
Submitted:
15 December 2023
Posted:
18 December 2023
You are already at the latest version
Abstract
APC mutation is the main driver mechanism of CRC development and leads to constitutively activated WNT signaling, overpopulation of ALDH+ stem cells (SCs), and incomplete differentiation. We previously reported that retinoic acid (RA) receptors are selectively expressed in ALDH+ SCs, which provides a way to target cancer SCs with retinoids to induce differentiation. Hypotheses: A functional link exists between WNT and RA pathways, and APC mutation generates a WNT:RA imbalance that decreases retinoid-induced differentiation and increases ALDH+ SCs. Accordingly, to restore parity in WNT:RA signaling, we induced wt-APC expression in APC-mutant CRC cells and assessed the ability of all-trans-retinoic acid (ATRA) to induce differentiation. Notably, ATRA substantially increased expression of the WNT-target gene, CYP26A1, and inducing wt-APC reduced this expression by 50%. Thus, RA and WNT pathways crosstalk to modulate CYP26A1 metabolism of retinoids. We found that inducing wt-APC augments ATRA-induced cell differentiation by: i) decreasing cell proliferation; ii) suppressing ALDH1A1 expression; iii) decreasing ALDH+ SCs; iv) increasing neuroendocrine cell differentiation. Furthermore, NanoString profiling/bioinformatics identified a novel CYP26A1-based network that links WNT and RA signaling. Moreover, CYP26A1 inhibitors sensitized CRC cells to the anti-proliferative effect of drugs that downregulate WNT signaling. Notably. in wt-APC-CRCs, decreased CYP26A1 improves patient survival. These findings have strong potential for clinical translation.
Keywords:
CYP26A1
; colorectal cancer
; cancer stem cells
; APC
; aldehyde dehydrogenase
; WNT
; retinoic acid
; enteroendocrine cells
1. Introduction
Our goal has been to understand how mutation in the APC gene causes overpopulation of aldehyde dehydrogenase-positive (ALDH+) cancer SCs (CSCs) and decreased differentiation of SCs into neuroendocrine cells (NECs). The APC tumor suppressor gene is a negative regulator of WNT-signaling, which controls SC population size [1,2]. ALDH is a key enzyme in retinoic acid (RA) signaling, which is a pathway that promotes SC differentiation [3,4,5,6,7]. Thus, we chose to investigate whether there is a functional connection between WNT and RA signaling, and if APC mutation generates an imbalance in a WNT:RA-linked mechanism that contributes to ALDH+ SC overpopulation by impeding retinoid-induced differentiation [8].
We previously reported that inducing wild type (wt)-APC in HT29 CRC cells, which contain homozygous mutant APC, leads to decreased cell proliferation and increased apoptosis [9,10,11]. In mice, inactivation of APC leads to intestinal adenoma formation which provided evidence for crypt SCs as the cells-of-origin of intestinal cancer [12,13]. Restoring Apc in murine tumors led to increased enterocyte differentiation, tumor regression, and re-established crypt-villus homeostasis in CRC [13].
Previously we found that RA receptors are selectively expressed in ALDH+ SCs, which indicates RA signaling mainly occurs in ALDH+ SCs [14]. Since ALDH is a key enzyme in the RA pathway and RA signaling occurs via ALDH+ SCs, it provides a mechanism to selectively target CSCs based on a therapeutic strategy involving retinoid-induced differentiation [15]. Indeed, retinoids have been previously studied as chemo-preventive drugs and systemic therapies in treatment of CRC and other cancers [16,17]. Furthermore, we found that RA ligands such as ATRA induce differentiation of ALDH+ SCs into NECs [8,18]. On the other hand, APC mutation leads to incomplete differentiation in CRC cells [8,19]. Altogether, these findings suggest that APC mutation attenuates RA signaling in ALDH+ SCs, which contributes to their overpopulation that drives CRC development [8].
To identify mechanisms that functionally link WNT and RA signaling, we investigated how inducing wt-APC expression to decrease WNT signaling affects ATRA’s ability to induce NEC differentiation and reduce ALDH+ CSCs in APC mutant CRC.
2. Materials and Methods
2.1. Cell Culture and Inducing wt-APC Expression
HT29 colon carcinoma cells containing a ZnCl2-inducible wt-APC or β-galactosidase (control) expression vector were provided by Dr. Bert Vogelstein’s lab (Johns Hopkins University) [9] We refer to the modified HT29 cells as HT29-APC or HT29-GAL cells. HT29 cells were chosen for most of the study because they contain a homozygous APC mutation and do not have any known retinoic acid receptor gene mutations. Modified cells were cultivated in McCoy’s 5A modified media (Gibco/Thermofisher Scientific) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals/R&D Systems), 1% penicillin:streptomycin (pen-strep; VWR), and 600 μg/mL hygromycin B (Thermofisher Scientific). All variations of HT29 cells grew equally well in terms of cell density. Respective expression plasmids were induced with 120 μM ZnCl2 (Sigma-Aldrich) for the times indicated. HT29-APC cells treated with ZnCl2 are referred to wt-APC and those not treated with ZnCl2 are referred to mt-APC. All cells were maintained in a 37°C incubator with 5% CO2.
2.2. Cell Proliferation
HT29 cells were plated at 12,000 cells per well into 96-well plates. Cells were allowed to adhere for 24 hrs and serum-starved for an additional 24 hrs. Cells were then treated for 48 hrs with ZnCl2 and/or ATRA (Stem Cell Technologies) as indicated. Controls involved no ZnCl2 or DMSO (0.1%; Fisher Scientific) vehicle. After indicated time, cells were washed in PBS and stained in 0.5% crystal violet (Sigma-Aldrich) solution for 20 minutes. Excess crystal violet was then washed off with deionized water and left to dry at room temperature for at least 24 hrs. Dried crystal violet stain was then re-suspended in 10% acetic acid solution and optical density measured at 570 nm using a Tecan Multi-Plate Reader, data was quantified using Tecan i-Control software, and analyzed using Microsoft Excel. Values reflect cell proliferation as our standard curve shows that absorbance values from crystal violet staining are directly proportional to cell number. The proliferation index is calculated by normalizing cell proliferation index of treated to untreated cells. Biological replicates were performed in triplicate with 6 technical replicates per condition, and the average plotted with standard error of the mean (SEM) indicated.
2.3. WNT/β-Catenin Activity
WNT/β-catenin activity was measured using the TCF/LEF Reporter (BPS Biosciences) and Two-Step or Dual Glo (Firefly and Renilla) Luciferase Assays (BPS Biosciences or Promega) according to manufacturer instructions. In brief, cells were plated onto clear bottom, white 96-well plates and adhered for 24 hrs. Next, cells were transfected with the TCF/LEF reporter plasmids using Lipofectamine 3000 (Thermofisher Scientific). Cells were then treated as indicated for 24 hrs. The Firefly and Renilla luciferase assays were performed, and luminescence recorded using a Tecan Multi-Plate Reader, i-Control software, and data analyzed using Microsoft Excel. Luciferase activity was measured by calculating the ratio of Firefly to Renilla luminescence signals. Biological replicates were performed in triplicate and with 6 technical replicates per condition. The average luciferase activity was plotted with SEM indicated.
2.4. NanoString Profiling
To measure the effects of manipulating WNT and RA signaling, we utilized the nCounter PanCancer Pathways Panel from NanoString to provide high-throughput data including differential gene expression and subsequent pathway analysis. The PanCancer Pathways Panel includes 770 genes involved in several cancer pathways including WNT, hedgehog, apoptosis, cell cycle, RAS, PI3K, STAT, MAPK, Notch, TGF-β, chromatin modification, transcriptional regulation, and DNA damage control [20] and we included an additional 55 genes of interest to evaluate SC marker, differentiation marker, ALDH isoforms, RA receptors, and HOX genes.
HT29-APC cells were plated at 700,000 cells per well onto 6-well plates and allowed to adhere for 24 hrs. Cells were then serum-starved for 24 hrs. Cells were then treated with 15 µM ATRA or 0.1% DMSO and 120 µM ZnCl2 or no ZnCl2 for 24 hrs. Total RNA was isolated from cell lysates using the Trizol method. Samples were frozen at -80°C and sent to Wistar Institute’s Genomic Core Facility for analyzing quality of RNA and subsequent NanoString profiling using the Pan-cancer Panel. Results were obtained in RCC file format and data analyzed using NanoString nSolver software. A heat map was then generated to show differential expression of 785 mRNAs amongst the four treatment conditions (+ZnCl2 & +ATRA, + ZnCl2 & -ATRA, - ZnCl2 & +ATRA, ZnCl2 & -ATRA). Ratios were determined using nSolver, significance assessed, and plotted in Microsoft Excel. Gene lists of interest were analyzed using the STRING database for protein-protein interaction networks and functional enrichment [21].
2.5. Western Blotting & Densitometry
Standard western blotting procedures were performed. Cells were plated, treated as indicated with 120 µM ZnCl2 (Millipore Sigma), ATRA, or DMSO and lysed with RIPA (Millipore Sigma) containing Halt Protease and Phosphatase Inhibitor (Thermofisher) and cysteine protease inhibitor E-64 (Millipore Sigma). Cells lysates were scraped from dishes and centrifuged at 12,000 rpm for 15 minutes and stored at -20°C. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermofisher) and 100 µg total protein was prepared in Laemmli buffer (Bio-Rad) and 2.5% or 5% β-mercaptoethanol (Bio-Rad). Samples were loaded onto 4-20% mini-precast gradient gels (Bio-Rad) for SDS-PAGE at 200V for 45 minutes. Proteins were then transferred to PVDF membrane (Thermofisher) at 90V for 2 hrs. Blots were then blocked in 5% BLOTTO (nonfat dried milk in tris buffered saline (TBS) with 0.1% Tween 20 [TBST]) for 1 hr. Next, blots were incubated in primary antibody prepared in BLOTTO overnight on a rocking platform at 4°C. Blots were then washed in TBST 3x for 10 minutes. Secondary antibody was prepared in BLOTTO and blots incubated for 1-2 hrs at room temperature. Blots were then washed in TBST 3x for 10 min and incubated in Super Signal West Dura Extended Duration Substrate (Thermofisher) for 5 minutes to detect horseradish peroxidase conjugation. Blots were then imaged using the LiCor Odyssey Fc developer and Image Studio software. Densitometry was done using Image Studio and graphs plotted using Microsoft Excel. Experiments were performed 3 times and standard error of the mean (SEM) was calculated and plotted as shown.
2.6. Flow Cytometry and Fluorescence Activated Cell Sorting
NEC and ALDH+ (ALDEFLUOR+) cells were quantified and/or isolated using GLP2R antibody conjugated to Alexafluor 594 (Novus Biologicals) with IgG control (R&D) or ALDEFLUOR Assay (Stem Cell Technologies) using manufacturer’s instructions. Flow cytometry was performed on a BD FACS Aria II and FACS performed on a BD LSR Fortessa. Data was processed using FACS Diva software and graphs plotted in Microsoft Excel with error bars indicating SEM. Experiments were done 3 times.
