
Submitted:
15 December 2023
Posted:
18 December 2023
You are already at the latest version
Abstract
One of the hallmarks of Alzheimer’s disease (AD) is the accumulation of beta-amyloid peptide (Aβ) leading to formation of soluble neurotoxic Aβ oligomers and insoluble amyloid plaques in various parts of the brain. Aβ undergoes post-translational modifications that alter its pathogenic properties. Aβ is produced not only in brain, but also in the peripheral tissues. Such Aβ, including its post-translationally modified forms, can enter the brain from circulation by binding to RAGE and contribute to the pathology of AD. However, the transport of modified forms of Aβ across the blood–brain barrier (BBB) has not been investigated. Here, we used a transwell BBB model as a controlled environment for permeability studies. We found that Aβ42 containing isomerized Asp7 residue (iso-Aβ42) and Aβ42 containing phosphorylated Ser8 residue crossed the BBB better than unmodified Aβ42, which correlated with different contribution of endocytosis mechanisms to the transport of these isoforms. Using microscale thermophoresis, we observed that RAGE binds to iso-Aβ42 an order of magnitude weaker than to Aβ42. Thus, post-translational modifications of Aβ increase the rate of its transport across the BBB and modify the mechanisms of the transport, which may be important for AD pathology and treatment.
Keywords:
Alzheimer’s disease
; blood–brain barrier
; beta-amyloid
; post-translational modifications
; RAGE
; caveolin-dependent endocytosis
; clathrin-dependent endocytosis
1. Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disease, accounting for 60-80% of all cases of dementia [1]. AD is characterized by various pathological markers in the brain, such as the accumulation of beta-amyloid peptide (Aβ), which can form senile plaques, intracellular accumulation of neurofibrillary tangles formed by hyperphosphorylated tau protein, and progressive loss of nerve cells [2]. Most cases of AD are sporadic and aging is considered a major risk factor for AD, but the pathways through which aging triggers the development of the disease are still unclear. It has been suggested that aging may induce post-translational modifications of Aβ (PTMs), which enhance its pathogenic properties. [3]. Thus, Aβ is capable of undergoing various PTMs that are triggered by enzymes or low molecular weight substances, as well as spontaneously [4]. Some of these modifications are isomerization of the aspartic acid residue at position 7 (iso-Aβ) and phosphorylation at serine 8 (pS8-Aβ). These modifications are located in the metal-binding domain of Aβ, which regulates its zinc-dependent oligomerization [5,6] and interaction with receptors [5,7]. In amyloid plaques, iso-Aβ was found to constitute more than 50% of all Aβ molecules [8]. Iso-Aβ has an increased ability to oligomerize [9], is more toxic [10] and demonstrates resistance to proteolysis [11]. At the same time, the level of iso-Aβ increases with age and in patients with AD [3]. PS8-Aβ was detected in brain tissue of both patients with AD and AD model mice. It is localized both in amyloid plaques and in the cytoplasm of neurons, and compared to unmodified Aβ has increased neurotoxicity in vitro [12] and higher resistance to degradation by an insulin-degrading enzyme [11]. Thus, pS8-Aβ and iso-Aβ are important isoforms that differ significantly in properties from intact Aβ. The changes in the homeostasis of these isoforms may trigger pathological events contributing to development of AD.
Numerous studies have shown that AD is accompanied by a disruption of the blood-brain barrier (BBB), which occurs at an early stage of the disease [13,14]. The BBB controls the entry of Aβ from plasma into the brain via the RAGE receptor, as well as the clearance of Aβ from the brain into the peripheral circulation via the LRP-1 receptor [15]. Disruption of these BBB functions can lead to pathological accumulation of Aβ in the brain and manifestation of AD symptoms. Increasing evidence indicates that Aβ from blood can enter the brain and serve as a trigger for the disease [16,17]. Interestingly, peripheral injection of synthetic Aβ42 into the bloodstream did not lead to the formation of amyloid plaques in the brains of mouse models of AD. However, intravenous injections of modified forms of Aβ altered the pathology of AD: the injection of iso-Aβ accelerated the amyloidogenesis [18], while injection of pS8-Aβ reduced the number of amyloid plaques in the brain of transgenic mice [5]. This evidence suggests that pathogenic isoforms of Aβ may arise in the circulatory system, after which they penetrate the brain and contribute to AD pathology [19]. However, the transport of modified forms of Aβ across the BBB has not been previously studied.
In this work, we compared the efficiency of transport of Aβ42, pS8-Aβ42, and iso-Aβ42 through a monolayer of BBB endothelial cells, and also established the contribution of clathrin- and caveolin-dependent mechanisms to this process. It was also determined how modifications of Aβ affect its affinity for RAGE.
2. Materials and Methods
2.1. Preparation of synthetic beta-amyloid peptides
[H2N]-DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA-[COOH] (Аβ42) and [H2N]-DAEFRH[isoD]SGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA-[COOH] (iso-Аβ42) were obtained from Lifetein (Somerset, NJ, USA). [H2N]-DAEFRHD[pS]GYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA-[COOH] (pS8-Аβ42) and [H2N]-DAEFRHDSGYEVHHQK-[COOH] (Aβ16) were obtained from Biopeptide (San Diego, CA, USA). [H2N]-LVFFAEDVGSNKGAIIGLMVGGVVIA-[COOH] (Aβ17-42) was obtained from Verta (Saint-Petersburg, Russia). Peptides were monomerized using hexafluoroisopropanol (Fluka), aliquoted and dried as described in Barykin et al [5]. An aliquot of Aβ was dissolved in 10 μl of dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Loius, MO, USA) at room temperature for an hour to obtain a 1.25 mM stock solution and then diluted in serum-free cell culture medium to the required concentrations.
2.2. Cell Culture
The murine bEnd.3 cell line, obtained from the American Type Culture Collection, was cultured at 37°C in an atmosphere of 5% CO2 in Dulbecco's Modified Eagles Medium (DMEM; Gibco, ThermoFisher Scientific, Waltham, MA, USA) containing 4.5 g/l glucose, 1% GlutaMax (Gibco, ThermoFisher Scientific, Waltham, MA, USA), 100 units/mL penicillin, 100 µg/mL streptomycin (Sigma, St. Louis, MO, USA) with the addition of 10% fetal bovine serum (FBS; Gibco, USA).
2.3. In vitro model of the BBB
2.3.1. Cell cultivation on transwell membrane
To simulate the BBB, bEnd.3 cells were cultured in transwell inserts (Greiner Bio-One, pore diameter 0.4 μm) submerged in the wells of a 12-well plate (Greiner Bio-One). BEnd.3 cells were seeded on the upper surface of the transwell membrane in an amount of 70 thousand per well and cultured for 7 days until confluence. The volume of DMEM medium (10% FBS) in the upper (luminal) compartment was 750 μl, in the lower (abluminal) compartment – 1 ml. Cell counting before seeding was carried out in a Goryaev chamber with preliminary staining of cells with damaged membranes with trypan blue (Invitrogen).
2.3.2. Measuring the passage of Aβ isoforms through a monolayer of bEnd.3 cells
The transport of Aβ and its isoforms was studied in the transwell model. Aβ is able to bind albumin and other serum proteins [20]. Therefore, before the experiment, the upper and lower sections of the transwell were washed with 500 and 1000 μL of serum-free DMEM, respectively. To study the transfer of Aβ from the luminal to abluminal compartment (modeling transport from the blood to the brain), the upper part of the transwell was filled with 300 μl of DMEM containing 1 μM Aβ. An appropriate amount of DMSO (0.08% DMSO) was added to control samples. The lower compartment was filled with 750 μl of DMEM. After adding the peptide, samples were taken from the lower part of the transwell in a volume of 200 μl after 2, 6, and 24 hours. Each time after sampling, 200 μl of DMEM medium was added to the lower compartment. After 6 hours of incubation, FBS was added to the upper compartment of each well to a final concentration of 5%. The concentration of Aβ in the samples was determined by enzyme-linked immunosorbent assay (ELISA). To account for dilution due to sampling and addition of DMEM medium, the concentration of Aβ was corrected using the following equation:
where Сt’ is the concentration of Aβ at time t, taking into account dilution; Сt is the concentration of Aβ measured with ELISA at time t; Сt-1 - concentration of Aβ measured at the previous time point; V is the volume added to the lower compartment after sampling; Vtotal is the total volume in the lower compartment.
The permeability coefficients obtained by incubating cells with 1 µM and 100 nM Aβ are presented in the Supplementary (Figure S1).
After each experiment, BBB permeability was assessed as described in section 3.3 to ensure that the cell monolayer was not disrupted by incubation with Aβ.
2.3.3. Endothelial permeability measurement
To assess paracellular permeability of the endothelium, the fluorescent label sodium fluorescein (Sigma-Aldrich) was used. The lower and upper compartments of the transwell were washed with PBS (Gibco) and filled with HBSS buffer (Gibco): 250 and 750 μl in the upper and lower compartments, respectively. Then, 50 μl of 60 μg/ml sodium fluorescein dissolved in HBSS was added to the upper compartment to a final concentration of 10 μg/ml. Samples (100 μl) were taken from the lower compartment at 0, 15, 30, 45, and 60 min after the addition of the fluorescent label. Each time after sampling, 100 μl of HBSS was added to the lower compartment. The fluorescence intensity in the samples was measured on a SPARK plate reader (Tecan, Switzerland) with an excitation wavelength of 485 nm and a fluorescence recording wavelength of 535 nm. Dilution of sodium fluorescein at each sampling step was taking into account using the following equation:
where It’ is the fluorescence intensity at time t after dilution correction; It is the fluorescence intensity measured at time t; It-1 is fluorescence intensity measured at the previous time point; V is the volume added to the lower compartment after sampling; Vtotal is the total volume in the lower compartment.
2.3.4. Study of the transport mechanisms of beta-amyloid and its isoforms
Various inhibitors were used to study the transport mechanisms of Aβ and its isoforms. The contribution of RAGE to the transport of Aβ across the endothelial monolayer was assessed using the antagonist of this receptor FPS-ZM1 (Sigma) at a concentration of 20 μM (to obtain a stock solution, FPS-ZM1 was dissolved in DMSO to a concentration of 305 mM). To study the caveolin-dependent transport of Aβ isoforms, the inhibitor filipin (Sigma) was used at a concentration of 3 μg/ml (to obtain a stock solution, filipin was dissolved in DMSO to a concentration of 5 mg/ml). An equivalent amount of DMSO was added to the control samples. The contribution of clathrin-dependent endocytosis was assessed using chlorpromazine (Merck) at a concentration of 5 μg/ml (to obtain a stock solution, chlorpromazine was dissolved in DMEM). These concentrations were selected based on literature data and tested for toxicity to bEnd.3 cells using MTT (Filipin) and WST (FPS-ZM1 and chlorpromazine) assays according to the manufacturer's protocol (Figure S2). Before experiments, cells were preincubated with inhibitors added to the upper transwell compartment for 1 hour, after which they were filled with solutions containing the inhibitor and 1 μM Aβ, and samples were taken from the lower compartment after 2, 6 and 24 hours.
