
Submitted:
15 December 2023
Posted:
18 December 2023
You are already at the latest version
Abstract
A defining feature of a productive infection is the co-opting of host cell resources for viral replication. Despite their repertoire of molecular functions, viruses subvert host defenses to take control of cellular factors such as RNA binding proteins (RBPs). RBPs are involved in virtually all steps of mRNA life forming ribonucleoprotein complexes (mRNPs) in a highly ordered and regulated process to control RNA fate and stability in the cell. Thus, the hallmark of this viral takeover is to reshape RNA fate in the cell to modulate host gene expression and evade immune responses. Here we provide an extensive review of work in this area, particularly how the host-viral interplay influences RBP functions to modulate the host cell. Overall, in this review, we highlight the myriad of ways RBPs can regulate RNA stability in either a pro-viral or antiviral manner by gathering novel insights gained from research studies in this field.
Keywords:
RNA binding proteins
; protein-RNA interactions
; RNA decay
; RNA granules
; viruses
1. Introduction
Viruses rely on intricate hijacking mechanisms to seize control of the host gene expression machinery for their own replication. Given their important regulatory roles in processes such as splicing, stability, localization, degradation, export, and translation, RNA binding proteins (RBPs) are at the center of this battle to control the gene expression resources [1,2,3]. Ribonucleoprotein complexes (RNPs) are assembled on mRNA, shaping the fate of transcripts and are recruited via structural elements, sequence motifs, or sequence-independent secondary and tertiary structures [1,4]. Thus, the mRNA-protein interface can vary widely from forming stable RNP complexes to transient interactions. In infected cells, this is further complicated by the presence of viral proteins that may disrupt any number of these post-transcriptional processes. For instance, localization of RBPs to different subcellular compartments has been shown to be highly hijacked by viruses in order to control host gene expression and reprogram the cell machinery towards viral transcripts [5,6,7]. Likewise, inhibition of 3′ end processing by viruses was shown to lead to production of read-through transcripts and failed transcription termination which can affect the repertoire of available mRNAs in the cell and therefore the extent of the host anti-viral defense arsenal [8,9]. Other mechanisms have also been shown to contribute to the viral-host interplay, such as manipulation of nuclear export to allow retention of host mRNAs in the nucleus leading to their targeted degradation [10,11] and therefore freeing up the translational machinery for viral mRNA processing [11,12]. Furthermore, the highly intricate functionality of membrane-less organelles containing RNAs and RBPs are formed as compartmentalized densities for host post-transcriptional regulation or as viral replication factories [13,14,15]. While an extensive amount of work in the field has shown the importance of RBPs in the regulation of gene expression, and as a particular crux of targeting during viral infection[5,6,16,17], one important gap remains to be explored: how can the same RNP complexes be pro-viral in some context while anti-viral in others? Studies portraying the impact of RNA-protein interactions during viral infection are an exciting avenue of research and will likely yield important information on transcript regulation both in pathogenic and non-pathogenic contexts. In this review we will explore the complex interplay between RNA and proteins and their role(s) in RNA stability during viral infections, shedding light on new avenues to explore RNA fate and RBP modulation during viral infections in the future.
2. RNA Elements and Viral Infection
To rapidly gain access to their host translational resources, certain viruses induce broad RNA degradation events orchestrated directly by their own viral endonucleases. These enzymes cleave processed transcripts, creating unprotected 5′ and 3′ ends that become targets for host exonucleases. Interestingly, recent work has begun to reveal RNA elements that are incorporated into select mRNAs that can drastically impact RNA stability in the face of these viral-induced decay events. Some of these RNA elements serve as target sites that recruit viral nucleases, while others can provide protective measures against viral-induced decay. For example, during Kaposi Sarcoma Associated Herpesvirus (KSHV) lytic replication, the virally encoded endonuclease SOX causes widespread decay of the mRNA transcriptome. However, a fraction of the transcriptome is spared from SOX-mediated degradation which likely ensures cell viability and proper progression viral replication. Thus, it appears that SOX targets are carefully selected and in fact, the Gaglia group has recently mapped SOX targeting motif that is preferentially used to during KSHV infection [18]. This targeting motif does not rely solely on sequence but rather on several key conserved residues alongside a potentially conserved structural motif [18,19]. Similarly, during Influenza A infection, the frameshift product PA-X functions as an endonuclease and contributes to the global viral-induced host shutoff. PA-X activity results in this decay event to free host resources for viral replication and negatively alter host defenses, but how does PA-X specifically locate the right transcripts to degrade? Through 5′ rapid amplification of complementary DNA ends coupled to high-throughput sequencing and predictions of RNA structures, Gaucherand et al. determined that PA-X targets GCUG tetramers in hairpin loops within transcripts for cleavage [20]. This specific element is widely abundant in the host genome but not in the viral genome, thus serving as a highly specific target to specifically differentiate host mRNAs [21]. It therefore emerges that using viral nucleases with a wide range of host transcripts is an effective way for viruses to remodel their host cell environment and that the careful picking of the nucleases targets is a key step in this process. The question then arises of whether the host fights back against this takeover and whether certain RNA elements could instead potentially restrict viral nuclease targeting? This is in fact what has been observed in the context of KSHV infection: the transcript for Interleukin-6 (IL-6) encodes an RNA element within its 3′ UTR shown to provide protection from SOX [22,23]. This RNA element of about 200nt was termed the Sox Resistant Element (SRE) [23]. This SRE appears to fold into a stemloop structure that likely served as a scaffold for recruiting RBPs and modulate the susceptibility to the viral endonuclease [23]. Several RBPs were identified as specifically binding this RNA element and together form a protective RNP. Intriguingly, this protective element appears to dominate over SOX targeting element, as a transcript that contains both the targeting and the protective elements will remain unaffected by SOX. This highlights that the fate of an mRNA and its stability in the face of viral infection is the result of a complex balance of its RBP landscape. This RNP environment on individual transcripts may serve as specific marker in the context of viral-induced decay and provide clues to understand how viruses can distinguish between host and viral genes.
3. Host - Viral RBP role during viral infections
Through evolutionary pressure and the imperative to fit through their host translational machinery, viruses have often found ways to mimic features of the host to hijack resources and facilitate their escape from the immune system [24,25,26]. By mimicking host features, especially on their transcripts, viruses also facilitate the hijacking of the RBP of the host and thus create RNP complexes that are indistinguishable from host RNP. For instance, during Dengue Virus (DENV) infection, the Receptor for activated C kinase 1 (RACK1) - a core component of the 40S ribosomal subunit- was shown to interact with host factors SERBP1 and Vigilin to promote viral replication [27]. The authors denoted RACK1 as a platform to bind and recruit SERBP1 and Vigilin to the 40S ribosomal subunit to then collectively connect viral RNAs to the translation machinery to facilitate DENV infection. Furthermore, Diosa-Toro and colleagues observed the host factor Y box binding protein 1 (YBX1) interaction with the structural protein E and the viral protein capsid C to facilitate viral assembly and egress in DENV [28]. These studies, among others, ascertain the role of cellular factors to aid processes that would promote viral replication and infection progression in the host cell [29,30,31].
However, not all cellular RBP favor viral replication, and mimicking host mRNA can be a double edge sword as some RBP can have potent antiviral effects. The host factor Shiftless (SHFL) for example is an RBP that has been shown to restrict expression of a number of viruses through its interaction with viral RNA, from inhibiting HIV frameshifts to manipulating cytoplasmic RNP granules [17,32,33]. Another example is the RNA binding protein polypyrimidine tract-binding protein 1 (PTBP1) which was shown by Qin et al. to target and degrade the viral nucleocapsid (N) protein by activating the MARCH8-NDP52 autophagosome pathway in Porcine epidemic diarrhea virus (PEDV) [34]. Moreover, in this study PTBP1 was also shown to activate type I interferon (IFN) pathway inhibiting PEDV replication [34].
And inevitably, some RBPs can have both pro or anti-viral roles depending on the context: for example, the RNA binding protein Ras GTPase-activating protein binding protein 1 (G3BP1) is known to mediate interactions with innate immune signaling molecules (RIG-I, cGAS, Caprin1) to facilitate the activation of immune pathways to counteract viral infections [7,35,36,37,38]. But G3BP1 can also be targeted to favor viral replication by different viruses [7,39] such as during ZIKV infection where G3BP1 is sequestered to disrupt stress granule formation and promote infection [39]. Collectively, these studies illustrate the profound impact cellular RBPs can have on the outcome of an infection. Interestingly, we can further ponder on the mechanisms by which cellular factors might discriminate between an RNA as self or non-self to exert its effects. Yet, we must take into consideration that the notion of what we try to designate as a “host specific RBP” and “viral specific RBP” can become a little obscure to define. Considering that cellular factors can reveal functionalities that are not restricted to viral RNAs or cellular RNAs but merely part of both. More studies in this area are needed to continue to elucidate the mechanisms by which cellular factors modulate processes in a pro-viral or antiviral way during viral infections.
