
Submitted:
15 December 2023
Posted:
18 December 2023
You are already at the latest version
Abstract
Glioblastoma (GB), even though not frequent, is an extremely aggressive brain tumor, with a strong impact on patient outcomes, in terms of both length and quality of life. The protocol estab-lished by Stupp and coworkers in 2005, based on radiotherapy and temozolomide chemotherapy, after maximum safe surgical resection, remains the gold standard for GB treatment. However, it is evident nowadays that the extreme GB intratumoral and intertumoral heterogeneity, invasive-ness and recurrence are not compatible with a routinary and unfortunately uneffective treatment. In this review article, we summarize the principal challenging issues in finding new valuable therapies for GB and focus on the impact that extracellular vesicle (EV) research and exploitation can have in the field. EVs are natural particles delimited by a lipidic bilayer and full of functional cellular content, released and uptaken by cells as key means of cell communications. Moreover, being stable in body fluids, well-immunotolerated, able to cross physiological, inter-species, and inter-kingdom barriers, and to target specific cells, releasing inherent or externally loaded functionally active molecules, EVs can be potentially ideal allies in fighting GB and assuring to GB patients a better prognosis. Here we report the main results obtained until now, focusing on both EV sources and molecular cargo used in the different functional studies, mainly in vivo. Finally, we perform a SWOT analysis reporting both the main advantages and the pitfalls of developing EV-based GB therapeutic strategies to highlight the main directions to explore for realizing the promise of EV exploitation for GB treatment.
Keywords:
glioblastoma
; extracellular vesicles
; tumor therapy
; drug-delivery
1. Introduction
Gliomas are the most frequent intracranial tumors in adults. The first reports of gliomas date back to the early 19th century thanks to the British Berns and Abernety, whereas the first comprehensive histomorphological description was made by the German pathologist Rudolf Virchow in 1865. Studying gliomas, Virchow first introduced the concept of “neuroglia”, from which gliomas derive as a connective tissue of the brain and the spinal cord, formed by star-shaped units, interconnected by fine fibers, in which the nervous elements are immersed” [1,2]. The term glioblastoma (GB) was first used in 1927 by the neuropathologist Percival Bailey and the neurosurgeon Harvey Cushing, who developed the first systematic classification and histological description of gliomas, giving the base for the glioma modern one [3]. Since then, glioma classification has been updated several times by the World Health Organisation (WHO), introducing different nomenclature and diagnostic criteria. In the fourth edition of the WHO Classification of CNS Tumors (WHO CNS4), the term “multiforme” was abolished [4]. Moreover, in WHO CNS4 and even more in the fifth and last edition of 2021 (WHO CNS5) besides the traditionally used histological and immunohistochemical features, molecular (genetic and expression data) parameters were also included, establishing a different approach to both CNS tumor nomenclature and grading, and emphasizing the importance of integrated diagnoses [5,6,7].
According to the WHO CNS tumor classification, GB is the highest and most severe prognostic grade, namely “grade IV” glioma, and the most aggressive and lethal among all primary brain tumors, with a median overall survival with no treatment at most 4 months, otherwise 14-17 months after diagnosis and a five-years survival rate of 5-6% [4,5,8,9]. Although considered a rare tumor, with an incidence rate of about 3 per 100,000 people, GB accounted for 14.5% of all CNS tumors and for 48.6% of the malignant ones, based on to the data collected in the central brain tumor registry of the United States (CBTRUS) since 2014 to 2018. [8]. The tumor has a slightly higher frequency in men than in women and a higher incidence in Caucasian than in African or Asian populations. The annual age adjusted incidence of GB increases with age from 0.15 per 100 000 in children to 15.03 per 100,000 in patients over 75, with a median age at diagnosis of 65 years In pediatric age, however, although rare, GB constitutes one of the groups of neoplasms with the worst prognosis [8,10]. Unfortunately, more recent studies indicate a rise in incidence of both GB and brain tumors in general, as highlighted by the Global Cancer Statistics of 2020 [11]. The etiology of GB is unknown with the only identifiable risk factor being exposure to ionizing radiation. Other possible contributing factors include aging, non ionizing radiation, and air pollution [12,13].
GB mostly develops in the cerebral hemispheres and, depending on the brain area affected and due to increased intracranial pressure, patients can show different clinical features, with a wide range of fast-developing and life-changing symptoms. These include neurological symptoms such as severe headache, loss of vision or alteration of the language, persistent weakness, as well as psychological and psychiatric symptoms, compprising unpredictable personality changes, that can severely compromise the quality of life and even the autonomy of patients, with strong impact on patient‘s family and entourage [14,15,16,17]. In most cases, diagnosis occurs only at the onset of symptoms, and is mostly based on magnetic resonance imaging (MRI), with the relative technical limitation in specificity (difficulties to exclude other diagnosis) and sensibility (spatial resolution of 2-3 mm) [18]. Since 2005 the most widely adopted therapy for GB consists of surgical removal of the tumor mass, followed by radiotherapy and chemotherapy, according to the protocol identified by Stupp and coworkers (Stupp et al., 2005). The surgical removal of GB is quite complex due to the high number of cells composing the tumor and their very high infiltration power in the surrounding healthy tissues, both causes of the high recurrence rate, and is strongly dependent on the location and accessibility of the tumor mass. Where possible, extensive surgical therapy is crucial to improve patients’ prognosis and lower the risk of recurrence, even though this might increase the risk of postoperative neurologic deficits [19]. After surgical resection and if this is not possible, patients receive radiotherapy in combination with chemotherapy, of which Temozolomide (TMZ), a DNA alkylating agent, is the chemotherapy of choice, [20].
More recently, alternative therapies, such as extended adjuvant TMZ treatment, use of the monoclonal anti-VEGFA antibody bevacizumab or the receptor tyrosine kinases inhibitor regorafenib, magnetic tumor-treating fields, and immunotherapies have been tested in clinical trials, alone or in combination with already established treatments [21,22,23,24]. Moreover, to clearly identify the most promising interventions, several meta-analysis studies have been comparing the efficacy of the different treatments across different randomized clinical trials, highlighting some encouraging results such as the ability of Bevacizumab in combination with TMZ to slightly increase progression-free survival (but not patient overall survival) serving as a salvage regimen for recurrent GB [25,26,27,28]. Until now, however, no breakthrough therapies leading to extensive and durable survival of patients have been found and survival rates for GB patients have shown no notable improvement in population statistic-based studies [10]. Therefore, there is a striking need for effective and less invasive diagnostic as well as therapeutic tools to early discover GB onset, and, even more, to block GB progression and avoid its recurrence after surgery.
Improving the knowledge of GB onset and progression at molecular, cellular, and systemic level is critical to unravel new key therapeutic targets and implement effective therapeutic strategies. In this scenario the discovery of natural and functional nanoparticles layered by a lipid membrane, called extracellular vesicles or EVs, both released and up-taken by cells in physiological and even more in pathological conditions, such as tumorigenesis, brings with it a strong potential for inspiring new, more effective and less invasive diagnostic and therapeutic approaches for GB patient treatment. In this review, we will focus on the significance of EVs for the development of new promising therapeutic strategies against GB.
2. Main issues in GB therapy
The poor prognosis of GB and the objective difficulties in developing successful therapies is due to the complex and peculiar set of features characterizing this type of tumor. In this paragraph we summarize the main GB attributes (Figure 1) to also highlight the direction towards basic investigation, research and development and technology transfer should look at.
2.1. Blood-brain barrier
The blood-brain barrier (BBB) is a very selective and specialized barrier, preserving the homeostasis of the CNS, protecting and isolating the brain from potentially dangerous molecules transported in the blood such as virus and other pathogens. It has a complex structure formed by multiple layers, including brain microvascular non-fenestrated endothelial cells sealed by junctional complex, surrounded on the outer face by the end feet of glial cells. The physiological barrier is the result of a series of physical, transport, and metabolic processes inside endothelial cells, highly coordinated by interactions with different vascular, immune, and neural cell types [29,30,31]. Therefore, the BBB is not only a physical barrier, but it actively acts through endothelial efflux transporters to pump back in the bloodstream small lipophilic molecules that can diffuse though the plasma membrane. Even though the BBB has an evolutionary precious role in preserving brain health and functions, unfortunately, in case of brain pathologies, such as GB, its existence has the disadvantage to severe hamper the access to the brain of therapeutic molecules, strongly limiting their clinical success [30,31,32]. Moreover, as the barrier works in both directions, the flow of molecules from the brain to the bloodstream is also restricted, reducing the diagnostic potential of liquid biopsies based on blood samples in brain diseases [18].
Interestingly, the integrity of the barrier is compromised in many brain pathologies, including GB, but this is not enough to enhance drug accumulation and efficacy. This is in part due to the extremely heterogeneous barrier disruption and the persistence of intact barrier besides tumor areas [31,33,34,35]. Moreover, in GB, the impaired BBB, also referred as blood-tumor barrier (BTB), is able to function synergistically with intact BBB to create a tumor-supportive adaptive environment, paradoxically permitting the transport of pro-tumoral molecules while limiting the entry of antitumoral ones [36,37].
2.2. Heterogeneity
GB is a highly dynamic and fast evolving tumor, characterized by a strong molecular and cellular heterogeneity inside the same tumor at a fixed time point, as well as at different times during tumor development and even more in different patients [38,39]. The high inter- and intra-tumoral heterogeneity can affect tumor diagnosis and characterization, being challenging to achieve a comprehensive description of the whole molecular signature in such a heterogeneous tumor through surgical biopsies. To have better chances of exact diagnosis, biopsies should be several and repeated during GB progression, a practice that is not feasible nor sustainable for the patient [18]. Moreover heterogeneity makes it exceptionally challenging to target all tumor cells inside the same patient as well as to cure different patients with the same drug [40]. Regarding GB, we can distinguish a molecular and a cellular heterogeneity.
- Molecular:
In 2016, the WHO CNS4 classified gliomas into low grade gliomas or LGGs and GB, further subdividing GB into primary and secondary tumors [4]. Primary and secondary GBs are histologically indistinguishable, but they develop from different genetic precursors and therefore show different genetic alterations. Primary or de novo GB is characterized by no mutation in isocitrate dehydrogenase 1 (IDH1), accounts for 90% of all GBs and affects older patients, showing a worse prognosis. Among the most frequent genetic alterations, loss of heterozygosity (LOH) at 10q chromosome, mutations or deletions in phosphatase and tensin homolog (PTEN) gene, mutation or amplification of the epidermazl growth factor receptor (EGFR) gene, and deletion of Cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B) gene have been identified. Secondary GB derive from LGGs or Astrocytomas, are rarer, usually diagnosed at a younger age and with a more favorable outcome. The most frequent genetic alterations include IDH1 gene mutations, LOH at 22q and 19q, and methylation of the O6 methylguanine-DNA methyltransferase (MGMT) promoter [41].