2.4. ELISA
The concentration of Aβ and its isoforms was measured using sandwich ELISA. BAM113cc antibodies (HyTest), which recognize the C-terminus of Aβ, were added to a 96-well ELISA plate (NEST) in a volume of 100 μl (0.5 ng/μl) and incubated overnight at +4°C. The wells of the plate were washed 2 times with 200 μl of PBST (0.05% Tween20) and blocked in 1% BSA (Dia-m) in PBST at room temperature and shaking for 3-4 hours. Then the plate was washed 2 times with 200 μl of PBST. BAM7cc antibodies (HyTest) conjugated to HRP were added (50 μl per well, 2 ng/μl), then standards and experimental samples were added (50 μl per well). Samples were incubated overnight at +4°C, washed 6 times with 200 µl PBST and analyzed using TMB (Merck). Absorbance was measured at 450 nm using a Multiskan FC Microplate Photometer (Thermo Fisher Scientific). The calibration curves are presented in the Supplementary (Figure S3).
2.5. Determination of parameters of interaction of Aβ and its isoforms with RAGE
The His-tag-containing sRAGE protein (Abcam) was stained with a fluorescent dye using the second-generation Monolith His-Tag Labeling Kit RED-tris-NTA according to the manufacturer's protocol. Aliquots of Aβ42, pS8-Aβ42, iso-Aβ42, and Aβ17-42 were dissolved in DMSO to a concentration of 5 mM, after which a series of dilutions were prepared to obtain solutions with Aβ concentrations from 6.1 nM to 200 μM. Aliquots of Aβ16 were dissolved in PBS to a concentration of 2.5 mM, after which a series of dilutions were prepared (the final concentration of Aβ16 in the samples varied from 38.1 nM to 1.25 mM). Samples were loaded into Monolith NT.115 Premium capillaries. The concentration of RED-tris-NTA-labeled sRAGE was constant (50 nM). All samples contained 4% DMSO and 20% glycerol. Microthermophoresis was performed using a Monolith NT.115 system (Nano Temper Technologies GmbH). Data analysis was performed using MO.Affinity Analysis v.2.3 software (Nano Temper Technologies GmbH).
2.6. Measurement of intracellular concentrations of Aβ and its isoforms
BEnd.3 cells were seeded into a 12-well plate (Greiner Bio-One) at 35 thousand per well and cultured for a week in DMEM (10% FBS), after which they were incubated with 1 μM Aβ42, pS8-Aβ42 or iso-Aβ42 within 24 hours in serum-free DMEM. Cells were washed three times with PBS, frozen in liquid nitrogen, and stored at −80°C overnight. Cells were lyzed on ice for 15 min with 250 μl of IP Lysis Buffer (Pierce) containing protease and phosphatase inhibitors (Roche) per well. The cells were removed using a scraper and placed in tubes, after which the cells were lysed for 1 hour at +4°C with shaking. The cell lysate was centrifuged for 10 minutes at 16000 g, +4°C, and the supernatant was collected. The amount of protein in the lysates was determined using a BCA assay kit (Sigma) according to the manufacturer's protocol. The concentration of Aβ and its isoforms in cell lysates was measured using ELISA as described above.
2.7. Statistical data processing
Experimental data are presented as the mean of independent experiments ± standard deviations (SD) or as a boxplot showing the median, lower and upper quartiles, minimum and maximum values of the sample. The number of independent experiments is indicated in the figure legends. The normality of the distribution was checked using the Kolmogorov-Smirnov test, and outliers were analyzed using the Q-test. Statistical differences between experimental groups for normally distributed samples were determined using Student's t test (when comparing two groups) or One-way ANOVA (when comparing multiple groups) using Tukey's test for multiple comparisons. Differences were considered statistically significant at p<0.05. Statistical analysis was performed using GraphPad Prism 8.0.2 software.
3. Results
3.1. Isomerized and phosphorylated Aβ pass through the BBB model more efficiently than unmodified Aβ.
The passage of Aβ42, pS8-Aβ42 and iso-Aβ42 across the BBB was measured in a transwell system. It was found that pS8-Aβ42 and iso-Aβ42 are transported by endothelial cells from the luminal (upper) transwell compartment to the abluminal (lower) compartment more efficiently than Aβ42 (Figure 1). Thus, the transport efficiency of pS8-Aβ42 was 1.8, 1.7 and 1.4 times higher than that of the unmodified peptide after 2, 6 and 24 hours of incubation, respectively. Transport of iso-Aβ42 through the endothelium was 1.9, 1.8 times (at 2 and 6 hours, respectively) and 1.4 times (at 24 hours) more efficient than Aβ42 transport. It can be seen that the transport rate is lower after 24 hours of incubation compared to 2 and 6 hours. A possible reason for this is that prolonged incubation can lead to degradation or aggregation of Aβ.
After the experiment, the integrity of the endothelium was checked using sodium fluorescein (Figure 2). The permeability of cell monolayer to sodium fluorescein did not differ between control cells and cells treated with amyloid peptides. This indicates that incubation with amyloid peptides did not affect the integrity of the bEnd.3 cell monolayer.
3.2. The mechanism of transport of Aβ42, pS8-Aβ42 and iso-Aβ42 across the BBB is different.
The different efficiency of passage of Aβ isoforms through the BBB model may indicate differences in the mechanisms of their transcellular transport. It is known that Aβ42 enters the brain from the blood through the mechanism of caveolin-dependent endocytosis, binding to RAGE, and Aβ42 is cleared from the brain to the blood mainly through the LRP-1 receptor via clathrin-dependent endocytosis [21] (Figure 3). In order to study the contribution of various mechanisms to the transport of Aβ and its isoforms, an inhibitor of caveolin-dependent endocytosis, filipin, and an inhibitor of clathrin-dependent endocytosis, chlorpromazine, were used.
It was found that filipin inhibits not only the transport of Aβ42, as reported previously [21], but also the passage of pS8-Аβ42 and iso-Аβ42 (Figure 4 A-C). The degree of inhibition for all of the isoforms was about 75% (Figure 4D).
Chlorpromazine inhibited the transport of Aβ42 and pS8-Aβ42 by 30% (Figure 5A,B). Surprisingly, the efficiency of iso-Aβ42 passage through the endothelium in the presence of chlorpromazine decreased by about 75% (Figure 5C). The degree of inhibition for iso-Aβ42 was different from other isoforms (Figure 5D). This indicates a difference in the contribution of clathrin and caveolin-dependent endocytosis to the transfer of Aβ isoforms across the endothelium in BBB model.
3.3. Aβ modifications affect the interaction with RAGE.
Differences in the efficiency of passage of Aβ42, pS8-Aβ42, and iso-Aβ42 through the BBB endothelium may be a result of the differences in interaction with RAGE. To study RAGE/Aβ interaction in our BBB model, inhibitor FPS-ZM1, which blocks the binding of Aβ42 to the V domain of the receptor [24], was used. FPS-ZM1 was found to significantly reduce the efficiency of transport of all Aβ isoforms through a monolayer of bEnd.3 cells (Figure 6). However, FPS-ZM1 inhibited the passage of Aβ42 and iso-Aβ42 more efficiently than that of pS8-Aβ42 for 2 and 24 hours of incubation (Figure 6D).
The dissociation constants (Kd) of Aβ42, pS8-Aβ42 and iso-Aβ42 with the soluble extracellular part of RAGE (sRAGE), determined using microscale thermophoresis (MST), were 1.0 ± 0.2 µM, 7 ± 2 µM and 23 ± 4 µM, respectively (Figure 7). In addition, it was found that the C-terminal domain of Aβ17-42 forms a complex with sRAGE with Kd = 10 ± 5 μM (Figure 8). The interaction of sRAGE with the N-terminal domain of Aβ1-16 was not detected. Thus, C-terminal domain is the main factor in the interaction of amyloid peptides with sRAGE, and the N-terminal domain modulates this interaction.
3.4. Aβ42, pS8-Aβ42 and iso-Aβ42 accumulate differently in bEnd.3 cells
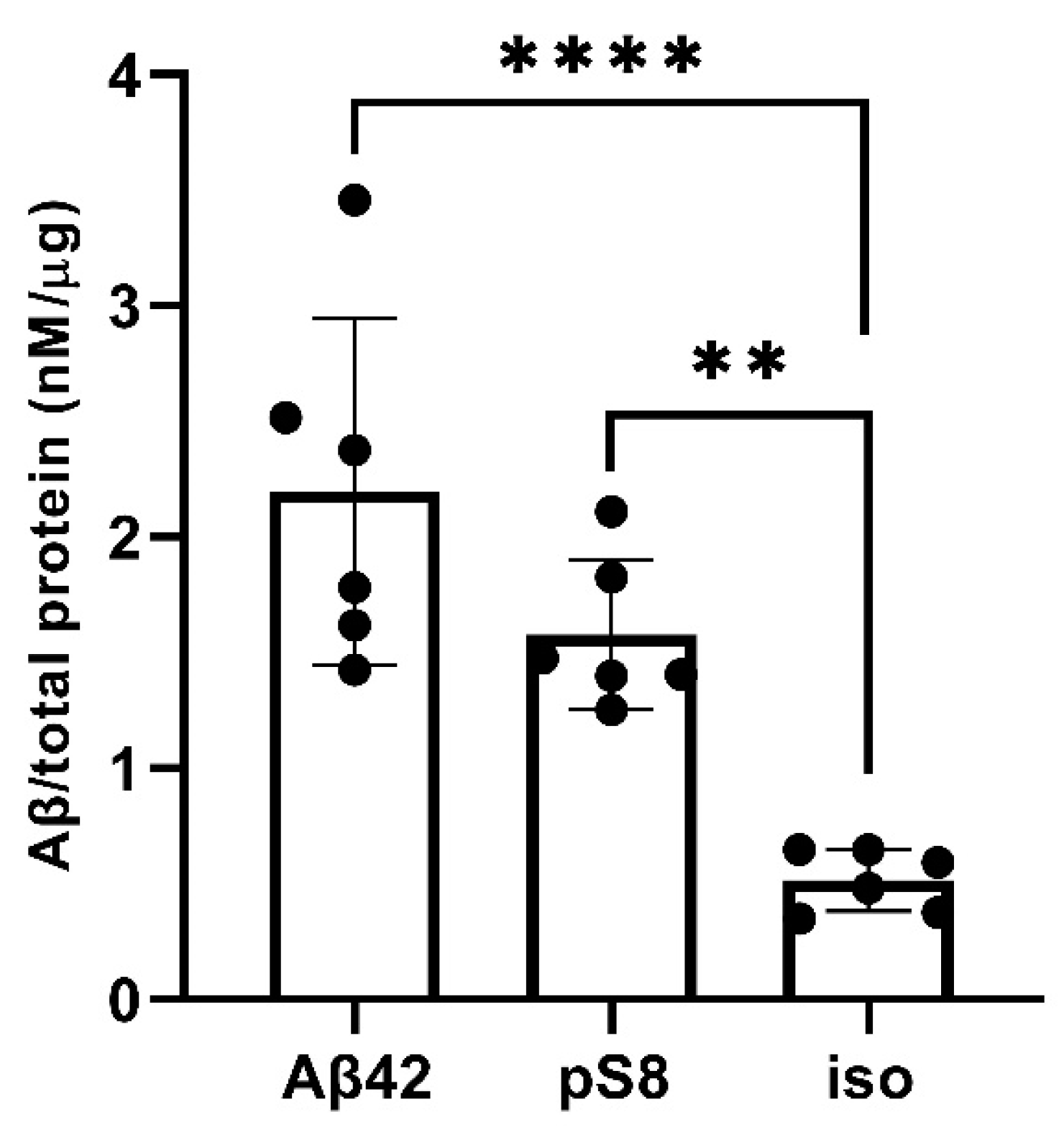
The reduced affinity of pS8-Aβ42 and iso-Aβ42 for RAGE compared to Aβ42 may cause lesser accumulation of these peptides inside cells and a more efficient transport to the abluminal side. To test this hypothesis, the intracellular levels of Aβ42, pS8-Aβ42, and iso-Aβ42 were measured after incubating cells with 1 μM of these peptides for 24 hours (Figure 9). It was shown that intact Aβ42 accumulates better inside cells, while the level of accumulation of iso-Aβ42 is minimal compared to other peptides.
