
Submitted:
18 December 2023
Posted:
19 December 2023
You are already at the latest version
Abstract
Genetic diversity is the foundation of evolutionary resilience, adaptive potential, and the flourishing vitality of living organisms, serving as the cornerstone for robust ecosystems and the continuous evolution of life on Earth. The landscape of directed evolution, a powerful biotechnological tool inspired by natural evolutionary processes, has undergone a transformative shift propelled by innovative strategies for generating genetic diversity. This shift is fuelled by several factors, encompassing the utilization of advanced toolkits like CRISPR-Cas and base editors, enhanced comprehension of biological mechanisms, cost-effective custom oligo pool synthesis, and the seamless integration of artificial intelligence and automation. This comprehensive review looks into the myriad methodologies employed for constructing gene libraries, both in vitro and in vivo, categorized into three major classes: random mutagenesis, focused mutagenesis, and DNA recombination. The objectives of this review are threefold: firstly, to present a panoramic overview of recent advances in genetic diversity creation, secondly, to inspire novel ideas for further innovation in the genetic diversity generation, and thirdly, to provide a valuable resource for individuals entering the field of directed evolution.
Keywords:
Protein engineering
; directed evolution
; genetic diversity
; random mutagenesis
; molecular cloning
1. Introduction
The year 2018 marked a significant milestone in the field of protein engineering, as it witnessed the well-deserved recognition of Prof. Frances H. Arnold with the Nobel Prize in Chemistry [1,2]. This prestigious accolade honoured her ground-breaking contributions to directed protein evolution, particularly through the utilization of a simple yet powerful algorithm. Directed evolution involves iterative cycles of genetic diversity creation, followed by screening and selection (Figure 1). The far-reaching applications and profound impacts of directed evolution are evident in various domains, including but not limited to, the development of highly active or stable enzymes, the creation of novel enzymatic functions and chemistries, and the engineering of biopharmaceutical proteins.
At the core of protein evolution lies the essential prerequisite of genetic diversity. Indeed, without this diversity, the evolutionary process becomes untenable. In recognition of this pivotal aspect, this review article is dedicated to examining methodologies deliberately designed for the creation of genetic diversity, with a focus on publications from 2014 onward. This article serves as an extension to our prior reviews on the same subject in 2006 [3] and 2013 [4].
The methodologies for generating genetic diversity can be categorized into three main classes: random mutagenesis, focused mutagenesis, and DNA recombination (Figure 2). There exist hybrid methods that integrate features derived from two or more of these classes, showcasing the versatility and adaptability of current approaches. In random mutagenesis, mutations are randomly introduced into the target gene of interest (GOI). Conversely, focused mutagenesis involves the randomization or semi-randomization of selected amino acid residues within the target gene. In DNA recombination, a collection of homologous or non-homologous genes undergo random fragmentation and subsequent reassembly to form chimeric constructs. The fundamentals of directed evolution and genetic diversity creation have been comprehensively documented in our recently published protein engineering textbook [5]. Therefore, this review primarily explores the significant advancements achieved in the past decade.
This review commences by discussing the latest advancements in molecular cloning, a crucial initial step in the process of generating genetic diversity. Subsequent to this, we delve into the assessment of gene library quality. Building upon this foundation, we offer succinct summaries of innovative methodologies within each category of genetic diversity creation, emphasizing their individual merits and limitations.
2. Molecular cloning
DNA cloning serves as the cornerstone for mutagenesis experiments, involving the insertion of a target GOI or a gene library into a cloning/expression vector. Molecular cloning also plays an integral role in all synthetic biology projects at various stages. Conventionally, this process relies on the use of restriction endonucleases, which, while fundamental in molecular biology, present certain limitations. Drawbacks such as sequence specificity, time-consuming restriction digestion and ligation reactions, and the creation of undesirable 'scars' underscore the necessity for more versatile DNA cloning techniques. To address contemporary demands and challenges and facilitate rapid advancements, DNA assembly methods must be robust, cost-effective, swift, user-friendly, and highly scalable.
In this section, we present a comprehensive overview of the latest advancements in DNA cloning technology over the past decade. These innovations are categorized into in vivo, in vitro, modular DNA assembly, and automated DNA assembly methods.
2.1. In vivo cloning
In vivo cloning techniques (Table 1) leverage the cellular machinery of a host organism, such as bacteria or yeast, to incorporate a DNA fragment into a vector. Recent descriptions of these techniques often rely on DNA recombination or repair systems, such as homologous recombination (HR) and non-homologous end joining (NHEJ). The appeal of in vivo cloning techniques lies in their distinct advantages: they obviate the need for in vitro ligation reactions and are not constrained by restriction endonuclease sites. Introducing a short homologous sequence (30 – 40 bp) to any vector or DNA fragment through PCR facilitates recombinational cloning, underscoring the simplicity and versatility of this approach.
An underutilized yet progressively popular in vivo technique involves harnessing the RecA-independent recombination (RAIR) machinery [6] found in commonly employed laboratory bacterial strains, such as Escherichia coli DH5α. RAIR is thought to be dependent on XthA, a 3'-to-5' exonuclease that resects the 3'-ends of linear DNA fragments introduced into E. coli cells, exposing the single-stranded 5'-overhangs [7]. Subsequently, the complementary single-stranded DNA ends hybridize with each other, and gaps are filled by DNA polymerase I and ligase LigA. Despite being originally described over three decades ago [8], the application of high-fidelity polymerases and low-cost oligonucleotides, coupled with insights into its molecular mechanisms [6,7,9,10,11,12,13], holds the promise of expanding its widespread adoption.
Traditionally, the budding yeast Saccharomyces cerevisiae has served as a platform for HR-based cloning [14,15,16], employing a shuttle vector to propagate plasmids in both yeast and E. coli. However, this approach limited the utility of yeast-based cloning exclusively to these two organisms. Recent breakthroughs in yeast recombination cloning [17,18,19,20,21] have transcended these limitations, allowing the technique to be applied universally for cloning multiple fragments of interest into any vector. Key innovation involves incorporating an origin of replication and a selection marker for yeast as a cassette or DNA fragment, which is co-inserted with other fragments for cloning. Assembled plasmids can then be selected in and purified from yeast, ready for transformation or transfection into desired host organisms. HR-based cloning in yeast can also be adapted for high-throughput DNA assembly and is amenable to automation [22].
While S. cerevisiae exhibits efficient HR machinery, its NHEJ activity is limited. By leveraging previous findings that the thermotolerant yeast Kluveromyces marxianus possesses a highly efficient NHEJ pathway [23], an NHEJ-mediated cloning method was developed [24]. This method employs a functional marker selection system (e.g., ura3) for the cloning of DNA fragments in K. marxianus. NHEJ-mediated cloning exploits the sequence-independent nature of NHEJ-based joining, allowing the cloning of DNA fragments without the need for homologous ends – a departure from the HR-based cloning approach.
CReasPy-Cloning harnesses the precision of Cas9 to cleave DNA at a user-specified locus, synergizing with the yeast's remarkably efficient homologous recombination [25]. This innovative approach enables the simultaneous cloning and engineering a bacterial genome in yeast. The successful application of CReasPy-Cloning was demonstrated through the cloning and engineering of the 0.816 Mbp genome of Mycoplasma pneumonia, showcasing its potential for bacterial genome manipulation.
Most in vivo cloning methods utilize bacteria or yeasts as hosts, necessitating transformation and plasmid extraction procedures for the introduction of extracted plasmids into other organisms. A recent advancement in this domain is the Phage Enzyme-Assisted In Vivo DNA Assembly (PEDA) method [26]. Simultaneous expression of phage-derived T5 DNA exonuclease and T4 DNA ligase facilitates in vivo DNA assembly in a diverse range of microorganisms such as Cupriavidus necator, Pseudomonas putida, Lactobacillus plantarum, and Yarrowia lipolytica. Another cutting-edge technique, the Yeast Life Cycle (YLC) assembly method [27], capitalizes on CRISPR-Cas9 and yeast meiosis to iteratively assemble large DNA fragments. The advantage of the YLC method lies in bypassing challenging in vitro steps associated with handling and importing large DNA fragments into a host system.
2.2. In vitro cloning
Gibson assembly, Seamless Ligation Cloning Extract (SLiCE), Sequence and Ligation Independent Cloning (SLIC), and In-Fusion cloning are all in vitro techniques. Their underlying principle relies on the presence of homology regions at the ends of vector and insert DNA fragments. These methods have gained widespread popularity due to their ability to overcome limitations associated with restriction endonuclease-based cloning. Recent advancements have integrated the flexibility of Gibson assembly with the precision of CRISPR-Cas9, allowing for the targeting of double-strand breaks at any desired location. This is particularly advantageous in situations where PCR amplification of vector fragments is challenging, and restriction endonuclease sites are unavailable. Guide RNAs guide the Cas9 endonuclease to target any double-stranded DNA sequence, effectively linearizing the vector. The linearized vector can then be employed in Gibson assembly or other cloning techniques [28,29]. CRISPR-Cas9 has also been utilized to excise large DNA fragments (~100kb) from bacterial chromosomes, a process that may be difficult to achieve through PCR amplification. The cut fragments are subsequently cloned using Gibson assembly [30]. Variations of the SLiCE technique, such as Zero-Background Redα (ZeBRα), leverage the ccdB gene to eliminate any background from uncut or re-ligated vectors during cloning [31].
Other recent advances, such as T5 Exonuclease-Dependent Assembly (TEDA), Single 3′-exonuclease-based multifragment DNA assembly (SENAX), and T5 exonuclease-mediated low-temperature DNA cloning (TLTC) [32,33,34], operate on the principle of exonuclease-generated overhangs within the homologous regions of vectors and DNA fragments. These methods offer a cost-effective alternative to Gibson assembly, as they only require the exonuclease enzyme for the in vitro reaction to create the necessary homologous overhangs. The subsequent steps of gap repair and DNA fragment ligation take place in vivo post-transformation into bacterial cells.
