
Submitted:
18 December 2023
Posted:
19 December 2023
You are already at the latest version
Abstract
Heterosis plays a significant role in enhancing variety, boosting yield, and raising economic value, but the molecular mechanism is still unclear. We analyzed the transcriptomes and 3D genomes of hybrid (F1) and its parents (w30 and 082). Analysis of the expression revealed a total of 485 specially expressed genes (SEGs), 173 differentially expressed genes (DEGs) above parental expression level, more actively expressed genes and up-regulated DEGs in F1. Further study revealed that the DEGs detected in F1 and its parents were mainly involved in response to auxin, plant hormone signal transduction, DNA metabolic process, Purine metabolism, starch and sucrose metabolism, which suggested that these biological processes may play a crucial role in the heterosis of B. rapa. The analysis of 3D genome data revealed that hybrid F1 tend to contain more transcriptionally active A compartments after hybridization. Additionally, F1 had a smaller TAD (topologically associated domain) genome length, but the number was the highest, and the expression change of activated TAD was higher than repressed TAD. More specific TAD boundaries were detected between parents and F1. Subsequently, 140 DEGs with genomic structural variants were selected as potential candidate genes. Among them, we found two DEGs with consistent expression changes in A/B compartments and TADs. Our findings suggest that genomic structural variants, such as TADs and A/B compartments, may affect gene expression and contribute to heterosis in B. rapa. This study provides further insight into the molecular mechanism of heterosis in B. rapa.
Keywords:
Heterosis
; Brassica rapa
; Hi-C
; A/B compartments
; TADs
; Integrative genomic analysis
1. Introduction
Heterosis is a widely observed biological phenomenon whereby hybrid offspring exhibit superior traits compared to their parents, including growth potential, viability, yield, and quality [1,2,3]. Given the increasing global population, the successful application of heterosis in agricultural production has dramatically contributed to augmenting crop yield, mitigating food crisis, and guaranteeing food security [4]. The concept of heterosis has been put forward and applied for more than 100 years [5]. At present, three hypotheses, namely dominance [6,7,8,9], over-dominance [9,10], and epistasis [11,12], have been proposed to explain the genetic basis of heterosis in most crops. However, these hypotheses remain theoretical and cannot fully explain the phenomenon of heterosis. The QTL mapping methods developed in the late 1980s laid a foundation for exploring heterosis, and extensive genetic analyses of hybrids in maize, rice, rape, and other plants led to the identification of numerous quantitative trait loci [13]. With the development of high-throughput sequencing technology, a series of dominant genes related to heterosis traits have been identified by integrating transcriptomics and other omics [14], thereby offering new insights into the molecular mechanisms underlying heterosis.
Genes encode genetic information in a one-dimensional linear sequence, but they are expressed by forming three-dimensional chromatin architecture. In addition, chromatin states and genome activity regulators also affect gene expression. And changes in gene expression and regulatory network are bound up with heterosis [3]. Chromatin conformation plays a crucial role in the execution of biological activities by many regulatory elements [15]. Upon sensing environmental and developmental cues, global and local chromatin rearrangements may occur, along with changes in gene transcription [16,17]. In Brassica napus, Studies have shown that dynamic three-dimensional chromatin structure helps the expression of plant hormone-related genes in hybrid F1 [18]. Chromosome organization can be dissected into multiple functional and structural domains [19], the units from high to low are chromosomal compartments (CTs), active (euchromatin, A-type) and inactive (heterochromatin, B-type) chromatin compartments, topologically associated domain (TADs), chromatin loops (CLs) and chromatin fibers. TAD regulates gene replication and epigenetic modification locally. The boundary region of TAD is genetically rich and conserved. Disruption of the TAD boundary can severely affect the regulation of gene expression and even lead to the occurrence of diseases [20]. The 3D genome and gene expression have complex relationships, with rapid dynamic changes in genome structure and gene expression accompanying hybridization. Nevertheless, little is known about the changes in 3D structure for heterosis traits in Brassica rapa.
Chinese cabbage (AA genome, 2n = 2x = 20), from the appearance can be roughly divided into two types: heading and non-heading Chinese cabbage. They are both highly nutritious, commercially viable, and easy to cultivate. But they also have many differences, such as nutrient contentions and stress resistance. To exploit the advantages of both and explore the mechanism of hybrid heterosis, we used non-heading Chinese cabbage 082 (P1) as the female parent and w30 (P2) as the male parent to generate the hybrid F1. We conducted a comparative analysis of Hi-C and RNA-seq data, identifying several genes with specifically and significantly different expression levels in hybrids, and discussed their potential functions and relationships. We also compared the 3D spatial structure of TADs and A/B compartments between the parents and hybrid F1, and observed conserved and altered chromatin structures between them. We further investigated the associations between gene expression and 3D chromatin structure. This study will contribute to our understanding of heterosis in Chinese cabbage, providing insight into how to achieve higher yields and better stress resistance through hybrid breeding.
2. Results
2.1. Heterosis is remarkable in the hybrid F1
A remarkable biomass heterosis was observed in hybrid F1 (see Figure 1a). To facilitate a quantitative comparison of their differences, we determined their fresh and dry weights at 21 days after sowing (Figure 1b). The values of MPH (mid-parent heterosis) and HPH (high-parent heterosis) both exceed 50% (Figure 1c and Figure 1d), and in terms of fresh weight, MPH is as high as 159.28%, while HPH is as high as 150.11% (see Figure 1c). It can be seen that F1 exhibited a remarkable degree of heterosis. Meanwhile, their obvious phenotypic differences suit the subsequent integrative genomic analysis.
2.2. More genes were actively expressed in the hybrid F1
High-quality transcriptome reference genomes of 124.57 Mb, 133.47 Mb, and 134.98 Mb were obtained for 082, w30, and F1 (Additional file 1: Table S1 and S2). Genes with FPKM ≥1 were defined as actively expressed. The number of actively expressed genes in the hybrid F1 (24,601) was more significant than those in the maternal line 082 (24,320) and paternal line w30 (24,524) (Additional file 2: Table S3–S5 and Figure 2a). However, no distinct differences were observed based on genome-wide FPKM expression profiles for the three varieties (Additional file 3: Table S6, Figure 2b and Figure S1). A pairwise analysis of these actively expressed genes further confirmed that many genes (more than 92%) are the common expression between the parents , and that the male parent exhibits greater specificity than the female parent (Figure 2c). F1 had more genes that were specifically expressed when compared to either parent alone (Figure 2d,e). More genes between F1 and P2 (23,226) shared common expression profiles (Figure 2e). Concurrently, we compared the correlation of all expressed genes with nine samples and found that the correlation coefficient between F1 and P2 (R=0.919) was higher than that between F1 and P1 (R= 0.888) (Figure 2g). Interestingly, when compared with both parents, F1 displayed the least specific expression of genes (485) compared to P1 (921) and P2 (891) (Figure 2f). We hypothesized that 485 genes with specific expressions in F1 may be involved in heterosis .
2.3. Transcriptome analysis of DEGs
To investigate the differential expression profiles of genes, we analyzed the expression patterns and expression levels of differentially expressed genes (DEGs). Firstly, we conducted a cluster analysis on the expression patterns. A portion of the genes showed the same expression pattern between the parents and F1, while others showed significant differences (Figure 3a), suggesting that these genes may be responsible for the heterosis of F1. Next, we analyzed DEGs expression levels. A total of 2,156 genes displayed significantly differential expression levels between the parents (P<0.05, |log2FC|>1) (Additional file 4: Table S8, Figure 3b and Table 1), and the ratio between maternal and paternal lines was similar. F1 and parents differed significantly in 2,568 genes, and most of those genes were expressed at higher levels in the hybrid F1 (Additional file 4: Table S8-9 and Table 1). Among these genes, we focused on 296 genes in F1 that had significantly different expression levels from both parents, and the expression level of most genes (173) in F1 is higher than its parents (Additional file 5: Table S10 and Table 1). We used the volcano map to summarize the significant DEGs (Figure 3b). These results further confirmed that hybridization not only activated the expression of more genes in F1, but also made the expression level of these genes in F1 significantly higher than in its parents (Table 1). In addition, Venn diagram analysis was conducted on the DEGs of the three groups (082 vs w30, F1 vs 082, F1 vs w30), and 102 shared DEGs were identified (Additional file 6: Table S11 and Figure 3c). Among them, 10 homologous hyperparental genes were identified in Arabidopsis, which enriched in developmental growth, stress and resistance, response to light, nitrogen compound biosynthetic process, carboxylic acid metabolic process, and lipid biosynthetic process (Additional file 7: Table S12).
