Submitted:
17 December 2023
Posted:
18 December 2023
You are already at the latest version
Abstract
Knowing that biomolecules, such as β-amyrin and α-amyrin, have some pharmacological effects, the aim of this study was directed towards exploring the protective effect of a Tomato Peel and Seed Extract (TPSE) for its soothing function but also for its capacity to modulate the adrenal axis, involved in stress response. Ex vivo tests were carried out on skin explants to evaluate the effectiveness of TPSE formulated at 0.5% on Calcitonin Gene-Related Peptide (CGRP) and IL-10 release, Kappa Opioid Receptor (KOR) and Caspase 14 expression. An in vivo study combined a clinical evaluation of skin homogeneity, psychological parameter as well as analysis of salivary cortisol and dehydroepiandrosterone concentrations. All measurements were carried out at the beginning and after 28 days of applying a TPSE face cream. TPSE regulated not only the release of CGRP and Il-10, reflecting an anti-neurogenic and anti-inflammatory properties, but also modulated KORs. Twenty height days of TPSE application induced a significant decrease in intensity and extent erythrosis, a lower output of salivary cortisol and a significant increase in pleasant emotions when compared to placebo.
These results provide encouragement to continue exploring the impact of cosmetic ingredient on psychophysiological parameters to improve skin health and well-being.
Keywords:
sensitive skin
; cortisol
; dehydroepiandrosterone
; saliva
; emotion
1. Introduction
Sensitive skin, which affects more than 50% of the world’s population is characterized by the International Forum for the Study of Itch (IFSI) as unpleasant sensations (stinging, burning, pain, pruritus, and tingling sensations) in response to stimuli that normally do not provoke such sensations [1]. Pathophysiological mechanisms are still being debated, such as the role of stratum corneum, transient receptor potential (TRP) channels, activation of TRP sensory proteins via triggering factors inducing the release of neurotransmitters, such as vasoactive intestinal polypeptide (VIP) and calcitonin gene-related polypeptide (CGRP), triggering vasodilatation and cell degranulation, and hyperreaction of the skin blood vessels [2]. Besides these hypotheses, role of neurotransmitters, endocrine factors, neuropeptides, and cytokines released from nerve endings seem to have a central role in the skin homeostasis [3]. Indeed, these molecules communicate amongst themselves and with corresponding systems in the dermis and hypodermis to secure the basic epidermal functions. Alterations in the functions in the cellular, molecular, and neural component systems may induce cutaneous pathologies such as psoriasis and atopic dermatitis. Among neuropeptides, kappa opioid receptor (KOR) has been extensively linked to numerous conditions such as anxiety, and depression [4]. Within the central nervous system, KOR plays a role in appropriate stress, which lead to negatives emotions, and discomfort in models related to pain and stress [5]. Opioid system is also present and functional in human skin. Indeed, the expression of δ-opioid (DOR), μ-opioid (MOR), and κ-opioid receptors (KOR) on mRNA level has been shown in human epidermal keratinocytes, fibroblasts, and melanocytes [6].
Simultaneously, there are psychological elements like stress and negative emotions that could potentially play a role in the emergence of itching sensations, a prevalent symptom among individuals with sensitive skin [7]. It has also been reported that individuals with sensitive skin are more likely to suffer from other skin conditions, inducing deleterious effects on psychological health, quality of life, and increasing chronic stress [8]. Physiological responses to stress are controlled by the autonomic nervous system (ANS) and the Hypothalamic-Pituitary-Adrenal (HPA) axis, inducing a secretion of corticotropin-releasing hormone (CRH), which induces the release of adrenocorticotropic hormone (ACTH) from the pituitary gland, culminating with the release of cortisol, a glucocorticoid, from the adrenal glands [9]. Cortisol release has systemic and cutaneous effects [10]. Secretion of cortisol follows a diurnal pattern, with lowest concentrations noted at bedtime, and highest in the early morning hours [11]. Among various aspects of the diurnal cycle that can been studied to investigate potential dysfunction in the HPA axis, is the cortisol awakening response (CAR), which represents a sharp increase in cortisol levels over the first 30-45 min following morning awakening [12], knowing that CAR is a distinct feature of the diurnal cycle [13]. It has been introduced as a measure of the HPA axis reactivity to awakening stress, possibly reflecting modifications in the HPA axis activity after stressors [14,15]. Area under the curve (AUC) is another measure to assess the overall secretion of homones over a specific time period [16]. Moreover, it has been shown a positive correlation between DHEA and mood, suggesting an opposite effect of this steroid as compared to cortisol, higher evening DHEA was significantly associated with lower negative mood in the morning, and lower anxiety, respectively [17]. DHEA concentration follows a similar circadian rhythm as cortisol, gradually declining as the day progresses. However, unlike cortisol, the post-awakening response is characterized by reduced DHEA concentration, as opposed to cortisol [18]. Salivary DHEA level and the cortisol/DHEA ratio in the morning are indicators of psychological and health status [19]. Both DHEA and cortisol can be readily measured in samples of saliva [20]. Salivary cortisol is representative of circulating, physiologically active free cortisol and salivary DHEA correlates very highly (r = 0.9) with blood measures [20]. Thus, assessing both salivary cortisol and DHEA appears to be useful to explore perturbations of the HPA axis, DHEA mirrors HPA axis dysfunction, whereas changes in cortisol could signify impairments in either the HPA axis drive and/or of the extra-pituitary regulation [21].
The activation of the HPA axis leads to the release of cortisol and dehydroepiandrosterone (DHEA). Cortisol concentrations decrease progressively throughout the day (Dehydroepiandrosterone (DHEA) is the other main adrenocortical products of secretory activity from the hypothalamic–pituitary–adrenal axis. It is a metabolic intermediate in the pathway for the synthesis of testosterone (nearly 90% of circulating T derives from the metabolism of peripheral precursors from DHEA (Labrie et al., 1997), estrone and estradiol (Filaire et al., 1998). It is the most abundant steroid hormones in women (and in men), with adult levels declining progressively by some two-thirds between the ages of 30 and 70 (Filaire et al., 1998). DHEA concentration presents a similar circadian rhythm as cortisol, with a progressive decreasing slope throughout the day, whereas postawakening response is characterized by reduced DHEA concentration, as opposed to cortisol (Wilcox et al., 2014).Given the widespread occurrence of sensitive skin worldwide, its management presents a promising focus for the inclusion of active ingredients in cosmetic products. A range of naturally derived complex blends, including botanical extracts, have been employed for this purpose. These include non-pharmacological methods, such as of moisturizers, specific bathing practices, and emollient devices. Additionally, topical pharmacotherapies like corticosteroids, wet wraps, and calcineurin inhibitors are commonly employed. Systemic immunosuppressants, including IL-32 gamma modulation, inhibition of the Janus kinase pathway, and spleen tyrosine kinase inhibitors, are also considered. Furthermore, non-pharmacological recommendations include treatments like ultraviolet light therapy and interventions involving prebiotic and probiotic approaches, as well as the utilization of botanical and biotechnological extracts (Filaire et al., 2023).
