
Submitted:
15 December 2023
Posted:
18 December 2023
You are already at the latest version
Abstract
Severe combined immunodeficiency disorders (SCID) are a genetically heterogeneous group of inherited defects characterized by severe abnormalities of immune system development and function that lead to a wide spectrum in clinical manifestations. A subgroup of patients presents a disabling and life-threatening clinical course. In these cases, allogeneic hematopoietic stem cell transplant provides a curative approach. The occurrence of SCID-associated lymphoproliferative disorders is rare and occur mostly in adenosine deaminase-deficient severe combined immunodeficiency disorders, after allogeneic hematopoietic stem cell transplant. Epstein Barr virus infection is present in the majority of individuals with primary immunodeficiency developing lymphoma, revealing compromised anti-tumour surveillance of virally transformed cells. Herein, we report a rare case of diffuse large B cell lymphoma occurring in an Epstein Barr virus negative, non-transplanted X-linked severe combined immunodeficiency 4-months-old infant, successfully treated with immunotherapy and allogeneic stem cell transplantation. This case suggests that Epstein Barr virus -independent mechanism of neoplastic transformation must take action in severe combined immunodeficiency associated lymphoma and unveils the curative potential of donor T cells after allogeneic transplantation.
Keywords:
DLBCL
; X-linked SCID
; allogeneic stem cell transplantation
; infant
; lymphoma
1. Introduction
Severe combined immunodeficiency disorders (SCID) are a genetically heterogeneous group of inherited defects that arise from monogenic defects comprising the adaptative immune system development and function, leading to a wide spectrum in clinical manifestations [1]. The most common mutations occur in the interleukin-2 receptor gamma chain (IL2RG) gene, resulting in a dramatic depletion of mature T and natural killer (NK) cells, and B cells, although present in normal numbers, are functionally disabled. This results in a severe humoral deficiency due to the defective T cell response [2,3]. Patients present within early-life with severe and recurrent opportunistic infections and failure to thrive [2]. The severity of the clinical and immunologic phenotype requires prompt intervention and for most patients, allogeneic hematopoietic stem cell transplant (allo-HSCT) remains the only curative treatment [4]. The occurrence of SCID-associated lymphoproliferative disorders (LPD) although rare, have been described, mostly in adenosine deaminase-deficient SCID and after transplant [5,6,7]. Patients with congenital and acquired immunodeficiency are more prone to develop lymphoproliferative disease associated with Epstein Barr virus (EBV), or less frequently with other viral infections. Histologically, SCID-associated LPD include indolent and aggressive non-Hodgkin lymphomas (NHL) and are commonly associated with EBV infection. Although rarely, EBV-negative lymphomas can develop in immunodeficient patients [5,8]. We describe an unique case of diffuse large B cell lymphoma (DLBCL) in an EBV-negative, non-transplanted X-linked SCID patient treated with immunotherapy and allo-HSCT.
2. Detailed Case Description
A 4-months-old infant was admitted to the hospital due to vomiting and diarrhea post-rotavirus vaccination, hereafter the patient presented urinary tract symptoms and urinary tract infection was diagnosed. Six days later, the patient presented a respiratory infection, associated with cough and progressive breathlessness, with rapid clinical deterioration. Mechanical ventilation was needed and a Pneumocystis jirovecii pneumonia was diagnosed. Clinical course of recurrent serious infections, lymphopenia and absence of thymic shadow on chest radiograph led to the suspicion of an immunodeficiency condition. Genetic studies identified a mutation in the IL2RG gene on chromosome X, confirming the diagnosis of X-linked SCID. The patient was promptly proposed for allo-HST. Meanwhile, the infant developed edema and inflammation of the left periorbital region with zygomatic bone destruction. Despite empiric antimicrobial with azithromycin, clarithromycin, rifampicin and ethambutol, aggravation of the periorbital lesion occurred and a bone biopsy from the left malar area was performed, suggesting osteomyelitis. However, periodic acid-Schiff (PAS) and Grocott methenamine silver (GMS) stains, for the diagnosis of fungal infections, were negative. Nevertheless, the patient commenced on antifungal treatment with amphotericin B and antimicrobials were adjusted to ceftriaxone, vancomycin and trimethoprim/Sulfamethoxazole. Thoracoabdominal computed tomography (CT) assessment showed aggravation of the periorbital lesion, multiple lung nodules, hepatomegaly with hepatic lesions and splenomegaly with splenic lesions (Figure 1A–C).