2.4. GO and KEGG analysis of DEGs
We conducted a gene ontology (GO) and KEGG pathway enrichment analysis to annotate the DEGs [21,22,23]. Using an over-represented p-value threshold of less than 0.018, we identified significant changes in the GO terms between F1 and its parents. The top GO terms for biological processes are as follows: “response to auxin”, “nucleoside transport”, and “nicotinamide riboside transport”, while the majority of DEGs were found in “DNA metabolic process” (78 DEGs). In terms of cell component, the most common GO term was “Golgi-associated vesicle,” while “Cytoplasmic membrane-bound vesicle” had the highest number of DEGs (11 DEGs). Regarding molecular function, the most common GO term was “ADP binding” with the most DEGs (Additional file 8: Table S13-14 and Figure 4a). These findings suggest that hybridization resulted in a substantial proportion of transcriptome alterations, and more genes were activated than repressed (Figure S2). We also identified the top 20 pathways assigned to our DEGs (Figure 4b). Most of the DEGs were involved in “Starch and sucrose metabolism” (12 DEGs), “Plant hormone signal transduction” (17 DEGs)”, and “Purine metabolism” (17 DEGs) (Additional file 9: Table S15-16).
2.5. Genome-wide interaction matrices of parents and F1
To explore the dynamic 3D spatial structural changes of P1, P2, and F1 genomes during B. rapa hybridization, we conducted Hi-C sequencing and obtained 117.8G, 141.1G, and 133.0G of raw data, respectively (Additional file 10: Table S17). We then mapped the clean Hi-C data to the high quality B. rapa genome (v2.5) reference genome [24] and filtered out invalid pairs for subsequent comparative 3D structural analysis (Additional file 10: Table S18). The genome-wide simulation images displayed that the nuclei of P1, P2 and F1 were nearly spherical, and the chromosomes were localized in a limited volume (Figure 5a). The results confirm previous findings that each chromosome occupies an exclusive region in the nucleus, a concept termed "chromosomal territory" [25]. At a resolution of 1 Mb, chromatin interaction analysis showed that the intra-chromosome interaction (cis interaction) of the three varieties was higher than the inter-chromosome interaction (trans interaction) (Figure S3). But F1 has a higher proportion of interchromosome/intrachromosome (trans/cis) contact than their parents (Additional file 10 : Table S19 and Figure S4). Therefore, each chromosome in the F1 nucleus may have a different space. To further explore the interaction patterns within chromosomes, we enhanced the resolution of the genome-wide interaction matrix to 100 kb (Figure S8). For example, among the interactions within chromosome 8 (Figure 5b), the dark red diagonal indicates the strongest interaction. Horizontal distance decreases the occurrence of intrachromosome interactions. Additionally, more refined components like TADs (Figure 5c) are available. Each diagonally distributed triangle corresponds to a topological association domain (TAD) (Figure 5e). Meanwhile, there was no significant difference in the distribution of compartments (Figure 5d) between F1 and parents.
2.6. Identification of A/B Compartment Shifts
To explore the changes in A/B Compartments between F1 and parents, each chromosome’s A/B compartment distribution of F1 and its parents were displayed (Figure 6a). F1 tends to contain more A compartments while parents tend to contain more B compartments (Additional file 11: Table S20 and Figure 6b). A total of 473 genes from P1 to F1 were identified as A-to-B shifts and 950 genes as B-to-A shifts. A combined analysis with transcriptomics revealed 15 DEGs in A-to-B shifts (2 DEGs down-regulated in F1) and 20 DEGs in B-to-A shifts (16 DEGs up-regulated in F1). Meanwhile, a total of 281 genes from P2 to F1 were identified as A-to-B shifts, and 115 genes were B-to-A shifts. Moreover, 7 DEGs in A-to-B shifts (1 DEGs down-regulated in F1) and 68 DEGs in B-to-A shifts (48 DEGs up-regulated in F1) (Additional file 11: Table S20). The inconsistent relationship resembles the results in Drosophila [26]. We found that hybridization can make it more inclined to transform into A compartments. However, by and large, there is almost no obvious difference between the A/B compartments of parents and F1 (Figure 6a and Additional file 11: Table S20). On the basis of this, we correlated the gene densities of 082-/w30-/F1 traid with A/B compartments. Results from F1 and its parents showed that A compartment had a higher gene density than B compartment (Figure S5). Further analysis of gene expression of A/B compartments found that there was no significant difference between F1 and parents, but the gene expression in A compartment was significantly higher than that in B compartment (Figure 6c). This is consistent with previous studies in Arabidopsis [27].
2.7. Changes in compartments during hybridization
During hybridization, 90.31% (A-toA-toA shifts are 72.96%, B-toB-toB shifts are 20.35%) of compartments did not change, suggesting that compartments in the nucleus are relatively stable. Furthermore, F1 showed a higher proportion of compartment A, which increased by 4.04% (including 3.14% for A-toB-toA shifts and 0.90% for B-toA-toA shifts), while compartment B decreased by 1.44%, compared to their parents. Moreover, very few compartments have completely different transformations from their parents, among which A-toA-toB shifts account for 0.06% and B-toB-toA shifts account for 1.15% (Figure 6d). Interestingly, 485 SEGs in F1 showed similar results, but A-toA-toB shifts did not occur. When parents' compartments were not aligned, the compartments in F1 were more inclined towards compartment A (Figure S6). Interestingly, the A/B compartment transformation analysis of 296 DEGs with significantly different expression levels of parents and F1 is similar to that of SEGs (Figure S6). These results suggest that different compartment activities between parents may influence compartment transitions in hybrids, ultimately impacting gene expression.
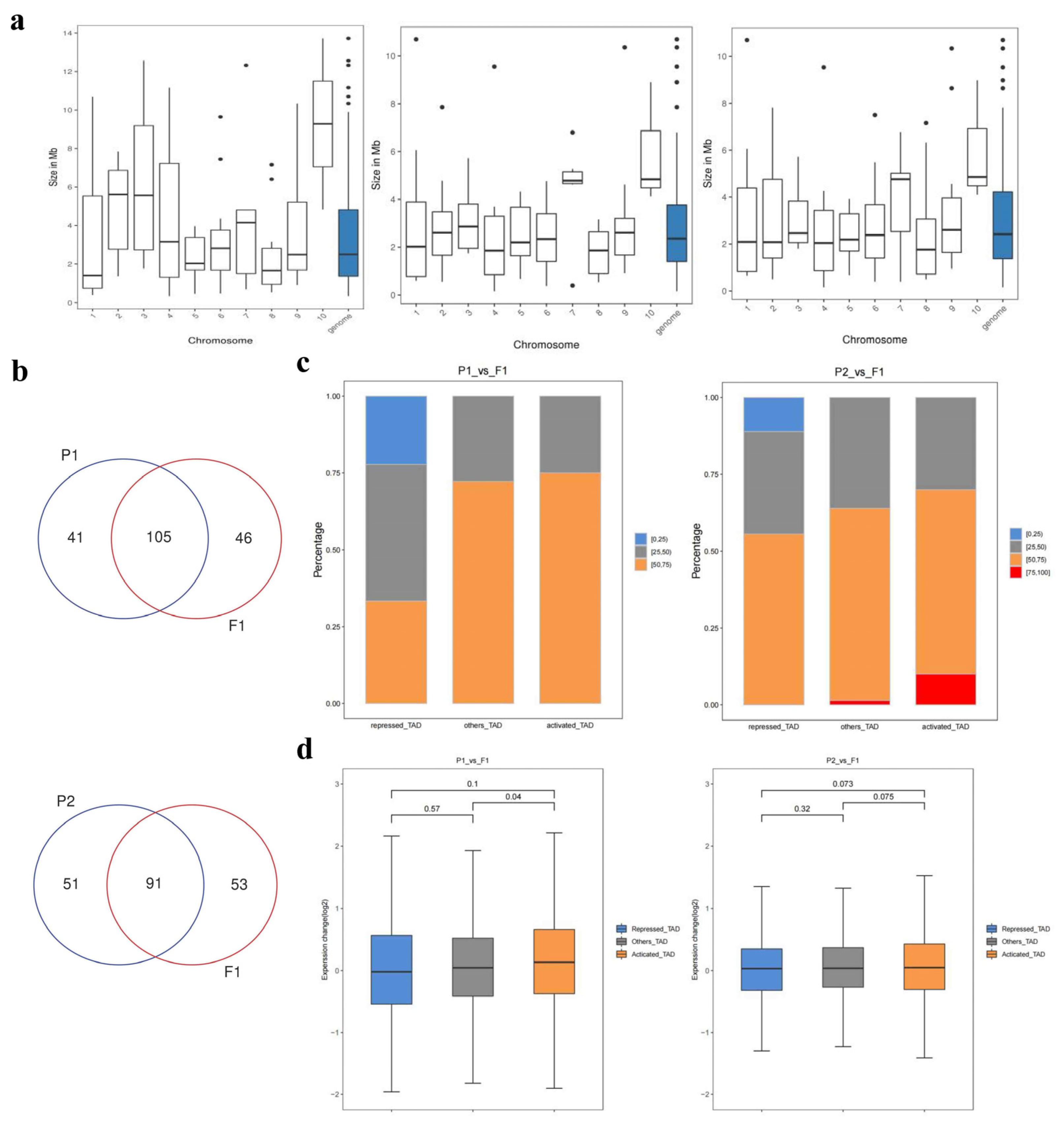
2.8. Identification of different kinds of topologically associating domains
TADs, as relatively static physical and regulatory domains, facilitate specific and intentional gene expression programs in various ways [28,29]. In order to more closely explore the 3D genomic differences between the parents and F1, we compared the change of TADs. F1 shared both similarities and differences with its parents in terms of TADs (Figure S7). Further statistical analyses on the number and length of TADs showed that the number of TADs in F1 was greater than that of its parents, and the average length of each TAD was smaller, so was the length of the whole genome (Additional 12: Table S21 and Figure 7a). Meanwhile, in comparison with P1, F1 exhibited 46 specific TADs and 105 conserved TADs (12 activated TADs; 10 repressed TADs; 83 other TADs) (Figure 7b). Similarly, Compared with P2, there were 91 conserved TADs (10 activated TADs; 9 repressed TADs; 72other TADs), and 53 specific TADs in F1 (Figure 7b). Besides, 141 DEGs were recognized in activated TADs (53 up-regulated DEGs in P1/P2) and 43 DEGs were identified in repressed TADs (42 down-regulated DEGs in P1/P2) (Additional file 12: Table S22). Together with transcriptome analysis, some DEGs were not expressed consistently with TAD types, and some DEGs were not detected in either active or repressed TADs. These observations that the formation of heterosis may be largely due to trans-regulatroy mechanisms. Figure 7c showed that activated TAD had a greater percentage of fold change (P1 or P2/F1) >1 than repressed TAD. Moreover, the expression change of activated TAD was higher than repressed TAD (Figure 7d). Based on the distribution of DI values in the upstream and downstream Windows, the correlation analysis of different boundary regions was carried out. We found more specific TAD boundaries between parents and F1, while less specific TAD boundaries between parents (Figure S9). This may be due to large-scale rearrangement of chromatin during hybridization. These results above indicate that the changes in 3D chromatin structure between the parents and F1 are greater than those between the parents.