4. Discussion
It is known that Aβ is expressed not only in the brain, but also in cells of other organs and tissues: kidneys and adrenal glands, heart, liver, spleen, pancreas, as well as in muscles, blood cells and endothelium [25]. Significant amounts of Aβ have been found in human red blood cells, and the Aβ42/Aβ40 ratio in red blood cells is higher than in plasma [26].
Increasing evidence indicates that peripheral Aβ can penetrate into the brain and play a significant role in the pathogenesis of AD. Thus, peripheral inoculation of brain extracts containing Aβ led to amyloidosis in the brain of mice [27,28,29]. It has also been shown that increasing the concentration of peripheral Aβ significantly reduces its removal from the brain [30]. Inhibition of RAGE-ligand interaction suppressed brain Aβ accumulation in a transgenic mouse model [31]. The important role of peripheral Aβ and its ability to enter the brain and trigger AD pathology was further highlighted in a parabiosis model in which the circulatory systems of a transgenic mouse with AD-like pathology and a wild-type mouse were connected. Using this model, the researchers demonstrated that human Aβ derived from a transgenic animal entered the wild-type mouse brain and initiated AD-like pathology, including tau hyperphosphorylation, neurodegeneration, neuroinflammation, impaired hippocampal long-term potentiation, and amyloid plaque formation [16]. Another study demonstrated the contribution of Aβ produced by blood cells to the pathogenesis of AD: when bone marrow was transplanted from transgenic mice to wild-type mice, the latter showed signs of AD pathology [17]. A number of data indicate that induction of AD requires not just an increase in the concentration of Aβ42, but the appearance of pathogenic forms carrying post-translational modifications [4,11]. Thus, intravenous administration of iso-Aβ42 accelerates amyloidogenesis in the brain of transgenic mice modeling AD [18], and introduction of pS8-Aβ42 into the blood, on the contrary, reduces the number of amyloid plaques [5]. At the same time, intravenous administration of the unmodified peptide does not affect the formation of amyloid plaques in the brain of model mice. It is possible that modified forms of Aβ arise in the circulatory system, after which they enter the brain and contribute to AD pathology [19].
In this work, we compared the efficiency of transport of Aβ isoforms in an in vitro model of the BBB, and also determined the contribution of different mechanisms of endocytosis to the passage of Aβ42, pS8-Aβ42 and iso-Aβ42 through the endothelium. It was found that pS8-Aβ42 and iso-Aβ42 are better transported by BBB endothelial cells than Aβ42 (Figure 1), which may be one of the factors determining the ability of modified forms of Aβ to influence cerebral amyloidogenesis when administered intravenously [5,18]. Notably, the concentrations used in our study are supraphysiological, and these findings should be validated in consecutive studies. Despite this, our data provide insight into the differences in the passage of Aβ isoforms across the BBB.
The main mechanism of transport of Aβ from the bloodstream to the brain is caveolin-dependent endocytosis [21]. Indeed, the inhibitor of this form of endocytosis, filipin, suppressed the transport of Aβ42, pS8-Aβ42, and iso-Aβ42 from the upper to lower compartment to the same extent (Figure 4D). Strikingly, the addition of chlorpromazine, which is an inhibitor of clathrin-dependent endocytosis, significantly suppressed the transport of iso-Aβ42 (Figure 5D). Thus, the transport of iso-Aβ42 may also be dependent on clathrin endocytosis. Also, the contribution of clathrin endocytosis was found for Aβ42 and pS8-Aβ42, but less pronounced than for iso-Aβ42. The involvement of clathrin-dependent endocytosis in transport of proteins from the bloodstream to the brain was previously shown for transferrin and insulin receptors [32,33,34,35], but not for beta-amyloid peptides.
It is assumed that RAGE plays a major role in the transfer of Aβ from the circulatory system to the brain. It was previously shown that in cells expressing RAGE an inhibitor of this receptor, FPS-ZM1, prevented oxidative stress induced by Aβ40 and Aβ42 [24]. However, the effect of FPS-ZM1 on the transport of Aβ and its isoforms across the BBB endothelium in vitro has not been studied. We found that FPS-ZM1 reduced the passage of Aβ42 through the endothelium of the BBB (Figure 6), which correlates well with data obtained previously for Aβ42 in vivo [24,31]. FPS-ZM1 also inhibited the transport of pS8-Aβ42 and iso-Aβ42, but the effect of this inhibitor on the passage of pS8-Aβ42 was less pronounced than for other isoforms (Figure 6D). Thus, it appears that RAGE is the major receptor in the transport of both Aβ and its modified forms across the BBB.
Since Aβ42, pS8-Aβ42, and iso-Aβ42 differed in their ability to penetrate the cell monolayer, we decided to compare the ability of these isoforms to interact with RAGE. There is relatively little data in the literature on the interaction parameters of Aβ with RAGE. Thus, in cell cultures, the dissociation constants of RAGE with Aβ40 and Aβ42 were 75 ± 5 nM [24] and 92 ± 40 nM [36], respectively. For purified RAGE, a dissociation constant with Aβ40 was shown to be 57 ± 14 nM [37]. Using the surface plasmon resonance method, it was revealed that sRAGE binds Aβ42 oligomers with a Kd of 17 nM [38], and the Kd for endogenous soluble RAGE (esRAGE) and Aβ42 was 44.9 nM [39]. Thus, direct measurements of the interaction of Aβ42 monomers and its isoforms with RAGE have not been previously carried out. The interaction constants obtained for Aβ42 are an order of magnitude higher compared to constants estimated in other systems. This may be due to the fact that in our experiments stabilizing agents and other additives that are far from physiological were used, which could affect the obtained constants. Nevertheless, this model allowed us to compare the binding of different isoforms with RAGE in the same conditions. Across the three Aβ42 isoforms, we found that RAGE demonstrates the highest affinity to Aβ42 and the lowest to iso-Aβ42 (Figure 7). These data are in good agreement with the results of computer modeling that we obtained earlier, according to which sRAGE has the lowest calculated Kd value with Aβ42 and the highest with iso-Aβ42 [40]. The obtained Kd values correlate with the accumulation of amyloid peptides inside cells (Figure 9). We also found that RAGE interacts with Aβ17-42, but not with Aβ1-16, and the binding constant of the receptor with Aβ17-42 was an order of magnitude smaller than the binding constant with the full-length Aβ42 peptide. Previously, we observed a similar pattern in the interaction of Aβ with Na+/K+-ATPase: binding to the enzyme was detected for Aβ17-42, but not for Aβ1-16 [5]. Probably, the hydrophobic C-terminal fragment Aβ17-42 makes a major contribution to the binding of Aβ42 to various protein molecules, while Aβ1-16 modulates this interaction.
Apparently, the high affinity of Aβ42 for RAGE is the reason for its accumulation in cells and lower transport efficiency compared to other isoforms, while the isoforms with lower affinity for the receptor are more easily transported across the endothelial cell and are able to dissociate from the receptor on the abluminal side. This mechanism was previously shown for the passage of antibodies to the transferrin receptor across the BBB. [35,41]. High-affinity antibodies against the transferrin receptor cause the antibody-receptor complex to be mainly directed to lysosomes, and those that undergo transcytosis remain associated with the receptor on the abluminal side. Low-affinity antibodies undergo transcytosis and dissociate on the abluminal side to a greater extent [35,41]. Similar studies focusing on drug delivery to the brain showed that transferrin-containing nanoparticles with high avidity for the transferrin receptor remained tightly associated with endothelial cells, whereas low avidity nanoparticles dissociated from the receptor after transcytosis [42]. In bEnd.3 cells, it was shown that strong binding of ligands to LRP-1 triggers internalization leading to endo-lysosomal sorting and degradation of ligand-receptor complex, while ligands with moderate binding strength to the receptor were transported across the endothelium [43]. Thus, the stronger binding of Aβ42 to RAGE may be the reason for its lowest transport efficiency of all isoforms across the bEnd.3 cell monolayer. Another factor influencing Aβ transport across the BBB may be different degrees of enzymatic degradation of Aβ isoforms. Thus, isomerization of the aspartate residue in Aβ has been shown to prevent its proteolysis in lysosomes [44]. PS8-Aβ is resistant to degradation by insulin degrading enzyme, unlike unmodified Aβ [11]. We found different affinities of Aβ isoforms for RAGE, which may affect enzymatic degradation.
5. Conclusions
According to our data, phosphorylated and isomerized Aβ are transported more efficiently across the endothelium of the BBB than the unmodified peptide. The RAGE receptor was found to be essential for the transport of both Aβ42 and its isoforms across the BBB. Differences in the transport of Aβ42, pS8-Aβ42, and iso-Aβ42 may be due to different mechanisms of endocytosis or different affinities of these isoforms for the RAGE receptor. The mechanisms of transport of Aβ42, pS8-Aβ42 and iso-Aβ42 across the BBB should be taken into account when developing agents for the treatment of AD. Thus, the data obtained may contribute to understanding the causes of the disease, as well as to the search for new drugs that prevent the accumulation of pathogenic Aβ isoforms in the brain.
