
Submitted:
15 December 2023
Posted:
18 December 2023
You are already at the latest version
Abstract
Pseudoxanthoma Elasticum (PXE) is an inherited disease characterized by elastic fibers calcification in the eyes, the skin and the cardiovascular system. PXE results from mutations ABCC6 that encodes an ABC transporter primarily expressed in liver and kidneys. It took nearly 15 years after identifying the gene to better understand the etiology of PXE. the ABCC6 functions facilitates the efflux of ATP (and other nucleotides) which is sequentially hydrolyzed by the ectonucleotidases ENPP1 and CD73 into pyrophosphate (PPi) and adenosine, both inhibitors of calcification. PXE, together with General Arterial Calcification of Infancy (GACI caused by ENPP1 mutations) as well as Calcification of Joints and Arteries (CALJA caused by NT5E/CD73 mutations) form a disease continuum with overlapping phenotypes and sharing steps of the same molecular pathway. The explanation of these phenotypes places ABCC6 as an upstream regulator of a purinergic pathway (ABCC6 → ENPP1 → CD73) that notably inhibits mineralization by maintaining a physiological Pi/PPi ratio in connective tissues. Based on a review of the literature and our recent experimental data, we suggest that PXE (and GACI/CALJA) be considered as an authentic “purinergic disease”. In this article, we recapitulate the pathobiology of PXE, review molecular and physiological data showing that, beyond PPi deficiency and ectopic calcification, PXE is associated with wide and complex alterations of purinergic systems. Finally, we speculate on future prospects regarding purinergic signaling and other aspects of this disease.
Keywords:
: PXE
; GACI
; calcification
; purinergic signaling
; adenosine
; ATP
1. Introduction
Physiological calcification is a metabolic process normally restricted to bones, teeth and primarily leading to the formation of hydroxyapatite crystals (also referred to as calcification or mineralization herein). Hydroxyapatite forms via the precipitation of calcium and phosphate salts and represents 65-70% of the mass of bone and 70-80% of dentin in teeth. Under normal physiological conditions, calcium and inorganic phosphate (Pi) concentrations are near saturation in most soft tissues, which necessitates strong calcification inhibition [1]. The prevention of calcification in soft tissues is an active mechanism that relies on multiple physiological mineralization inhibitors such as Matrix Gla protein (MGP), Fetuin-A, osteopontin and the inorganic pyrophosphate (PPi) [2] . Many host, environmental and genetic factors that contribute to the regulation of pro- vs anti-mineralization processes have been identified [3] but there are still important gaps in our mechanistic understanding of these pathologies. These gaps are critical barriers to developing comprehensive diagnostic and therapeutic strategies against ectopic calcification [4]. Ectopic calcification is seen in common cardiovascular diseases such as atherosclerosis, type-2 diabetes (T2D), chronic kidney disease (CKD), aortic stenosis (AS) and senile degeneration [3]. Abnormal mineralization produces arterial stiffness, valvular dysfunction and heart failure and represents a significant health concern [5]. There are several type of vascular calcification depending on the nature of the vascular layer affected and the associated risk factors [6]. Intimal calcification affects the endothelial layers of medium/large size elastic arteries and is primarily associated with atherosclerotic plaques. Mönckeberg’s media-sclerosis, or mediacalcosis, affects the media of arteries and is mostly seen in type 2 diabetes or kidney diseases [7].
The identification of disease-causing mutations in ABCC6 as the cause of pseudoxanthoma elasticum (PXE) [8,9,10] and the recent characterization of its function [11] has provided a wealth of new information on the molecular pathways regulating soft tissue calcification [12]. Indeed, ABCC6 facilitates the cellular efflux of nucleotides, notably ATP, which is sequentially converted at the cellular surface into the calcification inhibitors PPi and adenosine. This conversion is mediated by two membrane-bound nucleotidases, respectively ectonucleotide pyrophosphatase/phosphodiestherase 1 NPP1 (ENPP1) and the ecto-5’-nucleotidase (CD73, encoded by NT5E) [11,13,14,15]. ABCC6 deficiency mainly causes PXE (MIM#264800) [8,9,16], whereas defective ENPP1 (key enzyme for PPi production), leads to generalized arterial calcification of infancy (GACI, MIM#208000). Remarkably, some cases of GACI are caused by ABCC6 mutations (MIM#614473) while ENPP1 variants can be associated with PXE [17,18]. This duality highlights the phenotypic overlap between these two diseases, with GACI being a very severe form of PXE. Calcification of Joints and Arteries (CALJA, MIM#211800) is another related pathology due to mutations in NT5E (encoding CD73) [14]. The lack of CD73 indirectly leads to enhanced PPi degradation [14,15] from reduced adenosine signaling and activation of the tissue non-specific alkaline phosphatase (TNAP/ALPL) [15,19]. PXE, GACI and CALJA form a disease continuum with overlapping calcification phenotypes and clinical features with distinct yet related molecular mechanisms. The explanation of these phenotypes, places ABCC6 as an upstream regulator of a purinergic pathway that notably inhibits mineralization by maintaining a physiological Pi/PPi ratio in connective tissues (Figure 1).
In this review, we present arguments that ABCC6 is a purinergic signaling regulator, making PXE and by extension GACI and CALJA, authentic purinergic pathologies. We discuss how ABCC6 was linked to ATP efflux and PPi generation but also recent evidence that ABCC6 dysfunction leads to systemic imbalance in circulating nucleotides levels and ectonucleotidases expression and activities [20]. Because ABCC6, ENPP1, CD73 are functionally related, we propose that in addition to a PPi deficit, altered purinergic signaling also contributes to PXE manifestations.
2. The Genetics of PXE
2.1. Inheritance
The first formal description of PXE was performed almost 130 years ago [21]. PXE first appeared to be inherited as both autosomal recessive (AR) or autosomal dominant (AD) forms inheritance have also been described. The first attempt at defining the mode of transmission of PXE was done by Berlyne and co-workers [22]. A dual mode of inheritance was later suggested by Pope [23], generalizing the notion of different sub-types of PXE with two dominant and two recessive forms of PXE. In 1988, Neldner proposed a revised classification arguing that the various sub-types in fact corresponded to different stages of an age-dependent phenotype and he concluded that PXE was primarily inherited (97%) as AR [24]. Despite some dissonant publications in the following decade, researchers agreed that only 2 forms of PXE existed, a primarily recessively inherited disorder and a minor dominant form with incomplete penetrance [25]. The identification of PXE gene in 2000 [8,9] mostly settled the question of the inheritance. The generation of PXE knockout mouse models provided a “final nail in the coffin” of AD inheritance [26,27] and a publication by Dr. A. Bergen closed the debate declaring that “(these) findings mark the end of the autosomal dominant PXE segregation myth” [28].
With the most recent advances in molecular characterizations, we now understand that PXE, GACI, and also CALJA form a spectrum of similar AR diseases with overlapping calcification phenotypes but with vastly different prevalence, PXE being the most common of the three.
2.2. The PXE Gene
Using a systematic approach in a pre-genome sequence era, Struk et al. and van Soest and colleagues independently reported a significant linkage of the PXE phenotype to the chromosome 16p13.1 region [29,30] and screening of candidate genes led to the identification of disease-causing mutations in the ABCC6 gene that was then referred to as MRP6 in the old nomenclature [8,9,10]. ABCC6 belongs to the ATP-binding cassettes (ABC) superfamily, the largest family of transmembrane proteins. These proteins bind and hydrolyze ATP to drive the transport of a wide variety of molecules across cell membranes. Genetic alterations in at least 14 of these genes cause heritable diseases [31,32]. About a third of all these ABC transporter-related diseases are linked to genes from the C sub-family that includes the gene causing cystic fibrosis (ABCC7) [33] and now ABCC6. Today, more than 400 distinct mutations in ABCC6 have been reported in patients with classic PXE (https://www.ncbi.nlm.nih.gov/clinvar/?term=abcc6[gene]). Disease-causing variants lead to partial or total loss of function of the ABCC6 transporter and include missense and non-sense intronic mutations (causing miss splicing), insertions and deletions [16,34,35,36,37].
3. Disease manifestations
PXE manifestations affect the skin, the eye and the cardiovascular system with significant phenotypic and severity heterogeneity [24,38].
3.1. Skin
The manifestations result from elastorrhexis i.e. the fragmentation and calcification of elastic fibers in the dermis. The skin lesions are generally the first signs of PXE observed during childhood or adolescence and often progress slowly and unpredictably. Therefore, a dermatologist makes frequently the initial diagnosis. The accumulation of abnormal calcified elastic fibers in the mid-dermis produces these skin lesions, which consist of yellowish papules and plaques and laxity with loss of elasticity. These lesions can be seen on the face, neck, axilla, antecubital fossa, popliteal fossa, groin, and periumbilical area [24]. The diagnosis generally consists of skin biopsy of an affected area with von Kossa or Alizarin Red S histological staining that will show calcification and disrupted elastic fibers (elastorrhexia) in the mid- and lower dermis and molecular testing [39]. Beyond their cosmetic appearance, these lesions do not cause any pathological complications and cosmetic surgery (skin resection) is currently the only yet incomplete and transient treatment possible.
3.2. Eyes
Ocular lesions are another typical characteristic of PXE and are caused by the accumulation of calcified elastic fibers in the Bruch’s membrane, resulting in angioid streaks [40]. The majority of PXE patients will eventually develop ocular manifestations. Doyne was the first to describe these ocular streaks in 1889 [41], and Knapp introduced the term "Angioid streaks" for their resemblance to blood vessels [42]. Bilateral angioid streaks are normally seen as linear gray or dark red lines with irregular serrated edges lying beneath normal retinal blood vessels and represent breaks in the Bruch’s membrane. The combination of PXE and ocular manifestations was initially referred to as the Gronblad-Strandberg syndrome, after the names of two ophthalmologists who independently related the occurrence of angioid streaks to pseudoxanthoma elasticum in 1929 [43]. Angioid streaks are completely asymptomatic and can remain undetected late in life until retinal haemorrhages occur. The Bruch’s membrane, is not in a true sense a “membrane” but rather a heterogeneous layer separating the chorioid from the retina containing elastic fibers and collagen (type I, III and IV). Visual loss is one of the PXE manifestation that impact the most the life of patients. The have have a high risk of developing exudative macular neovascularisation secondary to breaks in a calcified and brittle Bruch’s membrane. Photodynamic therapy has been explored but was not found to be useful [44]. Anti-VEGF injections are effective as a palliative therapy [45] and this has become a common symptomatic treatment for PXE patients. However, a recent ophthalmology study revealed an age-dependent accumulation of optic nerve head drusen with high risk of severe inner retinal degeneration in patients with PXE, which seems independent of anti-VEGF injections [46].
3.3. Vasculature
In the vasculature, calcification leads to arteriosclerosis, which is frequently doubled with an accelerated atherosclerosis [47]. Common manifestations of PXE include peripheral arterial disease [48] with frequent intermittent claudication, stroke, uncommon gastrointestinal bleeding and occasionally other heterogeneous manifestations such as rete mirabile [49] and carotid hypoplasia [50]. Fragmentation and calcification of the elastic fibers is frequently observed in the media of middle-size arteries (Mönckeberg's media sclerosis)[51]. Even if PXE is not life threatening per se, growing evidences suggest that ABCC6 is part of functional hub of extracellular matrix homeostasis that can lead to major vascular manifestations in the presence of comorbidities [48].
3.3.1. Coronary artery disease
Although severe coronary artery disease (CAD) has sporadically been reported in PXE in a few case reports [52,53,54], myocardial infarction is an uncommon manifestation as most arterial lesions seem to affect primarily peripheral vessels [48,55,56]. The first large systematic characterization of PXE was actually published in 1988 by Dr. Neldner but it did not specifically focus on cardiac manifestations [24]. In this cohort of 100 PXE patients, Neldner reported only one patient with CAD. The in-depth cardiac evaluation of a cohort of 67 French patients in 2013 was remarkable as it was conducted in conjunction with the characterization of an Abcc6-/- mouse model [57]. In this cohort, only 3 patients (4%) had verified CAD and a single subject had an early severe case of CAD. Myocardial ischemia was also prospectively investigated. Treadmill tests were normal in 40 of the patients tested. Single Photon Emission Computed Tomography (SPECT) showed some limited perfusion defects in only two patients out of 27. Overall, the prevalence of CAD in this cohort was similar to that of the European population (7.3%) age of 40 to 70, thus coronary artery diseases cannot be specifically associated with ABCC6 deficiency. Post-hoc (10 years) analysis confirmed the long-term absence of significant PAD in this population (Bière et al. Personnal communication). Two other studies suggested an increased risk for CAD in carriers of p.Arg1141X variant [58,59] but a follow-up case-control study with a very large number of participants (66,831) did not any find any association between this p.Arg1141X (which had a frequency of 0.6%) and ischemic events [60].
3.3.2. Atherosclerosis
Multiple lines of evidence in the literature indicated a pathologic correlation between ABCC6 function and dyslipidemia [61,62,63,64]. To explore this possibility, we conducted a study presenting compelling evidence obtained with both mouse models and PXE patients that indeed ABCC6 deficiency promotes dyslipidemia and atherosclerosis in a haploinsufficient manner, with significant penetrance [47]. Although in this study we did not determine if dyslipidemia and atherosclerosis produced negative outcomes, myocardial ischemia does not seem to be a major complication of PXE.
3.4. Heart
If peripheral arterial disease is commonly associated with PXE, cardiac complications with various degree of severity have only been described in case reports until recently. The actual prevalence of cardiac complications has now been systematically investigated in few dedicated studies with limited cohorts. Nguyen et al. analyzed cardiac functions in 19 PXE patients and only found minor deviation from accepted values [65]. In another French cohort, cardiac volume, mass, and systolic and diastolic parameters were also within normal range with minor exceptions, suggesting that ABCC6 does not impact baseline heart function [57]. Aside from a few case reports [66,67,68,69], Vanakker et al. reported only one case (2%) of mitral valve prolapse in a cohort of 42 patients [70], which is consistent with the French cohort investigation (4%) [57] and these values do not differ significantly from the general population at ~2.4% [71].
3.5. Dystrophic Cardiac Calcification
In line with the French PXE cohort data, left ventricular (LV) size and function in aged 2-year-old mice were similar to controls. However, heart weight and cardiomyocyte size were significantly increased in the 24-month-old KO mice [57]. Mungrue et al. have reported an increased infarct size in Abcc6-/- mice after ischemia-reperfusion [72], which may also have clinical implications for patients, though myocardial events seem uncommon in PXE as stated above. Interestingly, Abcc6-/- mice develop dystrophic cardiac (DCC) phenotype has long been recognized in certain congenic strains of mice (C3H/HeJ, DBA/2J or 129S1/SvJ). These mice develop soft tissue calcifications sometimes reaching extensive proportions that it leads to congestive heart failure, especially when experimentally provoked with high fat diets or physical insults [73,74]. A single point mutation Abcc6 results in a constitutive decrease in hepatic ABCC6 protein levels and is directly responsible for the DCC phenotype [75,76]. C3H/HeJ mice develop a delayed PXE phenotype as compared to Abcc6-/- mice and DBA/2J and 129/SvJ mice also develop soft tissue mineralization [75,77,78,79]. The molecular basis on DCC, which depends on PPi and Abcc6 (not expressed in cardiomyocytes) [80,81,82] remains obscur. Unraveling its understudied pathomechanisms would greatly improve our understanding of PXE.
Over the years, it became apparent that aside from the development of passive ectopic calcification in Abcc6-/- mice, physiological challenges such as high fat diet, physical lesions can lead to an amplified pathophysiological response [47,72,80,83].
Overall, PXE does not appear to be associated outright with specific or frequent cardiac complications. Cardiac events reported in old US PXE cohorts might be in fact due the superimposition of usual cardiovascular risk factors. Indeed, the presence of cardiac hypertrophy in old Abcc6-/- mice and the enhanced susceptibility to atherosclerosis and dystrophic cardiac calcification in animal models suggest that the PXE patients could be susceptible to cardiopathy when comorbidities are present. A recent study adds further support to this possibility [83]. Experimental arterial hypertension was induced by deoxycorticosterone acetate (DOCA-salt) in uni-nephrectomised in Abcc6-/- mice. DOCA-salt induced an equivalent rise in blood pressure in Abcc6-/- mice and control mice. Calcification and significant fibrosis as well as gene expression profiles favoring calcification, fibrosis and extracellular matrix remodeling were seen in both the heart and aorta of Abcc6 KO animals by comparison to WT mice [83].
4. Animal models
Early after the ABCC6 gene discovery, Abcc6-/- mouse models were generated by two independent groups in 2005 by homologous recombination leading to generation of truncated Abcc6 transcript [26,27]. These mice breed with a Mendelian distribution of the KO allele and have a normal life span. Abcc6-/- mice lack the ABCC6 protein and develop an ectopic calcification phenotype analogous to the human PXE condition. They display spontaneous mineralization in vascular, ocular and renal tissues as well as in testis and vibrissae (whiskers). The calcification in vibrissae is quantifiable and is a reliable marker of disease progression [84]. These models have become very useful tools to investigate the PXE pathophysiology, specifically abnormal lipid metabolism and atherogenesis [47,61,85], excessive oxydative stress [86,87,88], increased arterial myogenic tone [89], cardiac fibrosis [72,83], dysregulated energy metabolism [20,90] and mitochondrial dysfunction [91,92]. The DCC phenotype of Abcc6-/-mice has been effectively used as a quick and quantifiable marker of ABCC6 function [37,80,82]. Of note, several of these pathophysiological manifestations involve extracellular nucleotides and nucleosides signaling [93,94,95,96].
5. The PXE pathophysiology is both metabolic and cellular
Even though PXE was initially considered as an elastic fiber disease [24], the identification of causative mutations in ABCC6, a gene primarily expressed in the liver and kidneys changed that perspective and suggested a metabolic origin to this pathology [97,98]. Several observations and experimentations lent credence to this possibility. PXE affects the vasculature, the skin and the eyes whereas ABCC6 expression is limited in the liver and kidneys. The first experiments to address the “metabolic” hypothesis showed that serum derived from patients altered elastic fiber formation from normal fibroblast in vitro [99]. This was followed by the demonstration that serum from Abcc6-/- mice had reduced ability to prevent calcification in vitro [100]. It was, however, the allogenic transplant of whisker tissues and the creation of parabiotic pairing of Abcc6-/- animals sharing circulation with control animals that provided the most compelling evidences that a circulating anti-calcifying factor regulated by ABCC6 caused the mineralization phenotype of PXE [101,102]. The identification of PPi as the circulating anti-calcifying factor has validated the notion of the systemic nature of the PXE pathophysiology [11].
An alternate cellular-based hypothesis was also proposed early on, giving affected tissue/resident cells such as fibroblasts or smooth muscle cells a major role in the development of the pathology. Because the detection of ABCC6 mRNA or its encoded protein was difficult mostly due to antibodies efficacy/specificity and very low mRNA expression levels, this hypothesis had few proponents. Most data in support of the cellular hypothesis derived from cultured dermal fibroblast which play a prominent role the homeostasis of the extracellular matrix (ECM) and its calcification [103]. Note that dermal fibroblast is the main human cell type that could be readily derived from PXE patients skin biopsies. Remarkably, many studies showed cellular and ECM characteristics specific to PXE fibroblasts, which were proposed as explanation to the dermal manifestations of the disease. These characteristics include enhanced synthesis of elastin and proteoglycan [104], raised MMP-2 degradative potential [105], dysregulated cholesterol metabolism [47,61], increased oxidative stress [88], altered mitochondrial structure and function [91] and enhanced cellular senescence [106].