The GB molecular signature has been identified and listed in The Cancer Genome Atlas (TCGA) project, reporting a large-scale-multi-dimensional analysis at genomic, epigenomic and transcriptomic level [42,43]. These studies highlighted frequent alterations in core oncogenic pathways, involving the tumor protein p53, the receptor tyrosine kinase/Ras/phosphoinositide 3-kinase, and retinoblastoma. Moreover, bulk gene expression profile analysis identified three main GB molecular subtypes, that are the proneural (TCGA-PN), classical (TCGA-CL) and mesenchymal (TCGA-MES), harboring mutations in PDGFRA and IDH1 (PN), EGFR (MES) or NF1 (CL), respectively, with a better prognosis for PN than MES subtype [39,44]. However, during time switching of a GB tumor from a subtype to another has been described, i.e. from PN to MES, being this one of the main cause of resisance to treatments [45,46]. Recently, single cell RNA sequencing pointed out to the high level of intratumoral complexity, by showing the presence of transcriptionally different subclones each one with peculiar features in the same tumor sample [47].
- Cellular:
A second layer of heterogeneity is due to the developmental state of GB cells in the tumor. GB contains subsets of GB stem cells (GSCs), that resemble ad could derive by neural stem cells (NSCs) [48,49,50,51]. GSCs are thought to represent the GB driving force, being able to maintain stemness and proliferation, but also to remain quiescent, and to escape both chemotherapy and radiotherapy, transferring this abilities to the other tumor cells, and condition the tumor microenvironment to support GB growth, progression and recurrency [52]. GSCs could be isolated thanks to the presence on the cell surface of CD133, CD44 and L1 cell adhesion molecule (L1CAM), but it is unknown whether different GSC markers isolate distinct cellular states or subtypes and whether tumors generated with different subpopulations of GSCs give rise to GB of comparable or diverse cellular composition. Several studies showed that the GSCs in recurrent GB differed from the ones that initiated and maintained the primary tumor, displaying different surface markers (CD15, BMI1, and SOX2, instead of CD133), and being more aggressive. Finally, GB single cell sequencing analysis suggest that tumor cell hierarchy arising from GSCs might account for the intratumoral cellular and molecular heterogeneity [37,53,54].
2.3. Invasiveness and Recurrency
GB cells have been shown to invade normal brain tissues, penetrating the brain parenchyma and the perivascular space by degrading the extracellular matrix through the release of proteolytic factors and by forming membrane-derived extensions known as invadopodia, involved in cell migration [55,56]. Patterns, directionality and mechanisms responsible for the invasive behavior comprise cell-to-cell and cell-to-ECM adhesion mechanisms, ECM and cytoskeletal remodelling and overall features of epithelial-mesenchymal transition (EMT), showing a preference towards specific brain regions (subventricular zone) despite others, such as hippocampus and cerebellum. Due to this aggressive migrative behavior, GB cells escape complete surgical resection, with following reoccurrence within a few centimeters from its original location. Almost all GB tumors return after surgery, radiotherapy, and chemotherapy, showing lower response rate to conventional treatments and extremely few options of treatment. To date, management protocols for recurrent GB patients are still missing and most of them are not eligible for surgical re-resection [57]. Multiple studies have identified in GSCs the tumor initiating and clonogenic potential as well as the origin of therapeutic resistance, being therefore the main actor in GB recurrency. The unremoved GSCs, after debulking, migrate within the resection cavity and initiate and recapitulate the whole tumor [58,59,60].
2.4. Therapeutic resistance
Therapeutic resistance is one of the main causes of poor prognosis for GB patients. This feature increases over time, weakening all the current therapeutic protocols for recurrent GB; in most cases surgery is no longer possible and the chemo and radiotherapy fail in counteracting tumor progression [37]. Many of the GB features here described have been also connected to radio-and/or chemo- resistance. First of all, the BBB presence limits chemotherapeutic access to the brain [31]. Moreover, therapeutic resistance increases over time due to the molecular changes that characterize GB progression, including the switching from the PN subtype to the MES one, and the accumulation of the O6-methylguanine-DNA methyltransferase (MGMT), which is in charge of DNA repair, whose increased levels are associated to increased TMZ chemoresistance [61]. GSCs also play a key role, showing low sensitivity to both chemo and radiotherapies, further decreasing following repeated chemoradiation treatments [62,63]. Therapeutic resistance is mediated by increase of MGMT, as well as of anti-apoptotic proteins, and drug-efflux transporters, which reduce drug sensitivity, and activation of both DNA damage checkpoints and DNA repair mechanism induced by radiation [64,65].
2.5. Immune escape
Immunosurveillance of the CNS is inherently adapted to maintain neuronal function and minimize non-specific immune responses. Additionally, GB cells contribute to generate an immunosuppressive microenvironment, by secreting soluble factors, interleukins, prostaglandins, and also EVs which target microglia, monocytes, and macrophages, inducing a tumor supportive M2 phenotype, as well as T lymphocytes, impairing their antitumoral activity. The complex GB microenvironment, conditioned by GB itself, collaborates in allowing the tumor to evade the immune response, progress and also recur [37,66,67,68].
3. Inquiring EV for GB treatment: strategies, advantages and future perspectives
GB cells communicate with each other and with the tumor microenvironment. In this respect EVs play key roles, transferring functional molecules (DNA, mRNAs, miRNAs, lnRNAs, proteins, lipids and metabolites) and their related properties between cells [69]. EVs are released by cells, mainly through two mechanisms that are budding from the cell membrane (microvesicles or ectosomes), or exocytosis of intraluminal vesicles formed inside the multivesicular bodies present in the cytoplasm (exosomes [70,71,72]. The first mechanism produces both small and large EVs, whereas the second mechanism gives rise more specifically to the smaller vesicles, due to physical cellular constraints [73]. EVs can travel through the bloodstream and other biological fluid and transport the messengers contained within at considerable distance from the site of origin. They are able to recognize specific cells through cell membrane interactions, and can be internalized in the target cells where EVs release their functional content [73,74,75]. EVs released by GB cells can condition non tumoral cells, such as macrophages, immune cells, astrocytes, endothelial cells to create a supportive microenvironment for GB growth and progression. On their side, tumor microenvironment cells release EVs that are uptaken by GB cells increasing their proliferation, invasiveness and malignancy. Several review articles have explored the role played by EVs in the cellular cross talk inside GB microenvironment [56,69,76,77]. Here we aim to address how EV biology can be exploited for bypassing current limitations in GB therapy and developing innovative and breakthrough therapeutic approaches (Figure 2).
4. Targeting EV biology and cargo
First, being EVs so much implicated in the establishment of a supportive microenvironment for GB, and more in general tumor, progression, and the induction of key malignant features, such as proliferation, invasiveness and chemoresistance, therapeutic strategies focused on blocking EV release, uptake, and circulation might improve patient outcome, irrespectively of the highly tumor heterogeneity.
Numerous strategies have been identified, based on targeting of key mechanisms for vesiculation, targeting of EVs on route through hemodialysis or blocking EV uptake, including the use of natural substances (i.e. the antifungal agent ketoconazol), as well as of repurposed drugs such as heparin and reserpine already used as anticoagulant and anti-hypertensive drugs, respectively [55,78]. In glioma cells heparan sulfate proteoglycans (HSPGs) have been identified as key modulators of EV uptake [79]. Decrease of HSPGs on the GB cell-membrane or the use of free heparin which competes with cell-membrane HSPGs for the binding to EVs, strongly reduces EV uptake [80]. However, heparin-mediated blocking of EV binding to target cells and following internalization is not specific of cancer cells [81,82]. The identification of selective strategies able to address tumor-derived EV biology, without affecting normal EVs, might be fundamental for the development of clinically suitable, effective and safe interventions, avoiding undesired off-target effects. As an example, recent studies have identified a specific Calcium dependent pathway inducing exosome release by cancer cells, involving Munc13-4, a Calcium-dependent soluble Nethylmaleimide–sensitive factor attachment protein (SNAP) receptor, upregulated in cancer cells [83]. In GB, increased levels of the mammalian Target of Rapamycin (mTOR) and consequent pathway activation is required for promoting EV release, through downregulation of the autophagy pathway, as well as for maintaining GSC tumor features, such as self-renewal, proliferation, migration and invasiveness [58]. Both mechanisms are potentially relevant for the development of novel approaches targeting GB-derived EV (GDEV) production.
In parallel, defining specific functionally relevant GB-associated EV cargo regulating the complex crosstalk inside GB environment, may also allow for specific and targeted therapeutic approaches. In particular, the last decade, gene/RNA therapy has significantly attracted the attention of the GB therapeutic field. Several long non coding RNAs (lncRNAs) with oncogenic activity have been found inside GDEVs [84]. Among these, the EV-associated lncRNAs POUF3F and TALC can remodel GB microenvironment, acting on endothelial cells and microglia respectively, inducing angiogenesis and GB progression (POU3F3) or M2 microglia polarization with consequent secretion of the complement components C5/C5a and induction of TMZ chemoresistance in GB cells (TALC) [85,86]. Other GDEV-associated lncRNAs, such as MALAT1, MEG3, NEAT1 and HOTAIR play key role in promoting EMT and possibly chemoresistance when uptaken by GB cells both in vitro and in vivo [84]. Accumulating evidence shows that miRNAs also play essential roles in GB pathogenesis with a high potentiality to be exploited in targeted therapeutic approaches [87]. Among the others, miR-9, upregulated in GSCs, has been found in EV isolated from both GB cell cultures and patients, strongly implied in the GB malignant phenotypic traits including cell proliferation and migration [88]. miR-9 is also involved in the expression of the drug efflux protein P-glycoprotein, which contributes to increase TMZ drug resistance into GB cells. In this respect, anti-miR-9 molecules encapsulated in MSC-EV- were shown to be successfully delivered to GB cells in vitro, causing the reduction of the levels of P-glycoprotein, and enhancing TMZ sensitivity [89]. Therefore, a promising direction is represented by therapeutic approaches based on EVs loaded with siRNAs to target GB cells and downregulate the expression of the inherent oncogenic lncRNAs or miRNAs, inhibiting their EV mediated transferring to other tumor cells as well as non-tumoral cells of the GB microenvironment
5. EVs as drug-delivery systems in GB therapy
Recently, nanomedicine has dominated the field of tumor therapy with respect to different approaches, thanks to the abilities of nanoparticles to be loaded with specific therapeutic agents as well as surface decorated with specific targeting molecules. First, synthetic nanocarriers such as liposomes, have been used, with the crucial advantage of high loading and surface functionalization efficiency, but important limitations due to low tolerability, relatively short circulation times in biological environments, fast body clearance and last but not least low capability for BBB. Even though different strategies have been developed to increase BBB permeability and enhance nanoparticle access to the brain tissue, low biocompatibility and high risk of off-target effects are still key issues to overcome [30,90]
EVs show key advantages in this respect. Several studies have shown their low immunogenicity and cytotoxicity, low clearance from the phagocytic system, prolonged circulation time, high biocompatibility and good cell uptake [91,92]. Interestingly, EVs are able to cross interspecies and even interkingdom boundaries: EVs isolated from different sources, including microalgae, bacteria and plants are easily uptaken by mammalian cells [93,94,95,96,97]. EVs can be loaded, through several physical/chemical/genetic strategies, with different functional cargoes, such as nucleic acids, proteins, natural substances or chemotherapeutics, that can be thus delivered to the cells. Moreover, the EV surface can be also functionalized with cell type-specific targeting ligands to enhance the interaction with specific cellular types [30,98]. EV encapsulation can enhance the solubility, stability, bioavailability, and in last instance the therapeutic effect of the molecule alone. Besides these features, EVs are also able to cross the BBB, being then ideal candidates for delivering therapeutic molecules to the brain to be used for treatment of brain pathologies [90].
