
Submitted:
17 December 2023
Posted:
18 December 2023
You are already at the latest version
Abstract
Background: Uncontrolled hemorrhage is a major preventable cause of death in patients with trauma. However, the majority of large animal models of hemorrhage have utilized controlled hemorrhage rather than uncontrolled hemorrhage to investigate the impact of immunopathy and coagulopathy on multi-organ failure (MOF) and mortality. This study evaluates these alterations in a severe porcine-controlled and uncontrolled hemorrhagic shock (HS) model.
Methods: Anesthetized female swine underwent controlled hemorrhage (22 ml/kg) and uncontrolled hemorrhage by partial splenic resection followed with or without lactated Ringer solution (LR) or Voluven® resuscitation. Blood chemistry, physiologic variables, systemic and tissue levels of complement proteins and cytokines, coagulation parameters, organ function, and damage were recorded and assessed.
Results: HS resulted in systemic and local complement activation, cytokine release, metabolic acidosis, hyperkalemia, MOF, and no animal survival. Resuscitation with LR and Voluven® after HS improved hemodynamic parameters (MAP and SI), metabolic acidosis, hyperkalemia, and survival but resulted in increased complement activation and worse coagulopathy. Compared with the LR group, the HS animal treated with Voluven® had worse dilutional anemia, coagulopathy, renal and hepatic dysfunction, increased myocardial complement activation and renal damage, and decreased survival rate.
Conclusions: Hemorrhagic shock triggers early immunopathy and coagulopathy that appear associated with MOF and death. This study indicates that immunopathy and coagulopathy are therapeutic targets that may be addressed with a high-impact adjunctive treatment to conventional resuscitation.
Keywords:
uncontrolled hemorrhagic shock
; immunopathy
; coagulopathy
; MOF
; mortality
; swine
; damage control resuscitation
1. Introduction
Most battlefield casualties still die in the pre-hospital phase before reaching a medical treatment facility [1]. Uncontrolled hemorrhage is the leading cause of preventable deaths on the battlefield and in civilian patients [1,2]. Management priorities are stopping bleeding and reversing shock by restoring circulating blood volume to prevent or reduce the risk of worsening trauma-induced coagulopathy (TIC) and multi-organ failure (MOF) [3]. However, managing trauma patients with hemorrhagic shock (HS) is complex and challenging, despite recent advances in treatment protocols, including tourniquets, permissive hypotension, point of care ultrasonography, tranexamic acid, massive transfusions, and all performed within the “golden hour.” Prehospital goal-directed HS resuscitation with low-volume crystalloid or colloid fluid seems a better option in resource-limited settings [4].
The optimal resuscitation fluid for the early treatment of severe bleeding patients remains highly debated. Roger et al. compared the rapidity of shock reversal with lactated Ringer (LR) or hydroxyethyl starch (HES) 130/0.4 at the early phase of controlled HS and found that HES restored MAP four times faster than LR [5]. A total of 35 randomized controlled trials (RCT) evaluating the management of traumatic HS within the first 24 hours of injury did not show a correlation between transfusion requirements and mortality [6]. Recently, damage control resuscitation (DCR) combines multiple methods to prevent or reverse HS-associated events such as acidosis, hypothermia, coagulopathy, and hypoperfusion [7]. Unlike observational studies, RCTs to support the DCR approach are currently lacking [6,8]. The lack and/or inconclusiveness of clinical trials evaluating traumatic HS urges animal studies. Despite the multifactorial pathogenesis of traumatic HS, coagulopathy is the focus of scientific discourse on animal models [2,9]. One-quarter to one-third of patients at major trauma centers have coagulopathy on admission [8,10]. A similar proportion of coagulopathy upon admission is seen in combat patients [9]. Early TIC is portrayed as hypocoagulability that results in bleeding. In contrast, later TIC is characterized by a hypercoagulable state associated with microvascular thrombosis and MOF [3]. TIC and the effectiveness of patients’ treatment are the subjects of ongoing multifaceted debate, including reverse causality [1,8,11]. Successful resuscitation of traumatic HS is unlikely without efficient hemostasis. Intracavitary, often incompressible, and overlooked bleeding is life-threatening [12,13].
Traumatic HS followed by fluid resuscitation results in global ischemia and reperfusion injury that can provoke a severe inflammatory response and worsen clinical outcomes. Systemic immune system activation is fundamental to developing MOF in this context and shares many features in common with systemic inflammatory response syndrome (SIRS). MOF remains a significant cause of morbidity and mortality in trauma patients, and current therapy is based on standard supportive care. Understanding the pathophysiology of HS and resuscitation will allow for developing targeted therapeutic strategies to minimize MOF and improve patient outcomes following traumatic HS.
An emerging body of evidence indicates that crosstalk between the complement, coagulation, and fibrinolytic cascades following traumatic hemorrhage leads to microthrombosis and thromboinflammation, thereby contributing to MOF and mortality [3,14]. Central to thromboinflammation is the loss of endothelial cells’ normal antithrombotic and anti-inflammatory functions, leading to coagulopathy, complementopathy, and immunopathy. Huber-Lang et al. discovered that thrombin in a proteolytic pattern activates C5 to generate C5a without C3 involvement [15]. Amara et al. found that coagulation/fibrinolysis factors activate complement components C3 and C5, which subsequently activate and trigger the complement pathway [16]. Similarly, Gulla et al. reported that the complement is a pro-coagulant factor leading to thrombin activation, which generates fibrin mesh [17]. We have noted the activation of complement and coagulation cascades and their interaction, impacting outcomes in preclinical animal models of traumatic hemorrhage [18,19] and clinical trauma patients [20]. Furthermore, our recent findings have shown that the synergistic effects of the traumatic triad (complementopathy, endotheliopathy, and coagulopathy) occurred early after trauma, contributed to poor clinical outcomes (MOF/death), and led to infectious complications; therefore, the triadic intercommunication model is proposed [21]. Additionally, biomarkers such as Bb, syndecan-1, and D-dimer are reliable early predictive biomarkers of clinical outcomes [21]. TIC involves many factors, including inflammatory response [9,22]. Inflammation and coagulation are reciprocally causally related processes [23]. Hemostatic resuscitation [11] may include pharmacological agents as potential adjuncts to fluid therapy to treat severe HS at or near the point of injury [24,25,26,27,28,29,30,31,32,33,34].
The purpose of this study is to characterize immune inflammatory responses, coagulation alterations, and their correlation with MOF and mortality in a lethal porcine model of controlled and uncontrolled HS with LR or HES permissive hypotensive resuscitation.
2. Results
2.1. Baseline Characterization and Mortality
Animals were subjected to controlled bleeding (22 ml/kg b.w.) and uncontrolled splenic hemorrhage followed with or without two fluid resuscitation arms ([LR (45 ml/kg) and Voluven® (15 ml/kg) at a rate of 1ml/kg/min]. No significant differences in the hemodynamic (Table 1) and metabolic (Table 2) parameters were observed during the pre-hemorrhagic phase (baseline) among the sham, H, H+LR, and H+Voluven®.
2.2. Physiological Responses to Hemorrhage
As shown in Table 1 and Table 2, the animals with HS experienced hemodynamic changes [decreased levels of pulse pressure (PP) and mean arterial pressure (MAP), and increased levels of shock index (SI)] and metabolic alterations [decreased base excess (BE) and venous oxygen saturation (SvO2), and increased lactate and potassium] starting at 30min and remaining up to 120min.
2.3. Fluid Resuscitation on Hemodynamic and Metabolic Parameters
As demonstrated in Table 1 and Table 2, uncontrolled shed blood volume (SBV) in swine resuscitated with LR was not significantly different from the H group. Resuscitation with Voluven® significantly increased uncontrolled SBV at 90, 120, and 360 minutes compared to LR (p<0.05), though equal uncontrolled blood loss at the pre-fluid resuscitation phase. There were no statistically significant differences between final resuscitation fluid consumption time and final urine output volume among the three groups (data not shown). PP, MAP, and BE increased, whereas shock index (SI), blood lactate, and potassium decreased in the resuscitated animals compared to those without fluid. There was no significant difference in the hemodynamic and metabolic parameters in the LR and Voluven® animals.
2.4. Fluid Resuscitation on Hemodilution and Coagulation Parameters
As shown in Table 2, hemoglobin (Hb) values remained constant in the sham group, decreased slightly in the H group at 90 and 120 minutes, and dropped significantly in other groups resuscitated with LR and Voluven® at 60, 90, and 120 min. Voluven® resuscitation further decreased Hb at 60, 90, and 120 min compared with the LR group. Hematocrit (Hct) values paralleled changes in Hb values (Table 2).
Thromboelastographic analysis determined whole blood coagulation function. The H+ Voluven® group drew out a reduction in the actual strength of the clot (G, Figure 1A) and the maximum amplitude (MA, Figure 1B), reaching significance in comparison with the H+LR group, indicating a relatively hypocoagulable state (p <0.05). Hemorrhaged animals resuscitated with fluids showed significantly lower fibrinogen concentrations in blood plasma (Figure 1C) and prolonged PT (Figure 1D) and aPTT compared with the H group (Figure 1E). Voluven® resuscitation resulted in a further increase in PT (Figure 1D) and aPTT (Figure 1E) and a further decrease in fibrinogen compared with the H+LR group (Figure 1C). Reaction time expressed as the ratio to the baseline value was not different when the H group was compared with fluid-resuscitated animals (p < 0.05 and p < 0.001 for H vs. H+LR, and H vs. H+Voluven®, respectively; Kruskal-Wallis test; data not shown). The same was valid for K-time. There was no apparent change in the angle of the respective groups.
2.5. Circulating Complement Activation and Cytokine Release
Total hemolytic complement (CH50) in Voluven®-treated hemorrhaged swine had a significantly increased level of complement function at 60, 90, and 120 minutes compared to hemorrhaged non-treated animals (Figure 2A). C3a desArg, a cleavage product of systemic C3 complement component activation, was significantly increased in LR-resuscitated animals at 90 minutes compared to those hemorrhaged swine (Figure 2B).
Some pro-inflammatory and anti-inflammatory cytokines were measured in the circulation. Normalization of tumor necrosis factor-α (TNF-α) values did not result in a significant difference between the hemorrhaged only and the H+LR group. There was no apparent difference in the TNF-α blood concentration between the H+Fluid groups nor between the H+LR and H+Voluven® groups (Figure 2C). Systemic IL-6 levels were also increased in Voluven®-treated animals at 120 minutes compared to LR-treated swine (Figure 2D). As a result of the significant differences among the individual IL-8 at the baseline, plasma levels of IL-8 are shown as a percent of the baseline to permit a direct comparison of relative changes after HS. When IL-8 concentration was normalized to the baseline values, a significant difference in its levels between respective groups could not be observed (Figure 2E). Anti-inflammatory cytokines such as IL-10 were undetected in the blood (data not shown).
2.6. End Organ Function
Creatinine (a biomarker of kidney function) blood levels were significantly elevated in Voluven®-treated hemorrhage swine at 60, 90, and 120 minutes after the onset of injury compared with the H+LR group (Figure 3A). Aspartate aminotransferase, a marker of hepatocellular damage, was significantly higher in injured and Voluven®-treated swine than in the H+LR group (Figure 3B) at 120 minutes. There was no statistically significant difference in muscle myocardium isoenzyme B (MMB) levels between hemorrhaged animals and those injured and fluid resuscitated (Figure 3C). Independently of experimental condition, creatine kinase (CK) levels did not show significant changes (Figure 3D).
2.7. Myocardial Inflammatory Responses to Hemorrhagic Shock and Fluid Resuscitation
As illustrated in Figure 4, increased deposition of C4d, C3, C5, C5b-9, and IL-6 at porcine hearts after exposure to hemorrhagic shock. Hemorrhaged animals with LR resuscitation did not affect these protein depositions compared to the H group. Voluven® resuscitated animals had significantly higher levels of C5b-9 in hearts than the groups of H and H+LR (p<0.05).
2.8. Pulmonary and Intestinal Inflammatory Responses to Hemorrhagic Shock and Fluid Resuscitation
As demonstrated in Figure 5, enhanced expression of C3 and C5b-9 in the jejunum tissue was observed in hemorrhaged animals with or without fluid resuscitation. There was no statistically significant difference among the three groups (H, H+LR, and H+Voluven®). Hemorrhage significantly increased intestinal deposition of C3 and IL-6. Fluid resuscitation had little impact on the intestinal tissue level of C3 and IL-6 except for LR resuscitation on C3 expression.
2.9. Effect of H and Fluid Resuscitation on Organ Histopathological Alterations
The exposure to the hemorrhagic shock w or w/o LR resuscitation resulted in pathological changes typical of mild myocarditis characterized by monocyte and neutrophil infiltration (white rectangle), whereas Voluven® resuscitation tends to be more inflammatory cell infiltration but not reach a statistically significant difference (Figure 6A&B). H, H+LR, and H+Voluven® induced moderate ~ severe pulmonary (septal thickening, inflammatory cell infiltration, alveolar hemorrhage, and edema) and intestinal (denuded villi with lamina propria exudate) damages. Still, there were no statistically significant differences among the three groups (Figure 6C~F). H and H+LR led to renal damage (proximal tubular epithelial cell hydropic degeneration, border brush loss, and interstitial inflammatory cell infiltration) (Figure 6G&H). Animals resuscitated with LR and Voluven® improved and worsened renal damage, respectively (Figure 6G&H).