Due to general aggravation with no response to treatment with antimicrobials, a new biopsy of the left periorbital lesion was performed which revealed the diagnosis of diffuse large B cell lymphoma (DLBCL), CD30+, negative for EBV (Figure 2A–D). Patient received one administration of the anti-CD20 monoclonal antibody Rituximab (375mg/m2) and 1 week later, underwent a matched unrelated donor allo-HSCT [Human leukocyte antigen (HLA) 10/10, cytomegalovirus matched, female donor], with reduced intensity conditioning [fludarabine 30mg/m²/day (d-7 to -3), treosulfan 12g/m2/day (d-7 to -5) and alentuzumab 0.2mg/kg/day (d-8 to -4)], followed by infusion of fresh peripheral blood stem cells (11.1x106 CD34+ cells/kg and 4.6x108 CD3+ cells/kg). Neutrophils and platelets engraftment was achieved by day +16, without major complications. Prophylactic immunosuppression consisted of cyclosporine A (initial dose 1.5mg/kg, twice daily) and mycophenolate mofetil (initial dose 15mg/kg, three times a day). On day +20 post-allo-HSCT, patient received a second administration of Rituximab (375mg/m2).
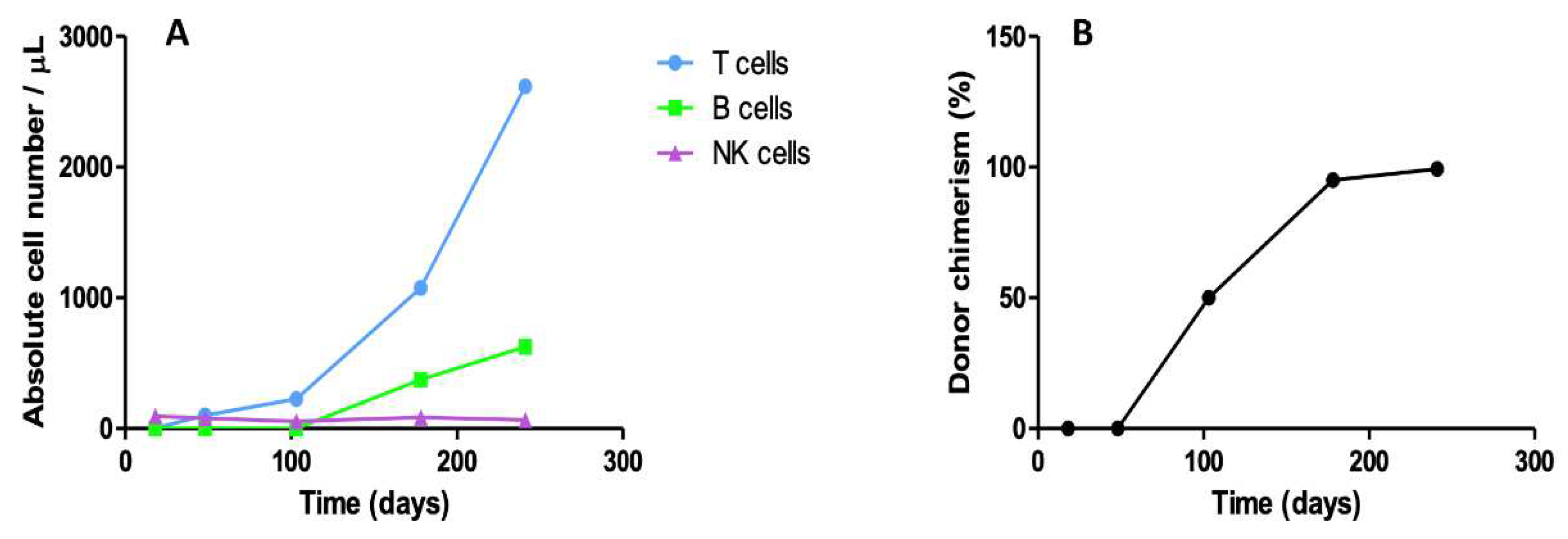
Since the infant did not show clinical signs of graft-versus-host disease (GvHD), immunosuppressors were tapered at 3 months after allo-HSCT. Two months post-HSCT, peripheral blood immunophenotyping revealed reconstitution of circulating CD3+ T (100/μL) and NK (78/μL) cells (Figure 3A). Bone marrow assessment at three months post-HSCT showed mixed chimerism of polymorphonuclear and T cells and no infiltration with DLBCL cells. Six months after transplant, patient presented engraftment of T (CD3+ 1077/μL), B (CD19+ 373/μL) and NK (87/μL) cells, as well as complete chimerism for T cells, in the peripheral blood (Figure 3A,B). Serial cranial and thoracoabdominal CT monitorization showed substantial reduction of the neoplastic lesion in the left zygomatic region and disappearance of the pulmonary, hepatic and splenic lesions (not shown). Nine months after allo-HSCT, patient had no serious infectious, presented with T, B and NK cell engraftment (2619/μL CD3+ T, 625/μL CD19+ B cell and 66/μL NK cells) and complete donor chimerism of T and NK cells (99.3% and 87%, respectively) (Figure 3A,B).
