Submitted:
19 December 2023
Posted:
20 December 2023
You are already at the latest version
Abstract
Cannabinoid metabolites have been reported to be more potent than the parent compounds. Among them, Ajulemic acid (AJA) is a synthetic analogue of Δ9-THC-11-oic which will be a good template structure for discovery of more potent analogues. Herein, we optimized the key allylic oxidation step to introduce the C-11 hydroxy group with high yield. And a series of compounds were prepared with this condition applied including HU-210, 11-Nor-Δ8-Tetrahydrocannabinol-9-carboxylic acid and Δ9-THC-COOH.
Keywords:
Phytocannabinoids
; Metabolites
; Ajulemic acid
; Riley oxidation
1. Introduction
Phytocannabinoids and their synthetic analogues are prime candidates for pharmaceutical innovation, exemplified by molecules such as Δ9 -tetrahydrocannabinol (Δ9-THC, 1, Figure 1) are known to possess potent analgesic and anti-inflammatory properties. And 1 was first identified in 1964 by Gaoni and Mechoulam as the principle bioactive component of marijuana (hashish), which has been used for centuries as both a therapeutic and recreational drug. More generally, cannabinoid-based chemical probes and leads are essential for continued exploration of the endocannabinoid system. [1] Prior to the 1980s, cannabinoids were hypothesized to produce their effects by nonspecific interactions with cell membranes. The absolute stereochemistry of 1 was established in 1967 [2], and more than 2 decades passed before it was identified as a modulator of the cannabinoid receptor [3].
The main metabolic pathways involved hydroxylations or oxygenations. It is well-known that the nature of substituents at the C-1, C-3, and C-9 positions play a critical role in efficient binding to the cannabinoid receptors [4]. SAR studies of the C-3 side chain demonstrated that a seven-carbon homologue was optimal for activity and that branched alkyl groups also led to improved binding affinity [5]. For example, the dimethylheptyl analogue 3 which is an oxygenation product of 2 exhibits an approximately 50-fold improvement in activity relative to 1 [6]. The relative importance of the C-1 hydroxyl differs between the two cannabinoid subtypes. Synthetic cannabinoid 3 [7]. possesses two hydrogen donors and exhibits significantly enhanced affinity for both the CB2 and the CB1 receptors, producing many of the same pharmacological effects as 1 [8].
Compound 4 is a synthetic analog of Δ9-THC-11-oic acid, the major metabolite of the psychoactive component of marijuana, Δ9-THC. Δ9-THC-11-oic acid [9] has no psychotropic activity and is present in the tissues of the millions of recreational users of Cannabis long after the mood altering effects are gone [10]. Its analgesic properties suggested that it would be a good template structure for discovery of more potent analogs. AJA is such an analog, and is a ‘first-in-class’ chemical entity designed to have increased anti-inflammatory properties and reduced psychotropic activity compared to its THC parent [11]. This compound was found to be well tolerated in a phase I clinical trial and subsequently completed a phase II studies wherein this molecule demonstrated efficacy in reducing chronic neuropathic pain without any major adverse effects [12].
There are few reports on the synthesis of cannabinoid metabolites. which vary from five to seven steps (Figure 2). Jiang et al. reported a synthetic route to synthesize the HHC analogues with seven steps in 24% overall yields [13]. And Tepper et al developed a synthetic route to synthesize the AJA within five steps but only in 13% overall yields [14]. Considering the rapid change in the illicit drug market, a concise synthetic where the combination of a simple replacement of the phenol protecting group (Figure 2) and the optimization of the riley oxidation condition give a better yield (43% overall). Therefore, a synthetic route for the synthesis of a key intermediate, which subsequently can be used to synthesize the cannabinoid metabolites was developed. The synthesis method to the intermediate is carried out on a multigram scale.
2. Results and discussion
The route to synthetic the ajulemic acid by Tepper was optimized with the change of the SeO2 mediated condition to improve the yield of the key allylic oxidation step. Our synthesis started with preparation of the tricyclic intermediate (5) [15] as shown in Scheme 1. Commercial available starting materials p-menthadienol (PMD) and 1,1-dimethylheptyl resorcinol (DMHR) were under the acid condition to promote the cyclization to obtain the tricyclic skeleton of the cannabinoid metabolites in 72% yield. Thus, further protection of the phenol group with a TBS group afforded the key tricyclic intermediate (6) in 97% yield. [11] To install the C11 hydroxyl group, (6) was subjected to a SeO2-mediated allylic oxidation, and product (7) bearing an aldehyde group at C11 was obtained in 65% yield. Then reduction of the new generated aldehyde group with NaBH4 afforded the hydroxy group in 89% yield, The resultant (8) was then reacted with 1M TBAF in THF to give product HU-210 in 93% yield. We then turned our attention to (7) for the total syntheses of ajulemic acid. To this end, (7) was first converted to acid (9) with treatment by Pinnick oxidation in 90% yield, further treatment of (9) with TBAF in THF to give product ajulemic acid in 92% yield.
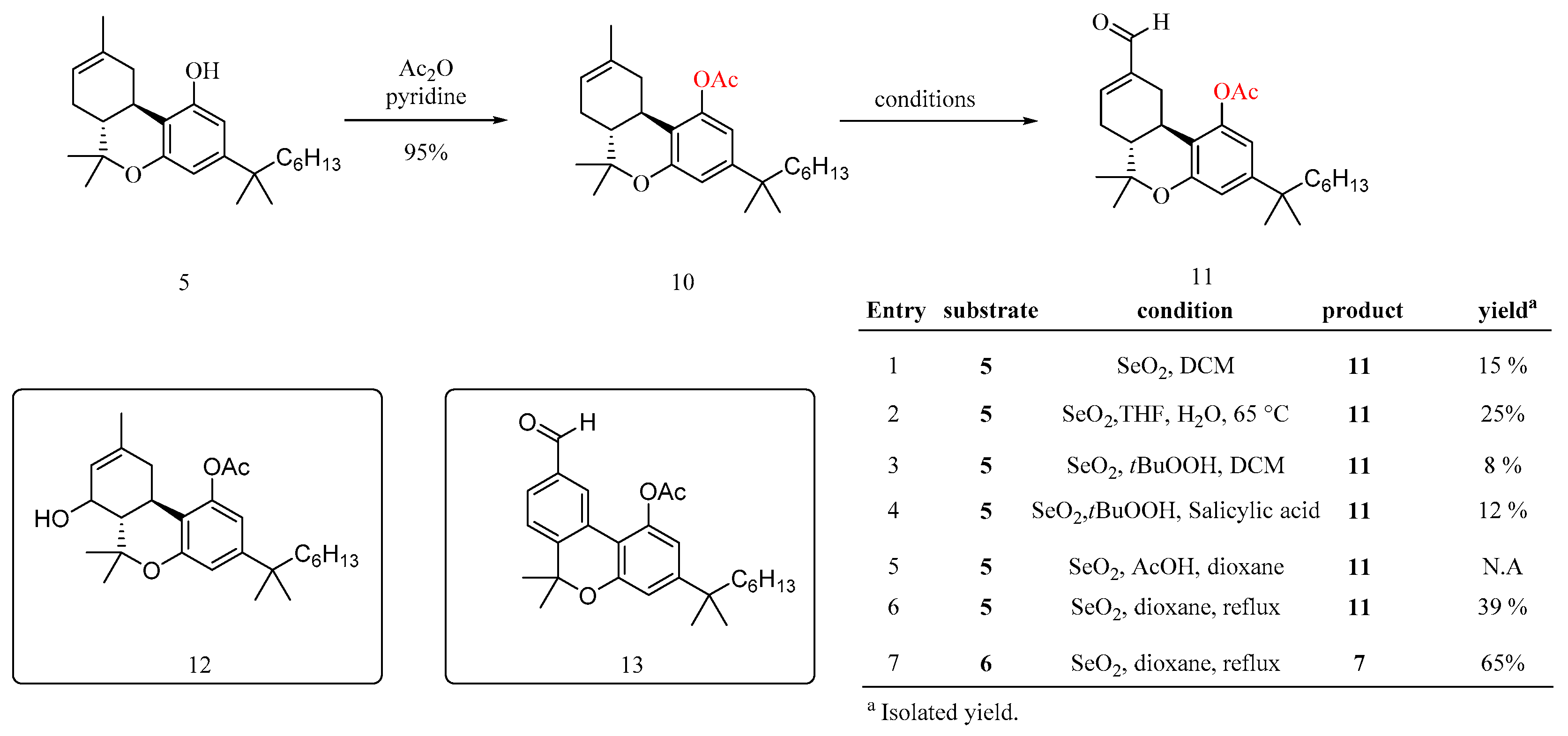
In the initial attempt, the acetyl protecting group of the tricyclic intermediate phenol 10 lead to a low yield of the allylic oxidation step. We tried a variety of conditions including SeO2/THF/H2O [14], SeO2/AcOH/DCM [16], SeO2/tBuOOH/salicylic acid/DCM [17], SeO2/tBuOOH/DCM [18] and SeO2/dioxane. The result shown that there was a byproduct (12) observed which is a regio-selected isomer. Also, there was another byproduct (13) shown in the Table 1 which is an aromatic product. Fortunately, SeO2 mediated riley oxidation in dioxane at 110 °C gave a moderate yield. When the acetyl group was replaced with the silyl ether protected group, the yield of the allylic oxidation was improved obviously due to the well decrease of the by-products.