2.10. Effect of Hemorrhage and Fluid Resuscitation on Survival
As you can see in Figure 7, six-hour survival for the Sham, H, H+LR, and H+Voluven® was 9/9 (360±0.0min), 0/14 (127.6±11.3min), 11/13 (353.3±6.4min), and 1/6 (282.2±27.9min), respectively. Better survival of hemorrhaged animals treated with LR than those treated with Voluven® was observed (p <0.05). A clear difference in the length of survival between hemorrhaged animals and those resuscitated with fluid was observed (p <0.001 and p <0.01, for the H vs. H+LR and H vs. H+Voluven®, respectively; the log-rank test). There was no correlation between the length of survival and SBV at 30 min or final blood loss.
3. Discussion
Hemorrhage after severe trauma remains the leading cause of potentially preventable death in young individuals (≤45 years) in traumatically injured civilians (~40%) and military populations (~50%) [35,36,37]. In combat, 87% of battlefield deaths after HS occur before reaching a medical facility. Outcomes of patients with HS and extremity bleeding have improved with the widespread use of tourniquets and early balanced transfusion therapy. However, point-of-injury and prehospital care of injured patients with uncontrolled truncal hemorrhage is a challenge and has had the same mortality over the last two decades despite recent advances in treatment protocols, including tourniquets, permissive hypotension, point-of-care ultrasonography, tranexamic acid, massive transfusions, and all performed within the “golden hour.” [38].
Hemorrhaged patients after severe trauma are particularly susceptible to the early development of coagulopathy, immunopathy [complementopathy, systemic inflammatory response syndrome (SIRS), immunoparesis], endotheliopathy, and metabopathy (tissue hypoperfusion-induced energy metabolic dysfunction and iron metabolic alterations) on admission to the hospital [14,20,30,33,34,39,40,41,42,43,44,45]. Moreover, trauma-induced exposure of tissue factor to flowing blood induces the activation of coagulation, which may trigger consumptive coagulopathy [3]. Uncontrolled bleeding after trauma initiates coagulopathy via loss of coagulation factors, red blood cells, and platelets, coagulation cascade activation, immunopathy, endotheliopathy, and metabopathy through global ischemia. Moreover, therapeutic approaches (e.g., fluid resuscitation, blood transfusion, surgical procedures, extracorporeal life support devices) can further worsen these multi-opathies and perpetuate bleeding [46].
Severe HS is a system failure, including coagulation, immunity, vital organs, vasculature, endothelium, mitochondria, and physiological barriers [47]. Post-HS systemic uncoupling of coagulation, immunity, vital organs vasculature, endothelium, mitochondria, and physiological barriers is critical in morbidity and mortality. Maintaining oxygen delivery to limit tissue hypoxia, inflammation, and organ function is essential for uncontrolled HS. Damage control resuscitation (DCR) recommends using more blood products and less clear fluids (crystalloid and colloid solution) as initial resuscitation in treating HS. However, prehospital resuscitation in austere environments largely relies on crystalloid and colloid intravascular expansion, as blood products are logistically arduous. Ordinary fluid resuscitation in such resource-limited conditions is the first therapeutic intervention to replace blood loss and preserve tissue perfusion until definite surgical control of bleeding can be achieved. Clear fluid resuscitation can worsen clinical outcomes by increasing blood loss by elevating blood pressure, dislodging blood clots, diluting coagulation factors and platelets, and causing inflammation and coagulopathy [48].
Our previous studies have shown controlled hemorrhagic shock in rats [33,34] and pigs [27], with typical fluid resuscitation, triggers tissue hypoperfusion, metabolic acidosis, systemic and local inflammation, and physiological barrier dysfunction, leading to multiple organ damage and death. Even though DCR with LR or Voluven® following HS improves some measures of hemodynamics, metabolism, hyperkalemia, and survival in this current study, these restricting fluid therapies result in further complement activation and worse coagulopathy. The most important advantages of using colloids are the rapid achievement of hemodynamic goals because of their slow diffusion into the extravascular space and logistical due to reducing the weight and volume of resuscitation. Some colloids have been attributed to additional harmful effects on hemostasis, with altered fibrin polymerization and decreased platelet adhesive and aggregating properties [5]. Clinical data from a series of large and randomized controlled trials in critically ill patients failed to show an outcomes advantage associated with colloidal fluid resuscitation and indicate that hydroxyethyl starch solutions may be related to significant adverse effects (e.g., acute kidney injury, need for renal replacement therapy, coagulopathies, and pathological tissue uptake) [49].
We previously demonstrated that Hextend® colloid infusion significantly contributes to dilutional anemia, tissue inflammation, complement activation, multiple-organ damage, and mortality, although it boosts hemodynamics after controlled HS in swine [27]. In this current study, Voluven® resuscitation led to more uncontrolled bleeding, worse dilutional anemia, systemic inflammation, coagulopathy, renal and hepatic dysfunction, increased myocardial complement activation, and renal damage. It decreased the survival rate compared to balanced LR crystalloid infusion. As such, developing optimal DCR by targeting immunopathy and/or coagulopathy may be a promising adjunctive strategy for prehospital settings.
In recent years, immunological damage control resuscitation has shown promise as a pharmacological agent for use in trauma [30,50,51,52,53,54], traumatic HS [55,56,57], and sepsis [58,59]. Our recent works have shown 1) the beneficial effects of complement inhibition as both a stand-alone and adjunctive treatment with HS [27,31,60,61]; and 2) the evident effectiveness of immunological damage control resuscitation even in complex animal models of combined HS and polytrauma [33,34,42]. Prehospital treatment of tranexamic acid (TXA) has been shown to reduce mortality in a large international trauma study [62], recommended in some pre-hospital systems, and included in the WHO list of essential medicines for treating trauma [World Health Organization. Summary of the Report of the 18th Meeting of the WHO Expert Committee on the Selection and Use of Essential Medicines. https://www.who.int/selection_medicines/committees/TRS_web_summary.pdf (2021)]. The prehospital use of TXA is still controversial because there was no significant difference in the mortality of TXA versus placebo in multicenter RCT [63,64]. However, patients with severe shock who received early prehospital administration of TXA had a significant reduction in 30-day mortality [65], suggesting the severe traumatic HS patient group would benefit the most from TXA. Altogether, a future hemostatic resuscitative crystalloid and colloid fluids in combination with pharmacological agents to deter coagulopathy and immunopathy may resolve some of these problems and show more outcome benefits.
Histological splenic examination indicated successful autotransfusion, at least in the early dead pig. The spleens from the pigs that survived the observation period (24 hours) had erythrocytes in the red pulp, but this fact cannot reflect the organ dynamics at the time close to injury. Unlike a human, a pig’s spleen sequesters up to 20-25% of an animal’s blood volume and can auto-transfuse blood at a high rate under severe hemorrhagic conditions [66]. Whether splenectomy is an essential procedure in porcine hemorrhage studies is an enduring issue [67,68], and caution needs to be exercised when bleeding is higher than 30% [69]. Vnuk et al. [70], using a volume-controlled hemorrhagic porcine model and specified anesthetics (azaperone, thiopental, and isoflurane), showed that sham-operated animals were hemodynamically more stable than splenectomized and those with an auto transplanted spleen. Fixed-volume models of hemorrhagic shock more closely simulate hemorrhage seen in accident victims or combat casualties [71]. We recognize potential differences among pigs with intact spleen that can transfuse irregular volumes of blood into circulation. The variation in the capacity of the spleen to permeate different volumes of blood is a categorical property of the animal’s body, independent of its environment. Therefore, this variability should be counted on despite the possible requirement of a relatively larger number of animals for testing. Pottecher et al. [72] underlined that the pre-hemorrhagic porcine splenectomy model only reproduces the situation when hemorrhagic shock follows elective surgical splenectomy.
4. Conclusions
Controlled and uncontrolled hemorrhage-triggered immunopathy and coagulopathy were associated with increased MOF and mortality. This study suggests that 1) LR crystalloid solution is a better agent than Voluven®, a colloid solution, and 2) the development of adjunctive strategies by targeting early immunopathy and/or coagulopathy may be a promising therapy for traumatic HS patients at the point-of-injury care and on-scene care.
5. Materials and Methods
5.1. Animal Study
The research complied with the Animal Welfare Act and the implementing Animal Welfare regulations, and the study was conducted in compliance with the Animal Welfare Act, the implementing Animal Welfare regulations, and the principles of the Guide for the Care and Use of Laboratory Animals, National Research Council. The facility’s Institutional Animal Care and Use Committee approved all research conducted in this study (approved code: A-10-017; approved date: 9/23/2012). The facility where this research was conducted is fully accredited by AAALAC International.
5.1.1. General Procedures
Yorkshire-cross female pigs weighing 38.9±2.9 kg (mean±SD) were obtained from Midwest Swine Research (Gibbon, MN), held in an AAALAC International-accredited facility, and their acclimatization lasted a minimum of 5 days. After this period, baseline hematologic and biochemical screening was performed.
5.1.2. Surgical Preparation
The pigs were fasted for 12 to 18 hours with free access to water before surgery. Pre-surgical procedures and instrumentation were applied as in our previous report [73]. In brief, pre-terminal anesthesia medication for secretion control (glycopyrrolate, Robinul, 0.01 mg/kg, Baxter Healthcare, Deerfield, IL) and sedation (tiletamine-zolazepam, Telazol®, 8 mg/kg, Wyeth, Fort Dodge, IA) were administered intramuscularly. After intubation and anesthesia, the animals were supine on a standard operating table. Surgical instrumentation was conducted under anesthesia with 1.5-2.5% isoflurane in 30% oxygen in the air using an automatic ventilator and monitor (Fabius GS gas anesthesia system and Infinity Delta XL monitoring system, Draeger Medical, Telford, PA). An end-tidal pCO2 of approximately 40 mm Hg was maintained. Core temperature was supported throughout by a homeothermic blanket and forced-air warming system. Urine was collected transurethrally using all silicone Foley catheters (10Fr., 3-mL balloon, Sherwood Medical, St. Louis, MO).
Vascular catheters were inserted via cut-downs. A micromanometer (SPC 330A; Millar Instruments, Inc., Houston, TX) was inserted non-occlusive into the left internal carotid artery for blood pressure and heart rate monitoring. A Swan-Ganz catheter was passed via the left jugular vein and positioned with its tip in the pulmonary artery, as confirmed by insertion tracings for continuous measurement of cardiac output and mixed venous oxygen saturation (Vigilance II Monitor, Edwards Lifesciences LLC, Irvine, CA). Occlusive catheters (8Fr. Sideport/percutaneous catheter introducer, Argon Medical Devices, Athens, TX) were inserted into the left femoral artery and left femoral vein for hemorrhage and fluid infusion, respectively. The arterial and venous blood samples were collected via these catheters. Another non-occlusive micromanometer was also put into the left femoral artery through the catheter introducer. These occlusive catheters were maintained patent by a slow continuous infusion of saline through an intra-flow adapter (3mL/hr, Intraflow continuous flush devices, Abbott Laboratories, Abbott Park, IL), which were connected to bags of normal saline pressurized to 300 mmHg. Access to the spleen was provided via a laparotomy, and a plastic sheet was placed between the spleen and the intestines.
5.1.3. Experimental Design
Once the instrumentation was completed and the vital parameters stabilized, the animals were subjected to somewhat modified procedures earlier reported [74]. A 10-minute baseline period began, and conducted hemodynamic measurements were collected from the analog and RS-232 signals on a data acquisition instrumentation rack (Dynamic Research Evaluation Workstation –DREW, US Army Institute of Surgical Research, San Antonio, TX).
Figure 8 shows the conduct of the experiments. The animals were enrolled in one of six experimental groups: 1) Control, sham-operated (cannulated but not hemorrhaged/injured), n =9; 2) H, hemorrhage + splenic injury, n=14; 3) H + LR, hemorrhage + splenic injury + lactated Ringer’s solution (LR), n= 13; and 4) H + Voluven®, hemorrhage + splenic injury + Voluven®, n= 6.
As previously described, a controlled hemorrhage (22 mL of blood/kg b.w. 100 mL/min) was performed using our custom error-sensing negative feedback computerized pump program [73]. After controlled hemorrhage, the animals were subjected to 1) partial splenic resection [74] or 2) soft tissue trauma induced on both sides of the right thigh and right femur fracture [75] immediately preceding splenic injury. The uncontrolled hemorrhage volume from the injured spleen was measured continuously by suctioning shed blood into canisters (Vac-Rite disposable suction system, Baxter Healthcare, Deerfield, IL) that had been placed on a balance (SR16000 Mettler Balance, Mettler-Toledo, Columbus, OH). The time the splenic injury was completed was marked as the zero-time point.
Resuscitation started with intravenous infusion crystalloid (lactated Ringer’s solution; LR) at 45 ml/kg body weight or colloid solution (Voluven®, 6% hydroxyethyl starch 130/0.4 in 0.9% sodium chloride injection; Fresenius Kabi Norge AS, Halden, Norway) at 15 ml/kg body weight at a rate of 1ml/kg/min using our servo-controlled computerized pump. Animals were observed for 6 hours after the injury or until death.