In addition, CT scan demonstrated only residual orbital alteration and no pulmonary, hepatic or splenic nodules (Figure 1D–F), compatible with complete remission (CR). Over 2 years after HSCT and upon discontinuation of immunosuppression, the infant shows an efficient reconstitution of the immune system, no signs of GvHD nor evidence of lymphoma recurrence, presenting satisfactory growth and development patterns.
3. Discussion
The overall frequency of SCID was for a long time estimated to be 1 in 50,000–100,000 live births. However, in recent years newborn screening programs have demonstrated that the frequency may actually be two- or more-fold higher with clear geographical and ethnic differences [1]. X-linked-SCID is one of the most common primary immunodeficiency states. The majority of patients have X-linked SCID caused by mutations in the IL2RG gene encoding the common γ chain (γc). The γc chain is shared by the IL2, IL4, IL7, IL9, IL15 and IL21 cytokine receptors. Cytokines mediate oligomerization of the γc chain with the appropriate cytokine receptor chain, which leads to Janus kinase 1 (JAK1) and Janus kinase 3 (JAK3) activation and phosphorylation of critical tyrosine residues in the receptor chains [2]. Upon phosphorylation there is dimerization and translocation to the nucleus where multiple genes are activated. High adenosine levels block the differentiation of thymocytes, induce thymic hypoplasia, and lead to apoptosis resulting in T, B, and NK depletion. Delayed and late onset forms have a wide spectrum of manifestations mostly involving immunological and autoimmune system [6]. Affected children generally appear well at birth, but within the first few months of life, demonstrate failure to clear infections and present with persistent respiratory tract or gastrointestinal infections and failure to thrive [1,6]. Persistent respiratory tract infection is common, accompanying persistent bronchiolitis-like signs. Insidiously progressive respiratory disease with radiological evidence of interstitial pneumonitis and hyperinflation suggests Pneumocystis jiroveci infection, which may be a co-pathogen with respiratory viruses. Severe invasive fungal infection is rare, but often fatal. There may be hepatomegaly, with or without splenomegaly. Rare presentations include Hodgkin-like polymorphous lymphoproliferative disorder, with rapidly growing extranodal tumours [2]. Our patient developed multiple infections, including Pneumocystis jiroveci pneumonia at SCID diagnosis, showing the typical presentation and the importance of prompt suspicion when. Survival in SCID patients relates with efficient T-cell reconstitution, and in the absence of successful HSCT most children die usually during the first year of life from overwhelming infection [1]. HSCT is the treatment of choice for patients with SCID. In these patients, the best results in transplantation are obtained using HLA-matched sibling donors, with survival of around 90%. The outcome is better in the absence of infection [2]. New chemotherapy conditioning regimens are gradually utilized, with improved outcome. An effective procedure is generally curative, with patients leading normal lives without medication, but few long-term studies have demonstrated long-term sequelae for some patients [2]. Particular problems associate to ongoing thymopoiesis, with failure leading to T lymphocyte senescence in the long term. Long-term immunoglobulin therapy is necessary for some B lymphocyte dysfunction or failure of donor engraftment [2]. Autologous stem cell gene therapy via vector-mediated transfer of healthy copies of an affected gene into autologous CD34+ cells has progressed from a highly experimental therapy to the first licensed gene therapy. In theory autologous stem cell gene therapy offers the appealing prospect of avoiding alloimmune reactions such as GVHD or rejection and a lower conditioning-related toxicity compared to allo-HSCT. Authors report that X-linked SCID gene therapy does not require chemotherapy and has led to complete immune reconstitution, but insertion of the retroviral vector close to oncogenes has led to the development