8A, light grey bars) as well as α-smooth muscle actin (αSMA, Figure 8B, light grey bars). TGFβ (5ng/ml) as well increased the expression of both molecules. In CCR6 KO cells CCL18 did not induce Col1A expression (Figure 8A, dark grey bars) whereas TGFβ induced a clear-cut up-regulation of Col1A. Interestingly, αSMA is even dose-dependently down-regulated when the CCR6 KO cells are stimulated with CCL18 (Figure 8B, dark grey bars). In contrast, TGFβ induced an up-regulation of αSMA in wild type cells as well as in the CCR6 KO cells.
4. Discussion
CCL18 was identified as a structural homolog of MIP1α (CCL3) and is primarily released by alternatively activated macrophages [44]. Just three years later it was found that CCL18 mRNA is up-regulated in the lungs of patients with hypersensitivity pneumonitis [45]. CCL18 gained interest as it stimulates collagen production in lung fibroblasts [28,43]. We demonstrated that CCL18 and collagen production are strongly connected [25,29] and that CCL18 is of prognostic usefulness in IPF [26,27]. However, although these results point to an important role of CCL18 in lung fibrosis, no information was available on the receptor responsible for its pro-fibrotic properties. Here we demonstrate that the pro-fibrotic effects of CCL18 are based on the expression of and the signal transduction by CCR6 in lung fibroblasts.
The role of CCR6 as a CCL18 receptor in fibrotic lung diseases was demonstrated by different approaches. Immunohistochemistry revealed CCR6 expression in human lung tissue from patients suffering from fibrotic lung diseases but not in tissue from lung cancer patients. The data show that different cells are affected including fibroblast as well as alveolar type II cells. Likewise, CCR6 was only expressed on fibroblasts from lungs of patients suffering from fibrotic lung diseases. In contrast, lung fibroblasts from lungs of controls (i.e. tumour free tissue from cancer patients) did not express CCR6. Thus, CCR6 expression in lung tissue might be a marker for an ongoing fibrotic process. In the current manuscript we focus on fibroblasts as these cells are the main drivers of pulmonary fibrosis by their capability to release large amount of matrix protein. In addition, expression of α-smooth muscle protein turns the fibroblasts into contractible so called myofibroblasts, a cell type frequently found in pulmonary fibrosis.
CCR6 was first described as a receptor for the chemokine CCL20 [46]. Our data demonstrate that indeed CCL18 and CCL20 both down-regulate CCR6 receptor expression in a dose dependent manner. However, the concentration required to reach a 50% downregulation are 10-fold higher for CCL18 compared with CCL20. It has been hypothesized that the binding of CCL20 is dependent on a “DCCL” motive present in the receptor binding region of CCL20 [47]. With a sequence of “LCCL” in this motive is different in CCL18 which might result in weaker binding of CCL18 to CCR6.
In fibroblast lines derived from fibrotic lungs CCL18 induces FGF2 release and increases collagen and αSMA expression which is inhibited by anti-CCR6 antibodies. This is of interest as current drugs like pirfenidone or nintedanib inhibit either the up-regulated release of FGF2 [48] or inhibit signal transduction of the FGF2-receptor [9]. As FGF2 release is upregulated by CCL18 it is tempting to speculate whether or not a therapy targeting CCL18/CCR6 is of additive value.
CCR6-dependent up-regulation of Col1A or αSMA was demonstrated by the fact that primary human fibroblasts without CCR6 did not react to CCL18 whereas CCR6 positive fibroblast lines up-regulate Col1A and αSMA after CCL18 stimulation. Blockade of CCR6 inhibited this up-regulation of both molecules.
Likewise, NIH 3T3 cells expressing high levels of CCR6 are comparable to human CCR6 positive fibroblast lines as they show a clear and dose dependent up-regulation of Col1A and αSMA after CCL18 stimulation. In contrast, upregulation of both markers is lost after knock-down of CCR6 on these cells. However, stimulation of the CCR6 KO cells with TGFβ induced a robust increase in αSMA and Col1A expression indicating that despite the CCR6 knock-down these cells are still able to increase αSMA and Col1A using other stimuli. These results strongly suggest that CCR6 expression on fibroblasts is crucial for the pro-fibrotic effects of CCL18.
Most interestingly, a mouse model of bleomycin-induced fibrosis showed that the administration of human CCL18 via an adenoviral vector increased TNF-, IFNγ-, MMP-2 and MMP-9 expression and increased lymphocytosis but attenuated unexpectedly the bleomycin induced collagen deposition [49]. However, our own preliminary analyses indicated that in the bleomycin model CCR6 is not expressed on lung tissue cells (data not shown) and thus, the responsible, pro-fibrotic receptor is missing. Moreover, the loss of CCR6 resulted in a dose dependent down-regulation of αSMA expression in NIH 3T3 cells. This might be an explanation for the more or less protective activity of CCL18 in the experiments presented by Pochetuhen and colleagues [49].
CCR6 is known to be expressed by CD4+ and CD8+ T-cells, by TNF activated granulocytes, immature dendritic cells, as well as by B cells and human intestinal cells but neither by macrophages nor by freshly isolated monocytes (summarized in [50]). CCR6 gained interest because it was found to be highly expressed on IL-17 producing cells [51] and on FoxP3 expressing IL-17 releasing regulatory T-cells [52]. Thus, the expression on fibroblasts and alveolar epithelial cells is remarkable. The reason for this expression currently is unknown, however, it has been reported, that in human umbilical vein vascular endothelial cells (HUVEC) CCR6 expression is up-regulated by the combined stimulation with HGF and VEGF [53]. In fibroblast-like synovial cells stimulation with IL-1β induced an up-regulation of CCR6 expression [54]. In our experiments, upregulation of CCR6 in normal human lung fibroblasts using IL-4, IL-10, IL-13 and TGFβ in various combinations failed (Figure S7). It is noteworthy that the increased CCR6 expression found in fibroblasts from patients suffering from IPF is stable over several culture passages of the fibroblast lines. Therefore, we conclude that the increased expression of CCR6 by fibroblast lines is not transiently induced by the surrounding cytokine milieu in the fibrotic lung but seems to be a key characteristic of fibroblasts from IPF lungs which is acquired during patho-mechanistic differentiation.
We found a concordant expression of CCR6 on lung fibroblasts demonstrated by immunohistochemistry of fibrotic lung and by flow cytometric analysis of fibroblast lines isolated from lungs of patients suffering from IPF and NSIP. The fact that not all fibroblast lines from IPF lungs express CCR6 might be caused by the heterogeneity of fibroblasts in fibrotic lungs as described earlier [55]. In addition, lung remodelling in IPF is not homogeneously distributed. Severely destructed areas with massive accumulation of fibroblasts and extracellular matrix build a patchwork with areas of almost normal or even completely normal lung histology [56]. In addition, evaluation of our fibroblast lines by the sampling source (explant, VATS, TBB) revealed a clear but insignificant difference between the different sampling methods. Explanted tissue derives from patients with a widely remodelled and dysfunctional end stage lung. In contrast VATS and TBB are diagnostic procedures and performed at less advanced lung remodelling. Thus, it is conceivable that fibroblasts although grown from an IPF lung sample might be CCR6 negative if this sample has been taken from a region of more or less normal histology. However, high CCR6 expression might also be a feature of an advanced disease reflecting the tremendous remodelling of the lung.
Lung tissue from patients with sarcoidosis and HP were used in the experiments described here and derived from explanted lungs after lung transplantation due to massive pulmonary fibrosis. Both diseases are based on an inflammatory background and are different from IPF/UIP [10]. Despite the fact that these patients suffered from lung fibrosis, the fibroblast lines isolated from these lungs showed no CCR6 expression. Although based on a small number of cell lines, this lack of expression is remarkable. Even though these diseases are also associated with increased CCL18 levels [25,45] a clear correlation of CCL18 levels with the natural course of the disease has not been shown. In contrast, in IPF CCL18 is a prognostic marker and phases of acute exacerbations are accompanied with increasing CCL18 levels [27]. Thus, we speculate that the interaction of CCR6 and CCL18 might be pivotal for the progress of IPF whereas in fibrosis due to sarcoidosis or HP these processes are obviously driven by other factors.