3. Materials and Methods
3.1. General Information
All solvents and reagents used in this study were purchased from commercial sources and used without further purification. Thin-layer chromatography (TLC) was performed using SIL G/UV 254 silica-glass plates. Flash chromatography was carried out using Silica Gel 60 (200-400 mesh) utilizing solvent systems defined in the experimental procedure for each synthesized molecule. NMR spectra were obtained using a JEOL JNM-ECZ600R 600 MHz spectrometer at room temperature. NMR spectra were calibrated using residual undeuterated solvent as an internal reference (CDCl3: 1H NMR = 7.26 ppm, 13CNMR = 77.16 ppm; Acetone- d6: 1H NMR = 2.05 ppm, 13C NMR = 206.3 ppm; DMSO-d6: 1H NMR =2.50 ppm, 13C NMR = 39.52 ppm; CD2Cl2: 1H NMR = 5.32 ppm, 13C NMR = 54.0 ppm; MeOD-d4 : 1H NMR = 3.31 ppm, 13CNMR = 49.0 ppm; The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad.High-resolution mass spectroscopy (HRMS) was carried out on a Vion IMS TOF-Q mass spectrometer. Deuterium incorporation was determined by HRMS.
3.2. Experimental Section
3.2.1. Synthesis of compound (5)
To a 3-neck round bottom flask was charged DMHR (5 g, 21.18 mmol, 1 equiv), p- toluenesulfonic acid (0.728 g, 3.83 mmol, 0.2 equiv) and toluene (150 ml). To this was added PMD (3.54 g, 23.29 mmol, 1.1 equiv) over 1 h, followed by a toluene (8 ml) rinse while maintaining the batch temperature at 15–30 °C. The batch was heated to 70–80 °C under partial vacuum, and a Dean–Stark trap filled with toluene was used to remove water azeotropically. And the reaction was quenched by addition of a saturated solution of NH4Cl (15 ml). and the mixture was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with brine (20 mL), and dried with Na2SO4.The solvent was removed in vacuum, and the residue was purified by a flash chromatography on silica gel (hexane/ethyl acetate = 50/1) to give 5 (6.45 g, 82% yield) as a yellow oil. Rf = 0.7 (silica gel, EtOAc/hexanes= 1/10). 1H NMR (600 MHz, Chloroform-d) δ 6.40 (d, J = 1.8 Hz, 1H), 6.23 (d, J = 1.8 Hz, 1H), 5.44 (dt, J = 5.0, 1.4 Hz, 1H), 3.20 (dt, J = 17.2, 3.0 Hz, 1H), 2.75 – 2.66 (m, 1H), 2.14 (d, J = 4.1 Hz, 1H), 1.95 – 1.77 (m, 3H), 1.71 (q, J = 1.3 Hz, 3H), 1.50 (ddd, J = 11.3, 6.1, 2.6 Hz, 2H), 1.40 (s, 3H), 1.26 – 1.16 (m, 13H), 1.12 (s, 3H), 1.07 (td, J = 8.9, 8.4, 4.1 Hz, 2H), 0.85 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 154.65, 154.55, 150.17, 134.90, 119.47, 110.29, 108.17, 105.54, 44.98, 44.61, 37.45, 36.10, 31.93, 31.63, 30.17, 28.88, 28.81, 28.01, 27.75, 24.76, 23.65, 22.82, 18.66, 14.24. IR (film, cm-1): 3384, 2958, 2927, 2856, 1622, 1413, 1034, 965, 838; HRMS (ESI) calcd for C25H38O2 [M+H]+: m/z 371.2945, found: 371.2944.
3.2.2. Synthesis of compound (6)
To a solution of 5 (5.79 g, 15.65 mmol, 1.0 equiv) in dry DMF (100 mL) at room temperature was added dry imidazole (4.75 g, 69.77 mmol, 4.46 equiv) and TBSCl (7.07 g, 46.9 mmol, 3.0 equiv) and the resultant mixture was stirred at the same temperature for 18 h. The mixture was quenched by addition of a saturated solution of NH4Cl (10 mL), and water (200 ml) was added to the mixture. The mixture was extracted with ethyl acetate (3 x 50 mL). Combined organic layers were washed with brine (25 mL), and dried over Na2SO4. The solvent of the organic phase was concentrated in vacuo, and the residue was purified by a flash column chromatography (hexane/EtOAc: 50/1) to give 6 (7.34 g, 97% yield) as a colorless oil. Rf =0.7 (hexane/EtOAc: 20/1). 1H NMR (600 MHz, Chloroform-d) δ 6.41 (d, J = 1.9 Hz, 1H), 6.35 (d, J = 1.9 Hz, 1H), 5.45 – 5.35 (m, 1H), 3.30 – 3.20 (m, 1H), 2.61 – 2.53 (m, 1H), 2.22 – 2.09 (m, 1H), 1.84 – 1.77 (m, 3H), 1.69 (d, J = 2.0 Hz, 3H), 1.50 (ddd, J = 10.3, 5.5, 1.2 Hz, 2H), 1.38 (s, 3H), 1.21 (d, J = 6.4 Hz, 8H), 1.19 (dd, J = 7.1, 3.5 Hz, 4H), 1.10 (s, 3H), 1.07 – 1.03 (m, 2H), 1.01 (s, 9H), 0.84 (t, J = 7.1 Hz, 3H), 0.26 (s, 3H), 0.13 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 154.71, 154.30, 149.40, 135.15, 119.36, 114.19, 109.83, 108.66, 45.34, 44.72, 37.44, 36.15, 32.24, 31.97, 31.74, 30.18, 29.06, 28.81, 28.19, 27.64, 26.13, 24.83, 23.54, 22.80, 18.47, 14.27, 14.23, -3.41, -4.24; HRMS (ESI) calcd for C31H52O2Si [M+H]+: m/z 485.3809, found: 485.3818.