6. The search for the substrate(s)
6.1. A restricted substrate specificity
Following the discovery of the PXE gene in 2000, significant milestones in PXE research quickly followed. After the identification of ABCC6 as the causative gene in PXE, Ilias et al, and Belinsky et al, used classic inverted vesicle methodology to demonstrate that ABCC6 is a genuine efflux pump able to transport measurable substrates across the plasma membrane such as glutathione conjugate of N-ethylmaleimide (NEM-GS), leukotriene C4 (LTC4) or the synthetic peptide BQ-123 [34,107]. Ilias et al, also reported that benzbromarone and indomethacin were effective inhibitors. These reports notably suggested a defined and probably restricted substrate specificity for ABCC6 but provided no clue as to the endogenous substrate(s).
6.2. Vitamin K: logical but no joy
The Matrix Gla Protein (MGP) is an important regulator of bone mineralization that requires carboxylation. Remarkably, a PXE-like phenotype was described in association with mutations in GGCX [108]. This gene encodes an enzyme that catalyzes the post-translational carboxylation of MGP and OC using vitamin K as a co-factor. As PXE patients have low serum levels of vitamin K [109], it was logical to suggest that ABCC6 transports vitamin K and that insufficient carboxylation of MGP was the cause of calcification in PXE [110]. However, three animal-based studies quickly ruled out vitamin K as having any prominent role in PXE [84,111,112]. Vitamin K was later shown not to be effectively transported by ABCC6 [113].
6.3. Adenosine
The main steps that linked ABCC6 dysfunction to altered purinergic signaling are summarized below.
A key publication in 2011 reported on the rare disease called CALJA. This recessive disease is caused mutations in NT5E, which encodes the ecto-5’-nucleotidase also referred to as CD73 [114]. CD73 catalyzes the hydrolysis of nucleotides monophosphate into their corresponding nucleoside and is the primary enzyme generating extracellular adenosine from AMP [115]. St Hilaire et al. demonstrated that CALJA is associated with defective adenosine signaling, which normally represses TNAP expression via the A2A receptor. In the absence of functional CD73, there is reduced adenosine signaling and the upregulation of TNAP accelerates the hydrolysis of PPi leading to vascular calcifications notably in the lower limb. Nt5e-/- mice only partially recapitulate the human phenotype. These animals have an elevated Pi/PPi ratio and develop skeletal hypermineralization at the costochondral junctions and stiffening of the joints but no vascular calcification or arterial tortuosity [116]. Moreover, we observed that these mice do not develop dystrophic cardiac calcification (Figure 2). The difference could be due to ½ life of adenosine, which is much longer (8 folds) in mice than in humans [117]. Because of the phenotypic overlap between CALJA and PXE, the authors suggested adenosine as a possible ABCC6 substrate [14]. However, Szabo et al, showed that adenosine is not effectively transported by ABCC6 [118].
6.4. PPi
One year later, Nitschke et al. added the missing link between ABCC6 and CD73 (Figure 1). Remarkably, this collaborative group of investigators reported that some cases of GACI are only associated with ABCC6 mutations, while some PXE patients carry disease-causing ENPP1 variants [17,119]. This paper revealed an important physiological overlap between PXE and GACI and more importantly a molecular pathway common to both pathologies.
It is in this context that Jansen and co-workers developed an untargeted metabolomics approach to identify the molecules transported by ABCC6 that could explain its role in the regulation of calcification [11]. The authors found that the expression of ABCC6 leads to a cellular efflux of nucleosides, nucleoside monophosphates, and nucleotide sugars, notably ATP, which is rapidly converted into PPi and adenosine at the cellular surface calcification. An in vivo follow-up study confirmed that ABCC6 is responsible for ~60% of plasma PPi levels [13]. Subsequent studies with Abcc6-/- and Enpp1-/- mice confirmed the pivotal role of PPi production in the control of soft tissues calcification in both PXE and GACI [81,82,120]. The report by Jansen et al. was a major breakthrough in the field of PXE and GACI as it not only paved the way towards credible therapeutic perspectives [19,82,120,121,122] with some already undergoing clinical trials [12] but it was also the first evidence that ABCC6 is positioned upstream of a purinergic signaling pathway: ABCC6 → ENPP1 → CD73 ⟞ TNAP (Figure1).
6.5. The question of ATP
Even if the Jansen et al model is very coherent [11], this work led to the question as to whether ABCC6 could directly transport ATP. Because ABCC1, a closely related homolog, can transport GTP and UTP, Jansen and co-workers measured the uptake of ATP, GTP, and UTP into ABCC6-containing vesicles. The results were negative, which suggested that ABCC6 doesn’t transport nucleoside triphosphates directly, but could do so indirectly via a different molecular mechanism and/or protein partner. Even though other ABC transporters, such as ABCB1 (P-glycoprotein) and ABCC7 (CFTR) have been associated with ATP release [123,124] these stories were controversial at the time [125] but have been revisited more recently [126]. Jansen et al. could not observe increased nucleotide release in a cellular model overexpressing ABCB1, ABCC1, ABCC3, ABCC4, or ABCC5. For now, the exact process by which ABCC6 facilitates ATP cellular efflux remains uncertain. Specific experiments could identify in the future the molecular mechanism implicated in this permeability but for the time being only speculation is possible. Because pannexin channels, connexin hemichannels, volume regulated anionic channels (VRAC) or ANKH have been associated with the cellular export of nucleotides [127], these could be possible molecular partners. ANKH is also of particular interest. Ank-/- mice develop progressive ankylosis, with progressive joint calcification and arthritis associated to an imbalanced of intra- vs extracellular PPi. These data were interpreted at that time as an impaired PPi extrusion [128]. In human, ANKH deficiency leads to bone metabolism dysfunction and craniometaphyseal dysplasia [129,130]. A recent study has shown that ANKH efflux nucleotides triphosphate from cells, including ATP as well as citrate but not PPi [131,132]. Therefore, depending on the type of cells or tissues, membrane channels and/or vesicular transport work in parallel to or instead of ABCC6 to efflux extracellular ATP (and other nucleotides) as precursors of anti-calcifying molecules. The relative importance and tissue distribution of these efflux mechanisms may account for the preponderance of calcification susceptibility of the joints, the vasculature, the skin, or cardiac valves (Table 1).
On a related note, we have shown in a recent study that PXE patients and Abcc6-/- mice have a highly significant 2-fold decrease in plasma ATP and ADP levels [20]. The origin of this deficit is not clear and needs to be investigated.
6.6. Plasma PPi does not fully explain calcification susceptibility
The central role of ABCC6 in PXE, GACI is now well established in humans [133] and DCC in animal models [78,134]. However, many aspects of the pathophysiology of ABCC6 dysfunction are still unexplained:
- If a deficit in PPi production is essential to the etiology of both PXE and GACI and compensation appears to be a credible therapeutic possibility [12], plasma PPi does not correlate with calcification heterogeneity in humans [18] and mice [77]. Similarly, in a recent report investigating 78 patients and 69 heterozygous, Van Gils et al, found that neither phenotype manifestation/severity nor genotype correlated with plasma PPi [135].
- The liver expression of ABCC6 is necessary but not sufficient for calcification inhibition [19,80]. The question of how peripheral tissues contribute to calcification inhibition still remains unresolved; however, we discovered that bone marrow-derived ABCC6 is necessary to achieve full calcification inhibition without affecting plasma PPi levels (Brampton et al, Submitted).
- Adding complexity to the relatively simplistic model shown on Figure 1, dermal fibroblasts of PXE patient also seem to display an impaired ability to generate PPi [136,137] and the crucial role of ANKH in the regulation of local PPi homeostasis [131] shows that in addition to ABCC6 keeping systemic PPi concentrations within the physiological range, extrahepatic PPi production (which can’t be assessed reliably as yet) is also a critical determinant of phenotypic outcome in PXE and GACI.
7. Altered ectonucleotidase activities associated with ABCC6 deficiency
Considering the clinical overlap of PXE with GACI and CALJA [114,119,133], it was not surprising that ABCC6 function relates to a cellular efflux of ATP, which is an important precursor to two of the main inhibitors of calcification i.e. PPi and adenosine. However, it is after a series of reports published within a few years of the seminal article of Jansen et al, [11] that the influences ABCC6 of on purinergic-related metabolism came into focus [15,19,136,137]. Miglionico et al, 2014 reported that silencing ABCC6 in HepG2 cells causes a downregulation of NT5E (CD73) and an increase in ALPL (TNAP) expression. Because TNAP transcription is repressed via an adenosine A2A/cyclic AMP/FOXO1-dependent mechanism [114,144] these data suggested that ABCC6 influence extracellular adenosine levels. As plasma adenosine levels are seemingly normal in Abcc6-/- mice [20], TNAP dysregulation in PXE is likely restricted to peripheral tissues as suggested by Ziegler et al. [19]. The role of TNAP in the calcification phenotype of PXE is of sufficient importance that its inhibition constitutes one of the most promising treatments to limit ectopic calcification in this disease [12].
To better determine the actual influence of ABCC6 deficiency on purinergic-related metabolism, we developed a study to measure circulating ATP and related metabolites and also soluble nucleotidase activities in both PXE patients and Abcc6-/- mice. This study assessed the expression of genes encoding ecto-cellular purinergic signaling proteins in liver with high Abcc6 expression and the aorta that has little to no expression [20]. Plasma ATP and pyrophosphate levels were markedly reduced in PXE patients and in Abcc6-/- mice, but surprisingly not adenosine. Moreover, soluble serum CD73 activity was increased in PXE patients and Abcc6-/- mice as compared to controls, which mirrors the increased levels in C3H/HeJ mice [145], a mouse strain that also harbors a naturally occurring Abcc6 mutation (Cf chapter 4, Animal Models) . Consistent with alterations of purinergic signaling, the expression of genes involved in purine and phosphate transport/metabolism was dramatically modified in Abcc6-/- mouse aorta, but much less so in liver. Only 8% of the investigated genes were altered in liver, whereas 38% were changed in aorta. What is remarkable, is that the most profound changes occurred in the vasculature, a tissue subjected to calcification but with very little Abcc6 expression, whereas the liver, which has the highest level of expression [98], presents no calcification and significantly less gene expression changes. In support of our observations, altered PPi metabolism was reported in skin fibroblasts isolated from PXE patients [136]. Our study provided the first systemic evidence that ABCC6 function regulates central and distal ectonucleotidase activities and purinergic signaling.
In a separate study, we found that both Enpp1 and Nt5e (CD73) genes are indeed differentially dysregulated in the liver of Abcc6-/- mice [146]. Interestingly, if Enpp1 seems to be downregulated in the tissues examined, CD73 is upregulated in the vasculature and kidneys of Abcc6-/- mice (Figure 3) and also shows increased activity in plasma (soluble form of CD73). This further suggests tissue-specific changes of CD73 expression/activity in response to ABCC6 deficiency [15,20,147]. As CD73 is the main enzyme generating extracellular adenosine, one would anticipate auto- and paracrine downstream effects, depending on the type of cells and tissues. Adenosine plays an immunomodulatory role in the liver and contributes to limit inflammation and tissue damages notably during ischemic preconditioning [148], in non-alcoholic fatty liver disease and steatohepatitis. Adenosine also promotes fibrosis on the long term [149,150,151,152]. Given the reduced CD73 expression in PXE, low levels of hepatic extracellular adenosine can reasonably be expected. If future studies confirm a deficit of adenosine signaling in PXE, this could provide potential therapeutic avenues targeting adenosine receptors or CD73 activity directly. Reduced CD73 activity doesn’t seem to cause obvious liver dysfunction but the work of Miglionico et al, suggested that the consequences likely manifest distally as both Fetuin A and osteopontin expression are also decreased [15]. Furthermore, Ziegler et al published strong evidences [19] for altered TNAP activity in peripheral tissue, which is consistent with altered adenosine signaling. If the consequence of reduced CD73 expression/activity is obvious with respect to PPi metabolism and ectopic calcification, the consequences of the overexpression of CD73 in renal, vascular and the circulation are less obvious. Adenosine promotes angiogenesis mostly through activation of A2A and A2B receptor on endothelial cells [153,154]. Because PXE patients experience abnormal choroidal neo-angiogenesis as well as vascular dysplasia (From carotid hypoplasia to carotid rete mirabile [48]), one could reasonably speculate that abnormal adenosine signaling could be responsible for these manifestations. Table 2 summarizes known altered purine metabolism associated with ABCC6 deficiency.
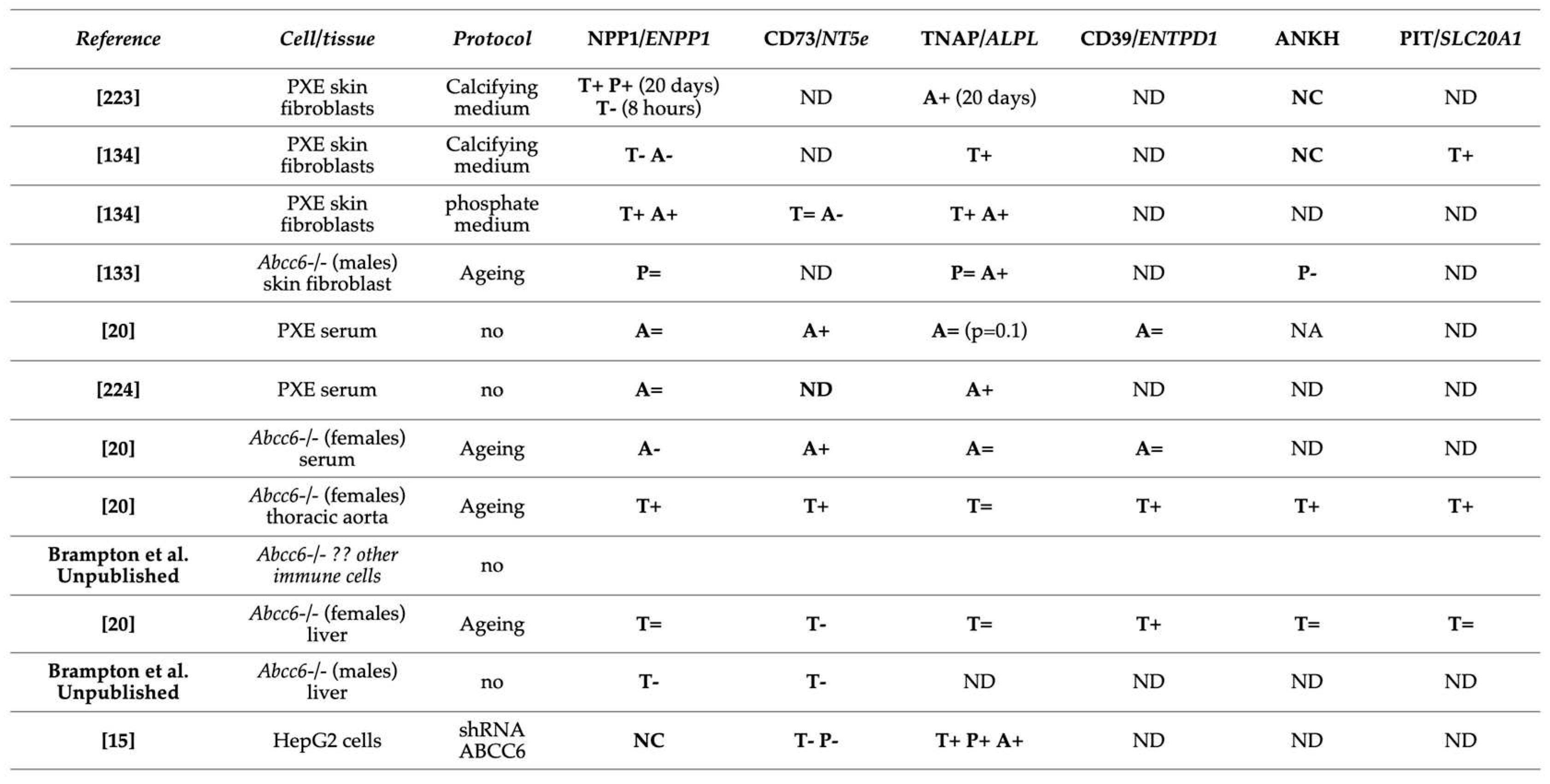
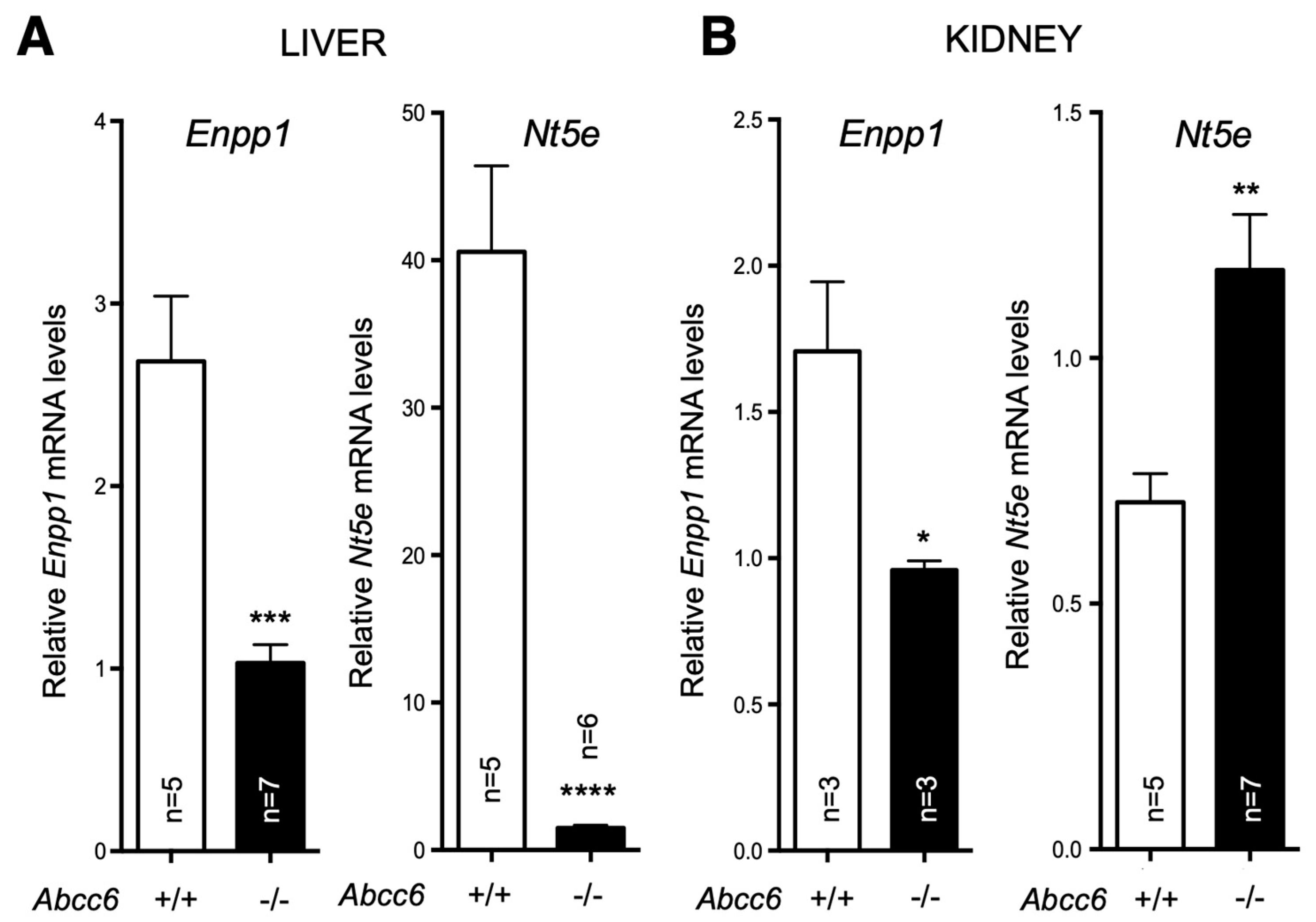

Table 2.