Concerning botanical extracts, polyunsaturated fatty acids (PUFAs), sterols, triterpenes among other natural molecules included in these extracts are recommended for soothing sensitive skin. More precisely, pentacyclic triterperne such as β-amyrin and α-amyrin exhibit various pharmacological effects, including immune-boosting, anti-inflammatory, antioxidant, anxiolytic and antidepressant properties [22]. Botanic oils, including oleic acid 18:1 (n-9) and linoleic acid 18:2 (n-6), used in topical skin have also been reported to have various effects, such as supporting the skin-barrier, promoting the rejuvenation of damage tissues [23]. Tomatoes (Solanum lycopersicum L., formerly Lycopersicon esculentum Mill.) are one of the most readily accessible vegetables worldwide. Its conversion into puree, juices, sauces, and dried powders results in a substantial volume of byproducts including seeds and wax [24]. α-amyrin and β-amyrin accumulate to relatively high concentrations in the lipid mixture coating the tomato [25]. Seeds are loaded with phenolic compounds, which have several bioactivities, such as antimicrobial and anti-inflammatory properties [24]. Consequently, the aim of this study was directed towards exploring the protective effect of a Tomato Peel and Seed Extract (TPSE) for its soothing function but also for its capacity to modulate the adrenal axis. For that ex vivo and in vivo studies were used. The aim of the ex vivo study was to evaluate the effectiveness of TPSE to modulate some main key markers implicated in the sensitive skin. In vivo study combined a clinical evaluation of skin homogeneity, psychological parameters as well as analysis of saliva samples regarding the concentration of cortisol and DHEA before and after 28 days of applying a TPSE face cream.
2. Materials and Methods
2.1. Active Ingredients
A mixture of Solanum lycopersicum L. wax and seed extracts from organic quality (INTELLIGENE® Defense batch number: ID121/001/A22) was supplied by PHENIX EN PROVENCE company (Tarascon, France). The extract composition was depleted in lycopene and enriched in amyrins content. Such composition was obtained using supercritical CO2 green extraction, high temperatures (45-60°C) and high pression (250-700 bars). Extractions were carried out separately for tomato peels and seeds. Then, a specific mixture of tomato seed oil and tomato peel wax was made to obtain a standardized composition in total amyrin content between 1.0 and 2.5%, linoleic acid (C18:2) content between 55-60% and oleic acid (C18:1) between 15 and 20% and lycopene < 0.1% (w/w). The total amyrin distribution showed that they consist 66% of δ-amyrin, 18 % of β-amyrin and 16% α-amyrin. The characterization of amyrins was determined by gas chromatography mass spectrometry (GC-MS) according to Bauer et al. [25]. Fatty acids and lycopene contents were determined by HPLC according to Gleize et al. [26].
2.2. Ex Vivo Experimentation
Experimentation was carried out on skin explants, a full-thickness skin biopsy embedded in a solid and nourishing matrix while its epidermal surface was left in contact with air. The aim of this investigation was to evaluate the effectiveness of TPSE at 0.5% (5 mg.ml-1) formulated in a vehicle on Calcitonin Gene-Related Peptide (CGRP), IL-10 release, Kappa Opioid Receptor (KOR) and Caspase 14 expression.
Nine explants of approximately 10 mm (± 1 mm) in diameter were prepared from an abdominoplasty procedure of a 36-year-old, phototype III, female donor. Explants were divided into batches and each experimental batch consisted of three replicates (n=3).
Three conditions were carried out as follows:
- -
- Untreated skin explants
- -
- Skin explant + vehicle 100%
- -
- Skin explant + vehicle (99.5%) + TPSE at 0.5%
Composition of the product
The composition of the vehicle was: 100% Caprylic/Capric Triglycerides (Dullberg Konzentra, Hamburg, Germany).
Every day, 5μl of vehicle or vehicle + TPSE 0.5% were applied on human skin explant. Skins were growing for 7 days. The specific medium was removed every two days.
Quantification of protein amount by ELISA
After 7 days of treatment, the supernatants were collected. The quantity of total proteins on each supernatant was measured by BCA test. This quantity allowed us to standardize each sample. IL-10 and CGRP were quantified by ELISA (pg.ml-1 reported to 1 mg of protein). The average amount of protein (pg.mg-1 of protein) ± standard error (SEM) was presented (four measures per sample).
Quantification of protein expression by Immunofluorescence
After 7 days of treatment, the skins were fixed and included in paraffin. The samples were microtome sectioned before stained. Caspase 14 and KOR were detected by immunofluorescence. The staining was observed with Zeiss Axio green fluorescence microscope. The average of protein fluorescence intensity (AU) ± SEM was presented (10 measures per sample).
2.3. In Vivo Trials
This single center randomized double-blinded interventional face study controlled clinical trial adhered to the principles of Good Clinical Practices and the declaration of Helsinki. According to local and European regulatory guidelines (Official Journal of EU of 10 March 2010 paragraph 1.2.9), this type of trial testing marketed cosmetics does not require approval from local ethics committees.
Population
Thirty-five female subjects, all aged between 40 and 65 years, were recruited according to skin types I to IV. Subjects received information regarding the aim and procedures of the experiment, and they gave their written informed consent to their participation. All were healthy and declared that they were neither pregnant nor breastfeeding at the time of the study. They were separated into two groups: Group A and Group B. The major difference in the treatment was that Group A was treated with placebo, whereas the Group B was treated with TPSE. Subjects with any facial skin diseases (i.e., acne, allergic dermatitis, glucocorticoid-dependent dermatitis, rosacea, infections), systemic disorders and ongoing pharmacological treatment were excluded. Exclusion criteria also included acute and/or chronic inflammation or infection of facial skin, exposure to sunlight or artificial UV rays within 15 days of the experiment. Pregnant women and women having benefited from facial injection were advised to avoid the application of other similar products during the whole study, which lasted 28 days.
Stinging Test
To determine if the volunteer had sensitive skin, a stinging test as described by Frosch and Duhring [27], was performed during the inclusion visit (D-7) by the dermatologist who recorded the subject’s sensations.
To induce a burning or stinging sensation, subjects were stimulated on both nasolabial folds with a 10% lactic acid aqueous solution for at least 2 min until a stinging sensation was triggered. The side of application of lactic acid was defined by randomization. The test was blinded for subjects. The intensity of the self-declared sensation of discomfort including stinging, tingling, itching, tightening, burning, or pain on the site of application was estimated using a 4-point scale (0 = none, 1 = mild, 2 = moderate, and 3 = severe). The cumulative scores at 2.5 and 5 min on the lactic acid side ≥ 3 were considered positive to the test and included in the experimentation. The stinging test was carried out under a controlled temperature and relative humidity (temperature: 21 ± 1 °C, hygrometry: 45 ± 5%). At D28, the same procedure was administered.
Test formulations
A face cream containing 0.5% of TPSE and a face cream placebo, which the following components, have been used:
Aqua, cyclopentasiloxane, isopropyl miristate, cetyl alcohol, glyceryl stereate, PEG-100 stereate/phenoxyethanol, dimethiconol, chlophenesin carbomer, sodium hydroxide. In addition to these, the “active”cream also contains 0.5% of TPSE.
Both study formulas were identical in appearance and supplied with two identical pump dispensers.
Treatment and evaluation visits
The TPSE face cream or the placebo face cream was applied twice daily to the face for 28 days. Each dose applied was 0.4 g. Dermatological evaluations were conducted at D28.