The first study on the ability of engineered EVs to cross the BBB and efficiently delivery functional molecules to the brain was performed by Alvarez-Erviti and coauthors using EVs derived from mouse bone-marrow dendritic cells [99]. To confer brain targeting capabilities, dendritic cells were previously engineered to express recombinant fusion proteins, containing the central nervous system–specific rabies viral glycoprotein (RVG) peptide, that specifically binds to the acetylcholine receptor, fused to the N terminus of murine dendritic cell (DC)-derived lysosome-associated membrane protein (Lamp2b), a highly EV-associated protein. Isolated EVs, exposing the RVG-Lamp2b fusion protein on the membrane, were electroporated with specific siRNA and then intravenously injected into mice, showing specific targeting and siRNA delivery to brain neurons, microglia, and oligodendrocytes [99]. Besides this study, a similar approach was used in mouse models for Parkinson’s disease [100], highlighted the great EV potential in penetrating the BBB and be exploited for the treatment of brain pathologies. More recently, Kim and coworkers [101] used the same Lamp2b-based approach to decorate EVs released form 293T cells with the Transferrin Receptor-binding peptide T7, able to target transferrin receptors naturally enriched on GB cell surface. The engineered T7-EVs were further modified to encapsulate antisense miRNA oligonucleotides against miR-21 (AMO-21). When injected intravenously into a intracranial GB xenotrasplanted rat model, T7-EVs resulted much more efficient of both unmodified and RVG-decorated EVs to reach the brain, target GB cells and deliver AMO21, causing a reduction of GB size [101].
Besides ad-hoc modified EVs, EVs from other cell types have been successfully tested for GB treatment. EVs derived from a mouse lymphoma cells line (EL-4), loaded with the anti-inflammatory drug curcumin or with an inhibitor of the signal transducer and activator of transcription 3 (Stat3), were efficiently and noninvasively delivered (via intranasal route) to mouse brain microglia cells, causing a significantly delayed brain tumor growth in a glioma mouse model [102]. Moreover, EVs from a brain endothelial cell line (b.END3), naturally enriched with the CD3 tetraspanin, loaded with Doxorubicin, were able to cross the BBB into a xenotrasplanted zebrafish brain tumor model (obtained by injecting GB cells into the zebrafish brain ventricle), significantly decreasing fluorescent intensity of xenotransplanted cancer cells and tumor growth markers [103].
MSCs are the better characterized source of EVs, used in several clinical trials, native or engineered, to target a plethora of different pathologies, including tumors [55,104]. MSCs can communicate with different cell types, and also GB cells, through direct contact by gap junctions and/or by contact-independent pathways (Biancone et al. 2012; Munoz et al. 2013). In fact stromal cells resembling MSCs are a key component of the GB microenvironment, where they can exert both tumor supportive and suppressive roles [105,106]. MSC-derived EVs carrying specific miRNA (miR-7, miR-34a, miR-124, miR-133b, miR-145, miR-146b miR-199a, miR-375 or miR-584-5p) were able to counteract GB tumor features, including cell proliferation, migration, and invasion in vitro by targeting specific molecules or pathways such as the Forkhead box (FOX)A2 (mir124a), Wnt pathway (miR133b), EGFR (miR-146b), SLC31A (miR-375) and MMP2 (miR-584-5p). Many of these MSC-EVs engineered with miRNAs were also tested in vivo, where they showed the ability to cross the BBB, promoting tumor regression in mouse xenotrasplanted models [87]. A very promising study is the one involving MSC-EVs loaded with miR124a and systemically delivered in mouse models xenotrasplanted intracranially with GB cells, being able to suppress tumor growth and prolong overall animal survival [107].
Tumor derived-EVs (TDEVs) can be also used as natural carriers for the delivery of antioncogenic molecules [55], thanks to their high tumor targeting and permeability, as a technological Trojan horse, as evoked by Simionescu and coauthors [87]. EVs isolated from engineered patient-derived GSCs carrying the miR-302-367 cluster, are internalized rapidly by neighboring GB cells, and are able to inhibit GB cell proliferation, stemness and invasion, through repression of Cyclin A, Cyclin D1, E2F1 and the CXCR4 pathways [108]. Moreover, GDEVs engineered with miR-124, miR-128, or miR-137 improved survival after injection in GB model mice when combined with chemotherapy [109], whereas GDEVs carrying miR-151a reduced chemoresistance in GB xenograft mouse models [110]. Cell treatment with miRNAs specific inductors can also be used, as valid alternative to external loading, for obtaining EVs carrying specific miRNA molecules. This is the approach used by Wang and coauthors (H. Wang et al., 2021), using traditional medicine substances to induce GDEV miR7-5p enrichment; GDEV carrying miR7-5p reduced GB formation and metastasis in GB nude mice models [111]. Obviously, the exploitation of GDEVs as drug carriers for GB therapy requires an extremely careful evaluation of the antioncogenic properties of the loaded therapeutic molecule vs the oncogenic potential of the GDEV intrinsic cargo and then of the relative cost-benefit assessment. Recently, Guo and coworkers [112] identified a saponin-mediated cargo elimination strategy to improve biosafety of GDEVs in GB therapeutic applications. Systematic analysis of the original proteins and RNAs together with functional in vitro and in vivo assays confirmed the high efficiency of the method in eliminating GB-EV cargo and its inherited abilities in promoting GB progression without affecting uptake by GB cells. Furthermore, saponin-treated GDEVs loaded with Doxorubycin displayed an effective tumor suppressive role in both subcutaneous and orthotopic GB mouse models [112]. These data are extremely promising and can be exploited for the development of novel visionary therapeutic pipelines which could involve the isolation of TDEVs directly from the patients, their oncogenic disarm through cargo elimination protocols, the loading and /or functionalization with anticancer molecules, and the final administration to the patient. Finally, even though TDEVs preferentially target (and are uptaken by) the cells of the same tumor type, TDEV surface can be functionalized with specific molecules involved in EV-cell interaction to improve or alter natural tumor tropisms. Geng and coauthors [113], have shown that GDEVs are more easily uptaken by GB cells than other type of tumor cells such as pancreatic cancer (PC) cells and viceversa, but EV functionalization with cyclic arginine-glycine-aspartic acid-tyrosine-cysteine (cRGDyC), a ligand for the integrin αvβ3 enriched in GB cells, is able to enhance ability of PC cells (and also Gb cells themselves) to target and be internalized into GB cells [113].
Other cells that naturally show a GB tropism are the neural stem cells (NSCs) and the teratocarcinoma cells. Both NSCs, from which GB might derive [48,49], and NTERA2 human teratocarcinoma cells, which resemble NSCs being able to differentiate into both glia and neurons [114,115], show GB tropism in vivo and were proposed for cell-based delivery systems in anti-GB therapy [116,117,118]. EVs released by NSCs and engineered with antisense oligonucleotides (ASOs) targeting oncogenic and tolerogenic signal transducer and activator of transcription 3 (STAT3) were effective after intracranial injection at distant site, in reaching glioma microenvironment, targeting and activating microglia to inhibit tumor growth [119]. Even more interestingly, native NTERA2-derived EVs, naturally carrying the oncodevelopmental factor Cripto [120,121,122], have been recently shown to impair GB cell migration, without inducing undesirable effects such as increased GB cell proliferation or enhanced TMZ drug resistance [123]. Cripto is a membrane-bound glycosylphosphatidyl inositol-anchored protein, that can be also cleaved and released as a soluble factor [124], and only recently found associated to EVs [123,125]. With respect to neural fate, mouse Cripto gene targeting was associated to neural differentiation in embryonic stem cell cultures, and specification of anterior neural identities in vivo, with Cripto null embryos mostly constituted by anterior neuroectoderm, including NSCs [126,127,128]. Cripto has been mostly identified associated to oncogenic features, also in GB, but it is also able to act as an antioncogene [129,130,131,132]. Even though these data have not yet been confirmed in vivo, they open the mind to new unobvious and even paradoxical paradigms in GB and more in general tumor progression and, subsequently, therapies. First, EV sorting and delivery might be an alternative route for regulating the spreading and activity of soluble and/or membrane-bound signaling molecules, modulating their final impact on target cells and then on cancer development and progression [55]. Second, specific subsets of TDEVs might possess an antitumoral potential to be used per se or eventually empowered through ad hoc modifications [55]. Therefore, molecularly identifying and isolating, or bio-mimicking, or even inducing these specific subsets of TDEVs is certainly a novel ambitious and promising therapeutic direction to explore.
6. Mitigating the pitfalls and realizing the potential of EV exploitation in GB treatment
Although these extremely promising results, no EV-based GB clinical trial is ongoing, even though EV-based therapies are under evaluation in clinical trials for treatment of other tumors. In this paragraph we will try to schematize the main issues and directions to explore to find the optimal solutions and realize the full EV potential as therapeutic advanced platform for GB treatment. The analysis of Strengths, Weaknesses, Opportunity and Threat (SWoT) of EV exploitation in novel GB therapeutic strategies is reported in Figure 3.
6.1. EV source
We have described above the main EV sources that have been used for GB therapeutic assays both in vitro an in vivo. They are all derived by mammalian cells, including cell lines of brain endothelial cells, tumor cells (GB but also teratocarcinoma), and stem cells (both neural and mesenchymal). To be exploited in patient therapy, EVs must be produced in high quantity and with an extremely high quality, satisfying compliance with good manufacturing practice (GMP), pharmaceutical biologics’ regulation and ongoing specific regulation for EV therapeutics [93]. The process needs careful scaling up and out, standardization, and quality assurance of the experimental pipeline, as well as quality control of the final products, requiring high commitment in terms of cost, resources and time. Alternative sources of EVs, such as plants, microalgae and bacteria have also been identified and their biocompatibility and biodistribution in animal models has been performing [93]. These alternative EV sources have lower cost of production, but unfortunately, until now no brain tropism has been proved. Therefore, the production pipeline must be extended with the insertion of an EV surface functionalization phase and successive isolation and characterization of the loaded EVs. A careful evaluation of the advantage and disadvantages of each strategy must be performed, in terms of therapeutic efficacy and efficiency as well as of cost and sustainability.
6.2. EV handling procedures
Different techniques have been used until now for EV isolation, including differential Ultracentrifugation (dUC) that is still the gold standard, different commercial kits, exclusion chromatography, tangential flow filtration [133], and loading, usually subdivided into endogenous or indirect (manipulation of the parental cell) and exogenous or direct (EV engineering), the latter one subdivided into passive (co-incubation of molecules and EVs) and active (i.e. electroporation, sonication) [134]. Standardized procedures should be identified to guarantee low inter batches variability and high reproducibility of the results. The international Society for EVs (ISEV) is strongly committed in this directions since years, providing the EV scientific community with the minimal information for studying EVs and several position papers dedicated to specific aspects of EV research and exploitation [135,136,137,138,139]. More recently, it is increasingly evident how quality management tools already used in other scientific context [140,141,142] can help in solve main issues in EV research and development [143,144]. In particular, application of the Failure Mode and Effect Analysis for risk assessment and management of scientific procedures [145] to the single steps of the EV production pipeline might help in avoiding and preventing pitfalls and failures in EV bioprocessing and manipulation [144,146], whereas application of the Design of Experiments mathematical model [147,148] might be a valid model to identify and optimize key factors in multivariable processes such as production as well as engineering or functional assays of the EVs [144,146,149]. More recently, we have proposed a new tool, inspired to multicriteria decision making, that is a multi-criteria EV decision-making grid (EV-DMG) as a novel, customizable, efficient and easy-to-use tool to support responsible EV research and innovation [150]. All together these instruments and guidelines can greatly improve the choice, optimization, standardization, quality assurance and control, and ultimately the reliability of the EV landscape.