Altered purine metabolism and transport in ABCC6 deficiency. Enzymes were evaluated through Protein (P) expression, Transcription (T) or their activity (A). NC mean no change and ND no determination.
Table 2.
Altered purine metabolism and transport in ABCC6 deficiency. Enzymes were evaluated through Protein (P) expression, Transcription (T) or their activity (A). NC mean no change and ND no determination.
Figure 3.
Expression of Enpp1 and Nt5e in liver and kidneys is dysregulated in a tissue-specific manner in Abcc6-/- mice. mRNA levels were determined by Real-Time PCR using specific TaqMan probes to Enpp1 and Nt5E. The data normalized to Gapdh is expressed as relative quantification. Results are +/- SEM. * p<0.05, ** p<0.01*** p<0.001, **** p<0.0001.
Figure 3.
Expression of Enpp1 and Nt5e in liver and kidneys is dysregulated in a tissue-specific manner in Abcc6-/- mice. mRNA levels were determined by Real-Time PCR using specific TaqMan probes to Enpp1 and Nt5E. The data normalized to Gapdh is expressed as relative quantification. Results are +/- SEM. * p<0.05, ** p<0.01*** p<0.001, **** p<0.0001.
8. Impaired purinergic signaling – a connection to other PXE manifestations
Nucleotide-based signaling is a primitive but highly conserved system in cells. Purine nucleotides (i.e. ATP, ADP...) and nucleosides (i.e. adenosine) play distinct roles as extracellular transmitters in a wide range of tissues [155,156]. Apart from neurogenic exocytosis, non-lytic release occurs by cellular efflux in response to various stimuli (shear stress, hypoxia…) and are often seen by cells as pro-inflammatory “danger signals” [157] and adenosine is considered as vasculoprotective [158]. Once in pericellular space, nucleotides bind purinergic P2X ion channels or P2Y G-coupled receptors whereas adenosine is recognized by P1 receptors (aka A1-3). Hydrolysis of extracellular nucleotides into adenosine is carried out by ectonucleotidases, which regulate agonist availability, P2 vs P1 activation and inflammatory status [149,158]. Among the many ectonucleotidases, the ENPP and ENTPD families and CD73 (i.e. NT5E) seem to be the most relevant and act in concert to attenuate ATP signaling and promote adenosine-signaling activation. Disruption of purinergic signaling is involved in many pathophysiological processes and its contribution to vascular remodeling, inflammation, thrombosis and bone mineralization, among others, is well documented [155,159,160,161,162]. Ectonucleotidases, in particular CD39 and CD73, have been shown to contribute to endothelium integrity preservation and prevention of vascular leakage and leucocytes extravasation, cytokine secretion, platelet aggregation and hemostasis. Their role in bone metabolism has been reviewed recently [163].
8.1. Immune cells and inflammation
We and others have previously reported a significant decrease in plasma purines and changes in ectonucleotidase expression in vivo and in vitro [15,20,47,80,147]. These findings indicate that ABCC6 deficiency not only reduces PPi production but also dysregulates nucleotide hydrolysis/metabolism. This dysregulation is likely to alter purinergic-related signaling and contribute to non-calcification manifestations in PXE. This assumption is based on the well-established role of extracellular nucleotide as proinflammatory molecule [164] vs the immunosuppressive function of adenosine [162].
Inflammation primarily promotes tissue calcification through macrophage-derived cytokines such as TNF-α, IL-1β, and IL-6 [165,166,167]. Although the specific mechanisms involved are not fully understood, an imbalance in the ATP/PPi ratio on the surface of innate immune cells impairs inflammatory and anti-calcification processes, as suggested by Villa-Bellosta et al. [168]. In macrophages, where ABCC6 is not expressed [47], pro-inflammatory polarization affects the expression of Enpp1 and Cd39 (i.e. Entpd1), leading to a reduction in the ATP/PPi ratio and promoting calcification.
The recent emergence of inflammation as a possible contributor to the progression of calcification in PXE [169,170,171] suggested that immune cells also contribute to calcification regulation. Moreover, the presence of IL-6 in circulation of mice and humans as reported by Brampton et al, [47] aligns well with the presence in PXE patients of spontaneous erythematous flareup associated with typical PXE skin papules [172]. Indeed, IL-6 is typically produced at sites of acute and chronic inflammation and is also essential for T-cell recruitment and triggers the expression of chemokines, including CCL5, which is also increased in patients [47]. Moreover, positron emission tomography combined with computed tomographic imaging has shown a positive correlation between calcification and inflammation in the skin and arteries of a 42-year-old PXE patient [169]. An unpublished study, which was recently submitted for publication (Rocour et al, Submitted), confirmed the presence of an inflammatory process with the presence of T-cell infiltrate with Th1 polarity and elevated expression of cytotoxicity markers (determined by RNAseq and RT-qPCR) in the skin lesions of a PXE patient. Another submitted yet unpublished study notably found that bone marrow-derived ABCC6 has significant impact on the calcification phenotype of Abcc6-/- mice (Brampton et al, Submitted). Together, these complementary studies provide further evidence that lymphocyte-mediated inflammation largely contribute to the calcification phenotype in PXE. At this point in time, the connection between ABCC6 function and IL-6 is not clear, however a recent study could provide some clues [173]. A very recent publication by Casemayo et al. showed that Abcc6 deficiency produced an immune modulation with positive effects on the renal manifestations of rhabdomyolysis [174]. In this study, the authors also demonstrated Abcc6 expression in CD45+ cells, which independently confirms a direct physiological role for this ABC transporter in immune cells.
Our previously published results showed that ABCC6 deficiency modifies the expression of purine metabolism and signaling-related genes in a tissue-specific manner, regardless of ABCC6 expression status [20]. Consistent with these results, we have observed variable gene expression between lymphocytes and macrophages isolated from Abcc6-/- mice. Remarkably, this dysregulation differs in some cases (cf Ank and Alpl, Ada on Figure 4) indicating that ABCC6 deficiency influence immune cells perhaps through specific autocrine and/or paracrine mechanisms. Interestingly, even in this limited experiment, the lack of ABCC6 seem to affects mostly genes related to PPi (Enpp1, Ank, and Alpl), while the CD73-encoding Nt5e gene is upregulated in both lymphocytes and macrophages. This suggested that the differential transcriptional regulations of purine metabolism-related genes are systemic, at least in Abcc6-/- animals and it is reasonable to suggest that these molecular pathways contribute to and/or aggravate the calcification phenotype. Considering the key role of P1 [162] and P2 [164] receptors in innate immunity and inflammatory process we anticipate that dysregulated immune responses and inflammation in PXE result from sub-nominal cytokines pattern linked to altered purinergic signaling. The connection between purinergic alterations, inflammatory processes and their physiological impact(s) should be a research priority in the coming years.
8.2. Vascular smooth muscle and endothelial cells – CD39
Interestingly, the decrease in plasma ATP that we documented in Abcc6-/- mice [20] was not mirrored in Entpd1-/- mice lacking NTPDase1/CD39, a major vascular ATPDase [175]. An imbalanced purinergic signaling may be important in disease-affected cells, particularly in vascular smooth muscle and endothelial cells.
Vascular smooth muscle cells (VSMCs) play a significant role in the development of vascular calcifications seen in PXE, GACI, CALJA, CKD and T2D [3]. Under normal conditions, VSMCs exhibit inherent anti-calcifying activity mediated by mineralization inhibitors such as fetuin-A, pyrophosphate (PPi), and matrix Gla protein (MGP) [3]. However, in pathological states, VSMCs undergo apoptotic and senescent processes, experience increased oxidative stress, and transdifferentiate into osteoblast-like cells [176,177]. As a consequence, they adopt an osteogenic phenotype expressing genes coding for the transcription factor RUNX-2, osteocalcin, sodium-dependent phosphate co-transporter (PiT-1), bone morphogenetic proteins (BMPs), and tissue-nonspecific alkaline phosphatase (TNAP) among others. The regulation of the Pi/PPi ratio is crucial in VSMC-associated mineralization [178]. This ratio depends on the transmembrane transports of Pi and PPi, as well as the transport and abundance of extracellular ATP, along with the activities of cell surface ectonucleotidases [168]. Yet, it remains unclear how the regulation of Pi/PPi ratio in local tissues relates to the systemic plasma PPi levels. This question is reminiscent of the ongoing debate surrounding the systemic versus cellular hypotheses of the PXE pathophysiology.
Under normal conditions, these cells primarily express NTPDase1/CD39, responsible for most of the surface ATPDase activity [175], and also ENPP1 [179], the latter being the bottleneck enzyme for PPi production. We observed a significant increase in the expression of genes encoding CD39, CD73, and ENPP1 in the vasculature of Abcc6-/- animals. Interestingly, we found no evidence of ABCC6 transporter expression in these cells from the micro- or macro-circulation (Kaufffenstein et al. Personal communication), although low expression levels could be present in gastrointestinal smooth muscle [180]. The possible expression of ABCC6 transporter in smooth muscle cells and its contribution to pathogenic processes, including calcification, certainly needs to be better characterized. However, no clear role of CD39 in soft tissue calcification has been reported and CD39-deficient (Entpd1-/-) mice did not exhibit significant vibrissae calcification (not shown) or DCC (Figure 2). It is surprising as one might have expected some anti-calcification activity as it competes with ENPP1 for ATP usage, and could possibly have had some influence on PPi production, which seems to depend on the ratio of ENPP1 to ENTPD1 activity [181]. Remarkably, it has recently been shown that hydrolysis of extracellular ATP by VSMCs undergoing chondrocyte-like differentiation generates Pi but not PPi [182] suggesting the loss of pyrophosphatase generation and/or an increase in NTPDase activity. While ENPP1 exert a clear anti-calcifying activity, the role of CD39 in SMCs calcification is not obvious and could depend on circumstances.
Therefore, CD39 probably doesn’t have a direct role in pathological calcification in PXE but its contribution to other PXE manifestations is a possibility. Indeed, in addition to the decrease in plasmatic adenylic nucleotides, we observed a significant increase in the expression of genes encoding CD39, CD73, and ENPP1 in the vasculature of Abcc6-/-animals, suggesting alterations in local nucleotide hydrolysis, Pi/PPi ratio and receptor-mediated purinergic signaling via P1 and P2-type of receptors. Local concentrations of ATP and ADP in the vasculature can impact inflammatory, thrombotic, and other cardiovascular manifestations associated with P2 purinergic receptor activation [160]. Aberrant P2 receptor activation may contribute to the vascular phenotype of PXE and underlie manifestations observed in patients and Abcc6-/- mice, such as cardiomyocyte hypertrophy [57], increased resistance arterial tone [89], thrombotic episodes, vascular occlusions [183], increased inflammatory infiltrate after ischemia-reperfusion [72], and fibrosis [184]. Interestingly, the P2X7 receptor was also overexpressed in these tissues. This receptor has recently been shown to limit calcification in dystrophic skeletal muscles [185]. In addition, PPi was found to have calcification-independent effects through activation of the P2X7 receptor [186]. The role of P2X7 in ectopic calcification in the context of ABCC6 deficiency certainly deserves another look.
8.3. How does ABCC6 function relates to cardiac manifestations?
The adenosine-generating extracellular pathway that ABCC6 regulates (ABCC6 → ENPP1 → CD73 → Adenosine, see Figure 1 and Figure 6) is likely to have a significant paracrine influence towards cardiac function in the presence of comorbidities. Remarkably, extracellular cAMP hydrolysis into adenosine by ENPP1 reduced cardiomyocytes hypertrophy through A1 adenosine receptors while providing antifibrotic signaling to cardiac fibroblasts via A2 adenosine receptors in a β-adrenergic-dependent model of heart failure [187]. Furthermore, the resolution of inflammation post myocardial infarction involves important changes in extracellular purine metabolism. For instance, the loss of CD73 dramatically alter cardiac function after ischemia/reperfusion, which is followed by a prolonged inflammatory response and enhanced fibrosis [188]. CD73 is highly expressed in T cells infiltrating the ischemic lesions. The resulting adenosine notably signals toward the inhibition of pro-inflammatory (IFN-γ) and pro-fibrotic (IL-17) cytokine production via the A2 receptor. This underlines the importance of adenosine in preventing adverse cardiac remodeling [189]. One should note that the loss ABCC6, an exporter of ATP, leads to significant changes in the expression of ENPP1 and CD73 ectonucleotidases in several distal tissues expressing or not ABCC6 (liver, kidneys) as well as in inflammatory cells (Figure 4) and we’ve recently discovered a significant role for T cells-mediated inflammation process in PXE patients (Brampton et al.; Rocour et al. Submitted). Myocardial mineralization is an under-reported form of ectopic calcification and is observed in the aging heart and in patients with diabetes, kidney disease, or myocardial injury secondary to ischemia or inflammation [190,191]. Calcification within the heart muscle is a common underlying causes of heart blocks where calcification and fibrosis interrupt the propagation of electrical impulses [192,193]. In pathological circumstances, cardiac fibroblasts adopt an osteoblast cell-like phenotype and contribute directly to the calcification of the heart muscle. ENPP1, which is induced upon cardiac injury, actively contributes to this phenotype [194].The presence of cardiac hypertrophy in old Abcc6-/- mice [57] and the well-characterized dystrophic cardiac calcification in animal models [75,76,80] suggest that the PXE patients could well be susceptible to cardiopathy when comorbidities are present as changes to the cardiac purine metabolism via elements of the ABCC6 ➞ ENPP1 ➞ CD73 ➞ Adenosine pathway could be responsible for the pathological cardiac manifestations reported in the many case reports involving PXE or GACI [195,196,197,198,199,200]
8.4. Compensatory mechanism?
The remodeling of the ectonucleotidase landscape also suggests long-term compensatory effects could result from ABCC6 deficiency. Genetic compensations ensure the survival and fitness of organisms in spite of genetic perturbations which is refer to as genetic robustness [201]. It is proposed that compensatory gene networks are established during embryonic development, enabling the organism to adapt to abnormal genetic variations. Recent studies investigating the effects of variation in maternal diets of Abcc6-/- mice during gestation revealed the influence of minerals, including PPi, on ectopic mineralization in the offspring [81,134]. Some of the results could be construed as possible genetic compensations. However, we have obtained more direct experimental evidence of potential compensatory mechanisms when investigating the impact of dietary PPi on dystrophic cardiac calcification (DCC) susceptibility in Abcc6-/- mice. Interestingly, mice fed a diet rich in PPi exhibited significant inhibition of calcification despite very low PPi bioavailability [120]. Contrary to expectations, reverting to a low PPi diet in adult Abcc6-/- animals not only failed to restore the DCC susceptibility of knockout mice but instead led to further calcification inhibition (2/3 additional decrease, Figure 5). This effect was reproducible up to 4 weeks following the dietary change and was consistent between the two existing Abcc6tm1Aabb and Abcc6tm1jfk mouse models [26,27]. Moreover, a new generation of mice bred and born on the low PPi diet regained a little more than half of the expected calcification susceptibility (Figure 5). Only the highly modified acceleration diet [202] was able to produce some normalization of the calcification susceptibility after a full month of treatment. These results are the strongest evidences to date for possible compensatory mechanisms with some possible heritable components perhaps in the form of epigenetic changes. Assessing transcriptomic and proteomic profiles as well as the epigenetic landscape in such animal models would provide additional insights into the range of pathophysiologic consequences of ABCC6 and PPi deficiencies.
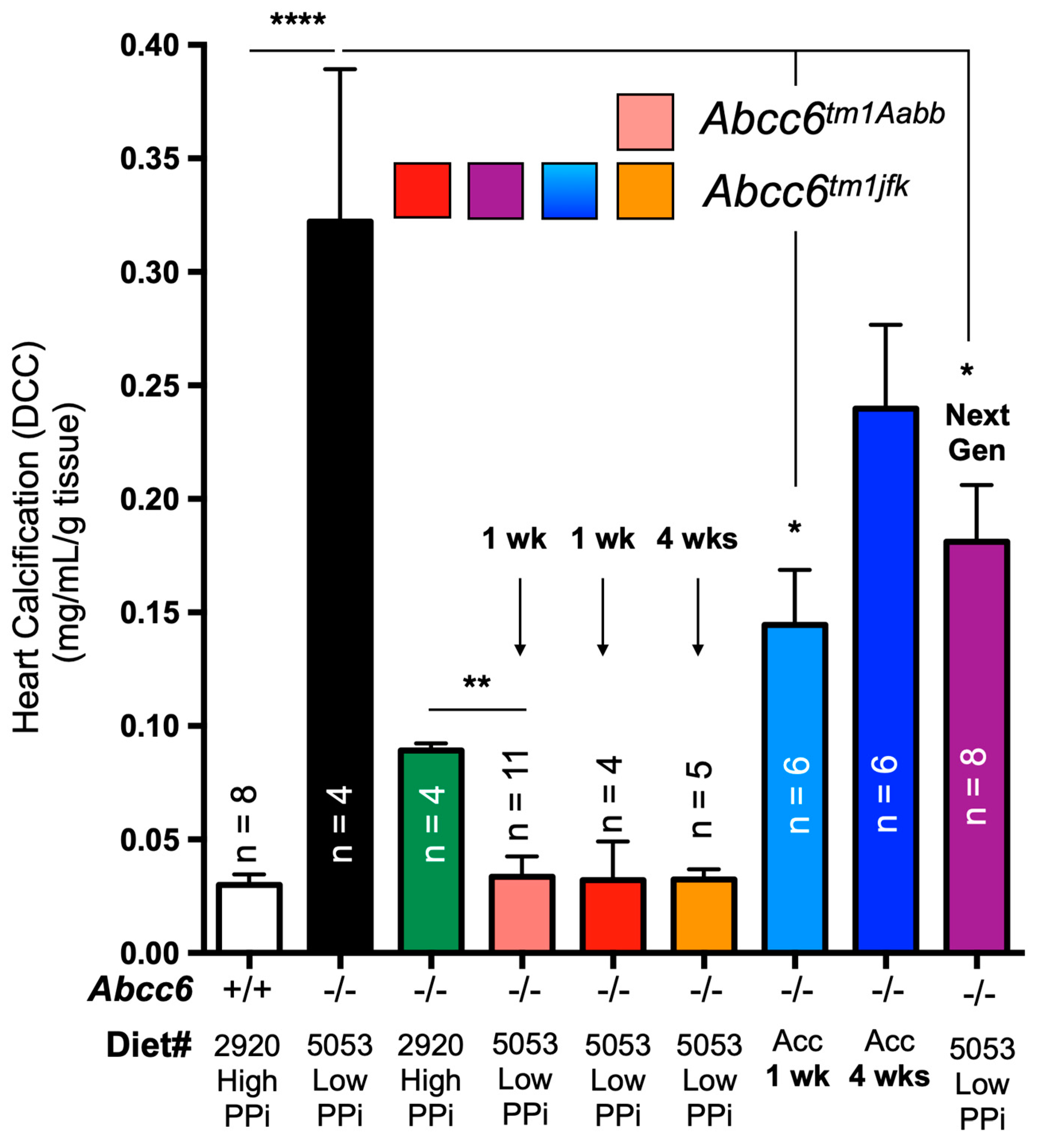
Figure 5.