5.1.4. Biosampling
As seen in Figure 8, an arterial (30 mL) blood sample was obtained before controlled hemorrhage (-30 min), after shock, and immediately before resuscitation (30 min), continued at 30-min intervals within the first 120 min and followed by hourly sampling. The acid-base balance in the blood was assayed using i-STAT cartridges (Abbott Laboratories, Abbott Park, IL). At the end of the observation or death (when end-tidal pCO2 ≤ 10 mmHg or flat-line ECG), each animal was euthanized with sodium pentobarbital (90 mg/kg IV, 10 mL Fatal Plus, Vortech Pharmaceuticals, LTD, Dearborn, MI) under surgical anesthesia.
Tissue harvest. The animals were euthanatized with pentobarbital sodium (90 mg/kg i.v.) at the end point following the procedures above. Tissue samples, including lung, small intestine, heart, and kidney, were removed and fixed with 10% formalin or 4% paraformaldehyde for histological and immunohistochemical analysis.
5.2. Assays
5.2.1. Reagents and Antibodies
CG4+ and CG8+ cartridges were purchased from Abbott (Princeton, NJ). Chicken anti-C3/C3a, mouse anti-C4d, mouse anti-C5b-9, and mouse anti-endothelial cell antibodies were obtained from Abcam Inc. (Cambridge, MA). Rabbit anti-C5 antibody was from Abbiotec, LLC (San Diego, CA). Mouse anti-IL-6 was purchased from R&D Systems (Minneapolis, MN). Mouse anti-porcine C3a, biotinylated anti-C3a, and porcine C3a standard were from Georg-August-Universität Göttingen Stiftung Öffentlichen Rechts (Göttingen, Germany). Goat anti-chicken Alexa Fluor 594, goat anti-mouse Alexa Fluor 488, goat anti-mouse Alexa Fluor 594 IgG (H+L) conjugated secondary antibodies, and ProLong Gold antifade reagent were from Invitrogen (Carlsbad, CA).
5.2.2. Histological Examination
Formalin-fixed tissues were embedded in paraffin, sectioned, and stained with hematoxylin-eosin as we described previously [18,27,42,60,61]; histological images were recorded under a light microscope (AX80; Olympus, Center Valley, Pa) by a pathologist blinded to the treatment group. Histological injury scores were graded according to the following: For lung injury, four parameters (alveolar fibrin edema, alveolar hemorrhage, septal thickening, and intra-alveolar inflammatory cells) were scored on each H&E-stained slide for 1) the severity (0: absent; 1, 2, and 3 for more severe changes) and 2) the extent of injury (0: absent; 1: <25%; 2: 25-50%; 3: >50%) for the lung tissue. Each slide’s mucosal damage of the small intestine was graded on a six-tiered scale. A score of 0 was assigned to a normal villus; villi with tip distortion were scored as 1; villi lacking goblet cells and containing Guggenheims’ spaces were scored as 2; villi with patch disruption of the epithelial cells were scored as 3; villi with exposed but intact lamina propria and epithelial cell sloughing were assigned a score of 4; villi in which the lamina propria was exuding were scored as 5; villi displaying hemorrhage or denuded were scored as 6.
Myocardial injury was assessed by using four parameters (edema, degeneration, inflammatory cell infiltration, congestion) were scored on each H&E-stained slide for 1) the severity (0: absent; 1, 2, and 3 for more severe changes) and 2) the extent of injury (0: absent; 1: <25%; 2: 25-50%; 3: >50%).
For the evaluation of the liver injury, four parameters were used. Vascular congestion/ thrombosis is defined as engorgement of portal venules, sinusoids, or terminal hepatic venules with erythrocytes, platelets, and/or fibrin material (score 0 for no change, score 1, 2, 3 for more extended and severe changes). Hepatocyte death is assessed by loss of nuclear detail and well-defined cellular borders (score 0 for no change, score 1, 2, 3 for more extended and severe changes). Degeneration is determined by cytoplasmic hydropic change, cytoplasmic vacuolization, and sinusoid derangement (score 0 for no change, score 1, 2, 3 for more extended and severe changes). Inflammation is evaluated by inflammatory cell infiltration such as macrophage, histocytes, and polymorphonuclear leukocyte (PMN) (score 0 for no change, score 1, 2, 3 for more extended and severe changes). The extent of liver damage (0: absent; 1: <25%; 2: 25-50%; 3: >50%) was also evaluated.
Kidney injury was evaluated by using the following scores: 0=normal histology; 1=slight alteration (loss of brush border, mild hydropic degeneration, mild congestion); 2=mild (intensive hydropic degeneration, mild vacuolization, and interstitial edema); 3=moderate (nuclear condensation, intensive vacuolization, modulate interstitial edema); 4=severe (necrotic/apoptotic cells, denudation/rupture of basement membrane); 5=necrosis (total necrosis of the tubule) are scored on each H&E stained slide for 1) the severity (0: absent; 1, 2, and 3 for more severe changes) and 2) the extent of injury (0: absent; 1: <25%; 2: 25-50%; 3: >50%).
5.2.3. Immunohistochemical Staining
As we described previously [27,42,60,61]. Paraformaldehyde-fixed lung and small intestine biopsies were snap-frozen at -70ºC; sections were cut at a 5µm-thickness with a cryostat and fixed in cold methanol for 20 minutes. The fixed sections were permeabilized with 0.2% Triton X-100 in PBS for 10 minutes and then blocked with 2% BSA in PBS for 30 minutes at room temperature. The sections were incubated with the primary antibodies (anti-C5b-9, C3a, and C5a) overnight at 4ºC, washed, and then incubated with the appropriate secondary antibodies labeled with Alexa Fluor 488 and 594 for 1 hour at room temperature. After washing, the sections were mounted with ProLong Gold antifade solution containing 4′, 6′-diamidino-2-phenylindole (DAPI) and visualized under a confocal laser scanning microscope (Radiance 2100; Bio-Rad, Hercules, NJ) at ×400 magnification. Negative controls were conducted by substituting the primary antibodies with corresponding immunoglobulin isotypes. Captured digital images were processed by Image J software (NIH, Bethesda, MD). As previously described, immunofluorescent intensity was quantified with a modified method [76]. Briefly, four to six images from each animal were opened using Adobe Photoshop software and adjusted until only the fluorescent deposits and no background tissue were visible. The image was changed to black and white pixels using Image J software, with black representing the target proteins’ deposits and white representing non-stained areas of the image. The image was then changed to red and white using the Adjust Threshold command, with fluorescent deposits being red. The image was analyzed to show the total red area in pixels squared. Values for the entire area for all animals in each group were averaged to give the average area of the fluorescent deposit.
5.2.4. Cytokine Assays
Serum TNF-α, IL (Interleukin) -6, and IL-8 levels were assessed by quantitative sandwich Enzyme-Linked Immunosorbent Assay (ELISA; R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
5.2.5. Quantitative Assessment of End Tissue Function
Creatinine, aspartate aminotransferase (AST), muscle myocardium isoenzyme B (MMB), and creatinine kinase (CK) were measured from blood samples using Dimension Xpand Plus Integrated Chemistry System (Siemens, Holliston, MA).
5.2.6. Measurement of Coagulation Parameters
Samples for coagulation assays were collected in citrated tubes. Prothrombin time (PT), Activated Partial Thromboplastin Time (aPTT), and fibrinogen concentrations were measured in platelet-poor plasma using the BCSTM XP system (Siemens, Deerfield, Illinois, USA). Whole-blood coagulation function was determined by Thrombelastography™ (TEG) (Haemoscope Corporation, Niles, IL).
5.2.7. Analysis of Complement Functional Activity
Serum complement activity was determined based on hemolytic activity [18,42]. Briefly, antibody-sensitized Gallus gallus domesticus red blood cells (Colorado Serum Company, Denver, CO) were incubated for one hour at 37°C with serial dilutions of serum samples in gelatin-Veronal buffer (pH 7.3). After centrifugation, the absorbance of the supernatant was determined at 405 nm, and the serum concentration inducing 50% of complement hemolytic activity was defined as a CH50 value.
5.2.8. Analysis of Plasma C3a
As we described previously [18,42], 96-well microplates (R&D Systems) were coated with anti-porcine C3a mAb (Georg-August-Universität Göttingen Stiftung Öffentlichen Rechts, Germany) in coating buffer (Na2CO3/NaHCO3 coating buffer, pH >10) overnight at 4°C. After blocking with 1% gelatin in coating buffer and washing three times with wash buffer (1× PBS containing 0.05% Tween 20), 100 µl of standards (porcine C3a) or porcine plasma samples were added into each well and incubated for 2 hours at room temperature. After incubation, the wells were washed three times with wash buffer. Then 100 µl of biotinylated anti-porcine-C3a IgG (K5/4, Georg-August-Universität Göttingen Stiftung Öffentlichen Rechts, Germany) was added and incubated for one hour at room temperature. Wells were washed and incubated with streptavidin-HRP for one hour at room temperature, followed by incubation with 100 μl of substrate (R&D Systems) for 20 minutes at room temperature. After incubation, 50 μl of stop solution (R&D Systems) was added to each well and read at 450nm with a plate reader.
5.3. Statistical Analysis
Data were analyzed using GraphPad Prism version 10 (GraphPad Software, La Jolla, CA) and are expressed as mean ± SEM. The log-rank (Mantel-Cox) test analyzed survival. Hemodynamic and metabolic parameters were analyzed using two-way ANOVA with Bonferroni posttests, while thromboelastographic data were reviewed with the Kruskal-Wallis test; P < 0.05 was considered significant.
Author Contributions
M.O.S, Y.L., B.L. performed the experiments, collected data, measured samples, and summarized data. Y.L. and M.O.S. conducted a statistical analysis of data. MOS and YL wrote the manuscript. Y.L., J.J.D., and J.B. reviewed and critically revised the manuscript. Y.L., J.J.D., and M.O.S. designed the overall study and interpreted data. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the DoD US Army Medical Research & Development Command FY18 Broad Agency Announcement (W81XWH190040/BA180313), and DoD Congressionally Directed Medical Research Programs, Peer Reviewed Medical Research Program, Technology/Therapeutic Development Award (W81XWH200040/PR190914).
Institutional Review Board Statement
All animal experiments were conducted according to the guidelines of the animal facility and with approval by the US Army Institute of Surgical Research Institutional Animal Care and Use Committee (approval protocol #: A10-017).
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated or analyzed during the current study are included in this published manuscript. The data presented in this manuscript are available on request from the corresponding author.
Acknowledgments
The authors thank the staff of the veterinary division for technical support.
Conflicts of Interest
The authors declare no conflicts of interest relevant to the manuscript submitted to the International Journal of Molecular Sciences. The opinions or assertions contained herein are the private views of the authors, and they are not to be construed as official or as reflecting the views of the Department of the Army or the Department of Defense.