3.2.3. Synthesis of compound (7)
To a stirred solution of 6 (5.74 g, 11.84 mmol, 1.0 equiv) in dry dioxane (200 mL) was added SeO2 (4.6 g, 41.46 mmol, 3.5 equiv) at room temperature, and resultant mixture was shielded from light and stirred at the 110 °C for 1 h. The mixture was quenched by filtration off through a celite pad, which was washed with DCM (50 mL). The combined filtrate was washed with saturated aq Na2S2O8 (3 x 20 mL), and the water phase was extracted with DCM (3 x 50 mL). Combined organic layers were washed with brine (50 mL) and dried over Na2SO4. The organic phase was concentrated in vacuo, and the residue was purified by a flash column chromatography (hexane/EtOAc: 10/1 to 4/1) to give 7 (3.82 g, 65% yield) as yellowish solid, Rf = 0.3. (hexane/EtOAc: 10/1). 1H NMR (600 MHz, Chloroform-d) δ 9.50 (s, 1H), 6.83 – 6.77 (m, 1H), 6.41 (d, J = 1.8 Hz, 1H), 6.37 (d, J = 1.8 Hz, 1H), 3.84 (ddd, J = 17.7, 4.3, 2.0 Hz, 1H), 2.60 – 2.48 (m, 2H), 2.19 – 2.10 (m, 1H), 1.90 (td, J = 11.6, 4.7 Hz, 1H), 1.80 (q, J = 2.1 Hz, 1H), 1.52 – 1.48 (m, 2H), 1.43 (s, 3H), 1.20 (dd, J = 15.9, 4.1 Hz, 13H), 1.13 (s, 3H), 1.04 (t, J = 4.0 Hz, 1H), 0.97 (s, 9H), 0.84 (t, J = 7.0 Hz, 3H), 0.28 (s, 3H), 0.13 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 193.42, 154.90, 154.06, 149.91, 148.69, 142.57, 112.86, 109.85, 108.43, 45.33, 44.70, 37.50, 31.95, 31.45, 30.16, 29.45, 28.98, 28.85, 27.69, 27.61, 26.08, 24.81, 22.79, 18.46, 18.31, 14.23, -3.43, -4.12. HRMS (ESI) calcd for C31H50O3Si [M+H]+: m/z 499.3602, found: 499.3615.
3.2.4. Synthesis of compound (8)
To a solution of 7 (2.80 g, 5.62 mmol, 1.0 equiv) in MeOH (30 mL) was added NaBH4 (256 mg, 6.74 mmol, 1.2 equiv) at 0 °C in one portion, and the resultant mixture was then stirred at 0 °C for 50 min. The reaction was quenched by addition of an aqueous solution of NH4Cl (10 mL), and the MeOH in resultant mixture was removed under vacuum, and the resultant residue was extracted with ethyl acetate (3 x 10 mL). The combined organic extracts were washed with brine (10 mL), and dried over Na2SO4. The solvent of the extract was concentrated under vacuum, and the residue was purified by a flash column chromatography (hexane/EtOAc: 10/1 to 4/1) to give 8 (2.5 g, 89% yield) as yellowish solid, Rf = 0.5. (hexane/EtOAc: 5/1). 1H NMR (600 MHz, Chloroform-d) δ 6.42 (d, J = 1.9 Hz, 1H), 6.35 (d, J = 1.9 Hz, 1H), 5.76 – 5.71 (m, 1H), 4.08 – 3.99 (m, 2H), 3.41 – 3.32 (m, 1H), 2.59 (td, J = 11.0, 4.5 Hz, 1H), 2.27 – 2.15 (m, 1H), 1.95 – 1.77 (m, 3H), 1.52 – 1.47 (m, 2H), 1.39 (s, 3H), 1.25 – 1.19 (m, 9H), 1.18 (q, J = 3.5 Hz, 3H), 1.11 (s, 3H), 1.07 – 1.02 (m, 2H), 0.99 (s, 9H), 0.84 (t, J = 7.1 Hz, 3H), 0.25 (s, 3H), 0.13 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 154.75, 154.23, 149.57, 138.57, 120.42, 113.78, 109.85, 108.64, 76.42, 66.91, 45.55, 44.70, 37.45, 32.00, 31.95, 30.16, 29.03, 28.80, 27.85, 27.62, 26.13, 24.82, 22.79, 18.45, 18.40, 14.23, -3.43, -4.17. HRMS (ESI) calcd for C31H52O3Si [M+H]+: m/z 501.3759, found: 501.3744.
3.2.5. Synthesis of compound (10)
To a solution of 5 (5.6 g, 13.5 mmol, 1.0 equiv) in pyridine (100 mL) was added Ac2O (5.11 ml, 6.74 mmol, 4 equiv) at 0 °C in one portion, and the resultant mixture was then stirred at room temperature for 4 h. The mixture was then added with water (100 mL) and the resultant residue was extracted with ethyl acetate (3 x 30 mL). The combined organic extracts were washed with H2O (3 x 10 mL) and brine (10 mL), and dried over Na2SO4. The solvent of the extract was concentrated under vacuum, and the residue was purified by a flash column chromatography (hexane/EtOAc: 10/1) to give 10 (5.93 g, 95% yield) as yellowish solid, Rf = 0.7. (hexane/EtOAc: 10/1). 1H NMR (600 MHz, Chloroform-d) δ 6.69 (d, J = 2.2 Hz, 1H), 6.52 (d, J = 2.2 Hz, 1H), 5.48 – 5.40 (m, 1H), 2.77 – 2.69 (m, 1H), 2.61 (td, J = 11.0, 5.0 Hz, 1H), 2.30 (s, 3H), 2.17 – 2.11 (m, 1H), 1.97 – 1.89 (m, 1H), 1.83 – 1.77 (m, 2H), 1.70 (s, 3H), 1.52 (dd, J = 8.0, 4.5 Hz, 2H), 1.39 (s, 3H), 1.21 (dd, J = 26.3, 3.2 Hz, 12H), 1.12 (s, 3H), 1.08 (ddd, J = 11.0, 5.2, 2.7 Hz, 2H), 0.85 (t, J = 7.0 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 169.06, 154.31, 150.27, 149.73, 134.01, 119.89, 115.83, 113.19, 112.27, 44.76, 44.57, 37.59, 36.20, 31.88, 31.86, 30.11, 28.75, 28.67, 27.85, 27.60, 24.68, 23.70, 22.80, 21.44, 18.68, 14.23. HRMS (ESI) calcd for C27H40O3 [M+H]+: m/z 413.6215, found: 413.6214.
3.2.6. Synthesis of compound (11)
To a 100-ml, 3-neck round bottom flask was charged compound 10 (1.23 g, 2.98 mmol, 1 equiv), Selenium dioxide (378 mg, 3.41 mmol, 1.25 equiv), tetrahydrofuran (12.2 ml, 4.3 equiv) and water (0.57 ml, 0.2 equiv). The reactor was heated to 55–65 °C, for 23.5 h. The mixture was quenched by filtration off through a celite pad, which was washed with DCM (20 mL). The combined filtrate was washed with saturated aq Na2S2O8 (3 x 10 mL), and the water phase was extracted with DCM (3 x 12 mL). Combined organic layers were washed with brine (15 mL) and dried over Na2SO4. The organic phase was concentrated in vacuo, and the residue was purified by a flash column chromatography (hexane/EtOAc: 10/1 to 4/1) to give 11 (0.32 g, 25% yield) as yellowish solid, Rf = 0.3. (hexane/EtOAc: 10/1), And also to give two byproducts 12 and 13.
Compound 11, 1H NMR (600 MHz, Chloroform-d) δ 9.50 (s, 1H), 6.90 – 6.81 (m, 1H), 6.70 (d, J = 1.7 Hz, 1H), 6.56 (d, J = 2.0 Hz, 1H), 3.43 – 3.34 (m, 1H), 2.57 – 2.54 (m, 1H), 2.30 (s, 3H), 2.16 – 2.10 (m, 1H), 1.91 (td, J = 11.5, 4.5 Hz, 2H), 1.53 – 1.49 (m, 2H), 1.44 (s, 3H), 1.23 (d, J = 2.9 Hz, 8H), 1.19 (t, J = 4.0 Hz, 5H), 1.16 (s, 3H), 1.07 (dq, J = 9.1, 3.7, 3.3 Hz, 2H), 0.84 (d, J = 7.3 Hz, 3H).13C NMR (151 MHz, Chloroform-d) δ 193.43, 169.28, 154.03, 150.83, 149.79, 148.81, 141.65, 114.59, 113.21, 112.67, 44.72, 44.54, 37.66, 31.87, 31.07, 30.09, 29.12, 28.75, 28.63, 27.55, 27.11, 24.67, 22.80, 21.46, 18.55, 14.26, 14.22. HRMS (ESI) calcd for C27H38O4 [M+H]+: m/z 427.2843, found: 427.2838.