Evaluation by clinical scoring of erythrosis/rosacea
Erythema was assessed using standardized three-dimensional images taken with the LifeViz® Micro stereophotogrammetric 3D imaging system (Quantificare, Sophia Antipolis, France), and analyzed using MySkin® software.
The redness obtained depends on the subcutaneous capillaries. A scoring of the intensity and extent of erythematous areas (0 to 4) was performed on the face as follow: 0: absence; 1: light; 2: moderate; 3: significant; 4: severe.
Clinical evaluation of skin homogeneity
To assess skin homogeneity, the dermatologist uses scores from 0 to 4 (score 0 corresponding to homogeneous skin and 4 corresponding to a very heterogeneous complexion with the presence of pigment spots).
Evaluation of dermatological tolerance of the 2 tested products
On D28, a clinical assessment of the condition of the skin in the treated area including erythema, edema, eczema (vesicles, edema, pruritus, desquamation) was carried out by indicating its nature, its intensity on a scale of 0 to 3 (0: absent; 1: mild; 2: moderate; 3: severe), the moment of its appearance in relation to the application of the product, its duration and the need for dermatological treatment. The medical examination made it possible to specify the possible occurrence of adverse events (AE) during the duration of the study. In the event of an undesirable event, a clinical investigation was carried out by the dermatologist.
Saliva sampling
The participants received the saliva sampling materials along with both spoken and written instructions. Saliva samples were collected in an adapted tube (Sarstedt, distributed by Salimetrics, Inc., State College, PA, USA) using a passive drool system. Briefly, participants were instructed to allow saliva to pool in the mouth and deposit it into 1.6 ml vials at three time points in the diurnal cycle at D0 and D28: at awakening, 40 min after waking, and at 19:00.
In between the two saliva collections (at awakening and 40 min after waking), participants were provided the opportunity to use the bathroom and may have gently moved around the room. To avoid contamination of saliva, participants were instructed to refrain from eating (particularly high protein foods), drinking beverages (other than water) containing alcohol, caffeine, or fruit juice, brushing teeth, flossing teeth, or engaging in activities that may involve placement of items in the mouth (i.e., smoking, chewing gum). It was also instructed not to be engaged in physical exercise during the post-awakening period (Stalder et al., 2016). Participants recorded the precise data and time of each saliva collection session on a Salivette® timesheet log.
The day after the last saliva collection, participants brought their samples to the laboratory in a small cooler bag. Samples were stored at -25°C until analysis.
The saliva volume was estimated by weighing to the nearest milligram, and the saliva density was assumed to be 1.0 ml-1 [28]. Each sample was frozen, thawed, and centrifuged at 1500g for 15 min to separate mucins. The supernatant was retrieved for measuring level of salivary cortisol (sCort), and sDHEA concentrations). Enzyme-linked immunosorbent assays (ELISA) were used for the saliva cortisol and DHEA analyses according to the manufacturer’s instruction (Salimetrics, Inc., State College, PA, USA) with a lower limit of sensitivity at 0.19 nmol.l−1 and 17.4 pmol.l−1, respectively.
All samples were tested in duplicate in the same series to avoid any variations between tests. The intra-assay maximum coefficient of variation was 2.6% for DHEA, and 2.6% for cortisol. The inter-assay maximum coefficient of variation was 2.7% for DHEA, and 7.4% for cortisol.
All samples were analysed according to a blind procedure and were decoded only after analyses were completed. All analyses were controlled for contraceptive use (“no” coded as “0” and “yes” coded as “1”).
Psychological parameter
Emotion and pleasure were characterized using the Brief Mood Introspection Scale (BMIS) originally developed by Mayer and Gaschke [29] and translated into French by Niedenthal and Dalle [30], which is widely used in psychological studies to assess pleasant emotions. This questionnaire is based on a set of sixteen emotional adjectives with four values ranging from "not at all" to "completely".
2.4. Statistical Analysis
IBM SPSS Statistics version 24.0 software package (SPSS Inc., Chicago, IL, USA) was used to analyze the data. Ex vivo data, cortisol and DHEA results are expressed as means and standard error of mean (SEM). Psychological characteristics are expressed as means and standard deviations (SD). Before analysis, the normality of the data distribution was checked using the Kolmogorov-Smirnov test. Nonparametric tests were used because of the absence of a Gaussian distribution. Considering ex vivo data, the non-parametric Kruskall wallis test following by Dunn’s test were used.
Considering in vivo data, non-parametric tests were used due to the absence of a Gaussian distribution. Data were analyzed by Wilcoxon signed-rank test or Wilcoxon signed-rank sum test.
Changes in DHEA (DHEA awakening response, DAR) and cortisol (cortisol awakening response, CAR) after awakening indexed as delta score between concentrations immediately after awakening and concentrations 40 min after awakening were measured thus yielding the relative change in concentrations from awakening to 40 min post awakening. The area under the total response curve with respect to the ground (AUC), using the trapezoid formula specified in Pruessner et al. (2003) was calculated for DHEA (AUC DHEA) and cortisol (AUC Cort). p-value ≤ 0.05 was statistically significant.
3. Results
3.1. Ex Vivo Experimentation
Effect on TPSE on CGRP release
Exposition to TPSE 0.5% significantly decreased CGRP release when compared to untreated sample (-53%; p < 0.01) or vehicle (Placebo) (-22%; p < 0.05), Figure 1).
Effect on TPSE on IL-10 release
The level of IL-10 significantly decreased in vehicle treated skin compared to untreated sample (-7.7%; p < 0.001) (Figure 2). There was also a significant difference between vehicle and TPSE treated skin (12.5%; p < 0.05), values noted after TPSE treated skin being higher.
Effect on TPSE on Caspase 14 expression
TPSE treated skin induced a significantly higher increase in the Caspase 14 expression (+ 27%; p < 0.005) compared to untreated skin and placebo treatment (+ 23%; p < 0.001).
Effect on TPSE on KOR expression
KOR expression significantly decreased with vehicle treatment compared to untreated (-16%; p < 0.01) (Figure 3). TPSE treatment also significantly decreased KOR expression compared to vehicle (-8.4%; p < 0.05).
3.2. Clinical Evaluation
No adverse events were reported during the study. All subjects have sensitive skin with a stinging test greater than 3 and dry skin.
Evaluation of intensity and extent of erythrosis/rosacea using the MySkin® report software
At D0, we noted that Group A (Group Placebo) and Group B (Group TPSE) presented the same erythrosis intensity and extent values. At D28, Group B presented a significant lower intensity value as compared to Group A (-50.6%; p < 0.05) (Table 1).
28 days of daily application of the TPSE induced a significant decrease in the intensity (-29%; p < 0.001) and extent of erythrosis (-14%; p < 0.01). This was not the case for the placebo application (Group A) (Table 1).
Clinical evaluation of skin homogeneity
At D0, values reflecting the level of skin homogeneity was similar in both groups. A significant decrease of 18% (p < 0.001) in clinical scoring, demonstrating an improvement of skin homogeneity, was observed after 28 days of TPSE 0.5% application. It was not the case with the placebo application (Table 2).
Score 0 corresponding to homogeneous skin and 4 corresponding to a very heterogeneous complexion with the presence of pigment spots.