We used fibroblast lines established from lungs of patients with squamous carcinoma as controls. The tissue was harvested far from the tumour and was therefore considered to be normal. It is of interest that in sera from patients with squamous carcinoma CCL18 levels are also increased, however, theses CCL18 levels have no impact on the survival time of these patients [33].
In summary, we conclude that CCR6 is an important receptor involved in pro-fibrotic activities of CCL18 and blocking of CCL18/CCR6 interaction or the signalling cascade induced by this interaction is an interesting therapeutic option for IPF.
5. Conclusions
In summary, we conclude that CCR6 is an important receptor involved in pro-fibrotic activities of CCL18 and blocking of CCL18/CCR6 interaction or the signalling cascade induced by this interaction is an interesting therapeutic option for IPF.
6. Patents
Supplementary Materials
Supporting figures and methods can be downloaded as a PDF document at the website of this paper posted on Preprints.org.
Author Contributions
G.Z. conceived the project. G.Z., K.H., S.J.S. and T.P. designed the study, performed the experiments and analysed data. M.T. designed the phage library. G.Z and A.W. performed the search in phage library. J.M-Q recruited the fibrosis patients, T.P. the control group. K.H. and P.E. performed culturing of fibroblasts. G.Z. performed primer design, C.M. PCR, C.M. and K.H. flow cytometry. H.E. designed transfection experiments. G.Z. and K.H. performed transfection. T.G. performed immunostaining. S.J.S. and K.H performed co-immunoprecipitation and western blot. G.Z., J.M-Q, and K.H. wrote the paper.
Funding
This work was supported by a grant from the German DPLD Network (GOLDnet; German Federal Ministry for Education and Research (BMBF), grant number 01GM0859). The article processing charge was funded by the Open Access Publishing funding programme of the Albert-Ludwigs-University Freiburg.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of the University Medical Centre Freiburg (vote number 276/02, renewed in 2007).
Informed Consent Statement
Written informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors like to thank Christel Heine, Olga Mayer, Sofia Kamenker, Nicole Wehrle, Stefanie Hahn and Hilke Rohde-Wagner for their technical help.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949-1961.
- American thoracic society/european respiratory society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. This joint statement of the american thoracic society (ats), and the european respiratory society (ers) was adopted by the ats board of directors, june 2001 and by the ers executive committee, june 2001. Am J Respir Crit Care Med 2002, 165, 277-304.
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A., et al. An official ats/ers/jrs/alat statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011, 183, 788-824. [CrossRef]
- Selman, M.; Pardo, A.; Richeldi, L.; Cerri, S. Emerging drugs for idiopathic pulmonary fibrosis. Expert Opin Emerg Drugs 2011, 16, 341-362. [CrossRef]
- Ley, B.; Collard, H.R.; King, T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011, 183, 431-440. [CrossRef]
- Travis, W.D.; Matsui, K.; Moss, J.; Ferrans, V.J. Idiopathic nonspecific interstitial pneumonia: Prognostic significance of cellular and fibrosing patterns: Survival comparison with usual interstitial pneumonia and desquamative interstitial pneumonia. Am J Surg Pathol 2000, 24, 19-33.
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y., et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014. [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L., et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014. [CrossRef]
- Chaudhary, N.I.; Roth, G.J.; Hilberg, F.; Muller-Quernheim, J.; Prasse, A.; Zissel, G.; Schnapp, A.; Park, J.E. Inhibition of pdgf vegf and fgf signalling attenuates fibrosis. Eur Respir J 2007, 29, 976-985. [CrossRef]
- Salvatore, M.; Ishikawa, G.; Padilla, M. Is it idiopathic pulmonary fibrosis or not? J Am Board Fam Med 2018, 31, 151-162.
- Wells, A.U. Patterns of progression in non-ipf fibrotic interstitial lung disease. Current Opinion in Pulmonary Medicine 2023, 29, 459-464. [CrossRef]
- Selman, M.; King, T.E.; Pardo, A. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001, 134, 136-151. [CrossRef]
- Spagnolo, P.; Semenzato, U. Revealing the pathogenic and ageing-related mechanisms of the enigmatic idiopathic pulmonary fibrosis (and chronic obstructive pulmonary disease). Curr Opin Pulm Med 2022, 28, 296-302. [CrossRef]
- Spagnolo, P.; Kropski, J.A.; Jones, M.G.; Lee, J.S.; Rossi, G.; Karampitsakos, T.; Maher, T.M.; Tzouvelekis, A.; Ryerson, C.J. Idiopathic pulmonary fibrosis: Disease mechanisms and drug development. Pharmacol Ther 2021, 222, 107798. [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic pulmonary fibrosis. New England Journal of Medicine 2018, 378, 1811-1823.
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941-1952.
- Daniil, Z.D.; Gilchrist, F.C.; Nicholson, A.G.; Hansell, D.M.; Harris, J.; Colby, T.V.; du Bois, R.M. A histologic pattern of nonspecific interstitial pneumonia is associated with a better prognosis than usual interstitial pneumonia in patients with cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 1999, 160, 899-905. [CrossRef]
- Schutyser, E.; Richmond, A.; Van Damme, J. Involvement of cc chemokine ligand 18 (ccl18) in normal and pathological processes. J Leukoc Biol 2005, 78, 14-26. [CrossRef]
- Schraufstatter, I.U.; Zhao, M.; Khaldoyanidi, S.K.; Discipio, R.G. The chemokine ccl18 causes maturation of cultured monocytes to macrophages in the m2 spectrum. Immunology 2012, 135, 287-298. [CrossRef]
- Porcheray, F.; Viaud, S.; Rimaniol, A.C.; Leone, C.; Samah, B.; Dereuddre-Bosquet, N.; Dormont, D.; Gras, G. Macrophage activation switching: An asset for the resolution of inflammation. Clin Exp Immunol 2005, 142, 481-489. [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat Rev Immunol 2003, 3, 23-35.
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 2004, 25, 677-686. [CrossRef]
- Tasaki, Y.; Fukuda, S.; Iio, M.; Miura, R.; Imai, T.; Sugano, S.; Yoshie, O.; Hughes, A.L.; Nomiyama, H. Chemokine parc gene (scya18) generated by fusion of two mip-1alpha/ld78alpha-like genes. Genomics 1999, 55, 353-357.
- Hieshima, K.; Imai, T.; Baba, M.; Shoudai, K.; Ishizuka, K.; Nakagawa, T.; Tsuruta, J.; Takeya, M.; Sakaki, Y.; Takatsuki, K., et al. A novel human cc chemokine parc that is most homologous to macrophage- inflammatory protein-1 alpha/ld78 alpha and chemotactic for t lymphocytes, but not for monocytes. J Immunol 1997, 159, 1140-1149.
- Prasse, A.; Pechkovsky, D.V.; Toews, G.B.; Jungraithmayr, W.; Kollert, F.; Goldmann, T.; Vollmer, E.; Muller-Quernheim, J.; Zissel, G. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via ccl18. Am J Respir Crit Care Med 2006, 173, 781-792. [CrossRef]
- Prasse, A.; Pechkovsky, D.V.; Toews, G.B.; Schafer, M.; Eggeling, S.; Ludwig, C.; Germann, M.; Kollert, F.; Zissel, G.; Muller-Quernheim, J. Ccl18 as an indicator of pulmonary fibrotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheum 2007, 56, 1685-1693. [CrossRef]
- Prasse, A.; Probst, C.; Bargagli, E.; Zissel, G.; Toews, G.B.; Flaherty, K.R.; Olschewski, M.; Rottoli, P.; Muller-Quernheim, J. Serum cc-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009, 179, 717-723. [CrossRef]
- Atamas, S.P.; Luzina, I.G.; Choi, J.; Tsymbalyuk, N.; Carbonetti, N.H.; Singh, I.S.; Trojanowska, M.; Jimenez, S.A.; White, B. Pulmonary and activation-regulated chemokine stimulates collagen production in lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2003, 29, 743-749. [CrossRef]
- Stahl, M.; Schupp, J.; Jager, B.; Schmid, M.; Zissel, G.; Muller-Quernheim, J.; Prasse, A. Lung collagens perpetuate pulmonary fibrosis via cd204 and m2 macrophage activation. PLoS One 2013, 8, e81382. [CrossRef]
- Luzina, I.G.; Papadimitriou, J.C.; Anderson, R.; Pochetuhen, K.; Atamas, S.P. Induction of prolonged infiltration of t lymphocytes and transient t lymphocyte-dependent collagen deposition in mouse lungs following adenoviral gene transfer of ccl18. Arthritis Rheum 2006, 54, 2643-2655. [CrossRef]
- Brand, S.; Olszak, T.; Beigel, F.; Diebold, J.; Otte, J.M.; Eichhorst, S.T.; Goke, B.; Dambacher, J. Cell differentiation dependent expressed ccr6 mediates erk-1/2, sapk/jnk, and akt signaling resulting in proliferation and migration of colorectal cancer cells. J Cell Biochem 2006, 97, 709-723. [CrossRef]
- Cambien, B.; Pomeranz, M.; Millet, M.A.; Rossi, B.; Schmid-Alliana, A. Signal transduction involved in mcp-1-mediated monocytic transendothelial migration. Blood 2001, 97, 359-366. [CrossRef]
- Plönes, T.; Krohn, A.; Burger, M.; Veelken, H.; Passlick, B.; Muller-Quernheim, J.; Zissel, G. Serum level of cc-chemokine ligand 18 is increased in patients with non-small-cell lung cancer and correlates with survival time in adenocarcinomas. PLoS One 2012, 7, e41746. [CrossRef]
- Trepel, M.; Arap, W.; Pasqualini, R. Modulation of the immune response by systemic targeting of antigens to lymph nodes. Cancer Research 2001, 61, 8110-8112.