Compound 12, 1H NMR (600 MHz, Chloroform-d) δ 6.99 (s, 1H), 6.43 (d, J = 1.9 Hz, 1H), 6.38 (d, J = 2.2 Hz, 1H), 5.68 (dt, J = 5.2, 1.7 Hz, 1H), 5.64 – 5.59 (m, 1H), 3.04 – 2.96 (m, 1H), 2.24 (s, 3H), 2.23 – 2.17 (m, 1H), 2.10 (td, J = 11.3, 3.8 Hz, 1H), 1.97 – 1.90 (m, 1H), 1.52 – 1.48 (m, 2H), 1.40 (s, 3H), 1.23 (s, 3H), 1.20 (s, 12H), 1.08 – 1.04 (m, 2H), 1.02 (s, 3H), 0.84 (d, J = 7.2 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 169.69, 155.11, 151.56, 134.68, 125.64, 108.70, 107.42, 107.09, 79.86, 75.64, 46.69, 44.43, 37.49, 37.35, 31.87, 30.10, 28.93, 28.80, 28.72, 27.55, 24.67, 22.77, 21.19, 19.19, 18.34, 14.22. HRMS (ESI) calcd for C27H40O4 [M+H]+: m/z 429.2999, found: 429.3000.
Compound 14, 1H NMR (600 MHz, Chloroform-d) δ 6.47 (d, J = 2.2 Hz, 1H), 6.33 (d, J = 2.2 Hz, 1H), 5.73 (d, J = 6.1 Hz, 1H), 4.39 – 4.32 (m, 1H), 2.80 (dd, J = 11.8, 8.5 Hz, 1H), 2.22 – 2.13 (m, 1H), 1.97 – 1.89 (m, 1H), 1.89 – 1.82 (m, 1H), 1.79 (d, J = 2.1 Hz, 3H), 1.51 (ddd, J = 11.3, 6.0, 2.4 Hz, 2H), 1.38 (s, 3H), 1.27 – 1.16 (m, 13H), 1.09 (s, 3H), 1.08 – 1.05 (m, 2H), 0.85 (t, J = 7.0 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 155.92, 154.69, 151.21, 134.70, 125.84, 108.23, 107.83, 107.60, 77.51, 77.37, 75.21, 44.89, 44.49, 40.95, 37.39, 31.95, 30.18, 28.78, 27.96, 27.87, 24.77, 22.83, 19.28, 18.64, 14.25. HRMS (ESI) calcd for C25H38O3 [M+H]+: m/z 387.2893, found: 387.2899.
Compound 15, 1H NMR (600 MHz, Chloroform-d) δ 9.94 (s, 1H), 8.57 (d, J = 1.4 Hz, 1H), 7.23 (d, J = 1.5 Hz, 1H), 6.56 (d, J = 1.7 Hz, 1H), 6.43 (d, J = 1.8 Hz, 1H), 5.97 (d, J = 5.0 Hz, 1H), 5.61 (d, J = 3.1 Hz, 1H), 1.76 (s, 6H), 1.55 (dd, J = 8.0, 4.3 Hz, 2H), 1.26 (s, 6H), 1.20 (q, J = 4.9, 4.0 Hz, 6H), 1.09 – 1.06 (m, 2H), 0.84 (s, 3H). HRMS (ESI) calcd for C25H32O4 [M+H]+: m/z 381.2424, found: 381.2428.
3.2.7. Synthesis of compound (3)
To a solution of 8 (2.20 g, 4.40 mmol, 1.0 equiv) in THF (60 mL) was added tetrabutylammonium fluoride (1M in THF, 4.84 mL, 1.1 equiv), and the resultant mixture was stirred at room temperature for 2 h. The reaction was quenched with saturated. aq. NH4Cl (10 mL), and the resultant mixture was extracted with Et2O (3 x 10 mL). The organic layers were sequentially washed with water (5 mL) and then with brine (5 mL), and dried over anhydrous MgSO4. The solvent of the extract was removed under vacuum, the residue was subjected to column chromatography on silica gel (hexane/EtOAc: 2/1) to afford 3 (1.57 mg, 93%) as a white solid. Rf = 0.4 (hexane/EtOAc: 2/1). 1H NMR (600 MHz, Chloroform-d) δ 6.39 (d, J = 1.8 Hz, 1H), 6.24 (d, J = 1.9 Hz, 1H), 5.75 (dd, J = 4.0, 2.4 Hz, 1H), 4.07 (q, J = 12.7 Hz, 2H), 3.43 (dd, J = 15.9, 4.5 Hz, 1H), 2.71 (d, J = 4.6 Hz, 1H), 2.22 (s, 1H), 1.92 – 1.80 (m, 3H), 1.49 (ddd, J = 11.3, 6.1, 2.7 Hz, 2H), 1.40 (s, 3H), 1.28 – 1.14 (m, 13H), 1.11 (s, 3H), 1.09 – 1.02 (m, 2H), 0.85 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 154.78, 154.50, 150.30, 138.33, 121.98, 110.03, 107.94, 105.75, 67.22, 45.12, 44.61, 37.42, 31.94, 31.50, 31.44, 30.19, 28.86, 28.79, 27.80, 27.71, 24.76, 22.83, 18.53, 14.24. IR (film, cm-1): 3413, 3223, 2924, 1624, 1580, 1415, 1186, 991, 842; HRMS (ESI) calcd for C25H38O3 [M+H]+: m/z 387.2894, found: 387.2891.
3.2.8. Synthesis of compound (9)
To a mixture of aldehyde 7 (2.69 g, 5.39 mmol, 1.0 equiv), NaH2PO4·2H2O (2.59 g, 648 mmol, 4.0 equiv), and 2-methyl-2-butene (3.79 g, 53.9 mmol, 10.0 equiv) in t-BuOH (100 mL) and H2O (25 mL) was added NaClO2 (1.95 g, 648 mmol, 4.0 equiv) at 0 °C. After being stirred for 1 h at that temperature, the mixture was extracted with EtOAc (3 × 40 mL) and H2O (15 mL). The organic layer was separated, dried over Na2SO4, and concentrated to give the residue, which was purified by column chromatography on silica gel (hexane/EtOAc: 20/1) to afford 9 (2.50 g, 90%) as a yellowish oil. Rf = 0.4 (hexane/EtOAc: 10/1). 1H NMR (600 MHz, Chloroform-d) δ 7.19 – 7.14 (m, 1H), 6.44 (d, J = 1.8 Hz, 1H), 6.40 (d, J = 1.9 Hz, 1H), 3.90 (ddd, J = 17.9, 4.4, 2.1 Hz, 1H), 2.57 (td, J = 11.2, 4.3 Hz, 1H), 2.50 – 2.40 (m, 1H), 2.04 (ddt, J = 16.4, 11.9, 2.4 Hz, 1H), 1.97 – 1.90 (m, 1H), 1.86 (td, J = 11.7, 4.6 Hz, 1H), 1.54 – 1.51 (m, 2H), 1.42 (s, 3H), 1.22 (dd, J = 13.7, 5.9 Hz, 13H), 1.13 (s, 3H), 1.10 – 1.04 (m, 2H), 1.01 (s, 9H), 0.86 (t, J = 7.0 Hz, 3H), 0.30 (s, 3H), 0.16 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 172.77, 154.84, 154.13, 149.74, 140.24, 130.98, 112.99, 109.81, 108.50, 75.98, 44.69, 44.53, 37.45, 31.93, 31.79, 30.15, 30.06, 29.01, 28.88, 28.85, 27.57, 26.07, 24.80, 22.77, 18.46, 18.26, 14.21, -3.44, -4.11. HRMS (ESI) calcd for C31H50O4Si [M+H]+: m/z 515.3551, found: 515.3559.