of lymphoproliferation in some patients [2]. The exact role of gene therapy in treatment algorithms still needs to be defined in the absence of comparative studies [1]. LPD associated with X-linked SCID are rare and the most frequently reported histotype in primary immune deficiencies is diffuse large B-cell lymphoma, which mostly occurs in extra nodal sites [7]. Our patient presented diffuse large B cell lymphoma, CD20+, D30+, negative for EBV. Drivers for increased risk of lymphoma in immunodeficiency conditions is unclear and most likely multifactorial, where genetics, immune dysregulation and infections play a role [9]. EBV is a relevant factor for lymphomas and LPD in patients with an underlying immune deficiency, being rare LPD with negative EBV [5,6]. It has been suggested that an abnormal activation of the immune system, namely lymphocytes, may trigger chronic antigen stimulation in these patients, predisposing for a higher risk of LPD, predominantly NHL, mostly extranodal, affecting sinuses, orbital cavity, stomach and almost unvaryingly, lungs [10]. SCID-associated NHL occur mainly during adulthood and post-alloHSCT, since these children undergo early HSCT or die precociously due to severe opportunistic infections [11]. Thymus transplant and HSCT offer promising results for patients with SCID. Two cases were described to have undergone thymus transplantation, and the authors concluded that it had normalized T-cell proliferative response and improved clearance of disseminated [13]. To date, only 4 cases of NHL in children with SCID have been described, all morphologically aggressive. One patient died soon after diagnosis, despite management of a severe infection and directed lymphoma treatment. Two patients were treated with Rituximab associated with chemotherapy and one patient with Rituximab alone, followed by allo-HSCT. All three patients achieved CR with uneventful follow-up [5,6,12,13]. Other previously published case series described two LPD EBV-negative and one with unknown EBV status, which received Rituximab and/or chemotherapy prior to HSCT with complete response [14]. Our patient presented remarkable reduction of the orbital tumour lesion in less than one month after the first administration of Rituximab followed by HSCT (data not shown). He received another dose of Rituximab on day +20 post-alloHSCT, and, over 2 years of follow-up, the patient is in CR, immunologically reconstituted and without HSCT-related complications. Two cases were described to have undergone thymus transplantation, and the authors concluded that it had normalized T-cell proliferative response and improved clearance of disseminated infections in the patients. EBV infection is present in the majority of individuals with primary immunodeficiency developing lymphoma, suggesting that tumour surveillance for virally transformed cells is compromised in these patients. LPD not associated with EBV has been reported, though much less frequently [15,16]. Our patient was EBV negative, supporting that LPD in SCID patients may also be dependent of non-immunological mechanisms, namely defects in DNA repair, compromised intracellular surveillance implicated in epigenetics and apoptosis, and microenvironmental changes that allow neoplastic cells to proliferate [17]. However, the lack of cellular immune recognition of viral encoded proteins on the tumor cells has been postulated as the most important factor responsible for preventing and controlling lymphoproliferation in the immunodeficient host [5]. Over the past years, the outcome for SCID has improved dramatically, and a number of large retrospective registry studies have documented the success in improving overall survival (OS) and immunologic recovery. The improvements are due to a number of different factors, including earlier diagnosis, better pre- and post-HSCT supportive care, improved HLA typing, the availability of compatible donors from unrelated volunteer and cord blood banks, and less toxic chemotherapy regimens to prepare patients for HSCT [4]. Long-term follow-up of SCID children after HSCT or gene therapy is of paramount importance [2,7].