Limitations: The use of higher than physiological concentrations of Aβ (1 μM) to study its transport is a limitation of this article. There are currently no commercial kits for measuring all Aβ isoforms, so we needed to develop this type of ELISA. However, the ELISA we developed does not have sufficient sensitivity to detect lower concentrations of Aβ and its isoforms. There are also a number of studies that use high concentrations of Aβ to study its transport across the BBB [45,46,47,48]. To our knowledge our work is the first study of the transport of Aβ42, pS8-Aβ42, and iso-Aβ42 across the BBB, as well as the first work where all isoforms of Aβ were measured by a single ELISA
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1. Permeability coefficient of amyloid beta isoforms after incubation with 1 µM or 0.1 µM for 6 and 24 hours. Figure S2. Testing the toxicity of filipin (A), chlorpromazine (B) and FPS-ZM1 (C) for bEnd.3 cells. Figure S3. ELISA calibration curves for Aβ42, pS8-Аβ42 and iso-Аβ42.
Author Contributions
Conceptualization, I.Y.P., V.A.M. and E.P.B.; methodology, K.B.V., I.Y.P., and E.P.B..; validation, I.Y.P. and V.A.M.; formal analysis, K.B.V., I.Y.P. and E.P.B.; investigation, K.B.V., I.Y.P. and E.P.B.; resources, V.A.M. and A.A.M.; writing—original draft preparation, K.B.V. and E.P.B.; writing—review and editing, I.Y.P., V.A.M. and A.A.M.; supervision, I.Y.P., V.A.M. and A.A.M.; project administration, V.A.M. and A.A.M., funding acquisition, V.A.M. and A.A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Higher Education of the Russian Federation (grant agreement № 075-15-2020-795, state contract № 13.1902.21.0027 of 29 September 2020, unique project ID: RF-190220X0027).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sonkusare, S.K.; Kaul, C.L.; Ramarao, P. Dementia of Alzheimer’s Disease and Other Neurodegenerative Disorders--Memantine, a New Hope. Pharmacol Res 2005, 51, 1–17. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s Disease. The Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Moro, M.L.; Phillips, A.S.; Gaimster, K.; Paul, C.; Mudher, A.; Nicoll, J.A.R.; Boche, D. Pyroglutamate and Isoaspartate Modified Amyloid-Beta in Ageing and Alzheimer’s Disease. Acta Neuropathologica Communications 2018, 6, 3. [Google Scholar] [CrossRef]
- Barykin, E.P.; Mitkevich, V.A.; Kozin, S.A.; Makarov, A.A. Amyloid β Modification: A Key to the Sporadic Alzheimer’s Disease? Front Genet 2017, 8, 58. [Google Scholar] [CrossRef]
- Barykin, E.P.; Petrushanko, I.Y.; Kozin, S.A.; Telegin, G.B.; Chernov, A.S.; Lopina, O.D.; Radko, S.P.; Mitkevich, V.A.; Makarov, A.A. Phosphorylation of the Amyloid-Beta Peptide Inhibits Zinc-Dependent Aggregation, Prevents Na,K-ATPase Inhibition, and Reduces Cerebral Plaque Deposition. Front Mol Neurosci 2018, 11, 302. [Google Scholar] [CrossRef]
- Zirah, S.; Kozin, S.A.; Mazur, A.K.; Blond, A.; Cheminant, M.; Segalas-Milazzo, I.; Debey, P.; Rebuffat, S. Structural Changes of Region 1-16 of the Alzheimer Disease Amyloid Beta-Peptide upon Zinc Binding and in Vitro Aging. J Biol Chem 2006, 281, 2151–2161. [Google Scholar] [CrossRef]
- Forest, K.H.; Alfulaij, N.; Arora, K.; Taketa, R.; Sherrin, T.; Todorovic, C.; Lawrence, J.L.M.; Yoshikawa, G.T.; Ng, H.-L.; Hruby, V.J.; et al. Protection against β-Amyloid Neurotoxicity by a Non-Toxic Endogenous N-Terminal β-Amyloid Fragment and Its Active Hexapeptide Core Sequence. Journal of Neurochemistry 2018, 144, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Perez, K.A.; Lago, L.C.; Klatt, S.; McLean, C.A.; Birchall, I.E.; Barnham, K.J.; Masters, C.L.; Roberts, B.R. Quantification of N-Terminal Amyloid-β Isoforms Reveals Isomers Are the Most Abundant Form of the Amyloid-β Peptide in Sporadic Alzheimer’s Disease. Brain Commun 2021, 3, fcab028. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Matsuoka, Y.; Shirasawa, T. Biological Significance of Isoaspartate and Its Repair System. Biol Pharm Bull 2005, 28, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Mitkevich, V.A.; Petrushanko, I.Y.; Yegorov, Y.E.; Simonenko, O.V.; Vishnyakova, K.S.; Kulikova, A.A.; Tsvetkov, P.O.; Makarov, A.A.; Kozin, S.A. Isomerization of Asp7 Leads to Increased Toxic Effect of Amyloid-Β42 on Human Neuronal Cells. Cell Death Dis 2013, 4, e939. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Heneka, M.T. Truncated and Modified Amyloid-Beta Species. Alz Res Therapy 2014, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Jamasbi, E.; Separovic, F.; Hossain, M.A.; Ciccotosto, G.D. Phosphorylation of a Full Length Amyloid-β Peptide Modulates Its Amyloid Aggregation, Cell Binding and Neurotoxic Properties. Mol Biosyst 2017, 13, 1545–1551. [Google Scholar] [CrossRef]
- Barisano, G.; Montagne, A.; Kisler, K.; Schneider, J.A.; Wardlaw, J.M.; Zlokovic, B.V. Blood–Brain Barrier Link to Human Cognitive Impairment and Alzheimer’s Disease. Nat Cardiovasc Res 2022, 1, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-Brain Barrier Breakdown Is an Early Biomarker of Human Cognitive Dysfunction. Nat Med 2019, 25, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The Blood-Brain Barrier in Alzheimer’s Disease. Neurobiol Dis 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Bu, X.-L.; Xiang, Y.; Jin, W.-S.; Wang, J.; Shen, L.-L.; Huang, Z.-L.; Zhang, K.; Liu, Y.-H.; Zeng, F.; Liu, J.-H.; et al. Blood-Derived Amyloid-β Protein Induces Alzheimer’s Disease Pathologies. Mol Psychiatry 2018, 23, 1948–1956. [Google Scholar] [CrossRef]
- Sun, H.-L.; Chen, S.-H.; Yu, Z.-Y.; Cheng, Y.; Tian, D.-Y.; Fan, D.-Y.; He, C.-Y.; Wang, J.; Sun, P.-Y.; Chen, Y.; et al. Blood Cell-Produced Amyloid-β Induces Cerebral Alzheimer-Type Pathologies and Behavioral Deficits. Mol Psychiatry 2021, 26, 5568–5577. [Google Scholar] [CrossRef]
- Kozin, S.A.; Cheglakov, I.B.; Ovsepyan, A.A.; Telegin, G.B.; Tsvetkov, P.O.; Lisitsa, A.V.; Makarov, A.A. Peripherally Applied Synthetic Peptide isoAsp7-Aβ(1-42) Triggers Cerebral β-Amyloidosis. Neurotox Res 2013, 24, 370–376. [Google Scholar] [CrossRef]
- Kozin, S.A.; Makarov, A.A. The Convergence of Alzheimer’s Disease Pathogenesis Concepts. Mol Biol 2019, 53, 896–903. [Google Scholar] [CrossRef]
- Biere, A.L.; Ostaszewski, B.; Stimson, E.R.; Hyman, B.T.; Maggio, J.E.; Selkoe, D.J. Amyloid Beta-Peptide Is Transported on Lipoproteins and Albumin in Human Plasma. J Biol Chem 1996, 271, 32916–32922. [Google Scholar] [CrossRef]
- Zhu, D.; Su, Y.; Fu, B.; Xu, H. Magnesium Reduces Blood-Brain Barrier Permeability and Regulates Amyloid-β Transcytosis. Mol Neurobiol 2018, 55, 7118–7131. [Google Scholar] [CrossRef]
- Abulrob, A.; Sprong, H.; Van Bergen en Henegouwen, P.; Stanimirovic, D. The Blood-Brain Barrier Transmigrating Single Domain Antibody: Mechanisms of Transport and Antigenic Epitopes in Human Brain Endothelial Cells. J Neurochem 2005, 95, 1201–1214. [Google Scholar] [CrossRef]
- Daniel, J.A.; Chau, N.; Abdel-Hamid, M.K.; Hu, L.; von Kleist, L.; Whiting, A.; Krishnan, S.; Maamary, P.; Joseph, S.R.; Simpson, F.; et al. Phenothiazine-Derived Antipsychotic Drugs Inhibit Dynamin and Clathrin-Mediated Endocytosis. Traffic 2015, 16, 635–654. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A Multimodal RAGE-Specific Inhibitor Reduces Amyloid β–Mediated Brain Disorder in a Mouse Model of Alzheimer Disease. J Clin Invest 2012, 122, 1377–1392. [Google Scholar] [CrossRef]
- Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Castaño, E.M.; Van Vickle, G.D.; Kalback, W.M.; Patton, R.L.; Luehrs, D.C.; Daugs, I.D.; Kuo, Y.-M.; et al. Amyloid Beta Peptides in Human Plasma and Tissues and Their Significance for Alzheimer’s Disease. Alzheimer’s & Dementia 2009, 5, 18–29. [Google Scholar] [CrossRef]
- Kiko, T.; Nakagawa, K.; Satoh, A.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. Amyloid β Levels in Human Red Blood Cells. PLoS One 2012, 7, e49620. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Obermüller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally Applied Abeta-Containing Inoculates Induce Cerebral Beta-Amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Fritschi, S.K.; Hamaguchi, T.; Obermüller, U.; Füger, P.; Skodras, A.; Schäfer, C.; Odenthal, J.; Heikenwalder, M.; Staufenbiel, M.; et al. Multiple Factors Contribute to the Peripheral Induction of Cerebral β-Amyloidosis. J Neurosci 2014, 34, 10264–10273. [Google Scholar] [CrossRef]
- Burwinkel, M.; Lutzenberger, M.; Heppner, F.L.; Schulz-Schaeffer, W.; Baier, M. Intravenous Injection of Beta-Amyloid Seeds Promotes Cerebral Amyloid Angiopathy (CAA). Acta Neuropathol Commun 2018, 6, 23. [Google Scholar] [CrossRef]
- Marques, M.A.; Kulstad, J.J.; Savard, C.E.; Green, P.S.; Lee, S.P.; Craft, S.; Watson, G.S.; Cook, D.G. Peripheral Amyloid-Beta Levels Regulate Amyloid-Beta Clearance from the Central Nervous System. J Alzheimers Dis 2009, 16, 325–329. [Google Scholar] [CrossRef]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE Mediates Amyloid-Beta Peptide Transport across the Blood-Brain Barrier and Accumulation in Brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Ayloo, S.; Gu, C. Transcytosis at the Blood–Brain Barrier. Current Opinion in Neurobiology 2019, 57, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, S.; Galindo, D.C.; Schwartz, M.W.; Banks, W.A.; Rhea, E.M. Endocytosis of Insulin at the Blood-Brain Barrier. Frontiers in Drug Delivery 2022, 2. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.; Sandra, A.; Siek, G.C.; Lucas, J.J.; Fine, R.E. Studies of the Mechanism of Iron Transport across the Blood-Brain Barrier. Annals of Neurology 1992, 32, S43–S50. [Google Scholar] [CrossRef] [PubMed]
- Goulatis, L.I.; Shusta, E.V. Protein Engineering Approaches for Regulating Blood–Brain Barrier Transcytosis. Current Opinion in Structural Biology 2017, 45, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Chellappa, R.C.; Lukose, B.; Rani, P. Correction: G82S RAGE Polymorphism Influences Amyloid-RAGE Interactions Relevant in Alzheimer’s Disease Pathology. PLOS ONE 2021, 16, e0248252. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Stern, D.; Kane, M.D.; Kuo, Y.M.; Lampert, H.C.; Roher, A.E. RAGE-Abeta Interactions in the Pathophysiology of Alzheimer’s Disease. Restor Neurol Neurosci 1998, 12, 167–173. [Google Scholar] [PubMed]
- Chen, X.; Walker, D.G.; Schmidt, A.M.; Arancio, O.; Lue, L.-F.; Yan, S.D. RAGE: A Potential Target for Abeta-Mediated Cellular Perturbation in Alzheimer’s Disease. Curr Mol Med 2007, 7, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, T.; Munesue, S.; Yamamoto, Y.; Sakurai, S.; Akhter, N.; Kitamura, Y.; Shiba, K.; Watanabe, T.; Yonekura, H.; Hayashi, Y.; et al. Endogenous Secretory Receptor for Advanced Glycation End-Products Inhibits Amyloid-β 1-42 Uptake into Mouse Brain. Journal of Alzheimer’s Disease 2012, 28, 709–720. [Google Scholar] [CrossRef]
- Tolstova, A.P.; Adzhubei, A.A.; Mitkevich, V.A.; Petrushanko, I.Y.; Makarov, A.A. Docking and Molecular Dynamics-Based Identification of Interaction between Various Beta-Amyloid Isoforms and RAGE Receptor. International Journal of Molecular Sciences 2022, 23, 11816. [Google Scholar] [CrossRef]
- Yu, Y.J.; Zhang, Y.; Kenrick, M.; Hoyte, K.; Luk, W.; Lu, Y.; Atwal, J.; Elliott, J.M.; Prabhu, S.; Watts, R.J.; et al. Boosting Brain Uptake of a Therapeutic Antibody by Reducing Its Affinity for a Transcytosis Target. Sci Transl Med 2011, 3, 84ra44. [Google Scholar] [CrossRef]
- Wiley, D.T.; Webster, P.; Gale, A.; Davis, M.E. Transcytosis and Brain Uptake of Transferrin-Containing Nanoparticles by Tuning Avidity to Transferrin Receptor. Proceedings of the National Academy of Sciences 2013, 110, 8662–8667. [Google Scholar] [CrossRef]
- Tian, X.; Leite, D.M.; Scarpa, E.; Nyberg, S.; Fullstone, G.; Forth, J.; Matias, D.; Apriceno, A.; Poma, A.; Duro-Castano, A.; et al. On the Shuttling across the Blood-Brain Barrier via Tubule Formation: Mechanism and Cargo Avidity Bias. Sci Adv 2020, 6, eabc4397. [Google Scholar] [CrossRef]
- Lambeth, T.R.; Riggs, D.L.; Talbert, L.E.; Tang, J.; Coburn, E.; Kang, A.S.; Noll, J.; Augello, C.; Ford, B.D.; Julian, R.R. Spontaneous Isomerization of Long-Lived Proteins Provides a Molecular Mechanism for the Lysosomal Failure Observed in Alzheimer’s Disease. bioRxiv 2019, 605626. [Google Scholar] [CrossRef] [PubMed]
- Zinchenko, E.; Klimova, M.; Mamedova, A.; Agranovich, I.; Blokhina, I.; Antonova, T.; Terskov, A.; Shirokov, A.; Navolokin, N.; Morgun, A.; et al. Photostimulation of Extravasation of Beta-Amyloid through the Model of Blood-Brain Barrier. Electronics 2020, 9, 1056. [Google Scholar] [CrossRef]
- Shubbar, M.H.; Penny, J.I. Therapeutic Drugs Modulate ATP-Binding Cassette Transporter-Mediated Transport of Amyloid Beta(1–42) in Brain Microvascular Endothelial Cells. European Journal of Pharmacology 2020, 874, 173009. [Google Scholar] [CrossRef] [PubMed]
- Dal Magro, R.; Simonelli, S.; Cox, A.; Formicola, B.; Corti, R.; Cassina, V.; Nardo, L.; Mantegazza, F.; Salerno, D.; Grasso, G.; et al. The Extent of Human Apolipoprotein A-I Lipidation Strongly Affects the β-Amyloid Efflux Across the Blood-Brain Barrier in Vitro. Frontiers in Neuroscience 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, B.; Crawford, F.; Bachmeier, C. Inhibition of ADAM10 Promotes the Clearance of Aβ across the BBB by Reducing LRP1 Ectodomain Shedding. Fluids and Barriers of the CNS 2016, 13, 14. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Passage of 1 μM Aβ42, pS8-Aβ42 and iso-Aβ42 through a monolayer of bEnd.3 cells from the upper transwell compartment to the lower compartment at 2, 6 and 24 hours. The amounts (pmol) of Aβ42, pS8-Aβ42 and iso-Aβ42 in the lower compartment measured by sandwich ELISA normalized by incubation time (min) and transwell area (cm2) are presented. Number of values in each group n=15-19 representing 6 independent experiments. ** - p < 0.01, **** - p < 0.0001.
Figure 1.
Passage of 1 μM Aβ42, pS8-Aβ42 and iso-Aβ42 through a monolayer of bEnd.3 cells from the upper transwell compartment to the lower compartment at 2, 6 and 24 hours. The amounts (pmol) of Aβ42, pS8-Aβ42 and iso-Aβ42 in the lower compartment measured by sandwich ELISA normalized by incubation time (min) and transwell area (cm2) are presented. Number of values in each group n=15-19 representing 6 independent experiments. ** - p < 0.01, **** - p < 0.0001.
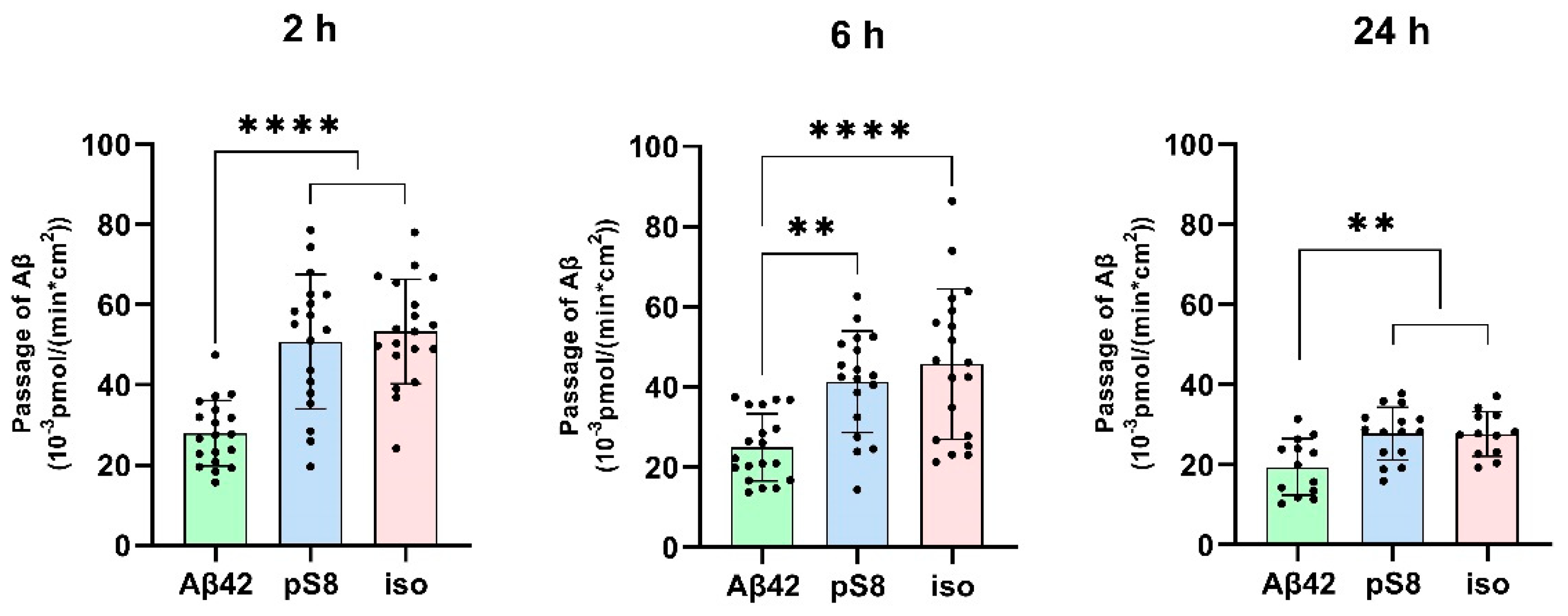
Figure 2.
Efficiency of sodium fluorescein passage through a monolayer of bEnd.3 cells. Prior to the measurement, the cells were incubated with 0.08% DMSO, or with 1 μM Aβ42, pS8-Aβ42, or iso-Aβ42 for 24 hours. Fluorescence intensity values in the lower transwell compartment are shown. Number of independent replicates n=3.
Figure 2.
Efficiency of sodium fluorescein passage through a monolayer of bEnd.3 cells. Prior to the measurement, the cells were incubated with 0.08% DMSO, or with 1 μM Aβ42, pS8-Aβ42, or iso-Aβ42 for 24 hours. Fluorescence intensity values in the lower transwell compartment are shown. Number of independent replicates n=3.