References
- Hooper, T. J.; De Pasquale, M.; Strandenes, G.; Sunde, G.; Ward, K. R. Challenges and Possibilities in Forward Resuscitation. Shock 2014, 41 Suppl 1, 13–20. [CrossRef]
- Gruen, R. L.; Brohi, K.; Schreiber, M.; Balogh, Z. J.; Pitt, V.; Narayan, M.; Maier, R. V. Haemorrhage Control in Severely Injured Patients. The Lancet 2012, 380 (9847), 1099–1108. [CrossRef]
- Moore, E. E.; Moore, H. B.; Kornblith, L. Z.; Neal, M. D.; Hoffman, M.; Mutch, N. J.; Schöchl, H.; Hunt, B. J.; Sauaia, A. Trauma-Induced Coagulopathy. Nat Rev Dis Primers 2021, 7 (1), 30. [CrossRef]
- Ramesh, G. H.; Uma, J. C.; Farhath, S. Fluid Resuscitation in Trauma: What Are the Best Strategies and Fluids? Int J Emerg Med 2019, 12 (1), 38. [CrossRef]
- Roger, C.; Muller, L.; Deras, P.; Louart, G.; Nouvellon, E.; Molinari, N.; Goret, L.; Gris, J. C.; Ripart, J.; De La Coussaye, J. E.; Lefrant, J. Y. Does the Type of Fluid Affect Rapidity of Shock Reversal in an Anaesthetized-Piglet Model of near-Fatal Controlled Haemorrhage? A Randomized Study. British Journal of Anaesthesia 2014, 112 (6), 1015–1023. [CrossRef]
- Curry, N.; Hopewell, S.; Dorée, C.; Hyde, C.; Brohi, K.; Stanworth, S. The Acute Management of Trauma Hemorrhage: A Systematic Review of Randomized Controlled Trials. Crit Care 2011, 15 (2), R92. [CrossRef]
- Leibner, E.; Andreae, M.; Galvagno, S. M.; Scalea, T. Damage Control Resuscitation. Clin Exp Emerg Med 2020, 7 (1), 5–13. [CrossRef]
- Holcomb, J. B.; Del Junco, D. J.; Fox, E. E.; Wade, C. E.; Cohen, M. J.; Schreiber, M. A.; Alarcon, L. H.; Bai, Y.; Brasel, K. J.; Bulger, E. M.; Cotton, B. A.; Matijevic, N.; Muskat, P.; Myers, J. G.; Phelan, H. A.; White, C. E.; Zhang, J.; Rahbar, M. H.; Prommtt Study Group, F. T. The Prospective, Observational, Multicenter, Major Trauma Transfusion (PROMMTT) Study: Comparative Effectiveness of a Time-Varying Treatment With Competing Risks. JAMA Surg 2013, 148 (2), 127. [CrossRef]
- Parr, M. J.; Bouillon, B.; Brohi, K.; Dutton, R. P.; Hauser, C. J.; Hess, J. R.; Holcomb, J. B.; Kluger, Y.; Mackway-Jones, K.; Rizoli, S. B.; Yukioka, T.; Hoyt, D. B. Traumatic Coagulopathy: Where Are the Good Experimental Models? Journal of Trauma: Injury, Infection & Critical Care 2008, 65 (4), 766–771. [CrossRef]
- Curry, N.; Davis, P. W. What’s New in Resuscitation Strategies for the Patient with Multiple Trauma? Injury 2012, 43 (7), 1021–1028. [CrossRef]
- Snyder, C. W.; Weinberg, J. A.; McGwin, G.; Melton, S. M.; George, R. L.; Reiff, D. A.; Cross, J. M.; Hubbard-Brown, J.; Rue, L. W.; Kerby, J. D. The Relationship of Blood Product Ratio to Mortality: Survival Benefit or Survival Bias? Journal of Trauma: Injury, Infection & Critical Care 2009, 66 (2), 358–364. [CrossRef]
- MacLeod, J. B. A.; Lynn, M.; McKenney, M. G.; Cohn, S. M.; Murtha, M. Early Coagulopathy Predicts Mortality in Trauma. J Trauma 2003, 55 (1), 39–44. [CrossRef]
- Maegele, M.; Lefering, R.; Yucel, N.; Tjardes, T.; Rixen, D.; Paffrath, T.; Simanski, C.; Neugebauer, E.; Bouillon, B. Early Coagulopathy in Multiple Injury: An Analysis from the German Trauma Registry on 8724 Patients. Injury 2007, 38 (3), 298–304. [CrossRef]
- Huber-Lang, M.; Lambris, J. D.; Ward, P. A. Innate Immune Responses to Trauma. Nat Immunol 2018, 19 (4), 327–341. [CrossRef]
- Huber-Lang, M.; Sarma, J. V.; Zetoune, F. S.; Rittirsch, D.; Neff, T. A.; McGuire, S. R.; Lambris, J. D.; Warner, R. L.; Flierl, M. A.; Hoesel, L. M.; Gebhard, F.; Younger, J. G.; Drouin, S. M.; Wetsel, R. A.; Ward, P. A. Generation of C5a in the Absence of C3: A New Complement Activation Pathway. Nat Med 2006, 12 (6), 682–687. [CrossRef]
- Amara, U.; Flierl, M. A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Brückner, U. B.; Nilsson, B.; Gebhard, F.; Lambris, J. D.; Huber-Lang, M. Molecular Intercommunication between the Complement and Coagulation Systems. The Journal of Immunology 2010, 185 (9), 5628–5636. [CrossRef]
- Gulla, K. C.; Gupta, K.; Krarup, A.; Gal, P.; Schwaeble, W. J.; Sim, R. B.; O’Connor, C. D.; Hajela, K. Activation of Mannan-binding Lectin-associated Serine Proteases Leads to Generation of a Fibrin Clot. Immunology 2010, 129 (4), 482–495. [CrossRef]
- Simovic, M. O.; Yang, Z.; Jordan, B. S.; Fraker, T. L.; Cancio, T. S.; Lucas, M. L.; Cancio, L. C.; Li, Y. Immunopathological Alterations after Blast Injury and Hemorrhage in a Swine Model of Prolonged Damage Control Resuscitation. Int J Mol Sci 2023, 24 (8), 7494. [CrossRef]
- Yang, Z.; Aderemi, O. A.; Zhao, Q.; Edsall, P. R.; Simovic, M. O.; Lund, B. J.; Espinoza, M. D.; Woodson, A. M.; Li, Y.; Cancio, L. C. Early Complement and Fibrinolytic Activation in a Rat Model of Blast-Induced Multi-Organ Damage. Mil Med 2019, 184 (Suppl 1), 282–290. [CrossRef]
- Yang, Z.; Simovic, M. O.; Liu, B.; Burgess, M. B.; Cap, A. P.; DalleLucca, J. J.; Li, Y. Indices of Complement Activation and Coagulation Changes in Trauma Patients. Trauma Surg Acute Care Open 2022, 7 (1), e000927. [CrossRef]
- Yang, Z.; Le, T. D.; Simovic, M. O.; Liu, B.; Fraker, T. L.; Cancio, T. S.; Cap, A. P.; Wade, C. E.; DalleLucca, J. J.; Li, Y. Traumatized Triad of Complementopathy, Endotheliopathy, and Coagulopathy - Impact on Clinical Outcomes in Severe Polytrauma Patients. Front Immunol 2022, 13, 991048. [CrossRef]
- Dutton, R. P. Current Concepts in Hemorrhagic Shock. Anesthesiology Clinics 2007, 25 (1), 23–34. [CrossRef]
- Niles, S. E.; McLaughlin, D. F.; Perkins, J. G.; Wade, C. E.; Li, Y.; Spinella, P. C.; Holcomb, J. B. Increased Mortality Associated With the Early Coagulopathy of Trauma in Combat Casualties. Journal of Trauma: Injury, Infection & Critical Care 2008, 64 (6), 1459–1465. [CrossRef]
- Bermudez, T.; Sammani, S.; Song, J. H.; Hernon, V. R.; Kempf, C. L.; Garcia, A. N.; Burt, J.; Hufford, M.; Camp, S. M.; Cress, A. E.; Desai, A. A.; Natarajan, V.; Jacobson, J. R.; Dudek, S. M.; Cancio, L. C.; Alvarez, J.; Rafikov, R.; Li, Y.; Zhang, D. D.; Casanova, N. G.; Bime, C.; Garcia, J. G. N. eNAMPT Neutralization Reduces Preclinical ARDS Severity via Rectified NFkB and Akt/mTORC2 Signaling. Sci Rep 2022, 12 (1), 696. [CrossRef]
- Campbell, J. C.; Li, Y.; van Amersfoort, E.; Relan, A.; Dubick, M.; Sheppard, F.; Pusateri, A.; Niemeyer, D.; Tsokos, G. C.; Dalle Lucca, J. J. C1 Inhibitor Limits Organ Injury and Prolongs Survival in Swine Subjected to Battlefield Simulated Injury. Shock 2016, 46 (3 Suppl 1), 177–188. [CrossRef]
- Carey, M. E. Analysis of Wounds Incurred by U.S. Army Seventh Corps Personnel Treated in Corps Hospitals during Operation Desert Storm, February 20 to March 10, 1991: The Journal of Trauma: Injury, Infection, and Critical Care 1996, 40 (Supplement), 165S-169S. [CrossRef]
- Dalle Lucca, J. J.; Li, Y.; Simovic, M. O.; Slack, J. L.; Cap, A.; Falabella, M. J.; Dubick, M.; Lebeda, F.; Tsokos, G. C. Decay-Accelerating Factor Limits Hemorrhage-Instigated Tissue Injury and Improves Resuscitation Clinical Parameters. J Surg Res 2013, 179 (1), 153–167. [CrossRef]
- Huber-Lang, M.; Gebhard, F.; Schmidt, C. Q.; Palmer, A.; Denk, S.; Wiegner, R. Complement Therapeutic Strategies in Trauma, Hemorrhagic Shock and Systemic Inflammation – Closing Pandora’s Box? Seminars in Immunology 2016, 28 (3), 278–284. [CrossRef]
- Karasu, E.; Nilsson, B.; Köhl, J.; Lambris, J. D.; Huber-Lang, M. Targeting Complement Pathways in Polytrauma- and Sepsis-Induced Multiple-Organ Dysfunction. Front Immunol 2019, 10, 543. [CrossRef]
- Lupu, L.; Horst, K.; Greven, J.; Mert, Ü.; Ludviksen, J. A. K.; Pettersen, K.; Lau, C.; Li, Y.; Palmer, A.; Qin, K.; Zhang, X.; Mayer, B.; van Griensven, M.; Huber-Lang, M.; Hildebrand, F.; Mollnes, T. E. Simultaneous C5 and CD14 Inhibition Limits Inflammation and Organ Dysfunction in Pig Polytrauma. Front. Immunol. 2022, 13, 952267. [CrossRef]
- Simovic, M. O.; Falabella, M. J.; Le, T. D.; DalleLucca, J. J.; Li, Y. Decay-Accelerating Factor Creates an Organ-Protective Phenotype after Hemorrhage in Conscious Rats. Int J Mol Sci 2022, 23 (21), 13563. [CrossRef]
- Yang, Z.; Nicholson, S. E.; Cancio, T. S.; Cancio, L. C.; Li, Y. Complement as a Vital Nexus of the Pathobiological Connectome for Acute Respiratory Distress Syndrome: An Emerging Therapeutic Target. Front Immunol 2023, 14, 1100461. [CrossRef]
- Yang, Z.; Nunn, M. A.; Le, T. D.; Simovic, M. O.; Edsall, P. R.; Liu, B.; Barr, J. L.; Lund, B. J.; Hill-Pryor, C. D.; Pusateri, A. E.; Cancio, L. C.; Li, Y. Immunopathology of Terminal Complement Activation and Complement C5 Blockade Creating a Pro-Survival and Organ-Protective Phenotype in Trauma. Br J Pharmacol 2023, 180 (4), 422–440. [CrossRef]
- Yang, Z.; Simovic, M. O.; Edsall, P. R.; Liu, B.; Cancio, T. S.; Batchinsky, A. I.; Cancio, L. C.; Li, Y. HMGB1 Inhibition to Ameliorate Organ Failure and Increase Survival in Trauma. Biomolecules 2022, 12 (1), 101. [CrossRef]
- Holcomb, J. B.; McMullin, N. R.; Pearse, L.; Caruso, J.; Wade, C. E.; Oetjen-Gerdes, L.; Champion, H. R.; Lawnick, M.; Farr, W.; Rodriguez, S.; Butler, F. K. Causes of Death in U.S. Special Operations Forces in the Global War on Terrorism: 2001–2004. Annals of Surgery 2007, 245 (6), 986–991. [CrossRef]
- Maughon, J. S. An Inquiry into the Nature of Wounds Resulting in Killed in Action in Vietnam. Mil Med 1970, 135 (1), 8–13. [CrossRef]
- Sauaia, A.; Moore, F. A.; Moore, E. E.; Moser, K. S.; Brennan, R.; Read, R. A.; Pons, P. T. Epidemiology of Trauma Deaths: A Reassessment. J Trauma 1995, 38 (2), 185–193. [CrossRef]
- Holcomb, J. B. Transport Time and Preoperating Room Hemostatic Interventions Are Important: Improving Outcomes After Severe Truncal Injury. Critical Care Medicine 2018, 46 (3), 447–453. [CrossRef]
- Barry, M.; Trivedi, A.; Vivona, L. R.; Chui, J.; Pathipati, P.; Miyazawa, B.; Pati, S. RECOVERY OF ENDOTHELIOPATHY AT 24 HOURS IN AN ESTABLISHED MOUSE MODEL OF HEMORRHAGIC SHOCK AND TRAUMA. Shock 2022, 58 (4), 313–320. [CrossRef]
- Bunch, C. M.; Chang, E.; Moore, E. E.; Moore, H. B.; Kwaan, H. C.; Miller, J. B.; Al-Fadhl, M. D.; Thomas, A. V.; Zackariya, N.; Patel, S. S.; Zackariya, S.; Haidar, S.; Patel, B.; McCurdy, M. T.; Thomas, S. G.; Zimmer, D.; Fulkerson, D.; Kim, P. Y.; Walsh, M. R.; Hake, D.; Kedar, A.; Aboukhaled, M.; Walsh, M. M. SHock-INduced Endotheliopathy (SHINE): A Mechanistic Justification for Viscoelastography-Guided Resuscitation of Traumatic and Non-Traumatic Shock. Front. Physiol. 2023, 14, 1094845. [CrossRef]