- Müller, O.J.; Kaul, F.; Weitzman, M.D.; Pasqualini, R.; Arap, W.; Kleinschmidt, J.A.; Trepel, M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nature Biotechnology 2003, 21, 1040-1046. [CrossRef]
- Michelfelder, S.; Lee, M.K.; deLima-Hahn, E.; Wilmes, T.; Kaul, F.; Muller, O.; Kleinschmidt, J.A.; Trepel, M. Vectors selected from adeno-associated viral display peptide libraries for leukemia cell-targeted cytotoxic gene therapy. Exp Hematol 2007, 35, 1766-1776. [CrossRef]
- Binder, M.; Vögtle, F.; Michelfelder, S.; Müller, F.; Illerhaus, G.; S, S.; Mertelsmann, R.; Trepel, M. Identification of their epitope reveals the structural basis for the mechanism of action of the immunosuppressive antibodies basiliximab and daclizumab. Cancer Res 2007, 67, 3518-3523. [CrossRef]
- Binder, M.; Müller, F.; Jackst, A.; Pantic, M.; Bacher, U.; Zu Eulenburg, C.; Veelken, H.; Mertelsmann, R.; Pasqualini, R.; Arap, W., et al. B-cell receptor epitope recognition correlates with the clinical course of chronic lymphocytic leukemia. Cancer 2011, 117, 1891-1900. [CrossRef]
- Marwitz, S.; Abdullah, M.; Vock, C.; Fine, J.; Visvanathan, S.; Gaede, K.; Hauber, H.; Zabel, P.; Goldmann, T. Hope-bal: Improved molecular diagnostics by application of a novel technique for fixation and paraffin-embedding. J Histochem Cytochem 2011 in press.
- Goldmann, T.; Vollmer, E.; Gerdes, J. What's cooking? Detection of important biomarkers in hope-fixed, paraffin-embedded tissues eliminates the need for antigen retrieval. Am J Pathol 2003, 163, 2638-2640. [CrossRef]
- Jullien, N. Amplifx 1.7.0. https://inp.univ-amu.fr/en/amplifx-manage-test-and-design-your-primers-for-pcr 2004-2008.
- El Agha, E.; Thannickal, V.J. The lung mesenchyme in development, regeneration, and fibrosis. J Clin Invest 2023, 133. [CrossRef]
- Luzina, I.G.; Tsymbalyuk, N.; Choi, J.; Hasday, J.D.; Atamas, S.P. Ccl18-stimulated upregulation of collagen produc,tion in lung fibroblasts requires sp1 signaling and basal smad3 activity. J Cell Physiol 2006, 206, 221-228.
- Kodelja, V.; Muller, C.; Politz, O.; Hakij, N.; Orfanos, C.E.; Goerdt, S. Alternative macrophage activation-associated cc-chemokine-1, a novel structural homologue of macrophage inflammatory protein-1 alpha with a th2-associated expression pattern. J Immunol 1998, 160, 1411-1418.
- Pardo, A.; Smith, K.M.; Abrams, J.; Coffman, R.; Bustos, M.; McClanahan, T.K.; Grein, J.; Murphy, E.E.; Zlotnik, A.; Selman, M. Ccl18/dc-ck-1/parc up-regulation in hypersensitivity pneumonitis. J Leukoc Biol 2001, 70, 610-616.
- Baba, M.; Imai, T.; Nishimura, M.; Kakizaki, M.; Takagi, S.; Hieshima, K.; Nomiyama, H.; Yoshie, O. Identification of ccr6, the specific receptor for a novel lymphocyte-directed cc chemokine larc. J Biol Chem 1997, 272, 14893-14898. [CrossRef]
- Perez-Canadillas, J.M.; Zaballos, A.; Gutierrez, J.; Varona, R.; Roncal, F.; Albar, J.P.; Marquez, G.; Bruix, M. Nmr solution structure of murine ccl20/mip-3alpha, a chemokine that specifically chemoattracts immature dendritic cells and lymphocytes through its highly specific interaction with the beta-chemokine receptor ccr6. J Biol Chem 2001, 276, 28372-28379.
- Oku, H.; Shimizu, T.; Kawabata, T.; Nagira, M.; Hikita, I.; Ueyama, A.; Matsushima, S.; Torii, M.; Arimura, A. Antifibrotic action of pirfenidone and prednisolone: Different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol 2008, 590, 400-408. [CrossRef]
- Pochetuhen, K.; Luzina, I.G.; Lockatell, V.; Choi, J.; Todd, N.W.; Atamas, S.P. Complex regulation of pulmonary inflammation and fibrosis by ccl18. Am J Pathol 2007, 171, 428-437. [CrossRef]
- Schutyser, E.; Struyf, S.; Van Damme, J. The cc chemokine ccl20 and its receptor ccr6. Cytokine Growth Factor Rev 2003, 14, 409-426. [CrossRef]
- Singh, S.P.; Zhang, H.H.; Foley, J.F.; Hedrick, M.N.; Farber, J.M. Human t cells that are able to produce il-17 express the chemokine receptor ccr6. J Immunol 2008, 180, 214-221. [CrossRef]
- Voo, K.S.; Wang, Y.H.; Santori, F.R.; Boggiano, C.; Wang, Y.H.; Arima, K.; Bover, L.; Hanabuchi, S.; Khalili, J.; Marinova, E., et al. Identification of il-17-producing foxp3+ regulatory t cells in humans. Proc Natl Acad Sci U S A 2009, 106, 4793-4798. [CrossRef]
- Gerritsen, M.E.; Tomlinson, J.E.; Zlot, C.; Ziman, M.; Hwang, S. Using gene expression profiling to identify the molecular basis of the synergistic actions of hepatocyte growth factor and vascular endothelial growth factor in human endothelial cells. Br J Pharmacol 2003, 140, 595-610. [CrossRef]
- Alaaeddine, N.; Hilal, G.; Baddoura, R.; Antoniou, J.; Di Battista, J.A. Ccl20 stimulates proinflammatory mediator synthesis in human fibroblast-like synoviocytes through a map kinase-dependent process with transcriptional and posttranscriptional control. J Rheumatol 2011, 38, 1858-1865. [CrossRef]
- Jordana, M.; Schulman, J.; McSharry, C.; Irving, L.B.; Newhouse, M.T.; Jordana, G.; Gauldie, J. Heterogeneous proliferative characteristics of human adult lung fibroblast lines and clonally derived fibroblasts from control and fibrotic tissue. Am Rev Respir Dis 1988, 137, 579-584. [CrossRef]
- Kaarteenaho, R. The current position of surgical lung biopsy in the diagnosis of idiopathic pulmonary fibrosis. Respir Res 2013, 14, 43. [CrossRef]
- Zissel, G.; Müller-Quernheim, J.; Prasse, A. Blockade of ccl18 signaling via ccr6 as a therapeutic option in fibrotic diseases and cancer. 2011.