4. RBP modulation of RNA stability in subcellular localizations during viral infections
The physical separation between the processes of transcription and translation enables RNA-Protein complexes (RNPs) to adapt and diversify throughout the life of an mRNA contingent on its subcellular localization. However, during viral infections, viral RNAs (vRNAs) compete with cellular RNAs to take over resources from the cell. This can lead to the disruption of common RBPs function and localization between cellular compartments or their reallocation towards vRNAs. To date, several studies have investigated many of the mechanisms in which RBPs modulate key pathways in response to environmental changes, particularly during viral infection [40,41,42,43]. In this section, we will review how RBPs regulate RNA stability between cell compartments during viral infections.
4.1. Nuclear and Cytoplasmic Regulation
RNA fate is highly controlled in the cell and RBPs are at the core of the complex process that regulates targeting between cellular compartments. In particular, in the context of viral infection where resources are re-allocated towards viral needs, the dynamic process of nuclear-cytoplasmic shuttling can be drastically altered. For example, in the context of gamma-herpesvirus infection where host shutoff takes the form of widespread mRNA decay, many RBP find themselves without an RNA target. Hence this massive RNA decay results in a broad scale relocalization of RBPs, many of them finding their way back to the nucleus. This thus creates a measurable “sensing” of this large-scale RNA decay in the cytoplasm, and the message can be relayed to the nucleus through the shuttling of RBPs. It was shown that this creates a feedback mechanism, akin to informing the transcription machinery of the state of RNA decay in the cytoplasm, and resulting in a halt of host transcription. Taken together, studies uncovered an intriguing interconnection between transcription rates in the nucleus and the stability of mRNAs in the cytoplasm [44,45,46,47]. Therefore, trafficking of RBPs between cellular compartments during events of stress such as viral-induced RNA decay, could be fundamental to relaying information about cellular mRNA abundance within the cell [44,47,48]. Several studies have detailed the modulation of RBPs during stress and between cellular compartments [48,49,50,51]. Differences on compartmentalization and functionality are observed primarily dependent on the pathway; compartment specific, shuttling compartments, RNA abundance or stress-regulation [49]. Interestingly, from these studies we can further inquire if host mRNA availability is reduced during targeted viral host mRNA degradation; to what extent protein abundance levels are regulated in subcellular compartments to somehow maintain “homeostasis” when re-localization of RBPs occur to different compartments? And to what extent does the cellular feedback response toward viral targeted RNA influence viral replication?
Another study by Garcia-Moreno and colleagues observed similar dynamics of RBP response reflecting RNA availability during Sindbis Virus (SINV) infection. The authors used a system wide approach known as RNA-interactome capture (RIC) to determine the distribution of RBPs in cells infected with SINV. In the context of high loss of cellular RNAs and concomitant high levels of viral RNAs, they observed a remodeling of the RBP interactome. In particular, host cellular RBPs EIFD3 (cap binding protein) SRPK1(alternative splicing, RNA export and stability), IF16 (enhances RIG-I transcription and activation), IFT5 (antiviral response), TRIM25, TRIM56 (E3 ubiquitin ligase), PPIA (regulation, signaling, apoptosis), HSP90AB1 (RNP remodeling), RTCB, DDX1 and FAM98A (form the tRNA ligase complex) were re-localized to viral factories [29]. Moreover, another affected RBP, GEMIN5, which usually localized to both the nucleus and cytoplasm mediating the assembly of small nuclear RNPs (snRNPs) was re-localized to viral factories and co-localizes with SINV RNA [29].
From these studies and others, we can perceive the battle between host and viruses to take control of cellular gene expression. Notably, RBP function and localization appears to be pivotal during viral infections to co-opt cellular processes. Many questions continue to mount surrounding the mechanisms that regulate the shuttling of RBPs during viral infection, especially during viral-induced RNA decay. With the advent of novel protein labeling methods, we anticipate that we will learn more about the dynamics of these processes in the coming years.
4.2. Nuclear Export
Messenger RNAs (mRNAs) undergo extensive processing before their export from the nucleus to the cytoplasm. Once mature, mRNAs are transported to the cytoplasm through nuclear pore complexes (NPCs) embedded in the nuclear envelope [10,11,52,53]. Unsurprisingly, transport of mRNAs is mediated by mRNP complexes composed of shuttling proteins. Commonly, the transcriptional export (TREX) complex binds mRNAs and recruits other export factors such as the THO complex, UAP56 (also known as HEL), and ALY/REF (also known as THOC4). Subsequently, the ALY/REF complex then interacts with nuclear export factor 1 (NXF1 also known as TAP)-Ntf2 like export factor 1(NXT1 also known as TAP-p15) among other factors to facilitate export of mRNAs from the nucleus to the cytoplasm [11]. NXF1 is known to assist in the export of bulk mRNA, while a subset of transcripts (including unspliced and partially spliced), are exported via the chromosomal region maintenance 1 (CRM1 also known as XPO1) [10]. Interestingly, several studies have shown how viruses target the export machinery to cause a reduction in host gene expression and downregulation of host antiviral responses [11,54,55,56]. For instance, in Hepatitis B Virus (HBV), the viral core protein HBc contains an NLS sequence and an arginine rich domain (ARD) that allows its physical interaction with NFX1 [57]. This interaction between HBc and NFX1 is suggested to mediate the shuttling of HBc between cellular compartments (Figure 1) and facilitate the export of HBV transcripts [57]. Furthermore, Gong and colleagues in their study identified the interaction of the KSHV encoded ORF10 protein with nuclear export proteins Rae1 and Nup98. This interaction blocked mRNA export leading to nuclear accumulation of transcripts [56]. The accumulation of mRNAs and inhibition of export serves to downregulate expression of host proteins and ensure viral access to the translational machinery [56,58]. Meanwhile, in Murine Leukaemia Virus (MLV) both nuclear export routes (NFX1 and CRM1) are proposed to be exploited by the virus [59]. Mougel et al., show in their study the interaction between MLV full length RNA and NFX1 allowing export of viral RNAs to further load factors for translation initiation. Simultaneously, in their study export by CRM1 marked MLV FL RNAs for viral packaging in the cytoplasm [59]. The use of these two export pathways by MLV highlights the ability of the viral RNA to assemble two different mRNP complexes and control RNA fate. Strikingly, the nature of an mRNP complex could be changed by the strategies viruses use to exploit cellular pathways such as nuclear export. Ongoing and future studies should continue to elucidate the processes that regulate the fate of viral and cellular mRNAs in these pathways.
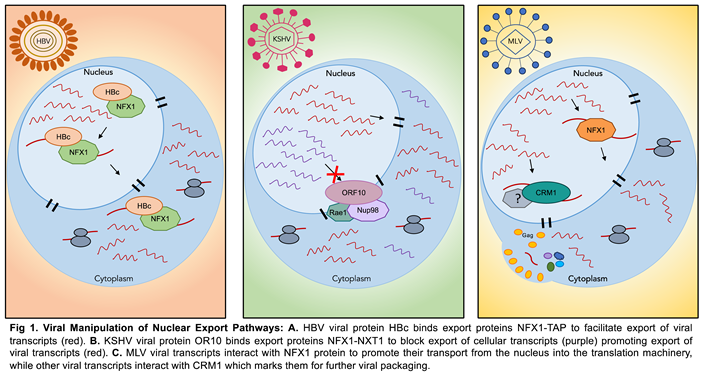 |
5. RNA Granules: A Nexus in the Viral-Host Struggle over RNA
RNA granules are biomolecular condensates of RNA and protein that exist within the cytoplasm. Though these densities are often described to undergo liquid-liquid phase separation (LLPS), the story is more complex. The description of LLPS helps to better illustrate what these foci are: sequestered conglomerates of biomolecules that are separated from the surrounding cytoplasmic matrix without any phospholipid barrier. However, the distinction can be quite gray in such a dynamic system; granules readily exchange materials with their surroundings [60,61,62]. Granules are not simply defined as viscous liquids but more precisely viscoelastic densities [63,64,65,66,67,68]. This distinction implies a duality and fluctuation of solid and liquid elements, which helps to better highlight how these subcellular compartments themselves are multifarious along with their functionality. Two classifications of cytoplasmic granules have emerged as important regulatory mechanisms within cells and both heavily rely on complex and dynamic interactions between RBP and RNA: Processing bodies (P-bodies) and Stress Granules (SGs). P-bodies are often comprised of mRNA and RBPs such as DDX6, ET-4, PAT1B, LSM14A, EDC4, DCP1/DCP2, and CPEB1 [69,70,71,72,73,74,75,76,77,78,79,80]. Along with ribonucleic acids, SGs often contain proteins such as G3BP1/G3BP2, TIA-1, TIAR, Atx2, eIF2, eIF3, eIF4A, eIF4B, eIF4E, eIF4G, and eIF5 [80,81,82,83,84,85,86,87,88]. Some granules, such as P-bodies, are constitutive, where treatment with a translation elongation inhibitor like cycloheximide will lead to their cytoplasmic loss [69,89,90]. Conversely, Stress Granules must be induced by treatment with a translation terminator such as puromycin [89,91] or via cellular stressors like viral agents [13]. Due to their dynamic nature, diversity in composition, and temporal/environmental sensitivity, RNA granules’ functionality varies broadly from mRNA storage, degradation, triage to assisting in cellular processes such as energetic conservation, the stress response, or the inflammatory response. Of note, this multitude of processes can readily be taken advantage of by viruses. Battle wages between virus and host over RNA granules’ RBPs to ultimately influence gene expression and arrest pathways, specifically over the control of RNAs.