4. Conclusions
SCID is one of the most sever forms of Primary immunodeficiencies and is life-threatening pediatric emergency. We report a rare case of a 4-months-old infant with concomitant X-linked SCID and aggressive EBV-negative B cell lymphoma, successfully treated with anti-CD20 monoclonal antibody and allo-HSCT, unveiling EBV-independent mechanism of neoplastic transformation and the curative potential of donor T cells in SCID-occurring lymphoma. The present work increases the evidence that cancer development in immunodeficient hosts may not be a specific immune defect in recognition of viral specific antigens, but possibly a defect in immune surveillance necessary for elimination of cells with abnormalities in proliferation, function and/or apoptosis. The present case demonstrates a correlation between donor T cells reconstitution and complete resolution of the lymphoma, indicating a forceful graft-versus-lymphoma effect. HSCT-adjuvant treatment with Rituximab produced a complete decrease in the size of the tumor prior to HSCT and after fludarabine, with complete resolution. This approach had no harmful influence on engraftment or GvHD occurrence. Further case reports may help understanding these processes and may contribute to the improvement of effective cancer immune therapies.
Author Contributions
Dina Rochate, performed patient ́s care, collected and analyzed the data, and revised the manuscript; Sara Duarte, performed patient ́s care, collected and analysed the data, wrote and revised the manuscript; Ana Rita Gomes and Rita Peixeiro performed patient ́s care; João Vaz Silva and Ângelo Rodrigues performed histological studies, Joana Guimarães performed biopsy of the left periorbital lesion; Luís Leite, Rosa Branca, Susana Roncon, Sónia Lemos, Ana Maia Ferreira and Carlos Pinho Vaz reviewed and edited the manuscript; all authors have read and approved the final version to be published.
Funding
This research received no external funding.
Informed Consent Statement
Written informed consent has been obtained from the patient to publish this paper.
Data Availability Statement
Not applicable.
Acknowledgments
No acknowledgments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- M. Albert, A. Lankester, A. Gennery, Primary Immunodeficiencies, in: The EBMT Handbook, Springer International Publishing, Cham, (2019): pp. 663–670. [CrossRef]
- M. van der Burg, A.R. Gennery, The expanding clinical and immunological spectrum of severe combined immunodeficiency. Educational paper, Eur J Pediatr. 170 (2011) 561–571. [CrossRef]
- W.J. Leonard, J.-X. Lin, J.J. O’Shea, The γc Family of Cytokines: Basic Biology to Therapeutic Ramifications, Immunity. 50 (2019) 832–850. [CrossRef]
- H.B. Gaspar, W. Qasim, E.G. Davies, K. Rao, P.J. Amrolia, P. Veys, How I treat severe combined immunodeficiency, Blood. 122 (2013) 3749–3758. [CrossRef]
- P. Mustillo, R.P.S. Bajwa, A.M. Termuhlen, K. Nicol, R. Scherzer, R. Jaffe, A.H. Filipovich, T.G. Gross, Tumor immune surveillance defect of X-linked severe combined immunodeficiency is not Epstein-Barr virus specific, Pediatr Blood Cancer. 51 (2008) 706–709. [CrossRef]
- C. Maas, R. Lüftinger, W. Krois, S. Matthes-Martin, G. Bayer, K. Boztug, M. Metzelder, EBV-positive B-cell lymphoma manifestation of the liver in an infant with RAG1 severe combined immunodeficiency disease, Pediatr Blood Cancer. 65 (2018) e27258. [CrossRef]
- M. Migliavacca, A. Assanelli, M. Ponzoni, R. Pajno, F. Barzaghi, F. Giglio, F. Ferrua, M. Frittoli, I. Brigida, F. Dionisio, R. Nicoletti, M. Casiraghi, M.G. Roncarolo, C. Doglioni, J. Peccatori, F. Ciceri, M.P. Cicalese, A. Aiuti, First Occurrence of Plasmablastic Lymphoma in Adenosine Deaminase-Deficient Severe Combined Immunodeficiency Disease Patient and Review of the Literature, Front Immunol. 9 (2018). [CrossRef]