- Burk, A.-M.; Martin, M.; Flierl, M. A.; Rittirsch, D.; Helm, M.; Lampl, L.; Bruckner, U.; Stahl, G. L.; Blom, A. M.; Perl, M.; Gebhard, F.; Huber-Lang, M. Early Complementopathy after Multiple Injuries in Humans. Shock 2012, 37 (4), 348–354. [CrossRef]
- Campbell, J. C.; Li, Y.; van Amersfoort, E.; Relan, A.; Dubick, M.; Sheppard, F.; Pusateri, A.; Niemeyer, D.; Tsokos, G. C.; Dalle Lucca, J. J. C1 Inhibitor Limits Organ Injury and Prolongs Survival in Swine Subjected to Battlefield Simulated Injury. Shock 2016, 46 (3 Suppl 1), 177–188. [CrossRef]
- Ganter, M. T.; Brohi, K.; Cohen, M. J.; Shaffer, L. A.; Walsh, M. C.; Stahl, G. L.; Pittet, J.-F. Role of the Alternative Pathway in the Early Complement Activation Following Major Trauma. Shock 2007, 28 (1), 29–34. [CrossRef]
- Li, Y.; Zhao, Q.; Liu, B.; Dixon, A.; Cancio, L.; Dubick, M.; Dalle Lucca, J. Early Complementopathy Predicts the Outcomes of Patients with Trauma. Trauma Surg Acute Care Open 2019, 4 (1), e000217. [CrossRef]
- Lord, J. M.; Midwinter, M. J.; Chen, Y.-F.; Belli, A.; Brohi, K.; Kovacs, E. J.; Koenderman, L.; Kubes, P.; Lilford, R. J. The Systemic Immune Response to Trauma: An Overview of Pathophysiology and Treatment. Lancet 2014, 384 (9952), 1455–1465. [CrossRef]
- Huber-Lang, M. S.; Ignatius, A.; Köhl, J.; Mannes, M.; Braun, C. K. Complement in Trauma-Traumatised Complement? Br J Pharmacol 2021, 178 (14), 2863–2879. [CrossRef]
- Dobson, G. P.; Morris, J. L.; Letson, H. L. Why Are Bleeding Trauma Patients Still Dying? Towards a Systems Hypothesis of Trauma. Front Physiol 2022, 13, 990903. [CrossRef]
- Medby, C. Is There a Place for Crystalloids and Colloids in Remote Damage Control Resuscitation? Shock 2014, 41 (Supplement 1), 47–50. [CrossRef]
- Cazzolli, D.; Prittie, J. The Crystalloid-colloid Debate: Consequences of Resuscitation Fluid Selection in Veterinary Critical Care. J Vet Emergen Crit Care 2015, 25 (1), 6–19. [CrossRef]
- Fleming, S. D.; Phillips, L. M.; Lambris, J. D.; Tsokos, G. C. Complement Component C5a Mediates Hemorrhage-Induced Intestinal Damage. Journal of Surgical Research 2008, 150 (2), 196–203. [CrossRef]
- Horstick, G.; Kempf, T.; Lauterbach, M.; Bhakdi, S.; Kopacz, L.; Heimann, A.; Malzahn, M.; Horstick, M.; Meyer, J.; Kempski, O. C1-Esterase-Inhibitor Treatment at Early Reperfusion of Hemorrhagic Shock Reduces Mesentry Leukocyte Adhesion and Rolling. Microcirculation 2001, 8 (6), 427–433. [CrossRef]
- Hoth, J. J.; Wells, J. D.; Jones, S. E.; Yoza, B. K.; McCall, C. E. Complement Mediates a Primed Inflammatory Response after Traumatic Lung Injury. Journal of Trauma and Acute Care Surgery 2014, 76 (3), 601–609. [CrossRef]
- Peckham, R. M.; Handrigan, M. T.; Bentley, T. B.; Falabella, M. J.; Chrovian, A. D.; Stahl, G. L.; Tsokos, G. C. C5-Blocking Antibody Reduces Fluid Requirements and Improves Responsiveness to Fluid Infusion in Hemorrhagic Shock Managed with Hypotensive Resuscitation. J Appl Physiol (1985) 2007, 102 (2), 673–680. [CrossRef]
- Wang, P.; Ba, Z. F.; Reich, S. S.; Zhou, M.; Holme, K. R.; Chaudry, I. H. Effects of Nonanticoagulant Heparin on Cardiovascular and Hepatocellular Function after Hemorrhagic Shock. American Journal of Physiology-Heart and Circulatory Physiology 1996, 270 (4), H1294–H1302. [CrossRef]
- Relja, B.; Wagner, N.; Franz, N.; Dieteren, S.; Mörs, K.; Schmidt, J.; Marzi, I.; Perl, M. Ethyl Pyruvate Reduces Acute Lung Damage Following Trauma and Hemorrhagic Shock via Inhibition of NF-κB and HMGB1. Immunobiology 2018, 223 (3), 310–318. [CrossRef]
- Wagner, N.; Dieteren, S.; Franz, N.; Köhler, K.; Mörs, K.; Nicin, L.; Schmidt, J.; Perl, M.; Marzi, I.; Relja, B. Ethyl Pyruvate Ameliorates Hepatic Injury Following Blunt Chest Trauma and Hemorrhagic Shock by Reducing Local Inflammation, NF-kappaB Activation and HMGB1 Release. PLoS One 2018, 13 (2), e0192171. [CrossRef]
- Yang, R.; Harada, T.; Mollen, K. P.; Prince, J. M.; Levy, R. M.; Englert, J. A.; Gallowitsch-Puerta, M.; Yang, L.; Yang, H.; Tracey, K. J.; Harbrecht, B. G.; Billiar, T. R.; Fink, M. P. Anti-HMGB1 Neutralizing Antibody Ameliorates Gut Barrier Dysfunction and Improves Survival after Hemorrhagic Shock. Mol Med 2006, 12 (4–6), 105–114. [CrossRef]
- Keshari, R. S.; Silasi, R.; Popescu, N. I.; Patel, M. M.; Chaaban, H.; Lupu, C.; Coggeshall, K. M.; Mollnes, T. E.; DeMarco, S. J.; Lupu, F. Inhibition of Complement C5 Protects against Organ Failure and Reduces Mortality in a Baboon Model of Escherichia Coli Sepsis. Proc Natl Acad Sci U S A 2017, 114 (31), E6390–E6399. [CrossRef]
- Morrison, A. M.; Wang, P.; Chaudry, I. H. A NOVEL NONANTICOAGULANT HEPARIN PREVENTS VASCULAR ENDOTHELIAL CELL DYSFUNCTION DURING HYPERDYNAMIC SEPSIS: Shock 1996, 6 (1), 46–51. [CrossRef]
- Dalle Lucca, J. J.; Simovic, M.; Li, Y.; Moratz, C.; Falabella, M.; Tsokos, G. C. Decay-Accelerating Factor Mitigates Controlled Hemorrhage-Instigated Intestinal and Lung Tissue Damage and Hyperkalemia in Swine. J Trauma 2011, 71 (1 Suppl), S151-160. [CrossRef]
- Dalle Lucca, J. J.; Li, Y.; Simovic, M.; Pusateri, A. E.; Falabella, M.; Dubick, M. A.; Tsokos, G. C. Effects of C1 Inhibitor on Tissue Damage in a Porcine Model of Controlled Hemorrhage. Shock 2012, 38 (1), 82–91. [CrossRef]
- CRASH-2 trial collaborators; Shakur, H.; Roberts, I.; Bautista, R.; Caballero, J.; Coats, T.; Dewan, Y.; El-Sayed, H.; Gogichaishvili, T.; Gupta, S.; Herrera, J.; Hunt, B.; Iribhogbe, P.; Izurieta, M.; Khamis, H.; Komolafe, E.; Marrero, M.-A.; Mejía-Mantilla, J.; Miranda, J.; Morales, C.; Olaomi, O.; Olldashi, F.; Perel, P.; Peto, R.; Ramana, P. V.; Ravi, R. R.; Yutthakasemsunt, S. Effects of Tranexamic Acid on Death, Vascular Occlusive Events, and Blood Transfusion in Trauma Patients with Significant Haemorrhage (CRASH-2): A Randomised, Placebo-Controlled Trial. Lancet 2010, 376 (9734), 23–32. [CrossRef]
- Guyette, F. X.; Brown, J. B.; Zenati, M. S.; Early-Young, B. J.; Adams, P. W.; Eastridge, B. J.; Nirula, R.; Vercruysse, G. A.; O’Keeffe, T.; Joseph, B.; Alarcon, L. H.; Callaway, C. W.; Zuckerbraun, B. S.; Neal, M. D.; Forsythe, R. M.; Rosengart, M. R.; Billiar, T. R.; Yealy, D. M.; Peitzman, A. B.; Sperry, J. L.; STAAMP Study Group. Tranexamic Acid During Prehospital Transport in Patients at Risk for Hemorrhage After Injury: A Double-Blind, Placebo-Controlled, Randomized Clinical Trial. JAMA Surg 2020, 156 (1), 11–20. [CrossRef]
- Rowell, S. E.; Meier, E. N.; McKnight, B.; Kannas, D.; May, S.; Sheehan, K.; Bulger, E. M.; Idris, A. H.; Christenson, J.; Morrison, L. J.; Frascone, R. J.; Bosarge, P. L.; Colella, M. R.; Johannigman, J.; Cotton, B. A.; Callum, J.; McMullan, J.; Dries, D. J.; Tibbs, B.; Richmond, N. J.; Weisfeldt, M. L.; Tallon, J. M.; Garrett, J. S.; Zielinski, M. D.; Aufderheide, T. P.; Gandhi, R. R.; Schlamp, R.; Robinson, B. R. H.; Jui, J.; Klein, L.; Rizoli, S.; Gamber, M.; Fleming, M.; Hwang, J.; Vincent, L. E.; Williams, C.; Hendrickson, A.; Simonson, R.; Klotz, P.; Sopko, G.; Witham, W.; Ferrara, M.; Schreiber, M. A. Effect of Out-of-Hospital Tranexamic Acid vs Placebo on 6-Month Functional Neurologic Outcomes in Patients With Moderate or Severe Traumatic Brain Injury. JAMA 2020, 324 (10), 961. [CrossRef]
- Li, S. R.; Guyette, F.; Brown, J.; Zenati, M.; Reitz, K. M.; Eastridge, B.; Nirula, R.; Vercruysse, G. A.; O’Keeffe, T.; Joseph, B.; Neal, M. D.; Zuckerbraun, B. S.; Sperry, J. L. Early Prehospital Tranexamic Acid Following Injury Is Associated With a 30-Day Survival Benefit: A Secondary Analysis of a Randomized Clinical Trial. Annals of Surgery 2021, 274 (3), 419–426. [CrossRef]
- Hannon, J. P.; Bossone, C. A.; Rodkey, W. G. Splenic Red Cell Sequestration and Blood Volume Measurements in Conscious Pigs. Am J Physiol 1985, 248 (3 Pt 2), R293-301. [CrossRef]
- Bebarta, V. S.; Daheshia, M.; Ross, J. D. The Significance of Splenectomy in Experimental Swine Models of Controlled Hemorrhagic Shock. J Trauma Acute Care Surg 2013, 75 (5), 920. [CrossRef]
- Boysen, S. R.; Caulkett, N. A.; Brookfield, C. E.; Warren, A.; Pang, J. M. Splenectomy Versus Sham Splenectomy in a Swine Model of Controlled Hemorrhagic Shock. Shock 2016, 46 (4), 439–446. [CrossRef]
- Kheirabadi, B. S.; Sandeen, J. L.; Dubick, M. A. Re: The Significance of Splenectomy in Experimental Swine Models of Hemorrhagic Shock. J Trauma Acute Care Surg 2013, 75 (5), 920–921. [CrossRef]
- Vnuk, D.; Lemo, N.; Nesek-Adam, V.; Maticić, D.; Radisić, B.; Kos, J.; Rumenjak, V.; Dohan Ehrenfest, D. M. Cardiopulmonary Effects of Hemorrhagic Shock in Splenic Autotransplanted Pigs: A New Surgical Model. Coll Antropol 2010, 34 (3), 923–930.
- Bellamy, R. F.; Maningas, P. A.; Wenger, B. A. Current Shock Models and Clinical Correlations. Ann Emerg Med 1986, 15 (12), 1392–1395. [CrossRef]
- Pottecher, J.; Chemla, D.; Xavier, L.; Liu, N.; Chazot, T.; Marescaux, J.; Fischler, M.; Diemunsch, P.; Duranteau, J. Re: The Significance of Splenectomy in Experimental Swine Models of Hemorrhagic Shock. J Trauma Acute Care Surg 2013, 75 (5), 921–922. [CrossRef]
- Sondeen, J. L.; Dubick, M. A.; Holcomb, J. B.; Wade, C. E. Uncontrolled Hemorrhage Differs from Volume- or Pressure-Matched Controlled Hemorrhage in Swine. Shock 2007, 28 (4), 426–433. [CrossRef]
- Sondeen, J. L.; Prince, M. D.; Kheirabadi, B. S.; Wade, C. E.; Polykratis, I. A.; de Guzman, R.; Dubick, M. A. Initial Resuscitation with Plasma and Other Blood Components Reduced Bleeding Compared to Hetastarch in Anesthetized Swine with Uncontrolled Splenic Hemorrhage. Transfusion 2011, 51 (4), 779–792. [CrossRef]
- Batchinsky, A. I.; Jordan, B. S.; Necsoiu, C.; Dubick, M. A.; Cancio, L. C. DYNAMIC CHANGES IN SHUNT AND VENTILATION-PERFUSION MISMATCH FOLLOWING EXPERIMENTAL PULMONARY CONTUSION. Shock 2010, 33 (4), 419–425. [CrossRef]
- Weeks, C.; Moratz, C.; Zacharia, A.; Stracener, C.; Egan, R.; Peckham, R.; Moore, F. D.; Tsokos, G. C. Decay-Accelerating Factor Attenuates Remote Ischemia-Reperfusion-Initiated Organ Damage. Clin Immunol 2007, 124 (3), 311–327. [CrossRef]
Figure 1.