6.3. Biodistribution
Assessment of EV biodistribution mainly detected as preferential sites spleen, liver, lungs, kidneys and gastrointestinal tract [151]. Moreover, as already discussed TDEVs have a high tropism towards tumor cells, especially of the same type. The source and handling of EVs can influence the final biodistribution, as well as the “ad hoc” cell surface functionalization [152]. EV size is also an important variable to consider: smaller EVs are easily eliminated by blood circulation, whereas larger EV can pass through the BBB with difficulty, but seem to transfer their cargo content more efficiently [30,89]. Several EVs have shown to be able to penetrate the BBB and deliver therapeutic agents into the brain, being suitable drug-delivery vectors to be administered intravenously. Identifying the amount of EVs to inject and the frequency of administration requires the assessment of the amount of EVs which passes the barrier and enter to the brain and the amount of EV cargo that is effectively delivered to the GB cells. However, since GB therapy often comprises as first step surgical tumor resection, concomitant intracranial injection at tumor site of therapeutic EVs able to counteract reoccurrence would also be a suitable strategy.
6.4. Designing Biomimetic particles
Very recently, another important research direction is exploring the possibility to obtain biomimetic nanocarriers, joining the advantages of both artificial and natural delivery vectors through the development of technological advanced ad hoc nanoplatforms. Recently, a nanoplatform termed EV-DNs, in which doxorubicin loaded heparin nanoparticles (DNs) were attached to the surface of native grapefruit EVs, was shown to penetrate the BBB in vivo, reducing the GB cell proliferation and increasing animal model survival [95]. Another biomimetic system, named RGE-Exo-SPION/Cur was implemented by loading curcumin (Cur) and superparamagnetic iron oxide nanoparticles (SPIONs) into EVs further functionalized by click chemistry to carry on the surface the neuropilin-1-targeted peptide (RGERPPR, RGE). RGE-Exo-SPION/Cur were efficient in targeting GB cells in orthotopic GB-bearing nude mouse models, accumulating in correspondence of the tumor, as well as in reducing the GB size, being relevant for simultaneous GB MRI-based diagnosis and therapy [153].
6.5. GB models
One of the main reasons for the limited success of novel therapeutic molecules entering clinical trials is the poor correlation between their efficacy in in vitro, ex vivo or in vivo tumor models and the patient treatment scenarios More and more sophisticated ex vivo models able to mimic the GB microenvironment have been developed to fill this gap, starting form glioma slice cultures directly derived from biopsies, neurosheres, hydrogel-coated scaffolds of different biomaterials, and arriving to complex 3D models such as GB organoids, and tumor-on-chips [7]. GB organoids are produced resembling brain organoids, show tumor heterogeneity, and can be extremely use to study drug sensitivity. Recently, a biobank of patient-derived glioblastoma organoids, each one showing the parental tumor heterogeneity has been developed, with a high value for identification of personalized target and treatment strategies [154]. However, to assess also the ability to cross the BBB and deliver efficiently the therapeutic molecule, without inducing immune reaction or other undesired effects, in vivo studies are absolutely needed. Several studies have shown how orthotopic xenotrasplanted immunocompetent mouse models, in which human GB cells are injected intracranially into brain parenchyma are able to recapitulate the patient GB-microenvironment interactions [155,156].
Funding
The work was funded within the framework of the project CEVITA of the AMICO 2 Programme of CNR – UVR supported by the PoC - PNRR measure of the Ministry of Enterprise and Made in Italy funded by the European Union - NextGenerationEU".
Acknowledgments
The author acknowledges Anna Maria Aliperti for English proofreading and Mariarosaria Aletta for bibliographic support.
Conflicts of Interest
The author has filed the patent PCT/IB2023/053735 relative to the use of lipidic vesicles for the treatment of aggressive tumors.
References
- Stoyanov, G.S.; Dzhenkov, D.L. On the Concepts and History of Glioblastoma Multiforme - Morphology, Genetics and Epigenetics. Folia Med. (Plovdiv). 2018, 60, 48–66. [Google Scholar] [CrossRef] [PubMed]
- Chvátal, A.; Verkhratsky, A. An Early History of Neuroglial Research: Personalities. Neuroglia 2018, 1, 16. [Google Scholar] [CrossRef]
- Bailey, P.; Cushing, H. A Classification of the Tumors of the Glioma Group on a Histogenetic Basis with a Correlated Study of Prognosis. J. Am. Med. Assoc. 1926, 87, 268. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Brandner, S.; Bertero, L.; Capper, D.; French, P.J.; Figarella-branger, D.; Giangaspero, F.; Haberler, C.; Hegi, M.E.; Kristensen, B.W.; et al. Molecular Diagnostic Tools for the World Health Organization (WHO) 2021 Classification of Gliomas, Glioneuronal and Neuronal Tumors; an EANO Guideline. 2023, 25, 1731–1749.
- Boccellato, C.; Rehm, M. Glioblastoma, from Disease Understanding towards Optimal Cell-Based in Vitro Models. Cell. Oncol. 2022, 45, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014-2018. Neuro. Oncol. 2021, 23, III1–III105. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Stetson, L.; Virk, S.; Barnholtz-Sloan, J.S. Epidemiology of Intracranial Gliomas. Intracranial Gliomas Part I - Surg. 2017, 30, 0. [Google Scholar]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. Glioblastoma 2017, 143–153. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Mathews, J.D.; Forsythe, A.V.; Brady, Z.; Butler, M.W.; Goergen, S.K.; Byrnes, G.B.; Giles, G.G.; Wallace, A.B.; Anderson, P.R.; Guiver, T.A.; et al. Cancer Risk in 680 000 People Exposed to Computed Tomography Scans in Childhood or Adolescence: Data Linkage Study of 11 Million Australians. BMJ 2013, 346, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Grech, N.; Dalli, T.; Mizzi, S.; Meilak, L.; Calleja, N.; Zrinzo, A. Rising Incidence of Glioblastoma Multiforme in a Well-Defined Population. Cureus 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Bender, S.; Jones, D.T.W.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and Adult Glioblastoma: Multiform (Epi)Genomic Culprits Emerge. Nat. Rev. Cancer 2014. [CrossRef] [PubMed]
- Golla, H.; Nettekoven, C.; Bausewein, C.; Tonn, J.C.; Thon, N.; Feddersen, B.; Schnell, O.; Böhlke, C.; Becker, G.; Rolke, R.; et al. Effect of Early Palliative Care for Patients with Glioblastoma (EPCOG): A Randomised Phase III Clinical Trial Protocol. BMJ Open 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Golla, H.; Ale Ahmad, M.; Galushko, M.; Hampl, J.; Maarouf, M.; Schroeter, M.; Herrlinger, U.; Hellmich, M.; Voltz, R. Glioblastoma Multiforme from Diagnosis to Death: A Prospective, Hospital-Based, Cohort, Pilot Feasibility Study of Patient Reported Symptoms and Needs. Support. Care Cancer 2014, 22, 3341–3352. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, M.; Osti, D.; Faletti, S.; Beznoussenko, G. V; Dimeco, F.; Surgery, N.; Hopkins, J.; Medicine, T.; Orientale, P. Extracellular Vesicles: The Key for Precision Medicine in Glioblastoma. 2022, 24, 184–196.
- Soliman, M.A.; Khan, A.; Azmy, S.; Gilbert, O.; Khan, S.; Goliber, R.; Szczecinski, E.J.; Durrani, H.; Burke, S.; Salem, A.A.; et al. Meta-Analysis of Overall Survival and Postoperative Neurologic Deficits after Resection or Biopsy of Butterfly Glioblastoma. Neurosurg. Rev. 2022, 45, 3511–3521. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Warren Mason, P.; Bent, M.J. van den B.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma in Elderly Patients. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib Compared with Lomustine in Patients with Relapsed Glioblastoma (REGOMA): A Multicentre, Open-Label, Randomised, Controlled, Phase 2 Trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Badruddoja, M.A.; Pazzi, M.; Sanan, A.; Schroeder, K.; Kuzma, K.; Norton, T.; Scully, T.; Mahadevan, D.; Ahmadi, M.M. Phase II Study of Bi-Weekly Temozolomide plus Bevacizumab for Adult Patients with Recurrent Glioblastoma. Cancer Chemother. Pharmacol. 2017, 80, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Ling, A.L.; Reardon, D.A.; Chiocca, E.A. Lessons Learned from Phase 3 Trials of Immunotherapy for Glioblastoma: Time for Longitudinal Sampling? 2023, 1–15.
- Kaka, N.; Hafazalla, K.; Samawi, H.; Simpkin, A.; Perry, J.; Sahgal, A.; Das, S. Progression-Free but No Overall Survival Benefit for Adult Patients with Bevacizumab Therapy for the Treatment of Newly Diagnosed Glioblastoma: A Systematic Review and Meta-Analysis. Cancers (Basel). 2019, 11, 1723. [Google Scholar] [CrossRef] [PubMed]
- Schritz, A.; Aouali, N.; Fischer, A.; Dessenne, C.; Adams, R.; Berchem, G.; Huiart, L.; Schmitz, S. Systematic Review and Network Meta-Analysis of the Efficacy of Existing Treatments for Patients with Recurrent Glioblastoma. Neuro-Oncology Adv. 2021, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gupta, T.; Mona, J.; Selvarajan, P.; Kannan, S.; Menon, N.; Tmh, A.; Memorial, T. Updated Systematic Review and Meta-Analysis of Extended Aduvant Temozolomide in Newly Diagnosed Glioblastoma. 2023, 5, 1–10.