The effect of dietary PPi on the acute dystrophic cardiac calcification (DCC) phenotype of WT (+/+) Abcc6-/- (-/-) mice. Mice consumed either a diet with high (2920) or low (5053) PPi content. Typically, Abcc6-/- mice develop limited DCC when fed high PPi diet as compared to animals fed low PPi diet. Remarkably, animals born and raised on high PPi diet for multiple generation failed to return to “normal” DCC susceptibility when exposed to low PPi chow, even when bred, born and raised for one generation on such diet. Only the use of an “accelerated diet” [202] restored calcification susceptibility to somewhat similar DCC susceptibility. The level of calcification was measured as total ventricular calcium and normalized to the weight of the tissue. The number of mice per group is shown and results are mean standard error of the mean. * p<0.05; ** p<0.01; **** p<0.0001. PPi: inorganic pyrophosphate.
Figure 5.
The effect of dietary PPi on the acute dystrophic cardiac calcification (DCC) phenotype of WT (+/+) Abcc6-/- (-/-) mice. Mice consumed either a diet with high (2920) or low (5053) PPi content. Typically, Abcc6-/- mice develop limited DCC when fed high PPi diet as compared to animals fed low PPi diet. Remarkably, animals born and raised on high PPi diet for multiple generation failed to return to “normal” DCC susceptibility when exposed to low PPi chow, even when bred, born and raised for one generation on such diet. Only the use of an “accelerated diet” [202] restored calcification susceptibility to somewhat similar DCC susceptibility. The level of calcification was measured as total ventricular calcium and normalized to the weight of the tissue. The number of mice per group is shown and results are mean standard error of the mean. * p<0.05; ** p<0.01; **** p<0.0001. PPi: inorganic pyrophosphate.
9. Pending questions and important future research directions
There are of course many questions remaining regarding the molecular and physiological aspects of PXE and GACI. Below are some of interrogations we deem most essential.
Despite the effective role of PPi in preventing of calcification [11,13,81,82,120,181,203,204,205,206,207,208], plasma PPi is not main determinant of ectopic calcification severity [19,209]. Therefore, establishing methods to determine local concentrations and the exact in situ molecular determinants seems essential to fully understand PXE, GACI and other pathological calcification such as Randall’s plaque and kidney stones as well as in arterial calcification resulting from liver failure [210,211,212].
Aside from its well-accepted contribution to ATP efflux, PPi and adenosine generation [11,13,213], ABCC6 also influences extracellular nucleotide metabolism in central and peripheral tissues [20], and cells ([15] and Figure 4), which creates a disruptive environment not only conducive to ectopic calcification but likely to other pathogenic processes such as vascular abnormalities [49,89], inflammation [169,170,171], atherosclerosis and dyslipidemia [47]. Further characterization of the impact of ABCC6 deficiency on extracellular nucleotide metabolism could help better understand the precise molecular underpinnings of PXE and GACI and define novel and/or complementary approach to treating these diseases more effectively (Table 3).
The precise molecular mechanism(s) by which ABCC6 function leads to ATP efflux [13] has remained a mystery. Initial in vitro vesicular experiments provided the evidence that ABCC6 was an authentic ABC transporter capable of effluxing organic anions in an ATP-dependent fashion such as BQ-123 cyclopentapeptide, leukotriene C4 (LTC4), N-ethylmaleimide S-glutathione (NEM-GS) [34], and S-(2,4-dinitrophenyl) glutathione [107]. However, none of the substrates seems relevant to calcification. For a time ABCC6 was thought to transport vitamin K [110], or adenosine [114] but these suggestions were quickly disproved by physiological experiments and direct transport assays [84,112,113,118]. ABCC6 function leads to ATP efflux but also many other nucleotides and nucleosides [11], which suggest a non-specific efflux. What contribution and effect(s), if any, these have on the PXE and GACI pathophysiology? Does ABCC6 exert the same physiological function in other cell-types?
The approximate 2 to 1 female-male ratio [48] in PXE patients has never been specifically studied or explained. Moreover, what clinical criteria should be used to reliably evaluate and quantify treatment outcomes for PXE patients? This question is constantly debated at PXE conferences but no firm answer has been proposed yet. This is due to the complexity of the PXE phenotype as multiple organ systems (skin, eyes, cardiovascular, renal etc…) are affected with various degree of distribution, severity and considerable inter- and intra-familial variability [141,214,215]. Indeed, identical mutations can cause the relatively mild PXE or severe GACI [18] in humans but also in mice [77]. The lack of genotype/phenotype correlation only compounds the issue. The presence of modifier genes that could be modulating calcification in PXE has been investigated in human and mice but their predictive values for clinical applications aren’t still clear [137,138,139,140,141,142,143]. The identification of such genes/proteins would give precious insights on the molecular partners of ABCC6.
10. Conclusions
ATP efflux is the identified molecular function of ABCC6, for now. ATP crosses the cell membrane via several tightly-regulated mechanisms and modulates a plethora of cellular functions under various pathophysiological conditions through P2 receptors activation. The hydrolysis of ATP generates adenosine which exerts wide vasculoprotective and anti-inflammatory effects through P1. Among the molecular candidates for ATP cellular release pathways, there are ATP-binding cassette (ABC) proteins including ABCB1, ABCC7 and now ABCC6. The molecular mechanisms underlying ABCC6-dependent ATP release remain largely unknown. Recent and compelling recent evidence (from our work and others) show that ABCC6 deficiency impact distal nucleotide metabolizing enzymes and its logical to assume downstream influence on P1 vs P2 receptors, whose action on cardiovascular homeostasis and other physiological functions are well documented (Figure 6).
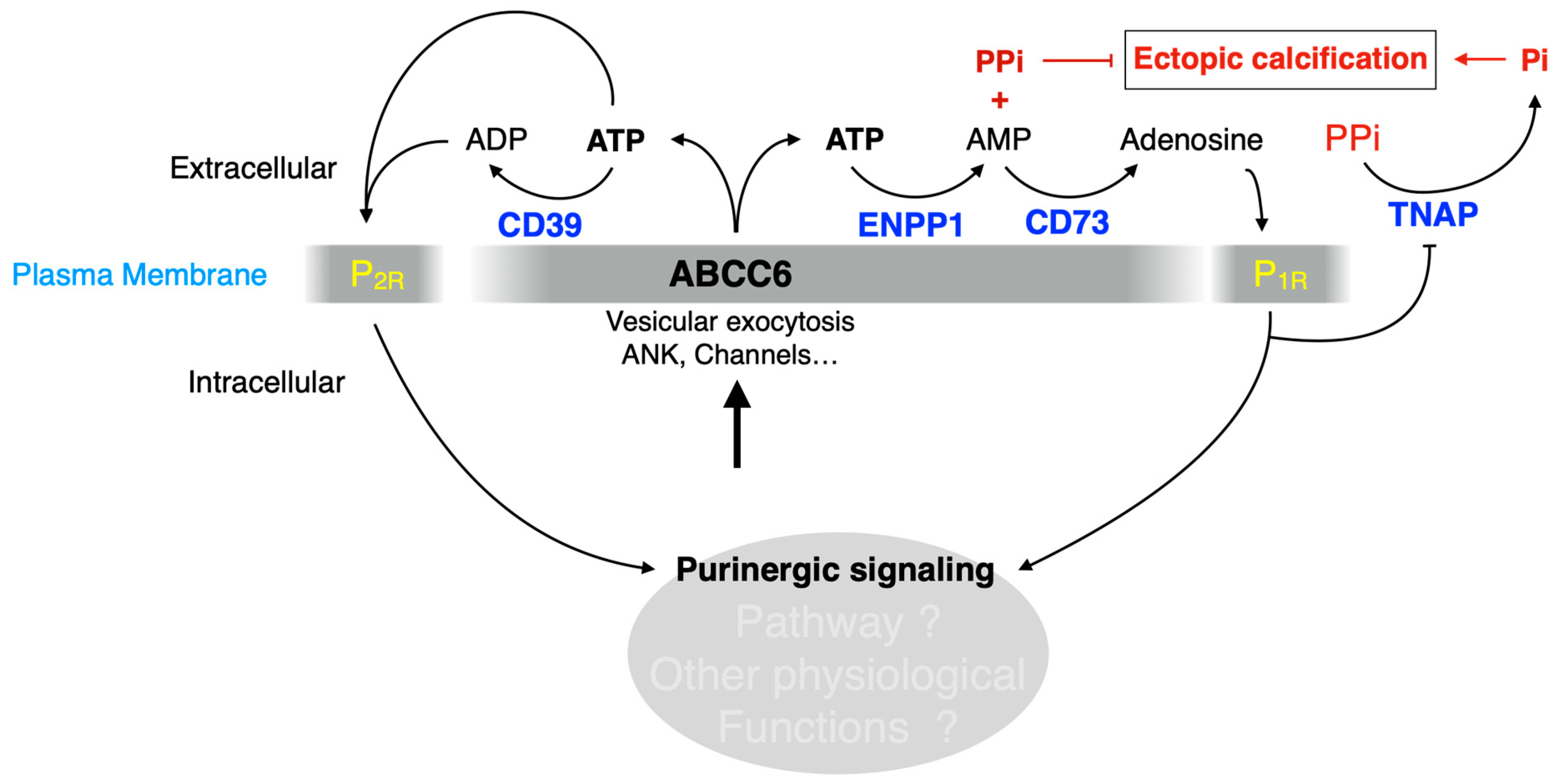
Figure 6.
ABCC6 deficiency likely modulates purinergic signaling causing calcification and other pathophysiological dysfunctions. ABCC6 contributes the cellular efflux of ATP from liver and other tissues/cells. This ATP is converted to AMP and PPi by ENPP1 activity, which contributes to calcification inhibition. We also suggest that ATP is also hydrolyzed to ADP by CD39 and signals towards P2 receptors (P2R). AMP is further dephosphorylated by CD73 (NT5E) to adenosine, which signals via the P1 receptors (P1R) to not only inhibit TNAP synthesis but likely also affect other molecular pathways. ABCC6 deficiency leads to significant decrease in plasma purines and changes in ectonucleotidases expression both in vivo and in vitro [15,20,47,80,147]. This literature along with original results presented in this review (Cf Figure 3, Figure 4 and Figure 5) strongly suggest that the lack of ABCC6 alters purinergic signaling in a variety of tissues and is the cause of pathologic manifestations other than calcification in PXE.
Figure 6.
ABCC6 deficiency likely modulates purinergic signaling causing calcification and other pathophysiological dysfunctions. ABCC6 contributes the cellular efflux of ATP from liver and other tissues/cells. This ATP is converted to AMP and PPi by ENPP1 activity, which contributes to calcification inhibition. We also suggest that ATP is also hydrolyzed to ADP by CD39 and signals towards P2 receptors (P2R). AMP is further dephosphorylated by CD73 (NT5E) to adenosine, which signals via the P1 receptors (P1R) to not only inhibit TNAP synthesis but likely also affect other molecular pathways. ABCC6 deficiency leads to significant decrease in plasma purines and changes in ectonucleotidases expression both in vivo and in vitro [15,20,47,80,147]. This literature along with original results presented in this review (Cf Figure 3, Figure 4 and Figure 5) strongly suggest that the lack of ABCC6 alters purinergic signaling in a variety of tissues and is the cause of pathologic manifestations other than calcification in PXE.
PXE and GACI are rare diseases that have been extensively studied over the last 2 decades and from which much has been learned. The gained knowledge is progressively being extended and applied to other diseases with ectopic calcification, for example the development Randall’s plaque and kidney stones and arterial calcification resulting from kidney or liver failure as well as systemic sclerosis [205,210,211,212,216]. We have described in this review a series of well-documented evidence reporting the dysregulation of purinergic signaling and related gene expression as a result of ABCC6 deficiency. This aspect of the pathobiology of PXE/GACI deserve a lot more attention as this undoubtedly contributes to ectopic calcification but most likely other manifestations of the diseases. Understanding the full molecular ramification of ABCC6 deficiency represents an opportunity to better treat PXE/GACI but also ectopic calcification when it manifests as co-morbidity in much more common pathologies such as diabetes, kidney failures, atherosclerosis or simply aging.
Data Availability Statement
No large datasets were generated or analyzed during the current study.
Acknowledgments
Financial support to OLS came from National Institutes of Health HL108249, P20GM113134, G12 MD007601. The funding agencies were not involved in the design or execution of this study.
Conflicts of Interest
The authors state no competing interests.