- Zissel, G.; Müller-Quernheim, J.; Prasse, A. Blockade of ccl18 signaling via ccr6 as a therapeutic option in fibrotic diseases and cancer. 2013.
Figure 1.
Expression of CCR6 in human lung tissue and lung fibroblast lines: A-E: Immunohistochemical staining of CCR6. CCR6 expression (red) is visible in bronchial epithelia (A), hyperplastic alveolar epithelial cells type II (B), fibroblasts (C) and probably macrophages (D) in a lung section from an IPF patient. In contrast, in a tumour-free lung from a cancer patient without COPD only a few cells, possibly macrophages, were found to express CCR6 (E) Magnifications A, B, and C: 200x, B and C: 400x.
Figure 1.
Expression of CCR6 in human lung tissue and lung fibroblast lines: A-E: Immunohistochemical staining of CCR6. CCR6 expression (red) is visible in bronchial epithelia (A), hyperplastic alveolar epithelial cells type II (B), fibroblasts (C) and probably macrophages (D) in a lung section from an IPF patient. In contrast, in a tumour-free lung from a cancer patient without COPD only a few cells, possibly macrophages, were found to express CCR6 (E) Magnifications A, B, and C: 200x, B and C: 400x.
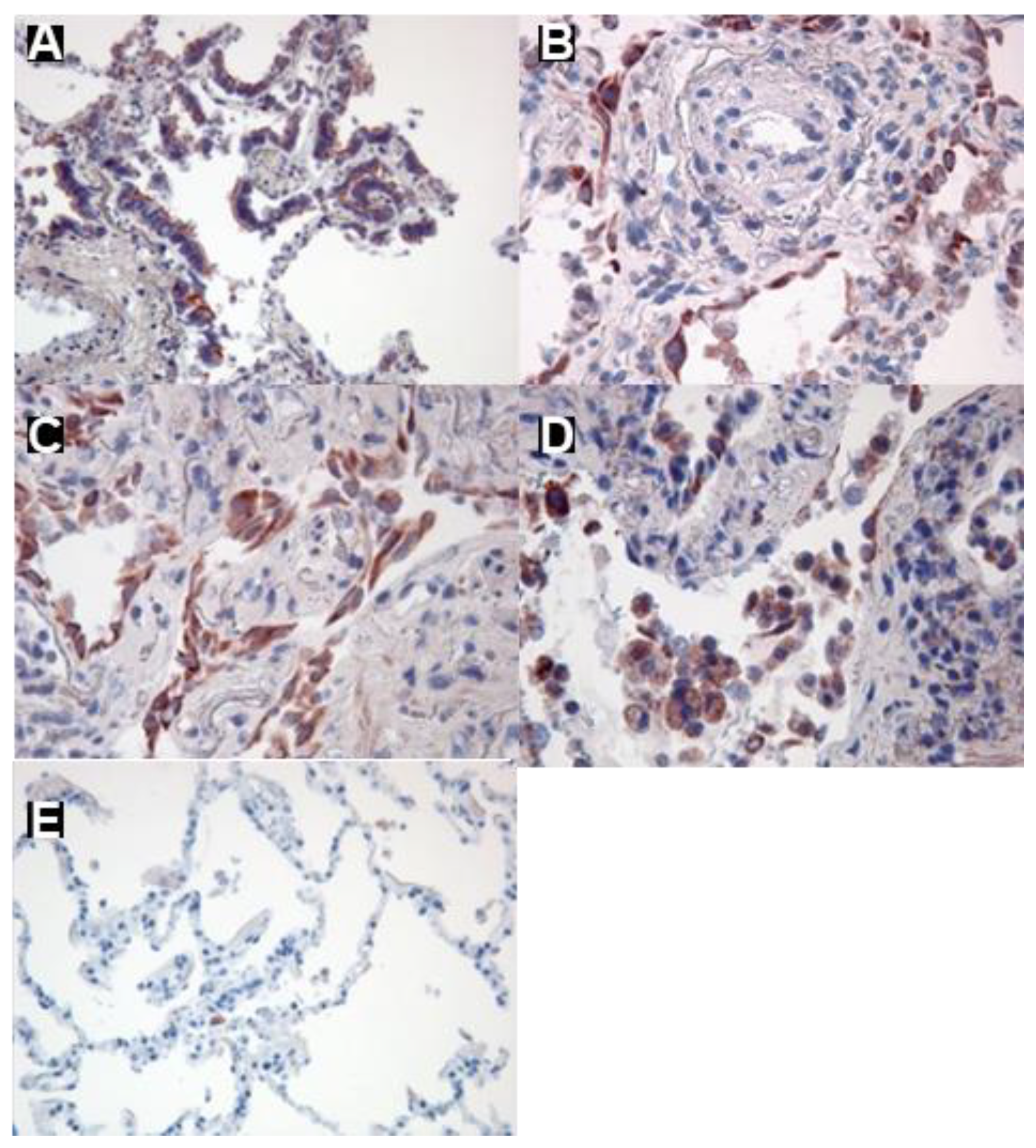
Figure 2.
CCR6 mRNA in human primary fibroblast lines. Quantitative PCR analysis of CCR6 expression by different fibroblast lines from patients suffering from squamous carcinoma (control; n=7) and pulmonary fibrosis (UIP: n=4, NSIP: n=3). Control fibroblasts did.
Figure 2.
CCR6 mRNA in human primary fibroblast lines. Quantitative PCR analysis of CCR6 expression by different fibroblast lines from patients suffering from squamous carcinoma (control; n=7) and pulmonary fibrosis (UIP: n=4, NSIP: n=3). Control fibroblasts did.
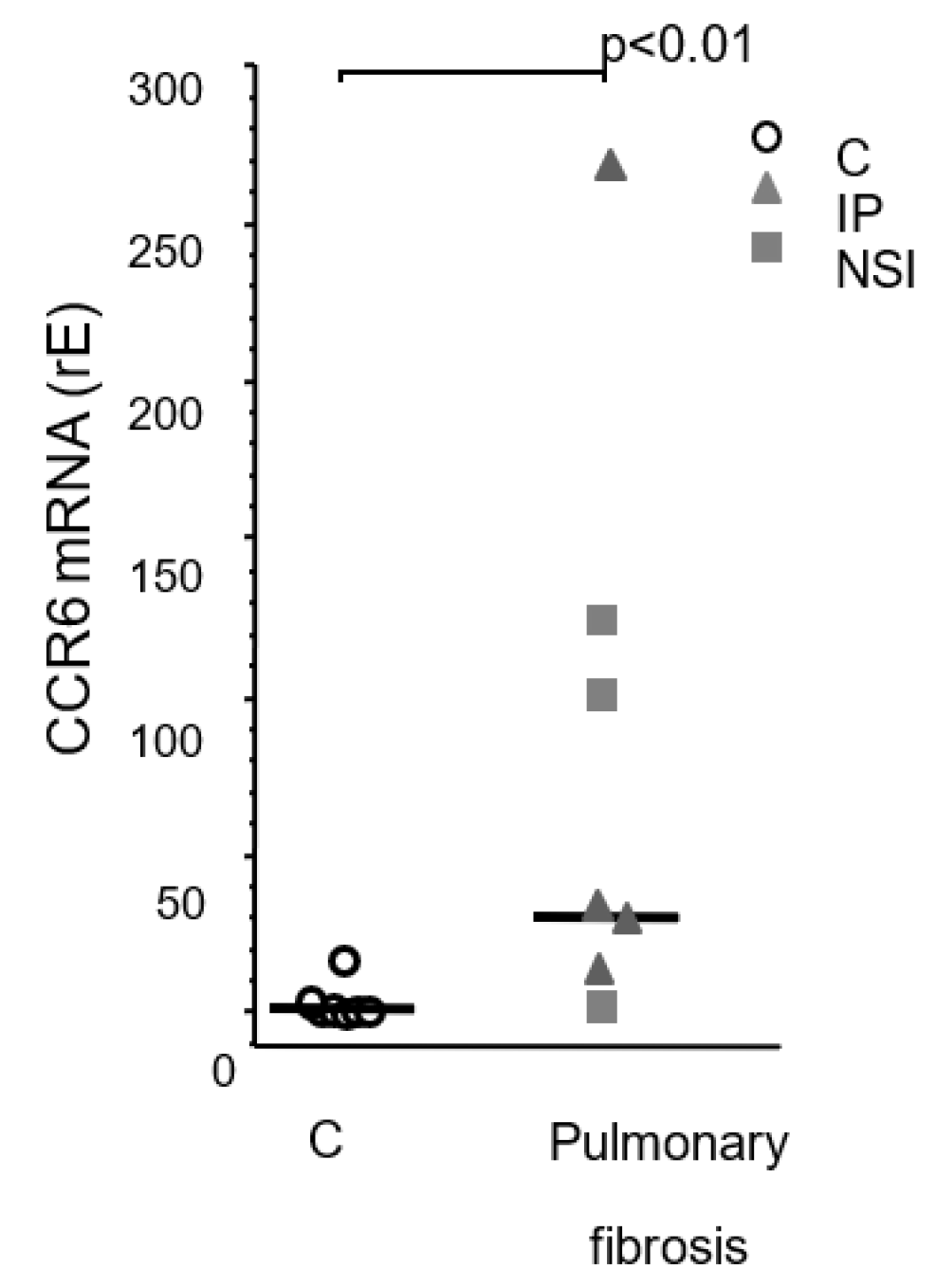
Figure 3.