2.9. Identification of candidate genes for heterosis and verification of qRT-PCR
Based on the above transcriptome and 3D genome joint analysis results, we identified 140 DEGs that are associated with genomic structural alterations, including 56 DEGs in A/B compartments, 87 DEGs in TADs (Additional file 14: Table S24). For example, the P1vs F1 group uses F1 as a reference, if the TAD type of P1 relative to F1 is repressed TAD, then P1 relative to F1 should be downregulated. And their functions are related to various processes, such as photosynthesis, plant cell size/division/cycle, resistance and stress, response to auxin, and carbohydrate metabolism. Notably, two candidate DEGs (BraA02003296 and BraA02003297) are shared between these categories. Besides, to verify the accuracy of RNA-seq data, we selected 10 DEGs between the parents and F1 in 173 DEGs mentioned above by Real-time quantitative RT-PCR (qRT-RCR) (Additional file 13: Table S23). The expression patterns of these 10 DEGs were highly consistent with the data obtained by RNA-seq (Figure S10).
Figure 7.
The analysis of the TADs in F1 and parents. (a). The TAD length genomic distribution of F1 and parents. From left to right: 082, F1, and w30. (b). Indicated P1 vs F1 venn diagrams of the TAD. Overlapping part represents conservative TAD, and vice versa represents specific TAD. (c). Proportional stacking maps of genes with different expression levels in different TADs. For each gene contained in the TADs, the change in expression level was calculated for both sets of samples, and the percentage of all genes corresponding to that TAD with Fold Change greater than 1. The percentages were divided into four categories, including 0-25%, 25-50%, 50-75%, and 75-100%. The results for the different categories of TADs were presented in the form of bar charts. (d). Gene expression of the three types of TADs (Repressed TAD/Others TAD/Actived TAD).
Figure 7.
The analysis of the TADs in F1 and parents. (a). The TAD length genomic distribution of F1 and parents. From left to right: 082, F1, and w30. (b). Indicated P1 vs F1 venn diagrams of the TAD. Overlapping part represents conservative TAD, and vice versa represents specific TAD. (c). Proportional stacking maps of genes with different expression levels in different TADs. For each gene contained in the TADs, the change in expression level was calculated for both sets of samples, and the percentage of all genes corresponding to that TAD with Fold Change greater than 1. The percentages were divided into four categories, including 0-25%, 25-50%, 50-75%, and 75-100%. The results for the different categories of TADs were presented in the form of bar charts. (d). Gene expression of the three types of TADs (Repressed TAD/Others TAD/Actived TAD).
3. Discussion
At present, the underlying molecular mechanism of heterosis remains elusive, and the effect of three-dimensional structural dynamics on heterosis is still in its infancy. Therefore, integrating 3D genomics and transcriptomics is imperative for gaining a more comprehensive understanding of the role of chromatin in 3D space in heterosis.
In this study, the transcriptome date confirmed that hybridization leads to activation of gene expression in F1 plants. However, only a small number of genes were specifically expressed or displayed significantly different levels of expression. These indicated that the genome-wide expression profiles of the hybrid F1 and parents are similar. Previous studies on heterosis produced by intraspecific hybridization also confirmed that most genes in hybrids and parents showed similar expression profiles, and the expression level of these genes is close to MPV [30,31,32]. Emerging genomic and epigenetic perspectives suggest that heterosis results from allelic interactions between parent genomes, resulting in changes in genetic programming that promote hybrid growth, stress tolerance, and fitness. For example, epigenetic modification of key regulatory genes in hybrids can alter complex physiological and metabolic regulatory networks to regulate biomass heterosis. GO function annotation indicated that DEGs were significantly enriched in "response to auxin", "nucleoside transport", " nicotinamide riboside transport ", while the largest number of DEGs are found in "DNA metabolic process". It is worth mentioning that in most studies on the molecular mechanism of heterosis, the metabolic process is unanimously recognized as related to growth or biomass heterosis [30,33,34]. In KEGG analysis, we also found DEGs enriched in "Starch and sucrose metabolism". The enrichment level of metabolic pathway in different tissues and development stages of different plants is different, and considerable studies believe that carbohydrate metabolism is related to the formation of heterosis [30,35,36]. In addition, "Plant hormone signal transduction" also be found. Studies have shown that dynamic three-dimensional chromatin structure helps the expression of plant hormone-related genes in F1, which is related to plant growth [18].
Furthermore, genome-wide chromosome simulations show that each chromosome occupies a dedicated region of the nucleus. This is also a good explanation for the higher intra-chromatin interactions than inter-chromatin interactions found at 1 Mb resolution. However, by calculating the interaction ratio of inter/intra, it was found that F1 is more active than that between the chromosomes of both parents. This may also be one of the reasons why F1 has more active genes and upregulated genes. In general, A compartments are in the transcriptional active region of euchromatin, and B compartments are in the transcriptional repressed region of heterochromatin. During hybridization, F1 have a greater number of A compartments, and the chromatin status of F1 is more inclined to A compartments when the parental compartments are different. Heterochromatin remodeling is critical for a variety of cellular processes [37]. Therefore, the B-toA compartments shift is considered beneficial to the formation of heterosis. In terms of TADs, we compared the specific TAD and TAD boundary, as well as the length and quantity of the TAD genomes. Parents and hybrids exhibit significant differences in terms of TAD. It has been appreciated that the existence of a preformed and stable topology (TADs) organizes the physical proximity between enhancers and their target genes. Although there is no obvious differences of A/B compartments, the change of TADs may be one of the reasons why hybridization activates more genes in F1. Subsequently, 140 DEGs with genomic structural variants were selected as potential candidate genes, including 3 photosynthetic genes (BraA02003217, BraA07001020,and BraA07001021), 8 plant cell size/division/cycle-related (BraA06003576, BraA05002844, BraA07001159, BraA03006134, BraA05001791, BraA02002099, BraA02003420, BraA02002337), 4 Carbohydrate metabolism genes (BraA07001318, BraA06002248, BraA07001149, BraA01003773), 9 resistance and stress related genes (BraA04000638, BraA07000080, BraA09003537, BraA09003725, BraA02003318, BraA07001184, BraA06002291, BraA07001349, BraA06002292), 3 genes response to auxin (BraA02003420, BraA06000631, BraA07001182), 1 development and cell death genes (BraA05002866), 1 senescence associated gene (BraA08002232). However, further functional verification is required for these candidate DEGs. In the future, we will obtain a mutant of the candidate DEGs through gene editing or other methods, observe its corresponding phenotype, and then obtain overexpressed plants to further determine the phenotype. In addition, expression pattern analysis of the candidate DEGs is also needed. For example, analyzing its expression levels in different tissues at different stages, or its expression sites in cells. If possible, we will further investigate its interacting proteins and the regulatory relationships between upstream and downstream.
Gene expression is precisely regulated by the multi-layered three-dimensional structure of chromatin [29]. Different layers of the 3D genome have various levels of regulatory control [38]. This study specifically focuses on the beneficial aspects of the 3D genome, however, further exploration is needed to understand the specific regulations. With the advancement of technology, it is believed that the impact of spatial structure changes of chromatin on heterosis will be further explored and interpreted in the future.