References
- Atzeni, F.; Sarzi-Puttini, P.; Bevilacqua, M. Calcium Deposition and Associated Chronic Diseases (Atherosclerosis, Diffuse Idiopathic Skeletal Hyperostosis, and Others). Rheum. Dis. Clin. North Am. 2006, 32, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Aranyi, T.; Cancela, M.L.; Carracedo, M.; Conceição, N.; Leftheriotis, G.; Macrae, V.; Martin, L.; Nitschke, Y.; Pasch, A.; et al. Endogenous Calcification Inhibitors in the Prevention of Vascular Calcification: A Consensus Statement From the COST Action EuroSoftCalcNet. Front. Cardiovasc. Med. 2019, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, F.; Nitschke, Y.; Terkeltaub, R. Genetics in arterial calcification: pieces of a puzzle and cogs in a wheel. Circ Res 2011, 109, 578–92. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.L.; Hutcheson, J.D.; Aikawa, E. Cardiovascular calcification: current controversies and novel concepts. Cardiovasc. Pathol. 2015, 24, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Shanahan, C.M. Medial Arterial Calcification: An Overlooked Player in Peripheral Arterial Disease. Arterioscler Thromb Vasc Biol 2016, 36, 1475–82. [Google Scholar] [CrossRef] [PubMed]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R. Vascular Calcification: Key Roles of Phosphate and Pyrophosphate. Int. J. Mol. Sci. 2021, 22, 13536. [Google Scholar] [CrossRef] [PubMed]
- Bergen, A.A.; Plomp, A.S.; Schuurman, E.J.; Terry, S.; Breuning, M.; Dauwerse, H.; Swart, J.; Kool, M.; Van Soest, S.; Baas, F.; et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Urban, Z.; Tschuch, C.; Csiszar, K.; Bacchelli, B.; Quaglino, D.; Pasquali-Ronchetti, I.; Pope, F.M.; Richards, A.; Terry, S.; et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 223–227. [Google Scholar] [CrossRef]
- Ringpfeil, F.; Lebwohl, M.G.; Christiano, A.M.; Uitto, J. Pseudoxanthoma elasticum: Mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc. Natl. Acad. Sci. USA 2000, 97, 6001–6006. [Google Scholar] [CrossRef]
- Jansen, R.S.; Küçükosmanoğlu, A.; de Haas, M.; Sapthu, S.; Otero, J.A.; Hegman, I.E.M.; Bergen, A.A.B.; Gorgels, T.G.M.F.; Borst, P.; van de Wetering, K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. USA 2013, 110, 20206–20211. [Google Scholar] [CrossRef] [PubMed]
- Shimada, B.K.; Pomozi, V.; Zoll, J.; Kuo, S.; Martin, L.; Le Saux, O. ABCC6, Pyrophosphate and Ectopic Calcification: Therapeutic Solutions. Int. J. Mol. Sci. 2021, 22, 4555. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.S.; Duijst, S.; Mahakena, S.; Sommer, D.; Szeri, F.; Váradi, A.; Plomp, A.; Bergen, A.A.; Elferink, R.P.O.; Borst, P.; et al. ABCC6–Mediated ATP Secretion by the Liver Is the Main Source of the Mineralization Inhibitor Inorganic Pyrophosphate in the Systemic Circulation—Brief Report. Arter. Thromb. Vasc. Biol. 2014, 34, 1985–1989. [Google Scholar] [CrossRef]
- Markello, T.C.; Pak, L.K.; Hilaire, C.S.; Dorward, H.; Ziegler, S.G.; Chen, M.Y.; Chaganti, K.; Nussbaum, R.L.; Boehm, M.; Gahl, W.A. Vascular pathology of medial arterial calcifications in NT5E deficiency: Implications for the role of adenosine in pseudoxanthoma elasticum. Mol. Genet. Metab. 2011, 103, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Miglionico, R.; Armentano, M.F.; Carmosino, M.; Salvia, A.M.; Cuviello, F.; Bisaccia, F.; Ostuni, A. Dysregulation of gene expression in ABCC6 knockdown HepG2 cells. Cell. Mol. Biol. Lett. 2014, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Beck, K.; Sachsinger, C.; Silvestri, C.; Treiber, C.; Göring, H.H.; Johnson, E.W.; De Paepe, A.; Pope, F.M.; Pasquali-Ronchetti, I.; et al. A Spectrum of ABCC6 Mutations Is Responsible for Pseudoxanthoma Elasticum. Am. J. Hum. Genet. 2001, 69, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Le Boulanger, G.; Labrèze, C.; Croué, A.; Schurgers, L.; Chassaing, N.; Wittkampf, T.; Rutsch, F.; Martin, L. An unusual severe vascular case of pseudoxanthoma elasticum presenting as generalized arterial calcification of infancy. Am. J. Med Genet. Part A 2009, 152A, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, Y.; Baujat, G.; Botschen, U.; Wittkampf, T.; du Moulin, M.; Stella, J.; Le Merrer, M.; Guest, G.; Lambot, K.; Tazarourte-Pinturier, M.-F.; et al. Generalized Arterial Calcification of Infancy and Pseudoxanthoma Elasticum Can Be Caused by Mutations in Either ENPP1 or ABCC6. Am. J. Hum. Genet. 2012, 90, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.G.; Ferreira, C.R.; MacFarlane, E.G.; Riddle, R.C.; Tomlinson, R.E.; Chew, E.Y.; Martin, L.; Ma, C.-T.; Sergienko, E.; Pinkerton, A.B.; et al. Ectopic calcification in pseudoxanthoma elasticum responds to inhibition of tissue-nonspecific alkaline phosphatase. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Kauffenstein, G.; Yegutkin, G.G.; Khiati, S.; Pomozi, V.; Le Saux, O.; Leftheriotis, G.; Lenaers, G.; Henrion, D.; Martin, L. Alteration of Extracellular Nucleotide Metabolism in Pseudoxanthoma Elasticum. J. Investig. Dermatol. 2018, 138, 1862–1870. [Google Scholar] [CrossRef]
- Darier, J. Pseudo-xanthome élastique. III ème congrès Intern de Dermat de Londres 1896, 289–295. [Google Scholar]
- Berlyne, G.M.; Bulmer, M.G.; Platt, R. The genetics of pseudoxanthoma elasticum1. Qjm: Int. J. Med. 1961, 30. [Google Scholar] [CrossRef]
- Pope, F.M. Two types of autosomal recessive pseudoxanthoma elasticum. Arch Dermatol 1974, 110, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Neldner, K.H. Pseudoxanthoma elasticum. Int J Dermatol 1988, 27, 98–100. [Google Scholar] [CrossRef]
- Uitto, J.; Boyd, C.D.; Lebwohl, M.G.; Moshell, A.N.; Rosenbloom, J.; Terry, S. International Centennial Meeting on Pseudoxanthoma Elasticum: Progress in PXE Research. J. Investig. Dermatol. 1998, 110, 840–842. [Google Scholar] [CrossRef]
- Gorgels, T.G.; Hu, X.; Scheffer, G.L.; van der Wal, A.C.; Toonstra, J.; de Jong, P.T.; van Kuppevelt, T.H.; Levelt, C.N.; de Wolf, A.; Loves, W.J.; et al. Disruption of Abcc6 in the mouse: novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum. Mol. Genet. 2005, 14, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Klement, J.F.; Matsuzaki, Y.; Jiang, Q.-J.; Terlizzi, J.; Choi, H.Y.; Fujimoto, N.; Li, K.; Pulkkinen, L.; Birk, D.E.; Sundberg, J.P.; et al. Targeted Ablation of the Abcc6 Gene Results in Ectopic Mineralization of Connective Tissues. Mol. Cell. Biol. 2005, 25, 8299–8310. [Google Scholar] [CrossRef]
- Bergen, A.A. Pseudoxanthoma Elasticum: the End of the Autosomal Dominant Segregation Myth. J. Investig. Dermatol. 2006, 126, 704–705. [Google Scholar] [CrossRef] [PubMed]
- Struk, B.; Neldner, K.H.; Rao, V.S.; Jean, P.S.; Lindpaintner, K. Mapping of Both Autosomal Recessive and Dominant Variants of Pseudoxanthoma Elasticum to Chromosome 16p13.1. Hum. Mol. Genet. 1997, 6, 1823–1828. [Google Scholar] [CrossRef]
- van Soest, S.; Swart, J.; Tijmes, N.; Sandkuijl, L.A.; Rommers, J.; Bergen, A.A. A Locus for Autosomal Recessive Pseudoxanthoma Elasticum, with Penetrance of Vascular Symptoms in Carriers, Maps to Chromosome 16p13.1. Genome Res. 1997, 7, 830–834. [Google Scholar] [CrossRef]
- Klein, I.; Sarkadi, B.; Varadi, A. An inventory of the human ABC proteins. Biochim Biophys Acta 1999, 1461, 237–62. [Google Scholar] [CrossRef] [PubMed]
- Stefková, J.; Poledne, R.; A Hubácek, J. ATP-binding cassette (ABC) transporters in human metabolism and diseases. Physiol Res 2004, 53, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.-S.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.-L.; et al. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Ilias, A.; Urban, Z.; Seidl, T.L.; Le Saux, O.; Sinko, E.; Boyd, C.D.; Sarkadi, B.; Varadi, A. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6). J Biol Chem 2002, 277, 16860–7. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Fülöp, K.; Yamaguchi, Y.; Iliás, A.; Szabó, Z.; Brampton, C.N.; Pomozi, V.; Huszár, K.; Arányi, T.; Váradi, A. Expression and In Vivo Rescue of Human ABCC6 Disease-Causing Mutants in Mouse Liver. PLOS ONE 2011, 6, e24738. [Google Scholar] [CrossRef]
- Pomozi, V.; Brampton, C.; Fülöp, K.; Chen, L.-H.; Apana, A.; Li, Q.; Uitto, J.; Le Saux, O.; Váradi, A. Analysis of Pseudoxanthoma Elasticum–Causing Missense Mutants of ABCC6 In Vivo ; Pharmacological Correction of the Mislocalized Proteins. J. Investig. Dermatol. 2014, 134, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Pomozi, V.; Brampton, C.; Szeri, F.; Dedinszki, D.; Kozak, E.; van de Wetering, K.; Hopkins, H.; Martin, L.; Varadi, A.; Le Saux, O. Functional Rescue of ABCC6 Deficiency by 4-Phenylbutyrate Therapy Reduces Dystrophic Calcification in Abcc6(-/-) Mice. J Invest Dermatol 2017, 137, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, N.; Martin, L.; Calvas, P.; Le Bert, M.; Hovnanian, A. Pseudoxanthoma elasticum: a clinical, pathophysiological and genetic update including 11 novel ABCC6 mutations. J. Med Genet. 2005, 42, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Plomp, A.S.; Toonstra, J.; Bergen, A.A.; van Dijk, M.R.; de Jong, P.T. Proposal for updating the pseudoxanthoma elasticum classification system and a review of the clinical findings. Am. J. Med Genet. Part A 2010, 152, 1049–1058. [Google Scholar] [CrossRef]
- Weenink, A.; Dijkman, G.; Demeijer, P. Pseudoxanthoma elasticum and its complications: two case reports. Neth. J. Med. 1996, 49, 24–29. [Google Scholar] [CrossRef]
- Doyne, R. Chorioidal and retinal changes the result of blows on the eye. Trans Ophthalmol Soc UK 1889, 9, 128. [Google Scholar]
- Knapp, H. On the formation of dark angioid streaksas an unusual metamorphosis of retinal hemorrhage. Arch Ophthalmol 1892, 21, 289–292. [Google Scholar]
- Groenblad, E. Angioid streaks: pseudoxanthoma elasticum: vorloeufige mitteilung. Acta Ophthalmol 1929, 7, 329. [Google Scholar] [CrossRef]
- Gass, J.D. “Comet” lesion: an ocular sign of pseudoxanthoma elasticum. Retina 2003, 23, 729–30. [Google Scholar] [CrossRef] [PubMed]
- Finger, R.P.; Issa, P.C.; Schmitz-Valckenberg, S.; Holz, F.G.; Scholl, H.N. Long-term effectiveness of intravitreal bevacizumab for choroidal neovascularization secondary to angioid streaks in pseudoxanthoma elasticum. Retina 2011, 31, 1268–1278. [Google Scholar] [CrossRef]
- Hess, K.; Raming, K.; Issa, P.C.; Herrmann, P.; Holz, F.G.; Pfau, M. Inner retinal degeneration associated with optic nerve head drusen in pseudoxanthoma elasticum. Br. J. Ophthalmol. 2021, 107, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Brampton, C.; Pomozi, V.; Chen, L.-H.; Apana, A.; McCurdy, S.; Zoll, J.; Boisvert, W.A.; Lambert, G.; Henrion, D.; Blanchard, S.; et al. ABCC6 deficiency promotes dyslipidemia and atherosclerosis. Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef]
- Lefthériotis, G.; Omarjee, L.; Le Saux, O.; Henrion, D.; Abraham, P.; Prunier, F.; Willoteaux, S.; Martin, L. The vascular phenotype in Pseudoxanthoma elasticum and related disorders: contribution of a genetic disease to the understanding of vascular calcification. Front. Genet. 2013, 4, 4. [Google Scholar] [CrossRef]
- Vasseur, M.; Carsin-Nicol, B.; Ebran, J.; Willoteaux, S.; Martin, L.; Lefthériotis, G. Carotid Rete Mirabile and Pseudoxanthoma Elasticum: An Accidental Association? Eur. J. Vasc. Endovasc. Surg. 2011, 42, 292–294. [Google Scholar] [CrossRef]
- Omarjee, L.; Fortrat, J.-O.; Larralde, A.; Le Pabic, E.; Kauffenstein, G.; Laot, M.; Navasiolava, N.; Mention, P.-J.; Linares, J.L.C.; Valdivielso, P.; et al. Internal Carotid Artery Hypoplasia: A New Clinical Feature in Pseudoxanthoma Elasticum. J. Stroke 2019, 21, 108–111. [Google Scholar] [CrossRef]
- Kranenburg, G.; De Jong, P.A.; Mali, W.P.; Attrach, M.; Visseren, F.L.J.; Spiering, W. Prevalence and severity of arterial calcifications in pseudoxanthoma elasticum (PXE) compared to hospital controls. Novel insights into the vascular phenotype of PXE. Atherosclerosis 2017, 256, 7–14. [Google Scholar] [CrossRef]
- Kevorkian, J.P.; Masquet, C.; Kural-Menasche, S.; Le Dref, O.; Beaufils, P. New report of severe coronary artery disease in an eighteen-year-old girl with pseudoxanthoma elasticum. Case report and review of the literature. Angiology 1997, 48, 735–41. [Google Scholar] [CrossRef] [PubMed]
- Kocaman, S.; Tavil, Y.; Yalcin, M. Severe coronary artery disease in a 21-year-old girl with pseudoxanthoma elasticum anomalous origin of the right coronary artery. Acta Cardiol. 2007, 62, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Miwa, K.; Higashikata, T.; Mabuchi, H. Intravascular ultrasound findings of coronary wall morphology in a patient with pseudoxanthoma elasticum. Heart 2004, 90, e61–e61. [Google Scholar] [CrossRef] [PubMed]
- Lefthériotis, G.; Abraham, P.; Le Corre, Y.; Le Saux, O.; Henrion, D.; Ducluzeau, P.H.; Prunier, F.; Martin, L. Relationship between ankle brachial index and arterial remodeling in pseudoxanthoma elasticum. J. Vasc. Surg. 2011, 54, 1390–1394. [Google Scholar] [CrossRef]
- Leftheriotis, G.; Kauffenstein, G.; Hamel, J.F.; Abraham, P.; Le Saux, O.; Willoteaux, S.; Henrion, D.; Martin, L. The Contribution of Arterial Calcification to Peripheral Arterial Disease in Pseudoxanthoma Elasticum. PLOS ONE 2014, 9, e96003. [Google Scholar] [CrossRef] [PubMed]
- Prunier, F.; Terrien, G.; Le Corre, Y.; Apana, A.L.Y.; Bière, L.; Kauffenstein, G.; Furber, A.; Bergen, A.A.B.; Gorgels, T.G.M.F.; Le Saux, O.; et al. Pseudoxanthoma Elasticum: Cardiac Findings in Patients and Abcc6-Deficient Mouse Model. PLOS ONE 2013, 8, e68700. [Google Scholar] [CrossRef] [PubMed]
- Köblös, G.; Andrikovics, H.; Prohászka, Z.; Tordai, A.; Váradi, A.; Arányi, T. The R1141X Loss-of-Function Mutation of the ABCC6 Gene Is a Strong Genetic Risk Factor for Coronary Artery Disease. Genet. Test. Mol. Biomarkers 2010, 14, 75–78. [Google Scholar] [CrossRef]
- Trip, M.D.; Smulders, Y.M.; Wegman, J.J.; Hu, X.; Boer, J.M.; Brink, J.B.T.; Zwinderman, A.H.; Kastelein, J.J.; Feskens, E.J.; Bergen, A.A.; et al. Frequent mutation in the ABCC6 gene (R1141X) is associated with a strong increase in the prevalence of coronary artery disease. Circ. 2002, 106, 773–775. [Google Scholar] [CrossRef]
- Hornstrup, L.S.; Tybjærg-Hansen, A.; Haase, C.L.; Nordestgaard, B.G.; Sillesen, H.; Grande, P.; Frikke-Schmidt, R.; Christoffersen, M.; Schnohr, P.; Jensen, G.B.; et al. Heterozygosity for R1141X in ABCC6 and Risk of Ischemic Vascular Disease. Circ. Cardiovasc. Genet. 2011, 4, 534–541. [Google Scholar] [CrossRef]
- Kuzaj, P.; Kuhn, J.; Dabisch-Ruthe, M.; Faust, I.; Götting, C.; Knabbe, C.; Hendig, D. ABCC6- a new player in cellular cholesterol and lipoprotein metabolism? Lipids Heal. Dis. 2014, 13, 118–118. [Google Scholar] [CrossRef] [PubMed]
- Peloso, G.M.; Demissie, S.; Collins, D.; Mirel, D.B.; Gabriel, S.B.; Cupples, L.A.; Robins, S.J.; Schaefer, E.J.; Brousseau, M.E. Common genetic variation in multiple metabolic pathways influences susceptibility to low HDL-cholesterol and coronary heart disease. J. Lipid Res. 2010, 51, 3524–3532. [Google Scholar] [CrossRef] [PubMed]
- Pisciotta, L.; Tarugi, P.; Borrini, C.; Bellocchio, A.; Fresa, R.; Guerra, D.; Quaglino, D.; Ronchetti, I.; Calandra, S.; Bertolini, S. Pseudoxanthoma elasticum and familial hypercholesterolemia: A deleterious combination of cardiovascular risk factors. Atherosclerosis 2010, 210, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Near, S.; Young, K.; Connelly, P.W.; Hegele, R.A. ABCC6 gene polymorphism associated with variation in plasma lipoproteins. J. Hum. Genet. 2001, 46, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-D.; Terbah, M.; Daudon, P.; Martin, L. Left Ventricular Systolic and Diastolic Function by Echocardiogram in Pseudoxanthoma Elasticum. Am. J. Cardiol. 2006, 97, 1535–1537. [Google Scholar] [CrossRef] [PubMed]
- Miki, K.; Yuri, T.; Takeda, N.; Takehana, K.; Iwasaka, T.; Tsubura, A. An autopsy case of pseudoxanthoma elasticum: histochemical characteristics. Med Mol. Morphol. 2007, 40, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Lopez, F.; Llorian, A.; Ferrer-Roca, O.; Betriu, A.; Sanz, G. Restrictive Cardiomyopathy in Pseudoxanthoma Elasticum. Chest 1980, 78, 113–115. [Google Scholar] [CrossRef]
- Lebwohl, M.G.; Distefano, D.; Prioleau, P.G.; Uram, M.; Yannuzzi, L.A.; Fleischmajer, R. Pseudoxanthoma Elasticum and Mitral-Valve Prolapse. New Engl. J. Med. 1982, 307, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Uno, K.; Fujii, T.; Mukai, M.; Handa, S. Mitral Stenosis in Pseudoxanthoma Elasticum. Chest 1992, 101, 1706–1707. [Google Scholar] [CrossRef]
- Vanakker, O.M.; Leroy, B.P.; Coucke, P.; Bercovitch, L.G.; Uitto, J.; Viljoen, D.; Terry, S.F.; Van Acker, P.; Matthys, D.; Loeys, B.; et al. Novel clinico-molecular insights in pseudoxanthoma elasticum provide an efficient molecular screening method and a comprehensive diagnostic flowchart. Hum. Mutat. 2007, 29, 205–205. [Google Scholar] [CrossRef]
- Basso, C.; Boschello, M.; Perrone, C.; Mecenero, A.; Cera, A.; Bicego, D.; Thiene, G.; De Dominicis, E. An echocardiographic survey of primary school children for bicuspid aortic valve. Am. J. Cardiol. 2004, 93, 661–663. [Google Scholar] [CrossRef]
- Mungrue, I.N.; Zhao, P.; Yao, Y.; Meng, H.; Rau, C.; Havel, J.V.; Gorgels, T.G.; Bergen, A.A.; MacLellan, W.R.; Drake, T.A.; et al. Abcc6 Deficiency Causes Increased Infarct Size and Apoptosis in a Mouse Cardiac Ischemia-Reperfusion Model. Arter. Thromb. Vasc. Biol. 2011, 31, 2806–2812. [Google Scholar] [CrossRef]
- Eaton, G.J.; Custer, R.P.; Johnson, F.N.; Stabenow, K.T. Dystrophic cardiac calcinosis in mice: genetic, hormonal, and dietary influences. Am J Pathol 1978, 90, 173–186. [Google Scholar]