Alternatives to generating compatible overhangs are techniques such as Uracil-Specific Excision Reagent (USER) Cloning. In USER Cloning, vector and insert fragments with short overlapping homology regions are produced through PCR. The primers used in this process introduce a single deoxyuracil (dU) residue, subsequently cleaved by the USER enzyme. This cleavage results in 3' overhangs, facilitating the annealing of multiple DNA fragments. Although USER cloning has a longstanding history [35], advancements in the past decade, such as optimizing the melting temperature of annealing DNA fragments [36] and employing in silico design tools like AMUSER [37], have enhanced its efficacy. Similar techniques that introduce compatible overhangs through PCR include QuickStep-Cloning [38], where two parallel asymmetrical PCR reactions generate overhangs. An improvement on this method is PTO-QuickStep cloning [39,40], where phosphorothioate (PTO) bonds, introduced via primers, are processed by iodine cleavage to generate overhangs. Nicks in the plasmids resulting from both USER cloning and QuickStep-Cloning are sealed following transformation into bacterial cells. For scarless and sequence-independent DNA assembly, the method using thermostable exonuclease and ligase (DATEL) [41,42] presents another alternative. This technique employs a combination of Taq and Pfu DNA polymerases to cleave single-stranded DNA flaps generated during the annealing of DNA fragments with overlaps. The resulting nicks are then ligated using a heat-stable DNA ligase.
In summary, the fundamental principle underlying the latest advancements in in vitro DNA assembly (Table 2) is to streamline the process, ensuring simplicity, cost-effectiveness, and versatility. Leveraging overlapping DNA sequences among fragments to be assembled allows for the insertion of any DNA fragment into any chosen vector without being confined by the limitations of restriction endonuclease sites. While each discussed technique presents its unique advantages and limitations, the optimal choice for a particular DNA assembly project will depend on specific requirements and contextual factors.
2.3. Modular DNA assembly
The in vivo and in vitro cloning techniques outlined above present numerous advantages compared to traditional methods relying on restriction endonucleases. However, they often involve custom-designed primers and constructs tailored for specific experiments or purposes. A paradigm shift occurred with modular DNA assembly in synthetic biology. This approach revolves around using standardized DNA 'parts' to construct complex assemblies, which, in turn, can serve as 'parts' for even more intricate constructions. Once these components are generated, sequence-verified, and validated, they become reusable and shareable among researchers. This fosters standardization, ultimately leading to significant time and cost savings in the field.
Initially, modular DNA assembly relied on type IIP restriction enzymes, leading to the development of the BioBrick system for standardization. More recent techniques have shifted to the Golden Gate cloning technology, leveraging type IIS restriction enzymes. Unlike type IIP enzymes, which recognize and cut within palindromic DNA sequences, type IIS enzymes identify non-palindromic sequences and cleave DNA outside of the recognition site. This innovation has emerged as a potent tool for seamless cloning, allowing user-defined DNA sequences to serve as cutting sites and facilitating the precise assembly of DNA fragments in a predetermined manner.
Golden Gate assembly has been embraced in the creation of two extensively utilized hierarchical modular DNA cloning methods: MoClo and Golden Braid. These methodologies employ DNA 'parts' organized in different hierarchical levels, each level being more intricate than the one preceding it. Comprehensive reviews [43,44] provide invaluable resources for users seeking to initiate projects with these techniques. While these methods excel in seamlessly cloning multiple DNA fragments, their application has been constrained by limited vector choices – specifically, the requirement for destination vectors designed to contain one (or more) appropriately oriented pairs of recognition sites for a type IIS restriction enzyme. Recent advancements [45] have addressed this limitation by introducing type IIS restriction sites that are compatible with type IIP restriction sites, significantly broadening the range of vectors compatible with modular DNA assembly. A summary of other recently developed modular assembly techniques is provided in Table 3.
2.4. Automated DNA assembly
Amidst numerous breakthroughs, advancements, and refinements, a diverse array of in vivo and in vitro DNA cloning methods now caters to a spectrum of scenarios – from straightforward insertion of a single DNA fragment into a vector to intricate assemblies involving large and/or multiple DNA fragments. As experimental designs grow increasingly complex, the norm is now the generation of hundreds, if not thousands, of library sequences. Consequently, there arises a demand for high-throughput methods capable of assembling a large number of DNA sequences to generate libraries.
The integration of automation into these processes (Table 4) offers several advantages, including the elimination of manual handling errors, enhanced reliability and reproducibility, and significant time and cost savings. This shift toward high-throughput methods represents a pivotal stride in accommodating the evolving demands of sophisticated experimental designs with large-scale library creation. Liquid handling robots like Opentrons play a pivotal role in enabling this shift, offering benefits such as cost-effectiveness in hardware, open-source software accessibility, and user-friendly operation. These robotic systems prove invaluable for automating a diverse array of workflows, ranging from nucleic acid extraction to PCR and various DNA assembly techniques.
3. Genetic diversity creation
The primary objective of creating genetic diversity is to generate various protein variants, with the hope that a subset of these variants, albeit often a small fraction, will exhibit favourable phenotypes compared to their parent or wildtype protein. The introduction of mutations into the GOI is a common approach in mutagenesis to achieve this diversity. The outcome of protein variants is influenced by several factors. Firstly, considering that there are 61 codons encoding 20 amino acids, altering the codon usage or the GC content of the DNA sequence can impact the mutagenesis outcome. This effect becomes particularly prominent when the mutational spectrum of the employed mutagenesis method is highly biased. Secondly, the organization of the genetic code imposes constraints on the mutagenesis outcome.
While protein engineers predominantly work with DNA, it's worth noting that RNA mutagenesis methods also exist. Fukuda et al. developed an RNA-mutagenesis method that leverages the intracellular RNA-editing mechanism [55]. In this approach, guide RNAs guide the editing enzyme, human adenosine deaminase acting on RNA (ADAR), inducing adenosine-to-inosine (A-to-I) mutations on RNA molecules. Other RNA-based mutagenesis strategies involve using Qβ replicase to generate complex mRNA libraries [56]. However, compared to DNA, RNA is a transient molecule, making it more challenging to track genotype-phenotype linkage. The conversion of RNA to cDNA is typically required to identify mutations introduced onto RNA. In the subsequent sections, our focus will primarily be on DNA mutagenesis methods.
3.1. Quality of a gene library
It is strongly advisable to assess the quality of a gene library, especially when employing a mutagenesis method for the first time on the target gene sequence. This evaluation is commonly achieved by sequencing a small subset, typically ranging from 10 to 20 randomly selected ‘variants’. If the gene library's quality is found to be suboptimal, proceeding to the expression of the protein library and subsequent selection/screening may not be prudent. The probability of identifying enhanced protein variants would be too minimal to justify the resources invested in such an undertaking.
Several key indicators are commonly employed to evaluate the quality of a gene library. Taking random DNA mutagenesis as an example, a high-quality gene library should exhibit the following characteristics:
- Mutations are precisely targeted to the GOI (i.e., no off-target mutations).
- Mutations are uniformly distributed along the entire GOI.
- All bases (A/T/G/C) experience mutations at the same frequency and are substituted with their three counterparts equally.
- The mutation frequency (number of errors per 1 kb of DNA) is not excessively high, preventing the predominance of non-functional protein variants.
- Duplicated sequences are avoided/eliminated.
- Wildtype sequences are absent in the gene library (i.e., no template carry-over).
Achieving an absolutely ideal gene library is practically elusive unless it exclusively comprises synthetic genes. The extent of deviation from this ideal serves as a practical measure of the gene library's quality. The Schwaneberg group conducted a comprehensive large-scale comparison among random mutagenesis libraries created using three approaches: error-prone polymerase chain reaction (epPCR) with low mutagenic conditions, epPCR with high mutagenic conditions, and Sequence Saturation Mutagenesis (SeSaM) [57,58,59]. After sequencing 1000 mutations for each library, the library quality was evaluated on both the DNA and protein levels [60,61]. The protein-level assessment primarily employed the Mutagenesis Assistant Program (MAP) [62,63,64]. The SeSaM library exhibits a preference for transversion mutations, contrasting with epPCR libraries that display a transition bias. Additionally, the SeSaM library demonstrates a significantly higher number of consecutive nucleotide substitutions. These characteristics result in a greater number and more diverse amino acid substitutions in the SeSaM library.
In directed evolution, preserving genotype-phenotype linkage is crucial for tracing an improved phenotype back to its genetic origins. During the library transformation process, transformants incorporating more than one plasmid could account for over 20% of the constructed library, thereby compromising the library quality. To mitigate the occurrence of multiple-plasmid transformants, it is possible to reduce their frequency by optimizing the amount of plasmid DNA used for transformation [65].
3.2. Random mutagenesis
Motivated by the technical simplicity of epPCR, several modified protocols have been developed. One adaptation is a modified epPCR protocol specifically optimized for small amplicons [66]. Another variation, known as Casting epPCR (cepPCR), directs random mutations to a specific region within the target GOI [66]. Furthermore, efforts are underway to extend epPCR methods beyond the conventional microbial hosts E. coli and S. cerevisiae, with attempts to optimize them for other microbial hosts. For example, in the transformation of Bacillus subtilis into a host for directed evolution, the epPCR product was fused with flanking regions and an antibiotic-resistant marker. This composite PCR product was then integrated into the chromosome through homologous recombination after transformation into the supercompetent cells of the B. subtilis strain SCK6 [67]. Additionally, a random mutagenesis protocol has been devised for the methylotrophic yeast Pichia pastoris [68]. This protocol involves the sequential amplification of plasmids using Phi29 DNA polymerase, encompassing error-prone rolling circle amplification (RCA) followed by multiple displacement amplification (MDA). Through these steps, it becomes feasible to obtain microgram amounts of plasmids for subsequent electroporation into Pichia cells, addressing a key challenge in employing Pichia for directed evolution.
In the following section, we will explore recent advancements in four key areas: (1) In vivo mutagenesis in E. coli, (2) virus-assisted mutagenesis, (3) random base editing, and (4) random insertion and deletion (InDel).
3.2.1. In vivo mutagenesis in E. coli
In vivo mutagenesis provides several distinct advantages over in vitro mutagenesis. This approach allows the integration of genetic diversity creation and selection, eliminating the need for a separate transformation step that often limits the size of gene libraries in in vitro mutagenesis. Additionally, it avoids the labour-intensive process associated with in vitro gene library creation.
Liu and colleague developed highly effective, inducible, broad-spectrum mutagenesis systems in E. coli, elevating mutation rates by over 320000 times compared to basal levels [69]. These plasmid systems rely on the induced and combinatorial expression of proteins associated with proofreading (dnaQ926), translesion synthesis (umuD’, umuC, reacA730), mismatch repair (dam, seqA), base excision (ugi, cda1), and base selection (emrR). Although efficient, this global mutagenesis strategy could introduce extensive mutations throughout the genome of the host, leading to undesirable issues such as toxicity, reduced library size, silencing of mutagenic plasmids, or the introduction of parasite variants into DNA libraries (mutations outside the target GOI that allow the host to circumvent the selection scheme).