Coagulation disturbances after hemorrhagic and fluid resuscitation. Whole blood Shear elastic modulus (G) parameter (A) and maximum amplitude (MA) were measured by thromboelastography. Plasma prothrombin time (PT), Activated Partial Thromboplastin Time (aPTT), and fibrinogen concentrations were assessed using the BCSTM XP system. * H+Voliuven/H+LR vs. H and † H+Voluven vs. H+LR, p<0.05 using two-way ANOVA followed by Bonferroni posttests. Data are presented as mean ± SEM.
Figure 1.
Coagulation disturbances after hemorrhagic and fluid resuscitation. Whole blood Shear elastic modulus (G) parameter (A) and maximum amplitude (MA) were measured by thromboelastography. Plasma prothrombin time (PT), Activated Partial Thromboplastin Time (aPTT), and fibrinogen concentrations were assessed using the BCSTM XP system. * H+Voliuven/H+LR vs. H and † H+Voluven vs. H+LR, p<0.05 using two-way ANOVA followed by Bonferroni posttests. Data are presented as mean ± SEM.
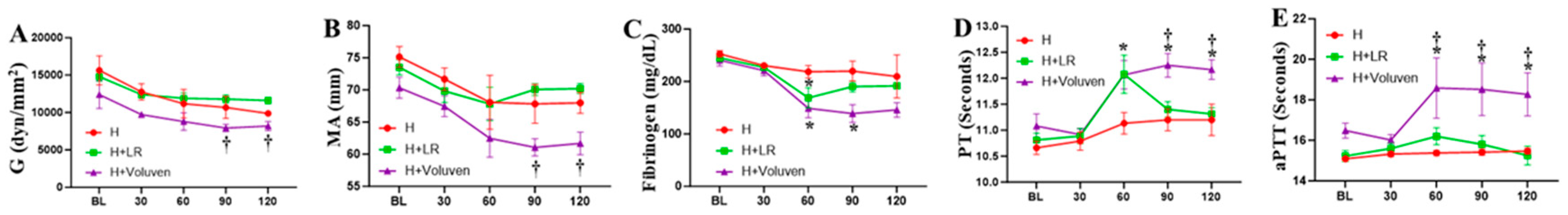
Figure 2.
Systemic inflammatory immune responses after hemorrhagic shock and fluid resuscitation. Serum hemolytic terminal complement activation was measured by CH50 assay (A), and blood levels of C3a (B), TNαF (C), IL-6 (D), and IL-8 (E) were assessed by ELISA. * vs. Sham, † vs. H, and ‡ vs. H+LR, p<0.05 using two-way ANOVA followed by Bonferroni posttests. Data are presented as mean ± SEM.
Figure 2.
Systemic inflammatory immune responses after hemorrhagic shock and fluid resuscitation. Serum hemolytic terminal complement activation was measured by CH50 assay (A), and blood levels of C3a (B), TNαF (C), IL-6 (D), and IL-8 (E) were assessed by ELISA. * vs. Sham, † vs. H, and ‡ vs. H+LR, p<0.05 using two-way ANOVA followed by Bonferroni posttests. Data are presented as mean ± SEM.
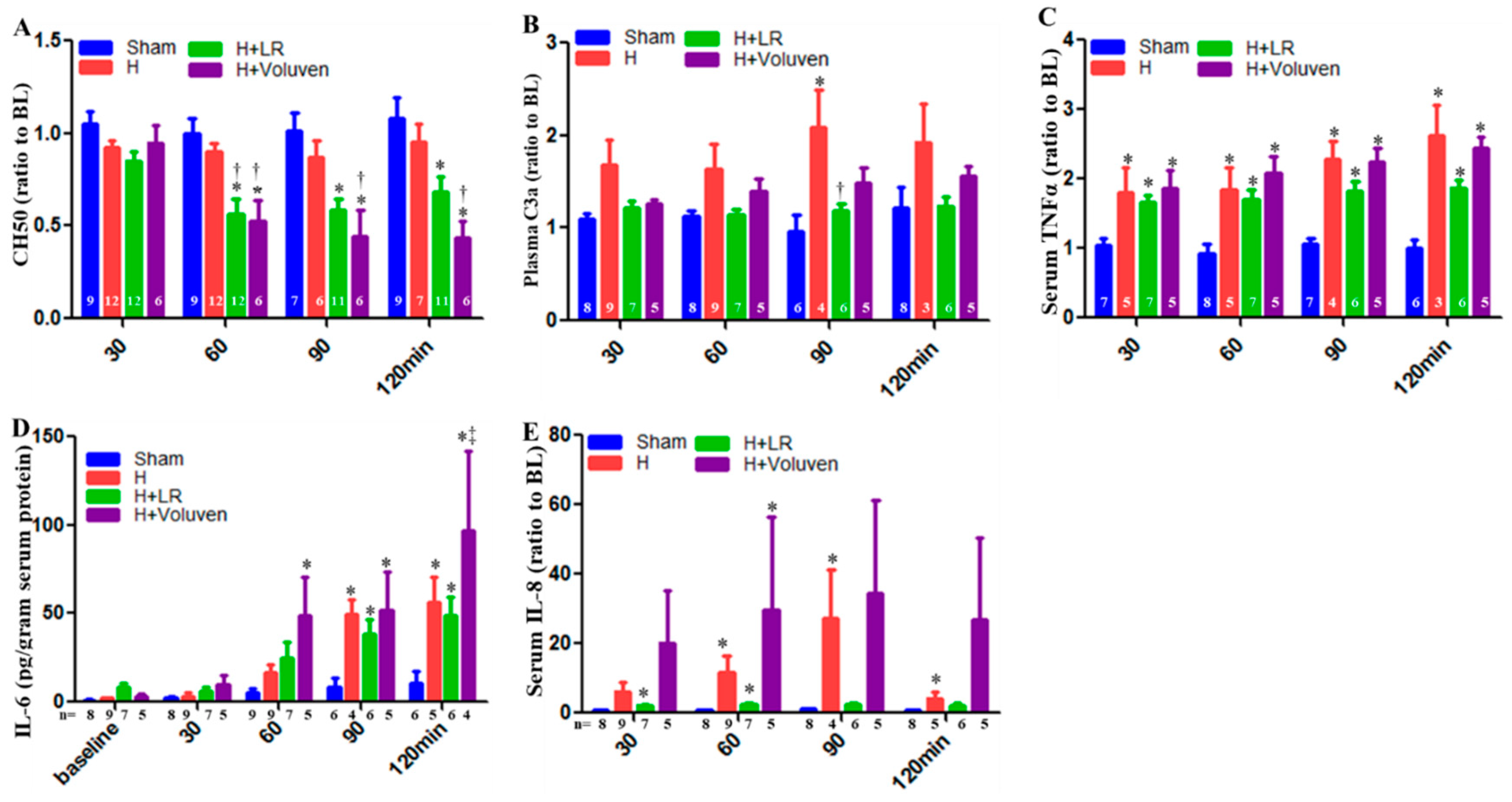
Figure 3.
Multiple organ dysfunctions in posthemorrhagic shock and fluid resuscitation. Blood creatinine (A), aspartate aminotransferase (AST, B), muscle myocardium isoenzyme B (MMB, C), and creatine kinase (CK, D) were determined by Siemens Dimension Xpand Plus Chemistry 6 Analyzer. * vs. Sham, † vs. H, ‡ vs. H+LR, p<0.05 using two-way ANOVA followed by Bonferroni posttests. Data are presented as mean ± SEM.
Figure 3.
Multiple organ dysfunctions in posthemorrhagic shock and fluid resuscitation. Blood creatinine (A), aspartate aminotransferase (AST, B), muscle myocardium isoenzyme B (MMB, C), and creatine kinase (CK, D) were determined by Siemens Dimension Xpand Plus Chemistry 6 Analyzer. * vs. Sham, † vs. H, ‡ vs. H+LR, p<0.05 using two-way ANOVA followed by Bonferroni posttests. Data are presented as mean ± SEM.
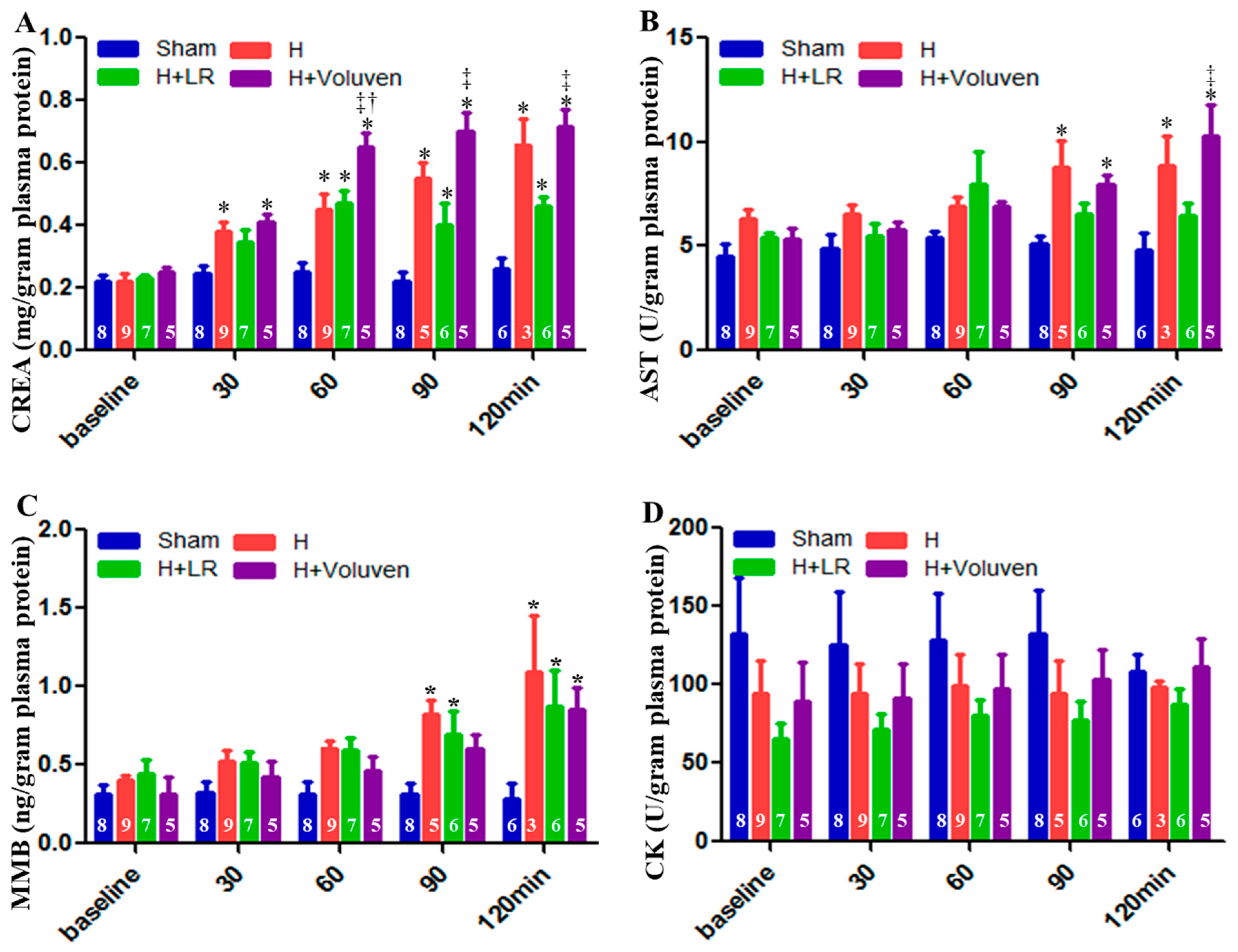
Figure 4.
Myocardial inflammatory responses after hemorrhagic shock and fluid resuscitation. Immunostaining and semiquantitative fluorescent intensity of C4d (A & B), C3 (C & D), C5 (E & F), C5b-9 (G & H), and IL-6 (I & J) in heart tissues were evaluated by immunohistochemistry. Scale bars = 50 μm. * vs. sham, † vs. H, and ‡ vs. H+LR, p<0.05 using one-way ANOVA followed by Bonferroni posttests. Data are presented as mean ± SEM. Vol, voluven®.
Figure 4.
Myocardial inflammatory responses after hemorrhagic shock and fluid resuscitation. Immunostaining and semiquantitative fluorescent intensity of C4d (A & B), C3 (C & D), C5 (E & F), C5b-9 (G & H), and IL-6 (I & J) in heart tissues were evaluated by immunohistochemistry. Scale bars = 50 μm. * vs. sham, † vs. H, and ‡ vs. H+LR, p<0.05 using one-way ANOVA followed by Bonferroni posttests. Data are presented as mean ± SEM. Vol, voluven®.