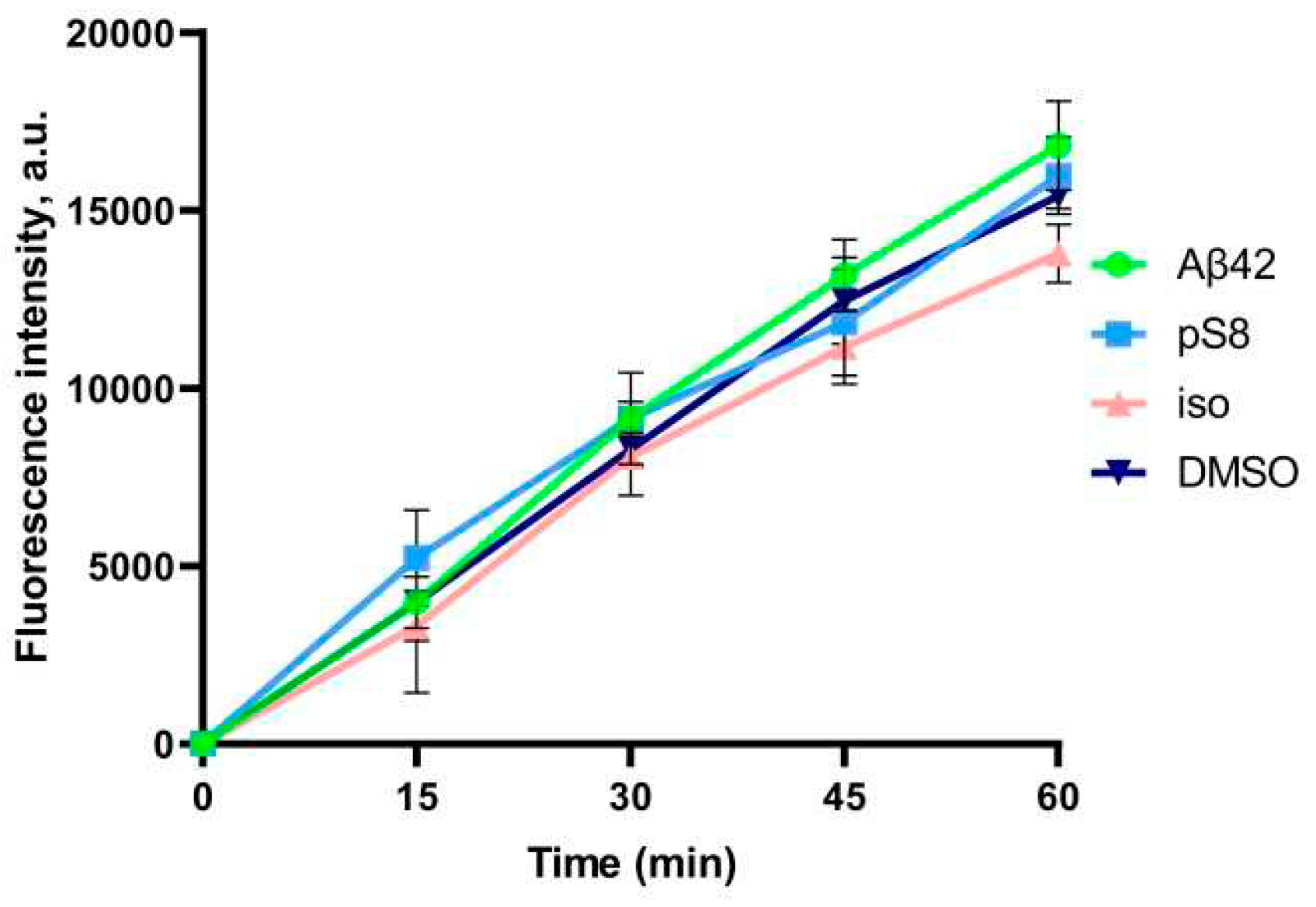
Figure 3.
Schematic representation of Aβ42 transport through the endothelium of the BBB and the underlying molecular mechanisms. Inhibitors affecting caveolin- and clathrin-dependent endocytosis are indicated in red. Filipin binds cholesterol in the membrane and interferes with caveolae formation [22]. Chlorpromazine affects the complex of adapter proteins AP-2 involved in clathrin-dependent endocytosis [23].
Figure 3.
Schematic representation of Aβ42 transport through the endothelium of the BBB and the underlying molecular mechanisms. Inhibitors affecting caveolin- and clathrin-dependent endocytosis are indicated in red. Filipin binds cholesterol in the membrane and interferes with caveolae formation [22]. Chlorpromazine affects the complex of adapter proteins AP-2 involved in clathrin-dependent endocytosis [23].
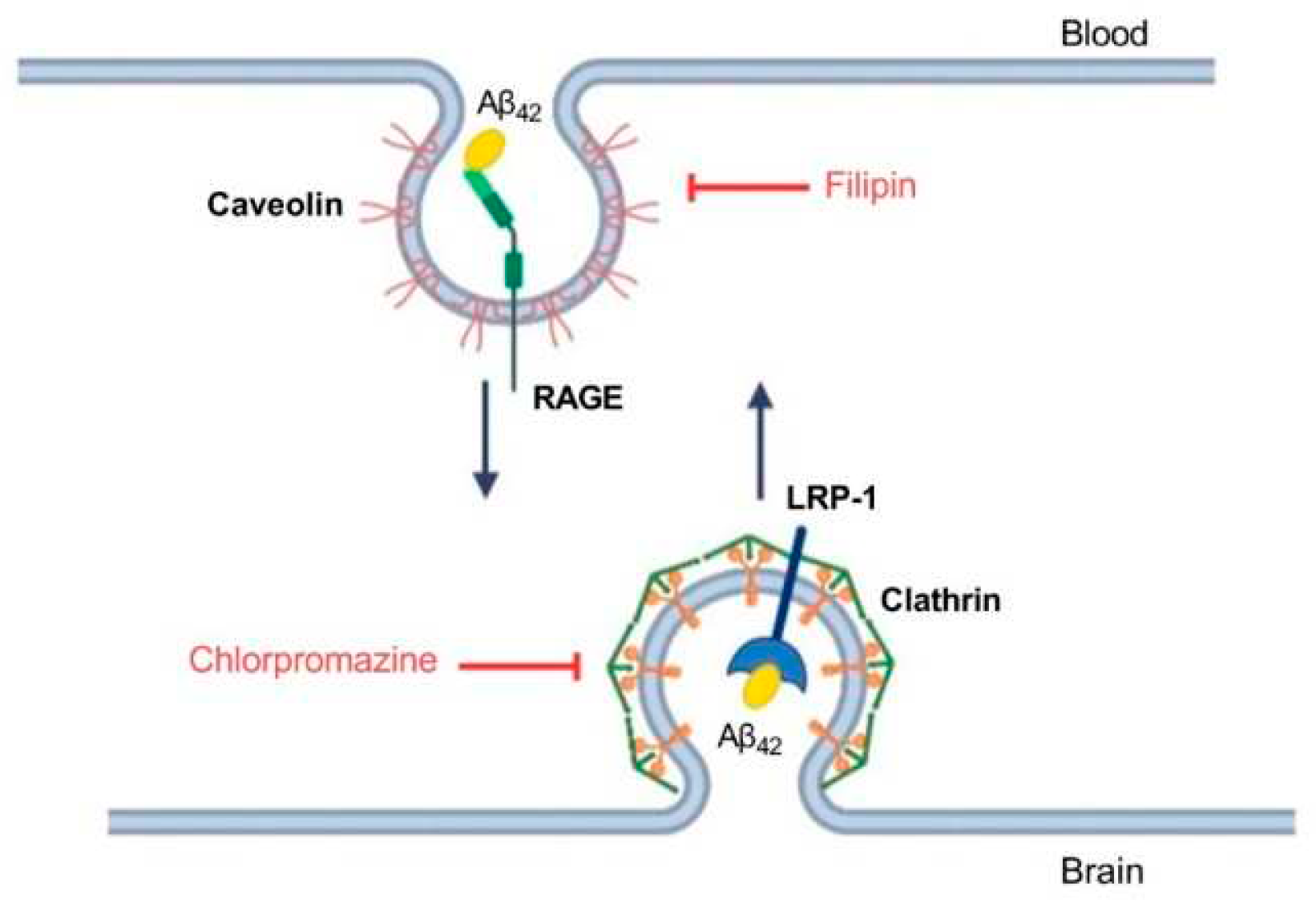
Figure 4.
Effects of filipin on the efficiency of Aβ42 (A), pS8-Aβ42 (B) and iso-Aβ42 (C) transport through a monolayer of bEnd.3 cells in the transwell model. The amounts (pmol) of Aβ42, pS8-Aβ42 and iso-Aβ42 in the lower compartment normalized by incubation time (min) and transwell area (cm2) after 2 and 6 hours of incubation with amyloid peptides in the absence or presence of filipin are shown. D) Comparison of the degree of inhibition of filipin Aβ42, pS8-Aβ42 and iso-Aβ42, where transport of the peptides in the absence of the inhibitor was taken as 100% (not shown). Summarized data from three independent experiments are presented, the number of values in each group n = 6-9, ** - p < 0.01, *** - p < 0.001, **** - p < 0.0001.
Figure 4.
Effects of filipin on the efficiency of Aβ42 (A), pS8-Aβ42 (B) and iso-Aβ42 (C) transport through a monolayer of bEnd.3 cells in the transwell model. The amounts (pmol) of Aβ42, pS8-Aβ42 and iso-Aβ42 in the lower compartment normalized by incubation time (min) and transwell area (cm2) after 2 and 6 hours of incubation with amyloid peptides in the absence or presence of filipin are shown. D) Comparison of the degree of inhibition of filipin Aβ42, pS8-Aβ42 and iso-Aβ42, where transport of the peptides in the absence of the inhibitor was taken as 100% (not shown). Summarized data from three independent experiments are presented, the number of values in each group n = 6-9, ** - p < 0.01, *** - p < 0.001, **** - p < 0.0001.
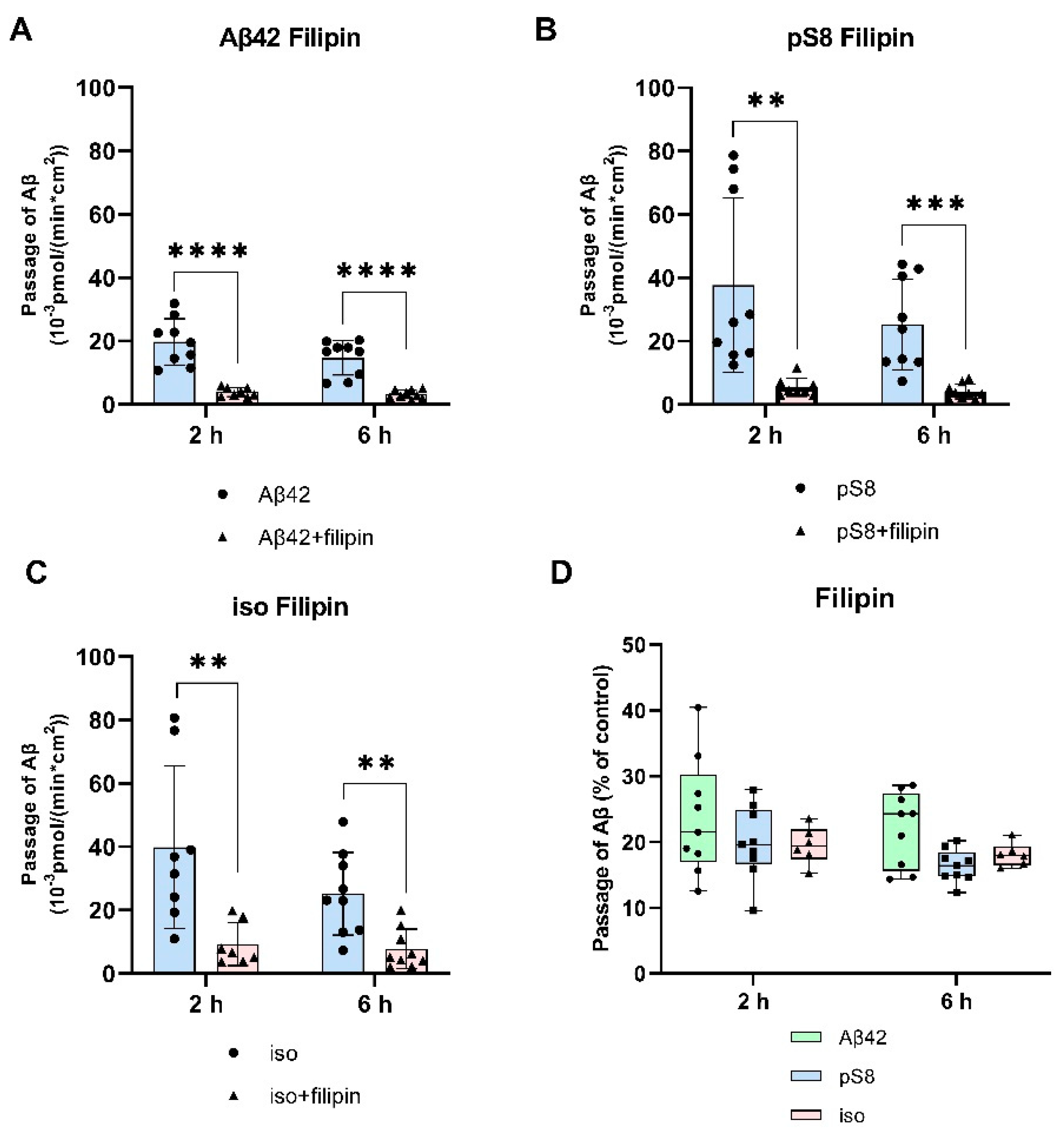
Figure 5.