4. Materials and Methods
4.1. Plant materials, growth conditions, and sample collection
The male parent w30(P2), female parent 082(P1) and their hybrid F1 used in this study were all provided by Nanjing Agricultural University. Besides, F1 was generated by hand pollination. For reducing bias, all plant material used for Hi-C and RNA-seq was grown in the same environment (24℃, 16 hours of light / 8 hours of darkness; vermiculite: peat = 1:1). Samples for Hi-C and RNA-seq were collected using fully expanded parental and hybrid F1 leaves (3rd upper leaf of a plant) with three replicates per sample. After being quick-frozen in liquid nitrogen, it was placed in a -80℃ refrigerator for later use. RNA-seq yielded three biological repeats.
4.2. Evaluation of heterosis
To measure phenotypic data, including fresh weight (FW) and dry weight (DW), 30 similar plants (10 plants per replicate) were collected at 21 days. The collected material was washed with distilled water immediately, then baked at 105°C for 30 minutes before further drying at 80℃ for 60 hours [18]. MPH (mid-parent heterosis) was calculated as follow: MPH = (the mean value of hybrid F1 - the average value of both parents) / the average value of both parents ×100%. HPH (high-parent heterosis) was calculated as follow: HPH = (the mean value of hybrid F1 - the optimal parental value) / the optimal parental value ×100%.
4.3. Hi-C libraries preparation and sequencing
The establishment of Hi-C librarires and completion of Illumina sequencing were conducted by Novogene Bioinformatics technology company (China). The libraries were built according to the standard protocol described previously with some modifications [39]. After fixing it with polyformaldehyde and DNA, use restriction endonuclease to make a gap at the cross-linking point, and use biotin marker at the same time. The adjacent DNA fragments were treated with nucleic acid ligase, then the protein at the junction was digested with protease, and finally the fragments with a length of 350 bp were broken by Covaris crusher for recovery. After the libraries were constructed, Qubit 2.0 and Q-PCR are used for preliminary and precise quantification respectively, while Agilent 2100 is used to detect the insertion size of the library. Finally, once the libraries are qualified, the different libraries are pooled and sequenced.
4.4. Hi-C read mapping
The quality of the obtained Hi-C sequencing data was controlled before output. Paired reads including adapter contaminated sequences, unknown nucleotide "N" ratios >10%, and more than 50% base Q <5 were filtered out. Next, the qualified reads obtained were processed using the HiCUP pipeline (version 0.57) [40]. For data comparison, Bowtie2 software (version 2.2.3) [41] was used to compare the obtained reads with B. rapa genome (version 2.5) [24]. The observed interaction matrices were constructed for the final effective contacts according to the statistical interaction matrices with a certain resolution interaction matrix. The maximum likelihood method is used to standardize the observation interaction matrix. The MDS algorithm of PASTIS software [42] was employed to imitate the 3D position of chromatin. On this basis, the constructed heat map of chromatin interaction is divided into A/B compartments with a resolution of 100 kb by principal component analysis (PCA). TadLib (hitad 0.1.1-r1) software was applied to estimate the TAD topology at a 40 Kb resolution with default parameters. RNA-seq was performed on the genes in TADs, and we classified TADs into three categories based on the proportion of genes with positive and negative FC in each TAD of P1/P2 vs F1: activated TADs, repressed TADs, and other TADs. Sort the foldchange from largest to smallest, with top 10% being considered activated TAD, bottom 10% being expressed TAD, and the rest being considered others TAD.
4.5. RNA-seq experiment and sequencings
In accordance with the instructions on the kit, total RNA of nine samples were extracted using the RNAprep Pure Plant Kit (Tiangen, Beijing, China). The purity, integrity, degradation, and contamination of RNA samples were assessed by Novogene Company, and RNA concentrations were accurately quantified by Qubit. In addition, the construction and sequencing of cDNA libraries were also completed by Novogene Company. The construction of cDNA libraries includes the synthesis, purification and end repair of double-stranded cDNA, adding A-tail, ligation of sequencing adapter, PCR enrichment and so more. After the completed libraries were qualified, different libraries were pooled according to the requirements of effective concentration and target data volume, and finally RNA sequencing was performed.
4.6. RNA-seq data analysis
Spliced reads were effectively compared to RNA-Seq data using HISAT 2 (version 2.1.0) [43]. In RNA-Seq analysis, in order to make different genes and sequencing data comparable, we introduced the concept of FPKM. FPKM represents the number of fragments per million from a gene per kilobase length [44]. DESeq2 R (version 1.18.0) software for differences in gene analysis (DEG), selection criteria for |log2 fold change| > 1, or error rate (FDR) < 0.05. The corrected P value was obtained by multiple hypothesis testing based on Benjamini and Hochberg's method, and the square of Pearson correlation coefficient between biological repetitions (R2 > 0.8) was used to test the experimental reliability. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases (www.kegg.jp/kegg/kegg1.html.) [21,22,23] were used to identify possible biological functions and pathways of DEGs. Among them, GOseq (Release2.12) software package was used to conduct GO enrichment analysis of DEG in R [45]. Cluster Profiler R package [46] was used to study the enrichment of DEGs in the KEGG pathway.
4.7. Quantitative reverse-transcription PCR
QRT-PCR was used to perform RNA-seq verification on 10 randomly selected DEGs. In qRT-PCR, cDNA derived from the reverse transcription of three biologically repetitive RNA of parents and hybrids. The extraction of total RNA was carried out according to the instructions of RNAprep Plant Kit (DP419; Tiangen, Beijing, China). For reverse transcription, Evo M-MLV RT Mix Kit with gDNA Clean [AG11728, Accurate Biotechnology (Hunan) Co., Ltd] was used, QuantStudio Q3 instrument was used for quantitative analysis, excel was used for mapping and data analysis. The primers of corresponding candidate DEGs are listed in Additional file 13. BrActin (Bra028615) was used as a quantitative reference [47]. The 2-△△Ct approach was applied to quantify relative gene expression levels [48].
5. Conclusions
Our study revealed the gene expression profiles and changes in 3D chromatin structure of B. rapa hybrids and parents. Several gene regulatory networks may play a crucial role in the heterosis of B. rapa, including those involved in “plant hormone signal transduction”, “Starch and sucrose metabolism”. The identification of 173 genes with higher expression in F1, as well as 485 genes with specific expression in F1, may contribute to the generation of heterosis traits. Additionally, we screened 140 heterosis candidate genes. Our results shed new light on the molecular mechanisms of B. rapa heterosis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Genome wide expression profile of the w30(P2)-/082(P1)-/F1 traid. FigureS2 The most enriched GO terms with up-regulated DEGs and down-regulated DEGs. Figure S3 Genome-wide Hi-C contact maps for 082, w30 and F1 at 1 Mb resolution. Figure S4 Ratio of inter/intra interactions in 082, w30 and F1.Figure S5 Gene density distribution under different genomic coordinates (bp) with the 1st (2nd, 3rd) principal component. With the y-axis 0 scale line as the reference, above is Compartment A and below is Compartment B. Figure S6 The percentage of the A/B compartments shift of SEGs and 296 DEGs. Figure S7 The distribution of TAD genome. Analysis of 10 pairs of intra-chromosome interaction of F1 and its parents samples. Figure S9 Indicated F1 and its parents’ venn diagrams of the TAD boundary; Figure S10 qRT-PCR verifies 10 DEGs between parents and F1. Additional file 1: Table S1 Output summary and quality control of RNA-Seq; Table S2 The summary of aligning results for P1(082), P2(w30) and F1; Additional file 2: Table S3 24320 genes were actively expressed in P1(082); Table S4 24524 genes were actively expressed in P2(w30); Table S5 24601 genes were actively expressed in the hybrid F1; Additional file 3:Table S6 FPKM values of all genes for P1(082), P2(w30)and F1; Additional file 4: Table S7 P1 vs P2 DEG; Table S8 P1 vs F1 DEG. Table S9 P2 vs F1 DEG; Additional file 5: Table S10 The expression levels of 296 DEGs were significantly different between F1 and its parents. Additional file 6:Table S11 102 DEGs shared by 082 vs W30, F1 vs 082, F1 vs W30. Additional file 7: Table S12 Description of homologous genes in Arabidopsis Thaliana. Additional file 8: Table S13 GO analysis of the DEGs between P1 (082) and F1; Table S14 GO analysis of the DEGs between P2(w30)and F1. Additional file 9: Table S15 KEGG analysis of DEGs between P1(082)and F1; Table S16 KEGG analysis of DEGs between P2(w30) and F1. Additional file 10: Table S17 Output summary and quality control of Hi-C sequencing; Table S18 Classification of filtered high quality HI-C data; Table S19 Cis and trans number statistics; Additional file 11: Table S20. The compartment type of genes in F1 and parents; Additional file 12:Table S21 Identification results of TAD; Additional file 13: Table S23 10 DEGs between parents and F1 were detected by qRT-PCR; Additional file 14: Table S24 140 DEGs uncovered from the integrative analysis of RNA-seq and Hi-C data.