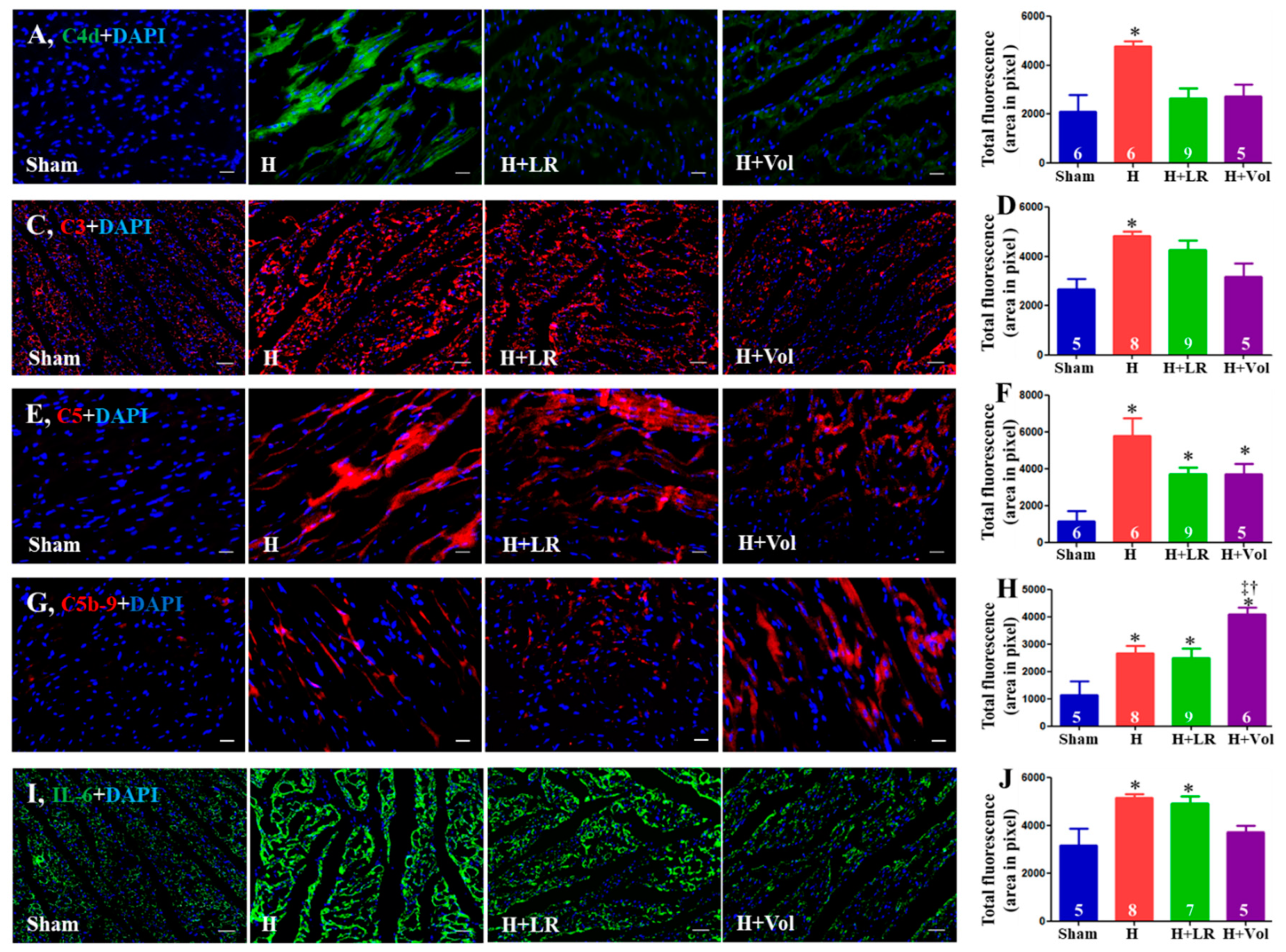
Figure 5.
Pulmonary/intestinal inflammatory responses after hemorrhagic shock and fluid resuscitation. Immunostaining and semiquantitative fluorescent intensity of C3 (A & B) and C5b-9 (C & D) in lungs, and C3 (E & F) and IL-6 (G & H) in jejunum were evaluated by immunohistochemistry. Scale bars = 50 μm. * vs. sham, p<0.05 using one-way ANOVA followed by Bonferroni posttests. Data are presented as mean ± SEM.
Figure 5.
Pulmonary/intestinal inflammatory responses after hemorrhagic shock and fluid resuscitation. Immunostaining and semiquantitative fluorescent intensity of C3 (A & B) and C5b-9 (C & D) in lungs, and C3 (E & F) and IL-6 (G & H) in jejunum were evaluated by immunohistochemistry. Scale bars = 50 μm. * vs. sham, p<0.05 using one-way ANOVA followed by Bonferroni posttests. Data are presented as mean ± SEM.
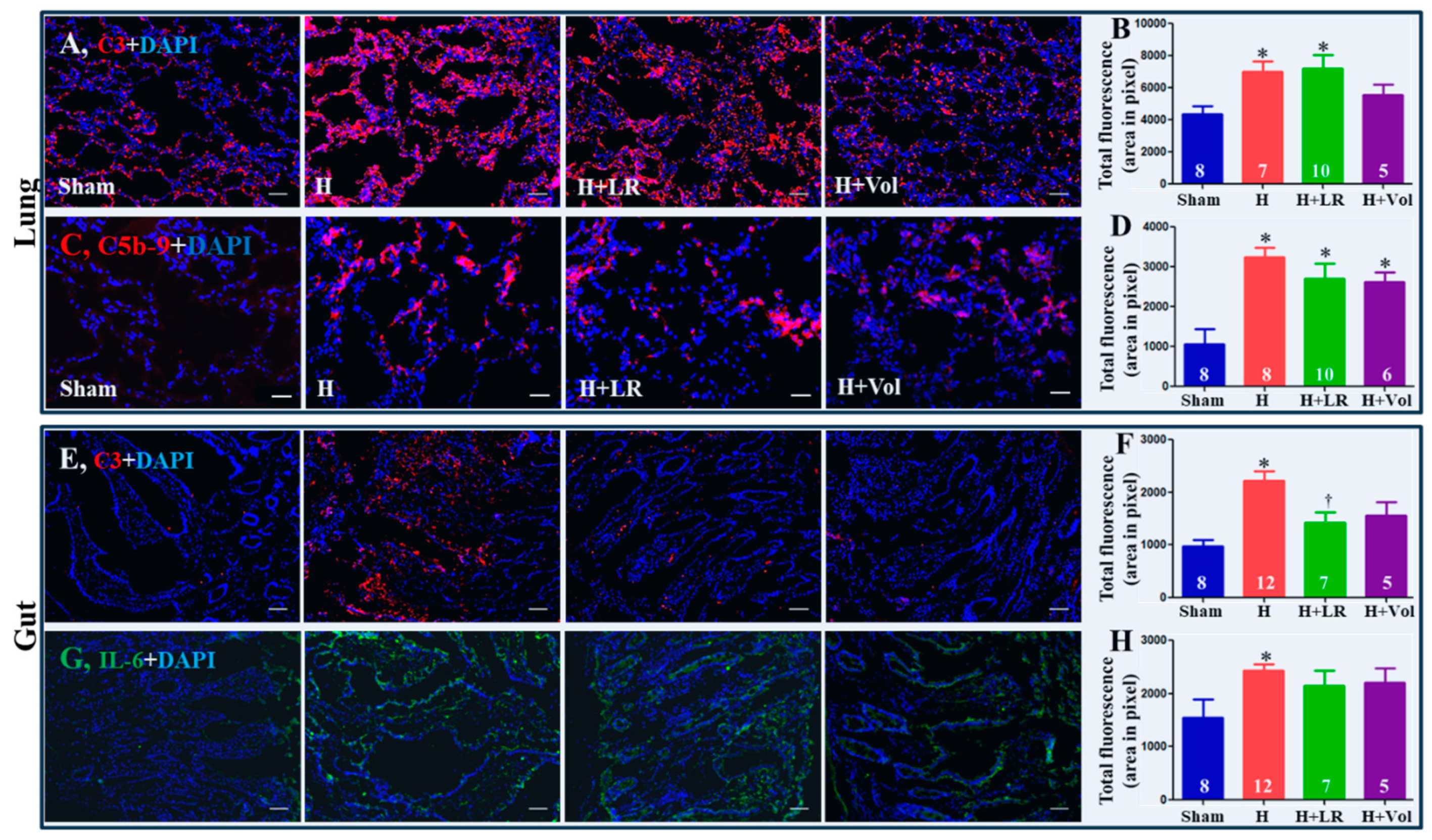
Figure 6.
Histopathological changes of vital organs after hemorrhagic shock and fluid resuscitation. Histopathological (H&E stain) photos and semiquantitative evaluations of the heart (A & B, scale bars=200µm), lung (C & D, scale bars=200µm), jejunum (E & F, scale bars=500µm), and kidney (G & H, scale bars=100µm). Myocarditis was marked with white arrows (A), and the insert in panel A magnifies the region of the indicated box to show the mononuclear cells and polymorphic nuclear cells in the corresponding inflammatory infiltration foci. Data are presented as mean ± SEM. * vs. sham, † vs. H, and ‡ vs. H+LR, p<0.05, using one-way ANOVA followed by Bonferroni posttests.
Figure 6.
Histopathological changes of vital organs after hemorrhagic shock and fluid resuscitation. Histopathological (H&E stain) photos and semiquantitative evaluations of the heart (A & B, scale bars=200µm), lung (C & D, scale bars=200µm), jejunum (E & F, scale bars=500µm), and kidney (G & H, scale bars=100µm). Myocarditis was marked with white arrows (A), and the insert in panel A magnifies the region of the indicated box to show the mononuclear cells and polymorphic nuclear cells in the corresponding inflammatory infiltration foci. Data are presented as mean ± SEM. * vs. sham, † vs. H, and ‡ vs. H+LR, p<0.05, using one-way ANOVA followed by Bonferroni posttests.
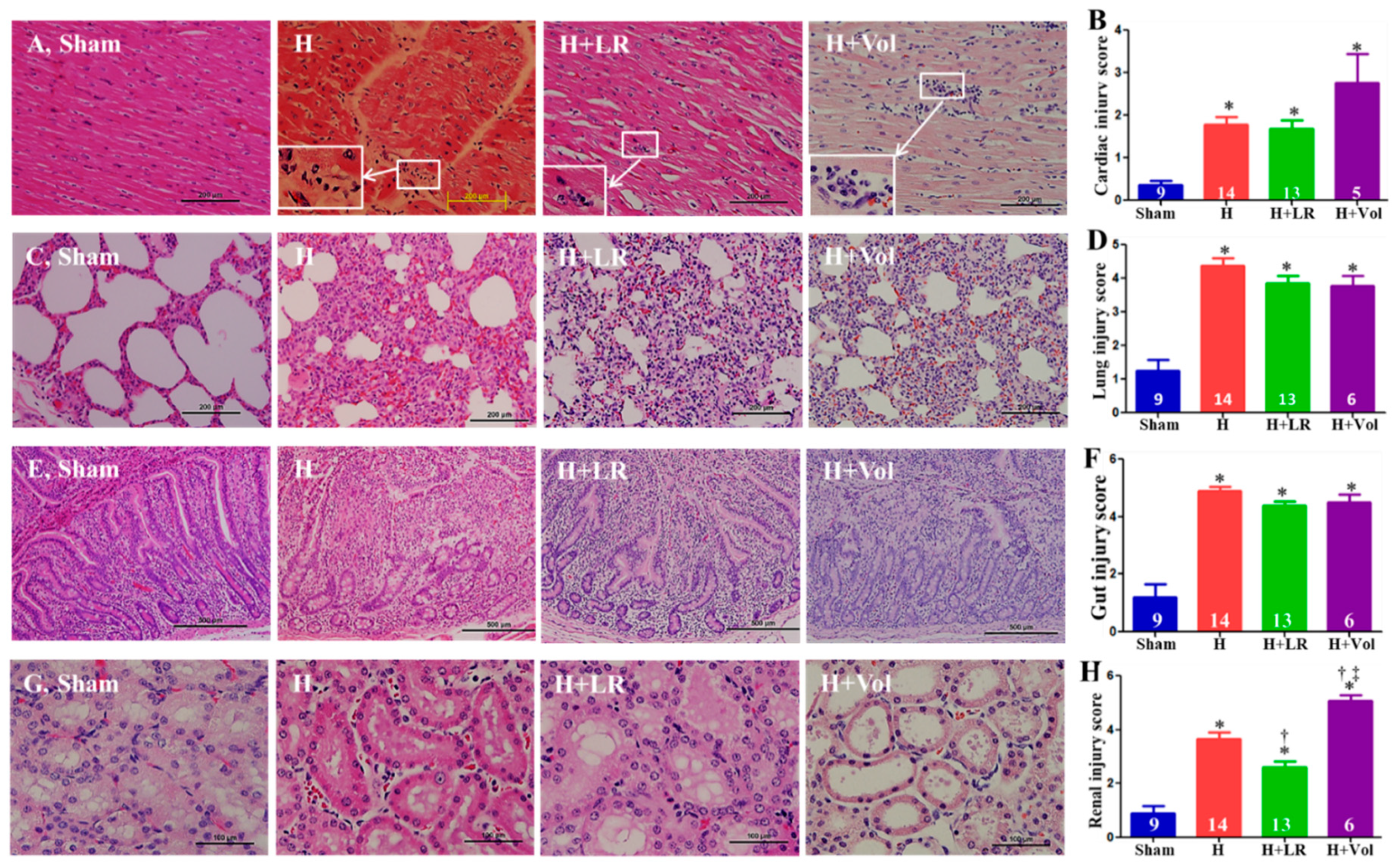
Figure 7.