2.7. Determining synergistic, Additive, or Antagonistic Anti-Proliferative Effects of CYP26A1 and WNT Signaling Inhibitors
We investigated the anti-proliferative effects of the CYP26A1 inhibitors Liarozole and Talarozole in combination with agents (Sulindac or Piroxicam) that have anti-tumor activity against APC-mutant tissues [reviewed in 16]. HT29 and HCT116 cells were cultivated in McCoy’s 5A media supplemented with 10% FBS and 1% pen-strep. SW480 cells were cultivated in Leibovitz L15 media supplemented with 10% FBS and 1% pen-strep. Cells were plated, allowed to adhere for 24 hrs, and serum-starved for 24 hrs. The cells were then treated for 48 hrs with the various agents as single drugs or in combination. For CYP26A1 inhibitors, IC50 doses of Liarozole and Talarozole are as follows: HT29 cells- 80 and 30 µM, HCT116 cells- 110 and 22 µM, and SW480 cells- 80 and 25 µM, respectively. For WNT inhibitors, IC50 doses of Sulindac and Piroxicam are as follows: HT29 cells- 680 µM and 550 µM, HCT116 cells- 450 µM and 600 µM, SW480 cells- 220 µM and 270 µM, respectively. In dose response experiments, Sulindac and Piroxicam doses were varied based on the IC50 dose (75% of IC50, IC50, 125% of IC50). For clarity, all concentrations are listed in Table S2. The proliferation index was determined using crystal violet assay (see above). Synergistic, additive, or antagonistic effects were calculated based on the Bliss independence dose-response model [22]. For example, these effects were calculated as the ratio of observed (observed values for the combination of drug a & b) divided by predicted (values for individual drug a multiplied by individual values for drug b), based on the Bliss Independence dose-response model. Further analyses were performed as described in main and supplementary figure legends.
2.8. Patient Survival Studies
RNA-seq, genotype, and clinical data on CRC patients were obtained from Genomic Data Commons (GDC) data portal. For individual patients, records of their APC genotypes, survival data, as well as CYP26A1 expression in both human CRC and normal human colon samples were paired together. These patients were then grouped based on whether they carry APC mutation and the change (either increase or decrease) of CYP26A1 expression between CRC and normal colon tissue samples.
2.9. Statistical Analyses
Statistics were calculated either in Excel using the t-test function or GraphPad Prism using one-way ANOVA, two-way ANOVA, or multiple unpaired t-tests as indicated in the figure legends. For patient survival studies, Kaplan-Meier survival curves of different groups were compared using log-rank tests.
3. Results
3.1. Inducing wt-APC Decreases WNT Signaling and Reduces Expression of WNT Target Genes
We first wanted to show that the HT29-APC cell line established by Morin et al [9] was functioning correctly in the ability of ZnCl2-treatment to induce wt-APC expression. Using two independent experimental approaches, we confirmed that expression of wt-APC occurs in response to ZnCl2 treatment (Figure S1). We initially measured WNT activity utilizing the TopFlash luciferase reporter assay which measures TCF/LEF transcriptional activity. There was a 90% reduction in WNT activity in ZnCl2-treated cells compared to untreated cells (Figure S1A). We then confirmed that reduced expression occurs in several proteins encoded by WNT/ β-catenin target genes [23] including MET [24], c-JUN [25], CD44 [26], and c-MYC [27] (Figure S1B).
3.2. ATRA Promotes WNT/β-Catenin Activity, wt-APC Attenuates ATRA’s Effect
Although ATRA reduces the number of ALDH+ SCs, reduces sphere-forming ability, and promotes differentiation of SCs in CRC [14], here we found that total cell proliferation in cells is reduced less than 20% at 48 hrs when treated with 20 µM ATRA (Figure 1A). However, when wt-APC expression was induced in HT29 cells, ATRA led to further reduction of total cell proliferation by 42% compared to untreated controls (Figure 1A). Dose response analysis also shows that cells with wt-APC are 2.4 times more responsive to the anti-proliferative effects of ATRA than cells with mt-APC (based on line slopes; Figure 1B). Additionally, we observed that WNT/β-catenin activity is increased by ATRA in HT29 cells by up to 10 times as compared to control (Figure 1C); but, this increase in WNT activity is attenuated with the expression of wt-APC (Figure 1C).
3.3. Inducing wt-APC Decreases ALDH+ Stem Cells and Increases NEC Differentiation
Here, we show an increase in the percentage of GLP2R+ NECs when wt-APC expression is induced (Figure 2A). The relative number of GLP2R+ cells quadruples when wt-APC is induced and increases 6-fold with combined wt-APC expression and ATRA as compared to mt-APC and no ATRA (Figure 2B). In parallel, the percentage of ALDH+ (ALDEFLUOR+) cells decreases by approximately 20% with ATRA alone and almost 60% with the combination of ATRA treatment and wt-APC induction (Figure 2C). In a similar manner, mRNA expression of ALDH1A1 decreased by 5-fold with the ATRA and wt-APC combination as compared to control (Figure 2D). These results further support the existence of an interplay between the WNT and RA signaling pathways.
We also investigated whether inducing wt-APC in HT29-APC cells changes the expression of NEC differentiation cell markers. Indeed, several NEC markers increase in expression with the expression of wt-APC in HT29 cells. Increased protein expression of CHGA, GLP2R, NSE (ENO2), and SSTR1 occurred as shown in (Figure 3). Overall, we found that inducing wt-APC in cells decreases ALDH+ CSCs and increases NEC differentiation.
3.4. Expression Profiling Identified CYP26A1 as a Link between WNT and RA Signaling Pathways
NanoString profiling revealed differential expression of 785 mRNAs across four treatment groups from results on ZnCl2 to induce wt-APC expression and ATRA to increase RA signaling (+ZnCl2 & +ATRA, +ZnCl2 & -ATRA, -ZnCl2 & +ATRA, -ZnCl2 & -ATRA). Hierarchical, agglomerative clustering was achieved and shown as a heat map that was generated using nSolver software (Figure 4A).
To examine the effect of inducing wt-APC expression and increasing RA signaling at the same time, we calculated the ratio of treated cells (+ZnCl2 & +ATRA) to corresponding control group (-ZnCl2 & -ATRA) using the nSolver software. A set of 248 genes was identified as being differentially expressed (Figure S2). To identify predicted signaling pathway interactions, we analyzed the set of 248 genes using the STRING database [21]. The large number of predicted interactions found in this network analysis (Figure S2) suggests that a substantial level of crosstalk occurs when the WNT and RA signaling pathways are modulated in HT29 cells.
By analyzing the list of 248 genes using the DAVID functional tool [29] and ranking the list of over 1500 hits, we identified those genes that are involved in RA signaling using the GOTERM_BP_DIRECT list. We then re-analyzed this list of 14 genes using the STRING database and found several predicted interactions. Of the 14 genes listed as involved in RA signaling, 12 were identified to have protein interactions: CYP26A1, RARA, HDAC2, KLF4, TNF, LEP, PDGFA, MYB, DKK1, WNT7B, WNT11, and FZD7 (Figure 4B). Notably, five WNT target genes were identified: CYP26A1 [30], WNT7B, WNT11, FZD7, and DKK1 (listed on NanoString’s Gene to Pathway Summary). However, CDKN2D and HOXA2 did not show any predicted connections with the other proteins identified in our current STRING network.
The highest change in expression occurred in CYP26A1 with a 15.52-fold increase when both wt-APC expression is induced and cells are treated with ATRA (Figure 4C). Moreover, HT29 cells treated with ATRA alone experience a 30.8-fold increase in CYP26A1 expression and inducing wt-APC attenuates this increase by 50% (Figure 5A). Western blot experiments validated the 50% decrease in CYP26A1 expression when cells are induced to express wt-APC and treated with 20 µM ATRA (Figure 5B,C).
3.5. CYP26A1 Inhibitor Agents Sensitize CRC Cells to the Anti-Proliferative Effect of Drugs That Downregulate WNT Signaling.
The anti-proliferative effects of CYP26A1 inhibitors, Liarozole and Talarozole, were evaluated alone and in combination with drugs that downregulate WNT signaling, Piroxicam and Sulindac (Figure 6 and Figure S3). Three CRC cell lines with mutations that activate WNT signaling were used, two with mutant APC (HT29 & SW480) and one with mutant β-catenin/CTNNB1 (HCT116). Each of the three cell lines have different RA receptor genotypes. HT29 cells have wild-type retinoid receptors, SW480 cells have mutations in RARA and RXRG, and HCT116 cells have mutant RARA [16]. Sulindac and Piroxicam have anti-tumor activity against APC mutant tissues, and Sulindac has been shown to target β-catenin and downregulate WNT signaling [reviewed in 16].
CRC cells were treated with the various agents as single drugs or in combination (Figure 6 and Figure S3). The Sulindac and Piroxicam doses were varied based on the IC50 (half-maximal inhibitory concentration) dose for treated cell lines. The data reveal that anti-tumor agents Sulindac and Piroxicam decrease the proliferation of CRC cells. The data also shows that the CYP26A1 inhibitors Liarozole and Talarozole alone reduce CRC proliferation. Moreover, when Sulindac or Piroxicam was combined with Liarozole or Talarozole, the drug combination produced additive or synergistic effects (Figure 6C,F). Further dose response analysis on the anti-proliferative effects of varying Piroxicam and Sulindac concentrations showed that Liarozole & Piroxicam, Liarozole & Sulindac, and Talarozole & Sulindac combinations produced the highest dose responsiveness in HT29, SW480, and HCT116 cells, respectively (Figure S4A,B).
3.6. Analysis of Human CRC Cases Indicates CYP26A1 Predicts Survival of Patients with Wild-type APC Tumors
To evaluate the effects of CYP26A1 on CRC patient survival, we conducted multi-omics bioinformatics analysis using RNA-seq, mutation, and clinical data obtained from Genomic Data Commons (GDC). Patients were grouped based on their APC genotypes and changes (increase or decrease) of CYP26A1 expression level between CRC and normal human colon samples. We found that the majority of CRC patients express increased levels of CYP26A1 in their CRC samples compared to normal colon tissue from the same patient, and this difference is statistically significant (Figure 7A). By itself, this change in CYP26A1 expression level is not a statistically significant predictor of patient survival (Figure 7B). However, when genotype of APC was used to further sub-group the patients, we found that CYP26A1 expression is a significant predictor of patient survival only when patient tumors are wild-type APC (Figure 7C). But estimated survival is not statistically significant between increased and decreased CYP26A1 expression groups among those patient tumors carrying APC mutations (Figure 7D). Similarly, when patients are sub-grouped based on CYP26A1 level, APC mutation is not a significant predictor of patient survival (Supplementary Figure 7A,B).