Salivary parameters
All data were in the normal range within the study for both groups [31]. Salivary flow rates did not change significantly over time. Values were between 0.531 and 0.540 mL.min-1. They presented a pronounced diurnal rhythm in accordance with the results of Seeman and Robbins [32]. Indeed, saliva cortisol concentrations increased after awakening and then progressively decreased towards the evening. Table 3 shows the results of CAR, DAR and AUC for cortisol and DHEA (means and SEM) obtained at D0 and D28 for the Group A (Placebo) and the Group B (TPSE). There were no significant differences between groups for the salivary parameters at D0.
Concerning the Group A, no significant difference was observed between D0 and D28 for AUC Cort, the measure of the total cortisol output in a day. However, CAR was significantly higher at D28 as compared to D0 (+10%, p < 0.05). At the same time, 28 days of placebo application induced a significant decrease in AUC DHEA (-9%, p < 0.01), whereas no significant difference was observed between D0 and D28 for DAR.
Concerning the Group B, while 28 days of TPSE application induced a significant decrease in AUC Cort, (-9.7%, p < 0.001), and CAR (-14.6%, p < 0.001), there was a significant increase in DAR (+100%, p < 0.001).
It is important to note that cortisol output in a day at D28 was significantly higher (+10%) in the Group A as compared to the AUC cortisol noted in the Group B (p < 0.01) (Table 3). The same observation was noted for CAR. As noted in the Table 3, using TPSE for 28 days induced a significant increase in the DHEA Awakening Response and DHEA secretion output as compared to the use of placebo.
Psychological parameter
The Brief Mood Introspection Scale (BMIS) used in this investigation; is based on eight dimensions: happiness, affection, calm, energy, fear, anger, fatigue and sadness, and specifically measures the mood changes of the subjects. Four subscores can be computed from the BMIS: pleasant–unpleasant, arousal–calm, positive–tired and negative–relaxed mood. We only focused on the subscores pleasant–unpleasant. At the beginning of the study, no differences in pleasant emotions evaluated by the BMIS were noted between the two groups (Table 4). Twenty-eight days of daily application of TPSE induced significant increase (+11%, p < 0.01) in positive emotions. This was not the case for the placebo application. At D28, pleasant emotions were significantly higher (+18%, p < 0.05) in Group B (TPSE) compared to Group A (Placebo).
4. Discussion
Skin conditions, such as sensitive skin, are linked to psychological symptoms, inducing anxiety and stress, and lower quality of life of affected people [33,34]. The increase in glucocorticoids linked to stress is well documented [9]. Other endogenous stress hormones, including DHEA, have the potential to modulate stress reactivity and contribute to the promotion of psychological well-being. Knowing that cosmetics usage has the capability to have some impact on self-perceived attractiveness and self-esteem [35]. The objective of this study was to evaluate the protective effects of TPSE for its soothing function but also for it potential to modulate adrenal axis. To our knowledge, this is the first study that has concomitantly assessed these two topics. The main results of this study were TPSE regulated release of CGRP (Figure 1), involved in the pathophysiology of sensitive skin reflecting an anti-neurogenic and anti-inflammatory properties. CGRP has been identified as a significant contributor to the mechanisms of skin reactivity (i.e., atopic dermatitis, psoriasis), these skin disturbances having emotional repercussions [36]. It plays a role in neurogenic inflammation, which can lead to uncomfortable sensations ranging from stinging, itching to severe pain. Uncomfortable sensations could likely be associated with negative emotions, and it has been shown a correlation between CGRP expression in the skin and anxiety on patients with atopic dermatitis [36]. This investigation also highlighted that cosmetic ingredients could modulate KORs (Figure 3), implicated in a variety of disturbances, such as anxiety. The opioidergic system, a complex intercellular signaling system composed of endogenous receptors and ligands, includes opioid receptors such as μ- (MOR), κ- (KOR), and δ- (DOR) receptors, all belonging to the G-protein-coupled 7-TM receptor superfamily. They are expressed in various cell types of healthy human skin [37]. In the central nervous system, KORs play a role in maladaptive stress responses, leading to negative emotions. This effect is believed to be linked to KORs located on monoaminergic neurons and axon terminals, which inhibit the release of neurotransmitters including dopamine and serotine [5].
Today, improving the appearance of the skin is no longer the only concern of cosmetic users: market analysis and consumer expectations show that the sensations provided by the application of a cosmetic product in the short and long term are now as important as the result obtained [38]. In this way, it has been shown that application of eyeshadows creating contrasting color stimulated emotional response [39]. Gervason et al. [40] reported a link between cosmetic application and mood in a population of women aged 60 and over. Emotional benefit of cosmetic camouflage in the treatment of facial conditions (i.e., acne, rosacea) were also reported [41]. Thus, data from the literature reported that it is possible to measure a positive change in mood profile, as well as a reduction in anxiety, after using a specific cosmetic product [42]. Application of a formulation with the active compound, including amyrins from a mixture of tomato seed oil and tomato peel wax, caused a considerable trend toward a significantly more positive mood, compared to the placebo (Table 4), our results being in line with the previous studies. The pharmacological effectiveness of α- and/or β-amyrin, which are representative members of triterpenoids, have been reported, particularly on mood via mechanisms involving GABA and monoamine oxidase. Thus, we showed that TPSE has a psychophysiological effect, namely a stress-relieving effect, that can be achieved if it is used in cosmetic products, particularly in face creams. These properties, in addition to other characteristics, make TPSE a promising cosmetic ingredient.
It is also widely acknowledged that emotional states can trigger a neuroendocrine response involving the release of salivary cortisol and DHEA. Hence, the hypothalamic–pituitary–adrenal (HPA) axis responds to a variety of physiological and psychological challenges that may or may not give rise to negative affect [15]. Nevertheless, while several reviews have explored the link between cortisol and mood, Joseph et al. [43] pointed out that there isn’t a singular theoretical framework or model that comprehensively elucidates the relationship between negative emotions and cortisol, as well as the link between, positive emotions and cortisol. The relationship between salivary cortisol and DHEA, emotions, and cosmetics has received limited attention. During an experimental study involving 35 women inhaling saffron volatile oil aroma for twenty minutes, a decrease in both cortisol concentrations and anxiety was observed [44]. Watanabe et al. [45] established an association between mood states, salivary cortisol, and essential oil aromatherapy in a group of 41 healthy female participants. More recently, Cabannes et al. [35] reported that the evaluation of cortisol at the same time as that psychological indicators provides a scientific approach to examine the benefit of a cosmetic product. Our investigation is in line with these previous studies, showing subjects having a low overall output of salivary cortisol after 28 days of TPSE applications when compared to placebo applications (Table 3). At the same time, we noted an increase in pleasant emotions through the BMIS score after 28 days of TPSE application. Significant rise of DHEA activity is also reported in this period (Table 3). Studies have already shown that cosmetics can positively impact well-being and mood, the assessment having been carried out using interviews. Although our study has some limitations such as the restricted number of salivary samples collected within a relatively short timeframe and the absence of assessment for other non-invasive psychological parameters, our investigation confirms that the impact of cosmetic active ingredients extends beyond solely influencing the pathophysiology of sensitive skin.
5. Conclusions
Using a non-invasive psychophysiological approach which can be employed to evaluate the effect of cosmetic active ingredients, we noted that co-products of tomato (TPSE) improve sensitive skin condition and have a significant impact on emotional well-being. This approach was done through the assessment of salivary parameters that reflect the HPA axis and psychological evaluation. To our knowledge, this is the first study in the cosmetic field which has used this type of approach, which is useful for sensitive skin, knowing that this physiopathology is due to both physiological problems with main psychological repercussions.