- S. Sharma, R.K. Pilania, G. Anjani, M. Sudhakar, K. Arora, R. Tyagi, M. Dhaliwal, P. Vignesh, A. Rawat, S. Singh, Lymphoproliferation in Inborn Errors of Immunity: The Eye Does Not See What the Mind Does Not Know, Front Immunol. 13 (2022). [CrossRef]
- Chua, I. Quinti, B. Grimbacher, Lymphoma in common variable immunodeficiency: interplay between immune dysregulation, infection and genetics, Curr Opin Hematol. 15 (2008) 368–374. [CrossRef]
- C. Cunningham-Rundles, P. Lieberman, G. Hellman, R.S.K. Chaganti, Non-hodgkin lymphoma in common variable immunodeficiency, Am J Hematol. 37 (1991) 69–74. [CrossRef]
- C. Wehr, L. Houet, S. Unger, G. Kindle, S. Goldacker, B. Grimbacher, A. Caballero Garcia de Oteyza, R. Marks, D. Pfeifer, A. Nieters, M. Proietti, K. Warnatz, A. Schmitt-Graeff, Altered Spectrum of Lymphoid Neoplasms in a Single-Center Cohort of Common Variable Immunodeficiency with Immune Dysregulation, J Clin Immunol. 41 (2021) 1250–1265. [CrossRef]
- C. Schuetz, et al. An Immunodeficiency Disease with RAG Mutations and Granulomas, New England Journal of Medicine. 358 (2008) 2030–2038. [CrossRef]
- R. Albar, M. Mahdi, F. Alkeraithe, K.N. Almufarriji, Epstein-Barr virus associated with high-grade B-cell lymphoma in nude severe combined immunodeficiency, BMJ Case Rep. 12 (2019) e227715. [CrossRef]
- Cohen JM, Sebire NJ, Harvey J, et al. Successful treatment of lymphoproliferative disease complicating primary immunodefi- ciency/immunodysregulatory disorders with reduced-intensity allo- geneic stem-cell transplantation. Blood 2007;110:2209–2214. [CrossRef]
- Brandau O, Schuster V, Weiss M, et al. Epstein-Barrvirus-negative boys with non-Hodgkin lymphoma are mutated in the SH2D1A gene, as are patients with X-linked lymphoproliferative disease (XLP). Hum Mol Genet 1999;8:2407–2413. [CrossRef]
- Strahm B, Rittweiler K, Duffner U, et al. Recurrent B-cell non- Hodgkin’s lymphoma in two brothers with X-linked lymphoproli- ferative disease without evidence for Epstein-Barr virus infection. Br J Haematol 2000;108:377–382. [CrossRef]
- G. Klein, E. Klein, Surveillance against tumors—is it mainly immunological, Immunol Lett. 100 (2005) 29–33. [CrossRef]
- Slatter, M. A., Angus, B., Windebank, K., Taylor, A., Meaney, C., Lester, T., Norbury, G., Hambleton, S., Abinun, M., Flood, T. J., Cant, A. J., & Gennery, A. R. (2011). Polymorphous lymphoproliferative disorder with Hodgkin-like features in common γ-chain-deficient severe combined immunodeficiency. The Journal of allergy and clinical immunology, 127(2), 533–535. [CrossRef]
Figure 1.
CT imaging before and after treatment: (A-C) CT imaging before treatment with Rituximab (day -10 of HSCT) showed extensive periorbital lesion (arrow in A), multiple nodules in both lungs (B), hepatomegaly with hepatic lesions and splenomegaly with splenic lesions (C). (D-F) CT imaging assessment of tumor response after treatment with Rituximab followed by allo-HSCT (day +367 post-HSCT) showed complete remission of the left orbital (D), thoracic (E), hepatic and splenic lesions (F).
Figure 1.
CT imaging before and after treatment: (A-C) CT imaging before treatment with Rituximab (day -10 of HSCT) showed extensive periorbital lesion (arrow in A), multiple nodules in both lungs (B), hepatomegaly with hepatic lesions and splenomegaly with splenic lesions (C). (D-F) CT imaging assessment of tumor response after treatment with Rituximab followed by allo-HSCT (day +367 post-HSCT) showed complete remission of the left orbital (D), thoracic (E), hepatic and splenic lesions (F).