4. Discussion
We previously established that overpopulation of ALDH+ CSCs correlates with zygosity state of APC mutation during the stepwise tumor development in FAP patients [1]. We also found that APC mutation causes failure of ALDH+ SCs to mature into NECs [8,18]. Together, these findings provide a clue to a functional interaction between the WNT and RA signaling pathways that maintain stemness and promote differentiation, respectively [8,14,18]. Accordingly, we conjectured that a link exists between WNT and RA pathways, and, when APC is mutant, an imbalance in a WNT:RA-linked mechanism promotes CRC development [8]. Our main finding herein identified CYP26A1 as a link between WNT and RA signaling that can be targeted to decrease ALDH+ SCs and increase retinoid-induced differentiation of APC-mutant CRC cells. The functional relevance of this finding is that 1) CYP26A1 expression is controlled by WNT signaling via APC [30], and 2) CYP26A1 enzyme controls intracellular RA metabolism and regulates RA signaling [16,17]. Our previous finding that RA signaling mainly occurs via ALDH+ SCs [14] raises two key questions addressed below:
- Q1 How is RA signaling regulated in ALDH+ SCs?
- Q2 How does dysregulation of RA signaling due to APC mutation contribute to overpopulation of ALDH+ SCs that drives development of CRC?
Studying the effect of wt-APC on ATRA response indicates that WNT signaling, via its target gene CYP26A1, regulates RA signaling in the differentiation of ALDH+ SCs.
We found that inducing wt-APC expression, which decreases WNT-signaling, reduced expression of several WNT target genes and increased sensitivity of HT29 cells to the anti-proliferative effects of ATRA (Figure 1 and Figure S1). We also found that ATRA treatment of HT29 cells significantly increased CYP26A1 expression (30.8-fold) and inducing wt-APC attenuated this increase by 50% (Figure 5A). This increase of CYP26A1 by ATRA treatment of APC mutant HT29 cells may be explained by two factors 1) CYP26A1 is a RA target gene [31,32,33] which should be induced by ATRA. 2) CYP26A1 is also a WNT target gene [30] and ATRA increases WNT/β-catenin activity in HT29 cells even though they already have constitutively activated WNT due to mutated APC (Figure 1C). Notably, increased CYP26A1 expression in ATRA treated HT29 cells decreases when they are induced to express wt-APC (Figure 5). This finding indicates that the ability of wt-APC to reduce WNT/β-catenin activity is the main mechanism that counterbalances the ability of ATRA to increase CYP26A1 expression. Consequently, since wt-APC inhibits WNT signaling, it should maintain the balance between WNT and RA signaling. Indeed, that is precisely what we observed because induction of wt-APC reversed ATRA’s ability to increase: i) WNT/β-catenin activity (Figure 1C) and ii) CYP26A1 expression (Figure 5). These findings show that modulation of CYP26A1 levels by WNT signaling regulates RA signaling in APC mutant cells. Thus, our findings indicate that the anti-proliferative effect of ATRA is greatly enhanced by decreased expression of CYP26A1 due to the ability of wt-APC to reduce WNT signaling.
Since the RA pathway components are mainly expressed in ALDH+ SCs [14], WNT signaling, via its target gene CYP26A1, regulates RA signaling in ALDH+ SCs. Indeed, inducing wt-APC expression reduced expression of CYP26A1 and enhanced the ability of ATRA to decrease CRC cell proliferation (Figure 1 and Figure 5). Moreover, inducing wt-APC: i) suppressed ALDH1A1 expression, ii) decreased ALDH+ SCs, and iii) increased neuroendocrine cell differentiation. These findings show that APC’s role in the control of WNT signaling regulates, via its target gene CYP26A1, RA signaling in the differentiation of ALDH+ CSCs.
To further explore the mechanism by which WNT signaling might regulate RA signaling, we examined differential mRNA expression in HT29 cells that were induced to express wt-APC and that were treated with ATRA compared to their respective controls. NanoString profiling revealed that among the 248 genes showing a significant change in expression, CYP26A1 showed the greatest increase in expression (Figure 4C and Figure S2). Additionally, our NanoString and bioinformatics analyses identified a novel CYP26A1-based network of protein interactions which reveals how WNT and RA pathways are inter-connected. Specifically, we found that a unique signaling cascade that links components of the RA signaling pathway (CYP26A1, RARA) with components of the WNT pathway (DKK1, WNT7B, WNT11, FZD7) via HDAC2, KLF4, and TNF (Figure 4B). Thus, a CYP26A1-based network of both RA and WNT signaling components is predicted to play a role in the functional link between these two pathways.
Studying the effect of APC mutation on NEC differentiation indicates that decreased RA signaling contributes to overpopulation of ALDH+ SCs that drives development of CRC.
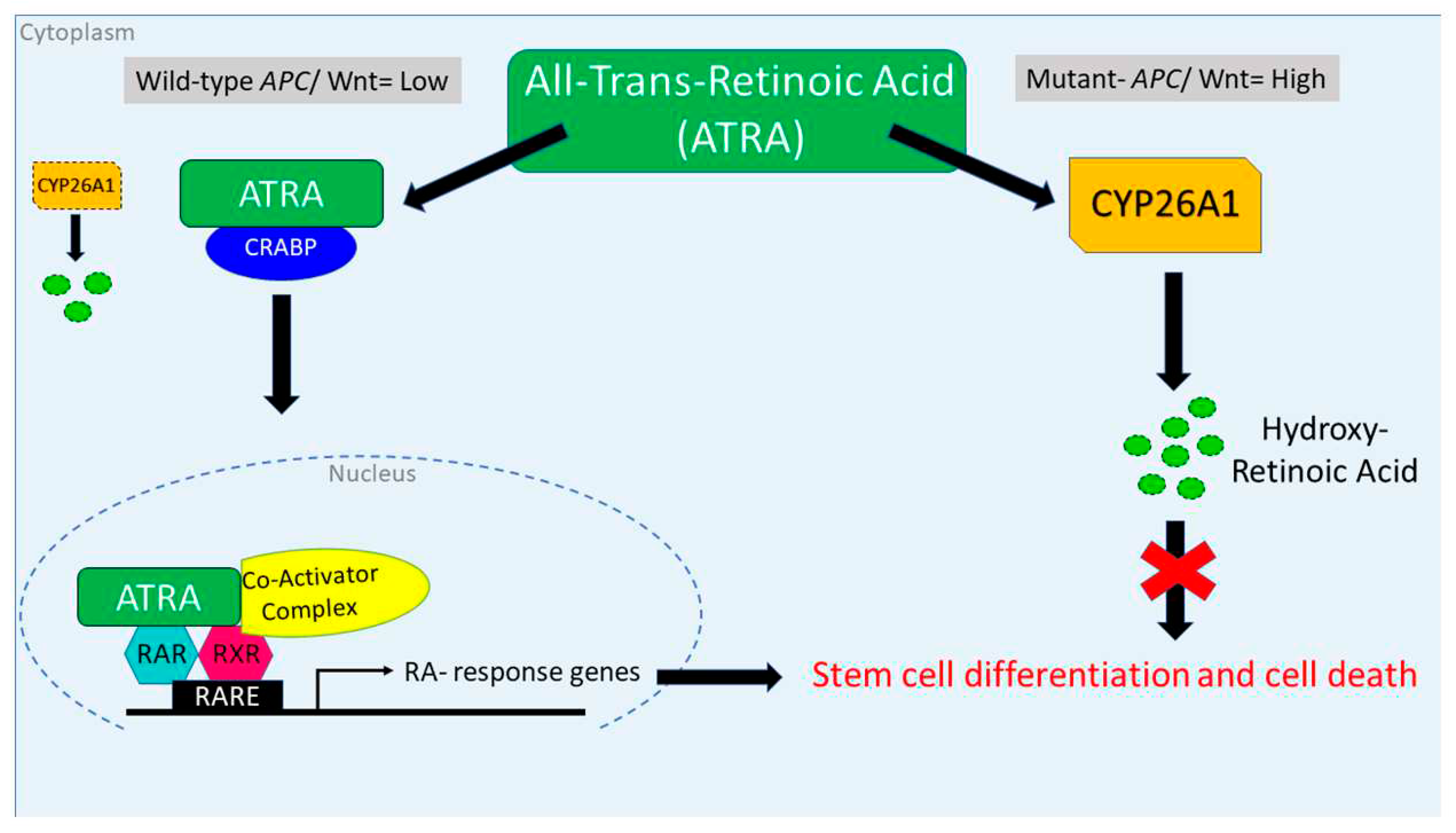
We found that inducing wt-APC expression, which decreases WNT-signaling and lowers CYP26A1 expression, leads to a decrease in cell proliferation (Figure 1A,B) and an increase in NEC differentiation (Figure 2A,B and Figure 3). Inducing wt-APC also decreased ALDH+ SC numbers and reduced expression of the ALDH1A1 SC marker (Figure 2C,D). Altogether, these findings show that elevated CYP26A1 levels prevent differentiation of CSCs in APC mutant cells by increasing RA clearance which reduces RA signaling (Figure 8). This mechanism provides an explanation for how decreased RA signaling contributes to ALDH+ SCs overpopulation that drives CRC development.
Our results concur with the finding in Shelton et al [30] in which CYP26A1 was upregulated in the intestine of APC mutant zebrafish embryos (and ApcMin/+ mouse adenomas, human FAP adenomas, and human sporadic CRCs) but not in zebrafish embryos with wt-APC. Defects in gut differentiation was also reversed with pharmacologic inhibition or knockdown of CYP26A1 in APC mutant zebrafish embryos [30]. Further studies on zebrafish showed that APC has a dual role in regulating Wnt and RA signaling [34,35]. Therefore, expression of wt-APC or pharmacologic inhibition of CYP26A1 may provide a novel therapeutic strategy to sensitize CRC cells to ATRA [36]. For example, a recent study by Penny et al. involved treating ApcMin/+ mice with the CYP26A1 inhibitor Liarozole [37]. The administration of Liarozole to ApcMin/+ mice increased endogenous RA signaling (by blocking ATRA metabolism), and dramatically reduced intestinal adenoma numbers in these Apc-mutant mice. We also found that the treatment of human CRC cells with Liarozole decreased proliferation, sphere formation, and the size of the ALDH+ SC population [14]. This suggests that decreasing the intracellular metabolism of ATRA using agents that inhibit CYP26A1 activity, might be a way to increase ATRA levels and therapeutically augment RA signaling in order to decrease CSC numbers in APC-mutant colon cancer tissues [16].
Clinical significance of our results that show CYP26A1 inhibitors, which block RA metabolism, sensitize CRC cells to anti-proliferative effect of drugs that downregulate WNT signaling.