MutaT7 is a targeted in vivo mutagenesis strategy that overcomes the challenges associated with global mutagenesis strategies [70]. It employs a DNA-damaging cytidine deaminase fused to a processive T7 RNA polymerase (T7RNAP), allowing continuous directed mutations to any DNA region downstream of a T7 promoter. To enhance mutation rates, eMutaT7 replaces the cytidine deaminase from rat APOBEC1 (rApo1) with Petromyzon marinus cytidine deaminase (PmCDA1), resulting in an increased mutation rate from 0.34 mutations/kb/day (MutaT7) to 4 mutations/kb/day [71]. The MutaT7 toolbox has recently been expanded further with the use of adenosine deaminase-T7 RNA polymerase fusion proteins [72], such as TadA8e fused to T7RNAP and TadA7.10 fused to T7RNAP. TadA8e and TadA7.10 are variants of E. coli tRNA adenosine deaminase, evolved to operate on DNA [73]. Despite its utility, the MutaT7 toolkit has limitations, including a restricted mutational spectrum, strand bias, and the necessity to prevent the repair of deoxyuridine (e.g., deleting uracil-DNA glycosylase in the host or using a uracil-DNA glycosylase inhibitor) for significant mutagenesis when using cytidine deaminase.
When employing a highly processive T7RNAP, mutations may extend beyond the intended target GOI. To confine the mutagenesis to a specific region, multiple copies of the T7 terminator are required, serving as a boundary to restrict mutagenesis [70]. Alternatively, in the T7-DIVA strategy [74], the catalytically dead Cas9 (dCas9), tethered to a custom-designed crRNA, acts as a 'roadblock' for the base deaminase-T7RNAP fusion proteins, effectively constraining the window of mutagenesis.
Instead of relying on base deaminase as a mutagenic agent and T7RNAP as a 'guide protein' (GP) to target mutations to specific loci, the EvolvR system offers continuous nucleotide diversification within a tunable window length at user-defined loci [75,76]. This is accomplished by directly generating mutations using engineered DNA polymerases (variant of E. coli PolI with reduced fidelity) targeted to specific loci through CRISPR-guided nickases (nCas9).
3.2.2. Virus-assisted mutagenesis
Viruses provide a distinctive avenue for designing rapid laboratory evolution experiments, capitalizing on their inherent capacity to evolve at a much faster pace than many living organisms [77]. This accelerated evolution is facilitated by their smaller genome size, which tolerates a high frequency of mutations, and a rapid rate of replication. These attributes present an excellent opportunity for the directed evolution of various biomolecules.
In the Viral Evolution of Genetically Actuating Sequences (VEGAS) method, the highly mutagenic RNA alphavirus Sindbis, belonging to the Togaviridae family and lacking known proof-reading capability, was employed to establish a mammalian-directed evolution system [78]. Estimates of RNA virus mutation frequencies range from 10-5 to 10-3 mutations per base replicated. The mutation rate of the Sindbis virus was quantified to be 1.0 × 10-4 ± 3.7 × 10-5 mutations/base/hr. To create a robust directed evolution platform that capitalizes on the replicative and mutagenic potential of the Sindbis virus, artificial selective pressure must be applied. Each Sindbis viral particle requires 240 copies of the structural proteins E1, E2, and capsid to form a functional viral particle capable of maturing and propagating; without this envelope, the virus is unable to mature and propagate. By engineering restrictions on structural genome transcription, it is possible to apply a selective pressure to transgenic Sindbis virus carrying a GOI. The VEGAS system therefore allows for the simultaneous operation of viral mutagenesis, selection, and heredity. It's noteworthy that other reports on virus-assisted directed evolution, which didn't utilize viruses for creating genetic diversity, are not included in this review.
3.2.3. Random base editing
We have introduced the use of base editors (BE; e.g., cytidine deaminase and adenosine deaminase) for random mutagenesis in E. coli in section 3.2.1. On first glance, this section on random base editing may seem redundant or repetitive. However, dedicating an additional section to random base editing is warranted for three reasons: (1) to discuss method variation, (2) to highlight the versatility of this approach, extending it to a wide range of organisms/cells beyond E. coli, and (3) to showcase its diverse applications. Indeed, the summary of methods in Table 5 emphasizes the popularity of this approach, further justifying a dedicated section on this topic.
Methods utilizing BEs for targeted random mutagenesis share a common chimeric protein design (Figure 3): a BE is tethered to a GP, with or without any accessory protein(s). The BE functions as the mutagenic agent by deaminating cytidine to uridine or adenosine to inosine. The GP directs the BE to specific gene loci for mutagenesis. Key variations among methods primarily reside in four areas: (1) the choice of BE, (2) the selection of GP, (3) the mechanisms of linkage between GP and BE, and (4) the utilization or absence of accessory protein(s).
The most commonly employed BEs include apolipoprotein B mRNA editing enzyme catalytic subunit 1 (rAPOBEC1) from rat (Rattus norvegicus), cytidine deaminase (PmCDA1) from sea lamprey (Petromyzon marinus), human activation-induced cytidine deaminase (hAID), E. coli tRNA adenosine deaminase (TadA), and their respective variants. CRISPR-Cas and T7RNAP represent the most popular choices for GPs. While gene fusion stands out as the most straightforward method for connecting a BE to its GP, alternative strategies have proven effective, including the utilization of the SRC homology domain 3 (SH3) and the MS2 bacteriophage coat protein. To augment base editing efficiency, the inclusion of accessory proteins is frequently essential. Uracil DNA glycosylase inhibitor (UGI), an 83-residue protein derived from Bacillus subtilis bacteriophage PBS1, is extensively utilized for this purpose [79]. Additionally, the incorporation of mismatch and base excision proteins, Apn2p and Msh6p, has been shown to enhance editing efficiency.
With the BE-GP approach demonstrating growing maturity and proven success across diverse microbes and mammalian cells, we foresee ongoing developments and expect to witness adaptations of this method to additional microbial systems.
3.2.4. Random insertion and deletion
Insertions and deletions in genomes occur naturally due to replication slippage or error-prone NHEJ of double-stranded breaks. While their utilization in protein engineering is infrequent due to their tendency to be highly deleterious, often resulting in frame-shift mutations that significantly alter the protein sequence or prematurely terminate translation, there is evidence suggesting potential benefits. Instances of altered protein functionality through insertions and deletions have been reported, such as the broadening of substrate specificity in β-lactamase [91] and the modification of coenzyme specificity in Rossman fold enzymes [92]. These findings underscore the need for further exploration and investigation into the potential applications of this approach in protein engineering.
Methods for insertion and deletion in protein engineering have been developed over the last two decades, as comprehensively outlined in a recent review [93]. Some approaches leverage the higher slippage rates of certain polymerases to insert or delete 1 to 2 bases, inducing frame-shift mutations. Alternatively, other methods involve the fragmentation of GOI using DNase I or cleavage through exonucleases, endonucleases, or chemicals. Subsequently, nucleotides are added through processes like terminal deoxynucleotide transferase [94] or rolling circle amplification [95], incorporating random numbers or blocks of nucleotides. This results in libraries containing both in-frame and frame-shift variants.
Another set of methods relies on transposons to generate insertion and deletion libraries, and these approaches can prevent frame-shift occurrences through careful transposon sequence design [96,97,98]. Typically, transposons are designed with recognition sites for selected restriction enzymes, enabling cleavage and subsequent religation to generate the insertion or deletion of nucleotide triplets. A notable example is the recently developed TRIAD method that randomly inserts or deletes one to three nucleotide triplets [99]. This method randomly inserts engineered mini-Mu transposons, defining the location of the insertion/deletion event. For deletion, the recognition site for the type II restriction enzyme MlyI is designed into both ends of the transposon. After MlyI digestion and religation, 3bp are deleted. Repeating this process using MlyI custom cassettes enables longer deletions of up to 9 bp. For insertion, an asymmetric transposon with NotI and MlyI restriction sites is used. Following restriction digest, a cassette carrying up to three NNN triplets can be inserted. The TRIAD method has been applied to evolve arylesterase activity in a phosphotriesterase [99] and enhance antibody affinity [100].
3.3. Focused mutagenesis
Advancement in focused mutagenesis is marked by five noteworthy trends. These trends encompass (1) a transition from site-directed mutagenesis to multi-site directed mutagenesis and even massive mutagenesis, (2) the incorporation of CRISPR/Cas9 in focused mutagenesis, mirroring trends seen in cloning and random mutagenesis, (3) the formulation of strategies to minimize library size, (4) the development of computational tools for automated oligo design, and (5) a shift from column-synthesized oligos to microarray-synthesized oligos for protein engineering. The subsequent sections will discuss each of these trends individually.
3.3.1. Multi-site directed mutagenesis
Significant progress in multi-site directed mutagenesis has predominantly emerged in methodologies utilizing single-stranded DNA (ssDNA) templates, as summarized in Table 6. A key example is Nicking Mutagenesis [101], leveraging a nicking enzyme (Nt.BbvCl) and exonucleases (ExoIII, ExoI) for the preparation of ssDNA templates. This approach involves a pool of mutagenic primers defining the targeted mutagenesis sites, which anneal to the ssDNA and undergo isothermal assembly. Following template strand removal, the complementary mutant strand is synthesized via PCR. Continuous protocol enhancements have been reported to bolster efficiency (Table 6). In contrast, the SLUPT method generates the ssDNA template by eliminating the phosphorylated strand in the linearly amplified GOI [102].
Another group of methods opts for the creation of gene fragments bearing the desired mutations, which are subsequently combined in a sequential assembly using techniques like megaprimer PCR, overlap extension PCR, or Golden Gate assembly. Drawing inspiration from genome recombineering methods such as MAGE, Higgins et al. devised a plasmid recombineering method for in vivo multi-site directed mutagenesis [103].
Another development involves leveraging solid-phase gene synthesis technologies to design libraries [104,105]. This approach minimizes the risk of stop codons and unintended mutations during PCR, thereby reducing screening efforts [104]. Notably, Öling et al. achieved an impressive 161 multisite mutations using this methodology [106].