5.1. Viral factors exercise control over P-body RBPs to promote viral replication
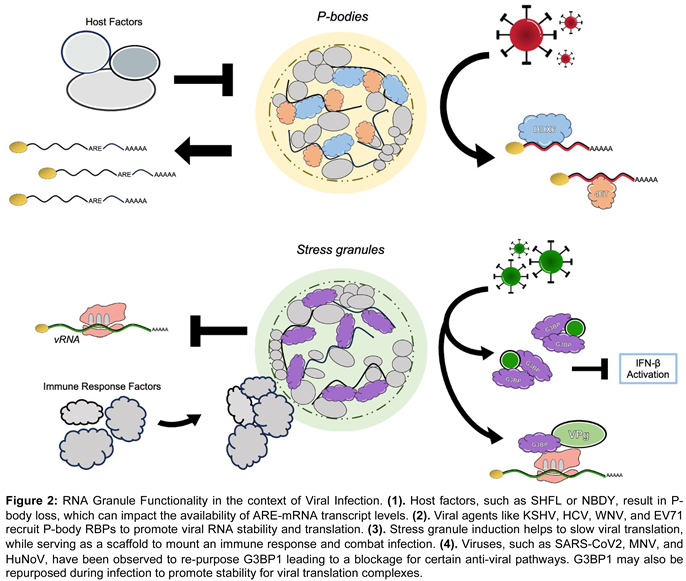
Processing bodies are thought to be sites of RNA metabolism within the cytoplasm of cells. P-bodies have been implicated in mRNA decay, translation repression, nonsense mediated decay (NMD), as well as mRNA silencing [76,92,93,94,95,96,97]. They have commonly been identified as sites of mRNA decay due several constituents’ ties to transcript degradation, namely DDX6, DCP1, DCP2, Xrn1, EDC3, EDC4, among others [69,70,71,72,73,74,75,76,77,78,79,80]. However, they can also serve as compartments to temporarily sequester some transcripts from the translation pool to control pathway activation/deactivation or for energetic conservation. P-bodies are thus prime targets for viruses that need to co-opt these pathways for their own replication. It has previously been shown that viruses such as KSHV, HCV, and WNV all utilize P-body constituents such as DDX6, Lsm-1, DDX3, Ago2, Xrn1 and others to promote viral RNA and protein stability [5,98,99,100,101,102,103,104,105]. Recently, investigation of Enterovirus 71 shows a similar strategy. This single stranded, positive sense RNA virus encodes for two proteases (2A and 3C) that help to cleave the viral polyprotein into 11 smaller proteins; however, some have speculated that these enzymes may be multifunctional especially during infection. Early characterization demonstrated that EV71 induces a loss of P-bodies within human cells; over the course of infection P-body microscopy and quantification revealed fewer foci that were generally smaller in size [106]. Using sodium arsenite (NaAs), an oxidative stress agent known to induce P-body formation, the researchers observed a significantly reduced amount of these granules within viral infected cells. Thus, Fan et al. were able to determine that EV71 blocks the formation of de novo P-bodies, where not even an extreme oxidative stressor can lead to their restoration. The group investigated whether the viral proteases played a role in this cytoplasmic granule loss. They ultimately unveiled that protease 2C leads to P-body loss and non-functional 2C has no effect on the granules [106]. Simultaneously, Fan et al. determined that certain scaffold proteins of P-bodies affected viral mRNA transcript levels. The absence of DDX6 and 4E-T lead to decreased viral transcript levels and viral mRNAs were elevated. Thus, they hypothesized that virally induced P-body loss reallocated and repurposed key P-body components for RNA stability and expression. During infection, a DDX6-VP1 (viral capsid protein) interaction was discovered and found to be facilitated by viral RNA; however, this interaction was lost in the absence of protease 2C [106]. The group’s final model highlighted that EV71′s protease 2C facilitates host RBP interactions with viral mRNA to promote viral gene expression, which ultimately leads to the blockage in formation of P-bodies. It emerges that viruses are masters at seizing control of P-bodies through their interactions with host RBPs and use them to promote virion production. Indirectly, this also likely suppresses host RNA expression and regulation which further remodels the host environment to be more favorable for viruses.
5.2. Hosts Manipulate P-body RBPs to Alter RNA Availability/Degradation to Combat Viral Infection
As previously mentioned, P-bodies are constitutively present RNA granules thought to exist for the purposes of RNA stasis along with some degradation capabilities. It has been widely reported that P-bodies often house transcripts with AU-rich elements (AREs) in their 3′ UTR for the purposes of degradation or translational repression [107,108,109,110,111,112] . Many ARE-bearing transcripts encode for chemokines and cytokines [113,114]. Several groups have demonstrated that following P-body loss, an increase in ARE-mRNAs follows [107,108,109,110,111,112]. These results suggest that decreases in P-body counts may in fact play an antiviral role with the increased expression of these effectors. To what extent does host and virus individually contribute to the loss of P-bodies during infection remains unclear. Are P-bodies altered to stimulate the host inflammatory response to mount an anti-viral response or to enhance environmental favorability for viral agents? A host RBP that we mentioned previously in this review - Shiftless (SHFL)- is a broad acting and potent antiviral factor [17,32,115,116,117,118,119,120] with known roles in regulating cytoplasmic granules. In the context of KSHV, SHFL appears to stringently restricts P-body foci [17]. The exact subcellular mechanism through which SHFL accomplishes this P-body loss remains unclear. SHFL localizes to P-bodies [121] and its overexpression causes their downregulation [17]. SHFL-mediated downregulation of P-bodies could subsequently alter the expression levels of certain ARE-mRNAs that are often included in these granules which likely contribute to SHFL overall ability to restrict viral agents. Outside of the context of infection, cellular loss of P-bodies typically occurs through two methods: 1. depletion of core protein components, or 2. expression/phosphorylation of the 68-amino acid microprotein: non-annotated P-body dissociating polypeptide (or “NBDY”). Loss of certain proteins such as LSm14a [71,75], DDX6 [73,75], or EDC4 have all been shown to lead to losses in P-bodies [70]. Otherwise, the expression and phosphorylation of the polypeptide NBDY has been shown to cause the loss of P-bodies, by potentially disrupting the electrostatic networks of these granules [122,123]. These cellular methods of P-body loss likely led to a translational increase in certain mRNAs. However, it has also been posited that this P-body regulation may also release many of these decapping and endonucleolytic enzymes leading to a translation suppression [124]. Therefore, it is possible that during viral infection, the host attempts to control the cytoplasmic gene expression environment by disassembling P-bodies. Viral agents often seek control over P-body RBPs as a tool to influence RNA expression within the cell. All the while, the host attempts to antagonize viral replication through a similar method: utilizing P-body associated RBPs to alter RNA expression for anti-viral pathways.
5.3. Viruses Influence Stress Granule RBPs to Suppress Host Immune Response Transcripts and Positively Regulate Viral RNA Fate
Stress granules (SGs) are thought to be cytoplasmic sites of translational regulation, where specific functionality can range from suppression of cell death pathways [125,126] to serving as a platform for an IFN response activation [127] to viral transcript sequestration foci [127,128,129]. It has been well characterized that SGs form upon viral infection, and these cytoplasmic granules are often targeted for downregulation by viruses [130,131,132,133,134]. Japanese Encephalitis Virus (JEV) has been shown to alter the localization of SG marker G3BP1 [135]; Dengue virus (DENV), West Nile virus (WNV), Murine Respirovirus (SeV), and the Zika virus (ZIKV) have all been shown to utilize viral biomolecules to sequester SG critical proteins to block granule formation [6,39,136]. Recently, the betacoronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV2), has been shown to have a similar effect on SG abundance and the re-purposing of the foci’s components. SARS-CoV2 encodes for 29 proteins, one of which is the nucleocapsid protein (NP) [137]. NP is responsible for packaging the viral RNA genome and helping virion egress from host cells [138]; it has also been found to interact with the structurally critical SG proteins G3BP1/G3BP2 [139]. Liu et al. observed that this NP-G3BP interaction limits the degree to which SGs can form in response to the viral infection, which in turn limits the host’s ability to stall translation and slow viral replication. The group went on the observe that SARS-CoV2′s NP sequestration of G3BPs not only resulted in the loss of SGs but also in the downregulation of IFN- β transcript levels and the RIG-I pathway (a critical piece of the innate immune response) as a whole [139]. In an uninhibited immune response system that combats viral infection, G3BP1 typically influences the RIG-I pathway, where IRF3 becomes phosphorylated ultimately causing an increase in IFN-β transcription [38]. The researchers were able to demonstrate how this virus exercises control over SG RBPs, such as G3BP1, to ultimately increase the propensity for viral mRNAs and translation. Through G3BP1 knockdown and knockout experiments, Liu et al. demonstrated that viral mRNAs were significantly elevated, identifying that NP-G3BP1 interactions lead to heightened viral replication states [38]. A genomic intermediate of the SARS-CoV2 genome appears to exist as dsDNA, which has an observable affinity for G3BP1; mechanically, this was hypothesized to be how the virus isolates this RBP from the host’s antiviral system [38]. Additionally, this SG component has been shown to be necessary for both murine norovirus (MNV) replication as well as Norwalk norovirus replication (a replicon used to model human norovirus, HuNoV) [140]. Following the knockout of G3BP1 within cells, both MNV and HuNoV were observed to have significantly low transcript levels and were no longer tied to cytotoxic outcomes; cells were seen to be virally resistant [140]. Interestingly, Hosmillo et al. were able to uncover that G3BP1 RNA binding domains directly impacted viral replication; the loss of these domains resulted in less viral yield. In addition, Hosmillo and colleagues discovered that G3BP1 interacts with the viral protein VPg and helps facilitate the loading of ribosomes and polysomes onto norovirus RNA. Thus, certain viruses take advantage of SG RBPs, such as G3BP1, to promote the stability and translation of viral RNAs. SGs present another fascinating focal point for where host and virus compete over RBPs to gain an advantage over the other.