Author Contributions
L.F. did the methodology. E.F. did the data analysis and wrote the manuscript. R.N.-K. participated in the methodology. All authors have read and agreed to the published version of the manuscript.
Funding
No findings.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author. The data are not publicly available due to privacy restrictions.
Acknowledgments
The authors would like to thank all subjects participating in the study.
Conflicts of Interest
All authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Misery, L.; Ständer, S.; Szepietowski, J.C. Definition of Sensitive Skin: An Expert Position Paper from the Special Interest Group on Sensitive Skin of the International Forum for the Study of Itch. Acta Derm. Venereol. 2017, 97, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Filaire, E.; Vialleix, C.; Cadoret, J.P.; Guénard, S.; Muller, C.; Dreux-Zhiga, A.; Berthon, J.Y. Characterization of reactive sensitive skin microbiota: effect of Halymenia durvillei (HD) extract treatment. Cosmetics. 2019, 6, 69. [Google Scholar] [CrossRef]
- Slominski, A.T.; Slominski, R.M.; Raman, C.; Chen, J.Y.; Athar, M.; Elmets, C. Neuroendocrine signaling in the skin with a special focus on the epidermal neuropeptides. A. J. Physiol. Cell Physiol. 2022, 1, C1757–C1776. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; Pickens, S.; Burma, N.E.; Ibarra-Lecue, I.; Yang, H.; Xue, L.; Cook, C.; Hakimian, J.K.; Severino, A.L.; Lueptow, L.; et al. Kappa Opioid Receptors Drive a Tonic Aversive Component of Chronic Pain. J. Neurosci. 2019, 39, 4162–4178. [Google Scholar] [CrossRef] [PubMed]
- West, A.M.; Holleran, K.M.; Jones, S.R. Kappa Opioid Receptors Reduce Serotonin Uptake and Escitalopram Efficacy in the Mouse Substantia Nigra Pars Reticulata. Int. J. Mol. Sci. 2023, 24, 2080. [Google Scholar] [CrossRef]
- Kauser, S.; Thody, A.J.; Schallreuter, K.U.; Gummer, C.L.; Tobin, D.J. Beta-endorphin as a regulator of human hair follicule melanocyte biology. J Invest Dermatol. 2004, 123, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Sanders, K.M.; Akiyama, T. The vicious cycle of itch and anxiety. Neurosci. Biobehav. Rev. 2018, 87, 17–26. [Google Scholar] [CrossRef]
- Farage, M.A. Psychological aspects of sensitive skin: a vicious cycle. Cosmetics. 2022, 9, 78. [Google Scholar] [CrossRef]
- Hua, J.; Le Scanff, C.; Larue, J.; José, F.; Martin, J.C.; Devillers, L.; Filaire, E. Global stress response during a social response during a social stress test: impact of alexithymia and its subfactors. Psychoneuroendocrinology. 2014, 50, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Graubard, R.; Perez-Sanchez, A.; Katta, R. Stress and skin: an overview of mind body therapies as a treatment strategy in dermatology. Dermatol. Pract. Concept. 2021, 11, e2021091. [Google Scholar] [CrossRef]
- Filaire, E.; Duche, P.; Lac, G.; Robert, A. Saliva cortisol, physical exercise and training: influences of swimming and handball on cortisol concentrations in women. Eur. J. Appl. Physiol. Occup. Physiol. 1996, 74, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Pruessner, J.C.; Wolf, O.T.; Hellhammer, D.H.; Buske-Kirschbaum, A.; von Auer, K.; Jobst, S.; Kaspers, F.; Kirschbaum, C. Free cortisol levels after awakening: a reliable biological marker for the assessment of adrenocortical activity. Life Sci. 1997, 61, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Clow, A.; Hucklebridge, F.; Thorn, L. The cortisol awakening response in context. Int. Rev. Neurobiol. 2010, 93, 153–175. [Google Scholar] [CrossRef]
- Durguerian, A.; Filaire, E.; Drogou, C.; Sauvet, F.; Bougard, C.; Chennaoui, M. Hyperactivity of the Sympatho-Adrenomedullary System Without Any Modification of the Hypothalamic-Pituitary-Adrenal Axis After Food Restriction Among High-Level Weightlifters. Strength Cond. Res. 2018, 32, 1643–1655. [Google Scholar] [CrossRef]
- Filaire, E.; Ferreira, J.P.; Oliveira, M.; Massart, A. Diurnal patterns of salivary alpha-amylase and cortisol secretion in female adolescent tennis players after 16 weeks of training. Psychoneuroendocrinology. 2013, 38, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Pruessner, J.C.; Kirschbaum, C.; Meinlschmid, G.; Hellhammer, D.H. Two formulas for computation of the area under the curve represent measures of total hormone concentration versus time-dependent change. Psychoneuroendocrinology. 2003, 28, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Van Niekerk, J.K.; Huppert, F.A.; Herbert, J. Salivary cortisol and DHEA: association with measures of cognition and well-being in normal older men, and effects of three months of DHEA supplementation. Psychoneuroendocrinology. 2001, 26, 591–612. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, R.R.; Granger, D.A.; Szanton, S.; Clark, F. Diurnal patterns and associations among salivary cortisol, DHEA and alpha-amylase in older adults. Physiol. Behav. 2014, 129, 11–16. [Google Scholar] [CrossRef]
- Hasegawa-Ohira, M.; Suguri, K.; Nomura, S. The Dehydroepiandrosterone Awakening Response as a Possible Index of Subjective Sleep Quality. Adv. Biomed. Engineering. 2016, 5, 132–136. [Google Scholar] [CrossRef]
- Lac, G. Saliva assays in clinical and research biology. Pathol. Biol. 2001, 49, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Oskis, A.; Clow, A.; Thorn, L.; Loveday, C.; Hucklebridge, F. Differences between diurnal patterns of salivary cortisol and dehydroepiandrosterone in healthy female adolescents. Stress. 2012, 15, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Viet, T.D.; Xuan, T.D.; Anh, L.H. 2. α-Amyrin and β-Amyrin Isolated from Celastrus hindsii Leaves and Their Antioxidant, Anti-Xanthine Oxidase, and Anti-Tyrosinase Potentials. Molecules. 2021, 26, 248. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.M.; Wagner, C.; Komarnytsky, S. The Enigma of Bioactivity and Toxicity of Botanical Oils for Skin Care. Front. Pharmacol. Sec. Ethnopharmacol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Tomar, M.; Bhuyan, D.J.; Punia, S.; Grasso, S. Tomato (Solanum lycopersicum L.) seed: A review on bioactives and biomedical activities. Biomed. Pharmacother. 2021, 142, 112018. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Schulte, E.; Their, H. Composition of the surface wax from tomatoes: II. Quantification of the components at the ripe red stage and during ripening. Eur. Food Res. Technol. 2004; 219, 487–491. [Google Scholar] [CrossRef]
- Gleize, B.; Steib, M.; André, E.; Reboul, E. Simple and fast HPLC method for simultaneous determination of retinol, tocopherols, coenzyme Q10 and carotenoids in complex samples. Food Chemistry. 2012, 134, 2560–2564. [Google Scholar] [CrossRef] [PubMed]
- Frosch, P.J.; Duhring, A.M.K. A method for appraising the stinging capacity of topically applied substances. J. Soc. Cosmet. Chem. 1977, 28, 197–209. [Google Scholar]
- Cole, A.S.; Eastao, J.E. Biochemistry and Oral Biology, 2nd ed.; Wright, London, 1988; p.476.