CCR6 protein expression in human primary fibroblast lines. Figure A depicts flow cytometry strategy: the cells were plotted in the forward/sideward scatter (SSC/FSC) and the population with the highest density was gated. Within this gate a second gate was defined using an isotype control IgG (left panel) and CCR6-stained cells within this gate were regarded as positive (right panel). (B) Analysis of the flow cytometry revealed a clear up regulation of CCR6 in fibroblasts lines established from pulmonary fibrosis patients (IPF and NSIP) compared to our controls (p<0.0001) but not in patients suffering from fibrosis due to sarcoidosis (SAR) or hypersensitivity pneumonitis (HP).
Figure 3.
CCR6 protein expression in human primary fibroblast lines. Figure A depicts flow cytometry strategy: the cells were plotted in the forward/sideward scatter (SSC/FSC) and the population with the highest density was gated. Within this gate a second gate was defined using an isotype control IgG (left panel) and CCR6-stained cells within this gate were regarded as positive (right panel). (B) Analysis of the flow cytometry revealed a clear up regulation of CCR6 in fibroblasts lines established from pulmonary fibrosis patients (IPF and NSIP) compared to our controls (p<0.0001) but not in patients suffering from fibrosis due to sarcoidosis (SAR) or hypersensitivity pneumonitis (HP).
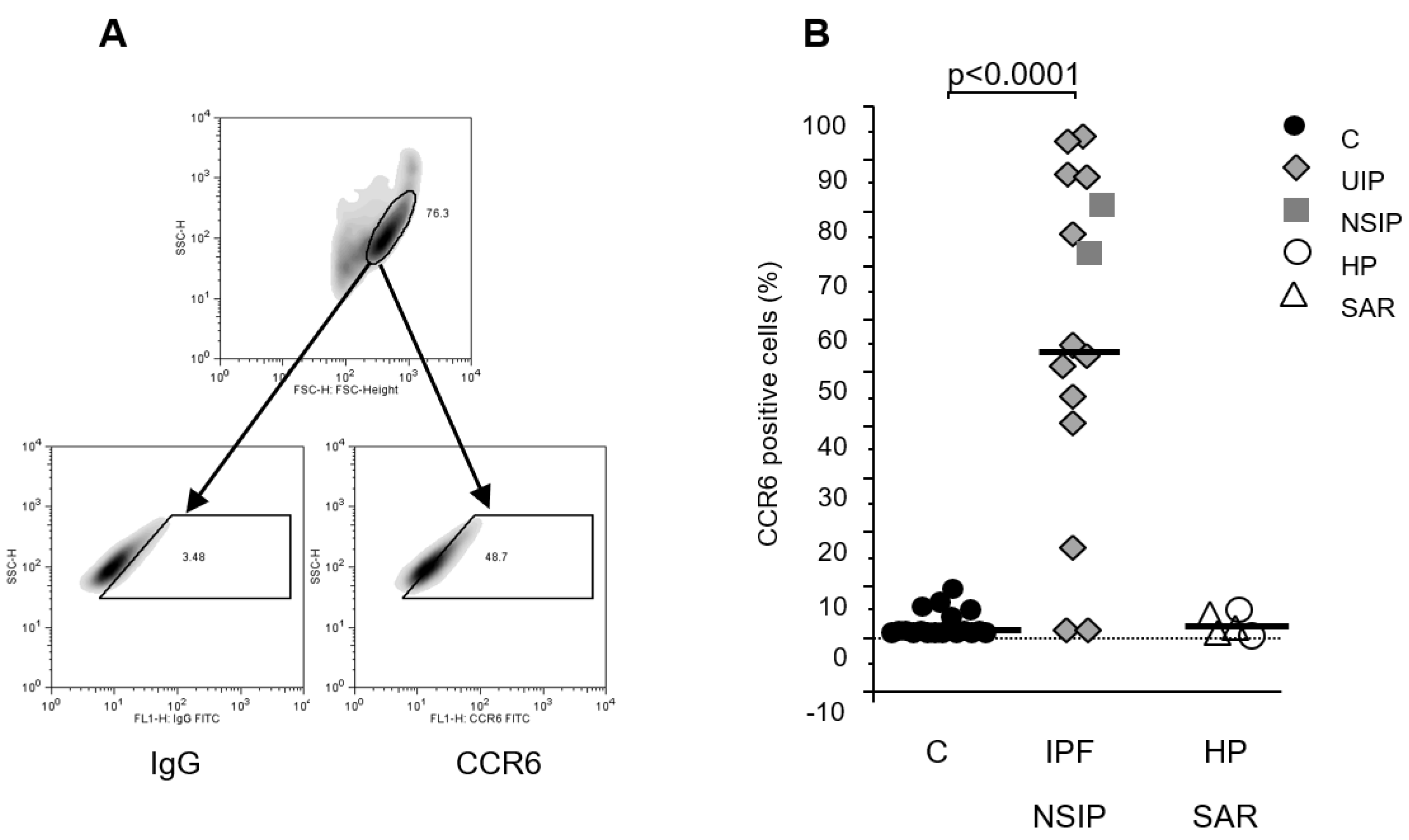
Figure 4.
Co-immunoprecipitation of CCR6*GFP (red) and CCL18 (green). RLE-6TN cells were loaded with 500 ng/mL recombinant CCL18 and the receptor/ligand complex was gently isolated from the membranes using anti-GFP beads. Lane 1: CCL18 (20ng); lane 2: lysed RLE-6TN cells only; M: marker; lane 3: CoIP after 10 minutes of CCL18 incubation; lane 4: CoIP after 30 minutes of incubation. CCR6*GFP is visible in all RLE-6TN preparations irrespective of the addition of CCL18 (lane 2, 3, and 4). CCL18 loaded directly on the gel forms a bright band at the lower end of lane 1 (green). CoIP after 10 minutes of CCL18 incubation did not reveal a visible CCL18 band. However, after 30minutes of incubation CoIP disclosed a small but visible CCL18 band (lane 4 bottom). The figure depicts one gel out of two independent experiments.
Figure 4.
Co-immunoprecipitation of CCR6*GFP (red) and CCL18 (green). RLE-6TN cells were loaded with 500 ng/mL recombinant CCL18 and the receptor/ligand complex was gently isolated from the membranes using anti-GFP beads. Lane 1: CCL18 (20ng); lane 2: lysed RLE-6TN cells only; M: marker; lane 3: CoIP after 10 minutes of CCL18 incubation; lane 4: CoIP after 30 minutes of incubation. CCR6*GFP is visible in all RLE-6TN preparations irrespective of the addition of CCL18 (lane 2, 3, and 4). CCL18 loaded directly on the gel forms a bright band at the lower end of lane 1 (green). CoIP after 10 minutes of CCL18 incubation did not reveal a visible CCL18 band. However, after 30minutes of incubation CoIP disclosed a small but visible CCL18 band (lane 4 bottom). The figure depicts one gel out of two independent experiments.