Author Contributions
LE: SWL, CWZ, YLW, XLH, YL, TKL and DX designed this study. LE and SWL performed the experiments. LE, SWL and CWZ prepared the article. All authors read and approved the final manuscript.
Funding
This study was financially supported by the Natural Science Foundation of Jiangsu Province (BK20200560) and National key R&D plan (2022YFD1200502).
Institutional Review Board Statement
Not appliacable.
Informed Consent Statement
Not appliacable.
Data Availability Statement
The data sets supporting the results of the present study are included within this article (and its additional files). All sequencing data generated for this study have been submitted to the NCBI Sequence Read Archive under accession number (PRJNA970959).
Conflicts of Interest
Not appliacable.
Abbreviations
| DEGs | Differentially expressed genes |
| FPKM | Fragments per kilobase of transcript sequence per million base pairs |
| GO | Gene ontology |
| MPV | Mid-parent value |
| qRT-PCR | Quantitative real-time PCR |
| QTL | Quantitative trait locus |
References
- Birchler, J.A.; Yao, H.; Chudalayandi, S.; Vaiman, D.; Veitia, R.A. Heterosis. Plant Cell 2010, 22, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Hochholdinger, F.; Hoecker, N. Towards the molecular basis of heterosis. Trends Plant Sci 2007, 12, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Birchler, J.A.; Auger, D.L.; Riddle, N.C. In search of the molecular basis of heterosis. Plant Cell 2003, 15, 2236–2239. [Google Scholar] [CrossRef]
- Hochholdinger, F.; Baldauf, J.A. Heterosis in plants. Curr Biol 2018, 28, R1089–R1092. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, M.; Zhang, Q.; Wei, X.; Huang, X. Exploring the molecular basis of heterosis for plant breeding. J Integr Plant Biol 2020, 62, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.F. Dominance of Linked Factors as a Means of Accounting for Heterosis. PNAS 1917, 3, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Bruce, A.B. THE MENDELIAN THEORY OF HEREDITY AND THE AUGMENTATION OF VIGOR. Science (New York, N.Y.) 1910, 32, 627–628. [Google Scholar] [CrossRef] [PubMed]
- Davenport, C.B. DEGENERATION, ALBINISM AND INBREEDING. Science (New York, N.Y.) 1908, 28, 454–455. [Google Scholar] [CrossRef] [PubMed]
- Crow, J.F. 90 years ago: The beginning of hybrid maize. Genetics 1998, 148, 923–928. [Google Scholar] [CrossRef]
- East, E.M. Heterosis. Genetics 1936, 21, 375–397. [Google Scholar] [CrossRef]
- Williams, W. Heterosis and the genetics of complex characters. Nature 1959, 184, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Richey, F.D. MOCK-DOMINANCE AND HYBRID VIGOR. Science 1942, 96, 280–281. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, R.; Uezone, K.; Ishikura, S.; Osabe, K.; Peacoc, W.J.; Dennis, E.S. Recent research on the mechanism of heterosis is important for crop and vegetable breeding systems. Breeding Sci 2018, 68, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Genomic and epigenetic insights into the molecular bases of heterosis. Nat. Rev. Genet 2013, 14, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol Cell 2016, 62, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.V.; Scheid, O.M. Stress-induced structural changes in plant chromatin. Curr Opin Plant Biol 2015, 27, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.; De Lucia, F.; Mylne, J.S.; Zhu, D.; Ohmido, N.; Pendle, A.; Kato, N.; Shaw, P.; Dean, C. Physical clustering of FLC alleles during Polycomb-mediated epigenetic silencing in vernalization. Gene Dev 2013, 27, 1845–1850. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xiong, J.; Shalby, N.; Zhuo, C.; Jia, Y.; Yang, Q.-Y.; Tu, J. Comparison of dynamic 3D chromatin architecture uncovers heterosis for leaf size in Brassica napus. J. Adv. Res 2022, 42, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.; Cavalli, G. The Role of Chromosome Domains in Shaping the Functional Genome. Cell 2015, 160, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Lupianez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Hom, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. NAR 2022. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Wang, X.; Liu, B.; Wu, J.; Liang, J.; Cui, Y.; Cheng, F.; Wang, X. Brassica rapa Genome 2.0: A Reference Upgrade through Sequence Re-assembly and Gene Re-annotation. Mol Plant 2017, 10, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, H.; Muller, S.; Neusser, M.; von Hase, J.; Calcagno, E.; Cremer, M.; Solovei, I.; Cremer, C.; Cremer, T. Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. PNAS 2002, 99, 4424–4429. [Google Scholar] [CrossRef] [PubMed]
- Ghavi-Helm, Y.; Jankowski, A.; Meiers, S.; Viales, R.R.; Korbel, J.O.; Furlong, E.E.M. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat Genet 2019, 51, 1272. [Google Scholar] [CrossRef] [PubMed]
- Grob, S.; Schmid, M.W.; Grossniklaus, U. Hi-C Analysis in Arabidopsis Identifies the KNOT, a Structure with Similarities to the flamenco Locus of Drosophila. Mol Cell 2014, 55, 678–693. [Google Scholar] [CrossRef]
- Gibcus, J.H.; Dekker, J. The Hierarchy of the 3D Genome. Molecular Cell 2013, 49, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome(vol 17, 661, 2016). Nat Rev Genet 2016, 17. [Google Scholar]
- Song, G.S.; Zhai, H.-L.; Peng, Y.G.; Zhang, L.; Wei, G.; Chen, X.-Y.; Xiao, Y.-G.; Wang, L.; Chen, Y.J.; Wu, B.; et al. Comparative Transcriptional Profiling and Preliminary Study on Heterosis Mechanism of Super-Hybrid Rice. Mol Plant 2010, 3, 1012–1025. [Google Scholar] [CrossRef]
- Wei, G.; Tao, Y.; Liu, G.; Chen, C.; Luo, R.; Xia, H.; Gan, Q.; Zeng, H.; Lu, Z.; Han, Y.; et al. A transcriptomic analysis of superhybrid rice LYP9 and its parents. PNAS 2009, 106, 7695–7701. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, H.; Diao, X.; Liu, Z.; Li, K.; Wu, Y.; Liang, Q.; Wang, H.; Huang, C. Transcriptome profiling and comparison of maize ear heterosis during the spikelet and floret differentiation stages. Bmc Genomics 2016, 17. [Google Scholar] [CrossRef]
- Gaertner, T.; Steinfath, M.; Andorf, S.; Lisec, J.; Meyer, R.C.; Altmann, T.; Willmitzer, L.; Selbig, J. Improved Heterosis Prediction by Combining Information on DNA- and Metabolic Markers. PLoS ONE 2009, 4. [Google Scholar] [CrossRef]
- Lisec, J.; Steinfath, M.; Meyer, R.C.; Selbig, J.; Melchinger, A.E.; Willmitzer, L.; Altmann, T. Identification of heterotic metabolite QTL in Arabidopsis thaliana RIL and IL populations. Plant J 2009, 59, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, B.; Guo, W.; Qin, Y.; Wang, L.; Zhang, Y.; Zhang, T. Quantitative trait loci mapping for yield and its components by using two immortalized populations of a heterotic hybrid in Gossypium hirsutum L. Mol Breeding 2012, 29, 297–311. [Google Scholar] [CrossRef]
- Zhang, H.Y.; He, H.; Chen, L.-B.; Li, L.; Liang, M.-Z.; Wang, X.-F.; Liu, X.-G.; He, G.-M.; Chen, R.-S.; Ma, L.-G.; et al. A genome-wide transcription analysis reveals a close correlation of promoter INDEL polymorphism and heterotic gene expression in rice hybrids. Mol Plant 2008, 1, 720–731. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, X.; Du, Z.; Wei, L.; Fang, H.; Dong, Q.; Niu, J.; Li, Y.; Gao, J.; Zhang, M.Q.; et al. The loss of heterochromatin is associated with multiscale three-dimensional genome reorganization and aberrant transcription during cellular senescence. Genome Res 2021, 31, 1121. [Google Scholar] [CrossRef]
- Kong, S.; Zhang, Y. Deciphering Hi-C: From 3D genome to function. Cell Biol Toxico 2019, 35, 15–32. [Google Scholar] [CrossRef] [PubMed]
- van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A method to study the three-dimensional architecture of genomes. JoVE 2010. [Google Scholar] [CrossRef]
- Wingett, S.; Ewels, P.; Furlan-Magaril, M.; Nagano, T.; Schoenfelder, S.; Fraser, P.; Andrews, S. HiCUP: Pipeline for mapping and processing Hi-C data. F1000Research 2015, 4, 1310–1310. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012, 9, 357–U354. [Google Scholar] [CrossRef] [PubMed]
- Szalaj, P.; Tang, Z.; Michalski, P.; Pietal, M.J.; Luo, O.J.; Sadowski, M.; Li, X.; Radew, K.; Ruan, Y.; Plewczynski, D. An integrated 3-Dimensional Genome Modeling Engine for data-driven simulation of spatial genome organization. Genome Res 2016, 26, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat Methods 2015, 12, 357–U121. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Dheda, K.; Huggett, J.F.; Bustin, S.A.; Johnson, M.A.; Rook, G.; Zumla, A. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 2004, 37, 112. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C-T method. Nat Protoc 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
Figure 1.