Effects chlorpromazine (CPZ) on the efficiency of Aβ42 (A), pS8-Aβ42 (B) and iso-Aβ42 (C) transport through a monolayer of bEnd.3 cells in the transwell model. The amounts (pmol) of Aβ42, pS8-Aβ42 and iso-Aβ42 in the lower compartment normalized by incubation time (min) and transwell area (cm2) after 2 and 6 hours of incubation with amyloid peptides in the absence or presence of CPZ are shown. D) Comparison of the degree of inhibition of CPZ Aβ42, pS8-Aβ42 and iso-Aβ42, where transport of the peptides in the absence of the inhibitor was taken as 100% (not shown). Summarized data from three independent experiments are presented, the number of values in each group n = 6-9, ns - not significant, * - p < 0.05, ** - p < 0.01, *** - p < 0.001, **** - p < 0.0001.
Figure 5.
Effects chlorpromazine (CPZ) on the efficiency of Aβ42 (A), pS8-Aβ42 (B) and iso-Aβ42 (C) transport through a monolayer of bEnd.3 cells in the transwell model. The amounts (pmol) of Aβ42, pS8-Aβ42 and iso-Aβ42 in the lower compartment normalized by incubation time (min) and transwell area (cm2) after 2 and 6 hours of incubation with amyloid peptides in the absence or presence of CPZ are shown. D) Comparison of the degree of inhibition of CPZ Aβ42, pS8-Aβ42 and iso-Aβ42, where transport of the peptides in the absence of the inhibitor was taken as 100% (not shown). Summarized data from three independent experiments are presented, the number of values in each group n = 6-9, ns - not significant, * - p < 0.05, ** - p < 0.01, *** - p < 0.001, **** - p < 0.0001.
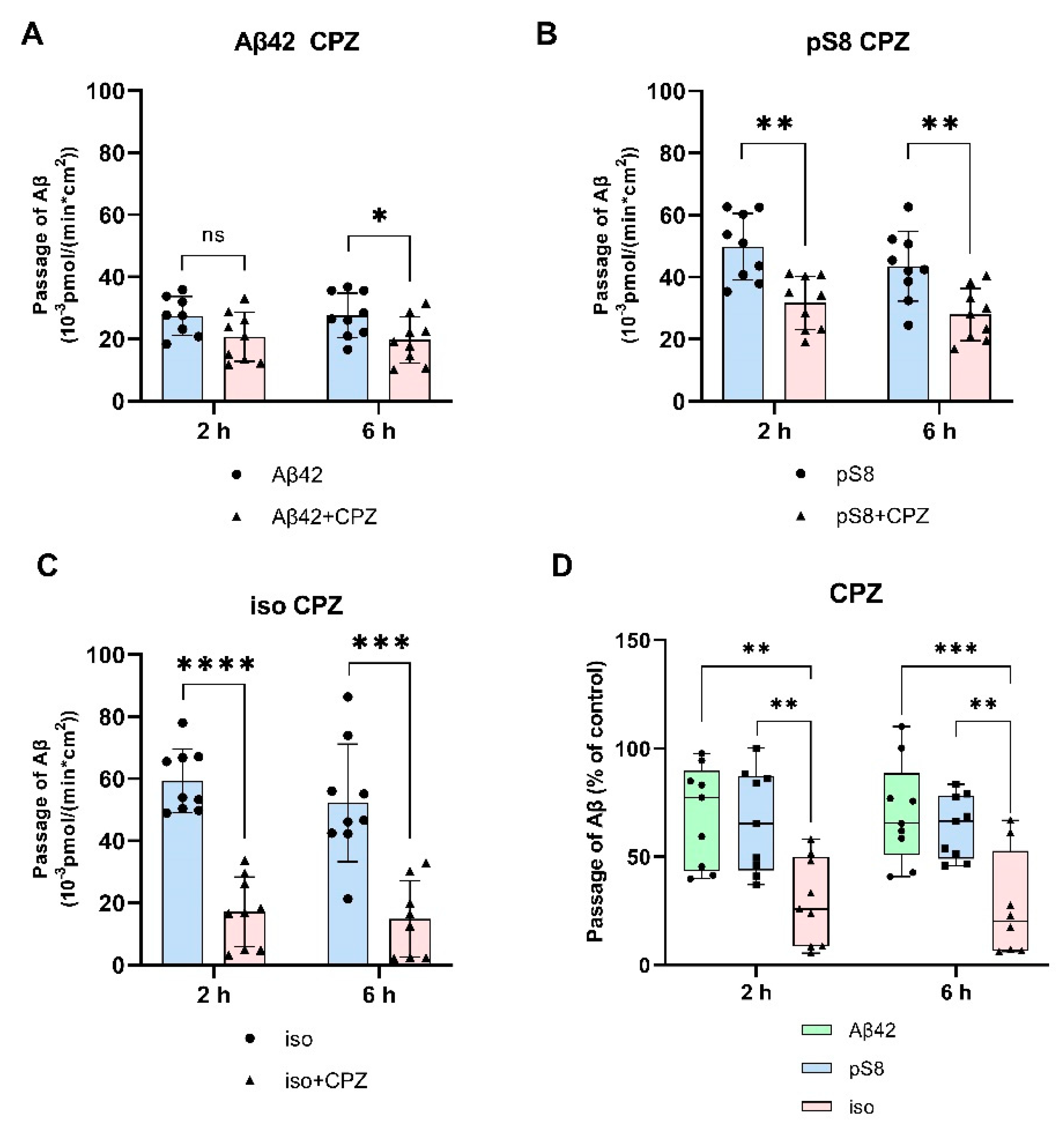
Figure 6.
Effect of the RAGE antagonist FPS-ZM1 on the efficiency of transport of Aβ42 (A), pS8-Aβ42 (B) and iso-Aβ42 (C) through a monolayer of bEnd.3 cells in the transwell model. The amounts (pmol) of Aβ42, pS8-Aβ42 and iso-Aβ42 in the lower compartment normalized by incubation time (min) and transwell area (cm2) after 2, 6 and 24 hours of incubation with amyloid peptides in the absence or presence of FPS-ZM1 are shown. D) Comparison of the degree of inhibition of FPS-ZM1 Aβ42, pS8-Aβ42 and iso-Aβ42, where transport of the peptides in the absence of the inhibitor was taken as 100% (not shown). Summarized data from three independent experiments are presented, the number of values in each group n = 6-9, * - p < 0.05, ** - p < 0.01, *** - p < 0.001, **** - p < 0.0001.
Figure 6.
Effect of the RAGE antagonist FPS-ZM1 on the efficiency of transport of Aβ42 (A), pS8-Aβ42 (B) and iso-Aβ42 (C) through a monolayer of bEnd.3 cells in the transwell model. The amounts (pmol) of Aβ42, pS8-Aβ42 and iso-Aβ42 in the lower compartment normalized by incubation time (min) and transwell area (cm2) after 2, 6 and 24 hours of incubation with amyloid peptides in the absence or presence of FPS-ZM1 are shown. D) Comparison of the degree of inhibition of FPS-ZM1 Aβ42, pS8-Aβ42 and iso-Aβ42, where transport of the peptides in the absence of the inhibitor was taken as 100% (not shown). Summarized data from three independent experiments are presented, the number of values in each group n = 6-9, * - p < 0.05, ** - p < 0.01, *** - p < 0.001, **** - p < 0.0001.