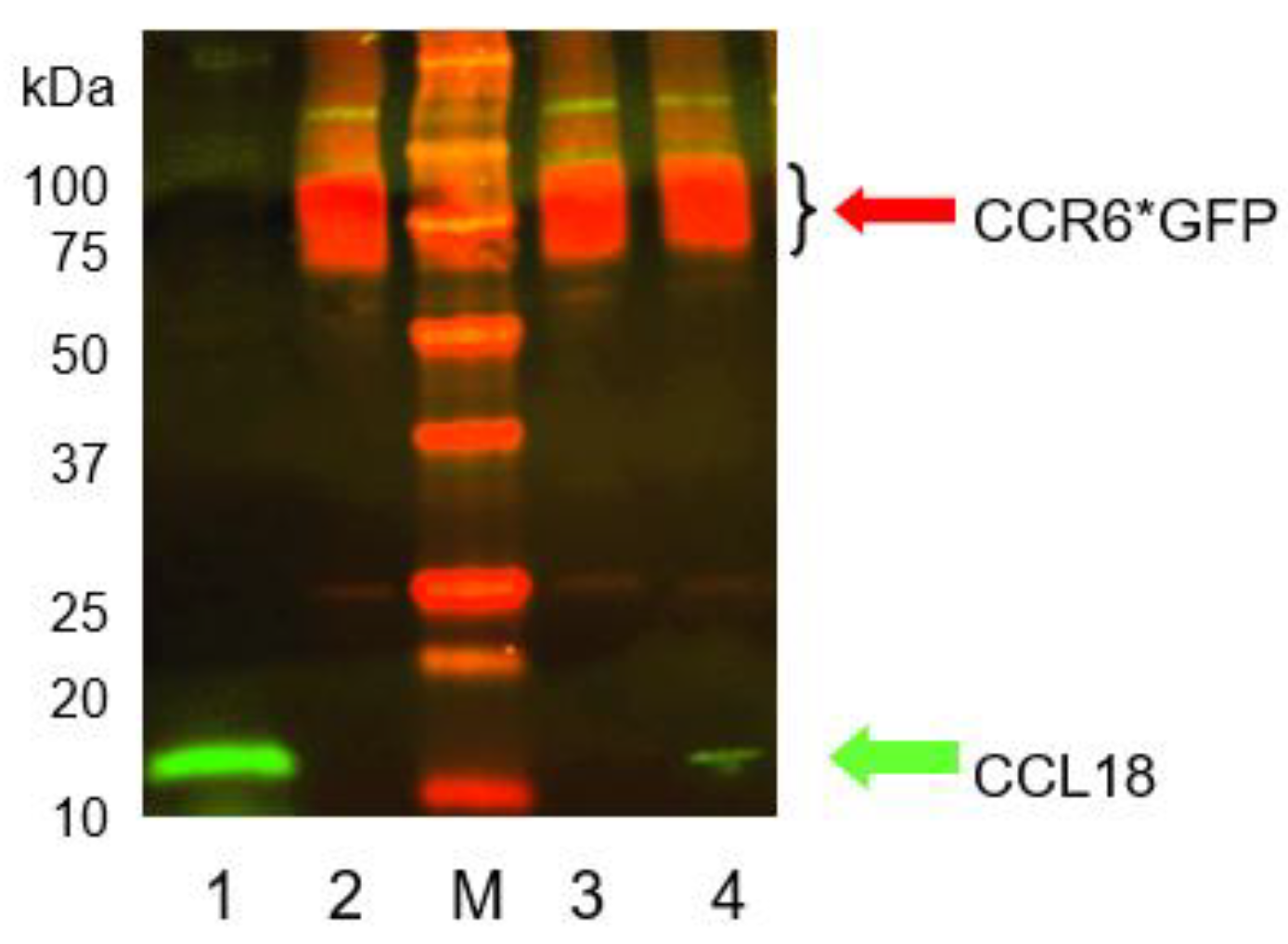
Figure 5.
A) CCL18 induces receptor internalization in CCR6*GFP transfected RLE-6TN cells. At time point 0 the receptor is located at the outer rim of the cells. Incubation with rhCCL18 (10 ng/mL) for 5 minutes induces receptor internalization visible as a ring or as bright dots within the cells (white arrows). After 20min of incubation with rhCCL18 the CCR6*GFP distribution is merely diffuse. In contrast, incubation with the irrelevant chemokine CXCL10 for 20min does not induce receptor internalization (magnification 10x40; 1 of 3 replicates). (B) Loading CCR6+ U937 cells with a wide concentration range of CCL18 or CCL20 reveals a clear dose-dependent internalization of CCR6. It is obvious that for the same effect approx. 10-fold more CCL18 is necessary compared with CCL20. The figure shows one illustrative series out of three.
Figure 5.
A) CCL18 induces receptor internalization in CCR6*GFP transfected RLE-6TN cells. At time point 0 the receptor is located at the outer rim of the cells. Incubation with rhCCL18 (10 ng/mL) for 5 minutes induces receptor internalization visible as a ring or as bright dots within the cells (white arrows). After 20min of incubation with rhCCL18 the CCR6*GFP distribution is merely diffuse. In contrast, incubation with the irrelevant chemokine CXCL10 for 20min does not induce receptor internalization (magnification 10x40; 1 of 3 replicates). (B) Loading CCR6+ U937 cells with a wide concentration range of CCL18 or CCL20 reveals a clear dose-dependent internalization of CCR6. It is obvious that for the same effect approx. 10-fold more CCL18 is necessary compared with CCL20. The figure shows one illustrative series out of three.
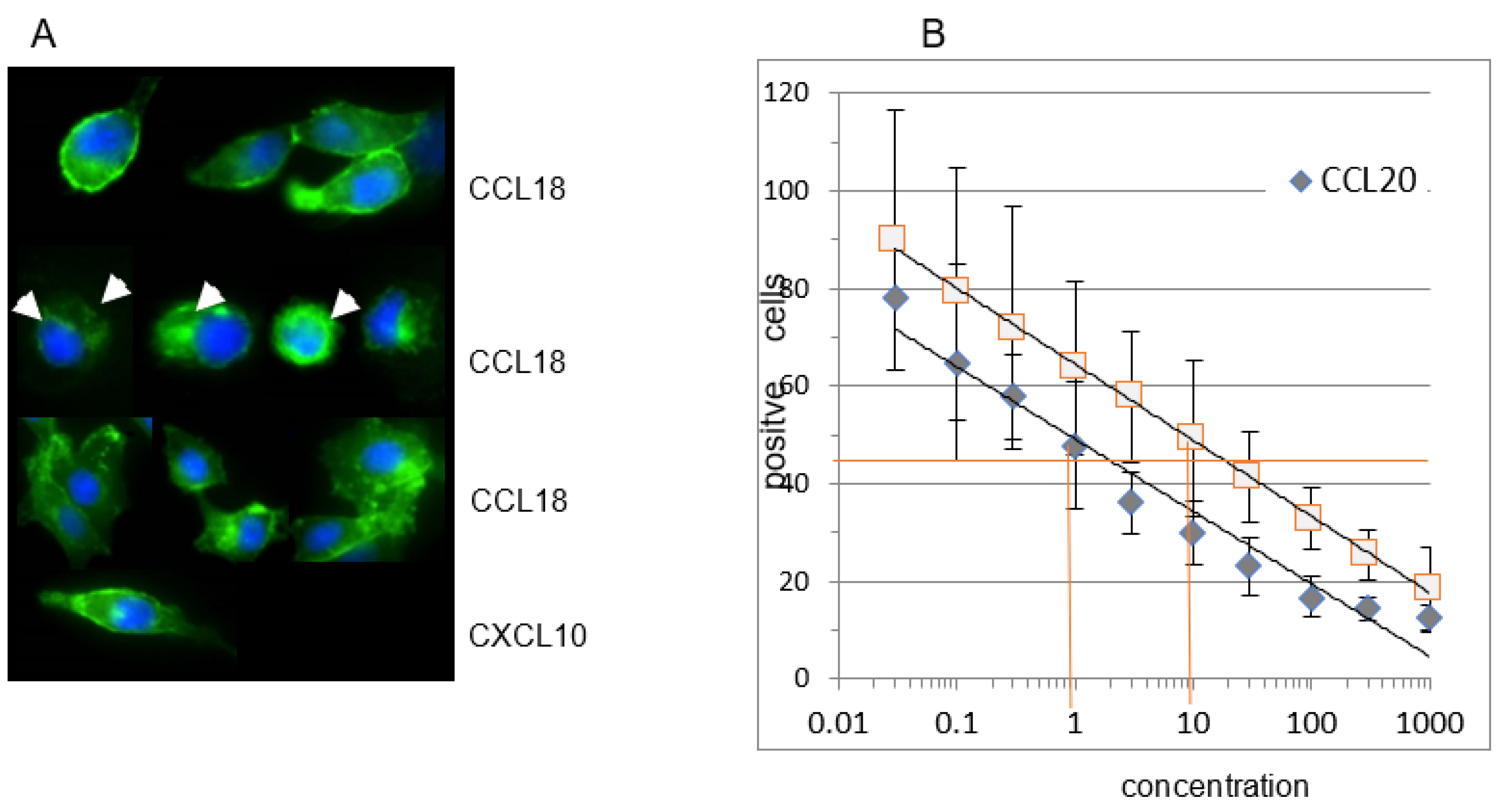
Figure 6.