3.3.2. CRISPR/Cas9-mediated mutagenesis
She et al. introduced a PCR-free, two-step In vitro CRISPR/Cas9-mediated Mutagenic (ICM) system designed for both single-site and multi-site directed mutagenesis [115]. In the first step, site-specific plasmid digestion is achieved by employing a complex of Cas9 with specific single guide RNA (sgRNA), followed by degradation with T5 exonuclease to create a 15-nucleotide homologous region. Subsequently, in step 2, primers containing the desired mutations are annealed to generate double-stranded DNA fragments, which are then ligated into the linearized plasmid. A distinct advantage of employing a PCR-free approach is the attainment of greater genetic diversity, attributed to the elimination of biases introduced by PCR, such as preferential primer binding to the template. Anticipating ongoing advancements, we foresee more methodologies integrating the use of CRISPR/Cas9 in the near future.
3.3.3. The numbers game
In focused mutagenesis, with the escalation of target sites, the library size (number of permutations) undergoes exponential expansion. When combined with oversampling to ensure certain degree of diversity coverage, this exerts significant strain on library screening, making the navigation of the entire diversity space practically challenging – a scenario often denoted as permutation explosion. Commonly, three strategies are employed to tackle this issue: the divide-and-conquer approach, the codon randomization strategy, and the use of machine learning (ML).
GeneORator [116] employs a divide-and-conquer approach. In this method, target sites are partitioned into subsets, such as subsets A and B. A permutation library is established for each subset, generating libraries A and B. Both libraries A and B are then independently screened for the desired phenotype. Another notable divide-and-conquer strategy is the iterative saturation mutagenesis (ISM) pioneered by Reetz [117], taking the approach one step further. Beneficial variants from each subset act as templates to randomize the other subset, leading to the creation of new libraries AB or BA for the subsequent round of screening. A potential limitation of the divide-and-conquer approach is that it may not capture some pairwise and higher-order effects, known as epistasis, between residues in the gene libraries.
M-ISM, a variant of the ISM method [118], employs a computational tool to design a 'small-intelligent' library for each subset. The term ‘small-intelligent’ denotes a minimal gene library size devoid of inherent amino acid biases, stop codons, or rare codons specific to the protein expression host [119]. Rather than sampling each target site with 19 amino acid alphabets, it is feasible to obtain improved protein variants using reduced amino acid alphabets, potentially even with just 1 or 2 alphabets [120].
DEgenerate Codon Optimization for Informed Libraries (DeCOIL) is a recently introduced ML method that optimizes degenerate codon libraries to sample protein variants likely to exhibit both high fitness and high diversity within the sequence search space [121]. Like all ML approaches, DeCOIL necessitates a training library to develop its ML model.
3.3.4. Automated oligo design
Mutation Maker [122] and GeneGenie [123] serve as oligo design platforms for protein engineering. Taking Mutation Maker as an example, the platform designs oligos for various applications, including site-scanning saturation mutagenesis, multi-site directed mutagenesis, and PCR-based accurate gene synthesis. The designed oligos can be assembled through overlap extension PCR, with the program optimizing melting temperature (Tm) and codon choice. The use of degenerate oligos remains popular in focused mutagenesis. Web-based applications like CodonGenie [124] facilitate the selection of degenerate codons. For high-throughput or massively parallel protein engineering, automated oligo design proves useful in minimizing errors and expediting experimental design.
3.3.5. Oligo pool (oPool) for cost-effective library construction
Rather than relying on oligonucleotides with degenerate codons synthesized through solid-phase phosphoramidite chemistry in a column-based synthesis, there is a growing preference for using microarray-synthesized oligonucleotides or oligo pools (oPools) [125]. oPools are mixes of thousands of individually designed polynucleotides, each up to 350 bases in length.
Individually designed oligos offer several distinct advantages, including the removal of redundancy from the gene library, extensive coverage of mutational space, precise selection of codons optimized for protein expression in a chosen host, and facilitation of studies such as deep mutational scanning (DMS) [126] that necessitate large oligonucleotide pools. Programmed Allelic Series (PALS) is a technique devised for massively parallel single amino acid mutagenesis [127]. It achieves this by integrating a low-cost oPool with overlap-extension mutagenesis. oPool has been demonstrated to yield higher quality gene libraries compared to degenerate oligos [128].
While oPools show significant promise for protein engineering, it is essential to acknowledge certain challenges [125]. First, although the number of individual user-defined oligo sequences in a pool is large, their individual concentrations are low. Second, in focused mutagenesis applications, some oligos might preferentially bind to the template. Exacerbated by low concentrations, certain mutations may not be present in the final constructed gene library. Third, as the length of the oligos in the pool increases, the percentage of truncated molecules also rises, further diminishing the expected concentration of full-length molecules. Fourth, the error rates for oPools are typically higher than those for column-synthesized oligos.
Irrespective of the method chosen for creating a gene library, effective target site identification is crucial for focused mutagenesis. A recent comprehensive review by the Dalby group offers an excellent overview of methodologies for identifying target sites based on sequence or structural information [129]. Additionally, the review provides a valuable list of computational tools designed for target site identification.
3.4. DNA recombination
While considerable strides have been made in random mutagenesis and focused mutagenesis categories, advancements in the DNA recombination category have been relatively modest. An article sought to tackle the challenge of recombining protein modules from distant parents with minimal disruption at crossover sites. To overcome this hurdle, an approach called key motif-directed recombination was introduced [130]. Members of the same protein superfamily often share common structural or sequence motifs. Validated through the creation of α/β-hydrolase chimeras, this method strategically conducted recombination at key sequence motif regions. These chimeras retained their biological functions and exhibited desirable properties.
4. Applications of genetic diversity creation and future prospects
Directed evolution has firmly established itself as a powerful tool in biotechnology, diversifying its applications from evolving single enzymes or proteins to fascinating areas such as metabolic pathway engineering, organismal engineering, viral engineering, and the engineering of molecular biology tools. In the section below, we provide recent examples of applications beyond engineering a single protein.
Biofuels have emerged as a promising alternative to fossil fuels in sustainable energy solutions. Agricultural waste, a rich source of cellulose, provides a valuable feedstock for biofuel production [131]. Cellobiose, an intermediate in the conversion of cellulose into glucose monomers for fermentative processes, is crucial for efficient biofuel production. Directed evolution has played a key role in engineering two essential proteins in the cellobiose utilization pathway in S. cerevisiae: β-glucosidase and cellodextrin transporter [132,133]. These advancements have significantly increased ethanol production, contributing to the development of a more sustainable biofuel industry.
In section 3.2.3 above, we extensively covered the use of the 'BE-GP' protein architecture for targeted random mutagenesis in a wide range of biological systems. By excluding the GP, the method transforms into a potent global mutagenesis approach suitable for adaptive laboratory evolution (ALE). Pan et al. showcased the application of BE for ALE of S. cerevisiae, enhancing resistance to isobutanol and acetate, and boosting the production of β-carotene [90].
In a recent study, directed evolution was leveraged to enhance the transduction capabilities of adeno-associated viruses (AAVs) [134]. Directed evolution of the AAV capsid protein resulted in variants exhibiting significantly improved transduction efficiencies. This breakthrough facilitated the genetic manipulation of microglia, specialized immune cells in the central nervous system, with unprecedented ease. Given the implication of microglial dysfunction in neurodegenerative disorders and brain cancers, this advancement opens avenues for a more profound understanding of their functions and potential therapeutic interventions.
In genome editing and targeting, the landscape has been revolutionized by the emergence of CRISPR-Cas9. The widely utilized Cas9 nuclease, originating from Streptococcus pyogenes, encounters challenges attributed to its substantial size, constraining its effectiveness in delivery into cells. A smaller alternative, the Cas9 orthologue from Campylobacter Jejuni (CjCas9), presents an appealing solution [135]. Nevertheless, the intricate PAM sequence recognized by CjCas9 imposes limitations on its versatility. Through successive rounds of directed evolution, a variant of CjCas9 named evoCjCas9 was found, boasting an altered PAM recognition sequence that is ten times more prevalent in the genome than the canonical PAM sequence [136]. Beyond this, evoCjCas9 demonstrates increased nuclease activity compared to its wildtype counterpart, thereby broadening the horizons of CRISPR-Cas9 technology. Throughout this review, CRISPR-Cas9 has been consistently emphasized for its role in cloning and genetic diversity creation. It's truly inspiring to witness the 'ripple effect' where proteins crafted through protein engineering contribute to the continual expansion of the capabilities of protein engineering itself.
The convergence of automation and the emergence of artificial intelligence (AI) is poised to bring about a transformative shift in the domain of directed evolution, promising substantial improvements in scale, efficiency, and precision of experiments. Automation stands to simplify labour-intensive processes like library construction, screening, and the identification of desired variants. AI tools, coupled with the capability of analysing extensive datasets, have the potential to steer experimental and variant design, expediting the discovery process.
Embarking upon this transformative era necessitates the unlocking of untapped biological potential. At the heart of this unlocking process lies the art and science of creating genetic diversity. It is in the diverse tapestry of our genetic landscape that the blueprint for the future of the bioeconomy is etched. Through the strategic exploration and manipulation of genetic diversity, we forge a path into an era where the frontiers of bioengineering unfold boundlessly, promising novel discoveries and innovations that will shape the landscape of tomorrow.
References
- Arnold FH (2019) Angew Chem Int Ed Engl 58: 14420. [CrossRef]
- Fasan R, Jennifer Kan SB, Zhao H (2019) ACS Catal 9: 9775. [CrossRef]
- Wong TS, Zhurina D, Schwaneberg U (2006) Comb Chem High Throughput Screen 9: 271. [CrossRef]
- Tee KL, Wong TS (2013) Biotechnol Adv 31: 1707. [CrossRef]
- Wong TS, Tee KL (2020) A practical guide to protein engineering. Springer Cham.
- Watson JF, Garcia-Nafria J (2019) J Biol Chem 294: 15271. [CrossRef]
- Nozaki S, Niki H (2019) J Bacteriol 201. [CrossRef]
- Jones DH, Howard BH (1991) Biotechniques 10: 62.