- Ivandic, B.T.; Qiao, J.H.; Machleder, D.; Liao, F.; A Drake, T.; Lusis, A.J. A locus on chromosome 7 determines myocardial cell necrosis and calcification (dystrophic cardiac calcinosis) in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 5483–5488. [Google Scholar] [CrossRef] [PubMed]
- Aherrahrou, Z.; Doehring, L.C.; Ehlers, E.-M.; Liptau, H.; Depping, R.; Linsel-Nitschke, P.; Kaczmarek, P.M.; Erdmann, J.; Schunkert, H. An Alternative Splice Variant in Abcc6, the Gene Causing Dystrophic Calcification, Leads to Protein Deficiency in C3H/He Mice. J. Biol. Chem. 2008, 283, 7608–7615. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Vera, I.; Che, N.; Wang, X.; Wang, S.S.; Ingram-Drake, L.; Schadt, E.E.; Drake, T.A.; Lusis, A.J. Identification of Abcc6 as the major causal gene for dystrophic cardiac calcification in mice through integrative genomics. Proc. Natl. Acad. Sci. USA 2007, 104, 4530–4535. [Google Scholar] [CrossRef]
- Le Corre, Y.; Le Saux, O.; Froeliger, F.; Libouban, H.; Kauffenstein, G.; Willoteaux, S.; Leftheriotis, G.; Martin, L. Quantification of the Calcification Phenotype of Abcc6-Deficient Mice with Microcomputed Tomography. Am. J. Pathol. 2012, 180, 2208–2213. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guo, H.; Chou, D.W.; Berndt, A.; Sundberg, J.P.; Uitto, J. Mouse Models for Pseudoxanthoma Elasticum: Genetic and Dietary Modulation of the Ectopic Mineralization Phenotypes. PLOS ONE 2014, 9, e89268. [Google Scholar] [CrossRef] [PubMed]
- Morikane, S.; Ishida, K.; Taniguchi, T.; Ashizawa, N.; Matsubayashi, M.; Kurita, N.; Kobashi, S.; Iwanaga, T. Identification of a DBA/2 Mouse Sub-strain as a Model for Pseudoxanthoma Elasticum-Like Tissue Calcification. Biol. Pharm. Bull. 2023, 46, 1737–1744. [Google Scholar] [CrossRef]
- Brampton, C.; Aherrahrou, Z.; Chen, L.-H.; Martin, L.; Bergen, A.A.; Gorgels, T.G.; Erdfdi, J.; Schunkert, H.; Szabó, Z.; Váradi, A.; et al. The Level of Hepatic ABCC6 Expression Determines the Severity of Calcification after Cardiac Injury. Am. J. Pathol. 2014, 184, 159–170. [Google Scholar] [CrossRef]
- Dedinszki, D.; Szeri, F.; Kozák, E.; Pomozi, V.; Tőkési, N.; Mezei, T.R.; Merczel, K.; Letavernier, E.; Tang, E.; Le Saux, O.; et al. Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol. Med. 2017, 9, 1463–1470. [Google Scholar] [CrossRef]
- Pomozi, V.; Brampton, C.; van de Wetering, K.; Zoll, J.; Calio, B.; Pham, K.; Owens, J.B.; Marh, J.; Moisyadi, S.; Váradi, A.; et al. Pyrophosphate Supplementation Prevents Chronic and Acute Calcification in ABCC6-Deficient Mice. Am. J. Pathol. 2017, 187, 1258–1272. [Google Scholar] [CrossRef]
- Omarjee, L.; Roy, C.; Leboeuf, C.; Favre, J.; Henrion, D.; Mahe, G.; Leftheriotis, G.; Martin, L.; Janin, A.; Kauffenstein, G. Evidence of Cardiovascular Calcification and Fibrosis in Pseudoxanthoma Elasticum Mouse Models Subjected to DOCA-Salt Hypertension. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Brampton, C.; Yamaguchi, Y.; Vanakker, O.; Van Laer, L.; Chen, L.-H.; Thakore, M.; De Paepe, A.; Pomozi, V.; Szabó, P.T.; Martin, L.; et al. Vitamin K does not prevent soft tissue mineralization in a mouse model of pseudoxanthoma elasticum. Cell Cycle 2011, 10, 1810–1820. [Google Scholar] [CrossRef]
- Ibold, B.; Tiemann, J.; Faust, I.; Ceglarek, U.; Dittrich, J.; Gorgels, T.G.M.F.; Bergen, A.A.B.; Vanakker, O.; Van Gils, M.; Knabbe, C.; et al. Genetic deletion of Abcc6 disturbs cholesterol homeostasis in mice. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Garcia-Fernandez, M.I.; Gheduzzi, D.; Boraldi, F.; Paolinelli, C.D.; Sanchez, P.; Valdivielso, P.; Morilla, M.J.; Quaglino, D.; Guerra, D.; Casolari, S.; et al. Parameters of oxidative stress are present in the circulation of PXE patients. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2008, 1782, 474–481. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, Q.; Uitto, J. Pseudoxanthoma elasticum: oxidative stress and antioxidant diet in a mouse model (Abcc6-/-). J Invest Dermatol 2008, 128, 1160–4. [Google Scholar] [CrossRef]
- Pasquali-Ronchetti, I.; Garcia-Fernandez, M.I.; Boraldi, F.; Quaglino, D.; Gheduzzi, D.; Paolinelli, C.D.V.; Tiozzo, R.; Bergamini, S.; Ceccarelli, D.; Muscatello, U. Oxidative stress in fibroblasts from patients with pseudoxanthoma elasticum: possible role in the pathogenesis of clinical manifestations. J. Pathol. 2005, 208, 54–61. [Google Scholar] [CrossRef]
- Kauffenstein, G.; Pizard, A.; Le Corre, Y.; Vessières, E.; Grimaud, L.; Toutain, B.; Labat, C.; Mauras, Y.; Gorgels, T.; Bergen, A.; et al. Disseminated Arterial Calcification and Enhanced Myogenic Response Are Associated With Abcc6 Deficiency in a Mouse Model of Pseudoxanthoma Elasticum. Arter. Thromb. Vasc. Biol. 2014, 34, 1045–1056. [Google Scholar] [CrossRef]
- Rasmussen, M.R.; Nielsen, K.L.; Laursen, M.R.; Nielsen, C.B.; Svendsen, P.; Dimke, H.; Christensen, E.I.; Johannsen, M.; Moestrup, S.K. Untargeted Metabolomics Analysis of ABCC6-Deficient Mice Discloses an Altered Metabolic Liver Profile. J. Proteome Res. 2016, 15, 4591–4600. [Google Scholar] [CrossRef]
- Lofaro, F.D.; Boraldi, F.; Garcia-Fernandez, M.; Estrella, L.; Valdivielso, P.; Quaglino, D. Relationship Between Mitochondrial Structure and Bioenergetics in Pseudoxanthoma elasticum Dermal Fibroblasts. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Martin, L.J.; Lau, E.; Singh, H.; Vergnes, L.; Tarling, E.J.; Mehrabian, M.; Mungrue, I.; Xiao, S.; Shih, D.; Castellani, L.; et al. ABCC6 Localizes to the Mitochondria-Associated Membrane. Circ. Res. 2012, 111, 516–520. [Google Scholar] [CrossRef]
- Gentile, D.; Natale, M.; Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F. The role of P2X7 receptors in tissue fibrosis: a brief review. Purinergic Signal. 2015, 11, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Kauffenstein, G.; Tamareille, S.; Prunier, F.; Roy, C.; Ayer, A.; Toutain, B.; Billaud, M.; Isakson, B.E.; Grimaud, L.; Loufrani, L.; et al. Central Role of P2Y 6 UDP Receptor in Arteriolar Myogenic Tone. Arter. Thromb. Vasc. Biol. 2016, 36, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, G.; Cronstein, B. Signaling pathways involving adenosine A2A and A2B receptors in wound healing and fibrosis. Purinergic Signal. 2016, 12, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-M.; Zhang, N.; Li, J.-S.; Yang, Z.-H.; Huang, X.-L.; Yang, X.-F. Purinergic receptors mediate endothelial dysfunction and participate in atherosclerosis. Purinergic Signal. 2022, 19, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.; Hayashi, K.; Dang, K.; Hayashi, M.; Boyd, C.D. Analysis of ABCC6 (MRP6) in normal human tissues. Histochem. 2005, 123, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.; Hayashi, K.; Nishiguchi, B.; Le Saux, O.; Hayashi, M.; Boyd, C.D. The Distribution of Abcc6 in Normal Mouse Tissues Suggests Multiple Functions for this ABC Transporter. J. Histochem. Cytochem. 2003, 51, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Bunda, S.; VanWart, C.M.; Douet, V.; Got, L.; Martin, L.; Hinek, A. Serum Factors from Pseudoxanthoma Elasticum Patients Alter Elastic Fiber Formation In Vitro. J. Investig. Dermatol. 2006, 126, 1497–1505. [Google Scholar] [CrossRef]
- Jiang, Q.; Li, Q.; Uitto, J. Aberrant Mineralization of Connective Tissues in a Mouse Model of Pseudoxanthoma Elasticum: Systemic and Local Regulatory Factors. J. Investig. Dermatol. 2007, 127, 1392–1402. [Google Scholar] [CrossRef]
- Jiang, Q.; Endo, M.; Dibra, F.; Wang, K.; Uitto, J. Pseudoxanthoma Elasticum Is a Metabolic Disease. J. Investig. Dermatol. 2009, 129, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Oldenburg, R.; Otsuru, S.; Grand-Pierre, A.E.; Horwitz, E.M.; Uitto, J. Parabiotic heterogenetic pairing of Abcc6-/-/Rag1-/- mice and their wild-type counterparts halts ectopic mineralization in a murine model of pseudoxanthoma elasticum. Am J Pathol 2010, 176, 1855–62. [Google Scholar] [CrossRef]
- Ronchetti, I.; Boraldi, F.; Annovi, G.; Cianciulli, P.; Quaglino, D. Fibroblast involvement in soft connective tissue calcification. Front. Genet. 2013, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Passi, A.; Albertini, R.; Contri, M.B.; de Luca, G.; de Paepe, A.; Pallavicini, G.; Ronchetti, I.P.; Tiozzo, R. Proteoglycan alterations in skin fibroblast cultures from patients affected with pseudoxanthoma elasticum. Cell Biochem Funct 1996, 14, 111–20. [Google Scholar] [CrossRef]
- Quaglino, D.; Sartor, L.; Garbisa, S.; Boraldi, F.; Croce, A.; Passi, A.; De Luca, G.; Tiozzo, R.; Pasquali-Ronchetti, I. Dermal fibroblasts from pseudoxanthoma elasticum patients have raised MMP-2 degradative potential. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2005, 1741, 42–47. [Google Scholar] [CrossRef]
- Tiemann, J.; Wagner, T.; Lindenkamp, C.; Plümers, R.; Faust, I.; Knabbe, C.; Hendig, D. Linking ABCC6 Deficiency in Primary Human Dermal Fibroblasts of PXE Patients to p21-Mediated Premature Cellular Senescence and the Development of a Proinflammatory Secretory Phenotype. Int. J. Mol. Sci. 2020, 21, 9665. [Google Scholar] [CrossRef]
- Belinsky, M.G.; Chen, Z.-S.; Shchaveleva, I.; Zeng, H.; Kruh, G.D. Characterization of the drug resistance and transport properties of multidrug resistance protein 6 (MRP6, ABCC6). Cancer Res. 2002, 62, 6172–6177. [Google Scholar]
- Vanakker, O.M.; Martin, L.; Gheduzzi, D.; Leroy, B.P.; Loeys, B.L.; Guerci, V.I.; Matthys, D.; Terry, S.F.; Coucke, P.J.; Pasquali-Ronchetti, I.; et al. Pseudoxanthoma Elasticum-Like Phenotype with Cutis Laxa and Multiple Coagulation Factor Deficiency Represents a Separate Genetic Entity. J. Investig. Dermatol. 2007, 127, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Vanakker, O.M.; Martin, L.; Schurgers, L.J.; Quaglino, D.; Costrop, L.; Vermeer, C.; Pasquali-Ronchetti, I.; Coucke, P.J.; De Paepe, A. Low serum vitamin K in PXE results in defective carboxylation of mineralization inhibitors similar to the GGCX mutations in the PXE-like syndrome. Mod. Pathol. 2010, 90, 895–905. [Google Scholar] [CrossRef]
- Borst, P.; van de Wetering, K.; Schlingemann, R. Does the absence of ABCC6 (multidrug resistance protein 6) in patients with Pseudoxanthoma elasticum prevent the liver from providing sufficient vitamin K to the periphery? Cell Cycle 2008, 7, 1575–9. [Google Scholar] [CrossRef]
- Gorgels, T.G.M.F.; Waarsing, J.H.; Herfs, M.; Versteeg, D.; Schoensiegel, F.; Sato, T.; Schlingemann, R.O.; Ivandic, B.; Vermeer, C.; Schurgers, L.J.; et al. Vitamin K supplementation increases vitamin K tissue levels but fails to counteract ectopic calcification in a mouse model for pseudoxanthoma elasticum. J. Mol. Med. 2011, 89, 1125–1135. [Google Scholar] [CrossRef]
- Jiang, Q.; Li, Q.; Grand-Pierre, A.E.; Schurgers, L.J.; Uitto, J. Administration of vitamin K does not counteract the ectopic mineralization of connective tissues in Abcc6 (-/-) mice, a model for pseudoxanthoma elasticum. Cell Cycle 2011, 10, 701–7. [Google Scholar] [CrossRef]
- Fülöp, K.; Jiang, Q.; Wetering, K.V.; Pomozi, V.; Szabó, P.T.; Arányi, T.; Sarkadi, B.; Borst, P.; Uitto, J.; Váradi, A. ABCC6 does not transport vitamin K3-glutathione conjugate from the liver: Relevance to pathomechanisms of pseudoxanthoma elasticum. Biochem. Biophys. Res. Commun. 2011, 415, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Hilaire, C.S.; Ziegler, S.G.; Markello, T.C.; Brusco, A.; Groden, C.; Gill, F.; Carlson-Donohoe, H.; Lederman, R.J.; Chen, M.Y.; Yang, D.; Siegenthaler, M.P.; Arduino, C.; Mancini, C.; Freudenthal, B.; Stanescu, H.C.; Zdebik, A.A.; Chaganti, R.K.; Nussbaum, R.L.; Kleta, R.; Gahl, W.A.; Boehm, M. NT5E mutations and arterial calcifications. N Engl J Med 2011, 364, 432–42. [Google Scholar] [CrossRef]
- Zimmermann, H. 5′-Nucleotidase: molecular structure and functional aspects. Biochem. J. 1992, 285, 345–365. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Price, T.P.; Sundberg, J.P.; Uitto, J. Juxta-articular joint-capsule mineralization in CD73 deficient mice: similarities to patients with NT5E mutations. Cell Cycle 2014, 13, 2609–15. [Google Scholar] [CrossRef]
- Joolharzadeh, P.; St. Hilaire, C. CD73 (Cluster of Differentiation 73) and the Differences Between Mice and Humans. Arter. Thromb. Vasc. Biol. 2019, 39, 339–348. [Google Scholar] [CrossRef]
- Szabó, Z.; Váradi, A.; Li, Q.; Uitto, J. ABCC6 does not transport adenosine — Relevance to pathomechanism of pseudoxanthoma elasticum. Mol. Genet. Metab. 2011, 104, 421. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, Y.; Rutsch, F. Generalized arterial calcification of infancy and pseudoxanthoma elasticum: two sides of the same coin. Front. Genet. 2012, 3, 302. [Google Scholar] [CrossRef]
- Pomozi, V.; Julian, C.B.; Zoll, J.; Pham, K.; Kuo, S.; Tőkési, N.; Martin, L.; Váradi, A.; Le Saux, O. Dietary Pyrophosphate Modulates Calcification in a Mouse Model of Pseudoxanthoma Elasticum: Implication for Treatment of Patients. J. Investig. Dermatol. 2018, 139, 1082–1088. [Google Scholar] [CrossRef]
- Cheng, Z.; O'Brien, K.; Howe, J.; Sullivan, C.; Schrier, D.; Lynch, A.; Jungles, S.; Sabbagh, Y.; Thompson, D. INZ-701 Prevents Ectopic Tissue Calcification and Restores Bone Architecture and Growth in ENPP1-Deficient Mice. J. Bone Miner. Res. 2020, 36, 1594–1604. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, I.J.; Cheng, Z.; Ralph, D.; O'Brien, K.; Flaman, L.; Howe, J.; Thompson, D.; Uitto, J.; Li, Q.; Sabbagh, Y. INZ-701, a recombinant ENPP1 enzyme, prevents ectopic calcification in an Abcc6(-/-) mouse model of pseudoxanthoma elasticum. Exp Dermatol 2022. [Google Scholar] [CrossRef] [PubMed]
- Roman, R.M.; Wang, Y.; Lidofsky, S.D.; Feranchak, A.P.; Lomri, N.; Scharschmidt, B.F.; Fitz, J.G. Hepatocellular ATP-binding Cassette Protein Expression Enhances ATP Release and Autocrine Regulation of Cell Volume. J. Biol. Chem. 1997, 272, 21970–21976. [Google Scholar] [CrossRef]
- Reisin, I.; Prat, A.; Abraham, E.; Amara, J.; Gregory, R.; Ausiello, D.; Cantiello, H. The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. J. Biol. Chem. 1994, 269, 20584–20591. [Google Scholar] [CrossRef] [PubMed]
- Sabirov, R.Z.; Okada, Y. ATP release via anion channels. Purinergic Signal 2005, 1, 311–28. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Le, G.; Ballard, H.J. Involvement of the cystic fibrosis transmembrane conductance regulator in the acidosis-induced efflux of ATP from rat skeletal muscle. J. Physiol. 2010, 588, 4563–4578. [Google Scholar] [CrossRef]
- Praetorius, H.A.; Leipziger, J. ATP release from non-excitable cells. Purinergic Signal. 2009, 5, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.M.; Johnson, M.D.; Kingsley, D.M. Role of the Mouse ank Gene in Control of Tissue Calcification and Arthritis. Science 2000, 289, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Nürnberg, P.; Thiele, H.; Chandler, D.; Höhne, W.; Cunningham, M.L.; Ritter, H.; Leschik, G.; Uhlmann, K.; Mischung, C.; Harrop, K.; et al. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat. Genet. 2001, 28, 37–41. [Google Scholar] [CrossRef]
- Reichenberger, E.; Tiziani, V.; Watanabe, S.; Park, L.; Ueki, Y.; Santanna, C.; Baur, S.T.; Shiang, R.; Grange, D.K.; Beighton, P.; et al. Autosomal Dominant Craniometaphyseal Dysplasia Is Caused by Mutations in the Transmembrane Protein ANK. Am. J. Hum. Genet. 2001, 68, 1321–1326. [Google Scholar] [CrossRef]
- Szeri, F.; Lundkvist, S.; Donnelly, S.; Engelke, U.F.H.; Rhee, K.; Williams, C.J.; Sundberg, J.P.; Wevers, R.A.; Tomlinson, R.E.; Jansen, R.S.; et al. The membrane protein ANKH is crucial for bone mechanical performance by mediating cellular export of citrate and ATP. PLOS Genet. 2020, 16, e1008884. [Google Scholar] [CrossRef] [PubMed]
- Szeri, F.; Niaziorimi, F.; Donnelly, S.; Fariha, N.; Tertyshnaia, M.; Patel, D.; Lundkvist, S.; van de Wetering, K. The Mineralization Regulator ANKH Mediates Cellular Efflux of ATP, Not Pyrophosphate. J. Bone Miner. Res. 2022, 37, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Martin, L.; Aherrahrou, Z.; Leftheriotis, G.; Váradi, A.; Brampton, C.N. The molecular and physiological roles of ABCC6: more than meets the eye. Front. Genet. 2012, 3, 289. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kingman, J.; van de Wetering, K.; Tannouri, S.; Sundberg, J.P.; Uitto, J. Abcc6 Knockout Rat Model Highlights the Role of Liver in PPi Homeostasis in Pseudoxanthoma Elasticum. J. Investig. Dermatol. 2017, 137, 1025–1032. [Google Scholar] [CrossRef]
- Van Gils, M.; Depauw, J.; Coucke, P.J.; Aerts, S.; Verschuere, S.; Nollet, L.; Vanakker, O.M. Inorganic Pyrophosphate Plasma Levels Are Decreased in Pseudoxanthoma Elasticum Patients and Heterozygous Carriers but Do Not Correlate with the Genotype or Phenotype. J. Clin. Med. 2023, 12, 1893. [Google Scholar] [CrossRef] [PubMed]
- Boraldi, F.; Bartolomeo, A.; Li, Q.; Uitto, J.; Quaglino, D. Changes in dermal fibroblasts from Abcc6(-/-) mice are present before and after the onset of ectopic tissue mineralization. J Investig. Dermatol 2014, 134, 1855–1861. [Google Scholar]
- Dabisch-Ruthe, M.; Kuzaj, P.; Götting, C.; Knabbe, C.; Hendig, D. Pyrophosphates as a major inhibitor of matrix calcification in Pseudoxanthoma elasticum. J. Dermatol. Sci. 2014, 75, 109–120. [Google Scholar] [CrossRef]
- Boraldi, F.; Costa, S.; Rabacchi, C.; Ciani, M.; Vanakker, O.; Quaglino, D. Can APOE and MTHFR polymorphisms have an influence on the severity of cardiovascular manifestations in Italian Pseudoxanthoma elasticum affected patients? Mol. Genet. Metab. Rep. 2014, 1, 477–482. [Google Scholar] [CrossRef] [PubMed]
- De Vilder, E.Y.G.; Hosen, M.J.; Martin, L.; De Zaeytijd, J.; Leroy, B.P.; Ebran, J.M.; Coucke, P.J.; De Paepe, A.; Vanakker, O.M. VEGFA variants as prognostic markers for the retinopathy in pseudoxanthoma elasticum. Clin Genet 2020, 98, 74–79. [Google Scholar] [CrossRef]