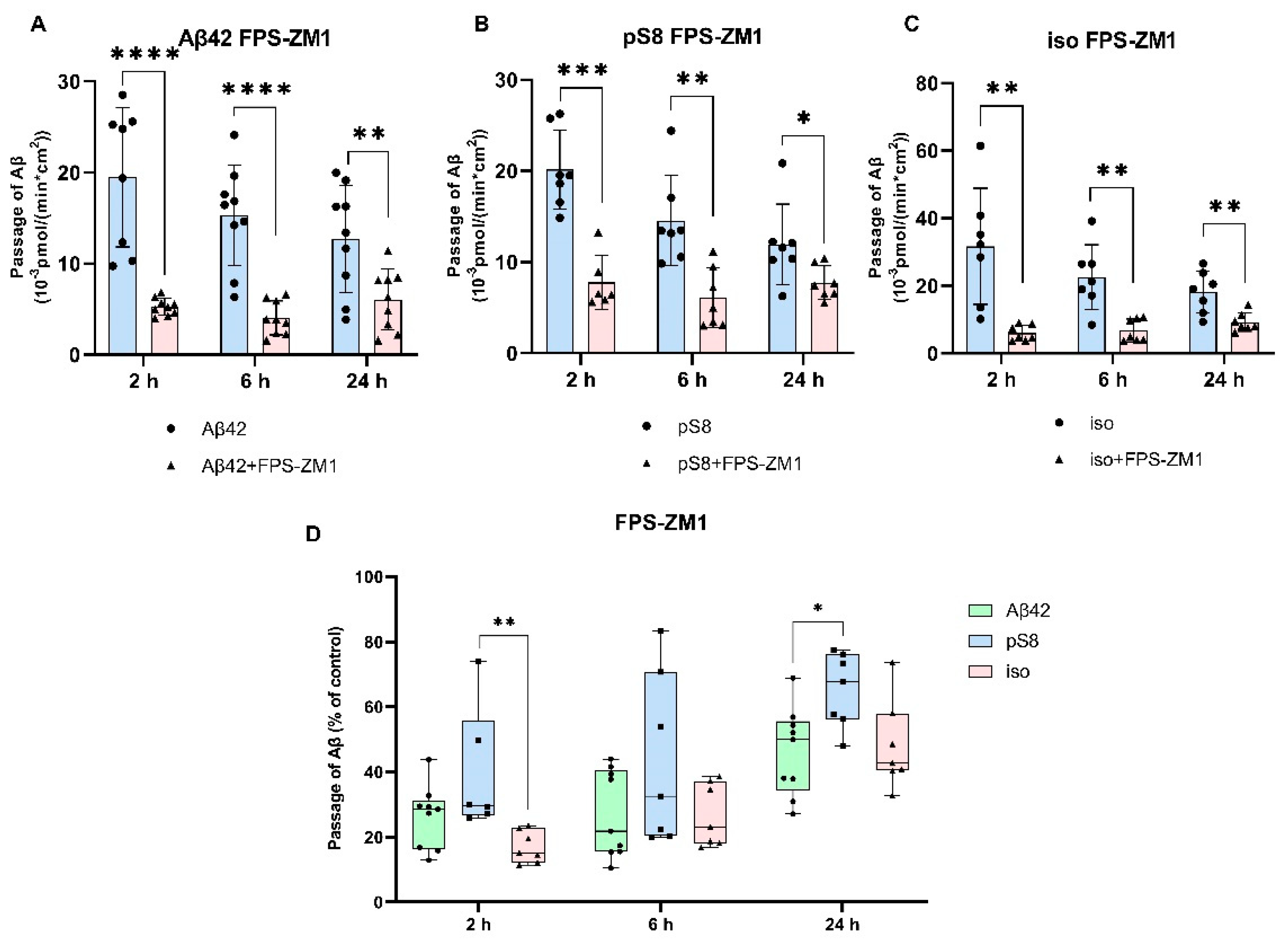
Figure 7.
Interaction of Aβ isoforms with sRAGE. A) MST curves showing the fraction of RAGE which is in the complex with the peptide at different concentrations of Aβ and its isoforms. B) Values of dissociation constants (Kd) for complexes of Aβ isoforms with sRAGE. Number of replicates in each group n = 4-5, * - p < 0.05, **** - p < 0.0001.
Figure 7.
Interaction of Aβ isoforms with sRAGE. A) MST curves showing the fraction of RAGE which is in the complex with the peptide at different concentrations of Aβ and its isoforms. B) Values of dissociation constants (Kd) for complexes of Aβ isoforms with sRAGE. Number of replicates in each group n = 4-5, * - p < 0.05, **** - p < 0.0001.
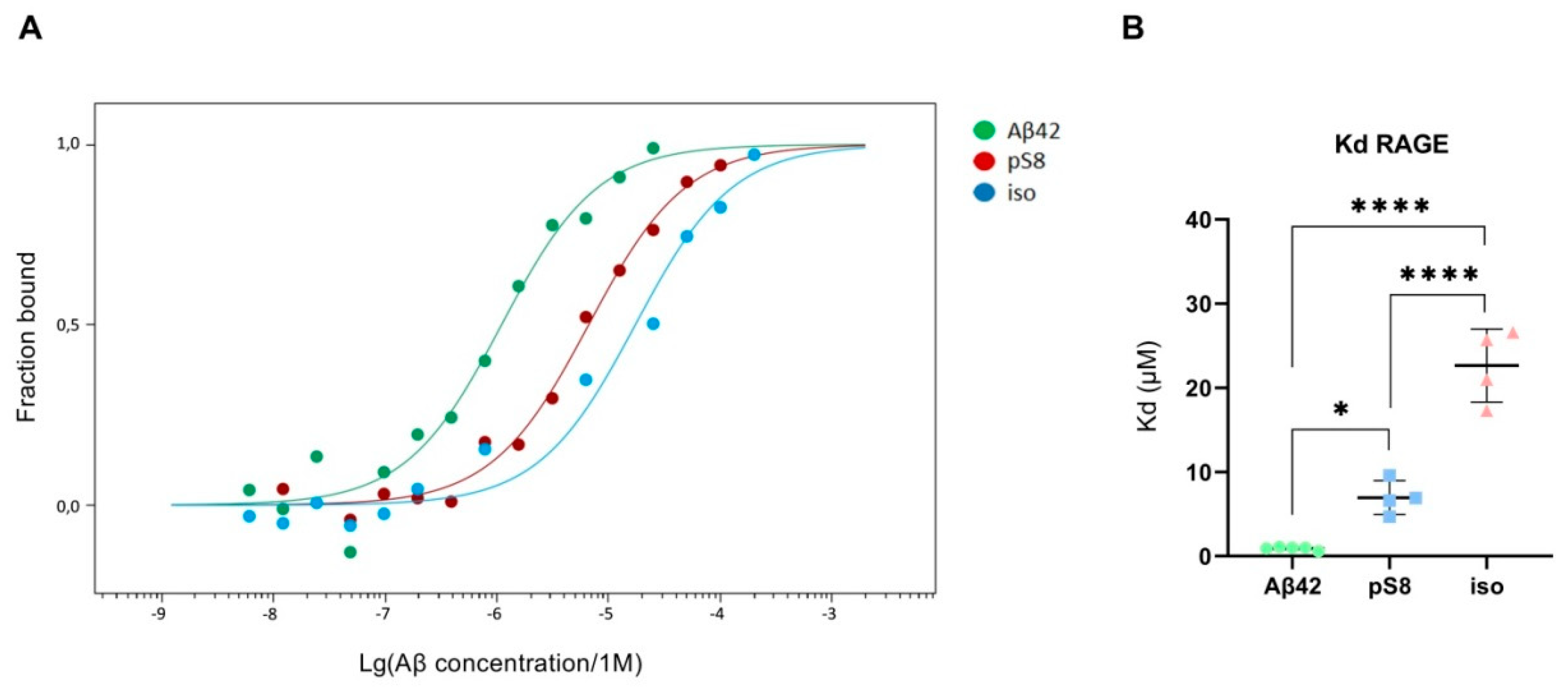
Figure 8.
MST curve illustrating the interaction of Aβ17-42 with sRAGE.
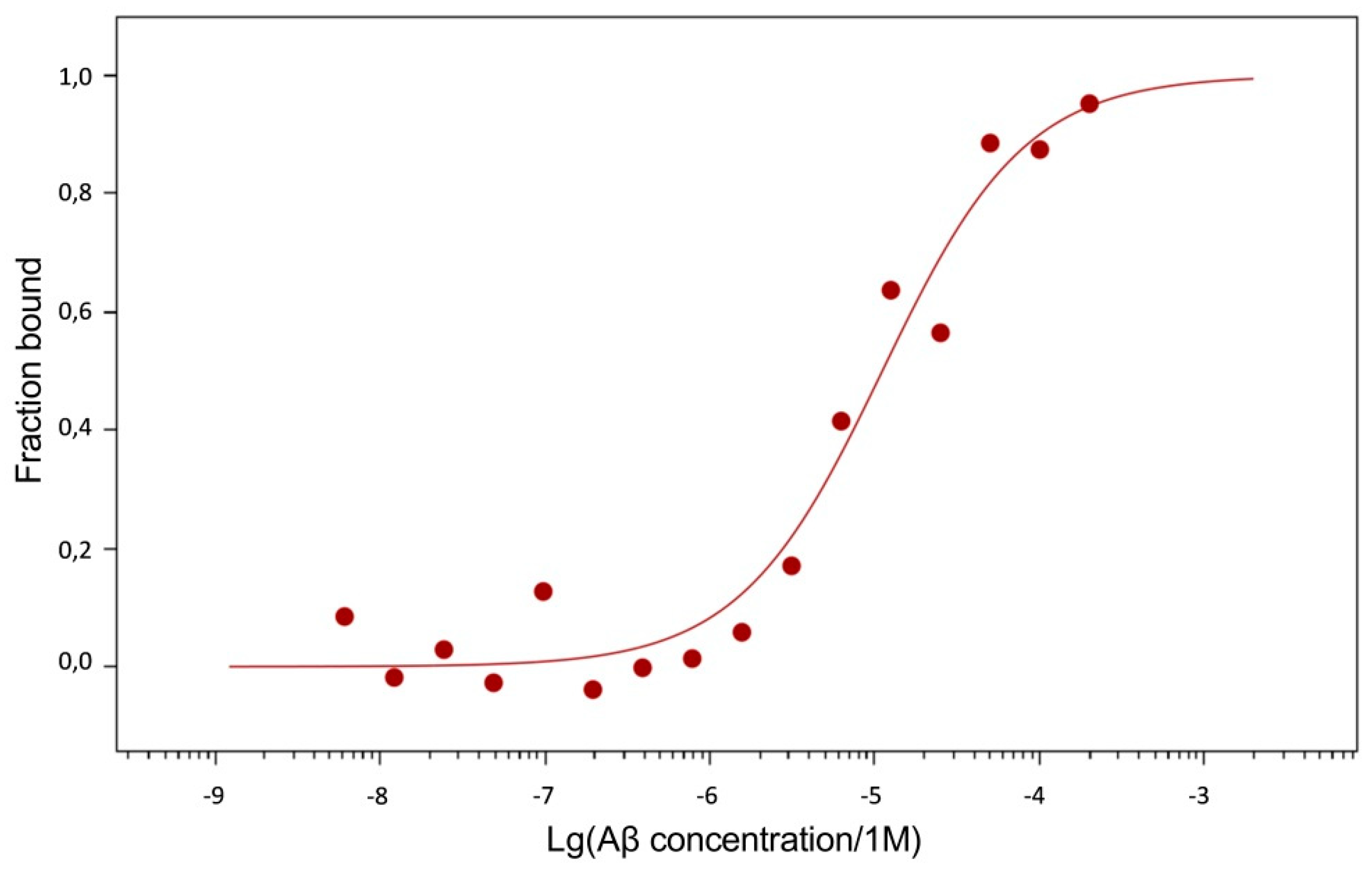
Figure 9.
Levels of Aβ42, pS8-Aβ42 and iso-Aβ42 in bEnd.3 cells after 24 h of incubation with these peptides (1 μM). The data were obtained on cell lysates using ELISA. The concentrations of peptides normalized to the total protein (μg) in the samples are presented. Number of replicates in each group n = 6, ** - p < 0.01, **** - p < 0.0001.
Figure 9.
Levels of Aβ42, pS8-Aβ42 and iso-Aβ42 in bEnd.3 cells after 24 h of incubation with these peptides (1 μM). The data were obtained on cell lysates using ELISA. The concentrations of peptides normalized to the total protein (μg) in the samples are presented. Number of replicates in each group n = 6, ** - p < 0.01, **** - p < 0.0001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