5.4. Host Agents Orchestrate Stress Granule RBPs to Effectively Quench Viral Replication
Stress Granules have been widely characterized as antiviral subcellular compartments; SG RBPs, such as G3BP1 and TIA-1, are non-traditional components of the host immune-response in the context of these granules. Functionally, SGs exist to help sequester transcripts out of the translation pool and otherwise slow protein synthesis. Liu et al. implicitly define this purpose. SARS-CoV2 must manipulate G3BP1 to improve viral replication, meaning hosts use of G3BP1 actively and effectively mutes RNA expression enough to hinder viral replication [38]. Through the knockdown of G3BP1, they observed a significant increase in viral mRNAs, again implicitly demonstrating this host RBP usefulness in gene expression control via granular formation [38]. Alternatively, the host-defense significance of SG RBPs may be seen in the case of the α-coronavirus porcine epidemic diarrhea virus (PEDV). PEDV threatens suckling piglets globally. Following PEDV infection, SGs form and eventually are lost towards the latter stages (i.e., around 36 hours) [141]. Sun et al. identified that overexpression of G3BP1 resulted in significantly decreased levels of viral mRNA [141]. They further discovered that PEDV encodes a protease that cleaves G3BP1 in the late stages of infection which causes this loss. They were able to pinpoint the specific cleavage sites that viral caspase-8 targets down to two aspartic acid residues. Interestingly, robust SGs with cleavage resistant G3BP1 stringently limited viral transcripts as well as overall viral titers, indicating that SGs can efficiently limit viral transcription and translation. Studies like these demonstrate the capability and anti-viral functionality of SGs and how the host attempts to use RBPs like G3BP1 to restrict viral agents.
 |
5.5. Granule Functionality/Affects are Convoluted; Both Virus and Host Wield SG RBPs for Their Own Benefit
The above sections detailing the great virus and host struggle over P-body and SG RBPs already outlines how ambiguous the functionality of granules can be. One other fascinating viral case study that depicts this murkiness can be seen in the context of Viral Hemorrhagic Septicemia Virus (VHSV); a member of Rhabdoviridae and threatens dozens of fish species globally. [142]. And appears to trigger the formation of Stress Granules [143]. Interestingly, G3BP1 was shown to be redistributed to viral replication complexes and prove to be essential for efficient virion production [143]. Even though viral infection results in SG formation, VHSV still influences G3BP1 to facilitate its own replication. However, SGs also seem to play an anti-viral role within the system. Though IFN output was markedly higher under G3BP1 knockdown conditions, viral mRNA levels were also significantly higher. This suggests that G3BP1 and SGs can limit viral replication to an extent, likely via translational arrest and sequestration [143]. VHSV utilizes SG RBPs such as G3BP1 to enhance replication, yet the host also exercises some control over these RBPs to simultaneously limit viral replication [143]. This case of viral infection and SG dynamics captures the convoluted subcellular environment and depicts how RNA granules exist at the axis where pro-viral and pro-host states teeter. Have viral agents robustly conquered this host defense and adapted to largely benefit from granules? Will host mechanisms adapt or can they be shaped to re-purpose the likes of P-bodies and SGs to effectively combat viral replication?
6. Conclusions and Future Perspectives
RNA binding proteins comprise the subcellular foundation for host survival and viral replicative strategies. While this review covers many of the most recent findings of how both sides of this conflict attempt to take advantage of these proteins, new studies continue to unveil different viral case studies and mechanisms of exploit. mRNP complexes present an exciting avenue of study to better understand viral replication and takeover. The first major component of these complexes, mRNA, directly influence gene expression and thus serve as a major target for viruses. Certain elements within transcripts can convey protection from nucleolytic activity or even help distinguish between virus and host. Proper gene expression requires fine-tuned RNA processing, and this multifaceted quality control system helps to regulate the cell homeostasis. However, it can prove to be disastrous when certain viral agents target one juncture of RNA processing and critical information is lost. The other component of these mRNP complexes, RBPs, also directly contribute to gene expression and are critical subcellular components that must be subjected to control for successful viral replication. Often these proteins are thought control stability to transcripts and even impact their localization. Certain host factors, such as NFX1 and CRM1, critically regulate nuclear egress of transcripts, which can largely impact host survival dependent upon whether virus or host control these export pathways. Interestingly, RBPs also can help regulate the dynamics of certain RNA granules, which can significantly impact gene expression. Through P-bodies and stress granules, RBPs influence mRNA sequestration, decay, and release into the translation pool.
Furthermore, from the studies reviewed here we can continue to interrogate how cellular pathways are affected by viral takeover, but also how the host responds to the viral attack. As mRNA abundance and availability can be drastically altered during viral infection. Cellular feedback pathways can potentially serve to relay stress signals during infection. This area is still ripe for further study since it is still unclear which cellular factors are involved and to what extent communication is carried out in the cell between cellular compartments. Further work to elucidate the effects on transcription and translation are also still needed. Since viral infection is an event characterized by unique mechanisms that vary from agent-to-agent; certain homologies and divergence would be expected. Moreover, more studies to discern RNA-Protein interactions when RBP expression levels fluctuate in cells during viral infections are imperative to shed light into the unknown biological significance of RBP localization to more than one cellular compartment, multifunctionality, and response to target availability. Studies in these areas could prove exceptionally fruitful, providing a deeper understanding of these processes which could lead to the discovery of new antiviral targets and the development of therapeutic agents.
Regarding the RBPs associated with RNA granules, many factors and mechanisms are yet to be discovered. Specifically, within the context of viral infection, granule RBPs and their contribution appear to be quite convoluted and dependent from virus to virus. For viral agents, such as EV71, that lead to P-body loss, it remains to be seen mechanistically how the virus utilizes and recruits RBPs like DDX6 to provide stability and promote viral gene expression. Further characterization also ought to be conducted on exactly how the host wields P-bodies and their biomolecules specifically to alter its own gene expression to ultimately combat viral gene expression. For stress granule RBPs, how prevalent is this viral hijacking strategy? What other viral agents have evolved mechanisms to disrupt SG formation for purposes other than a lack of transcript sequestration? Additionally, how may host defense counteract this disassembly of the widely regarded anti-viral granule type? The characterization of RNA granule functionality during viral infection has emerged as an exciting frontier for better understanding virus-host interactions at the subcellular level.