Our findings likely have vast clinical importance. For instance, given that RA receptor mutant CRC cells show similar response to the CYP26A1 inhibitors (Liarozole and Talarozole) as do non-mutant cells (Figure 6), this indicates that the inhibition of CYP26A1 leads to a high enough intracellular RA level to induce growth inhibition regardless of RA receptor genotype. We then investigated the effects of these CYP26A1 inhibitors in combination with agents (Sulindac or Piroxicam) that have anti-tumor activity against APC-mutant tissues and Sulindac is known to target β-catenin and downregulate WNT signaling [reviewed in 16]. Notably, when Sulindac or Piroxicam was combined with CYP26A1 inhibitors Liarozole or Talarozole, the drug combination produced additive or synergistic effects (Figure 6C,F).
That the drug combinations reduce CRC proliferation to a greater extent than the CYP26A1 inhibitors or anti-WNT agents alone indicates that inhibiting CYP26A1 to increase RA signaling combined with the effect of inhibiting WNT signaling holds great promise as a therapeutic approach in oncology.
For human CRC cases, CYP26A1 predicts patient survival according to APC genotype.
Bioinformatics analysis of human CRC cases provides further evidence that CYP26A1 has clinical significance (Figure 7). We found that the majority of CRC patients express an increased level of CYP26A1 in their tumor tissues compared to normal colon from the same patient. Considering that CYP26A1 is a key enzyme responsible for RA degradation, patients who express lower levels of CYP26A1 should have higher levels of RA in their tumor cells, which should up-regulate RA-signaling activity and endow CRC patients with better survival. This prediction is supported by our results showing that CYP26A1 is only a significant determinant of patient survival when patient tumors carry wild-type, but not mutant, APC. This finding also supports the existence of crosstalk between WNT and RA-signaling that is dependent upon CYP26A1 expression level (hence cellular RA level), and APC genotype of the patients.
5. Conclusions
This work has vast clinical significance because it shows how CRC SCs might be sensitized to retinoid-induced cellular differentiation. We present a mechanism and therapeutic strategy to explain how differentiation-inducing agents such as ATRA may be used to promote CSC differentiation. Indeed, RA is an effective therapeutic agent in the treatment of acute promyelocytic leukemia (APL) [38,39,40,41,42]. RA therapy has also been shown to improve the survival of neuroblastoma patients [43]. While using retinoids to treat other cancers has shown limited success [16,17], therapeutically inhibiting CYP26A1, as we have shown herein, to reduce RA metabolism may prove useful in designing approaches to promote differentiation of CSCs with retinoids. Therefore, our findings are important because CRC development is driven by overpopulation of CSCs which are resistant to conventional chemotherapies and radiation [44,45,46]. Thus, finding ways to promote retinoid-induced differentiation of CSCs in vivo may lead to new and more effective therapeutic strategies for CRC patients.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1. WNT/β- catenin activity is reduced with expression of wt-APC. Figure S2. Inducing wild-type APC and treating with ATRA to drive RA signaling activates a highly regulated network of 248 proteins.
Author Contributions
Conceptualization, (BMB, COBF); methodology (COBF, LMO); software (COBF, BO, CZ); validation (COBF, LMO, VOH, BO, CZ); formal analysis (COBF, LMO, VOH, BO, CZ); investigation (COBF, VOH, BO, CZ); resources (BMB, COBF, LMO); data curation (COBF, CZ); writing—original draft preparation (BMB, COBF); writing—review and editing (BMB, COBF, BO, CZ, VH); visualization (COBF); supervision (BBM); project administration (NBN, LMO); funding acquisition (BMB). All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported in part by The Lisa Dean Moseley Foundation (2022; BMB, COBF, LMO), Cancer B*Ware Foundation (2018; BMB.), The Cawley Center for Translational Cancer Research Fund (2021; BMB, COBF, LMO), The Carpenter Foundation (BMB), University of Delaware Department of Biological Sciences (2021; VOH, BO, CZ) and INBRE NIH/NIGMS GM103446 (BMB).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics#_bookmark21.
Acknowledgments
We would like to thank Dr. Nicholas Petrelli for his support at the Helen F. Graham Cancer Center and Research Institute. We also thank the Vogelstein Lab at John’s Hopkins University for providing the inducible wt-APC cell lines. Special thanks to Sandy Widura and Dr. Sonali Majumdar at the Wistar Institute for performing our NanoString profiling experiments. Thanks also to all members past and present of Dr. Boman’s lab: Dr. Shirin Modarai, Victoria Stark, and Molly Lausten for their thoughtful discussions and contributions. Lastly, we acknowledge Dr. Ahui Ma and Dr. Jennifer Sims-Mourtada for their thoughtful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
CRC: colorectal cancer, ALDH: aldehyde dehydrogenase, SC: stem cell, CSC: cancer stem cell, ATRA: all-trans-retinoic acid, APC: adenomatous polyposis coli, wt: wild type, mt: mutant, WNT: Wingless and Int-1 signaling, CYP26A1: Cytochrome P450 Family 26 Subfamily A Member 1, RA = retinoic acid, NEC: neuroendocrine cell, DMSO: dimethyl sulfoxide, IC50: half-maximal inhibitory concentration.
References
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde Dehydrogenase 1 Is a Marker for Normal and Malignant Human Colonic Stem Cells (SC) and Tracks SC Overpopulation during Colon Tumorigenesis. Cancer Res 2009, 69, 3382–3389. [Google Scholar] [CrossRef] [PubMed]
- Boman, B.M.; Walters, R.; Fields, J.Z.; Kovatich, A.J.; Zhang, T.; Isenberg, G.A.; Goldstein, S.D.; Palazzo, J.P. Colonic Crypt Changes during Adenoma Development in Familial Adenomatous Polyposis. Am J Pathol 2004, 165, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Ma, I.; Allan, A.L. The Role of Human Aldehyde Dehydrogenase in Normal and Cancer Stem Cells. Stem Cell Rev Rep 2010, 7, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Marchitti, S.A.; Brocker, C.; Stagos, D.; Vasiliou, V. Non-P450 Aldehyde Oxidizing Enzymes: The Aldehyde Dehydrogenase Superfamily. Expert Opin Drug Metab Toxicol 2008, 4, 697–720. [Google Scholar] [CrossRef] [PubMed]
- M., A.; Bagirova, M.; Nehir, O.; Yaman, S.; Sefik, E.; Cakir, R.; Canim, S.; Elcicek, S.; Yesilkir, S. Aldehyde Dehydrogenase: Cancer and Stem Cells. In Dehydrogenases; InTech, 2012. [Google Scholar] [CrossRef]
- Black, W.J.; Stagos, D.; Marchitti, S.A.; Nebert, D.W.; Tipton, K.F.; Bairoch, A.; Vasiliou, V. Human Aldehyde Dehydrogenase Genes: Alternatively Spliced Transcriptional Variants and Their Suggested Nomenclature. Pharmacogenet Genomics 2009, 19, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Das, B.C.; Thapa, P.; Karki, R.; Das, S.; Mahapatra, S.; Liu, T.-C.; Torregroza, I.; Wallace, D.P.; Kambhampati, S.; Veldhuizen, P. Van; et al. Retinoic Acid Signaling Pathways in Development and Diseases. Bioorg Med Chem 2014, 22, 673–683. [Google Scholar] [CrossRef]
- Zhang, T.; Ahn, K.; Emerick, B.; Modarai, S.R.; Opdenaker, L.M.; Palazzo, J.; Schleiniger, G.; Fields, J.Z.; Boman, B.M. APC Mutations in Human Colon Lead to Decreased Neuroendocrine Maturation of ALDH+ Stem Cells That Alters GLP-2 and SST Feedback Signaling: Clue to a Link between WNT and Retinoic Acid Signalling in Colon Cancer Development. PLoS One 2020, 15, e0239601. [Google Scholar] [CrossRef]
- Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Apoptosis and APC in Colorectal Tumorigenesis. Proc Natl Acad Sci USA 1996, 93, 7950–7954. [Google Scholar] [CrossRef]
- Zhang, T.; Fields, J.Z.; Opdenaker, L.; Otevrel, T.; Masuda, E.; Palazzo, J.P.; Isenberg, G.A.; Goldstein, S.D.; Brand, M.; Boman, B.M. Survivin-Induced Aurora-B Kinase Activation. Am J Pathol 2010, 177, 2816–2826. [Google Scholar] [CrossRef]
- Zhang, T.; Otevrel, T.; Gao, Z.; Gao, Z.; Ehrlich, S.M.; Fields, J.Z.; Boman, B.M. Evidence That APC Regulates Survivin Expression: A Possible Mechanism Contributing to the Stem Cell Origin of Colon Cancer. Cancer Res 2001, 61, 8664–8667. [Google Scholar]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt Stem Cells as the Cells-of-Origin of Intestinal Cancer. Nature 2008, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Dow, L.E.; O’Rourke, K.P.; Simon, J.; Tschaharganeh, D.F.; van Es, J.H.; Clevers, H.; Lowe, S.W. APC Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell 2015, 161, 1539–1552. [Google Scholar] [CrossRef]
- Modarai, S.R.; Gupta, A.; Opdenaker, L.M.; Kowash, R.; Masters, G.; Viswanathan, V.; Zhang, T.; Fields, J.Z.; Boman, B.M. The Anti-Cancer Effect of Retinoic Acid Signaling in CRC Occurs via Decreased Growth of ALDH Colon Cancer Stem Cells and Increased Differentiation of Stem Cells. Oncotarget 2018, 9, 34658–34669. [Google Scholar] [CrossRef]
- Clark, D.W.; Palle, K. Aldehyde Dehydrogenases in Cancer Stem Cells: Potential as Therapeutic Targets. Ann Transl Med 2016, 4, 518. [Google Scholar] [CrossRef]
- Hunsu, V.O.; Facey, C.O.B.; Fields, J.Z.; Boman, B.M. Retinoids as Chemo-Preventive and Molecular-Targeted Anti-Cancer Therapies. Int J Mol Sci 2021, 22, 7731. [Google Scholar] [CrossRef]
- Facey COB, Boman BM. Retinoids in Treatment of Colorectal Cancer [Online First], IntechOpen. Available online: https://www.intechopen.com/online-first/retinoids-in-treatment-of-colorectal-cancerRetinoids. [CrossRef]
- Modarai, S.R.; Opdenaker, L.M.; Viswanathan, V.; Fields, J.Z.; Boman, B.M. Somatostatin Signaling via SSTR1 Contributes to the Quiescence of Colon Cancer Stem Cells. BMC Cancer 2016, 16, 941. [Google Scholar] [CrossRef] [PubMed]
- Boman, B.M.; Fields, J.Z. An APC:WNT Counter-Current-Like Mechanism Regulates Cell Division Along the Human Colonic Crypt Axis: A Mechanism That Explains How APC Mutations Induce Proliferative Abnormalities That Drive Colon Cancer Development. Front Oncol 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://Nanostring.Com/Products/Ncounter-Assays-Panels/Oncology/Ncounter-Pancancer-Pathways-Panel/.