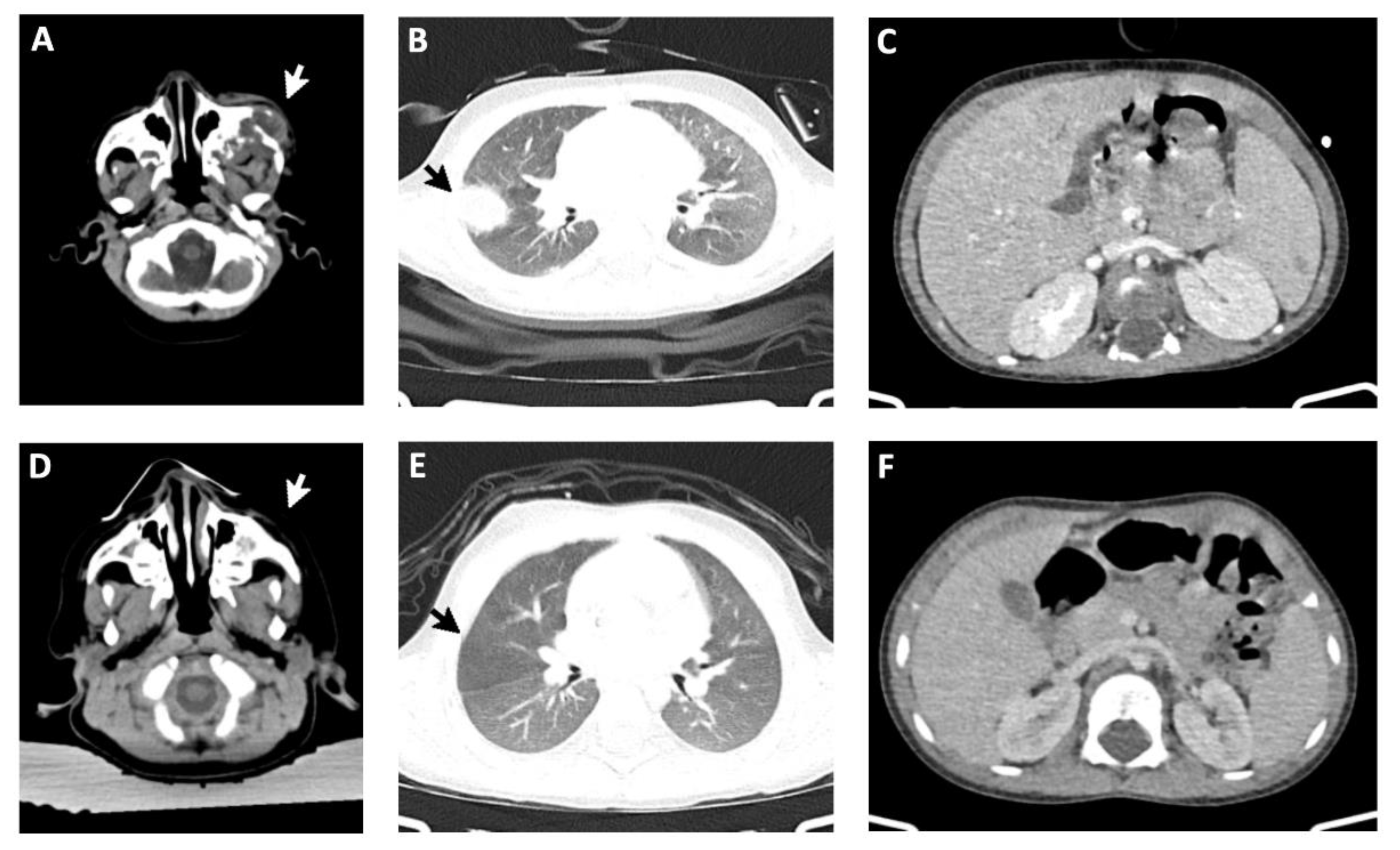
Figure 2.
Histological diagnosis of diffuse large B cell lymphoma: (A) The lesion is composed of large atypical cells interspersed in an inflammatory background, with destruction of bone tissue (H&E 40x); (B) At higher magnification, the neoplastic cells display irregular and somewhat hyperlobated nuclei, with moderate amounts of cytoplasm (H&E 400x); (C) The large cells exhibit CD20 positivity. The inset shows mitotic activity in the neoplastic cells (CD20 100x). (D) The neoplastic cells also display CD30 positivity. All the other immunostains performed were negative, as well as EBV-encoded small RNA – EBER (not shown).
Figure 2.
Histological diagnosis of diffuse large B cell lymphoma: (A) The lesion is composed of large atypical cells interspersed in an inflammatory background, with destruction of bone tissue (H&E 40x); (B) At higher magnification, the neoplastic cells display irregular and somewhat hyperlobated nuclei, with moderate amounts of cytoplasm (H&E 400x); (C) The large cells exhibit CD20 positivity. The inset shows mitotic activity in the neoplastic cells (CD20 100x). (D) The neoplastic cells also display CD30 positivity. All the other immunostains performed were negative, as well as EBV-encoded small RNA – EBER (not shown).