Funding
M.M is supported NIH grant R35GM138043, Y.B was supported by a training grant (T32 GM139789) and a smith Spaulding fellowship; D.H is supported by the United States Air Force. The views expressed in this submission are those of the author and do not reflect the official policy or position of the United States Air Force, Department of Defense, or the U.S. Government.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Singh, G.; Pratt, G.; Yeo, G.W.; Moore, M.J. The Clothes Make the mRNA: Past and Present Trends in mRNP Fashion. Annu Rev Biochem 2015, 84, 325–354. [Google Scholar] [CrossRef]
- Mitchell, S.F.; Parker, R. Principles and properties of eukaryotic mRNPs. Mol Cell 2014, 54, 547–558. [Google Scholar] [CrossRef]
- Dreyfuss, G.; Kim, V.N.; Kataoka, N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol 2002, 3, 195–205. [Google Scholar] [CrossRef]
- Corley, M.; Burns, M.C.; Yeo, G.W. How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol Cell 2020, 78, 9–29. [Google Scholar] [CrossRef]
- Chahar, H.S.; Chen, S.; Manjunath, N. P-body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology 2013, 436, 1–7. [Google Scholar] [CrossRef]
- Iseni, F.; Garcin, D.; Nishio, M.; Kedersha, N.; Anderson, P.; Kolakofsky, D. Sendai virus trailer RNA binds TIAR, a cellular protein involved in virus-induced apoptosis. EMBO J 2002, 21, 5141–5150. [Google Scholar] [CrossRef]
- Jayabalan, A.K.; Griffin, D.E.; Leung, A.K.L. Pro-Viral and Anti-Viral Roles of the RNA-Binding Protein G3BP1. Viruses 2023, 15. [Google Scholar] [CrossRef]
- Boreikaite, V.; Passmore, L.A. 3’-End Processing of Eukaryotic mRNA: Machinery, Regulation, and Impact on Gene Expression. Annu Rev Biochem 2023, 92, 199–225. [Google Scholar] [CrossRef]
- Vijayakumar, A.; Park, A.; Steitz, J.A. Modulation of mRNA 3’-End Processing and Transcription Termination in Virus-Infected Cells. Front Immunol 2022, 13, 828665. [Google Scholar] [CrossRef]
- Carmody, S.R.; Wente, S.R. mRNA nuclear export at a glance. J Cell Sci 2009, 122, 1933–1937. [Google Scholar] [CrossRef]
- Guo, J.; Zhu, Y.; Ma, X.; Shang, G.; Liu, B.; Zhang, K. Virus Infection and mRNA Nuclear Export. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Boyne, J.R.; Colgan, K.J.; Whitehouse, A. Recruitment of the complete hTREX complex is required for Kaposi’s sarcoma-associated herpesvirus intronless mRNA nuclear export and virus replication. PLoS Pathog 2008, 4, e1000194. [Google Scholar] [CrossRef]
- Lloyd, R.E. Regulation of stress granules and P-bodies during RNA virus infection. Wiley Interdiscip Rev RNA 2013, 4, 317–331. [Google Scholar] [CrossRef]
- Sagan, S.M.; Weber, S.C. Let’s phase it: viruses are master architects of biomolecular condensates. Trends Biochem Sci 2023, 48, 229–243. [Google Scholar] [CrossRef]
- Roden, C.; Gladfelter, A.S. RNA contributions to the form and function of biomolecular condensates. Nat Rev Mol Cell Biol 2021, 22, 183–195. [Google Scholar] [CrossRef]
- Cao, L.; Liu, S.; Li, Y.; Yang, G.; Luo, Y.; Li, S.; Du, H.; Zhao, Y.; Wang, D.; Chen, J.; et al. The Nuclear Matrix Protein SAFA Surveils Viral RNA and Facilitates Immunity by Activating Antiviral Enhancers and Super-enhancers. Cell Host Microbe 2019, 26, 369–384. [Google Scholar] [CrossRef]
- Rodriguez, W.; Mehrmann, T.; Hatfield, D.; Muller, M. Shiftless Restricts Viral Gene Expression and Influences RNA Granule Formation during Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication. J Virol 2022, 96, e0146922. [Google Scholar] [CrossRef]
- Gaglia, M.M.; Rycroft, C.H.; Glaunsinger, B.A. Transcriptome-Wide Cleavage Site Mapping on Cellular mRNAs Reveals Features Underlying Sequence-Specific Cleavage by the Viral Ribonuclease SOX. PLoS Pathog 2015, 11, e1005305. [Google Scholar] [CrossRef]
- Mendez, A.S.; Vogt, C.; Bohne, J.; Glaunsinger, B.A. Site specific target binding controls RNA cleavage efficiency by the Kaposi’s sarcoma-associated herpesvirus endonuclease SOX. Nucleic Acids Res 2018, 46, 11968–11979. [Google Scholar] [CrossRef]
- Gaucherand, L.; Iyer, A.; Gilabert, I.; Rycroft, C.H.; Gaglia, M.M. Cut site preference allows influenza A virus PA-X to discriminate between host and viral mRNAs. Nat Microbiol 2023, 8, 1304–1317. [Google Scholar] [CrossRef]
- Gaucherand, L.; Porter, B.K.; Levene, R.E.; Price, E.L.; Schmaling, S.K.; Rycroft, C.H.; Kevorkian, Y.; McCormick, C.; Khaperskyy, D.A.; Gaglia, M.M. The Influenza A Virus Endoribonuclease PA-X Usurps Host mRNA Processing Machinery to Limit Host Gene Expression. Cell Rep 2019, 27, 776–792 e777. [Google Scholar] [CrossRef]
- Hutin, S.; Lee, Y.; Glaunsinger, B.A. An RNA element in human interleukin 6 confers escape from degradation by the gammaherpesvirus SOX protein. J Virol 2013, 87, 4672–4682. [Google Scholar] [CrossRef]
- Muller, M.; Hutin, S.; Marigold, O.; Li, K.H.; Burlingame, A.; Glaunsinger, B.A. A ribonucleoprotein complex protects the interleukin-6 mRNA from degradation by distinct herpesviral endonucleases. PLoS Pathog 2015, 11, e1004899. [Google Scholar] [CrossRef]
- Greenbaum, B.D.; Levine, A.J.; Bhanot, G.; Rabadan, R. Patterns of evolution and host gene mimicry in influenza and other RNA viruses. PLoS Pathog 2008, 4, e1000079. [Google Scholar] [CrossRef]
- Vijaykrishna, D.; Mukerji, R.; Smith, G.J. RNA Virus Reassortment: An Evolutionary Mechanism for Host Jumps and Immune Evasion. PLoS Pathog 2015, 11, e1004902. [Google Scholar] [CrossRef]
- Smyth, R.P.; Negroni, M.; Lever, A.M.; Mak, J.; Kenyon, J.C. RNA Structure-A Neglected Puppet Master for the Evolution of Virus and Host Immunity. Front Immunol 2018, 9, 2097. [Google Scholar] [CrossRef]
- Brugier, A.; Hafirrassou, M.L.; Pourcelot, M.; Baldaccini, M.; Kril, V.; Couture, L.; Kummerer, B.M.; Gallois-Montbrun, S.; Bonnet-Madin, L.; Vidalain, P.O.; et al. RACK1 Associates with RNA-Binding Proteins Vigilin and SERBP1 to Facilitate Dengue Virus Replication. J Virol 2022, 96, e0196221. [Google Scholar] [CrossRef]
- Diosa-Toro, M.; Kennedy, D.R.; Chuo, V.; Popov, V.L.; Pompon, J.; Garcia-Blanco, M.A. Y-Box Binding Protein 1 Interacts with Dengue Virus Nucleocapsid and Mediates Viral Assembly. mBio 2022, 13, e0019622. [Google Scholar] [CrossRef]
- Garcia-Moreno, M.; Noerenberg, M.; Ni, S.; Jarvelin, A.I.; Gonzalez-Almela, E.; Lenz, C.E.; Bach-Pages, M.; Cox, V.; Avolio, R.; Davis, T.; et al. System-wide Profiling of RNA-Binding Proteins Uncovers Key Regulators of Virus Infection. Mol Cell 2019, 74, 196–211. [Google Scholar] [CrossRef]
- Takamatsu, Y.; Krahling, V.; Kolesnikova, L.; Halwe, S.; Lier, C.; Baumeister, S.; Noda, T.; Biedenkopf, N.; Becker, S. Serine-Arginine Protein Kinase 1 Regulates Ebola Virus Transcription. mBio 2020, 11. [Google Scholar] [CrossRef]
- Merino, V.F.; Yan, Y.; Ordonez, A.A.; Bullen, C.K.; Lee, A.; Saeki, H.; Ray, K.; Huang, T.; Jain, S.K.; Pomper, M.G. Nucleolin mediates SARS-CoV-2 replication and viral-induced apoptosis of host cells. Antiviral Res 2023, 211, 105550. [Google Scholar] [CrossRef]
- Wang, X.; Xuan, Y.; Han, Y.; Ding, X.; Ye, K.; Yang, F.; Gao, P.; Goff, S.P.; Gao, G. Regulation of HIV-1 Gag-Pol Expression by Shiftless, an Inhibitor of Programmed -1 Ribosomal Frameshifting. Cell 2019, 176, 625–635. [Google Scholar] [CrossRef]
- Rodriguez, W.; Muller, M. Shiftless, a Critical Piece of the Innate Immune Response to Viral Infection. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Qin, W.; Kong, N.; Zhang, Y.; Wang, C.; Dong, S.; Zhai, H.; Zhai, X.; Yang, X.; Ye, C.; Ye, M.; et al. PTBP1 suppresses porcine epidemic diarrhea virus replication via inducing protein degradation and IFN production. J Biol Chem 2023, 299, 104987. [Google Scholar] [CrossRef]
- Kedersha, N.; Panas, M.D.; Achorn, C.A.; Lyons, S.; Tisdale, S.; Hickman, T.; Thomas, M.; Lieberman, J.; McInerney, G.M.; Ivanov, P.; et al. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J Cell Biol 2016, 212, 845–860. [Google Scholar] [CrossRef]