3.2.9. Synthesis of compound (4)
To a solution of 9 (1.4 g, 2.72 mmol, 1.0 equiv) in THF (40 mL) was added tetrabutylammonium fluoride (1M in THF, 3.0 mL, 1.1 equiv), and the resultant mixture was stirred at room temperature for 2 h. The reaction was quenched with saturated. aq. NH4Cl (10 mL), and the resultant mixture was extracted with Et2O (3 x 10 mL). The organic layers were sequentially washed with water (5 mL) and then with brine (5 mL), and dried over anhydrous MgSO4. The solvent of the extract was removed under vacuum, the residue was subjected to column chromatography on silica gel (hexane/EtOAc: 10/1) to afford 4 (1.0 g, 92%) as a white solid. Rf = 0.6 (hexane/EtOAc: 10/1). 1H NMR (600 MHz, Chloroform-d) δ 7.17 (dt, J = 5.1, 2.3 Hz, 1H), 6.40 (d, J = 1.8 Hz, 1H), 6.24 (d, J = 1.8 Hz, 1H), 3.84 (ddd, J = 17.8, 4.8, 2.3 Hz, 1H), 2.68 (td, J = 11.3, 4.6 Hz, 1H), 2.50 – 2.38 (m, 1H), 2.10 – 1.95 (m, 2H), 1.85 (td, J = 11.7, 4.6 Hz, 1H), 1.50 (ddd, J = 8.3, 6.2, 3.5 Hz, 2H), 1.42 (s, 3H), 1.26 – 1.20 (m, 9H), 1.18 (d, J = 4.0 Hz, 3H), 1.14 (s, 3H), 1.09 – 1.01 (m, 2H), 0.85 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 172.30, 154.64, 154.48, 150.56, 140.54, 130.58, 109.23, 108.05, 105.72, 44.61, 44.14, 37.48, 31.92, 31.17, 30.16, 29.89, 28.91, 28.78, 27.68, 24.75, 22.82, 18.46, 14.24. IR (film, cm-1): 3384, 2958, 2927, 2856, 1622, 1412, 1185, 1032, 965, 838; HRMS (ESI) calcd for C25H36O4 [M+H]+: m/z 401.2686, found: 401.2675.
Δ8 -THC, 1H NMR (600 MHz, Chloroform-d) δ 6.29 (d, J = 1.6 Hz, 1H), 6.11 (d, J = 1.8 Hz, 1H), 5.44 (dd, J = 4.8, 2.6 Hz, 1H), 4.66 (s, 1H), 3.24 – 3.16 (m, 1H), 2.70 (d, J = 4.8 Hz, 1H), 2.45 (td, J = 7.7, 5.2 Hz, 2H), 2.18 – 2.11 (m, 1H), 1.89 – 1.78 (m, 3H), 1.71 (s, 3H), 1.60 – 1.54 (m, 2H), 1.38 (s, 3H), 1.34 – 1.29 (m, 4H), 1.11 (s, 3H), 0.89 (t, J = 7.0 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 155.00, 154.88, 142.87, 134.90, 119.47, 110.63, 110.26, 107.75, 45.00, 36.14, 35.57, 31.70, 30.75, 28.02, 27.71, 23.64, 22.69, 18.64, 14.17. HRMS (ESI) calcd for C21H30O2 [M+H]+: m/z 315.2319, found: 315.2330.
11-Nor-Δ8-Tetrahydrocannabinol-9-carboxylic Acid, 1H NMR (600 MHz, Methanol-d4) δ 7.03 (dd, J = 5.2, 2.6 Hz, 1H), 6.18 (d, J = 1.6 Hz, 1H), 6.10 (d, J = 1.6 Hz, 1H), 3.87 (ddd, J = 17.7, 4.5, 2.4 Hz, 1H), 2.60 (td, J = 11.2, 4.4 Hz, 1H), 2.43 (d, J = 8.6 Hz, 1H), 2.40 (d, J = 7.7 Hz, 2H), 2.10 – 1.96 (m, 1H), 1.85 – 1.72 (m, 2H), 1.59 – 1.53 (m, 2H), 1.37 (s, 3H), 1.36 – 1.28 (m, 4H), 1.09 (s, 3H), 0.91 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, Methanol-d4) δ 170.90, 157.83, 155.76, 143.73, 139.41, 132.43, 110.98, 109.75, 108.54, 77.00, 46.06, 36.62, 32.80, 32.66, 32.05, 31.52, 29.55, 27.91, 23.60, 18.43, 14.40. HRMS (ESI) calcd for C21H28O4 [M+H]+: m/z 345.2060, found: 345.2056.
Δ9 -THC, 1H NMR (600 MHz, Chloroform-d) δ 6.33 – 6.30 (m, 1H), 6.28 (d, J = 1.6 Hz, 1H), 6.15 (d, J = 1.6 Hz, 1H), 4.80 (s, 1H), 3.21 (dt, J = 10.9, 2.6 Hz, 1H), 2.44 (td, J = 7.5, 3.5 Hz, 2H), 2.21 – 2.14 (m, 2H), 1.95 – 1.89 (m, 1H), 1.70 (d, J = 2.0 Hz, 1H), 1.69 (dq, J = 2.3, 1.0 Hz, 3H), 1.59 – 1.54 (m, 2H), 1.42 (s, 3H), 1.41 (s, 1H), 1.33 – 1.28 (m, 4H), 1.10 (s, 3H), 0.90 – 0.87 (m, 3H). 13C NMR (151 MHz, Chloroform-d) δ 154.91, 154.30, 142.97, 134.56, 123.85, 110.23, 109.17, 107.68, 77.37, 45.93, 35.61, 33.70, 31.65, 31.30, 30.79, 27.71, 25.15, 23.51, 22.68, 19.41, 14.16. HRMS (ESI) calcd for C21H30O2 [M+H]+: m/z 315.2319, found: 315.2320.
Δ9-THC-COOH,1H NMR (400 MHz, Methanol-d4) δ 8.05 (d, J = 2.2 Hz, 1H), 6.20 (s, 1H), 6.11 (s, 1H), 3.35 (d, J = 3.4 Hz, 1H), 2.53 (dd, J = 18.5, 6.8 Hz, 1H), 2.42 (t, J = 7.6 Hz, 3H), 2.04 (dd, J = 12.8, 7.3 Hz, 1H), 1.69 – 1.51 (m, 3H), 1.41 (s, 4H), 1.32 (ddd, J = 12.7, 10.0, 5.6 Hz, 4H), 1.09 (s, 3H), 0.90 (t, J = 6.9 Hz, 3H). 13C NMR (151 MHz, Methanol-d4) δ 171.52, 157.20, 155.95, 144.78, 144.07, 130.08, 109.84, 108.39, 108.19, 77.92, 46.15, 36.64, 35.95, 32.65, 32.06, 27.92, 26.66, 25.50, 23.61, 19.25, 14.40. HRMS (ESI) calcd for C21H28O4 [M+H]+: m/z 345.2060, found: 345.2059.
4. Conclusion
In this study, a straightforward synthetic route was developed for the synthesis of a key intermediate on a multigram scale to be used for the synthesis of cannabinoid metabolites. Optimization of the riley oxidation of the key tricyclic intermediate is significantly improving the yield and make it achievable to apply this condition into the synthesis of different analogues of this skeleton. And this kind of analogue used as a product to meet the different needs in different labs for synthesis of potential cannabinoid metabolites. Similar synthesis strategies could be applied in the synthesis of similar metabolites of other cannabinoids metabolites like 11-Nor-Δ8-Tetrahydrocannabinol-9-carboxylic Acid and Δ9-THC-COOH.
Author Contributions
Conceptualization, W.L. (Wenbin Liu). and X.Z. (Xuejun Zhao).; investigation, W.S. (Wenbin Shao); P.L. (Pingyong Liao) and X.Z. (Xiaoyan Zhang); writing-original draft preparation, W.S. (Wenbin Shao); B.F. (Binbin Fan); X.C. (Xilong Chen) and R.C. (Ruijia Chen); supervision, W.S. (Wenbin Shao); All authors have read and agreed to the published version of the manuscript.