Survival in a porcine model of hemorrhagic shock and fluid resuscitation. Percent survival (A) and survival time are shown. * p<0.05 vs. sham, † p<0.05 vs. H, and ‡ vs. H+LR, p<0.05 using a log-rank test (A) and one-way ANOVA followed by Bonferroni posttests (B), respectively.
Figure 7.
Survival in a porcine model of hemorrhagic shock and fluid resuscitation. Percent survival (A) and survival time are shown. * p<0.05 vs. sham, † p<0.05 vs. H, and ‡ vs. H+LR, p<0.05 using a log-rank test (A) and one-way ANOVA followed by Bonferroni posttests (B), respectively.
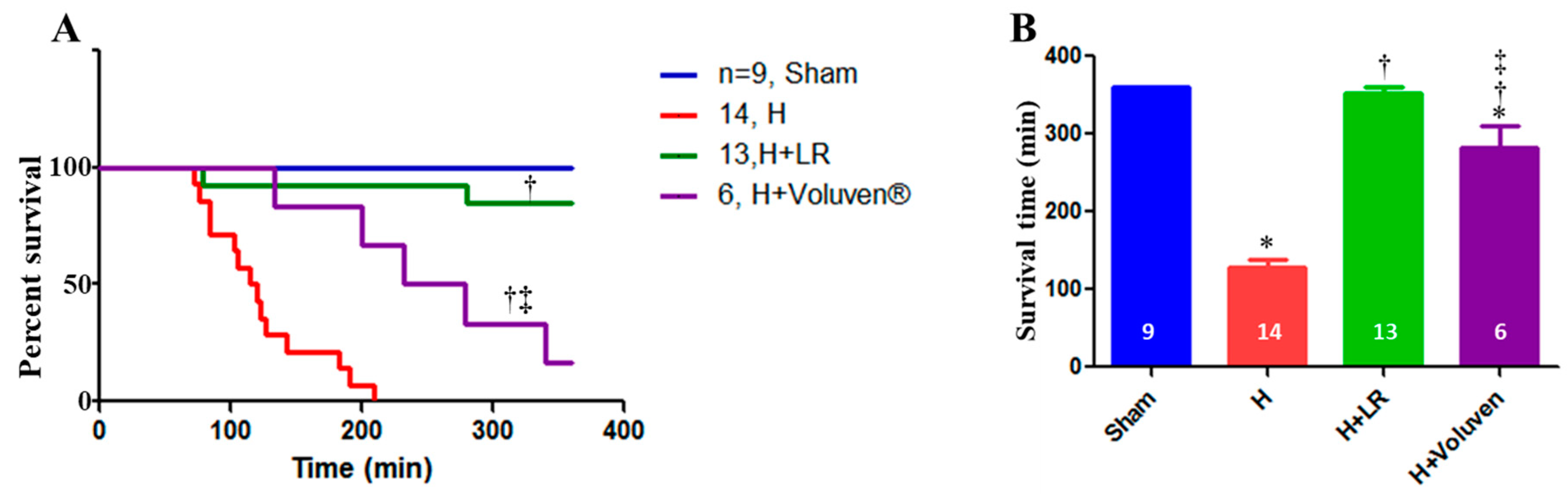
Figure 8.
Scheme of the experimental design.
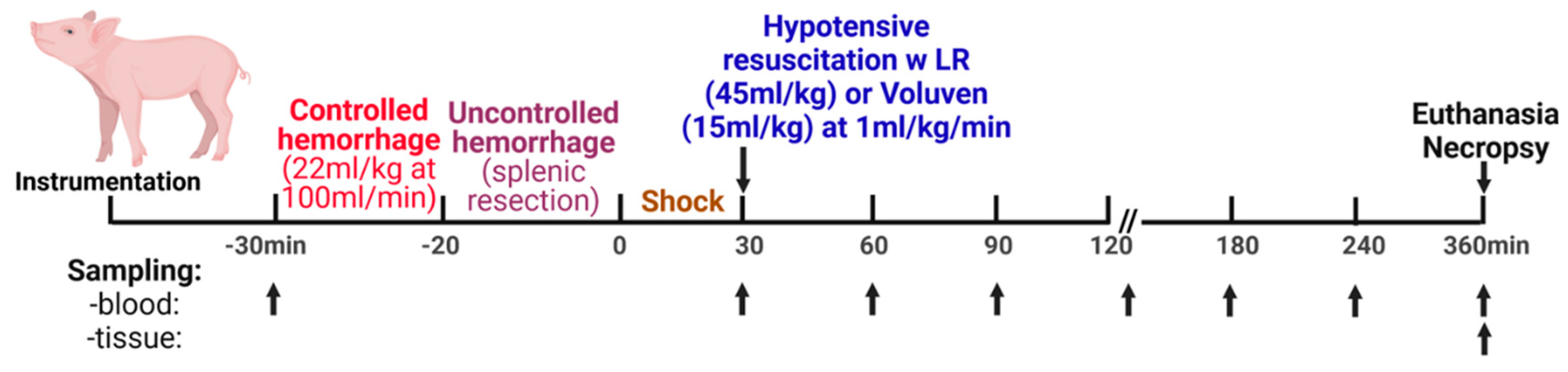

Table 1.
Hemodynamic responses to hemorrhagic shock and fluid resuscitation.
| Group | ||||
|---|---|---|---|---|
| Sham | H | H + LR | H+Voluven | |
| n | 9 | 13 | 12 | 6 |
| Body weight (kg) | 39.4±1.1 | 38.2±0.6 | 39.1±0.8 | 40.8±1.7 |
| Controlled SBV (ml/kg) | 22 | 22 | 22 | 22 |
| Uncontrolled SBV (ml/kg) | ||||
| At time 30 | N/A | 11.1±1.0 | 7.8±0.9 | 10.1±1.5 |
| 60 | N/A | 12.7±1.1 | 10.7±2.4 | 13.9±1.8 |
| 90 | N/A | 12.7±1.1 | 8.6±1.0 | 16.1±2.3‡ |
| 120 | N/A | 12.5±1.6 | 8.6±1.0 | 17.5±2.7‡ |
| 360min | N/A | 13.7±1.0 | 11.0±2.3 | 18.3±2.8‡ |
| PP (mmHg) | ||||
| Baseline | 26.7±2.1 | 28.7±3.2 | 29.1±2.8 | 28.8±1.8 |
| at time 30 | 28.5±1.7 | 20.1±2.9 | 17.7±2.7* | 19.7±4.0* |
| 60 | 29.1±1.3 | 21.5±3.1 | 22.0±2.6 | 27.8±3.1 |
| 90 | 27.2±1.5 | 14.3±1.5 | 22.0±3.3 | 28.0±2.6† |
| 120 min | 26.0±1.5 | 16.3±3.8 | 21.3±3 | 24.6±3.9 |
| MAP (mmHg) | ||||
| Baseline | 63.4±2.5 | 62.6±3.2 | 63.4±3.1 | 60.8±1.6 |
| at time 30 | 65.1±1.3 | 38.7±2.3* | 39.5±3.8* | 32.2±4.1* |
| 60 | 63.3±1.4 | 37.9±2.2* | 44.6±2.8* | 44.3±1.5* |
| 90 | 62.4±1.9 | 29.6±1.9* | 44.1±1.9*† | 43.0±1.3* |
| 120 min | 63.4±1.8 | 26.9±2.2* | 43.0±1.6*† | 38.1±3.7* |
| Shock index (bpm/mmHg) | ||||
| Baseline | 1.3±0.1 | 1.4±0.2 | 1.3±0.1 | 1.3±0.1 |
| at time 30 | 1.4±0.1 | 4.5±0.3* | 5.2±1.2* | 4.8±0.7* |
| 60 | 1.5±0.1 | 4.5±0.2* | 3.5±0.4* | 3.2±0.1*† |
| 90 | 1.5±0.1 | 5.4±0.5* | 3.2±0.2*† | 3.1±0.1*† |
| 120 min | 1.6±0.1 | 5.3±0.2* | 3.4±0.2*† | 3.3±0.1*† |
Notes: Data are expressed as mean ± SEM; n, number of samples; H, hemorrhage; LR, lactated Ringer’s solution; PP, pulse pressure; MAP, mean arterial pressure; SBV, shed blood volume. *p<0.05 vs. Sham; †p<0.05 vs. H; ‡p<0.05 vs. H+LR (two-way ANOVA, GraphPad Prism 5.03).There was no significant difference in the body weight between the groups (one-way ANOVA, GraphPad Prism 10).
Table 2.
Metabolic responses to hemorrhagic shock and fluid resuscitation.
| Group | ||||
|---|---|---|---|---|
| Sham | H | H + LR | H+Voluven | |
| n | 9 | 14 | 12 | 6 |
| pH: Baseline | 7.4±0.0 | 7.4±0.0 | 7.4±0.0 | 7.4±0.0 |
| at time 30 | 7.4±0.0 | 7.4±0.0 | 7.4±0.0 | 7.4±0.0 |
| 60 | 7.4±0.0 | 7.4±0.0 | 7.4±0.0 | 7.4±0.0 |
| 90 | 7.5±0.0 | 7.4±0.0 | 7.4±0.0 | 7.4±0.0 |
| 120 min | 7.5±0.0 | 7.4±0.0 | 7.4±0.02 | 7.4±0.1 |
| Base excess (mmol/L): Baseline | 5.3±0.8 | 6.2±0.6 | 5.2±1.0 | 6.7±0.8 |
| at time 30 | 6.3±0.9 | 2.5±0.6* | 2.8±0.7 | 0.4±1* |
| 60 | 5.9±0.8 | -0.7±1.0* | 1.8±1.0* | 1.6±0.9* |
| 90 | 6.1±1.0 | -3.4±2.2* | 3.5±1.0† | 3.2±1.2† |
| 120 min | 7.1±0.9 | -3.7±1.7* | 4.0±1.1† | 4.0±1.4† |
| Lactate (mmol/L): Baseline | 2.2±0.2 | 2.0±0.1 | 2.2±0.2 | 2.0±0.2 |
| at time 30 | 1.9±0.1 | 4.1±0.2* | 3.3±0.2 | 5.3±0.6* |
| 60 | 1.7±0.1 | 6.6±0.6* | 5.1±0.8* | 5.5±0.4* |
| 90 | 1.7±0.2 | 8.9±1.9* | 4.3±0.7*† | 4.9±0.4*† |
| 120 min | 1.5±0.2 | 9.3±1.5* | 3.8±0.6*† | 5.2±0.8*† |
| SvO2 (%): Baseline | 79.0±3.3 | 73.4±2.4 | 76.0±1.2 | 79.4±2.2 |
| at time 30 | 79.4±2.2 | 54.3±6.5* | 52.2±6.5* | 66.5±6.4 |
| 60 | 78.2±2.5 | 62.1±6.5 | 59.1±3.7* | 71.7±4.2 |
| 90 | 74.4±1.4 | 71.3±7.4 | 54.2±5.6* | 71.8±4.3 |
| 120 min | 77.1±2.6 | 49.4±10.4* | 56.5±4.8* | 67.3±6.1 |
| Hemoglobin (g/dl): Baseline | 8.3±0.3 | 8.4±0.17 | 8.6±0.2 | 8.1±0.2 |
| at time 30 | 8.1±0.4 | 8.31±0.23 | 8.6±0.4 | 7.8±0.4 |
| 60 | 8±0.4 | 8.25±0.27 | 6.9±0.6* | 4.5±0.4* |
| 90 | 7.9±0.3 | 6.6±1.0 | 6.3±0.6 | 4.6±0.5‡† |
| 120min | 8.3±0.3 | 7.1±0.7 | 6.7±0.37† | 4.4±0.4†* |
| Hct (%): Baseline | 24.5±1.06 | 24.7±0.5 | 25.4±0.6 | 23.8±0.8 |
| at time 30 | 25.2±1.2 | 24.5±0.7 | 25.3±1.2 | 23.0±1.2 |
| 60 | 24.2±1.4 | 23.2±1.3 | 19.4±1.8 | 13.2±1.0*†‡ |
| 90 | 23.5±1.2 | 19.4±2.8 | 18.4±1.6 | 12.8±1.4*†‡ |
| 120min | 25.2±1.2 | 21.0±2.1 | 19.7±1.1* | 12.4±1.1*†‡ |
| Potassium (mmol/L): Baseline | 4.1±0.0 | 4.0±0.1 | 3.9±0.1 | 3.9±0.1 |
| at time 30 | 4.2±0.1 | 4.7±0.1 | 4.6±0.3 | 5.1±0.3* |
| 60 | 4.2±0.1 | 5.1±0.2* | 4.4±0.3† | 3.8±0.1† |
| 90 | 4.4±0.1 | 5.2±0.4 | 4.4±0.1 | 4.3±0.1 |
| 120 min | 4.5±0.1 | 6.2±0.7* | 4.6±0.1† | 4.8±0.1† |
Notes: Data are expressed as mean ± SEM; n, number of samples; H, hemorrhage; LR, lactated Ringer’s solution; SvO2, mixed venous oxygen saturation. *=p<0.05 vs. Sham; †=p<0.05 vs. H; ‡=p<0.05 vs. H+LR (two-way ANOVA, GraphPad Prism 10).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