Comparisons of heterosis in w30(P2)-/082(P1)-/F1 traid. (a). Phenotypes of the hybrid F1 and its parents in Brassica rapa. (b). W30(P2)-/082(P1)-/F1 traid at 21 DAS. (c). Indicated the total fresh weight of the w30(P2)-/082(P1)-/F1 traid. (d). Indicated the total dry weight of the w30(P2)-/082(P1)-/F1 traid. MPH indicated the mid-parent heterosis in the w30(P2)-/082(P1)-/F1 traid, while HPH indicated over-parent heterosis. Values with different letters differ significantly (p<0.05).
Figure 1.
Comparisons of heterosis in w30(P2)-/082(P1)-/F1 traid. (a). Phenotypes of the hybrid F1 and its parents in Brassica rapa. (b). W30(P2)-/082(P1)-/F1 traid at 21 DAS. (c). Indicated the total fresh weight of the w30(P2)-/082(P1)-/F1 traid. (d). Indicated the total dry weight of the w30(P2)-/082(P1)-/F1 traid. MPH indicated the mid-parent heterosis in the w30(P2)-/082(P1)-/F1 traid, while HPH indicated over-parent heterosis. Values with different letters differ significantly (p<0.05).
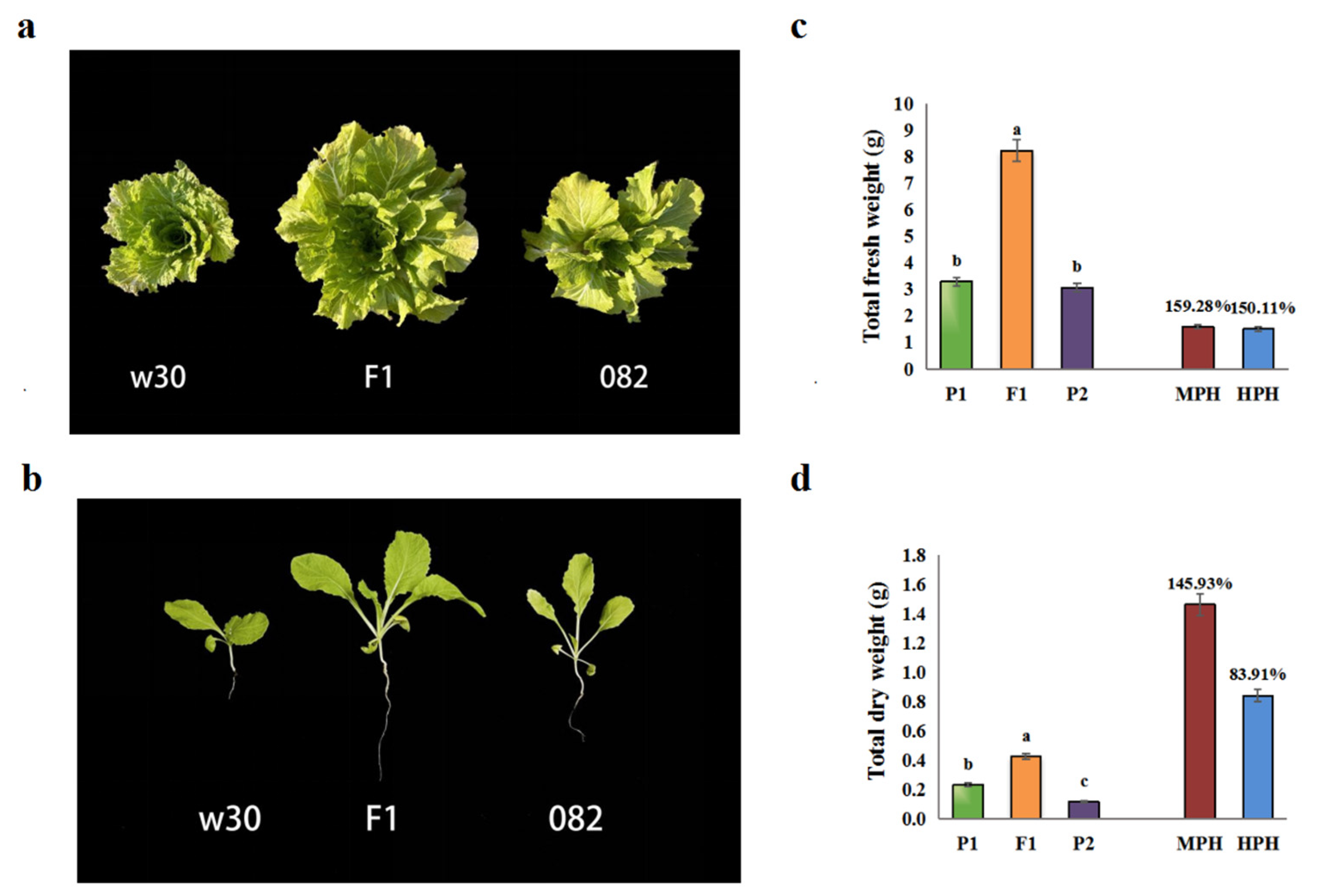
Figure 2.
Comparative analysis of the actively expressed genes detected in w30(P2)-/082(P1)-/F1 traid. (a). Number of actively expressed genes in F1 and its parents. (b). Genome wide expression profile of w30(P2)-/082(P1)-/F1 traid. (c).(d).(e).(f). Indicated venn diagrams of the actively expressed genes drew by comparative analysis. (g). Indicated the correlation of nine samples calculated by the expression profiles of all detected genes.
Figure 2.
Comparative analysis of the actively expressed genes detected in w30(P2)-/082(P1)-/F1 traid. (a). Number of actively expressed genes in F1 and its parents. (b). Genome wide expression profile of w30(P2)-/082(P1)-/F1 traid. (c).(d).(e).(f). Indicated venn diagrams of the actively expressed genes drew by comparative analysis. (g). Indicated the correlation of nine samples calculated by the expression profiles of all detected genes.
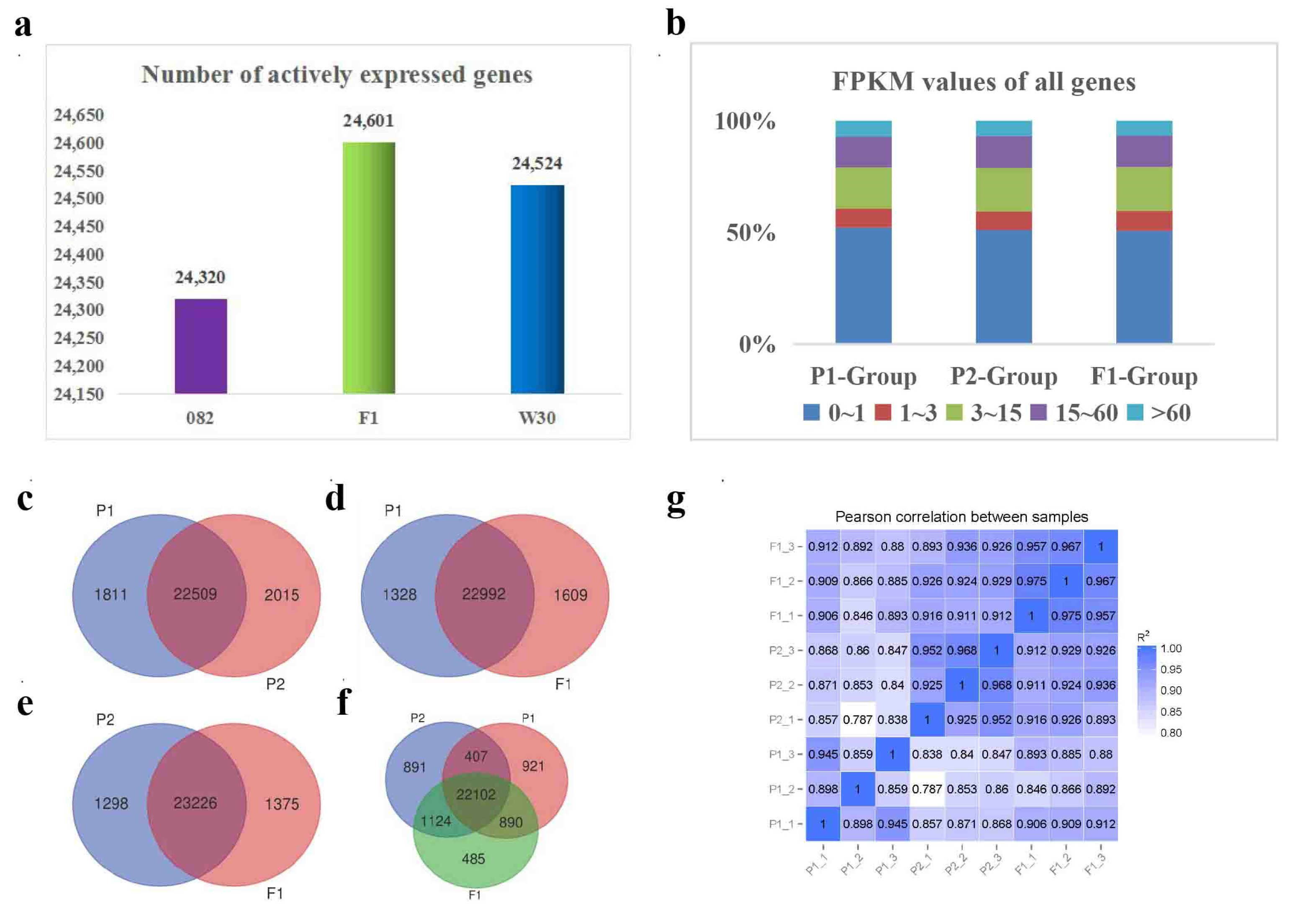
Figure 3.