- Mayer, J.D.; Gaschke, Y.N. The experience and meta-experience of mood. Int. J. Pers. Soc. Psychol. 1988, 55, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Niedenthal, P.; Dalle, N. Emotional response categorization during naturally induced emotions. Eur. J. Soc. Psychol. 2001, 31, 737–742. [Google Scholar] [CrossRef]
- Fiet, J.; Passat, P.; Guechot, J.; Gourmel, B.; Villette J., M.; Cathelineau, G. Interet du dosage du cortisol dans la salive. Nouvelle Presse Medicale. 1981, 10, 2664. [Google Scholar]
- Seeman, T.E.; Robbins, R.J. Aging and hypothalamic-pituitary-adrenal response to challenge in humans. Endocr. Rev. 1994, 15, 233–260. [Google Scholar]
- Manav, V.; Karaali, M.G.; Erdem, O.; Aksu, A.E.K. Association between biophysical properties and anxiety in patients with sensitive skin. Skin Res. Technol. 2022, 28, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, M.M.; Kamen, T.; Desai, S.R. The Psychosocial Burden of Skin Disease and Dermatology Care Insights Among Skin of Color Consumers. Dermatol. 2023, 1, 1027–1033. [Google Scholar] [CrossRef]
- Cabannes, M.; Risselada, C.; Chaisemartin, L.; Pasquet, J.; Couval, E.; Berthon, J.Y.; Filaire, E. Increase in subjective well-being and psychological health after application of C8-silk lipoamino acid functionalized pigments included in a foundation. Int. J. Cosm. Sci. 2019, 41, 489–495. [Google Scholar] [CrossRef]
- Abdelhadi, S.; Nordlind, K.; Johansson, B.; Theodorsson, E.; Holst, M.; Lönndahl, L. Expression of calcitonin gene-related peptide in atopic dermatitis and correlation with distress. Immunopharmacol. Immunotoxicol. 2023, 7, 1–6. [Google Scholar] [CrossRef]
- Bigliardi, P.L.; Dancik, Y.; Neumann, C.; Bigliardi-Qi, M. Opioids and Skin Homeostasis, Regeneration and Ageing-What’s the Evidence? Exp. Dermatol. 2016, 25, 586–591. [Google Scholar] [CrossRef]
- Sattayakhom, A.; Wichit, S.; Koomhin, P. The Effects of Essential Oils on the Nervous System: A Scoping Review. Molecules. 2023, 28, 3771. [Google Scholar] [CrossRef]
- Min-Kyung, K. Brain waves and emotional responses, according to color stimulation of eye shadows using contrast and similar color arrangement. As. J. Beauty Cosmetol. 2018, 16, 509–521. [Google Scholar]
- Gervason, S.; Napoli, M.; Dreux-Zhiga, A.; Lazzarelli, C.; Garcier, A.; Briand, A.; Thepot, A.; Berthon, J.Y.; Filaire, E. Attenuation of negative effects of senescence in human skin using an extract from Sphingomonas hydrophobicum: development of new skin care solution. Int. J. Cosmetic Sci. 2019, 41, 391–397. [Google Scholar] [CrossRef]
- Levy, L.; Emer, J. Emotional benefit of cosmetic camouflage in the treatment of facial skin conditions: personal experience and review. Clin. Cosmet. Investig. Dermatology. 2012, 5, 173–182. [Google Scholar]
- Rizzi, V.; Gubitosa, J.; Fini, P.; Cosma, P. Neurocosmetics in Skincare-the Fascinating World of Skin–Brain Connection: A Review to Explore Ingredients, Commercial Products for Skin Aging, and Cosmetic Regulation. Cosmetics. 2021, 8, 66. [Google Scholar] [CrossRef]
- Joseph, N.T.; Jiang, Y.; Zilioli, S. Momentary emotions and salivary cortisol: a systematic review and meta-analysis of ecological momentary assessment studies. Neurosc. Behav. Rev. 2021, 125, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Toyoshima, K.; Komaki, R. Psychological and neuroendocrinological effects of odour of saffron (Crocus sativus). Phytomed. 2011, 18, 726–730. [Google Scholar] [CrossRef]
- Watanabe, E.; Kuchta, K.; Kimura, M.; Rauwald, H.W.; Kamei, T.; Imanishi, J. Effects of bergamot (Citrus bergamia (Risso) Wright & Arn.) essential oil aromatherapy on mood states, parasympathetic nervous system activity, and salivary cortisol levels in 41 healthy females. Forsch. Komplement. Med. 2015, 22, 43–49. [Google Scholar]
Figure 1.
Release of calcitonin gene-related peptide (CGRP) from skin explants, untreated or treated with topical application of vehicle or TPSE 0.5%, after 7 days of treatment. Data are represented as mean ± SEM. * p < 0.05; ** p < 0.01) (Vehicle versus untreated or TPSE versus Vehicle).
Figure 1.
Release of calcitonin gene-related peptide (CGRP) from skin explants, untreated or treated with topical application of vehicle or TPSE 0.5%, after 7 days of treatment. Data are represented as mean ± SEM. * p < 0.05; ** p < 0.01) (Vehicle versus untreated or TPSE versus Vehicle).