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein–Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Liu Q, Yin X, Languino LR. Evaluation of drug combination effect using a Bliss independence dose-response surface model. Stat Biopharm Res 2018, 10, 112–122. [Google Scholar] [CrossRef]
- Available online: http://Web.Stanford.Edu/Group/Nusselab/Cgi-Bin/Wnt/Target_genes.
- Boon, E.M.J.; van der Neut, R.; van de Wetering, M.; Clevers, H.; Pals, S.T. Wnt Signaling Regulates Expression of the Receptor Tyrosine Kinase Met in Colorectal Cancer. Cancer Res 2002, 62, 5126–5128. [Google Scholar]
- Mann, B.; Gelos, M.; Siedow, A.; Hanski, M.L.; Gratchev, A.; Ilyas, M.; Bodmer, W.F.; Moyer, M.P.; Riecken, E.O.; Buhr, H.J.; et al. Target Genes of β-Catenin–T Cell-Factor/Lymphoid-Enhancer-Factor Signaling in Human Colorectal Carcinomas. Proc Natl Acad Sci USA 1999, 96, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Wielenga, V.J.M.; Smits, R.; Korinek, V.; Smit, L.; Kielman, M.; Fodde, R.; Clevers, H.; Pals, S.T. Expression of CD44 in APC and Tcf Mutant Mice Implies Regulation by the WNT Pathway. Am J Pathol 1999, 154, 515–523. [Google Scholar] [CrossRef] [PubMed]
- He, T.-C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C- MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Wiedenmann, B.; Franke, W.W.; Kuhn, C.; Moll, R.; Gould, V.E. Synaptophysin: A Marker Protein for Neuroendocrine Cells and Neoplasms. Proc Natl Acad Sci USA 1986, 83, 3500–3504. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://David.Ncifcrf.Gov/.
- Shelton, D.N.; Sandoval, I.T.; Eisinger, A.; Chidester, S.; Ratnayake, A.; Ireland, C.M.; Jones, D.A. Up-Regulation of CYP26A1 in Adenomatous Polyposis Coli-Deficient Vertebrates via a WNT-Dependent Mechanism: Implications for Intestinal Cell Differentiation and Colon Tumor Development. Cancer Res 2006, 66, 7571–7577. [Google Scholar] [CrossRef] [PubMed]
- Ozpolat B, Mehta K, Lopez-Berestein G. Regulation of a highly specific retinoic acid-4-hydroxylase (CYP26A1) enzyme and all-trans-retinoic acid metabolism in human intestinal, liver, endothelial, and acute promyelocytic leukemia cells. Leuk Lymphoma 2005, 46, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res 2002, 43, 1773–1808. [Google Scholar] [CrossRef] [PubMed]
- Rhinn M, Dollé P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Nadauld LD, Sandoval IT, Chidester S. Adenomatous polyposis coli control of retinoic acid biosynthesis is critical for zebrafish intestinal development and differentiation. J Biol Chem 2004, 279, 51581–51589. [Google Scholar] [CrossRef]
- Nadauld LD, Chidester S, Shelton DN. Dual roles for adenomatous polyposis coli in regulating retinoic acid biosynthesis and Wnt during ocular development. Proc Natl Acad Sci USA 2006, 103, 13409–13414. [Google Scholar] [CrossRef]
- Nelson, C.; Buttrick, B.; Isoherranen, N. Therapeutic Potential of the Inhibition of the Retinoic Acid Hydroxylases CYP26A1 and CYP26B1 by Xenobiotics. Curr Top Med Chem 2013, 13, 1402–1428. [Google Scholar] [CrossRef] [PubMed]
- Penny, H.L.; Prestwood, T.R.; Bhattacharya, N.; Sun, F.; Kenkel, J.A.; Davidson, M.G.; Shen, L.; Zuniga, L.A.; Seeley, E.S.; Pai, R.; et al. Restoring Retinoic Acid Attenuates Intestinal Inflammation and Tumorigenesis in ApcMin/ Mice. Cancer Immunol Res 2016, 4, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Idres, N.; Benoı̂t, G.; Flexor, M.A.; Lanotte, M.; Chabot, G.G. Granulocytic Differentiation of Human NB4 Promyelocytic Leukemia Cells Induced by All-Trans Retinoic Acid Metabolites. Cancer Res 2001, 61, 700–705. [Google Scholar] [PubMed]
- Zhu, J.-W.; Shi, X.G.; Chu, H.Y.; Tong, J.H.; Wang, Z.; Naoe, T.; Waxman, S.M.; Chen, S.; Chen, Z. Effect of retinoic acid isomers on proliferation, differentiation and PML relocalization in the APL cell line NB4. Leukemia 1995, 9, 302–309. [Google Scholar] [PubMed]
- Fang, J.; Chen, S.-J.; Tong, J.-H.; Wang, Z.-G.; Chen, G.-Q. Treatment of Acute Promyelocytic Leukemia with ATRA and As2O3: A Model of Molecular. Cancer Biol Ther 2002, 1, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Kantarjian, H.; Ravandi, F. Acute Promyelocytic Leukemia Current Treatment Algorithms. Blood Cancer J 2021, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Tallman, M.S. Acute Promyelocytic Leukemia (APL): Remaining Challenges towards a Cure for ALL. Leuk Lymphoma 2019, 60, 3107–3115. [Google Scholar] [CrossRef] [PubMed]
- Gudas, L.J.; Wagner, J.A. Retinoids Regulate Stem Cell Differentiation. J Cell Physiol 2011, 226, 322–330. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and Their Therapeutic Implications in Cancer Treatment. Stem Cells Int 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int J Mol Sci 2017, 18, 2574. [Google Scholar] [CrossRef]
- Boman, B.M.; Wicha, M.S. Cancer Stem Cells: A Step Toward the Cure. J Clin Oncol 2008, 26, 2795–2799. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Wt-APC expression sensitizes HT29 cells to ATRA’s ability to decrease cell proliferation and attenuates ATRA-induced WNT/β- catenin activity. A) Cell proliferation was reduced by 65% when wt-APC was induced by ZnCl2 and cells were treated with 20 µM ATRA for 48hrs. Values are normalized to mt-APC DMSO control. B) Cell proliferation was reduced by approximately 10% (10 μM) and 20% (20 μM) in mutant-APC (mt-APC) cells (black line). Cell proliferation was further reduced by approximately 36% (10 μM) and 42% (20 μM) in wildtype-APC (wt-APC) cells (dashed gray line). Similar results were seen at 24hrs, altogether indicating a time- and concentration- dependent effect. Mt-APC and wt-APC values were normalized to each of their respective DMSO controls. C) TCF/LEF Reporter Assay was performed to determine WNT/β- catenin activity. ATRA caused an increase in WNT- activity in mt-APC cells (black bars) however, this increase was partially attenuated when wt-APC expression was induced (gray bars). Experiments (n=3) were performed with error bars plotted as standard error of the mean (SEM). Multiple unpaired t-tests determined statistical significance where p=<0.05.
Figure 1.
Wt-APC expression sensitizes HT29 cells to ATRA’s ability to decrease cell proliferation and attenuates ATRA-induced WNT/β- catenin activity. A) Cell proliferation was reduced by 65% when wt-APC was induced by ZnCl2 and cells were treated with 20 µM ATRA for 48hrs. Values are normalized to mt-APC DMSO control. B) Cell proliferation was reduced by approximately 10% (10 μM) and 20% (20 μM) in mutant-APC (mt-APC) cells (black line). Cell proliferation was further reduced by approximately 36% (10 μM) and 42% (20 μM) in wildtype-APC (wt-APC) cells (dashed gray line). Similar results were seen at 24hrs, altogether indicating a time- and concentration- dependent effect. Mt-APC and wt-APC values were normalized to each of their respective DMSO controls. C) TCF/LEF Reporter Assay was performed to determine WNT/β- catenin activity. ATRA caused an increase in WNT- activity in mt-APC cells (black bars) however, this increase was partially attenuated when wt-APC expression was induced (gray bars). Experiments (n=3) were performed with error bars plotted as standard error of the mean (SEM). Multiple unpaired t-tests determined statistical significance where p=<0.05.
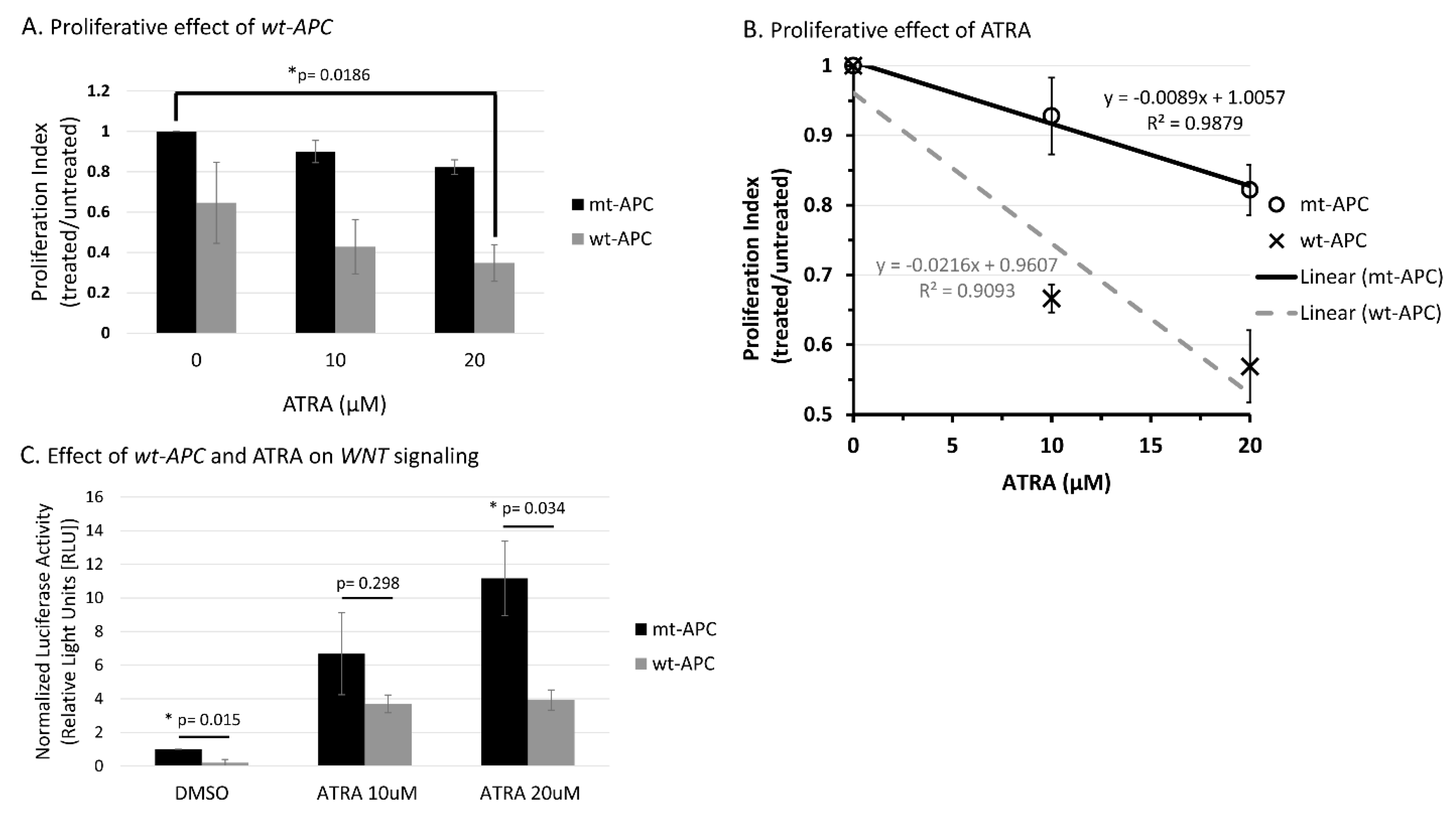
Figure 2.