Overview of the DEGs of the hybrid F1 and its parents. (a). Cluster analysis of differentially expressed genes for w30(P2)-/082(P1)-/F1 traid. The heat maps were constructed based on the log2 (fold-change of the normalized expression levels) of two arbitrary samples in w30(P2)-/082(P1)-/F1 traid. The color scale represents the log2 (fold-change of the normalized expression levels) of two arbitrary samples with blue denoting low expression and red denoting high expression. (b). Volcano map of DEGs. Red points denote up-regulated genes; green points denote down-regulated genes; blue points denote non-differentiated genes. Each group is referenced by the one after 'vs', for example, P1 vs P2 is referenced by P2. (c). Indicated Venn diagrams of the differentially expressed genes drew by comparative analysis of A, B and C group. Group A represents P1 vs P2, group B represents P1 vs F1, group C represents P1 vs P2.
Figure 3.
Overview of the DEGs of the hybrid F1 and its parents. (a). Cluster analysis of differentially expressed genes for w30(P2)-/082(P1)-/F1 traid. The heat maps were constructed based on the log2 (fold-change of the normalized expression levels) of two arbitrary samples in w30(P2)-/082(P1)-/F1 traid. The color scale represents the log2 (fold-change of the normalized expression levels) of two arbitrary samples with blue denoting low expression and red denoting high expression. (b). Volcano map of DEGs. Red points denote up-regulated genes; green points denote down-regulated genes; blue points denote non-differentiated genes. Each group is referenced by the one after 'vs', for example, P1 vs P2 is referenced by P2. (c). Indicated Venn diagrams of the differentially expressed genes drew by comparative analysis of A, B and C group. Group A represents P1 vs P2, group B represents P1 vs F1, group C represents P1 vs P2.
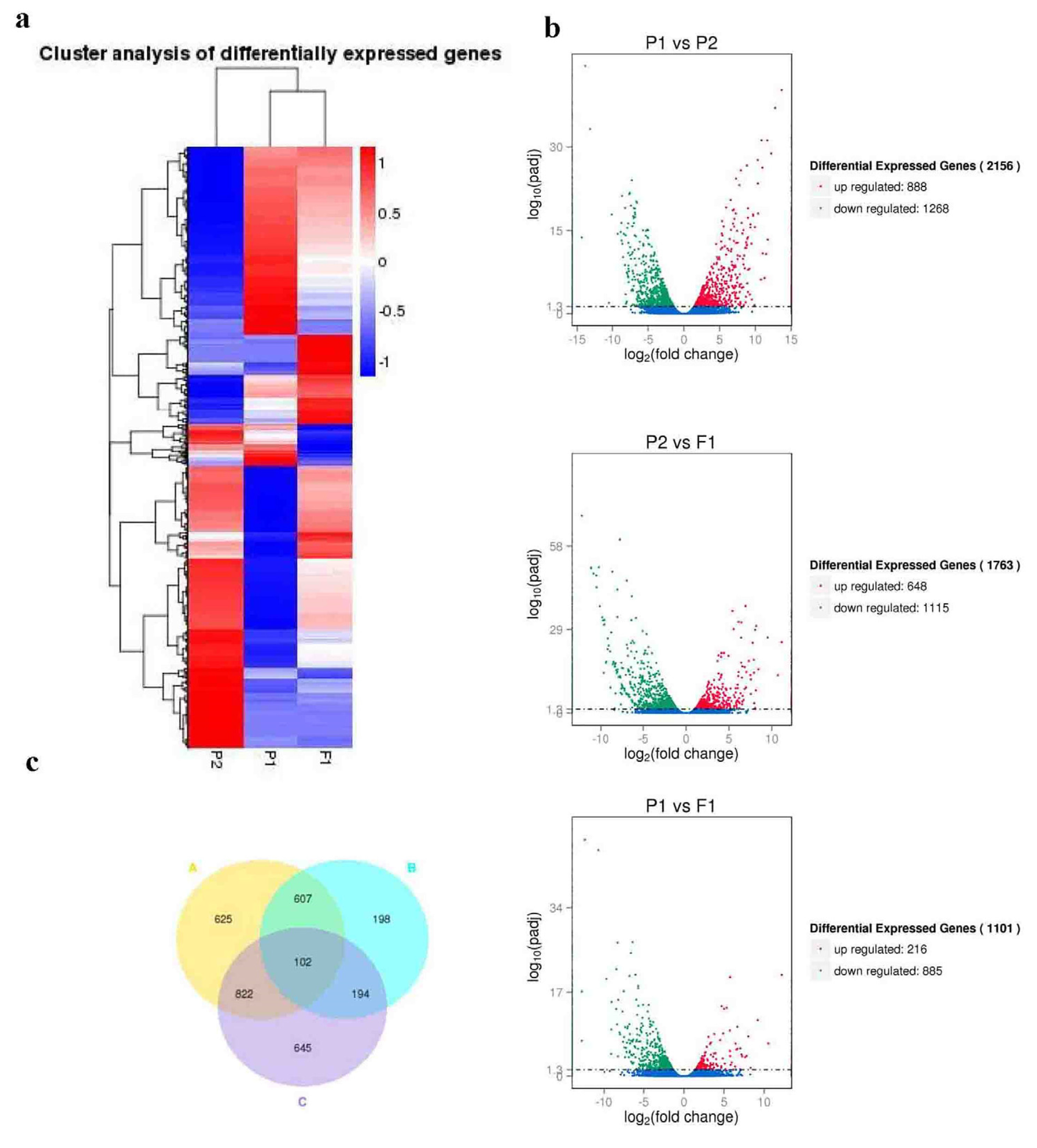
Figure 4.
Analysis of diferentially expressed genes (DEGs) between F1 and its parents. (a). The most enriched GO terms with all DEGs. The ordinate is the enriched GO term, the abscissa is the number of DEGs in the term. (b). The top 20 enriched KEGG pathways of DEGs. The pathway label is on the vertical axis, the Rich factor is on the horizontal axis, the size of the dots represents the number of DEGs in the pathway, and the color of the dots represents the distinct Q-value levels. Each group is referenced by the one after 'vs', for example, P1 vs F1 is referenced by F1. Kyoto Encyclopedia of Genes and Genomes (KEGG) databases (www.kegg.jp/kegg/kegg1.html.) were used .
Figure 4.
Analysis of diferentially expressed genes (DEGs) between F1 and its parents. (a). The most enriched GO terms with all DEGs. The ordinate is the enriched GO term, the abscissa is the number of DEGs in the term. (b). The top 20 enriched KEGG pathways of DEGs. The pathway label is on the vertical axis, the Rich factor is on the horizontal axis, the size of the dots represents the number of DEGs in the pathway, and the color of the dots represents the distinct Q-value levels. Each group is referenced by the one after 'vs', for example, P1 vs F1 is referenced by F1. Kyoto Encyclopedia of Genes and Genomes (KEGG) databases (www.kegg.jp/kegg/kegg1.html.) were used .