A FGF2 release of human lung fibroblasts. Fibroblasts from fibrotic tissue (dark bars) release significantly higher levels of FGF2 compared with control fibroblasts (light bars). CCL18 significantly increases FGF2 release in fibrotic fibroblasts but barely in control fibroblasts. The CCL18 induced up-regulation of FGF2 release is abrogated by the addition of an anti-CCR6 antibody. The isotype control induced an insignificant reduction of the FGF2 release (C = control: n=6; Fib = fibrosis: n=6). B CCL18 induced collagen 1 (upper panel) and alpha-smooth muscle actin (αSMA, (lower panel)) mRNA expression by human lung fibroblasts is blocked by anti-CCR6 (rE = relative Expression; n=4).
Figure 6.
A FGF2 release of human lung fibroblasts. Fibroblasts from fibrotic tissue (dark bars) release significantly higher levels of FGF2 compared with control fibroblasts (light bars). CCL18 significantly increases FGF2 release in fibrotic fibroblasts but barely in control fibroblasts. The CCL18 induced up-regulation of FGF2 release is abrogated by the addition of an anti-CCR6 antibody. The isotype control induced an insignificant reduction of the FGF2 release (C = control: n=6; Fib = fibrosis: n=6). B CCL18 induced collagen 1 (upper panel) and alpha-smooth muscle actin (αSMA, (lower panel)) mRNA expression by human lung fibroblasts is blocked by anti-CCR6 (rE = relative Expression; n=4).
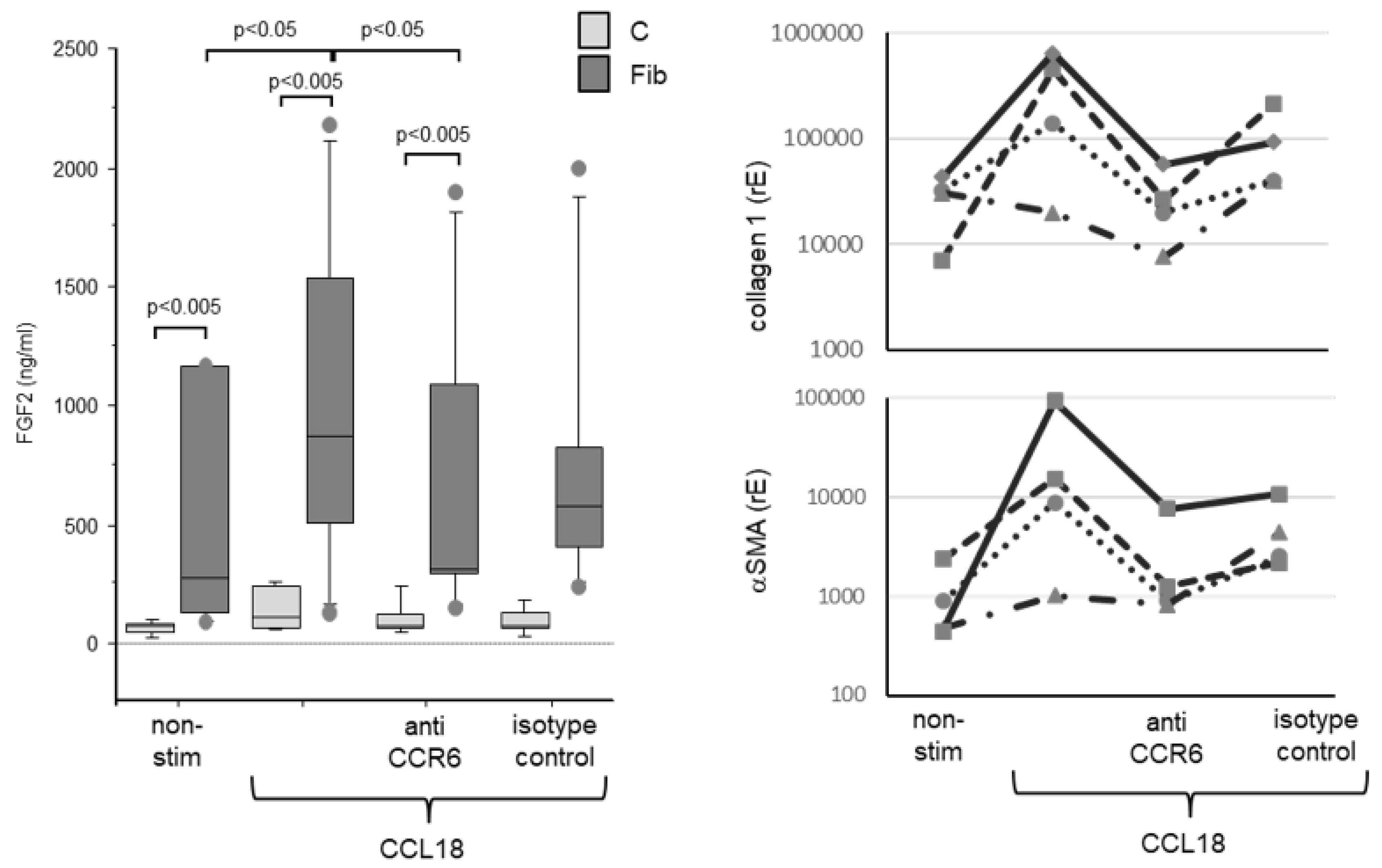
Figure 7.
In wild type NIH 3T3 cells CCL18 induces a dose dependent increase in Col1A and αSMA mRNA expression. TGFβ induced less Col1A but more αSMA mRNA expression compared with the highest dose of CCL18. In contrast, in the CCR6 KO clone no up-regulation of Col1A mRNA expression was seen. Interestingly, αSMA mRNA expression is down-regulated in this clone (rE = relative Expression; n=4).
Figure 7.
In wild type NIH 3T3 cells CCL18 induces a dose dependent increase in Col1A and αSMA mRNA expression. TGFβ induced less Col1A but more αSMA mRNA expression compared with the highest dose of CCL18. In contrast, in the CCR6 KO clone no up-regulation of Col1A mRNA expression was seen. Interestingly, αSMA mRNA expression is down-regulated in this clone (rE = relative Expression; n=4).