Funding
This work is funded by National Key Research and Development Program of China (2022YFC3300901), National Natural Science Foundation of China (52272087), Science and Technology Development Fund of Shanghai Municipal Public Security Bureau (2022013), Shanghai Scientific and Technological Innovation Project (22ZR1457400), Shanghai Scientific and Technological Innovation Project (21dz1200200).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Scotter, E. L.; Abood, M. E.; Glass, M. The Endocannabinoid System as a Target for the Treatment of Neurodegenerative Disease. Br. J. Pharmacol. 2010, 160, 480. [CrossRef]
- Mechoulam, R.; Gaoni, Y. The absolute configuration of Δ1-tetrahydrocannabinol, the major active constituent of hashish. Tetrahedron Lett. 1967, 8, 1109−1111. [CrossRef]
- Devane, W. A.; Dysarz, F. A., III; Johnson, M. R.; Melvin, L. S.; Howlett, A. C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988, 34, 605−613. [CrossRef]
- Seltzman, H. H. Structure and receptor activity for classical cannabinoids. Curr. Med. Chem. 1999, 6, 685−704. [CrossRef]
- Howlett, A. C.; Barth, F.; Bonner, T. I.; Cabral, G.; Casellas, P.; Devane, W. A.; Felder, C. C.; Herkenham, M.; Mackie, K.; Martin, B. R.; Mechoulam, R.; Pertwee, R. G. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161−202. [CrossRef]
- Mechoulam, R., Lander, N., Breuer, A., and Zahalka, J. Synthesis of the Individual, Pharmacologically Distinct, Enantiomers of a Tetrahydrocannabinol Derivative. Tetrahedron Asymmetry 1990, 1, 315−318. [CrossRef]
- Showalter, V. M.; Compton, D. R.; Martin, B. R.; Abood, M. E. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J. Pharmacol. Exp. Ther. 1996, 278, 989−999. https://www.researchgate.net/publication/14384810.
- Mechoulam, R.; Lander, N.; University, A.; Zahalka, J. Syntheis of the individual, pharmacologically distinct enantiomers of a tetrahydrocannabinol derivative. Tetrahedron: Asymmetry 1990, 1, 315−319. [CrossRef]
- Perez-Reyes, M. In Pharmacokinetics and Pharmacodynamics of Psychoactive Drugs; Barnett, G., Chiang, N. C., Eds.; Biomedical Publishers: Foster City, 1985; p287. [CrossRef]
- Nasrin, S.; Watson, C. J. W.; Perez-Paramo, Y. X.; Lazarus, P. Cannabinoid Metabolites as Inhibitors of Major Hepatic CYP450 Enzymes, with Implications for Cannabis-Drug Interactions. Drug Metab. Dispos. 2021, 49, 1070−1080. [CrossRef]
- Burstein, S. H.; Audette, C. A.; Breuer, A.; Devane, W. A.; Colodner, S.; Doyle, S. A.; Mechoulam, R. Synthetic nonpsychotropic cannabinoids with potent anti-inflammatory, analgesic and leukocyte antiadhesion activities. J. Med. Chem. 1992, 35, 3135−3141. [CrossRef]
- Rhee, M.-H.; Vogel, Z.; Barg, J.; Bayewitch, M.; Levy, R.; Hanu, L.; Breuer, A.; Mechoulam, R. Cannabinoid derivatives: binding to cannabinoid receptors and inhibition of adenylcyclase. J. Med. Chem. 1997, 40, 3228−3233. [CrossRef]
- Shan, J.; Christos I-T.; Fei, T.; Christina, A. B.; Catherine, M. K.; Jimit. G. R.; Tian. H.; Simiao. W.; Jo-Hao. H.;, Yiran. W.; Travis. W. G.; Nikolai. Z.; Ganesh. A. Thakur, Zhi-Jie L.; Keith. A. S.; Laura. M. B.; Spyros. P. N.; Alexandros. M. Novel Functionalized Cannabinoid Receptor Probes: Development of Exceptionally Potent Agonists. J. Med. Chem. 2021, 64, 3870−3884. [CrossRef]
- Mark A. T.; Robert B. Z.; Sumner H. B. Ultrapure ajulemic acid has improved CB2 selectivity with reduced CB1 activity. Bioorg. Med. Chem. 2014 22. 3245–3251. [CrossRef]
- John W. H.; Seon A. H.; Nataliya L.; Alicia L. S. T.; Jenny L. W.; Dana E. S.; Billy R. M. 1-Bromo-3-(10,10-dimethylalkyl)-1-deoxy-Δ8-tetrahydrocannabinols: New selective ligands for the cannabinoid CB2 receptor. Bioorg. Med. Chem. 2010 18. 7809–7815. [CrossRef]
- Mehta. G.; Shinde. H. M. Enantiospecific total synthesis of 6-epi-(-)-hamigeran B. Intramolecular Heck reaction in a sterically constrained environment. Tetrahedron Lett. 2003, 44, 7049-7053. [CrossRef]
- Xu. P.-F.; Chen. Y.-S.; Lin. S.-I.; Lu. T.-J. Chiral Tricyclic Iminolactone Derived from (1R)-(+)-Camphor as a Glycine Equivalent for theAsymmetric Synthesis of α-Amino Acids. J. Org. Chem. 2002, 67, 2309-2314. [CrossRef]
- Fürstner, A.; Gastner, T. Total Synthesis of Cristatic Acid. Org. Lett. 2000, 2, 2467-2470. [CrossRef]
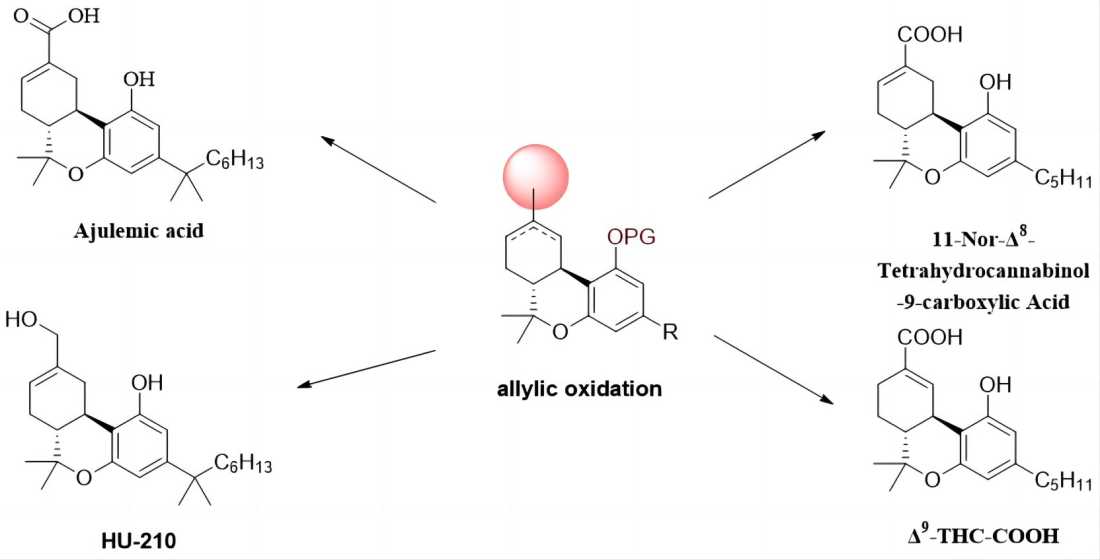
Figure 1.
General structural information of cannabinoid metabolites with oxygen at C-11 cite.