- Kim, S.S.; Sze, L.; Liu, C.; Lam, K.P. The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance interferon-beta response. J Biol Chem 2019, 294, 6430–6438. [Google Scholar] [CrossRef]
- Liu, Z.S.; Cai, H.; Xue, W.; Wang, M.; Xia, T.; Li, W.J.; Xing, J.Q.; Zhao, M.; Huang, Y.J.; Chen, S.; et al. G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol 2019, 20, 18–28. [Google Scholar] [CrossRef]
- Yang, W.; Ru, Y.; Ren, J.; Bai, J.; Wei, J.; Fu, S.; Liu, X.; Li, D.; Zheng, H. G3BP1 inhibits RNA virus replication by positively regulating RIG-I-mediated cellular antiviral response. Cell Death Dis 2019, 10, 946. [Google Scholar] [CrossRef]
- Bonenfant, G.; Williams, N.; Netzband, R.; Schwarz, M.C.; Evans, M.J.; Pager, C.T. Zika Virus Subverts Stress Granules To Promote and Restrict Viral Gene Expression. J Virol 2019, 93. [Google Scholar] [CrossRef]
- Davis, Z.H.; Verschueren, E.; Jang, G.M.; Kleffman, K.; Johnson, J.R.; Park, J.; Von Dollen, J.; Maher, M.C.; Johnson, T.; Newton, W.; et al. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol Cell 2015, 57, 349–360. [Google Scholar] [CrossRef]
- Trendel, J.; Schwarzl, T.; Horos, R.; Prakash, A.; Bateman, A.; Hentze, M.W.; Krijgsveld, J. The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 2019, 176, 391–403 e319. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Yang, H.; Liu, P.; Hamiti, M.; Zhang, X.; Xu, Y.; Quan, W.; Zhang, Y.; Yu, W.; Jiao, L.; et al. Splicing factor SF3B3, a NS5-binding protein, restricts ZIKV infection by targeting GCH1. Virol Sin 2023, 38, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Lunitz, V.; Ruiz-Orera, J.; Hubner, N.; van Heesch, S. Multifunctional RNA-binding proteins influence mRNA abundance and translational efficiency of distinct sets of target genes. PLoS Comput Biol 2021, 17, e1009658. [Google Scholar] [CrossRef] [PubMed]
- Timmers, H.T.M.; Tora, L. Transcript Buffering: A Balancing Act between mRNA Synthesis and mRNA Degradation. Mol Cell 2018, 72, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, E.; Gilbertson, S.; Alla, R.; Glaunsinger, B. Viral Nucleases Induce an mRNA Degradation-Transcription Feedback Loop in Mammalian Cells. Cell Host Microbe 2015, 18, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Schwalb, B.; Schulz, D.; Pirkl, N.; Etzold, S.; Lariviere, L.; Maier, K.C.; Seizl, M.; Tresch, A.; Cramer, P. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res 2012, 22, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Hartenian, E.; Glaunsinger, B.A. Feedback to the central dogma: cytoplasmic mRNA decay and transcription are interdependent processes. Crit Rev Biochem Mol Biol 2019, 54, 385–398. [Google Scholar] [CrossRef]
- Gilbertson, S.; Federspiel, J.D.; Hartenian, E.; Cristea, I.M.; Glaunsinger, B. Changes in mRNA abundance drive shuttling of RNA binding proteins, linking cytoplasmic RNA degradation to transcription. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Backlund, M.; Stein, F.; Rettel, M.; Schwarzl, T.; Perez-Perri, J.I.; Brosig, A.; Zhou, Y.; Neu-Yilik, G.; Hentze, M.W.; Kulozik, A.E. Plasticity of nuclear and cytoplasmic stress responses of RNA-binding proteins. Nucleic Acids Res 2020, 48, 4725–4740. [Google Scholar] [CrossRef]
- Park, R.; El-Guindy, A.; Heston, L.; Lin, S.F.; Yu, K.P.; Nagy, M.; Borah, S.; Delecluse, H.J.; Steitz, J.; Miller, G. Nuclear translocation and regulation of intranuclear distribution of cytoplasmic poly(A)-binding protein are distinct processes mediated by two Epstein Barr virus proteins. PLoS One 2014, 9, e92593. [Google Scholar] [CrossRef]
- Soto-Rifo, R.; Rubilar, P.S.; Ohlmann, T. The DEAD-box helicase DDX3 substitutes for the cap-binding protein eIF4E to promote compartmentalized translation initiation of the HIV-1 genomic RNA. Nucleic Acids Res 2013, 41, 6286–6299. [Google Scholar] [CrossRef]
- Sandri-Goldin, R.M. Nuclear export of herpes virus RNA. Curr Top Microbiol Immunol 2001, 259, 2–23. [Google Scholar]
- Wente, S.R.; Rout, M.P. The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol 2010, 2, a000562. [Google Scholar] [CrossRef] [PubMed]
- Vogt, C.; Bohne, J. The KSHV RNA regulator ORF57: target specificity and its role in the viral life cycle. Wiley Interdiscip Rev RNA 2016, 7, 173–185. [Google Scholar] [CrossRef]
- Gales, J.P.; Kubina, J.; Geldreich, A.; Dimitrova, M. Strength in Diversity: Nuclear Export of Viral RNAs. Viruses 2020, 12. [Google Scholar] [CrossRef]
- Gong, D.; Kim, Y.H.; Xiao, Y.; Du, Y.; Xie, Y.; Lee, K.K.; Feng, J.; Farhat, N.; Zhao, D.; Shu, S.; et al. A Herpesvirus Protein Selectively Inhibits Cellular mRNA Nuclear Export. Cell Host Microbe 2016, 20, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Li, H.C.; Huang, E.Y.; Su, P.Y.; Wu, S.Y.; Yang, C.C.; Lin, Y.S.; Chang, W.C.; Shih, C. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog 2010, 6, e1001162. [Google Scholar] [CrossRef]
- Pardamean, C.I.; Wu, T.T. Inhibition of Host Gene Expression by KSHV: Sabotaging mRNA Stability and Nuclear Export. Front Cell Infect Microbiol 2021, 11, 648055. [Google Scholar] [CrossRef] [PubMed]
- Mougel, M.; Akkawi, C.; Chamontin, C.; Feuillard, J.; Pessel-Vivares, L.; Socol, M.; Laine, S. NXF1 and CRM1 nuclear export pathways orchestrate nuclear export, translation and packaging of murine leukaemia retrovirus unspliced RNA. RNA Biol 2020, 17, 528–538. [Google Scholar] [CrossRef]
- Dundr, M.; Hebert, M.D.; Karpova, T.S.; Stanek, D.; Xu, H.; Shpargel, K.B.; Meier, U.T.; Neugebauer, K.M.; Matera, A.G.; Misteli, T. In vivo kinetics of Cajal body components. J Cell Biol 2004, 164, 831–842. [Google Scholar] [CrossRef]
- Phair, R.D.; Misteli, T. High mobility of proteins in the mammalian cell nucleus. Nature 2000, 404, 604–609. [Google Scholar] [CrossRef]
- Weidtkamp-Peters, S.; Lenser, T.; Negorev, D.; Gerstner, N.; Hofmann, T.G.; Schwanitz, G.; Hoischen, C.; Maul, G.; Dittrich, P.; Hemmerich, P. Dynamics of component exchange at PML nuclear bodies. J Cell Sci 2008, 121, 2731–2743. [Google Scholar] [CrossRef] [PubMed]
- Mittag, T.; Pappu, R.V. A conceptual framework for understanding phase separation and addressing open questions and challenges. Mol Cell 2022, 82, 2201–2214. [Google Scholar] [CrossRef] [PubMed]
- Alshareedah, I.; Moosa, M.M.; Pham, M.; Potoyan, D.A.; Banerjee, P.R. Programmable viscoelasticity in protein-RNA condensates with disordered sticker-spacer polypeptides. Nat Commun 2021, 12, 6620. [Google Scholar] [CrossRef] [PubMed]
- Bergeron-Sandoval, L.P.; Kumar, S.; Heris, H.K.; Chang, C.L.A.; Cornell, C.E.; Keller, S.L.; Francois, P.; Hendricks, A.G.; Ehrlicher, A.J.; Pappu, R.V.; et al. Endocytic proteins with prion-like domains form viscoelastic condensates that enable membrane remodeling. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Kota, D.; Zhou, H.X. Shear relaxation governs fusion dynamics of biomolecular condensates. Nat Commun 2021, 12, 5995. [Google Scholar] [CrossRef] [PubMed]
- Jawerth, L.; Fischer-Friedrich, E.; Saha, S.; Wang, J.; Franzmann, T.; Zhang, X.; Sachweh, J.; Ruer, M.; Ijavi, M.; Saha, S.; et al. Protein condensates as aging Maxwell fluids. Science 2020, 370, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Ruggeri, F.S.; Vigolo, D.; Kamada, A.; Qamar, S.; Levin, A.; Iserman, C.; Alberti, S.; George-Hyslop, P.S.; Knowles, T.P.J. Biomolecular condensates undergo a generic shear-mediated liquid-to-solid transition. Nat Nanotechnol 2020, 15, 841–847. [Google Scholar] [CrossRef]
- Andrei, M.A.; Ingelfinger, D.; Heintzmann, R.; Achsel, T.; Rivera-Pomar, R.; Luhrmann, R. A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA 2005, 11, 717–727. [Google Scholar] [CrossRef]
- Yu, J.H.; Yang, W.H.; Gulick, T.; Bloch, K.D.; Bloch, D.B. Ge-1 is a central component of the mammalian cytoplasmic mRNA processing body. RNA 2005, 11, 1795–1802. [Google Scholar] [CrossRef]