- Hendig, D.; Knabbe, C.; Götting, C. New insights into the pathogenesis of pseudoxanthoma elasticum and related soft tissue calcification disorders by identifying genetic interactions and modifiers. Front. Genet. 2013, 4, 114. [Google Scholar] [CrossRef]
- Hosen, M.J.; Van Nieuwerburgh, F.; Steyaert, W.; Deforce, D.; Martin, L.; Leftheriotis, G.; De Paepe, A.; Coucke, P.J.; Vanakker, O.M. Efficiency of Exome Sequencing for the Molecular Diagnosis of Pseudoxanthoma Elasticum. J. Investig. Dermatol. 2015, 135, 992–998. [Google Scholar] [CrossRef]
- Luo, H.; Faghankhani, M.; Cao, Y.; Uitto, J.; Li, Q. Molecular Genetics and Modifier Genes in Pseudoxanthoma Elasticum, a Heritable Multisystem Ectopic Mineralization Disorder. J. Investig. Dermatol. 2020, 141, 1148–1156. [Google Scholar] [CrossRef]
- Vanakker, O.M.; Hosen, M.J.; De Paepe, A. The ABCC6 transporter: what lessons can be learnt from other ATP-binding cassette transporters? Front. Genet. 2013, 4, 203. [Google Scholar] [CrossRef] [PubMed]
- Moorhead, W.J.; Chu, C.C.; Cuevas, R.A.; Callahan, J.T.; Wong, R.; Regan, C.; Boufford, C.K.; Sur, S.; Liu, M.; Gomez, D.; MacTaggart, J.N.; Kamenskiy, A.; Boehm, M.; Hilaire, C.S. Dysregulation of FOXO1 (Forkhead Box O1 Protein) Drives Calcification in Arterial Calcification due to Deficiency of CD73 and Is Present in Peripheral Artery Disease. Arterioscler Thromb Vasc Biol 2020, 40, 1680–1694. [Google Scholar] [CrossRef] [PubMed]
- Snider, N.T.; Griggs, N.W.; Singla, A.; Moons, D.S.; Weerasinghe, S.V.; Lok, A.S.; Ruan, C.; Burant, C.F.; Conjeevaram, H.S.; Omary, M.B. CD73 (ecto-5′-nucleotidase) hepatocyte levels differ across mouse strains and contribute to mallory-denk body formation. J. Hepatol. 2013, 58, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Pomozi, V.; Brampton, C.; van de Wetering, K.; Zoll, J.; Calio, B.; Pham, K.; Owens, J.B.; Marh, J.; Moisyadi, S.; Váradi, A.; et al. Pyrophosphate Supplementation Prevents Chronic and Acute Calcification in ABCC6-Deficient Mice. Am. J. Pathol. 2017, 187, 1258–1272. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, F.; Cuviello, F.; Pace, M.C.; Armentano, M.F.; Miglionico, R.; Ostuni, A.; Bisaccia, F. Extracellular ATP Regulates CD73 and ABCC6 Expression in HepG2 Cells. Front. Mol. Biosci. 2018, 5, 75. [Google Scholar] [CrossRef]
- Hart, M.L.; Much, C.; Gorzolla, I.C.; Schittenhelm, J.; Kloor, D.; Stahl, G.L.; Eltzschig, H.K. Extracellular Adenosine Production by Ecto-5′-Nucleotidase Protects During Murine Hepatic Ischemic Preconditioning. Gastroenterology 2008, 135, 1739–1750. [Google Scholar] [CrossRef]
- Burnstock, G.; Knight, G.E.; Greig AV, H. Purinergic Signaling in Healthy and Diseased Skin. J. Investig. Dermatol. 2012, 132, 526–546. [Google Scholar] [CrossRef]
- Burnstock, G.; Pelleg, A. Cardiac purinergic signalling in health and disease. Purinergic Signal. 2014, 11, 1–46. [Google Scholar] [CrossRef]
- Burnstock, G.; Vaughn, B.; Robson, S.C. Purinergic signalling in the liver in health and disease. Purinergic Signal. 2013, 10, 51–70. [Google Scholar] [CrossRef]
- Jain, S.; Jacobson, K.A. Purinergic signaling in diabetes and metabolism. Biochem. Pharmacol. 2020, 187, 114393–114393. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does it Exert its Protective Effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Linden, J. Adenosine in Tissue Protection and Tissue Regeneration: TABLE 1. Mol. Pharmacol. 2005, 67, 1385–1387. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef]
- Burnstock, G.; Knight, G.E. Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol 2004, 240, 31–304. [Google Scholar]
- Idzko, M.; Ferrari, D.; Riegel, A.-K.; Eltzschig, H.K. Extracellular nucleotide and nucleoside signaling in vascular and blood disease. Blood 2014, 124, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic Signaling during Inflammation. New Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef]
- Agrawal, A.; Jørgensen, N.R. Extracellular purines and bone homeostasis. Biochem. Pharmacol. 2021, 187, 114425. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic Signaling during Inflammation. New Engl. J. Med. 2013, 368, 1260–1260. [Google Scholar] [CrossRef]
- Hechler, B.; Gachet, C. Purinergic Receptors in Thrombosis and Inflammation. Arter. Thromb. Vasc. Biol. 2015, 35, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.; Koch-Nolte, F.; Dahl, G. Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu Rev Immunol 2019, 37, 325–347. [Google Scholar] [CrossRef]
- Zuccarini, M.; Giuliani, P.; Caciagli, F.; Ciccarelli, R.; Di Iorio, P. In Search of a Role for Extracellular Purine Enzymes in Bone Function. Biomolecules 2021, 11, 679. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Henaut, L.; Sanchez-Nino, M.D.; Castillo, G.A.-E.; Sanz, A.B.; Ortiz, A. Targeting local vascular and systemic consequences of inflammation on vascular and cardiac valve calcification. Expert Opin. Ther. Targets 2015, 20, 89–105. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, I.-K.; Jeon, J.-H. Vascular Calcification—New Insights into Its Mechanism. Int. J. Mol. Sci. 2020, 21, 2685. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, Z.; Zhang, L.; Yan, J.; Shao, C.; Jing, L.; Li, L.; Wang, Z. Role of Macrophages in the Progression and Regression of Vascular Calcification. Front. Pharmacol. 2020, 11, 661. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V. Alternatively activated macrophages exhibit an anticalcifying activity dependent on extracellular ATP/pyrophosphate metabolism. Am. J. Physiol. Physiol. 2016, 310, C788–C799. [Google Scholar] [CrossRef]
- Mention, P.-J.; Lacoeuille, F.; Leftheriotis, G.; Martin, L.; Omarjee, L. 18F-Flurodeoxyglucose and 18F-Sodium Fluoride Positron Emission Tomography/Computed Tomography Imaging of Arterial and Cutaneous Alterations in Pseudoxanthoma Elasticum. Circ. Cardiovasc. Imaging 2018, 11, e007060. [Google Scholar] [CrossRef]
- Omarjee, L.; Mention, P.J.; Janin, A.; Kauffenstein, G.; Pabic, E.L.; Meilhac, O.; Blanchard, S.; Navasiolava, N.; Leftheriotis, G.; Couturier, O.; Jeannin, P.; Lacoeuille, F.; Martin, L. Assessment of Inflammation and Calcification in Pseudoxanthoma Elasticum Arteries and Skin with 18F-FluroDeoxyGlucose and 18F-Sodium Fluoride Positron Emission Tomography/Computed Tomography Imaging: The GOCAPXE Trial. J Clin Med 2020, 9. [Google Scholar] [CrossRef]
- Peng, S.; Sun, X.; Wang, X.; Wang, H.; Shan, Z.; Teng, W.; Li, C. Myeloid related proteins are up-regulated in autoimmune thyroid diseases and activate toll-like receptor 4 and pro-inflammatory cytokines in vitro. Int. Immunopharmacol. 2018, 59, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Bowen, A.R.; Götting, C.; LeBoit, P.E.; McCalmont, T.H. Pseudoxanthoma elasticum-like fibers in the inflamed skin of patients without pseudoxanthoma elasticum. J. Cutan. Pathol. 2007, 34, 777–781. [Google Scholar] [CrossRef]
- Rabin, J.; Zhao, Y.; Mostafa, E.; Al-Suqi, M.; Fleischmann, E.; Conaway, M.R.; Mann, B.J.; Chhabra, P.; Brayman, K.L.; Krupnick, A.; et al. Regadenoson for the treatment of COVID-19: A five case clinical series and mouse studies. PLOS ONE 2023, 18, e0288920. [Google Scholar] [CrossRef] [PubMed]
- Casemayou, A.; Belliere, J.; Letavernier, E.; Colliou, E.; El Hachem, H.; Zarowski, J.; Bazin, D.; Kounde, C.; Piedrafita, A.; Feuillet, G.; et al. Abcc6 deficiency prevents rhabdomyolysis-induced acute kidney injury. Sci. Rep. 2023, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kauffenstein, G.; Drouin, A.; Thorin-Trescases, N.; Bachelard, H.; Robaye, B.; D'Orléans-Juste, P.; Marceau, F.; Thorin. ; Sévigny, J. NTPDase1 (CD39) controls nucleotide-dependent vasoconstriction in mouse. Cardiovasc. Res. 2009, 85, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, Y.; Rutsch, F. Genetics in Arterial Calcification: Lessons Learned From Rare Diseases. Trends Cardiovasc. Med. 2012, 22, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Tyson, K.L.; Reynolds, J.L.; McNair, R.; Zhang, Q.; Weissberg, P.L.; Shanahan, C.M. Osteo/Chondrocytic Transcription Factors and Their Target Genes Exhibit Distinct Patterns of Expression in Human Arterial Calcification. Arter. Thromb. Vasc. Biol. 2003, 23, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Towler, D.A. Inorganic pyrophosphate: a paracrine regulator of vascular calcification and smooth muscle phenotype. Arterioscler Thromb Vasc Biol 2005, 25, 651–4. [Google Scholar] [CrossRef] [PubMed]
- Prosdocimo, D.A.; Douglas, D.C.; Romani, A.M.; O'Neill, W.C.; Dubyak, G.R.; Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V.; Trueblood, K.E.; Mohr, S.; et al. Autocrine ATP release coupled to extracellular pyrophosphate accumulation in vascular smooth muscle cells. Am. J. Physiol. Physiol. 2009, 296, C828–C839. [Google Scholar] [CrossRef]
- Beck, K.; Dang, K.; Boyd, C.D. The tissue distribution of murine Abcc6 (Mrp6) during embryogenesis indicates that the presence of Abcc6 in elastic tissues is not required for elastic fiber assembly. Histochem. J. 2005, 36, 167–170. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; O’neill, W.C. Pyrophosphate deficiency in vascular calcification. Kidney Int. 2018, 93, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Buchet, R.; Tribes, C.; Rouaix, V.; Doumeche, B.; Fiore, M.; Wu, Y.; Magne, D.; Mebarek, S. Hydrolysis of Extracellular ATP by Vascular Smooth Muscle Cells Transdifferentiated into Chondrocytes Generates P(i) but Not PP(i). Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Pingel, S.; Pausewang, K.S.; Passon, S.G.; Blatzheim, A.K.; Gliem, M.; Issa, P.C.; Hendig, D.; Horlbeck, F.; Tuleta, I.; Nickenig, G.; et al. Increased vascular occlusion in patients with pseudoxanthoma elasticum. Vasa 2017, 46, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Rau, C.D.; Wang, J.; Avetisyan, R.; Romay, M.C.; Martin, L.; Ren, S.; Wang, Y.; Lusis, A.J. Mapping genetic contributions to cardiac pathology induced by Beta-adrenergic stimulation in mice. Circ. Cardiovasc. Genet. 2015, 8, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Rumney, R.M.H.; Róg, J.; Chira, N.; Kao, A.P.; Al-Khalidi, R.; Górecki, D.C. P2X7 Purinoceptor Affects Ectopic Calcification of Dystrophic Muscles. Front. Pharmacol. 2022, 13, 935804. [Google Scholar] [CrossRef]
- Pelegrin, P.; Surprenant, A. Dynamics of macrophage polarization reveal new mechanism to inhibit IL-1beta release through pyrophosphates. EMBO J 2009, 28, 2114–27. [Google Scholar] [CrossRef]
- Sassi, Y.; Ahles, A.; Truong, D.-J.J.; Baqi, Y.; Lee, S.-Y.; Husse, B.; Hulot, J.-S.; Foinquinos, A.; Thum, T.; Müller, C.E.; et al. Cardiac myocyte–secreted cAMP exerts paracrine action via adenosine receptor activation. J. Clin. Investig. 2014, 124, 5385–5397. [Google Scholar] [CrossRef] [PubMed]
- Bonner, F.; Borg, N.; Jacoby, C.; Temme, S.; Ding, Z.; Flogel, U.; Schrader, J. Ecto-5'-nucleotidase on immune cells protects from adverse cardiac remodeling. Circ Res 2013, 113, 301–12. [Google Scholar] [CrossRef]
- Borg, N.; Alter, C.; Görldt, N.; Jacoby, C.; Ding, Z.; Steckel, B.; Quast, C.; Bönner, F.; Friebe, D.; Temme, S.; et al. CD73 on T Cells Orchestrates Cardiac Wound Healing After Myocardial Infarction by Purinergic Metabolic Reprogramming. Circ. 2017, 136, 297–313. [Google Scholar] [CrossRef]
- Rostand, S.G.; Sanders, C.; Kirk, K.A.; Rutsky, E.A.; Fraser, R.G. Myocardial calcification and cardiac dysfunction in chronic renal failure. Am J Med 1988, 85, 651–7. [Google Scholar] [CrossRef]
- Shackley, B.S.; Nguyen, T.P.; Shivkumar, K.; Finn, P.J.; Fishbein, M.C. Idiopathic massive myocardial calcification: a case report and review of the literature. Cardiovasc. Pathol. 2011, 20, e79–e83. [Google Scholar] [CrossRef] [PubMed]
- A Bridge, J.; McManus, B.M.; Remmenga, J.; Cuppage, F.P. Complete heart block in the 18p--syndrome. Congenital calcification of the atrioventricular node. Arch Pathol Lab Med 1989, 113, 539–541. [Google Scholar] [PubMed]
- Henderson, R.R.; Santiago, L.M.; Spring, D.A.; Harrington, A.R. Metastatic Myocardial Calcification in Chronic Renal Failure Presenting as Atrioventricular Block. New Engl. J. Med. 1971, 284, 1252–1253. [Google Scholar] [CrossRef] [PubMed]
- Pillai, I.C.; Li, S.; Romay, M.; Lam, L.; Lu, Y.; Huang, J.; Dillard, N.; Zemanova, M.; Rubbi, L.; Wang, Y.; et al. Cardiac Fibroblasts Adopt Osteogenic Fates and Can Be Targeted to Attenuate Pathological Heart Calcification. Cell Stem Cell 2016, 20, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Bertulezzi, G.; Paris, R.; Moroni, M.; Porta, C.; Nastasi, G.; Amadeo, A. Atrial septal aneurysm in a patient with pseudoxanthoma elasticum. Acta Cardiol 1998, 53, 223–225. [Google Scholar] [PubMed]
- Fang, M.L.; Astarita, R.W.; Steinman, H.K. Cardiac Calcifications and Yellow Papules in a Young Man. Arch. Dermatol. 1988, 124, 1563–1564. [Google Scholar] [CrossRef]
- Farmakis, D.; Vesleme, V.; Papadogianni, A.; Tsaftaridis, P.; Kapralos, P.; Aessopos, A. Aneurysmatic dilatation of ascending aorta in a patient with beta-thalassemia and a pseudoxanthoma elasticum-like syndrome. Ann Hematol 2004, 83, 596–9. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, I.; Callea, F.; Travaglini, L.; Amodeo, A.; Cogo, P.; Secinaro, A.; Bizzarri, C.; Cutrera, R.; El Hachem, M.; Francalanci, P. Heart transplant and 2-year follow up in a child with generalized arterial calcification of infancy. Eur. J. Pediatr. 2014, 173, 1735–1740. [Google Scholar] [CrossRef]
- Nolte, K. Sudden cardiac death owing to pseudoxanthoma elasticum: A case report. Hum. Pathol. 2000, 31, 1002–1004. [Google Scholar] [CrossRef]
- Przybojewski, J.Z.; Maritz, F.; A Tiedt, F.; Van Der Walt, J.J. Pseudoxanthoma elasticum with cardiac involvement. A case report and review of the literature. S Afr Med J. 1981, 59, 268–275. [Google Scholar]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Uitto, J. The mineralization phenotype in Abcc6 ( -/- ) mice is affected by Ggcx gene deficiency and genetic background--a model for pseudoxanthoma elasticum. J Mol Med 2010, 88, 173–81. [Google Scholar] [CrossRef] [PubMed]
- Fleisch, H.; Russell, R.G.G.; Straumann, F. Effect of Pyrophosphate on Hydroxyapatite and Its Implications in Calcium Homeostasis. Nature 1966, 212, 901–903. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, J.K. Biological Role of Inorganic Pyrophosphate; Springer Science and Business Media LLC: Dordrecht, GX, Netherlands, 2001; ISBN 9781461355519. [Google Scholar]
- Hsu, V.M.; Kozák, E.; Li, Q.; Bocskai, M.; Schlesinger, N.; Rosenthal, A.; McClure, S.T.; Kovács, L.; Bálint, L.; Szamosi, S.; et al. Inorganic pyrophosphate is reduced in patients with systemic sclerosis. Rheumatology 2021, 61, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- O'Neill, W.C.; Lomashvili, K.A.; Malluche, H.H.; Faugere, M.-C.; Riser, B.L. Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int. 2011, 79, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Orriss, I.R.; Arnett, T.R.; Russell, R.G.G. Pyrophosphate: a key inhibitor of mineralisation. Curr. Opin. Pharmacol. 2016, 28, 57–68. [Google Scholar] [CrossRef]
- Väärämäki, S.; Pelttari, S.; Uusitalo, H.; Tökési, N.; Váradi, A.; Nevalainen, P.I. Pyrophosphate Treatment in Pseudoxanthoma Elasticum (PXE)-Preventing ReOcclusion After Surgery for Critical Limb Ischaemia. Surgical Case Reports 2019, 2, 1–3. [Google Scholar] [CrossRef]
- Ralph, D.; Nitschke, Y.; Levine, M.A.; Caffet, M.; Wurst, T.; Saeidian, A.H.; Youssefian, L.; Vahidnezhad, H.; Terry, S.F.; Rutsch, F.; et al. ENPP1 variants in patients with GACI and PXE expand the clinical and genetic heterogeneity of heritable disorders of ectopic calcification. PLOS Genet. 2022, 18, e1010192. [Google Scholar] [CrossRef]
- Laurain, A.; Rubera, I.; Razzouk-Cadet, M.; Bonnafous, S.; Albuquerque, M.; Paradis, V.; Patouraux, S.; Duranton, C.; Lesaux, O.; Lefthériotis, G.; et al. Arterial Calcifications in Patients with Liver Cirrhosis Are Linked to Hepatic Deficiency of Pyrophosphate Production Restored by Liver Transplantation. Biomedicines 2022, 10, 1496. [Google Scholar] [CrossRef]
- Letavernier, E.; Bouderlique, E.; Zaworski, J.; Martin, L.; Daudon, M. Pseudoxanthoma Elasticum, Kidney Stones and Pyrophosphate: From a Rare Disease to Urolithiasis and Vascular Calcifications. Int. J. Mol. Sci. 2019, 20, 6353. [Google Scholar] [CrossRef]
- Letavernier, E.; Kauffenstein, G.; Huguet, L.; Navasiolava, N.; Bouderlique, E.; Tang, E.; Delaitre, L.; Bazin, D.; de Frutos, M.; Gay, C.; et al. ABCC6 Deficiency Promotes Development of Randall Plaque. J. Am. Soc. Nephrol. 2018, 29, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Addison, W.N.; Azari, F.; Sørensen, E.S.; Kaartinen, M.T.; McKee, M.D. Pyrophosphate Inhibits Mineralization of Osteoblast Cultures by Binding to Mineral, Up-regulating Osteopontin, and Inhibiting Alkaline Phosphatase Activity. J. Biol. Chem. 2007, 282, 15872–15883. [Google Scholar] [CrossRef] [PubMed]
- Plomp, A.S.; Bergen, A.A.; Florijn, R.J.; Terry, S.F.; Toonstra, J.; van Dijk, M.R.; de Jong, P.T. Pseudoxanthoma elasticum: Wide phenotypic variation in homozygotes and no signs in heterozygotes for the c.3775delT mutation in ABCC6. Anesthesia Analg. 2009, 11, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Uitto, J.; Jiang, Q.; Váradi, A.; Bercovitch, L.G.; Terry, S.F. Pseudoxanthoma elasticum: diagnostic features, classification and treatment options. Expert Opin. Orphan Drugs 2014, 2, 567–577. [Google Scholar] [CrossRef]
- Lau, W.L.; Liu, S.; Vaziri, N.D. Chronic Kidney Disease Results in Deficiency of ABCC6, the Novel Inhibitor of Vascular Calcification. Am. J. Nephrol. 2014, 40, 51–55. [Google Scholar] [CrossRef]
Figure 1.