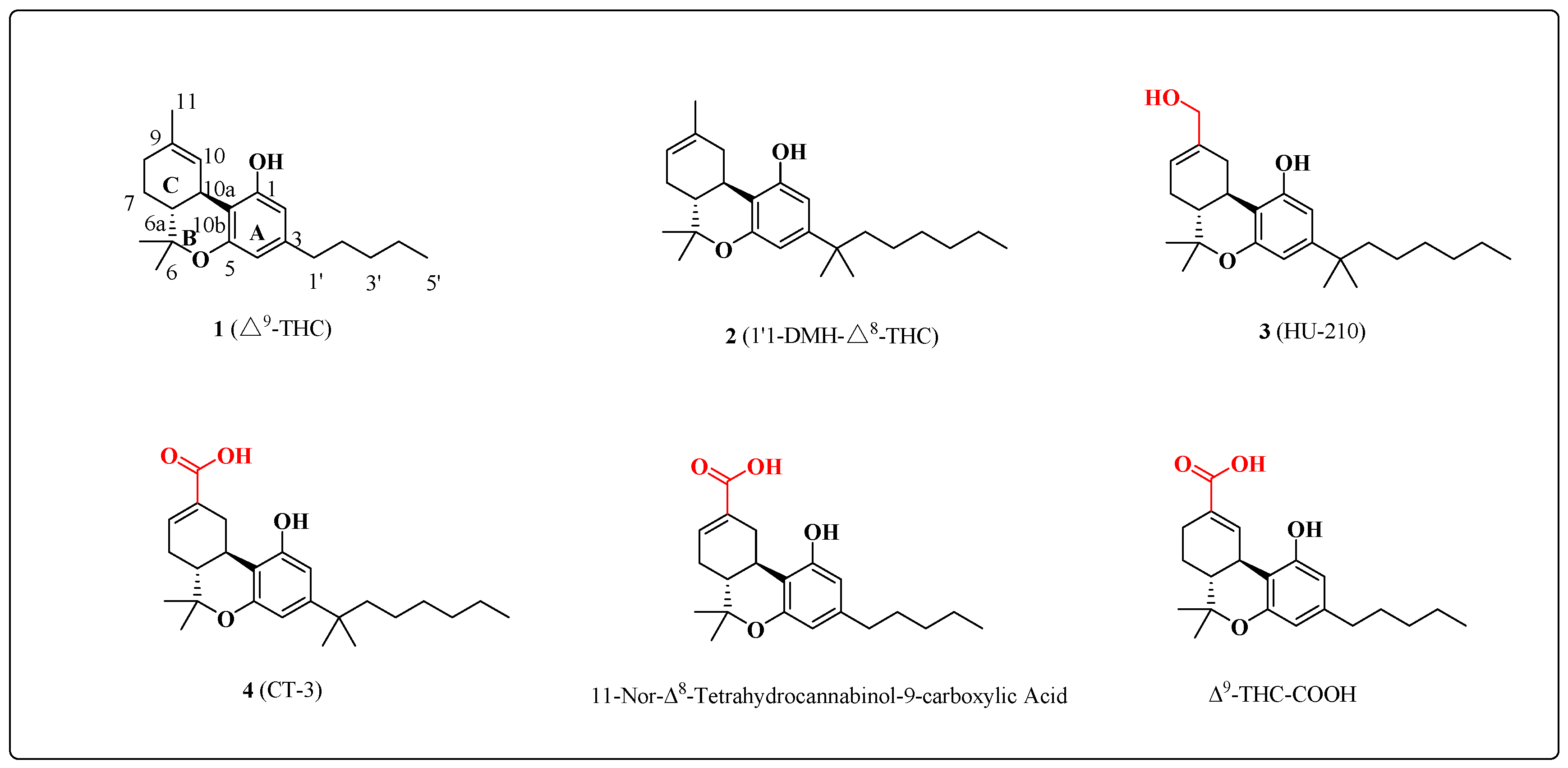
Figure 2.
Overview of the Syntheses of the C-11 oxygenation intermediates.
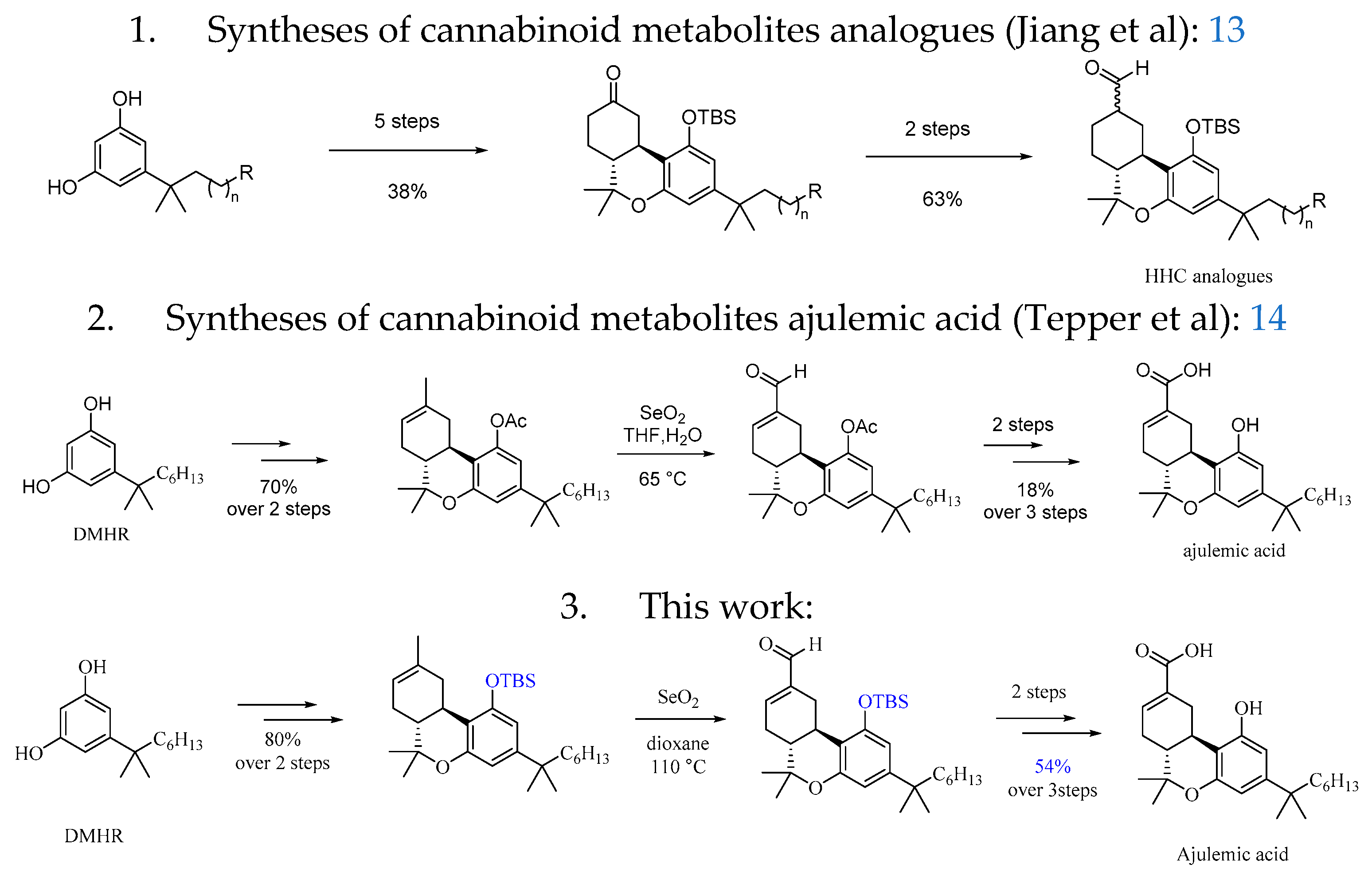
Scheme 1.
Synthesis of ajulemic acid and HU-210. Reagents and conditions: (a) 1. p-TSA, toluene; 80 °C for 1h; yield of 82%. (b) TBSCl, imidazole, and N, N-dimethylformamide (DMF); room temperature for overnight duration; yield of 97%; (c) SeO2, dioxane; 110 °C for 1 h; yield of 65%. (d) NaBH4, and MeOH; 0 °C for 50 min; yield of 89%. (e) TBAF, and tetrahydrofuran (THF); room temperature for 2 h; yield of 93%; (f) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH and H2O(4:1); room temperature for 1 h; yield of 90%; (g) TBAF, and tetrahydrofuran (THF); room temperature for 2 h; yield of 92%.
Scheme 1.
Synthesis of ajulemic acid and HU-210. Reagents and conditions: (a) 1. p-TSA, toluene; 80 °C for 1h; yield of 82%. (b) TBSCl, imidazole, and N, N-dimethylformamide (DMF); room temperature for overnight duration; yield of 97%; (c) SeO2, dioxane; 110 °C for 1 h; yield of 65%. (d) NaBH4, and MeOH; 0 °C for 50 min; yield of 89%. (e) TBAF, and tetrahydrofuran (THF); room temperature for 2 h; yield of 93%; (f) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH and H2O(4:1); room temperature for 1 h; yield of 90%; (g) TBAF, and tetrahydrofuran (THF); room temperature for 2 h; yield of 92%.