- Yang, W.H.; Yu, J.H.; Gulick, T.; Bloch, K.D.; Bloch, D.B. RNA-associated protein 55 (RAP55) localizes to mRNA processing bodies and stress granules. RNA 2006, 12, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Wilczynska, A.; Aigueperse, C.; Kress, M.; Dautry, F.; Weil, D. The translational regulator CPEB1 provides a link between dcp1 bodies and stress granules. J Cell Sci 2005, 118, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Ozgur, S.; Chekulaeva, M.; Stoecklin, G. Human Pat1b connects deadenylation with mRNA decapping and controls the assembly of processing bodies. Mol Cell Biol 2010, 30, 4308–4323. [Google Scholar] [CrossRef] [PubMed]
- Marnef, A.; Maldonado, M.; Bugaut, A.; Balasubramanian, S.; Kress, M.; Weil, D.; Standart, N. Distinct functions of maternal and somatic Pat1 protein paralogs. RNA 2010, 16, 2094–2107. [Google Scholar] [CrossRef] [PubMed]
- Ayache, J.; Benard, M.; Ernoult-Lange, M.; Minshall, N.; Standart, N.; Kress, M.; Weil, D. P-body assembly requires DDX6 repression complexes rather than decay or Ataxin2/2L complexes. Mol Biol Cell 2015, 26, 2579–2595. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Behm-Ansmant, I.; Schweizer, D.; Izaurralde, E. P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol Cell Biol 2007, 27, 3970–3981. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.; Parton, R.M.; Rabouille, C.; Weil, T.T.; Davis, I. Localized Translation of gurken/TGF-alpha mRNA during Axis Specification Is Controlled by Access to Orb/CPEB on Processing Bodies. Cell Rep 2016, 14, 2451–2462. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.J.; Marchand, V.; Ephrussi, A. Drosophila Ge-1 promotes P body formation and oskar mRNA localization. PLoS One 2011, 6, e20612. [Google Scholar] [CrossRef] [PubMed]
- Eystathioy, T.; Jakymiw, A.; Chan, E.K.; Seraphin, B.; Cougot, N.; Fritzler, M.J. The GW182 protein colocalizes with mRNA degradation associated proteins hDcp1 and hLSm4 in cytoplasmic GW bodies. RNA 2003, 9, 1171–1173. [Google Scholar] [CrossRef]
- Sharma, N.R.; Zheng, Z.M. RNA Granules in Antiviral Innate Immunity: A Kaposi’s Sarcoma-Associated Herpesvirus Journey. Front Microbiol 2021, 12, 794431. [Google Scholar] [CrossRef]
- Buchan, J.R.; Muhlrad, D.; Parker, R. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J Cell Biol 2008, 183, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.; Xu, Y.; Wang, B.; David, M.D.; Schubert, P.; Kennedy, D.; Schrader, J.W. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol Cell Biol 2007, 27, 2324–2342. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Chen, S.; Gilks, N.; Li, W.; Miller, I.J.; Stahl, J.; Anderson, P. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell 2002, 13, 195–210. [Google Scholar] [CrossRef]
- Mazroui, R.; Sukarieh, R.; Bordeleau, M.E.; Kaufman, R.J.; Northcote, P.; Tanaka, J.; Gallouzi, I.; Pelletier, J. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2alpha phosphorylation. Mol Biol Cell 2006, 17, 4212–4219. [Google Scholar] [CrossRef]
- Bolster, D.R.; Kubica, N.; Crozier, S.J.; Williamson, D.L.; Farrell, P.A.; Kimball, S.R.; Jefferson, L.S. Immediate response of mammalian target of rapamycin (mTOR)-mediated signalling following acute resistance exercise in rat skeletal muscle. J Physiol 2003, 553, 213–220. [Google Scholar] [CrossRef]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol 1999, 147, 1431–1442. [Google Scholar] [CrossRef]
- White, J.P.; Lloyd, R.E. Regulation of stress granules in virus systems. Trends Microbiol 2012, 20, 175–183. [Google Scholar] [CrossRef]
- Ivanov, P.; Kedersha, N.; Anderson, P. Stress Granules and Processing Bodies in Translational Control. Cold Spring Harb Perspect Biol 2019, 11. [Google Scholar] [CrossRef]
- Hirose, T.; Ninomiya, K.; Nakagawa, S.; Yamazaki, T. A guide to membraneless organelles and their various roles in gene regulation. Nat Rev Mol Cell Biol 2023, 24, 288–304. [Google Scholar] [CrossRef]
- Kedersha, N.; Cho, M.R.; Li, W.; Yacono, P.W.; Chen, S.; Gilks, N.; Golan, D.E.; Anderson, P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol 2000, 151, 1257–1268. [Google Scholar] [CrossRef]
- Sheth, U.; Parker, R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 2003, 300, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Wilby, E.L.; Weil, T.T. Relating the Biogenesis and Function of P Bodies in Drosophila to Human Disease. Genes (Basel) 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, N.; Ebert, J.; Unterholzner, L.; Lindner, D.; Izaurralde, E.; Conti, E. SMG7 is a 14-3-3-like adaptor in the nonsense-mediated mRNA decay pathway. Mol Cell 2005, 17, 537–547. [Google Scholar] [CrossRef]
- Unterholzner, L.; Izaurralde, E. SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol Cell 2004, 16, 587–596. [Google Scholar] [CrossRef]
- Sen, G.L.; Blau, H.M. Argonaute 2/RISC resides in sites of mammalian mRNA decay known as cytoplasmic bodies. Nat Cell Biol 2005, 7, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Behm-Ansmant, I.; Rehwinkel, J.; Doerks, T.; Stark, A.; Bork, P.; Izaurralde, E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev 2006, 20, 1885–1898. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.A.; Singh, G.K.; Kleer, M.; Katsademas, T.; Castle, E.L.; Boudreau, B.Q.; Corcoran, J.A. Kaposi’s sarcoma-associated herpesvirus (KSHV) utilizes the NDP52/CALCOCO2 selective autophagy receptor to disassemble processing bodies. PLoS Pathog 2023, 19, e1011080. [Google Scholar] [CrossRef]
- Sharma, N.R.; Majerciak, V.; Kruhlak, M.J.; Yu, L.; Kang, J.G.; Yang, A.; Gu, S.; Fritzler, M.J.; Zheng, Z.M. KSHV RNA-binding protein ORF57 inhibits P-body formation to promote viral multiplication by interaction with Ago2 and GW182. Nucleic Acids Res 2019, 47, 9368–9385. [Google Scholar] [CrossRef]
- Ariumi, Y.; Kuroki, M.; Kushima, Y.; Osugi, K.; Hijikata, M.; Maki, M.; Ikeda, M.; Kato, N. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J Virol 2011, 85, 6882–6892. [Google Scholar] [CrossRef]
- Pager, C.T.; Schutz, S.; Abraham, T.M.; Luo, G.; Sarnow, P. Modulation of hepatitis C virus RNA abundance and virus release by dispersion of processing bodies and enrichment of stress granules. Virology 2013, 435, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Perez-Vilaro, G.; Scheller, N.; Saludes, V.; Diez, J. Hepatitis C virus infection alters P-body composition but is independent of P-body granules. J Virol 2012, 86, 8740–8749. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y.; Kuroki, M.; Abe, K.; Dansako, H.; Ikeda, M.; Wakita, T.; Kato, N. DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J Virol 2007, 81, 13922–13926. [Google Scholar] [CrossRef] [PubMed]
- Jangra, R.K.; Yi, M.; Lemon, S.M. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J Virol 2010, 84, 6810–6824. [Google Scholar] [CrossRef] [PubMed]
- Scheller, N.; Mina, L.B.; Galao, R.P.; Chari, A.; Gimenez-Barcons, M.; Noueiry, A.; Fischer, U.; Meyerhans, A.; Diez, J. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc Natl Acad Sci U S A 2009, 106, 13517–13522. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Xu, Z.; Liu, P.; Qin, Y.; Chen, M. Enterovirus 71 2A Protease Inhibits P-Body Formation To Promote Viral RNA Synthesis. J Virol 2021, 95, e0092221. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, J.A.; Johnston, B.P.; McCormick, C. Viral activation of MK2-hsp27-p115RhoGEF-RhoA signaling axis causes cytoskeletal rearrangements, p-body disruption and ARE-mRNA stabilization. PLoS Pathog 2015, 11, e1004597. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.F.; Sanduja, S.; Deane, N.G.; Blackshear, P.J.; Dixon, D.A. Transforming growth factor beta regulates P-body formation through induction of the mRNA decay factor tristetraprolin. Mol Cell Biol 2014, 34, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Franks, T.M.; Lykke-Andersen, J. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes Dev 2007, 21, 719–735. [Google Scholar] [CrossRef]