- Begagić, E.; Pugonja, R.; Bečulić, H.; Čeliković, A.; Tandir Lihić, L.; Kadić Vukas, S.; Čejvan, L.; Skomorac, R.; Selimović, E.; Jaganjac, B.; et al. Molecular Targeted Therapies in Glioblastoma Multiforme: A Systematic Overview of Global Trends and Findings. Brain Sci. 2023, 13, 1602. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Liu, F.; Gan, S.; Roy, S.; Hasan, I.; Zhang, B.; Guo, B. Emerging Extracellular Vesicle-Based Carriers for Glioblastoma Diagnosis and Therapy. Nanoscale 2023, 15, 10904–10938. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Shusta, E.V. Blood–Brain Barrier Modulation to Improve Glioma Drug Delivery. Pharmaceutics 2020, 12, 1085. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Sane, R.; Oberoi, R.; Ohlfest, J.R.; Elmquist, W.F. Delivery of Molecularly Targeted Therapy to Malignant Glioma, a Disease of the Whole Brain. Expert Rev. Mol. Med. 2011, 13, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Santarosa, C.; Castellano, A.; Conte, G.M.; Cadioli, M.; Iadanza, A.; Terreni, M.R.; Franzin, A.; Bello, L.; Caulo, M.; Falini, A.; et al. Dynamic Contrast-Enhanced and Dynamic Susceptibility Contrast Perfusion MR Imaging for Glioma Grading: Preliminary Comparison of Vessel Compartment and Permeability Parameters Using Hotspot and Histogram Analysis. Eur. J. Radiol. 2016, 85, 1147–1156. [Google Scholar] [CrossRef]
- Law, M.; Yang, S.; Babb, J.S.; Knopp, E.A.; Golfinos, J.G.; Zagzag, D.; Johnson, G. Comparison of Cerebral Blood Volume and Vascular Permeability from Dynamic Susceptibility Contrast-Enhanced Perfusion MR Imaging with Glioma Grade. Am. J. Neuroradiol. 2004, 25, 746–755. [Google Scholar]
- Onda, K.; Tanaka, R.; Takahashi, H.; Takeda, N.; Ikuta, F. Cerebral Glioblastoma with Cerebrospinal Fluid Dissemination: A Clinicopathological Study of 14 Cases Examined by Complete Autopsy. Neurosurgery 1989, 25. [Google Scholar] [CrossRef]
- Noch, E.K.; Ramakrishna, R.; Magge, R. Challenges in the Treatment of Glioblastoma: Multisystem Mechanisms of Therapeutic Resistance. World Neurosurg. 2018, 116, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Yekula, A.; Taylor, A.; Beecroft, A.; Kang, K.M.; Small, J.L.; Muralidharan, K.; Rosh, Z.; Carter, B.S.; Balaj, L. The Role of Extracellular Vesicles in Acquisition of Resistance to Therapy in Glioblastomas. Cancer Drug Resist. 2021, 4, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science (80-. ). 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Wen, P.Y. Glioma in 2014: Unravelling Tumour Heterogeneity-Implications for Therapy. Nat. Rev. Clin. Oncol. 2015, 12, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, N. Glioblastoma Multiforme and Genetic Mutations: The Issue Is Not over YetAn Overview of the Current Literature. J. Neurol. Surgery, Part A Cent. Eur. Neurosurg. 2020, 81, 64–70. [Google Scholar] [CrossRef] [PubMed]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Yong, R.L.; Paddison, P.; Zhu, J. Comparison of Glioblastoma (GBM) Molecular Classification Methods. Semin. Cancer Biol. 2018, 53, 201–211. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal Differentiation Mediated by NF-ΚB Promotes Radiation Resistance in Glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Riester, M.; Cheng, Y.K.; Huse, J.T.; Squatrito, M.; Helmy, K.; Charles, N.; Michor, F.; Holland, E.C. Most Human Non-GCIMP Glioblastoma Subtypes Evolve from a Common Proneural-like Precursor Glioma. Cancer Cell 2014, 26, 288–300. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human Glioblastoma Arises from Subventricular Zone Cells with Low-Level Driver Mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Almengló, C.; Caamaño, P.; Fraga, M.; Devesa, J.; Costoya, J.A.; Arce, V.M.; José Costoya, C.A. From Neural Stem Cells to Glioblastoma: A Natural History of GBM Recapitulated in Vitro. J Cell Physiol 2021, 236, 7390–7404. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Yang, K.; Rich, J.N. The Evolving Landscape of Glioblastoma Stem Cells. Curr. Opin. Neurol. 2013, 26. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Parada, L.F.; Dirks, P.B.; Wechsler-Reya, R.J. Brain Tumor Stem Cells Remain in Play. J. Clin. Oncol. 2017, 35, 2428–2431. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Reimand, J.; Lan, X.; Head, R.; Zhu, X.; Kushida, M.; Bayani, J.; Pressey, J.C.; Lionel, A.C.; Clarke, I.D.; et al. Single Cell-Derived Clonal Analysis of Human Glioblastoma Links Functional and Genomic Heterogeneity. Proc. Natl. Acad. Sci. 2015, 112, 851–856. [Google Scholar] [CrossRef]
- Huang, Q.; Zhang, Q.B.; Dong, J.; Wu, Y.Y.; Shen, Y.T.; Zhao, Y.D.; Zhu, Y.D.; Diao, Y.; Wang, A.D.; Lan, Q. Glioma Stem Cells Are More Aggressive in Recurrent Tumors with Malignant Progression than in the Primary Tumor, and Both Can Be Maintained Long-Term in Vitro. BMC Cancer 2008, 8, 1–11. [Google Scholar] [CrossRef]
- Liguori, G.L.; Kralj-Iglič, V. Pathological and Therapeutic Significance of Tumor-Derived Extracellular Vesicles in Cancer Cell Migration and Metastasis. Cancers (Basel). 2023, 15, 4425. [Google Scholar] [CrossRef]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Cifola, I.; Caratelli, S.; Sconocchia, G.; D’Agnano, I.; Cenciarelli, C. Glioma Extracellular Vesicles for Precision Medicine: Prognostic and Theragnostic Application. Discov. Oncol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Benson, R.; Hakim, A.; Rath, G.K. Management of Glioblastoma after Recurrence: A Changing Paradigm. J. Egypt. Natl. Canc. Inst. 2016, 28, 199–210. [Google Scholar] [CrossRef]
- Ryskalin, L.; Biagioni, F.; Lenzi, P.; Frati, A.; Fornai, F. MTOR Modulates Intercellular Signals for Enlargement and Infiltration in Glioblastoma Multiforme. Cancers (Basel). 2020, 12, 2486. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Hassiotou, F.; Nowak, A. Glioblastoma Stem-like Cells: At the Root of Tumor Recurrence and a Therapeutic Target. Carcinogenesis 2014, 36, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A Restricted Cell Population Propagates Glioblastoma Growth after Chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Molecular Sciences Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. 2019. [CrossRef]
- Osuka, S.; Sampetrean, O.; Shimizu, T.; Saga, I.; Onishi, N.; Sugihara, E.; Okubo, J.; Fujita, S.; Takano, S.; Matsumura, A.; et al. IGF1 Receptor Signaling Regulates Adaptive Radioprotection in Glioma Stem Cells. Stem Cells 2013, 31, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Qazi, M.A.; Vora, P.; Venugopal, C.; McFarlane, N.; Subapanditha, M.K.; Murty, N.K.; Hassell, J.A.; Hallett, R.M.; Singh, S.K. A Novel Stem Cell Culture Model of Recurrent Glioblastoma. J. Neurooncol. 2016, 126, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of Gene Expression and Chemoresistance of CD133+ Cancer Stem Cells in Glioblastoma. Mol. Cancer 2006, 5, 1–12. [Google Scholar] [CrossRef]
- Jain, P.; Vashist, S.; Panjiyar, B.K. Navigating the Immune Challenge in Glioblastoma: Exploring Immunotherapeutic Avenues for Overcoming Immune Suppression. 2023, 15. [CrossRef]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Options Oncol. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Cuzzubbo, S.; McArthur, S.; Durrant, L.G.; Adhikaree, J.; Tinsley, C.J.; Pockley, A.G.; McArdle, S.E.B. Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Tankov, S.; Walker, P.R. Glioma-Derived Extracellular Vesicles – Far More Than Local Mediators. Front. Immunol. 2021, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The Biology and Function of Exosomes in Cancer. J. Clin. Invest. 2016. [CrossRef]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367. [Google Scholar] [CrossRef]
- Mantile, F.; Franco, P.; Stoppelli, M.P.; Liguori, G.L. Biological Role and Clinical Relevance of Extracellular Vesicles as Key Mediators of Cell Communication in Cancer. In Biological Membrane Vesicles: Scientific, Biotechnological and Clinical Considerations. Advances in Biomembranes and Lipid Self-Assembly; Elsiever, 2020; Vol. 33.
- Kahlert, C.; Kalluri, R. Exosomes in Tumor Microenvironment Influence Cancer Progression and Metastasis. J. Mol. Med. 2013. [CrossRef] [PubMed]
- McAndrews, K.M.; Kalluri, R. Mechanisms Associated with Biogenesis of Exosomes in Cancer. Mol. Cancer 2019. [CrossRef] [PubMed]
- Catalano, M.; Serpe, C.; Limatola, C. Microglial Extracellular Vesicles as Modulators of Brain Microenvironment in Glioma. Int. J. Mol. Sci. 2022, 23, 3165. [Google Scholar] [CrossRef]
- Kopper, T.J.; Yu, X.; Graner, M.W. Immunopathology of Extracellular Vesicles in Macrophage and Glioma Cross-Talk. J. Clin. Med. 2023, 12, 3430. [Google Scholar] [CrossRef]
- Wang, S.E. Extracellular Vesicles in Cancer Therapy. Semin. Cancer Biol. 2022, 86, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Christianson, H.C.; Svensson, K.J.; Van Kuppevelt, T.H.; Li, J.P.; Belting, M. Cancer Cell Exosomes Depend on Cell-Surface Heparan Sulfate Proteoglycans for Their Internalization and Functional Activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef] [PubMed]