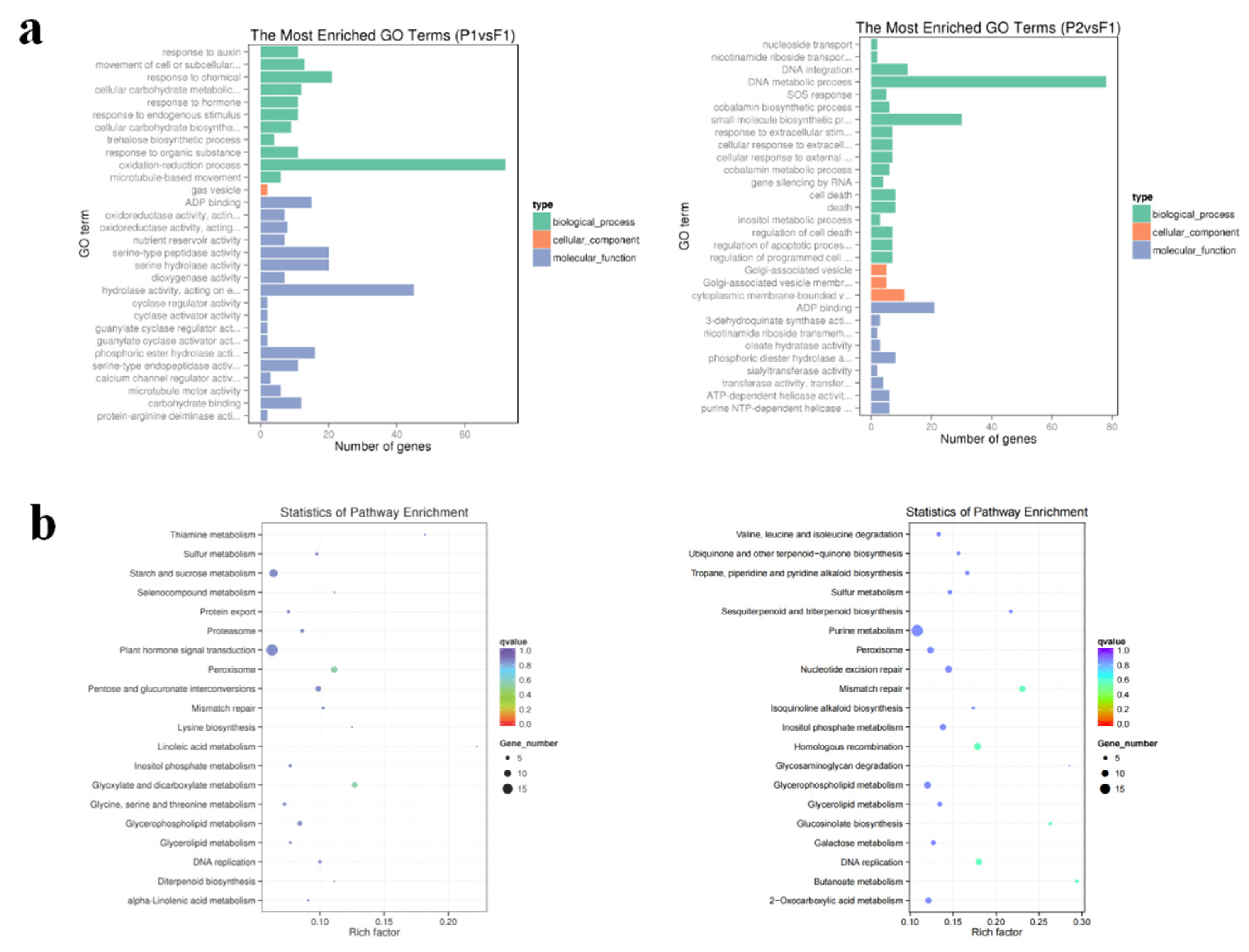
Figure 5.
Hi-C analysis of chromatin contacts in F1 and parents. (a). 3D model of whole chromosomes. From left to right: 082, F1, and w30. Each chromosome is represented by a different hue. (b). Intrachromosomal interactions of chromosome at 100 Kb resolution. (c). Each triangle distributed diagonally is represented as a topologically associated domain (TAD). (d).First principal component values showing A/B compartment status. (e). Distribution of TADs.
Figure 5.
Hi-C analysis of chromatin contacts in F1 and parents. (a). 3D model of whole chromosomes. From left to right: 082, F1, and w30. Each chromosome is represented by a different hue. (b). Intrachromosomal interactions of chromosome at 100 Kb resolution. (c). Each triangle distributed diagonally is represented as a topologically associated domain (TAD). (d).First principal component values showing A/B compartment status. (e). Distribution of TADs.
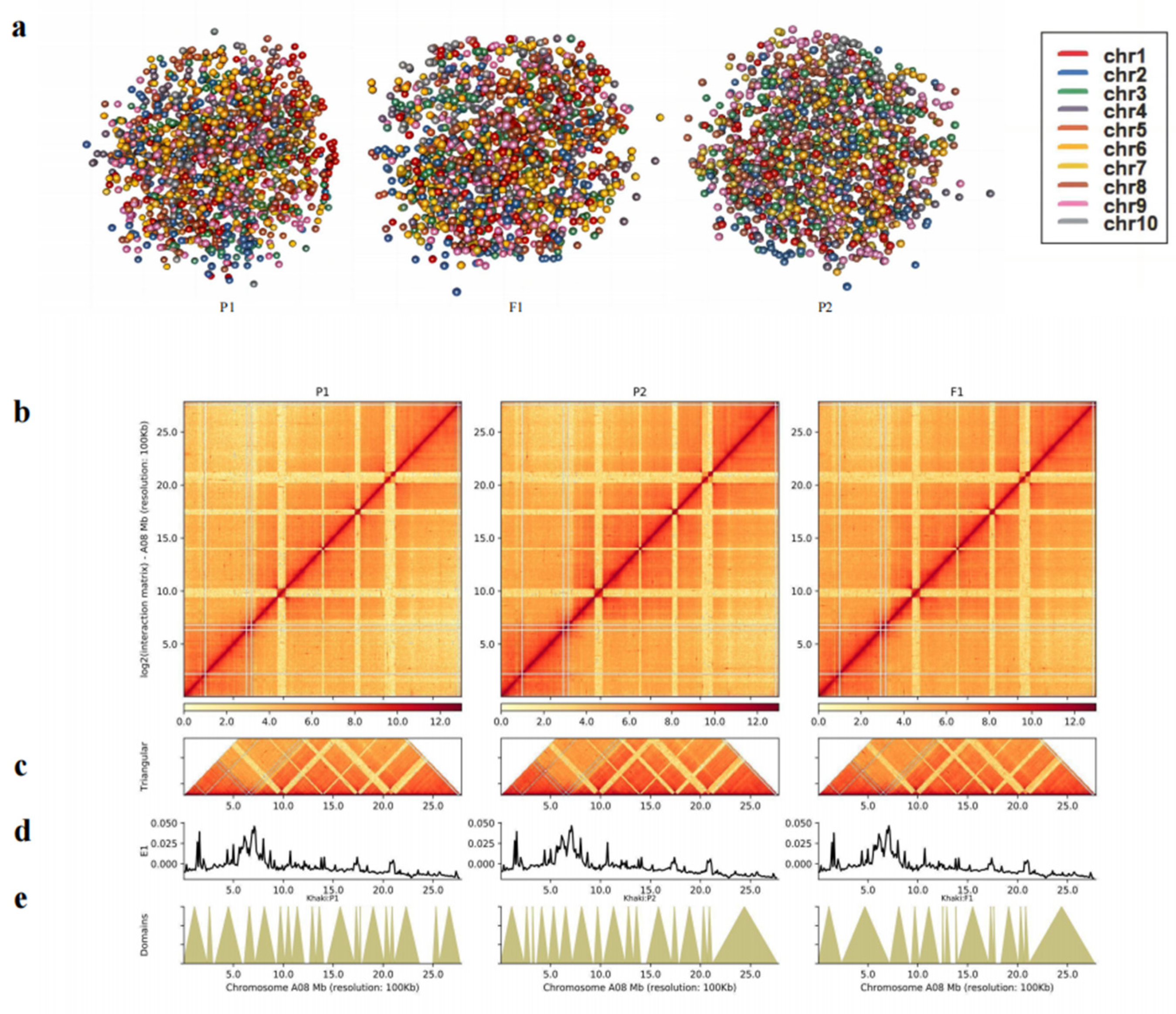
Figure 6.
The analysis of the A/B compartments in F1 and parents. (a). The distributions of the A/B compartment on each chromosome. With the y-axis 0 scale line as the reference, above is Compartment A and below is Compartment B. (b).The number of genes contained in the A and B compartments. (c). Box plots representing the gene expression on the A and B compartments. (d). The percentage of the A/B compartment shifts between F1 and its parents.
Figure 6.
The analysis of the A/B compartments in F1 and parents. (a). The distributions of the A/B compartment on each chromosome. With the y-axis 0 scale line as the reference, above is Compartment A and below is Compartment B. (b).The number of genes contained in the A and B compartments. (c). Box plots representing the gene expression on the A and B compartments. (d). The percentage of the A/B compartment shifts between F1 and its parents.

Table 1.
Differentially expressed genes detected in the hybrid F1 and their parents.
| Hybrid set samples |
Up | Down | B2P | Total |
|---|---|---|---|---|
| 082 vs w30 | 888 (41.19%) | 1268(58.81%) | 2156 | |
| F1 vs 082 | 885(80.38%) | 216(19.62%) | 1101 | |
| F1 vs w30 | 1115(63.24%) | 648(36.76%) | 1763 | |
| F1 vs (082 and w30) | 173 (58.45%) | 32(10.81%) | 91(30.74%) | 296 |
Note: DGPP indicated the differentially expressed genes (DEGs) between two parents. DGHP indicated the DEGs between the hybrid and their parents. B2P indicated the expression levels of the DEGs between the two parental lines. Each group is referenced by the one after 'vs', for example, 082 vs w30 is referenced by w30.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