- Garcia-Nafria J, Watson JF, Greger IH (2016) Sci Rep 6: 27459. [CrossRef]
- Huang F, Spangler JR, Huang AY (2017) PLoS One 12: e0183974. [CrossRef]
- Jacobus AP, Gross J (2015) PLoS One 10: e0119221. [CrossRef]
- Kostylev M, Otwell AE, Richardson RE, Suzuki Y (2015) PLoS One 10: e0137466. [CrossRef]
- Wang Y, Liu Y, Chen J, Tang MJ, Zhang SL, Wei LN, Li CH, Wei DB (2015) Genet Mol Res 14: 12306. [CrossRef]
- Chen X, Yuan H, He W, Hu X, Lu H, Li Y (2005) Sci China C Life Sci 48: 330. [CrossRef]
- Gibson DG (2009) Nucleic Acids Res 37: 6984. [CrossRef]
- Oldenburg KR, Vo KT, Michaelis S, Paddon C (1997) Nucleic Acids Res 25: 451. [CrossRef]
- Ding W, Weng H, Jin P, Du G, Chen J, Kang Z (2017) Bioengineered 8: 296. [CrossRef]
- Joska TM, Mashruwala A, Boyd JM, Belden WJ (2014) J Microbiol Methods 100: 46. [CrossRef]
- Kuijpers NG, Solis-Escalante D, Bosman L, van den Broek M, Pronk JT, Daran JM, Daran-Lapujade P (2013) Microb Cell Fact 12: 47. [CrossRef]
- Mashruwala AA, Boyd JM (2016) Methods Mol Biol 1373: 33. [CrossRef]
- van Leeuwen J, Andrews B, Boone C, Tan G (2015) Cold Spring Harb Protoc 2015: pdb prot085100. [CrossRef]
- Ip K, Yadin R, George KW (2020) Methods Mol Biol 2205: 79. [CrossRef]
- Abdel-Banat BM, Nonklang S, Hoshida H, Akada R (2010) Yeast 27: 29. [CrossRef]
- Hoshida H, Murakami N, Suzuki A, Tamura R, Asakawa J, Abdel-Banat BM, Nonklang S, Nakamura M, Akada R (2014) Yeast 31: 29. [CrossRef]
- Ruiz E, Talenton V, Dubrana MP, Guesdon G, Lluch-Senar M, Salin F, Sirand-Pugnet P, Arfi Y, Lartigue C (2019) ACS Synth Biol 8: 2547. [CrossRef]
- Pang Q, Ma S, Han H, Jin X, Liu X, Su T, Qi Q (2022) ACS Synth Biol 11: 1477. [CrossRef]
- He B, Ma Y, Tian F, Zhao GR, Wu Y, Yuan YJ (2023) Nucleic Acids Res 51: 8283. [CrossRef]
- Jeong YK, Yu J, Bae S (2019) Scientific Reports 9. [CrossRef]
- Wang JW, Wang A, Li K, Wang B, Jin S, Reiser M, Lockey RF (2015) Biotechniques 58: 161. [CrossRef]
- Jiang W, Zhao X, Gabrieli T, Lou C, Ebenstein Y, Zhu TF (2015) Nat Commun 6: 8101. [CrossRef]
- Richter D, Bayer K, Toesko T, Schuster S (2019) Sci Rep 9: 2980. [CrossRef]
- Dao VL, Chan SA, Zhang JY, Ngo RKJ, Poh CL (2022) Scientific Reports 12. [CrossRef]
- Xia Y, Li K, Li J, Wang T, Gu L, Xun L (2019) Nucleic Acids Res 47: e15. [CrossRef]
- Yu F, Li X, Wang F, Liu Y, Zhai C, Li WQ, Ma LX, Chen WP (2023) Frontiers in Bioengineering and Biotechnology 11. [CrossRef]
- Nisson PE, Rashtchian A, Watkins PC (1991) PCR Methods Appl 1: 120. [CrossRef]
- Cavaleiro AM, Kim SH, Seppala S, Nielsen MT, Norholm MH (2015) ACS Synth Biol 4: 1042. [CrossRef]
- Genee HJ, Bonde MT, Bagger FO, Jespersen JB, Sommer MO, Wernersson R, Olsen LR (2015) ACS Synth Biol 4: 342. [CrossRef]
- Jajesniak P, Wong TS (2015) J Biol Eng 9: 15. [CrossRef]
- Jajesniak P, Tee KL, Wong TS (2019) Int J Mol Sci 20. [CrossRef]
- Jajesniak P, Tee KL, Wong TS (2022) Methods Mol Biol 2461: 123. [CrossRef]
- Jin P, Ding W, Du G, Chen J, Kang Z (2016) ACS Synth Biol 5: 1028. [CrossRef]
- Kang Z, Ding W, Jin P, Du G, Chen J (2018) Methods Mol Biol 1772: 421. [CrossRef]
- Bird JE, Marles-Wright J, Giachino A (2022) ACS Synth Biol 11: 3551. [CrossRef]
- Marillonnet S, Grutzner R (2020) Curr Protoc Mol Biol 130: e115. [CrossRef]
- Sorida M, Bonasio R (2023) Cell Reports Methods 3. [CrossRef]
- Storch M, Casini A, Mackrow B, Fleming T, Trewhitt H, Ellis T, Baldwin GS (2015) ACS Synth Biol 4: 781. [CrossRef]
- van Dolleweerd CJ, Kessans SA, Van de Bittner KC, Bustamante LY, Bundela R, Scott B, Nicholson MJ, Parker EJ (2018) Acs Synthetic Biology 7: 1018. [CrossRef]
- Lin D, O'Callaghan CA (2018) Nucleic Acids Res 46: e113. [CrossRef]
- Taylor GM, Heap JT (2020) Methods Mol Biol 2205: 219. [CrossRef]
- Taylor GM, Mordaka PM, Heap JT (2019) Nucleic Acids Res 47: e17. [CrossRef]
- Trubitsyna M, Honsbein A, Jayachandran U, Elfick A, French CE (2020) Methods Mol Biol 2205: 161. [CrossRef]
- Storch M, Haines MC, Baldwin GS (2020) Synthetic Biology 5. [CrossRef]
- Enghiad B, Xue P, Singh N, Boob AG, Shi CY, Petrov VA, Liu R, Peri SS, Lane ST, Gaither ED, Zhao HM (2022) Nature Communications 13. [CrossRef]
- Bryant JA, Kellinger M, Longmire C, Miller R, Wright RC (2023) Synthetic Biology 8. [CrossRef]
- Fukuda M, Umeno H, Nose K, Nishitarumizu A, Noguchi R, Nakagawa H (2017) Sci Rep 7: 41478. [CrossRef]
- Kopsidas G, Carman RK, Stutt EL, Raicevic A, Roberts AS, Siomos MA, Dobric N, Pontes-Braz L, Coia G (2007) BMC Biotechnol 7: 18. [CrossRef]
- Wong TS, Roccatano D, Loakes D, Tee KL, Schenk A, Hauer B, Schwaneberg U (2008) Biotechnol J 3: 74. [CrossRef]
- Wong TS, Tee KL, Hauer B, Schwaneberg U (2004) Nucleic Acids Res 32: e26. [CrossRef]
- Wong TS, Tee KL, Hauer B, Schwaneberg U (2005) Anal Biochem 341: 187. [CrossRef]
- Zhao J, Frauenkron-Machedjou VJ, Kardashliev T, Ruff AJ, Zhu L, Bocola M, Schwaneberg U (2017) Appl Microbiol Biotechnol 101: 3177. [CrossRef]
- Zhao J, Kardashliev T, Joelle Ruff A, Bocola M, Schwaneberg U (2014) Biotechnol Bioeng 111: 2380. [CrossRef]
- Verma R, Wong TS, Schwaneberg U, Roccatano D (2014) Methods Mol Biol 1179: 279. [CrossRef]
- Wong TS, Roccatano D, Schwaneberg U (2007) Biotechnol J 2: 133. [CrossRef]
- Wong TS, Roccatano D, Zacharias M, Schwaneberg U (2006) J Mol Biol 355: 858. [CrossRef]
- Li H, Li J, Jin R, Chen W, Liang C, Wu J, Jin JM, Tang SY (2018) Biotechnol Lett 40: 1101. [CrossRef]
- Yang J, Ruff AJ, Arlt M, Schwaneberg U (2017) Biotechnol Bioeng 114: 1921. [CrossRef]
- Ye B, Li Y, Tao Q, Yao X, Cheng M, Yan X (2020) Front Microbiol 11: 570280. [CrossRef]
- Tachioka M, Sugimoto N, Nakamura A, Sunagawa N, Ishida T, Uchiyama T, Igarashi K, Samejima M (2016) Biotechnol Biofuels 9: 199. [CrossRef]
- Badran AH, Liu DR (2015) Nat Commun 6: 8425. [CrossRef]
- Moore CL, Papa LJ, 3rd, Shoulders MD (2018) J Am Chem Soc 140: 11560. [CrossRef]
- Park H, Kim S (2021) Nucleic Acids Res 49: e32. [CrossRef]
- Mengiste AA, Wilson RH, Weissman RF, Papa Iii LJ, Hendel SJ, Moore CL, Butty VL, Shoulders MD (2023) Nucleic Acids Res 51: e31. [CrossRef]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR (2017) Nature 551: 464. [CrossRef]
- Alvarez B, Mencia M, de Lorenzo V, Fernandez LA (2020) Nat Commun 11: 6436. [CrossRef]
- Halperin SO, Tou CJ, Wong EB, Modavi C, Schaffer DV, Dueber JE (2018) Nature 560: 248. [CrossRef]
- Sadanand S (2018) Nat Biotechnol 36: 819. [CrossRef]
- Jewel D, Pham Q, Chatterjee A (2023) Curr Opin Chem Biol 76: 102375. [CrossRef]
- English JG, Olsen RHJ, Lansu K, Patel M, White K, Cockrell AS, Singh D, Strachan RT, Wacker D, Roth BL (2019) Cell 178: 748. [CrossRef]
- Wang L, Xue W, Yan L, Li X, Wei J, Chen M, Wu J, Yang B, Yang L, Chen J (2017) Cell Res 27: 1289. [CrossRef]
- Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, Mochizuki M, Miyabe A, Araki M, Hara KY, Shimatani Z, Kondo A (2016) Science 353. [CrossRef]
- Hess GT, Fresard L, Han K, Lee CH, Li A, Cimprich KA, Montgomery SB, Bassik MC (2016) Nat Methods 13: 1036. [CrossRef]
- Ma Y, Zhang J, Yin W, Zhang Z, Song Y, Chang X (2016) Nat Methods 13: 1029. [CrossRef]
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR (2016) Nature 533: 420. [CrossRef]
- Gehrke JM, Cervantes O, Clement MK, Wu Y, Zeng J, Bauer DE, Pinello L, Joung JK (2018) Nat Biotechnol 36: 977. [CrossRef]
- Chen H, Liu S, Padula S, Lesman D, Griswold K, Lin A, Zhao T, Marshall JL, Chen F (2020) Nat Biotechnol 38: 165. [CrossRef]
- Cravens A, Jamil OK, Kong D, Sockolosky JT, Smolke CD (2021) Nat Commun 12: 1579. [CrossRef]
- Volke DC, Martino RA, Kozaeva E, Smania AM, Nikel PI (2022) Nat Commun 13: 3026. [CrossRef]
- Skrekas C, Limeta A, Siewers V, David F (2023) ACS Synth Biol 12: 2278. [CrossRef]
- Zimmermann A, Prieto-Vivas JE, Cautereels C, Gorkovskiy A, Steensels J, Van de Peer Y, Verstrepen KJ (2023) Nat Commun 14: 3389. [CrossRef]
- Pan Y, Xia S, Dong C, Pan H, Cai J, Huang L, Xu Z, Lian J (2021) ACS Synth Biol 10: 2440. [CrossRef]
- Hwang J, Cho KH, Song H, Yi H, Kim HS (2014) Antimicrob Agents Chemother 58: 6265. [CrossRef]
- Toledo-Patiño S, Pascarelli S, Uechi G-i, Laurino P (2022) bioRxiv: 2022.05.16.491946. [CrossRef]
- Savino S, Desmet T, Franceus J (2022) Biotechnol Adv 60: 108010. [CrossRef]
- Fujii R, Kitaoka M, Hayashi K (2006) Nucleic Acids Res 34: e30. [CrossRef]
- Kipnis Y, Dellus-Gur E, Tawfik DS (2012) Protein Eng Des Sel 25: 437. [CrossRef]
- Liu SS, Wei X, Ji Q, Xin X, Jiang B, Liu J (2016) J Biotechnol 227: 27. [CrossRef]
- Hayes F, Hallet B (2000) Trends Microbiol 8: 571. [CrossRef]
- Jones DD (2005) Nucleic Acids Res 33: e80. [CrossRef]
- Emond S, Petek M, Kay EJ, Heames B, Devenish SRA, Tokuriki N, Hollfelder F (2020) Nat Commun 11: 3469. [CrossRef]
- Skamaki K, Emond S, Chodorge M, Andrews J, Rees DG, Cannon D, Popovic B, Buchanan A, Minter RR, Hollfelder F (2020) Proc Natl Acad Sci U S A 117: 27307. [CrossRef]
- Wrenbeck EE, Klesmith JR, Stapleton JA, Adeniran A, Tyo KE, Whitehead TA (2016) Nat Methods 13: 928. [CrossRef]
- Meinke G, Dalda N, Brigham BS, Bohm A (2021) Synth Biol (Oxf) 6: ysaa030. [CrossRef]
- Higgins SA, Ouonkap SVY, Savage DF (2017) ACS Synth Biol 6: 1825. [CrossRef]
- Hoebenreich S, Zilly FE, Acevedo-Rocha CG, Zilly M, Reetz MT (2015) ACS Synth Biol 4: 317. [CrossRef]
- Li A, Sun Z, Reetz MT (2018) Chembiochem 19: 2023. [CrossRef]
- Oling D, Lawenius L, Shaw W, Clark S, Kettleborough R, Ellis T, Larsson N, Wigglesworth M (2018) ACS Synth Biol 7: 2317. [CrossRef]
- Cozens C, Pinheiro VB (2018) Nucleic Acids Res 46: e51. [CrossRef]
- Kirby MB, Medina-Cucurella AV, Baumer ZT, Whitehead TA (2021) Protein Eng Des Sel 34. [CrossRef]
- Mighell TL, Toledano I, Lehner B (2023) PLoS One 18: e0288158. [CrossRef]
- Belsare KD, Andorfer MC, Cardenas FS, Chael JR, Park HJ, Lewis JC (2017) ACS Synth Biol 6: 416. [CrossRef]
- Bloom JD (2014) Mol Biol Evol 31: 2753. [CrossRef]
- Chung DH, Potter SC, Tanomrat AC, Ravikumar KM, Toney MD (2017) Protein Eng Des Sel 30: 347. [CrossRef]
- Pullmann P, Ulpinnis C, Marillonnet S, Gruetzner R, Neumann S, Weissenborn MJ (2019) Sci Rep 9: 10932. [CrossRef]
- Hejlesen R, Fuchtbauer EM (2020) Biotechniques 68: 345. [CrossRef]
- She W, Ni J, Shui K, Wang F, He R, Xue J, Reetz MT, Li A, Ma L (2018) ACS Synth Biol 7: 2236. [CrossRef]
- Currin A, Kwok J, Sadler JC, Bell EL, Swainston N, Ababi M, Day P, Turner NJ, Kell DB (2019) ACS Synth Biol 8: 1371. [CrossRef]
- Reetz MT, Carballeira JD (2007) Nat Protoc 2: 891. [CrossRef]
- Wang X, Zheng K, Zheng H, Nie H, Yang Z, Tang L (2014) J Biotechnol 192 Pt A: 102. [CrossRef]
- Tang L, Gao H, Zhu X, Wang X, Zhou M, Jiang R (2012) Biotechniques 52: 149. [CrossRef]
- Sun Z, Lonsdale R, Li G, Reetz MT (2016) Chembiochem 17: 1865. [CrossRef]
- Yang J, Ducharme J, Johnston KE, Li FZ, Yue Y, Arnold FH (2023) ACS Synth Biol 12: 2444. [CrossRef]
- Hiraga K, Mejzlik P, Marcisin M, Vostrosablin N, Gromek A, Arnold J, Wiewiora S, Svarba R, Prihoda D, Clarova K, Klempir O, Navratil J, Tupa O, Vazquez-Otero A, Walas MW, Holy L, Spale M, Kotowski J, Dzamba D, Temesi G, Russell JH, Marshall NM, Murphy GS, Bitton DA (2021) ACS Synth Biol 10: 357. [CrossRef]
- Swainston N, Currin A, Day PJ, Kell DB (2014) Nucleic Acids Res 42: W395. [CrossRef]
- Swainston N, Currin A, Green L, Breitling R, Day PJ, Kell DB (2017) PeerJ Comput. Sci. 3: e120. [CrossRef]
- Kuiper BP, Prins RC, Billerbeck S (2022) Chembiochem 23: e202100507. [CrossRef]
- Fowler DM, Fields S (2014) Nat Methods 11: 801. [CrossRef]
- Kitzman JO, Starita LM, Lo RS, Fields S, Shendure J (2015) Nat Methods 12: 203. [CrossRef]
- Medina-Cucurella AV, Steiner PJ, Faber MS, Beltran J, Borelli AN, Kirby MB, Cutler SR, Whitehead TA (2019) Protein Eng Des Sel 32: 41. [CrossRef]
- Yu H, Ma S, Li Y, Dalby PA (2022) Biotechnol Adv 56: 107926. [CrossRef]
- Zhou X, Gao L, Yang G, Liu D, Bai A, Li B, Deng Z, Feng Y (2015) Chembiochem 16: 455. [CrossRef]
- Kumar JA, Sathish S, Prabu D, Renita AA, Saravanan A, Deivayanai VC, Anish M, Jayaprabakar J, Baigenzhenov O, Hosseini-Bandegharaei A (2023) Chemosphere 331: 138680. [CrossRef]
- Hu ML, Zha J, He LW, Lv YJ, Shen MH, Zhong C, Li BZ, Yuan YJ (2016) Front Microbiol 7: 241. [CrossRef]
- Kim H, Oh EJ, Lane ST, Lee WH, Cate JHD, Jin YS (2018) J Biotechnol 275: 53. [CrossRef]
- Lin R, Zhou Y, Yan T, Wang R, Li H, Wu Z, Zhang X, Zhou X, Zhao F, Zhang L, Li Y, Luo M (2022) Nat Methods 19: 976. [CrossRef]
- Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, Song DW, Lee KJ, Jung MH, Kim S, Kim JH, Kim JH, Kim JS (2017) Nat Commun 8: 14500. [CrossRef]
- Schmidheini L, Mathis N, Marquart KF, Rothgangl T, Kissling L, Bock D, Chanez C, Wang JP, Jinek M, Schwank G (2023) Nat Chem Biol. [CrossRef]
Figure 1.
The directed evolution cycle. The parental gene of interest (GOI) undergoes mutagenesis to generate a diverse pool of genetic variants. This pool is then subjected to a selection process targeting the desired phenotype, enabling the identification of improved variant(s). This iterative cycle is repeated until the desired trait is successfully achieved.
Figure 1.
The directed evolution cycle. The parental gene of interest (GOI) undergoes mutagenesis to generate a diverse pool of genetic variants. This pool is then subjected to a selection process targeting the desired phenotype, enabling the identification of improved variant(s). This iterative cycle is repeated until the desired trait is successfully achieved.