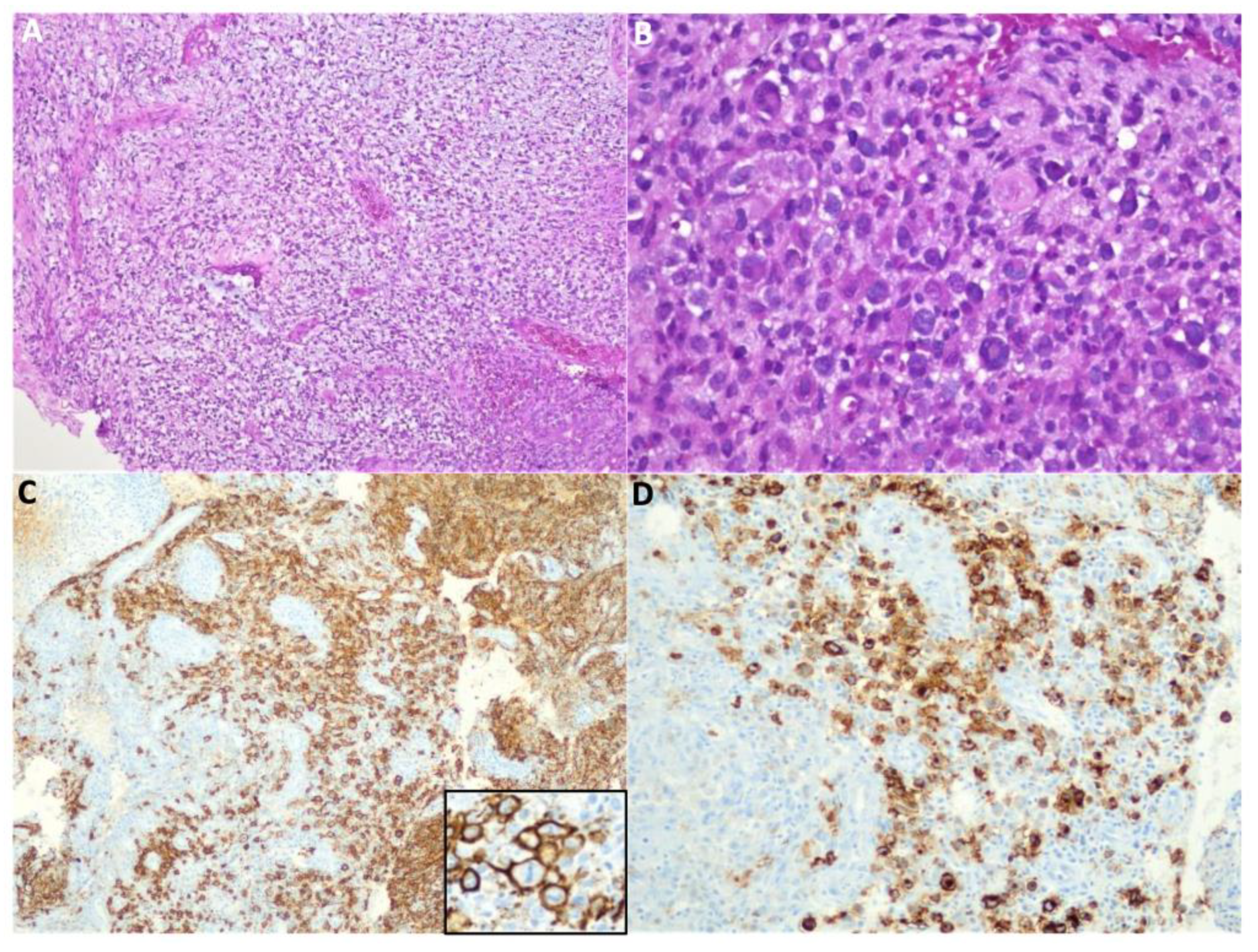
Figure 3.
Donor lymphocyte and natural killer (NK) recovery and chimerism: (A) Reconstitution of T, B and NK cells, and (B) donor T cell chimerism, at various time points after allo-HSCT.
Figure 3.
Donor lymphocyte and natural killer (NK) recovery and chimerism: (A) Reconstitution of T, B and NK cells, and (B) donor T cell chimerism, at various time points after allo-HSCT.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