- Atai, N.A.; Balaj, L.; Van Veen, H.; Breakefield, X.O.; Jarzyna, P.A.; Van Noorden, C.J.F.; Skog, J.; Maguire, C.A. Heparin Blocks Transfer of Extracellular Vesicles between Donor and Recipient Cells. J. Neurooncol. 2013, 115, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Maguire, C.A.; Balaj, L.; Sivaraman, S.; Crommentuijn, M.H.W.; Ericsson, M.; Mincheva-Nilsson, L.; Baranov, V.; Gianni, D.; Tannous, B.A.; Sena-Esteves, M.; et al. Microvesicle-Associated AAV Vector as a Novel Gene Delivery System. Mol. Ther. 2012, 20, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Couchman, J.R.; Multhaupt, H.; Sanderson, R.D. Recent Insights into Cell Surface Heparan Sulphate Proteoglycans and Cancer [Version 1; Referees: 3 Approved]. F1000Research 2016, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Messenger, S.W.; Woo, S.S.; Sun, Z.; Martin, T.F.J. A Ca2+-Stimulated Exosome Release Pathway in Cancer Cells Is Regulated by Munc13-4. J. Cell Biol. 2018, 217, 2877–2890. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Pillai, P.P. Current Insights on Extracellular Vesicle-Mediated Glioblastoma Progression: Implications in Drug Resistance and Epithelial-Mesenchymal Transition. Biochim. Biophys. Acta - Gen. Subj. 2022, 1866, 130065. [Google Scholar] [CrossRef] [PubMed]
- Lang, H.L.; Hu, G.W.; Chen, Y.; Liu, Y.; Tu, W.; Lu, Y.M.; Wu, L.; Xu, G.H. Glioma Cells Promote Angiogenesis through the Release of Exosomes Containing Long Non-Coding RNA POU3F3. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 959–972. [Google Scholar] [PubMed]
- Li, Z.; Meng, X.; Wu, P.; Zha, C.; Han, B.; Li, L.; Sun, N.; Qi, T.; Qin, J.; Zhang, Y.; et al. Glioblastoma Cell-Derived LncRNA-Containing Exosomes Induce Microglia to Produce Complement C5, Promoting Chemotherapy Resistance. Cancer Immunol. Res. 2021, 9, 1383–1399. [Google Scholar] [CrossRef]
- Simionescu, N.; Zonda, R.; Petrovici, A.R.; Georgescu, A. The Multifaceted Role of Extracellular Vesicles in Glioblastoma: Microrna Nanocarriers for Disease Progression and Gene Therapy. Pharmaceutics 2021, 13, 988. [Google Scholar] [CrossRef]
- Geng, L.; Xu, J.; Zhu, Y.; Hu, X.; Liu, Y.; Yang, K.; Xiao, H.; Zou, Y.; Liu, H.; Ji, J.; et al. Targeting MiR-9 in Glioma Stem Cell-Derived Extracellular Vesicles: A Novel Diagnostic and Therapeutic Biomarker. Transl. Oncol. 2022, 22, 101451. [Google Scholar] [CrossRef]
- Munoz, J.L.; Bliss, S.A.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Delivery of Functional Anti-MiR-9 by Mesenchymal Stem Cell-Derived Exosomes to Glioblastoma Multiforme Cells Conferred Chemosensitivity. Mol. Ther. - Nucleic Acids 2013, 2, e126. [Google Scholar] [CrossRef]
- Shahjin, F.; Chand, S.; Yelamanchili, S.V. Extracellular Vesicles as Drug Delivery Vehicles to the Central Nervous System. J. Neuroimmune Pharmacol. 2020, 15, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Macedo-Pereira, A.; Martins, C.; Lima, J.; Sarmento, B. Digging the Intercellular Crosstalk via Extracellular Vesicles: May Exosomes Be the Drug Delivery Solution for Target Glioblastoma? J. Control. Release 2023, 358, 98–115. [Google Scholar] [CrossRef]
- Benecke, L.; Coray, M.; Umbricht, S.; Chiang, D.; Figueiró, F.; Muller, L. Exosomes: Small Evs with Large Immunomodulatory Effect in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 3600. [Google Scholar] [CrossRef]
- Chen, Y.S.; Lin, E.Y.; Chiou, T.W.; Harn, H.J. Exosomes in Clinical Trial and Their Production in Compliance with Good Manufacturing Practice. Tzu Chi Med. J. 2020, 32, 113–120. [Google Scholar] [CrossRef]
- Picciotto, S.; Santonicola, P.; Paterna, A.; Rao, E.; Raccosta, S.; Romancino, D.P.; Noto, R.; Touzet, N.; Manno, M.; Schiavi, E.D.; et al. Extracellular Vesicles From Microalgae: Uptake Studies in Human Cells and Caenorhabditis Elegans. Front. Bioeng. Biotechnol. 2022, 10, 830189. [Google Scholar] [CrossRef]
- Niu, W.; Xiao, Q.; Wang, X.; Zhu, J.; Li, J.; Liang, X.; Peng, Y.; Wu, C.; Lu, R.; Pan, Y.; et al. A Biomimetic Drug Delivery System by Integrating Grapefruit Extracellular Vesicles and Doxorubicin-Loaded Heparin-Based Nanoparticles for Glioma Therapy. Nano Lett. 2021, 21, 1484–1492. [Google Scholar] [CrossRef]
- Giancaterino, S.; Boi, C. Alternative Biological Sources for Extracellular Vesicles Production and Purification Strategies for Process Scale-Up. Biotechnol. Adv. 2023, 63, 108092. [Google Scholar] [CrossRef]
- Adamo, G.; Fierli, D.; Romancino, D.P.; Picciotto, S.; Barone, M.E.; Aranyos, A.; Božič, D.; Morsbach, S.; Raccosta, S.; Stanly, C.; et al. Nanoalgosomes: Introducing Extracellular Vesicles Produced by Microalgae. J. Extracell. Vesicles 2021, 10, e12081. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Gu, J.; Zhang, J.; Shi, H.; Qian, H.; Wang, D.; Xu, W.; Pan, J.; Santos, H.A. Engineered Extracellular Vesicles for Cancer Therapy. Adv. Mater. 2021, 33, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of SiRNA to the Mouse Brain by Systemic Injection of Targeted Exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.M.; Wiklander, P.B.O.; Nordin, J.Z.; Al-Shawi, R.; Wood, M.J.; Vithlani, M.; Schapira, A.H.V.; Simons, J.P.; El-Andaloussi, S.; Alvarez-Erviti, L. Systemic Exosomal SiRNA Delivery Reduced Alpha-Synuclein Aggregates in Brains of Transgenic Mice. Mov. Disord. 2014, 29, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Kim, M.; Lee, Y.; Byun, J.W.; Hwang, D.W.; Lee, M. Systemic Delivery of MicroRNA-21 Antisense Oligonucleotides to the Brain Using T7-Peptide Decorated Exosomes. J. Control. Release 2020, 317, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of Brain Inflammatory Diseases by Delivering Exosome Encapsulated Anti-Inflammatory Drugs from the Nasal Region to the Brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome Delivered Anticancer Drugs across the Blood-Brain Barrier for Brain Cancer Therapy in Danio Rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef]
- Xunian, Z.; Kalluri, R. Biology and Therapeutic Potential of Mesenchymal Stem Cell-Derived Exosomes. Cancer Sci. 2020, 111, 3100–3110. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; von der Ohe, J.; Hass, R. Msc-Derived Extracellular Vesicles in Tumors and Therapy. Cancers (Basel). 2021, 13, 5212. [Google Scholar] [CrossRef]
- Pavon, L.F.; Sibov, T.T.; De Souza, A.V.; Da Cruz, E.F.; Malheiros, S.M.F.; Cabral, F.R.; De Souza, J.G.; Boufleur, P.; De Oliveira, D.M.; De Toledo, S.R.C.; et al. Tropism of Mesenchymal Stem Cell toward CD133+ Stem Cell of Glioblastoma in Vitro and Promote Tumor Proliferation in Vivo. Stem Cell Res. Ther. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Lang, F.M.; Hossain, A.; Gumin, J.; Momin, E.N.; Shimizu, Y.; Ledbetter, D.; Shahar, T.; Yamashita, S.; Parker Kerrigan, B.; Fueyo, J.; et al. Mesenchymal Stem Cells as Natural Biofactories for Exosomes Carrying MiR-124a in the Treatment of Gliomas. Neuro. Oncol. 2018, 20, 380–390. [Google Scholar] [CrossRef]
- Fareh, M.; Almairac, F.; Turchi, L.; Burel-Vandenbos, F.; Paquis, P.; Fontaine, D.; Lacas-Gervais, S.; Junier, M.P.; Chneiweiss, H.; Virolle, T. Cell-Based Therapy Using MiR-302-367 Expressing Cells Represses Glioblastoma Growth. Cell Death Dis. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, V.; Nowicki, M.O.; Idriss, M.; Jimenez, M.A.; Lugli, G.; Hayes, J.L.; Mahmoud, A.B.; Zane, R.E.; Passaro, C.; Ligon, K.L.; et al. The Functional Synergism of MicroRNA Clustering Provides Therapeutically Relevant Epigenetic Interference in Glioblastoma. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zeng, A.; Wei, Z.; Yan, W.; Yin, J.; Huang, X.; Zhou, X.; Li, R.; Shen, F.; Wu, W.; Wang, X.; et al. Exosomal Transfer of MiR-151a Enhances Chemosensitivity to Temozolomide in Drug-Resistant Glioblastoma. Cancer Lett. 2018, 436, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, J.; Ao, F.; Tang, Y.; Xu, P.; Wang, M.; Huang, M. Tumor-Derived Exosomal MicroRNA-7-5p Enhanced by Verbascoside Inhibits Biological Behaviors of Glioblastoma in Vitro and in Vivo. Mol. Ther. - Oncolytics 2021, 20, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Hu, G.; Xia, Y.; Li, H.Y.; Yuan, J.; Zhang, J.; Chen, Y.; Guo, H.; Yang, Y.; Wang, Y.; et al. Eliminating the Original Cargos of Glioblastoma Cell-Derived Small Extracellular Vesicles for Efficient Drug Delivery to Glioblastoma with Improved Biosafety. Bioact. Mater. 2022, 16, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Geng, T.; Leung, E.; Chamley, L.W.; Wu, Z. Functionalisation of Extracellular Vesicles with Cyclic-RGDyC Potentially for Glioblastoma Targeted Intracellular Drug Delivery. Biomater. Adv. 2023, 149, 213388. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W. Retinoic Acid Induces Neuronal Differentiation of a Cloned Human Embryonal Carcinoma Cell Line in Vitro. Dev. Biol. 1984, 103, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Langlois, A.; Duval, D. Differentiation of the Human NT2 Cells into Neurons and Glia. 1997, 219, 213–219.