The ABCC6 pathway influences calcification regulation extracellular purinergic metabolism. ABCC6 facilitates the cellular efflux of ATP from liver and other tissues/cells, which is quickly converted to pyrophosphate (PPi), a potent inhibitor of mineralization. Decreased plasma PPi levels cause calcification in PXE and GACI. NT5E activity leads to adenosine production, which affects many biological activities including inflammation, and inhibition of TNAP synthesis. TNAP degrades PPi into inorganic phosphate (Pi), an activator of calcification which leads to vascular calcification in CALJA patients.
Figure 1.
The ABCC6 pathway influences calcification regulation extracellular purinergic metabolism. ABCC6 facilitates the cellular efflux of ATP from liver and other tissues/cells, which is quickly converted to pyrophosphate (PPi), a potent inhibitor of mineralization. Decreased plasma PPi levels cause calcification in PXE and GACI. NT5E activity leads to adenosine production, which affects many biological activities including inflammation, and inhibition of TNAP synthesis. TNAP degrades PPi into inorganic phosphate (Pi), an activator of calcification which leads to vascular calcification in CALJA patients.
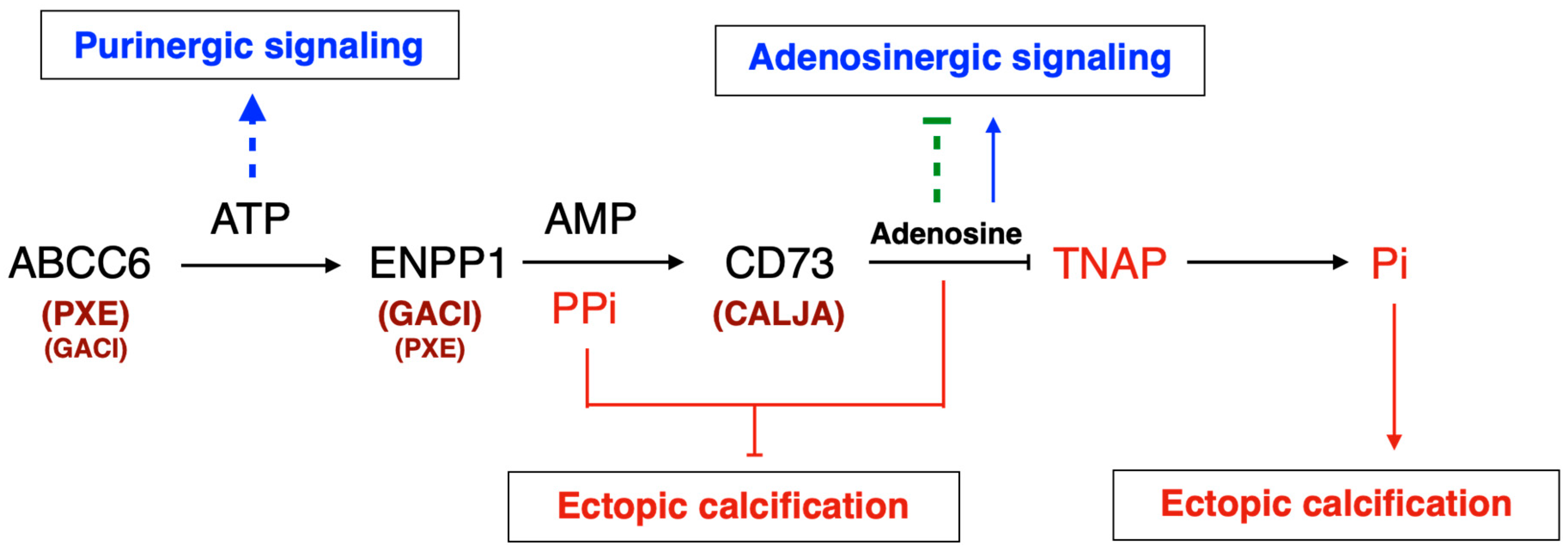
Figure 2.
Quantification of the dystrophic cardiac calcification (DCC) phenotype of Abcc6-/-, Nt5e-/- and Entpd1-/- mouse models vs wild type (WT) animals. The calcium content in the murine hearts after cryoinjury, described previously [80], was expressed as a ratio between the injured ventral and uninjured dorsal sections. Results were expressed as mean ± SEM and considered significant for. ** p<0.01.
Figure 2.
Quantification of the dystrophic cardiac calcification (DCC) phenotype of Abcc6-/-, Nt5e-/- and Entpd1-/- mouse models vs wild type (WT) animals. The calcium content in the murine hearts after cryoinjury, described previously [80], was expressed as a ratio between the injured ventral and uninjured dorsal sections. Results were expressed as mean ± SEM and considered significant for. ** p<0.01.
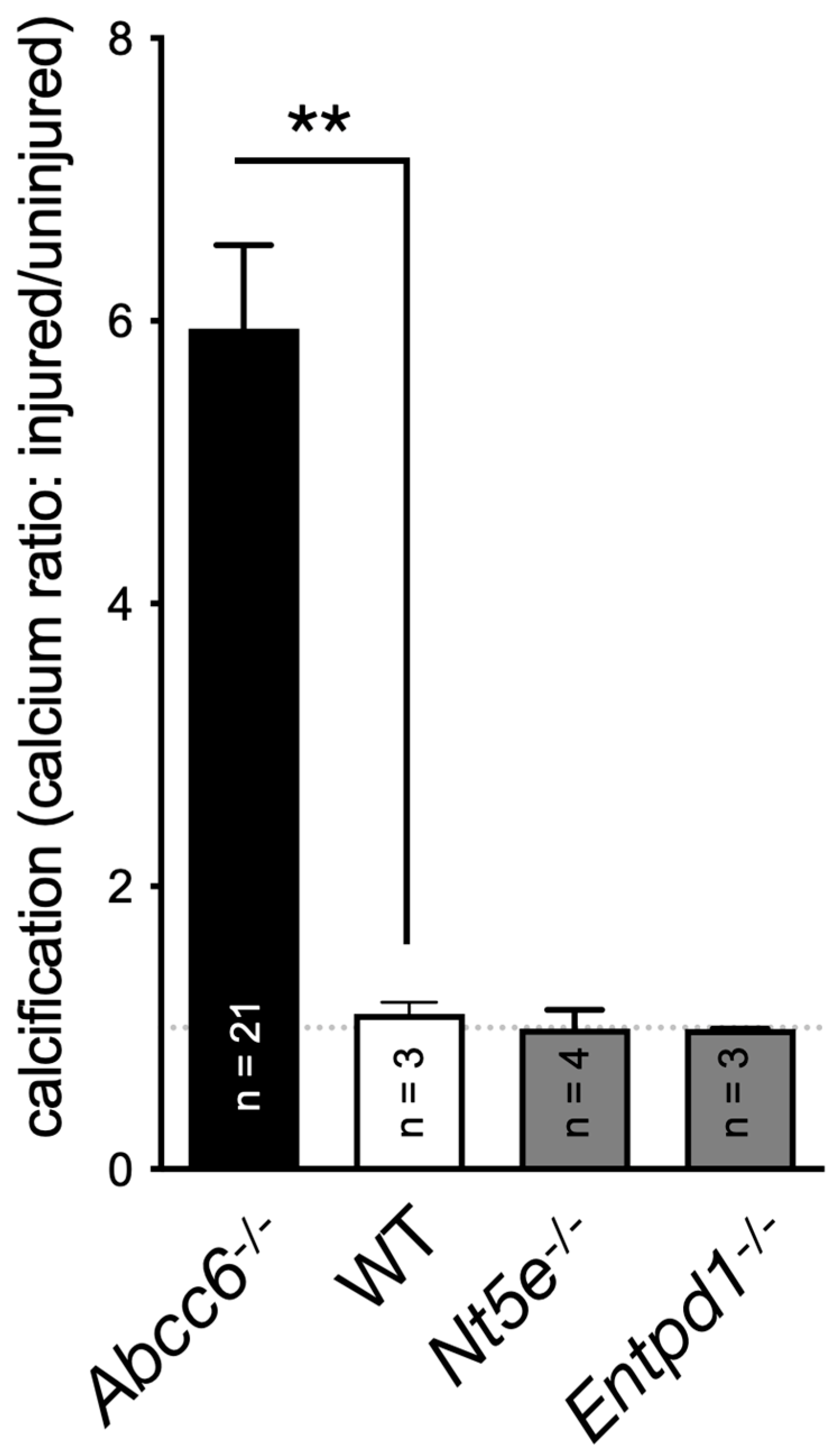
Figure 4.
The expression of a few selected purinergic signaling-related genes in lymphocytes (A) and macrophages (B) isolated from WT (+/+) Abcc6-/- (-/-) mice. The expression profile is differentially dysregulated in lymphocytes (Abcc6 expression) and in non-polarized macrophages (no Abcc6 expression). The data shows relative gene expression profiles normalized to Gapdh. Mouse numbers are 5 vs 4 in lymphocytes and 6 vs 6 in macrophages. Results are show +/- SEM. * p<0.05.
Figure 4.
The expression of a few selected purinergic signaling-related genes in lymphocytes (A) and macrophages (B) isolated from WT (+/+) Abcc6-/- (-/-) mice. The expression profile is differentially dysregulated in lymphocytes (Abcc6 expression) and in non-polarized macrophages (no Abcc6 expression). The data shows relative gene expression profiles normalized to Gapdh. Mouse numbers are 5 vs 4 in lymphocytes and 6 vs 6 in macrophages. Results are show +/- SEM. * p<0.05.

Table 1.
Mutations associated to human diseases and mouse calcifying phenotype. OMIM (Online Mendelian Inheritance in Man® reference); NR, not reported.
Table 1.
Mutations associated to human diseases and mouse calcifying phenotype. OMIM (Online Mendelian Inheritance in Man® reference); NR, not reported.
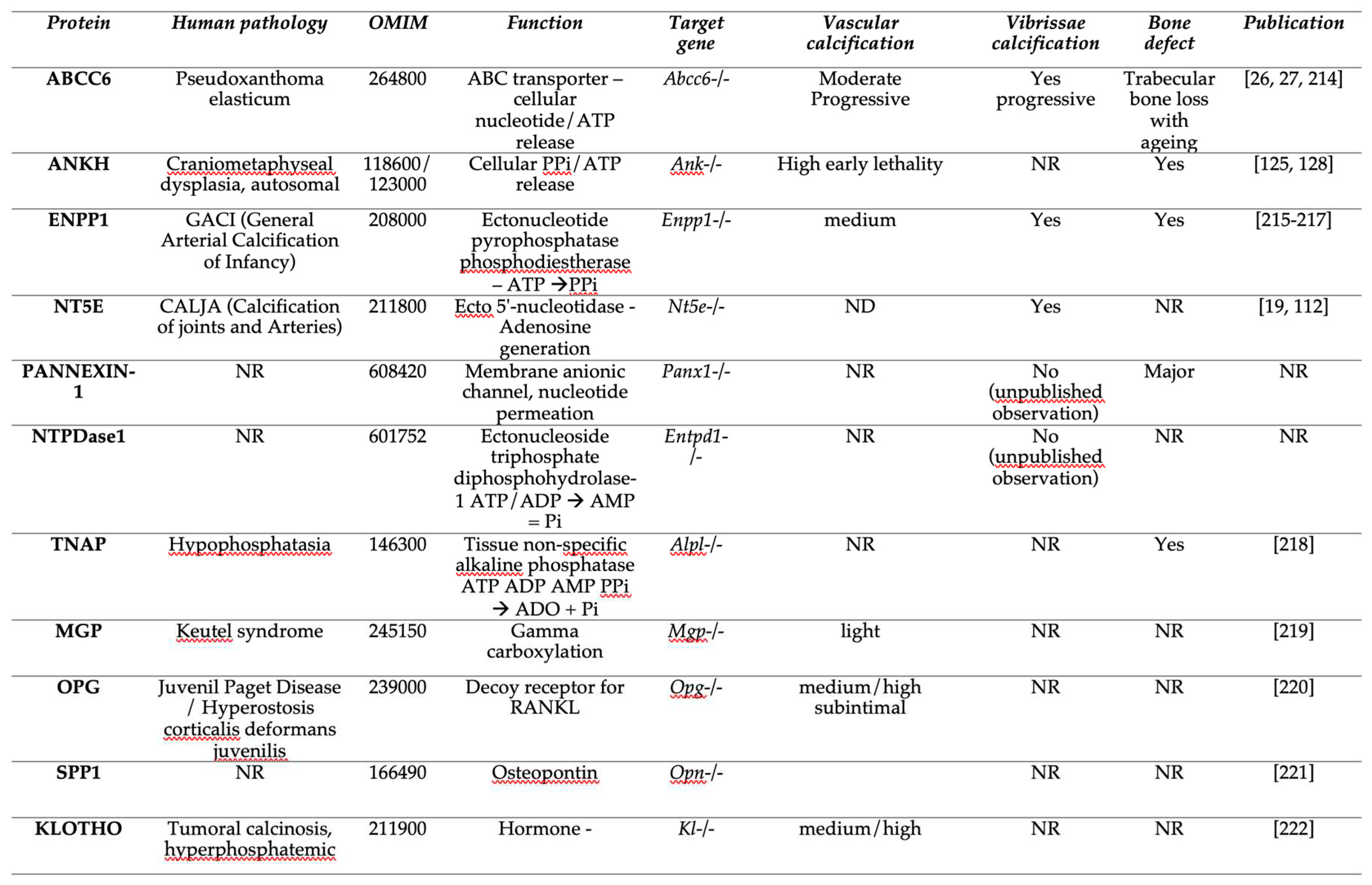
Table 3.
Potential purine homeostasis-related treatment in PXE and related diseases.
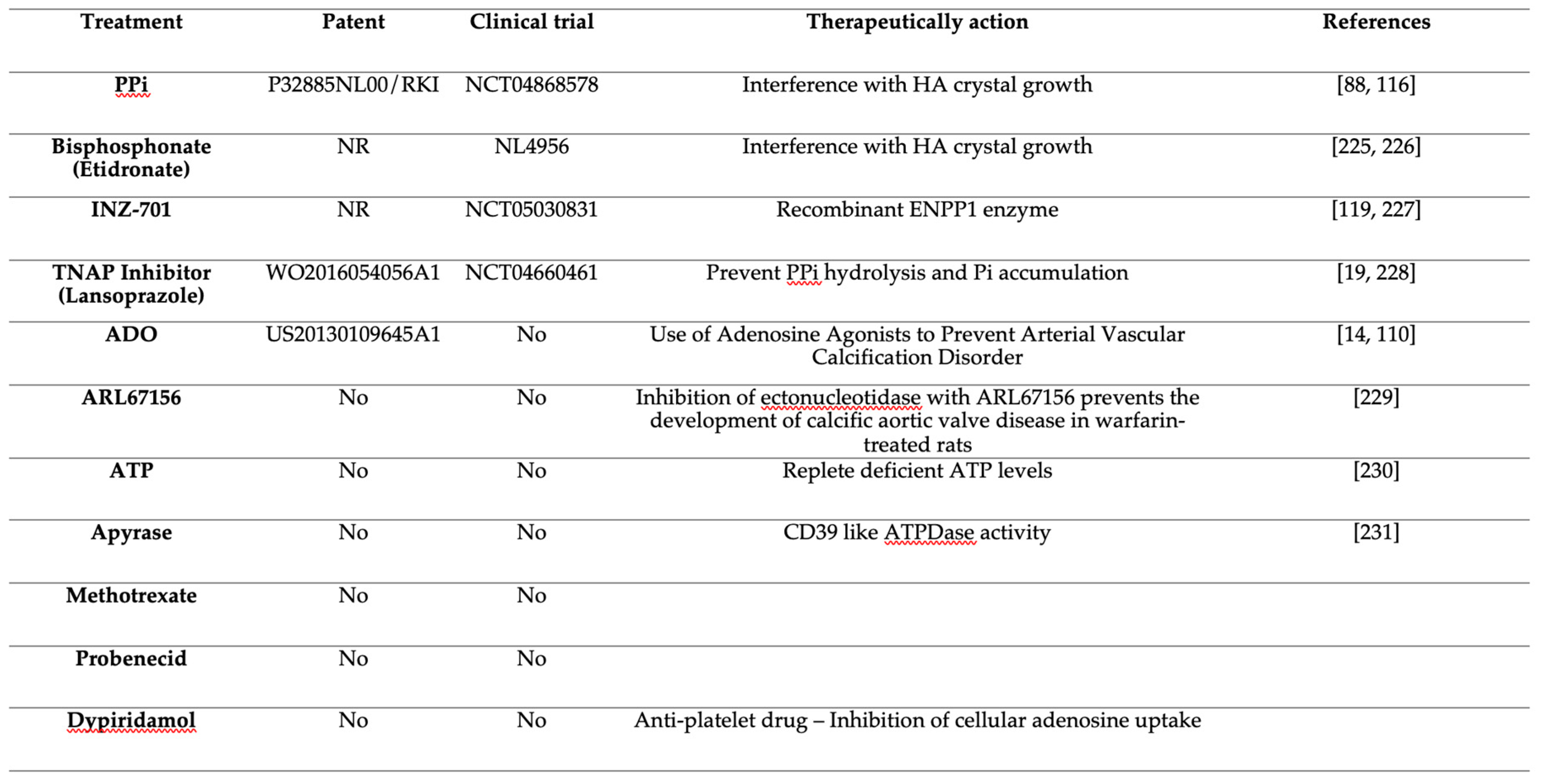
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