Inducing wt-APC enhances differentiation of neuroendocrine cells: GLP2R + cells increase and ALDH+ cells decrease. A) GLP2R+ and GLP2R- cells were sorted from parent HT29 cells (mt-APC) and from cells induced to express wt-APC. The percentage of GLP2R+ subpopulation is shown. B) Cells were stained for GLP2R, and relative number calculated by normalizing to untreated mt-APC cells. The relative number of GLP2R+ cells quadruple when wt-APC is induced (black bars) and increases by 6-fold with the combination of wt-APC and ATRA (comparing mt-APC black bar and wt-APC gray bar). C) The percentage of ALDH+ SCs cells decreased by ~20% with ATRA alone and up to ~60% when HT29 cells were induced to express wt- APC. D) Results from NanoString profiling show a similar decrease in ALDH1A1 mRNA in response to ATRA and wt-APC. Experiments were performed 3 times and statistical significance was determined by two-way ANOVA with multiple comparisons.
Figure 2.
Inducing wt-APC enhances differentiation of neuroendocrine cells: GLP2R + cells increase and ALDH+ cells decrease. A) GLP2R+ and GLP2R- cells were sorted from parent HT29 cells (mt-APC) and from cells induced to express wt-APC. The percentage of GLP2R+ subpopulation is shown. B) Cells were stained for GLP2R, and relative number calculated by normalizing to untreated mt-APC cells. The relative number of GLP2R+ cells quadruple when wt-APC is induced (black bars) and increases by 6-fold with the combination of wt-APC and ATRA (comparing mt-APC black bar and wt-APC gray bar). C) The percentage of ALDH+ SCs cells decreased by ~20% with ATRA alone and up to ~60% when HT29 cells were induced to express wt- APC. D) Results from NanoString profiling show a similar decrease in ALDH1A1 mRNA in response to ATRA and wt-APC. Experiments were performed 3 times and statistical significance was determined by two-way ANOVA with multiple comparisons.
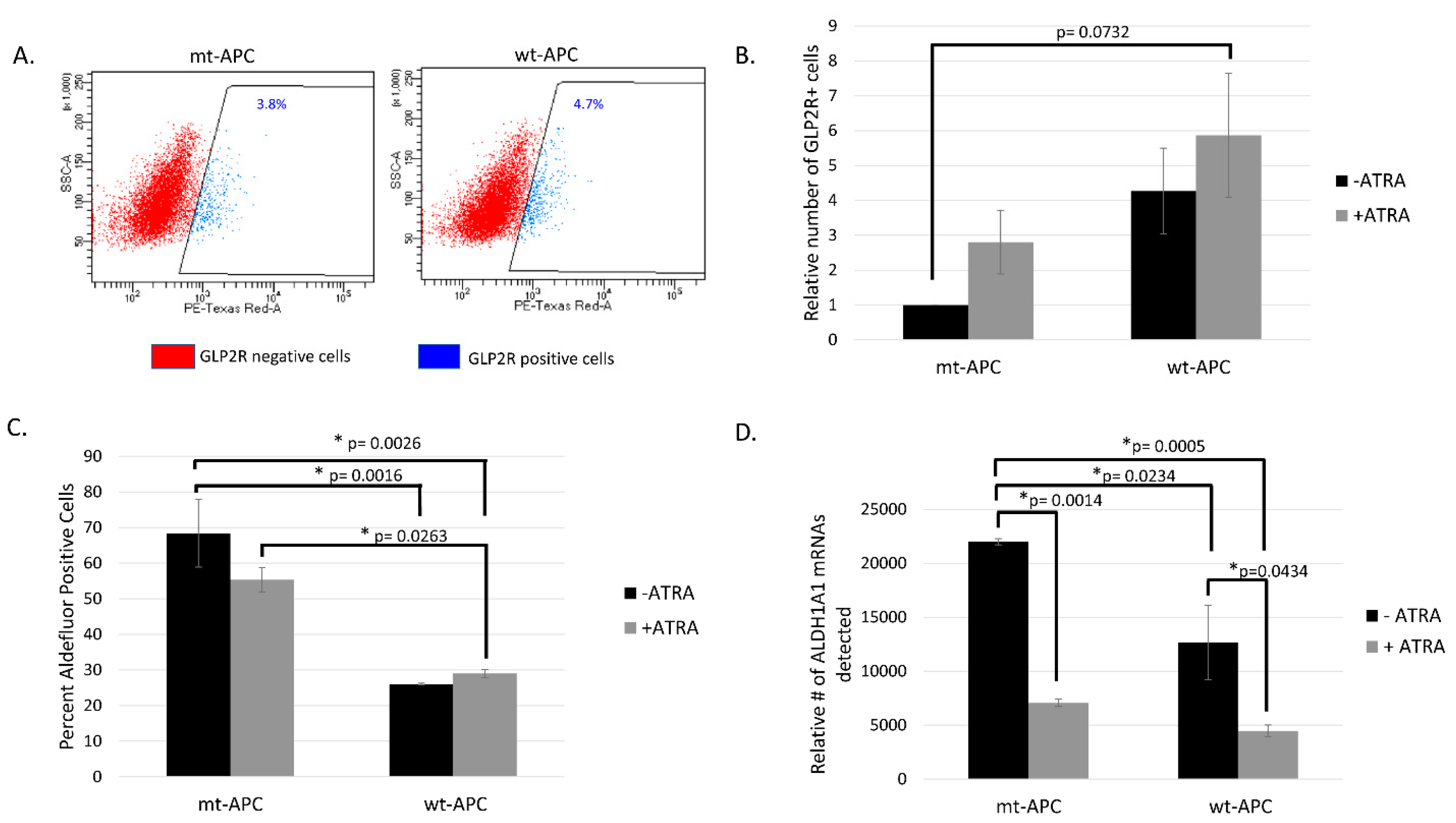
Figure 3.
Protein expression of neuroendocrine markers is increased with induction of wt-APC. HT29 cells were induced to express wt-APC. Western blot (A) and densitometry (B) were performed to analyze protein expression of neuroendocrine markers. Expression of full- length APC is shown and verified in the top right panel of (A). Chromogranin A (CHGA), GLP2R, and NSE/ enolase are increased with wt-APC but, SSTR1 is increased to a lesser extent. *The lower CHGA band was analyzed for densitometry, higher bands may indicate post-translational modification such as glycosylation of the protein. The experiment (n=3) was performed with averages plotted in (B) and representative blot in (A). Statistical significance was determined by multiple unpaired t-tests.
Figure 3.
Protein expression of neuroendocrine markers is increased with induction of wt-APC. HT29 cells were induced to express wt-APC. Western blot (A) and densitometry (B) were performed to analyze protein expression of neuroendocrine markers. Expression of full- length APC is shown and verified in the top right panel of (A). Chromogranin A (CHGA), GLP2R, and NSE/ enolase are increased with wt-APC but, SSTR1 is increased to a lesser extent. *The lower CHGA band was analyzed for densitometry, higher bands may indicate post-translational modification such as glycosylation of the protein. The experiment (n=3) was performed with averages plotted in (B) and representative blot in (A). Statistical significance was determined by multiple unpaired t-tests.
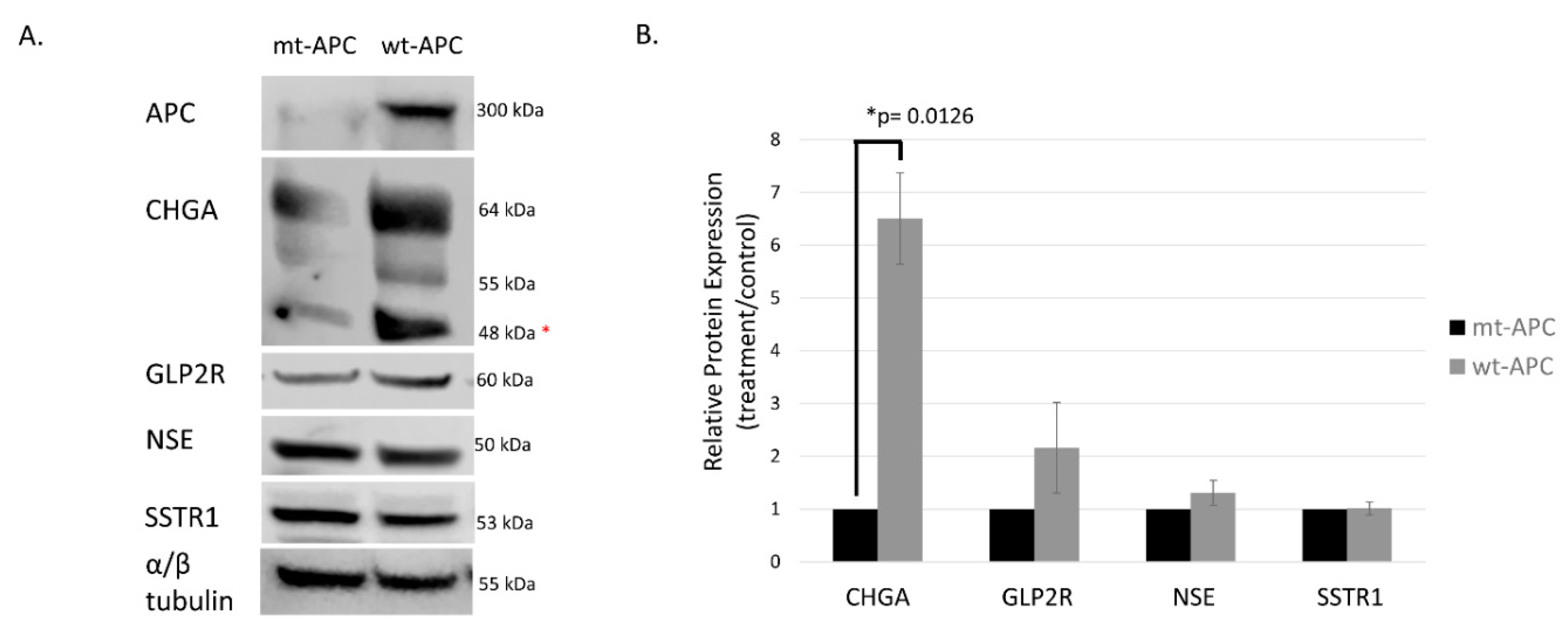
Figure 4.