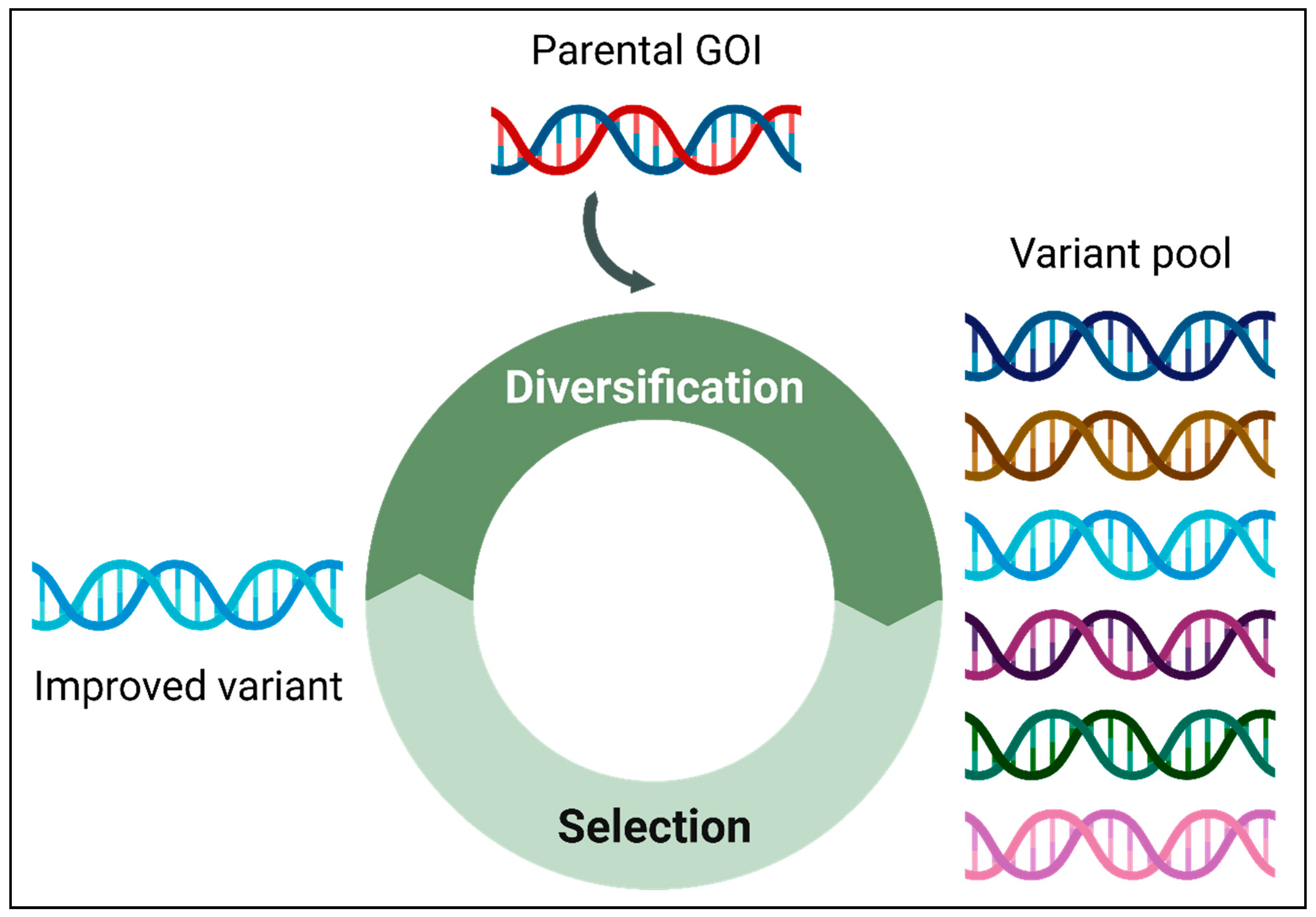
Figure 2.
Classification of genetic diversity creation methods. The diverse methods for generating a genetically varied gene pool can be systematically categorized into three main classes: random mutagenesis, focused mutagenesis, and DNA recombination. Random mutagenesis involves the introduction of random mutations throughout the starting parental gene sequence. Focused mutagenesis targets mutations to specific pre-selected region(s) or amino acid residue(s) within the starting parental gene sequence. DNA recombination generates chimeric sequences by combining segments from a set of either homologous or non-homologous parental sequences. .
Figure 2.
Classification of genetic diversity creation methods. The diverse methods for generating a genetically varied gene pool can be systematically categorized into three main classes: random mutagenesis, focused mutagenesis, and DNA recombination. Random mutagenesis involves the introduction of random mutations throughout the starting parental gene sequence. Focused mutagenesis targets mutations to specific pre-selected region(s) or amino acid residue(s) within the starting parental gene sequence. DNA recombination generates chimeric sequences by combining segments from a set of either homologous or non-homologous parental sequences. .
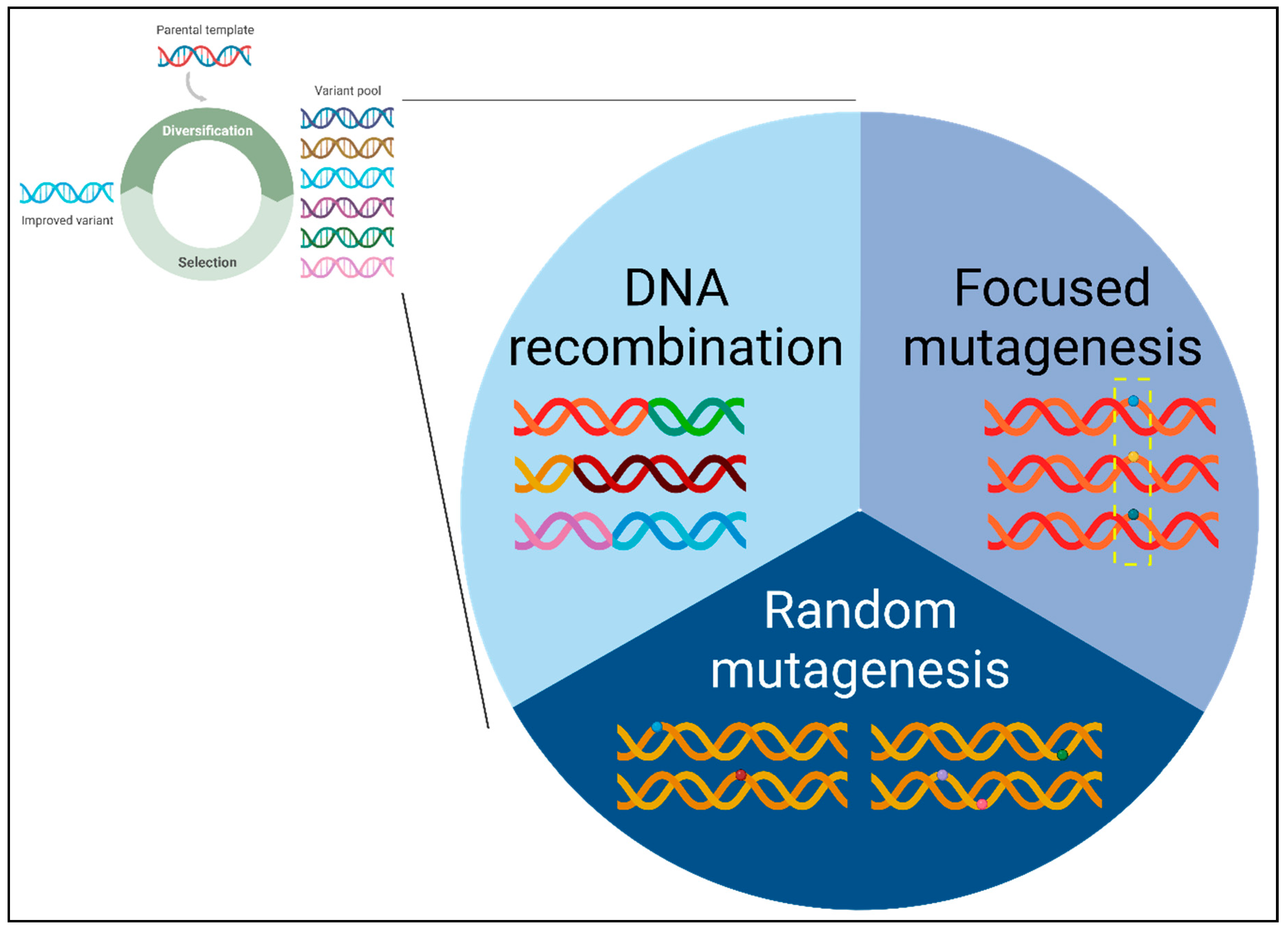
Figure 3.
Targeted random mutagenesis using chimeric proteins comprising a base editor (BE) and a guide protein (GP), following the general BE-GP protein architecture. BE is the mutagenic agent, introducing random mutations through its base editing activity (e.g., cytidine and adenosine deamination). GP, with DNA-binding capability, guides or leads the BE to its target locus within the gene of interest (GOI) to effect mutagenesis. Typical BE choices include cytidine deaminase (CDA) or error-prone DNA polymerase (Pol). Frequently used GP candidates are T7 RNA polymerase (RNAP) or catalytically dead Cas9 (dCas9)/Cas9 nickase (nCas9). BE is tethered to GP via gene fusion or protein-protein or protein/RNA interactions through the utilization of the SRC homology domain 3 (SH3) and the MS2 bacteriophage coat protein. In some methods, an accessory protein such as uracil DNA glycosylase inhibitor (UGI) is required.
Figure 3.
Targeted random mutagenesis using chimeric proteins comprising a base editor (BE) and a guide protein (GP), following the general BE-GP protein architecture. BE is the mutagenic agent, introducing random mutations through its base editing activity (e.g., cytidine and adenosine deamination). GP, with DNA-binding capability, guides or leads the BE to its target locus within the gene of interest (GOI) to effect mutagenesis. Typical BE choices include cytidine deaminase (CDA) or error-prone DNA polymerase (Pol). Frequently used GP candidates are T7 RNA polymerase (RNAP) or catalytically dead Cas9 (dCas9)/Cas9 nickase (nCas9). BE is tethered to GP via gene fusion or protein-protein or protein/RNA interactions through the utilization of the SRC homology domain 3 (SH3) and the MS2 bacteriophage coat protein. In some methods, an accessory protein such as uracil DNA glycosylase inhibitor (UGI) is required.