- Tyler, M.A.; Ulasov, I.V.; Sonabend, A.M.; Nandi, S.; Han, Y.; Marler, S.; Roth, J.; Lesniak, M.S. Neural Stem Cells Target Intracranial Glioma to Deliver an Oncolytic Adenovirus in Vivo. Gene Ther. 2009, 16, 262–278. [Google Scholar] [CrossRef] [PubMed]
- Attia, N.; Mashal, M.; Grijalvo, S.; Eritja, R.; Puras, G.; Pedraz, J.L. Cationic Niosome-Based HBMP7 Gene Transfection of Neuronal Precursor NT2 Cells to Reduce the Migration of Glioma Cells in Vitro. J. Drug Deliv. Sci. Technol. 2019, 53. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, S. Human NT2 Neural Precursor-Derived Tumor-Infiltrating Cells as Delivery Vehicles for Treatment of Glioblastoma. Hum. Gene Ther. 2010, 21, 683–694. [Google Scholar] [CrossRef]
- Adamus, T.; Hung, C.-Y.; Yu, C.; Kang, E.; Hammad, M.; Flores, L.; Nechaev, S.; Zhang, Q.; Gonzaga, J.M.; Muthaiyah, K.; et al. Glioma-Targeted Delivery of Exosome-Encapsulated Antisense Oligonucleotides Using Neural Stem Cells. Mol. Ther. - Nucleic Acids 2022, 27, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Persico, M.G.; Liguori, G.L.; Parisi, S.; D’Andrea, D.; Salomon, D.S.; Minchiotti, G. Cripto in Tumors and Embryo Development. Biochim. Biophys. Acta - Rev. Cancer 2001, 1552. [Google Scholar] [CrossRef]
- Minchiotti, G.; Parisi, S.; Liguori, G.L.; D’Andrea, D.; Persico, M.G. Role of the EGF-CFC Gene Cripto in Cell Differentiation and Embryo Development. Gene 2002, 287. [Google Scholar] [CrossRef] [PubMed]
- de Castro, N.P.; Rangel, M.C.; Nagaoka, T.; Salomon, D.S.; Bianco, C. Cripto-1: An Embryonic Gene That Promoted Tumorigeneis. Futur. Oncol. 2010, 6, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Mantile, F.; Kisovec, M.; Adamo, G.; Romancino, D.P.; Hočevar, M.; Božič, D.; Bedina Zavec, A.; Podobnik, M.; Stoppelli, M.P.; Kisslinger, A.; et al. A Novel Localization in Human Large Extracellular Vesicles for the EGF-CFC Founder Member CRIPTO and Its Biological and Therapeutic Implications. Cancers (Basel). 2022, 14, 3700. [Google Scholar] [CrossRef]
- Minchiotti, G.; Parisi, S.; Liguori, G.; Signore, M.; Lania, G.; Adamson, E.D.; Lago, C.T.; Persico, M.G. Membrane-Anchorage of Cripto Protein by Glycosylphosphatidylinositol and Its Distribution during Early Mouse Development. Mech. Dev. 2000, 90. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, Y.; Zhang, M.; Li, T.; Zheng, X.; Guo, Q.; Zhang, X. Exosomal Cripto-1 Serves as a Potential Biomarker for Perihilar Cholangiocarcinoma. Front. Oncol. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Parisi, S.; D’Andrea, D.; Lago, C.T.; Adamson, E.D.; Persico, M.G.; Minchiotti, G. Nodal-Dependent Cripto Signaling Promotes Cardiomyogenesis and Redirects the Neural Fate of Embryonic Stem Cells. J. Cell Biol. 2003, 163, 303–314. [Google Scholar] [CrossRef]
- Liguori, G.L.; Echevarría, D.; Improta, R.; Signore, M.; Adamson, E.; Martínez, S.; Persico, M.G. Anterior Neural Plate Regionalization in Cripto Null Mutant Mouse Embryos in the Absence of Node and Primitive Streak. Dev. Biol. 2003, 264, 537–549. [Google Scholar] [CrossRef]
- Liguori, G.L.; Echevarria, D.; Bonilla, S.; D’Andrea, D.; Liguoro, A.; Persico, M.G.; Martinez, S. Characterization of the Functional Properties of the Neuroectoderm in Mo Use Cripto-/- Embryos Showing Severe Gastrulation Defects. Int. J. Dev. Biol. 2009, 53. [Google Scholar] [CrossRef]
- Pilgaard, L.; Mortensen, J.H.; Henriksen, M.; Olesen, P.; Sørensen, P.; Laursen, R.; Vyberg, M.; Agger, R.; Zachar, V.; Moos, T.; et al. Cripto-1 Expression in Glioblastoma Multiforme. Brain Pathol. 2014, 24, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, B.B.; Saetran, H.A.; Mørk, S.J.; Margaryan, N. V; Eide ¶,#, G.E.; Petersen, K.; Strizzi, L.; Hendrix, M.J.C. Age-Dependent Association between Protein Expression of the Embryonic Stem Cell Marker Cripto-1 and Survival of Glioblastoma Patients 1,2. Transl. Oncol. 2013, 6, 732. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Rangel, M.C.; Castro, N.P.; Nagaoka, T.; Rollman, K.; Gonzales, M.; Salomon, D.S. Role of Cripto-1 in Stem Cell Maintenance and Malignant Progression. Am. J. Pathol. 2010, 177, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, E.; Liguoro, A.; D’Orsi, L.; Mancinelli, S.; Barbieri, A.; Palma, G.; Arra, C.; Liguori, G.L. Cripto Haploinsufficiency Affects in Vivo Colon Tumor Development. Int. J. Oncol. 2014, 45. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.T.; Witwer, K.W.; VVan Balkim, B.W.M.; Rohde, E.; KIANG, Li.S. Concise Review: Developing Best-Practice Models for the Therapeutic Use of Extracellular Vesicles. STEMCELLS Transl. Med. 2017;61730–1739 2017, 6, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, V.V.; Svistunov, A.A.; Chubarev, V.N.; Dostdar, S.A.; Sokolov, A.V.; Brzecka, A.; Sukocheva, O.; Neganova, M.E.; Klochkov, S.G.; Somasundaram, S.G.; et al. Extracellular Vesicles in Cancer Nanomedicine. Semin. Cancer Biol. 2019, 69, 212–225. [Google Scholar] [CrossRef]
- Welsh, J.A.; Van Der Pol, E.; Arkesteijn, G.J.; Bremer, M.; Brisson, A.; Coumans, F.; Dignat-George, F.; Duggan, E.; Ghiran, I.; Giebel, B.; et al. MIFlowCyt-EV: A Framework for Standardized Reporting of Extracellular Vesicle Flow Cytometry Experiments; MIFlowCyt-EV: A Framework for Standardized Reporting of Extracellular Vesicle Flow Cytometry Experiments. 2020. [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018. [Google Scholar] [CrossRef]
- Witwer, K.W.; Soekmadji, C.; Hill, A.F.; Wauben, M.H.; Buzás, E.I.; Di Vizio, D.; Falcon-Perez, J.M.; Gardiner, C.; Hochberg, F.; Kurochkin, I.V.; et al. Updating the MISEV Minimal Requirements for Extracellular Vesicle Studies: Building Bridges to Reproducibility. J. Extracell. Vesicles 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Boilard, E.; Buzas, E.I.; Cheng, L.; Falcón-Perez, J.M.; Gardiner, C.; Gustafson, D.; Gualerzi, A.; Hendrix, A.; Hoffman, A.; et al. Considerations towards a Roadmap for Collection, Handling and Storage of Blood Extracellular Vesicles. J. Extracell. Vesicles 2019, 8. [Google Scholar] [CrossRef]
- Nieuwland, R.; Siljander, P.R.M.; Falcón-Pérez, J.M.; Witwer, K.W. Reproducibility of Extracellular Vesicle Research. Eur. J. Cell Biol. 2022, 101. [Google Scholar] [CrossRef]
- Bongiovanni, A.; Colotti, G.; Liguori, G.L.; Di Carlo, M.; Digilio, F.A.; Lacerra, G.; Mascia, A.; Cirafici, A.M.; Barra, A.; Lanati, A.; et al. Applying Quality and Project Management Methodologies in Biomedical Research Laboratories: A Public Research Network’s Case Study. Accredit. Qual. Assur. 2015, 20, 203–213. [Google Scholar] [CrossRef]
- Hollmann, S.; Regierera, B.; D’Elia, D.; Kisslinger, A.; Liguori, G.L. Toward the Definition of Common Strategies for Improving Reproducibility, Standardization, Management, and Overall Impact of Academic Research. Adv. Biomembr. Lipid Self-Assembly 2022, 35, 2–24. [Google Scholar]
- Digilio, F.A.; Lanati, A.; Bongiovanni, A.; Mascia, A.; Di Carlo, M.; Barra, A.; Cirafici, A.M.; Colotti, G.; Kisslinger, A.; Lacerra, G.; et al. Quality-Based Model for Life Sciences Research Guidelines. Accredit. Qual. Assur. 2016, 21. [Google Scholar] [CrossRef]
- Liguori, G.L.; Kisslinger, A. Standardization and Reproducibility in EV Research: The Support of a Quality Management System. In Biological Membrane Vesicles: Scientific, Biotechnological and Clinical Considerations. Advances in Biomembranes and Lipid Self-Assembly; Elsiever, 2021.
- Liguori, G.L.; Kisslinger, A. Quality Management Tools on the Stage: Old but New Allies for Rigor and Standardization of Extracellular Vesicle Studies. Front. Bioeng. Biotechnol. 2022, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mascia, A.; Cirafici, A.M.; Bongiovanni, A.; Colotti, G.; Lacerra, G.; Di Carlo, M.; Digilio, F.A.; Liguori, G.L.; Lanati, A.; Kisslinger, A. A Failure Mode and Effect Analysis (FMEA)-Based Approach for Risk Assessment of Scientific Processes in Non-Regulated Research Laboratories. Accredit. Qual. Assur. 2020, 25, 311–322. [Google Scholar] [CrossRef]
- Herwig, C.; Pörtner, R.; Möller, J. Digital Twins Tools and Concepts for Smart Biomanufacturing; 2022. ISBN 9783030716592.
- Mancinelli, S.; Zazzu, V.; Turcato, A.; Lacerra, G.; Digilio, F.A.; Mascia, A.; Di Carlo, M.; Cirafici, A.M.; Bongiovanni, A.; Colotti, G.; et al. Applying Design of Experiments Methodology to PEI Toxicity Assay on Neural Progenitor Cells. In Mathematical Models in Biology: Bringing Mathematics to Life; Zazzu, V., Ferraro, M.B., Guarracino, M.R., Eds.; Springer International Publishing: Cham, 2015; pp. 45–63. ISBN 978-3-319-23497-7. [Google Scholar]
- Mancinelli, S.; Turcato, A.; Kisslinger, A.; Bongiovanni, A.; Zazzu, V.; Lanati, A.; Liguori, G.L. Design of Transfections: Implementation of Design of Experiments for Cell Transfection Fine Tuning. Biotechnol. Bioeng. 2021, 118, 4488–4502. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Faruqu, F.N.; Liam-or, R.; Abu Abed, O.; Li, D.; Venner, K.; Errington, R.J.; Summers, H.; Wang, J.T.-W.; Al-Jamal, K.T. Design of Experiment (DoE)-Driven in Vitro and in Vivo Uptake Studies of Exosomes for Pancreatic Cancer Delivery Enabled by Copper-Free Click Chemistry-Based Labelling. J. Extracell. Vesicles 2020, 9, 1779458. [Google Scholar] [CrossRef]
- Loria, F.; Picciotto, S.; Adamo, G.; Zendrini, A.; Raccosta, S.; Manno, M. A Decision-Making Tool to Navigate through Extracellular Vesicle Research and Product Development. 2023.
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.A.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic Biodistribution of Extracellular Vesicles in Vivo Using a Multimodal Imaging Reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular Vesicle in Vivo Biodistribution Is Determined by Cell Source, Route of Administration and Targeting. J. Extracell. Vesicles 2015, 4, 1–13. [Google Scholar] [CrossRef]
- Jia, G.; Han, Y.; An, Y.; Ding, Y.; He, C.; Wang, X.; Tang, Q. NRP-1 Targeted and Cargo-Loaded Exosomes Facilitate Simultaneous Imaging and Therapy of Glioma in Vitro and in Vivo. Biomaterials 2018, 178, 302–316. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-Tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.; Dubois, G.G.; Xavier, L.L.; Geraldo, H.H.; da Fonseca, C.C.C.; Correia, H.H.; Meirelles, F.; Ventura, G.; Romão, L.; Canedo, S.H.S.; et al. The Orthotopic Xenotransplant of Human Glioblastoma Successfully Recapitulates Glioblastoma-Microenvironment Interactions in a Non-Immunosuppressed Mouse Model. BMC Cancer 2014, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; García-Bernal, D.; Bueno, C.; Ródenas, M.; Moraleda, J.M.; Macian, F.; Martínez, S. Glioblastoma Progression Is Assisted by Induction of Immunosuppressive Function of Pericytes through Interaction with Tumor Cells. Oncotarget 2017, 8, 68614–68626. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Main features of Glioblastoma responsible of his high malignancy and poor prognosis.