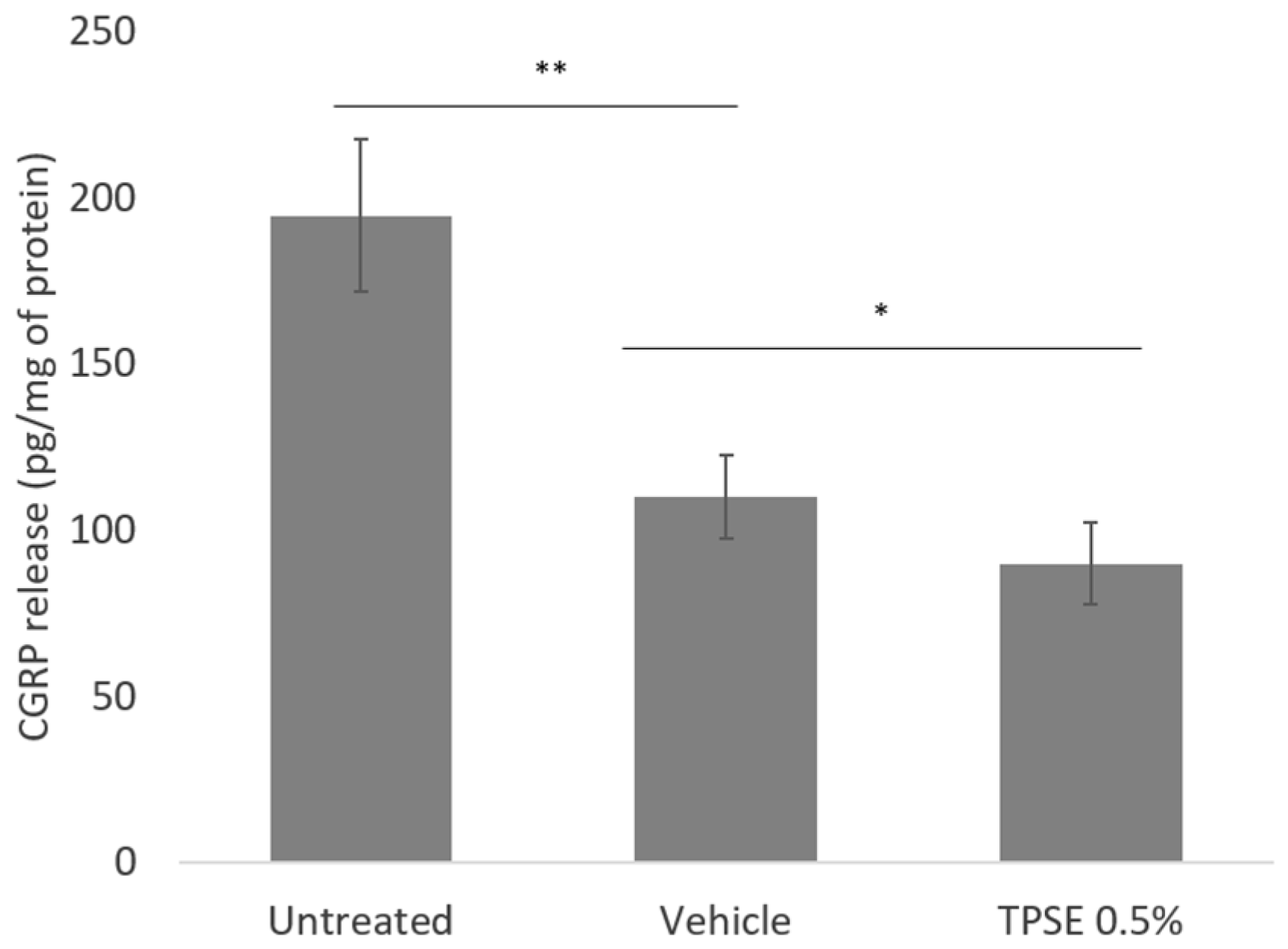
Figure 2.
Release of IL-10 from skin explants, untreated or treated with topical application of vehicle or TPSE 0.5%, after 7 days of treatment. Data are represented as mean ± SEM. *** p < 0.001; * p < 0.05) (Vehicle versus untreated or TPSE versus Vehicle).
Figure 2.
Release of IL-10 from skin explants, untreated or treated with topical application of vehicle or TPSE 0.5%, after 7 days of treatment. Data are represented as mean ± SEM. *** p < 0.001; * p < 0.05) (Vehicle versus untreated or TPSE versus Vehicle).
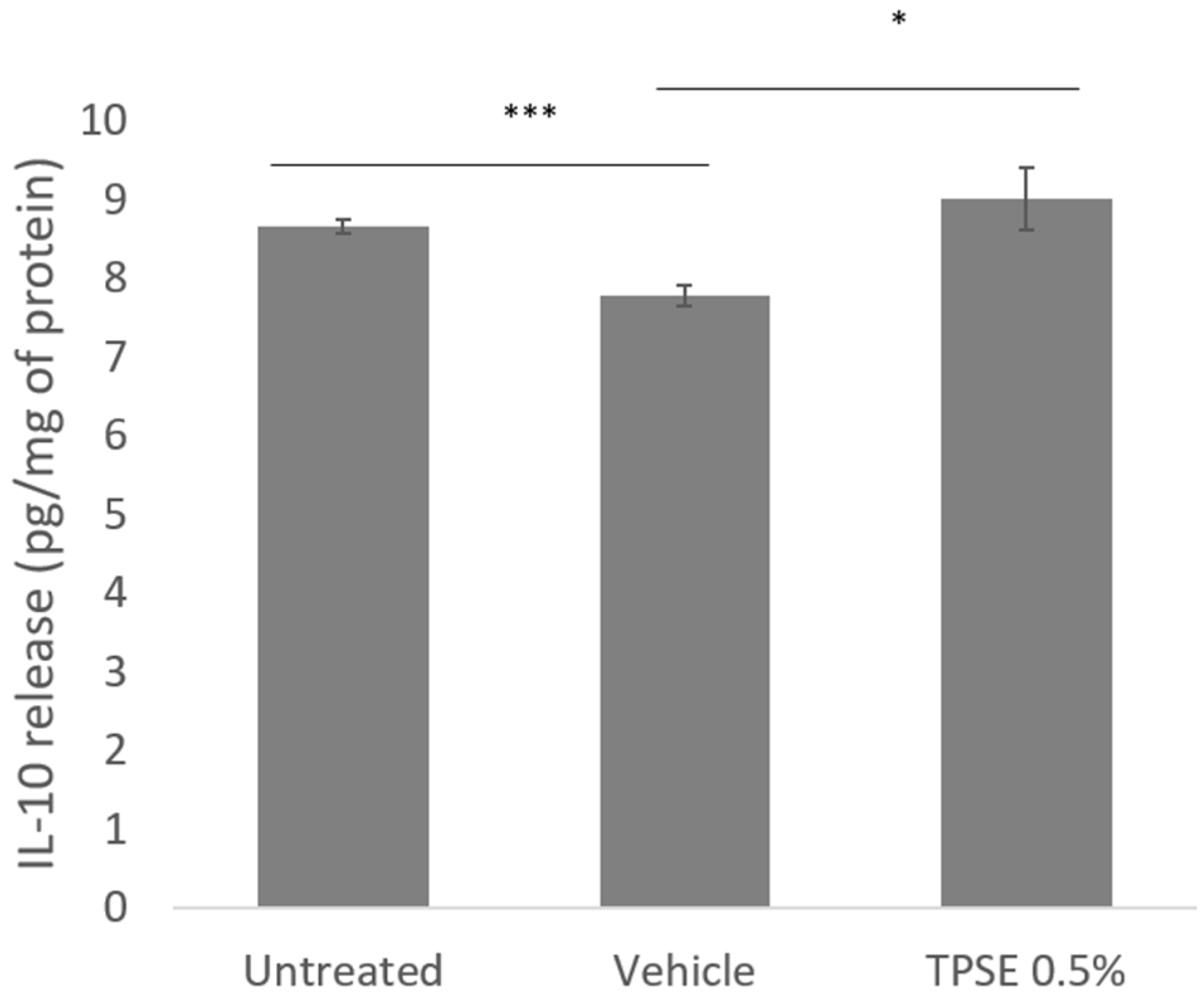
Figure 3.
Quantification of KOR expression in skin explants untreated or treated with topical application of vehicle or TPSE, after 7 days of treatment. Data are represented as mean ± SEM. ** p < 0.01; * p < 0.05) (Vehicle versus untreated or TPSE versus Vehicle).
Figure 3.
Quantification of KOR expression in skin explants untreated or treated with topical application of vehicle or TPSE, after 7 days of treatment. Data are represented as mean ± SEM. ** p < 0.01; * p < 0.05) (Vehicle versus untreated or TPSE versus Vehicle).