A predicted protein network linking RA and WNT pathways. A) Hierarchical, agglomerative clustering using NanoString nSolver software generated a heatmap showing differential expression of 785 mRNAs amongst four treatment conditions. Yellow = low expression, blue = high expression, black = negligible expression at or below background level. B) A subset of 248 genes was identified with significant fold-change in expression between wt-APC + ATRA versus mt-APC -ATRA (Figure S2). DAVID analysis identified a list of 14 genes involved in cellular response to RA. STRING software analysis of the 14 genes (C) identified network interactions (B).
Figure 4.
A predicted protein network linking RA and WNT pathways. A) Hierarchical, agglomerative clustering using NanoString nSolver software generated a heatmap showing differential expression of 785 mRNAs amongst four treatment conditions. Yellow = low expression, blue = high expression, black = negligible expression at or below background level. B) A subset of 248 genes was identified with significant fold-change in expression between wt-APC + ATRA versus mt-APC -ATRA (Figure S2). DAVID analysis identified a list of 14 genes involved in cellular response to RA. STRING software analysis of the 14 genes (C) identified network interactions (B).
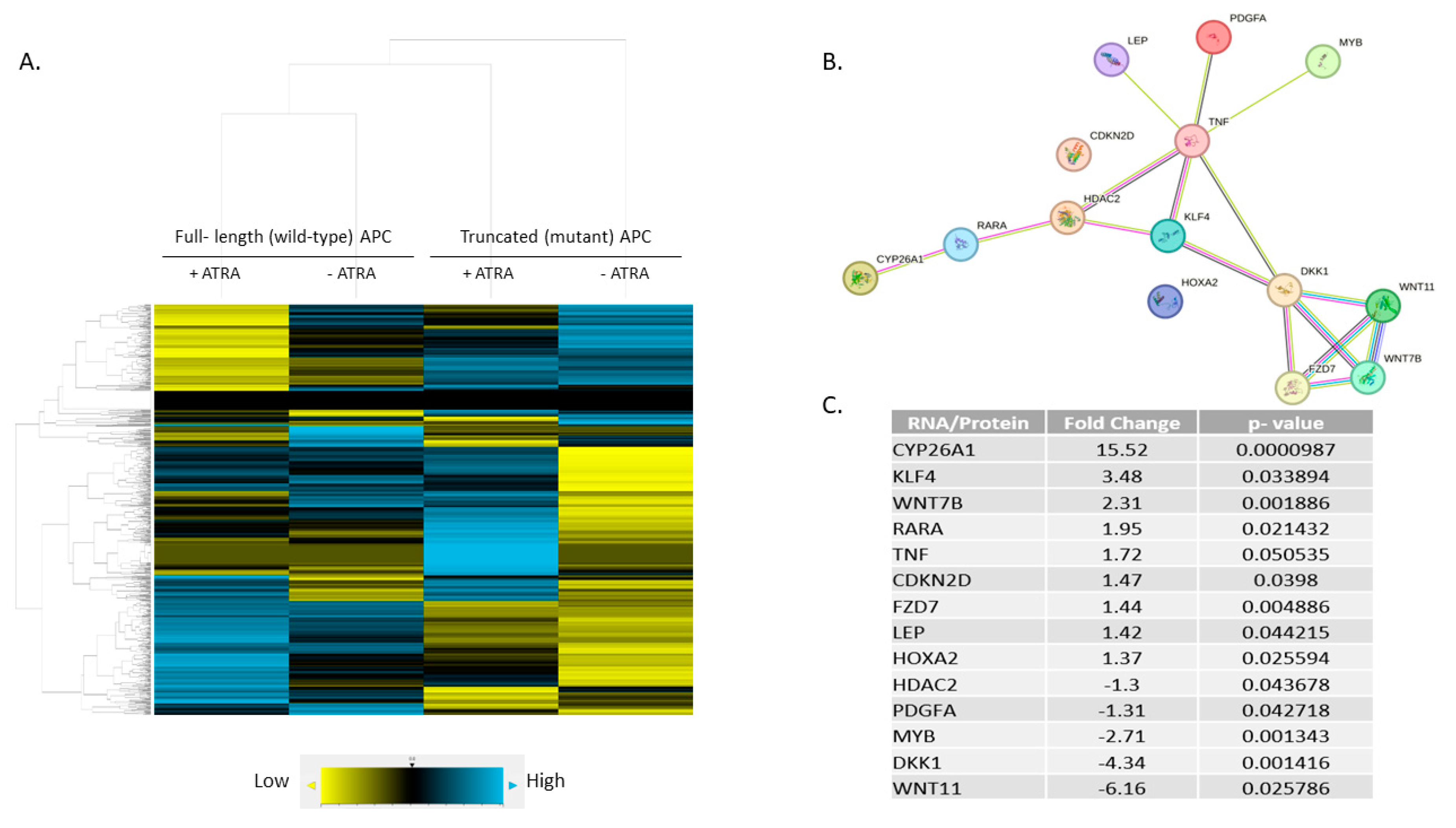
Figure 5.
Inducing wt-APC expression reduces ATRA induced CYP26A1 expression. (A) NanoString mRNA profiling and western blot analysis of protein expression (B, C) revealed a 50% decrease in CYP26A1 expression when wt-APC expression was induced in ATRA-treated HT29 cells. Experiments were performed in triplicate and a representative blot is shown in (B). Statistical significance was determined by multiple unpaired t-test (A) and one-way ANOVA (C).
Figure 5.
Inducing wt-APC expression reduces ATRA induced CYP26A1 expression. (A) NanoString mRNA profiling and western blot analysis of protein expression (B, C) revealed a 50% decrease in CYP26A1 expression when wt-APC expression was induced in ATRA-treated HT29 cells. Experiments were performed in triplicate and a representative blot is shown in (B). Statistical significance was determined by multiple unpaired t-test (A) and one-way ANOVA (C).
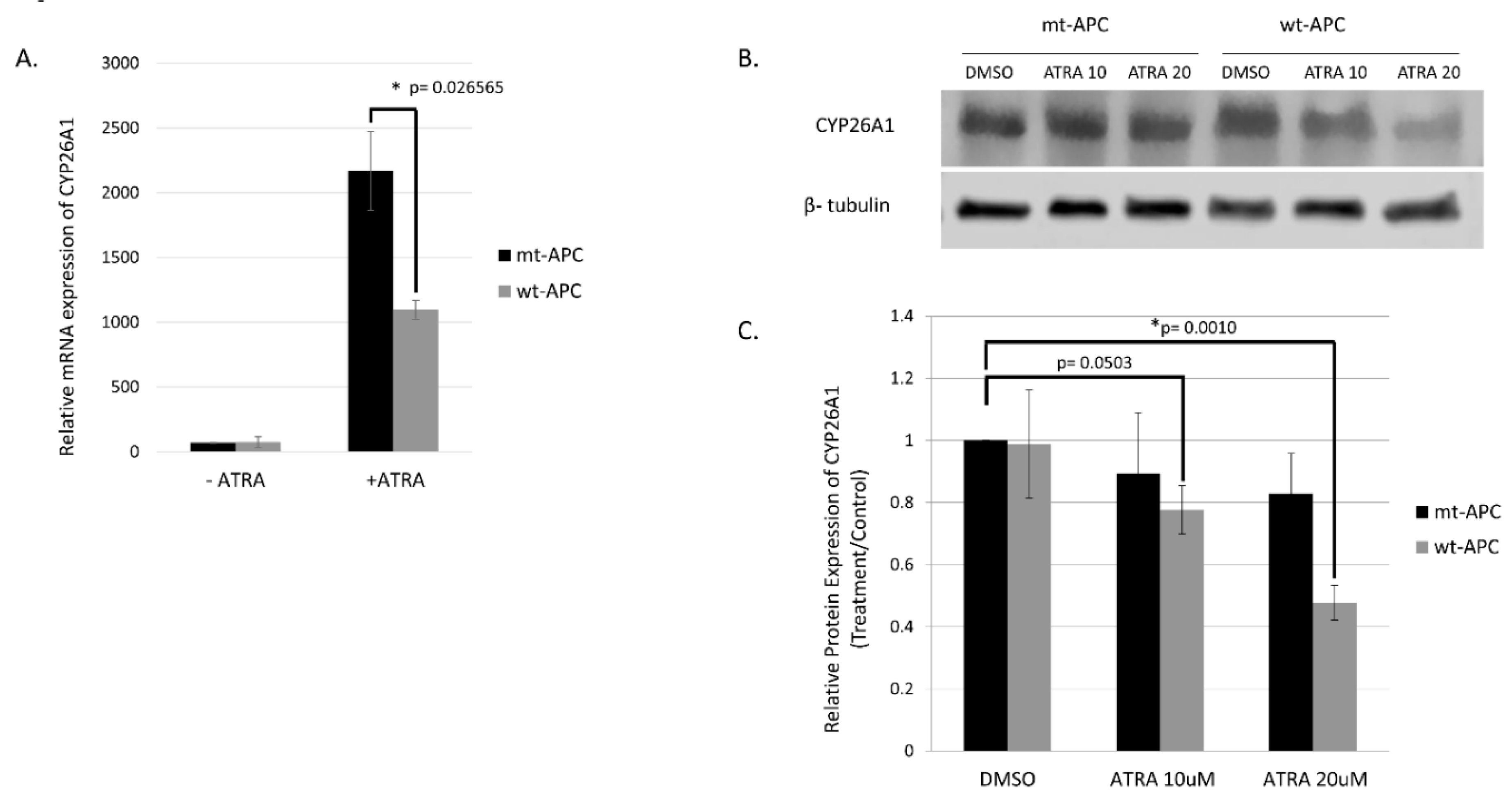
Figure 6.