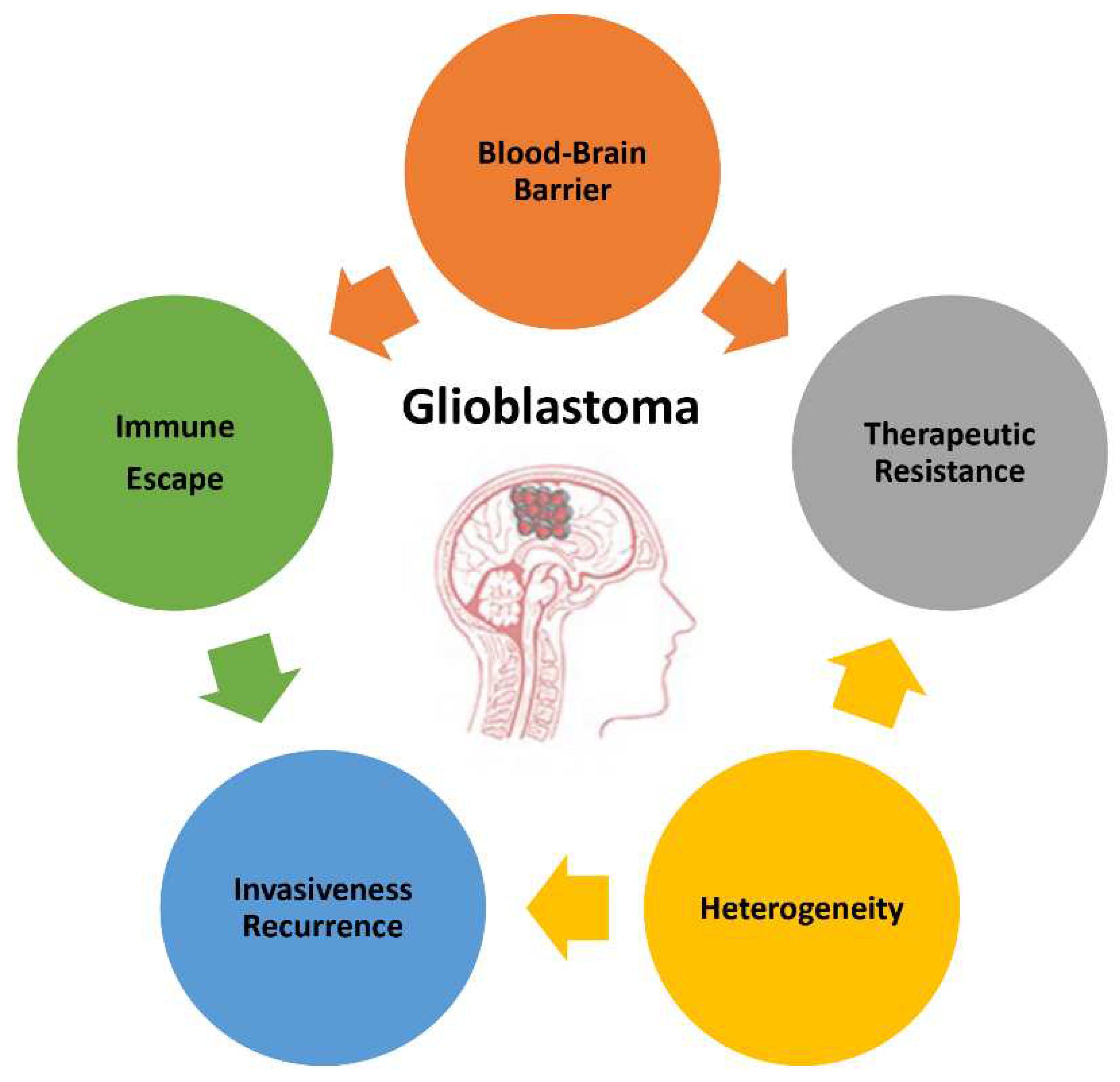
Figure 2.
EV exploitation strategy for Glioblastoma treatment. The Figure schematizes on the left the identified targets to block EV release and uptake or impair specific oncogenic glioblastoma features, in the middle the EV sources and on the right the therapeutic molecules used in functional assays in Gliobastoma models (both in vitro and in vivo).
Figure 2.
EV exploitation strategy for Glioblastoma treatment. The Figure schematizes on the left the identified targets to block EV release and uptake or impair specific oncogenic glioblastoma features, in the middle the EV sources and on the right the therapeutic molecules used in functional assays in Gliobastoma models (both in vitro and in vivo).
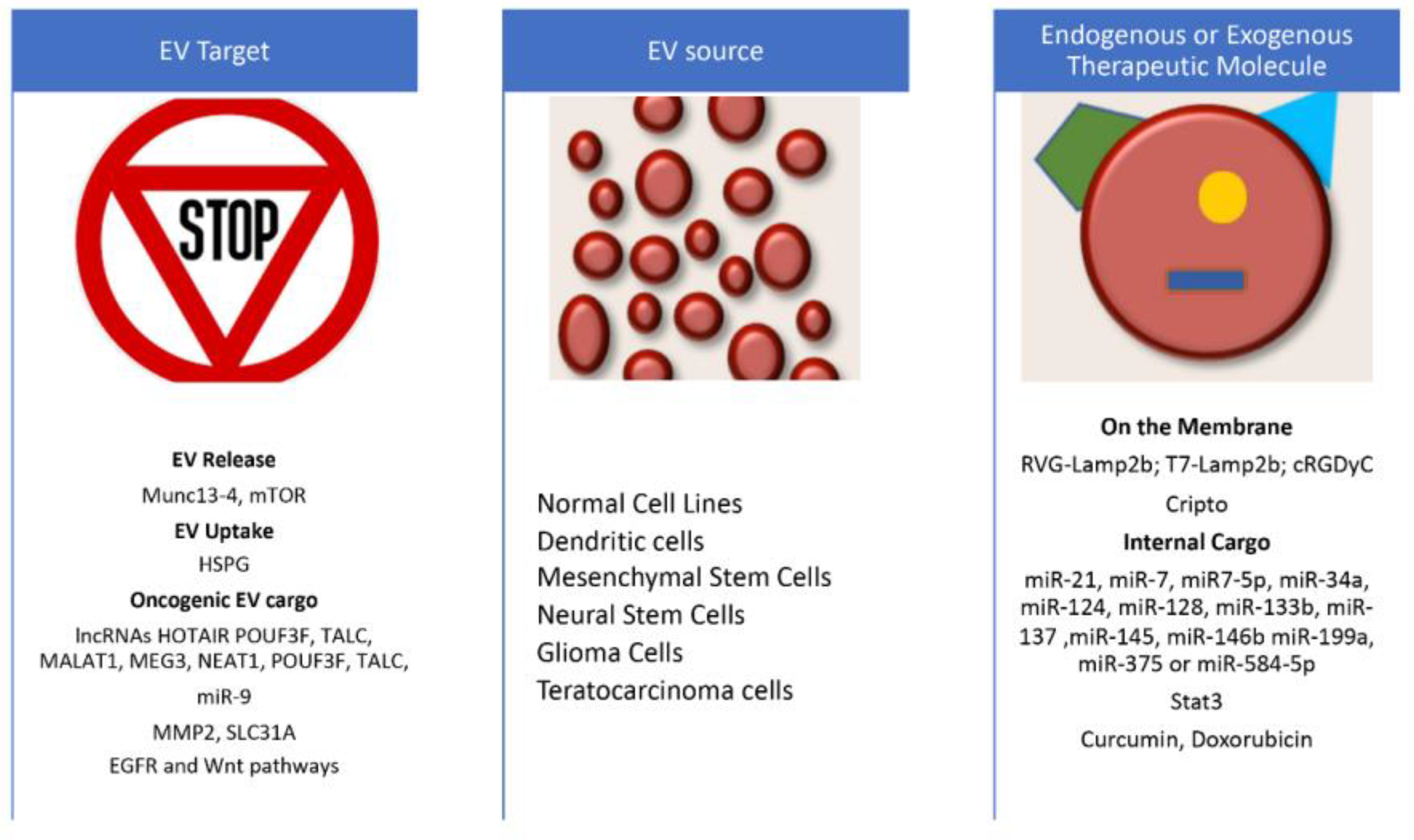
Figure 3.
SWOT analysis of the EV exploitationin GB treatment, highlighting the relative Strenghts (S), Weaknesses (W), Opportunities (O), and Threats (T).
Figure 3.
SWOT analysis of the EV exploitationin GB treatment, highlighting the relative Strenghts (S), Weaknesses (W), Opportunities (O), and Threats (T).
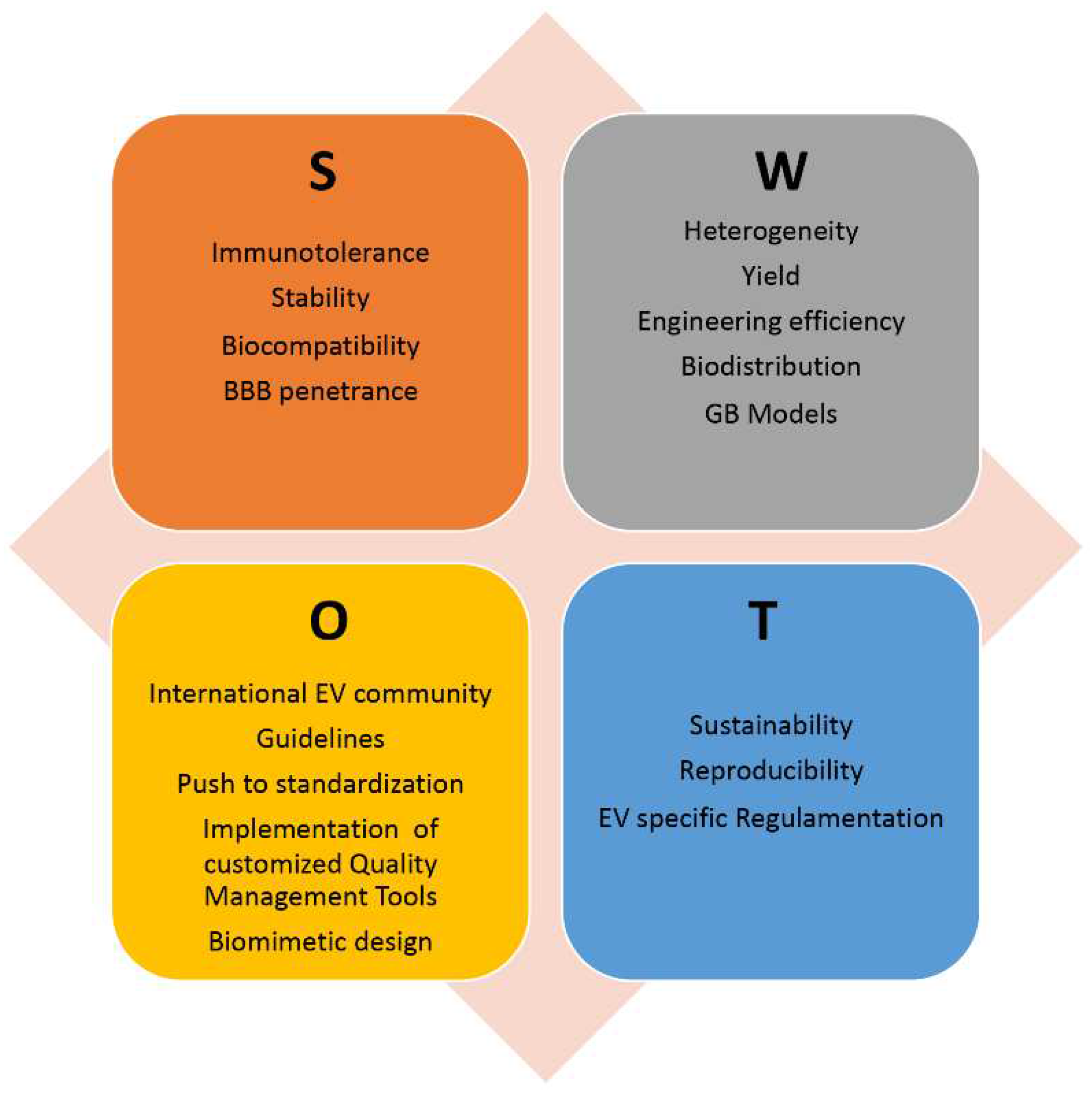
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