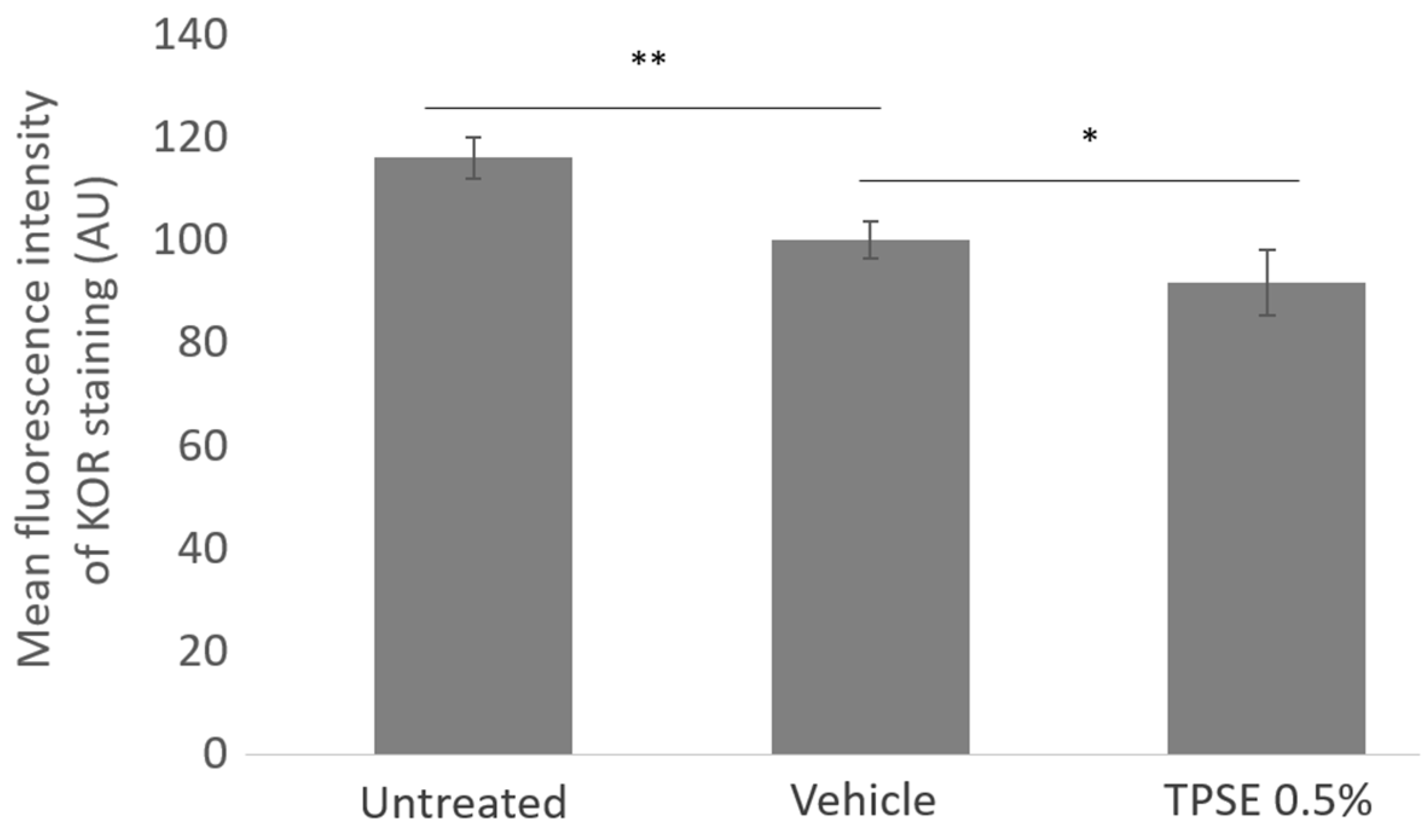

Table 1.
Mean ± SD of erythrosis intensity and extent in Group A (Placebo) and Group B (TPSE) at the beginning of the study (D0) and after 28 days of application (D28). ** p < 0.01; *** p < 0.001 (D28 versus D0).
Table 1.
Mean ± SD of erythrosis intensity and extent in Group A (Placebo) and Group B (TPSE) at the beginning of the study (D0) and after 28 days of application (D28). ** p < 0.01; *** p < 0.001 (D28 versus D0).
| Group A (Placebo) | Group B (TPSE) | |
|---|---|---|
|
Erythrosis Intensity D0 D28 |
2.10 ± 0.97 1.95 ± 1.01 |
1.62 ± 0.77 1.15 ± 0.58*** |
|
Erythrosis extent D0 D28 |
1.65 ± 0.71 1.60 ± 0.70 |
1.60 ± 0.82 1.38 ± 0.77** |
Table 2.
Mean ± SD of skin homogeneity in Group A (Placebo) and Group B (TPSE) at the beginning of the study (D0) and after 28 days of application (D28). ***: p < 0.001 (D28 versus D0).
Table 2.
Mean ± SD of skin homogeneity in Group A (Placebo) and Group B (TPSE) at the beginning of the study (D0) and after 28 days of application (D28). ***: p < 0.001 (D28 versus D0).
| Clinical scoring for skin homogeneity analysis | D0 | D28 |
|---|---|---|
|
Group A (Placebo) Group B (TPSE) |
1.45 ± 1.07 1.36 ± 0.73 |
1.30 ± 0.95 1.12 ± 0.70*** |
Table 3.
Results (means and SEM) on the cortisol and DHEA release obtained at D0 (beginning of protocol), and D28 (end of protocol) in Group A (Placebo) and Group B (TPSE). **: p < 0.01; ***: p < 0.001: Group B versus Group A).
Table 3.
Results (means and SEM) on the cortisol and DHEA release obtained at D0 (beginning of protocol), and D28 (end of protocol) in Group A (Placebo) and Group B (TPSE). **: p < 0.01; ***: p < 0.001: Group B versus Group A).
| Group A (Placebo) | Group B (TPSE) | |
|---|---|---|
| D0 | ||
|
CAR AUC Cort DAR AUC DHEA |
5.95 ± 0.18 38.95 ± 0.85 0.18 ± 0.03 7.82 ± 0.10 |
6.34 ± 0.15 39.04 ± 0.61 0.18 ± 0.01 7.86 ± 0.07 |
| D28 | ||
|
CAR AUC Cort DAR AUC DHEA |
6.56 ± 0.16 39.26 ± 0.79 0.27 ± 0.06 7.58 ± 0.15 |
5.43 ± 0.20*** 35.36 ± 0.62** 0.36 ± 0.04** 8.08 ± 0.09** |
CAR: Cortisol awakening response (nmol-1); DAR: DHEA awakening response (nmol-1); AUC: Area under the curve Cortisol (arbitrary units).
Table 4.
Means (SEM) of BMIS in Group A (Placebo) and Group B (TPSE) at the beginning of the study (D0) and after 28 days of application (D28). **: p < 0.01; (D28 versus D0).
Table 4.
Means (SEM) of BMIS in Group A (Placebo) and Group B (TPSE) at the beginning of the study (D0) and after 28 days of application (D28). **: p < 0.01; (D28 versus D0).
| Group A (Placebo) | Group B (TPSE) | |
|---|---|---|
| D0 D28 |
47.3 ± 2.5 41.4 ± 2.6 |
44.0 ± 1.349.0 ± 1.4** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