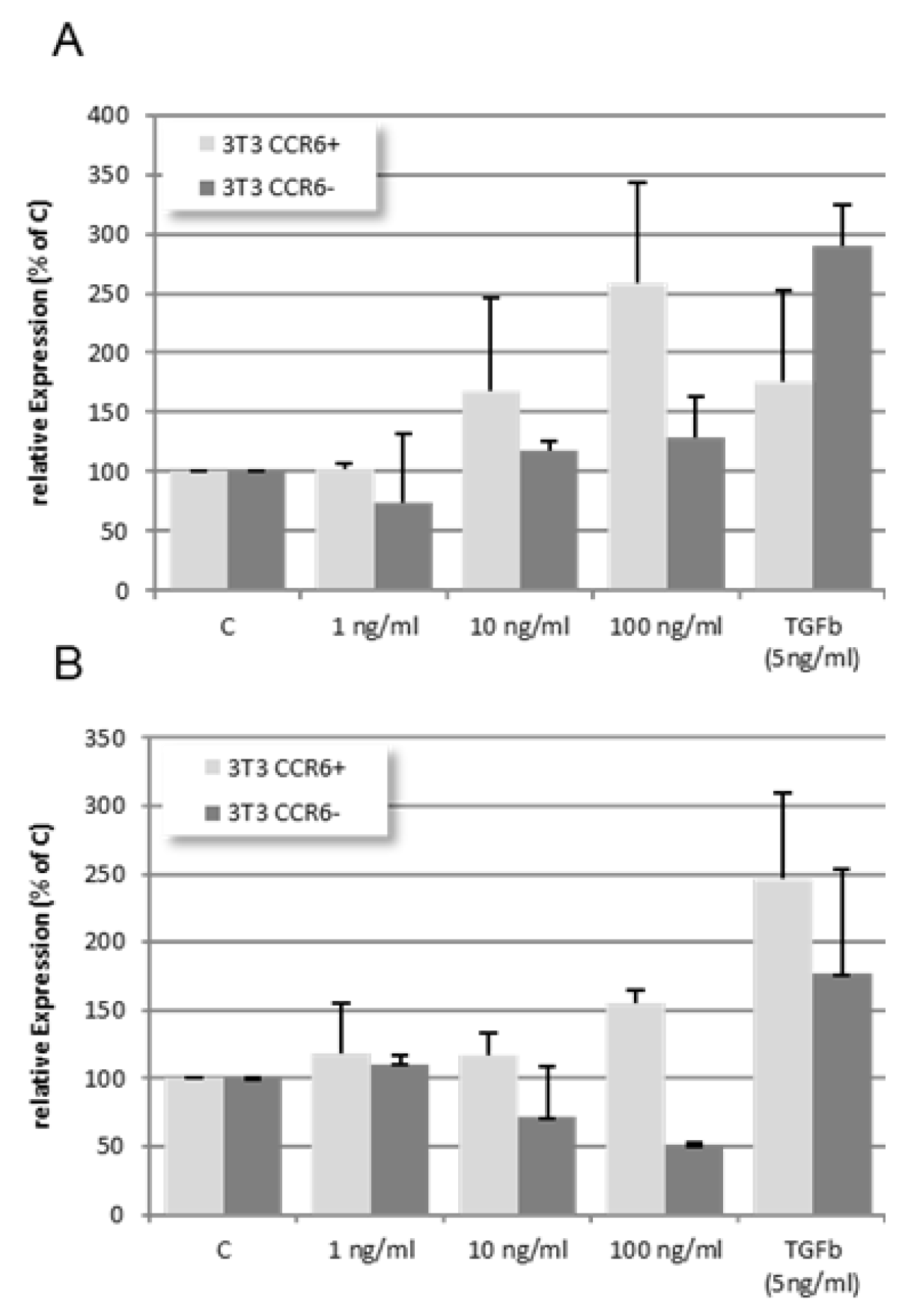
Figure 8.
Wild type and CCR6 KO NIH 3T3 cells were harvested and stained using isotype and anti-CCR6 antibodies as described. Gating as well as overlay of the flow cytometric analyses demonstrate that wild type NIH 3T3 cells clearly express CCR6 on the surface (upper panel). In contrast, the 3T3 CCR6 KO clone does not express CCR6 anymore (lower panel).
Figure 8.
Wild type and CCR6 KO NIH 3T3 cells were harvested and stained using isotype and anti-CCR6 antibodies as described. Gating as well as overlay of the flow cytometric analyses demonstrate that wild type NIH 3T3 cells clearly express CCR6 on the surface (upper panel). In contrast, the 3T3 CCR6 KO clone does not express CCR6 anymore (lower panel).
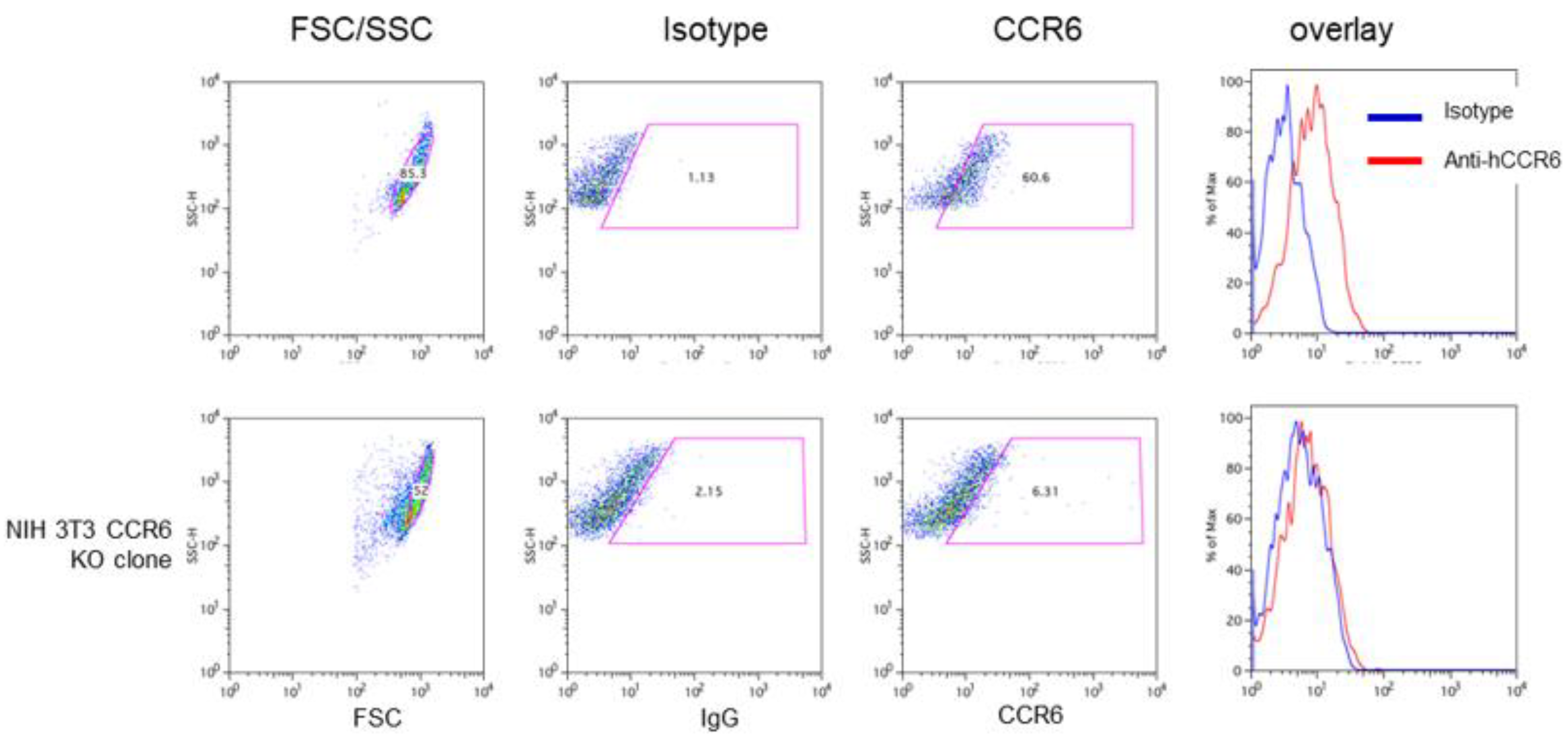

Table 1.
List of primers used in qPCR together with the accession numbers used for generating the primers.
Table 1.
List of primers used in qPCR together with the accession numbers used for generating the primers.
| Gene | accession number |
forward primer | reverse primer |
|---|---|---|---|
| hGAPDH | NM_002046 | CACCAGGGCTGCTTTTAACT | GATCTCGCTCCTGGAAGATG |
| hCCR6 | NM_004367 NM_031409 |
GCACAAAATGATGGCAGTGG | CCGAAGCACTTCCAGGTTGT |
| hCollagen 1A1 | NM_000088 | CCCTGTCTGCTTCCTGTAAACT | CATGTTCGGTTGGTCAAAGATA |
| hαSMA | NM_001141945 | CATCATGCGTCTGGATCTGG | GGACAATCTCACGCTCAGCA |
| mGAPDH | NM_001289726 | GCGAGACCCCACTAACATCAAA | cttttggctccacccttcaagt |
| mCollagen 1A1 | NM_007742 | TGCTGGGAAACATGGAAACCGA | AGGTTCTCCTTTGTCACCTCGGAT |
| mαSMA | NM_007392 | cacccagcaccatgaagatcaagA | CCTGTTTGCTGATCCACATCTGCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