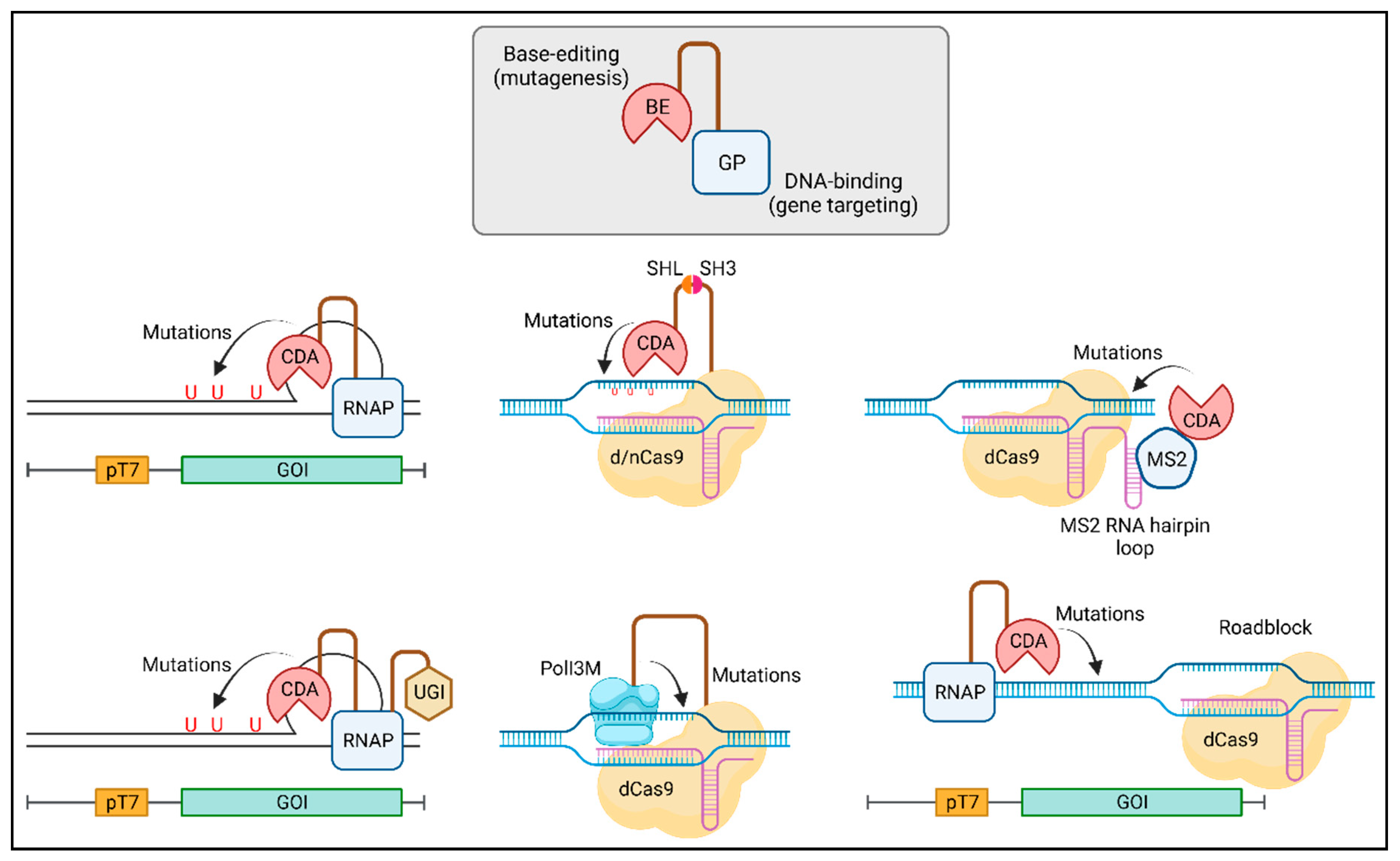

Table 1.
In vivo cloning methods developed in the past decade (HR, homologous recombination; NHEJ, non-homologous end joining).
Table 1.
In vivo cloning methods developed in the past decade (HR, homologous recombination; NHEJ, non-homologous end joining).
| In vivo cloning method | Method description | Reference(s) |
|---|---|---|
| Bacterial in vivo cloning (HR-based) | Depends on RecA-independent recombination (RAIR) pathway. DNA fragments generated via PCR or restriction enzyme digestion with overlapping homologous sequences can be used to directly transform bacteria. | [6,7,9,10,11,12,13,14,15,16] |
| Yeast in vivo cloning (HR-based) | Highly efficient HR pathway in yeast (S. cerevisiae) can be used for assembling multiple DNA fragments with homologous sequences directing the order of assembly. | [18,19,20,21] |
| Yeast in vivo cloning (NHEJ-based) | Highly efficient NHEJ pathway in thermotolerant yeast (K. marxianus) allows directly joining two DNA fragments without the need for homologous ends. | [24] |
| CReasPy-cloning | Combines ability of CRISPR-Cas9 and HR pathway of yeast to clone and edit large bacterial genomes at multiple loci. | [25] |
| Phage Enzyme-Assisted In Vivo DNA Assembly (PEDA) method | Simultaneous expression of exonuclease and ligase allows in vivo cloning in wide range of microorganisms. | [26] |
| Yeast Life Cycle (YLC) assembly method | Combines CRISPR-Cas9 and meiosis of yeast to iteratively assemble large DNA fragments. | [27] |
Table 2.
In vitro cloning methods developed in the past decade.
| In vitro cloning method | Method description | Reference(s) |
|---|---|---|
| T5 Exonuclease-Dependent Assembly (TEDA) | Exonuclease generates homologous overhangs, gap repair and ligation completed in vivo. | [33] |
| Single 3′-exonuclease-based multifragment DNA assembly (SENAX) | [32] | |
| T5 exonuclease-mediated low-temperature DNA cloning (TLTC) | [34] | |
| Uracil-Specific Excision Reagent (USER) | Uracil-specific endonuclease generates homologous overhangs by digesting deoxyuracil introduced by primers. | [36,37] |
| PTO-QuickStep cloning | Phosphorothioate bonds introduced by primers are processed by iodine cleavage to generate homologous overhangs. | [39,40] |
| Scarless and sequence-independent DNA assembly method using thermostable exonuclease and ligase (DATEL) | Thermostable exonuclease generates homologous overhangs and thermostable ligase joins DNA fragments. | [41,42] |
Table 3.
Modular DNA assembly methods developed in the past 10 years.
| Modular DNA assembly method | Method description | Reference(s) |
|---|---|---|
| Biopart Assembly Standard for Idempotent Cloning (BASIC) | Makes use of reusable linkers and parts. Orthogonal oligonucleotide linkers with single stranded overhangs are used to assemble DNA parts. Flexibility with the order of various DNA parts. | [46] |
| Modular Idempotent DNA Assembly System (MIDAS) | Requires three type IIS restriction enzymes and more complex than other modular DNA assemblies. Advantages include the ability to add new parts between existing parts rather than at the end. | [47] |
| MetClo Assembly | Controlling methylation (which cuts or blocks the recognition site) of a single type IIS recognition enzyme allows for a simpler hierarchical DNA assembly system. | [48] |
| Start-Stop Assembly | 3-bp overhangs corresponding to start and stop codons allow for scarless assembly of coding sequences. | [49,50] |
| PaperClip DNA Assembly | Unlike most other modular DNA assembly methods, does not require restriction enzymes. Four oligos per DNA part allow flexible ordering of DNA parts and reuse of oligos. | [51] |
Table 4.
Automated DNA assembly methods reported in the past decade.
| Automated DNA assembly methods | Method description | Reference(s) |
|---|---|---|
| DNA assembly with BASIC on Opentrons (DNA-BOT) | Modular DNA assembly technique BASIC has been automated using robotic liquid handler Opentrons | [52] |
| Plasmid Maker | Combining the use of artificial restriction enzymes, custom software and robotic systems, an end-to-end system designed for automated DNA assembly. | [53] |
| AssemblyTron | Golden Gate and HR-dependent in vivo assemblies were automated using Opentrons | [54] |
Table 5.
Targeted and non-targeted (global) random base editing methods, reported in the past 10 years.
Table 5.
Targeted and non-targeted (global) random base editing methods, reported in the past 10 years.
| Method/first author’s name (year of publication) |
Guide protein (GP) | Base editor (BE) | Linkage between GP and BE | Organisms/cells validated | Ref |
|---|---|---|---|---|---|
| Targeted mutagenesis | |||||
| Nishida et al. (2016) | dCas91 or nCas92 | PmCDA1 | Gene fusion or interaction between SH3 (SRC homology domain 3) and SHL (SH3 interaction ligand) | S. cerevisiae and CHO | [80] |
| CRISPR-X (2016) | dCas91 | hAID*Δ4 | MS2 bacteriophage coat protein (MCP) binding to the MS2 RNA stem-loop | K-562 cell | [81] |
| TAM (2016) | dCas91 | hAID, hAID CD5, hAID P182X6 or hAID R190X7 | Gene fusion | K-562 cell and HEK293T cell | [82] |
| Komor et al. (2016) | dCas91 or nCas92 | hAID, rAPOBEC1, hAPOBEC3G or PmCDA1 | Gene fusion | U2OS cell, HEK293T cell and HCC1954 cell | [83] |
| Gehrke et al. (2018) | nCas92 | rAPOBEC1 or engineered hAPOBEC3A | Gene fusion | U2OS cell and HEK293T | [84] |
| TRACE (2020) | T7 RNA polymerase | rAPOBEC1 or hAID*Δ4 | Gene fusion | HEK293T cell | [85] |
| TRIDENT (2021) | T7 RNA polymerase | PmCDA1 or yeTadA1.08 | Gene fusion | S. cerevisiae | [86] |
| Volke et al. (2022) | nCas92 | rAPOBEC1 | Gene fusion | P. putida and P. aeruginosa | [87] |
| Skrekas et al. (2023) | dCas91 | hAID*Δ4, TadA8e9 or TadA8e V106W10 | Gene fusion | S. cerevisiae | [88] |
| CoMuTER (2023) | dCas33 | PmCDA1 or rAPOBEC1 | Gene fusion | S. cerevisiae | [89] |
| Global mutagenesis | |||||
| Pan et al. (2021) | rAPOBEC1 | N/A | S. cerevisiae | [90] | |
1Catalytically dead Cas9, containing D10A and H840A mutations; 2Cas9 nickase, containing D10A mutation; 3Nuclease-deficient Cas3, containing H74A and D75A mutations; 4Truncated AID, i.e., AIDΔ196-198, containing mutations K10E, T82I and E156G; 5Truncated AID retaining only the catalytic domain, i.e., AIDΔ94-198; 6Truncated AID, i.e., AIDΔ182-198; 7Truncated AID, i.e., AIDΔ190-198; 8Variant of E. coli TadA, evolved in yeast; 9Variant of E. coli TadA; 10Variant of E. coli TadA.
Table 6.
Multi-site directed mutagenesis reported in the last decade.
| Method/first author (year of publication) |
Method description and mutagenic agent |
Template | Maximum mutated sites per gene | Ref |
|---|---|---|---|---|
| ssDNA template | ||||
| Nicking Mutagenesis (2016) |
|
Plasmid ssDNA | 7 | [101] |
| Darwin Assembly (2018) |
|
Plasmid ssDNA | 19 | [107] |
| Nicking Mutagenesis (2021) |
|
Plasmid ssDNA | 15 | [108] |
| SLUPT (2021) |
|
Linear GOI ssDNA | 17 | [102] |
| SUNi Mutagenesis (2023) |
|
Plasmid ssDNA | n.r* | [109] |
| Assembly of mutated gene fragments | ||||
| Combinatorial Codon mutagenesis (CCM) (2014, 2017) |
|
Plasmid dsDNA | 7 | [110,111] |
| Chung et al. (2017) |
|
Linear GOI dsDNA | 14 | [112] |
| Golden Mutagenesis (2019) |
|
Plasmid dsDNA | 5 | [113] |
| Hejlesen et al. (2020) |
|
Plasmid dsDNA | 3 | [114] |
| In vivo method | ||||
| Plasmid recombineering (2017) |
|
Plasmid dsDNA | 2 | [103] |
*n.r. not reported.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