Effect of CYP26A1 inhibitor and WNT inhibitor drug combinations on HT29, HCT116, and SW480 CRC cells. Panels A, B, D, and E show that anti-tumor agents Sulindac and Piroxicam alone decrease the proliferation of the CRC cell lines having mt-APC. The data also shows that the CYP26A1 inhibitors Liarozole and Talarozole alone reduce CRC proliferation. Panels C and F shows that when Sulindac or Piroxicam was combined with Liarozole or Talarozole, the drug combination produced additive or synergistic effects. These effects were calculated as the ratio of observed divided by predicted response (see methodes) based on the Bliss Independence dose-response model [22]. Statistical significance was determined by two-way ANOVA. Extended data showing additional concentrations are shown in Figure S3 and concentrations listed in Table S2.
Figure 6.
Effect of CYP26A1 inhibitor and WNT inhibitor drug combinations on HT29, HCT116, and SW480 CRC cells. Panels A, B, D, and E show that anti-tumor agents Sulindac and Piroxicam alone decrease the proliferation of the CRC cell lines having mt-APC. The data also shows that the CYP26A1 inhibitors Liarozole and Talarozole alone reduce CRC proliferation. Panels C and F shows that when Sulindac or Piroxicam was combined with Liarozole or Talarozole, the drug combination produced additive or synergistic effects. These effects were calculated as the ratio of observed divided by predicted response (see methodes) based on the Bliss Independence dose-response model [22]. Statistical significance was determined by two-way ANOVA. Extended data showing additional concentrations are shown in Figure S3 and concentrations listed in Table S2.

Figure 7.
CYP26A1 expression is a significant predictor of patient survival when patient tumors carry wild-type, but not mutant, APC. RNA-seq, mutation, and survival data of CRC patients were obtained from the GDC data portal and analyzed. (A) CYP26A1 expression in both human CRC and normal human colon samples were plotted. Dots representing paired samples from the same patient are connected by lines. Particularly, blue lines indicate increased CYP26A1 expression in human patient CRC samples compared to normal colon samples from the same patient; red lines indicate decreased expression of CYP26A1 in human CRC samples compared to normal colon samples from the same patient. Shaded area indicates 95% confidence interval. Results show that the majority of CRC patients have increased expression of CYP26A1 in their CRC samples compared to normal colon samples, and this increase is statistically significant. (B) This difference in CYP26A1 expression level between CRC and normal colon sample is not a statistically significant predictor of patient survival. (C) When patients are additionally grouped based on whether they carry APC mutations or not, results show that change in CYP26A1 expression between paired-tumor and normal samples is a significant predictor of patient survival only when their tumors carry wt-APC. (D) Such significance is not seen when patient tumors carry mutant APC (mt-APC).
Figure 7.
CYP26A1 expression is a significant predictor of patient survival when patient tumors carry wild-type, but not mutant, APC. RNA-seq, mutation, and survival data of CRC patients were obtained from the GDC data portal and analyzed. (A) CYP26A1 expression in both human CRC and normal human colon samples were plotted. Dots representing paired samples from the same patient are connected by lines. Particularly, blue lines indicate increased CYP26A1 expression in human patient CRC samples compared to normal colon samples from the same patient; red lines indicate decreased expression of CYP26A1 in human CRC samples compared to normal colon samples from the same patient. Shaded area indicates 95% confidence interval. Results show that the majority of CRC patients have increased expression of CYP26A1 in their CRC samples compared to normal colon samples, and this increase is statistically significant. (B) This difference in CYP26A1 expression level between CRC and normal colon sample is not a statistically significant predictor of patient survival. (C) When patients are additionally grouped based on whether they carry APC mutations or not, results show that change in CYP26A1 expression between paired-tumor and normal samples is a significant predictor of patient survival only when their tumors carry wt-APC. (D) Such significance is not seen when patient tumors carry mutant APC (mt-APC).
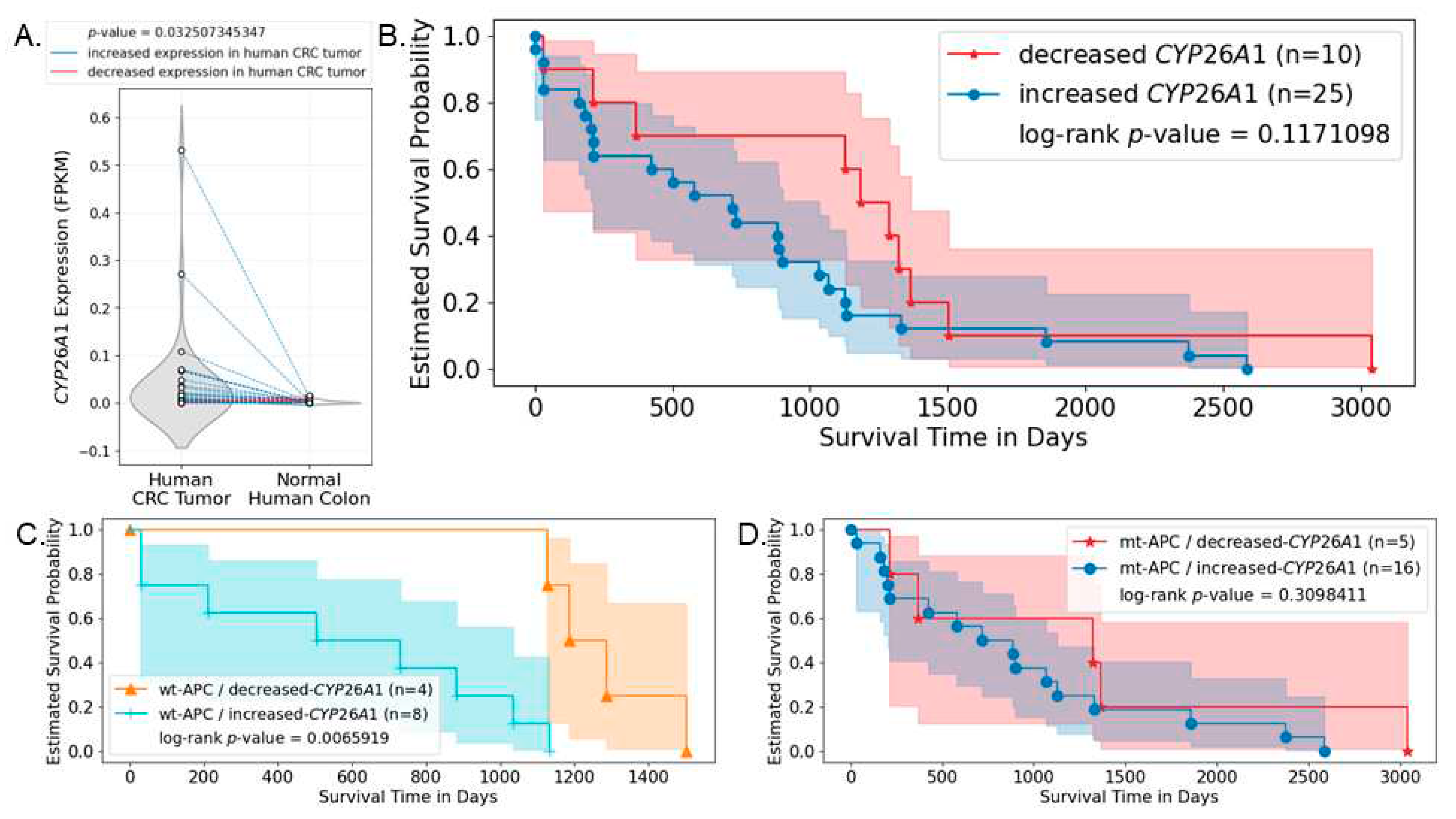
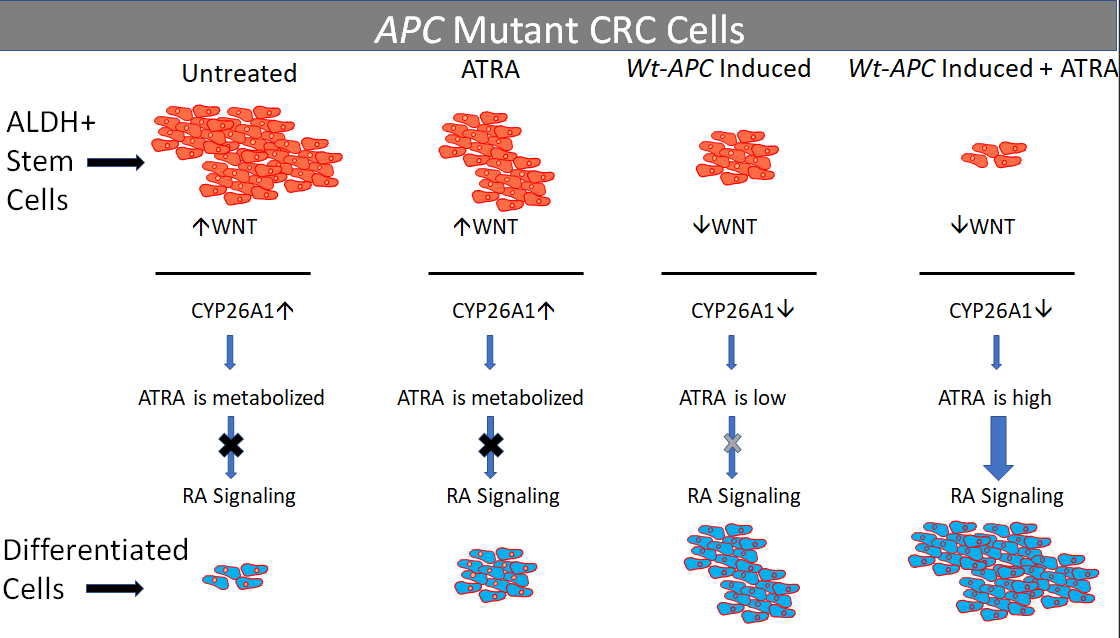
Figure 8.
Attenuation of WNT signaling by induced wt-APC expression decreases CYP26A1 and increases progression through the RA pathway. High WNT signaling levels correlate with a mutant APC genotype. In this case, when ATRA is administered, CYP26A1 is overexpressed which increases metabolism of ATRA into hydroxy-retinoic acid, thereby preventing differentiation of SCs. When wt-APC expression is induced, components of the WNT signaling pathway, including CYP26A1, are not highly expressed. In this case, when ATRA is administered, it binds to CRABP to enter the nucleus and RA response genes are transcribed and then translated in the cytoplasm to allow differentiation of SCs and subsequent cell death.
Figure 8.
Attenuation of WNT signaling by induced wt-APC expression decreases CYP26A1 and increases progression through the RA pathway. High WNT signaling levels correlate with a mutant APC genotype. In this case, when ATRA is administered, CYP26A1 is overexpressed which increases metabolism of ATRA into hydroxy-retinoic acid, thereby preventing differentiation of SCs. When wt-APC expression is induced, components of the WNT signaling pathway, including CYP26A1, are not highly expressed. In this case, when ATRA is administered, it binds to CRABP to enter the nucleus and RA response genes are transcribed and then translated in the cytoplasm to allow differentiation of SCs and subsequent cell death.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