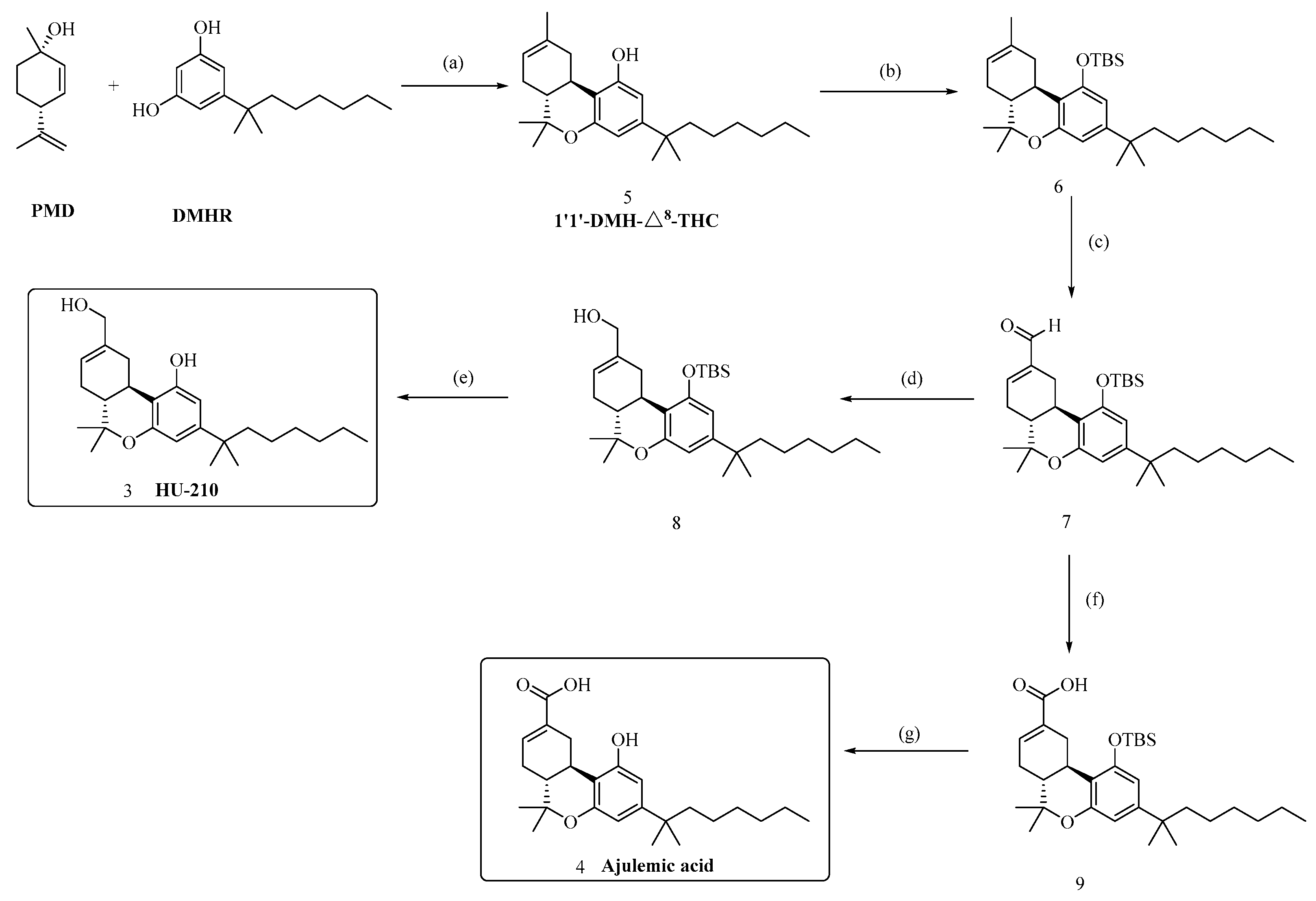
Scheme 2.
Syntheses of 11-Nor-Δ8-Tetrahydrocannabinol-9-carboxylic Acid and Δ9-THC-COOH with the same operation.
Scheme 2.
Syntheses of 11-Nor-Δ8-Tetrahydrocannabinol-9-carboxylic Acid and Δ9-THC-COOH with the same operation.
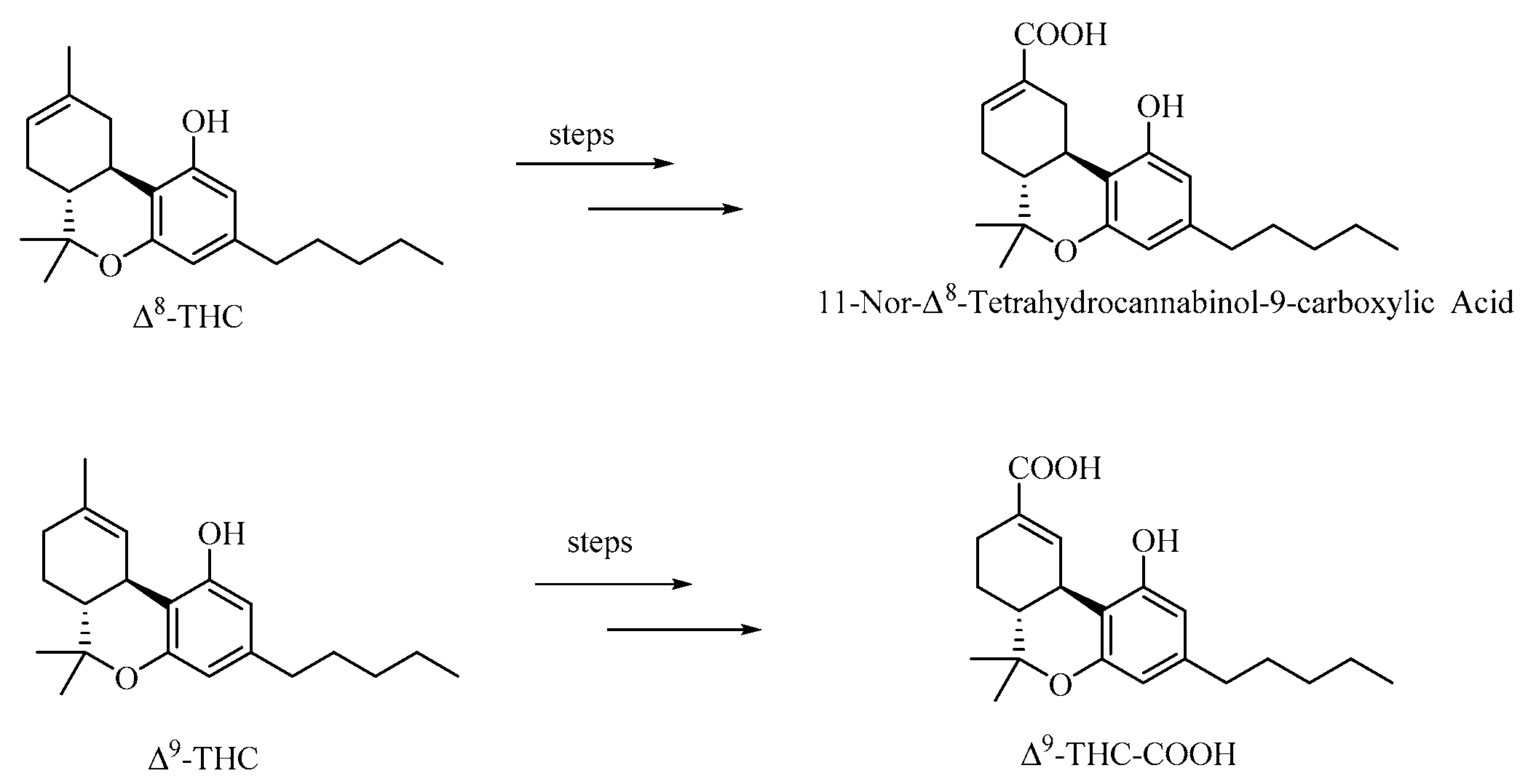

Table 1.
The optimization of the riley oxidation (see SI).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