- Vindry, C.; Marnef, A.; Broomhead, H.; Twyffels, L.; Ozgur, S.; Stoecklin, G.; Llorian, M.; Smith, C.W.; Mata, J.; Weil, D.; et al. Dual RNA Processing Roles of Pat1b via Cytoplasmic Lsm1-7 and Nuclear Lsm2-8 Complexes. Cell Rep 2017, 20, 1187–1200. [Google Scholar] [CrossRef]
- Hubstenberger, A.; Courel, M.; Benard, M.; Souquere, S.; Ernoult-Lange, M.; Chouaib, R.; Yi, Z.; Morlot, J.B.; Munier, A.; Fradet, M.; et al. P-Body Purification Reveals the Condensation of Repressed mRNA Regulons. Mol Cell 2017, 68, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, J.A.; Khaperskyy, D.A.; Johnston, B.P.; King, C.A.; Cyr, D.P.; Olsthoorn, A.V.; McCormick, C. Kaposi’s sarcoma-associated herpesvirus G-protein-coupled receptor prevents AU-rich-element-mediated mRNA decay. J Virol 2012, 86, 8859–8871. [Google Scholar] [CrossRef] [PubMed]
- Bakheet, T.; Williams, B.R.; Khabar, K.S. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res 2006, 34, D111–114. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Shyu, A.B. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci 1995, 20, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.C., D.; Irudayam, J.I.; Ali, A.; Yang, O.O.; Arumugaswami, V. . C19ORF66 is an interferon-stimulated gene (ISG) which Inhibits human immunodeficiency virus-1. Journal of Hepatology, 2020; 116. [Google Scholar]
- Suzuki, Y.; Chin, W.X.; Han, Q.; Ichiyama, K.; Lee, C.H.; Eyo, Z.W.; Ebina, H.; Takahashi, H.; Takahashi, C.; Tan, B.H.; et al. Characterization of RyDEN (C19orf66) as an Interferon-Stimulated Cellular Inhibitor against Dengue Virus Replication. PLoS Pathog 2016, 12, e1005357. [Google Scholar] [CrossRef] [PubMed]
- Kinast, V.; Plociennikowska, A.; Anggakusuma; Bracht, T.; Todt, D.; Brown, R.J.P.; Boldanova, T.; Zhang, Y.; Bruggemann, Y.; Friesland, M.; et al. C19orf66 is an interferon-induced inhibitor of HCV replication that restricts formation of the viral replication organelle. J Hepatol 2020, 73, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, X.; Yao, Z.; Dong, X.; Zhang, D.; Hu, Y.; Zhang, S.; Lin, J.; Chen, J.; An, S.; et al. C19orf66 interrupts Zika virus replication by inducing lysosomal degradation of viral NS3. PLoS Negl Trop Dis 2020, 14, e0008083. [Google Scholar] [CrossRef] [PubMed]
- Hanners, N.W.; Mar, K.B.; Boys, I.N.; Eitson, J.L.; De La Cruz-Rivera, P.C.; Richardson, R.B.; Fan, W.; Wight-Carter, M.; Schoggins, J.W. Shiftless inhibits flavivirus replication in vitro and is neuroprotective in a mouse model of Zika virus pathogenesis. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef]
- Yu, D.; Zhao, Y.; Pan, J.; Yang, X.; Liang, Z.; Xie, S.; Cao, R. C19orf66 Inhibits Japanese Encephalitis Virus Replication by Targeting -1 PRF and the NS3 Protein. Virol Sin 2021, 36, 1443–1455. [Google Scholar] [CrossRef]
- Balinsky, C.A.; Schmeisser, H.; Wells, A.I.; Ganesan, S.; Jin, T.; Singh, K.; Zoon, K.C. IRAV (FLJ11286), an Interferon-Stimulated Gene with Antiviral Activity against Dengue Virus, Interacts with MOV10. J Virol 2017, 91. [Google Scholar] [CrossRef]
- Na, Z.; Luo, Y.; Schofield, J.A.; Smelyansky, S.; Khitun, A.; Muthukumar, S.; Valkov, E.; Simon, M.D.; Slavoff, S.A. The NBDY Microprotein Regulates Cellular RNA Decapping. Biochemistry 2020, 59, 4131–4142. [Google Scholar] [CrossRef] [PubMed]
- Na, Z.; Luo, Y.; Cui, D.S.; Khitun, A.; Smelyansky, S.; Loria, J.P.; Slavoff, S.A. Phosphorylation of a Human Microprotein Promotes Dissociation of Biomolecular Condensates. J Am Chem Soc 2021, 143, 12675–12687. [Google Scholar] [CrossRef] [PubMed]
- Bloch, D.B.; Sinow, C.O.; Sauer, A.J.; Corman, B.H.P. Assembly and regulation of the mammalian mRNA processing body. PLoS One 2023, 18, e0282496. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.P.; Wei, L.N. RhoA/ROCK1 signaling regulates stress granule formation and apoptosis. Cell Signal 2010, 22, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Samir, P.; Kesavardhana, S.; Patmore, D.M.; Gingras, S.; Malireddi, R.K.S.; Karki, R.; Guy, C.S.; Briard, B.; Place, D.E.; Bhattacharya, A.; et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 2019, 573, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 2012, 7, e43031. [Google Scholar] [CrossRef]
- Burgess, H.M.; Mohr, I. Defining the Role of Stress Granules in Innate Immune Suppression by the Herpes Simplex Virus 1 Endoribonuclease VHS. J Virol 2018, 92. [Google Scholar] [CrossRef]
- Mok, B.W.; Song, W.; Wang, P.; Tai, H.; Chen, Y.; Zheng, M.; Wen, X.; Lau, S.Y.; Wu, W.L.; Matsumoto, K.; et al. The NS1 protein of influenza A virus interacts with cellular processing bodies and stress granules through RNA-associated protein 55 (RAP55) during virus infection. J Virol 2012, 86, 12695–12707. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, Y.; Tang, Q.; Yang, X.; Qin, Y.; Chen, M. Inclusion bodies of human parainfluenza virus type 3 inhibit antiviral stress granule formation by shielding viral RNAs. PLoS Pathog 2018, 14, e1006948. [Google Scholar] [CrossRef]
- Ng, C.S.; Jogi, M.; Yoo, J.S.; Onomoto, K.; Koike, S.; Iwasaki, T.; Yoneyama, M.; Kato, H.; Fujita, T. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J Virol 2013, 87, 9511–9522. [Google Scholar] [CrossRef]
- White, J.P.; Cardenas, A.M.; Marissen, W.E.; Lloyd, R.E. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2007, 2, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Visser, L.J.; Medina, G.N.; Rabouw, H.H.; de Groot, R.J.; Langereis, M.A.; de Los Santos, T.; van Kuppeveld, F.J.M. Foot-and-Mouth Disease Virus Leader Protease Cleaves G3BP1 and G3BP2 and Inhibits Stress Granule Formation. J Virol 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Sadasivan, J.; Vlok, M.; Wang, X.; Nayak, A.; Andino, R.; Jan, E. Targeting Nup358/RanBP2 by a viral protein disrupts stress granule formation. PLoS Pathog 2022, 18, e1010598. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Okamoto, T.; Fukuhara, T.; Kambara, H.; Morita, E.; Mori, Y.; Kamitani, W.; Matsuura, Y. Japanese encephalitis virus core protein inhibits stress granule formation through an interaction with Caprin-1 and facilitates viral propagation. J Virol 2013, 87, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Emara, M.M.; Brinton, M.A. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc Natl Acad Sci U S A 2007, 104, 9041–9046. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: open the door for novel therapies. Signal Transduct Target Ther 2022, 7, 26. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Liu, H.; Bai, Y.; Zhang, X.; Gao, T.; Liu, Y.; Li, E.; Wang, X.; Cao, Z.; Zhu, L.; Dong, Q.; et al. SARS-CoV-2 N Protein Antagonizes Stress Granule Assembly and IFN Production by Interacting with G3BPs to Facilitate Viral Replication. J Virol 2022, 96, e0041222. [Google Scholar] [CrossRef]
- Hosmillo, M.; Lu, J.; McAllaster, M.R.; Eaglesham, J.B.; Wang, X.; Emmott, E.; Domingues, P.; Chaudhry, Y.; Fitzmaurice, T.J.; Tung, M.K.; et al. Noroviruses subvert the core stress granule component G3BP1 to promote viral VPg-dependent translation. Elife 2019, 8. [Google Scholar] [CrossRef]
- Sun, L.; Chen, H.; Ming, X.; Bo, Z.; Shin, H.J.; Jung, Y.S.; Qian, Y. Porcine Epidemic Diarrhea Virus Infection Induces Caspase-8-Mediated G3BP1 Cleavage and Subverts Stress Granules To Promote Viral Replication. J Virol 2021, 95. [Google Scholar] [CrossRef]
- Ammayappan, A.; Vakharia, V.N. Molecular characterization of the Great Lakes viral hemorrhagic septicemia virus (VHSV) isolate from USA. Virol J 2009, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Ramnani, B.; Powell, S.; Shetty, A.G.; Manivannan, P.; Hibbard, B.R.; Leaman, D.W.; Malathi, K. Viral Hemorrhagic Septicemia Virus Activates Integrated Stress Response Pathway and Induces Stress Granules to Regulate Virus Replication. Viruses 2023, 15. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



